


CLPB is a mitochondrial intermembrane space AAA+ domain–containing disaggregase. CLPB mutations are associated with 3-methylglutaconic aciduria and neutropenia; however, the molecular mechanism underscoring disease and the contribution of CLPB substrates to disease pathology remains unknown. Interactions between CLPB and mitochondrial quality control (QC) factors, including PARL and OPA1, have been reported, hinting at dysregulation of organelle QC in disease. Utilizing proteomic and biochemical approaches, we show a stress-specific aggregation phenotype in a CLPB-null environment and define the CLPB substrate profile. We illustrate an interplay between intermembrane space proteins including CLPB, HAX1, HTRA2, and the inner membrane quality control proteins (STOML2, PARL, YME1L1; SPY complex), with CLPB deficiency impeding SPY complex function by virtue of protein aggregation in the intermembrane space. We conclude that there is an interdependency of mitochondrial QC components at the intermembrane space/inner membrane interface, and perturbations to this network may underscore CLPB disease pathology.
Introduction
Caseinolytic peptidase B, or CLPB, is a mitochondrial AAA+ domain–containing intermembrane space (IMS) disaggregase (Cupo and Shorter, 2020; Thevarajan et al., 2020) that is associated with the onset of 3-methylglutaconic aciduria type 7 with both autosomal dominant (MGCA7A, Mendelian Inheritance in Man [MIM] #619835) and recessive (MGCA7B, MIM #616271) presentations (Wortmann et al., 2015, 2021). Pathogenic CLPB mutations are also linked to a form of autosomal dominant severe congenital neutropenia (SCN; SCN9, MIM #619813). The molecular details underscoring disease pathology and if or how CLPB substrates contribute to pathology remain unclear.
Unlike the Hsp100/Clp disaggregase family of yeast and bacteria, human CLPB lacks an N-terminal domain and middle domain and has exchanged a nucleotide-binding domain (NBD) for a novel ankyrin repeat domain (ANK; Cupo and Shorter, 2020; Li et al., 2006). Human CLPB engages substrates as a homo-hexamer, which prompts rapid assembly into a dodecameric species via end-to-end ANK domain stacking (Cupo et al., 2022; Spaulding et al., 2022). This alternative domain architecture, quaternary structure, and intraorganellar localization of human CLPB compared with bacterial and yeast orthologs (Abrahão et al., 2017; Cupo et al., 2022; Rizo et al., 2019) suggests the human disaggregase has assumed novel functions. Indeed, interaction between CLPB and HAX1 (Chen et al., 2019; Fan et al., 2022; Wakula et al., 2020) is implicated in intracellular modulation of mitochondrial protein synthesis and neutrophil granulocyte differentiation (Fan et al., 2022). HAX1 is a highly disordered protein (Suzuki et al., 1997) with weak sequence similarity to Bcl-2 domain-containing proteins (Fadeel and Grzybowska, 2009). Pathogenic mutation of HAX1 causes autosomal recessive SCN or Kostmann Syndrome (SCN3, MIM #610738; Klein et al., 2007), and granulopoiesis is frequently arrested at the same stage in both MGCA7B and Kostmann Syndrome patients (Roques et al., 2014; Wortmann et al., 2015), hinting toward common features of mitochondrial dysfunction in both diseases.
Both CLPB and HAX1 functionally interact with the inner membrane (IM) SPY complex (STOML2, PARL, YME1L1) via the PARL and YME1L1 proteases, respectively (MacVicar et al., 2019; Wai et al., 2016). PARL cleavage liberates CLPB precursor from the IM following protein import into the organelle (Cupo and Shorter, 2020; Saita et al., 2017). However, stable interactions between CLPB and wild-type PARL (Saita et al., 2017; Wai et al., 2016), or proteolytically inactive PARL (Saita et al., 2017), suggest this interaction may have a function beyond substrate processing. CLPB–HAX1 cooperation with components of the mitochondrial quality control (QC) network, including SPY or prohibitin complexes, has been suggested (Wai et al., 2016; Wortmann et al., 2021) but unexplored at a functional level.
We interrogated the CLPB protein interaction landscape to gain insight into the CLPB substrate profile and pathomechanism(s) associated with CLPB dysregulation. We find stress-specific aggregation of mitochondrial proteins in CLPB-deficient cells align with phenotypic observations of CLPB-deficient patient cells. ATPase-dead and substrate-trap mutants reveal CLPB interacting proteins, which include IMS proteins, HAX1 and HTRA2, as well as the IM SPY complex. Our data suggest the absence of CLPB compromises the solubility of proteins in the IMS and at outer membrane (OM)/IM boundaries, impacting SPY complex integrity and function. We conclude that aberrant CLPB activity broadly impedes proteolytic operations in mitochondria, compromising mitochondrial QC and health, providing a molecular insight into the mitochondrial disease linked to CLPB dysfunction.
Results
Loss of CLPB impacts mitochondrial health
To investigate the biological consequences of CLPB deficiency, we generated a HEK CLPB knockout (KO) cell line (CLPBKO; Flp-In T-REx 293) using CRISPR-Cas9 genome editing (Fig. S1 A). Label-free quantitative (LFQ) mass spectrometry (MS) on isolated mitochondria from wild-type control and CLPBKO cells revealed HAX1, in addition to the pro-apoptotic factors ENDOG, PABPN1, and BAX, was significantly increased in abundance (Fig. 1 A and Table S1). The IM OMA1 protease was significantly depleted in CLPBKO cells, while various complex I (CI) and complex IV (CIV) subunits and assembly factors were reduced in abundance. These trends align with published proteomics from MGCA7B patient lymphoblasts (Tucker et al., 2022), validating this CLPBKO cell line as an accurate model of CLPB deficiency. Assessment of oxidative phosphorylation (OXPHOS) complexes using blue native (BN)-PAGE in control and CLPBKO mitochondria solubilized in digitonin showed CIV (assembly or stability) was mildly impacted in the absence of CLPB (Fig. 1 B, lanes 5 and 6). Consistent with this observation, CLPBKO cells have significantly lower respiration (both basal and maximal; Fig. 1 C); a higher extracellular acidification rate (ECAR) to oxygen consumption rate (OCR) ratio (Fig. 1 D); and accumulate reactive oxygen species (ROS) under both basal conditions and when stimulated by menadione (Fig. 1 E).
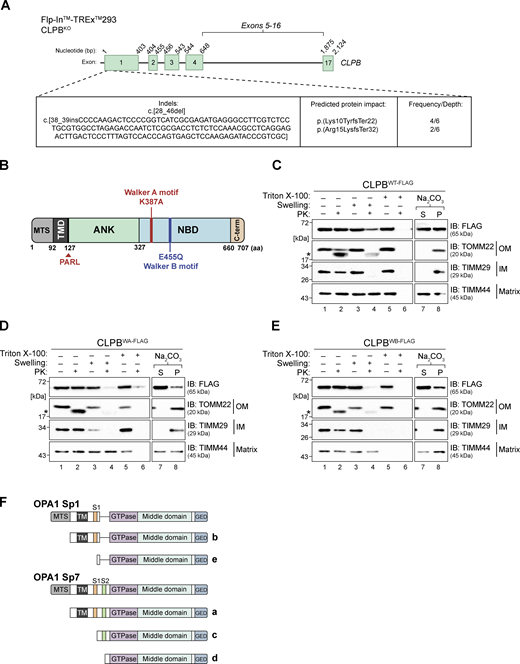
Mutation of Walker A or B motifs may impact CLPB localization. (A) Schematic representation of indels present in the CLPBKO HEK FlpIn-TREx cell line (Clone #4), introduced using CRISPR-Cas9 genome editing, according to CLPB transcript variant 1 (NM_030813.6). Exon 1 is shared by all CLPB variants. (B) Schematic representation of CLPB domain architecture, with relative positions of Walker A (K387A) and Walker B (E455Q) mutant loci indicated within the NBD. MTS, mitochondrial targeting signal; TMD, transmembrane domain. (C–E) Mitochondria were isolated from (C) CLPBWT-FLAG, (D) CLPBWA-FLAG, and (E) CLPBWB-FLAG stable cell lines and treated for sub-fractionation of the mitochondrial compartments (left), and carbonate extraction (right). Samples were run on SDS-PAGE and analyzed via immunoblotting (IB). * = protease protected protein. S = supernatant, P = pellet. (F) Schematic depicting processing of L-OPA1 splice variants 1 (b) and 7 (a) (Sp1/7). OMA1 cleavage of L-OPA1 a or b at S1 yields S-OPA1 c and e splice variants, respectively. Subsequent processing by YME1L1 at S2 of c-form S-OPA1 yields d-form S-OPA1. MTS = mitochondrial targeting sequence. TM = transmembrane domain. GED = GTPase effector domain. Figure adapted from Baker et al. (2014). Source data are available for this figure: SourceData FS1.
Mutation of Walker A or B motifs may impact CLPB localization. (A) Schematic representation of indels present in the CLPBKO HEK FlpIn-TREx cell line (Clone #4), introduced using CRISPR-Cas9 genome editing, according to CLPB transcript variant 1 (NM_030813.6). Exon 1 is shared by all CLPB variants. (B) Schematic representation of CLPB domain architecture, with relative positions of Walker A (K387A) and Walker B (E455Q) mutant loci indicated within the NBD. MTS, mitochondrial targeting signal; TMD, transmembrane domain. (C–E) Mitochondria were isolated from (C) CLPBWT-FLAG, (D) CLPBWA-FLAG, and (E) CLPBWB-FLAG stable cell lines and treated for sub-fractionation of the mitochondrial compartments (left), and carbonate extraction (right). Samples were run on SDS-PAGE and analyzed via immunoblotting (IB). * = protease protected protein. S = supernatant, P = pellet. (F) Schematic depicting processing of L-OPA1 splice variants 1 (b) and 7 (a) (Sp1/7). OMA1 cleavage of L-OPA1 a or b at S1 yields S-OPA1 c and e splice variants, respectively. Subsequent processing by YME1L1 at S2 of c-form S-OPA1 yields d-form S-OPA1. MTS = mitochondrial targeting sequence. TM = transmembrane domain. GED = GTPase effector domain. Figure adapted from Baker et al. (2014). Source data are available for this figure: SourceData FS1.
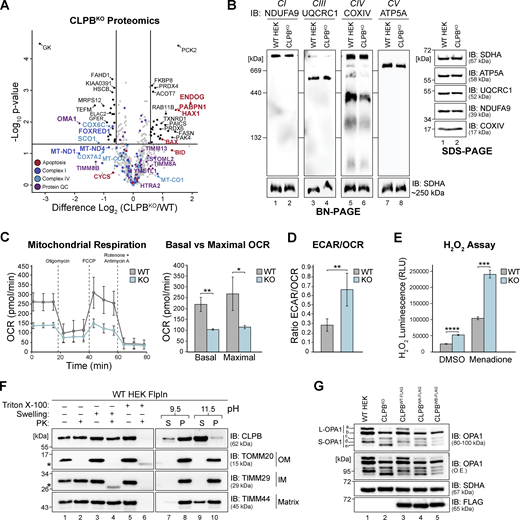
Loss of CLPB influences mitochondrial health. (A) Mitochondria were isolated from control and CLPBKO cells and subjected to LFQ MS. Data are presented as a volcano plot depicting the level of proteins in CLPBKO relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (B) Mitochondria were isolated from control and CLPBKO cells and prepared for BN-PAGE (left) and SDS-PAGE (right). For BN-PAGE analysis, mitochondria were solubilized in digitonin-containing buffer and separated on a 4–16% acrylamide BN-PAGE gel, followed by immunoblotting (IB) with the indicated antibodies. For SDS-PAGE, mitochondrial pellets were resuspended in SDS-containing loading buffer and run on a 10–16% acrylamide tris-tricine gel followed by immunoblotting with the indicated antibodies. (C) OCRs were quantified in control and CLPBKO cells using a Seahorse analyzer. Left: Oligomycin, FCCP, and antimycin A/rotenone were added at the indicated time points to measure basal, maximal, and non-mitochondrial respiration rates. Right: Calculated basal and maximal OCR rates. Data averaged over three independent experiments. Error bars represent mean ± SD (n = 3), two-sample t test; *P < 0.05, **P < 0.01. (D) ECAR (mpH/min)/OCR (pmol/min) ratio calculations as obtained in C. Error bars represent mean ± SD (n = 3), two-sample t test; **P < 0.01. (E) Measurement of H2O2 ROS in control and CLPBKO cell lines after a 2 h vehicle (DMSO) or 10 µM menadione pre-treatment. Error bars represent mean ± SD (n = 3 for both vehicle and menadione treated). Two-sample t test; ***P < 0.001. ****P < 0.0001. (F) Mitochondrial sub-fractionation (lanes 1–6) and carbonate extraction (lanes 7–10) on mitochondria isolated from HEK wild-type cells to examine endogenous CLPB localization. Samples were run on a 10–16% acrylamide tris-tricine gel and immunoblotted with the indicated antibodies. S = supernatant, P = pellet. TOMM20 = OM localized control, TIMM29 = IM localized control, TIMM44 = matrix localized control, * = protease protected protein. (G) OPA1 processing in control, CLPBKO, and induced CLPBWT-FLAG, CLPBWA-FLAG, and CLPBWB-FLAG cell lines. Isolated mitochondria were run on a 8–10% acrylamide tris-tricine gel and and immunoblotted with antibodies as indicated. Stable cell lines were induced with 0.1 µg/ml tetracycline for 14 h prior to mitochondrial isolation. Source data are available for this figure: SourceData F1.
