


The establishment of long-lasting immunity against pathogens is facilitated by the germinal center (GC) reaction, during which B cells increase their antibody affinity and differentiate into antibody-secreting cells (ASC) and memory cells. These events involve modifications in chromatin packaging that orchestrate the profound restructuring of gene expression networks that determine cell fate. While several chromatin remodelers were implicated in lymphocyte functions, less is known about SMARCA5. Here, using ribosomal pull-down for analyzing translated genes in GC B cells, coupled with functional experiments in mice, we identified SMARCA5 as a key chromatin remodeler in B cells. While the naive B cell compartment remained unaffected following conditional depletion of Smarca5, effective proliferation during B cell activation, immunoglobulin class switching, and as a result GC formation and ASC differentiation were impaired. Single-cell multiomic sequencing analyses revealed that SMARCA5 is crucial for facilitating the transcriptional modifications and genomic accessibility of genes that support B cell activation and differentiation. These findings offer novel insights into the functions of SMARCA5, which can be targeted in various human pathologies.
Introduction
A hallmark of the protective adaptive immune response lies in its capacity to generate rapidly responding memory cells and plasma cells (PC) that secrete protective antibodies, thereby fostering long-lasting and effective immunity (Corcoran and Tarlinton, 2016). Antigen-specific long-lived PCs predominantly originate within germinal centers (GC), which are specialized microanatomical structures located within secondary lymphoid organs. These structures are divided into the dark zone (DZ) and the light zone (LZ) based on lymphocyte density, cellular assemblies, and cell surface markers (Allen et al., 2004; Victora et al., 2010). In the LZ, B cells encounter antigens presented on follicular dendritic cells followed by uptake and presentation of peptides on major histocompatibility complex class II (MHCII) to cognate helper T cells (Batista and Harwood, 2009; Heesters et al., 2014; Song and Craft, 2023). Subsequently, B cells receive T cell help signals, such as the engagement of the B cell CD40 receptor with the T cell CD40 ligand (Liu et al., 2015). B cells that acquire sufficient T cell-derived signals are selected to migrate into the DZ, where extensive proliferation, clonal expansion, and insertion of somatic hypermutations (SHM) into the immunoglobulin genes occur. This cyclic sequence of events within GCs leads to the generation of memory and antibody-secreting cells (ASCs) that carry relatively high-affinity immunoglobulins (Stoler-Barak and Shulman, 2022; Cyster and Allen, 2019; Victora and Nussenzweig, 2022; Kwak et al., 2019; Shlomchik and Weisel, 2012; Steele et al., 2024).
The packaging of DNA has a significant impact on gene expression since this dynamic modulation of chromatin structure allows or restricts access of the transcriptional machinery to the DNA. Chromatin remodelers are enzymatic complexes that use the energy of ATP hydrolysis to dynamically regulate and modify the structure of the chromatin by sliding, repositioning, ejecting, or restructuring nucleosomes (Narlikar et al., 2013). GC formation depends on significant chromatin reorganization to allow gene and enhancer accessibility (Doane et al., 2021). Several families of chromatin remodelers and their functions have been described including the SWI/SNF (switch/sucrose non-fermentable), ISWI (imitation switch), CHD (chromodomain helicase DNA-binding), and INO80 (inositol-requiring 80) complexes (Bowman, 2010; Clapier et al., 2017; Sokpor et al., 2018; Tyagi et al., 2016; Jiang et al., 2023). Whereas in the context of B cell immune responses, several chromatin remodelers have been examined, such as SWI/SNF, INO80, and CHD, less is known about the functions of the ISWI complex (Schmiedel et al., 2021; Bossen et al., 2015; Zikmund et al., 2019; Holley et al., 2014; Papin et al., 2022; Kracker et al., 2015; Hagman et al., 2022; Arends et al., 2019; Deng et al., 2024). SMARCA5, also known as SNF2H, is the core catalytic unit of the ISWI family of remodelers and is involved in sliding, spacing, and repositioning of nucleosomes along the DNA, rather than evicting them (Bomber et al., 2023; Barisic et al., 2019). SMARCA5 contributes to the regulation of the expression of genes associated with cell cycle progression, DNA replication, recombination, and repair processes (Barisic et al., 2019; Corona and Tamkun, 2004; Clapier and Cairns, 2009; Collins et al., 2002; Bozhenok et al., 2002; Toiber et al., 2013). Proper control of gene expression is required for normal cellular function, and dysregulation of chromatin accessibility can lead to a wide range of pathologies including GC-derived lymphoma (Papin et al., 2022). Recent studies have implicated SMARCA5 in the progression of several cancer types including blood cancer (Jin et al., 2015; Thakur et al., 2022; Stopka et al., 2000; Zikmund et al., 2020; Radzisheuskaya et al., 2023; Bayona-Feliu et al., 2023; Gigek et al., 2011). SMARCA5 is also involved in various developmental processes including embryonic, hematopoietic stem and progenitor cell, and neurological differentiation (Ding et al., 2021; Kokavec et al., 2017; Zikmund et al., 2019; Li et al., 2021; Alvarez-Saavedra et al., 2014). With regards to immune cells, it was demonstrated that SMARCA5 plays an essential role in thymocyte development during selection, and has a repressive or activating effect on cytokine gene expression in activated T cells (Zikmund et al., 2019; Precht et al., 2010). B cells undergo extensive genomic reorganization during their differentiation into GC and ASCs to allow the expression of genes that promote cell cycle progression, state transition, and dictate cellular identity (Bunting et al., 2016; Vilarrasa-Blasi et al., 2021; Papin et al., 2022; Nutt et al., 2011). Nonetheless, the role of SMARCA5 in B lymphocyte activation and differentiation has not been characterized.
Here, using Smarca5-deficient B cells, we find that SMARCA5 is essential for the effective proliferation of activated B cells, GC formation, immunoglobulin class-switching, and ASC differentiation. Using high-throughput multiomic sequencing techniques, we demonstrate that SMARCA5 binds and controls the chromatin accessibility of activated B cells, pre-GC B cells, and ASCs differentiation-promoting genes. Thus, our study demonstrates the significance of chromatin remodeling by SMARCA5 in ensuring proper gene expression during the emergence of the humoral immune response.
Results
SMARCA5 is strongly translated in GC B cells
Transcriptomic profiling is a well-established technique for revealing the cellular state of particular subsets of cells. Nevertheless, it provides a snapshot of the total mRNA within the cells, rather than characterizing the mRNA that serves as a template for protein translation at a given time point. Thus, complementing transcriptomic approaches with protein translation profiling introduces an additional layer of insight, enhancing our understanding of biological processes. To study the translational state of GC B cells, we crossed genetically modified RiboTag mice carrying a transgene that expresses a hemagglutinin (HA)-tagged ribosomal protein, with mice expressing B cell–specific Cre, under the Aicda promoter (Aicda.Cre.Rpl22HA) (Sanz et al., 2009; Haimon et al., 2018). In combination with markers detected by flow cytometry, this mouse strain allows the isolation of ribosomes specifically from LZ and DZ B cells, providing a tool to capture the actively translating mRNAs (translatome), and offering insights into the genes and pathways that are actively translated in the GC (Fig. 1 A). DZ and LZ B cells sorted from lymph nodes (LNs) extracted from mice, 7 days after immunization with NP-KLH, exhibited distinct gene expression patterns. Specifically, 524 and 425 (DZ and LZ, respectively) genes were enriched after immunoprecipitation (IP) of HA-tagged ribosomes (translatome) compared to the total mRNA (transcriptome) (Fig. 1, B and C). Using this screening approach, we found multiple transcripts that were enriched in the translatome, some of which are already known as key regulators in GC cells, including Bcl6, Rgs13, Cd74, Junb, Pax5, Basp1, and Cd79a (Fig. 1, D and E; and Table S1). Biological pathway analysis of the translatome data revealed primarily changes in mRNA pathways, and changes in cell cycle, antigen presentation, lymphocyte activation, and metabolic processes (Fig. S1). Furthermore, this profiling enabled us to detect a significant enrichment of Smarca5 ribosomal-associated transcripts both in DZ and LZ B cells, which was never previously studied in the context of activated B lymphocytes (Fig. 1, D and E). SMARCA5 plays an important role in supporting the DNA damage response and repair of double-strand breaks. Since these events occur extensively during the B cell immune response, we focused on this chromatin remodeler (Toiber et al., 2013; Lan et al., 2010; Roco et al., 2019). Thus, the RiboTag-expressing mice enabled us to gain a deeper understanding of gene expression at the translational level within GC B cells in vivo after immunization and specifically highlighted the importance of SMARCA5.

