


Phosphoinositide signaling lipids (PIPs) are key regulators of membrane identity and trafficking. Of these, PI(3,5)P2 is one of the least well-understood, despite key roles in many endocytic pathways including phagocytosis and macropinocytosis. PI(3,5)P2 is generated by the phosphoinositide 5-kinase PIKfyve, which is critical for phagosomal digestion and antimicrobial activity. However PI(3,5)P2 dynamics and regulation remain unclear due to lack of reliable reporters. Using the amoeba Dictyostelium discoideum, we identify SnxA as a highly selective PI(3,5)P2-binding protein and characterize its use as a reporter for PI(3,5)P2 in both Dictyostelium and mammalian cells. Using GFP-SnxA, we demonstrate that Dictyostelium phagosomes and macropinosomes accumulate PI(3,5)P2 3 min after engulfment but are then retained differently, indicating pathway-specific regulation. We further find that PIKfyve recruitment and activity are separable and that PIKfyve activation stimulates its own dissociation. SnxA is therefore a new tool for reporting PI(3,5)P2 in live cells that reveals key mechanistic details of the role and regulation of PIKfyve/PI(3,5)P2.
Introduction
The inositol phospholipids are a family of interconvertible signaling molecules that define distinct endocytic compartments. Differential phosphorylation at three positions on the inositol headgroup allows eight different signaling species to be generated. These are used to recruit specific effector proteins to membranes and are interconverted by a large family of lipid kinases and phosphatases. Transitions between different phosphoinositides (PIPs) are thus central components of the identity, sorting, and progression of almost all membrane compartments.
One of the least understood PIP transitions is the generation of PI(3,5)P2 by the phosphoinositide 5-kinase PIKfyve (Fab1p in yeast). PIKfyve is recruited to early endosomes via its FYVE (Fab1, YOTB, Vac1, and EEA1) domain that binds PI(3)P and subsequently phosphorylates it at the 5-position to produce PI(3,5)P2 (Cabezas et al., 2006; Sbrissa et al., 1999; Yamamoto et al., 1995). PIKfyve is in a complex with the scaffolding regulator Vac14 (ArPIKfyve) and the 5-phosphatase Fig4 (Sac3), which catalyzes the reverse reaction (Botelho et al., 2008; Rudge et al., 2004; Sbrissa et al., 2007). This complex is under intricate regulation, and while genetic, biochemical, and structural studies have provided some mechanistic insight (Berwick et al., 2004; Ikonomov et al., 2009; Lees et al., 2020; Sbrissa et al., 2000), the dynamics of PI(3,5)P2 formation in vivo have remained elusive due to the lack of a reliable live-cell reporter.
Disruption of PIKfyve causes severe and widespread defects in endocytic trafficking in all organisms studied, typified by the formation of swollen endosomes and defects in lysosomal degradation (Buckley et al., 2019; Choy et al., 2018; de Lartigue et al., 2009; Dove et al., 2009; Ikonomov et al., 2001; Kim et al., 2014; Krishna et al., 2016; Nicot et al., 2006). PIKfyve activity is therefore critical for several physiologically important pathways such as the digestion of autophagic and phagocytic cargo and endocytosed receptors (Buckley et al., 2019; de Lartigue et al., 2009; Demirsoy et al., 2017; Ferguson et al., 2009; Kim et al., 2014; McCartney et al., 2014b; Rusten et al., 2006). Unsurprisingly, disrupted PI(3,5)P2 signaling is implicated in a broad range of diseases (reviewed in McCartney et al., 2014a; Shisheva, 2012).
How these phenotypes result from PI(3,5)P2 deficiency is not clear. Several studies have indicated that PI(3,5)P2 can directly regulate lysosomal ion channels such as mucolipin (TRPML), two-pore calcium channels, and the chloride channel ClC7 (Dong et al., 2010; Leray et al., 2022; Samie et al., 2013; Wang et al., 2012). PI(3,5)P2 has also been shown to regulate association of the two vacuolar ATPase subcomplexes on the yeast vacuole, although this has not yet been confirmed in other organisms (Li et al., 2014). Loss of PI(3,5)P2 can also affect lysosomal transport and delivery to the compartments targeted for degradation (Li et al., 2016). Each mechanism can potentially contribute to the phenotypes observed, but our understanding is limited by poor knowledge of where and when PI(3,5)P2 is generated.
We and others have previously demonstrated a key and evolutionarily conserved role for PIKfyve during phagosome maturation. Phagocytosis is used by cells to engulf large extracellular particles such as microbes, dead cells, and debris, and is highly related to the process of macropinocytosis, whereby cells take up bulk extracellular fluid (King and Kay, 2019). After engulfment, both pathways follow a complex series of maturation steps to deliver antimicrobial and digestive components to defend against potential pathogens, recycle nutrients, or generate antigens for presentation (Buckley et al., 2016; Chen et al., 2010; Levin et al., 2016).
PIKfyve is important for phagosome–lysosome fusion in macrophages, neutrophils, and the amoeba Dictyostelium discoideum (Buckley et al., 2019; Dayam et al., 2015, 2017; Isobe et al., 2019; Kim et al., 2014). We recently showed that loss of PIKfyve in Dictyostelium severely disrupts phagosomal proteolysis and killing, rendering them hypersensitive to infection with Legionella pneumophila (Buckley et al., 2019). Whilst this demonstrates the importance of PIKfyve in innate immunity, its regulation remains poorly understood.
A major obstacle to understanding PI(3,5)P2 function and regulation has been the lack of a reliable reporter to visualize when and where it is generated in cells. Although a tandem repeat of the N-terminal lipid-binding region of TRPML showed PI(3,5)P2 specificity in vitro and looked promising in cells (Li et al., 2013), others found clear evidence of non-specific endosomal recruitment and retention after PI(3,5)P2 depletion (Hammond et al., 2015). We therefore set out to identify and validate an alternative reporter to faithfully describe PI(3,5)P2 dynamics within live cells. Here, we identify and characterize Dictyostelium SnxA as a highly selective PI(3,5)P2-binding protein. Using phagosome and macropinosomes maturation as accessible endocytic pathways, we use fluorescent SnxA fusions as reporters to visualize and describe PI(3,5)P2 dynamics in live cells for the first time and uncover key regulatory steps in both PIKfyve recruitment and activity.
Results
Identification of SnxA as a PI(3,5)P2-binding protein in Dictyostelium
To identify a specific PI(3,5)P2-binding protein that could be used as a reporter, we started with the assumption that such proteins would only be recruited to membranes in the presence of PIKfyve. As PX (Phox homology) domains are frequently used by proteins to bind specific PIPs, we searched the Dictyostelium genome for proteins containing a PX domain and screened C-terminally tagged GFP fusions of these for PIKfyve-dependent localization by expression in both WT and ΔPIKfyve cells.
Of the proteins cloned, most localized to membranes irrespective of the presence or absence of PIKfyve (e.g., Fig. 1 A). However, one (DDB_G0289833) clearly localized to large vesicles with the appearance of macropinosomes in WT but was completely cytosolic in ΔPIKfyve cells (Fig. 1 B). DDB_G0289833 is previously unstudied and contains a coiled-coil domain in addition to the PX domain. As this domain combination is found in many mammalian sorting nexins, we named the Dictyostelium protein SnxA. To further confirm the dependence of SnxA recruitment on PIKfyve, we applied the PIKfyve inhibitor apilimod (Cai et al., 2013) to WT cells expressing SnxA-GFP. This caused rapid dissociation of SnxA-GFP from all membranes, becoming completely cytosolic within 10 min (Fig. 1, C and D).