Loss of CLPB influences mitochondrial health. (A) Mitochondria were isolated from control and CLPBKO cells and subjected to LFQ MS. Data are presented as a volcano plot depicting the level of proteins in CLPBKO relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (B) Mitochondria were isolated from control and CLPBKO cells and prepared for BN-PAGE (left) and SDS-PAGE (right). For BN-PAGE analysis, mitochondria were solubilized in digitonin-containing buffer and separated on a 4–16% acrylamide BN-PAGE gel, followed by immunoblotting (IB) with the indicated antibodies. For SDS-PAGE, mitochondrial pellets were resuspended in SDS-containing loading buffer and run on a 10–16% acrylamide tris-tricine gel followed by immunoblotting with the indicated antibodies. (C) OCRs were quantified in control and CLPBKO cells using a Seahorse analyzer. Left: Oligomycin, FCCP, and antimycin A/rotenone were added at the indicated time points to measure basal, maximal, and non-mitochondrial respiration rates. Right: Calculated basal and maximal OCR rates. Data averaged over three independent experiments. Error bars represent mean ± SD (n = 3), two-sample t test; *P < 0.05, **P < 0.01. (D) ECAR (mpH/min)/OCR (pmol/min) ratio calculations as obtained in C. Error bars represent mean ± SD (n = 3), two-sample t test; **P < 0.01. (E) Measurement of H2O2 ROS in control and CLPBKO cell lines after a 2 h vehicle (DMSO) or 10 µM menadione pre-treatment. Error bars represent mean ± SD (n = 3 for both vehicle and menadione treated). Two-sample t test; ***P < 0.001. ****P < 0.0001. (F) Mitochondrial sub-fractionation (lanes 1–6) and carbonate extraction (lanes 7–10) on mitochondria isolated from HEK wild-type cells to examine endogenous CLPB localization. Samples were run on a 10–16% acrylamide tris-tricine gel and immunoblotted with the indicated antibodies. S = supernatant, P = pellet. TOMM20 = OM localized control, TIMM29 = IM localized control, TIMM44 = matrix localized control, * = protease protected protein. (G) OPA1 processing in control, CLPBKO, and induced CLPBWT-FLAG, CLPBWA-FLAG, and CLPBWB-FLAG cell lines. Isolated mitochondria were run on a 8–10% acrylamide tris-tricine gel and and immunoblotted with antibodies as indicated. Stable cell lines were induced with 0.1 µg/ml tetracycline for 14 h prior to mitochondrial isolation. Source data are available for this figure: SourceData F1.
CLPB is an established IMS resident (Antonicka et al., 2020; Fan et al., 2022; Thevarajan et al., 2020). Subfractionation of wild-type mitochondria followed by western blotting using CLPB antibodies (raised against the N-terminal 350 amino acids) showed resistance of endogenous CLPB to Proteinase K (PK) in mitoplasts (Fig. 1 F, lanes 3 and 4). This was unlike TIMM29, an IM protein that has a PK-accessible domain in the IMS (Fig. 1 F), and suggests that CLPB can adopt a conformation or exist in a complex that protects it from PK. Carbonate extraction showed that endogenous CLPB is present in the pellet (membrane) fraction at pH 9.5, but is released from the membrane at higher alkalinity (Fig. 1 F, lanes 7–10), implying peripheral association with the IM in agreeance with existing literature (Fan et al., 2022; Thevarajan et al., 2020). Next, we ascertained the behavior of endogenous and stably expressed CLPB in the cell models created for use in this study. This included stable cell lines expressing C-terminal 3×FLAG: (i) wild-type CLPB (CLPBWT-FLAG); (ii) Walker A K387A mutant (CLPBWA-FLAG); or (iii) Walker B E455Q mutant (CLPBWB-FLAG; Fig. S1 B). The K387A Walker A mutation prevents ATP binding and CLPB oligomerization, while the E455Q Walker B motif mutation prevents ATP hydrolysis and substrate release, creating a “substrate trap” (Cupo and Shorter, 2020). The profile of the stable cell line expressing CLPBWT-FLAG was consistent with that of endogenous CLPB (Fig. S1 C). Conversely, non-functional CLPBWA-FLAG and CLPBWB-FLAG mutants were accessible to PK degradation in mitoplasts (Fig. S1, D and E, lanes 3 and 4), suggesting ATP hydrolysis may facilitate alternate conformation or interactions of the protein, lack of which render it accessible to external protease.
The significant reduction of OMA1 protein level in CLPBKO cells (Fig. 1 A) prompted us to monitor OPA1 processing, as OMA1 and YME1L1 proteases cooperate to cleave long-form OPA1 (L-OPA1; denoted as a and b) into short-form OPA1 (S-OPA1; denoted as, c, d, and e; Anand et al., 2014; Fig. S1 F). Indeed, excessive OPA1 processing was apparent in CLPBKO cells (Fig. 1 G, compare lanes 1 and 2), with S-OPA1 forms c and e accumulating in the absence of CLPB. This is consistent with what is observed in CLPB short guide RNA (sgRNA) knockdown in THP-1 cells (Chen et al., 2019) and with stress-induced OMA1 proteolytic processing of OPA1 in mouse embryonic fibroblasts (Baker et al., 2014). The OPA1 processing phenotype was reverted upon re-expression of CLPBWT-FLAG (Fig. 1 G, lane 3) but not mutant CLPBWA-FLAG or CLPBWB-FLAG, implying functional CLPB is required for efficient OPA1 processing. These results suggest that loss of CLPB has an indirect impact on OXPHOS function, leading to excessive ROS accumulation, damaging electron transport chain components and inducing localized stress within the IMS as indicated by changes to OPA1 processing and depletion of OMA1 abundance.
Mitochondrial protein aggregation profile in the CLPB-null environment
To interrogate the function and substrate pool of CLPB in mitochondria, we developed an experimental pipeline to monitor the solubility of mitochondrial proteins based on published approaches (Fig. 2 A; Cupo and Shorter, 2020; Mahat et al., 2016; Wilkening et al., 2018). Following solubilization in TX-100, mitochondrial samples were partitioned into a pellet (insoluble) and supernatant (soluble) fraction for analysis via SDS-PAGE (Fig. S2, A and B) or LFQ MS (Fig. 2, B and C). SDS-PAGE and immunoblotting shows depletion of HTRA2 from the soluble fraction, and moderate accumulation of OPA1, YME1L1, and HTRA2 in the pellet fraction of CLPBKO mitochondria (Fig. S2, A and B). Mitochondria isolated from CLPBKO cells under endogenous conditions (Fig. 2 B) or heat stress (Fig. 2 C) were fractionated and prepared for proteomic analysis. Protein abundance in pellet and supernatant fractions was normalized against CLPBKO mitochondrial proteomics (Fig. 1 A) to account for the steady-state levels of proteins. Proteins with >1.5-fold increased abundance in the pellet (after normalization) were considered “insoluble,” or more prone to aggregation in CLPBKO mitochondria (Fig. 2, B and C; and Tables S2 and S3). Under non-stress conditions, multiple proteins accumulated in the pellet fraction, implying an impact on their solubility (Fig. 2 B; and Tables S2 and S3). Agreeing with existing literature (Cupo and Shorter, 2020), HAX1 and OPA1 became predominantly insoluble in the absence of CLPB, and HAX1 was strongly depleted from the corresponding supernatant fraction (Fig. S2 C). SAMM50, TOMM40, AFG3L2, NNT, MT-CO2, and IMMT are newly identified aggregation-prone proteins in the absence of CLPB. Insolubility of mitochondrial DNA encoded MT-CO2 aligns with the observed OXPHOS defect in CLPBKO cells (Fig. 1, B and C). The influence of external heat stress on mitochondrial protein solubility was assessed in control and CLPBKO cells by applying heat shock (HS) in cell culture for 2 h prior to mitochondrial isolation. HS resulted in an additional 11 proteins becoming prone to aggregation in the absence of CLPB (Fig. 2 C; and Tables S2 and S3). This included mitochondrial calcium uniporter regulator MICU2, import components TIMM50 and TIMM13, metabolite carriers SLC25A13 and SLC25A10, and the serine protease HTRA2. Notably, HAX1, HTRA2, and PARL protease were all significantly depleted from the corresponding supernatant fraction (Fig. S2 D).

CLPB deficiency triggers excessive protein aggregation. (A) Schematic representation of the aggregation assay pipeline utilized in this study. (B and C) Protein insolubility in CLPBKO without (B) and with (C) heat shock (+HS). Isolated mitochondria from control and CLPBKO cells (untreated or following a 2 h HS at 42°C) were prepared for LFQ MS as outlined in A. Data is presented as a volcano plot depicting the level of proteins in the CLPBKO pellets relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. To correct for anticipated differences in protein abundance within CLPBKO, data were normalized against CLPBKO proteomics (Fig. 1 A and Table S3). Proteins labeled in red had a fold difference >1.5 following normalization and are regarded as most strongly insoluble. Proteins labeled in dark red in C are also identified as insoluble in B. (D) Fold change comparison between proteins identified in the pellet fraction of: CLPBKO (−HS) (B), CLPBWT-FLAG (−HS), CLPBKO (+HS) (C), and CLPBWT-FLAG (+HS) following normalization to CLPBKO proteomics (Fig. 1 A and Table S3). The CLPBWT-FLAG stable cell line was induced with 0.1 µg/ml tetracycline for 12 h prior to immediate HS, or 14 h without HS. Values in bold are “strongly insoluble,” with fold difference >1.5 following normalization to CLPBKO proteomics. ND = not detected. (E) Immunofluorescence imaging reveals discrete HTRA2 foci in HeLa CLPBKO cells. HeLa control, CLPBKO, and CLPBKO cells stably expressing CLPBWT-FLAG or CLPBWB-FLAG were stained with MitoTracker Deep Red (magenta) and subjected to immunofluorescence analysis with antibodies directed against HTRA2 (green) and FLAG (cyan). Scale bar is equivalent to 20 or 5 µM (magnified) as indicated. (F) Monitoring HTRA2 foci clearance following CLPBWT-FLAG re-expression in HeLa CLPBKO background. CLPBWT-FLAG was transiently expressed in control and CLPBKO HeLa for indicated time points. Cells were stained with MitoTracker Deep Red (magenta) and subjected to immunofluorescence analysis with antibodies directed against HTRA2 (green) and FLAG (cyan). Scale bar is equivalent to 5 µM.
CLPB deficiency triggers excessive protein aggregation. (A) Schematic representation of the aggregation assay pipeline utilized in this study. (B and C) Protein insolubility in CLPBKO without (B) and with (C) heat shock (+HS). Isolated mitochondria from control and CLPBKO cells (untreated or following a 2 h HS at 42°C) were prepared for LFQ MS as outlined in A. Data is presented as a volcano plot depicting the level of proteins in the CLPBKO pellets relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. To correct for anticipated differences in protein abundance within CLPBKO, data were normalized against CLPBKO proteomics (Fig. 1 A and Table S3). Proteins labeled in red had a fold difference >1.5 following normalization and are regarded as most strongly insoluble. Proteins labeled in dark red in C are also identified as insoluble in B. (D) Fold change comparison between proteins identified in the pellet fraction of: CLPBKO (−HS) (B), CLPBWT-FLAG (−HS), CLPBKO (+HS) (C), and CLPBWT-FLAG (+HS) following normalization to CLPBKO proteomics (Fig. 1 A and Table S3). The CLPBWT-FLAG stable cell line was induced with 0.1 µg/ml tetracycline for 12 h prior to immediate HS, or 14 h without HS. Values in bold are “strongly insoluble,” with fold difference >1.5 following normalization to CLPBKO proteomics. ND = not detected. (E) Immunofluorescence imaging reveals discrete HTRA2 foci in HeLa CLPBKO cells. HeLa control, CLPBKO, and CLPBKO cells stably expressing CLPBWT-FLAG or CLPBWB-FLAG were stained with MitoTracker Deep Red (magenta) and subjected to immunofluorescence analysis with antibodies directed against HTRA2 (green) and FLAG (cyan). Scale bar is equivalent to 20 or 5 µM (magnified) as indicated. (F) Monitoring HTRA2 foci clearance following CLPBWT-FLAG re-expression in HeLa CLPBKO background. CLPBWT-FLAG was transiently expressed in control and CLPBKO HeLa for indicated time points. Cells were stained with MitoTracker Deep Red (magenta) and subjected to immunofluorescence analysis with antibodies directed against HTRA2 (green) and FLAG (cyan). Scale bar is equivalent to 5 µM.

Aggregation-prone proteins are lost from corresponding CLPBKO soluble fractions. (A) Mitochondria (50 µg pellets) isolated from control and CLPBKO were solubilized in Triton X-100 containing buffer (in duplicate) and incubated prior to fractionation. Samples were run on a 10–16% tris-tricine gel followed by immunoblotting (IB) with indicated antibodies. S = supernatant, P = pellet. (B) Quantification of protein abundance in the respective soluble fraction as in A. Calculated as mean percentage ± SD of total for each protein, normalized to the level of succinate dehydrogenase (complex II) subunit A (SDHA). Significance determined by Student’s two-sample t test (n = 3). ****P < 0.0001, NS = not significant, P > 0.05. (C and D) Supernatant fractions corresponding to (C) basal and (D) + HS aggregation assay pellet data from Fig. 2, B and C, respectively. Data are presented as a volcano plot depicting the level of proteins in the CLPBKO supernatants relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. To correct for anticipated differences in protein abundance within the CLPBKO cell line, data were normalized against CLPBKO proteomics (Fig. 1 A and Table S3). Proteins labeled in red had a fold difference >1.5 following normalization and are regarded as most depleted from the soluble fraction. Source data are available for this figure: SourceData FS2.