GC B cells are enriched in SMARCA5 mRNA transcripts during translation. (A) Scheme illustrating the experimental design. (B and C) Volcano plots showing differential gene expression following ribosomal IP (Sort-IP) in DZ (B) and LZ (C) B cells (n = 4; two independent experiments, P value < 0.05, and log2 FC >0.58 or less than −0.58; raw P values were adjusted for multiple testing using the procedure of Benjamini and Hochberg). (D and E) Graphs showing normalized reads of selected enriched genes in the Sort-IP group derived from DZ (D) or LZ (E) B cells (n = 4; two independent experiments, two-tailed Student’s t test). Each dot in the graphs represents a single mouse; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
GC B cells are enriched in SMARCA5 mRNA transcripts during translation. (A) Scheme illustrating the experimental design. (B and C) Volcano plots showing differential gene expression following ribosomal IP (Sort-IP) in DZ (B) and LZ (C) B cells (n = 4; two independent experiments, P value < 0.05, and log2 FC >0.58 or less than −0.58; raw P values were adjusted for multiple testing using the procedure of Benjamini and Hochberg). (D and E) Graphs showing normalized reads of selected enriched genes in the Sort-IP group derived from DZ (D) or LZ (E) B cells (n = 4; two independent experiments, two-tailed Student’s t test). Each dot in the graphs represents a single mouse; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.

Translatome biological processes. Pathway analysis of enriched genes in DZ or LZ B cells following ribosome IP (Sort-IP) (n = 4; two independent experiments, adjusted P value < 0.05, and log2 FC > 0.58 or less than −0.58; raw P values were adjusted for multiple testing using the procedure of Benjamini and Hochberg).
Translatome biological processes. Pathway analysis of enriched genes in DZ or LZ B cells following ribosome IP (Sort-IP) (n = 4; two independent experiments, adjusted P value < 0.05, and log2 FC > 0.58 or less than −0.58; raw P values were adjusted for multiple testing using the procedure of Benjamini and Hochberg).
SMARCA5 is essential for GC and ASC formation, but not for B cell homeostasis
To investigate whether SMARCA5 plays a role in B cell homeostasis and immune responses in vivo, we crossed transgenic mice carrying a conditional inactivation of the Smarca5 gene with mice expressing B cell–specific Cre under the CD23 promoter (CD23.Cre.Smarca5fl/fl) (Kwon et al., 2008; Kokavec et al., 2017). Protein expression analysis revealed that naive B cells express SMARCA5, whereas only residual levels of protein were detected in Smarca5-deficient B cells (Fig. S2 A).
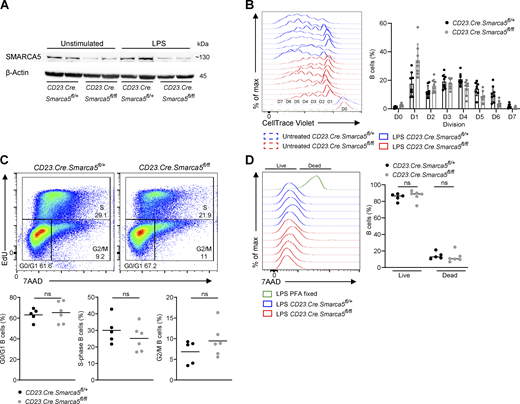
SMARCA5 is required for proper cell division and not for cell viability. (A) SMARCA5 protein expression was determined by western blot analysis of B cells that were either left unmanipulated or stimulated with LPS. Blots show two independent biological repeats. (B) Flow cytometry histograms of all repetitions related to Fig. 3 A, and quantification of CTV in each cell division of the in vitro proliferation of B cells treated with LPS (n = 8; two independent experiments). (C) Analysis of the different cell-cycle stages in LPS stimulated B cells by EdU incorporation and 7AAD DNA staining (n = 5–6; two independent experiments, two-tailed Student’s t test; ns, not significant). (D) Flow cytometry histograms and the fraction of live or dead cells measured by the viability dye 7AAD, of LPS-stimulated splenic B cells (n = 5–6; two independent experiments, one-way ANOVA; ns, not significant). Each dot in the graph represents a single mouse. Source data are available for this figure: SourceData FS2.
SMARCA5 is required for proper cell division and not for cell viability. (A) SMARCA5 protein expression was determined by western blot analysis of B cells that were either left unmanipulated or stimulated with LPS. Blots show two independent biological repeats. (B) Flow cytometry histograms of all repetitions related to Fig. 3 A, and quantification of CTV in each cell division of the in vitro proliferation of B cells treated with LPS (n = 8; two independent experiments). (C) Analysis of the different cell-cycle stages in LPS stimulated B cells by EdU incorporation and 7AAD DNA staining (n = 5–6; two independent experiments, two-tailed Student’s t test; ns, not significant). (D) Flow cytometry histograms and the fraction of live or dead cells measured by the viability dye 7AAD, of LPS-stimulated splenic B cells (n = 5–6; two independent experiments, one-way ANOVA; ns, not significant). Each dot in the graph represents a single mouse. Source data are available for this figure: SourceData FS2.
First, the immunoglobulin titers in the serum of unmanipulated CD23.Cre.Smarca5fl/fl and control mice were determined by ELISA. Serum immunoglobulins from Smarca5-deficient mice exhibited reduced levels of both IgM and class-switched IgA and IgG1 compared with the control mice (Fig. 2 A). Nonetheless, under homeostatic conditions, no differences were detected in the frequencies of B cell subsets in the bone marrow (BM), suggesting that B cell development and homeostasis remained intact (Fig. 2 B). Yet, the frequencies of ASCs in the BM of B cell–specific Smarca5-deficient mice were significantly reduced compared to their control counterparts (Fig. 2 C). Given that the GC reaction is the predominant source of ASCs, we sought to determine whether impaired GC formation is responsible for the reduced frequency of ASCs in the BM and reduced levels of immunoglobulins in the serum. To this end, CD23.Cre.Smarca5fl/fl mice and control CD23.Cre.Smarca5fl/+ mice were immunized subcutaneously in the hind footpads with 4-hydroxy-3-nitrophenyl (NP) coupled to keyhole limpet hemocyanin (KLH) in alum (Stoler-Barak and Shulman, 2022). Flow cytometric analysis of popliteal LN-derived cells 7 days following immunization revealed a major and significant reduction in the frequency of both ASCs and GC B cells in the absence of SMARCA5 (Fig. 2 D). To examine if this effect is B cell–intrinsic or mediated through additional B cell–independent factors, we immunized chimeric mice hosting 50% WT and 50% Smarca5-deficient B cells. GC recovered from these mice contained only WT B cells (Fig. 2 E). These results suggest that SMARCA5 plays a role in the GC and ASC formation.

GC and ASC formation are dependent on SMARCA5. (A) Serum IgM, IgA, and IgG1 titers as determined by ELISA in control or Smarca5-deficient mice (n = 7–10 unmanipulated mice; two independent experiments, two-tailed Student’s t test; **P ≤ 0.01, ***P ≤ 0.001). (B) Representative flow cytometry plots and frequencies of B cell subsets in the BM of unmanipulated control versus Smarca5-deficient mice (n = 5; two independent experiments, two-tailed Student’s t test; ns, not significant). (C) Representative flow cytometry plots and frequencies of total ASCs in the BM of unmanipulated mice (n = 5; two independent experiments, two-tailed Student’s t test; *P ≤ 0.05). (D) Representative flow cytometry plots and frequencies of total B cells, ASCs, and GC B cells in popliteal LNs 7 days after NP-KLH immunization (n = 10–11; four independent experiments, two-tailed Student’s t test; ***P ≤ 0.001). (E) Representative flow cytometry plot and frequencies of GC B cells in popliteal LNs 7 days after NP-KLH immunization of chimeric mice consisting of 50% tdTomato CD45.2 WT and 50% CD45.2 Smarca5-deficient BM cells (n = 5; two independent experiments, two-tailed Student’s t test; ***P ≤ 0.001). (F) Representative flow cytometry plots and frequencies of ASCs and GC B cells in popliteal LNs 7 days after NP-KLH immunization of control and AID.Cre.Smarca4fl/fl mice (n = 7–12; three independent experiments, two-tailed Student’s t test; *P ≤ 0.05, ***P ≤ 0.001). Each dot in the graphs represents a single mouse.