Identification of SnxA as a PI(3,5)P2reporter. (A and B) Expression of GFP-fusions of PX-domain-containing proteins in WT and ΔPIKfyve Ax3 Dictyostelium. DDB_G0285711 (A) localized to large vesicles in both cells, whereas DDB_G0289833 (B) was completely cytosolic in ΔPIKfyve. (C) Timelapse of DDB_G0289833 (SnxA)-GFP localization in Ax2 cells after addition of the PIKfyve inhibitor apilimod (3 μM). (D) Quantification of the number of SnxA-GFP vesicles per cell. >90 cells measured per time-point; data show mean ± SEM of three independent experiments. (E) Representative PIP array using lysates from WT cells expressing SnxA-GFP and probed with anti-GFP antibody. (F) Schematics of SnxA-GFP, 1xPX-GFP, and 2xPX-GFP. (G) Representative images of their localization in the cells indicated. 1xPX-GFP remained predominately cytosolic in WT cells, whereas 2xPX-GFP localized strongly to large vesicles, which was lost in ΔPIKfyve and apilimod treated cells. All scale bars = 2 μm. Source data are available for this figure: SourceData F1.
Identification of SnxA as a PI(3,5)P2reporter. (A and B) Expression of GFP-fusions of PX-domain-containing proteins in WT and ΔPIKfyve Ax3 Dictyostelium. DDB_G0285711 (A) localized to large vesicles in both cells, whereas DDB_G0289833 (B) was completely cytosolic in ΔPIKfyve. (C) Timelapse of DDB_G0289833 (SnxA)-GFP localization in Ax2 cells after addition of the PIKfyve inhibitor apilimod (3 μM). (D) Quantification of the number of SnxA-GFP vesicles per cell. >90 cells measured per time-point; data show mean ± SEM of three independent experiments. (E) Representative PIP array using lysates from WT cells expressing SnxA-GFP and probed with anti-GFP antibody. (F) Schematics of SnxA-GFP, 1xPX-GFP, and 2xPX-GFP. (G) Representative images of their localization in the cells indicated. 1xPX-GFP remained predominately cytosolic in WT cells, whereas 2xPX-GFP localized strongly to large vesicles, which was lost in ΔPIKfyve and apilimod treated cells. All scale bars = 2 μm. Source data are available for this figure: SourceData F1.
PIKfyve activity is required for the formation of both PI(3,5)P2 and PI(5)P. To determine the lipid-binding specificity of SnxA, we examined binding to PIP arrays using lysates from SnxA-GFP expressing Dictyostelium cells. In this assay, SnxA-GFP bound with at least a 20-fold preference to PI(3,5)P2 over all other PIPs with almost no non-specific binding at even the highest lipid concentrations (Fig. 1 E).
To optimize lipid binding and reporter characteristics, we then tested GFP-fusions of either the PX domain alone or a tandem repeat of this domain (1xPX and 2xPX-GFP, respectively, Fig. 1 F). 1xPX-GFP had low contrast, localizing poorly to large vesicles in some cells, but was completely cytosolic in many others. Contrast was significantly enhanced in the 2xPX construct, which localized with similar characteristics and PIKfyve-dependence as full-length SnxA-GFP (Fig. 1 G). The 2xPX-GFP construct also bound similarly to PIP arrays, localizing specifically to PI(3,5)P2 (Fig. S1 A).
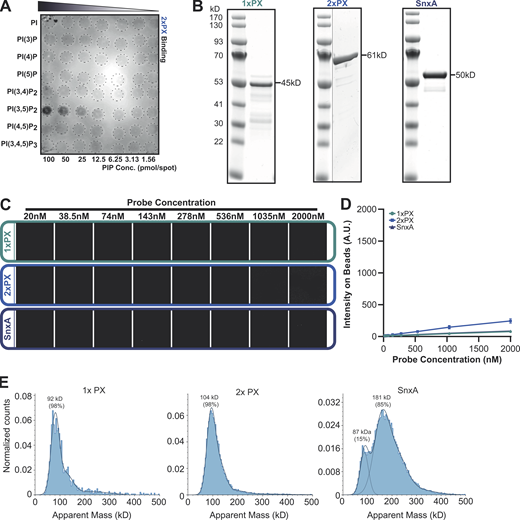
SnxA does not bind PI(5)P. (A) PIP array performed using lysates from WT cells expressing 2xPX-GFP and probed with anti-GFP antibody. (B) Coomassie-stained SDS-PAGE gels of recombinant GFP fusion SnxA probes used for lipid binding assays. (C) Representative images of SnxA probe titrated against PI(5)P-containing membrane-coated beads. (D) This is quantified and plotted as the mean ± SD from three independent experiments. (E) Mass photometry analysis of recombinant SnxA fusions. Note that exact molecular weights of native proteins are inaccurate below ∼100 kD using this technique. Source data are available for this figure: SourceData FS1.

SnxA does not bind PI(5)P. (A) PIP array performed using lysates from WT cells expressing 2xPX-GFP and probed with anti-GFP antibody. (B) Coomassie-stained SDS-PAGE gels of recombinant GFP fusion SnxA probes used for lipid binding assays. (C) Representative images of SnxA probe titrated against PI(5)P-containing membrane-coated beads. (D) This is quantified and plotted as the mean ± SD from three independent experiments. (E) Mass photometry analysis of recombinant SnxA fusions. Note that exact molecular weights of native proteins are inaccurate below ∼100 kD using this technique. Source data are available for this figure: SourceData FS1.
To confirm PI(3,5)P2 specificity using more physiologically relevant membranes, all three GFP fusions were produced recombinantly (Fig. S1 B) and screened for binding to beads coated with bilayers consisting of 5% phosphoinositide and 95% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine. Lipid binding and affinities were then determined by measuring GFP recruitment by microscopy (Maib and Murray, 2022). As anticipated, both SnxA-GFP and 2xPX-GFP probes showed high specificity for PI(3,5)P2, while 1xPX showed very little binding (Fig. 2, A–D). By titrating the probe concentration at a fixed amount of membrane, we determined an apparent Kd of 187.3 ± 13 nM for full-length SnxA and 217.5 ± 7 nM for 2xPX (Fig. 2, E and F). Importantly, no binding to PI(5)P, the other product of PIKfyve, was observed under any conditions (Fig. S1, C and D).

PI(3,5)P 2 binding specificity. (A–D) Representative images and quantification of binding of recombinant GFP-SnxA probes to membrane-coated beads containing the indicated PIPs. (E) Binding of each SnxA probe titrated against beads harboring PI(3,5)P2. (F) Intensities measured under identical imaging conditions and apparent Kds. All graphs show the mean ± SD from three independent experiments; scale bars indicate 5 μm.
PI(3,5)P 2 binding specificity. (A–D) Representative images and quantification of binding of recombinant GFP-SnxA probes to membrane-coated beads containing the indicated PIPs. (E) Binding of each SnxA probe titrated against beads harboring PI(3,5)P2. (F) Intensities measured under identical imaging conditions and apparent Kds. All graphs show the mean ± SD from three independent experiments; scale bars indicate 5 μm.
These data demonstrate that the PX domain of SnxA is a highly selective PI(3,5)P2-binding domain. Although a tandem repeat of this domain increases binding affinity, this unexpectedly provides no improvement over the full-length protein. We therefore tested whether the full-length protein dimerizes by measuring the mass distribution of the recombinant fusion proteins by mass photometry (Sonn-Segev et al., 2020). While the 1x and 2xPX GFP fusions existed as a single population with apparent molecular weights of 92 and 104 kD, respectively, 85% of the full-length SnxA protein had an apparent molecular weight of 181 kD—approximately double that expected for a monomer. Full-length SnxA likely dimerizes via its coiled-coil domain, providing an avidity similar to the 2xPX construct. This may be advantageous for imaging as it will incorporate two GFP-moieties into each dimer and increase brightness, although it is also possible that full-length SnxA could dimerize with other proteins within cells.
Validation of SnxA as a PI(3,5)P2 reporter in mammalian cells
Our data in Dictyostelium indicate that SnxA has high selectivity for both PI(3,5)P2 and reversible binding, indicated by rapid dissociation upon PIKfyve inhibition. To test whether it could be universally used as a PI(3,5)P2 reporter in other organisms, we made human-codon-optimized versions of both the full-length SnxA and the 2xPX construct to express as N-terminal GFP-fusions in a panel of mammalian cell lines.
In all cell lines examined, both full-length SnxA and 2xPX GFP fusions localized clearly to large intracellular vesicles (Fig. 3). The localization was almost completely lost upon inhibition of PIKfyve with apilimod, and although a small number of GFP-positive puncta were observed in some cells, these were much smaller and resembled aggregates, with no visible lumen (see Fig. 3, A, C and E, zoom panels). Effective inhibition of PIKfyve was confirmed by the presence of typically swollen vacuoles visible in these cells.

