


To distinguish pathogens from commensals, the intestinal epithelium employs cytosolic innate immune sensors. Activation of the NAIP–NLRC4 inflammasome initiates extrusion of infected intestinal epithelial cells (IEC) upon cytosolic bacterial sensing. We previously reported that activation of the inflammasome in tuft cells, which are primarily known for their role in parasitic infections, leads to the release of prostaglandin D2 (PGD2). We observe that NAIP–NLRC4 inflammasome activation in tuft cells leads to an antibacterial response with increased IL-22 and antimicrobial protein levels within the small intestine, which is dependent on PGD2 signaling. A NKp46+ subset of ILC3 expresses the PGD2 receptor CRTH2 and is the source of the increased IL-22. Inflammasome activation in tuft cells also leads to better control of Salmonella Typhimurium in the distal small intestine. However, tuft cells in the cecum and colon are dispensable for antibacterial immunity. These data support that intestinal tuft cells can also induce antibacterial responses, possibly in a tissue-specific manner.

Introduction
The gastrointestinal tract as a barrier tissue is home to many complex interactions between host tissue, the microbiota, and potential pathogenic microbes. One challenge intestinal epithelial cells (IECs) must overcome is to distinguish commensals from pathogens. An innate pathway that IECs use to specifically recognize bacterial pathogens is the NAIP–NLRC4 inflammasome. It is activated when sensor NAIP proteins recognize their respective ligands, bacterial T3SS components or flagellin, within the cytosol of the host cell (Tenthorey et al., 2014; Kofoed and Vance, 2011; Zhao et al., 2011, 2016; Rauch et al., 2016; Rayamajhi et al., 2013; Yang et al., 2013). This leads to a conformational change allowing binding of the adaptor NLRC4 and results in the formation of the full inflammasome and activation of Caspase-1. The strictly cytosolic location of this process ensures a distinction between commensal bacteria, which do not invade IEC or use secretion systems to inject effectors, and pathogens.
Induction of the NAIP–NLRC4 inflammasome in IECs is important for host defense against several pathogenic bacteria. By inducing extrusion of the infected epithelial cell, activation of the NAIP–NLRC4 inflammasome in IEC decreases Salmonella Typhimurium tissue invasion at acute timepoints, protects against Citrobacter rodentium, and renders mice virtually resistant to Shigella invasion (Sellin et al., 2014; Rauch et al., 2017; Mitchell et al., 2020; Hausmann et al., 2020; Fattinger et al., 2021; Nordlander et al., 2014). This extrusion of infected cells does not affect luminal bacterial levels measurably but prevents tissue pathology caused by an immune reaction to the invading bacteria (Fattinger et al., 2021; Sellin et al., 2014) In conjunction with extrusion, inflammasome activation in epithelial cells releases IL-18 and prostaglandin E2 (PGE2), a fast-acting lipid inflammatory mediator (Rauch et al., 2017).
These studies determined the impacts of activating the NAIP–NLRC4 inflammasome in all IECs. Inflammasomes are a group of innate sensors activated by different pathogenic insults that all lead to the same canonical outcome of Caspase-1 activation, cytokine cleavage, and pore formation (Churchill et al., 2022). However, it was reported that NLRP6 inflammasome activation in specialized goblet cells causes mucus release (Birchenough et al., 2016), suggesting that inflammasome activation in specific IEC subpopulations can activate different signaling cascades and downstream outcomes.
One rare subset of IEC, called tuft cells, is critical for sensing parasites and signals on immune cells to initiate antiparasitic responses within the small intestine (Von Moltke et al., 2016; McGinty et al., 2020; Schneider et al., 2018; Nadjsombati et al., 2018; Gerbe et al., 2016; Howitt et al., 2016; Lei et al., 2018). Tuft cell activation during parasite infection leads to a signaling circuit of tuft cell–derived IL-25 and the eicosanoid LTC4 to innate lymphoid cells type 2 (ILC2), inducing tuft cell amplification and other extensive remodeling of the intestinal epithelium required for an efficient antiparasitic response.
The involvement of tuft cells in the intestinal epithelial sensing of bacterial pathogens and ensuing immune reactions is less understood. Within the lung, the release of Pseudomonas aeruginosa quorum sensing molecules activates the bitter sensing taste receptors, Tas2R105 and Tas2R108, on tuft cells to induce Ca2+ influx through Trpm5 (Hollenhorst et al., 2023). Tas2R108 also allows for urethral tuft cells to sense heat-killed uropathogenic Escherichia coli (Kandel et al., 2018). Within the gastrointestinal tract, tuft cells have been shown to be activated by succinate produced by microbiota (Lei et al., 2018; Banerjee et al., 2020), a ligand that can also be produced by protists and helminths that leads to a type II immune response after tuft cell sensing (Nadjsombati et al., 2018). However, little is known about whether tuft cells specifically respond to pathogenic bacteria. It has been suggested that tuft cells in the intestine sense a specific bacterial ligand, N-C11-G, secreted by the pathogen Shigella flexneri, in an invasion-independent manner, and this sensing leads to the release of eicosanoids such as PGD2 (Xiong et al., 2022), a finding that recently has been questioned (Billipp et al., 2024). Synthesis of PGD2 occurs when arachidonic acid is metabolized by PTGS1/2 into PGH2, which is further metabolized into PGD2 by hematopoietic prostaglandin D synthase (HPGDS) (Funk, 2001). Tuft cells express high levels of eicosanoid synthesis enzymes, including PTGS1 and HPGDS (Gerbe et al., 2016; Haber et al., 2017; DelGiorno et al., 2020; Bezençon et al., 2008).
Tuft cells also express the NAIP–NLRC4 inflammasome, and we have previously shown that activation of this inflammasome specifically in tuft cells mediates the release of PGD2 (Oyesola et al., 2021). In the conditions tested, we observe that tuft cells are the only IECs of the small intestine that release PGD2 upon inflammasome activation, suggesting an active participation and perhaps tuft cell–specific signaling in epithelial bacterial pathogen sensing. During parasitic infection, mice lacking one of the receptors for PGD2, CRTH2, expel the helminth parasite Nippostrongylus brasiliensis more efficiently than wildtype (WT) control animals. This observation demonstrates that PGD2–CRTH2 has the potential to suppress the anti-helminth response (Oyesola et al., 2021). It is unknown what downstream impacts PGD2–CRTH2 signaling might have after NAIP–NLRC4 inflammasome activation during a bacterial infection.
One hint to the function of PGD2 during antibacterial responses is that in mice, RNA for the PGD2 receptor CRTH2 is expressed by ILC3s (Nagashima et al., 2019; Gury-BenAri et al., 2016; Xu et al., 2019). ILC3s are tissue-resident cells within the lamina propria that can release the cytokines IL-17 and IL-22, which are important for early antibacterial responses, and upregulate antimicrobial protein transcription in IECs (Valeri and Raffatellu, 2016).
Herein, we test the hypothesis that NAIP–NLRC4 inflammasome activation in tuft cells activates a PGD2–ILC3 signaling cascade to induce a host antibacterial response. We observe increased IL-22 levels after NAIP–NLRC4 inflammasome activation in tuft cells in a PGD2-dependent manner, specifically in the small intestine. Inconsistent with Xiong et al. (2022), we find no changes in CD45+ tuft cells after S. flexneri infection or protection from Shigella infection by tuft cells. Activation of the PGD2–ILC3 signaling cascade in the small intestine is sufficient to control S. Typhimurium in this tissue, but tuft cells and CRTH2 on ILC3 are not necessary, highlighting the complex layers of protection in innate defense from bacterial infection. Our findings support that tuft cells not only initiate antiparasitic immunity, but through activation of the NAIP–NLRC4 inflammasome, tuft cells mediate a signaling circuit that activates antibacterial responses.
Results and discussion
Tuft cell inflammasome activation leads to a PGD2-dependent antibacterial tissue response specifically in the small intestine
Our original observation of tuft cell inflammasome-induced PGD2 release was in small intestinal tissue (Oyesola et al., 2021). To assess our hypothesis of tuft cell signaling to ILC3 in small intestinal versus colon tissue, we used a system that allows specific activation of the NAIP–NLRC4 inflammasome in all tuft cells. iNLRC4 mice (Rauch et al., 2017; Mitchell et al., 2020; Roncaioli et al., 2023; Oyesola et al., 2021) were crossed with a tamoxifen-inducible tuft cell–specific Cre recombinase line (Pou2f3-CreERT2) (Fig. 1 A). iNLRC4 mice harbor the Nlrc4 gene and an IRES-GFP after a floxed Stop cassette in the Rosa26 locus and are on an Nlrc4−/− background. This crossing therefore yields mice that, upon tamoxifen treatment, express the Nlrc4 gene only in tuft cells, which we refer to as iNLRC4-Pou2f3-CreERT2. The dependence of the NLRC4 adaptor on the sensor NAIP proteins ensures that this system to study the NAIP–NLRC4 inflammasome only works in cells that naturally express NAIPs. Tamoxifen diet alone did not cause inflammasome activation and change the number of tuft cells in our mice (Fig. S1 A) and led to GFP (i.e., construct) expression in around 30–40% of tuft cells (Fig. S1 B).