GC and ASC formation are dependent on SMARCA5. (A) Serum IgM, IgA, and IgG1 titers as determined by ELISA in control or Smarca5-deficient mice (n = 7–10 unmanipulated mice; two independent experiments, two-tailed Student’s t test; **P ≤ 0.01, ***P ≤ 0.001). (B) Representative flow cytometry plots and frequencies of B cell subsets in the BM of unmanipulated control versus Smarca5-deficient mice (n = 5; two independent experiments, two-tailed Student’s t test; ns, not significant). (C) Representative flow cytometry plots and frequencies of total ASCs in the BM of unmanipulated mice (n = 5; two independent experiments, two-tailed Student’s t test; *P ≤ 0.05). (D) Representative flow cytometry plots and frequencies of total B cells, ASCs, and GC B cells in popliteal LNs 7 days after NP-KLH immunization (n = 10–11; four independent experiments, two-tailed Student’s t test; ***P ≤ 0.001). (E) Representative flow cytometry plot and frequencies of GC B cells in popliteal LNs 7 days after NP-KLH immunization of chimeric mice consisting of 50% tdTomato CD45.2 WT and 50% CD45.2 Smarca5-deficient BM cells (n = 5; two independent experiments, two-tailed Student’s t test; ***P ≤ 0.001). (F) Representative flow cytometry plots and frequencies of ASCs and GC B cells in popliteal LNs 7 days after NP-KLH immunization of control and AID.Cre.Smarca4fl/fl mice (n = 7–12; three independent experiments, two-tailed Student’s t test; *P ≤ 0.05, ***P ≤ 0.001). Each dot in the graphs represents a single mouse.
We next examined whether SMARCA5 plays a role in B cell immune responses after initial B cell activation. For this purpose, we generated AID.Cre.Smarca5fl/fl mice and compared their ability to generate GC and ASC B cells with control mice in response to immunization. In this mouse model, the frequency of GC B cells was lower compared to control mice but a significant number of GC and ASC B cells were still detected (Fig. 2 F). We conclude that SMARCA5 plays a major early role during the initiation of the B cell response and to some extent after B cell activation and activation-induced cytidine deaminase (AID) expression.
Proper B cell proliferation, class-switch recombination, and ASC differentiation in vitro require SMARCA5
Improper GC seeding could be a result of diminished B cell early activation and cell expansion events. Thus, to examine whether SMARCA5 plays a role in B cell proliferation, splenic B cells were labeled with CellTrace Violet (CTV) and subjected to LPS stimulation for 4 days in vitro (Stoler-Barak et al., 2023). Smarca5-deficient B cells exhibited about 1.3-fold reduced cell division and proliferation relative to their control counterparts (Fig. 3 A). Although this difference appears minor, a more detailed quantification of specific cell cycle stages revealed a major defect in entry into the early and late cell cycle events resulting in a fourfold decreased number of Smarca5-deficient B cells after 4 days in culture (Fig. 3 A and Fig. S2 B). Cell cycle analysis using EdU and 7AAD staining did not reveal any significant defect at a specific stage in B cells that were stimulated with LPS (Fig. S2 C). Additionally, a cell viability assay did not detect enhanced cell death in Smarca5-deficient B cells (Fig. S2 D). We further explored CD86 upregulation as a marker of B cell activation. Early CD86 expression in response to LPS stimulation after 16 h was unaffected by the absence of SMARCA5, whereas late activation 4 days after stimulation was significantly compromised compared with the control group (Fig. 3 B). These observations suggest that Smarca5-deficient B cells can partially respond to initial stimulation, but proper activation and further downstream events are severely impaired. Moreover, class-switch recombination (CSR) was examined in vitro by stimulating splenic B cells with LPS in the presence of IL-4, followed by flow cytometry analysis after 4 days. Whereas a population of B cells that expressed IgG1 in response to the stimulation was clearly detected in the control cells, Smarca5-deficient B cells displayed almost no CSR to IgG1 (Fig. 3 C). Consistent with the in vivo findings (Fig. 2, C and D), Smarca5-deficient B cells also demonstrated impaired ability to differentiate into ASCs 4 days after LPS stimulation in vitro (Fig. 3 D). Collectively, these experiments indicate that SMARCA5 plays a crucial role in initial B cell activation and expansion, which greatly affects their proper differentiation and CSR.

SMARCA5 is required for B cell expansion, class-switch recombination, and ASC formation. (A) Representative flow cytometry histograms and quantification of CTV indicating the in vitro proliferation and absolute number of splenic B cells treated with LPS for 4 days (n = 5–8; two independent experiments, two-tailed Student’s t test; **P ≤ 0.01). (B) Representative flow cytometry histograms and frequencies of untreated B cells, and B cells stimulated with LPS for 16 h (top) or 4 days (bottom) (n = 5–6; two independent experiments, one-way ANOVA; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ns, not significant). (C and D) Representative flow cytometry plots and frequencies of IgG1+ B cells (C) or total B cells and ASCs (D) derived from naive spleens that were either left unmanipulated or stimulated in vitro with LPS+IL-4 for 4 days (n = 4–6; two independent experiments, one-way ANOVA; ***P ≤ 0.001, ns, not significant). Each dot in the graphs represents a single mouse.
SMARCA5 is required for B cell expansion, class-switch recombination, and ASC formation. (A) Representative flow cytometry histograms and quantification of CTV indicating the in vitro proliferation and absolute number of splenic B cells treated with LPS for 4 days (n = 5–8; two independent experiments, two-tailed Student’s t test; **P ≤ 0.01). (B) Representative flow cytometry histograms and frequencies of untreated B cells, and B cells stimulated with LPS for 16 h (top) or 4 days (bottom) (n = 5–6; two independent experiments, one-way ANOVA; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ns, not significant). (C and D) Representative flow cytometry plots and frequencies of IgG1+ B cells (C) or total B cells and ASCs (D) derived from naive spleens that were either left unmanipulated or stimulated in vitro with LPS+IL-4 for 4 days (n = 4–6; two independent experiments, one-way ANOVA; ***P ≤ 0.001, ns, not significant). Each dot in the graphs represents a single mouse.
The formation of antigen-specific GC B cells and ASCs depends on SMARCA5 functions
Our findings highlight a crucial role for SMARCA5 in orchestrating polyclonal immune responses. Since in the absence of SMARCA5, GC structures were not formed, resulting in a significant reduction in both ASC numbers and antibody titers in the serum, further direct analysis of SMARCA5 functions was not feasible (Fig. 2 D). To bypass these constraints and enable the detection of antigen-specific GC B cells and ASCs, we crossed GFP-expressing mice, which carry a transgenic BCR specific for 4-hydroxy-3-nitrophenyl (NP) when coupled to Igλ, known as B1-8hi (Shih et al., 2002) with either control or CD23.Cre.Smarca5fl/fl mice. Subsequently, we utilized these mice as donors for the adoptive transfer of antigen-specific B cells into WT host mice. The mice were then immunized with NP-KLH for 3, 5, or 7 days, followed by LN imaging and flow cytometry quantification (Fig. 4 A). Utilizing intravital two-photon laser scanning microscopy (TPLSM) (Stoler-Barak et al., 2019) revealed that Smarca5-deficient B cells were unable to generate GC structures on day 7, though a clear B cell response was observed at earlier time points (Fig. 4 B).

SMARCA5 is required to establish an appropriate antigen-specific B cell immune response. (A) Scheme illustrating the experimental design. (B) Representative TPLSM images of intact popliteal LNs removed at the indicated time points after NP-KLH immunization (n = 3–4; three independent experiments). Scale bar: 200 µm. (C–E) Representative flow cytometry plots and frequencies of total transferred (C), activated (D; left and middle), GC (D; right), or ASCs (E), antigen-specific B1-8hi B cells in popliteal LNs at the indicated time points after NP-KLH immunization (n = 7; two independent experiments, two-tailed Student’s t test; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). Each dot in the graph represents a single mouse.
