


Impaired pulmonary angiogenesis plays a pivotal role in the progression of pulmonary arterial hypertension (PAH) and patient mortality, yet the molecular mechanisms driving this process remain enigmatic. Our study uncovered a striking connection between mitochondrial dysfunction (MD), caused by a humanized mutation in the NFU1 gene, and severely disrupted pulmonary angiogenesis in adult lungs. Restoring the bioavailability of the NFU1 downstream target, lipoic acid (LA), alleviated MD and angiogenic deficiency and rescued the progressive PAH phenotype in the NFU1G206C model. Notably, significant NFU1 expression and signaling insufficiencies were also identified in idiopathic PAH (iPAH) patients’ lungs, emphasizing this study’s relevance beyond NFU1 mutation cases. The remarkable improvement in mitochondrial function of PAH patient-derived pulmonary artery endothelial cells (PAECs) following LA supplementation introduces LA as a potential therapeutic approach. In conclusion, this study unveils a novel role for MD in dysregulated pulmonary angiogenesis and PAH manifestation, emphasizing the need to correct MD in PAH patients with unrecognized NFU1/LA deficiency.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive life-threatening disease. The uncontrolled growth of pulmonary vascular cells and subsequent vascular remodeling is a primary cause of elevated pulmonary vascular resistance (PVR) and right ventricle (RV) dysfunction, ultimately leading to the patient’s death (Rabinovitch, 2012; Wilkins, 2012). The research spanning basic, translational, and clinical studies has consistently identified impaired mitochondrial function as a hallmark feature in both preclinical and clinical PAH (Archer et al., 2010a; Paulin and Michelakis, 2014; Rafikov et al., 2015; Paulin et al., 2014; Sproule et al., 2008). However, despite this accumulating evidence, the causative link between mitochondrial dysfunction (MD) and PAH was not established. Our team has created a new rat model with a human point mutation in the NFU1 protein (NFU1G206C) (Niihori et al., 2020). NFU1 is an iron–sulfur cluster scaffold protein that assembles and transfers 4Fe–4S clusters to the target proteins, such as succinate dehydrogenase (SDH) and lipoyl synthase (LAS) (Navarro-Sastre et al., 2011; Ahting et al., 2015). As the adequate activity of LAS is required to ensure a sufficient supply of lipoic acid (LA), a critical cofactor for multiple mitochondrial enzymes, LAS inhibition secondary to NFU1 insufficiency severely disrupts mitochondrial homeostasis. In particular, it was described that it affects the activities of pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase, proteins of the glycine cleavage system (GCS), and branched-chain α-ketoacid dehydrogenase (Navarro-Sastre et al., 2011; Solmonson and DeBerardinis, 2018). Together with the directly impaired activity of SDH, the enzyme that participates in both the tricarboxylic acid cycle and the electron transport chain, the inhibitory NFU1 mutation leads to severe mitochondrial disease and multiple MD syndrome 1 described in human carriers (Navarro-Sastre et al., 2011; Ahting et al., 2015; Kollberg et al., 2009; Legati et al., 2017; Mochel et al., 2008). Moreover, 70% of patients with NFU1 mutation develop PAH (Navarro-Sastre et al., 2011), which is 14,000–70,000 times more often compared with a rate reported for the general population (10–50 cases per million) (Ruopp and Cockrill, 2022; Waxman and Zamanian, 2013). Introducing this mutation in the rat genome has also initiated a spontaneous PAH phenotype in otherwise healthy Sprague–Dawley (SD) rats (Niihori et al., 2020). Aside from increased RV systolic pressure (RVSP) and RV hypertrophy, this model is characterized by severe pulmonary vascular remodeling and vaso-obliterative disease. Examination of isolated pulmonary artery (PA) smooth muscle cells (PASMCs) from NFU1 rats revealed a metabolic shift favoring enhanced glycolysis, resistance to apoptosis, and a markedly proliferative phenotype (James et al., 2021). These previous discoveries suggested the causative role of MD in PAH, although the specific mechanisms responsible for the MD-mediated PAH manifestation remained unclear.
Angiogenesis, or the formation of new blood vessels from preformed vessels, is responsible for the 35-fold expansion of pulmonary capillary volume between birth and adulthood (Gao et al., 2016) and also increases vessel length and branching in adult lungs in response to stimulation (Hopkins and McLoughlin, 2002). In PAH, however, angiogenesis is described as impaired or dysfunctional (Tuder and Voelkel, 2002; Humbert et al., 2019; Yuan et al., 2019). Despite increased proliferative potential and apoptosis resistance, PA endothelial cells (PAECs) isolated from PAH patients or animals have less order branching network (Masri et al., 2005), decreased migration capacity (Tielemans et al., 2018), and severe angiogenic defects confirmed in vitro and in vivo (Vattulainen-Collanus et al., 2016). Furthermore, the reduced density of the distal pulmonary vessels is a typical radiological manifestation of clinical PAH (Moledina et al., 2011), which correlates with increased PA pressure and RV dysfunction, as demonstrated by right heart catheterization and cardiac magnetic resonance imaging (Synn et al., 2021b). Overexpression of angiogenic factors, in contrast, prevents PAH initiation or reverses established PAH (Campbell et al., 2001; Miao et al., 2021; Zhao et al., 2006). Thus, the ongoing loss of angiogenic capacity throughout the disease represents one of the central pathogenic events in PAH responsible for disease progression and patient mortality (Moledina et al., 2011; Synn et al., 2021a). However, while the contribution of inadequate angiogenesis to PAH is confirmed, the initiating mechanisms driving angiogenic deficiency remain not fully defined (Voelkel et al., 2007).
Thus, the disturbances in pulmonary angiogenesis were never directly connected to an impaired mitochondrial function, mainly because the metabolism of endothelial cells (ECs) is known to rely on glycolysis. However, this is relevant for the quiescent EC only (Coutelle et al., 2014). In contrast, growing ECs increasingly depend on mitochondrial respiration to meet their elevated energy demands. To address this knowledge gap and investigate whether MD impacts pulmonary angiogenesis in PAH, we used the NFU1G206C rat model. Rats with MD were compared with wild types (WTs) to evaluate progressive changes in the pulmonary vasculature and capillary network and analyze the angiogenic capacity of PAEC isolated from both genotypes. Our results demonstrate that rats with MD exhibit a significantly reduced complexity of the pulmonary vascular morphology with a severe loss of small PAs, decreased arterial branching, reduced capillary density, and diminished overall angiogenic capacity of pulmonary ECs (PECs). Notably, improved mitochondrial function following LA supplementation was sufficient to rescue the deficiencies in vascular morphology and attenuate the PAH phenotype. Furthermore, we uncovered the presence of NFU1 insufficiency in the pulmonary tissue of idiopathic PAH (iPAH) patients and the potential of LA to rescue the MD in PAECs from human PAH lungs. Collectively, these findings suggest the high relevance of the discovered mechanisms to PAH patients with and without NFU1 mutation. As the level of vascular pruning is linked to PAH severity and patient mortality (Moledina et al., 2011; Synn et al., 2021a), the intervention that could preserve adequate pulmonary angiogenesis holds great promise for improving patient outcomes.
Results
NFU1G206C mutation induces a spontaneous and progressive PAH phenotype
The assessment of rat hemodynamics revealed that rats with the mutation in the NFU1 protein (NFU1G206C rats) develop a spontaneous increase in RVSP (Fig. 1 A) at the age of 6 wk, with a further progression in 10-wk-old rats. These changes were accompanied by an increased RV dP/dtmax, as a measure of RV contractility (Fig. 1 B), and decreased RV dP/dtmin, a marker of RV relaxation (Fig. 1 C), both reaching significance by 10 wk. In contrast to the pressure, significant RV hypertrophy (RV/ left ventricle + septum [LV+S] ratio) was observed in both younger and older NFU1G206C rats (Fig. 1 D), suggesting the direct contribution of MD to RV hypertrophy. The PAH phenotype was also characterized by severe remodeling of small PAs (Fig. 1, E–G), also persisting starting from 6 wk of age. The extent of PA remodeling was comparable with an advanced Sugen/Hypoxia (Su/Hx) model with RVSP ∼100 mmHg (Rafikova et al., 2015; Rafikov et al., 2019).

Rats with NFU1 G206C mutation develop a spontaneous and progressive PAH phenotype. At the age of 6 or 10 wk, WT and NFU1G206C rats were subjected to the non-survival hemodynamic assessment using a Millar pressure transducer catheter inserted into the RV and histological evaluation of pulmonary tissues. (A–C) Changes in pulmonary hemodynamics were evaluated using RVSP (A), RV dP/dt max as a measure of cardiac contractility (B), and dP/dt min as a measure of RV relaxation (C). (D) RV hypertrophy was assessed by the Fulton index, or the ratio between the wet weight of the RV and the LV+S. Compiled from three independent experiments with a total of 10 rats per group. (E) Small PA morphometry in H&E-stained pulmonary tissues was quantified in 10 random transversely sectioned PAs by an investigator unaware of group assignment. (F) Vascular occlusion score was defined as (total vessel area – lumen area)/total vessel area × 100. (G) Media thickness was quantified as an average of four measurements of media size in the same vessel. Scale bar = 100 µm. Panels A–D are compiled from three independent experiments with N = 10 rats per group. Panels E–G are compiled from two independent experiments with N = 5 rats per group. For all panels, the statistical analysis was performed using one-way ANOVA followed by Bonferroni post hoc multiple comparison test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
Rats with NFU1 G206C mutation develop a spontaneous and progressive PAH phenotype. At the age of 6 or 10 wk, WT and NFU1G206C rats were subjected to the non-survival hemodynamic assessment using a Millar pressure transducer catheter inserted into the RV and histological evaluation of pulmonary tissues. (A–C) Changes in pulmonary hemodynamics were evaluated using RVSP (A), RV dP/dt max as a measure of cardiac contractility (B), and dP/dt min as a measure of RV relaxation (C). (D) RV hypertrophy was assessed by the Fulton index, or the ratio between the wet weight of the RV and the LV+S. Compiled from three independent experiments with a total of 10 rats per group. (E) Small PA morphometry in H&E-stained pulmonary tissues was quantified in 10 random transversely sectioned PAs by an investigator unaware of group assignment. (F) Vascular occlusion score was defined as (total vessel area – lumen area)/total vessel area × 100. (G) Media thickness was quantified as an average of four measurements of media size in the same vessel. Scale bar = 100 µm. Panels A–D are compiled from three independent experiments with N = 10 rats per group. Panels E–G are compiled from two independent experiments with N = 5 rats per group. For all panels, the statistical analysis was performed using one-way ANOVA followed by Bonferroni post hoc multiple comparison test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
Impaired mitochondrial function in NFU1G206C PECs severely alters pulmonary vascular morphology, replicating the main pathogenic features of patient PAH
Our previous studies discovered a significant reduction in mitochondrial respiration in PASMCs isolated from NFU1G206C rats (James et al., 2021). In this study, we investigated the functionality of mitochondria in PECs, the critical contributors to angiogenesis. The Seahorse analysis revealed that PECs from 10-wk-old NFU1G206C rats had a significantly lower rate of basal and maximal respiration (Fig. 2, A–C) and the attenuated levels of ATP production (Fig. 2 D) compared to cells from the age-matched WTs, which was compensated by a significant upregulation of glycolysis (Fig. S1). In addition, by assessment of mitochondrial morphology, it was discovered that NFU1 mutation disrupted the normal mitochondrial network and induced mitochondrial accumulation in the perinuclear area (Fig. 2 E). This recruitment of mitochondria to the nucleus is a known response to impaired mitochondrial function aimed to restore mitochondrial homeostasis by better regulation of mitochondrial activities controlled by the nucleus (Eisenberg-Bord and Schuldiner, 2017). Measuring the balance between two dynamin-related protein 1 (Drp1) phosphorylation sites—Ser616, which promotes Drp1-mediated mitochondrial fission (Fig. 2 F), and Ser637, which reverses this process (Fig. 2 G), showed that both were significantly elevated in NFU1G206C versus WT PECs, although the intensity of the signal induced mitochondrial fragmentation was higher. Assessing the levels of peroxisome proliferator-activated receptor γ coactivator 1 α (PGC-1α) provided no evidence of impaired mitochondrial biogenesis in the NFU1 model (Fig. 2 H). Taken together, these results confirm a severely impaired mitochondrial respiration in NFU1G206C PECs, which appear to be due to the functional changes in already preexisting mitochondria and an imbalanced mitochondrial fission/fusion process shifted toward mitochondrial fragmentation. This outcome provides a solid rationale for studying whether an impaired MD in NFU1G206C PECs affects their angiogenic capacity and contributes to developing a spontaneous PAH phenotype.
