


TNFα and IFNγ (TNF/IFNγ) synergistically induce caspase-8 activation and cancer cell death. However, the mechanism of IFNγ in promoting TNF-initiated caspase-8 activation in cancer cells is poorly understood. Here, we found that in addition to CASP8, CYLD is transcriptionally upregulated by IFNγ-induced transcription factor IRF1. IRF1-mediated CASP8 and CYLD upregulation additively mediates TNF/IFNγ-induced cancer cell death. Clinically, the expression levels of TNF, IFNγ, CYLD, and CASP8 in melanoma tumors are increased in patients responsive to immune checkpoint blockade (ICB) therapy after anti–PD-1 treatment. Accordingly, our genetic screen revealed that ELAVL1 (HuR) is required for TNF/IFNγ-induced caspase-8 activation. Mechanistically, ELAVL1 binds CASP8 mRNA and extends its stability to sustain caspase-8 expression both in IFNγ-stimulated and in basal conditions. Consequently, ELAVL1 determines death receptors–initiated caspase-8–dependent cell death triggered from stimuli including TNF and TRAIL by regulating basal/stimulated caspase-8 levels. As caspase-8 is a master regulator in cell death and inflammation, these results provide valuable clues for tumor immunotherapy and inflammatory diseases.
Introduction
The pleiotropic cytokine TNFα (shortened as TNF) exerts its inflammatory or cell death functions by binding to the plasma membrane death domain–containing receptor TNFR1 (Xu et al., 2021; He and Wang, 2018; Martens et al., 2020). This binding causes the trimerization of TNFR1 to recruit death domain proteins TNFR1-associated death domain protein (TRADD) and RIPK1 to the cytoplasmic side of the plasma membrane. TRADD recruits E3 ligases TRAF2 and cIAP1/2 to add K11- and K63-linked polyubiquitin chains on RIPK1; these will further recruit the linear ubiquitin chain assembly complex (LUBAC) complex, composed of HOIL-1–interacting protein (HOIP), heme-oxidized IRP2 ubiquitin ligase 1 (HOIL), and Sharpin, to add M1-linked polyubiquitin chain on RIPK1 (Green, 2019; Zhang et al., 2018). These polyubiquitin chains act as scaffolds; these scaffolds recruit NEMO-IKKα-IKKβ and TAB2-TAB3-TAK1, which (through forming membrane-bound complex I) activate NF-κB and MAPK signaling, respectively, and cause prosurvival cell fate (Grootjans et al., 2017).
A variety of small molecules are used to switch TNF-initiated complex I to dissociate from the membrane to form a cytosolic death-activating complex II (Martens et al., 2020). There, TRADD, RIPK1, FAS-associated death domain protein (FADD), and caspase-8 interact with each other bringing multiple caspase-8 in proximity and leading to caspase-8 auto-cleavage and activation (Vandenabeele et al., 2023). For example, the IκB kinase (IKK) inhibitors, TAK1 inhibitor (5Z-7-oxozeaenol, 5Z7), and small molecule Smac mimetic (Smac in brief), which was used to induce degradation of cIAP1/2, therefore decreasing the polyubiquitination of RIPK1, are common reagents to cause cell death when combined with TNF (Van Loo and Bertrand, 2023; Grootjans et al., 2017). cFLIP is a negative regulator of caspase-8. Blocking cFLIP synthesis by cycloheximide (CHX) also promotes caspase-8 activation and cell death in the presence of TNF (Vandenabeele et al., 2023). In addition, CYLD deubiquitinates components on complex I (including RIPK1) and is therefore required for TNF plus 5Z7-/Smac-/CHX-induced complex II formation and caspase-8 activation (Peltzer et al., 2016). Activation of the apical caspase-8 further cleaves and activates downstream functionally redundant caspase-3 and caspase-7 (and/or other substrates) to cause cell death (Galluzzi et al., 2018).
Like TNF, IFNγ is also a cytokine that exerts diverse cellular functions. Extracellular IFNγ binds to the membrane receptors IFNGR1 and IFNGR2 to activate the intracellular tyrosine kinases JAK1 and JAK2 to induce the phosphorylation of the transcription factor STAT1 (Lee and Ashkar, 2018). Phosphorylated STAT1, in a form of homodimer, translocates to the nucleus to drive the expression of target genes, including the transcription factor IRF1. IRF1 in turn regulates expression of hundreds of genes by binding to the IFN-stimulated response element (ISRE) motifs in the promoters of downstream genes (Lee and Ashkar, 2018).
IFNγ is known to synergize with TNF to cause cell death in multiple cancer cell lines (Shen et al., 2018). Recent work evidenced the essential role of tumor microenvironment secreted TNF and IFNγ (TNF/IFNγ) in inducing cancer cell death, which may successfully contribute to tumor immunotherapy (Freeman et al., 2021; Sun et al., 2023). Inhibiting LUBAC complex or TBK1 increases TNF/IFNγ-induced cancer cell death and improves the efficacy of tumor immunotherapy in multiple models. However, the administration of TNF and IFNγ (either individually or in combination) causes systemic toxicity in animals and humans, which hampers the use of natural TNF and IFNγ in cancer therapy (Shen et al., 2018). The fusion of peptides (or antibodies) targeting tumor-associated antigens (tumor-homing peptides/antibodies) to TNF and/or IFNγ are actively explored to specifically enrich TNF and IFNγ to the tumor, which can kill cancer cells without bringing systemic toxicity (Shen et al., 2018). Further, the combination of TNF and IFNγ was more powerful than stimulation of either cytokine alone both in vitro and in vivo, resulting from the synergistic effect of IFNγ in promoting TNF-initiated caspase-8 activation and cell death (Buntinx et al., 2004; Chang et al, 2004; Selleri et al., 1995; Karki et al., 2021a; Malireddi et al., 2021).
The canonical IFNγ signaling-mediated IRF1 upregulation is required for TNF/IFNγ-induced caspase-8 activation and cell death, suggesting downstream genes of IRF1 promote TNF-initiated caspase-8 activation (Karki et al., 2021a; Malireddi et al., 2021). NF-κB pathway was proposed to be inhibited or, on the contrary, activated by TNF/IFNγ stimulation to affect downstream gene expression and induce cell death (Shen et al., 2018). Furthermore, other works have also proposed that IFNγ does not interfere with TNF-stimulated NF-κB activation (Karki et al., 2021a). The CASP8 gene (encoding caspase-8) itself is a downstream target of IRF1 (Ruiz-Ruiz et al., 2000). However, it still remains to be elucidated if IRF1-upregulated caspase-8 levels play a mechanistic role in synergizing with TNF to induce cell death. In addition, it is not yet known whether another downstream target(s) of IRF1 is required for TNF/IFNγ-induced caspase-8 activation. Recently, two studies indicated that TNF/IFNγ and TLR agonists/IFNγ synergize in inducing inducible nitric oxide synthase (iNOS) expression in murine bone marrow–derived macrophages (BMDMs; Simpson et al., 2022; Karki et al., 2021a). This synergy activates PANoptosis (Karki et al., 2021a) and apoptosis (Simpson et al., 2022) and is involved in cytokine storm-induced tissue damage, including COVID-19. However, iNOS was proposed not to be required for TNF/IFNγ-induced death in cancer cells (Malireddi et al., 2021). Therefore, the exact target(s) of IRF1 in promoting TNF-initiated caspase-8 activation in cancer cells remains elusive.
In this study, we performed RNA sequencing (RNA-seq) analysis upon IFNγ treatment of cancer cells and found that, in addition to CASP8, CYLD is a previously uncharacterized target gene of IRF1. By combining CRISPR-Cas9–mediated knockin technology to edit the ISRE motif in CASP8 and CYLD promoters with other genetical approaches, we found IRF1-mediated transcriptional upregulation of CASP8 and CYLD additively promote TNF/IFNγ-induced cancer cell death independent of iNOS expression. Consistent with our findings in cells, we detected a positive correlation between TNF/IFNγ/IRF1/CYLD/CASP8 expression and the therapeutic efficacy of immunotherapy in advanced melanoma patients. In addition, an unbiased whole genome–wide CRISPR-Cas9 screen identified the mRNA binding protein ELAVL1 (HuR) is required to bind the 3′UTR of CASP8 mRNA and extend its half-life. This ELAVL1-mediated stabilization of CASP8 mRNA facilitates TNF/IFNγ-, TNF/Smac-, TNF/CHX-, and TRAIL-induced cell death, establishing a role of ELAVL1 in the regulating of the master cell death component caspase-8.
Results
iNOS undergoes differential regulation and exerts distinct functions during TNF/IFNγ-induced death of murine BMDMs and cancer cells
Initially, we experimentally confirmed the previous reports that IFNγ synergizes with TNF to induce the death of murine BMDMs and cancer cells (Malireddi et al., 2021; Karki et al., 2021a). We considered cancer cells from multiple origins, including cervical cancer, colon cancer, colorectal adenocarcinoma, osteosarcoma, lymphoma, acute monocytic leukemia, and melanoma (HeLa, HCT-116, HT-29, U2OS, U937, THP-1, and B16F0 cells) and found that the cell death induction occurred in an IFNγ dose-dependent manner (Fig. S1, A and B). We also confirmed that treatment of murine BMDMs with TNF and IFNγ (TNF/IFNγ) increased iNOS expression and NO production, and that the iNOS inhibitor 1400W blocked TNF/IFNγ-induced NO production and cell death (Fig. 1, A–D) (Karki et al., 2021a).
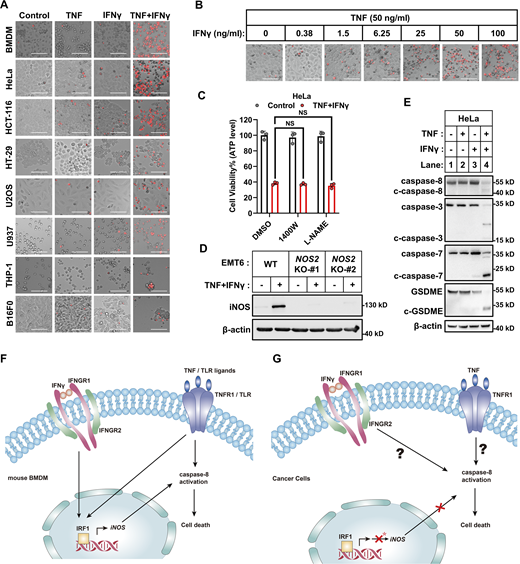
iNOS is dispensable in TNF/IFNγ-induced human and murine origin cancer cell death. (A) Representative images of cell death in indicated cancer cell lines with TNF, IFNγ, or TNF/IFNγ treatments for 36 h (n = 3). PI staining was used to show dead cells. Scale bar, 275 μm. (B) Representative images of PI staining in HeLa cells after 36 h treatment with TNF alone or plus indicated concentrations of IFNγ. Scale bar, 275 μm. (C) HeLa cells were treated with TNF/IFNγ in the presence or absence of 1400W, or L-NAME for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: NS. (D) EMT6 cells (parental and Nos2 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 12 h. Protein expression of iNOS was analyzed by immunoblotting (n = 3). (E) HeLa cells were treated with TNF, IFNγ, or TNF/IFNγ for 36 h. The expression of cleaved and full-length caspase-3/7/8 and GSDME proteins were analyzed by immunoblotting (n = 3). (F and G) Schematic of the biochemical mechanism of TNF or TLR ligands plus IFNγ induced cell death in murine BMDMs (F) or cancer cells (G). Asterisk: TNF/IFNγ failed to induce the expression of iNOS in human cancer cells, or the induced iNOS in murine cancer cells has no effect on cell death. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS1.
iNOS is dispensable in TNF/IFNγ-induced human and murine origin cancer cell death. (A) Representative images of cell death in indicated cancer cell lines with TNF, IFNγ, or TNF/IFNγ treatments for 36 h (n = 3). PI staining was used to show dead cells. Scale bar, 275 μm. (B) Representative images of PI staining in HeLa cells after 36 h treatment with TNF alone or plus indicated concentrations of IFNγ. Scale bar, 275 μm. (C) HeLa cells were treated with TNF/IFNγ in the presence or absence of 1400W, or L-NAME for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: NS. (D) EMT6 cells (parental and Nos2 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 12 h. Protein expression of iNOS was analyzed by immunoblotting (n = 3). (E) HeLa cells were treated with TNF, IFNγ, or TNF/IFNγ for 36 h. The expression of cleaved and full-length caspase-3/7/8 and GSDME proteins were analyzed by immunoblotting (n = 3). (F and G) Schematic of the biochemical mechanism of TNF or TLR ligands plus IFNγ induced cell death in murine BMDMs (F) or cancer cells (G). Asterisk: TNF/IFNγ failed to induce the expression of iNOS in human cancer cells, or the induced iNOS in murine cancer cells has no effect on cell death. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS1.
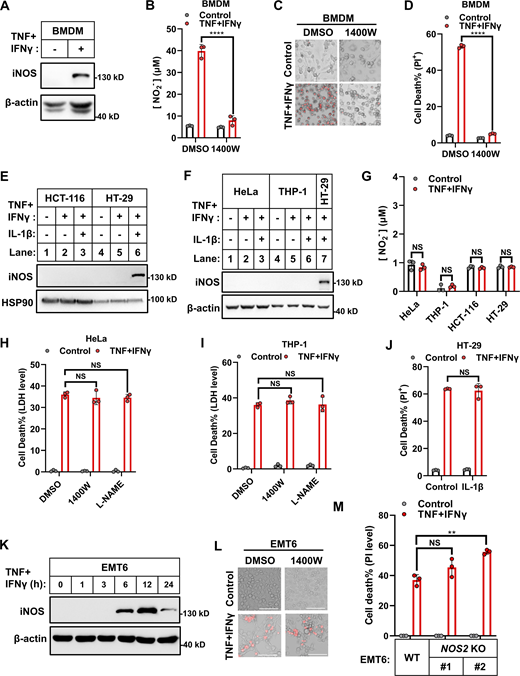
iNOS exerts distinct functions during TNF/IFNγ-induced death of diverse cell lines. (A) Murine BMDMs were treated with TNF/IFNγ for 24 h. Protein expression of iNOS was analyzed by immunoblotting (n = 3). (B) Murine BMDMs were treated with TNF/IFNγ in the presence or absence of the iNOS inhibitor 1400W (100 μM) for 24 h (n = 3). Nitrite (NO2−) production in the culture medium was measured by the Griess assay. Two-tailed t test was performed: ****P < 0.0001. (C) Representative images of cell death in murine BMDMs after 48 h treatments of TNF/IFNγ in the presence or absence of 1400W. PI staining was used to show dead cells. Scale bar, 275 μm. (D) Murine BMDMs were treated with TNF/IFNγ in the presence or absence of 1400W for 48 h (n = 3). Cell death was measured by PI staining followed by flow cytometry. Two-tailed t test was performed: ****P < 0.0001. (E and F) HCT-116 and HT-29 cells (E) and HeLa and THP-1 cells (F) were treated as indicated for 24 h. Protein expression of iNOS was analyzed by immunoblotting (n = 4). (G) HeLa, THP-1, HCT-116, and HT-29 cells were treated with TNF/IFNγ for 24 h (n = 3). Nitrite (NO2−) production in the culture medium was measured. Two-tailed t test was performed: NS (not significant). (H and I) HeLa (H) or THP-1 (I) cells were treated with TNF/IFNγ in the presence or absence of 1,400 W, or the NOS inhibitor L-NAME (1 mM) for 48 h (n = 3). Cell death was measured by LDH levels released in the culture medium. One-way analysis of variance (ANOVA) was performed: NS. (J) HT-29 cells were treated with TNF/IFNγ in the presence or absence of IL-1β (100 ng/ml) for 48 h (n = 3). Cell death was measured by PI staining followed by flow cytometry. A two-tailed t test was performed: NS. (K) EMT6 cells were treated with TNF/IFNγ for indicated time points. Protein expression of iNOS was analyzed by immunoblotting (n = 2). (L) Representative images of PI staining in EMT6 cells after 36 h treatments of TNF/IFNγ in the presence or absence of 1400W; scale bar, 275 μm. (M) Parental EMT6 and NOS2 KO cells (#1 and #2 clones with different gRNA) were treated with TNF/IFNγ for 36 h (n = 3). Cell death was measured by PI staining followed by quantification. One-way ANOVA was performed: NS; **P = 0.0036. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F1.