SMARCA5 is required to establish an appropriate antigen-specific B cell immune response. (A) Scheme illustrating the experimental design. (B) Representative TPLSM images of intact popliteal LNs removed at the indicated time points after NP-KLH immunization (n = 3–4; three independent experiments). Scale bar: 200 µm. (C–E) Representative flow cytometry plots and frequencies of total transferred (C), activated (D; left and middle), GC (D; right), or ASCs (E), antigen-specific B1-8hi B cells in popliteal LNs at the indicated time points after NP-KLH immunization (n = 7; two independent experiments, two-tailed Student’s t test; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). Each dot in the graph represents a single mouse.
To gain further insight into the magnitude of the antigen-specific response in the absence of SMARCA5, we used flow cytometry to quantify B cell subsets during the immune response. Analysis of popliteal LNs at 3, 5, and 7 days after immunization revealed a significantly lower fraction of total Smarca5-deficient B1-8hi B cells compared with control (Fig. 4 C). The fraction of the cells that showed expression of activation and GC cell markers was smaller compared to the control cells (Fig. 4 D). Further analysis of both resting or activated populations revealed minor changes in the viability of Smarca5-deficient antigen-specific B cells (Fig. S3, A and B). Additionally, on days 5 and 7 after immunization, while the majority of ASCs in the control group were antigen-specific B1-8hi B cells, almost no B1-8hi ASCs were detected in the absence of SMARCA5 (Fig. 4 E). These findings confirmed the utility of the adoptive cell transfer model to examine the role of SMARCA5 at later stages of the response.

The viability of resting and activated antigen-specific B cells is not substantially impaired following SMARCA5 depletion. (A and B) Flow cytometry plots and frequencies of resting (A) and activated (B) antigen-specific B1-8hi GFP B cells in popliteal LNs 5 days after NP-KLH immunization (n = 6; two independent experiments, one-way ANOVA; *P ≤ 0.05, **P ≤ 0.01; ns, not significant). Each dot in the graph represents a single mouse.
The viability of resting and activated antigen-specific B cells is not substantially impaired following SMARCA5 depletion. (A and B) Flow cytometry plots and frequencies of resting (A) and activated (B) antigen-specific B1-8hi GFP B cells in popliteal LNs 5 days after NP-KLH immunization (n = 6; two independent experiments, one-way ANOVA; *P ≤ 0.05, **P ≤ 0.01; ns, not significant). Each dot in the graph represents a single mouse.
SMARCA5 is essential for the genetic rewiring of activated B cells during the early stages of the immune response
The small fraction of early responding and GC B cells that was detected following adoptive transfer, provided an opportunity to probe deeper into the molecular mechanisms of SMARCA5 during the B cell immune response. To determine the impact of SMARCA5 depletion on gene expression profiles during the early stages preceding GC seeding, we conducted bulk RNA-sequencing (RNA-seq) experiments using transferred GFP+ B1-8hi B cells sorted 5 days after immunization (Fig. 5 A). Our analysis revealed 1,353 genes that were upregulated and 1,323 genes that were downregulated when comparing the control cells to the Smarca5-deficient B cells (log2 FC greater than or equal to ±0.58, adjusted P < 0.05) (Fig. 5 B). Consistent with previous literature related to the functions of this chromatin-remodeler (Barisic et al., 2019; Corona and Tamkun, 2004; Clapier and Cairns, 2009; Collins et al., 2002; Bozhenok et al., 2002), Smarca5-deficient B cells exhibited downregulation of cell cycle and DNA repair genes. Furthermore, we report for the first time that genes known to activate the GC and ASC differentiation programs (Shi et al., 2015) were also downregulated in the absence of SMARCA5 (Fig. 5 C) providing an additional explanation for the diminished GC and ASC phenotypes that we observed (Fig. 2, Fig. 3, and Fig. 4). In addition, by using the Metascape tool to determine the biological processes affected by SMARCA5 depletion, we found that cell cycle, cell division, DNA repair, and DNA recombination were significantly impaired in the deficient cells and upregulated in the control population (Fig. 5 D). To identify the signaling pathways adversely affected in Smarca5-deficient B cells, we conducted gene set enrichment analysis (GSEA). This analysis revealed a significant downregulation of genes related to the E2F and G2M pathways, known as entry checkpoints to phases of mitosis and DNA synthesis (Stark and Taylor, 2004; Dimova and Dyson, 2005) (Fig. 5 E). Collectively, these findings reveal the pivotal involvement of SMARCA5 in the processes of cell division, DNA replication and repair in B cells, in accordance with data previously published for other cell types (Lan et al., 2010; Collins et al., 2002; Corona and Tamkun, 2004; Kokavec et al., 2017). Although we compared RNA from control and Smarca5-deficient cells that were present on day 5 in the immunized mice, some of these differences may represent the inability of the latter to become fully activated. Collectively, our results identify for the first time the novel role of SMARCA5 in shaping the transcriptome landscape and cell activation events governing the formation of both pre-GC and ASCs.

SMARCA5 mediates proper genetic re-programming of activated B cells. (A) Scheme illustrating the experimental design. (B) Volcano plots showing differential gene expression in transferred B1-8hi B cells 5 days after immunization (n = 3–6; three independent experiments, adjusted P value <0.05, and log2 FC > 0.58 or less than −0.58; raw P values were adjusted for multiple testing using the procedure of Benjamini and Hochberg). (C) Heatmap of differentially expressed genes. (D) GO term biological pathway analysis of upregulated genes in control B1-8hi B cells. (E) GSEA analysis of differentially expressed genes. NES, normalized enrichment score; FDR, false discovery rate.
SMARCA5 mediates proper genetic re-programming of activated B cells. (A) Scheme illustrating the experimental design. (B) Volcano plots showing differential gene expression in transferred B1-8hi B cells 5 days after immunization (n = 3–6; three independent experiments, adjusted P value <0.05, and log2 FC > 0.58 or less than −0.58; raw P values were adjusted for multiple testing using the procedure of Benjamini and Hochberg). (C) Heatmap of differentially expressed genes. (D) GO term biological pathway analysis of upregulated genes in control B1-8hi B cells. (E) GSEA analysis of differentially expressed genes. NES, normalized enrichment score; FDR, false discovery rate.
SMARCA5 controls chromatin accessibility of GC and ASC-determining genes
The bulk RNA-seq data may include cells that escaped the Smarca5-deletion and responded to the antigen, although they were unable to become fully activated and form GCs (Fig. 4 B). To gain a more comprehensive understanding of how SMARCA5 regulates B cell activation before GC formation, we performed an integrated analysis using multiple high-throughput sequencing approaches. To this end, we employed the single-nucleus multiome technique, which combines ATAC (assay for transposase-accessible chromatin) and GEX (gene expression of RNA transcripts) (Wang et al., 2022) sequencing analysis of transferred GFP+ B1-8hi B cells, sorted 5 days after immunization. Since only a small number of cells were recovered from individual mice, the multiome procedure was applied to single nuclei from B cells pooled from 10 mice. The ATAC-seq analysis of total cells identified 79,332 peaks, of which 11,882 peaks showed differential accessibility in the control cells, and only 1,168 peaks exhibited differential accessibility in the absence of SMARCA5 (log2 FC greater than or equal to ±0.3, adjusted P < 0.1). Additionally, Gene Ontology (GO) analysis for biological pathways revealed that the genes associated with these differential accessibility peaks were enriched for leukocyte activation and differentiation processes (Fig. 6 A). An analysis of each individual cluster revealed changes in chromatin accessibility in most of them (Fig. S4 A). This analysis demonstrates the key role of SMARCA5 in influencing the accessibility of multiple genomic areas in antigen-specific early-responding B cells.

Chromatin accessibility of GC- and ASC-promoting genes is regulated by SMARCA5. (A) MA plot of the log average (A) on the X-axis, and log ratio (M) on the Y-axis representing the changes in accessibility peaks under SMARCA5 deficiency (left); GO term biological pathway analysis of genes associated with differential accessibility peaks (right) upregulated in either control or Smarca5-deficient transferred B1-8hi B cells, 5 days after immunization. Upregulated peaks in the control B cells are marked in pink and the upregulated peaks in the Smarca5-deficient B cells are marked in blue (log2 FC greater than or equal to ±0.3, adjusted P < 0.1). (B) Distribution of SMARCA5 accessibility peaks (ATAC-seq) and binding sites (using the CUT&RUN protocol) across genomic regions. TTS: transcription termination site. (C) Box plots representing the median, quartiles, and 5th and 95th percentiles of changes in gene expression of control compared to Smarca5-deficient B cells in genes linked to ATAC peaks or both ATAC and CUT&RUN peaks compared with total genes. P value was calculated by a two-sided Wilcoxon rank sum test. (D) UMAP projections of Multiome profiles with color coding according to the different clusters. Top and bottom UMAPs represent the control and Smarca5-deficient groups, respectively. (E) Dot plots depicting the RNA expression of selected marker genes presented by average expression and percent expression per cluster. The top and bottom plots represent the control and deficient groups, respectively. (F) Gene tracks depicting chromatin accessibility and expression of selected marker genes from specific clusters. Each track also represents the DNA binding sites of SMARCA5, as shown by CUT&RUN peaks.