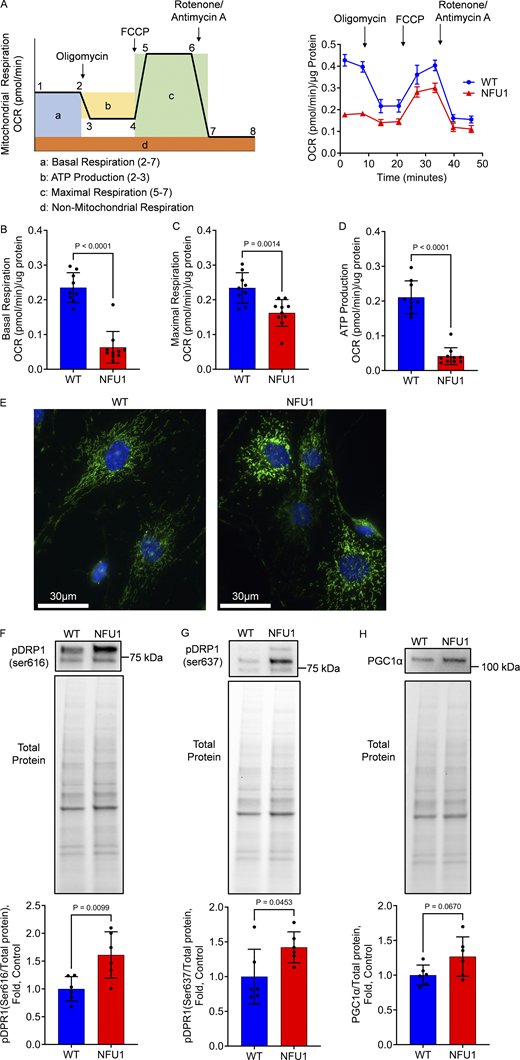
PECs isolated from NFU1G206Crats show impaired mitochondrial function and mitochondrial morphology.(A) The mitochondrial respiration of PECs isolated from 10-wk-old WT and NFU1G206C rats was evaluated by a standard mito-stress Seahorse assay (A, left schematic graph depicturing the curve generated by Seahorse XF Cell Mito Stress Test, and the analyzed parameters of mitochondrial function: basal and maximal respiration, and ATP production. OCR - oxygen consumption rate; right graph represents the original mitostress test profile observed in PAECs isolated from WT and NFU1G206C rats). (B–D) NFU1G206C genotype is associated with a consistently reduced basal (B) and maximal (C) mitochondrial respiration and attenuated ATP production rate (D). (E) To assess the changes in mitochondrial morphology, the isolated PECs were stained with antibodies against the mitochondrial marker mitoNEET located on the outer mitochondrial membrane. (F) The observed fractionation of the mitochondria in NFU1G206C PECs was confirmed by an increased signal from phospo-DRP1 at Ser616, controlling mitochondrial fission. (G) This effect was accompanied by a slight increase in the compensatory phosphorylation of DRP1 at Ser637, which inhibits mitochondrial division. (H) Mitochondrial biogenesis controlled by PGC1α remained unaffected. Panels A–D: N = 9 and 10 for WT and NFU1 groups, correspondingly. Panel E represents at least 100 random fields imaged per each experimental group. Panels F–H: N = 6/group. All panels are representative of two independent experiments. For all panels, the analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data in A are presented as mean ± SEM. All the rest data are shown as mean ± SD. Source data are available for this figure: SourceData F2.
PECs isolated from NFU1G206Crats show impaired mitochondrial function and mitochondrial morphology.(A) The mitochondrial respiration of PECs isolated from 10-wk-old WT and NFU1G206C rats was evaluated by a standard mito-stress Seahorse assay (A, left schematic graph depicturing the curve generated by Seahorse XF Cell Mito Stress Test, and the analyzed parameters of mitochondrial function: basal and maximal respiration, and ATP production. OCR - oxygen consumption rate; right graph represents the original mitostress test profile observed in PAECs isolated from WT and NFU1G206C rats). (B–D) NFU1G206C genotype is associated with a consistently reduced basal (B) and maximal (C) mitochondrial respiration and attenuated ATP production rate (D). (E) To assess the changes in mitochondrial morphology, the isolated PECs were stained with antibodies against the mitochondrial marker mitoNEET located on the outer mitochondrial membrane. (F) The observed fractionation of the mitochondria in NFU1G206C PECs was confirmed by an increased signal from phospo-DRP1 at Ser616, controlling mitochondrial fission. (G) This effect was accompanied by a slight increase in the compensatory phosphorylation of DRP1 at Ser637, which inhibits mitochondrial division. (H) Mitochondrial biogenesis controlled by PGC1α remained unaffected. Panels A–D: N = 9 and 10 for WT and NFU1 groups, correspondingly. Panel E represents at least 100 random fields imaged per each experimental group. Panels F–H: N = 6/group. All panels are representative of two independent experiments. For all panels, the analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data in A are presented as mean ± SEM. All the rest data are shown as mean ± SD. Source data are available for this figure: SourceData F2.

The severe glycolytic shift in NFU1 PEC s is efficiently attenuated by LA supplementation. (A–C) PECs isolated from 10-wk-old WT and untreated and LA-treated NFU1G206C rats were used in Seahorse XF glycolysis stress test (A). The glycolysis (Extracellular acidification rate [ECAR] after adding the saturating amounts of glucose, B) and glycolytic capacity (maximum ECAR after shutting down mitochondrial respiration by oligomycin, C) were quantified for each experimental group. For all panels, N = 20–28 per group. Representative of two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SEM.
The severe glycolytic shift in NFU1 PEC s is efficiently attenuated by LA supplementation. (A–C) PECs isolated from 10-wk-old WT and untreated and LA-treated NFU1G206C rats were used in Seahorse XF glycolysis stress test (A). The glycolysis (Extracellular acidification rate [ECAR] after adding the saturating amounts of glucose, B) and glycolytic capacity (maximum ECAR after shutting down mitochondrial respiration by oligomycin, C) were quantified for each experimental group. For all panels, N = 20–28 per group. Representative of two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SEM.
The structural analysis of the pulmonary vascular tree by three-dimensional high-resolution micro computed tomography (micro-CT) revealed that the WT rats undergo an age-related increase in the pulmonary vascular network (Fig. 3 A). These findings support previous reports demonstrating ongoing angiogenesis in postnatal and adult lungs (Razavi et al., 2012). In contrast, the NFU1G206C genotype exhibited a severely impaired ability to form new vessels, particularly evident in 10-wk-old rats (Fig. 3 A), which reproduces the diminished density of the distal pulmonary vessels observed in clinical PAH (Moledina et al., 2011) and associated with an increased risk of patient death (Synn et al., 2021a; Moledina et al., 2011). Alongside impaired angiogenesis, the “dead-tree” pulmonary vasculature in PAH patients can also be attributed to vascular remodeling. Indeed, the enlarged micro-CT scans of the NFU1G206C rats demonstrated partial or complete vaso-obliteration, visible as extra-slim contrast sections or complete breaks in PA integrity in 10-wk-old NFU1G206C animals (Fig. 3 A, red arrows). These findings confirm the correlation between vascular loss and PA remodeling.

The deficient morphology of pulmonary vascular tree in NFU1 G206C rats. The pulmonary vasculature of 6- and 10-wk-old WT and NFU1G206C rats filled with Microfill compound was scanned on an Inveon micro-CT scanner (440 projections converted into a 360° rotation view or every 10° image) to reveal the morphology of the pulmonary vascular tree. (A) Representative micro-CT scans demonstrate the progressive increase in the complexity of the pulmonary vascular network in elder WTs, which is absent in 10-wk-old NFU1 mutant rats (A, scale bars in original scans are 5 mm). The enlarged areas corresponded to the areas inside the red square (scale bar = 1 mm), confirming the difference in the distal PA network across the groups and the presence of vasoobliterative disease in the 10-wk-old NFU1G206C rats (red arrows). (B–D) The custom-built MATLAB code used to analyze the density of the entire pulmonary vasculature (B), or the separated images of the large vessels with a diameter above 49.5 µm (corresponding to the branches of 1–4 order, C), and the PAs with a diameter between 49.5 and 9 µm (corresponding to the branches of 5–9 order, D) revealed a significant difference in peripheral PA density of 10-wk-old rats. (E–J) The length of the right middle lobar artery, segmented from the surrounding vasculature (E, scale bar = 1 mm), and the changes in the luminal cross-sectional diameter measured every 1 mm (F) and plotted as a function of distance from the branching point (G–J) were used to characterize the morphological changes in the specific PA. For all panels, N = 5/group is compiled from two independent experiments. A comparison between four animal groups (A–E) was done using one-way ANOVA followed by a Bonferroni post-hoc multiple comparison test. Two group analysis (G–J) was performed using the Student’s t test. For all panels, P < 0.05 was considered significant. Data are presented as mean ± SD.
The deficient morphology of pulmonary vascular tree in NFU1 G206C rats. The pulmonary vasculature of 6- and 10-wk-old WT and NFU1G206C rats filled with Microfill compound was scanned on an Inveon micro-CT scanner (440 projections converted into a 360° rotation view or every 10° image) to reveal the morphology of the pulmonary vascular tree. (A) Representative micro-CT scans demonstrate the progressive increase in the complexity of the pulmonary vascular network in elder WTs, which is absent in 10-wk-old NFU1 mutant rats (A, scale bars in original scans are 5 mm). The enlarged areas corresponded to the areas inside the red square (scale bar = 1 mm), confirming the difference in the distal PA network across the groups and the presence of vasoobliterative disease in the 10-wk-old NFU1G206C rats (red arrows). (B–D) The custom-built MATLAB code used to analyze the density of the entire pulmonary vasculature (B), or the separated images of the large vessels with a diameter above 49.5 µm (corresponding to the branches of 1–4 order, C), and the PAs with a diameter between 49.5 and 9 µm (corresponding to the branches of 5–9 order, D) revealed a significant difference in peripheral PA density of 10-wk-old rats. (E–J) The length of the right middle lobar artery, segmented from the surrounding vasculature (E, scale bar = 1 mm), and the changes in the luminal cross-sectional diameter measured every 1 mm (F) and plotted as a function of distance from the branching point (G–J) were used to characterize the morphological changes in the specific PA. For all panels, N = 5/group is compiled from two independent experiments. A comparison between four animal groups (A–E) was done using one-way ANOVA followed by a Bonferroni post-hoc multiple comparison test. Two group analysis (G–J) was performed using the Student’s t test. For all panels, P < 0.05 was considered significant. Data are presented as mean ± SD.
To quantify the changes in the pulmonary arterial bed of the NFU1 mutant rats, we established a method of measuring PA density using a custom-built MATLAB code. The quantitative analysis of the total PA density (Fig. 3 B) confirmed the progressive increase in pulmonary vascular complexity of elder WTs compared to the younger rats, while NFU1 mutants showed a severely decreased pulmonary PA density with no progression in pulmonary vascular complexity upon aging. To separate the contribution of the large and small PAs to the total PA density, we repeated the same analysis in the large vessels with a diameter above 49.5 µm (which represented the PAs of 1–4 order, Fig. 3 C) and small vessels with a diameter between 49.5 and 9 µm, the smallest size visible on the micro-CT scan (Fig. 3 D). This approach confirmed that the difference in the PA network comes from the small vessels, while the large vessels showed a comparable morphology across all the animal groups.
For a more comprehensive evaluation of micro-CT images, we measured the length of the right middle lobar artery. In WT rats, this artery was visible and quantifiable (i.e., contrast-permeable) throughout the entire lobe (Fig. 3 E). Conversely, the NFU1G206C rats exhibited an early decline in contrast flow. The luminal cross-sectional area of the right middle lobar artery was evaluated as a function of distance from the branching point (Fig. 3 F). The ongoing pulmonary angiogenesis in WT rats correlated with increased artery length and diameter in the 10-wk-old animals (Fig. 3 G). In contrast, the morphology of the same artery in NFU1G206C rats showed minimal age-related changes (Fig. 3 H). Within the same age group, the NFU1G206C animals showed a significant vascular deficiency, particularly in the elderly rats (Fig. 3, I and J). Furthermore, the steeper slope of the length/diameter curves in the NFU1G206C rats suggests that the altered pulmonary morphology associated with the NFU1G206C genotype results from co-occurrence of impaired angiogenesis and vascular remodeling.
