


IKKα, encoded by CHUK, is crucial in the non-canonical NF-κB pathway and part of the IKK complex activating the canonical pathway alongside IKKβ. The absence of IKKα causes fetal encasement syndrome in humans, fatal in utero, while an impaired IKKα-NIK interaction was reported in a single patient and causes combined immunodeficiency. Here, we describe compound heterozygous variants in the kinase domain of IKKα in a female patient with hypogammaglobulinemia, recurrent lung infections, and Hay–Wells syndrome-like features. We showed that both variants were loss-of-function. Non-canonical NF-κB activation was profoundly diminished in stromal and immune cells while the canonical pathway was unexpectedly partially impaired. Reintroducing wt CHUK restored non-canonical NF-κB activation. The patient had neutralizing autoantibodies against type I IFN, akin to non-canonical NF-κB pathway deficiencies. Thus, this is the first case of biallelic CHUK mutations disrupting IKKα kinase function, broadening non-canonical NF-κB defect understanding, and suggesting IKKα’s role in canonical NF-κB target gene expression in humans.

Introduction
The NF-κB pathway is divided into the canonical (or classical) and the non-canonical (or alternative) pathways. They both lead in fine to the translocation in the nucleus of homo or hetero dimers formed by the particular association of five transcription factors including RelA (also called p65), c-Rel, RelB, NF-κB1 (the precursor p105 of p50), and NF-κB2 (the precursor p100 of p52) (Smale, 2012). These dimers are sequestered in the cytoplasm by inhibitors of NF-κB (IκB family), such as the IκΒα inhibitor for the canonical pathway or the unprocessed form of NFKB2 p100 for the non-canonical one (Zandi et al., 1997; Baeuerle and Baltimore, 1988a, 1988b, 1989). The canonical pathway provides a rapid response to the activation of various receptors (Toll-like receptors, tumor necrosis factor receptor 1, ectodysplasin A receptor, B cell receptor, and T cell receptor) and leads to the transcription of multiple proinflammatory and prosurvival genes (Schnappauf and Aksentijevich, 2020; Baltimore, 2009). The “IKK complex” containing the catalytic subunits IKKα and IKKβ and a regulatory subunit IKKγ (NEMO) (Zandi et al., 1997; Mercurio et al., 1997; Ea et al., 2006; Israël, 2010) is essential to this process. Once activated, the effector serine/threonine kinases of the IKK complex phosphorylates the IκΒα inhibitor on two serines (and other IκΒ members such as IκΒε or IκΒβ), leading to their polyubiquitination and degradation by the 26S proteasome within seconds. Free from inhibition, canonical dimers containing p65 or c-Rel can translocate to the nucleus where they exert their transcriptional activities. In contrast, the non-canonical NF-κB pathway is fully activated after hours of receptor engagement. These receptors are all members of the TNFR superfamily and include, for example, CD40, LTβR, and BAFF-R, all of which have been implicated in the development of lymphoid organs, normal maturation, and activation of B and T cells. The non-canonical pathway is dependent on IKKα (Dejardin et al., 2002). Before receptor activation, the activating kinase of IKKα called NIK is constantly degraded by a cIAP1-2/TRAF2/TRAF3 complex (Sun, 2017; Bista et al., 2010). After ligation of receptors, TRAF3 is degraded, and NIK accumulates and phosphorylates IKKα at serine 176/180 (Dejardin et al., 2002). Once activated, NIK and IKKα phosphorylate NF-κB2 (p100), tagging it for polyubiquitination (Dejardin et al., 2002; Senftleben et al., 2001). The partial cleavage by the proteasome of p100 into p52 results in RelB:p52 dimers translocation to the nucleus and transcription of target genes. Depending on the engaged receptor, the non-canonical pathway regulates the development of secondary lymphoid organs and their organization (via LTβ receptor), the medullary thymic epithelial cell differentiation and AIRE expression (via RANK), thymocyte emigration (LTβ receptor), B cell survival, development, and maturation (via CD40 and BAFF receptor), and finally controls the bone formation and homeostasis (via RANK) (Sun, 2012, 2017). While murine models have revealed the relative importance of each component of the NF-κB signaling pathways, the study of patients with inborn errors of immunity and deleterious mutations in NF-κB transcription factors or signaling components reveals their non-redundant roles and their implication in humans (Bista et al., 2010; Hu et al., 2001; Li et al., 1999; Takeda et al., 1999; Franzoso et al., 1998; Tucker et al., 2007; Yin et al., 2001; Bousfiha et al., 2020; Tangye et al., 2022; Zhang et al., 2017; Barnabei et al., 2021). Regarding the non-canonical pathway, biallelic NIK deficiency, as well as complete RELB deficiency, has been associated with a combined immune deficiency with immune dysregulation (Willmann et al., 2014; Merico et al., 2015; Ovadia et al., 2017; Sharfe et al., 2015; Le Voyer et al., 2024). Heterozygous NFKB2 variants have been reported with various mechanisms from complete deficiency in both p100 and the processed p52 to uncleavable p100 leading to p52 loss-of-function (LOF) and p100 gain-of-function (GOF) phenotype (Le Voyer et al., 2023; Chen et al., 2013; Meissner et al., 2024). Patients with heterozygous NFKB2 variants exhibit a wide variety of symptoms due to immune dysregulation associated with various levels of immunodeficiency, from hypogammaglobulinemia to combined immunodeficiency. Until recently, no living patients were described with CHUK (coding IKKα) deleterious mutations. Homozygous nonsense null CHUK mutations led to the fetal encasement syndrome that is lethal in utero, similar to mice knock-out for this gene who died quickly after birth (Lahtela et al., 2010). Of note, parents carrying heterozygous nonsense variants were reported to be healthy, suggesting tolerance to haploinsufficiency for IKKα (Lahtela et al., 2010). Recently, a single patient presenting with a combined immunodeficiency and immune dysregulation was reported carrying a homozygous missense mutation in CHUK (p. Y580C). This mutation led to the selective impairment of the non-canonical NF-κB pathway by impairing the ability of IKKα to interact with its activator NIK (Bainter et al., 2021).
In this report, we describe the case of a patient who suffered from syndromic immunodeficiency and various immune dysregulation due to an autosomal recessive form of IKKα kinase deficiency.
Results
A patient with immunodeficiency, lymphoma, and compound heterozygous missense mutations in CHUK
The patient was first reported in a clinical description by Khandelwal et al. and was found associated with a heterozygous mutation (Khandelwal et al., 2017). Briefly, the patient (P) was the third child of an otherwise healthy non-consanguineous kindred (Fig. 1 A). The patient’s presentation was reminiscent of Hay–Wells syndrome with Pierre Robin sequence, ectodermal dysplasia including alopecia, blepharophimosis, and abnormal dental enamel (Cole et al., 2009; Celli et al., 1999). She also had skeletal malformations of feet and hands requiring surgery in infancy (see Clinical case report in Materials and methods). In addition, she had profound hypogammaglobulinemia associated with recurrent otitis and pneumonia requiring immunoglobulin replacement therapy from the age of 9 mo. She also experienced chronic onychomycosis of the fingernails. A diagnosis of celiac disease was made based on villous atrophy, following the exploration of growth failure without growth hormone deficiency. This villous atrophy did not respond well to the gluten-free regimen, and it was hypothesized to be a consequence of a chronic enteric viral infection or immune dysregulation and T cell infiltration rather than a bona fide celiac disease (Riller et al., 2023; Strohmeier et al., 2022; Klocperk et al., 2022). Peripheral blood immunophenotyping showed a lack of memory B cells and an excess of naïve T cells as well as an excess of effector memory CD8+ T cells and a CD4+ lymphocytosis (Table S1). Biological follow-up showed fluctuant cytolytic hepatitis with reactional unspecific hepatitis on liver biopsies and mild sinusoidal fibrosis. At 15 years of age, she was diagnosed with diffuse large B cell lymphoma (DLBCL) involving the lungs, associated with pleural effusion (Fig. 1 B). Immunohistochemistry was consistent with non-germinal center (non-GC) DLBCL, and Epstein–Barr–virus–encoded small RNAs were negative. The patient died of a massive pulmonary hemorrhage a few days after the second infusion of chemotherapy. Sanger sequencing of TP63, the known gene to be mutated in Hay–Wells syndrome, showed no mutation, and an array comparative genomic hybridization (CGH) did not show any abnormality (Khandelwal et al., 2017). The initial description reported a de novo heterozygous missense variant in CHUK (encoding IKKα) leading to the substitution of a histidine into arginine at position 142 of the amino-acid sequence, close to the catalytic site of the kinase domain of IKKα (CHUK c. 425A>G, p.H142R, Fig. 1 C). However, we sequenced the CHUK complementary DNA (cDNA) from fibroblasts and peripheral blood mononuclear cells (PBMCs), and we identified a second missense variant (CHUK c.569 C>T, Fig. 1 C) leading to the substitution of a proline into a leucine at position 190 (p.P190L) within the kinase domain of IKKα. Sanger sequencing of the proband’s SV40-fibroblasts’ and activated T cells’ genomic DNA and SV40-fibroblasts’ cDNA confirmed both heterozygous variants (Fig. S1 A). Both variants were absent from public databases (gnomAD v4) and were predicted deleterious by in silico scores with a CADD score of 25.1 and 30, respectively, well above the mean significant cutoff (MSC) threshold of 3.13 (Fig. 1 E) (Zhang et al., 2018; Itan et al., 2016). This patient was thus carrying two germline-predicted deleterious variants in the kinase domain of IKKα.
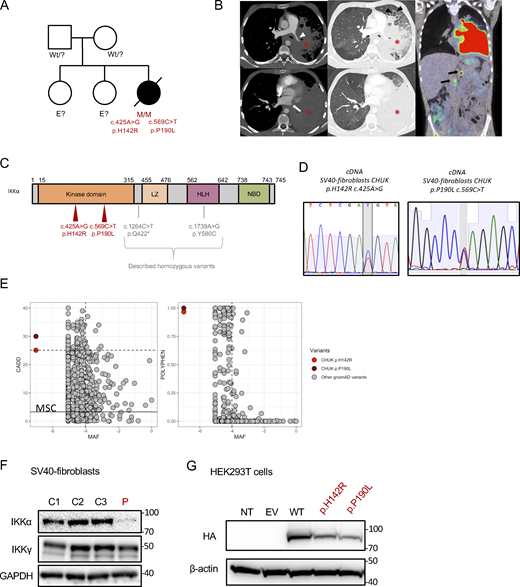
Pedigree, clinical manifestations, and mutations of a patient with immunodeficiency. (A) Pedigree of the family; “wt” indicates a wt genotype for CHUK, E? indicates that we don’t have the genotype of this family member. (B) Aspect of primary pulmonary lymphoma on the CT scan: consolidation of the lower two-thirds of the left lung (asterisk) with the invasion of the mediastinum, infiltration of the left pulmonary artery (white arrowhead), and complete thrombosis of the left pulmonary veins (white arrow). Thickening of the interlobular septa in the remainder of the ventilated lung, consistent with venous stasis or carcinomatous lymphangitis (black arrowhead). No normal lymph nodes or adenomegaly were individualized in the mediastinum or in the pulmonary hilum. A few small lymph nodes were visible in the axillary areas. The PET-CDFDG 18F performed within 48 h showed hypermetabolism of the lesions and in three coeliomesenteric lymphadenopathies (black arrows). (C) Localization of the mutation on the protein sequence of IKKα. The patient mutations are shown in red, while previously in vitro validated reported variants are in gray. (D) Sanger sequencing of RT-PCR products (cDNA) from the indicated cells. (E) Combined annotation-dependent depletion score versus minor allele frequency (CADD-MAF) (left panel) and polyphen score versus MAF (right panel). The patient variants are in red and dark red, and the other gnomAD variants are in gray. MSC: mean significance cutoff for CHUK CADD score (3.13). (F) Protein expression of IKKα and IKKγ οn protein lysates from healthy controls SV40-fibroblasts and the patient (P, in red). GAPDH was used as a protein loading control. (G) Protein expression of HA-tagged IKKα on protein lysates from transfected HEK293T cells with the different variants, the wt, or an EV. The patient variants are displayed in red. Source data are available for this figure: SourceData F1.
Pedigree, clinical manifestations, and mutations of a patient with immunodeficiency. (A) Pedigree of the family; “wt” indicates a wt genotype for CHUK, E? indicates that we don’t have the genotype of this family member. (B) Aspect of primary pulmonary lymphoma on the CT scan: consolidation of the lower two-thirds of the left lung (asterisk) with the invasion of the mediastinum, infiltration of the left pulmonary artery (white arrowhead), and complete thrombosis of the left pulmonary veins (white arrow). Thickening of the interlobular septa in the remainder of the ventilated lung, consistent with venous stasis or carcinomatous lymphangitis (black arrowhead). No normal lymph nodes or adenomegaly were individualized in the mediastinum or in the pulmonary hilum. A few small lymph nodes were visible in the axillary areas. The PET-CDFDG 18F performed within 48 h showed hypermetabolism of the lesions and in three coeliomesenteric lymphadenopathies (black arrows). (C) Localization of the mutation on the protein sequence of IKKα. The patient mutations are shown in red, while previously in vitro validated reported variants are in gray. (D) Sanger sequencing of RT-PCR products (cDNA) from the indicated cells. (E) Combined annotation-dependent depletion score versus minor allele frequency (CADD-MAF) (left panel) and polyphen score versus MAF (right panel). The patient variants are in red and dark red, and the other gnomAD variants are in gray. MSC: mean significance cutoff for CHUK CADD score (3.13). (F) Protein expression of IKKα and IKKγ οn protein lysates from healthy controls SV40-fibroblasts and the patient (P, in red). GAPDH was used as a protein loading control. (G) Protein expression of HA-tagged IKKα on protein lysates from transfected HEK293T cells with the different variants, the wt, or an EV. The patient variants are displayed in red. Source data are available for this figure: SourceData F1.