iNOS exerts distinct functions during TNF/IFNγ-induced death of diverse cell lines. (A) Murine BMDMs were treated with TNF/IFNγ for 24 h. Protein expression of iNOS was analyzed by immunoblotting (n = 3). (B) Murine BMDMs were treated with TNF/IFNγ in the presence or absence of the iNOS inhibitor 1400W (100 μM) for 24 h (n = 3). Nitrite (NO2−) production in the culture medium was measured by the Griess assay. Two-tailed t test was performed: ****P < 0.0001. (C) Representative images of cell death in murine BMDMs after 48 h treatments of TNF/IFNγ in the presence or absence of 1400W. PI staining was used to show dead cells. Scale bar, 275 μm. (D) Murine BMDMs were treated with TNF/IFNγ in the presence or absence of 1400W for 48 h (n = 3). Cell death was measured by PI staining followed by flow cytometry. Two-tailed t test was performed: ****P < 0.0001. (E and F) HCT-116 and HT-29 cells (E) and HeLa and THP-1 cells (F) were treated as indicated for 24 h. Protein expression of iNOS was analyzed by immunoblotting (n = 4). (G) HeLa, THP-1, HCT-116, and HT-29 cells were treated with TNF/IFNγ for 24 h (n = 3). Nitrite (NO2−) production in the culture medium was measured. Two-tailed t test was performed: NS (not significant). (H and I) HeLa (H) or THP-1 (I) cells were treated with TNF/IFNγ in the presence or absence of 1,400 W, or the NOS inhibitor L-NAME (1 mM) for 48 h (n = 3). Cell death was measured by LDH levels released in the culture medium. One-way analysis of variance (ANOVA) was performed: NS. (J) HT-29 cells were treated with TNF/IFNγ in the presence or absence of IL-1β (100 ng/ml) for 48 h (n = 3). Cell death was measured by PI staining followed by flow cytometry. A two-tailed t test was performed: NS. (K) EMT6 cells were treated with TNF/IFNγ for indicated time points. Protein expression of iNOS was analyzed by immunoblotting (n = 2). (L) Representative images of PI staining in EMT6 cells after 36 h treatments of TNF/IFNγ in the presence or absence of 1400W; scale bar, 275 μm. (M) Parental EMT6 and NOS2 KO cells (#1 and #2 clones with different gRNA) were treated with TNF/IFNγ for 36 h (n = 3). Cell death was measured by PI staining followed by quantification. One-way ANOVA was performed: NS; **P = 0.0036. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F1.
However, unlike the observations in murine BMDMs, treatment of the tested human cancer cells with TNF/IFNγ did not induce expression of iNOS or production of NO, and iNOS inhibitor 1400W and the NOS inhibitor L-NAME did not block TNF/IFNγ-induced death in multiple cancer cell lines (Fig. 1, E–I; and Fig. S1 C). Since IL-1β (a potent proinflammatory cytokine) is known to synergize with TNF or IFNγ in inducing iNOS in some cell types (Jana et al., 2005), we cotreated HT-29 cells with IL-1β/TNF/IFNγ and found that such treatment increased iNOS expression (Fig. 1, E and F). However, this treatment failed to augment TNF/IFNγ-induced cell death (Fig. 1 J). Results indicated that iNOS was dispensable for TNF/IFNγ-induced cell death in human cancer cells.
We explored whether this discrepancy about iNOS in TNF/IFNγ-induced cell death of murine BMDMs versus human cancer cells was due to differences in cell types, or murine/human species. We tested the murine breast cancer cell line (EMT6) and found that TNF/IFNγ treatment increased iNOS expression (Fig. 1 K). However, neither pharmaceutical inhibition nor genetic knockout (KO) of iNOS blocked TNF/IFNγ-induced EMT6 cell death (Fig. 1, L and M; and Fig. S1 D). Collectively, iNOS induction is not required for TNF/IFNγ-induced cell death in the tested cancer cells.
TNF/IFNγ was proposed to cause PANoptosis in the cell types simultaneously incorporated with the pyroptosis, apoptosis, and necroptosis machinery upon activating the central apical caspase-8 (Karki et al., 2021a; Chen et al., 2023; Karki and Kanneganti, 2022). We confirmed that TNF/IFNγ treatment of HeLa cells caused cleavage of caspase-8, caspase-3, caspase-7, and the caspase-3 substrate GSDME (Fig. S1 E). Thus, our results support the notion that some iNOS-independent mechanism accounts for IFNγ’s synergism with TNF to induce caspase-8 activation and cell death in cancer cells (Fig. S1, F and G).
IFNγ induces CYLD and caspase-8 expression in cancer cell lines
As the transcription factor IRF1 but not the IRF1-inducible iNOS is required for TNF/IFNγ-induced cancer cell death (Malireddi et al., 2021), results indicate that the upregulation of some other IRF1 downstream gene(s) might account for IFNγ’s synergism with TNF to induce cancer cell death. If this is the case, then these genes should fulfill at least two criteria: (i) these genes are transcriptionally upregulated by the IFNγ-IRF1 pathway; (ii) KO of these genes blocks TNF/IFNγ-induced cell death. We combined two strategies, whole genome–wide CRISPR-Cas9 screen and RNA-seq technology (Fig. 2, A and B), to identify the potential candidates.
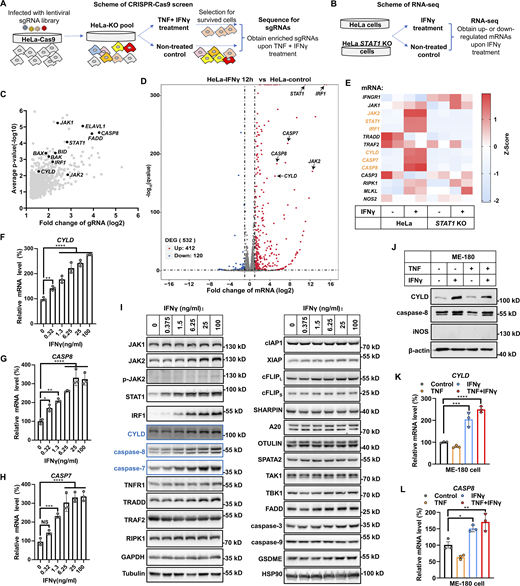
IFNγ induces CYLD/caspase-8 expression in cancer cells. (A) Schematic of the CRISPR-Cas9 screen strategy to identify regulators in TNF/IFNγ-induced cell death. (B) Schematic of the RNA-seq strategy to identify genes’ response to IFNγ treatment. (C) HeLa-Cas 9 cells were transfected with gRNA library and treated with TNF/IFNγ for 48 h, followed by DNA isolation and sequencing. Log2-transformed average fold change (FC) for enrichment of sgRNAs in HeLa-Cas 9 cells treated with TNF/IFNγ versus control cells is shown by volcano plot. Significant enriched genes identified in this paper are labeled. (D) HeLa cells were treated with IFNγ for 12 h, followed by RNA isolation and 3′mRNA-sequencing. Differentially expressed genes (DEGs) that are up- or downregulated in IFNγ-treated HeLa cells compared with untreated HeLa cells are shown by a volcano plot. Adjusted P < 0.05, cut-off values log2(FC) > 1 or log2(FC) < −1. Labeled genes were the top elevated genes involved in TNF/IFNγ signaling pathway. (E) HeLa cells (parental and STAT1 KO) were treated with IFNγ for 24 h, followed by RNA isolation and 3′mRNA-sequencing. The effect of IFNγ treatment on indicated genes was assessed by a heatmap. (F–H) HeLa cells were treated with IFNγ at the indicated concentration for 24 h. The mRNA expression of CYLD (F), CASP8 (G), and CASP7 (H) was measured by qPCR (n = 3). One-way ANOVA was performed: **P = 0.0084, ****P < 0.0001 (F); *P = 0.0298, **P = 0.0011, ****P < 0.0001 (G); NS, ***P = 0.0004, ****P < 0.0001 (H). (I) HeLa cells were treated with the indicated concentration of IFNγ for 24 h. The expression of indicated proteins involved in the IFNγ and TNF signaling pathways was analyzed by immunoblotting (n = 4). (J) ME-180 cells were treated with TNF, IFNγ, or TNF/IFNγ for 24 h. The protein expression of CYLD, caspase-8, and iNOS was analyzed by immunoblotting (n = 3). (K and L) ME-180 cells were treated with TNF, IFNγ, or TNF/IFNγ for 24 h (n = 3). The mRNA level of CYLD (K) and CASP8 (L) was measured by qPCR. One-way ANOVA was performed: ***P = 0.0005, ****P < 0.0001 (K); *P = 0.0276, **P = 0.0042 (L). Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F2.
IFNγ induces CYLD/caspase-8 expression in cancer cells. (A) Schematic of the CRISPR-Cas9 screen strategy to identify regulators in TNF/IFNγ-induced cell death. (B) Schematic of the RNA-seq strategy to identify genes’ response to IFNγ treatment. (C) HeLa-Cas 9 cells were transfected with gRNA library and treated with TNF/IFNγ for 48 h, followed by DNA isolation and sequencing. Log2-transformed average fold change (FC) for enrichment of sgRNAs in HeLa-Cas 9 cells treated with TNF/IFNγ versus control cells is shown by volcano plot. Significant enriched genes identified in this paper are labeled. (D) HeLa cells were treated with IFNγ for 12 h, followed by RNA isolation and 3′mRNA-sequencing. Differentially expressed genes (DEGs) that are up- or downregulated in IFNγ-treated HeLa cells compared with untreated HeLa cells are shown by a volcano plot. Adjusted P < 0.05, cut-off values log2(FC) > 1 or log2(FC) < −1. Labeled genes were the top elevated genes involved in TNF/IFNγ signaling pathway. (E) HeLa cells (parental and STAT1 KO) were treated with IFNγ for 24 h, followed by RNA isolation and 3′mRNA-sequencing. The effect of IFNγ treatment on indicated genes was assessed by a heatmap. (F–H) HeLa cells were treated with IFNγ at the indicated concentration for 24 h. The mRNA expression of CYLD (F), CASP8 (G), and CASP7 (H) was measured by qPCR (n = 3). One-way ANOVA was performed: **P = 0.0084, ****P < 0.0001 (F); *P = 0.0298, **P = 0.0011, ****P < 0.0001 (G); NS, ***P = 0.0004, ****P < 0.0001 (H). (I) HeLa cells were treated with the indicated concentration of IFNγ for 24 h. The expression of indicated proteins involved in the IFNγ and TNF signaling pathways was analyzed by immunoblotting (n = 4). (J) ME-180 cells were treated with TNF, IFNγ, or TNF/IFNγ for 24 h. The protein expression of CYLD, caspase-8, and iNOS was analyzed by immunoblotting (n = 3). (K and L) ME-180 cells were treated with TNF, IFNγ, or TNF/IFNγ for 24 h (n = 3). The mRNA level of CYLD (K) and CASP8 (L) was measured by qPCR. One-way ANOVA was performed: ***P = 0.0005, ****P < 0.0001 (K); *P = 0.0276, **P = 0.0042 (L). Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F2.
Our results led us to consider three categories of candidate genes: (i) the IFNγ signaling components JAK2, STAT1, and IRF1; (ii) the TNF signaling components CYLD and CASP8; and (iii) the mRNA binding protein ELAVL1 (Fig. 2, C–E). IFNγ signaling is required for TNF/IFNγ-induced cell death by upregulating IRF1. This category validated the reliability of both assays. TNF signaling is (unsurprisingly) found in the CRISPR-Cas9 screen (Fig. 2 C). However, it is not known whether CYLD is a downstream gene of IFNγ, although CASP8 is known to be a downstream gene of IFNγ (Ruiz-Ruiz et al., 2000). Further, whether IFNγ-upregulated CASP8 and CYLD promote TNF-initiated cell death is not genetically validated. ELAVL1 is only enriched in the CRISPR-Cas9 screen (Fig. 2 C) but not in the RNA-seq analysis. We included ELAVL1 in our analysis as it functions to regulate the stability or translational efficiency of target mRNAs (Schultz et al., 2020). It is possible that ELAVL1 plays a role in stabilizing IFNγ-upregulated genes and supports TNF/IFNγ-induced cell death.
We experimentally confirmed that IFNγ stimulation results in the upregulation of CYLD, CASP8, and CASP7 (another positive control) mRNA in HeLa cells (Fig. 2, F–H). Immunoblotting showed that the CYLD, caspase-8, and caspase-7 proteins are constitutively expressed and that their levels are increased by IFNγ doses to an extent more obvious than that of many other components in the TNF and/or apoptosis signaling (Fig. 2 I). Testing of another human cervical cancer cell line (ME180) (Fig. 2, J–L), and murine BMDMs, EMT6 cells, and B16F0 cell lines (Fig. S2) showed that IFNγ stimulation similarly led to increased CYLD and CASP8 levels. Notably, the synergistic effect of TNF on IFNγ-induced CYLD and caspase-8 expression was not as robust as in elevating iNOS expression (Fig. 2, J–L; and Fig. S2).
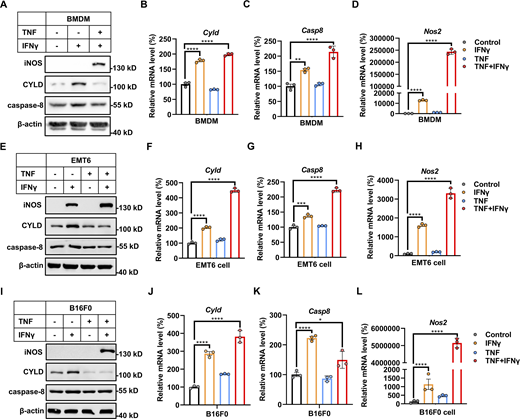
IFNγ induces CYLD and caspase-8 expression in murine cell lines. (A, E, and I) BMDMs (A), EMT6 (E), and B16F0 (I) cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h. The protein expression of CYLD, caspase-8, and iNOS was analyzed by immunoblotting (n = 3). (B–D) BMDM cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h (n = 3). The mRNA expression of Cyld (B), Casp8 (C), and Nos2 (D) was measured by qPCR. One-way ANOVA was performed: ****P < 0.0001 (B); **P = 0.0029, ****P < 0.0001 (C); ****P < 0.0001 (D). (F–H) EMT6 cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h (n = 3). The mRNA levels of Cyld (F), Casp8 (G), and Nos2 (H) were measured by qPCR. One-way ANOVA was performed: ****P < 0.0001 (F); ***P = 0.0003, ****P < 0.0001 (G); ****P < 0.0001 (H). (J–L) B16F0 cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h (n = 3). The mRNA expression of Cyld (J), Casp8 (K), and Nos2 (L) were measured by qPCR. One-way ANOVA was performed: ****P < 0.0001 (J); *P = 0.03, ****P < 0.0001 (K); ****P < 0.0001 (L). Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS2.