After evaluating the changes in the entire pulmonary vasculature, we examined the density and morphology of small PAs and capillaries. The pulmonary microangiography confirmed the age-dependent increase in the complexity of peripheral pulmonary vasculature in WTs and the absence of microvascular expansion in NFU1G206C rats (Fig. 4 A). In contrast to the larger scale angiogenic assessment based on CT scans, the analysis of microvasculature showed angiogenic defects in NFU1G206C rats as early as 6 wk after birth, which continued to persist with aging. Thus, the number of vascular brunches (Fig. 4 B) and junctions (Fig. 4 C), as well as the total vascular length (Fig. 4 D), were significantly reduced in either 6- or 10-wk-old NFU1G206C rats compared to the age-matched WTs. In addition, the histological analysis of lungs stained with Griffonia simplicifolia isolectin B4 (GS-IB4), a lectin specific for rodent microvascular endothelium (Allen et al., 2015; King et al., 2004), showed that 10-wk-old NFU1G206C rats also have a significantly reduced pulmonary capillary network (Fig. 4 E).

NFU1 G206C genotype impairs pulmonary microvascular density and homeostasis. The complexity and network of pulmonary microvasculature in 6- and 10-wk-old WT and NFU1G206C rats were visualized by microangiography of peripheral PA. (A) Representative microangiography images show the age-dependent progression in the vascular network of distal arteries in WTs and deficient microvasculature of NFU1G206C rats (scale bar for the original top images = 1 mm). The enlarged areas (scale bar = 0.5 mm), which correspond to the areas inside the red squares, were quantitively assessed by manually tracing the vessels and converting them into binary images. (B–D) The number of arterial branches (B) and junctions (C) and the total length of the arterial segments (D) within the given area (0.5 mm of the peripheral vessels along the lobe) were compared between the groups. Histological assessment of the pulmonary capillary density was performed by staining paraffinized lung tissue slides with GS-IB4, a lectin specific for rodent microvascular endothelium (Allen et al., 2015). (E) Representative images from 10-wk-old rats show a loss of pulmonary capillaries in NFU1 mutant rodents (bar scales for original and enlarged images are 150 and 50 µm, correspondingly), which was confirmed by counting the number of capillaries in 10 random fields per animal, performed in a blinded manner. (F–I) For the endothelial/smooth muscle cell co-staining, the lung tissues of WT and NFU1G206C rats were treated with anti-vWF, anti-αSMA antibodies, and DAPI staining (F, scale bar = 50 µm), and 20 random fields per animal were used to quantify the vWF/DAPI (G), αSMA/DAPI (H), or vWF/αSMA (I) ratios. Homeostasis of microvasculature was compared between the genotypes by measuring the level of endothelial–pericyte interaction. 10-µm-thick pulmonary slides were stained with GS-IB4 (green), the antibodies against the NG2 proteoglycan, a specific marker of pericytes (red), and DAPI nuclear stain (blue). (J) Representative 3D confocal images show the presence of a tight interaction between these two cell types in the 10-wk-old WT rats and the loss of direct contacts between capillary endothelium and pericytes in the age-matched NFU1G206C rats (scale bar = 3 µm). (K) The quantitative evaluation of this cell–cell communication, assessed as a yellow area resulted from an overlap between the green and red signal and normalized per number of nuclei in the field, confirmed a drastic loss of interaction between the microvascular endothelium and mural cells (scale bar = 100 µm). Panels A–K: N = 5/group; panel E: N = 4/group; panels F–K: N = 6/group. All panels are compiled from two to three independent experiments. A comparison between four animal groups (B–D) was performed using one-way ANOVA followed by a Bonferroni post-hoc multiple comparison test. Two groups (E–K) were analyzed using the Student’s t test. For all panels, P < 0.05 was considered significant. Data are presented as mean ± SD.
NFU1 G206C genotype impairs pulmonary microvascular density and homeostasis. The complexity and network of pulmonary microvasculature in 6- and 10-wk-old WT and NFU1G206C rats were visualized by microangiography of peripheral PA. (A) Representative microangiography images show the age-dependent progression in the vascular network of distal arteries in WTs and deficient microvasculature of NFU1G206C rats (scale bar for the original top images = 1 mm). The enlarged areas (scale bar = 0.5 mm), which correspond to the areas inside the red squares, were quantitively assessed by manually tracing the vessels and converting them into binary images. (B–D) The number of arterial branches (B) and junctions (C) and the total length of the arterial segments (D) within the given area (0.5 mm of the peripheral vessels along the lobe) were compared between the groups. Histological assessment of the pulmonary capillary density was performed by staining paraffinized lung tissue slides with GS-IB4, a lectin specific for rodent microvascular endothelium (Allen et al., 2015). (E) Representative images from 10-wk-old rats show a loss of pulmonary capillaries in NFU1 mutant rodents (bar scales for original and enlarged images are 150 and 50 µm, correspondingly), which was confirmed by counting the number of capillaries in 10 random fields per animal, performed in a blinded manner. (F–I) For the endothelial/smooth muscle cell co-staining, the lung tissues of WT and NFU1G206C rats were treated with anti-vWF, anti-αSMA antibodies, and DAPI staining (F, scale bar = 50 µm), and 20 random fields per animal were used to quantify the vWF/DAPI (G), αSMA/DAPI (H), or vWF/αSMA (I) ratios. Homeostasis of microvasculature was compared between the genotypes by measuring the level of endothelial–pericyte interaction. 10-µm-thick pulmonary slides were stained with GS-IB4 (green), the antibodies against the NG2 proteoglycan, a specific marker of pericytes (red), and DAPI nuclear stain (blue). (J) Representative 3D confocal images show the presence of a tight interaction between these two cell types in the 10-wk-old WT rats and the loss of direct contacts between capillary endothelium and pericytes in the age-matched NFU1G206C rats (scale bar = 3 µm). (K) The quantitative evaluation of this cell–cell communication, assessed as a yellow area resulted from an overlap between the green and red signal and normalized per number of nuclei in the field, confirmed a drastic loss of interaction between the microvascular endothelium and mural cells (scale bar = 100 µm). Panels A–K: N = 5/group; panel E: N = 4/group; panels F–K: N = 6/group. All panels are compiled from two to three independent experiments. A comparison between four animal groups (B–D) was performed using one-way ANOVA followed by a Bonferroni post-hoc multiple comparison test. Two groups (E–K) were analyzed using the Student’s t test. For all panels, P < 0.05 was considered significant. Data are presented as mean ± SD.
To investigate whether the PH phenotype in NFU1G206C rats is associated with impaired microvascular density, vascular remodeling, or both, we also co-stained lung tissues with the von Willebrand Factor (vWF), produced exclusively by endothelium (Steffes et al., 2022), and αSMA to stain PASMCs (Fig. 4 F), and quantified each signal (Fig. 4, G and H) or their ratio (Fig. 4 I). The results confirmed the co-presence of both angiogenic deficiency and media hypertrophy of the developed small PAs.
ECs require direct interaction with pericytes to facilitate blood vessel formation. Pericytes, which are adventitial cells located in the capillary basement membrane, play a critical role in guiding capillary sprouts and maintaining vessel stability and maturity (Stapor et al., 2014; Bergers and Song, 2005). To evaluate the effectiveness of pericyte–endothelial interaction in the NFU1G206C model, we performed a co-staining of pulmonary tissues with Griffonia and NG2, a commonly used pericyte marker (Stallcup, 2018). The 3D confocal imaging showed a tight interaction between endothelial and pericyte cells in WTs, with direct interdigitations protruding from pericytes into ECs. In contrast, NFU1G206C rats showed a noticeable separation between endothelial and pericyte cells (Fig. 4 J). Quantitative analysis of this interaction, assessed as the intensity of the yellow signal resulting from the overlap between green Griffonia and red NG2 staining, confirmed a significant drop in endothelial/pericyte association in the NFU1G206C rats (Fig. 4 K).
The restored LA bioavailability is sufficient to preserve pulmonary vascular morphology and angiogenic capacity and represents a potentially beneficial approach for PAH patients with an impaired NFU1/LA-PDH axis
The mutation in the NFU1 gene impairs the ability of the NFU1 protein to transfer iron-sulfur clusters, leading to the compromised activity of LAS, one of its known targets. This, in turn, affects the functioning of several crucial mitochondrial proteins that rely on LA as a co-factor. To investigate if restoring LA bioavailability would be sufficient to improve mitochondrial function and alleviate angiogenic deficiencies in NFU1 rats, we conducted a long-term supplementation of LA to NFU1G206C rats beginning during prenatal development and continuing into adulthood (until 10 wk of age, Fig. 5 A). The increased LA bioavailability upon its supplementation significantly elevated pulmonary and PEC PDH lipoylation impaired in this model (Niihori et al., 2020) (Fig. 5, B and C). Analysis of mitochondrial function in PECs isolated from LA-treated and untreated NFU1G206C rats showed significant improvements in basal and maximal respiration and increased ATP production (Fig. 5, D–G). This effect was accompanied by a normalized glycolytic rate in LA-treated NFU1G206C PECs (Fig. S1). LA supplementation has also preserved mitochondrial morphology (Fig. 5 H), decreased the pDRP1(Ser 616) signal that initiates mitochondrial fission (Fig. 5 I), and increased the levels of pDRP1(Ser 637) that inhibits mitochondrial fragmentation (Fig. 5 J). The mitochondrial biogenesis was not affected by either NFU1 mutation or LA treatment (Fig. 5 K).
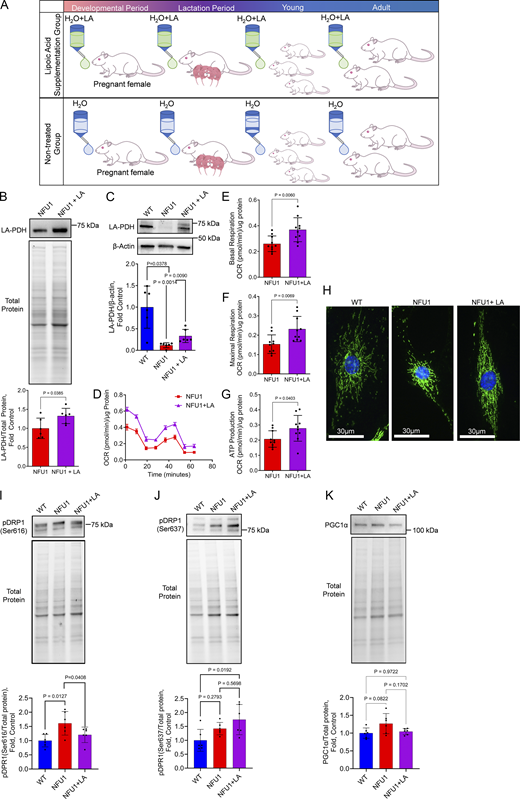
Chronic LA supplementation restores mitochondrial function in NFU1G206CPECs. Inhibitory mutation in the NFU1 protein affects LA synthesis and the activity of LA-dependent mitochondrial enzymes. (A) To investigate where the restored LA bioavailability will be sufficient to prevent MD and spontaneous PAH phenotype, the LA-containing water (1.70 mM) was supplied to pregnant and lactating mothers and then to the pups until 10 wk of age when the animals were instrumented to measure pulmonary hemodynamics. (B and C) This LA treatment was sufficient to significantly increase the level of PDH lipoylation in the lungs or isolated PECs. (D–J) PECs isolated from LA-supplied NFU1G206C rats showed a significant improvement in mitochondrial function compared to untreated NFU1 mutants (D), with basal (E) and maximal (F) respiration and ATP production rate (G) being significantly elevated. LA supplementation has also preserved the mitochondrial morphology of PECs (H), decreased mitochondrial fission signaling (pSer616DRP1, I), and increased the pSer637DRP1 signal inhibiting mitochondrial fractionation (J). (K) Mitochondrial biogenesis was not affected by the LA (K). Panels B and C: N = 6/group; panels D–G: N = 10/group; H represents at least 100 random fields imaged per each experimental group; panels I–K: N = 6/group; the WT and NFU1 data were obtained from Fig. 2, F–H with addition of NFU1+LA group. All panels represent two independent experiments. Statistical analysis was performed using the Student’s t test (B and D–F) or one-way ANOVA followed by a Bonferroni post-hoc multiple comparison test (C and I–K). NFU1 versus NFU1+LA in C were compared by t test; in I, by one-way ANOVA selected columns (Bonferroni). Data in D are presented as mean ± SEM. For the rest of the panels, data are presented as mean ± SD. P < 0.05 considered significant. Source data are available for this figure: SourceData F5.