Sanger sequencing on different cell types from the patient and healthy controls. (A) Sanger sequencing of the patient mutation on genomic DNA. (B) Example of TOPO-TA cloning results from RT-PCR products encompassing the two mutations of the patient; the frequency of the corresponding sequence is written on the right. (C) Relative quantity of CHUK mRNA in SV40-fibroblast from patient and controls assessed by RT-qPCR.
Sanger sequencing on different cell types from the patient and healthy controls. (A) Sanger sequencing of the patient mutation on genomic DNA. (B) Example of TOPO-TA cloning results from RT-PCR products encompassing the two mutations of the patient; the frequency of the corresponding sequence is written on the right. (C) Relative quantity of CHUK mRNA in SV40-fibroblast from patient and controls assessed by RT-qPCR.
Given that the parents’ DNAs were not available, we performed a TOPO-TA cloning of an RT-PCR product encompassing the two mutations on cDNA from the patient’s SV40-fibroblasts. The sequencing of colonies showed that the mutations were biallelic (Fig. S1 B). Therefore, we concluded that the patient was compound heterozygous for these predicted deleterious variants in CHUK. No quantitative difference was found by RT-qPCR of CHUK mRNA (Fig. S1 C) in fibroblasts, suggesting that both alleles were equally expressed. Conversely, total IKKα protein expression was lower in SV40-fibroblasts than in the controls, suggesting a potential instability of the mutated proteins (Fig. 1 F). The coding sequence of CHUK was cloned into a plasmid containing an N-terminal HA-tag sequence, and site-directed mutagenesis was performed to obtain the patient’s variants (Bainter et al., 2021). Transfection experiments of these variants or the wt in HEK293T cells showed a slightly decreased expression for both variants as compared with the wt, thereby confirming that the IKKα p.H142R and p.P190L mutants could be less stable than the wt IKKα (Fig. 1 G).
Perturbed immune phenotype in PBMCs
We analyzed the immune phenotype from thawed PBMCs sampled and cryopreserved 5 years before the patient’s death using cytometry by time of flight (CyTOF). Unsupervised clustering allowed the identification of 35 immune subsets (Fig. 2 A). On this single time point analysis, we observed a profound naïve and memory B cells lymphopenia (Fig. 2 B). In addition, plasmablasts were absent and a subclustering of B cells revealed an excess of CD19+CD20+CD27−IgD+CD38high consistent with transitional B cells, a result in agreement with previously described immune phenotype in the context of non-canonical NF-κB defects (Fig. S2 A) (Willmann et al., 2014; Schlechter et al., 2017; Le Voyer et al., 2023; Chen et al., 2013; Bainter et al., 2021). The T cell compartment was characterized by a relative excess of naïve CD4+ and CD8+ T cells as previously observed in patients with a defective canonical NF-κB pathway (Fig. 2 B). This patient had an excess of highly activated HLA-DR+ CD38high CD127low effector memory CD8+ T cells, a cluster that we and others have already described to be associated with chronic viral infections (Fig. 2 B) (Riller et al., 2023; Klocperk et al., 2022; Paiardini et al., 2005). In contrast, we observed a trend toward a reduced proportion of Th2 and Th17 polarized memory CD4+ T cells according to the involvement of IKKα in the generation of Th17 in the recently reported biallelic LOF CHUK variant p.Y580C (Fig. S2 B) (Bainter et al., 2021).

Mass-cytometry analysis of PBMC s from the patient and controls. (A) Uniform manifold approximation and projection (UMAP) of patient cells (P) and two representative controls (CTL1 and CTL2) with colors corresponding to the identified clusters (below the UMAPs). Arrows show clusters with obvious different proportions compared with controls on the patient UMAP. (B) Boxplot showing the relative abundance (proportion of all cells) of each identified cluster. The patient is in red, and controls are in black.
Mass-cytometry analysis of PBMC s from the patient and controls. (A) Uniform manifold approximation and projection (UMAP) of patient cells (P) and two representative controls (CTL1 and CTL2) with colors corresponding to the identified clusters (below the UMAPs). Arrows show clusters with obvious different proportions compared with controls on the patient UMAP. (B) Boxplot showing the relative abundance (proportion of all cells) of each identified cluster. The patient is in red, and controls are in black.

Heatmap and relative abundances of B cells and T-helper CD4 + T cells. (A) B cells were subjected to reclustering based on the type_markers shown on the heatmap (left panel) in 10 clusters. The heatmap shows the median scaled expression of each marker on each subset. The right panel shows the relative abundance of each cluster between the patient (in red) and the controls (in black). (B) The memory CD4+ T cells were subjected to reclustering to identify T-helper cell subsets. The relative abundance of each identified cluster is shown (patient in red, controls in black).
Heatmap and relative abundances of B cells and T-helper CD4 + T cells. (A) B cells were subjected to reclustering based on the type_markers shown on the heatmap (left panel) in 10 clusters. The heatmap shows the median scaled expression of each marker on each subset. The right panel shows the relative abundance of each cluster between the patient (in red) and the controls (in black). (B) The memory CD4+ T cells were subjected to reclustering to identify T-helper cell subsets. The relative abundance of each identified cluster is shown (patient in red, controls in black).
Both IKKα variants were unable to activate non-canonical NF-κB in vitro and are LOF for kinase activity
To specifically assess the non-canonical NF-κB pathway activity in an ectopic expression model without interference with the canonical one, we generated a HEK293T RELA−/− cell line in which we co-expressed IKKα, RELB, and NFKB2, in addition to a κB-dependent firefly luciferase expression plasmid and a thymidine kinase-dependent renilla luciferase plasmid (Fig. 3 A). By transfecting increasing amounts of IKKα wt, we observed a dose-dependent increase of the relative luciferase intensity, showing that this model was relevant to assess IKKα activity in the non-canonical NF-κB pathway (Fig. 3 B). Transfection of each of the patient’s variants (IKKα p.H142R or IKKα p.P190L), or a biochemical kinase-dead variant (p.K44A), or a p.Q422* variant, a known IKKα null variant (Lahtela et al., 2010), significantly failed to induce NF-κB activity. Therefore, this experiment suggested a LOF for both CHUK variants (Fig. 3 B). Of note the expression of the IKKα p.Y580C variant, known to have a preserved kinase activity but an impaired binding to NIK (Bainter et al., 2021), led to a luciferase activity comparable with that of the wt IKKα. These results indicated that this experimental setting was NIK independent and allowed us to evaluate the intrinsic kinase activity of IKKα, for which both studied variants were at least hypomorphic.
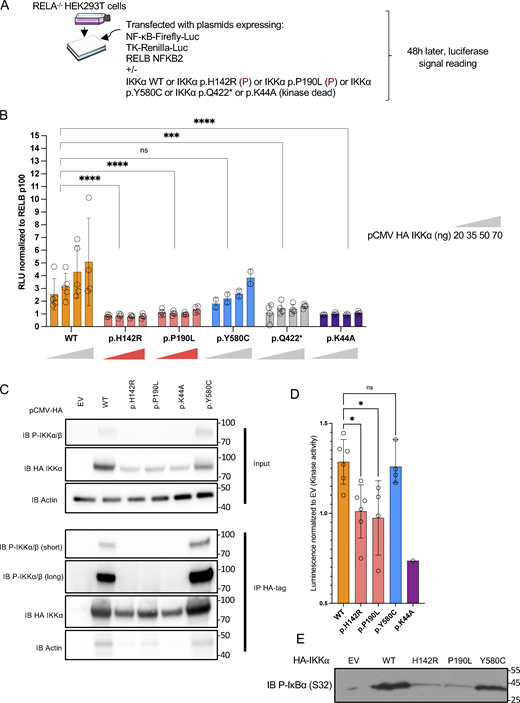
Non-canonical NF-κB activity in an ectopic expression system and kinase assays. (A) Schematic description of the experiment. RELA−/− HEK293T cells were transfected with an NF-κB firefly luciferase reporter and thymidine-kinase Renilla luciferase reporter plasmid together with RELB and NFKB2 coding plasmids. Additional transfection of either an EV plasmid or containing the coding sequence of wt IKKα or mutants IKKα allowed the assessment of their own activity toward the activation of the non-canonical NF-κB pathway. (B) Relative luciferase activity resulting from the transfection of an increasing dose of a plasmid coding IKKα wt, p.H142R, p.P190L, p.Y580C, p.Q422*, or p.K44A (from 20 to 70 ng as indicated), co-transfected with RELB and p100. The NF-κB Firefly signal was normalized to the Renilla signal and then to transfection with RELB p100 only. An EV was co-transfected in each condition to reach a constant amount of plasmid. Statistical analysis was performed using a One-Way ANOVA with multiple comparisons by Dunn’s test (****P < 0.0001, ***P < 0.001). Representative of at least three experiments. (C) Western blot of phosphorylated IKKα/β before (whole cell lysate/input) and after immunoprecipitation of HA-tag IKKα transfected in HEK293T cells (IP HA-Tag). Representative of three experiments. (D) IKKα kinase activity after pulldown of the different alleles expressed as the relative luminescence of the wt allele or the variants to the EV. Each point represents one experiment. Statistical analysis was performed using a one-Way ANOVA with multiple comparisons by Dunn’s test (*P < 0.05). (E) Western blot showing the phosphorylated form (S32) of a GST-IκΒα protein after incubation with the pulled-down IKKα (or EV). All figures are representative of at least two independent experiments. Error bars represent mean ± SD in all barplots. Source data are available for this figure: SourceData F3.
Non-canonical NF-κB activity in an ectopic expression system and kinase assays. (A) Schematic description of the experiment. RELA−/− HEK293T cells were transfected with an NF-κB firefly luciferase reporter and thymidine-kinase Renilla luciferase reporter plasmid together with RELB and NFKB2 coding plasmids. Additional transfection of either an EV plasmid or containing the coding sequence of wt IKKα or mutants IKKα allowed the assessment of their own activity toward the activation of the non-canonical NF-κB pathway. (B) Relative luciferase activity resulting from the transfection of an increasing dose of a plasmid coding IKKα wt, p.H142R, p.P190L, p.Y580C, p.Q422*, or p.K44A (from 20 to 70 ng as indicated), co-transfected with RELB and p100. The NF-κB Firefly signal was normalized to the Renilla signal and then to transfection with RELB p100 only. An EV was co-transfected in each condition to reach a constant amount of plasmid. Statistical analysis was performed using a One-Way ANOVA with multiple comparisons by Dunn’s test (****P < 0.0001, ***P < 0.001). Representative of at least three experiments. (C) Western blot of phosphorylated IKKα/β before (whole cell lysate/input) and after immunoprecipitation of HA-tag IKKα transfected in HEK293T cells (IP HA-Tag). Representative of three experiments. (D) IKKα kinase activity after pulldown of the different alleles expressed as the relative luminescence of the wt allele or the variants to the EV. Each point represents one experiment. Statistical analysis was performed using a one-Way ANOVA with multiple comparisons by Dunn’s test (*P < 0.05). (E) Western blot showing the phosphorylated form (S32) of a GST-IκΒα protein after incubation with the pulled-down IKKα (or EV). All figures are representative of at least two independent experiments. Error bars represent mean ± SD in all barplots. Source data are available for this figure: SourceData F3.
To confirm the loss of kinase activity of the patient’s alleles, we transfected the variants in HEK293T cells and performed immunoprecipitation using an anti-HA-tag antibody (Fig. 3 C). We observed a complete loss of autophosphorylation of IKKα upon transfection of the patient’s variants or the kinase-dead variant p.K44A as compared with the wt or the p.Y580C published variant (Fig. 3 C). A kinase activity was measured using an ADP-Glo Luciferase system (see Materials and methods). While we observed an increased kinase activity after pulling-down the wt IKKα, the activity was significantly impaired for both studied variants while IKKα p.Y580C showed a normal activity (Fig. 3 D). This was also true by looking at the phosphorylation at serine 32 of a GST-tagged IκBα (Fig. 3 E). Finally, co-transfection of NIK and the different variants showed that, in contrast to the wt IKKα, the variants were less able to induce the shift of NIK as assessed by western blot, a phenomenon known to be linked to IKKα’s kinase activity which phosphorylates NIK to mediate its degradation in a retrocontrol loop (Fig. 4 A) (Razani et al., 2010). These experiments strongly supported that both the IKKα p.H142R and IKKα p.P190L proteins displayed a profound kinase activity defect. We cannot exclude that this observation was partially linked to an impaired interaction with NIK as assessed by co-immunoprecipitation after transfection where both the patient’s variants were less able to interact with NIK (Fig. 4 B).