Chromatin accessibility of GC- and ASC-promoting genes is regulated by SMARCA5. (A) MA plot of the log average (A) on the X-axis, and log ratio (M) on the Y-axis representing the changes in accessibility peaks under SMARCA5 deficiency (left); GO term biological pathway analysis of genes associated with differential accessibility peaks (right) upregulated in either control or Smarca5-deficient transferred B1-8hi B cells, 5 days after immunization. Upregulated peaks in the control B cells are marked in pink and the upregulated peaks in the Smarca5-deficient B cells are marked in blue (log2 FC greater than or equal to ±0.3, adjusted P < 0.1). (B) Distribution of SMARCA5 accessibility peaks (ATAC-seq) and binding sites (using the CUT&RUN protocol) across genomic regions. TTS: transcription termination site. (C) Box plots representing the median, quartiles, and 5th and 95th percentiles of changes in gene expression of control compared to Smarca5-deficient B cells in genes linked to ATAC peaks or both ATAC and CUT&RUN peaks compared with total genes. P value was calculated by a two-sided Wilcoxon rank sum test. (D) UMAP projections of Multiome profiles with color coding according to the different clusters. Top and bottom UMAPs represent the control and Smarca5-deficient groups, respectively. (E) Dot plots depicting the RNA expression of selected marker genes presented by average expression and percent expression per cluster. The top and bottom plots represent the control and deficient groups, respectively. (F) Gene tracks depicting chromatin accessibility and expression of selected marker genes from specific clusters. Each track also represents the DNA binding sites of SMARCA5, as shown by CUT&RUN peaks.

Changes in chromatin accessibility in the absence of SMARCA5. (A) MA plots of the log average (A) on the X-axis, and log ratio (M) on the Y-axis representing the changes in accessibility peaks under SMARCA5 deficiency for each expression cluster. (B) Venn diagrams indicating the intersection and total number of peaks of the ATAC and CUT&RUN datasets. (C) Dot plots depicting the RNA expression of the most expressed genes in each cluster presented by average expression and percent expression. Left and right plots represent the control and deficient groups, respectively. (D) Gene tracks of chromatin accessibility and SMARCA5 DNA binding sites as shown by CUT&RUN peaks for selected marker genes.
Changes in chromatin accessibility in the absence of SMARCA5. (A) MA plots of the log average (A) on the X-axis, and log ratio (M) on the Y-axis representing the changes in accessibility peaks under SMARCA5 deficiency for each expression cluster. (B) Venn diagrams indicating the intersection and total number of peaks of the ATAC and CUT&RUN datasets. (C) Dot plots depicting the RNA expression of the most expressed genes in each cluster presented by average expression and percent expression. Left and right plots represent the control and deficient groups, respectively. (D) Gene tracks of chromatin accessibility and SMARCA5 DNA binding sites as shown by CUT&RUN peaks for selected marker genes.
We next wished to characterize the genome-wide distribution of SMARCA5 open chromatin peaks. Approximately, 42% of peaks were mainly localized to introns, 38% to intergenic regions, 13% to gene promoters, and <5% were localized to exons. Notably, the overall distribution of accessible genomic sites remained comparable between control and Smarca5-deficient B cells (Fig. 6 B). CUT&RUN (Cleavage Under Targets & Release Using Nuclease) is a modified protocol derived from chromatin IP, followed by sequencing (ChIP-seq) used to analyze protein DNA interactions on a genome-wide scale (Janssens and Henikoff, 2019). To further examine if SMARCA5 binding sites are specifically associated with gene accessibility, we performed CUT&RUN using GC B cells from LNs of WT mice. This analysis revealed that SMARCA5 predominantly binds at promoters (34%), introns (31.4%), and intergenic regions (25.9%) (Fig. 6 B). To examine those chromatin changes and SMARCA5 binding sites that are associated with gene regulation (i.e., only genes that were differentially expressed between the control and the Smarca5-deficient cells), the ATAC-seq and CUT&RUN results were integrated with the bulk RNA-seq data. This integration demonstrated that differentially expressed genes upregulated in antigen-specific control B cells were also significantly associated with detectable overlapping ATAC and CUT&RUN peaks (Fig. 6 C and Fig. S4 B). Thus, SMARCA5 primarily regulates the accessibility and expression of bound genes rather than overall genome-wide changes. To explore the transcriptomic landscape promoting B cell activation and differentiation prior to GC seeding and after antigen encounter, we examined the cell cluster annotation of B cell subsets using the integrated ATAC and GEX multiome profiles.
Uniform weighted nearest neighbor manifold approximation and projection (wnnUMAP) dimensionality reduction analysis is designed to combine diverse datasets obtained from the same cells to establish a unified singular portrayal of single-cell multimodal data (Hao et al., 2021). Using wnnUMAP of 5,993 control and 7,041 Smarca5-deficient individual B cell nuclei, we identified 11 B cell clusters (Fig. 6, D and E). Although this sequencing procedure and subsequent analysis allowed the detection of activated pre-GC B cells and ASC subsets, the identity of the cells in other clusters was less clear (Fig. S4 C). Among the different clusters, most of the changes occurred in clusters 0 and 1 (Fig. S4 A), suggesting that these represent initially activated B cells that cannot progress to other stages of B cell differentiation in the absence of SMARCA5. Furthermore, the abundance of pre-GC and ASC clusters was reduced in the Smarca5-deficient B cells, demonstrating that this cell analysis provides similar results, as obtained by flow cytometry (Fig. 6 D). The gene expression profiles reveal that B cells deficient in Smarca5 exhibit a notable impairment in their capacity to undergo class-switching to IgG1 isotype (Fig. 6, D and E) and to express genes that are associated with pre-GC B cells (cluster 11; Bcl6, Aicda, S1pr2, FAS, Basp1) and ASCs (cluster 10; Mzb1, Xbp1, Irf4, Prdm1, Sdc1) (Fig. 6, D and E). Collectively, we conclude that SMARCA5 is essential for the expression of B cell differentiation genes.
Given our results demonstrating that SMARCA5 plays a crucial role in regulating GC and ASC genetic programs, we aimed to investigate whether this protein can directly target the critical genes that determine the fate of these B cells (Nutt et al., 2011). Gene track analysis of open chromatin coverage alongside ATAC and CUT&RUN peaks indicated that SMARCA5 CUT&RUN DNA binding was enriched at genomic regions of GC and ASC-promoting genes, as exemplified by Aicda, Basp1, Bcl6, and Xbp1 tracks. Moreover, open chromatin tracks showed markedly altered accessibility signals in these GC and ASC-promoting genes in the absence of SMARCA5 (Fig. 6 F and Fig. S4 D). These signals with differential accessibility could be attributed to variable regulatory elements such as enhancers or suppressors. Thus, we conclude that SMARCA5 is required for driving cellular responses and B cell proliferation and fate decisions through rewiring gene expression by modifying gene accessibility.
Discussion
The establishment of effective long-lasting protection from pathogens requires the differentiation of naive B cells into GC cells and antibody-forming cells that secrete neutralizing antibodies (Victora and Nussenzweig, 2022; Biram et al., 2019). This process involves remarkable changes in the expression of genes that control cell fate and specialized B cell subset functions (Papin et al., 2022; Bunting et al., 2016; Vilarrasa-Blasi et al., 2021). To identify novel genes that control cell differentiation or cell states, we examined the translatome of the GC rather than using typical transcriptome analyses (Haimon et al., 2018; Sanz et al., 2009). Furthermore, our multiomic approach that combined three different data sets allowed us to detect a Smarca5-deficient B cell response on day 5, and these cells showed profound changes in expression and accessibility of SMARCA5-bound genes to those detected in WT mice. These data sets were compared with the findings obtained by bulk mRNA-seq to provide robust and reliable results.