Aggregation-prone proteins are lost from corresponding CLPBKO soluble fractions. (A) Mitochondria (50 µg pellets) isolated from control and CLPBKO were solubilized in Triton X-100 containing buffer (in duplicate) and incubated prior to fractionation. Samples were run on a 10–16% tris-tricine gel followed by immunoblotting (IB) with indicated antibodies. S = supernatant, P = pellet. (B) Quantification of protein abundance in the respective soluble fraction as in A. Calculated as mean percentage ± SD of total for each protein, normalized to the level of succinate dehydrogenase (complex II) subunit A (SDHA). Significance determined by Student’s two-sample t test (n = 3). ****P < 0.0001, NS = not significant, P > 0.05. (C and D) Supernatant fractions corresponding to (C) basal and (D) + HS aggregation assay pellet data from Fig. 2, B and C, respectively. Data are presented as a volcano plot depicting the level of proteins in the CLPBKO supernatants relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. To correct for anticipated differences in protein abundance within the CLPBKO cell line, data were normalized against CLPBKO proteomics (Fig. 1 A and Table S3). Proteins labeled in red had a fold difference >1.5 following normalization and are regarded as most depleted from the soluble fraction. Source data are available for this figure: SourceData FS2.
Re-expression of CLPBWT-FLAG in CLPBKO cells cleared endogenous aggregation, as 8 of the 10 previously insoluble proteins (Fig. 2 B) were absent from the pellet fraction (Fig. 2 D). HAX1 remained strongly insoluble despite CLPBWT-FLAG re-expression, though markedly reduced from its initial fold change in CLPBKO cells (Fig. 2 D and Table S3). Aggregate accumulation following HS in CLPBWT-FLAG complemented cells moderately protected against aggregation (Fig. 2 D and Table S3), with SLC25A10, SLC25A13, STOML2 TIMM13, TIMM50, and YME1L1 returning to basal levels. MT-CO2, NDUFA13, OPA1, and HAX1 persisted in the insoluble fraction upon HS (Fig. 2 D and Table S3), suggesting these proteins may be highly susceptible to aggregation or take longer to resolve. Strong insolubility of SPY complex components (STOML2, PARL, YME1L1) in the absence of CLPB could compromise proteolytic clearance in the IMS and IM, further exacerbating aggregate load in mitochondria. Proteins identified as significantly more insoluble in CLPBKO cells were either IMS residents or flank the OM/IM, supporting the IMS localization and function of CLPB. Consequently, CLPB functional dysregulation may indirectly impact broader mitochondrial processes including OXPHOS, protein import, and apoptotic induction, as observed in the mitochondrial proteomic dataset (Table S1).
We were drawn to the strong insolubility of both HAX1 and HTRA2 in CLPBKO cells (Fig. 2 D), as autosomal recessive mutations in HTRA2 and HAX1 cause 3-methylglutaconic aciduria type 8 (MIM #617248; Mandel et al., 2016; Oláhová et al., 2017) and Kostmann Syndrome (SCN3, MIM #610738; Fadeel and Grzybowska, 2009; Klein et al., 2007), respectively, which share phenotypic similarities with CLPB-deficiency (MGCA7A/B). HAX1 has been observed to form discrete puncta in CLPB-deficient HeLa cells (Fan et al., 2022). Similarly, immunofluorescence imaging of HTRA2 localization in control and CLPBKO HeLa cells revealed HTRA2 puncta in the absence of CLPB (Fig. 2 E). Re-expression of single FLAG-tagged CLPBWT-FLAG cleared these puncta and HTRA2 distribution returned to homogeneous staining of the mitochondrial network, while re-expression of CLPBWB-FLAG mutant could not effectively mitigate aggregate accumulation to the same extent (Fig. 2 E). The rate of HTRA2 aggregate clearance with CLPBWT-FLAG re-expression was rapid, occurring within 12 h (Fig. 2 F). Overall, these findings suggest CLPB disaggregase activity maintains the solubility of a subset of proteins located within the IMS or OM/IM, in particular the IMS residents HAX1 and HTRA2 and members of the IM proteolytic hub SPY.
Surveying the CLPB interactome and substrate profile
The strong insolubility of IMS and OM/IM resident proteins in the absence of CLPB prompted us to explore the CLPB interaction network and identify potential CLPB substrates. Isolated interactions between CLPB and HAX1, HTRA2, OPA1, and PARL have been reported (Botham et al., 2019; Chen et al., 2019; Saita et al., 2017; Wakula et al., 2020); however, complete coverage of the CLPB interactome has not yet been captured. We performed native affinity enrichment immunoprecipitation (IP) MS using cells stably expressing CLPBWT-FLAG or ATPase-dead CLPBWA-FLAG and substrate trap CLPBWB-FLAG as disaggregase-substrate engagement is typically transient (Arhar et al., 2021). CLPBWT-FLAG IP revealed a single predominant interactor, HAX1 (Fig. 3 A and Table S4), supporting previous findings (Fan et al., 2022; Wakula et al., 2020). Comparatively, CLPBWA-FLAG (Fig. 3 B) and CLPBWB-FLAG (Fig. 3 C) IP enriched common interactors of protein QC and apoptosis proteins, including STOML2, YME1L, PARL, and ENDOG. Significantly enriched across either data set were protein biogenesis factors, TIMM8B, TIMM13, and AIFM1; the apoptosis-related CHCHD2; and OXPHOS assembly factors TTC19, SCO2, and COA7 (Fig. 3 D). These data suggest that CLPB has a stable interaction with HAX1, in addition to other transient interactions captured by substrate-trap CLPB that function in QC, apoptosis, and OXPHOS.

Defining the CLPB interactome and substrate profile. (A–C) Mitochondria were isolated from control, (A) CLPBWT-FLAG, (B) CLPBWA-FLAG, or (C) CLPBWB-FLAG stable cell lines following 14 h induction with 0.1 µg/ml tetracycline. Samples were processed for FLAG IP and eluates were prepared for LFQ MS. Data are presented as a volcano plot depicting the level of enrichment relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and to the right of the vertical line (eight-fold absolute change) are regarded as significantly enriched. Functional annotations correspond to MitoCarta 3.0 categorization. (D) Table containing all significantly enriched proteins across CLPBWT-FLAG (WT), CLPBWA-FLAG (WA), and CLPBWB-FLAG (WB) IP datasets with corresponding fold change across each experiment. Mitochondrial compartmentalization is listed according to MitoCarta 3.0 categorization. HAX1, HTRA2, TIMM13, TIMM8A, and TIMM8B were manually assigned to the IMS. (E) Venn diagram of all detected CLPBWA-FLAG and CLPBWB-FLAG interacting proteins against all proteins that were predominantly more insoluble in CLPBKO with or without HS. Proteins common to both groups are defined as CLPB interactors. (F) Visualization of common interacting partners between CLPBWA-FLAG and CLPBWB-FLAG IP datasets using the STRING database webtool (Szklarczyk et al., 2019). All active interaction sources were utilized. Functional annotations correspond to MitoCarta 3.0 categorization, and proteins with “import” or “OXPHOS” annotations have been excluded for clarity.
Defining the CLPB interactome and substrate profile. (A–C) Mitochondria were isolated from control, (A) CLPBWT-FLAG, (B) CLPBWA-FLAG, or (C) CLPBWB-FLAG stable cell lines following 14 h induction with 0.1 µg/ml tetracycline. Samples were processed for FLAG IP and eluates were prepared for LFQ MS. Data are presented as a volcano plot depicting the level of enrichment relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and to the right of the vertical line (eight-fold absolute change) are regarded as significantly enriched. Functional annotations correspond to MitoCarta 3.0 categorization. (D) Table containing all significantly enriched proteins across CLPBWT-FLAG (WT), CLPBWA-FLAG (WA), and CLPBWB-FLAG (WB) IP datasets with corresponding fold change across each experiment. Mitochondrial compartmentalization is listed according to MitoCarta 3.0 categorization. HAX1, HTRA2, TIMM13, TIMM8A, and TIMM8B were manually assigned to the IMS. (E) Venn diagram of all detected CLPBWA-FLAG and CLPBWB-FLAG interacting proteins against all proteins that were predominantly more insoluble in CLPBKO with or without HS. Proteins common to both groups are defined as CLPB interactors. (F) Visualization of common interacting partners between CLPBWA-FLAG and CLPBWB-FLAG IP datasets using the STRING database webtool (Szklarczyk et al., 2019). All active interaction sources were utilized. Functional annotations correspond to MitoCarta 3.0 categorization, and proteins with “import” or “OXPHOS” annotations have been excluded for clarity.
Consolidating the CLPBWA-FLAG and CLPBWB-FLAG IP data (Fig. 3, A–D) and CLPBKO protein solubility data (Fig. 2, B–D; and Fig. S2, C and D) revealed 14 interacting proteins that were also significantly enriched in the CLPBKO pellet fraction (basal or with HS; Fig. 3 E). These CLPB interactors include CPOX, GPD2, HAX1, HTRA2, MICU2, OPA1, PARL, SLC25A10, SLC25A13, STOML2, TIMM13, TIMM50, TOMM40, and YME1L1. STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) analysis applied to the significant hits across all three IP experiments (CLPBWT-FLAG, CLPBWA-FLAG, and CLPBWB-FLAG; Fig. 3 F) revealed that CLPB interactors cluster in defined groups. The SPY complex sits at the center of the cluster with other proteins involved in homeostasis or cell health such as CLPB, PMPCA, HTRA2, AIFM1, and ENDOG being peripherally associated. Thus, CLPB disaggregase action is fundamental to maintaining the solubility of IMS and OM/IM proteins. Proteins that aggregate in CLPBKO cells but fail to interact with either CLPBWA-FLAG or CLPBWB-FLAG, for example, AFGL3L2, may be prone to aggregation in a CLPB-null environment but are not necessarily CLPB interactors. This suggests CLPB contributes to broader proteostatic control, and indirectly supports mitochondrial health and function.
Lack of CLPB compromises SPY complex function
We sought to further investigate any functional interplay between the CLPB, HAX1, and SPY. First, we interrogated the proteomic consequences of either STOML2 or HAX1 depletion to dissect the implications of their aggregation in CLPBKO cells. Isolated mitochondria from HEK STOML2 (STOML2KO) and HAX1 (HAX1KO) KO cells (Fig. S3, A and B) were analyzed using LFQ proteomics. STOML2KO mitochondria showed similar proteomic perturbations as in CLPBKO cells, specifically: (i) upregulation of HAX1; (ii) depletion of OMA1; and (iii) moderate depletion of various CI/CIV components (Fig. S3 C and Table S1). Conversely, HAX1KO proteomics varied greatly from CLPBKO and STOML2KO, aside from common enrichment of ENDOG (Fig. S3 D and Table S1). KO of STOML2 or HAX1 had no impact on endogenous CLPB localization to the IMS (Fig. S3, E and F) or on CLPB disaggregase activity based on protein aggregation profiles (Fig. S3, G and H; and Tables S2 and S3). This data suggests the disaggregase activity of CLPB does not require STOML2 or HAX1.

Neither STOML2KO or HAX1KO impede CLPB localization or disaggregase activity. (A) Schematic representation of indels present in the STOML2KO HEK FlpIn-TREx cell line, introduced using CRISPR-Cas9 genome editing, according to STOML2 transcript variant 1 (NM_013442.3). Exon 3 is shared by all STOML2 variants. (B) Schematic representation of the homozygous indel present in the HAX1KO HEK FlpIn-TREx cell line, introduced using CRISPR-Cas9 genome editing, according to HAX1 transcript variant 1 (NM_006118.4). This mutation impacts both HAX1 isoforms a and b. (C and D) STOML2KO and HAX1KO HEK mitochondrial proteomics. Mitochondria were isolated from control and (C) STOML2KO or (D) HAX1KO HEK cell lines and subjected to LFQ MS. Data are presented as a volcano plot depicting the level of proteins in (C) STOML2KO or (D) HAX1KO relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (E and F) Mitochondrial sub-fractionation and carbonate extraction on mitochondria isolated from (E) STOML2KO cells and (F) HAX1KO cells to examine endogenous CLPB localization. Samples were run a 10–16% acrylamide tris-tricine gel and analyzed via immunoblotting (IB). * = protease protected protein. S = supernatant, P = pellet. (G and H) STOML2KO or (H) HAX1KO cells were exposed to heat shock (HS) for 2 h at 42°C and pellet fractions (insoluble proteins) were prepared for LFQ MS as outlined in Fig. 2 A. Data is presented as a volcano plot depicting the level of proteins in the STOML2KO pellet relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. To correct for anticipated differences in protein abundance within the STOML2KO or HAX1KO, data were normalized against STOML2KO mitochondrial proteomics (Fig. S3 C and Table S3) or HAX1KO mitochondrial proteomics (Fig. S3 D and Table S3). Proteins labeled in red had a fold difference >1.5 following normalization and are regarded as most strongly insoluble. Source data are available for this figure: SourceData FS3.
Neither STOML2KO or HAX1KO impede CLPB localization or disaggregase activity. (A) Schematic representation of indels present in the STOML2KO HEK FlpIn-TREx cell line, introduced using CRISPR-Cas9 genome editing, according to STOML2 transcript variant 1 (NM_013442.3). Exon 3 is shared by all STOML2 variants. (B) Schematic representation of the homozygous indel present in the HAX1KO HEK FlpIn-TREx cell line, introduced using CRISPR-Cas9 genome editing, according to HAX1 transcript variant 1 (NM_006118.4). This mutation impacts both HAX1 isoforms a and b. (C and D) STOML2KO and HAX1KO HEK mitochondrial proteomics. Mitochondria were isolated from control and (C) STOML2KO or (D) HAX1KO HEK cell lines and subjected to LFQ MS. Data are presented as a volcano plot depicting the level of proteins in (C) STOML2KO or (D) HAX1KO relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (E and F) Mitochondrial sub-fractionation and carbonate extraction on mitochondria isolated from (E) STOML2KO cells and (F) HAX1KO cells to examine endogenous CLPB localization. Samples were run a 10–16% acrylamide tris-tricine gel and analyzed via immunoblotting (IB). * = protease protected protein. S = supernatant, P = pellet. (G and H) STOML2KO or (H) HAX1KO cells were exposed to heat shock (HS) for 2 h at 42°C and pellet fractions (insoluble proteins) were prepared for LFQ MS as outlined in Fig. 2 A. Data is presented as a volcano plot depicting the level of proteins in the STOML2KO pellet relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. To correct for anticipated differences in protein abundance within the STOML2KO or HAX1KO, data were normalized against STOML2KO mitochondrial proteomics (Fig. S3 C and Table S3) or HAX1KO mitochondrial proteomics (Fig. S3 D and Table S3). Proteins labeled in red had a fold difference >1.5 following normalization and are regarded as most strongly insoluble. Source data are available for this figure: SourceData FS3.