Chronic LA supplementation restores mitochondrial function in NFU1G206CPECs. Inhibitory mutation in the NFU1 protein affects LA synthesis and the activity of LA-dependent mitochondrial enzymes. (A) To investigate where the restored LA bioavailability will be sufficient to prevent MD and spontaneous PAH phenotype, the LA-containing water (1.70 mM) was supplied to pregnant and lactating mothers and then to the pups until 10 wk of age when the animals were instrumented to measure pulmonary hemodynamics. (B and C) This LA treatment was sufficient to significantly increase the level of PDH lipoylation in the lungs or isolated PECs. (D–J) PECs isolated from LA-supplied NFU1G206C rats showed a significant improvement in mitochondrial function compared to untreated NFU1 mutants (D), with basal (E) and maximal (F) respiration and ATP production rate (G) being significantly elevated. LA supplementation has also preserved the mitochondrial morphology of PECs (H), decreased mitochondrial fission signaling (pSer616DRP1, I), and increased the pSer637DRP1 signal inhibiting mitochondrial fractionation (J). (K) Mitochondrial biogenesis was not affected by the LA (K). Panels B and C: N = 6/group; panels D–G: N = 10/group; H represents at least 100 random fields imaged per each experimental group; panels I–K: N = 6/group; the WT and NFU1 data were obtained from Fig. 2, F–H with addition of NFU1+LA group. All panels represent two independent experiments. Statistical analysis was performed using the Student’s t test (B and D–F) or one-way ANOVA followed by a Bonferroni post-hoc multiple comparison test (C and I–K). NFU1 versus NFU1+LA in C were compared by t test; in I, by one-way ANOVA selected columns (Bonferroni). Data in D are presented as mean ± SEM. For the rest of the panels, data are presented as mean ± SD. P < 0.05 considered significant. Source data are available for this figure: SourceData F5.
After confirming that LA supplementation improves mitochondrial function in PECs, we investigated whether LA could prevent angiogenic deficiency in the lungs. Micro-CT scans of LA-supplied NFU1G206C rats showed a drastic difference in the morphology of the pulmonary vasculature compared to untreated NFU1G206C rats, with the fully preserved complexity of the vascular network and no evidence of vaso-obliterative events (Fig. 6 A). Quantitative analysis confirmed that LA supplementation did not affect total (Fig. 6 B) or large (Fig. 6 C) vessel density. However, it effectively prevented the loss of small PAs (Fig. 6 D). LA treatment also normalized the length and diameter of the right middle lobar artery (Fig. 6, E and F) and preserved the complexity of peripheral PAs (Fig. 6, H–J). Furthermore, the restored bioavailability of LA significantly improved the interaction between capillary endothelium and pericytes (Fig. 6, K and L). These findings suggest that disrupted mitochondrial function is critical in altering the pulmonary endothelial landscape.

Increased LA bioavailability is sufficient to prevent angiogenic defects in NFU1 G206C rats. (A) Representative micro-CT scans of LA-treated and untreated NFU1G206C rats demonstrate a radical difference in the pulmonary vascular network. The deficient pulmonary vascular morphology and obliteration of small PAs (red arrows inside the enlarged areas) associated with the NFU1G206C genotype do not manifest in NFU1 mutant rats supplied with LA. (B–D) The custom-built MATLAB code was used to quantify the density of the entire pulmonary vascular tree (B), and vessels separated on the large (>49.5 µm, which correspond to the branches of 1–4 order, C) and smaller vasculature (D) revealed that the increased pool of bioavailable LA preserves angiogenesis of smaller pulmonary vessels without affecting larger vasculature. (E and F) LA treatment has also increased the length of the right middle lobar artery (E) and restored the luminal cross-sectional diameter (F). (G–J) Characterization of the microvascular network (G) additionally confirmed that the numbers of branches (H), junctions (I), and the total length of peripheral PAs (J) in LA-treated NFU1G206C rats are free of defects associated with NFU1 mutation. The bar scale for the original and enlarged representative angiographic images is 1 and 0.5 mm, correspondingly. The degree of the direct interaction between capillary ECs stained with GS-IB4 (green) and pericyte cells stained with antibodies against pericyte marker NG2 proteoglycan (red) served as a measure of microvascular homeostasis. (K and L) The apparent disconnection between these cells in the untreated NFU1 rats, visualized by 3D confocal microscopy (K, scale bar = 3 µm), was also observed as the absence of the green and red signal overlap on the fluorescent microscopy (L, scale bar = 100 µm). LA supplementation has efficiently preserved endothelial/pericyte association, sustaining a healthy environment for the pulmonary capillary cells. Panels A–F: N = 5/group; panels G–J: N = 5 in NFU1 and N = 6 in NFU1+LA groups. Panels K and L: N = 6/group. For all panels, the data is compiled from two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
Increased LA bioavailability is sufficient to prevent angiogenic defects in NFU1 G206C rats. (A) Representative micro-CT scans of LA-treated and untreated NFU1G206C rats demonstrate a radical difference in the pulmonary vascular network. The deficient pulmonary vascular morphology and obliteration of small PAs (red arrows inside the enlarged areas) associated with the NFU1G206C genotype do not manifest in NFU1 mutant rats supplied with LA. (B–D) The custom-built MATLAB code was used to quantify the density of the entire pulmonary vascular tree (B), and vessels separated on the large (>49.5 µm, which correspond to the branches of 1–4 order, C) and smaller vasculature (D) revealed that the increased pool of bioavailable LA preserves angiogenesis of smaller pulmonary vessels without affecting larger vasculature. (E and F) LA treatment has also increased the length of the right middle lobar artery (E) and restored the luminal cross-sectional diameter (F). (G–J) Characterization of the microvascular network (G) additionally confirmed that the numbers of branches (H), junctions (I), and the total length of peripheral PAs (J) in LA-treated NFU1G206C rats are free of defects associated with NFU1 mutation. The bar scale for the original and enlarged representative angiographic images is 1 and 0.5 mm, correspondingly. The degree of the direct interaction between capillary ECs stained with GS-IB4 (green) and pericyte cells stained with antibodies against pericyte marker NG2 proteoglycan (red) served as a measure of microvascular homeostasis. (K and L) The apparent disconnection between these cells in the untreated NFU1 rats, visualized by 3D confocal microscopy (K, scale bar = 3 µm), was also observed as the absence of the green and red signal overlap on the fluorescent microscopy (L, scale bar = 100 µm). LA supplementation has efficiently preserved endothelial/pericyte association, sustaining a healthy environment for the pulmonary capillary cells. Panels A–F: N = 5/group; panels G–J: N = 5 in NFU1 and N = 6 in NFU1+LA groups. Panels K and L: N = 6/group. For all panels, the data is compiled from two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
The angiogenic potential of PECs isolated from WT, NFU1G206C, or LA-treated NFU1G206C rats was evaluated ex vivo using Matrigel, migration, and scratch assays. All complementary approaches showed a severely impaired angiogenesis of NFU1G206C PECs compared to WTs, with a diminished ability to form the tubes or nodes (Fig. 7, A–C) and migrate (Fig. 7, D and E). However, upon the LA supplementation, the NFU1G206C PECs regained the angiogenic properties and showed a significantly improved ability to develop an endothelial network.
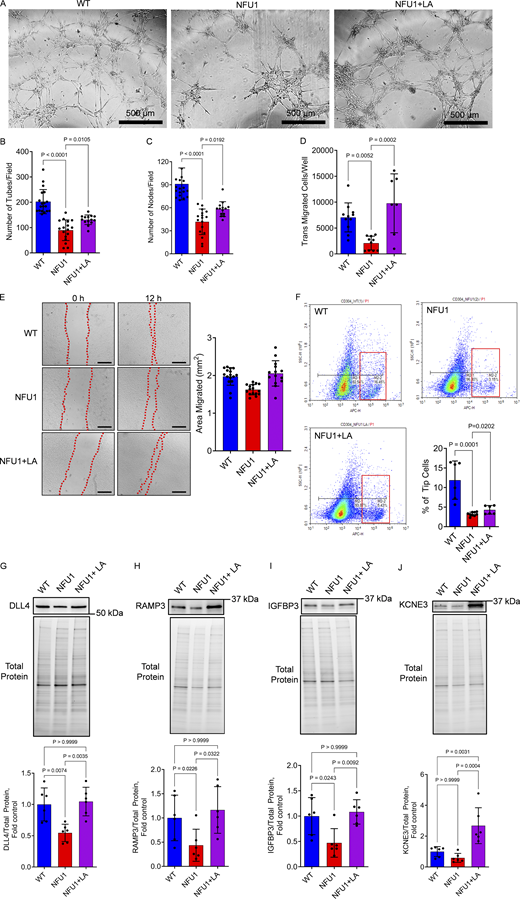
LA restores the angiogenic capacity of NFU1G206CPECs. PECs, isolated from WT and NFU1G206C rats treated or not with LA, were plated on the dish pre-coated with extracellular matrix solution, and the number of tubes and junctions was counted by an investigator blinded to the experimental setup. (A–C) PECs isolated from untreated NFU1 mutant rats displayed a disturbed ability to sprout and form tubes and nodes in the Matrigel (scale bar = 500 µm), while LA supplementation significantly improved the angiogenic profile of isolated PECs. (D and E) LA has also restored the migratory capacity of PECs, assessed by either transwell migration (D) or scratch assays (E, scale bar = 500 µm), confirming the importance of mitochondrial respiration in the energy-consuming migratory phenotype. (F) Measuring the CD304 positive tip cells population revealed the deficiency in this angiogenic cell population among the PECs isolated from untreated NFU1G206C rats and significantly improved CD304+ cell numbers in the LA-treated animals. (G–J) The expression profile of established markers and regulators of tip cell functionality DLL4 (G), RAMP3 (H), IGFBP3 (I), and KCNE3 (J) additionally confirm the deficiency in the tip cell population and function in the untreated and regained activity of tip cells in LA-treated NFU1G206C PECs. Panels A–C: N = 19 in WT, N = 17 in NFU1, and N = 14 in NFU1+LA groups; panel D: N = 11 in WT, N = 10 in NFU1, and N = 7 in NFU1+LA groups; panel E: N = 16 in WT and NFU1 groups, N = 14 in NFU1+LA groups; panel F: N = 6 in WT and NFU1+LA groups, N = 9 in NFU1 group; Panels G–J: N = 6/group. For all panels, the data is compiled from two independent experiments. Statistical analysis was performed using one-way ANOVA followed by the Bonferroni post-hoc multiple comparison test. NFU1 and NFU1+LA groups in B, C, and F were compared by the Student’s t test. WT versus NFU1 in H were compared by one-way ANOVA selected columns (Bonferroni). P < 0.05 was considered significant. Data are presented as mean ± SD. Source data are available for this figure: SourceData F7.
LA restores the angiogenic capacity of NFU1G206CPECs. PECs, isolated from WT and NFU1G206C rats treated or not with LA, were plated on the dish pre-coated with extracellular matrix solution, and the number of tubes and junctions was counted by an investigator blinded to the experimental setup. (A–C) PECs isolated from untreated NFU1 mutant rats displayed a disturbed ability to sprout and form tubes and nodes in the Matrigel (scale bar = 500 µm), while LA supplementation significantly improved the angiogenic profile of isolated PECs. (D and E) LA has also restored the migratory capacity of PECs, assessed by either transwell migration (D) or scratch assays (E, scale bar = 500 µm), confirming the importance of mitochondrial respiration in the energy-consuming migratory phenotype. (F) Measuring the CD304 positive tip cells population revealed the deficiency in this angiogenic cell population among the PECs isolated from untreated NFU1G206C rats and significantly improved CD304+ cell numbers in the LA-treated animals. (G–J) The expression profile of established markers and regulators of tip cell functionality DLL4 (G), RAMP3 (H), IGFBP3 (I), and KCNE3 (J) additionally confirm the deficiency in the tip cell population and function in the untreated and regained activity of tip cells in LA-treated NFU1G206C PECs. Panels A–C: N = 19 in WT, N = 17 in NFU1, and N = 14 in NFU1+LA groups; panel D: N = 11 in WT, N = 10 in NFU1, and N = 7 in NFU1+LA groups; panel E: N = 16 in WT and NFU1 groups, N = 14 in NFU1+LA groups; panel F: N = 6 in WT and NFU1+LA groups, N = 9 in NFU1 group; Panels G–J: N = 6/group. For all panels, the data is compiled from two independent experiments. Statistical analysis was performed using one-way ANOVA followed by the Bonferroni post-hoc multiple comparison test. NFU1 and NFU1+LA groups in B, C, and F were compared by the Student’s t test. WT versus NFU1 in H were compared by one-way ANOVA selected columns (Bonferroni). P < 0.05 was considered significant. Data are presented as mean ± SD. Source data are available for this figure: SourceData F7.
To investigate the impact of MD on endothelial sprouting, we quantified the number of CD304-positive tip ECs in each genotype (Fig. 7 F). CD304, also known as neuropilin-1, is a transmembrane receptor that plays a crucial role in normal angiogenesis by allowing ECs to attain the tip cell position, enable filopodia formation, and protrude in a new direction (Fantin et al., 2013; Gerhardt et al., 2004). We found that PECs isolated from untreated NFU1G206C rats show the loss of the CD304+ cell population. Assessment of the additional tip cell markers such as DLL4, RAMP3, IGFBP3, and KCNE3 (Fig. 7, G–J) confirmed the drop in tip cell population in the lungs of NFU1 mutant rats. However, LA supplementation improved or fully restored the profile of endothelial tip cells to levels comparable to those found in WT animals.