IFNγ induces CYLD and caspase-8 expression in murine cell lines. (A, E, and I) BMDMs (A), EMT6 (E), and B16F0 (I) cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h. The protein expression of CYLD, caspase-8, and iNOS was analyzed by immunoblotting (n = 3). (B–D) BMDM cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h (n = 3). The mRNA expression of Cyld (B), Casp8 (C), and Nos2 (D) was measured by qPCR. One-way ANOVA was performed: ****P < 0.0001 (B); **P = 0.0029, ****P < 0.0001 (C); ****P < 0.0001 (D). (F–H) EMT6 cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h (n = 3). The mRNA levels of Cyld (F), Casp8 (G), and Nos2 (H) were measured by qPCR. One-way ANOVA was performed: ****P < 0.0001 (F); ***P = 0.0003, ****P < 0.0001 (G); ****P < 0.0001 (H). (J–L) B16F0 cells were treated with IFNγ, TNF, or TNF/IFNγ for 24 h (n = 3). The mRNA expression of Cyld (J), Casp8 (K), and Nos2 (L) were measured by qPCR. One-way ANOVA was performed: ****P < 0.0001 (J); *P = 0.03, ****P < 0.0001 (K); ****P < 0.0001 (L). Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS2.
Higher TNF/IFNγ expression is associated with higher CYLD/CASP8 in melanoma tumors
Further, we analyzed publicly available tumor gene expression data of two advanced melanoma patient cohorts treated with anti–PD-1 antibodies (Fig. S3, A and J) (Riaz et al., 2017; Roh et al., 2017). Our aim was to test whether higher TNF/IFNγ expression is correlated with higher CYLD/CASP8 in tumors. In the first cohort, we chose patients who had increased IFNγ expression after anti–PD-1 therapy (Riaz et al., 2017). In the response group, patients who received anti–PD-1 treatment showed increased expression of TNF, IFNγ, and downstream genes IRF1, CYLD, and CASP8, supporting the hypothesis that higher TNF/IFNγ expression is associated with higher CYLD/CASP8 levels in vivo (Fig. S3, A–I). In non-responding patients, the increase in TNF and IFNγ levels was less dramatic compared with that in the responding patients after anti–PD-1 treatment, and correspondingly, the levels of IRF1, CYLD, and CASP8 did not achieve a noteworthy increase (Fig. S3, A–I). The contribution of increased TNF/IFNγ/CYLD/CASP8 levels to the therapeutic efficacy of immunotherapy in this cohort is uncertain, as a statistical difference between the responding and non-responding groups was not obtained due to the relatively low number of analyzed patients (Fig. S3, B–F). However, in the second cohort of advanced melanoma patients who received anti–PD-1 therapy (Fig. S3 J) (Roh et al., 2017), higher TNF, IFNγ, IRF1, and CYLD levels were detected in responding patients compared with non-responding patients (Fig. S3, K–N). Therefore, increased TNF and IFNγ levels were associated with higher expression of CYLD and CASP8 in tumors and might contribute to a better therapeutic efficacy of immunotherapy.

TNF/IFNγ/CYLD/CASP8 expression is positively associated with responses in ICB therapy. (A) A cohort of advanced melanoma patients having received the anti–PD-1 antibody Nivolumab. Tumors from these patients before and after therapy were analyzed by RNA-seq. Patients were split into responding (R) and not responding (NR) groups according to Response Evaluation Criteria in Solid Tumors (RECIST) criteria. (B–H) Normalized TNF (B), IFNγ (C), IRF1 (D), CYLD (E), CASP8 (F), CASP7 (G), and NOS2 (H) levels before and after therapy for indicated patient populations in patient cohort from A. Wilcoxon signed-rank test was used to determine the significance: NS, ***P = 0.0004 (B); **P = 0.0078, ****P < 0.0001 (C); *P = 0.0391, ****P < 0.0001 (D); NS, **P = 0.0032 (E); NS, ***P = 0.0004 (F); NS, *P = 0.0403 (G); NS, *P = 0.0325 (H). Whiskers of the boxplots indicate 1.5× the interquartile ranges. (I) Kaplan–Meier survival curves for melanoma patient cohort from A which have received anti–PD-1 therapy with induced (n = 17) or not induced (n = 8) TNF/IFNγ signature. A log-rank test was performed: **P = 0.0072. (J) A cohort of metastatic melanoma patients who received anti–PD-1 therapy were analyzed. Patient tumors before and after therapy were analyzed by RNA-seq. Patients were split into R and NR groups according to RECIST criteria. (K–N) Normalized TNF (K), IFNγ (L), IRF1 (M), and CYLD (N) levels for indicated patient populations in the patient cohort from J. Mann–Whitney test was used to determine the significance: NS, ***P = 0.0006 (K); NS, ***P = 0.0006 (L); NS, ***P = 0.0003 (M); NS, ***P = 0.0003 (N). Whiskers of the boxplots indicate 1.5× the interquartile ranges.
TNF/IFNγ/CYLD/CASP8 expression is positively associated with responses in ICB therapy. (A) A cohort of advanced melanoma patients having received the anti–PD-1 antibody Nivolumab. Tumors from these patients before and after therapy were analyzed by RNA-seq. Patients were split into responding (R) and not responding (NR) groups according to Response Evaluation Criteria in Solid Tumors (RECIST) criteria. (B–H) Normalized TNF (B), IFNγ (C), IRF1 (D), CYLD (E), CASP8 (F), CASP7 (G), and NOS2 (H) levels before and after therapy for indicated patient populations in patient cohort from A. Wilcoxon signed-rank test was used to determine the significance: NS, ***P = 0.0004 (B); **P = 0.0078, ****P < 0.0001 (C); *P = 0.0391, ****P < 0.0001 (D); NS, **P = 0.0032 (E); NS, ***P = 0.0004 (F); NS, *P = 0.0403 (G); NS, *P = 0.0325 (H). Whiskers of the boxplots indicate 1.5× the interquartile ranges. (I) Kaplan–Meier survival curves for melanoma patient cohort from A which have received anti–PD-1 therapy with induced (n = 17) or not induced (n = 8) TNF/IFNγ signature. A log-rank test was performed: **P = 0.0072. (J) A cohort of metastatic melanoma patients who received anti–PD-1 therapy were analyzed. Patient tumors before and after therapy were analyzed by RNA-seq. Patients were split into R and NR groups according to RECIST criteria. (K–N) Normalized TNF (K), IFNγ (L), IRF1 (M), and CYLD (N) levels for indicated patient populations in the patient cohort from J. Mann–Whitney test was used to determine the significance: NS, ***P = 0.0006 (K); NS, ***P = 0.0006 (L); NS, ***P = 0.0003 (M); NS, ***P = 0.0003 (N). Whiskers of the boxplots indicate 1.5× the interquartile ranges.
IFNγ-upregulated CYLD and caspase-8 additively promote TNF/IFNγ-induced cell death
Next, we genetically investigated whether IFNγ-upregulated CYLD and caspase-8 are required for TNF/IFNγ-induced cancer cell death. First, individually KO of CYLD, caspase-8, and FADD (required for caspase-8 activation) in HeLa cells (Fig. 3 A) and KO of CYLD and caspase-8 in HCT-116 cells (Fig. 3 B) blocked TNF/IFNγ-induced cell death. As CYLD and caspase-8 are constitutively expressed in the tested human cancer cells and IFNγ stimulation further increases their expression (Fig. 2), we use shRNAs for CASP8 and CYLD to decrease the expression levels of caspase-8 and CYLD upon IFNγ stimulation to achieve a comparable level as that in untreated control cells (Fig. 3, C and E, lanes 4 versus lanes 1). Individual knockdown of caspase-8 significantly inhibited TNF/IFNγ-induced cell death (Fig. 3, C and D). CYLD knockdown also significantly inhibited TNF/IFNγ-induced cell death (Fig. 3, E and F), and combined knockdown of caspase-8 and CYLD inhibited TNF/IFNγ-induced cell death in an additive manner (Fig. 3, G and H). Similarly, knockdown CYLD in EMT6 cells and in murine BMDMs inhibited TNF/IFNγ-induced death (Fig. S4, A–D).
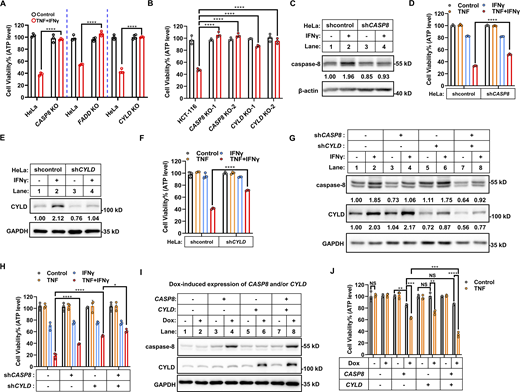
CYLD and caspase-8 are required for TNF/IFNγ-induced cell death. (A) HeLa cells (parental, caspase-8 KO, FADD KO, and CYLD KO) cells were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: ****P < 0.0001. (B) HCT-116 cells (parental, CASP8 KO clones #1 and #2 with different gRNA, CYLD KO clones #1 and #2 with different gRNA) were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (C) HeLa cells were stably expressed with control (shcontrol) or CASP8 shRNA (shCASP8) and treated with IFNγ for 24 h. The protein expression of caspase-8 was analyzed by immunoblotting (n = 3). (D) HeLa shcontrol and shCASP8 cells were treated with TNF, IFNγ, or TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: ****P < 0.0001. (E) HeLa cells were stably expressed with control (shcontrol) or CYLD shRNA (shCYLD) and treated with IFNγ for 24 h. The protein expression of CYLD was analyzed by immunoblotting (n = 3). (F) HeLa shcontrol and shCYLD cells were treated with TNF, IFNγ, or TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: ****P < 0.0001. (G) HeLa control (shcontrol) and CASP8 knockdown (shCASP8) cells were further infected with shcontrol or shCYLD lentivirus, and treated with IFNγ for 24 h. The protein expression of CYLD and caspase-8 was analyzed by immunoblotting (n = 4). (H) HeLa cells (control, CASP8 knockdown, CYLD knockdown, CASP8 and CYLD double knockdown) were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: *P = 0.0321; ****P < 0.0001. (I and J) HeLa cells were infected with lentivirus express caspase-8 or empty vector. Then the cells were further infected with lentivirus express CYLD or empty vector. The protein expression was dox inducible. The expression of indicated proteins from these cells in the presence or absence of dox was analyzed by immunoblotting (I) (n = 3). Cells treated with TNF for 48 h in the presence or absence of dox (n = 3). Cell viability was determined by ATP levels (J). Two-tailed t test was performed: NS, **P = 0.0011, 0.0013 (left to right), ***P = 0.0002, 0.0004 (left to right), ****P < 0.0001. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F3.
CYLD and caspase-8 are required for TNF/IFNγ-induced cell death. (A) HeLa cells (parental, caspase-8 KO, FADD KO, and CYLD KO) cells were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: ****P < 0.0001. (B) HCT-116 cells (parental, CASP8 KO clones #1 and #2 with different gRNA, CYLD KO clones #1 and #2 with different gRNA) were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (C) HeLa cells were stably expressed with control (shcontrol) or CASP8 shRNA (shCASP8) and treated with IFNγ for 24 h. The protein expression of caspase-8 was analyzed by immunoblotting (n = 3). (D) HeLa shcontrol and shCASP8 cells were treated with TNF, IFNγ, or TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: ****P < 0.0001. (E) HeLa cells were stably expressed with control (shcontrol) or CYLD shRNA (shCYLD) and treated with IFNγ for 24 h. The protein expression of CYLD was analyzed by immunoblotting (n = 3). (F) HeLa shcontrol and shCYLD cells were treated with TNF, IFNγ, or TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: ****P < 0.0001. (G) HeLa control (shcontrol) and CASP8 knockdown (shCASP8) cells were further infected with shcontrol or shCYLD lentivirus, and treated with IFNγ for 24 h. The protein expression of CYLD and caspase-8 was analyzed by immunoblotting (n = 4). (H) HeLa cells (control, CASP8 knockdown, CYLD knockdown, CASP8 and CYLD double knockdown) were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: *P = 0.0321; ****P < 0.0001. (I and J) HeLa cells were infected with lentivirus express caspase-8 or empty vector. Then the cells were further infected with lentivirus express CYLD or empty vector. The protein expression was dox inducible. The expression of indicated proteins from these cells in the presence or absence of dox was analyzed by immunoblotting (I) (n = 3). Cells treated with TNF for 48 h in the presence or absence of dox (n = 3). Cell viability was determined by ATP levels (J). Two-tailed t test was performed: NS, **P = 0.0011, 0.0013 (left to right), ***P = 0.0002, 0.0004 (left to right), ****P < 0.0001. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F3.
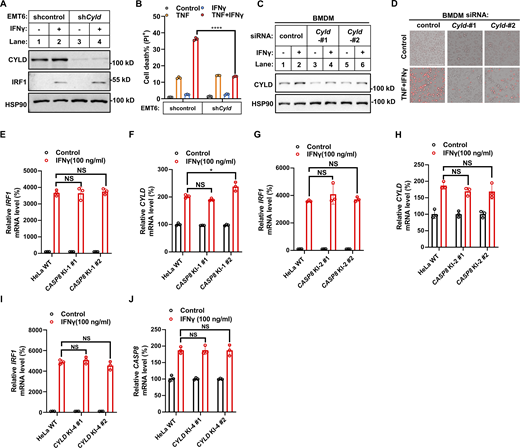
IFNγ induces CYLD levels to synergize with TNF to induce cell death. (A) EMT6 cells were infected with lentivirus with control (shcontrol) or Cyld shRNA (shCyld) and treated with IFNγ for 24 h. The protein expression of CYLD and IRF1 was analyzed by immunoblotting (n = 3). (B) EMT6 shcontrol and shCyld cells were treated with TNF, IFNγ, or TNF/IFNγ for 48 h (n = 3). Cell death was measured by PI staining. Two-tailed t test was performed: ****P < 0.0001. (C) Murine BMDMs were transfected with control or Cyld siRNAs (#1 and #2 targeting different regions of Cyld) for 48 h, after which cells were treated with IFNγ for 24 h. The protein expression of CYLD was analyzed by immunoblotting (n = 3). (D) BMDMs (Control and Cyld knockdown) were treated with TNF/IFNγ for 48 h (n = 3). PI staining was used to show dead cells. Scale bar, 275 μm. (E and F) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The mRNA levels of IRF1 (E) and CYLD (F) were determined by qPCR. One-way ANOVA was performed: NS (E); NS, *P = 0.0133 (F). (G and H) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The mRNA levels of IRF1 (G) and CYLD (H) were determined by qPCR. One-way ANOVA was performed: NS. (I and J) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated with IFNγ or vehicle control for 24 h (n = 3). IRF1 (I) and CASP8 (J) mRNA levels were determined by qPCR (n = 3). One-way ANOVA was performed: NS (I); NS (J). Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS4.