Mitochondria isolated from control, CLPBKO, HAX1KO, and STOML2KO cells were solubilized in digitonin and analyzed using BN-PAGE and immunoblotting to assess SPY complex integrity. This revealed a strong reduction in the level of assembled SPY complex in CLPBKO cells, while the loss of HAX1 (a stable CLPB interactor, Fig. 3 A) had no impact on SPY complex integrity (Fig. S4 A). This observed reduction in SPY complex was consistent across multiple CLPBKO clones (Fig. S4 B) and re-expression of functional CLPBWT-FLAG restored SPY complex levels, while non-functional CLPBWB-FLAG failed to do so (Fig. 4, A and B). SPY complex abundance returned to wild-type levels with 6 h of CLPBWT-FLAG re-expression (Fig. S4 C), while longer expression time of either CLPBWA-FLAG or CLPBWB-FLAG mutants did not aid recovery (Fig. S4, D and E). As the steady-state levels of SPY complex components are not reduced in the absence of CLPB (Fig. 4 A, right panel), this suggests that either SPY complex assembly or integrity is impacted. We asked if the loss of SPY complex on BN-PAGE was due to the insolubility of its constitutive components in CLPBKO cells (as shown in Fig. 2, B and C) during the processing of samples for BN-PAGE. Therefore, mitochondria isolated from control, CLPBKO, CLPBWT-FLAG, and CLPBWB-FLAG were solubilized in digitonin-containing buffer, and soluble (supernatant) and insoluble (pellet) fractions were separated by SDS-PAGE. Indeed, STOML2 was significantly reduced from the CLPBKO soluble fraction (typically loaded onto BN-PAGE) and accumulated within the pellet (discarded for BN-PAGE analysis; Fig. 4, C and D). Further, STOML2 solubility could only be restored to wild-type levels with re-expression of functional CLPBWT-FLAG and not mutant CLPBWB-FLAG (Fig. 4, C and D). Therefore, reduced levels of SPY complex on BN-PAGE in cells lacking CLPB is a consequence of SPY components aggregating in the absence of the disaggregase.
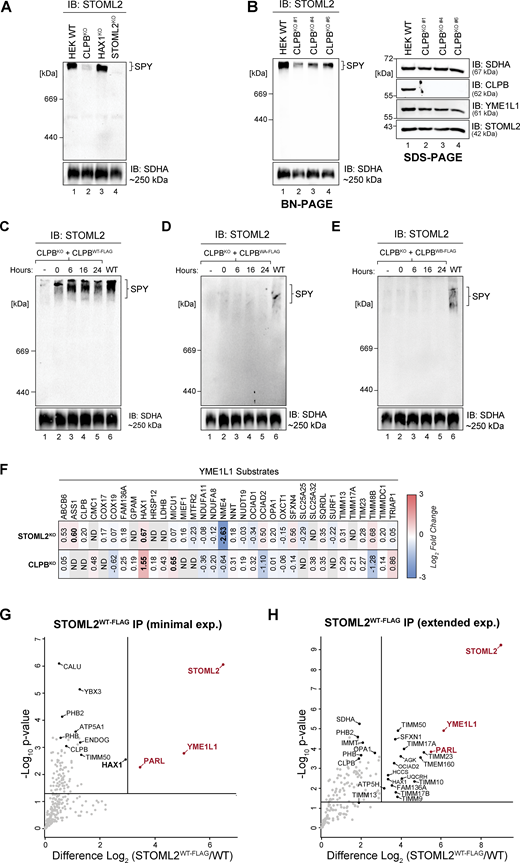
SPY complex instability is specific to CLPBKO. (A) Mitochondria from control, CLPBKO, HAX1KO, and STOML2KO were resuspended in solubilization buffer containing 1% digitonin. Lysates were run on a 4–16% acrylamide BN-PAGE gel and analyzed by immunoblotting (IB) with an anti-STOML2 antibody to assess integrity of the SPY complex. (B) Mitochondria isolated from control and CLPBKO clones #1, #4, and #6 were analyzed by BN-PAGE (left) and SDS-PAGE (right). Mitochondria were solubilized in 1% digitonin for BN-PAGE prior to electrophoresis or resuspended in SDS-loading dye prior to SDS-PAGE. Gels were analyzed by immunoblotting with antibodies as indicated. (C–E) SPY complex integrity was assessed by BN-PAGE following induction of CLPBWT-FLAG, CLPBWA-FLAG, and CLPBWB-FLAG with 1 µg/ml tetracycline for the indicated duration. Isolated mitochondria were resuspended in solubilization buffer containing 1% digitonin and lysates were run on a continuous 4–10% acrylamide BN-PAGE gel followed by immunoblotting with an anti-STOML2 antibody to assess SPY complex recovery over time. (F) Table of Log2 fold changes of known YME1L1 substrates from CLPBKO (Fig. 1 A and Table S1) and STOML2KO (Fig. S3 C and Table S1) isolated mitochondrial proteomics. YME1L1 substrates listed are as defined in MacVicar et al. (2019). (G and H) Mitochondria were isolated from control and STOML2WT-FLAG stable cell lines following (G) 4 h (minimal expression) or (H) 16 h (extended expression) induction with 1 µg/ml tetracycline. STOML2WT-FLAG interactors were captured via IP and eluates were then prepared for LFQ MS. Data is presented as a volcano plot depicting the level enrichment relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and to the right of the vertical line (eight-fold change) are regarded as significantly enriched. Proteins in red are components of the SPY complex. Source data are available for this figure: SourceData FS4.
SPY complex instability is specific to CLPBKO. (A) Mitochondria from control, CLPBKO, HAX1KO, and STOML2KO were resuspended in solubilization buffer containing 1% digitonin. Lysates were run on a 4–16% acrylamide BN-PAGE gel and analyzed by immunoblotting (IB) with an anti-STOML2 antibody to assess integrity of the SPY complex. (B) Mitochondria isolated from control and CLPBKO clones #1, #4, and #6 were analyzed by BN-PAGE (left) and SDS-PAGE (right). Mitochondria were solubilized in 1% digitonin for BN-PAGE prior to electrophoresis or resuspended in SDS-loading dye prior to SDS-PAGE. Gels were analyzed by immunoblotting with antibodies as indicated. (C–E) SPY complex integrity was assessed by BN-PAGE following induction of CLPBWT-FLAG, CLPBWA-FLAG, and CLPBWB-FLAG with 1 µg/ml tetracycline for the indicated duration. Isolated mitochondria were resuspended in solubilization buffer containing 1% digitonin and lysates were run on a continuous 4–10% acrylamide BN-PAGE gel followed by immunoblotting with an anti-STOML2 antibody to assess SPY complex recovery over time. (F) Table of Log2 fold changes of known YME1L1 substrates from CLPBKO (Fig. 1 A and Table S1) and STOML2KO (Fig. S3 C and Table S1) isolated mitochondrial proteomics. YME1L1 substrates listed are as defined in MacVicar et al. (2019). (G and H) Mitochondria were isolated from control and STOML2WT-FLAG stable cell lines following (G) 4 h (minimal expression) or (H) 16 h (extended expression) induction with 1 µg/ml tetracycline. STOML2WT-FLAG interactors were captured via IP and eluates were then prepared for LFQ MS. Data is presented as a volcano plot depicting the level enrichment relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and to the right of the vertical line (eight-fold change) are regarded as significantly enriched. Proteins in red are components of the SPY complex. Source data are available for this figure: SourceData FS4.

CLPB disaggregase is required to maintain SPY integrity. (A) Mitochondria isolated from control, CLPBKO, CLPBWT-FLAG, and CLPBWB-FLAG cells were prepared for BN-PAGE (left) and SDS-PAGE (right) analysis. For BN-PAGE, mitochondrial pellets were solubilized in digitonin-containing buffer and lysates were analyzed on a 4–16% gradient BN-PAGE gel and immunoblotted (IB) with the indicated antibodies. SDS-PAGE samples were solubilized in SDS-loading buffer prior to tris-tricine electrophoresis and immunoblotting with the indicated antibodies. *, stably expressed CLPB band lacking FLAG. (B) Quantification of STOML2 signal on BN-PAGE from A. Calculated as mean percentage ± SD of STOML2 signal, normalized to the level of SDHA (n = 4). (C) Mitochondria from control, CLPBKO, CLPBWT-FLAG, or CLPBWB-FLAG were solubilized in digitonin-containing buffer and separated into insoluble and soluble fractions by centrifugation and analyzed by SDS-PAGE and immunoblotting. (D) Quantification of STOML2 abundance in “Soluble” fraction shown in C. Calculated as mean percentage ± SD of “Total solubilized” for each cell line, normalized to SDHA level (n = 4). (E) Control, CLPBKO, and STOML2KO cell pellets were harvested following CHX treatment and solubilized in RIPA lysis buffer prior to SDS-PAGE and immunoblotting with antibodies as indicated. p, precursor form; m, mature form. (F) Quantification of PGAM5 processing as shown in E. Calculated as mean percentage ± SD of total PGAM5 signal (sum of p-PGAM5 and m-PGAM5 signals) across each time point per cell line, normalized to acylglycerol kinase (AGK) level (n = 4). (G) Mitochondria were isolated from control STOML2WT-FLAG (STOML2KO + STOML2WT-FLAG) and STOML2/CLPBdKO stable cell lines expressing STOML2WT-FLAG. Interactors of STOML2WT-FLAG were captured via IP and eluates were analyzed by LFQ MS. Data are presented as a volcano plot depicting the levels of interacting partners in CLPBKO as compared with control (n = 3). Proteins on the right of the volcano are more enriched in CLPBKO and proteins on the left of the volcano are more enriched in the control. Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of the vertical lines (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (H) Mitochondria were isolated from control CLPBWB-FLAG (CLPBKO + CLPBWB-FLAG) and CLPB/STOML2dKO stable cell line expressing CLPBWB-FLAG and processed as described in G. Throughout this figure, statistical significance was determined by Student’s two-sample t test = *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS = not significant, P > 0.05. Source data are available for this figure: SourceData F4.
CLPB disaggregase is required to maintain SPY integrity. (A) Mitochondria isolated from control, CLPBKO, CLPBWT-FLAG, and CLPBWB-FLAG cells were prepared for BN-PAGE (left) and SDS-PAGE (right) analysis. For BN-PAGE, mitochondrial pellets were solubilized in digitonin-containing buffer and lysates were analyzed on a 4–16% gradient BN-PAGE gel and immunoblotted (IB) with the indicated antibodies. SDS-PAGE samples were solubilized in SDS-loading buffer prior to tris-tricine electrophoresis and immunoblotting with the indicated antibodies. *, stably expressed CLPB band lacking FLAG. (B) Quantification of STOML2 signal on BN-PAGE from A. Calculated as mean percentage ± SD of STOML2 signal, normalized to the level of SDHA (n = 4). (C) Mitochondria from control, CLPBKO, CLPBWT-FLAG, or CLPBWB-FLAG were solubilized in digitonin-containing buffer and separated into insoluble and soluble fractions by centrifugation and analyzed by SDS-PAGE and immunoblotting. (D) Quantification of STOML2 abundance in “Soluble” fraction shown in C. Calculated as mean percentage ± SD of “Total solubilized” for each cell line, normalized to SDHA level (n = 4). (E) Control, CLPBKO, and STOML2KO cell pellets were harvested following CHX treatment and solubilized in RIPA lysis buffer prior to SDS-PAGE and immunoblotting with antibodies as indicated. p, precursor form; m, mature form. (F) Quantification of PGAM5 processing as shown in E. Calculated as mean percentage ± SD of total PGAM5 signal (sum of p-PGAM5 and m-PGAM5 signals) across each time point per cell line, normalized to acylglycerol kinase (AGK) level (n = 4). (G) Mitochondria were isolated from control STOML2WT-FLAG (STOML2KO + STOML2WT-FLAG) and STOML2/CLPBdKO stable cell lines expressing STOML2WT-FLAG. Interactors of STOML2WT-FLAG were captured via IP and eluates were analyzed by LFQ MS. Data are presented as a volcano plot depicting the levels of interacting partners in CLPBKO as compared with control (n = 3). Proteins on the right of the volcano are more enriched in CLPBKO and proteins on the left of the volcano are more enriched in the control. Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of the vertical lines (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (H) Mitochondria were isolated from control CLPBWB-FLAG (CLPBKO + CLPBWB-FLAG) and CLPB/STOML2dKO stable cell line expressing CLPBWB-FLAG and processed as described in G. Throughout this figure, statistical significance was determined by Student’s two-sample t test = *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS = not significant, P > 0.05. Source data are available for this figure: SourceData F4.
We assessed if the function of the SPY complex was impacted in the CLPB-null environment, specifically that of the proteases PARL and YME1L1. PGAM5 phosphatase is a substrate of PARL protease (Sekine et al., 2012), and cleaved PGAM5 regulates mitophagy and apoptosis via BCL-xL and FUNDC1 dephosphorylation, respectively, at the mitochondrial OM (Chen et al., 2014; Ma et al., 2020). PGAM5 processing was delayed significantly in CLPBKO cells following cycloheximide (CHX) treatment to block protein synthesis (Fig. 4, E and F). This is contrasted by STOML2KO cells where accelerated PGAM5 processing and delayed TIMM17A turnover demonstrate dysregulation of PARL and YME1L1 proteases, respectively (Wai et al., 2016). YME1L1 activity was surveyed by looking at the steady-state levels of known YME1L1 substrates (MacVicar et al., 2019) in the mitochondrial proteomics data of control versus CLPBKO (Fig. 1 A) and STOML2KO (Fig. S3 C). This showed no accumulation of YME1L1 substrates in the absence of CLPB or STOML2, suggesting protease activity may not be affected (Fig. S4 F). Together, these data demonstrate that the SPY complex is sensitive to aggregation in the CLPB-null environment, which compromises SPY complex integrity as observed by BN-PAGE, and function as evidenced by delayed PGAM5 processing. We reason this could be due to (i) a regulatory role of CLPB in the maintenance of the SPY complex or (ii) the accumulation of aggregated CLPB substrates at SPY, which is overwhelming this proteolytic hub causing and downstream consequences.