In the next step, we investigated whether the preserved angiogenic profile was sufficient to prevent the development of a spontaneous PAH phenotype in NFU G206C rats. Our findings demonstrate that replenishing the bioactive LA pool attenuates changes in RVSP (Fig. 8 A), RV contractility (dP/dt max, Fig. 8 B), and RV relaxation (dP/dt min, Fig. 8 C), and fully preserves the size of the RV in NFU1 mutants (Fig. 8 D). This finding supports the central role of mitochondria in the RV remodeling, which was proposed but not directly confirmed (Agrawal et al., 2020; Hwang et al., 2021). Furthermore, the histological analysis revealed that LA treatment rescued rats from the severe vascular remodeling induced by the NFU1G206C genotype (Fig. 8, E–G). Notably, no changes in hemodynamic parameters were seen in WTs supplemented with LA (Fig. S2), indicating that additional LA has no impact when LA availability is not compromised.

L A supplementation averts the PAH phenotype in NFU1 G206C rats. (A– G ) LA prevented PAH-promoting effects of NFU1 mutation on pulmonary hemodynamics (RVSP, A; RV contractility, B; RV relaxation, C) and RV hypertrophy (D). LA therapy has also prevented changes in PA morphology by protecting small PAs from severe vascular remodeling (E–G). Panels A–D: N = 10 in WT and untreated NFU1 groups; N = 12 in LA-supplied NFU1G206C rats; panels E–G: N = 5 in WT and untreated NFU1 groups; N = 6 in LA-supplied NFU1G206C rats. Compiled from two to three independent experiments, statistical analysis was performed using one-way ANOVA followed by Bonferroni post-hoc multiple comparison test, and P < 0.05 considered significant. Data are presented as mean ± SD.
L A supplementation averts the PAH phenotype in NFU1 G206C rats. (A– G ) LA prevented PAH-promoting effects of NFU1 mutation on pulmonary hemodynamics (RVSP, A; RV contractility, B; RV relaxation, C) and RV hypertrophy (D). LA therapy has also prevented changes in PA morphology by protecting small PAs from severe vascular remodeling (E–G). Panels A–D: N = 10 in WT and untreated NFU1 groups; N = 12 in LA-supplied NFU1G206C rats; panels E–G: N = 5 in WT and untreated NFU1 groups; N = 6 in LA-supplied NFU1G206C rats. Compiled from two to three independent experiments, statistical analysis was performed using one-way ANOVA followed by Bonferroni post-hoc multiple comparison test, and P < 0.05 considered significant. Data are presented as mean ± SD.

Lack of effect of LA supplementation on pulmonary hemodynamics or RV weight in WT rats. To investigate whether the chronic LA supplementation will produce any changes in pulmonary hemodynamic or RV function in WTs, the WT rats were treated with LA using the same protocol as in NFU1 mutants (Fig. 4 A—1.70 mM of LA was supplied in drinking water starting from the prenatal period and continued till the age of 10 wk age, when the animals were instrumented for the hemodynamic assessment). (A–D) None of the parameters, including RVSP (A), RV dP/dt max as a measure of cardiac contractility (B), dP/dt min as a measure of RV relaxation (C), and Fulton index (D) as a marker of RV hypertrophy, were altered in WT rats upon LA supplementation. For all panels, N = 10 in WT and N = 8 in WT+LA groups. Compiled from two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
Lack of effect of LA supplementation on pulmonary hemodynamics or RV weight in WT rats. To investigate whether the chronic LA supplementation will produce any changes in pulmonary hemodynamic or RV function in WTs, the WT rats were treated with LA using the same protocol as in NFU1 mutants (Fig. 4 A—1.70 mM of LA was supplied in drinking water starting from the prenatal period and continued till the age of 10 wk age, when the animals were instrumented for the hemodynamic assessment). (A–D) None of the parameters, including RVSP (A), RV dP/dt max as a measure of cardiac contractility (B), dP/dt min as a measure of RV relaxation (C), and Fulton index (D) as a marker of RV hypertrophy, were altered in WT rats upon LA supplementation. For all panels, N = 10 in WT and N = 8 in WT+LA groups. Compiled from two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
To test whether NFU1 insufficiency is a contributing factor to the classical preclinical PH model, we investigated the levels of NFU1 expression and function in the lungs of rats exposed to the Su/Hx using our standard lab protocol (Rafikov et al., 2019). The results revealed that Su/Hx induces a significant loss of NFU1 protein (Fig. 9 A) and reduced PDH lipoylation (Fig. 9 B), confirming an impaired LAS activity in Su/Hx rats similar to what we observed in the NFU1G206C animals (Niihori et al., 2020). This discovery suggests a potential role of NFU1 dysfunction in the impaired mitochondrial function reported in this animal model (Colon Hidalgo et al., 2022).
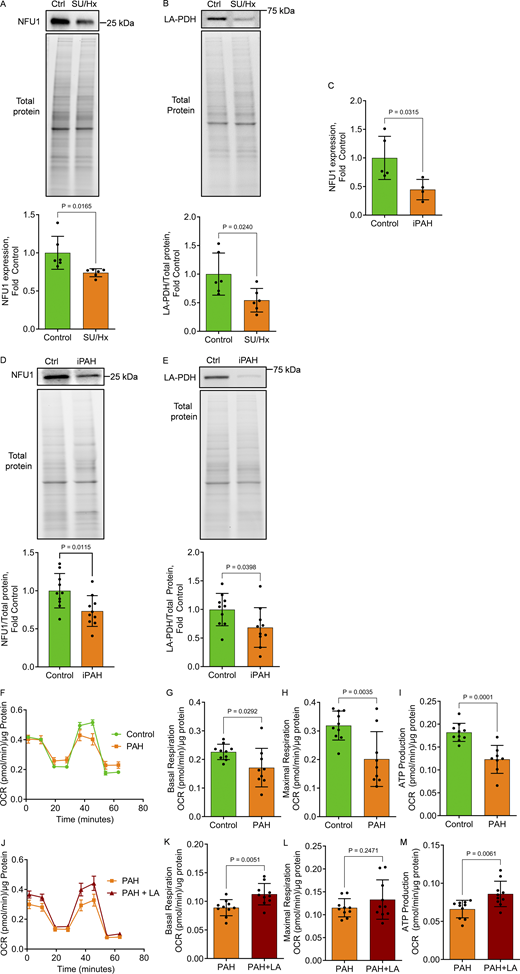
NFU1 deficiency in Su/Hx rat model and iPAH patients and the protective potential of LA supplementation to mitigate MD in patient PAECs. (A–E) The analysis of pulmonary tissues obtained from Su/Hx rats revealed a previously unrecognized decrease in NFU1 protein levels (A) and binding of LA to PDH (B) in this classical PAH model. In addition to animals, the translational aspect of this study was confirmed by revealing a similar insufficiency in the NFU1 expression (C), protein levels (D), and signaling, assessed as PDH lipoylation (E) in the lung samples collected from iPAH patients, the result consistent with the findings observed in NFU1G206C rats (Niihori et al., 2020). (F–I) The mito-stress Seahorse assay revealed an inhibited mitochondrial respiration in PAECs isolated from PAH patients (F) with a significantly attenuated mitochondrial basal (G) and maximal (H) respiration and decreased rate of ATP production (I). (J–M) Notably, acute exposure (12 h) of PAH PAECs to LA was sufficient to significantly improve mitochondrial function in patient PAECs (J) and restore basal (K) but not maximal (L) mitochondrial respiration and increase ATP levels (M), highlighting the critical role of insufficient LA bioavailability in the impaired mitochondrial function observed in PAH patient PAECs. Panels A and B: N = 6/group; panel C: N = 5 in Control and N = 4 in iPAH group; panel D and E: N = 10/group; panels F–H: N = 10 in Control and N = 9 in PAH group; panel I: N = 10 in Control, N = 8 in PAH group; panels J–M: N = 10/group. Representative of one or two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data in F and J are presented as mean ± SEM. For the rest panels, data are presented as mean ± SD. Source data are available for this figure: SourceData F9.
NFU1 deficiency in Su/Hx rat model and iPAH patients and the protective potential of LA supplementation to mitigate MD in patient PAECs. (A–E) The analysis of pulmonary tissues obtained from Su/Hx rats revealed a previously unrecognized decrease in NFU1 protein levels (A) and binding of LA to PDH (B) in this classical PAH model. In addition to animals, the translational aspect of this study was confirmed by revealing a similar insufficiency in the NFU1 expression (C), protein levels (D), and signaling, assessed as PDH lipoylation (E) in the lung samples collected from iPAH patients, the result consistent with the findings observed in NFU1G206C rats (Niihori et al., 2020). (F–I) The mito-stress Seahorse assay revealed an inhibited mitochondrial respiration in PAECs isolated from PAH patients (F) with a significantly attenuated mitochondrial basal (G) and maximal (H) respiration and decreased rate of ATP production (I). (J–M) Notably, acute exposure (12 h) of PAH PAECs to LA was sufficient to significantly improve mitochondrial function in patient PAECs (J) and restore basal (K) but not maximal (L) mitochondrial respiration and increase ATP levels (M), highlighting the critical role of insufficient LA bioavailability in the impaired mitochondrial function observed in PAH patient PAECs. Panels A and B: N = 6/group; panel C: N = 5 in Control and N = 4 in iPAH group; panel D and E: N = 10/group; panels F–H: N = 10 in Control and N = 9 in PAH group; panel I: N = 10 in Control, N = 8 in PAH group; panels J–M: N = 10/group. Representative of one or two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data in F and J are presented as mean ± SEM. For the rest panels, data are presented as mean ± SD. Source data are available for this figure: SourceData F9.
Mutations in the NFU1 gene were initially described in humans; however, these mutations are rare and are associated with a very low lifespan for carriers. To investigate whether NFU1 insufficiency is present in adult PAH patients without NFU1 mutation, we repeated the measurements of NFU1 expression and LA bioavailability in the lung samples obtained from iPAH and non-PAH control subjects. The results confirmed that even patients without NFU1 mutation demonstrate a ∼50% reduction in NFU1 expression (Fig. 9 C) and a ∼30% decrease in NFU1 protein levels (Fig. 9 D) compared to non-PAH controls. The observed diminished lipoylation of PDH as a marker of impaired NFU1 signaling in iPAH pulmonary tissue (Fig. 9 E) additionally validated the relevance of NFU1 insufficiency-induced MD to human iPAH. Indeed, the additional evaluation of mitochondrial function in human PAECs (hPAECs) isolated from PAH and non-PAH subjects showed signs of impaired mitochondrial respiration in PAH hPAECs similar to those seen in rat NFU1G206C PECs (Fig. 9, F–I). Most importantly, the acute treatment of PAH hPAECs with LA significantly improved basal mitochondrial respiration and ATP production (Fig. 9, J–M), highlighting the importance of LA deficiency as one of the critical contributors to inadequate mitochondrial function in PAH hPAECs.
To examine whether the beneficial effects of LA were, at least in part, due to its antioxidant properties, we repeated some critical experiments in the presence of N-Acetyl L-Cysteine (NAC), another established antioxidant. NAC in the dose of 100 µM was reported efficient in sustaining intracellular glutathione synthesis and providing cytoprotection against oxidative stress (Whillier et al., 2009; Zhou et al., 2020). However, the same dose failed to improve mitochondrial respiration or ATP production (Fig. S3, A–D) in NFU1G206C PECs. NAC supplementation has also worsened the defective angiogenic properties of NFU1G206C PECs, as indicated by a significant reduction in the formation of tubes and nodes to a non-quantifiable level (Fig. S3 E). These findings indicate that the beneficial effects of LA on the angiogenic properties of NFU1G206C PECs cannot be attributed solely to its antioxidant activity.