Expression of SnxA fusions in mammalian cells. (A–F) Representative images and quantification of either full-length SnxA or 2xPX GFP fusions expressed in either HEK293, MIA PaCa-2, or RPE-1 cells. Top panels of A, C, and E are maximum intensity projections of a spinning-disc Z-stack, with single sections and enlargements of the boxed regions below. Cells were either left untreated (−) or pretreated with 1 μM apilimod (Ap; +) for 1 h prior to imaging. Arrowheads indicate the GFP-positive structures that were quantified over three independent experiments in B, D, and F. 30 cells per experiment for three independent experiments were scored for each condition and P values were calculated by unpaired T test (n = 3, *P < 0.05, **P < 0.01); scale bars represent 10 μm. (G) Shows an image from a timeseries (Video 1) of a HEK293 cell expressing GFP-SnxA just after addition of Texas-Red dextran. Scale bars represent 2 μm.
Expression of SnxA fusions in mammalian cells. (A–F) Representative images and quantification of either full-length SnxA or 2xPX GFP fusions expressed in either HEK293, MIA PaCa-2, or RPE-1 cells. Top panels of A, C, and E are maximum intensity projections of a spinning-disc Z-stack, with single sections and enlargements of the boxed regions below. Cells were either left untreated (−) or pretreated with 1 μM apilimod (Ap; +) for 1 h prior to imaging. Arrowheads indicate the GFP-positive structures that were quantified over three independent experiments in B, D, and F. 30 cells per experiment for three independent experiments were scored for each condition and P values were calculated by unpaired T test (n = 3, *P < 0.05, **P < 0.01); scale bars represent 10 μm. (G) Shows an image from a timeseries (Video 1) of a HEK293 cell expressing GFP-SnxA just after addition of Texas-Red dextran. Scale bars represent 2 μm.
We also noted that upon paraformaldehyde fixation, GFP-SnxA was also partly enriched near the plasma membrane (Fig. S2 A). This was insensitive to apilimod treatment and did not occur with the GFP-2xPX probe. A similar localization was also never visible in unfixed cells (e.g., Fig. 3). Plasma membrane enrichment is therefore due to non-specific binding via the coiled-coil domain rather than an additional pool of PI(3,5)P2. As this localization is sufficiently distinct from endosomal PI(3,5)P2 pools it does not prevent use of GFP-SnxA as a probe for experiments where fixation is necessary; however, we advocate caution interpreting any observations at or near the cell surface and the use of appropriate controls.

Characterization of SnxA probes. (A) Confocal images of HEK293 cells transfected with either full-length or 2xPX-GFP fusions ± apilimod after fixation with paraformaldehyde. (B) Phagosomal proteolysis of both Ax2 (WT) and ΔPIKfyve Dictyostelium cells expressing different SnxA-derived probes, measured by digestion of bead-conjugated DQ-BSA. Data show mean ± SD of three independent experiments. (C) Images of macropinosomes in Ax2 cells expressing each probe or ΔPIKfyve cells after overnight incubation in Texas-Red dextran. The bright vesicles indicate concentration of macropinosomes contents. All scale bars represent 2 μm.

Characterization of SnxA probes. (A) Confocal images of HEK293 cells transfected with either full-length or 2xPX-GFP fusions ± apilimod after fixation with paraformaldehyde. (B) Phagosomal proteolysis of both Ax2 (WT) and ΔPIKfyve Dictyostelium cells expressing different SnxA-derived probes, measured by digestion of bead-conjugated DQ-BSA. Data show mean ± SD of three independent experiments. (C) Images of macropinosomes in Ax2 cells expressing each probe or ΔPIKfyve cells after overnight incubation in Texas-Red dextran. The bright vesicles indicate concentration of macropinosomes contents. All scale bars represent 2 μm.
Although both fusion proteins were recruited to vesicles of comparable size, significantly fewer structures were often observed in cells expressing GFP-2xPX than those expressing the full-length GFP-SnxA. This varied between cell lines; both reporters behaved similarly in MIA PaCa-2 pancreatic cancer cells, whereas the 2xPX-GFP was recruited to very few structures in both retinal pigment epithelial (RPE-1) and human embryonic kidney (HEK293) cells. As some cell lines were unaffected, the differences in others must lie in negative effects of reporter expression rather than different PI(3,5)P2 binding dynamics. We also noticed that cells expressing GFP-2xPX grew more slowly and generally appeared less healthy than untransfected or GFP-SnxA–expressing cells. Therefore, while a tandem PX domain construct may be a less complex reporter for PI(3,5)P2, it has a greater potential to disrupt cellular function than full-length SnxA. The degree of disruption will depend on factors such as expression levels and cell type and should be assessed for each specific experimental context, as for all such probes.
As the large vesicles observed resemble macropinosomes, we tested this by incubating HEK293 cells with 70 kD Texas-Red dextran, which is predominantly taken up by this route. Dextran clearly labeled all GFP-SnxA positive vesicles, indicating that they are macropinocytic in origin. However, not all dextran-containing vesicles had GFP-SnxA, indicating that PI(3,5)P2 is only transiently present (Fig. 3 G and Video 1). Although PI(3,5)P2 is thought to be a general component of late endosomal/lysosomal pathways, we did not observe small puncta that could represent classical endocytic compartments. See the discussion for potential explanations.
Macropinocytosis in HEK293 cells expressing GFP-SnxA (white) directly following addition of 2.5 mg/ml Texas-Red dextran (red), related toFig. 3 G . Images were captured every 60 s by spinning-disc microscopy and displayed at four frames per second. Scale bar = 2 μm.
Macropinocytosis in HEK293 cells expressing GFP-SnxA (white) directly following addition of 2.5 mg/ml Texas-Red dextran (red), related toFig. 3 G . Images were captured every 60 s by spinning-disc microscopy and displayed at four frames per second. Scale bar = 2 μm.
SnxA-probes do not disrupt PIKfyve signaling in Dictyostelium
To validate SnxA as a PI(3,5)P2 reporter in detail we returned to the Dictyostelium model system. As disruption of PIKfyve blocks both phagosomal proteolysis and the concentration of macropinosome content, we started by testing whether expression of SnxA-GFP fusions also affected these processes (Buckley et al., 2019). Neither full-length SnxA, 1xPX, nor 2xPX GFP-fusions had deleterious effects in either assay, and we did not notice any obvious changes to cell morphology or growth (Fig. S2, C and D). Therefore none of the SnxA probes appear to disrupt PI(3,5)P2 signaling in Dictyostelium.
We also generated multiple independent SnxA knockouts and asked whether SnxA played an important function in phagosome maturation itself. Unlike ΔPIKfyve cells, ΔSnxA mutants had no significant defects in bacterial growth, bulk fluid uptake, phagocytosis, phagosome proteolysis, or acidification (Fig. S3, A–G). Following an extensive investigation, the only difference we could observe between WT and ΔSnxA cells was a subtle but reproducible alteration in the dynamics of macropinosome shrinkage after internalization: whilst macropinosomes in WT cells transiently enlarge after internalization before shrinking and concentrating their content, the expansion phase does not occur in ΔSnxA cells and can be rescued by re-expression of SnxA-GFP (Fig. S3, H and I). Despite this, bulk fluid uptake is unaffected and no axenic growth defect was observed. We speculate that the transient swelling indicates SnxA-dependent fusion of another endocytic compartment to very early macropinosomes, but this requires further investigation. In any case, SnxA is not necessary for maturation of either phagosomes or macropinosomes and, therefore, other effectors of PIKfyve must exist.

Loss of SnxA has little effect on phagosome or macropinosome maturation. (A) Images of WT and ΔsnxA-1 colonies growing on a lawn of K. aerogenes. Average colony diameter over time is shown below. Scale bar = 10 mm. (B) Growth of mutant cells in different bacteria. Numbers indicate the number of Dictyostelium cells seeded on each well. (C) Summary of growth on a range of bacteria screened as in B. (D) Quantification of endocytosis calculated by FITC dextran uptake. (E) Phagocytosis of 1 μm fluorescent beads, measured by flow cytometry and normalized to WT cells at 60 min. (F and G) Phagosomal proteolysis (F) and pH (G) over time, measured by fluorescence changes after engulfment of either DQ-BSA or FITC/Alexa594 reporter beads, respectively. (H) Representative images of macropinosomes shrinking after a 2-min (mins) pulse of TRITC-dextran; scale bar = 2 μm. (I) Size is quantified. All data shown are the mean ± SD of at least three independent experiments. ΔsnxA-1, 2, and 3 indicate independent knockout clones.

Loss of SnxA has little effect on phagosome or macropinosome maturation. (A) Images of WT and ΔsnxA-1 colonies growing on a lawn of K. aerogenes. Average colony diameter over time is shown below. Scale bar = 10 mm. (B) Growth of mutant cells in different bacteria. Numbers indicate the number of Dictyostelium cells seeded on each well. (C) Summary of growth on a range of bacteria screened as in B. (D) Quantification of endocytosis calculated by FITC dextran uptake. (E) Phagocytosis of 1 μm fluorescent beads, measured by flow cytometry and normalized to WT cells at 60 min. (F and G) Phagosomal proteolysis (F) and pH (G) over time, measured by fluorescence changes after engulfment of either DQ-BSA or FITC/Alexa594 reporter beads, respectively. (H) Representative images of macropinosomes shrinking after a 2-min (mins) pulse of TRITC-dextran; scale bar = 2 μm. (I) Size is quantified. All data shown are the mean ± SD of at least three independent experiments. ΔsnxA-1, 2, and 3 indicate independent knockout clones.
PI(3,5)P2 dynamics during phagosome and macropinosome maturation
To test whether the large SnxA-GFP–positive vesicles observed in Dictyostelium were also macropinosomes and determine at which point PI(3,5)P2 is present, we labeled macropinosomes with a 2-min pulse of Texas-Red dextran and then imaged different fields of view at time intervals after dextran removal. For optimal imaging and uniform expression, we generated a stably integrated SnxA-GFP cell line selecting for a clone with low expression/high contrast for microscopy experiments. This allows the percentage of macropinosomes positive for GFP to be measured for up to 10 min of maturation (Buckley et al., 2016). This demonstrated that SnxA-GFP is transiently recruited to macropinosomes within 2 min of engulfment, peaking at 4 min before removal after 5–6 min (Fig. 4, A and B).