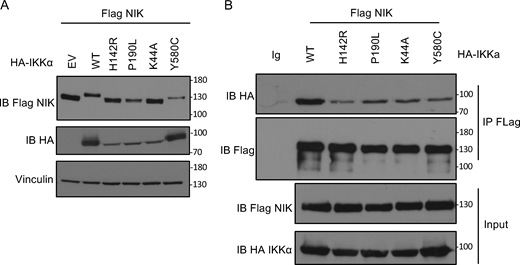
Co-immunoprecipitation between IKKα and NIK. (A) Whole cell lysate of HEK293T cells co-transfected with Flag-NIK and EV or HA-IKKa wt or mutants (as indicated) were subjected to western blot. Immunoblot of Flag-NIK, HA-IKKα, and vinculin as a housekeeping gene. (B) Immunoprecipitation of Flag-NIK after transfection of HEK293T cells with Flag-NIK and HA-IKKα wt or mutants (as indicated). Immunoblot of Flag-NIK and HA-IKKα. All figures are representative of at least two independent experiments. Both panels are representative of two independent experiments. Source data are available for this figure: SourceData F4.
Co-immunoprecipitation between IKKα and NIK. (A) Whole cell lysate of HEK293T cells co-transfected with Flag-NIK and EV or HA-IKKa wt or mutants (as indicated) were subjected to western blot. Immunoblot of Flag-NIK, HA-IKKα, and vinculin as a housekeeping gene. (B) Immunoprecipitation of Flag-NIK after transfection of HEK293T cells with Flag-NIK and HA-IKKα wt or mutants (as indicated). Immunoblot of Flag-NIK and HA-IKKα. All figures are representative of at least two independent experiments. Both panels are representative of two independent experiments. Source data are available for this figure: SourceData F4.
Impaired non-canonical NF-κB pathway activation in patient’s cells
IKKα is involved in the phosphorylation of p100 that induces its processing into p52, leading to the translocation of a p52:RelB dimer in the nucleus (Senftleben et al., 2001). LTβR (stimulated with LTα1β2) and Fn14 (stimulated with Tweak) are receptors known to engage the non-canonical pathway (Dejardin et al., 2002; Roos et al., 2010; Saitoh et al., 2003). We previously showed that both variants were kinase LOF (Figs. 3 and 4). We assessed the cleavage of p100 to p52 in SV40-fibroblasts from the patient and a patient with a previously described heterozygous variant in NFKB2 (p.R853*) known to impair the cleavage of p100 (Le Voyer et al., 2023). In both cases, the cleavage of p100 to p52 was nearly abolished at different time points after stimulation, indicating an impaired activation of the non-canonical NF-κB pathway (Fig. 5 A).
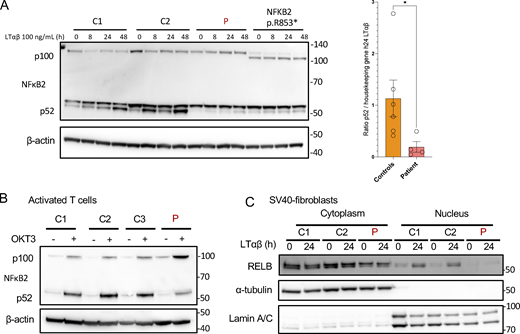
Non-canonical NF-κB signaling in patient cells. (A) Western blot of NFκB2 p100/p52 on cell lysates from SV40-fibroblasts after activation of the non-canonical pathway with LTαβ for 8, 24, or 48 h, as indicated. β-Actin was used as a loading control. Representative of at least three experiments. *: P-value <0.05 after Mann-Whitney Wilcoxon test. (B) Western blot of NFκB2 p100/p52 on cell lysates from activated T cells starved 24 h before stimulation with OKT3 for 48 h. β-Actin was used as a loading control. Representative of two experiments. (C) Western blot of cytoplasmic and nuclear protein extracts from SV40-fibroblasts activated with LTαβ for 24 h assessing the translocation of RELB in the nucleus. α-Tubulin was used the loading control of cytoplasmic extracts while Lamin A/C was used for nuclear extracts. Representative of three experiments. Source data are available for this figure: SourceData F5.
Non-canonical NF-κB signaling in patient cells. (A) Western blot of NFκB2 p100/p52 on cell lysates from SV40-fibroblasts after activation of the non-canonical pathway with LTαβ for 8, 24, or 48 h, as indicated. β-Actin was used as a loading control. Representative of at least three experiments. *: P-value <0.05 after Mann-Whitney Wilcoxon test. (B) Western blot of NFκB2 p100/p52 on cell lysates from activated T cells starved 24 h before stimulation with OKT3 for 48 h. β-Actin was used as a loading control. Representative of two experiments. (C) Western blot of cytoplasmic and nuclear protein extracts from SV40-fibroblasts activated with LTαβ for 24 h assessing the translocation of RELB in the nucleus. α-Tubulin was used the loading control of cytoplasmic extracts while Lamin A/C was used for nuclear extracts. Representative of three experiments. Source data are available for this figure: SourceData F5.
The same impairment in p100 cleavage was observed in anti-CD3–activated T cells from the patient (Fig. 5 B). This was associated with an impaired proliferation of activated T cells (Fig. S3 A) upon anti-CD3 stimulation and an impaired activation assessed by the upregulation of CD69 (Fig. S3 B). Accordingly, the translocation of RELB into the nucleus was nearly abolished in SV40-fibroblasts as assessed by western blots after LTα1β2, Tweak, or TNFα stimulation or by electrophorectif mobility shift assay (EMSA) experiments (Fig. 5 C; and Fig. S3, C and D). We also observed that VCAM1 expression, a known target gene of the NF-κB dimer RELB:p52 in fibroblasts, was lower at the transcriptomic level upon LTα1β2 stimulation of SV40-fibroblast (Fig. S3 E). These results were consistent with those reported with the homozygous p.Y580C variant (Bainter et al., 2021). Altogether, we showed that the defective kinase activity of both the IKKα p.H142R and the IKKα p.P190L mutants resulted in a profound impairment of the non-canonical NF-κB pathway activation.
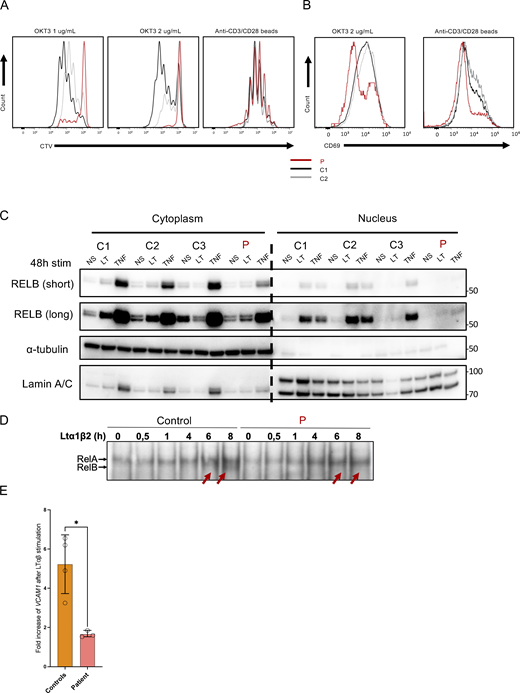
Functional impact of the variant on non-canonical and canonical NF-κB pathways. (A) Dilution histogram of Cell Trace Violet dye (CTV) after stimulation of activated T cells with OKT3 at 1 or 2 µg/ml for 4 days or anti-CD3/CD28 beads for 4 days. Representative of two independent experiments. (B) Histogram of CD69 activation marker upregulation on activated T cells after 4 days of stimulation with either OKT3 or anti-CD3/CD28 beads. Patient is in red, and controls are in gray. Representative of two independent experiments. (C) Western blot of cytoplasmic and nuclear extracts from SV40-fibroblasts after 48 h of LTαβ (LT) or TNFα (TNF) stimulation. RELB was revealed using a specific antibody to assess RELB production and nuclear translocation. α-Tubulin was used the loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. The patient is indicated in red, and the controls are C1, C2, and C3. Representative of two independent experiments. (D) Electromobility shift assay on nuclear extracts from healthy control and patient SV40-fibroblasts after different time points of LTα1β2 stimulation showing the absence of RELB binding (red arrows) in patient as compared to controls (lower thin band) and the normal binding of RELA (upper band). Representative of three independent experiments. (E) Relative VCAM1 mRNA expression after 24 h of LTαβ relative to non-stimulated condition. These experiments have been performed on healthy controls and patient SV40-fibroblasts. *: P-value <0.05 after Mann-Whitney-Wilcoxon test. All figures are representative of at least two independent experiments. Source data are available for this figure: SourceData FS3.
Functional impact of the variant on non-canonical and canonical NF-κB pathways. (A) Dilution histogram of Cell Trace Violet dye (CTV) after stimulation of activated T cells with OKT3 at 1 or 2 µg/ml for 4 days or anti-CD3/CD28 beads for 4 days. Representative of two independent experiments. (B) Histogram of CD69 activation marker upregulation on activated T cells after 4 days of stimulation with either OKT3 or anti-CD3/CD28 beads. Patient is in red, and controls are in gray. Representative of two independent experiments. (C) Western blot of cytoplasmic and nuclear extracts from SV40-fibroblasts after 48 h of LTαβ (LT) or TNFα (TNF) stimulation. RELB was revealed using a specific antibody to assess RELB production and nuclear translocation. α-Tubulin was used the loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. The patient is indicated in red, and the controls are C1, C2, and C3. Representative of two independent experiments. (D) Electromobility shift assay on nuclear extracts from healthy control and patient SV40-fibroblasts after different time points of LTα1β2 stimulation showing the absence of RELB binding (red arrows) in patient as compared to controls (lower thin band) and the normal binding of RELA (upper band). Representative of three independent experiments. (E) Relative VCAM1 mRNA expression after 24 h of LTαβ relative to non-stimulated condition. These experiments have been performed on healthy controls and patient SV40-fibroblasts. *: P-value <0.05 after Mann-Whitney-Wilcoxon test. All figures are representative of at least two independent experiments. Source data are available for this figure: SourceData FS3.
Reconstitution of the patient’s SV40-fibroblasts restores a normal non-canonical NF-κB phenotype
To further confirm that the identified CHUK variants were responsible for the observed in vitro phenotype, we transduced fibroblasts from the patient with an empty lentivirus or a lentivirus containing the wt CHUK coding sequence. The transduced cells were stimulated by LTα1β2 or Tweak, and we assessed the cleavage of p100. We still observed the impaired cleavage of p100 to p52 as well as the impaired RELB nuclear translocation after 24 or 48 h of LTα1β2 or Tweak stimulation in the cells transduced with the empty lentivirus (Fig. 6). In contrast, transduction of the wt IKKα rescued the p100 cleavage as well as the nuclear translocation of RELB as compared with healthy control SV40-fibroblasts (Fig. 6, A–C). These results supported the causality of the two CHUK variants and confirmed an autosomal recessive form of IKKα deficiency.
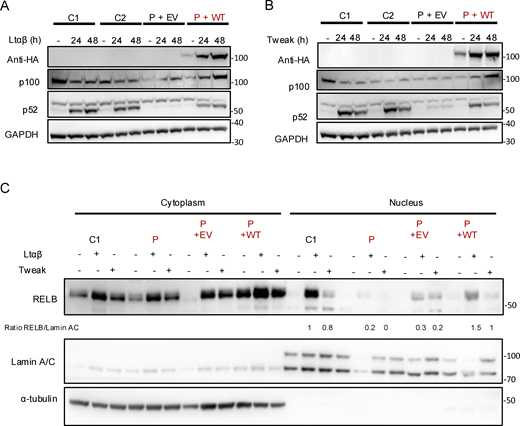
Reconstitution of patient cells with IKKα restored the non-canonical NF-κB defect in vitro. (A and B) Patient cells were transduced with an EV (P+EV) lentivirus or containing the coding sequence of wt IKKα (P+wt). GFP-positive cells were sorted, and cell lysate after activation of the non-canonical pathway by LTαβ (A) or Tweak (B) were subjected to western blot. p100 and p52 were revealed using specific antibodies and GAPDH was used as a loading control. Representative of three independent experiments. (C) Patient cells were transduced with an EV (P+EV) lentivirus or containing the coding sequence of wt IKKα (P+wt). GFP-positive cells were sorted. After activation with either LTαβ or Tweak, cytoplasmic and nuclear protein separation was performed and subjected to western blot. RELB was assessed using a specific C-terminal antibody to assess its nuclear translocation after activation in transduced patient’s cells compared with controls. α-Tubulin was used as a loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. RELB protein expression was quantified and normalized to the quantity of Lamin A/C in arbitrary units. Representative of three independent experiments. Source data are available for this figure: SourceData F6.