IFNγ induces CYLD levels to synergize with TNF to induce cell death. (A) EMT6 cells were infected with lentivirus with control (shcontrol) or Cyld shRNA (shCyld) and treated with IFNγ for 24 h. The protein expression of CYLD and IRF1 was analyzed by immunoblotting (n = 3). (B) EMT6 shcontrol and shCyld cells were treated with TNF, IFNγ, or TNF/IFNγ for 48 h (n = 3). Cell death was measured by PI staining. Two-tailed t test was performed: ****P < 0.0001. (C) Murine BMDMs were transfected with control or Cyld siRNAs (#1 and #2 targeting different regions of Cyld) for 48 h, after which cells were treated with IFNγ for 24 h. The protein expression of CYLD was analyzed by immunoblotting (n = 3). (D) BMDMs (Control and Cyld knockdown) were treated with TNF/IFNγ for 48 h (n = 3). PI staining was used to show dead cells. Scale bar, 275 μm. (E and F) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The mRNA levels of IRF1 (E) and CYLD (F) were determined by qPCR. One-way ANOVA was performed: NS (E); NS, *P = 0.0133 (F). (G and H) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The mRNA levels of IRF1 (G) and CYLD (H) were determined by qPCR. One-way ANOVA was performed: NS. (I and J) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated with IFNγ or vehicle control for 24 h (n = 3). IRF1 (I) and CASP8 (J) mRNA levels were determined by qPCR (n = 3). One-way ANOVA was performed: NS (I); NS (J). Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS4.
On the contrary, we individually or simultaneously expressed CYLD and caspase-8 in HeLa cells employing the doxycycline (dox)-inducible system. Dox-induced expression of CYLD did not cause cell death, and the administration of TNF (at a concentration that alone would not kill HeLa cells) induced cell death (Fig. 3, I and J), indicating that overexpression of CYLD sensitizes cells to TNF-induced cell death. Dox-induced expression of caspase-8 in cells is, on the contrary, toxic, and the administration of TNF further potentiated cell death (Fig. 3, I and J). Moreover, although the toxicity of simultaneously expressing CYLD and caspase-8 is comparable with that of overexpressing caspase-8 alone, TNF-induced cell death is more severe when the two are overexpressed together (Fig. 3, I and J). Therefore, dox-induced expression of CYLD, caspase-8, or both in combination is sufficient to sensitize cells to TNF-induced killing, and this effect occurs in an additive manner.
Previous studies found that IRF1 bound to the ISRE (consensus motif GTTTCXXTTTC/T) in the promoter of target genes to increase their expression (Ng et al., 2011; Antonczyk et al., 2019). We analyzed the CASP8 promoter sequence and found two potential ISREs (Fig. 4 A, CASP8 ISRE1 and ISRE2). Employing CRISPR-Cas9–mediated DNA double-strand breakage and homologous recombination technology, we mutated the ISRE1 and ISRE2 motifs in CASP8 promoter in HeLa cell to generate knockin mutations CASP8-KI-1 and CASP8-KI-2 (Fig. 4 A), respectively, and chose two clones of each mutation. HeLa CASP8-KI-1 clones showed similar basal caspase-8 expression and retained the responsiveness to IFNγ stimulation when compared with parental HeLa cells (Fig. 4, B and C; and Fig. S4, E and F). Both HeLa CASP8-KI-1 clones were similarly sensitive to TNF/IFNγ-induced cell death as parental HeLa cells (Fig. 4 D). In contrast, HeLa CASP8-KI-2 clones nearly totally abolished IFNγ-induced caspase-8 upregulation (Fig. 4, E and F), although the upregulation of IRF1 and CYLD remained the same (Fig. S4, G and H). As a result, TNF/IFNγ-induced cell death was dramatically inhibited in the HeLa CASP8-KI-2 clones (Fig. 4 G).
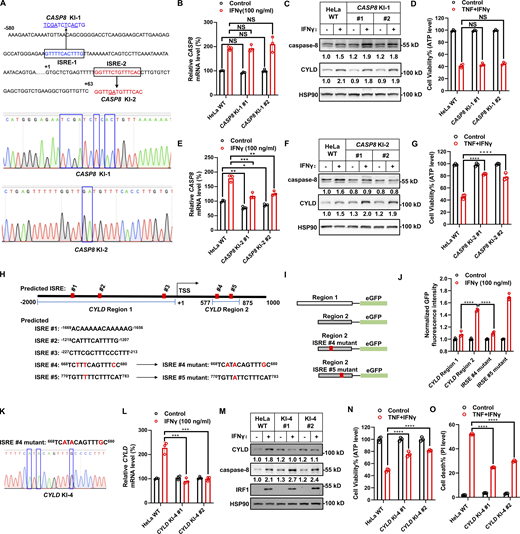
Upregulation of caspase-8 and CYLD levels is required for TNF/IFNγ-induced cell death. (A) Homology-directed repair facilitates mutating endogenous predicted ISRE motif 1 and 2 (ISRE-1 and ISRE-2) in CASP8 promoter region. The designed mutant bases are underlined. Sanger sequencing confirms the successful knockin of mutated ISRE-1 and ISRE-2. (B) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The CASP8 mRNA level was determined by qPCR. One-way ANOVA was performed: NS. (C) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with IFNγ for 24 h. The protein expression of caspase-8 and CYLD was analyzed by immunoblotting (n = 2). (D) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: NS. (E) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The CASP8 mRNA level was determined by qPCR. One-way ANOVA was performed: *P = 0.0208; **P = 0.0014, 0.002 (left to right); ***P = 0.001. (F) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with IFNγ for 24 h. The protein expression of caspase-8 and CYLD was analyzed by immunoblotting (n = 4). (G) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (H) The sequences of predicted ISREs at CYLD promoter region. (I) Schematic diagram of the reporter constructs. (J) The GFP fluorescence intensity derived from the indicated CYLD promoter reporters in the presence or absence of IFNγ. The GFP fluorescence intensity was quantified by flow cytometry (n = 3), and the values in the groups without IFNγ stimulation are set as 1.0. Two-tailed t test was performed: ****P < 0.0001. (K) Homology-directed repair facilitates mutating ISRE #4 in CYLD promoter region. Representative sequencing results from CYLD KI-4 clones confirm the success in mutation. (L) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated with IFNγ or vehicle control for 24 h. CYLD mRNA levels were determined by qPCR (n = 3). One-way ANOVA was performed: NS, ***P = 0.0001, 0.0002 (from left to right). (M) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated as indicated for 24 h. The protein expression of caspase-8 and CYLD were analyzed by immunoblotting (n = 3). (N and O) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated as indicated for 36 h. Cell viability was determined by ATP levels (N) (n = 3). Cell death was measured by PI staining followed by flow cytometry (O) (n = 3). One-way ANOVA was performed: ****P < 0.0001. Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F4.
Upregulation of caspase-8 and CYLD levels is required for TNF/IFNγ-induced cell death. (A) Homology-directed repair facilitates mutating endogenous predicted ISRE motif 1 and 2 (ISRE-1 and ISRE-2) in CASP8 promoter region. The designed mutant bases are underlined. Sanger sequencing confirms the successful knockin of mutated ISRE-1 and ISRE-2. (B) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The CASP8 mRNA level was determined by qPCR. One-way ANOVA was performed: NS. (C) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with IFNγ for 24 h. The protein expression of caspase-8 and CYLD was analyzed by immunoblotting (n = 2). (D) HeLa cells (parental, mutated ISRE-1 knock-in clones #1 and #2) were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: NS. (E) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with IFNγ for 24 h (n = 3). The CASP8 mRNA level was determined by qPCR. One-way ANOVA was performed: *P = 0.0208; **P = 0.0014, 0.002 (left to right); ***P = 0.001. (F) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with IFNγ for 24 h. The protein expression of caspase-8 and CYLD was analyzed by immunoblotting (n = 4). (G) HeLa cells (parental, mutated ISRE-2 knock-in clones #1 and #2) were treated with TNF/IFNγ (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (H) The sequences of predicted ISREs at CYLD promoter region. (I) Schematic diagram of the reporter constructs. (J) The GFP fluorescence intensity derived from the indicated CYLD promoter reporters in the presence or absence of IFNγ. The GFP fluorescence intensity was quantified by flow cytometry (n = 3), and the values in the groups without IFNγ stimulation are set as 1.0. Two-tailed t test was performed: ****P < 0.0001. (K) Homology-directed repair facilitates mutating ISRE #4 in CYLD promoter region. Representative sequencing results from CYLD KI-4 clones confirm the success in mutation. (L) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated with IFNγ or vehicle control for 24 h. CYLD mRNA levels were determined by qPCR (n = 3). One-way ANOVA was performed: NS, ***P = 0.0001, 0.0002 (from left to right). (M) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated as indicated for 24 h. The protein expression of caspase-8 and CYLD were analyzed by immunoblotting (n = 3). (N and O) HeLa cells (parental, CYLD KI-4 clones #1 and #2) were treated as indicated for 36 h. Cell viability was determined by ATP levels (N) (n = 3). Cell death was measured by PI staining followed by flow cytometry (O) (n = 3). One-way ANOVA was performed: ****P < 0.0001. Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F4.
We performed a similar analysis from −2,000 to +1,000 on the CYLD promoter and identified five predicted ISREs (Fig. 4 H). eGFP reporter constructs containing region 1 (−2,000 to +1, +1 denoting the transcriptional start site) and region 2 (577–875) encompassing three and two predicted ISREs, respectively, were generated (Fig. 4, H and I), and lentiviral stable cell lines were established. Reporter assays showed that region 2, but not region 1, could further activate the eGFP reporter upon IFNγ stimulation (Fig. 4 J), suggesting that the ISRE lies in region 2. Therefore, the predicted ISRE #4 and ISRE #5 were mutated to generate region 2 ISRE #4 mutant and region 2 ISRE #5 mutant, respectively (Fig. 4, H and I). The region 2 ISRE #4 mutant, but not the region 2 ISRE #5 mutant, lost responsiveness to IFNγ stimulation, indicating that ISRE #4 was the ISRE responding to IFNγ (Fig. 4 J). To confirm this, we generated CYLD-KI cells with ISRE #4 mutated (Fig. 4 K) and selected two independent clones. HeLa CYLD-KI clones completely abolished IFNγ-induced CYLD upregulation (Fig. 4, L and M), while the upregulation of IRF1 and CASP8 remained the same as in parental cells (Fig. S4, I and J). As expected, TNF/IFNγ-induced cell death was significantly inhibited in the two HeLa CYLD-KI clones (Fig. 4, N and O). Collectively, our results indicated that IFNγ-stimulated caspase-8 and CYLD upregulation additively promoted TNF/IFNγ-induced cell death in cancer cells.
The JAK1-JAK2-STAT1-IRF1 axis mediates induction of CYLD and caspase-8
IFNγ binds IFNGR1/2 to activate the JAK1-JAK2-STAT1-IRF1 axis to regulate the transcription of target genes (Lee and Ashkar, 2018), and we tested whether this axis is required for IFNγ-induced expression of CYLD and caspase-8. KO IFNGR1, JAK1, JAK2, STAT1, and IRF1 individually in HeLa cells blocked IFNγ-induced CYLD and caspase-8 expression (Fig. 5 A), as well as TNF/IFNγ-induced caspase-8 auto-cleavage and cell death (Fig. 5, B and C). Consistent with this, experiments with multiple cancer cell lines showed that the JAK1/JAK2 inhibitors PF-04965842 and Upadacitinib blocked IFNγ-induced expression of CYLD and caspase-8, and TNF/IFNγ-induced caspase-8 activation and cell death (Fig. 5, D–I).
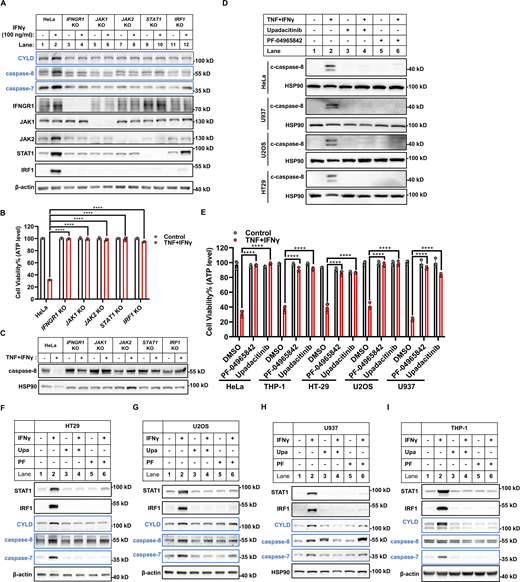
IFNγ induces CYLD and caspase-8 expression via the JAK1/JAK2/STAT1/IRF1 signaling. (A) HeLa cells (parental, IFNGR1 KO, JAK1 KO, JAK2 KO, STAT1 KO, and IRF1 KO) were treated with IFNγ for 24 h. The expression of indicated proteins was analyzed by immunoblotting (n = 4). (B) Indicated cells were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (C) Indicated cells were treated with TNF/IFNγ for 36 h. The protein expression of full-length caspase-8 was analyzed by immunoblotting (n = 3). (D and E) Indicated cells were treated with TNF/IFNγ in the presence or absence of the JAK inhibitors PF-004965842 (3 μM) or Upadacitinib (3 μM) (n = 3). The protein expression of cleaved caspase-8 was analyzed by immunoblotting (D). Cell viability was determined by ATP levels (E). One-way ANOVA was performed: ****P < 0.0001. (F–I) HT-29 (F), U2OS (G), U937 (H), and THP-1 (I) cells were treated with IFNγ in the presence or absence of PF-004965842 (3 μM) or Upadacitinib (3 μM) for 24 h (n = 3). The expression of indicated proteins was analyzed by immunoblotting. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F5.
IFNγ induces CYLD and caspase-8 expression via the JAK1/JAK2/STAT1/IRF1 signaling. (A) HeLa cells (parental, IFNGR1 KO, JAK1 KO, JAK2 KO, STAT1 KO, and IRF1 KO) were treated with IFNγ for 24 h. The expression of indicated proteins was analyzed by immunoblotting (n = 4). (B) Indicated cells were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (C) Indicated cells were treated with TNF/IFNγ for 36 h. The protein expression of full-length caspase-8 was analyzed by immunoblotting (n = 3). (D and E) Indicated cells were treated with TNF/IFNγ in the presence or absence of the JAK inhibitors PF-004965842 (3 μM) or Upadacitinib (3 μM) (n = 3). The protein expression of cleaved caspase-8 was analyzed by immunoblotting (D). Cell viability was determined by ATP levels (E). One-way ANOVA was performed: ****P < 0.0001. (F–I) HT-29 (F), U2OS (G), U937 (H), and THP-1 (I) cells were treated with IFNγ in the presence or absence of PF-004965842 (3 μM) or Upadacitinib (3 μM) for 24 h (n = 3). The expression of indicated proteins was analyzed by immunoblotting. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F5.
Given that both STAT1 and IRF1 are transcription factors, we systematically investigated the role of STAT1 and IRF1 in the IFNγ-induced transcription of CYLD and CASP8. WT IRF1 and IRF1 W11R (the mutation abolishes the DNA binding and transactivating activity of IRF1) (Eason et al., 1999) were stably expressed in HeLa WT cells and STAT1 KO HeLa cells using dox-inducible system. We found that dox-induced expression of IRF1, but not of the IRF1 W11R mutant variant, induced the expression of CYLD and caspase-8 in HeLa WT cells and induced cell death in the presence of TNF (Fig. 6 A); these outcomes mimicked those from IFNγ treatment. In STAT1 KO HeLa cells, dox-induced expression of IRF1 also induced expression of CYLD and caspase-8 and caused cell death in the presence of TNF (Fig. 6 B), indicating that IRF1 is sufficient for the IFNγ-induced expression of CYLD and caspase-8.