We observed that SMARCA5 is highly translated in the GC, and by using a conditional knock-out mouse, we show that its functions are essential for an intact humoral immune response. Typically, naive B cells do not require profound changes in their chromatin structure to support their functions under homeostasis, and indeed, deletion of SMARCA4 or SMARCA5 does not affect the naive B cell compartment (Schmiedel et al., 2021; Choi et al., 2012). Nonetheless, upon encounter with cognate antigens or danger signals such as TLR ligands, B cells begin to express a series of genes required for their proper activation. SMARCA5 plays a role in initial B cell proliferation, suggesting that cell cycle progression prior to robust differentiation depends on SMARCA5-chromatin remodeling activity. Accordingly, SMARCA5 was essential for B cell immune responses in vivo, demonstrating that this chromatin remodeler is required for genetic programs that mediate cellular proper activation, expansion, and differentiation. In addition to SMARCA5’s role in cell proliferation, we found that Xbp1, a gene essential for ASC formation (Klein et al., 2006; Nutt et al., 2011), is controlled also by SMARCA5, most likely by regulating its gene accessibility, and indeed, ASCs were not formed by Smarca5-deficient B cells in vitro or in vivo. Interestingly, early B cell development, a process that depends on profound chromatin remodeling, proliferation, and differentiation, was shown to depend on SMARCA5 as well (Zikmund et al., 2019). CTCF facilitates promoter–enhancer interactions, which are essential for GC formation and ASC differentiation (Pérez-García et al., 2017; Ong and Corces, 2014), and its interactions with SMARCA5 might be essential for GC seeding since deletion of either of these components perturbs B cell differentiation (Bomber et al., 2023; Wiechens et al., 2016; Dluhosova et al., 2014).
In agreement with its role in cell cycle progression in other cell types (He et al., 2016; Alvarez-Saavedra et al., 2014; Zikmund et al., 2019; Zhang et al., 2020; Kokavec et al., 2017), we demonstrate that SMARCA5 is required for proper expression of cell cycle genes during GC seeding, and indeed, activated B cells rapidly proliferate during both this stage and the GC response (Glaros et al., 2021; Grenov et al., 2022). Specifically, SMARCA5 was previously shown to play a role in pre-DNA-replication complex formation (Sugimoto et al., 2011). We showed that DNA-bound SMARCA5 is enriched in gene promoters in agreement with studies that examined other cell types (Morris et al., 2014; Sala et al., 2011). In Smarca5-deficient B cells, the Aicda locus was less accessible, Aicda mRNA was transcribed to a lesser degree, and CSR to IgG1 was severely impaired in vitro and in vivo. Nonetheless, additional DNA damage–related genes such as Msh3, Msh6, and Ung were also dependent on SMARCA5, suggesting that this chromatin remodeler controls a wide range of genetic programs that support CSR, and perhaps SHM (Stoler-Barak et al., 2023; Saha et al., 2021; Chi et al., 2020). Furthermore, SMARCA5 is recruited to double-strand DNA breaks and plays a role in their proper repair (Lan et al., 2010; Pinto Jurado et al., 2024; Aydin et al., 2014; Min et al., 2014). These events prevent genomic instability through chromatin remodeling (Toiber et al., 2013). Mutations related to AID functions were correlated with transcription–replication conflicts, which depend on chromatin remodeling functions, including SMARCA5 (Bayona-Feliu et al., 2023). Thus, in vitro, CSR depends on SMARCA5 activity both for proper AID expression, and regulation of other factors associated with the DNA-damage pathway, suggesting that SMARCA5 acts as a master regulator of CSR in addition to other cellular functions.
Collectively, we found that SMARCA5 plays a critical role in the B cell immune response through the control of gene expression that mediates proper B cell activation and cell cycle progression during vectorial differentiation (Glaros et al., 2021). Chromatin remodeler subunits are frequently mutated in lymphomas that arise from GCs (Papin et al., 2022; Mlynarczyk et al., 2019; Victora et al., 2012), and some of these can be targeted by drugs for cancer treatment (Jevtic et al., 2022; Oyama et al., 2021; Kaur et al., 2019). The essential role of SMARCA5 for B cell expansion in vivo indicates that specific inhibitors could serve as a target for the treatment of B cell lymphomas and antibody-mediated autoimmune diseases.
Materials and methods
Mice
The transgenic Smarca5flox/flox mice were generated and provided by T. Stopka (First Faculty of Medicine, Charles University, Vestec, Czech Republic). Rpl22HA mice were provided by S. Jung (Department of Immunology and Regenerative Biology, Weizmann Institute of Science, Rehovot, Israel). CD23cre mice were generated and provided by M. Busslinger (Research Institute of Molecular Pathology, Vienna, Austria). B1-8hi transgenic mice expressing the NP-specific B cell receptor were a kind gift from M. Nussenzweig (Rockefeller University, New York, NY). AicdaCre/+, GFP, CD45.1, and tdTomato mice were purchased from the Jackson Laboratory. WT (C57BL/6) mice were purchased from Envigo. For the generation of chimeric mice, CD45.1 hosts were irradiated with 950 rad and then reconstituted with BM cells isolated from WT tdTomato CD45.2 (Smarca5+/+) and CD45.2 CD23.Cre.Smarca5fl/fl mice at a 1:1 ratio. In all experiments, both male and female mice were used at the age of 6–15 wk. This research complied with all relevant ethical regulations, and all mouse experiments were approved by the Weizmann Institute Animal Care and Use Committee (IACUC number 01040123-3).
Immunizations
Mice were injected with 25 μl PBS containing 10 µg NP-KLH (BioSearch Technologies) in Alum adjuvant (Thermo Fisher Scientific) into each hind footpad.
Adoptive cell transfer
For B cell isolation, donor splenic tissue was removed and forced through a 70-µm mesh in PBS. B cells were purified using negative selection anti-CD43 beads and magnetic-activated cell sorting (MACS) (Miltenyi Biotec) according to the manufacturer’s instructions. For adoptive transfer, 3–5 × 106 total B1-8hi B cells were transferred by intravenous injection to host mice 1 day before the immunization.
Flow cytometry
Popliteal LNs were removed at the indicated time points after immunization and forced through a 70-µm mesh into PBS containing 2% fetal calf serum (FCS) and 1 mM EDTA (FACS wash buffer) to attain single-cell suspensions. To block nonspecific binding to Fc receptors, 1 µg/ml anti-CD16/32 (BioLegend) was added to the single cell suspension on ice for 5 min. For staining surface markers, cells were incubated with fluorescently labeled antibodies (Table 1) at a 1:400 dilution for 30 min on ice. Cells were gated as lymphocytes (FSC-A versus SSC-A) and single cells (FSC-A versus FSC-H). GC cells were gated as live/single, B220+ CD38Lo FASHi. Activated cells were gated as live/single, GL7Hi and FASHi. ASCs were gated as live/single and CD138+ cells. Antigen-specific B1-8hi B cells were gated as GFP+ cells. For viability assessment, 7AAD (BD Biosciences) was added at a 1:50 dilution to the resuspended cells for 5 min before analysis. To determine the absolute number of cells, 4 μl of precision count beads (BioLegend) was added to 200 μl cell suspension (half of the total sample). Stained cell suspensions were analyzed using a CytoFlex flow cytometer (Beckman Coulter).