PI(3,5)P 2 dynamics during macropinosome and phagosome maturation. (A) Recruitment of SnxA-GFP in Dictyostelium cells following a 2-min dextran pulse-chase to label macropinosomes. Different fields of view were taken at time points after dextran washout. (B) The proportion of macropinosomes positive for SnxA-GFP at each time after dextran addition. >40 vesicles were scored per time point for each of three independent experiments. (C and D) Images from timelapse microscopy of SnxA-GFP during phagocytosis of pHrodo-yeast (red) in WT (C) and ΔPIKfyve (D) cells. See Video 2 for the complete sequence. (E) Quantification of SnxA-GFP enrichment at the phagosomal membrane averaged over a total of 51 WT and 31 ΔPIKfyve phagosomes across three independent experiments (**P < 0.01, ***P < 0.005. Fold enrichment normalized to 10 s before engulfment. Graph shows the mean ± SEM across independent experiments; thin lines show each the individual repeats. (F) Example long-term timelapse of SnxA-GFP recruitment to phagosomes. (G) Quantification of 20 phagocytic events followed in this way over three experiments and scored for SnxA-GFP localization over time. All scale bars = 2 μm and all graphs show the mean ± SEM of three independent experiments unless otherwise stated.
PI(3,5)P 2 dynamics during macropinosome and phagosome maturation. (A) Recruitment of SnxA-GFP in Dictyostelium cells following a 2-min dextran pulse-chase to label macropinosomes. Different fields of view were taken at time points after dextran washout. (B) The proportion of macropinosomes positive for SnxA-GFP at each time after dextran addition. >40 vesicles were scored per time point for each of three independent experiments. (C and D) Images from timelapse microscopy of SnxA-GFP during phagocytosis of pHrodo-yeast (red) in WT (C) and ΔPIKfyve (D) cells. See Video 2 for the complete sequence. (E) Quantification of SnxA-GFP enrichment at the phagosomal membrane averaged over a total of 51 WT and 31 ΔPIKfyve phagosomes across three independent experiments (**P < 0.01, ***P < 0.005. Fold enrichment normalized to 10 s before engulfment. Graph shows the mean ± SEM across independent experiments; thin lines show each the individual repeats. (F) Example long-term timelapse of SnxA-GFP recruitment to phagosomes. (G) Quantification of 20 phagocytic events followed in this way over three experiments and scored for SnxA-GFP localization over time. All scale bars = 2 μm and all graphs show the mean ± SEM of three independent experiments unless otherwise stated.
We then observed SnxA-GFP recruitment to phagosomes using timelapse confocal microscopy of cells engulfing pHrodo-labeled yeast. Unlike macropinosomes, individual yeast containing phagosomes could be reliably followed over time, so we also developed an automated image analysis pipeline to measure the fluorescence intensity in a ring around the yeast and average over multiple events (Fig. S4, A–C). This showed that phagosomes also acquire SnxA-GFP 2 min after engulfment, with no enrichment observed in ΔPIKfyve controls (Fig. 4, C–E; and Video 2). In contrast to the rapid loss from macropinosomes, longer-term time lapses indicated that SnxA-GFP then remained on the phagosomal membrane for a further 20 min (Fig. 4, F and G). Therefore, although the early stages of macropinosome and phagosome maturation appear identical, the timing then appears to diverge.

Quantification of phagosomal recruitment and comparison of SnxA and 2xPX. (A) Shows automated methodology to quantify fluorescent reporter recruitment to a phagocytic event. Channel containing yeast (red) is thresholded and particles larger than 1.5 μm2 are segmented (white circles). (B) This area is used to monitor fluorescence change of pHrodo-labeled-yeast, a proxy for phagosomal pH. (C) The perimeter is enlarged by 0.2 μm to generate a ring of 0.3 μm thickness over the phagosomal membrane (inner ring). As a control, an additional ring further away from the yeast (outer ring, expanded by 0.85 μm from the yeast) was also generated to give a measure of background cytosol fluorescence that should remain constant. (D) Timelapse of 2xPX-GFP recruitment to a pHrodo-labeled yeast containing phagosome in WT cells. (E) An identical experiment in ΔPIKfyve mutants. (F) Quantification of phagosomal enrichment in both cell lines. N indicates the total number of phagosomes measured, across three independent experiments. *P < 0.05, ***P < 0.005. Graph shows the mean ± SEM across independent experiments; thin lines show each of the individual repeats. (G) Timelapse of phagocytosis in WT cells coexpressing SnxA-GFP and 2xPX-RFP (see Video 3 for full timeseries). (H) Quantification of probe intensities over time in this video.

Quantification of phagosomal recruitment and comparison of SnxA and 2xPX. (A) Shows automated methodology to quantify fluorescent reporter recruitment to a phagocytic event. Channel containing yeast (red) is thresholded and particles larger than 1.5 μm2 are segmented (white circles). (B) This area is used to monitor fluorescence change of pHrodo-labeled-yeast, a proxy for phagosomal pH. (C) The perimeter is enlarged by 0.2 μm to generate a ring of 0.3 μm thickness over the phagosomal membrane (inner ring). As a control, an additional ring further away from the yeast (outer ring, expanded by 0.85 μm from the yeast) was also generated to give a measure of background cytosol fluorescence that should remain constant. (D) Timelapse of 2xPX-GFP recruitment to a pHrodo-labeled yeast containing phagosome in WT cells. (E) An identical experiment in ΔPIKfyve mutants. (F) Quantification of phagosomal enrichment in both cell lines. N indicates the total number of phagosomes measured, across three independent experiments. *P < 0.05, ***P < 0.005. Graph shows the mean ± SEM across independent experiments; thin lines show each of the individual repeats. (G) Timelapse of phagocytosis in WT cells coexpressing SnxA-GFP and 2xPX-RFP (see Video 3 for full timeseries). (H) Quantification of probe intensities over time in this video.
PI(3,5)P2 dynamics during phagosome maturation. WT (left) and ΔPIKfyve (right) cells expressing the SnxA-GFP probe for PI(3,5)P2 (white). Red particles are pHrodo-labeled yeast. Note the complete loss of localization in the mutants. Relates to Fig. 4, C and D. Images were captured every 5 s by spinning-disc microscopy and displayed at seven frames per second. Scale bar = 2 μm.
PI(3,5)P2 dynamics during phagosome maturation. WT (left) and ΔPIKfyve (right) cells expressing the SnxA-GFP probe for PI(3,5)P2 (white). Red particles are pHrodo-labeled yeast. Note the complete loss of localization in the mutants. Relates to Fig. 4, C and D. Images were captured every 5 s by spinning-disc microscopy and displayed at seven frames per second. Scale bar = 2 μm.
The quantitative analysis of phagosome maturation also allowed us to compare recruitment of 2xPX-GFP to full-length SnxA-GFP. This showed that 2xPX-GFP was recruited to phagosomes with identical dynamics to the full-length SnxA, but whereas SnxA-GFP remained stably associated with the phagosome (Fig. 4 E), the intensity of 2xPX-GFP slowly decreased after 3 min (Fig. S4, D–F). Co-expression of 2xPX-RFP in cells expressing SnxA-GFP showed that this effect was dominant as both reporters were then lost with comparable dynamics (Fig. S4, G and H; and Video 3). Therefore, 2xPX expression again exhibited a greater tendency to interfere with PI(3,5)P2 signaling than SnxA-GFP, consistent with our observations in mammalian cells. However, in Dictyostelium at least, this was relatively minor and insufficient to detectably perturb phagosomal digestion. As 2xPX had superior contrast to full-length SnxA as an RFP fusion, with indistinguishable initial recruitment, it is therefore still useful in some circumstances, with appropriate caution.
Colocalization of SnxA-GFP (green) and 2xPX-RFP (magenta) to a newly formed phagosome containing an Alexa405-labeled yeast (blue) in a WTDictyostelium cell. Images were captured every 20 s by spinning-disc microscopy and displayed at a rate of seven frames per second. Scale bar = 2 μm.
Colocalization of SnxA-GFP (green) and 2xPX-RFP (magenta) to a newly formed phagosome containing an Alexa405-labeled yeast (blue) in a WTDictyostelium cell. Images were captured every 20 s by spinning-disc microscopy and displayed at a rate of seven frames per second. Scale bar = 2 μm.
The SnxA reporter reveals complex coordination of PIKfyve recruitment and activity
PI(3,5)P2 is generated by phosphorylation of PI(3)P, which is both the substrate of PIKfyve and responsible for its recruitment via the FYVE domain. SnxA allows us to observe for the first time PI(3,5)P2 accumulation in live cells, so we next asked how this relates to both PI(3)P and PIKfyve dynamics.
To compare the relative dynamics of PI(3)P and PI(3,5)P2, we co-expressed SnxA-GFP with the PI(3)P reporter RFP-2xFYVE (Gillooly et al., 2000) and followed early phagosome maturation. Although RFP-2xFYVE expression caused a delay in SnxA-GFP recruitment compared with our previous experiments, it was clearly recruited first, immediately following engulfment (Fig. 5, A and B; and Video 4). SnxA-GFP was then enriched on the phagosome several minutes later (Fig. 5, A and B; and Video 4). In this experiment, this was accompanied by a partial drop in RFP-2xFYVE intensity; this is consistent with PIKfyve depleting PI(3)P as indicated in mammalian cells (Hazeki et al., 2012; Kim et al., 2014), although our previous studies show that PI(3)P dynamics are unaffected by loss of PIKfyve in Dictyostelium (Buckley et al., 2019). Either way, our observations confirm the early formation of PI(3)P and subsequent conversion to PI(3,5)P2 by phagosomal PIKfyve (Ellson et al., 2001; Hazeki et al., 2012).