Antioxidant N AC shows no improvement in the mitochondrial function or angiogenic capacity of PECs isolated from NFU1 G206C rats. To investigate whether the antioxidant activity of LA is essential for the LA-induced improvements in mitochondrial function and angiogenesis, we treated NFU1G206C PECs with NAC, a well-established antioxidant. At dose 100 µM, NAC is known to provide cytoprotection against oxidative stress by sustaining intracellular glutathione synthesis. (A–D) However, the NFU1G206C PECs exposed to NAC for 24 h show no significant improvement in mitochondrial function (A) assessed by measuring the mitochondrial basal (B) and maximal (C) respiration and the rate of ATP production (D). (E) Moreover, NAC treatment further impaired the angiogenic ability of NFU1G206C PEC to sprout and form an endothelial network in the Matrigel, rendering the changes unquantifiable. For all panels, N = 5 per group. Representative of one to two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
Antioxidant N AC shows no improvement in the mitochondrial function or angiogenic capacity of PECs isolated from NFU1 G206C rats. To investigate whether the antioxidant activity of LA is essential for the LA-induced improvements in mitochondrial function and angiogenesis, we treated NFU1G206C PECs with NAC, a well-established antioxidant. At dose 100 µM, NAC is known to provide cytoprotection against oxidative stress by sustaining intracellular glutathione synthesis. (A–D) However, the NFU1G206C PECs exposed to NAC for 24 h show no significant improvement in mitochondrial function (A) assessed by measuring the mitochondrial basal (B) and maximal (C) respiration and the rate of ATP production (D). (E) Moreover, NAC treatment further impaired the angiogenic ability of NFU1G206C PEC to sprout and form an endothelial network in the Matrigel, rendering the changes unquantifiable. For all panels, N = 5 per group. Representative of one to two independent experiments. Statistical analysis was performed using the Student’s t test, and P < 0.05 was considered significant. Data are presented as mean ± SD.
Discussion
Two types of angiogenesis, sprouting and intussusceptive, are known as pivotal contributors to the formation of new vessels in the adult lung (Eldridge and Wagner, 2019; Ackermann et al., 2014, 2020; Konerding et al., 2012). Sprouting angiogenesis, discovered nearly 200 years ago, is better studied and represents a process by which ECs form sprouts that grow towards an angiogenic stimulus to create new blood vessels. It requires the phenotypic transformation of quiescent ECs into the actively respiring migratory tip cells that “sprout” filopodia and direct the new vessel formation or into proliferative stalk cells that form tubes and branches of the new vessels. In contrast, intussusceptive angiogenesis occurs when a preformed vessel splits in two through an extension of the vessel wall into the lumen. While sprouting angiogenesis is well characterized and involves a rewiring of endothelial metabolism to cope with the increased energetic demands of migrating and proliferating cells (Fitzgerald et al., 2018), the more recently discovered intussusceptive angiogenesis is less understood (Eldridge and Wagner, 2019) and has not been explored in terms of its interconnection with changes in metabolism. Therefore, this study focuses on the sprouting component of pulmonary angiogenesis.
In PAH, the decrease in pulmonary vasculature complexity strongly and invertedly correlates with an increase in the patients’ mean PA pressure (Boxt et al., 1986; Rabinovitch et al., 1981). Furthermore, the degree of pulmonary vascular loss is the only prognostic marker that significantly correlates with patient mortality, while World Health Organization (WHO) functional class, percentage of predicted 6-min walk distance, pulmonary blood flow, or PVR are not significantly associated with this outcome (Moledina et al., 2011). Being a dynamic and energy-consuming process, angiogenesis requires increased energy support. Many critical steps, including remodeling of the actin cytoskeleton, EC migration, acquiring the tip cell phenotype, and sprout formation, are highly ATP dependent (Lamalice et al., 2007). Thus, while the metabolism of quiescent EC mainly relies on glycolysis, the motile phenotype, which operates close to its respiratory limits, increasingly demands adequate activity of mitochondrial oxidative phosphorylation (Coutelle et al., 2014). This substantial change in the metabolism of pro-angiogenic ECs renders them more vulnerable to perturbations in mitochondrial function. Indeed, it was noticed that in the systemic vasculature, the MD disrupts normal angiogenesis (Coutelle et al., 2014). However, there is a stark contrast in angiogenesis regulation between systemic and pulmonary circulation since the same hypoxic stimulation induces marked angiogenesis in the systemic vasculature and a loss of blood vessels in the lungs (Hopkins and McLoughlin, 2002). Therefore, the contribution of MD to the dysfunctional angiogenesis in PAH remains unknown and requires clarification.
In this study, we aimed to bridge this gap in knowledge by conducting a detailed investigation of pulmonary angiogenesis in rats with impaired mitochondrial function. Unlike other established animal models of PAH, such as chronic hypoxia, monocrotaline, or Su/Hx, which also have MD as a part of a complex sequence of pathogenic events, the NFU1G206C model produces a primary MD due to the inhibitory mutation in the NFU1 (Niihori et al., 2020), a critical regulator of mitochondrial homeostasis. This “pure” MD results in the impaired activity of several critical mitochondrial enzymes on an otherwise healthy background. Using this model, we have previously addressed a crucial question regarding the primary versus secondary role of MD in the complex pathogenesis of PAH. Our findings demonstrate that even in the absence of any co-existing pathogenic mechanisms, MD is sufficient to initiate a spontaneous PAH phenotype (Niihori et al., 2020). The current study is focused on evaluating these changes in progression. This approach allows for the elucidation of whether PAH is present in younger animals or develops later in adulthood. More importantly, following the age-mediated changes in the lung morphology helped to discover a tight association between progressive impairment in pulmonary angiogenesis and increased severity of the PAH phenotype.
As shown in Fig. 2, E and H, we found that the level of pulmonary angiogenesis in the 6-wk-old rats was comparable between NFU1G206C rats and WTs, which corresponded to the minimal difference in pulmonary hemodynamics (Fig. 1, A–C). These results suggest that, until a certain age, the level of pulmonary angiogenesis in NFU1 mutants remains adequate to the demand. Upon aging, though, the growing body requires an additional oxygen supply, and WT rats respond to this stimulation by activating the angiogenesis of small PA. In contrast, the presence of MD in NFU1 mutant animals disrupts this adaptation and limits angiogenic capacity. Indeed, we discovered that rats with MD show a reduced differentiation and functionality of tip cells and the loss of interaction between capillary ECs and pericytes, known to compromise microvasculature formation and maturation and results in the impaired density of pulmonary capillaries and reduced complexity of peripheral PAs. Given the critical importance of the local hypoxic milieu for the initiation and progression of vascular remodeling, we propose that these conspicuous deficiencies of pulmonary morphology drive the PAH phenotype in NFU1G206C rats.
Diminished microvascular density is known as a key factor contributing to local hypoxia in cardio-metabolic and renal disorders (Katsuumi et al., 2016; Zhang et al., 2023). In PAH, this hypoxic microenvironment triggers severe biochemical and functional changes in all vascular cells, including endothelial, smooth muscle cells, and adventitial fibroblasts, leading to a highly proliferative state of the pulmonary vasculature and subsequent vascular remodeling (Stenmark et al., 2006). The appearance of a classical “dead tree” picture in the patient pulmonary vasculature is thought to result from a combination of progressive vascular loss and obliteration of precapillary PA in the patient’s lungs. The co-occurrence of the same angiogenic defects and severe vascular remodeling in the NFU1G206C rats supports this interconnection. Moreover, it could explain the observed correlations between the degree of the vascular pruning and the severity of the PAH or patient outcomes, as the loss of pulmonary vasculature appears to be an upstream event that drives the entire course of the disease. Therefore, targeting inadequate angiogenesis should yield significant protection.
Indeed, our approach of restoring mitochondrial function by addressing the insufficiency in LA bioavailability proved to be highly effective. Supplementation of LA to NFU1G206C rats completely preserved pulmonary vascular tree morphology at the level of healthy age-matched WTs. Furthermore, the abolished or significantly attenuated deficiencies in the pulmonary vascular network, microvascular density, and pericyte–endothelial interaction, along with the restored angiogenic capacity of PECs ex vivo, indicate that our approach targets the correct factor responsible for the angiogenic deficiency in this model. The absence of signs of vascular remodeling in LA-treated rats prevented RV hypertrophy and almost entirely preserved RVSP, which supports the critical importance of adequate angiogenesis in protecting against PAH initiation.
It is important to note the discourse surrounding the capacity of free LA to directly facilitate protein lipoylation. Thus, it was reported that LA supplementation in pregnant mice heterozygous by LAS failed to prevent the prenatal deaths of homozygous embryos (Yi and Maeda, 2005). Based on these results, it was concluded that eukaryotic cells could not use exogenously supplied LA and depend on its intramitochondrial synthesis. Indeed, the bacterial enzyme lipoate ligase (LplA) directly lipoylates proteins using free exogenous LA in a two-stage reaction involving lipoyl-AMP formation and the subsequent transfer of the lipoyl moiety to target proteins. However, the mammalian analog of LplA, LIPT1, is described as lacking the activity to form lipoyl-AMP (Cronan, 2020).
Nevertheless, several studies are challenging this prevailing view that mammals do not produce lipoyl-AMP from free LA by providing evidence that the crystal structure of bovine LIPT1 contains endogenous lipoyl-AMP (Fujiwara et al., 2007). Additionally, the mammalian LIPT1 has demonstrated its ability to conjugate the lipoyl-AMP moiety to both GCS H protein and the E2 subunits, suggesting the existence of the salvage pathway in mammals (Fujiwara et al., 1996, 1997). However, despite these findings, the specific enzyme responsible for activating free lipoate to form lipoyl-AMP in mammals remains unidentified, prompting active scientific debates (Cao et al., 2018).
Furthermore, some previous studies noticed the apparent compensatory effects in the models with impaired LA metabolism, which may suggest the ability of LIPT1 or other mammalian enzymes to process free lipoate (Soreze et al., 2013). Most importantly, the exogenous LA supplementation to aged mice was recently reported to induce a significantly elevated PDH lipoylation (Tajima et al., 2019). In line with these previous findings, we reported a substantial increase in E2 lipoylation in the whole lung tissues and pulmonary ECs isolated from the LA-supplemented rats (Fig. 5, B and C).
Further research is necessary to comprehensively understand the biochemical pathways involved in LA metabolism and identify the enzymes in the mammalian salvage pathway. However, while gaps in current knowledge regarding the origins of lipoyl-AMP in mammalian systems still exist and limit the conclusions about the mechanism of LA-induced protection, our findings suggest a highly beneficial effect of LA supplementation in patients with LA synthesis deficiency.
To avoid the excess in vessel formation, angiogenesis is counterbalanced by the regression of unnecessary vessels or pruning (Ricard and Simons, 2015). The balance between angiogenesis and pruning is tightly regulated by VEGF/VEGFR2 signaling, which promotes angiogenesis upon upregulation and stimulates pruning when suppressed. Although the NFU1G206C model does not directly target VEGF/VEGFR2 signaling and reduces, rather than increases, apoptosis of the vascular cells, promoting apoptosis resistance (James et al., 2021), the contribution of vascular pruning to the decreased density of pulmonary vessels in NFU1G206C rats cannot be excluded entirely. Following WT and NFU1G206C rats at different ages allowed us to observe no loss of already preformed pulmonary vessels, providing no evidence of enhanced vessel regression. Nevertheless, more precise studies are required to address the contribution of MD, if any, to the different aspects of vascular pruning and its potential contribution to the deficient vascular network in this model.
NFU1 mutations in humans are rare and typically fatal in infancy. However, the analysis of lung tissues from iPAH patients performed in this study provides strong evidence of NFU1 insufficiency even in adult non-carriers. Thus, we observed that both NFU1 expression and protein levels were significantly reduced in the lungs of iPAH versus non-PAH subjects, indicating a diminished NFU1 synthesis. Furthermore, the decreased PDH lipoylation in iPAH patients’ lungs, similar to that seen in NFU1G206C rats (Niihori et al., 2020), provides additional evidence of reduced NFU1 activity. These findings, together with the discovery of insufficiency in the NFU1 expression and signaling in the classical Su/Hx rat model of PAH, underscore the substantial relevance of the NFU1G206C model to PAH induced in animals or humans without NFU1 mutation.
Thus, the reduced activity of PDH is an accepted contributor to human PAH. It is believed that the hypoxic activation of pyruvate dehydrogenase kinase (PDK) is responsible for PDH inactivation (Archer et al., 2010b), although the clinical trial with PDK inhibitor dichloroacetate (DCA) was beneficial only in a fraction of iPAH patients (Michelakis et al., 2017), while the rest were not sensitive to the therapy. The authors conclude that DCA fails to restore PDH activity in patients whose PDH is inhibited through a PDK-independent pathway. Indeed, in this study, we provide evidence of an alternative mechanism of PDH regulation, emphasizing the role of MD associated with NFU1 or LA deficiency in preclinical or clinical PAH.
The metabolic theory of PAH, developed by several research groups (Paulin and Michelakis, 2014; Culley and Chan, 2018; Archer et al., 2008; Xu et al., 2021), has already put forward the concept of a central role of MD in PAH pathobiology. In particular, they emphasize that various molecular pathways involved in PAH, seemingly unrelated, share a common denominator, namely, the inhibition of mitochondrial function. MD also produces a spectrum of pathogenic outcomes known as essential players of PAH pathogenesis, including metabolic disturbance, apoptosis resistance, oxidative stress, and impaired energy balance. There are different flavors of MD that range from impaired mitochondrial numbers due to the disbalance in mitochondrial biogenesis, mitophagy, or a misguided mitochondrial fission/fusion to the dysfunctional mitochondrial respiratory chain, mitochondrial DNA damage, and uncontrolled production of respiratory oxygen species. However, the impaired LA biogenesis secondary to NFU1 insufficiency represents an entirely new mechanism of severe MD that has been previously overlooked. We found that even an acute exposure of PAECs isolated from PAH patients to LA significantly improves altered mitochondrial respiration in patient cells, emphasizing the critical importance of LA bioavailability and the insufficiency of LA biosynthesis in PAH PAECs. These beneficial effects of LA supplementation seen in the phenotypically modified PAH patient cells suggest the high relevance of the research performed in the NFU1G206C rat model to PAH patients.