Reconstitution of patient cells with IKKα restored the non-canonical NF-κB defect in vitro. (A and B) Patient cells were transduced with an EV (P+EV) lentivirus or containing the coding sequence of wt IKKα (P+wt). GFP-positive cells were sorted, and cell lysate after activation of the non-canonical pathway by LTαβ (A) or Tweak (B) were subjected to western blot. p100 and p52 were revealed using specific antibodies and GAPDH was used as a loading control. Representative of three independent experiments. (C) Patient cells were transduced with an EV (P+EV) lentivirus or containing the coding sequence of wt IKKα (P+wt). GFP-positive cells were sorted. After activation with either LTαβ or Tweak, cytoplasmic and nuclear protein separation was performed and subjected to western blot. RELB was assessed using a specific C-terminal antibody to assess its nuclear translocation after activation in transduced patient’s cells compared with controls. α-Tubulin was used as a loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. RELB protein expression was quantified and normalized to the quantity of Lamin A/C in arbitrary units. Representative of three independent experiments. Source data are available for this figure: SourceData F6.
Partially impaired transcription of canonical NF-κB target genes
IKKα belongs to the NEMO complex along with the catalytic subunit IKKβ and the regulatory subunit IKKγ. Although previous publications showed that IKKα was dispensable in the IKK complex to phosphorylate IκBα (Hu et al., 1999, 2001; Polley et al., 2016), a major checkpoint of the canonical NF-κB pathway activation, we had several arguments in favor of a partial impairment of the canonical pathway activation in cells from the patient. The presence of ectodermal dysplasia and the results of the immunophenotype, showing an excess of naïve CD4+ and CD8+ T cells, are clinical and biological arguments for the involvement of the canonical NF-κB pathway (Puel et al., 2004). We thus performed bulk RNA sequencing of SV40-fibroblasts from the patient and controls before and after 6 h of stimulation with TNFα or Poly I:C (TLR3 agonist), two potent activators of the canonical NF-κB pathway (Fig. 7). We also included NEMOY/− SV40-fibroblasts (NEMO null), known to be unresponsive to TNFα stimulation (Smahi et al., 2000), as well as the SV40-fibroblasts from a patient carrying the heterozygous NFKB2 p.R853* variant (Le Voyer et al., 2023). Upon TNFα stimulation, TNFα induced–genes in control cells or genes from the TNFα signaling via NF-κB signature were significantly less upregulated in fibroblasts from the patient than in fibroblasts from controls (Fig. 7 A and Fig. S4 A). Accordingly, pathway activity inference based on previous knowledge (PROGENy) showed a profound defect in the activation of the TNFα signaling (Fig. 7 B) (Badia-I-Mompel et al., 2022). This was confirmed by Ingenuity Pathway Analysis (IPA) (Qiagen) showing that TNFα, IL1β, and lipopolysaccharide, three potent activators of the canonical NF-κB pathway, were in the top three molecules predicted to be inhibited based on the differentially expressed genes (DEGs) between the patient’ cells and control cells (Fig. 7 C). Although the DEG analysis for the NF-κB target genes induced after TLR3 stimulation suggested similar activation between the patient and controls (Fig. S4 B), a list of genes was consistently downregulated after both TNFα and TLR3 stimulation (RELB, NFKB2, NFKBIA, ICAM1, and BIRC3 among others; Fig. S4 C). Moreover, molecules predicted to be inhibited were mostly NF-κB inducers (Fig. S4 D). Of note, IRF3 target genes after TLR3 stimulation were normally upregulated in the patient cells (Fig. S5 A). In contrast to the patient’s cells, the NFKB2 p.R853* patient’s cells were in the range of controls both after TNFα and TLR3 stimulation for NF-κB target genes (Fig. 7 A and Fig. S5 B) while NEMO null cells were unable or less able to upregulate genes upon TNFα and TLR3 stimulation, as previously reported (Fig. 7 A and Figs. S4 and S5) (Audry et al., 2011). RT-qPCR confirmed these results on a subset of canonical NF-kB target genes (Fig. S5 C). We then assessed the phosphorylation of p65 and IκBα along with IκBα degradation. We observed that these functions were not affected in the patient’s cells, suggesting a normal activity of the NEMO complex and confirming in humans the redundant role of the kinase activity of IKKα in this complex (Fig. S5 D). In contrast, we observed that the nuclear translocation of p65 and p50 was significantly diminished in SV40-fibroblasts from the patient upon TNFα stimulation. The levels of p65 and p50 translocation in the patient’s cells were intermediate when compared with the level of translocation in the NEMO null SV40-fibrobalsts (no translocation) and in the NFKB2 p.R853* heterozygous SV40-fibroblasts (normal translocation) (Fig. 8, A and B). This observation was also true for poly I:C stimulation (Fig. S5 E). To validate our findings, we used two different cellular models. In the first one, in HEK293T IKKα KO, we observed a reduced translocation of p65 and p50 upon TNFα stimulation, thus suggesting that the canonical pathway impairment observed in the patient’s cells is linked to the defect in IKKα (Fig. 8, C and D). Of note, in this model, as in the patient’s cells, we did not observe any impairment in the degradation of IκBα, confirming an equivalent activation of the NEMO complex (Fig. S5 D and Fig. 8 C). In the second one, we stably transduced the 3T3-NIH IKKα KO cells with a lentivirus empty vector (EV) or a lentivirus containing the wt human IKKα or the kinase-dead mutant (p.K44A) or one variant of the patient (p.H142R). We observed an increased p65 nuclear translocation when the cells were transduced with the wt IKKa but not with the p.K44A nor with the p.H142R variants as compared with the EV condition (Fig. 8, E and F).

Exploration of the canonical NF-κB signaling in patient cells. (A) Heatmap showing the normalized and scaled expression (vst) of genes significantly upregulated in control SV40-fibroblasts after stimulation with TNFα 10 ng/ml for 6 h. Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (B) Pathway enrichment score of DEGs between the patient and controls SV40-fibroblasts stimulated with TNFα. A negative blue bar indicates a downregulation of the indicated pathway based on the PROGENy database while a red bar indicates an upregulated pathway. (C) Z-score of the different “molecules” predicted to be inhibited (top 10 based on the z-score) based on IPA (Qiagen Inc.) for the mentioned conditions. A negative score indicate that the molecule is predicted to be inhibited based on the DEGs that have been processed. The RNA sequencing experiment has been performed once in duplicate.
Exploration of the canonical NF-κB signaling in patient cells. (A) Heatmap showing the normalized and scaled expression (vst) of genes significantly upregulated in control SV40-fibroblasts after stimulation with TNFα 10 ng/ml for 6 h. Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (B) Pathway enrichment score of DEGs between the patient and controls SV40-fibroblasts stimulated with TNFα. A negative blue bar indicates a downregulation of the indicated pathway based on the PROGENy database while a red bar indicates an upregulated pathway. (C) Z-score of the different “molecules” predicted to be inhibited (top 10 based on the z-score) based on IPA (Qiagen Inc.) for the mentioned conditions. A negative score indicate that the molecule is predicted to be inhibited based on the DEGs that have been processed. The RNA sequencing experiment has been performed once in duplicate.

Bulk RNA sequencing of SV40-fibroblasts. (A) Heatmap showing the normalized and scaled expression (vst) of genes significantly upregulated in control SV40-fibroblasts after stimulation with TNFα 10 ng/ml for 6 h and significantly downregulated in SV40-fibroblasts from the patient compared with the controls after TNFα stimulation (from the TNFα via NF-κB signaling list of genes, MSIgDB). Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (B) Heatmap showing the normalized and scaled expression (vst) of genes significantly upregulated in control SV40-fibroblasts after stimulation with Poly I:C 1 µg/ml for 6 h from the TNFα via NF-κB signaling list of genes, MSIgDB. Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (C) Heatmap showing the list of genes significantly downregulated in patient’s cells compared to control cells after poly I:C 1 µg/ml 6 h stimulation, from the list of genes TNFα via NF-κB signaling list of genes, MSIgDB. (D) Z-score of the different “molecules” predicted to be inhibited (top 10 based on the z-score) based on IPA (Qiagen Inc.) after poly I:C stimulation. A negative score indicate that the molecule is predicted to be inhibited based on the DEGs that have been processed.
Bulk RNA sequencing of SV40-fibroblasts. (A) Heatmap showing the normalized and scaled expression (vst) of genes significantly upregulated in control SV40-fibroblasts after stimulation with TNFα 10 ng/ml for 6 h and significantly downregulated in SV40-fibroblasts from the patient compared with the controls after TNFα stimulation (from the TNFα via NF-κB signaling list of genes, MSIgDB). Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (B) Heatmap showing the normalized and scaled expression (vst) of genes significantly upregulated in control SV40-fibroblasts after stimulation with Poly I:C 1 µg/ml for 6 h from the TNFα via NF-κB signaling list of genes, MSIgDB. Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (C) Heatmap showing the list of genes significantly downregulated in patient’s cells compared to control cells after poly I:C 1 µg/ml 6 h stimulation, from the list of genes TNFα via NF-κB signaling list of genes, MSIgDB. (D) Z-score of the different “molecules” predicted to be inhibited (top 10 based on the z-score) based on IPA (Qiagen Inc.) after poly I:C stimulation. A negative score indicate that the molecule is predicted to be inhibited based on the DEGs that have been processed.
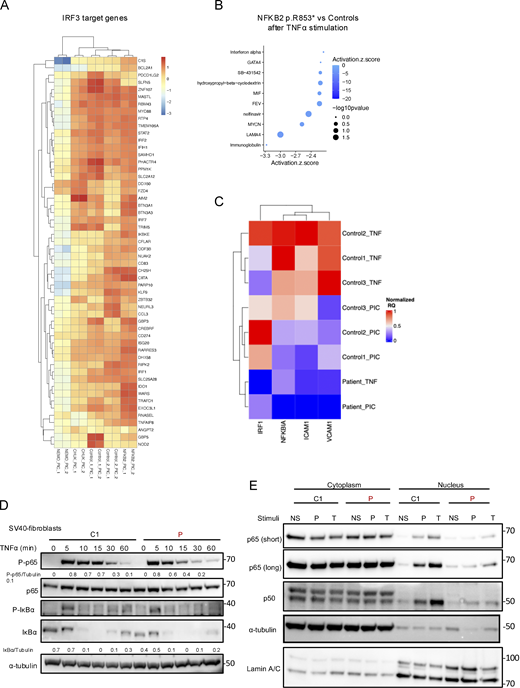
Bulk RNA sequencing of SV40-fibroblasts and RT-qPCR and western blot of the canonical NF-κB pathway. (A) Heatmap showing the normalized and scaled expression (vst) of IRF3 target genes significantly upregulated in controls after stimulation with poly I:C 10 µg/ml for 6 h. Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (B) Z-score of the different “molecules” predicted to be inhibited (top 10 based on the z-score) based on IPA (Qiagen Inc.) after poly TNFα stimulation for the NFKB2 p.R853* cells. A negative score indicates that the molecule is predicted to be inhibited based on the DEGs that have been processed. (C) Heatmap showing the relative quantity normalized to GAPDH of each genes (scaled between 0 and 1, in column), for each control or the patient’s cells (in row) after stimulation with TNFα 10 ng/ml for 6 h or poly I:C 1 µg/ml for 6 h (as indicated by TNF or PIC respectively), as assessed by RT-qPCR. (D) Western blot assessing the quantity of phospho-p65 (S536), p65, phospho-IκBα (S32), and IκBα on cell lysates from SV40-fibroblasts stimulated with TNFα at indicated time points. Tubulin was used as a loading control. (E) Western blot of cytoplasmic and nuclear extracts from SV40-fibroblasts after 2 h of TNFα (T) (10 ng/ml) or poly I:C (P) (1 µg/ml) stimulation. RELA/p65 and p50 were revealed using a specific antibody to assess for translocation in the nucleus. α-tubulin was used as a loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. Source data are available for this figure: SourceData FS5.
Bulk RNA sequencing of SV40-fibroblasts and RT-qPCR and western blot of the canonical NF-κB pathway. (A) Heatmap showing the normalized and scaled expression (vst) of IRF3 target genes significantly upregulated in controls after stimulation with poly I:C 10 µg/ml for 6 h. Each column represents a patient or a control. RNA sequencing has been performed in biological duplicates for each control or patient (as indicated by _1 or _2 at the end of each column name). (B) Z-score of the different “molecules” predicted to be inhibited (top 10 based on the z-score) based on IPA (Qiagen Inc.) after poly TNFα stimulation for the NFKB2 p.R853* cells. A negative score indicates that the molecule is predicted to be inhibited based on the DEGs that have been processed. (C) Heatmap showing the relative quantity normalized to GAPDH of each genes (scaled between 0 and 1, in column), for each control or the patient’s cells (in row) after stimulation with TNFα 10 ng/ml for 6 h or poly I:C 1 µg/ml for 6 h (as indicated by TNF or PIC respectively), as assessed by RT-qPCR. (D) Western blot assessing the quantity of phospho-p65 (S536), p65, phospho-IκBα (S32), and IκBα on cell lysates from SV40-fibroblasts stimulated with TNFα at indicated time points. Tubulin was used as a loading control. (E) Western blot of cytoplasmic and nuclear extracts from SV40-fibroblasts after 2 h of TNFα (T) (10 ng/ml) or poly I:C (P) (1 µg/ml) stimulation. RELA/p65 and p50 were revealed using a specific antibody to assess for translocation in the nucleus. α-tubulin was used as a loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. Source data are available for this figure: SourceData FS5.