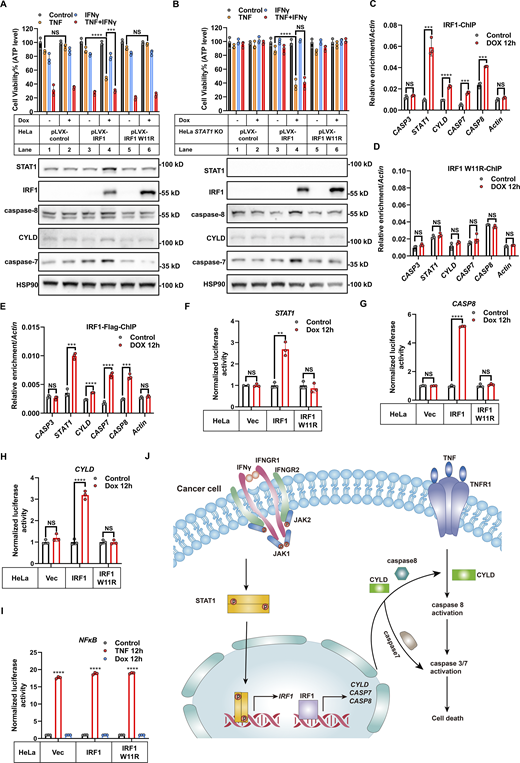
IRF1 binds to the promoters of CYLD/CASP8 and activates their transcription. (A and B) HeLa parental (A) and STAT1 KO cells (B) were infected with lentivirus-expressed IRF1, IRF1 W11R mutant variant, or the empty vector. Cells were then treated with TNF, IFNγ, or TNF/IFNγ for 48 h in the presence or absence of dox (n = 3). The expression of indicated proteins from these cells in the presence or absence of dox was analyzed by immunoblotting (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: NS, ***P = 0.0001, ****P < 0.0001 (A); NS, ****P < 0.0001 (B). (C and D) ChIP-qPCR analysis of IRF1 (C) or IRF1 W11R mutant variant (D) stably expressed in HeLa IRF1 KO cells binding to the promoter region of the indicated genes using an anti-IRF1 antibody (n = 3). Values are a percentage of the input. Two-tailed t test was performed: NS, ***P = 0.000488, 0.000172, 0.000173 (from left to right), ****P < 0.0001 (C); NS (D). (E) ChIP-qPCR analysis of IRF1-Flag binding to the promoter region of indicated genes in HeLa IRF1 KO cells stably expressed IRF1-Flag using an anti-Flag antibody (n = 3). Two-tailed t test was performed: NS, ***P = 0.000166, 0.000138 (from left to right), ****P < 0.0001. (F–I) HeLa cells stably expressed IRF1, IRF1 W11R mutant variant, or an empty vector were transfected with STAT1 (F), CASP8 (G), CYLD (H), and NF-κB (I) promoter region containing TA-Firefly luciferase reporter plasmids and Renilla luciferase plasmid. After 24 h transfection, dox was added to induce protein expression for 12 h (n = 2 or 3). The reporter activities were determined by firefly luciferase signals normalized to the Renilla luciferase signal. Two-tailed t test was performed: NS, **P = 0.001097 (F); NS, ****P < 0.0001 (G); NS, ****P < 0.0001 (H). One-way ANOVA was performed: ****P < 0.0001 (I). (J) Schematic of the biochemical mechanism of TNF/IFNγ-induced cell death in cancer cells. IFNγ induces the expression of CYLD/CASP8/CASP7 by JAK1/JAK2/STAT1/IRF1 pathway to promote TNF/IFNγ-induced cell death. Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F6.
IRF1 binds to the promoters of CYLD/CASP8 and activates their transcription. (A and B) HeLa parental (A) and STAT1 KO cells (B) were infected with lentivirus-expressed IRF1, IRF1 W11R mutant variant, or the empty vector. Cells were then treated with TNF, IFNγ, or TNF/IFNγ for 48 h in the presence or absence of dox (n = 3). The expression of indicated proteins from these cells in the presence or absence of dox was analyzed by immunoblotting (n = 3). Cell viability was determined by ATP levels. Two-tailed t test was performed: NS, ***P = 0.0001, ****P < 0.0001 (A); NS, ****P < 0.0001 (B). (C and D) ChIP-qPCR analysis of IRF1 (C) or IRF1 W11R mutant variant (D) stably expressed in HeLa IRF1 KO cells binding to the promoter region of the indicated genes using an anti-IRF1 antibody (n = 3). Values are a percentage of the input. Two-tailed t test was performed: NS, ***P = 0.000488, 0.000172, 0.000173 (from left to right), ****P < 0.0001 (C); NS (D). (E) ChIP-qPCR analysis of IRF1-Flag binding to the promoter region of indicated genes in HeLa IRF1 KO cells stably expressed IRF1-Flag using an anti-Flag antibody (n = 3). Two-tailed t test was performed: NS, ***P = 0.000166, 0.000138 (from left to right), ****P < 0.0001. (F–I) HeLa cells stably expressed IRF1, IRF1 W11R mutant variant, or an empty vector were transfected with STAT1 (F), CASP8 (G), CYLD (H), and NF-κB (I) promoter region containing TA-Firefly luciferase reporter plasmids and Renilla luciferase plasmid. After 24 h transfection, dox was added to induce protein expression for 12 h (n = 2 or 3). The reporter activities were determined by firefly luciferase signals normalized to the Renilla luciferase signal. Two-tailed t test was performed: NS, **P = 0.001097 (F); NS, ****P < 0.0001 (G); NS, ****P < 0.0001 (H). One-way ANOVA was performed: ****P < 0.0001 (I). (J) Schematic of the biochemical mechanism of TNF/IFNγ-induced cell death in cancer cells. IFNγ induces the expression of CYLD/CASP8/CASP7 by JAK1/JAK2/STAT1/IRF1 pathway to promote TNF/IFNγ-induced cell death. Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F6.
Next, we performed IRF1 chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR) and found that IRF1, but not the IRF1 W11R mutant variant, bound to the promoter regions of the CYLD and CASP8 loci (Fig. 6, C and D). These results were further confirmed using cells expressing IRF1-Flag (Fig. 6 E). Further, dox-induced expression of IRF1, but not of the IRF1 W11R mutant variant, increased the expression of a luciferase reporter driven by either STAT1, CYLD, or CASP8 promoters, but not by the NF-κB promoter (Fig. 6, F–I). Collectively, these results demonstrated that IRF1 is bound to the promoters of CYLD and CASP8 to activate their transcription (Fig. 6 J).
ELAVL1 is required for TNF/IFNγ-induced cancer cell death and caspase-8 expression
From the above set of results, we learned that IFNγ-induced CYLD and caspase-8 upregulation additively underlie the mechanism of IFNγ’s synergism to promote TNF-initiated cell death. Following this, we tested the third category of candidate gene ELAVL1, which itself is not transcriptionally regulated by IFNγ (Fig. 2 D). Instead, ELAVL1 acts as a posttranscriptional regulator (Schultz et al., 2020) and might mediate TNF/IFNγ-induced cell death by stabilizing IFNγ-upregulated CYLD and/or CASP8.
First, we found that KO ELAVL1 both in HeLa cells and in HCT-116 cells blocked TNF/IFNγ-induced cell death, indicating an indispensable role of ELAVL1 in TNF/IFNγ-induced cell death in these two cancer cell lines (Fig. 7, A–D). Re-expression of 3xFlag tagged ELAVL1 in ELAVL1-KO cells restored the sensitivity to TNF/IFNγ-induced cell death (Fig. 7, E–H). Mutation of the amino acids required for the RNA-binding function of ELAVL1 (Fig. 7 I) (Wang et al., 2013) lost its cell death function (Fig. 7, E–H), indicating the RNA binding activity of ELAVL1 is required for TNF/IFNγ-induced cell death. Then, we tested whether ELAVL1 mediates TNF/IFNγ-induced cell death by regulating the levels of IFNγ upregulated CYLD and/or CASP8.
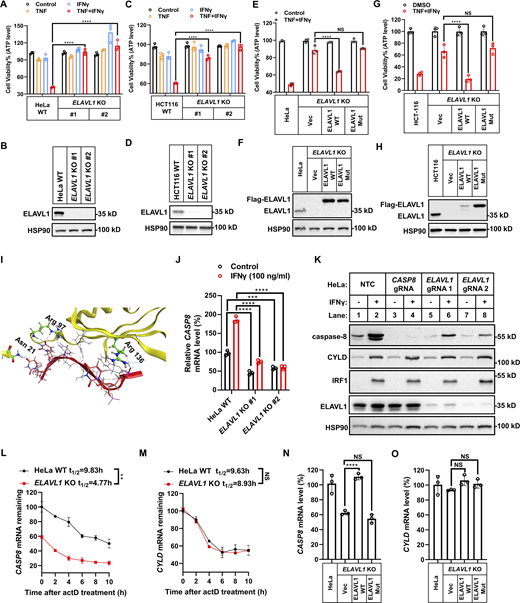
ELAVL1 is required for TNF/IFNγ-induced cell death. (A and C) HeLa (A) or HCT-116 (C) cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (B and D) The protein expression of ELAVL1 in indicated cells was analyzed by immunoblotting (n = 2). (E and G) HeLa (E) or HCT-116 (G) cells (parental, ELAVL1 KO, ELAVL1 KO stably expressed with WT ELAVL1 or mutated ELAVL1) were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: NS, ****P < 0.0001. (F and H) The protein expression of ELAVL1 in indicated cells was analyzed by immunoblotting (n = 2). (I) The binding interface between ELAVL1 and RNA in the ELAVL1-RNA complex (PDB 4ED5) is shown. Amino acids critical for the RNA binding of ELAVL1 are highlighted. (J) HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h (n = 3). The mRNA levels of CASP8 were measured by qPCR. One-way ANOVA was performed: ***P = 0.0003, ****P < 0.0001. (K) HeLa cells (parental, CASP8 KO, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h. The expression of indicated proteins was analyzed by immunoblotting (n = 4). (L and M) HeLa cells (parental and ELAVL1 KO) were treated with actinomycin D for the indicated time. The mRNA levels of CASP8 (L) and CYLD (M) were measured by qPCR (n = 3). The mRNA levels in HeLa cells before the addition of actinomycin D (0 h) were set as 100%. Two-tailed t test was performed: **P = 0.0024 (L); NS (M). (N and O) The mRNA levels of CASP8 (N) and CYLD (O) in HeLa cells (parental, ELAVL1 KO, ELAVL1 KO stably expressed with WT ELAVL1 or mutated ELAVL1) were measured by qPCR (n = 3). One-way ANOVA was performed: NS, ****P < 0.0001 (N); NS (O). Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F7.
ELAVL1 is required for TNF/IFNγ-induced cell death. (A and C) HeLa (A) or HCT-116 (C) cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001. (B and D) The protein expression of ELAVL1 in indicated cells was analyzed by immunoblotting (n = 2). (E and G) HeLa (E) or HCT-116 (G) cells (parental, ELAVL1 KO, ELAVL1 KO stably expressed with WT ELAVL1 or mutated ELAVL1) were treated with TNF/IFNγ for 48 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: NS, ****P < 0.0001. (F and H) The protein expression of ELAVL1 in indicated cells was analyzed by immunoblotting (n = 2). (I) The binding interface between ELAVL1 and RNA in the ELAVL1-RNA complex (PDB 4ED5) is shown. Amino acids critical for the RNA binding of ELAVL1 are highlighted. (J) HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h (n = 3). The mRNA levels of CASP8 were measured by qPCR. One-way ANOVA was performed: ***P = 0.0003, ****P < 0.0001. (K) HeLa cells (parental, CASP8 KO, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h. The expression of indicated proteins was analyzed by immunoblotting (n = 4). (L and M) HeLa cells (parental and ELAVL1 KO) were treated with actinomycin D for the indicated time. The mRNA levels of CASP8 (L) and CYLD (M) were measured by qPCR (n = 3). The mRNA levels in HeLa cells before the addition of actinomycin D (0 h) were set as 100%. Two-tailed t test was performed: **P = 0.0024 (L); NS (M). (N and O) The mRNA levels of CASP8 (N) and CYLD (O) in HeLa cells (parental, ELAVL1 KO, ELAVL1 KO stably expressed with WT ELAVL1 or mutated ELAVL1) were measured by qPCR (n = 3). One-way ANOVA was performed: NS, ****P < 0.0001 (N); NS (O). Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F7.
Surprisingly, KO expression of ELAVL1 blocked IFNγ-stimulated upregulation of CASP8 mRNA levels (Fig. 7 J) but did not block upregulation of CYLD mRNA levels (Fig. S5 A). Furthermore, ELAVL1 KO significantly decreased the basal CASP8 mRNA levels (Fig. 7 J) by shortening the half-life of CASP8 mRNA (Fig. 7, L and M) in an RNA-binding activity-dependent manner (Fig. 7, N and O). The decrease in CASP8 mRNA levels in ELAVL1 KO cells both in basal and IFNγ-stimulated conditions resulted in the decrease of caspase-8 protein levels in the tested cancer cell lines (Fig. 7 K and Fig. S5 B). This decrease of caspase-8 levels in ELAVL1 KO cells did not result from a compromise of IFNγ signaling, as (i) KO expression of ELAVL1 decreased basal caspase-8 expression in the absence of IFNγ stimulation; and (ii) ELAVL1 KO did not block IFNγ-stimulated IRF1 and the downstream CYLD expression (Fig. 7 K and Fig. S5 B).
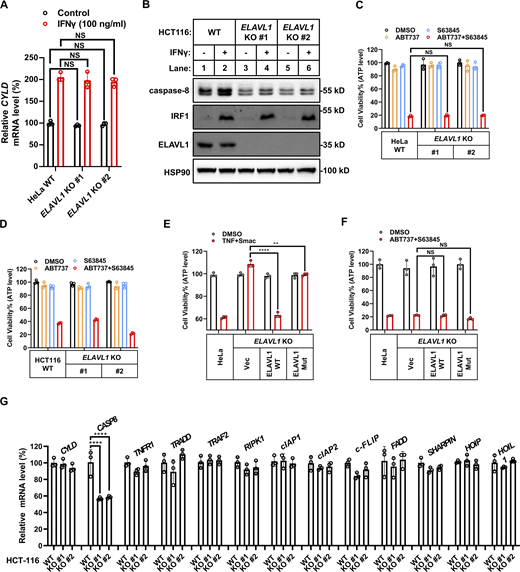
ELAVL1 is a general regulator involved in TNF-mediated cell death. (A) HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h (n = 3). The mRNA levels of CYLD were measured by qPCR. One-way ANOVA was performed: NS. (B) HCT-116 cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h. The expression of indicated proteins was analyzed by immunoblotting (n = 3). (C and D) HeLa (C) or HCT-116 (D) cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with ABT737/S63845 for 12 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed. (E and F) HeLa cells (parental, ELAVL1 KO, ELAVL1 KO stably expressed with WT ELAVL1 or mutated ELAVL1) were treated with TNF/Smac (E) or ABT737/S63845 (F) (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: **P = 0.0053, ****P < 0.0001 (E); NS (F). (G) The mRNA levels of indicated genes involved in TNF signaling were measured in HCT-116 cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) by qPCR (n = 3). One-way ANOVA was performed: ****P < 0.0001. Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS5.