In conclusion, this study introduces a novel molecular mechanism of NFU1 insufficiency and subsequent LA deficiency as a trigger for disturbed pulmonary angiogenesis, which, in turn, contributes to the initiation and progression of preclinical PAH. Moreover, based on the protective effects of LA for alleviating MD in PAEC isolated from PAH patients, the supplementation of LA to patients with unrecognized NFU1 insufficiency may become a novel therapeutic approach. Given the importance of adequate mitochondrial respiration for pulmonary angiogenic homeostasis found in this study, we propose that not only the NFU1/LA axis but also other mechanisms responsible for the MD in PAECs can significantly contribute to preclinical and clinical PAH. Elucidating these mechanisms will provide efficient therapeutic targets for improving pulmonary angiogenesis tightly associated with PAH severity and patient outcome.
Materials and methods
Animals
The SD rat model with a point mutation (G206C, GGC to TGC, homologous to human G208C) at rat NFU1 locus engineered by CRISPR/CAS9-mediated genome editing was created by Cyagen Biosciences, Inc. as described before (Niihori et al., 2020). SD rats purchased from Charles River were used as WT or to induce Su/Hx PAH. WT and homozygous NFU1G206C SD rats were bred in the University Animal Care facilities at the University of Arizona. The University of Arizona’s Institutional Animal Care and Use Committee committee has approved the breeding protocol and all experimental procedures. Animals were kept in a 12-h light–dark cycle and received standard rodent food and water ad lib. Each experimental group consisted of rats of both sexes (at a 50/50 ratio). LA supplementation was given in the drinking water at a concentration of 1.70 mM. The Su/Hx model was created as previously described (Rafikov et al., 2019; Rafikova et al., 2018). Briefly, the PAH was initiated by a single dose of the VEGFR2 antagonist, SU5416, 50 mg/kg s.c., followed by 3 wk of hypoxia exposure (10% ± 0.5% of O2, using a hypoxic chamber (BioSpherix), PROOX 110 BioSpherix oxygen controller, and LB-2 CO2 analyzer; Sensormedics) and 2 wk of normoxia.
Hemodynamic measurements and organ harvest
For hemodynamic assessments, the animals were anesthetized (Inactin, T133, 100 mg/kg i.p.; Sigma-Aldrich) and instrumented as previously described (Rafikova et al., 2013). Briefly, a customized pressure transducer catheter (SPR-513; Millar Instruments), connected to a Millar Transducer Control Unit TC-510 and PL3504 PowerLab 4/35 data acquisition system (ADInstruments), was inserted into RV via the right jugular vein and right atrium. After a short stabilization period, RV pressure was recorded for up to 30 min. At the end of the pressure recording, the PE-240 polyethylene tube (Beckton Dickinson) was inserted into the trachea and connected to a Harvard Rodent Ventilator (Model 683; Harvard Apparatus) to facilitate the breeding. The thorax was opened, the left atrium was cut, and the lungs were flushed with saline (0.9% sodium chloride) via a needle inserted into the RV. Animals were euthanized by removing heart/lung block. Lungs, RV, and LV+S were dissected and weighed. Half of the left lung was fixed in formalin and embedded in paraffin for histological examination.
Analysis of PA remodeling
For the morphometric assessment of pulmonary vessels, 5-μm lung tissue sections were dewaxed and stained with hematoxylin and eosin (H&E) using a standard protocol developed by the University of Arizona Tissue Acquisition and Cellular/Molecular Analysis Shared Resource histology core. 10 small transversely sectioned pulmonary arteries per animal were randomly selected. The morphometric analysis was done blindly by an investigator unaware of the animal group using FIJI Image J software (Fiji.sc). Pulmonary vascular remodeling was assessed as previously described (Niihori et al., 2020). Briefly, for the occlusion score, the total vessel area, defined as the area within the lamina elastica externa, and the lumen area, defined as the area within the lamina elastica interna, were traced. The vascular occlusion score (%) was quantified as follows: (total vessel area − lumen area)/total vessel area × 100. For the media thickness, the size of the media layer was measured in the four different areas (corresponding to 3, 6, 9, and 12 h) of transversely sectioned vessels and the average media thickness per vessel was calculated. Additionally, the pulmonary vascular remodeling was assessed by measuring the alpha-smooth muscle actin (αSMA)–positive signal normalized over the DAPI signal in the small PAs of WT and NFU1G206C rats. A minimum of 20 PAs per animal were randomly selected and the mean value for each animal was obtained.
micro-CT of pulmonary vasculature
Anesthetized rats placed in a supine position were intubated and connected to the rodent ventilator (Harvard Apparatus). The abdominal cavity was cut and opened and the heart and lungs were exposed. The vena cava was cut to prevent blood circulation and ensure euthanasia. PE 160 connected to a 60 ml syringe (Beckton Dickinson) filled with warm (37°C) saline was inserted into the RV through a small cut in the free RV wall, advanced to the main PA, and secured with 4-0 silk suture to keep the position. The left atrium was removed and saline was pumped at a constant rate of 2 ml/min for 5 min using a syringe pump (Genie Touch; Kent Scientific Corp.). The MICROFIL compound (MV-122; Flow Tech, Inc.) was freshly prepared according to the manufacturer’s protocol and delivered at the rate of 0.05 ml/min (for 10 wk old animals) and 0.03 ml/min (for 6 wk old animals) to uniformly fill the lung. After MICROFIL polymerization at 4°C overnight, the lung/heart block was removed and washed in cold PBS for 2 h. Images (440 projections over 220° of rotation) were acquired using the Inveon micro-CT scanner (Siemens Medical Solutions USA, Inc.). The medium magnification with a 2 × 2 binning was used, the transaxial field of view was set at 2,048 px (34.13 mm), and the axial scan length was set at 45 mm. Exposure settings were 50 kV, with 500 micro-A current and an acquisition time of 1,800 ms. Image reconstruction was done using a Deldkamp algorithm, a Shepp-Logan filter, and noise reduction.
PA density, length, and lumen diameter analysis
DICOM files from a micro-CT scan were converted into jpg images every 10° to create a 360° rotation view using Inveon Research Workplace (Siemens). A custom-built software tool on MATLAB R2019b (The MathWorks, Inc.) was used to analyze these images. Transverse plane grayscale images were converted into black-and-white images using histogram-based thresholding. Morphological opening (Haralilck et al., 1987) was used to extract whole lung, large-sized (with a diameter >49.55 µm), and small-sized (9–49.55 µm) PAs. Image deconvolution was achieved by repeating the morphological opening operation using structuring elements of radii 2, and 11 pixels to achieve the target sizes, where each pixel in the image corresponds to 4.5 µm. Subtracting branches of successive thicknesses from the larger branches produces residuals that correspond to tertiary branches of smaller diameter. PA axial length and the changes in PA diameter per vascular length were evaluated in the right middle lobe using the modified method of morphologic evaluation of pulmonary arteries (Boxt et al., 1986). Briefly, the PAs were segmented from the surrounding vasculature, the inflection points were identified, and the “inflection distance” was calculated as the axial length of the PA starting from the main branching point and to the visualized tip of the artery. The serial arterial cross-sectional diameters in 1 mm intervals were measured to evaluate the changes in PA diameter as a function of vascular length.
Pulmonary microangiography
After the micro-CT scanning, the heart was removed from the MICROFIL-infused lung/heart block, and the samples were prepared as previously described (Smith et al., 2015). Briefly, the lungs were dehydrated at 50%, 70%, 80%, 90%, and 100% ethanol and immersed in methyl salicylate (M6752; Sigma-Aldrich) overnight at room temperature. Small pulmonary arteries were imaged with the Zeiss Axiozoom 16 fluorescent stereo microscope (Carl Zeiss SBE, LLC.) equipped with a Hamamatsu Flash 4.0 camera (Hamamatsu Photonics) using 40× magnification. The images were processed with Zen Blue software (Carl Zeiss SBE, LLC.). The vasculature tree in a given area (0.5 mm from the peripheral area along the edge of the lobe) was selected and traced with Photoshop CS software (Adobe, Inc.) and converted into binary images using FIJI Image J software (fiji.sc) software. The number of arterial branches, arterial junctions, and the total length of arterial segments in a given area were analyzed using FIJI Image J software (fiji.sc), as previously published (Smith et al., 2015). The data were normalized by the area (1 mm2) in each lung.
Analysis of pulmonary capillary density and endothelial–pericyte interaction
Lungs embedded in paraffin block were cut 4 µm thick, deparaffinized, and underwent antigen retrieval. Slides blocked with 10% bovine serum albumin (BSA) for 15 min at room temperature were stained with Isolectin GS-IB4 from Griffonia simplicifolia, Alexa Fluor 488 conjugate (I21411; Thermo Fisher Scientific) at 1:50 dilution for 1 h in the dark, as published (King et al., 2004). Slides were washed four times and sealed with Prolong (P36962; Invitrogen). Microscopy was performed using a Leica DMI6000 multifunction motorized inverted microscope (Buffalo Grove) at 20×. The number of capillaries in the field was quantified by an investigator blinded to the experimental setup by analyzing 10 random fields of each slide using Fiji ImageJ (Version-1.52p; National Institutes of Health, Washington, DC, USA). Alternatively, the slides were stained with vWF (1:50, Cat#27186-1-AP; Proteintech) and αSMA (1:100, Cat#14-9760-82; Invitrogen), incubated with secondary antibodies conjugated with Dylight 650 for vWF (red) and Alexa Fluor 488 for αSMA (green), sealed with mounting medium with DAPI, and visualized under an inverted microscope (Revolve; ECHO). The quantification was performed by normalizing the vWF and αSMA signals per DAPI signal or by measuring the vWF/αSMA ratio in 20 random fields per animal.
For endothelial/pericyte costaining, slides underwent permeabilization using 10% Triton X-100 for 10 min at room temperature, blocked with 10% goat serum for 1 h at room temperature, and probed with anti-NG2 chondroitin sulfate proteoglycan antibody (AB5320; Millipore Sigma) at 1:100 dilution overnight at +4°C. On the next day, slides were washed in TBS with Tween 20 and probed with donkey anti-rabbit IgG (H+L) secondary antibody conjugated with DyLight 650 fluorophore (NBP1-75644; Novus) at 1:100 dilution for 1 h at room temperature. Slides were counterstained with Isolectin GS-IB4 from Griffonia simplicifolia, Alexa Fluor 488 Conjugate (I21411; Thermo Fisher Scientific) at 1:100 dilution, and DAPI solution (564907; BD Pharmingen) at 1:10,000 dilution for 10 min at room temperature and sealed with ProLong Gold antifade mountant (P10144; Invitrogen). Microscopy was performed using an ECHO RVL2-K Revolve microscope at 20×. The area of overlap between the red and green channels was quantified using Fiji ImageJ software (version 2.9; National Institutes of Health, Washington, DC, USA) and normalized per blue (nuclei) area to account for the amount of tissue in the field. To obtain confocal 3D images, paraffin block embedded lungs were cut into 10-µm-thick sections and processed and stained in the same manner as previously described. Microscopy was performed using the Zeiss LSM880 confocal microscope with Airyscan at 60× oil immersion and accompanying ZenBlack software (version 2.3; Carl Zeiss Microscopy). Z-stacked images were processed into 3D renders using Imaris software (version 10.0; Oxford Instruments).