Antibodies used in the study
| Target antigen | Clone | Catalog number | Manufacturer |
|---|---|---|---|
| Flow cytometry | |||
| CD45R/B220 APC | RA3-6B2 | 103212 | BioLegend |
| CD45R/B220 APC-e780 | RA3-6B2 | 47-0452-82 | eBioscience |
| CD19 PB | 6D5 | 115523 | BioLegend |
| CD38 AF700 | 90 | 56-0381-82 | eBioscience |
| CD95/FAS PE/Cy7 | Jo2 | 557653 | BD Biosciences |
| GL7 AF647 | GL7 | 144606 | BioLegend |
| CD138 BV605 | 281-2 | 142516 | BioLegend |
| CXCR4-BV421 | I276FI2 | 146511 | BioLegend |
| CD86-APC | GL-1 | 105012 | BioLegend |
| IgM PerCP-e710 | II/41 | 46-5790-82 | eBioscience |
| IgD APC-e780 | 11-26 | 47-5993-82 | eBioscience |
| IgG1-FITC | RMG1-1 | 406606 | Biolegend |
| CD4 APC-e780 | GK1.5 | 47-0041-82 | eBioscience |
| CD8 APC-e780 | 53-6.7 | 47-0081-82 | eBioscience |
| F4/80 APC-e780 | BM8 | 47-4801-82 | eBioscience |
| Gr-1 APC-e780 | RB6-8C5 | 47-5931-82 | eBioscience |
| CD45.2-BV421 | 104 | 109831 | BioLegend |
| CUT&RUN | |||
| SNF2H | - | ab3749 | Abcam |
| Rabbit IgG isotype control | DA1E | 3900S | Cell Signaling Technology |
| Western blotting | |||
| SNF2H | - | ab3749 | Abcam |
| β-actin | - | 3700 | Cell Signaling |
| Anti-rabbit-HRP | - | 7074 | Cell Signaling |
| Anti-mouse-HRP | - | 7076 | Cell Signaling |
ELISA
Serum was collected from unmanipulated mice, and IgM, IgA, and IgG1 titers were determined by ELISA using goat anti-mouse IgM (ab97230), IgA (ab97235), and IgG1 (ab97240) conjugated to horseradish peroxidase (Abcam) at 1:2,500.
In vitro activation, proliferation, and CSR assays
Spleens were removed and forced through a 70-µm mesh into FACS washing buffer to obtain single-cell suspensions. For the proliferation assay, splenic cells were stained with CTV (Invitrogen) according to the manufacturer’s instructions. Cells were seeded at 1 × 106/ml in a 24-well plate and incubated at 37°C in B-cell medium (RPMI-1640 medium supplemented with 10% FBS, 100 µg/ml penicillin/streptomycin, 50 µg/ml gentamycin, 2 mM glutamine and pyruvate, nonessential amino acids, and 50 µM β-mercaptoethanol). For activation and proliferation assays, cells were stimulated with 50 µg/ml LPS for 4 days, and CTV dilution was assessed by flow cytometry. For CSR assay, cells were stimulated with 50 µg/ml LPS + 50 ng/ml mouse IL-4 for 4 days.
Western blotting
Unstimulated or LPS-stimulated cell pellets were lysed using radioimmunoprecipitation assay buffer (Sigma-Aldrich). Lysates were clarified at 21,000 g for 15 min at 4°C, and protein concentration was determined using the BCA protein assay (Thermo Fisher Scientific). Samples containing 20 μg of total protein were prepared with 4×LDS sample buffer (NuPAGE; Thermo Fisher Scientific) and were then resolved on a 4–20% Bis-Tris gel (GeneScript SurePAGE). Proteins were separated by electrophoresis and were then transferred to a nitrocellulose membrane (Bio-Rad) using the Trans-Blot Turbo system (Bio-Rad). The membrane was blocked with 5% BSA in PBS-T (wt/vol) for 1 h at room temperature, washed four times for 5 min with PBS-T, and incubated with the following primary antibodies: rabbit anti-SNF2H (ab-3749, 1:500; Abcam, overnight at 4°C) and mouse anti-β-actin (#3700, 1:1,000; Cell Signaling, overnight at 4°C). Membranes were washed four times for 5 min with PBS-T and incubated with the corresponding HRP-linked secondary antibody (mouse #7076/rabbit #7074; Cell Signaling) for 1 h at room temperature. Super-Signal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific) was used to detect HRP activity. The membrane was stripped using Restore stripping buffer (Thermo Fisher Scientific) between incubation with the two antibodies.
EdU proliferation assay
For cell cycle analysis, 100 µM of the nucleoside analog 5-ethynyl-2′-deoxyuridine (EdU; Molecular Probes) in PBS was added to LPS-stimulated cells in culture. After 1 h, the cells were stained for the surface marker B220, followed by EdU detection using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Molecular Probes) according to the manufacturer’s instructions. 7AAD (BD Biosciences) was added at a 1:50 dilution 5 min before analysis by flow cytometry.
TPLSM image acquisition
A Zeiss LSM 880 upright microscope fitted with a Coherent Chameleon Vision laser was used for imaging experiments. Whole LNs were dissected and images were acquired with a femtosecond-pulsed two-photon laser tuned to 940 nm. The microscope was fitted with a filter cube containing 505 LPXR to split the emission to a detector (with a 500–550 nm filter for GFP fluorescence). Tile images were acquired as 100–200 μm Z stacks with 5-μm intervals between each Z plane. The zoom was set to 1.5 and images were acquired at 512 × 512 x–y resolution. Images were processed using Imaris software (Bitplane).
Ribosome IP
For IP of tagged ribosomes (RiboTag) from cell lysates, followed by the extraction and sequencing of the associated mRNAs, four mice were immunized subcutaneously with 50 μl KLH (45 µg per mouse) emulsified in CFA. After 7 days, the inguinal LNs were collected and filtered through a mesh with FACS wash buffer. Fluorescently labeled antibodies were added for 30 min on ice. GC B cells were identified as live/single, B220+, CD38−, FAS+, F4/80−, Gr-1−, CD4−, CD8− cells. DZ and LZ cells were gated as CXCR4hi CD86lo and CXCR4lo CD86hi. A total of 3 × 105 DZ and 2 × 105 LZ B cells per mouse were sorted into FACS wash buffer and subjected to the ribosomal IP protocol. After sorting, the sample was divided to one third (“sort” sample) and two thirds (“sort-IP” sample) into separate tubes. The samples were centrifuged at 400 g for 10 min at 4°C. Supernatants were removed, and the pellet from the “sort” tube was resuspended in 100 μl Dynabeads mRNA direct kit lysis/binding buffer (Life Technologies). The pellet of the “sort-IP” tube was lysed in 800 μl of ice-cold lysis buffer (50 mM Tris, pH 7.4, 100 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM dithiothreitol [DTT], 1:100 protease inhibitor cocktail [Sigma-Aldrich], 200 U/ml RNasin [Promega], and 0.1 mg/ml cycloheximide [Sigma-Aldrich] in RNase-free double distilled water (DDW). 10 µg of anti-HA antibody (H9658; Sigma-Aldrich) were added followed by an overnight incubation with slow rotation in a cold room. In parallel, 100 μl per sample of Dynabeads Protein G (Thermo Fisher Scientific) were washed three times with lysis buffer and kept overnight at 4°C. The next day, the beads were added to each sample, followed by incubation for 2 h on a rotator in a cold room. Next, the samples were washed three times with high-salt buffer (50 mM Tris, 300 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM DTT, 1:200 protease inhibitor, 100 U/ml RNasin, and 0.1 mg/ml cycloheximide in RNase-free DDW) for 5 min per wash in a cold room on a rotator. Following the washes, beads were magnetically selected and the excess buffer was removed. Finally, 100 μl Dynabead mRNA direct kit lysis/binding buffer (Life Technologies) was added to the beads and RNA was extracted according to the manufacturer’s instructions for bulk RNA-seq.
Bulk RNA-seq and data processing
Mice were injected subcutaneously with NP-KLH in alum into the hind footpad (10 µg in 25 μl per side) for 5 days. Popliteal LNs were sorted for CD4−, CD8−, GR-1−, F4/80−, and GFP+ expression for antigen-specific B cells. Next, 3 × 104 cells were sorted directly into 100 μl Dynabeads mRNA direct kit lysis/binding buffer (Life Technologies) using a FACS ARIA cell sorter (BD) and immediately frozen on dry ice. mRNA was isolated using the Dynabeads mRNA direct kit according to the manufacturer’s instructions (Life Technologies). A bulk adaptation of the massively parallel single-cell RNA-seq protocol (MARS-seq) was used (Jaitin et al., 2014; Keren-Shaul et al., 2019) to generate RNA-seq libraries for transcriptomic analysis. Alignment and differential expression analysis was performed using the UTAP pipeline v1.10.280 (Kohen et al., 2019). The pipeline quantifies the genes annotated in RefSeq (extended by 1,000 bases toward the 5′ edge and 100 bases in the 3′ direction). The threshold for significant differential expression was log2FC > 0.58 or less-than −0.58, P < 0.05. Volcano plots were created using Prism Version 10.0 (GraphPad). GSEA was performed using GSEA 4.3.2 with the GSEA preranked tool (Subramanian et al., 2005). The Molecular Signature Database hallmark gene sets were used to perform pathway enrichment analysis. The Metascape v3.5.20240101 tool (Zhou et al., 2019) was used to define unique pathways that were significantly modified based on the threshold indicated above.