Activation of the NAIP–NLRC4 inflammasome in tuft cells induces an antimicrobial response in small intestinal tissue. (A) Schematic representation of the crossing of iNLRC4 mice with Pou2f3-creERT2 mice to yield iNLRC4-Pou2f3-creERT2 mice. (B) DCLK-1 (tuft cells: green), Phalloidin (actin: yellow), propidium iodide (pyroptosing cells: red), and DAPI (nuclei: blue) in small intestines from C57BL/6 mice were treated with 0.2 µg/g of PA and 0.1 µg/g LFn-Fla for 60 min. Scale bar = 20 µm; representative image; n = 3 mice. (C) iNLRC4-Pou2f3-CreERT2 and Nlrc4−/− littermate controls were retro-orbitally injected with Flatox (LFn-FlaA [0.4 µg/g] and PA [0.8 µg/g]) and small intestines were harvested 4 h later. Epithelial cells were isolated from tissues and the percentage of tuft cells among epithelial cells was quantified by flow cytometry (n = 6–7, data combined from two experiments). (D–L) iNLRC4-Pou2f3-CreERT2 and Nlrc4−/− littermate controls were retro-orbitally injected with two FlaTox dosages (LFn-FlaA [0.1 µg/g] and PA [0.2 µg/g]) 48 h apart. (J–L) Animals were injected 30 min before FlaTox with AZD1981 or vehicle intraperitoneally. Tissues were harvested 24 h after the last injection. IL-22 protein levels within the small intestine (D and J) and colon (G) were quantified by ELISA. Downstream targets of IL-22 receptor signaling, Reg3γ (E, H, and K), and Reg3β (F, I, and L) were measured by qPCR. Data pooled from three (D–I) or two (J–L) experiments (n = 13–16 [D and G], n = 9 [E, F, H, and I], n = 6–12 [J–L]) LOD = limit of detection. Mann–Whitney test was performed to determine significance. *, P < 0.05, **, P < 0.01, ****, P < 0.0001, ns = not significant.
Activation of the NAIP–NLRC4 inflammasome in tuft cells induces an antimicrobial response in small intestinal tissue. (A) Schematic representation of the crossing of iNLRC4 mice with Pou2f3-creERT2 mice to yield iNLRC4-Pou2f3-creERT2 mice. (B) DCLK-1 (tuft cells: green), Phalloidin (actin: yellow), propidium iodide (pyroptosing cells: red), and DAPI (nuclei: blue) in small intestines from C57BL/6 mice were treated with 0.2 µg/g of PA and 0.1 µg/g LFn-Fla for 60 min. Scale bar = 20 µm; representative image; n = 3 mice. (C) iNLRC4-Pou2f3-CreERT2 and Nlrc4−/− littermate controls were retro-orbitally injected with Flatox (LFn-FlaA [0.4 µg/g] and PA [0.8 µg/g]) and small intestines were harvested 4 h later. Epithelial cells were isolated from tissues and the percentage of tuft cells among epithelial cells was quantified by flow cytometry (n = 6–7, data combined from two experiments). (D–L) iNLRC4-Pou2f3-CreERT2 and Nlrc4−/− littermate controls were retro-orbitally injected with two FlaTox dosages (LFn-FlaA [0.1 µg/g] and PA [0.2 µg/g]) 48 h apart. (J–L) Animals were injected 30 min before FlaTox with AZD1981 or vehicle intraperitoneally. Tissues were harvested 24 h after the last injection. IL-22 protein levels within the small intestine (D and J) and colon (G) were quantified by ELISA. Downstream targets of IL-22 receptor signaling, Reg3γ (E, H, and K), and Reg3β (F, I, and L) were measured by qPCR. Data pooled from three (D–I) or two (J–L) experiments (n = 13–16 [D and G], n = 9 [E, F, H, and I], n = 6–12 [J–L]) LOD = limit of detection. Mann–Whitney test was performed to determine significance. *, P < 0.05, **, P < 0.01, ****, P < 0.0001, ns = not significant.

Tuft cell inflammasome expression and activation effects. (A) Tuft cell numbers are not affected by genotype or tamoxifen diet. Colony C57BL/6 or Nlrc4–/–Pou2f3-creERT2+ and iNLRC4 Pou2f3-creERT2+ littermates were fed normal chow or tamoxifen chow for 7 days and the percentage tuft cells (CD24+SiglecF+) among EpCam+ cells was determined by flow cytometry (n = 3–7). (B) Percentage GFP positive (iNLRC4 expressing) tuft- and non-tuft cells from mice treated as in A (n = 3–4). (C) Epithelial monolayers were treated with FlaTox alone or FlaTox and BABTA-AM or Disulfiram and supernatants were analyzed for eicosanoid release. n = 4–5 combined data from two experiments; ****, P < 0.0001. (D and E) iNLRC4-Pou2f3-CreERT2 and Nlrc4−/−Pou2f3-CreERT2 littermate controls were retro-orbitally injected with two FlaTox dosages (LFn-FlaA [0.1 µg/g] and PA [0.2 µg/g]) 48 h apart and tissues were harvested 24 h after the last injection. IL-18 (D) and IL-1α (E) protein levels within the small were quantified by ELISA, data pooled from multiple experiments (n = 12–18).
Tuft cell inflammasome expression and activation effects. (A) Tuft cell numbers are not affected by genotype or tamoxifen diet. Colony C57BL/6 or Nlrc4–/–Pou2f3-creERT2+ and iNLRC4 Pou2f3-creERT2+ littermates were fed normal chow or tamoxifen chow for 7 days and the percentage tuft cells (CD24+SiglecF+) among EpCam+ cells was determined by flow cytometry (n = 3–7). (B) Percentage GFP positive (iNLRC4 expressing) tuft- and non-tuft cells from mice treated as in A (n = 3–4). (C) Epithelial monolayers were treated with FlaTox alone or FlaTox and BABTA-AM or Disulfiram and supernatants were analyzed for eicosanoid release. n = 4–5 combined data from two experiments; ****, P < 0.0001. (D and E) iNLRC4-Pou2f3-CreERT2 and Nlrc4−/−Pou2f3-CreERT2 littermate controls were retro-orbitally injected with two FlaTox dosages (LFn-FlaA [0.1 µg/g] and PA [0.2 µg/g]) 48 h apart and tissues were harvested 24 h after the last injection. IL-18 (D) and IL-1α (E) protein levels within the small were quantified by ELISA, data pooled from multiple experiments (n = 12–18).
We first sought to determine whether tuft cells undergo extrusion after inflammasome activation as enterocytes do (Sellin et al., 2014; Rauch et al., 2017). WT mice were retro-orbitally injected with FlaTox (Rauch et al., 2017; Von Moltke et al., 2012), which consists of the NAIP5/6 ligand, flagellin, conjugated to the N-terminal fragment of lethal factor from Bacillus anthracis, and protective antigen (PA). PA forms a pore that allows the flagellin conjugated to the N-terminus of the lethal factor to be transported in the cytosol of the host cell and thus selectively activate the NAIP–NLRC4 inflammasome. We observe tuft cells (DCLK1+) positive for propidium iodide, signifying pore formation after inflammasome activation, and purse-like remodeling of actin demonstrating extrusion around tuft cells in intestines of mice injected with FlaTox, confirming our previous observations that normal WT tuft cells can activate NAIP–NLRC4 (Oyesola et al., 2021) (Fig. 1 B). Comparing iNLRC4-Pou2f3-CreERT2 mice to inflammasome deficient mice, the percentage of tuft cells detectable by flow cytometry among IECs is significantly decreased 4 h after inflammasome activation specifically in tuft cells (Fig. 1 C). Taken together, these data support that tuft cells are extruded from the epithelial layer after tuft cell activation of the NAIP–NLRC4 inflammasome.