Rat PEC isolation, culture, and mitochondrial staining
Rat PECs were isolated from WT and NFU1G206C rats using the Miltenyi Rat Endothelial Cell isolation kit (130-109-679; Miltenyi Biotec). In brief, the saline-flushed rat lungs were finely chopped and digested in collagenase/diaspase (10 mg/ml). Following this procedure, the digestion was halted by adding 10% FBS to DMEM. The cells were filtered in series through a 70- and 40-µM sieve and washed three times in Dulbecco's PSB (DPBS) containing 0.5% BSA. The purification of ECs was carried out according to the manufacturer’s protocol. The isolated PEC’s purity was confirmed by staining for CD31 (FAB3628P-025; R&D Systems) and measured using the flow cytometer (Novocyte; ACEA Biosciences, Inc.). The experiments were performed only on cells with purity >90%. Rat PECs were used for western blotting at P = 0 or plated in complete EC media (1011; Sciencell) and used for further assays. For assays with LA treatment, a basal concentration of 1 µM of LA was maintained in the media unless specifically mentioned. For the tip cell analysis, the cells were stained using the CD304 antibody (MA5-32179; Invitrogen) and counted by the flow cytometer (Novocyte; ACEA Biosciences, Inc.). To visualize the mitochondrial network, rat PECs were cultured on the coverslips (Neuvitro GG-18-15-PLL) until reaching 60–70% confluency, fixed in 4% paraformaldehyde for 15 min at room temperature, and permeabilized with 0.5% Triton X-100 in PBS for 10 min. Blocking was performed with 5% BSA in PBS for 1 h at room temperature. Following blocking, cells were incubated overnight at 4°C with the antibody against MitoNEET (83775S, diluted 1:100 in DPBS containing 1% BSA; Cell Signaling), the outer mitochondrial membrane protein regulating mitochondrial network connectivity (Vernay et al., 2017). After staining, cells were washed three times with PBS to remove unbound antibodies and the coverslips were mounted onto glass slides using ProLong Diamond Antifade Mountant with DAPI (P36962; Thermo Fisher Scientific). Images were acquired at 60× magnification using a fluorescence microscope with DAPI, FITC, and Cy5 filters (ECHO Revolve). At least 100 random fields per experimental group were imaged.
hPAEC isolation and maintenance
Deidentified lung tissue specimens from patients with PAH and non-diseased donors (Controls) were obtained from the University of Pittsburgh Tissue Medical Center Lung Transplant Program and Tissue Donation Program at the University of Pittsburgh Medical Center and through Pulmonary Hypertension Breakthrough Initiative (PHBI). Prior to tissue collection, informed consent was obtained from all subjects and/or their legal guardian(s) by the University of Pittsburgh Medical Center Lung Transplant Program and Tissue Donation Program in accordance with the University of Pittsburgh Institutional Review Boards (IRB) policies. The PHBI study protocol was approved by the IRB of the participating sites in the network. PAECs from small (<1.5 mm outer diameter) muscularized PAs were isolated and cultured as published (Xu et al., 2007). Briefly, PAs were extensively cleaned from the surrounding tissue and perfused with 0.5% Trypsin (Gibco) and 2.5 mg/ml Collagenase I (Worthington Biochemical Corporation). The purity of the resulting culture maintained in Endothelial Cell Growth Medium MV (PromoCell) was confirmed by a combination of vWF immunofluorescent staining (ab6994; Abcam) and visual identification. Lung tissue samples were used for measuring NFU1 expression and protein levels and PDH lipoylation (Niihori et al., 2020), as indicated below.
Seahorse assay
Agilent Seahorse XFp cell mitostress and glycolysis stress tests were performed as previously described (James et al., 2021). In brief, isolated PECs were seeded in a 24- or 96-well Seahorse cell culture microplate and left overnight to form a monolayer. On the day of the assay, the media was aspirated and cells were incubated at 37°C in a non-CO2 incubator for 1 h with 0.5 ml XF base medium (102353-100; Agilent) supplemented with pyruvate (1 mmol/L), glutamine (2 mmol/L), and glucose (10 mmol/L). Oligomycin (56 μl, 10 µmol/L), carbonyl cyanide 4-[trifluoromethoxy] phenylhydrazone (62 μl, 10 µmol/L), and rotenone+antimycin-A (69 μl, 5 µmol/L) were added to the flux pack wells. The oxygen consumption rate was then determined according to the manufacturer’s instructions. For LA or NAC treatment, rat and human ECs were treated with 15 or 20 µM LA or 100 µM NAC for 12 or 24 h, respectively. The oxygen consumption rates were measured as described above.
Tube formation assay
Tube formation assay was performed using the angiogenesis assay kit (ab204726; Abcam). Briefly, after PECs were isolated from WT, untreated NFU1G206C rats (NFU1 group), or NFU1G206C rats supplied with LA (NFU1+LA group) and grown to 70–80% confluence. They were trypsinized and 20,000 cells/well were plated with complete EC medium (1101; ScienCell) in 96-well plates precoated with extracellular matrix solution provided in the kit. The plates were incubated for 18 h and visualized under an inverted microscope (Revolve; ECHO). The number of tubes and junctions was counted as a function of tube formation capacity by an investigator blinded to the experimental groups. For the NAC treatment, rat PECs were incubated with 100 µM NAC for 12 h.
Cell transwell migration assay
Cell migration was analyzed using Cell Migration/Chemotaxis Assay Kit (ab235673; Abcam). Briefly, after isolated PECs attained a confluence of 70–80%, the cells were trypsinized and 50,000 cells/well were plated in the upper chamber of the kit in basal EC medium. Next, the chemotaxis agent was added to the lower chamber. After 48 h, the upper chamber was swabbed with a sterile cotton swab to remove non-migrated cells. The lower chamber was washed twice in assay washing buffer and centrifuged at 1,000 g for 5 min. The cell dissociation solution with the dye was added and the cells were incubated at 37°C for 1 h. The number of migrated cells was estimated according to the manufacturer’s instructions.
Scratch wound healing assay
The scratch healing capacity was measured using a previously published protocol with minor modifications (Liang et al., 2007). Briefly, ECs were seeded into a 24-well plate precoated with poly-D-lysine. Once the cells reached ∼90% confluence, a sterile 200-μl pipette tip was used to create a wound by dragging it perpendicularly across the cell monolayer. After washing the wells with serum-free EC media (1101b; ScienCell) to remove debris and floating cells, fresh complete EC media (1101; ScienCell) was added. The wound area was imaged at 0 and 12 h after scratching using an inverted microscope (Revolve; ECHO). The images of the wound area at each time point were manually traced and measured using ImageJ software by an investigator blinded to the experimental setup. The area of wound closure was determined as a difference between the initial wound area at 0 h and the wound area 12 h after scratching.
RNA extraction
Human lung samples obtained from PHBI were stored at −80°C. RNA isolation and purification were performed using the RNeasy Kit (#74104; Qiagen) according to the manufacturer’s protocol. In brief, the 30 mg of lung tissue powder prepared in liquid nitrogen was homogenized in the provided lysis buffer by passing the lysate through a 20-gauge needle attached to a sterile plastic syringe 5–10 times until a homogeneous lysate was achieved. The lysate was centrifuged for 3 min at 10,000 g and the supernatant was mixed with an equal volume of 70% ethanol. The mixture transferred to the RNeasy spin column was centrifuged for 15 s at ≥8,000 g, the column was washed twice with the kit washing buffer, and the 30–50 μl of RNAse-free water was added to the spin column membrane and centrifuged for another 1 min to elute the RNA. 1 µg of RNA was converted to C-DNA using the iScript Reverse Transcription Supermix for quantitative RT-PCR (RT-qPCR) (1708841; Bio-Rad) according to the manufacturer’s protocol. qPCR was performed using IDT primetime primers designed for human NFU1 (5′-TCTTCAGATCCTGCTTCTCCT-3′) (5′-ACCAGATTTCATCACTGTCACA-3′) and B2M (5′-ACCTCCATGATGCTGCTTAC-3′) (5′-GGACTGGTCTTTCTATCTCTTGT-3′) as the housekeeping gene using the iTaq Universal SYBR Green Supermix (#1725122; Bio-Rad).
Western blotting
For the protein analysis, EC lysate was prepared using radioimmunoprecipitation assay buffer (89900; Thermo Fisher Scientific) with protease and phosphatase inhibitor cocktail (78441; Thermo Fisher Scientific). The lysates were centrifuged at 10,000 g for 10 min and the protein concentrations were determined using the Pierce BCA Protein Assay Kit (23225; Thermo Fisher Scientific). The samples were incubated in 6X Laemmli sample buffer for 5 min at 95°C (Boston Bioproducts, Inc.). The proteins were separated electrophoretically on a 4–20% Mini-PROTEAN TGX stain-free gels (Bio-Rad Laboratories, Inc.) and transferred to a membrane using the PowerPac Universal power supply and Trans-Blot Turbo transferring system (Bio-Rad Laboratories, Inc.). Membranes blocked with 5% BSA were probed at 4°C overnight using the following antibodies: anti-DLL4 (1:1,000, NB600-892; Novus Biologicals), anti-RAMP3 (1:1,000, sc-365313; Santa Cruz Biotechnology, Inc.), anti-IGFBP3 (1:1,000, sc-365936; Santa Cruz Biotechnology, Inc.), anti-KCNE3 (1:1,000, sc-393841; Santa Cruz Biotechnology, Inc.), anti-pSer616DRP1 (1:1,000, PA5-64821; Thermo Fisher Scientific), anti-pSer637DRP1 (4867s, anti-pSer616DRP1; Cell Signaling), anti-PGC1α (1:1,000, sc-517380; Santa Cruz Biotechnology, Inc.), anti-LA (1:1,000, 437695; Millipore), and anti-β-actin (1:1,000, sc-47778; Santa Cruz Biotechnology, Inc.) antibodies. Anti-rabbit IgG, HRP-linked antibodies (1:5,000, 7074S; Cell Signaling Technology) were used as secondary antibodies. Chemiluminescent bands were visualized using the ChemiDoc MP Imaging System (Bio-Rad Laboratories, Inc.) and the reactive bands were analyzed with the Image Lab software (Bio-Rad Laboratories, Inc.). Protein loading normalization was performed using free stain gels as described previously (Rivero-Gutiérrez et al., 2014). Some membranes were stripped and reprobed for more than one protein or used in more than one figure for the better logic flow.
Statistical analysis
Animals were randomly assigned for the experiments from the animal database. The morphology and density of large and small pulmonary vessels, histological examination, and cell migration data were analyzed in a blinded manner; other experiments were performed by investigators unaware of the tested hypothesis and expected outcomes. The data are reported as mean values (±SD). Significance was determined by either the unpaired t test or by one-way analysis of variance (ANOVA) followed by the Bonferroni multiple comparison test to compare all columns or selected pairs of columns using GraphPad Prism 7. For all statistical tests, P values <0.05 were considered statistically significant. The Grubbs’ test (extreme studentized deviate) was used to determine the significant outliers. This criterion was predetermined before the initiation of the data analysis.
Online supplemental material
Fig. S1 shows the result of Seahorse XF glycolysis stress test in PECs isolated from WT, NFU1, and NFU1 rats treated with LA. The experiment confirms that a significant glycolytic shift observed in NFU1 PECs is efficiently downregulated back to the control levels by LA supplementation. Fig. S2 shows RVSP, dP/dt max, dP/dt min, and Fulton index in WT rats supplemented or not with LA. This result provides the evidence of no pulmonary hemodynamic changes in the LA-treated WT rats. Fig. S3 investigates the effects of NAC on the mitochondrial respiration and tubing formation PECs isolated from NFU1 mutant rats. Contrary to LA, NAC treatment produces no significant improvement in NFU1 PEC’s mitochondrial respiration and further impaired the angiogenic ability of NFU1G206C PECs to form an endothelial network in the Matrigel.
Data availability
All data generated in this study are presented in the manuscript and/or supplementary information. Any further information required for reproducing results or replicating the procedures will be made available by the corresponding author upon reasonable request.
Acknowledgments
We thank the PHBI for providing pulmonary tissue samples from iPAH and non-PAH subjects.
This work was supported by National Institutes of Health (NIH) R01 grants HL133085 and HL160666, and the American Heart Association grant 969574 to O. Rafikova; NIH R01 grants HL132918 and HL151447 to R. Rafikov; NIH R01 grants HL150638, HL130261, and HL172488 to E.A. Goncharova; NIH R01 grants HL139664, HL160018, HL134776, and HL59886 to V. de Jesus Perez; NIH K99 grant 1K99HL171869 to J. James; the American Heart Association grant 23CDA1050843 to M. Niihori; and the American Heart Association grant 23DIVSUP1065885 to O.S. Lawal. Funding for the PHBI is provided under an National Heart, Lung, and Blood Institute R24 grant, HL123767, and by the Cardiovascular Medical Research and Education Fund.
Author contributions: M. Niihori: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Visualization, Writing—original draft, J. James: Formal analysis, Investigation, Methodology, Validation, Visualization, Writing—original draft, M.V. Varghese: Data curation, Investigation, Methodology, Validation, N. McClain: Investigation, O.S. Lawal: Investigation, Validation, Visualization, R.C. Philip: Formal analysis, Methodology, Software, Visualization, Writing—review & editing, B.K. Baggett: Data curation, Investigation, D.A. Goncharov: Investigation, Resources, V. de Jesus Perez: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, E.A. Goncharova: Resources, Writing—original draft, Writing—review & editing, R. Rafikov: Conceptualization, Funding acquisition, Investigation, Supervision, O. Rafikova: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Writing—original draft, Writing—review & editing.