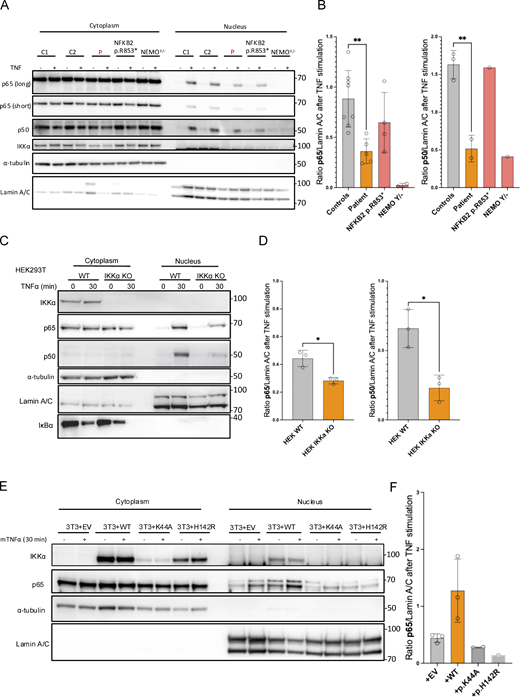
Defective translocation of p65 and p50 in SV40-fibroblasts and in IKKα-deficient cellular models. (A) Western blot of cytoplasmic and nuclear protein extracts from SV40-fibroblasts activated with TNFα for 30 min assessing the translocation of p65/RELA and p50 in the nucleus. α-Tubulin was used as a loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. (B) Quantification of nuclear translocation for p65/RELA and p50. Statistical analysis was performed using an unpaired t test (**P < 0.01). Representative of at least three independent experiments. (C) Western blot of cytoplasmic and nuclear extracts from HEK293T cells (IKKα ΚΟ or not as indicated) after 30 min of TNFα (10 ng/ml) stimulation. RELA/p65 and p50 were revealed using a specific antibody to assess for translocation in the nucleus. α-Tubulin was used the loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. IκΒα was probed as a control of stimulation. (D) Barplots show the quantification of translocation normalized to the quantity of Lamin A/C for RELA/p65 and p50 after stimulation. An unpaired t test was used to perform statistical analysis (*P < 0.05). (E) Western blot of cytoplasmic and nuclear extracts from 3T3-NIH IKKα knocked-out transduced back with a lentivirus coding for the human IKKα (or empty) with or without indicated mutations (p.K44A, kinase dead, p.H142R, patient variant). Cells were subjected to mouse TNFα stimulation (30 min, 10 ng/ml) before protein extraction. RELA/p65 was assessed as well as α-tubulin (used as a loading control of cytoplasmic extracts) and Lamin A/C (used as a loading control for nuclear extracts). (F) Barplots show the quantification of p65 translocation after stimulation for each transduced cell line. All figures are representative of at least two independent experiments. Source data are available for this figure: SourceData F8.
Defective translocation of p65 and p50 in SV40-fibroblasts and in IKKα-deficient cellular models. (A) Western blot of cytoplasmic and nuclear protein extracts from SV40-fibroblasts activated with TNFα for 30 min assessing the translocation of p65/RELA and p50 in the nucleus. α-Tubulin was used as a loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. (B) Quantification of nuclear translocation for p65/RELA and p50. Statistical analysis was performed using an unpaired t test (**P < 0.01). Representative of at least three independent experiments. (C) Western blot of cytoplasmic and nuclear extracts from HEK293T cells (IKKα ΚΟ or not as indicated) after 30 min of TNFα (10 ng/ml) stimulation. RELA/p65 and p50 were revealed using a specific antibody to assess for translocation in the nucleus. α-Tubulin was used the loading control of cytoplasmic extract while Lamin A/C was used for nuclear extracts. IκΒα was probed as a control of stimulation. (D) Barplots show the quantification of translocation normalized to the quantity of Lamin A/C for RELA/p65 and p50 after stimulation. An unpaired t test was used to perform statistical analysis (*P < 0.05). (E) Western blot of cytoplasmic and nuclear extracts from 3T3-NIH IKKα knocked-out transduced back with a lentivirus coding for the human IKKα (or empty) with or without indicated mutations (p.K44A, kinase dead, p.H142R, patient variant). Cells were subjected to mouse TNFα stimulation (30 min, 10 ng/ml) before protein extraction. RELA/p65 was assessed as well as α-tubulin (used as a loading control of cytoplasmic extracts) and Lamin A/C (used as a loading control for nuclear extracts). (F) Barplots show the quantification of p65 translocation after stimulation for each transduced cell line. All figures are representative of at least two independent experiments. Source data are available for this figure: SourceData F8.
Altogether, these results suggested that the two IΚΚα missense variants negatively impacted the canonical NF-κB pathway, at least by reducing the translocation of p65 and p50, but without affecting the cytosolic function of the NEMO complex. Of note, this negative impact is milder than the complete loss-of-activation observed in the NEMO null cells.
Neutralizing autoantibodies against type I IFNs are detectable in the patient serum
This patient suffered from a severe deficiency of the non-canonical NF-κB pathway. Recently, neutralizing autoantibodies against type I IFN were described as a hallmark of non-canonical NF-κB deficiency in patients with p52 LOF/p100 IκBδ GOF NFKB2 variants, and biallelic LOF NIK and RELB variants (Le Voyer et al., 2023; Bodansky et al., 2023). Using a previously described luciferase assay, we questioned the presence of such autoantibodies in the context of this autosomal recessive IKKα deficiency (Le Voyer et al., 2023; Bastard et al., 2021). Interestingly, serum from the patient (5 years before her death, well before the SARS-CoV-2 pandemic) neutralized IFN-α2 both at 10 ng/ml (Fig. 9 A) and 100 pg/ml (more physiological concentration, Fig. 9 B) and IFN-ω at 100 pg/ml but did not neutralize IFN-β (Fig. 9, A and B).

Neutralizing autoantibodies against type I IFNs. (A) Luciferase-based neutralization assay to detect auto-Abs neutralizing 10 ng/ml IFNα2, IFN-ω, or IFN-β in an individual previously known to have neutralizing anti-IFN-I auto-Abs (C+), healthy donors (HD), and the patient, and the patient with the variants in CHUK (P). (B) Luciferase-based neutralization assay to detect auto-Abs neutralizing 100 pg/ml IFNα2, IFN-ω in an individual previously known to have neutralizing anti-IFN-I auto-Abs (C+), healthy donors (HD), and the patient with the variants in CHUK (P). Relative luciferase unit (RLU) is calculated by normalizing Firefly luciferase activity against the Renilla luciferase activity, before normalization against non-stimulated conditions. An induction factor of >5 relative to non-stimulated conditions was considered to correspond to neutralizing activity (dashed line). These experiments have been performed twice.
Neutralizing autoantibodies against type I IFNs. (A) Luciferase-based neutralization assay to detect auto-Abs neutralizing 10 ng/ml IFNα2, IFN-ω, or IFN-β in an individual previously known to have neutralizing anti-IFN-I auto-Abs (C+), healthy donors (HD), and the patient, and the patient with the variants in CHUK (P). (B) Luciferase-based neutralization assay to detect auto-Abs neutralizing 100 pg/ml IFNα2, IFN-ω in an individual previously known to have neutralizing anti-IFN-I auto-Abs (C+), healthy donors (HD), and the patient with the variants in CHUK (P). Relative luciferase unit (RLU) is calculated by normalizing Firefly luciferase activity against the Renilla luciferase activity, before normalization against non-stimulated conditions. An induction factor of >5 relative to non-stimulated conditions was considered to correspond to neutralizing activity (dashed line). These experiments have been performed twice.
Discussion
In this study, we reported the first compound heterozygous LOF mutation in CHUK leading to a profound loss of IKKα kinase activity and a reduced IKKα protein expression. In humans, the complete absence of the protein IKKα (CHUK null) was shown to be lethal in utero, a phenotype reminiscent of complete IKKα deficiency (IKKα−/−) in mice that die early after birth (Hu et al., 1999, 2001; Takeda et al., 1999; Lahtela et al., 2010). Only one patient presenting with a combined immunodeficiency and carrying a homozygous missense mutation in CHUK has been so far described (Bainter et al., 2021). This mutation (p.Y580C) changed a residue in the helix-loop-helix domain of IKKα and was shown to abrogate the interaction between IKKα and its activating kinase NIK. Consequently, cells from that patient exhibited a profound defect of the non-canonical NF-κB pathway activation with an impaired cleavage of p100 to p52 despite a normal kinase activity of the mutant in vitro. The phenotype was reproduced in a mouse model. Importantly, the canonical NF-κB pathway was not reported to be affected in the patient carrying the homozygous IKKα p.Y580C (Bainter et al., 2021).
The patient herein studied suffered from a syndromic combined immunodeficiency including hypogammaglobulinemia, recurrent infections, digestive and liver inflammation, along with bone malformation and ectodermal dysplasia. Two heterozygous private missense mutations modifying highly conserved residues in the kinase domain of IKKα were identified. We showed that both alleles were kinase LOF in vitro and that this was leading to a complete deficiency in the activation of the non-canonical NF-κB pathway in the patients’ cells. We also observed a reduced interaction with NIK upon co-immunoprecipitation with IKKα in an overexpression model. However, this result could also reflect the instability of kinase-dead variants. In any case, these results were consistent with the patient’s phenotype which included hypogammaglobulinemia, low B cells, absence of memory B cells and plasmablasts, and related recurrent infections as well as the presence of autoantibodies against type I IFNs. All these manifestations were previously reported in other defects affecting the non-canonical NF-κB pathway (Willmann et al., 2014; Schlechter et al., 2017; Sharfe et al., 2015; Le Voyer et al., 2023; Bainter et al., 2021). Moreover, mice knocked-in for a kinase-dead IKKα (IKKα p.K44A, IKKαKA/KA) showed an early arrest of B cell development associated with an impaired non-canonical NF-κB signaling and impaired formation of secondary lymphoid organs (Balkhi et al., 2012). We therefore concluded that this patient suffered from a very severe deficiency of IKKα kinase activity.
Interestingly, we also observed a partial defect in the transcription of target genes of the canonical pathway in the SV40-fibroblasts from the patient upon TNFα stimulation. In agreement with previous publications showing a redundant role of IKKα in the NEMO complex, we did not observe differences in the phosphorylation or degradation of IκBα in cells from the patient (Hu et al., 1999). In contrast, bulk RNA sequencing of SV40-fibroblasts before and after stimulation with TNFα or poly I:C showed a partial defect in the transcription of the canonical NF-κB pathway target genes. Whether this result was linked to an excess of the uncleaved p100 exerting an IκΒδ inhibitory effect on p65-containing dimers was a possibility (Tao et al., 2014; Razani et al., 2010). A reduced level of nuclear p65 was also described in the IKKαKA/KA mouse model, and the authors suggested that p100 could inhibit its translocation (Balkhi et al., 2012). Consistently, we observed a significant defect in the nuclear translocation of p65 and p50 after TNFα stimulation in the patient’s cells. Moreover, we reproduced this observation in two cellular models reproducing either the absence of IKKα or the presence of a kinase-dead mutant. The exact mechanism of this observation remains to be determined. In patients with autosomal recessive NIK deficiency, the canonical pathway was impacted but the experiments did not specifically look at the role of the p100 excess (Willmann et al., 2014). In the context of NFKB2 deficiency, the relative excess of p100 is a hallmark of the mutations affecting the C-terminal processing inhibitory domain (PID) (Le Voyer et al., 2023). In an ectopic expression model of those NFkB2 mutations, it was shown that p100 accumulation was associated with cytoplasmic retention of p65 (Lee et al., 2014). In our report, cells from an NFKB2 mutated patient (p52 LOF/p100 GOF) did not exhibit any defect of the canonical NF-κB pathway activation as assessed by bulk RNA sequencing after TNFα or poly I:C stimulation. Moreover, we observed a normal p65/p50 nuclear translocation upon TNFα stimulation. Therefore, the defect observed for particular canonical NF-κB target genes activation in the patient carrying two IKKα LOF mutations could be a direct consequence of the defective kinase activity rather than the relative excess of p100 or an association of both mechanisms. A possible explanation for the defective induction of canonical target genes may rely on the nuclear role of IKKα (Swaminathan, 2003). It was previously shown that IKKα shuttle into the nucleus where it phosphorylates histone H3 on Serine 10 thereby promoting the transcription of NF-κB target genes (Anest et al., 2003, 2004; Yamamoto et al., 2003). The defective kinase activity observed for both mutants could therefore lead to less histone H3 phosphorylation upon canonical stimuli, potentially dampening the transcription of some NF-κB target genes as observed in our RNA sequencing experiment. Whether this also explained the reduction in the activation of the JAK-STAT pathway as observed by inference analysis or if this was an indirect consequence of the lower level of transcription of IL-6 and IFNγ remains to be determined. In any case, the defective activation of the NF-κB canonical pathway could account for the phenotype of the patient who displayed many features of canonical defect such as dental enamel abnormalities, a skin disease resembling ectodermal dysplasia and elevated naive CD4+ and CD8+ T cell, all being clinical and biological features known to occur in patient with autosomal dominant GOF mutations in NFKBIA or hypomorphic X-linked mutations in NEMO (Smahi et al., 2000; Courtois et al., 2003; Courtois and Smahi, 2006). Of note, TP63, the gene known to be mutated in Hay–Wells syndrome for which the patient was first sequenced due to similar clinical skin and bones phenotype was found downregulated in patient fibroblasts upon TNFα stimulation in our RNA sequencing experiment. Whether this result could explain the severity of the skin and bone phenotype was beyond the scope of this study and would require further investigations.