We have previously shown that PGD2 is released rapidly after NAIP–NLRC4 inflammasome activation in tuft cells (Oyesola et al., 2021). Since we observed dye uptake by tuft cells while still in the epithelium, and Gasdermin pores have recently been reported as necessary for eicosanoid release in macrophages (Mehrotra et al., 2024), we wondered whether Gasdermin pores and the resulting calcium flux would be necessary for tuft cells to activate the calcium-dependent phospholipase A2 and therefore the eicosanoid synthesis cascade. In vitro–cultured primary epithelial cells release significant amounts of prostaglandin into the supernatant after inflammasome activation, and this can be blocked with the Gasdermin inhibitor disulfiram or the calcium chelator BABTA-AM, supporting this hypothesis (Fig. S1 C). Since lipid mediators are released quickly after synthesis, we hypothesized that PGD2 release would occur proceeding or during cell extrusion, allowing signaling to nearby cells. Small intestinal ILC3 from the lamina propria express CRTH2 by RNA sequencing analysis (Nagashima et al., 2019; Gury-BenAri et al., 2016; Xu et al., 2019). Therefore, we hypothesized that tuft cell release of PGD2 would signal to activate ILC3 during acute bacterial infection. To study a potential tuft cell–PGD2–ILC3 communication axis, we first analyzed levels of one of the ILC3 signature cytokines, IL-22, in intestinal tissue after tuft cell inflammasome activation. After two doses of Flatox spaced 48 h apart, we observed a significant increase in IL-22 protein levels within the small intestine in iNLRC4-Pou2f3-CreERT2 mice compared with Nlrc4−/− littermates (Fig. 1 D). In conjunction, transcription of antimicrobial proteins downstream of IL-22 signaling, Reg3γ (Fig. 1 E), and Reg3β (Fig. 1 F) is significantly increased in iNLRC4-Pou2f3-CreERT2 mice compared with Nlrc4−/− littermates after inflammasome activation. Inflammasome-associated proteins IL-18 and IL-1α were not significantly different between groups (Fig. S1, D and E), presumably due to low expression in tuft cells (Haber et al., 2017), and IL-1β and IL-25 were not detectable. IL-22 protein levels, Reg3γ, and Reg3β were however not significantly different in the colon (Fig. 1, G–I), consistent with previous literature that demonstrates that small intestinal and colonic tuft cells can have different functions (Strine and Wilen, 2022). These results demonstrate that IL-22, an ILC3 and Th17 signature cytokine, and antimicrobial transcripts within the small intestine are increased after NAIP–NLRC4 inflammasome activation in tuft cells.
To determine if the observed IL-22 increase after tuft cell inflammasome activation within the small intestine is dependent on CRTH2 signaling, we administered an inhibitor specific to this receptor, AZD1981. Treatment with AZD1981 completely abrogated the increase in small intestine IL-22 in iNLRC4-Pou2f3-CreERT2 mice after tuft cell inflammasome activation (Fig. 1 J). AZD1981 also blocks increases in Reg3γ (Fig. 1 K) and Reg3β (Fig. 3 L) within the small intestine of iNLRC4-Pou2f3-CreERT2 mice, confirming that the observed antimicrobial response is dependent on PGD2 signaling.
Tuft cells play no role in protection from Shigella in the colon
Our observation on tuft cell inflammasome activation and PGD2 effects were specific to the small intestine. A previous report suggested that tuft cells can sense a secreted extracellular bacterial ligand (as opposed to bacterial invasion) leading to tuft cell proliferation and suppression of bacterial growth during infection with S. flexneri with effects observed both in the small intestine and colon (Xiong et al., 2022). A more recent report could find no stimulation of tuft cells by this ligand (Billipp et al., 2024). Shigella activates the NAIP–NLRC4 inflammasome, which leads to complete restriction of colonization with Shigella strain 2457T in WT mice in the colon and cecum, but small intestinal colonization was not assessed (Mitchell et al., 2020). We therefore wondered whether the Shigella restricting effect observed by Xiong et al. was due to the tuft cell inflammasome sensing bacterial invasion rather than a surface receptor sensing a secreted ligand. To test this, we crossed Pou2f3−/− to an Nlrc4-deficient background to generate Shigella susceptible littermate mice with or without tuft cells, with or without the NAIP–NLRC4 inflammasome. When we analyzed large intestinal infection and inflammation, Shigella epithelial replication was dependent on NLRC4, whereas Shigella CFUs were largely undetectable in cecum and colon IECs in Pou2f3–/– mice, similar to WT controls. Weight loss (Fig. S2 A), inflammation as determined by tissue shrinkage (Fig. S2, B and C), and epithelial colonization (Fig. S2 D) were also dependent on NLRC4 status and unchanged between Nlrc4−/− and Nlrc4−/−Pou2f3−/− littermates. However, even in the absence of NLRC4, we could not observe consistent colonization of small intestinal tissue by Shigella. In addition, we did not observe the previously reported increase in total tuft cells (Fig. S2, E and F) or CD45+ tuft cells after S. flexneri infection of WT mice (Fig. S2, G and H). Thus, our observations disagree with the previously reported role of tuft cells for defense against Shigella infection in the colon or cecum. However, as we observed no colonization of the small intestine, the role of the tuft cell inflammasome in this tissue during bacterial infection was unclear.

The tuft cell inflammasome does not protect from colon Shigella infection. (A–E) C57BL/6, Pou2f3−/−, Nlrc4−/−, and Pou2f3−/−Nlrc4−/− mice were pretreated with streptomycin orally and then infected with 5 × 107–8 CFUs of Shigella flexneri by oral gavage. Body weight was measured throughout the experiment (A), and 2 days after infection, tissues were harvested and analyzed for caecum length (B), colon length (C), CFUs (D), and fecal MPO (E). (F–I)Nlrc4+/− mice were pretreated with oral gavage of streptomycin sulfate and then infected with 5 × 106 CFUs of Shigella flexneri by oral gavage. 2 days after infection, the small intestine was harvested, and epithelial cells were isolated to be analyzed by flow cytometry. (F and G) (F) Representative flow plots of total tuft cells from naive and infected animals, which is enumerated in G. (H) Representative flow plots of CD45+ and CD45− tuft cells, numbers are enumerated in I. Each data point represents one mouse, pooled data from multiple experiments (n = 6–11 [A–E], n = 11–12 [G], n = 7–8 [I]), and bars shown as median. Mann–Whitney test was performed to determine significance. *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001.
The tuft cell inflammasome does not protect from colon Shigella infection. (A–E) C57BL/6, Pou2f3−/−, Nlrc4−/−, and Pou2f3−/−Nlrc4−/− mice were pretreated with streptomycin orally and then infected with 5 × 107–8 CFUs of Shigella flexneri by oral gavage. Body weight was measured throughout the experiment (A), and 2 days after infection, tissues were harvested and analyzed for caecum length (B), colon length (C), CFUs (D), and fecal MPO (E). (F–I)Nlrc4+/− mice were pretreated with oral gavage of streptomycin sulfate and then infected with 5 × 106 CFUs of Shigella flexneri by oral gavage. 2 days after infection, the small intestine was harvested, and epithelial cells were isolated to be analyzed by flow cytometry. (F and G) (F) Representative flow plots of total tuft cells from naive and infected animals, which is enumerated in G. (H) Representative flow plots of CD45+ and CD45− tuft cells, numbers are enumerated in I. Each data point represents one mouse, pooled data from multiple experiments (n = 6–11 [A–E], n = 11–12 [G], n = 7–8 [I]), and bars shown as median. Mann–Whitney test was performed to determine significance. *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001.
A subset of ILC3 express the PGD2 receptor and increase IL-22 after tuft cell inflammasome activation in the small intestine
We turned our focus on the signaling leading to the IL-22 increase after tuft cell inflammasome activation in the small intestine. To confirm the observation of PGD2 receptor expression on ILC3s by single-cell RNA sequencing, we determined if the Gpr44 locus, which encodes the CRTH2 protein, is active in ILC3s. We used a novel Gpr44 transcriptional reporter mouse. These mice have a floxed coding sequence of Gpr44 with proximal GFP, which will be expressed once the Gpr44 locus is deleted (Fig. S3, A and B). Using these mice crossed to Rorγt-Cre mice, we observed GFP+CD4− cells in the villus lamina propria of all regions of the healthy, unperturbed small intestine (Fig. 2 A and Fig. S3 C). Rare CD4+GFP+ cells were also observable. Receptor-positive cells are on average distanced similarly to tuft cells as CD4+GFP− cells (Fig. S3 D).