To explore the notion of a regulatory role of CLPB at SPY, we generated an inducible STOML2WT-FLAG cell line (in the STOML2KO background) and performed affinity enrichment IP MS. STOML2WT-FLAG strongly enriched YME1L1 and PARL, constituting the core SPY complex (Fig. S4 G and Table S4) agreeing with previous finding (Wai et al., 2016). HAX1 was one of the first interactions to be re-established upon introduction of STOML2WT-FLAG expression, aside from PARL and YME1L1 (Fig. S4 G and Table S4) and is significantly enriched following longer STOML2WT-FLAG re-expression (Fig. S4 H and Table S4). CLPB is not a stable interactor as expected given CLPBWT-FLAG did not stably interact with SPY components. As HAX1 is a joint substrate of both CLPB and YME1L1 (MacVicar et al., 2019), we monitored the turnover of HAX1 following CHX treatment of control, CLPBKO and STOML2KO cells to delineate connections between CLPB and the SPY complex. Mitochondria were isolated following 0 or 20 h of CHX treatment and residual HAX1 levels were quantified (Fig. S5 A). In wild-type mitochondria, HAX1 levels were reduced by 59% after 20 h of CHX treatment, indicative of efficient turnover. HAX1 turnover could still occur in CLPBKO and STOML2KO cells; however, this turnover was significantly slower compared to wild type; reduced by only 40% and 28%, respectively (Fig. S5 A). This data suggests some level of cooperation between CLPB and the SPY complex for joint substrates like HAX1. Insolubility of SPY components within CLPBKO cells (Fig. 4, C and D) may partially contribute to the delayed turnover of HAX1, supporting a degree of functional interplay between these two QC components.

STOML2/CLPBdKO has a cumulative impact on OXPHOS integrity. (A) Quantification of HAX1 turnover (left) and residual HAX1 remaining in isolated mitochondria following 20 h CHX treatment (right). HAX1 processing was calculated as mean percentage ± SD of HAX1 signal across each time point per cell line, relative to control signal and normalized to AGK level. Residual HAX1 was calculated as mean percentage ± SD of HAX1 signal remaining following CHX treatment. Significance determined by Student’s t test (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. (B) Mitochondria were isolated from control and uninduced STOML2/CLPBdKO HEK cells and subjected to LFQ MS. Volcano plot depicts the level of proteins in STOML2/CLPBdKO relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (C) Topographical heatmap showing Log2 fold difference values of CI subunits mapped against CI and supercomplex (SC) PDB structures from STOML2/CLPBdKO mitochondria, as determined in B (Table S1). N = NADH oxidation module (N-module). Q = ubiquinone reduction module (Q-module). CI: PDB 5LDW, CIII: PDB 5XTE, CIV: PDB 5Z62. (D) Left: Mitochondria isolated from control, CLPBKO, STOML2KO, and STOML2/CLPBdKO (no induction) were resuspended in solubilization buffer containing 1% digitonin. Lysates were run on a 4–16% acrylamide BN-PAGE gel, followed by immunoblotting (IB) with anti-NDUFA9 and anti-COXIV antibodies to assess OXPHOS supercomplex integrity. Right: Isolated mitochondria were resuspended in SDS-containing buffer and run on a 10–16% tris tricine gel followed by immunoblotting with indicated antibodies. Source data are available for this figure: SourceData FS5.
STOML2/CLPBdKO has a cumulative impact on OXPHOS integrity. (A) Quantification of HAX1 turnover (left) and residual HAX1 remaining in isolated mitochondria following 20 h CHX treatment (right). HAX1 processing was calculated as mean percentage ± SD of HAX1 signal across each time point per cell line, relative to control signal and normalized to AGK level. Residual HAX1 was calculated as mean percentage ± SD of HAX1 signal remaining following CHX treatment. Significance determined by Student’s t test (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. (B) Mitochondria were isolated from control and uninduced STOML2/CLPBdKO HEK cells and subjected to LFQ MS. Volcano plot depicts the level of proteins in STOML2/CLPBdKO relative to control (n = 3). Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of either vertical line (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (C) Topographical heatmap showing Log2 fold difference values of CI subunits mapped against CI and supercomplex (SC) PDB structures from STOML2/CLPBdKO mitochondria, as determined in B (Table S1). N = NADH oxidation module (N-module). Q = ubiquinone reduction module (Q-module). CI: PDB 5LDW, CIII: PDB 5XTE, CIV: PDB 5Z62. (D) Left: Mitochondria isolated from control, CLPBKO, STOML2KO, and STOML2/CLPBdKO (no induction) were resuspended in solubilization buffer containing 1% digitonin. Lysates were run on a 4–16% acrylamide BN-PAGE gel, followed by immunoblotting (IB) with anti-NDUFA9 and anti-COXIV antibodies to assess OXPHOS supercomplex integrity. Right: Isolated mitochondria were resuspended in SDS-containing buffer and run on a 10–16% tris tricine gel followed by immunoblotting with indicated antibodies. Source data are available for this figure: SourceData FS5.
We then asked if the SPY complex interaction network is altered in the absence of CLPB to evaluate a direct action of CLPB at SPY. We created a STOML2/CLPB double KO (dKO) HEK cell line, where the expression of STOML2WT-FLAG could be induced (STOML2/CLPBdKO + STOML2WT-FLAG). LFQ proteomics on isolated mitochondria from wild-type HEK cells versus uninduced STOML2/CLPBdKO demonstrates that the loss of both STOML2 and CLPB strongly impacts CI/CIV integrity (Fig. S5, B and C; and Table S1). The defect is recapitulated on BN-PAGE, where CI and CIV assemblies were reduced compared with either CLPBKO or STOML2KO cell lines (Fig. S5 D). IP of STOML2WT-FLAG from cells with or without CLPB revealed changes to the SPY complex interactome in a CLPB-null environment (Fig. 4 G and Table S5). Specifically, interactions between STOML2WT-FLAG and HAX1 or HTRA2 were significantly reduced. Loss of interaction between SPY and HAX1 and HTRA2 upon CLPB depletion is likely due to excessive aggregation of these substrates (hence removal from the supernatant fraction used for IP due to clarifying spin). As a complementary approach, we knocked-out STOML2 in the background of the CLPBWB-FLAG stable cell line (CLPB/STOML2dKO + CLPBWB-FLAG) to monitor how the interaction profile of substrate trap CLPBWB-FLAG changed in the absence of assembled SPY complex. This showed that CLPBWB-FLAG interaction with YME1L1 is reduced in the absence of STOML2, suggesting that colocalization of YME1L1 with PARL and STOML2 could aid disaggregase action at the SPY complex (Fig. 4 H and Table S5). CLPBWB-FLAG-HAX1 interaction was also moderately enhanced upon loss of SPY. Together these data suggest a HAX1-HTRA2-SPY axis dependent upon the presence of CLPB. The current findings do not suggest a direct regulatory role of CLPB at the SPY complex, but we cannot dismiss this possibility.
To investigate the idea that protein aggregates could be overwhelming SPY in the absence of CLPB, we expressed HAX1WT-FLAG in control and CLPBKO cells and looked for colocalization of HAX1 with STOML2 or TIMM44 (IM control) using immunofluorescence (anti-FLAG and anti-STOML2 staining or anti-FLAG and anti-TIMM44 staining; Fig. 5, A and B). Pearson correlation was performed on individual line scans of mitochondrial tubules expressing HAX1WT-FLAG. This showed a significant localization of HAX1WT-FLAG puncta with STOML2 in CLPBKO cells, but not in wild-type cells (Fig. 5 C), suggesting that HAX1 aggregates are accumulating at STOML2-containing sites. This was not observed when the analysis was done against TIMM44. As a final measure, we assessed if the interaction between CLPB and the SPY complex is mediated by aggregation-prone substrates like HAX1. We asked if CLPBWB-FLAG still interacts with the SPY complex in the absence of HAX1. HAX1 was depleted in the background of the CLPBWB-FLAG stable cell line using CRISPR/Cas9 (polyclonal HAX1KO cell line), and CLPBWB-FLAG IP was performed. Depletion of HAX1 levels reduced the interaction between CLPBWB-FLAG and HTRA2, but the interaction between CLPBWB-FLAG and the SPY components was not impacted (Fig. 5 D and Table S5). This suggests the interaction between CLPB and SPY can occur independently of the key CLPB interactor, HAX1, although we cannot eliminate the possibility that other substrates are present and could be facilitating this interaction. This data supports the notion that in the absence of CLPB, aggregate proteins accumulate at SPY leading to dysfunction of the proteolytic hub. Overloading of the supporting mitochondrial QC network in CLPBKO may drive dysregulated proteostasis, manifesting in the diverse phenotypic presentation observed upon CLPB deficiency (Fig. 5 E).

HAX1 aggregates colocalize with SPY complex in CLPB KO . (A and B) Control and CLPBKO HEK cells were transiently transfected with HAX1WT-FLAG and treated for immunofluorescence with anti-FLAG (magenta) and (A) anti-STOML2 or (B) anti-TIM44 (green) antibodies. Arrowheads denote areas of increased signal intensity consistent with the formation of HAX1-containing punctae. Scale bar is equivalent to 10 or 2 µM (zoom) as indicated. Dashed box indicates the zoom region shown on the far right. (C) Pearson’s correlation coefficient (r) analysis of 1 μm linescans of HAX1WT-FLAG and endogenous STOML2 or TIMM44 signal in HEK control or CLPBKO cell lines. n = ≥160 measurements per condition from three independent experiments, ≥28 images/condition. ****P <0.0001, two tailed Mann-Whitney test. (D) Mitochondria were isolated from control CLPBWB-FLAG (CLPBKO + CLPBWB-FLAG) and CLPB/HAX1dKO stable cell line expressing CLPBWB-FLAG. Interactors were captured via IP and eluates were then prepared for LFQ MS. Data are presented as a volcano plot depicting the levels of CLPBWB-FLAG interacting partners in HAX1KO as compared with control (n = 3). Proteins on the right of the volcano are more enriched in the absence of HAX1. Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of the vertical lines (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (E) Schematic depicting the relationship between IMS and IM protein QC mechanisms. CLPB maintains HAX1 solubility thus enabling HAX1-HTRA2 interactivity and promoting their association with SPY (left). In the absence of CLPB, HAX1 is vulnerable to aggregation, colocalizing with SPY in discrete punctae (right). Functional (soluble) interactions between HAX1-HTRA2 and SPY are lost in the absence of CLPB. Excessive aggregation in a CLPB-null environment likely drives overloading at IMS QC machinery, leading to dysregulated proteostasis. The broad phenotypic outcomes of CLPB deficiency, such as reduced OXPHOS capacity, overabundance of apoptotic factors, and extreme ROS accumulation, may be due to an inefficiency of supporting QC factors in the face of acute proteotoxic stress.
HAX1 aggregates colocalize with SPY complex in CLPB KO . (A and B) Control and CLPBKO HEK cells were transiently transfected with HAX1WT-FLAG and treated for immunofluorescence with anti-FLAG (magenta) and (A) anti-STOML2 or (B) anti-TIM44 (green) antibodies. Arrowheads denote areas of increased signal intensity consistent with the formation of HAX1-containing punctae. Scale bar is equivalent to 10 or 2 µM (zoom) as indicated. Dashed box indicates the zoom region shown on the far right. (C) Pearson’s correlation coefficient (r) analysis of 1 μm linescans of HAX1WT-FLAG and endogenous STOML2 or TIMM44 signal in HEK control or CLPBKO cell lines. n = ≥160 measurements per condition from three independent experiments, ≥28 images/condition. ****P <0.0001, two tailed Mann-Whitney test. (D) Mitochondria were isolated from control CLPBWB-FLAG (CLPBKO + CLPBWB-FLAG) and CLPB/HAX1dKO stable cell line expressing CLPBWB-FLAG. Interactors were captured via IP and eluates were then prepared for LFQ MS. Data are presented as a volcano plot depicting the levels of CLPBWB-FLAG interacting partners in HAX1KO as compared with control (n = 3). Proteins on the right of the volcano are more enriched in the absence of HAX1. Proteins above the horizontal cutoff (P value <0.05, Student's t test) and outside of the vertical lines (1.5-fold absolute change) are regarded as significantly altered in abundance. Functional annotations correspond to MitoCarta 3.0 categorization. (E) Schematic depicting the relationship between IMS and IM protein QC mechanisms. CLPB maintains HAX1 solubility thus enabling HAX1-HTRA2 interactivity and promoting their association with SPY (left). In the absence of CLPB, HAX1 is vulnerable to aggregation, colocalizing with SPY in discrete punctae (right). Functional (soluble) interactions between HAX1-HTRA2 and SPY are lost in the absence of CLPB. Excessive aggregation in a CLPB-null environment likely drives overloading at IMS QC machinery, leading to dysregulated proteostasis. The broad phenotypic outcomes of CLPB deficiency, such as reduced OXPHOS capacity, overabundance of apoptotic factors, and extreme ROS accumulation, may be due to an inefficiency of supporting QC factors in the face of acute proteotoxic stress.