ELAVL1 is a general regulator involved in TNF-mediated cell death. (A) HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h (n = 3). The mRNA levels of CYLD were measured by qPCR. One-way ANOVA was performed: NS. (B) HCT-116 cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with IFNγ for 24 h. The expression of indicated proteins was analyzed by immunoblotting (n = 3). (C and D) HeLa (C) or HCT-116 (D) cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with ABT737/S63845 for 12 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed. (E and F) HeLa cells (parental, ELAVL1 KO, ELAVL1 KO stably expressed with WT ELAVL1 or mutated ELAVL1) were treated with TNF/Smac (E) or ABT737/S63845 (F) (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: **P = 0.0053, ****P < 0.0001 (E); NS (F). (G) The mRNA levels of indicated genes involved in TNF signaling were measured in HCT-116 cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) by qPCR (n = 3). One-way ANOVA was performed: ****P < 0.0001. Data represent the mean value ± SD from n biological replicates or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData FS5.
ELAVL1 is required for death receptors–initiated cell death by stabilizing caspase-8
As ELAVL1 KO decreased caspase-8 expression even under basal conditions in the absence of IFNγ stimulation, the results indicated that ELAVL1 would be required for general death receptors–initiated caspase-8–dependent cell death, not limited to only one situation, which is TNF/IFNγ-induced cell death. Indeed, KO ELAVL1 decreased the generation of cleaved (therefore activated) caspase-8 in cells upon TNF/IFNγ, TNF/SMAC, and TNF/CHX stimulation (Fig. 8, A–D). Accordingly, KO ELAVL1 in two cancer cell lines significantly inhibited TNF/SMAC-, TNF/CHX-, and TRAIL-induced cell death (Fig. 8, E–H, and N), but did not inhibit caspase-8–independent intrinsic apoptosis initiated by ABT737 plus S63845 (Fig. S5, C–F). Therefore, results indicated that ELAVL1 is required for the general death receptors–initiated caspase-8–dependent death in cancer cell lines.
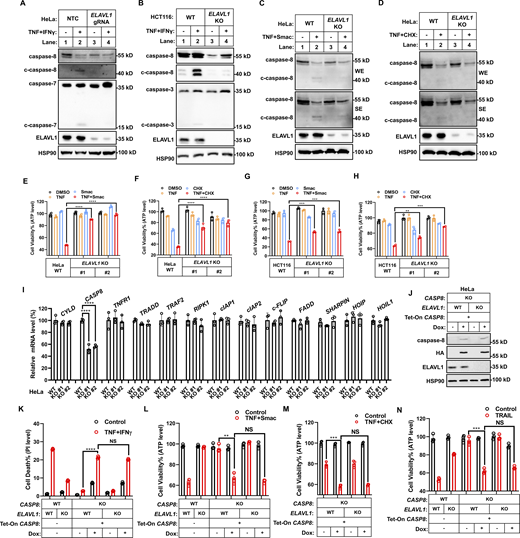
ELAVL1 regulates TNF- and TRAIL-mediated cell death by targeting caspase-8. (A and B) HeLa (A, n = 4) or HCT-116 (B, n = 3) cells (parental and ELAVL1 KO) were treated with TNF/IFNγ for 36 h. NTC is short for non-targeting control. The protein expression of cleaved and full-length caspase-3/7/8 was analyzed by immunoblotting. (C and D) HeLa cells (parental and ELAVL1 KO) were treated with TNF/Smac for 24 h (C) or TNF/CHX for 12 h (D). The protein expression of cleaved and full-length caspase-8 was analyzed by immunoblotting (n = 3). (E and F) HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with TNF/Smac (E) or TNF/CHX (F) for 24 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001 (E); ****P < 0.0001 (F). (G and H) HCT-116 cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with TNF/Smac (G) or TNF/CHX (H) for 24 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ***P = 0.0004, 0.0003 (from left to right, G); **P = 0.0024, ***P = 0.0001 (H). (I) The mRNA levels of indicated genes involved in TNF signaling were measured in HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) by qPCR (n = 3). One-way ANOVA was performed: ****P < 0.0001. (J) HeLa cells (CASP8 KO, CASP8, and ELAVL1 double KO) were infected with lentivirus-expressed CASP8 coding sequence with HA tag. The expression of indicated proteins from these cells in the presence or absence of dox was analyzed by immunoblotting (n = 4). (K–N) HeLa cells (parental, CASP8 KO, ELAVL1 KO, CASP8, and ELAVL1 double KO) were infected with lentivirus-expressed CASP8 or the empty vector. Cells were then treated with TNF/IFNγ (K), TNF/Smac (L), TNF/CHX (M), or TRAIL (N) in the presence or absence of dox (n = 3). Cell death was measured by PI staining followed by flow cytometry. Cell viability was determined by ATP levels. Two-tailed t test was performed: NS, ****P < 0.0001 (K); NS, **P = 0.0015 (L); NS, ***P = 0.0004 (M); NS, ***P = 0.0004 (N). Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F8.
ELAVL1 regulates TNF- and TRAIL-mediated cell death by targeting caspase-8. (A and B) HeLa (A, n = 4) or HCT-116 (B, n = 3) cells (parental and ELAVL1 KO) were treated with TNF/IFNγ for 36 h. NTC is short for non-targeting control. The protein expression of cleaved and full-length caspase-3/7/8 was analyzed by immunoblotting. (C and D) HeLa cells (parental and ELAVL1 KO) were treated with TNF/Smac for 24 h (C) or TNF/CHX for 12 h (D). The protein expression of cleaved and full-length caspase-8 was analyzed by immunoblotting (n = 3). (E and F) HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with TNF/Smac (E) or TNF/CHX (F) for 24 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ****P < 0.0001 (E); ****P < 0.0001 (F). (G and H) HCT-116 cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) were treated with TNF/Smac (G) or TNF/CHX (H) for 24 h (n = 3). Cell viability was determined by ATP levels. One-way ANOVA was performed: ***P = 0.0004, 0.0003 (from left to right, G); **P = 0.0024, ***P = 0.0001 (H). (I) The mRNA levels of indicated genes involved in TNF signaling were measured in HeLa cells (parental, ELAVL1 KO clones #1 and #2 with different gRNA) by qPCR (n = 3). One-way ANOVA was performed: ****P < 0.0001. (J) HeLa cells (CASP8 KO, CASP8, and ELAVL1 double KO) were infected with lentivirus-expressed CASP8 coding sequence with HA tag. The expression of indicated proteins from these cells in the presence or absence of dox was analyzed by immunoblotting (n = 4). (K–N) HeLa cells (parental, CASP8 KO, ELAVL1 KO, CASP8, and ELAVL1 double KO) were infected with lentivirus-expressed CASP8 or the empty vector. Cells were then treated with TNF/IFNγ (K), TNF/Smac (L), TNF/CHX (M), or TRAIL (N) in the presence or absence of dox (n = 3). Cell death was measured by PI staining followed by flow cytometry. Cell viability was determined by ATP levels. Two-tailed t test was performed: NS, ****P < 0.0001 (K); NS, **P = 0.0015 (L); NS, ***P = 0.0004 (M); NS, ***P = 0.0004 (N). Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F8.
As ELAVL1 is a posttranscriptional regulator that affects the stability and/or translational efficiency of multiple target mRNAs through binding to their 3′UTR, its KO would in theory affect mRNA and/or protein expression levels of multiple downstream genes (Schultz et al., 2020). To test whether the resistance to TNF- and TRAIL-initiated cell death in ELAVL1 KO cells mainly resulted from decreased caspase-8 expression, we performed qPCR analysis to map whether the mRNA levels of other known genes involved in this cell death pathway were decreased in ELAVL1 KO cells. Results indicated that the mRNA level of CASP8 but no other related genes decreased in ELAVL1 KO cells (Fig. 8 I and Fig. S5 G).
In addition, as ELAVL1 binds 3′UTR of target mRNAs to function, the deficiency of CASP8 3′UTR in the exogenously expressed CASP8 mRNA would lose its responsiveness to ELAVL1. Therefore, we expressed HA-tagged caspase-8 without its native 3′UTR portion using a Tet-On system (Tet-On CASP8) in caspase-8 KO cells. Dox treatment induced expression of caspase-8 in the Tet-On CASP8 cell, and KO expression of ELAVL1 did not cause a decrease of caspase-8 expression level (Fig. 8 J). Accordingly, KO expression of ELAVL1 in the Tet-On CASP8 cell could not block TNF/IFNγ-, TNF/SMAC-, TNF/CHX-, and TRAIL-induced cell death (Fig. 8, K–N). Collectively, results indicate that ELAVL1 is required for multiple death receptors–initiated cell death by stabilizing caspase-8 expression.
ELAVL1 binds CASP8 mRNA to increase caspase-8 expression
It is suggested that ELAVL1 traffics between cell nucleus and cytosol, and the cytosolic ELAVL1 binds mature mRNAs to stabilize the bound mRNAs and/or increase their translational efficiency (Schultz et al., 2020). We confirmed that ELAVL1 localized both in nuclear and cytosol fractions in HeLa extracts, with a higher portion in the nucleus (Fig. 9, A and B). IP of 3xFlag tagged ELAVL1 combined with qPCR analysis indicated that ELAVL1 binds CASP8 mRNA (Fig. 9, C and D). RNA binding deficient ELAVL1 variant significantly compromised in its ability to bind CASP8 mRNA compared with its WT counterpart (Fig. 9, C and D).
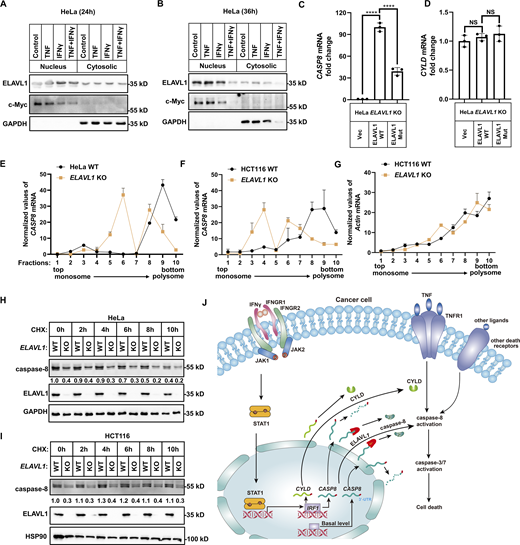
ELAVL1 increases the translational efficiency of caspase-8. (A and B) HeLa cells were treated with TNF, IFNγ, or TNF/IFNγ for 24 h (A) or 36 h (B). Cell lysates were separated into nucleus and cytosolic components. Subcellular localization of ELAVL1 was analyzed by immunoblotting (n = 3). (C and D) HeLa ELAVL1 KO cells were infected with lentivirus express Flag-ELAVL1, Flag-ELAVL1 mutant, or the empty vector. Then cells were lysed and subjected to RNA IP using anti-FLAG antibody. Relative enrichment of CASP8 (C) and CYLD (D) mRNA levels were measured by qPCR (n = 3). The mRNA levels in HeLa cells were set as 1. One-way ANOVA was performed: ****P < 0.0001 (C); NS (D). (E) HeLa cells (parental, ELAVL1 KO) were fractionated into 10 gradient fractions by sucrose gradient centrifugation. Polysome fraction analysis of the distribution of CASP8 mRNAs was measured by qPCR (mean value from two independent experiments). (F and G) HCT-116 cells (parental, ELAVL1 KO) were fractionated into 10 gradient fractions by sucrose gradient centrifugation. Polysome fraction analysis of the distribution of CASP8 (F) or CYLD (G) mRNAs was measured by qPCR (mean value from two independent experiments). (H and I) HeLa (H, n = 4) or HCT-116 (I, n = 3) cells (parental, ELAVL1 KO) were treated with CHX for the indicated time. The expression of indicated proteins was analyzed by immunoblotting. (J) The biochemistry mechanism model of TNF/IFNγ-induced cell death. IFNγ activates IFNGR1/JAK1/STAT1/IRF1 pathway, which then induces the transcription of CYLD/CASP8/CASP7. When CASP8 mRNA translocates from nucleus to cytosol, ELAVL1 promotes the mRNA stability and translation of CASP8. Finally, increased expression of caspase-8 and CYLD can synergistically promote TNF-mediated caspase-8 activation and cell death. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F9.
ELAVL1 increases the translational efficiency of caspase-8. (A and B) HeLa cells were treated with TNF, IFNγ, or TNF/IFNγ for 24 h (A) or 36 h (B). Cell lysates were separated into nucleus and cytosolic components. Subcellular localization of ELAVL1 was analyzed by immunoblotting (n = 3). (C and D) HeLa ELAVL1 KO cells were infected with lentivirus express Flag-ELAVL1, Flag-ELAVL1 mutant, or the empty vector. Then cells were lysed and subjected to RNA IP using anti-FLAG antibody. Relative enrichment of CASP8 (C) and CYLD (D) mRNA levels were measured by qPCR (n = 3). The mRNA levels in HeLa cells were set as 1. One-way ANOVA was performed: ****P < 0.0001 (C); NS (D). (E) HeLa cells (parental, ELAVL1 KO) were fractionated into 10 gradient fractions by sucrose gradient centrifugation. Polysome fraction analysis of the distribution of CASP8 mRNAs was measured by qPCR (mean value from two independent experiments). (F and G) HCT-116 cells (parental, ELAVL1 KO) were fractionated into 10 gradient fractions by sucrose gradient centrifugation. Polysome fraction analysis of the distribution of CASP8 (F) or CYLD (G) mRNAs was measured by qPCR (mean value from two independent experiments). (H and I) HeLa (H, n = 4) or HCT-116 (I, n = 3) cells (parental, ELAVL1 KO) were treated with CHX for the indicated time. The expression of indicated proteins was analyzed by immunoblotting. (J) The biochemistry mechanism model of TNF/IFNγ-induced cell death. IFNγ activates IFNGR1/JAK1/STAT1/IRF1 pathway, which then induces the transcription of CYLD/CASP8/CASP7. When CASP8 mRNA translocates from nucleus to cytosol, ELAVL1 promotes the mRNA stability and translation of CASP8. Finally, increased expression of caspase-8 and CYLD can synergistically promote TNF-mediated caspase-8 activation and cell death. Data represent the mean value ± SD from n biological replicates, or a representative immunoblot from n independent experiments. Source data are available for this figure: SourceData F9.
Upon binding to target mRNA, ELAVL1 was found to stabilize and/or regulate the translational efficiency of target mRNAs. We performed polysome profile analysis in cytoplasmic lysates to evaluate whether ELAVL1 KO alters the polysome distribution of CASP8 mRNA. Results showed that CASP8 mRNA was localized mainly to the gradient bottom where polysomes reside in the parental cells of two different cancer cell lines (Fig. 9, E–G). This indicates that the CASP8 mRNA had been actively translated. In contrast, the distribution of CASP8 mRNA shifted from polysomal to prepolysomal fractions in ELAVL1 KO cells, which would lower the translational efficiency (Fig. 9, E–G). However, ELAVL1 KO did not affect caspase-8 protein stability posttranslation (Fig. 9, H and I). Collectively, results indicated that ELAVL1 binds CASP8 mRNA to both increase its stability and enhance its translation to increase caspase-8 protein levels and mediate multiple death receptors–initiated cell death (Fig. 9 J).
Discussion
TNF-initiated cell death signaling is a complex process (Silke, 2011). This is in part due to the exquisite balance between cell survival and cell death and to the different consequences upon caspase-8 activation from different triggers in the presence of different downstream molecules (Vandenabeele et al., 2023). TNF plus small molecules like Smac, CHX, or 5Z7 are among the extensively used, well-accepted combinations to trigger cell death in multiple types of cells. Smac targets inhibitor of apoptosis proteins (IAPs) to induce degradation of cIAP1/2 or inhibition of XIAP. CHX blocks cellular translation and therefore blocks the synthesis of the fast turnover protein cFLIP. 5Z7 inhibits kinase activity of TAK1 to decrease MAPK and NF-κB activation. Like other approaches, for example, inhibiting the NF-κB activation or inhibiting export of nuclear proteins (Karki et al., 2021b, 2022), these canonical strategies activate TNF-initiated cell death by “blocking” an activity.