Recruitment of SnxA relative to PI(3)P and PIKfyve. (A) Representative timelapse of cells co-expressing 2xFYVE-RFP (magenta) and SnxA-GFP (green) during phagocytosis of an unlabeled yeast. Engulfed yeast is indicated with an asterisk; scale bar = 2 μm. See Video 4 for full-time series. (B) Quantification of the fluorescence intensities of both channels around the yeast in this video. (C and D) Recruitment of GFP-PIKfyve to a newly formed macropinosome and phagosome, respectively. The full timelapse is shown in Videos 5 and 6. (E) Timelapse of cells co-expressing 2xPX-RFP (magenta) and PIKfyve-GFP (green) during phagocytosis of Alexa405-yeast (blue). (F) Quantification of the fluorescence intensities around the phagosome in E. Data shown is the mean intensity ± the SD around the phagosomal membrane normalized between minimum and maximum intensities observed.
Recruitment of SnxA relative to PI(3)P and PIKfyve. (A) Representative timelapse of cells co-expressing 2xFYVE-RFP (magenta) and SnxA-GFP (green) during phagocytosis of an unlabeled yeast. Engulfed yeast is indicated with an asterisk; scale bar = 2 μm. See Video 4 for full-time series. (B) Quantification of the fluorescence intensities of both channels around the yeast in this video. (C and D) Recruitment of GFP-PIKfyve to a newly formed macropinosome and phagosome, respectively. The full timelapse is shown in Videos 5 and 6. (E) Timelapse of cells co-expressing 2xPX-RFP (magenta) and PIKfyve-GFP (green) during phagocytosis of Alexa405-yeast (blue). (F) Quantification of the fluorescence intensities around the phagosome in E. Data shown is the mean intensity ± the SD around the phagosomal membrane normalized between minimum and maximum intensities observed.
Simultaneous imaging of PI(3)P (RFP-2xFYVE, magenta) and PI(3,5)P2 (SnxA-GFP, green) recruitment to a yeast-containing phagosome (unlabeled) in WT cells. Images were captured every 10 s by spinning-disc microscopy and displayed at five frames per second. The yeast is annotated by an asterisk. Scale bar = 2 μm.
Simultaneous imaging of PI(3)P (RFP-2xFYVE, magenta) and PI(3,5)P2 (SnxA-GFP, green) recruitment to a yeast-containing phagosome (unlabeled) in WT cells. Images were captured every 10 s by spinning-disc microscopy and displayed at five frames per second. The yeast is annotated by an asterisk. Scale bar = 2 μm.
We then investigated how the dynamics of PIKfyve related to PI(3,5)P2 accumulation by expression of PIKfyve-GFP. Although PIKfyve-GFP expressed poorly in WT cells, it was clearly visible on large vesicles that resembled macropinosomes (Fig. S5 A). A diffuse perinuclear cluster of presumably subresolution vesicles could also be observed in some cells. Contrast was improved by expression in ΔPIKfyve cells due to loss of competition with the endogenous protein, which also rescued the swollen endosomal phenotype (Fig. S5 B). This allowed PIKfyve-GFP dynamics to be observed over time, demonstrating recruitment to both macropinosomes and phagosomes within 60 s of engulfment (Fig. S5, C and D; and Videos 5 and 6). This is consistent with the role of PIKfyve in the early maturation and its initial recruitment by PI(3)P. Although low signal and movement in the Z-axis meant it was not possible to reliably follow individual macropinosomes much beyond ∼2 min, we observed removal of PIKfyve-GFP from phagosomes ∼3 min after engulfment.

Expression of PIKfyve-GFP in Dictyostelium. (A) Localization of PIKfyve-GFP in WT cells. Boxes indicate regions enlarged in the panels on the right; arrowhead indicates perinuclear cluster of vesicles. (B) Rescue of the macropinosome concentration defect by PIKfyve-GFP expression in ΔPIKfyve cells. The cell lines indicted were incubated overnight in Texas-Red dextran overnight prior to confocal microscopy. Scale bar = 2 μm.

Expression of PIKfyve-GFP in Dictyostelium. (A) Localization of PIKfyve-GFP in WT cells. Boxes indicate regions enlarged in the panels on the right; arrowhead indicates perinuclear cluster of vesicles. (B) Rescue of the macropinosome concentration defect by PIKfyve-GFP expression in ΔPIKfyve cells. The cell lines indicted were incubated overnight in Texas-Red dextran overnight prior to confocal microscopy. Scale bar = 2 μm.
Recruitment of PIKfyve-GFP to a newly formed macropinosome (asterisk). PIKfyve-GFP expressed in a ΔPIKfyve cell. Images were captured every 5 s by spinning-disc microscopy and displayed at five frames per second. Scale bar = 2 μm.
Recruitment of PIKfyve-GFP to a newly formed macropinosome (asterisk). PIKfyve-GFP expressed in a ΔPIKfyve cell. Images were captured every 5 s by spinning-disc microscopy and displayed at five frames per second. Scale bar = 2 μm.
Recruitment of GFP-PIKfyve to phagosomes. Engulfment of an Alexa405-labeled yeast (blue) by a GFP-PIKfyve (white) expressing ΔPIKfyve cell. Images were captured every 20 s by spinning-disc microscopy and displayed at five frames per second. Scale bar = 2 μm.
Recruitment of GFP-PIKfyve to phagosomes. Engulfment of an Alexa405-labeled yeast (blue) by a GFP-PIKfyve (white) expressing ΔPIKfyve cell. Images were captured every 20 s by spinning-disc microscopy and displayed at five frames per second. Scale bar = 2 μm.
To examine how PIKfyve recruitment and dissociation relate to PI(3,5)P2 production, we coexpressed PIKfyve-GFP with the SnxA probe (RFP-2xPX) in cells engulfing Alexa-405–labeled yeast. This confirmed that PIKfyve is recruited 1–2 min prior to PI(3,5)P2 accumulation but also showed that they only colocalize fleetingly, with PIKfyve-GFP dissociating immediately after the SnxA probe arrives (Fig. 5, E and F; and Video 7). PIKfyve is therefore recruited early during phagosome maturation but dissociates rapidly upon activation and accumulation of PI(3,5)P2.
Simultaneous imaging of PIKfyve-GFP (green) and PI(3,5)P2 formation (RFP-2xPX, in magenta) in WT cells engulfing an Alexa405-labeled yeast (blue). Images were captured every 10 s by spinning-disc microscopy and displayed at five frames per second. Asterisk indicates the engulfed yeast. Scale bar = 2 μm.
Simultaneous imaging of PIKfyve-GFP (green) and PI(3,5)P2 formation (RFP-2xPX, in magenta) in WT cells engulfing an Alexa405-labeled yeast (blue). Images were captured every 10 s by spinning-disc microscopy and displayed at five frames per second. Asterisk indicates the engulfed yeast. Scale bar = 2 μm.
As PI(3)P is present throughout the first 10 min of both macropinosome and phagosome maturation (Buckley et al., 2019; King and Kay, 2019), dissociation of PIKfyve at 2–3 min indicates an additional layer of regulation. As PIKfyve dissociation coincides with PI(3,5)P2 accumulation, we used the inhibitor apilimod to test whether PIKfyve activity is required for its release. Consistent with this, PIKfyve-GFP appeared to accumulate more strongly on phagosomes in aplimod-treated cells. Timelapse microscopy then confirmed that apilmod treatment caused PIKfyve to be retained on phagosomes for as long as we could follow them (∼15 min), compared to 4 min in untreated controls (Fig. 6, A–C). This demonstrates that PIKfyve dissociation requires its catalytic activity, providing a mechanism to couple activation and release.