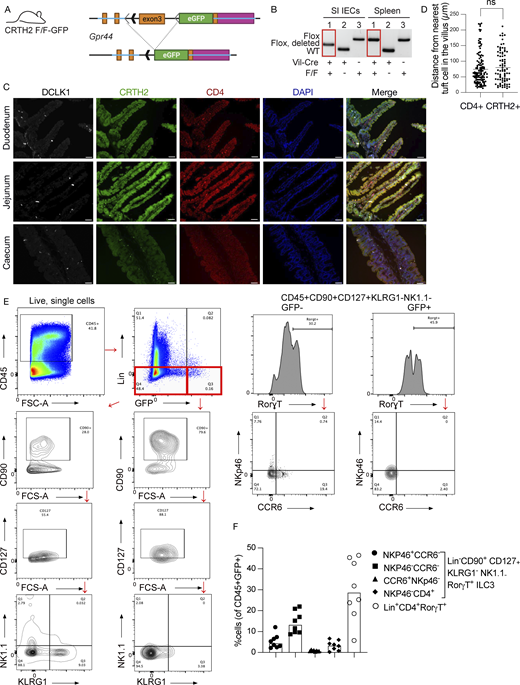
Validation of Gpr44fl/fl-GFP mice and determination of ILC3s within the lamina propria. (A) Schematic of mouse design. LoxP sites were inserted into the Gpr44 exon 3, followed by the sequence encoding eGFP. The presence of Cre recombinase excises the targeted segment of Gpr44 exon 3, resulting in deletion and allowing for transcription of eGFP from the endogenous Gpr44 promoter. (B) Genotyping PCR showing cell lineage-specific deletion of the targeted Gpr44 exon 3 sequence. Recombination occurs only in the presence of Cre in a cell lineage-specific manner in small intestinal (SI) epithelial cells (IECs) but not splenocytes in Vil1-Cre Gpr44fl/fl-GPF mice. WT allele = 239 bp; Flox allele = 348 bp; Flox allele after recombination = 279 bp. (C and D) Representative images of duodenum, jejunum, and cecum sections from naive Gpr44fl/+Rorγt-Cre+ mice showing cells positive for DCLK-1 (white), CRTH2-GFP (green), and CD4 (red); blue color indicates DAPI counterstained nuclei. Scale bar = 50 µm (C); representative image (D) quantification of CD4+ cells or CRTH2+ cells from nearest tuft cells. n = 3 mice; ns = not significant. (E) Lamina propria cells were isolated from Gpr44fl/flRorγt-Cre or Gpr44fl/+Rorγt-Cre and stained with a flow panel as described in Materials and methods to identify ILC3s. To analyze the ILC3s, CD45+ cells were gated and lineage negative (CD3−, CD19−, and CD14−) cells were classified into two groups: GFP high and GFP low. Both groups were then further gated into CD90+, CD127+, NK1.1−, KLRG1−, and Rorγt+. Then they were further analyzed for the presence or absence of CCR6 and NKp46. (F) Quantification of different subtypes of ILC3s out of total GFP (CRTH2)-expressing cells. Source data are available for this figure: SourceData FS3.
Validation of Gpr44fl/fl-GFP mice and determination of ILC3s within the lamina propria. (A) Schematic of mouse design. LoxP sites were inserted into the Gpr44 exon 3, followed by the sequence encoding eGFP. The presence of Cre recombinase excises the targeted segment of Gpr44 exon 3, resulting in deletion and allowing for transcription of eGFP from the endogenous Gpr44 promoter. (B) Genotyping PCR showing cell lineage-specific deletion of the targeted Gpr44 exon 3 sequence. Recombination occurs only in the presence of Cre in a cell lineage-specific manner in small intestinal (SI) epithelial cells (IECs) but not splenocytes in Vil1-Cre Gpr44fl/fl-GPF mice. WT allele = 239 bp; Flox allele = 348 bp; Flox allele after recombination = 279 bp. (C and D) Representative images of duodenum, jejunum, and cecum sections from naive Gpr44fl/+Rorγt-Cre+ mice showing cells positive for DCLK-1 (white), CRTH2-GFP (green), and CD4 (red); blue color indicates DAPI counterstained nuclei. Scale bar = 50 µm (C); representative image (D) quantification of CD4+ cells or CRTH2+ cells from nearest tuft cells. n = 3 mice; ns = not significant. (E) Lamina propria cells were isolated from Gpr44fl/flRorγt-Cre or Gpr44fl/+Rorγt-Cre and stained with a flow panel as described in Materials and methods to identify ILC3s. To analyze the ILC3s, CD45+ cells were gated and lineage negative (CD3−, CD19−, and CD14−) cells were classified into two groups: GFP high and GFP low. Both groups were then further gated into CD90+, CD127+, NK1.1−, KLRG1−, and Rorγt+. Then they were further analyzed for the presence or absence of CCR6 and NKp46. (F) Quantification of different subtypes of ILC3s out of total GFP (CRTH2)-expressing cells. Source data are available for this figure: SourceData FS3.

ILC3s are the source of increased IL-22 within the small intestine after NAIP–NLRC4 inflammasome activation in tuft cells. (A) Representative images of ileum sections from naive Gpr44fl/+Rorγt-Cre+ mice showing cells positive for DCLK-1 (white), CRTH2-GFP (green), and CD4 (red); blue color indicates DAPI counterstained nuclei. Scale bar = 50 µm; representative image; n = 3 mice. (B) Flow cytometry quantification of CRTH2 (GFP)-positive cells among Lin−ILC3 and Lin+CD4 T cells. (C–H) Flow cytometry analysis of IL-22 producing ILC3s after inflammasome activation specifically in tuft cells. iNLRC4-Pou2f3-creERT2 mice or littermate controls were administered two FlaTox dosages (LFn-FlaA [0.1 µg/g] and PA [0.2 µg/g]) 48 h apart and the small intestine was harvested 24 h after last Flatox injection. Cells from the lamina propria were isolated and intracellular IL-22 was measured by flow cytometry. (C) Representative flow plots of intracellular IL-22 in NKp46+CCR6− ILC3s from Nlrc4−/− and iNLRC4-Pou2f3-creERT2 after Flatox administration. (D–H) Quantification of the frequency of IL-22+ cells among different subtypes of ILC3s. Each data point represents one mouse, pooled data from multiple experiments (n = 8 [B], n = 13–14 [D–H]), and bars shown as median. Mann–Whitney test was performed to determine significance. *, P < 0.05, ns = not significant.
ILC3s are the source of increased IL-22 within the small intestine after NAIP–NLRC4 inflammasome activation in tuft cells. (A) Representative images of ileum sections from naive Gpr44fl/+Rorγt-Cre+ mice showing cells positive for DCLK-1 (white), CRTH2-GFP (green), and CD4 (red); blue color indicates DAPI counterstained nuclei. Scale bar = 50 µm; representative image; n = 3 mice. (B) Flow cytometry quantification of CRTH2 (GFP)-positive cells among Lin−ILC3 and Lin+CD4 T cells. (C–H) Flow cytometry analysis of IL-22 producing ILC3s after inflammasome activation specifically in tuft cells. iNLRC4-Pou2f3-creERT2 mice or littermate controls were administered two FlaTox dosages (LFn-FlaA [0.1 µg/g] and PA [0.2 µg/g]) 48 h apart and the small intestine was harvested 24 h after last Flatox injection. Cells from the lamina propria were isolated and intracellular IL-22 was measured by flow cytometry. (C) Representative flow plots of intracellular IL-22 in NKp46+CCR6− ILC3s from Nlrc4−/− and iNLRC4-Pou2f3-creERT2 after Flatox administration. (D–H) Quantification of the frequency of IL-22+ cells among different subtypes of ILC3s. Each data point represents one mouse, pooled data from multiple experiments (n = 8 [B], n = 13–14 [D–H]), and bars shown as median. Mann–Whitney test was performed to determine significance. *, P < 0.05, ns = not significant.
To further confirm this immunohistochemistry observation, we assessed GFP expression in cells of the small intestinal lamina propria by flow cytometry (Fig. 2 B; and Fig. S3, E and F). By parent population, the biggest percentage of Gpr44-GFP expressing cells was in the group of NKp46+CCR6− ILC3 (∼7%), followed by NKp46−CCR6−CD4+ cells. As a percentage of GFP+ cells, on average, most cells were lineage+CD4+Rorγt+ cells, albeit with large mouse-to-mouse variability, followed by NKp46−CCR6− cells (Fig. S3 F).
To determine if a specific subgroup of these receptor-expressing cell types is the source of increased IL-22 within the small intestine after tuft cell inflammasome activation, we used flow cytometry to measure intracellular IL-22 levels in the cell types that have been shown to produce IL-22 (Perusina Lanfranca et al., 2016) and that we observed to express CRTH2. There was a significant increase of IL-22 expressing ILC3s in iNLRC4-Pou2f3-CreERT2 mice compared with Nlrc4–/–-Pou2f3-CreERT2 littermates after inflammasome activation only in the NKp46+CCR6− ILC3 population (Fig. 2, C and D), which are the ILC3 subtype with the most CRTH2 expressing cells. We observed no significant differences in the number of IL-22+ NKp46−CCR6− or NKp46−CCR6+ or NKP46−CD4+ ILC3 or CD4+Lineage+Rorγt+ T cells (Fig. 2, E–H). Since <4% of the other ILC3 subtypes express the receptor, it is possible that an increase in IL22 is not detected in their parent population. These data suggest that ILC3s contain a subpopulation of CRTH2-expressing cells responsible for increased IL-22 after tuft cell–specific inflammasome activation.
Tuft cells are necessary for the induction of an antimicrobial response in the small intestine but not Salmonella defense
To address whether the observed PGD2-ILC3 signaling is necessary for defense during infection, we bred the Gpr44fl-reporter mice to obtain littermates sufficient or deficient in CRTH2 in Rorγt expressing cells. When we analyzed small intestinal tissue of naive mice, we did not notice changes in the populations of reporter-expressing cells in receptor-deficient animals compared with mice where ILC3 can express the CRTH2 receptor (not shown), suggesting there is no role for this receptor in the development of an ILC3 subtype.