CUT&RUN analysis
Two WT mice were immunized subcutaneously with 50 μl KLH (45 µg per mouse) emulsified in CFA. After 7 days, the inguinal LNs were collected and filtered through a mesh with FACS wash buffer. Fluorescently labeled antibodies were added for 30 min on ice. GC B cells were identified as B220+, CD38−, FAS+, GL7+, F4/80−, Gr-1−, CD4−, CD8−, and CD138− cells. A total of 5 × 105 GC B cells per mouse were sorted into PBS, washed twice, and subjected to the CUT&RUN protocol version 3, as previously described (Janssens and Henikoff, 2019). Briefly, GC B cells were added to a concanavalin A–coated bead slurry that was preincubated with CaCl2 and MnCl2 to activate the beads. The cells and beads were rotated for 10 min at room temperature to ensure that all cells were coated onto the beads. The cells of each mouse were divided into two samples. Tubes were then placed into a magnetic stand to separate the cells from the liquid and the supernatants were discarded. The cell-bead mixtures were resuspended in 100 μl of antibody buffer containing 0.05% digitonin for permeabilization. Next, 1 μl of monoclonal rabbit-anti mouse SNF2H antibody (Abcam) or a rabbit monoclonal isotype antibody (Cell Signaling Technologies) was added to the permeabilized samples and the mixture was incubated overnight on a rotator. The following day, the beads were washed twice, and then incubated for 1 h in 150 μl of digitonin-containing wash buffer with 700 ng/ml of pA/G-MNase (kindly provided by the lab of Steven Henikoff, Howard Hughes Medical Institute, Seattle, WA, USA). After incubation, the beads were washed twice and resuspended in 100 μl of digitonin-containing wash buffer. The beads were then chilled to 0°C in a metal block resting in ice. Finally, 2 μl of 100 mM CaCl2 was added to activate the MNase activity, resulting in a final concentration of 2 mM. The reaction was stopped after 30 min. The bead–cell mixture was incubated for at least 30 min at 37°C to release restricted DNA fragments. Then, the tubes were placed into a magnetic stand and the supernatants containing DNA fragments of interest were collected. DNA fragments were used directly for library preparation by performing joint end repair and poly A tailing, Truseq adapter ligation and PCR enrichment with the KAPA HiFi master mix (Roche) for 10–14 cycles. CUT&RUN libraries were then analyzed with a Nova-Seq 6000 sequencer (Illumina) using paired end sequencing.
CUT&RUN data processing
Reads were aligned to the mm10 genome assembly using Bowtie (Langmead et al., 2009) and peaks were called using MACS2 (Zhang et al., 2008). Peaks from both replicates were combined using bedtools (Quinlan and Hall, 2010) and only peaks that overlapped with ATAC-seq peaks were considered for downstream analysis (14,574 peaks). The peaks were annotated using HOMER (Heinz et al., 2010).
Multiome analysis
Isolated B cells from five pooled control or CD23.Cre.Smarca5fl/fl B1-8hi GFP+ donor animals were adoptively transferred (3–5 × 106) to ten recipient WT mice. After 4 h, the mice were injected subcutaneously with NP-KLH in alum into the hind footpad (10 µg per side) and intraperitoneally (25 µg per side). Inguinal and popliteal LNs were removed 5 days later, and cells from all ten recipient mice were sorted together as a single pooled sample. 7 × 104 GFP+ cells were sorted into FACS wash buffer, and nuclei were isolated using the Chromium Nuclei Isolation Kit (10x Genomics) according to the manufacturer’s instructions. Nuclei were counted using trypan blue and hemocytometer and imaged at 40× magnification to assess their quality. Nuclei were diluted in nuclei buffer supplemented with 1 mM DTT and 1 U/μl RNAse inhibitor to a final concentration of 3,700 nuclei/μl and immediately processed with the Chromium Next GEM Single Cell Multiome ATAC + Gene Expression kit (10x Genomics) according to the manufacturer’s protocol. Final libraries were quantified by qPCR with the NEB-next Library Quant Kit (New England Biolabs) and with Qubit and TapeStation. Sequencing was done with a Nova-Seq 6000 (Illumina) using two SP100 cycle kits (for the ATAC-seq library and the gene expression library), allocating 25,000 read pairs per nucleus for the ATAC-seq library and 20,000 read pairs per nucleus for gene expression libraries.
Multiome data processing
CellRanger-arc pipeline (Zheng et al., 2017; Satpathy et al., 2019) (v2.0.2; 10x Genomics) with default parameters was used for demultiplexing, alignment (mm10 reference genome, 2020-A version, downloaded from 10x website), filtering, barcode counting, peak calling, and counting of both ATAC and GEX molecules. Downstream analysis was done using Seurat (Hao et al., 2021) (v4.9) and Signac (Stuart et al., 2021) (v1.9) packages. First, each modality (ATAC or GEX) was analyzed separately. For ATAC data, cells with the percentage of reads in peaks below 40%, or with ATAC fragments below 1,000 or above 100,000 were filtered, as well as cells with nucleosome enrichment >4, transcriptional start site enrichment <2, or black list fraction >10%. Cellranger peaks of both KO and control samples were merged using the GenomicRanges package (Lawrence et al., 2013) in R, and the samples were integrated using the “FindIntegrationAnchors” function after normalization using the “RunTFIDF” function, and Data reduction using the “RunSVD” function. For GEX data, cells that had <500 UMIs, <250 or >6,500 genes, and >10% of mitochondrial reads were removed. GEX samples were integrated using the SCTransform workflow in Seurat (Lawrence et al., 2013; Hafemeister and Satija, 2019). Both ATAC and GEX data were integrated using the weighted nearest neighbor (WNN) method in Seurat. The WNN graph was used for UMAP visualization and clustering. Peaks were re-called using the “CallPeaks” function with group by cluster. A total of 79,332 peaks were identified. The peaks were exported in BED format and were annotated using HOMER. Differential accessible (DA) peaks between conditions were found using the “RunPresto” function from the “SeuratWrappers” package with default settings (test.use = “wilcox”). Peaks with avg_log2FC above 0.3 or below −0.3 and with FDR <0.1 were considered as DA. The “LinkPeaks” function with default settings was used to find peaks that are correlated with the expression of nearby genes. Overlap between ATAC seq peaks and SMARCA5 CUT&RUN peaks was evaluated using the GenomicRanges package. Specific genomics regions showing ATAC-seq coverage, peaks, and gene expression were plotted using the “CoveragePlot” function from the Signac package. Dot plots of specific genes were plotted using the “DotPlot” function from Seurat with custom-made modifications using the “ggplot” (Wickham, 2016) package and “ggnewscale” R package, version 0.4.3.
Statistical analysis
Statistical significance was determined using GraphPad Prism Version 10.0 using the statistical tests and number of repetitions indicated in each figure.
Online supplemental material
Fig. S1 shows an analysis of the biological processes in DZ and LZ B cells affected by ribosomal IP. Fig. S2 presents an extended analysis of the proliferation and viability of LPS-stimulated B cells in the absence of SMARCA5. Fig. S3 shows the effect of Smarca5 deficiency on the viability of resting and activated antigen-specific B cells 5 days after immunization. Fig. S4 exhibits an extended analysis of the chromatin differential accessibility in the absence of SMARCA5. Table S1 contains the list of translated genes (translatome) for DZ and LZ B cells.
Data availability
The data are available in the main article, its supplemental material, source data file, and in GEO dataset GSE261015.
Acknowledgments
Z. Shulman is supported by the European Research Council grant no. 101001613, Israel Science Foundation grant no. 1272/23, and Morris Kahn Institute for Human Immunology. Z. Shulman is a member of the European Molecular Biology Organization Young Investigator Program. T. Stopka received grant support from Grantová Agentura České Republiky (24-10435S, 24-10353S), Next Generation EU, and Programme EXCELES (LX22NPO5102).
Author contributions: L. Stoler-Barak: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Visualization, Writing—original draft, Writing—review & editing, D. Schmiedel: Investigation, Methodology, Writing—review & editing, A. Sarusi-Portuguez: Formal analysis, A. Rogel: Investigation, R. Blecher-Gonen: Investigation, Resources, Z. Haimon: Methodology, T. Stopka: Investigation, Z. Shulman: Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing—original draft, Writing—review & editing.