PIKfyve activity is required for its dissociation from phagosomes. (A) Timelapses of PIKfyve-GFP expressing cells during phagocytosis of pHrodo-yeast (red) before (top) and after (bottom) addition of 5 μM apilimod. (B) The PIKfyve-GFP intensity around the phagosome in these movies. (C) PIKfyve-GFP dynamics over multiple events by manually scoring the time of PIKfyve-GFP delivery and release. 14 untreated events and 10 apilimod-treated events were scored over three experiments, each with the thick line indicating the proportion of GFP-positive phagosomes at each time after engulfment. Thin lines represent individual events. All scale bars = 2 μm.
PIKfyve activity is required for its dissociation from phagosomes. (A) Timelapses of PIKfyve-GFP expressing cells during phagocytosis of pHrodo-yeast (red) before (top) and after (bottom) addition of 5 μM apilimod. (B) The PIKfyve-GFP intensity around the phagosome in these movies. (C) PIKfyve-GFP dynamics over multiple events by manually scoring the time of PIKfyve-GFP delivery and release. 14 untreated events and 10 apilimod-treated events were scored over three experiments, each with the thick line indicating the proportion of GFP-positive phagosomes at each time after engulfment. Thin lines represent individual events. All scale bars = 2 μm.
Discussion
In this study, we identify the Dictyostelium protein SnxA as a highly selective PI(3,5)P2-binding protein and demonstrate its use as a reporter for PIKfyve signaling in both Dictyostelium and mammalian cells. Using SnxA-GFP fusions, we have been able to visualize PI(3,5)P2 dynamics in live cells for the first time and provide insight into the complex regulation of PIKfyve and the early regulation of phagosome and macropinosome maturation.
Whilst we find that both full-length SnxA and tandem PX domain constructs are selective for PI(3,5)P2, we note several caveats that should be considered with their use. First, the full-length protein contains a coiled-coil domain in addition to the PI(3,5)P2-binding PX domain. The primary function of this appears to be mediating homodimerization, which is highly advantageous in increasing binding avidity, but we cannot completely exclude interactions with other proteins. Nonetheless, with the exception of the moderate PI(3,5)P2-independent enrichment at the plasma membrane observed upon paraformaldehyde fixation, full-length SnxA is recruited to the same compartments with comparable dynamics to a 2xPX construct lacking the coiled-coil domain. Therefore, any additional interactions appear to have minor or no effects on localization.
The 2xPX construct is theoretically superior as it has the same Kd as a SnxA dimer and should be more specific. However, we observed tandem PX domain fusions also have a greater tendency to disrupt PI(3,5)P2 signaling in both Dictyostelium and mammalian cells. Though disruption was minor in Dictyostelium, cell morphology, growth, and macropinocytosis were clearly perturbed in some mammalian cell lines. This is most likely due to the 2xPX probe outcompeting endogenous PI(3,5)P2 effectors or regulators. We speculate this is due to a slower off-rate of a constitutive tandem fusion compared with a SnxA dimer where the domains are more spaced and connected by transient electrostatic interactions.
Despite this caveat, the 2xPX probe often had better contrast for microscopy. This was especially evident when expression levels varied over a large range or when imaging parameters were challenging, such as in co-expression experiments. In Dictyostelium cells expressing SnxA-GFP from an extrachromosomal vector, vesicular localization was only visible above the background signal in lower expressing cells, whereas 2xPX-GFP and particularly 2xPX-RFP performed much better than their counterparts. Deleterious effects of 2xPX expression in mammalian cells were also not evident in all cell lines tested and are likely to be minimized by low expression, using weaker promoters than those used in this study. It is also possible to bypass probe expression artifacts altogether by using recombinant probes to stain fixed cells (Hammond et al., 2009; Maib and Murray, 2022). The choice of full-length SnxA or 2xPX probes, therefore, depends on individual experimental conditions. We recommend future users carefully test both using appropriate controls including treatment with PIKfyve inhibitors and functional readouts of the pathways under investigation where possible.
The ability to observe the localization and dynamics of both the PIKfyve enzyme and its product has provided several new mechanistic insights. Combined, our data support a model where PIKfyve is regulated in a series of coordinated steps: Initially, inactive PIKfyve is recruited by binding PI(3)P via its FYVE domain before subsequent activation causes rapid PI(3,5)P2 accumulation. Finally, PIKfyve dissociates depending on its own activity.
Complex regulation is not unexpected. PIKfyve is unusual in that its catalytic substrate (PI(3)P) also mediates its recruitment via additional interactions with the FYVE domain. While PIKfyve could be released from the membrane by simply using up all the PI(3)P (Kerr et al., 2010), this is unlikely to be the case in Dictyostelium as significant PI(3)P is retained on phagosomes long after PIKfyve dissociates and loss of PIKfyve does not affect PI(3)P dynamics (Buckley et al., 2019). There is therefore likely to be additional regulation of the FYVE domain binding. This is supported by a recent study demonstrating recruitment of the yeast PIKfyve ortholog (Fab1) is regulated by phosphorylation near its FYVE domain by TORC1, causing translocation between the yeast vacuole and signaling endosomes (Chen et al., 2021). Although it is unclear whether this mechanism is conserved in other endocytic pathways or organisms, the finding that PIKfyve recruitment to PI(3)P is also conditionally regulated during Dictyostelium phagosome maturation highlights strong similarities.
A further complication is that PIKfyve is complexed with the phosphatase Fig4, which can potentially break down PI(3,5)P2 as soon as it is generated. As PI(3,5)P2 accumulates in a burst almost 2 min after PIKfyve arrival, either PIKfyve or Fig4 catalytic activity must also be regulated. The partial structure of the PIKfyve complex suggests that PIKfyve can be inhibited by autophosphorylation, requiring protein dephosphorylation activity of Fig4 for activation (Lees et al., 2020). Fig4 may therefore also be temporally regulated during phagosome maturation or an additional unknown regulatory mechanism may exist. Our observations that PIKfyve dissociates as soon as PI(3,5)P2 accumulates but is retained if pharmacologically inactivated indicates that catalytic activity also affects membrane association. This is consistent with a previous study in mammalian cells, where catalytically inactive PIKfyve was also retained on macropinosomes (Kerr et al., 2010). Although the FYVE domain is not well resolved in the published structure, it is suggested to lie near the kinase domain (Lees et al., 2020). This may provide potential for an additional level of regulation to explain the coordination of recruitment and activity in vivo but needs more investigation.
Many aspects of maturation are shared between phagosomes, macropinosomes, and other endocytic pathways, such as classical clathrin-mediated endosomes. These also transition from a Rab5/PI(3)P-positive early form to later compartments demarked by Rab7, vacuolar ATPase, and lysosomal components. Furthermore, degradation of classically endocytosed receptors, autophagosomes, and entotic vesicles are also sensitive to PIKfyve inhibition (de Lartigue et al., 2009; Krishna et al., 2016; Qiao et al., 2021). A significant limitation of SnxA as a PI(3,5)P2 probe is that we were not able to see any clear recruitment to these smaller endocytic compartments. There are several potential explanations for this. For example, PI(3,5)P2 may not be present in sufficient abundance in other endocytic pathways due to their limited membrane surface, or a limited pool of PI(3,5)P2 may already be fully occupied by endogenous binding proteins. Low levels of SnxA recruitment onto small compartments may also simply not be visible over the cytosolic background with standard confocal imaging.
Whatever the reason, it is likely that our observations with phagosomes and macropinosomes are applicable to other endocytic routes and overcome the technical challenges of following ∼100 nm vesicles in sufficient detail over time. Using Dictyostelium SnxA to report PI(3,5)P2 dynamics in live cells, we have harnessed an experimentally tractable approach to define the complex sequence of events that occur in the first minutes of phagosome maturation with key mechanistic insights into regulation of PIKfyve and PI(3,5)P2.
Materials and methods
Dictyostelium culture