We did not observe small intestinal tissue colonization by Shigella or, another pathogen often used in the study of ILC3, C. rodentium. Therefore, to study the role of CRTH2+ ILC3 during infection and the resulting increase in PGD2 tissue levels through tuft cells, we infected mice with Salmonella Typhimurium ΔSsaR. These bacteria lack a component of the SPI-2 T3SS and therefore cannot successfully evade the phagosome of myeloid cells (Pfeifer et al., 1999), which leads to productive infection of only epithelial cells via the SPI-I T3SS. In contrast to WT Salmonella, which is lethal in C57BL/6 mice, this strain is cleared over a week. Infected tuft cells will sense Salmonella via the NAIP–NLRC4 inflammasome and release PGD2. Thus, Salmonella ΔSsaR allows us to study a model inflammasome-activating gastrointestinal pathogen in the distal small intestine without the complications of systemic spread and mortality at later time points (Matheis et al., 2020; Gül et al., 2023). Tissue colony-forming units (CFUs) were determined from gentamicin-treated and washed samples to eliminate luminal bacteria. At the time point assessed, we did not observe changes in Salmonella small intestinal or cecum tissue invasion when Rorγt-expressing cells were lacking CRTH2 (Fig. 3, A and B). Similarly, we did not see a difference between receptor expressing Gpr44fl/+ Rorγt-Cre+ and receptor-deficient Gpr44fl/fl Rorγt-Cre+ IL22 expression (Fig. 3 C). This is reminiscent of previously observed redundancies between ILC3 subsets and IL-22–inducing signals in the infection of otherwise immune-sufficient hosts (Song et al., 2015; Rankin et al., 2016).

Tuft cells are necessary for an antimicrobial signature but tuft cell–PGD2–ILC3 signaling is not required for Salmonella defense. (A–C) Gpr44 fl/flRorγt-Cre or Gpr44fl/+Rorγt-Cre mice were infected through oral gavage with 5 × 107 CFUs of Salmonella Typhimurium ΔSsaR. All mice were pretreated with streptomycin 24 h prior to the infection. Tissue CFUs from small intestine (ileum) (A) or cecum (B) 4 days after infection. (C) Flow cytometry of Gpr44-GFP Lin−CD90+CD127+KLRG1−NK1.1−Rorγt+ IL22 expression. (D–F) Mice deficient or sufficient in tuft cells were infected through oral gavage with 5 × 107 CFUs of Salmonella Typhimurium ΔSsaR. Tissue CFUs from the small intestine (ileum) (D) or caecum (E) as well as quantification of tissue expression of Reg3γ mRNA (F) 5 days after infection. Each data point represents one mouse, pooled data from multiple experiments (n = 10–13 [A and B], n = 8 [C], n = 14–18 [D and E], n = 6–18 [F]), and bars shown as median. Mann–Whitney test was performed to determine significance. *, P < 0.05.
Tuft cells are necessary for an antimicrobial signature but tuft cell–PGD2–ILC3 signaling is not required for Salmonella defense. (A–C) Gpr44 fl/flRorγt-Cre or Gpr44fl/+Rorγt-Cre mice were infected through oral gavage with 5 × 107 CFUs of Salmonella Typhimurium ΔSsaR. All mice were pretreated with streptomycin 24 h prior to the infection. Tissue CFUs from small intestine (ileum) (A) or cecum (B) 4 days after infection. (C) Flow cytometry of Gpr44-GFP Lin−CD90+CD127+KLRG1−NK1.1−Rorγt+ IL22 expression. (D–F) Mice deficient or sufficient in tuft cells were infected through oral gavage with 5 × 107 CFUs of Salmonella Typhimurium ΔSsaR. Tissue CFUs from the small intestine (ileum) (D) or caecum (E) as well as quantification of tissue expression of Reg3γ mRNA (F) 5 days after infection. Each data point represents one mouse, pooled data from multiple experiments (n = 10–13 [A and B], n = 8 [C], n = 14–18 [D and E], n = 6–18 [F]), and bars shown as median. Mann–Whitney test was performed to determine significance. *, P < 0.05.
To further probe a role for tuft cell–ILC3 signaling during bacterial infection, we infected Pou2f3-sufficient and -deficient littermates with Salmonella ΔSsaR. Similar to our results with CRTH2 deficient ILC3, we did not observe differences in bacterial tissue invasion between mice sufficient or deficient in tuft cells (Fig. 3, D and E). However, a significant difference in the induction of the IL-22–induced antimicrobial gene Reg3γ in the small intestine (Fig. 3 F) indicates that tuft cell presence is required for this response in the small intestine.
The tuft cell inflammasome is sufficient to protect from infection in the small intestine
We did not observe a necessity for ILC3 CRTH2 expression in defense against Salmonella invasion, but a necessity for tuft cells in mounting a full antimicrobial response. Many innate defense pathways, especially in mucosal barrier tissues where pathogens have evolved to enter and circumvent host defenses, are redundant (Van Es et al., 2019; Songhet et al., 2010; Broz et al., 2010; Rankin et al., 2016). We therefore wondered whether in the absence of other potential ILC3-activating signals such as IL-18 (Valle-Noguera et al., 2023) and PGE2 (Duffin et al., 2016), released by enterocytes upon inflammasome activation and extrusion, tuft cell inflammasome sensing and the resulting signaling is sufficient to control pathogenic bacteria. We infected iNLRC4-Pou2f3-CreERT2 mice and littermate Nlrc4−/− controls. At 18 h or 5 days after infection, the ileum and cecum were harvested. We hypothesized that, as tuft cells are rare in specific pathogen free mice, their extrusion will not significantly affect pathogen numbers; however, PGD2-induced antimicrobial signaling could influence pathogen clearance at later time points in the absence of the strong early restriction through inflammasome-mediated cell extrusion (Fattinger et al., 2021; Sellin et al., 2014; Rauch et al., 2017). Indeed, there is no significant difference in CFUs in either the ileum (Fig. 4 A) or cecum (Fig. 4 B) at 18 h after infection, a timepoint where enterocyte inflammasome-induced extrusion shows the strongest effect in invasion prevention. A trend for a decrease in CFUs is, however, appreciable in the small intestine (Fig. 4 A). At 5 days after infection, iNLRC4-Pou2f3-CreERT2 mice show a significantly decreased CFU count compared with Nlrc4−/− within the small intestine (Fig. 4 C). No differences were observed in the cecum (Fig. 4 D). 50% of iNLRC4-Pou2f3-CreERT2 animals completely cleared the bacteria from their small intestine at day 5 after infection compared with only 25% of Nlrc4−/− mice (Fig. 4 E). Consistent with our earlier result with FlaTox induced inflammasome activation, we observed increased IL-22 protein levels within the small intestine of iNLRC4-Pou2f3-CreERT2 mice compared with Nlrc4−/− littermate controls at 5 days after infection (Fig. 4 F). No differences in IL-22 were observed in the cecum (Fig. 4 G). Based on these data, we conclude that PGD2–ILC3 signaling after tuft cell–specific inflammasome activation can contribute to bacterial pathogen clearance in the small intestine.

Tuft cell–specific NAIP–NLRC4 inflammasome activation mediates control of Salmonella Typhimurium infection in the small intestine. iNLRC4-Pou2f3-creERT2mice or Nlrc4−/− littermate controls were pretreated with oral gavage of streptomycin, then 24 h later infected with 5 × 107 CFUs of Salmonella Typhimurium ΔSsaR by oral gavage. (A–D) Tissue CFUs from the small intestine (A) and cecum (B) 18 h after infection and small intestine (C) and cecum (D) 5 days after infection. (E) Number of Nlrc4−/− or iNLRC4-Pou2f3-creERT2 mice that had no detectable CFUs at day 5 after infection. (F and G) IL-22 protein levels were determined by ELISA in small intestine (F) or cecum (G). Each data point represents one mouse, pooled data from multiple experiments (n = 9 [A and B], n = 14–20 [C–G]), bars shown as the median. Significance was determined by Mann–Whitney U test (A–D, F, and G). *, P < 0.05, **, P < 0.01.
Tuft cell–specific NAIP–NLRC4 inflammasome activation mediates control of Salmonella Typhimurium infection in the small intestine. iNLRC4-Pou2f3-creERT2mice or Nlrc4−/− littermate controls were pretreated with oral gavage of streptomycin, then 24 h later infected with 5 × 107 CFUs of Salmonella Typhimurium ΔSsaR by oral gavage. (A–D) Tissue CFUs from the small intestine (A) and cecum (B) 18 h after infection and small intestine (C) and cecum (D) 5 days after infection. (E) Number of Nlrc4−/− or iNLRC4-Pou2f3-creERT2 mice that had no detectable CFUs at day 5 after infection. (F and G) IL-22 protein levels were determined by ELISA in small intestine (F) or cecum (G). Each data point represents one mouse, pooled data from multiple experiments (n = 9 [A and B], n = 14–20 [C–G]), bars shown as the median. Significance was determined by Mann–Whitney U test (A–D, F, and G). *, P < 0.05, **, P < 0.01.