Intriguingly, the patient died from complications related to a DLBCL of the lung, a malignancy frequently driven by an excess of NF-κB activation (Eluard et al., 2022). It was thus surprising that this type of lymphoma developed in the context of this IKKα deficiency. However, a similar, although rare, observation was made in other primary immune deficiency and dysregulation such as the monoallelic NFKB2 deficiency, with only one case of non-Hodgkin lymphoma (non-EBV related) (Klemann et al., 2019). Interestingly, mouse models have suggested a role of IKKα in preventing skin tumor development by the suppression of EGFR and c-Myc pathways while another model showed that IKKαKA/KA mice were prone to develop systemic inflammation and lung tumors (lung squamous cell carcinomas) (Descargues et al., 2008; Xiao et al., 2013). Whether similar mechanisms (e.g., non-NF-κB roles of IKKα) explain the development of a B cell lymphoma in this patient remains to be determined, and the contribution of additional somatic genetic drivers was not investigated (Leveille and Johnson, 2021). We also could not exclude the contribution of defective immunosurveillance in this disease as the patient’s T cells failed to proliferate in vitro upon non-specific TCR stimulation (Schreiber et al., 2011; Afshar-Sterle et al., 2014). In contrast, the patient did not suffer from gross cellular immunity deficiency except for the onychomycosis that could be linked to a defect in Th17, as previously described in the other patient with IKKα deficiency (Bainter et al., 2021). Interestingly, both the patient herein described and the other patient with the homozygous p.Y580C variant had liver complications and villous atrophy, the latter being more severe as it required liver transplantation (Bainter et al., 2021). Such clinical manifestations were already described in NIK and RELB deficiencies (Willmann et al., 2014; Ovadia et al., 2017). A possibility could be that the non-canonical NF-κB pathway defect was at the origin of a global immune dysregulation and infiltration of organs with autoreactive T cells although this was not demonstrated. In line with this was the autoimmunity and uncharacterized immune dysregulations with lymphocytic organ infiltration widely reported in NFKB2 mutated patients (Klemann et al., 2019). Among several possible mechanisms, the break of central tolerance due to the impaired expression of AIRE by medullary epithelial thymic cells, as previously described in mice and recently in humans, was an appealing hypothesis (Le Voyer et al., 2023; Zhu et al., 2006). Accordingly, the studied patient displayed neutralizing autoantibodies against type I IFNs as recently shown in humans with other germline deleterious mutations of the non-canonical NF-κB pathway (Le Voyer et al., 2023; Bodansky et al., 2023). Interestingly, a recent description showing the presence of auto-antibodies neutralizing type I IFNs in female patients with incontinentia pigmenti (carrying a hemizygous LOF variant in NEMO) emphasizes a role for the canonical NF-κB pathway in the involution of the thymus in those patients (Rosain et al., 2024). Whether the patient described in this study developed autoimmunity because of both a non-canonical and canonical NF-κB defect remains to be determined. Another possibility could be that the patient herein studied suffered from a recently described entity called EVAH, for enteric viral-associated hepatitis, where chronic infections with enteric viruses in immunodeficient patients were associated with chronic hepatitis (Riller et al., 2023, 2024; Fourgeaud et al., 2023; Bucciol et al., 2018).
Overall, we described the first human biallelic mutations in CHUK leading to a severe deficiency of the IKKα kinase activity. As expected, the NIK-IKKα dependent non-canonical NF-κB pathway was severely compromised. Our findings also showed that induction of particular canonical NF-κB target genes was partially impaired, which may explain the severity of the patient’s phenotype, including its skin abnormalities, and emphasize the role of IKKα kinase activity in the human canonical NF-κB pathway activation. The study of this single patient emphasized that a severe deficiency of IKKα kinase activity was compatible with life in humans, although associated with immunodeficiency and malformations, while the complete loss of protein expression was lethal (Lahtela et al., 2010).
Materials and methods
Study participants
Before the study, all patients signed informed consents approved by the CERAPH-Centre (IRB: #00011928). The biological samples are part of Inserm UMR1163/Imagine collection declared to the French Ministère de la Recherche (CODECOH no. DC-2020-3994).
Clinical case report
The patient was born to healthy, non-consanguineous Caucasian parents, with no family history of genetic diseases. She has been monitored since pregnancy and birth (at 38.5 wk) for a polymalformative syndrome, which includes ankyloblepharon, buccal synechia, retrognathia, ectrodactyly of the feet, hypoplasia of both thumbs, cleft palate, ectodermal dysplasia with sparse hair, hypoplastic external genitalia, and maxillomandibular adhesions (Khandelwal et al., 2017). She underwent surgery for ankyloblepharon, ectrodactyly, and cleft palate. The deciduous teeth were conical, and agenesis of permanent teeth was observed. The patient experienced severe gastroesophageal reflux and growth retardation without growth hormone deficiency, requiring tube feeding and eventually a gastrostomy. A diagnosis of ceoliac disease was made based on the presence of villous atrophia without detected auto-antibodies, gluten-free regimen was not enough to reverse growth retardation. She also suffered from recurrent lower respiratory tract infections associated with hypogammaglobulinemia from 5 mo of age, requiring immunoglobulin replacement therapy since then. These recurrent episodes of infections led to the development of bronchiectasis in the lower and middle right lobes and atelectasis. Liver enzymes were chronically mildly elevated without specific diagnosis made, the liver biopsy showing mild lymphocytic inflammation. At 15 years old, the patient developed a pulmonary lesion that turned out to be a localization of DLBCL. Immunohistochemistry was consistent with non-GC DLBCL. The patient died of a massive pulmonary hemorrhage few days after the second infusion of chemotherapy.
Genetic analysis
Next-generation sequencing was previously described in the publication of one mutation of the patient (Khandelwal et al., 2017). Sanger sequencing was performed to confirm the next-generation sequencing results and to analyze cDNA and genomic DNA from primary cell lines of the patient. Purified PCR products were directly sequenced using BigDye Terminators (version 1.1) and a 3500xL Genetic Analyzer (Applied Biosystems).
Primary fibroblasts isolation
Skin biopsy was separated in two parts by using scalpel and introduce in a 25 cm2 culture flask (Thermo Fisher Scientific). After 15 min without medium at 37°C in a humidified 5% CO2 incubator, skin biopsies attached to the plastic of the flask and culture medium was added (RPMI 1640 from Life Technologies supplemented with 20% Fetal Bovin Serum (FBS; Life Technologies), 1% penicillin-streptomycin (PS; Thermo Fisher Scientific), 1% amphotericin B (Thermo Fisher Scientific), 50 µg/ml gentamicin (Thermo Fisher Scientific). Fibroblasts usually starts to grow after 1 or 2 wk of culture and are trypsinized (Trypsin; Thermo Fisher Scientific) and cultured in 25 cm2 culture flasks (passage 1) and then in 75 cm2 culture flasks (passage 2). After these two passages, cells are frozen in FBS 10% dimethylsulfoxide (DMSO; Thermo Fisher Scientific).
Cell culture
SV40-fibroblasts, HEK293T, and RELA−/− HEK293T cells were maintained in DMEM (Life Technologies) supplemented with 10% FBS (Thermo Fisher Scientific Gibco) and 1% PS (Thermo Fisher Scientific). B-EBV cell lines were maintained in complete RPMI 1640 (RPMI supplemented with 10% FBS, 1% PS; Life Technologies). T cells activation was carried out from the PBMC of patients or of healthy donors. T cells were activated by binding CD3/CD28 receptors with Dynabeads (#11141D; Thermo Fisher Scientific). Cells were cultured at 1.106/ml/10 μl of Dynabeads in complete Panserin medium (Panserin 401 medium [# P04-710401; PAN BIOTECH]), supplemented with 5% human AB serum, 1% PS (15140122; Thermo Fisher Scientific), and 1% L-glutamine (25030081; Thermo Fisher Scientific) and filtered with a unit Nalgene Rapid-Flow single-use sterile filtration unit with polyethersulfone (PES) membrane (#568-0010; Thermo Fisher Scientific). After 3 days of culture, cells were centrifuged on Ficoll and then recultured in complete Panserin with interleukin-2 (IL-2) at 100 international units/ml (U/ml). Every 2 days, cell expansion was continued by adding Panserin complete with IL-2. 3T3-NIH IKKα KO cells were a gift from Véronique Baud laboratory.
Transformation of primary fibroblasts with SV40
Primary fibroblasts were immortalized by lipofection of 2.5 µg of pBSSVD2005 (#21826; Addgene) coding the large-T antigen of Simian virus-40, using Lipofectamine 2000 as previously described in the Materials and methods. After three to four cycles of cultures, fibroblasts changed their shape and growing rate and were considered transformed and called SV40-fibroblasts.
CRISPRCas9 in HEK293T cells
CRISPR-Cas9 technology was used to knock-out RELA from wt HEK293T cells. RNA guides were designed using the online tool CRISPOR. Single guides cDNA were cloned into MLM3636 plasmid (plasmid #43860; Addgene). Sequences of the plasmids were verified by Sanger sequencing. wt HEK293T cells were transfected using lipofectamine 2000 (Life Technologies) with the plasmid MLM3636 containing the cDNA sequence of the single-guide and a plasmid coding the Cas9 (pCas9_GFP, plasmid #44719; Addgene). 48 h later, GFP-positive cells were single-cell sorted in flat-bottom 96 wells plates (Falcon). Growing clones of transfected HEK293T cells were screened by Sanger sequencing for genomic alteration around the single-guide target. Homozygous clones for frameshift deletion or insertion in RELA were selected and subjected to western blot for the N-terminal part of p65. Loss-of-expression clones were selected for further experiments. Response to TNFα was null in NF-κB luciferase experiments. These cells are called RELA KO HEK293T cells in the manuscript. DNA sequence of the single guides targeting exon 4 of RELA are as follow: Fw 5′-ACACCGCTTTCCTACAAGCTCGTGGG-3′; Rv 5′-AAAACCCACGAGCTTGTAGGAAAGCG-3′. IKKα KO was performed in HEK293T cells RELA KO following the same protocol by using the following guides targeting exon 3: Fw 5′-CACTGTGTGATCTTGTTACTTAGCC-3′ Rv 5′-TCTCTCTCATCTCAAAACATCCACA-3′.
Subcloning in mammalian expression vectors
CHUK cDNA was amplified using primers encompassing the 5′UTR (forward) and the 3′UTR (Reverse) from control PBMC cDNA. The agarose gel band at the expected size was purified and amplified by using primers adding a restriction site at each end of the coding sequence (from ATG to TAA). The purified band was subcloned into the mammalian expression vector pCMV-HA-tagged (Clontech). Directed mutagenesis was performed following the Q5-site directed mutagenesis kit protocol (NEB).
pCMV-MYK-tagged NIK and pEF1A-p100 were obtained by subcloning NIK and p100 from available backbones (pCMV4-p100 #23287; Addgene, EGFP-NIK #111197; Addgene, respectively) using the NEBuilder HiFi DNA Assembly Cloning kit following manufacturer instructions (NEB). pCMV6-RELB was kindly provided by the team of Pr. J.-L. Casanova. Sanger sequencing was used to verify the sequence of the plasmids from the promoter to the end of the insert after subcloning and directed mutagenesis.
HA-CHUK coding sequence was also transferred to a lentiviral plasmid backbone (pMSCV-MCS-EF1-GFP-T2A-Puro) using the NEBuilder HiFi DNA Assembly Cloning kit.
All plasmid production was performed by transforming DH5α E. coli competent cells (NEB) or Stbl3 for the lentivirus plasmids (NEB). Extraction and purification of plasmid DNA was performed on PureLink HiPure Plasmid Midiprep Kit (Thermo Fisher Scientific) or Wizard Plus SV Minipreps DNA Purification Systems (Promega).
Transfection of HEK293T cells
HEK293T cells were seeded in plates 24 h before the transfection. wt or RELA−/− HEK293T cells were transiently transfected using Lipofectamine 2000 (Life Technologies) with the different plasmids as indicated in the figures (CHUK, RELB, p100, NIK). The DNA and Lipofectamine 2000 dilution were performed in optiMEM (Life Technologies). 24 or 48 h later, cells were harvested to perform western blot or luciferase assays.
Non-canonical NF-κB pathway luciferase in HEK293T cells
To activate specifically the non-canonical pathway in an ectopic expression model of CHUK we used the RELA−/− HEK293T cells. RELA−/− HEK293T cells were seeded in 96 flat-bottom compatible with luciferase reading plates (655098; Greiner) and transfected 24 h later by using Lipofectamine 2000. Cells were transfected with pCMV-HA-tagged EV or CHUK wt or mutants, pEF1A-P100 wt and pCMV6-RELB depending on the experiments and two reporter plasmids, one Firefly Luciferase reporting NF-κΒ activity pGL4.32[luc2P/NF-κB-RE/Hygro] (Promega) and one pTK Renilla Luciferase as an internal control (Thermo Fischer Scientific). 48 h later, luciferase signal was revealed and measured with the Dual-Glo Luciferase Assay System (Promega). The quantity of plasmid was kept constant in all conditions by adding the remaining dose of EV.