Discussion
We explored the function and interaction network of the mitochondrial disaggregase CLPB under steady-state and stress conditions. Our cell models lacking CLPB show strong phenotypic overlap with published MGCA7B patient data (Tucker et al., 2022), revealing an enrichment of HAX1, depletion of the OMA1 protease, and reduction of CI and CIV subunits. IP data coupled with protein solubility profiles using CLPB mutants allowed us to catalog a discrete list of CLPB substrates, which become prone to aggregation in the absence of the disaggregase. This has uncovered a close relationship between CLPB and all three members of the SPY complex, which becomes strongly insoluble in the absence of CLPB, ultimately influencing the integrity and functionality of the complex. We conclude that these impacts on the SPY by virtue of CLPB dysfunction affect protein QC mechanisms in the IMS and IM, exacerbating respiratory, morphological, and proteostatic defects, and fundamentally perturbing mitochondrial health.
A reoccurring feature in our data was an impact on OXPHOS systems in conditions of compromised protein QC. CI and CIV subunits were destabilized in both CLPBKO and STOML2KO mitochondrial proteomics, with a cumulative effect evident in STOML2/CLPBdKO. CLPBKO cells showed defects in basal and maximal oxygen consumption; however, IP data did not support a role for CLPB in OXPHOS assembly or regulation, outside of a moderate interaction between CLPBWA-FLAG and COA7, which has been previously described (Formosa et al., 2022). We presume that these defects are secondary consequences of other dysregulated processes in the absence of CLPB, most likely impacts the SPY components STOML2 and YME1L1. Within the SPY complex, STOML2 modulates the activity of PARL, facilitating PINK1 and PGAM5 processing to manage mitophagy, mitochondrial dynamics, and survival (Wai et al., 2016). STOML2 can also mitigate OMA1 proteolytic activity under stress (Wai et al., 2016) and is implicated in cardiolipin biogenesis and modulation, as well as OXPHOS complex stability and supercomplex formation (Christie et al., 2011; Mitsopoulos et al., 2015). YME1L1 is an IMS-facing AAA+ domain–containing protease implicated in the maintenance of protein import, lipid metabolism, and mitochondrial morphology via OMA1 proteolytic regulation and OPA1 processing (Anand et al., 2014; Consolato et al., 2018; Ohba et al., 2020). Loss of SPY complex integrity and function in CLPBKO appears to reflect these diverse proteostatic functions; S-OPA1 accumulates in processed subtypes concordant with OMA1 overactivation, PGAM5 processing following CHX treatment is limited, pro-apoptotic factors accumulate in the absence of CLPB, and OXPHOS stability is disturbed.
Overlapping profiles of the CLPBKO and STOML2KO mitochondrial proteomics support loss of the SPY complex being a primary component of the CLPBKO phenotype. Overactivation of OMA1 triggers autocatalytic degradation of the protease (Baker et al., 2014), and so dysregulation upon loss of SPY (either in STOML2KO or CLPBKO) could manifest in excessive OPA1 processing and reduced OMA1 abundance in mitochondrial proteomics. Processed PGAM5 dephosphorylates BCL-xL, limiting BAX-BAK accumulation at the mitochondrial OM (Ma et al., 2020), and so restricted PGAM5 processing may partly explain an enrichment of pro-apoptotic factors in CLPBKO cells. We propose that there is a functional interplay between CLPB and SPY in the maintenance of IMS/IM protein QC. In the absence of CLPB, protein aggregates accumulate at SPY likely exacerbating aggregation and obstructing SPY functionality. Nonetheless, we cannot rule out the possibility that CLPB directly regulates SPY complex solubility independent of aggregation-prone substrates.
CLPB and HAX1 are frequently enriched alongside SPY complex members (Fielden et al., 2021; Kang et al., 2019; Wai et al., 2016). Specifically, PARLFLAG IP strongly enriched both CLPB and HAX1 in addition to STOML2 and YME1L1, while all other known PARL substrates (aside from CLPB) were absent (Wai et al., 2016). HAX1 is a 279-aa protein containing a single, C-terminal transmembrane domain with a highly disordered N-terminal domain with weak Bcl-2 domain homology (Fadeel and Grzybowska, 2009; Fan et al., 2022). The CLPB binding domain of HAX1 has been refined to amino acids 126–136 (Fan et al., 2022; Wakula et al., 2020), and reciprocal interaction is dependent on the CLPB ANK domains (Fan et al., 2022). In vitro, CLPB is a stand-alone disaggregase (Cupo and Shorter, 2020), which exists in either hexameric or dodecameric populations in the absence of substrate, but preferentially assumes dodecameric confirmation upon substrate engagement (Cupo et al., 2022; Spaulding et al., 2022). As disaggregase substrate affinity can be weak, the degree of HAX1 enrichment with CLPBWT-FLAG IP was notable and infers functional relevance. HTRA2 is an IMS serine protease and within murine mitochondria, Htra2 is a neuroprotective, antiapoptotic modulator (Martins et al., 2004), and is activated by Parl via Hax1 (Chao et al., 2008). Loss of HAX1/HTRA2 interaction with SPY upon CLPBKO may indirectly disrupt apoptotic regulation and cristae maintenance, contributing to MGCA7A/B pathogenesis. Further, loss of CLPB-YME1L1 interaction in the absence of STOML2 suggests that colocalization with SPY components could be functionally beneficial and may support a novel HAX1-CLPB-SPY regulatory mechanism at the IM.
In summary, we have characterized the broad impacts of CLPB deficiency, refined a list of putative CLPB substrates, and delineated a novel interaction network between the SPY complex and a CLPB-HAX1-HTRA2 axis. CLPB is required to maintain protein solubility in the IMS and IM, and failure to do so has consequences at the SPY complex, leading to broader impacts on mitochondrial protein homeostasis and cell health. Our results suggest interconnectivity between QC components at the mitochondrial IMS and IM and provide a rational for the overlapping phenotypic presentations across pathologies associated with CLPB, HAX1, and HTRA2 deficiency.
Materials and methods
Cell lines, transient transfection, and stable cell generation
HEK293 Flp-In T-REx (Thermo Fisher Scientific) wild type and CRISPR/Cas9 KO CLPB, STOML2, and HAX1 cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 5% or 10% [vol/vol] fetal calf serum (In vitro Technologies) and 0.01% [vol/vol] penicillin–streptomycin. Transient transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Tetracycline-inducible HEK293 Flp-In T-REx cell lines stably expressing 3×FLAG-tagged proteins were generated using the T-REx Flp-In system (pcDNA5/FRT vector; Thermo Fisher Scientific) according to the manufacturer’s instructions. HeLa wild type and CRISPR/Cas9 KO CLPB cell lines were cultured in DMEM (Gibco) with 10% [vol/vol] tetracycline-reduced fetal calf serum. HeLa cells stably expressing 1×FLAG-tagged CLPBWT-FLAG and CLPBWB-FLAG were generated by lentiviral transduction. Briefly, lentiviral particles were generated by transiently transfecting HEK293T cells with pLVX-puro (containing either CLPBWT-FLAG or CLPBWB-FLAG) for 24 h using Lenti-X Packaging Single Shots (VSV-G; Takara). Following transfection, medium was replaced with fresh DMEM, and cells were incubated for a further 24 h. Virus-containing culture supernatant was collected and filtered, before being applied to HeLa cell cultures with 4 µg/ml polybrene. Cells were selected in 3 µg/ml puromycin for at least three passages before manual single-cell sorting and screening. All cell lines were grown at 37°C in a humidified atmosphere containing 5% CO2. Cell lines were confirmed to be mycoplasma negative using MycoAlert Mycoplasma detection kit (Lonza). Cell lines were not authenticated but parental lines were derived from commercial sources.
Gene editing and screening
Editing of the CLPB gene in the HEK293 Flp-In T-REx cell line was performed using a pSpCas9(CLPB)-2A-GFP plasmid targeting exon 1 of the CLPB gene (generated and gifted by the lab of Professor Mike Ryan, Monash University, Melbourne, Australia). sgRNAs against exon 3 of the STOML2 gene and exon 1 and 2 of the HAX1 gene were designed using CHOP-CHOP (Labun et al., 2019) and ligated into either pSpCas9(BB)-2A-GFP or pSpCas9(BB)-2A-Puro (Addgene). The pSpCas9(BB)-2A-GFP plasmids were independently transfected into HEK293 Flp-In T-REx cells using Lipofectamine 3000 according to the manufacturer’s instructions 24 h prior to single-cell sorting by GFP expression via fluorescence-activated cell sorting. HAX1 sgRNA-containing pSpCas9(BB)-2A-Puro plasmids were cotransfected into CLPBWB-FLAG cells using Lipofectamine 3000 according to manufacturer’s instructions 24 h prior to puromycin selection (1 µg/ml). After 72 h of selection, surviving cells were combined to establish a polyclonal population. Individual cell lines were expanded and screened via western blotting (STOML2WTCLPBdKO and CLPBWBSTOML2dKO and CLPBWBHAX1dKO cell lines using CLPB, STOML2, or HAX1 specific antibodies, respectively) or Sanger sequencing of the CRISPR target site (CLPBKO, STOML2KO, and HAX1KO cell lines). Complete loss of target gene expression was further verified via tandem MS.
For CRISPR/Cas9-mediated gene KO in HeLa cells, the pX335 vector (Addgene) was used based on a nicked Cas9 enzyme (Ran et al., 2013). For targeting the CLPB gene, two sgRNAs were synthesized. These pX335 plasmids were transfected into HeLa cells using GeneJuice-mediated transfections (Novagen) according to the manufacturer’s guidelines. Prior to transfection, 4 × 105 cells were seeded on one well of a 6-well plate. Transfection was repeated three times. 24 h after the last transfection, cells were counted and diluted to a concentration of 1,000 cells per ml, and 100 µl of this dilution was added to each well of two 96-well plates. After 72 h, 100 µl DMEM was added to each well, and emerging single-cell colonies (potential KO cells) were marked and transferred to one well of a 12-well plate when a confluency of 50% was reached. Individual clones were expanded and screened via western blotting using an anti-CLPB antibody.
Primers used in this study
Primers used in this study are listed in Table 1.
Primers used in this study
| Oligonucleotides | Source | Identifier |
|---|---|---|
| CLPB cDNA seq FWD: 5′-ATTAGGTACCAATGCTGGGGTCCCTGGTG-3′ | This study | N/A |
| CLPB cDNA seq REV: 5′-GTCAGGATCCGATGGTGTTGCACACCTT-3′ | This study | N/A |
| STOML2 cDNA seq FWD: 5′-AAAAAAGCTTAATGCTGGCGCGCGCGGC-3′ | This study | N/A |
| STOML2 cDNA seq REV: 5′-GGGAGGTACCACTCATCTTGACTCGATCAA-3′ | This study | N/A |
| STOML2 sgRNA FWD: 5′-CACCGTATCGGATCCGGTCTAACAC-3′ | This study | N/A |
| STOML2 sgRNA REV: 5′-AAACGTGTTAGACCGGATCCGATAC-3′ | This study | N/A |
| HAX1 sgRNA FWD #1: 5′-CACCGAAAGAGGCTCATTCCCGTAC-3′ | This study | N/A |
| HAX1 sgRNA REV #1: 5′-AAACGTACGGGAATGAGCCTCTTTC-3′ | This study | N/A |
| HAX1 sgRNA FWD #2: 5′-CACCGCATCAAAGCCGAAGTTATCG-3′ | This study | N/A |
| HAX1 sgRNA REV #2: 5′-CGTAGTTTCGGCTTCAATAGCCAAA-3′ | This study | N/A |
| CLPB indel seq FWD: 5′-ATTAGGATCCGGTATCGAAGCGTCCTCCCT-3′ | This study | N/A |
| CLPB indel seq REV: 5′-ACTGAAGCTTGCTTAGGGCTGTGGACTACC-3′ | This study | N/A |
| STOML2 indel seq FWD: 5′-ATATGGTACCGGGTTTTCTTACCAGTGACACC-3′ | This study | N/A |
| STOML2 indel seq REV: 5′-GAGTAAGCTTGCAACCACATCCTAATAGCCTC-3′ | This study | N/A |
| HAX1 indel seq FWD: 5′-GACGGGATCCTTGGTCTGCAGAAAGAAGAACA-3′ | This study | N/A |
| HAX1 indel seq REV: 5′-CGCGAAGCTTGCTCTTTATTTCACCCACCAAC-3′ | This study | N/A |
| CLPB K387A FWD: 5′-TCATCTGGAATAGGAGCAACAGAGCTGGCCAAGCAG-3′ | This study | N/A |
| CLPB K387A REV: 5′-CTTGGCCAGCTCTGTTGCTCCTATTCCAGATGATCC-3′ | This study | N/A |
| CLPB E455Q FWD: 5′-GTGGTGCTCTTTGATCAAGTAGACAAGGCCCATCCA-3′ | This study | N/A |
| CLPB E455Q REV: 5′-ATGGGCCTTGTCTACTTGATCAAAGAGCACCACAGC-3′ | This study | N/A |
| CLPB sgRNA FWD #1 (HeLa): 5′-CACCGGCCGGAATGTGACTAC-3′ | This study | N/A |
| CLPB sgRNA REV #1 (HeLa): 5′-AAACGTAGTCACATTCCGGCC-3′ | This study | N/A |
| CLPB sgRNA FWD #2 (HeLa): 5′-CACCGCCATGGCCCCGGAGCGT-3′ | This study | N/A |
| CLPB sgRNA REV #2 (HeLa): 5′-AAACACGCTCCGGGGCCATGGC-3′ | This study | N/A |
Mitochondrial treatments and cell lysate preparation for SDS-PAGE
Mitochondria were isolated from cells via differential centrifugation as described previously (Kang et al., 2017, 2019). The protein concentration of clarified mitochondria was determined using Pierce BCA protein assay kit (Thermo Fisher Scientific). For sub-fractionation experiments, 50 µg mitochondrial pellets (in duplicate) were resuspended in either isolation buffer (20 mM HEPES-KOH [pH 7.6], 220 mM mannitol, 70 mM sucrose, 1 mM EDTA), Swelling Buffer (10 mM HEPES, pH 7.4) or 0.5% [vol/vol] Triton X-100 at 1 mg/ml and incubated on ice for 10 min. One sample of each buffer set was treated with 50 µg/ml PK, followed by a 10 min incubation on ice. PMSF was added to all tubes at a final concentration of 1 mM, with an additional 10 min incubation on ice prior to trichloroacetic acid (TCA) precipitation. For carbonate extraction experiments, 100 µg of mitochondrial protein was resuspended in 100 mM Na2CO3 (prepared fresh at indicated pH) at 1 mg/ml and incubated on ice for 30 min. Membrane (pellet) and soluble (supernatant) fractions were separated via ultracentrifugation at 100,000 g for 30 min at 4°C prior to TCA precipitation. In both sub-fractionation and carbonate extraction experiments, TCA precipitation was performed by adding 72% [wt/vol] TCA to each sample to a final concentration of 12.5%. Carbonate-extracted pellet samples were resuspended in 12.5% TCA directly. For protein solubility assays, 50 µg mitochondrial pellets (in duplicate) solubilized in either Triton X-100-containing buffer (0.5% [vol/vol] Triton X-100, 200 mM KCl, 30 mM Tris/HCl [pH 7.4], 0.5 mM PMSF, 5 mM EDTA, and 1× complete protease inhibitor [Roche]) or digitonin containing buffer (1% [wt/vol] digitonin, 20 mM Tris-Cl, pH 7.4, 50 mM NaCl, 0.1 mM EDTA and 10% [vol/vol] glycerol). SDS-PAGE loading dye was added directly to one sample, constituting the “Total” fraction. The remaining sample was clarified at 20,000 g for 20 min at 4°C, and SDS-PAGE loading dye was added to both supernatant (“Soluble”) and pellet (“Insoluble”) fractions. For whole-cell lysis, cells were isolated and washed in PBS prior to resuspension and lysis in house-made radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% [vol/vol] Triton-X100, 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] SDS, 50 mM Tris-Cl, pH 8.0). Lysate was clarified at 16,000 g prior to preparation for SDS-PAGE. For cycloheximide treatment experiments, cells were treated with cycloheximide at 50 µg/ml for the indicated duration prior to either whole-cell lysis or mitochondrial isolation.