In this study, both IRF1-mediated transcriptional increase of CASP8 and CYLD levels and ELAVL1-mediated posttranscriptional stabilization of CASP8 mRNA increase CYLD and caspase-8 expression to additively promote TNF/IFNγ-induced cell death. It seems likely that IFNγ lowers the threshold of cells to TNF-induced cell death by “activating” an activity (in this case increasing caspase-8 and CYLD protein levels) but not by blocking an activity (IAP or cFLIP levels) as canonical triggers do. Further, this activating mechanism is supported by the synergistic induction of iNOS upon TNF/TLR agonists plus IFNγ stimulation in murine BMDMs, which is required for caspase-8 activation and cell death (Simpson et al., 2022; Karki et al., 2021a). Therefore, TNF/IFNγ represents a novel possibility to activate cell death, and this type of activation might be extensive in pathological situations, as both cytokines are highly expressed and associated with cell death in tumor immunotherapy and inflammatory diseases (including cytokine storm-induced tissue damage) (Karki and Kanneganti, 2022; Chen et al., 2023).
Previously, activation of caspase-8–induced cell death was solely recognized as apoptosis, including the direct activation of caspase-3/7 by caspase-8, and the cleavage of Bid or modulation of BCL2 family proteins to activate intrinsic apoptosis (Kesavardhana et al., 2020; Simpson et al., 2022). Lately, caspase-8 was found to modulate necroptosis and pyroptosis, two programmed necrosis pathways with strong inflammatory consequences (Vandenabeele et al., 2023). Further, it has been recently shown that in some cell types, especially in immune cells, a protein complex PANoptosome forms, which consists of multiple key components from pyroptosis, apoptosis, and necroptosis (Chen et al., 2023). This protein complex activates multiple cell death executioners after caspase-8 activation and causes the cell death named PANoptosis. In our case, our focus was on the processes upstream of the activation of the apical caspase-8. In HeLa cells where GSDME expresses, we could detect the activation of pyroptosis marker. However, RIPK3 is not expressed in multiple cancer cells. Therefore, we propose that in different cell lines, where different key components are expressed, the exact molecules directly causing cell death downstream of caspase-8 differ.
Combined results from this study and previous observations suggest that the underlying mechanism of synergy between TNF and IFNγ in inducing cell death was different between murine BMDMs and cancer cells. Our results indicate that murine BMDMs undergo TNF/IFNγ-iNOS-CYLD-caspase-8–mediated cell death, whereas cancer cells undergo TNF/IFNγ-CYLD-caspase-8–mediated cell death. The reason why iNOS is specifically required for murine BMDM death, but not for cancer cell death, is not yet clear. We speculate that in murine BMDMs, there is an unknown negative regulator of extrinsic apoptosis, which could be removed by iNOS-mediated NO production. Furthermore, both NO and reactive oxygen species (ROS) are signaling molecules with critical and complicated roles in processes including cell death, metabolism, development, and other biological events. Although the current results ruled out the role of iNOS and, therefore, NO in TNF/IFNγ-induced cell death in cancer cells, it remains to be determined whether ROS plays a role in this cancer cell death.
CYLD, a deubiquitinase that is known to convert the membrane-bound proinflammatory signaling complex (complex I) to the apoptosis-inducing cytosolic complex (complex II) (Moquin et al., 2013), and caspase-8, the apical apoptosis-inducing caspase in complex II, are key prodeath proteins. It is thus not surprising that their elevation by IFNγ contributed to TNF-induced death. Consistent with our results, overexpression of CYLD in the lung cancer cells (A549 and H460) synergizes with TNF to activate cell death (Lin et al., 2016). Although combined knockdown of CYLD and caspase-8 dramatically protected cell death from TNF/IFNγ treatment, the existence of other IFNγ downstream factors cannot be excluded. For example, the expression of TNFR1 and FADD, which are required for TNF-initiated cell death, and XIAP and cFLIP proteins, which negatively regulate caspase-8 activation, are also marginally increased in an IFNγ dose-dependent manner (Fig. 2 I). Consistent with the observation that upregulation of the complex I deubiquitinase CYLD promotes TNF-initiated cell death, decreasing the activity of E3 ligase LUBAC complex sensitizes cells to TNF/IFNγ-induced cell death (Freeman et al., 2021).
ELAVL1 is a “multifaceted molecule” that regulates both protective and detrimental pathways by regulating different targets (Schultz et al., 2020). The inhibitor of apoptosome—prothymosin α—was identified to be upregulated by ELAVL1 and inhibit intrinsic apoptosis (Lal et al., 2005). Moreover, previous studies found ELAVL1 stabilizes Bcl-2 mRNA and increases Bcl-2 protein translation to inhibit intrinsic apoptosis (Ishimaru et al., 2009). Therefore, ELAVL1 was previously recognized to be tumorigenic and potentially acted as a target for anticancer therapy. By contrast, a recent study proposed that ELAVL1 inhibits Bcl-2 translation rather than decreasing its mRNA level (Gao et al., 2022). Further, studies also proposed that ELAVL1 modulates p53 and c-Myc expression and/or associates with PHAPI to promote intrinsic apoptosis (Schultz et al., 2020). Noteworthy, when using ABT737 plus S63845 to activate intrinsic apoptosis (Fig. S5), ELAVL1 KO cells were similarly sensitive as their parental cells, which might reflect a comprehensive effect of regulation of multiple substrates by ELAVL1 in the intrinsic apoptosis.
In contrast, seldom does research report on the targets of ELAVL1 in TNF-initiated extrinsic pathways and its regulation on caspase-8 activation. Our results, based on an unbiased genetic screen, indicated that in contrast to the previous protumorigenic role, ELAVL1 might promote antitumor immunotherapy by activating TNF/IFNγ- and other death receptors–induced cell death. Furthermore, the role of ELAVL1 in increasing caspase-8 expression and consequently caspase-8 function is striking. Caspase-8 deficiency in mice activates RIPK3- and MLKL-dependent necroptosis in intestinal epithelial cells, hematopoietic colony-forming cells, and many other cell types, leading to embryonic lethality in mice (Fritsch et al., 2019). Similarly, ELAVL1 KO mice exhibited embryonic lethality with cell death in the hematopoietic and intestinal systems (Ghosh et al., 2009). It is of great interest to test whether caspase-8 levels were decreased in the ELAVL1 KO mice, leading to the activation of RIPK3- and MLKL-dependent necroptosis in the hematopoietic, intestinal, and other systems, causing the failure of embryonic development in mice. Furthermore, any environmental or pathological cues that cause the decrease in ELAVL1 expression and tissue damage might also activate necroptosis by decreasing caspase-8 expression. Given the hub role of caspase-8 in multiple cell death and inflammation pathways and its indispensable function in inflammatory diseases (Kesavardhana et al., 2020), the different roles of ELAVL1-mediated regulation of caspase-8 in healthy or in disease states need deeper investigation. For example, TNF/TLR agonist plus IFNγ-induced cell death is representative of the inflammatory phenotype in diseases, including sepsis and SARS-CoV-2–induced pathologies (Karki et al., 2021a; Simpson et al., 2022). Therefore, inhibiting ELAVL1 might represent an additional target to intervene in such pathologies.
Materials and methods
Cell culture
All mammalian cells were cultured at 37°C with 5% CO2. HCT-116, HeLa, and 293T cells were cultured in DMEM (C11995500BT; Gibco). U937, THP-1, and B16F0 cells were cultured in RPMI-1640 (C11875500BT; Gibco); U2OS and HT-29 cells were cultured in McCoy’s 5A (16600082; Gibco). DMEM, RPMI-1640, and McCoy’s 5A were supplemented with 10% FBS (10099141C; Gibco) and 1% penicillin and streptomycin. HeLa, HCT-116, U2OS, HT-29, U937 and THP-1 were obtained from the American Type Culture Collection. 293T and B16F0 were obtained from the Cell Resource Center, Peking Union Medical College. All cell lines were free of mycoplasma contamination. Primary BMDMs were generated from bone marrow derived from C57BL/6J WT mice. The tibia and femur ends were cut, and the bones were flushed with sterile PBS. Bone marrow was collected, centrifuged at 1,000 g for 5 min, and transferred to non-tissue culture–treated 10-cm dishes (1 × 107/plate). Cells were grown for 5–6 days in DMEM supplemented with 10% FBS (GIBCO), 30% L929 conditioned media, and 1% penicillin and streptomycin. BMDMs were resuspended with cold PBS and then seeded into 24-well plates at 5 × 105 cells per well for experiments.
Treatment of cancer cells with cytokines and compounds
Different cytokine and compound treatments were used to treat various cancer cell lines. HeLa, U2OS, B16F0, and THP-1 cells were treated with 50 ng/ml TNF (10602-HNAE; Sino Biological) plus 100 ng/ml IFNγ (11725-HNAS; Sino Biological). In HCT-116 cells, 2 ng/ml TNF plus 100 ng/ml IFNγ were used, while 50 ng/ml TNF plus 12.5 ng/ml IFNγ were used in HT-29 cells. Finally, 20 ng/ml TNF plus 50 ng/ml IFNγ were used in U937 cells. The cytokine IL-1β (10139-HNAE; Sino Biological) was used at 100 ng/ml. Cancer cells were also treated with 100 ng/ml TNF plus 500 nM Smac (CAS No.: 411230-24-5; synthesized in NIBS), or 100 ng/ml TNF plus 12.5 μg/ml CHX (239763-M; Sigma-Aldrich). Intrinsic apoptosis was induced in cancer cells with 5 μM ABT-737 (197333; Sigma-Aldrich) plus 2 μM S63845 (HY-100741; MedChemExpress). The inhibition of iNOS was achieved using 1 mM L-NAME (HY-18729A; MedChemExpress) or 100 µM 1400W (HY-18731; MedChemExpress). JAKs inhibitors, such as 3 μM upadacitinib (HY-19569; MedChemExpress) and 3 μM PF-04965842 (HY-107429; MedChemExpress), were also used.
Cell viability assay
To determine cell viability, the cellular ATP level was measured using the Cell Titer-Glo Luminescent Cell Viability Assay kit (G7573; Promega), as per the manufacturer’s instructions. Luminescence intensity was measured using a microplate reader (PerkinElmer EnSpire), and data were analyzed using GraphPad Prism (GraphPad Software, Inc.). A non-linear regression model with a sigmoidal dose response was used to fit the curves.
Cell death assay by propidium iodide (PI) staining
A total of 1 × 105 cells per well were seeded on 24-well plates 1 day before the experiment. The cells were treated with the appropriate treatments for the designated time. For flow cytometry, the cells were harvested by trypsinization and resuspended in fresh PBS containing PI (1 μM, 537059; Sigma-Aldrich). After incubation for 10 min, the cells were analyzed by a flow cytometer. The data were analyzed using FlowJo Software. For microscopy imaging, the cells were washed with PBS three times and then incubated with fresh culture medium containing PI (1 μM) for 10 min. Live cell images were obtained using the EVOS M7000 image system (AMF7000; Thermo Fisher Scientific). Transmitted light and RFP channels were chosen. Images were taken at RT with a 10× objective lens.
Cell death assay by lactate dehydrogenase (LDH) release
Cell death was measured by the release of LDH from the cells into the culture medium using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (G1780; Promega), as per the manufacturer’s instructions. The absorbance was read using a microplate reader (PerkinElmer EnSpire), and the data were analyzed using GraphPad Prism (GraphPad Software, Inc.).
Western blotting
Cells were lysed either in radioimmunoprecipitation assay (RIPA)-lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% TritonX-100, 1% sodium deoxycholate, 0.1% SDS, and cOmplete EDTA-free Protease Inhibitor Cocktail [05892791001; Roche]) or directly solubilized in 1× SDS buffer, then subjected to SDS-PAGE. Cells lysed with RIPA-lysis buffer were set on ice for 30 min, and then centrifuged at 20,000 g for 10 min. The supernatants were then mixed with 4× SDS buffer and subjected to SDS-PAGE. Then the proteins were transferred to nitrocellulose membranes (66485; PALL) under constant 400 mA current for 90 min. The membranes were further blocked in tris buffered saline with tween-20 (TBST) buffer with 5% milk for 1 h and incubated with primary antibodies, which were also diluted in TBST buffer with 5% milk, at 4°C overnight. After washing with TBST buffer three times and 5 min for each time, membranes were incubated with horseradish peroxidase (HRP) conjugated anti-rabbit or anti-mouse secondary antibody for 1 h at RT. Then the membranes were washed three times with TBST buffer and incubated with chemiluminescence substrate (NEL105001EA; Perkin Elmer). The chemiluminescent signals were captured by a chemiluminescence imaging system (MiniChemi 610; SINSAGE). The quantification analysis of western blot band was conducted by ImageJ and normalized to the internal control. The following antibodies were used in western blotting: anti-iNOS Rabbit mAb (13120; Cell Signaling Technology), anti-HSP90 Rabbit mAb (13171-1-AP; Proteintech), anti–caspase-8 Rabbit mAb (4790; Cell Signaling Technology), anti–cleaved caspase-8 Rabbit mAb (9496; Cell Signaling Technology), anti–caspase-3 Rabbit mAb (9662; Cell Signaling Technology), anti–caspase-7 Rabbit mAb (9492; Cell Signaling Technology), anti-GSDME Rabbit mAb (ab215191; Abcam), anti–β-actin Rabbit mAb (4970; Cell Signaling Technology), anti-GAPDH Rabbit mAb (HRP-60004; Proteintech), anti-RIP1 Rabbit mAb (3493; Cell Signaling Technology), anti-CYLD Rabbit mAb (8462; Cell Signaling Technology), anti-FADD Rabbit mAb (14906-1-AP; Proteintech), anti-IRF1 Rabbit mAb (8478; Cell Signaling Technology), anti-JAK1 Rabbit mAb (ab138005; Abcam), anti-JAK2 Rabbit mAb (ab108596; Abcam), anti–p-JAK1 Rabbit mAb (3331; Cell Signaling Technology), anti-STAT1 Rabbit mAb (ab23400; Abcam), anti-TNFR1 Rabbit mAb (21574-1-AP; Proteintech), anti-TRADD Mouse mAb (ab280374; Abcam), anti-TRAF2 Rabbit mAb (ab126758; Abcam), anti-cIAP1 Rabbit mAb (ab108361; Abcam), anti-XIAP Rabbit mAb (2045; Cell Signaling Technology), anti-cFLIP Rabbit mAb (56343; Cell Signaling Technology), anti-Tubulin Rabbit mAb (10068-1-AP; Proteintech), anti-A20 Rabbit mAb (5630; Cell Signaling Technology), anti-OTULIN Rabbit mAb (ab211328; Abcam), anti-SPATA2 Mouse mAb (sc-515283; Santa Cruz), anti-TAK1 Rabbit mAb (5206; Cell Signaling Technology), anti-TBK1 Rabbit mAb (3504; Cell Signaling Technology), anti–caspase-9 Mouse mAb (9508; Cell Signaling Technology), anti-IFNGR1 Rabbit mAb (ab134070; Abcam), anti–p-STAT1 Rabbit mAb (ab109457; Abcam), anti-ELAVL1 Rabbit pAb (11910-1-AP; Proteintech), anti-HA-tag mAb-HRP-DirecT (M180-7; Medical and Biological Laboratories), anti-Rabbit HRP (A0545; Sigma-Aldrich), anti-Mouse HRP (A9044; Sigma-Aldrich).