The functions of intestinal tuft cells are not completely known. Understanding how different cell types, including tuft cells within the gastrointestinal tract, contribute to the host inflammatory response will provide more foundational knowledge to understand how inflammatory responses are initiated, maintained, or changed within the gastrointestinal tract. It may also uncover new potential therapies for enteric disease.
We demonstrate that NAIP–NLRC4 inflammasome activation in tuft cells induces a PGD2–ILC3 signaling pathway within the small intestine and promotes clearance of a gastrointestinal bacterial pathogen. While not demonstrated definitively, expression of the receptor for PGD2, CRTH2, on ILC3 makes a direct signaling effect likely, possibly in redundancy with other signals such as IL-18 (Victor et al., 2017) when the inflammasome is activated in enterocytes as well. We demonstrate that tuft cells are not solely parasitic sensors but also participate in host defense responses against bacteria. We have previously shown that tuft cells release PGD2 after NAIP–NLRC4 inflammasome activation (Oyesola et al., 2021). While PGD2 is detrimental to antiparasitic responses, our data support that PGD2 release during bacterial infection is beneficial to the host and leads to better control of the pathogenic bacteria. It is tempting to speculate that PGD2 signaling after inflammasome activation in the intestine leads to a balance between antiparasitic responses and antibacterial responses and that tuft cells are the orchestrators of this balance. Tuft cells are also preferentially infected by murine norovirus (Wilen et al., 2018). It is conceivable that tuft cells integrate many different pathogen signals and have the potential to act as a rheostat for the ensuing host inflammatory response.
Interestingly, this response is highly regional as we only observe it in the small but not the large intestine. Future studies will have to address if there is also regionality in parts of the small intestines, as has been observed for CCR6+ILC3 (Guendel et al., 2020).
Only a subset of ILC3 expresses the receptor for PGD2, CRTH2, in mice. Interestingly, in humans, CRTH2 is expressed on ILC2 (Spits and Mjösberg, 2022), whereas the second known PGD2 receptor, DP1, is expressed on intestinal ILC3 (Elmentaite et al., 2021). CRTH2–ILC3 signaling or tuft cells were not required to restrict Salmonella in the small intestine of otherwise immunocompetent hosts. However, tuft cells were required for the induction of antimicrobial signals following Salmonella infection. Thus, during infection, PGD2 signaling to ILC3 might go beyond CRTH2. Whether DP1 can be upregulated on ILC3 during infection in mice and compensate for the loss of CRTH2 will be the subject of future studies. Additionally, Salmonella is known to be quite resistant to IL-22–induced antimicrobial factors such as Reg3γ or lipocalin (Behnsen et al., 2014), which we used as additional readouts to demonstrate ILC3 activation. Thus, other restricting factors such as cell extrusion might be dominant during Salmonella infection and explain why we observed sufficiency but not necessity of this pathway. Unfortunately, C. rodentium, a bacterial pathogen very susceptible to the effects of IL-22 often used to study the role of ILC3, does not infect the small intestine, and we are not aware of other bacterial infection models of the small intestine that activate the inflammasome. Nevertheless, the marked clearance of Salmonella from mice with tuft cell–restricted NLRC4 expression compared with Nlrc4-deficient mice, combined with the lack of Reg3γ induction in infected tuft cell–deficient mice, reveals a role for tuft cells in bacterial host defense. We speculate that this response may have evolved in response to bacterial pathogens of the small intestine that can evade epithelial cell extrusion or specifically infect tuft cells.
A previous report demonstrated tuft cell proliferation and PGD2 release upon sensing of a bacterial metabolite, N-C11-G, during S. flexneri infection and this promoted tuft cell amplification and protection (Xiong et al., 2022). While we also observed that small intestinal tuft cells release PGD2 (Oyesola et al., 2021), the Shigella study did not rule out the involvement of the NAIP–NLRC4 inflammasome during infection with NAIP ligand expressing pathogens as we have shown. In our hands, we however, do not observe protection from Shigella by tuft cells even in the colon or an increase in CD45+ tuft cells during Shigella infection. This is consistent with previous observations that Shigella cannot efficiently colonize inflammasome-competent mice due to the extremely efficient restriction of these bacteria by the acute epithelial extrusion response (Mitchell et al., 2020). In agreement with our observation, a recent study reported that administration of N-C11-G to intestinal epithelial monolayers did not result in the release of leukotrienes or cause ion flux, indicating that tuft cells did not respond to this ligand (Billipp et al., 2024). Thus, the recognition of pathogenic bacteria–specific extracellular ligands by tuft cells warrants further study.
Here, we link inflammasome-triggered tuft cell PGD2 release to the activation of ILC3s. The crosstalk between lipid mediator release from tuft cells and ILC is reminiscent of signaling observed during antiparasitic responses. During Heligmosomoides polygyrus infection, a type of leukotriene, LTC4, is released by tuft cells and induces ILC2s to proliferate and release IL-13 (McGinty et al., 2020). Using more sensitive methods rather than RNA sequencing, Gpr44 expression is also detectable on goblet cells and tuft cells (Oyesola et al., 2021). Therefore, additional effects of PGD2 released by tuft cells, especially during type 2 inflammation, where goblet and tuft cells are increased, are conceivable. Our findings suggest that tuft cells have a unique ability to sense multiple different pathogens, and by release of specific lipid mediators they can manipulate downstream inflammatory responses.
Materials and methods
Mice, FlaTox injections, and inhibitor treatment
iNLRC4 (Rauch et al., 2017), Nlrc4−/− (Tenthorey et al., 2020), Pou2f3-CreERT2 (McGinty et al., 2020), Gpr44fl/fl-GFP mice, and B6.FVB-Tg(Rorc-cre)1Litt/J (common name: Rorγt-Cre Strain # 022791 [McGinty et al., 2020; Eberl and Littman, 2004]) breeding was maintained in specific pathogen–free facilities at Oregon Health and Science University (OHSU). iNLRC4-Pou2f3-creERT2 mice were generated by crossing iNLRC4 mice with Pou2f3-CreERT2, both on a Nlrc4−/− background. To determine if the Gpr44 locus is active in ILC3s, Gpr44fl/fl mice were crossed with Rorγt-Cre mice. To induce CreERT2, iNLRC4-Pou2f3creERT2 and Nlrc4−/− littermate mice were given ad libitum access to tamoxifen-impregnated food (500 mg/kg, TD.130857; Envigo) for 1 wk before the experiment began and remained for the duration of the experiment. 1 wk after tamoxifen food administration, mice were retro-orbitally injected with FlaTox comprising of LFn-FlaA from Vibrio parahemolyticus or Legionella pneumophila (0.1 μg/g) and PA (0.2 μg/g) given at 4 μl/g of body weight twice in the period of 48 h. AZD1981 or vehicle (1:5 DMSO:PBS) was given 5 μg/g body weight.
Pou2f3−/− mice (McGinty et al., 2020) were crossed to C57BL/6 or Nlrc4−/− (Tenthorey et al., 2020) to generate single or double knockouts and heterozygous controls and maintained in specific pathogen–free conditions at the University of Washington.
For all in vivo studies, age- and sex-matched, cohoused 8–20 wk littermate or littermate parent (Robertson et al., 2019) mice were used, and all experiments were repeated at least twice.
For detection of pyroptotic epithelial cells, mice were retro-orbitally injected with 100 µg propidium iodide 10 min before euthanasia.
All experiments were performed as approved by the Institutional Animal Care and Use Committee of the OHSU and the University of Washington.
Generation and validation of CRTH2fl/fl(or Gpr44fl/fl)-GFP mice
The exon 3 coding region of the Gpr44 gene (NCBI Reference Sequence: NM_009962.3) on mouse chromosome 19 was targeted for conditional deletion. To construct the targeting vector, mouse genomic fragments containing homology arms were amplified from a bacterial artificial chromosome (BAC) clone using high-fidelity Taq DNA polymerase and were sequentially assembled into a targeting vector with the polyA-loxP-endogenous splice acceptor of intron 2–5′UTR of exon 3-EGFP-SV40 polyA cassette. In the targeting vector, the Neo cassette was flanked by self-deletion anchor sites and diptheria toxin A was used for negative selection. The targeting construct was linearized by restriction digestion, purified, and transfected into C57BL/6 embryonic stem (ES) cells according to Taconic Inc. proprietary methods. The transfected ES cells were selected with G418 (200 μg/ml). G418-resistant clones (188) were picked, amplified, and PCR-screened for homologous recombination, identifying 11 potential targeted clones. Six were further confirmed by Southern blot analysis to be correctly targeted. Targeted ES cell clones were injected into C57BL/6 albino embryos, which were then reimplanted into CD-1 pseudopregnant females. Founder animals were identified by their coat color, and germline transmission was confirmed by breeding with C57BL/6 females and subsequent genotyping of the offspring.