Gel electrophoresis and immunoblotting
Tris-tricine SDS-PAGE was performed as previously described (Kang et al., 2019). As noted in each experiment, acrylamide solution (49.5% [wt/vol] acrylamide, 1.5% [wt/vol] bis-acrylamide) was diluted in tricine buffer (3 M Tris-Cl, pH 8.45, 0.2% [wt/vol] SDS) to create 10 and 16% or 8 and 10% acrylamide solutions, with 13% [vol/vol] glycerol added to the 16% mix (of 10–16%) or 10% mix (of 8–10%). Gels were poured using a gradient mixer and a 4% acrylamide stacker mix was poured over polymerized 10–16% and 8–10% gradient gels. Samples were resuspended in SDS-PAGE loading dye (100 mM Tris-Cl, pH 6.8, 200 mM 1,4-dithiothreitol, 4% [wt/vol] SDS, 20% [wt/vol] glycerol, 0.1% [wt/vol] bromophenol blue) and boiled at 95°C for 15 min prior to loading. Electrophoresis was performed overnight in tris-tricine SDS-PAGE cathode buffer (0.1 M Tris, 0.1 M Tricine, pH 8.45, 0.1% SDS) and anode buffer (0.2 M Tris-Cl, pH 8.9).
BN-PAGE was performed as previously described (Kang et al., 2019). Acrylamide solutions were diluted in BN gel buffer (200 mM α-amino n-caproic acid, 150 mM Bis-Tris, pH 7.0) to create acrylamide solutions for the desired gradients, with 13% [vol/vol] glycerol added to the 16% and 10% mixes. Gels were poured using a gradient mixer, and either a 4% acrylamide stacker mix was poured over polymerized gels or a continuous gradient was employed where specified. Isolated mitochondria (80 µg) were solubilized in solubilization buffer (20 mM Tris-Cl, pH 7.4, 50 mM NaCl, 0.1 mM EDTA, and 10% [vol/vol] glycerol) with 1% [wt/vol] digitonin and incubated on ice for 30 min. Solubilized samples were clarified at 16,000 g for 30 min at 4°C and BN-PAGE loading dye (0.5% [wt/vol] Coomassie blue G250, 50 mM α-amino n-caproic acid, 10 mM Bis-Tris, pH 7.0) was added to the clarified supernatant prior to loading. Electrophoresis was performed overnight at 4°C using BN-PAGE anode (50 mM Bis-Tris, pH 7.0) and “blue” cathode buffer (50 mM tricine, 15 mM Bis-Tris, pH 7.0, 0.02% [wt/vol] Coomassie blue G250) for 1.5 h before being swapped to “clear” buffer (50 mM tricine, 15 mM Bis-Tris, pH 7.0) for the remainder of the run.
Following electrophoresis, SDS-PAGE and BN-PAGE gels were transferred onto 0.45 µm polyvinylidene fluoride membranes using an Owl HEP-1 Semidry Electroblotting system (Thermo Fisher Scientific). Immunoblotting was performed using primary and secondary antibodies as outlined in Table 2. Imaging was performed using a ChemiDoc XRS+ or MP imaging machine (BioRad) using Clarity Western ECL Substrate (BioRad) to generate the signal. Image processing and quantification were carried out in Image Lab software (BioRad).
Cellular respiration measurements
Mitochondrial OCR and ECAR were measured simultaneously in live cells using a Seahorse Bioscience XF24-3 analyzer according to the manufacturer’s instructions. Briefly, 50,000 cells were plated per well in 24-well plates treated with poly-D-lysine and allowed to settle overnight. For each independent experiment (n = 3), 8–10 replicate wells were utilized for both control and CLPBKO cell lines. Prior to assay start, cell media was changed to non-buffered Seahorse DMEM base medium (102353-100; Agilent) containing 25 mM glucose, 1 mM sodium pyruvate, and 2 mM glutamine. Each assay cycle comprised a 2-min mix, 2-min wait, and 3-min measure using the following inhibitors: 1 μM oligomycin; 0.7 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP); 0.5 μM rotenone; and 0.3 μM antimycin A. Basal and maximal OCR were calculated as the average of the three data points prior to oligomycin or rotenone/antimycin A addition, respectively, across each replicate plate. ECAR/OCR ratio was calculated from the average of the first ECAR point (measured concurrently with OCR) across each replicate plate divided by the average basal OCR value.
ROS measurement
Levels of ROS were measured by monitoring H2O2 accumulation using the ROS-Glo H2O2 Assay kit (Promega) according to the manufacturer’s instructions. Briefly, 5 × 103 cells were seeded in two triplicate sets onto a white-bottom 96-well plate and allowed to recover for 24 h. The provided H2O2 dilution buffer and luciferin precursor substrate were then added to cells with either vehicle control (DMSO) or 10 μM menadione and incubated at 37°C for 2 h. After incubation, ROS-Glo detection solution was added to all wells and the plate was incubated for 20 min at RT in the dark. Luminescence intensity was measured on a FLUOstar OPTIMA microplate reader (BMG LABTECH) and data were statistically analyzed using an unpaired two-tailed Student’s t test.
Immunofluorescence imaging
HeLa cells were seeded onto sterile glass coverslips, and if applicable, transfected with expression plasmids 24 h. For MitoTracker Deep Red (Invitrogen) staining, cells were incubated in medium containing the fluorescent dye (20 nM MitoTracker Deep Red) for 30 min. Following incubation, cells were washed three times with medium without dye and incubated for a further 30 min in dye-free medium at 37°C. Cells were washed in PBS and fixed in 3.7% [wt/vol] paraformaldehyde for 15 min, washed twice with PBS, and then permeabilized in 0.2% [vol/vol] Triton X-100 in PBS for 5 min. Cells were washed three times in PBS and blocked in 0.1% [wt/vol] BSA in PBS for 30 min followed by overnight incubation at 4°C with primary antibody diluted in 0.1% [wt/vol] BSA in PBS. Cells were washed four times with PBS prior to incubation with secondary fluorescent antibodies for 1 h in 0.1% [wt/vol] BSA in PBS (goat anti-mouse AlexaFluor 546 conjugate [Invitrogen] and goat anti-rabbit AlexaFluor 488 [Invitrogen]). Cells were washed four times with PBS and coverslips were then mounted onto glass microscope slides with ProLong Gold antifade reagent (Invitrogen). Images were acquired at RT on a Zeiss LSM Meta 710 confocal microscope (Zeiss). Samples were mounted on a 63×/1.4 PL APO DIC oil lens and images were captured using Zen (Zeiss) software, and images were processed using the ImageJ/FIJI software package (Schindelin et al., 2012).
HEK cells were seeded onto sterile glass coverslips pretreated with 0.01% poly-L-lysine (Sigma-Aldrich) prior to induction and fixation. Cells were fixed in paraformaldehyde (4% [vol/vol] in PBS containing 5% [wt/vol] sucrose) for 10 min, washed three times with PBS, and then permeabilized in 0.1% [vol/vol] Triton X-100 in PBS for 10 min. Cells were then washed three times in PBS and incubated for 1 h with primary antibody diluted in 3% [wt/vol] BSA in PBS. Cells were again washed three times with PBS prior to secondary antibody incubation for 1 h in 3% [wt/vol] BSA in PBS (goat anti-mouse AlexaFluor 488 conjugate [Invitrogen] and goat anti-rabbit AlexaFluor 568 conjugate [Invitrogen]). Cells were washed five times with PBS, with 2.5 ng/µl Hoechst stain (Invitrogen) added in the second wash. Coverslips were then mounted to glass microscope slides with mounting media (90% [vol/vol] glycerol, 205 mM 1,4-diazabicyclo[2.2.2]octane, 80 mM Tris-Cl [pH 8.0]). Images were acquired at RT using a Lecia SP8 confocal microscope. Samples were mounted on a 63×/1.4 HC PL APO CS2 oil lens, images were captured using Leica LAS × SP8 software and processed using the ImageJ/FIJI software package (Schindelin et al., 2012). Line scan analysis was completed using Plot Profile in Fiji (Schindelin et al., 2012). For each cell, the fluorescent intensity (a.u.) of 488 and 552 nm channels of multiple 1 µm lines were recorded from mitochondrial tubules expressing HAX1WT-FLAG. Measurements were obtained from ≥28 cells, from three independent experiments. Pearson correlation (r) analysis was performed using Microsoft Excel; statistical analysis was performed using Prism software.
Antibodies
Antibodies used in this study are listed in Table 2.
Antibodies used in this study
| Antibodies | Source | Identifier |
|---|---|---|
| Rabbit polyclonal anti-AGK | GeneTex | AB_1949602 |
| Mouse monoclonal anti-ATP5A | Abcam | AB_301447 |
| Mouse monoclonal anti-ATP5β | Thermo Fisher Scientific | AB_221512 |
| Rabbit polyclonal anti-CLPB | Abcam | AB_235349 |
| Rabbit polyclonal anti-COXIV | Cell Signaling Technology | AB_2085424 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | AB_262044 |
| Rabbit polyclonal anti-HAX1 | Abcam | AB_2847902 |
| Mouse monoclonal anti-HTRA2 | R&D Systems | CAT NO. MAB1458 |
| Rabbit polyclonal anti-HTRA2 | Proteintech | AB_2122835 |
| Rabbit polyclonal anti-NDUFA9 | Mike Ryan, Monash University, Melbourne, Australia | N/A |
| Mouse monoclonal anti-OPA1 | BD Biosciences | AB_399888 |
| Mouse monoclonal anti-SDHA | Abcam | AB_301433 |
| Rabbit polyclonal anti-STOML2 | Proteintech | AB_2286822 |
| Rabbit polyclonal anti-TIMM29 | Sigma-Aldrich | AB_10963429 |
| Rabbit polyclonal anti-TIMM44 | Proteintech | AB_2204679 |
| Mouse monoclonal anti-TOMM20 | Santa Cruz Biotechnology | AB_628381 |
| Mouse monoclonal anti-TOMM22 | Santa Cruz Biotechnology | AB_2287717 |
| Mouse monoclonal anti-UQCRC1 | Abcam | AB_110252 |
| Rabbit polyclonal anti-YME1L1 | Proteintech | AB_2217459 |
Aggregation assay for MS
Mitochondrial protein aggregation was assessed using a pipeline formulated from previously described methods (Cupo and Shorter, 2020; Mahat et al., 2016; Wilkening et al., 2018). Cells were heat-shocked in cell culture at 42°C for 2 h in an atmosphere of 5% CO2 immediately prior to mitochondrial isolation. Following isolation, 250 µg mitochondrial pellets were resuspended in lysis buffer (0.5% [vol/vol] Triton X-100, 200 mM KCl, 30 mM Tris/HCl [pH 7.4], 0.5 mM PMSF, 5 mM EDTA, and 1× complete protease inhibitor [Roche]) at 10 mg/ml and incubated for 10 min at 4°C. Samples were then clarified at 20,000 g for 20 min at 4°C and supernatant was collected. The pellet was washed again in lysis buffer and the second supernatant was discarded. Supernatant and pellet samples were then prepared using the suspension-trapping (S-TRAP) system (ProtiFi; HaileMariam et al., 2018). Pellet fractions were resuspended in S-TRAP 1× solubilization buffer (5% [vol/vol] SDS, 50 mM triethylammonium bicarbonate [TEAB] pH 7.55) and 2× solubilization buffer was added to supernatant samples. Samples were then reduced and alkylated according to the manufacturer’s instructions before being loaded onto S-TRAP micro columns and digested with trypsin at 37°C overnight. Peptides were then eluted from the columns according to the manufacturer’s instructions and dried using a CentriVap Benchtop Vacuum Concentrator (Labconco) prior to reconstitution in 0.1% [vol/vol] trifluoroacetic acid (TFA), 2% [vol/vol] acetonitrile (ACN) for MS analysis.