Co-immunoprecipitation between NIK and IKKα
For the co-immunoprecipitation NIK/IKKα experiments, HEK 293T cells were transfected with a Flag-NIK kinase dead expression vector along with a vector encoding HA-IKKα either wt, H142R, P190L. 2 days after transfection, the cells were lysed (50 mM Tris pH 7,5, 1% NP-40, 1 mM EDTA, 6 mM EGTA, 120 mM NaCl, 15 mM sodium pyrophosphate, PhosSTOP [4906845001; Sigma-Aldrich] and Compete [4906837001; Sigma-Aldrich] and 20 mM NEM [N-ethylmaleimide, E1271; Sigma-Aldrich]) and the protein extract was subjected to an immunoprecipitation with either control IgG-beads (A0919; Sigma-Aldrich) or Anti-Flag-M2 beads (F2426; Sigma-Aldrich). Half of the immunoprecipitates were used for an immunoblot for detecting Flag-NIK and the other half for the detection of HA-IKKα.
Kinase assay
HEK293T cells were transfected with pCMV-HA EV, pCMV-HA CHUK p.H142R, p.P190L, p.K44A, or p.Y580C. After 24 h, cells were lysed in cold lysis buffer (containing Tris HCl pH 7 50 mM, NaCl 150 mM, EDTA 1 mM, Triton 100-X 1%) and proteins were subjected to immunoprecipitation using an anti-HA tag monoclonal antibody (Sepharose Bead Conjugate, clone C29F4; Cell Signaling) following manufacturer instructions, overnight at 4°C. The following day, sepharose beads were washed five times in cold lysis buffer and two times in room temperature 1× kinase buffer (V4068; Promega IKKα Kinase Enzyme System). Kinase reaction was performed for 1 h at room temperature by adding 2× kinase reaction buffer containing ATP and IKKtide as a substrate on the sepharose pellet. The supernatant was used to measure the kinase activity using the ADP-Glo Luciferase-based kinase assay following manufacturer instructions (V6930; Promega). Sepharose beads were washed again two times in lysis buffer and subjected to immunoblot after denaturation. Results are normalized to the EV condition.
Kinase assay using GST-IκBα
For the kinase assay of IKKα, HEK 293T cells were transfected with a vector encoding HA-IKKα (wt, H142R, P190L, or Y580C) or an EV. 2 days after transfection, the cells were lysed (50 mM Tris pH 7.5, 1% NP-40, 1 mM EDTA, 6 mM EGTA, 120 mM NaCl, 15 mM sodium pyrophosphate, PhosSTOP [4906845001; Sigma-Aldrich] and Compete [4906837001; Sigma-Aldrich] and the protein extract was subjected to an immunoprecipitation with an anti-HA AB [901514; BioLegend] coupled to A/G Agarose beads [SC-2003; Santa-Cruz]). Kinase reaction was performed for 1 h at 30°C in presence of 200 ng GST-IκBα in a kinase assay buffer (20 mM HEPES pH 7,5, 10 mM MgCl2, 2 mM DTT, 100 µM ATP, PhosSTOP [4906845001; Sigma-Aldrich] and Compete [4906837001; Sigma-Aldrich]). Immunoblots were then performed for the detection of p-IκBα S32.
Lentiviral production
HEK293T Lenti-X cells were cultured in DMEM 10% FBS 1% PS. The day before the experiment, cells were seeded in 75 cm2 flasks (Falcon) for cell culture. At Day 0 (D0), cells were transfected by Lipofectamine 2000 with 10 µg of the plasmid containing the coding sequence of HA-CHUK (with mutation or not, pMSCV-HA-CHUK-EF1A-GFP-T2A-Puro), 7.5 µg of psPAX2 (plasmid #12260; Addgene) and 5 µg of pMD2.G (plasmid #12259; Addgene). 60 h later, the media was collected, centrifuge at 3,000 rpm at 4°C for 10 min to pellet cell debris. The supernatant was filtered through a 0.22 µm low protein binding membrane (Millipore) and aliquots were stored at −80°C before use. The same protocol was used to produce lentiviral particles coding for RELA using a homemade pMSCV-HA-RELA-EF1A-GFP-T2A-Puro plasmid.
Lentiviral transduction
SV40-fibroblasts were seeded in 6-well plates (Falcon) 24 h before the transduction to reach 70–90% confluence. The day of transduction 1 ml of lentiviral production was added to 500 µl of optiMEM (Life Technologies) and transferred in each well of fibroblasts after adding 8 µg/ml polybrene (Sigma-Aldrich). Medium was changed 6 h after and cells were sorted based on the positivity of GFP after one passage. HEK293T cells RELA KO and RELA/IKKα KO were transduced using the same protocol (lentiviral plasmid pMSCV-HA-RELA-EF1A-GFP-T2A-Puro). Cells were sorted based on their GFP expression on a MA900 Sony cell sorter cytometer.
Cell stimulation
Patient’s and control SV40-fibroblasts were stimulated with 100 ng/ml LTα1β2 (8884-LY-025/CF; R&D Systems) or 10 ng/ml Tweak (310-06; PeproTech) to stimulate the non-canonical pathway of NF-κB and 10 ng/ml TNFα (#210-TA-020; R&D Systems) to stimulate the canonical NF-κB pathway. Poly(I:C) (HMW) from Invivogen was used in the bulk RNA sequencing experiment. Mouse recombinant TNF (#575206; BioLegend) was used at the concentration of 10 ng/ml to stimulate 3T3-NIH cells.
RT-qPCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen), and cDNA was prepared using the Quantitect Reverse Transcription Kit (Qiagen) after the depletion of genomic DNA. SYBR Select Master Mix was used to perform Real-time quantitative PCR using the LightCycler VIIA7 System (Roche), following manufacturer’s instructions. The following primers were used to quantify VCAM1 Fw 5′-GATTCTGTGCCCACAGTAAGGC-3′ Rv 5′-TGGTCACAGAGCCACCTTCTTG-3′; TNFAIP3 Fw 5′-TCCTCAGGCTTTGTATTTGAGC-3′ Rv 5′-TGTGTATCGGTGCATGGTTTTA-3′; CHUK Fw 5′-CTCGGAAACCAGCCTCTCAATG-3′ Rv 5′-GATAAACTTCTGGAAGCAAATGGC-3′, ICAM1 Fw 5′-AGCGGCTGACGTGTGCAGTAAT-3′ Rv 5′-TCTGAGACCTCTGGCTTCGTCA-3′; IRF1 Fw 5′-GAGGAGGTGAAAGACCAGAGC-3′; NFKBIA Fw 5′-GAGACCTGGCCTTCCTCAAC-3′ Rv 5′-TCTCGGAGCTCAGGATCACA-3′. Quantification was normalized to GAPDH. RNA expression was analyzed using the ∆∆Ct method. All primers were checked for linearity by serial dilution of a control cDNA. A R2 > 0.99 was obtained for each set of primers and deltaCT between 10-fold dilution was always 3.3 Ct.
Bulk RNA sequencing
RNA was extracted from SV40-fibroblasts stimulated or not with TNFα 10 ng/ml or poly I:C 10 µg/ml (High Molecular Weight; Invivogen) for 6 h. Extraction was performed using the Zymo Quick-RNA 96 Kit (R1052) following manufacturer instructions with the DNAse step. RNA integrity and concentration were assessed by capillary electrophoresis using a Fragment Analyzer (Agilent Technologies). RNA sequencing libraries were prepared starting from 500 ng of total RNA using the Universal Plus mRNA-Seq kit (Nugen) as recommended by the manufacturer. The oriented cDNA produced from the poly-A+ fraction was sequenced on a NovaSeq6000 from Illumina (Paired-End reads 100 bases + 100 bases). A total of ∼50 million passing-filters paired-end reads were produced per library. Paired-end RNA sequencing reads were aligned to the human Ensembl genome GRCh38.91 using STAR (v2.7.5a) and counted using featureCounts from the Subread R package. The raw count matrix was analyzed using DESeq2 (version 21.38.3). Prefiltering to keep only rows that have a count of at least 10 for a minimal number of samples was performed. Differential expression analysis was performed using the “DESeq” function with default parameters. For visualization and clustering, the data were normalized using the “variant stabilizing transformation” method implemented in the “vst” function. Plots were generated using ggplot2 (version 3.4.4) and pheatmap (version 1.0.12).
Pathway activity inference analysis of bulk RNA sequencing data was performed using decoupleR package (version 2.5.3), using default parameters. Visualization of results was performed using ggplot2 (version 3.4.4). IPA (Qiagen Inc.) was also used to perform pathway activity inference and decipher molecules predicted to be inhibited or activated. All results of the DEGs between groups, the decoupleR, and IPA analysis are available in Table S2.
Whole-cell lysates
Whole-cell lysates from SV40-fibroblasts, activated T cells, and HEK293T cells were prepared by using radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific) supplemented with protease inhibitor cocktail (#87785; Thermo Fisher Scientific) and phosphatase inhibitors cocktails 2 and 3 (P5726-1ML; Sigma-Aldrich, P0044-1ML; Sigma-Aldrich) following manufacturer instructions (complete RIPA buffer). For adherent cells, culture wells were washed three times with cold PBS and complete RIPA buffer was added in the well. After 5 min, all cells were lysed in the well and the cell lysate was transferred in aliquots and sonicated using the Bioruptor sonication device previously cooled at 4°C (#B01060010; Diagenode) with 10 cycles of 30 s on, 30 s off. Cell lysates were centrifugated at full speed for 5 min on a microcentrifuge at 4°C and supernatants (protein extracts) were collected. Proteins were quantified using the Pierce BCA Protein Assay kit (#23225; Thermo Fisher Scientific) just before use. Protein extracts were used immediately or frozen at −20°C for short-term conservation and −80°C for long-term conservation.
Cytonuclear extractions
Cells were harvested, washed three times with cold PBS, and centrifuged at 600 g for 5 min. The NE-PER protein extraction system was used to extract cytoplasmic and nuclear protein fractions (#78833; Thermo Fisher Scientific) following manufacturer instructions. Briefly, after centrifugation, the CER 1 buffer supplemented with protease inhibitor cocktail (#87785; Thermo Fisher Scientific) and phosphatase inhibitors cocktails 2 and 3 (P5726-1ML; Sigma-Aldrich, P0044-1ML; Sigma-Aldrich) was added to the cell pellet, vortexed, and placed on ice for 10 min. After 10 min, CER 2 was added for 1 min and cells were centrifugated at >14,000 g on a microcentrifuge a 4°C. The supernatant was collected (cytoplasmic fraction) and frozen. The remaining pellet of the aliquot was washed two times with cold PBS and NER buffer supplemented as previously described was added to the remaining pellet. During 40 min, the aliquots were sonicated for 10 min and vortexed every 10 min. The nucleus lysate was centrifugated at maximum speed (>14,000 g) at 4°C for 10 min and the supernatant was collected and frozen (nuclear fraction).
Western blots
Samples were denatured and reduced with Bolt LDS Sample buffer 4× (Thermo Fisher Scientific) and Bolt sample reducing agent 10× (Thermo Fisher Scientific). The migration was carried out on 8% Bis-Tris gels (Thermo Fisher Scientific) at 150V. The transfer was done on polyvinylidene fluoride (PVDF) membranes (#IB24002; Thermo Fisher Scientific) with the iBlot 2 dry transfer system (#IB21001; Thermo Fisher Scientific) using the P0 program. The membranes were blocked with 5% bovine serum albumin (BSA) in TBS (Tris Buffer saline) Tween 0.1% for 1 h at room temperature with shaking and then incubated with the primary antibody with stirring at 4°C overnight. After three washes with TBS Tween 0.1% (TBST), the anti-rabbit or anti-mouse secondary antibody coupled to HRP (Horse Radish Peroxidase) was incubated for 1 h at room temperature with stirring at a dilution of 1:10,000 in 5% BSA TBST. After three washes with TBST, the membrane was incubated for 5 min, at room temperature, in the dark, with the HRP substrate, contained in the commercial solution SuperSignal West Pico PLUS Chemiluminescent Substrate (# 34580; Thermo Fisher Scientific), then developed on a ChemiDoc (Biorad) or Fusion FX (Vilber). The antibodies used for western blot were as follows: rabbit anti-p-65 (E379; Abcam), rabbit anti-RELB (33907; Abcam), rabbit anti-phospho-IkBa (14D4; Cell Signaling), rabbit anti-phospho-p65 (93H1; Cell Signaling), rabbit anti-IkBa (44D4; Cell Signaling), rabbit anti-NFkB2 (18D10; Cell Signaling), rabbit anti-α-tubulin (11H10; Cell Signaling), mouse anti-lamin A/C (4C11; Cell Signaling), mouse anti-p50 (E10; Santa Cruz), mouse anti-β-actin (8H10D10; Cell Signaling), rabbit anti-GAPDH (14C10; Cell Signaling), and mouse anti-HA-tag (6E2; Cell Signaling) were used for detection by immunoblot of HA-tagged proteins, and rabbit anti-HA-tag (C29F4; Cell Signaling) was used for immunoprecipitation of HA-tagged proteins.