All D. discoideum cells were derived from the Ax2 (Kay) laboratory strain background (strain ID: DBS0235521) and grown in adherent culture in filter-sterilized HL5 medium (Formedium) at 22°C. Cells expressing extrachromosomal plasmids were transformed by electroporation and grown in appropriate antibiotic selection by addition of either 20 µg/ml hygromycin (Invitrogen) or 10 µg/ml G418 (Sigma-Aldrich). The PIKfyve knockout strain in Ax2 background was generated as previously described (Buckley et al., 2019). Live cell imaging was performed in defined SIH medium (Formedium).
Growth on bacterial lawns was performed by seeding ∼20 Dictyostelium cells with suspension of Klebsiella aerogenes on Sussman's Media agar plates (Formedium). Colonies were visible after 5, and colony size was measured each day. Tests of growth on a panel of bacteria were performed as described by Froquet et al. (2009). Dilutions from 1 to 1 × 104 cells were seeded on agarose in wells of a 96-well plate containing the relevant bacteria. After several days (dependent on bacterial strain), wells were scored for the presence of Dictyostelium colonies. Bacteria used were a gift from Pierre Cosson (University of Geneva, Geneva, Switzerland) and were Klebsiella pneumoniae laboratory strain and 52145 isogenic mutant (Benghezal et al., 2006), Micrococcus luteus (Wilczynska and Fisher, 1994), the isogenic Pseudomonas aeruginosa strains PT5 and PT531 (rhlR-lasR avirulent mutant; Cosson et al., 2002), Escherichia coli B/r (Gerisch, 1959), non-sporulating Bacillus subtilis 36.1 (Ratner and Newell, 1978), and E. coli DH5α (Thermo Fisher Scientific). An avirulent strain of K. pneumoniae was obtained from ATCC (Strain no. 51697).
Mammalian cell culture
HEK293, Mia-PaCa-2, and RPE-1 cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, and 50 μg/ml penicillin/streptomycin at 37°C in a humidified incubator with 5% CO2. For transient transfection with SnxA-GFP or 2xPX-GFP probes, cells were seeded in glass-bottomed dishes or coverslips at 200,000 cells/ml and grown overnight before treatment with Lipofectamine 3000 Transfection Reagent as per manufactures guidelines. For apilimod treatment, 1 μM apilimod was added 24 h after transfection, for at least 1 h prior to imaging.
To score the number of GFP-positive vesicles in live cells, 10 images were captured per condition per repeat. Cells were scored for the number of identifiable vesicles within the cell. A Z-stack was used to help identify vesicles, although single Z-stacks are shown in figures for clarity. Untransfected cells or those with extremely high levels of expression were not counted. To see dextran uptake in live cells, media containing 2.5 mg/ml 70 kD Texas-Red dextran (Life Technologies) was added. Images were captured every 60 s for up to an hour. For fixation, cells on coverslips were first washed twice in PBS and treated with 4% PFA for 10 min before another wash and mounting. All mammalian cell images were captured using a Nikon W1 Spinning Disk confocal using a Plan Apo λ 100×1.4NA oil objective using a Prime 95B A19F203018 camera. Acquisition was controlled via Nikon NIS elements. Live cells were imaged at 37°C and 5% CO2 in their DMEM growth medium.
Cloning
All gene sequences used for cloning were obtained from dictybase (http://www.dictybase.org/; Fey et al., 2013). PX-domain-containing proteins encoded within the Dictyostelium genome were identified by Basic Local Alignment Search Tool. Full-length SnxA-GFP (pJSK619) and PIKfyve-GFP (pJV0025) plasmids were generated by PCR of the respective coding sequences from cDNA using primeSTAR max DNA polymerase (Clontech), subcloning into Zero Blunt TOPO II vector (Life Technologies), and then cloning into the BglII/SpeI sites of the pDM1045 expression vector (Paschke et al., 2018; Veltman et al., 2009). To generate 1x and 2xPX expression plasmids, the sequence for the PX domain was synthesized by DC Biosciences with a 5′BglII sites and a 5′BamHI and SpeI sites. This was cloned into the Bglll/SpeI sites of pDM1043 to give the 1xPX-GFP expression vector pJV0010. A second copy of the PX domain was then inserted into the BglII sites of this as a BglII/BamHI fragment of the synthesized gene to give the 2xPX-GFP construct pJV0016. The C-terminal mCherry fusion expression vector pDM1097 was used to generate the 2xPX-RFP expression vector. The same procedure was used to generate the mammalian expression vectors. The full-length and PX-domain-coding sequences were human codon optimized, synthesized, and cloned as BamHI/EcoRI fragments into pEGFP-C1. The full-length, 1xPX, and 2xPX constructs are pJSK659, 663, and 664, respectively. These will be available at http://Addgene.com.
2xFYVE-GFP (pJSK418) uses the sequence from the human Hrs gene and was previously described (Calvo-Garrido et al., 2014). Co-expression plasmids were created by first cloning the RFP-fusion sequence into the pDM series shuttle vectors (pDM1042 and pDM1121) before cloning as an NgoMIV fragment into the appropriate GFP-expression vector. The SnxA-GFP integrating vector was made by cloning full-length SnxA into the integrating C-terminal GFP vector pDM1053 (Paschke et al., 2018). Prior to transformation, the plasmid was linearized with BamHI and transformation was performed with 100 U of BamHI, producing a 5–10-fold increase in the number of colonies compared with controls. Colonies were selected and screened by confocal microscopy for a cell line with low expression and high contrast.
PIP arrays
PIP arrays were performed using whole cell lysates from Dictyostelium cells expressing the GFP-fusion constructs. Approximately, 2 × 108 cells were lysed in radioimmunoprecipitation assay buffer (50 mM Tris HCL, pH 7.5, 150 mM NaCl, 0.1% SDS, 2 mM EDTA, 0.5% sodium deoxycholate, 1× Halt protease inhibitor cocktail [Pierce]). PIP arrays (Echelon Biosciences) were blocked for 1 h at room temperature in 3% fatty-acid-free BSA in Tris-buffered saline with 0.1% tween and then incubated with lysates for 1 h. Membranes were incubated with a custom rabbit polyclonal anti-GFP antibody (gift from Andrew Peden, University of Sheffield, Sheffield, UK) for 1 h at room temperature before washing and then Alexa Fluor 800–conjugated anti-rabbit secondary antibody (A32735; Life Technologies) was added before imaging in a LiCor Odyssey SA fluorescent imager.
Purification of recombinant proteins
All SnxA probes were expressed as N-terminal 6xHis and eGFP fusion proteins in BL21 cells in Lysogeny Broth media containing ampicillin and 1.7 g lactose per liter for 7 h at 37°C before decreasing to 18°C for ∼10–12 h. Cells were pelleted and lysed by ultrasound before clearing by centrifugation at 60,000 × g for 1 h. Cleared lysates were filtered through a 0.22-µm filter and purified using a 5 ml His trap HP column followed by anion exchange using a 5 ml Capto-Q column and size exclusion chromatography using a Superdex 200 16/60 pg column. All probes were snap-frozen in liquid nitrogen and stored at −80°C.
Lipid-binding experiments
The experiments were performed as previously described (Maib and Murray, 2022). 1 mg liposomes, containing each of the seven different phosphatidylinositols were produced by mixing 95 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine with 5 mol % of the respective phosphatidylinositol together with 0.1% Atto647N-DOPE. The mixtures were evaporated under nitrogen and dried overnight in a vacuum extruder. Dried lipids were resuspended in 1 ml buffer containing 150 mM NaCl, 20 mM Hepes, and 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and subjected to six freeze–thaws in liquid nitrogen. Liposomes ∼100 nm diameter were generated by passing the lipid mixture 11 times through a 100-nm filter (Whatman Nuclepore). Liposomes were aliquoted, snap-frozen, and stored at −20°C.
Membrane-coated beads were generated by adding 10 µg of liposomes to ∼0.5 × 106 10 µm silica beads (Whitehouse Scientific) in 100 μl of 200 mM NaCl for 30 min rotation at room temperature. Beads were washed twice and resuspended in a buffer containing 150 mM NaCl, 20 mM Hepes, and 0.5 mM TCEP and blocked with 10% goat serum for 30 min. Drops of 7.5 μl beads were added into the corners of uncoated µ-Slide 8-well chambers (Ibidi) and 7.5 μl of the purified SnxA probes was added and mixed by pipetting at indicated final concentrations. Lipid-binding kinetics were allowed to equilibrate for 30 min at room temperature before imaging close to the equator of the beads.
Confocal images were acquired using a Leica SP8 Confocal Microscope with a Leica HC PL APO CS2 63×/1.40 Oil objective at 0.75 base zoom with 1,024 × 1,024 pixels scan. GFP and Atto647N-DOPE fluorescence were imaged simultaneously without any measurable bleed-through. Data were analyzed using a custom ImageJ script that segments the Atto647N-DOPE channel to create a mask around the outer circumference of each bead. Segmented masks were then used to measure GFP fluorescence around each bead.
Mass photometry