Gene silencing by shRNAs
293T cells were co-expressed with shRNA-containing pLKO.1 lentiviral plasmid, pMD.2 G, and psPAX2 for using Lipofectamine 3000 (Invitrogen) 48 h, and the supernatant containing virus particles was used to infect cells. Cells were selected with puromycin 48 h after viral transduction before use in experiments. The shRNAs used were from Sigma-Aldrich shRNA clone library identified by the RNAi consortium (TRC) numbers.
RT-qPCR
Total RNA was prepared using the TRIzol Reagent (TOYOBO). After reverse transcription, Takara SYBR Premix Ex Taq kits (RR420A; Takara) were used to detect the mRNA expression levels of indicated genes. The expression levels of target genes were normalized by subtracting the corresponding β-actin/GAPDH threshold cycle (CT) value.
Genome-wide CRISPR/Cas9 screen
The screen was conducted as previously described (Yan et al., 2021). Briefly, the pooled small guide RNA (sgRNA) library was obtained commercially from Addgene (#1000000049). Lentiviral sgRNA library amplification, packaging, and viral titer measurement were performed according to the manufacturer’s instructions. To conduct the genome-wide screen, HeLa-Cas9 cells were constructed and seeded in four 15-cm dishes (5 × 106 cells/dish) with a total number of 2 × 107 cells. After 12 h, cells were infected with the lentiviral sgRNA library at an MOI of 0.3. Cells were treated with 8 μg/ml puromycin 36 h later to eliminate uninfected cells. Then, the infected cells were amplified and seeded into 12 × 15-cm dishes (1 × 107 cells/dish). When the cells reached a confluency above 90%, half of them were collected as the control group. The other six dishes (6 × 107 cells in total) were treated with TNF/IFNγ for 72 h. The surviving cells were reseeded and treated as above for another two rounds. Subsequently, the surviving cells were harvested and lysed to extract genomic DNAs, which served as templates for amplifying the sgRNA sequences. The PCR products were sequenced and quantified using a NovaSeq 6000 (Illumina) by ANNOROAD company. The sequencing data were further processed and analyzed using the MAGeCK (model-based analysis of genome-wide CRISPR/Cas9 KO) algorithm (Li et al., 2014). The forward and reverse compressed FASTQ files of reads were aligned with STAR-aligner (Dobin et al., 2013). Then the aligned FASTQ files were initially input. The command mageck count was typed in and then run to align reads onto the reference sgRNA library files. The read counts of each sgRNA, which represented the number of reads mapped to original sequence of each sgRNA, were obtained. To identify screen hits, the mageck test subcommand was performed with input of count tables generated in the previous mageck count step. The abundance changes between the control and treatment conditions were calculated after the process. P values for positive and negative selection were assigned.
Knockin mutation of ISRE motifs in the CASP8/CYLD promoter region
To knock in mutated ISRE motifs in the CASP8 promoter region, different fragments of the promoter were generated using PCR amplification with genomic DNA from HeLa cells as the template. Primers were used as follows:
Repair#1: F1: 5′-GAGCACTCAAGCTGTCTGCAGTCAG-3′,
R1: 5′-AACAGTGAGATCGATTCTCCCATGGCCTCTTCAA-3′,
F2: 5′-AGAATCGATCTCACTGTTAAAAAATCAGTCCTTC-3′,
R2: 5′-GCTTAATCTCACAGACAGCAG-3′.
Repair#2: F1: 5′-GCAGGAATCATTATAGCTACTTTATGAATG-3′,
R1: 5′-ACTCAGAGCACATGACTTACTTCCTCCAAACCTTTGCTCC-3′,
F2: 5′-GTAAGTCATGTGCTCTGAGTTTTTGGTTGATGTTTCACCT-3′,
R2: 5′-CTCTGGAAATCCCAGGCTTA-3′.
Mutated positions are underlined. The resulting PCR products were cloned into a pTOPO-T vector. To introduce the desired mutations, sgRNAs targeting CASP8 (#1 5′-TGGGAGAAGTTTTCACTTT-3′, #2 5′-TGGAGCAAAGGTTTGGAGG-3′) were cloned into the pX458 vector. The cells were transfected with the pX458-CASP8 sgRNA and repair DNA, and single cells were selected and sequenced to confirm the presence of the expected mutations.
To mutate ISRE motif at the CYLD promoter region, single-stranded oligodeoxynucleotide (ssODN) was designed to introduce the desired mutations and had 40 nt homology arms. ssODN was used as follows:
5′-GTCGGGGTTCTGGCCTTGTTAATGGCGTGTTCTGCTTTTTCATACAGTTTGCCCCTT TCTAGGGTGAGGATGGTTCTACACAGCCACCCGGAGTTCCT-3′. Mutated positions are underlined. To introduce the desired mutations, sgRNA targeting CYLD promoter (5′-CATCCTCACCCTAGAAAGG-3′) was cloned into the pX458 vector. The cells were transfected with the pX458-CYLD sgRNA (10 nM) and ssODN (10 nM) using Lipofectamine 3000. To improve the editing efficiency of homology-directed repair, 20 µM NU7026 (N1537; Sigma-Aldrich) was added during transfection to inhibit non-homologous end joining. Single cells were selected and sequenced to confirm the presence of the expected mutations.
Generation of dox-inducible protein expression cell lines
Dox-inducible protein expression cell lines were generated using lentiviral particles produced by transfecting vectors (pLVX-IRF1-EGFP, pCW-CYLD-BSD, or pLenti-CASP8-Puro), psPAX2, and pMD2.G into 293T cells with Lipofectamine 3000. The IRF1 inducible cells were selected based on GFP expression with a BD FACSAria Fusion flow cytometer. The CYLD inducible cells were selected with blasticidin (10 μg/ml). The caspase-8 inducible cells were selected with puromycin (8 μg/ml). Protein expression was induced with a concentration of 0.1 μg/ml dox.
GFP-based reporter system
ISRE motifs were predicted according to the open-access database JASPAR (http://jaspar.genereg.net). Different fragments of the CYLD promoter region containing predicted ISRE motifs were amplified by PCR using HeLa genomic DNA as template and inserted into the lentiviral reporter plasmids to control GFP expression. HeLa cells were stably expressed with the reporter systems. Cells were treated with 100 ng/ml IFN-γ for 16 h, and the GFP fluorescence intensity was quantified by flow cytometry.
CRISPR-Cas9–mediated gene KOs
Gene KO cell lines were generated using CRISPR-Cas9 technology. HeLa, HCT-116, or U2OS cells were transfected with sgRNA-pX458 plasmids using Lipofectamine 3000, and GFP-positive cells were selected with a BD FACSAria Fusion flow cytometer. Single clones were grown in 96-well plates and transferred to 24-well plates, and positive clones were selected by immunoblotting.
NO measurement
NO content was measured in cancer cells or in the supernatant of BMDMs treated with various compounds using the Griess reagent kit (G7921; Invitrogen).
Bioinformatic and RNA-seq analysis
RNA-seq analysis was performed using raw read counts from patients before and after treatment with anti–PD-1 therapy, which were downloaded from the NCBI GEO database (GSE91061; Riaz et al., 2017). The raw read counts were normalized using the rLog-transformation. Nanostring data of patients with anti–PD-1 therapy were obtained from Table S8 of a published paper (Roh et al., 2017).
ChIP assay
To perform ChIP assays, HeLa PLVX-IRF1 cells were seeded in 10-cm plates at 90% confluency. After 24 h, IRF1 expression was induced by adding dox. After 12 h, the cells were cross-linked with 1% formaldehyde for 10 min at 37°C and quenched with glycine. The samples were then washed with ice-cold PBS and scraped in PBS containing protease inhibitors. Cell pellets were incubated with 3% SDS lysis buffer (50 mM Tris-HCl, pH 8, 20 mM EDTA) for 10 min on ice and sonicated on an ultrasonic homogenizer for 3 min at 20% power on ice to shear DNA to an average fragment size of 100–1,000 bp. 30 μl of each sonicated sample was used to determine the DNA concentration and fragment size. Samples were immunoprecipitated overnight with 20 μg of anti-IRF1 antibody (8478T; Cell Signaling Technology) using 50 μl of ChIP-grade Protein A + G Magnetic Beads (26162; Thermo Fisher Scientific) at 4°C. The beads were collected and washed, and then RNase A was added and incubated for 1 h at 37°C. The samples were then treated with proteinase K overnight at 65°C to reverse the crosslinks and destroy the proteins. DNA fragments were recovered by phenol/chloroform extraction and ethanol precipitation and used as a template for subsequent qPCR. Samples were normalized to input DNA.
Dual-luciferase reporter assays
To measure promoter activity, CYLD, CASP8, and STAT1 promoter fragments were amplified using HeLa cell genomic DNA as a template and inserted into the luciferase reporter plasmid pGL6 basic vector (D2102; Beyotime) at the MluI/HindⅢ sites using Gibson cloning. HeLa PLVX-IRF1 cells were cotransfected with the constructed pGL6 reporter vector and pRL-TK-Renilla luciferase (D2760; Beyotime) using Lipofectamine 3000 (Thermo Fisher Scientific) in 6-well plates. After 6 h of transfection, 0.1 μg/ml of dox was added to induce IRF1 expression. After 12 h, the activities of firefly luciferase and Renilla luciferase in transfected cells were measured sequentially from a single sample using the luciferase reporter assay kit (E1980; Promega). Results were presented as firefly luciferase activity normalized to Renilla luciferase activity.
Protein stability measurement
To block protein translation, cells were treated with 100 ng/ml CHX for the times indicated in the figures. After treatment, cells were lysed using 1× SDS sample buffer for immunoblotting analysis.
Subcellular fractionation
Cells were collected and washed twice with cold PBS to separate cytosolic and nuclear fractions. The cell pellets were resuspended in buffer C (10 mM HEPES, pH 7.5, 5 mM MgCl2, 10 mM KCl, 1 mM DTT, protease inhibitor cocktail) and placed on ice for 15 min. 0.1% NP-40 was added and the samples were centrifuged at 4,000 g for 10 min at 4°C to obtain the cytosolic fraction. The pellets were resuspended in buffer N (20 mM HEPES, pH 7.5, 5 mM MgCl2, 0.5 M NaCl, 1 mM DTT, 0.1 mM EDTA, 20% glycerol) at 4°C for 30 min. The samples were then centrifuged at 12,000 g for 15 min at 4°C to obtain the nuclear fraction.
Polysome fractionation
To isolate polysomes, cells were treated with 100 μg/ml CHX at 37°C for 30 min and washed twice with cold PBS supplemented with 100 μg/ml CHX. Cells were then scraped and resuspended in hypotonic lysis buffer (5 mM Tris-HCl, pH 7.6, 1.5 mM KCl, 2.5 mM MgCl2, 100 μg/ml CHX, 0.2 U/μl RNase inhibitor, and 2 mM DTT). 1% Triton X-100 and 1% sodium deoxycholate were added and gently mixed with cells. The samples were then centrifuged at 12,000 g for 10 min at 4°C. The supernatants were layered onto a sucrose gradient and centrifuged in a Beckman SW-41Ti rotor at 36,000 rpm for 3 h at 4°C. RNA was isolated from the collected samples for qPCR analysis.
RNA IP
To perform RNA IP, cells were seeded in 10-cm plates and collected when they reached 90% confluency. The cells were then suspended in polysome lysis buffer (10 mM HEPES, pH 7, 100 mM KCl, 5 mM MgCl2, 0.5% NP-40, 1 mM DTT, 200 units/ml RNase OUT [10777019; Invitrogen], and protease inhibitor cocktail). The suspension was stored at −80°C to enhance cell lysis. After thawing, the lysate was centrifuged at 12,000 g for 10 min at 4°C and a 10 μl aliquot of the cell lysate supernatant was collected as input. Anti-Flag M2 agarose beads (A2220; Sigma-Aldrich) were used to precipitate the RNA, and the beads were washed five times with NT-2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40). RNA was isolated from the samples for subsequent qPCR analysis.
Ethics declarations
All animal experiments were conducted following protocols approved by the Ethics Committee of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences (CAS), Beijing, China.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.0 software. The results are presented as mean ± SD from three independent experiments. All the statistical details can be found in the figure legends.
Online supplemental material
Fig. S1 serves as a complement to Fig. 1. It shows that TNF/IFNγ synergistically induces cell death in diverse human and murine cancer cells. It also demonstrates that Nos2 KO has no effect on TNF/IFNγ-induced cell death in murine EMT6 cells. Fig. S2 shows that IFNγ induces increased expression of Cyld, Casp8, and Nos2 in murine cells and complements Fig. 2. Fig. S3 shows the correlation between TNF/IFNγ/CYLD/CASP8 expression and immune checkpoint blockade (ICB) therapy efficiency based on the published database of two cohorts of melanoma patients. Fig. S4 demonstrates that Cyld knockdown significantly inhibits TNF/IFNγ-induced cell death and complements Fig. 3. Fig. S5 shows that ELAVL1 KO significantly inhibits TNF/Smac-induced cell death and complements Fig. 7. Table S1 shows siRNA, qPCR, and CRISPR/Cas9 sgRNA primer sequences, and shRNA TRC numbers used in this study.
Data availability
All data generated in this study are included in the manuscript and its supplementary information files. All data are available from the corresponding authors upon reasonable request.
Acknowledgments
Y. Ai and B. Yan express thanks to Dr. Xiaodong Wang (National Institute of Biological Sciences [NIBS]) and Dr. Zhiyuan Zhang (NIBS) for their continuous encouragement and support. We thank Dr. Xuejun Jiang (Memorial Sloan Kettering Cancer Center), Dr. Bing Zhu (Institute of Biophysics, CAS), Dr. Liming Sun (Center for Excellence in Molecular Cell Science, CAS), Dr. Ting Han (NIBS), and Dr. Xing Liu (Institut Pasteur of Shanghai, CAS) for helpful suggestions. We thank Xiaoyan Jiang (worked at Institute of Genetics and Developmental Biology from July to December 2021) for her contribution to the generation of KO cell lines and cell viability assays. We thank Dr. Nan Zhao, Dr. Wenbin Zhang, Dr. Zhangcheng Ding, and Dr. Bo Cui from NIBS; and members of Dr. Feng Shao’s lab (NIBS) and Dr. Ting Han’s lab (NIBS) for their assistance with protocols, reagents, and discussions. We thank Dr. Keqiong Ye and Dr. Jia Qi from the Institute of Biophysics for their assistance with polysome fractionation.
This work was supported by the National Key R&D Program of China (2022YFA1304500 to Y. Ai), the National Natural Science Foundation of China (32321004 to Y. Ai and 22307139 to B. Yan), the State Key Laboratory of Molecular Developmental Biology and CAS Pioneer Hundred Talents Program startup funding to Y. Ai, Tianjin Medical University General Hospital startup funding to B. Yan, and the Special Research Assistant Program of the Chinese Academy of Sciences to Z. Deng. The funders had no role in study design, data collection, and/or interpretation.
Author contributions: Y. Ai and B. Yan conceived and supervised the study and wrote the manuscript. B. Deng, J. Wang, T. Yang, and Z. Deng performed most of the experiments. J. Yuan, L. Fang, and C. Liang helped analyze the patient’s data from the NCBI GEO database. F. Chen, B. Zhang, and Z. Zhou helped with cell culture and cell death analysis. Y. Ai, B. Yan, B. Deng, J. Wang, T. Yang, Z. Deng, and F. Chen analyzed and interpreted the results.