Native IP
Isolated mitochondria were solubilized at 2 mg/ml in digitonin-containing solubilization buffer (20 mM Bis-Tris, 50 mM NaCl, 10% [vol/vol] glycerol, pH 7.4 and 1% [wt/vol] digitonin) supplemented with 1× complete protease inhibitor (Roche) and incubated on a rotary wheel for 45 min at 4°C. Insoluble debris was pelleted at 16,000 g for 30 min at 4°C and clarified supernatant was diluted 1:10 [vol/vol] in dilution buffer (20 mM Bis-Tris, 50 mM NaCl, 10% [vol/vol] glycerol, pH 7.4) to achieve a final digitonin concentration of 0.1%. Samples were then applied to M2 anti-FLAG resin (Sigma-Aldrich) that had been pre-equilibrated with wash buffer (20 mM Bis-Tris, 50 mM NaCl, 10% [vol/vol] glycerol, pH 7.4, 0.1% [wt/vol] digitonin) and mixed end-over-end for 2 h at 4°C. Following incubation, clarified supernatant (unbound protein) was removed and the resin was washed four times with wash buffer. Bound proteins were eluted from the resin in two successive 100 µl volumes with elution buffer (0.2 M glycine, pH 2.5), restored to neutral pH (1/10 vol 1 M Tris, pH 8.0), and precipitated with the addition of 1 ml ice-cold 100% acetone overnight, prior to preparation for MS.
Sample preparation for MS
For isolated mitochondria MS analysis, 50 µg mitochondrial pellets were solubilized, reduced, and alkylated in SDC buffer (1% [wt/vol] sodium deoxycholate [SDC], 100 mM Tris-Cl, pH 8.1, 40 mM chloroacetamide [CAA], and 10 mM tris(2-carboxyethyl)phosphine [TCEP]) by boiling for 5 min at 99°C with shaking followed by 15 min of sonication (Powersonic 603 Ultrasonic Cleaner, 40 KHz on high power) at RT. Proteins were digested with 1 µg trypsin (in 50 mM acetic acid) overnight at 37°C. Insoluble debris was removed via centrifugation at 20,000 g for 5 min at RT and clarified supernatant was loaded onto stage tips (Kulak et al., 2014) containing 3× styrene-divinylbenzene reversed phase sulfonate substrate (3M Empore [Sigma-Aldrich]) plugs. Tryptic peptides were extracted upon addition of 100 µl isopropanol, 1% [vol/vol] TFA to stage tips, and bound following centrifugation at 3,000 g at RT. Stage tips were washed with 100 µl isopropanol, 1% [vol/vol] TFA, then 100 µl 0.2% [vol/vol] TFA at 3,000 g at RT, followed by elution in 100 µl 80% [vol/vol] ACN, 1% [vol/vol] ammonium hydroxide at 3,000 g at RT. Samples were acidified to 1% [vol/vol] TFA and dried using a CentriVap Benchtop Vacuum Concentrator (Labconco) and reconstituted in 40 µl 0.1% [vol/vol] TFA, 2% [vol/vol] ACN. Reconstituted samples were sonicated for 15 min as above and clarified via centrifugation at 20,000 g for 10 min at RT before being transferred to sample vials for analysis.
For acetone-precipitated IP eluates, samples were resuspended in urea/ABC buffer (8 M urea, 50 mM ammonium bicarbonate [ABC], pH 8.0) and solubilized via sonication for 15 min at RT. Proteins were reduced and alkylated with 5 mM TCEP, 50 mM CAA, respectively, followed by incubation at 37°C for 30 min with shaking. 50 mM ABC buffer was added to samples to dilute urea concentration to 2 M, and proteins were digested with 1 µg trypsin (in 50 mM acetic acid) overnight at 37°C. Stage tips (Kulak et al., 2014) containing 2× styrene-divinylbenzene cross-linked copolymer substrate (3M Empore [Sigma-Aldrich]) plugs were made and activated with 50 µl 100% ACN and equilibrated with 50 µl 0.1% [vol/vol] TFA, 2% [vol/vol] ACN, both centrifuged at 2,500 g at RT. Following digest, samples were acidified to 1% [vol/vol] TFA and centrifuged at 20,000 g for 5 min at RT to pellet insoluble debris before supernatant was loaded onto stage tips. Tryptic peptides were bound to substrate via centrifugation at 2,500 g at RT, followed by a wash spin with 100 µl 0.1% [vol/vol] TFA, 2% [vol/vol] can, and elution in 100 µl 0.1% [vol/vol] TFA, 2% [vol/vol] ACN. Eluted peptides were dried using a CentriVap Benchtop Vacuum Concentrator (Labconco) and reconstituted in 15 µl 0.1% [vol/vol] TFA, 2% [vol/vol] ACN before being prepared for analysis as above.
Liquid chromatography and tandem MS (LC-MS/MS) specifications and data analysis
LC-MS/MS analysis of isolated mitochondria, IP, and aggregation assay samples was performed on a QExactive plus Orbitrap mass spectrometer (Thermo Fisher Scientific) with a nanoESI interface in combination with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC system was equipped with an Acclaim Pepmap nano-trap column (Dinoex-C18, 100 Å, 75 µm × 2 cm) and an Acclaim Pepmap RSLC analytical column (Dinoex-C18, 100 Å, 75 µm × 50 cm). Samples were injected into the enrichment column at an isocratic flow of 5 µl/min of 2% [vol/vol] ACN containing 0.1% vol/vol formic acid for 5 min applied before the enrichment column was switched in-line with the analytical column. The eluents were 5% [vol/vol] DMSO in 0.1% [vol/vol] formic acid (solvent A) and 5% DMSO in 100% [vol/vol] ACN and 0.1% [vol/vol] formic acid (solvent B). For isolated mitochondria and aggregation assay samples, the flow gradient was (i) 0–6 min at 2% B, (ii) 6–35 min, 2–23% B, (iii) 35–45 min 23–40% B, (iv) 45–50 min, 40–80% B, (v) 50–55 min, 80-80% B, and (vi) 55–65 min, 80-2% and was equilibrated at 2% B for 10 min before the next sample injection. For IP samples, the flow gradient was (i) 0–6 min at 2% B, (ii) 6–65 min, 2–23% B, (iii) 65–75 min, 23–40% B, (iv) 75–80 min, 40–80% B, (v) 80–85 min, 80-80% B, and (vi) 85–95 min, 80-2% and was equilibrated at 2% B for 10 min before the next sample injection. The mass spectrometer was operated in the data-dependent mode, full MS spectra were acquired in positive mode, 70,000 resolution, an automatic gain control (AGC) target of 3e6, and maximum injection time (IT) of 50 ms. The top 15 most intense peptide ions with z ≥ 2 and intensity threshold of 1.7e4 were subject to MS/MS using high-energy collision dissociation. The isolation window was set at 1.2 m/z and precursors were fragmented using a normalized collision energy of 30%. MS/MS resolution was set at 17,500 with AGC target of 1e5 and maximum IT of 100 ms. Dynamic exclusion was set at 30 s.
Raw files were analyzed using the MaxQuant platform (version 1.6.5.0; Tyanova et al., 2016a) and were searched against the Uniprot human protein database (canonical and isoforms [reviewed], downloaded March 2021). Default LFQ search parameters were applied, with a minimum LFQ ratio count of 2 and “match between runs” enabled in all cases. Subsequent statistical analysis was performed using the Perseus platform (version 1.6.15.0; Tyanova et al., 2016b). Briefly, the “proteinGroups.txt” output file from MaxQuant was loaded into Perseus, and hits labeled as “Only identified by site,” “Reverse,” or “Potential contaminant” was removed. LFQ intensities were Log2 transformed, replicates were labeled according to the experimental group (i.e., wild type or KO), and mitochondrial proteins were annotated using the MitoCarta 3.0 database (Rath et al., 2021; matching by Protein ID). At this point, subsequent analysis differed depending on the experiment. For isolated mitochondria and aggregation experiments, matrix rows were filtered for ≥2 valid values in each experimental group, and rows were globally normalized against proteins annotated as “mitochondrial” (designated “+” in MitoCarta 3.0) using the “Subtract row cluster” function. Non-mitochondrial proteins were excluded, and mitochondrial (“+”) and mito-interacting proteins were retained prior to statistical testing. For IP experiments, the “Summary statistics (rows)” function was used to calculate the number of valid values in the experimental (e.g., FLAG-tagged) protein eluate, and rows were filtered for ≥2 valid values in the experimental group alone. Control group (e.g., wild type or no tag) LFQ values were then imputed from the normal distribution to fill missing or non-valid values (1.8 σ [SD] downshift, 0.3 σ width). As above, the matrix was then filtered for non-mitochondrial proteins prior to statistical testing. In both cases, unpaired two-sample t tests were performed with statistical significance determined by P value (P value <0.05), displayed as a volcano plot using the “Scatter plot” function with “Student’s t test difference” on the x-axis and “−Log Student’s t test P value” on the y-axis.
Topographical mapping of Log2-transformed LFQ intensities to OXPHOS complexes I, III, and IV Protein Data Bank (PDB) structures (CI: PDB 5LDW, CIII: PDB 5XTE, CIV: PDB 5Z62) was performed using python scripts as described previously (Stroud et al., 2016).
Quantification and statistical analysis
Statistical analysis of LFQ MS data was performed in the Perseus software suit, and unpaired two-sample t tests were utilized to determine statistical significance (P value <0.05). Statistical significance throughout was performed in either Microsoft Excel or Prism software and calculated using either two-tailed Mann-Whitney tests or unpaired two-sample t tests where data is presented as mean ± SD. Quantification of HTRA2 foci was carried out in the ImageJ/FIJI software package (Schindelin et al., 2012). Data distribution in proteomics datasets was visualized as a histogram to assess normality, but no formal testing was applied.
Online supplemental material
Fig. S1 contains CLPBKO HEK FlpIn-TREx indels, a schematic representation of CLPB domain structure, mitochondrial sub-fractionation and carbonate extraction analysis of HEK CLPBWT-FLAG, CLPBWA-FLAG, and CLPBWB-FLAG stable cell lines and a schematic representation of OPA1 splicing variants. Fig. S2 demonstrates HTRA2 insolubility in CLPBKO via western blot and contains CLPBKO ± HS supernatant proteomics data. Fig. S3 contains STOML2KO and HAX1KO HEK FlpIn-TREx indels, isolated mitochondrial proteomics data, mitochondrial sub-fractionation and carbonate extraction analysis, and + HS pellet proteomics data. Fig. S4 demonstrates the specificity and reproducibility of the CLPBKO SPY defect, compares YME1L1 substrate abundance between CLPBKO and STOML2KO, and clarifies the STOML2WT-FLAG interactome. Fig. S5 shows that the rate of HAX1 turnover is dampened in both CLPBKO and STOML2KO and demonstrates a severe CI/CIV defect in STOML2/CLPBdKO cells via isolated mitochondrial proteomics and BN-PAGE analysis. Table S1 contains HEK CLPBKO, STOML2KO, HAX1KO, and STOML2/CLPBdKO proteomic datasets. Table S2 contains ± HS supernatant and pellet proteomics data for CLPBKO, CLPBWT-FLAG, STOML2KO, and HAX1KO. Table S3 contains normalized solubility data for CLPBKO, CLPBWT-FLAG, STOML2KO, and HAX1KO ± HS supernatant and pellet proteomics. Table S4 contains CLPBWT-FLAG, CLPBWA-FLAG, CLPBWB-FLAG, and STOML2WT-FLAG native IP datasets. Table S5 contains STOML2/CLPBdKO, CLPB/STOML2dKO, and CLPB/HAX1dKO native IP datasets.
Data availability
All MS proteomics data have been deposited to the ProteomeXchange Consortium, via the PRIDE partner repository, with the dataset identifier PXD042313 (https://www.ebi.ac.uk/pride/archive/projects/PXD042313).
Acknowledgments
We thank the Bio21 Mass Spectrometry and Proteomics Facility for the provision of instrumentation, training, and technical support.
This research was supported by Medical Research Future Fund Genomics Health Futures Mission—2020 Genomics Health Futures Mission Grant Opportunity (2007959) to D. Stojanovski and D.R. Thorburn; National Health and Medical Research Council Ideas Grant (APP2021085) to D. Stojanovski, D.R. Thorburn, and A.E. Frazier; and Mito Foundation funding (Incubator Grant to D. Stojanovski; Large equipment grant to A.E. Frazier and D.R. Thorburn). T. Langer was supported by grants from the Deutsche Forschungsgemeinschaft (German Research Foundation)—F’OR2848 (A01) and the German-Israeli-Project (RA1028/10-2). M.J. Baker, A.J. Anderson, and J.J. Crameri are supported by Australian Government Research Training Program scholarships and by Mito Foundation PhD Top-Up scholarships. The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program.
Author contributions: Conceptualization: T. Langer and D. Stojanovski. Data Curation: N/A. Formal Analysis: M.J. Baker, A.J. Anderson, C.S. Palmer, J.J. Crameri, and A.E. Frazier. Funding Acquisition: D.R. Thorburn, T. Langer, D. Stojanovski, and A.E. Frazier. Investigation: M.J. Baker, K.U. Blau, A.J. Anderson, C.S. Palmer, L.F. Fielden, J.J. Crameri, D. Milenkovic, and A.E. Frazier. Methodology: M.J. Baker, K.U. Blau, A.J. Anderson, C.S. Palmer, L.F. Fielden, J.J. Crameri, D. Milenkovic, and A.E. Frazier. Project Administration: D. Stojanovski. Resources: D.R. Thorburn, A.E. Frazier, T. Langer, and D. Stojanovski. Software: N/A. Supervision: A.E. Frazier, T. Langer, and D. Stojanovski. Visualization: M.J. Baker, K.U. Blau, C.S. Palmer, J.J. Crameri, and A.E. Frazier. Validation: M.J. Baker, and K.U. Blau. Writing—Original Draft: M.J. Baker and D. Stojanovski. Writing—Review and Editing: All authors.