Gpr44fl/fl-GFP mice were then bred at the University of Washington to Vil1-Cre mice to generate Vil1-Cre x Gpr44fl/fl-GFP animals. To test cell lineage–specific deletion of Gpr44 in Vil1-expressing IECs, small IECs and splenocytes were isolated as described previously (Oyesola et al., 2021) from Vil1-Cre+Gpr44fl/fl-GFP, Vil1-Cre+Gpr44WT/WT, and Vil1-Cre−Gpr44fl/fl-GFP mice. DNA was extracted using the GenCatchPCR Purification kit (Epoch Life Science) according to manufacturer instructions. A genotyping PCR was used to amplify genomic DNA from the WT allele (239 bp); the targeted, non-recombined allele (348 bp); and the targeted, recombined allele (279 bp) to confirm recombination only in Vil1-Cre Gpr44fl/fl-GFP mice in small intestine IECs (Fig. S2). Primers used were F1: 5′-GTTGGATCCACTGGAACTGAATTAT-3′ and R1: 5′-TGTAATCTCAGTGCTGGAGAAAGAC-3′.
All animal procedures were approved and performed in accordance with the Institutional Animal Care and Use Committees at OHSU and the University of Washington.
Bacterial infections
The day before infection, mice were food-restricted for 4 h and then orally gavaged with 100 μl of 250 mg/ml streptomycin sulfate in H2O to deplete the microbiome (Barthel et al., 2003). On the day of infection, mice were again food-restricted for 4 h and then infected by oral gavage with either 5 × 107 CFUs of Salmonella Typhimurium ΔSsaR in 100 μl PBS or 5 × 107–8 CFUs of S. flexneri serovar 2a 2457T in 100 μl PBS. Ileum and ceca were collected from Salmonella Typhimurium ΔSsaR or Shigella-infected mice 18 or 24 h or 5 days after infection and processed for CFU enumeration, flow cytometry, or protein analysis as described below.
FlaTox component production
LFn-FlaA V. parahaemolyticus or L. pneumophila was produced in insect cells as previously described (Tenthorey et al., 2020). Briefly, using the BaculoGold system, a recombinant protein for cytosolic delivery of V. parahaemolyticus or L. pneumophila FlaA was produced. The FlaA coding sequence was subcloned into pAcSG2-6xHis-LFn and then cotransfected with the BestBac linearized baculovirus DNA (Expression systems) into SF9 cells. Infectious baculovirus was then incubated with High Five cells to generate recombinant proteins. Cells were lysed on ice using a dounce homogenizer in a lysis buffer. Supernatants from clarified samples were bound to nickel resin, then recombinant proteins were column purified by gravity.
PA was purified as described before (Tenthorey et al., 2020). Briefly, PA was expressed in E. coli periplasm and purified using periplasmic lysis, followed by dialysis and anion exchange chromatography. Endotoxin was removed using Pierce endotoxin removal columns.
Harvesting tissue
Upon completion of the experimental timeline, mice were euthanized by CO2 asphyxiation, and cervical dislocation was performed to ensure euthanasia. An incision was made in the peritoneum of mice and the small intestine was cut from the cecum. For flow cytometry, the whole small intestine was collected, fileted open, rinsed in PBS, and cut into 2-cm pieces. For protein and RNA quantification, the whole small intestine was removed and then cut into three pieces (separating the proximal 2 cm for the duodenum, middle 2 cm for jejunum, and very distal 2 cm for ileum). Colon and cecum tissue was collected separately. Feces were removed and two representative pieces from each region were flash-frozen and processed for either protein or quantitative PCR (qPCR) analysis. Another two pieces from each region were fileted open and pinned flat into shallow dishes containing 4% paraformaldehyde for 2 h at room temperature (RT). The fixed tissue was placed into 30% sucrose overnight and then rolled into a Swiss roll and placed into cassettes containing OCT and frozen until ready for cutting.
CFU enumeration
To quantify Salmonella tissue invasion, 2-cm pieces of distal small intestine and half of the cecum were collected from Salmonella Typhimurium–infected mice and washed in 400 μg/ml gentamicin in PBS for 30 min at RT. To ensure that gentamicin was sufficiently washed away, tissue was rinsed with ice-cold PBS five times. Tissue was then homogenized in 1 ml ice-cold PBS with a Kinematica Polytron. Serial dilutions were made of cecum and three or four 10 μl droplets from each serial dilution were plated on MacConkey agar plates containing streptomycin. 100 μl of undiluted small intestine homogenate were plated directly onto MacConkey agar using bead plating. 18–20 h later, colonies were enumerated and final CFUs were determined for each tissue type.
To enumerate intracellular Shigella CFU from IECs, tissue was imaged and measured and then prepared as above, incubated in RPMI with 5% FBS, 2 mM L-glutamine, and 400 mg/ml of gentamicin for 1–2 h, washed five times in PBS, cut into pieces and placed in 15 ml of stripping solution (HBSS, 10 mM HEPES, 1 mM DTT, 2.6 mM EDTA), and incubated at 37°C for 25 min with gentle shaking. Tissue was filtered through a 100-µm filter, washed, and filtered again. The epithelial cell fraction was incubated in 50 mg/ml gentamicin for 20 min, spun at 300 × g at 4°C for 8 min, and washed twice in PBS. After the second wash, the pellet was resuspended and lysed in 1 ml of 1% Triton X-100. Serial dilutions were made from this solution and plated on tryptic soy broth agar plates containing 0.01% Congo red and 100 µg/ml streptomycin, and Congo red–positive colonies were counted after overnight incubation at 37°C.
Cryostat and immuno fluorescence staining
OCT-embedded frozen tissues from different regions of the small intestine and caecum were taken and cut into 7-μm sections. The tissue sections were placed in PBST (PBS + 0.5% Tween20) for 3–4 min for OCT removal. The sections were then permeabilized with PBS + 0.5% Triton X-100 for 10 min and blocked with 10% donkey serum or 10% goat serum for 1 h at RT. The sections were then incubated with respective primary antibodies in PBST (rabbit-anti-DCLK1 [1:1,000, ab31704; Abcam], rat-anti-CD4 [1:50, MA1-146; Thermo Fischer Scientific], GFP-boost [1:200, gb2AF488; Chromotek]) overnight at 4°C. Following day, the sections were rinsed with PBST three times, 3–4 min each and incubated with respective secondary antibodies in PBST (donkey-anti-rabbit AF647 [1:800, AB_2492288; Jackson ImmunoResearch Labs], goat anti-rabbit AF488 [1:500, AB_2338046; Jackson ImmunoResearch Labs], and donkey-anti-rat Dylight755 [1:100, SA5-10031; Invitrogen; Thermo Fisher Scientific]) for 1 h at RT. When needed, Phalloidin-647 (A22287, 0.165 μM; Thermo Fisher Scientific) was applied on tissue sections for 30 min at RT. Sections were then rinsed with PBST three times, 3–4 min each, and incubated with DAPI (0.1 μg/ml in PBS) for 10 min at RT. After rinsing the sections once in PBS, Vectashield Hardset mounting media was applied and the coverslip was placed on top. The sections were then imaged with a Zeiss Apotome. For the sections with anti-CD4 antibodies, imaging was done in 3 × 3 binning. For better visualization, some channels were pseudocolored to the colors as mentioned in the respective figures. Imaging was done using three biological replicates with tissues from three different mice. For each section, three random sites were selected and three images were taken. All analysis was done using Fiji. DCLK1, CD4, and CRTH2 (GFP) positive cells were located, and for each of the CD4 and CRTH2 (GFP) positive cells, the distance was measured from the nearest DCLK1 positive cells in the villus using the ruler function of Fiji.