EMSA experiments
Control and patient fibroblasts were stimulated with 100 ng/ml LTα1β2 (8884-LY-025/CF; R&D Systems) at the indicated time points. Nuclear extracts were prepared and analyzed for DNA-binding activity by using the HIV-LTR tandem kB oligonucleotide as a kB probe.
Proliferation assay of activated T cells
Day 10 of culture-activated T cells were starved of IL-2 for 72 h (after three washes in PBS and cultivated in complete Panserin without IL-2), washed, incubated with CellTrace violet reagent (Invitrogen) for 8 min at 37 °C in the dark, and washed twice more. A total of 2 × 105 cells were seeded into 96-well plates and subjected to different stimuli (plate-bound anti-CD3 antibody (Invitrogen) and soluble anti-CD3/CD28 beads (#11141D; Thermo Fisher Scientific). The cells were cultured for 4 days, washed with PBS, and stained with anti-CD3 (PE-Cy7; BioLegend), CD4 (PerCP5.5; BioLegend), CD8 (APC; BioLegend), CD25 (BV650; BioLegend), and CD69 (PE, Thermo Fisher Scientific) antibodies prior to flow cytometry measurement on a Novocyte (Agilent). Analysis was carried out on the online software OMIQ (OMIQ by Dotmatics) as well as data visualization.
Detection of autoantibodies using a luciferase reporter assay
The blocking activity of anti-IFN-α2, anti-IFN-ω, and anti-IFNβ auto-antibodies (auto-Abs) was determined with a reporter luciferase assay. Briefly, HEK293T cells were transfected with a plasmid containing the firefly luciferase gene under the control of the human ISRE promoter in the pGL4.45 backbone and a plasmid constitutively expressing Renilla luciferase for normalization (pRL-SV40). Cells were transfected in the presence of the X-tremeGene 9 transfection reagent (ref. number 6365779001; Sigma-Aldrich) for 24 h. Cells in Dulbecco’s modified Eagle medium (DMEM, Thermo Fisher Scientific) supplemented with 2% fetal calf serum (FCS) and 10% healthy control or patient serum/plasma were either left unstimulated or were stimulated with IFN-α2 (ref. number 130-108-984; Miltenyi Biotec) or IFN-ω (ref. number SRP3061; Merck) at 10 ng/ml or 100 pg/ml, or with IFN-β (ref. number: 130-107-888; Miltenyi Biotech) at 10 ng/ml for 16 h at 37°C. Each sample was tested once for each cytokine and dose in at least two independent experiments. Finally, cells were lysed for 20 min at room temperature and luciferase levels were measured with the Dual Luciferase Reporter 1000 assay system (ref. number E1980; Promega) according to the manufacturer’s protocol. Luminescence intensity was measured with a VICTOR X Multilabel Plate Reader (PerkinElmer Life Sciences). Firefly luciferase activity values were normalized against Renilla luciferase activity values. These values were then normalized against the plasma used in non-stimulated conditions. Samples were considered to be neutralizing if the luciferase activity signal, normalized against non-stimulated conditions, was below 5.
CyTOF staining
PBMCs from the patient and controls were analyzed using CyTOF (Maxpar Direct Immune Profiling System Fluidigm, Les Ulus, France) including a 30-marker antibody panel (ref. #201325; Fluidigm). 3 × 106 PBMCs resuspended in 300 μl of MaxPar Cell Staining Buffer were incubated for 10 min at room temperature after the addition of 5 μl of Human TruStain FcX (BioLegend Europ). Then the 300 μl were directly added to the dry antibody cocktail containing 30 antibodies for 30 min. All samples were washed three times in MaxPar Cell Staining Buffer and then fixed in 1.6% paraformaldehyde (Sigma-Aldrich) for 10 min. After centrifugation, the PBMCs were incubated overnight in Fix and Perm Buffer with 1:1,000 of iridium intercalator (pentamethylcyclopentadienyl-Ir (III)-dipyridophenazine; Fluidigm) and frozen at −80°C before acquisition.
CyTOF acquisition
After thawing, cells were washed and resuspended at a concentration of 1 million cells per ml in Maxpar Cell Acquisition Solution, a high-ionic-strength solution, and mixed with 10% EQ Beads immediately before acquisition. Samples were acquired on the Helios mass cytometer and CyTOF software version 6.7.1014 (Fluidigm, Inc.) at the “Plateforme de Cytométrie de la Pitié-Salpétriêre (CyPS).” An average of 500,000 events were acquired per sample. Dual count calibration, noise reduction, cell length threshold between 10 and 150 pushes, and a lower convolution threshold equal to 10 were applied during acquisition. Mass cytometry standard files produced by the HELIOS were normalized using CyTOF Software v. 6.7.1014. Four parameters (center, offset, residual, and width) were used for data cleaning to resolve ion fusion events (doublets) from single events from the Gaussian distribution generated by each event. After data cleaning, the program produces new FCS files consisting of only intact live singlet cells.
CyTOF analysis
FCS files containing intact live singlet cells were uploaded in R version 4.0.3 using the flowCore package. All file were concatenated in a SingleCellExperiment with the SingleCellExperiment package. After quality control (number of cells per sample, expression pattern of all markers across samples), all cells were submitted to clustering using the cluster function (FlowSOM and ConsensusMetaClustering) following the recommendation from the CATALYST packages (Crowell et al., 2020). For clustering, the k parameter was set to 60 to detect small immune populations. Clusters were then identified based on their expression of type markers according to previous knowledge of immune cell phenotype and following the manufacturer’s instructions from Fluidigm Maxpar Immune Profiling. Clusters expressing the same cell type markers were merged into one immune cell population. The proportion of clusters among all intact viable cells was then defined using the appropriate function from CATALYST and compared between groups using the Mann–Whitney Wilcoxon test in R. For visualization purposes, dimensionality reduction in UMAPs was performed using the runDR function from CATALYST with neighbors set to 15 and minimum distance to 0.4. All other visualizations were performed using in-house and the ggplot2 function. Subclustering of memory T cells was performed on all memory CD4+ T cells (excluding Tregs) based on the markers CXCR3, CXCR5, CCR6, and CCR4 (defined as type markers in a new panel file). Subclustering of B cells was performed based on the following markers: CD19, CD11c, CD27, CXCR5, CD38, CD20, HLA-DR, and IgD.
Statistical analysis and data representation
All statistical analyses were performed with R version 4.2.3. As indicated in the figures, results were analyzed with appropriate parametric or non-parametric statistical tests. For all analyses, the threshold for statistical significance was set to P < 0.05. Data visualization was made either in R with ggplot2 and ggpubr packages or GraphPad Prism.
Online supplemental material
Fig. S1 shows Sanger sequencing on genomic DNA, the sequences of TOPO-TA cloning of the patient CHUK variants, and the relative quantity of CHUK mRNA in the patient’s fibroblasts. Fig. S2 shows the clustering of B cells from the CyTOF data, the relative abundance of each cluster identified, and the relative frequency of T-helper subsets of memory CD4+ T cells. Fig. S3 shows the proliferation capacity and activation status of activated T cells from the patient compared with controls, the translocation of RELB into the nucleus of control and patient’s SV40-fibroblasts after stimulation with TNF or lymphotoxin, an EMSA showing RELA and RELB binding to a κB consensus sequence after stimulation with lymphotoxin, and the relative quantity of VCAM1 mRNA after lymphotoxin stimulation of patient and control SV40-fibroblasts. Fig. S4 shows heatmaps displaying the normalized abundance of transcripts from different signatures and the activation z-score of different molecules predicted to be inhibited based on bulk RNA sequencing data. Fig. S5 shows heatmaps displaying the normalized abundance of transcripts from different signatures and the activation z-score of different molecules predicted to be inhibited based on bulk RNA sequencing data, the relative abundance of transcripts based on RT-qPCR, and western blots of the canonical NF-κB pathway activation cascade upon TNF stimulation. Table S1 shows routine immune phenotype of the patient’ whole blood. Table S2 shows DEGs from the bulk RNA sequencing analysis.
Data availability
RNA sequencing data generated in this article are available in the Gene Expression Omnibus (GEO-NCBI) under repository number GSE267600. All data are available upon reasonable request to the corresponding authors.
Acknowledgments
We would like to thank the core facilities of the Imagine Institute (bioinformatics, genomics, flow cytometry) and the Flow Cytometry Platform of the Pitié-Salpêtrière Hospital (CyPS) for CyTOF acquisition and technical advice.
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and by government grants managed by the Agence National de la Recherche (ANR) as part of the “Investment for the Future” program (Institut Hospitalo-Universitaire Imagine, grant no. ANR-10-IAHU-01; Recherche Hospitalo-Universitaire, grant no. ANR-18-RHUS-0010), the Centre de Référence Déficits Immunitaires Héréditaires (CEREDIH), the Agence National de la Recherche (grant nos. ANR-18-CE17-0001 “Action”; and ANR-22-CE15-0047-02 “BREAK-ITP”), the Fondation pour la Recherche Médicale (FRM: EQU202103012670), by the Agence Nationale de Recherches sur le Sida et les hépatites virales (ANRS grant number ECTZ275762), and by a grant from Pharming. The Laboratory of Human Genetics of Infectious Diseases is supported by the Howard Hughes Medical Institute, the Rockefeller University, the St Giles Foundation, the National Institutes of Health (R01AI127564), the Fisher Center for Alzheimer’s Research Foundation, the Meyer Foundation, the JBP Foundation, the ANR, the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), ANR GENVIR (ANR-20-CE93-003), ANR AAILC (ANR-21-LIBA-0002) and ANR AI2D (ANR-22-CE15-0046) projects, the ANR-RHU programme (ANR-21-RHUS-08-COVIFERON), the HORIZON-HLTH-2021-DISEASE-04 programme under grant agreement 101057100 (UNDINE), the Square Foundation, Grandir—Fonds de solidarité pour l’enfance, the SCOR Corporate Foundation for Science, the Battersea & Bowery Advisory Group, Stavros Niarchos Foundation (SNF) as part of its grant to the SNF Institute for Global Infectious Disease Research at The Rockefeller University, William E. Ford, General Atlantic’s Chairman and Chief Executive Officer, Gabriel Caillaux, General Atlantic’s Co-President, Managing Director and Head of Business in EMEA, and the General Atlantic Foundation, the French Ministry of Higher Education, Research, and Innovation (MESRI-COVID-19), INSERM, and Paris Cité University. T. Le Voyer was supported by the Bettencourt-Schueller Foundation and the MD-PhD program of Imagine Institute. This work was also supported by grants from Institut National du Cancer, Fondation ARC pour la Recherche sur le Cancer, University Paris Cité, 12 Rounds contre le Cancer (V. Baud), and doctoral funding from the French Ministry of Higher Education and Research (T. Becquard). I. Meyts is a senior clinical investigator at Fonds Wetenschappelijk Onderzoek (FWO) Vlaanderen and is supported by the Jeffrey Modell Foundation and by FWO grant G0B5120N. J.-C. Debray is supported by the Fonds National de la Recherche Scientifique under grant no. 40007512. This work is supported by ERN-RITA. Q. Riller received an Institut Imagine MD-PhD fellowship and a Société Nationale Française de Médecine Interne fellowship as well as the Advancing Science and Practice in the Retail Environment Award 2024.
Author contributions: Q. Riller: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing - original draft, Writing - review & editing, B. Sorin: Conceptualization, Investigation, Methodology, Writing - review & editing, C. Courteille: Investigation, D. Ho-Nhat: Methodology, T. Le Voyer: Investigation, J.-C. Debray: Investigation, M.-C. Stolzenberg: Validation, M. Schmutz: Data curation, Investigation, O. Pellé: Data curation, Investigation, T. Becquard: Investigation, M. Rodrigo Riestra: Investigation, L. Berteloot: Data curation, Resources, Validation, M. Migaud: Investigation, L. Delage: Methodology, Writing - review & editing, M. Jeanpierre: Investigation, C. Boussard: Methodology, C. Brunaud: Investigation, A. Magérus: Project administration, Resources, Supervision, Writing - review & editing, C. Bretot: Resources, V. Michel: Investigation, C. Roux: Investigation, C. Picard: Resources, Validation, Writing - original draft, C. Masson: Formal analysis, C. Bole-Feysot: Methodology, N. Cagnard: Software, A. Corneau: Data curation, Resources, I. Meyts: Conceptualization, Funding acquisition, Methodology, Resources, Writing - review & editing, V. Baud: Conceptualization, Formal analysis, Investigation, J.-L. Casanova: Funding acquisition, Writing - original draft, Writing - review & editing, A. Fischer: Conceptualization, Writing - original draft, Writing - review & editing, E. Dejardin: Conceptualization, Funding acquisition, Methodology, Validation, Writing - review & editing, A. Puel: Resources, Writing - review & editing, C. Boulanger: Resources, Writing - review & editing, B. Neven: Conceptualization, Investigation, Resources, Supervision, Writing - review & editing, F. Rieux-Laucat: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing - review & editing.