Protein analysis
Flash-frozen tissue was placed in PBS with complete mini protease inhibitor cocktail (#4693124001; Millipore-Sigma) and homogenized with a handheld Kinematica Polytron. Samples were frozen and then thawed fully and vortexed before they were spun down at 10,000 × g to pellet cellular debris. The protein concentration of the supernatant was determined by following the manufacturer’s instructions for the Pierce BCA Protein Assay Kit (#23227; Thermo Fisher Scientific). Once total protein concentration was determined, 10 µg of protein were loaded into manually coated IL-22 96-well High Affinity plates, and IL-22 levels were determined by IL-22 ELISA kit (#88-7422-22; Thermo Fisher-Scientific) following the manufacturer’s protocol. The 96-well plate was read using a plate reader (CLARIOstar). To assess fecal mouse myeloperoxidase (MPO) levels, pelleted fecal homogenates were analyzed with sandwich ELISA (R&D Systems) according to the manufacturer’s instructions. MPO concentrations were interpolated from an MPO standard curve using GraphPad Prism 10 for macOS (v. 10.2.0). Fecal CFU and MPO levels were normalized to fecal mass.
qPCR analysis
RNA from tissue was purified using the TRIZOL method by manufacturer’s protocol (Thermo Fisher Scientific). Samples were measured for RNA concentration and purity. To synthesize the cDNA, 1 µg of RNA was incubated with 1 ml of DNase in 10 ml total and allowed to incubate for 30 min at 37°C. DNase stop solution was added and incubated for 10 min at 65°C. dNTPs were incubated for 5 min at 65°C and then cooled to 4°C. Master mix containing Superscript IV reverse transcriptase (#18090050; Thermo Fisher Scientific) was added to samples and incubated at 55°C for 10 min, followed by 80°C for 10 min qPCR analysis: Real-Time PCR on cDNA was performed using PowerUp SYBR Green Master mix (#A25742; Thermo Fisher Scientific) and analyzed with an Quantstudio3 (Applied Biosystems). Relative quantities were determined by normalization to housekeeping genes Rps17 or Actin β. Fold-increase in expression was calculated by normalizing expression data to control values of individual experiments.
Primer sequences: Reg3γ F: 5′-TTCCTGTCCTCCATGATCAAAA-3′ R: 5′-CATCCACCTCTGTTGGGTTCA-3′, Reg3β F: 5′-ATGCTGCTCTCCTGCCTGATG-3′ R: 5′-CTAATGCGTGCGGAGGGTATATTC-3′, Rps17 (housekeeping gene) F: 5′-CGCCATTATCCCCAGCAAG-3′ R: 5′-TGTCGGGATCCACCTCAATG-3′.
Actin β (housekeeping gene) F: 5′-CGCAGCCACTGTCGAGTC-3′ R: 5′-CCTTCTGACCCATTCCCACC-3′.
Eicosanoid analysis
Crypts were isolated from the small intestine of mice and monolayer cultures were generated as previously described (McGinty et al., 2020). Briefly, ∼12 cm of the proximal small intestine was isolated and the villi scraped off with a glass slide. Following three PBS washes, the tissue was cut into 2-mm sections and then incubated in 2 mM EDTA for 30 min at 4°C. After removing the EDTA solution, PBS was added and the intestinal pieces were pipetted up and down with a 10-ml serological pipet three times and then filtered through a 70-μm strainer. 48-well plates were coated with 125 μl of a 2% Matrigel solution in cold DMEM and incubated at 37°C for at least 30 min. Pelleted crypts were resuspended in complete organoid media (DMEM/F12 with penicillin and streptomycin, 1 mM HEPES, 1x N2 and B27 supplement, 5 mM N-acetylcysteine, 50 mg/ml EGF, 1/20 volume Noggin and R-spondin conditioned media) with 10 μM Y27632. Crypts were plated and incubated at 37°C overnight. Media was replaced with complete organoid media without Y27632 and incubated for at least 1 h prior to treatment.
Monolayers were pretreated for 45 min with DMSO, 10 μM BAPTA-AM, or 30 μM Disulfiram, followed by FlaTox (4 µg/ml PA combined with 2 µg/ml LFn-flagellin) for 30 min at 37°C. Stimuli were added in HBSS with Ca2+ and Mg2+. Supernatants were collected and used to perform ELISA for PGE2 (Item No 514010; Cayman Chemical). Cells were lysed in radioimmunoprecipitation assay buffer and total protein content was determined by the Pierce BCA Protein Assay Kit (23227; Thermo Fisher Scientific).
Flow cytometry
For epithelial cell isolations
Isolated small intestines were placed into 15 ml of DTT wash solution (5% FCS, 10 mM HEPES, 5 mM DTT, and HBSS [no Ca/Mg]) gently shaking at 37°C for 10 min. Samples were vortexed and then placed into EDTA wash solution (5% FCS, 10 mM HEPES, 5 mM EDTA, and HBSS [no Ca/Mg]) gently shaking for 15 min at 37°C. The supernatant was collected through a 70-mm cell strainer. Another wash of EDTA wash solution was added to small intestines and incubated for 5 min while gently shaking at 37°C. The second epithelial cell isolation was collected and added to the previous epithelial cell flow through. Samples were then stained for: Live dead (Zombie Aqua), EpCam-AF647, CD24-PE, C-kit- PerCp, CD45+-AF700, and SiglecF-BV421. Tuft cells were identified as EpCam+, SiglecF+, and CD24+ cells.
For lamina propria isolations
Lamina propria single-cell suspensions were collected using the lamina propria isolation protocol from Miltenyi Biotech with modifications. Briefly, isolated small intestines were washed in ice-cold PBS and then placed into strip buffer (5% FBS, 10 mM HEPES, 5 mM DTT, 5 mM EDTA, and 1% Pen/Strep/Glu in PBS) gently shaking at 37°C for 20 min. Samples were vortexed and then placed into a second wash with strip buffer gently shaking for 20 min. Samples were rinsed with D10 media (10% FBS, 10 mM HEPES, and 1% Pen/Strep/Glu in DMEM) and then digested with the Lamina Propria Isolation kit from Miltenyi Biotech following the manufacturer’s protocol while gently shaking for 30 min. Samples were then placed into C tubes and mechanically digested using the Miltenyi Biotech tissue homogenizer—small intestine program. The digested samples were then put through a 70-mm cell strainer, cells were counted, and the required number of cells were incubated for 4 h at 37°C in a CO2 incubator in DMEM with 1 mM sodium pyruvate, 100 μM 2-mercaptoethanol, and 2 mM L-glutamine with 1 µl/ml golgi plug and then stained. Staining was done in a 96-well V-bottom plate in 100 µl total volume for 4–5 million cells per well. Surface staining was done for 30 min at 4°C using the following antibodies: CD3-BV650, CD19-BV650, CD14-SB645, NK1.1-SB780, KLRG1-BUV395, CD45-BUV496, CD90-APC, CD127-BUV737, CD4-SB600, NKp46-APC-eflour780, CCR6-Pe-dazzle594, and lime viability dye. After surface staining, fixation was done using Foxp3 transcription factor staining buffer set (Invitrogen) or BD Cytofix/Cytoperm fixation/permeabilization kit when staining for GFP. Intracellular staining was done using Rorγt-PerCP-efluor710, IL-22-PE, and Rabbit-anti-GFP antibodies either 1 h at RT or overnight at 4°C. Anti-GFP antibody was stained overnight at 4°C and the next day secondary antibody (anti-rabbit-Alexa Fluor488) staining was done for 2 h at RT. ILC3s were identified as Lineage−CD90+CD127+KLRG1−NK1.1−Rorγt+ population. TH17-cells were identified as CD3+CD4+ Rorγt+.
Online supplemental material
Fig. S1 is related to Fig. 1 and shows validation and phenotyping of iNLRC4-Pou2f3-creERT2 mice and requirements for IEC eicosanoid release. Fig. S2 demonstrates no role for tuft cells in an anti-Shigella response in the colon and cecum. Fig. S3 is related to Fig. 2 and shows validation of Gpr44fl/fl mice and additional data on CRTH2 expressing cell location and identity in the intestine.
Data availability
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank members of the Rauch laboratory and Jakob von Moltke for their input and Leigh Knodler (The University of Vermont, Burlington, VT, USA) for reagents. The authors further wish to thank members of the OHSU flow cytometry and microscopy cores (RRID:SCR_009961) for their support and advice.
This work was supported by the National Institutes of Health, the National Institute of Allergy and Infectious Diseases (R01 AI167974), and the Rainin Foundation Synergy Award to I. Rauch; the National Institutes of Health (R01 AI130379 and R01 AI167974) to E.D. Tait Wojno; OHSU funds to I. Rauch; and University of Washington startup funds to E.D. Tait Wojno. S. Krug is a Cystic Fibrosis Foundation RDP Fellow supported SINGH19R0. P.S. Mitchell is a Howard Hughes Medical Institute Freeman Hrabowski Scholar and is supported by the National Institutes of Health grant DP2 AI154432.
Author contributions: M.J. Churchill: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing - original draft, Writing - review & editing, A. Pandeya: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing - review & editing, R. Bauer: Data curation, Formal analysis, Investigation, Visualization, Writing - review & editing, T. Christopher: Data curation, Formal analysis, Investigation, Methodology, S. Krug: Formal analysis, Investigation, Validation, Visualization, R. Honodel: Investigation, S. Smita: Resources, L. Warner: Investigation, B.M. Mooney: Investigation, Methodology, Resources, Supervision, Writing - review & editing, A.R. Gibson: Investigation, Writing - review & editing, P.S. Mitchell: Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing - review & editing, E.D. Tait Wojno: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing - review & editing, I. Rauch: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing.