Mass photometry measurements were performed as described in Sonn-Segev et al. (2020). Briefly, coverslips were cleaned in 50% isopropanol, and 5 μl of sample was applied to a culture well gasket. Samples were diluted to a concentration between 25 and 50 nM in buffer containing 250 mM NaCl, 20 mM Hepes, and 0.5 M TCEP immediately before measurements. Data were acquired using OneMP mass photometer (Refeyn Ltd) AcquireMP (Refeyn Ltd), and data analysis was performed using DiscoverMP (Refeyn Ltd). Calibration was carried out using standards of known molecular weight. For each sample, between 1,315 and 10,380 individual events were recorded and analyzed.
Preparation of fluorescent yeast and dextran
Saccharomyces cerevisiae were used for phagocytosis assays. Yeast were grown for 3 d at 37°C in standard yeast peptone dextrose media (Formedium) until stationary phase before centrifugation at 1,000 × g for 5 min, resuspended at a final concentration of 1 × 109 cells/ml in PBS, pH 7, and frozen until use. Yeast were fluorescently labeled using either pHrodo red succinimidyl ester (Life Technologies) or Alexa-405 succinimidyl ester (Life Technologies) at a final concentration of 0.25 and 2.5 mM, respectively. 0.5 × 109 yeast were resuspended in 200 μl of PBS at pH 8.5 and incubated with 10 μl of prepared dye for 30 min at 37°C with gentle shaking. After, cells were pelleted and sequentially washed in 1 ml of PBS, pH 8.5, 1 ml of 25 nM Tris-HCl, pH 8.5, and 1 ml of PBS, pH 8.5, again. Finally, cells were resuspended in 500 μl of KK2, pH 6.1, and kept at −20°C. Yeast were diluted in SIH to a working concentration of 1 × 108 yeast/ml before use. Texas-Red 70 kD Dextran (Life Technologies) was resuspended in water and diluted to a working concentration of 2 mg/ml.
Microscopy and image analysis
For fluorescence microscopy, ∼1 × 106Dictyostelium cells were seeded in 35-mm glass-bottomed microscopy dishes (Mat tek P35G-1.5-14-C) and left to grow overnight in SIH medium (Formedium). Cells were imaged on a Zeiss LSM880 AiryScan Confocal with 63×1.4NA Plan Apochromat oil objective and processed using the Zeiss Zen microscope software unless otherwise stated. All Dictyostelium imaging was performed at room temperature.
For timelapse microscopy of phagocytosis, most of the medium was aspirated immediately before imaging and 20 μl of 1 × 108 labeled yeast added. After 60 s, cells were overlaid with a thin layer of 1% agarose in SIH and excess medium removed. For normal time lapses, images were captured every 10 s for up to 10 min. For extended time lapses, images were captured every 60 s. Automated analysis of the fluorescent intensity around each phagosomal membrane was performed using the Python plugin pyimagej (https://pypi.org/project/pyimagej/). Images where phagosomal uptake could be seen were manually selected. To segment out the phagosome, the yeast channel was thresholded and particles larger than 1.5 μm2 selected. Particles on time-adjacent frames were automatically scored for similarity based on position and size, and high-similarity particles were categorized into the same event. Then, throughout each event, the phagosome area was monitored for fluorescence change of pHrodo-labeled yeast as a proxy for phagosomal pH. The selection perimeter was enlarged into two rings of ∼0.35 μm thickness: an inner ring 0.2 μm enlarged, to measure enrichment at the phagosomal membrane, and an outer ring 0.85 μm enlarged, to monitor background cytosol fluorescence. Each event was then manually screened for errors before mean fluorescence intensity was recorded for each frame, normalized to intracellular background, and fold-enrichment from −10 s calculated. Because addition of an agarose overlay appeared to suppress the effects of apilimod, cells treated with 5 µM apilimod (added >2 h before imaging; USBiological) were imaged without overlay through a larger Z range every 10 s using a Nikon W1 Spinning Disk confocal with a Plan Apo λ 100×1.4NA oil objective using a Prime 95B A19F203018 camera. Acquisition was controlled via Nikon NIS elements. The automated analysis pipeline was not sensitive enough to detect PIKfyve-GFP enrichment, so events were manually followed and scored as either GFP-positive or -negative using ImageJ.
Dextran pulse-chases were performed by aspirating most of the medium and then adding 50 μl of 2 mg/ml 70 kD Texas-Red dextran (11580226; Life Technologies) for the indicated time. Dextran was then removed by washing thrice in SIH medium, leaving ∼1 ml of SIH in the dish. Cells were immediately imaged, with multiple fields of view taken every 2 min for 10 min. GFP colocalization at each time point was then scored manually.
Generating SnxA knockouts
The SnxA (DDB_G0289833) locus was disrupted by homologous recombination, inserting a blasticidin resistance cassette within the gene and deleting 550 bp of coding sequence. The knockout construct was made using a one-step recombination method into the pKOSG-IBA-Dicty vector as previously described (Wiegand et al., 2011). The 5′ recombination arm was generated with the primers 5′-AGCGCGTCTCCAATGACCACCCAGGTTAAAAATAATTCC-3′/5′-AGCGCGTCTCCCTTCTTCCTTTTCTAAGAGAATATATTTGG-3′, and the 3′ arm with 5′-AGCGCGTCTCCGTTGCCTTAAAAGAGACGAAGGT-3′/5′-AGCGCGTCTCCTCCCCTGGCTTTGTTTTTATAAAACAG-3′. PCR products were recombined with the vector using StarCombinase (IBA GmbH). The resulting vector was used to transform Ax2 Dictyostelium, and the resulting blasticidin-resistant clones were screened for successful gene disruption by PCR using the primers 5′-GCACTGGGAGTTCCAATATCAATATCATC-3′/5′-ATAATTAATTCAACATCTTGCAAAT-3′. Multiple independent clones were isolated and used for functional characterization.
Endocytosis and fluid-phase proteolysis
Macropinocytosis and exocytosis were measured, as previously described (Maniak, 2001), by incubating 5 × 106 cells/ml in HL5 medium containing 2 mg/ml 70 kD FITC dextran (FD705; Sigma-Aldrich). 500 μl of cells were then removed at each time point and added to 1 ml ice-cold KK2 (0.1 M potassium phosphate, pH 6.1). Cell pellets were then washed once and lysed in 50 mM Na2HPO4, pH 9.3, 0.2% Triton X-100 before FITC fluorescence was measured in a fluorimeter. Exocytosis was measured in the same way, except cells were preincubated with FITC-dextran for at least 2 h, before washing out and measuring the decrease in fluorescence over time.
Phagocytosis assays
Bead uptake, proteolysis, and acidification were performed as previously described in detail in Sattler et al. (2013). Briefly, phagocytosis was measured by uptake of 1 μm YG-green beads (Polysciences Inc.). Cells were incubated with beads for the times indicated and fluorescence was measured by flow cytometry. pH and proteolysis were measured using 3 μm silica beads (Kisker Biotech) conjugated to either FITC or BSA, respectively, as well as Alexa594 as a reference dye. Synchronous phagocytosis was caused by centrifuging beads onto confluent cells in a 96-well plate (1,200 RPM for 10 s) before washing and measuring changes in the fluorescence intensities over time. All samples were measured in triplicate.
Statistics
Statistical analysis was performed using Graphpad Prism 9. Biological replicate numbers and statistical tests used for each experiment are detailed in each figure legend. P values were calculated by unpaired Students T tests assuming a normal distribution, although this was not formally tested. P < 0.05 was deemed significant with * indicating P < 0.05, **P < 0.01, and ***P < 0.005 throughout. All images shown are representative of at least three independent experiments.
Online supplementary material
Fig. S1 shows additional in vitro characterization of the reporter, including confirmation of recombinant protein purification, absence of binding to PI(5)P, and mass photometry analysis of dimerization. Fig. S2 shows non-specific cortical localization of SnxA-GFP in mammalian cells and lack of dominant effects of probe expression in Dictyostelium cells. Fig. S3 shows phenotypic analysis of snxA knockout cells. Fig. S4 outlines our automated image analysis pipeline, as well as additional quantification of 2xPX-GFP dynamics in Dictyostelium. Fig. S5 provides confirmation of GFP-PIKfyve localization in WT cells and its ability to rescue PIKfyve mutant phenotypes. Video 1 shows recruitment of GFP-snxA to macropinosomes in mammalian cells. Video 2 shows PI(3,5)P2 dynamics during Dictyostelium phagosome maturation, with a direct comparison between SnxA-GFP and 2xPX-RFP in Video 3. Simultaneous imaging of PI(3)P and PI(3,5)P2 probes during phagosome maturation is shown in Video 4. Videos 5 and 6 show examples of PIKfyve-GFP recruitment to a newly formed macropinosome and phagosome, respectively. Video 7 shows simultaneous imaging of PIKfyve-GFP and PI(3,5)P2 dynamics during Dictyostelium phagosome maturation.
Data availability
The raw data and code are available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank Douwe Veltman and Peggy Paschke for their continued development of their Dicytostelium expression system, and Robert Insall for his support in the early days of this project. Cell imaging work was performed in the Wolfson Light Microscopy Facility at Sheffield.
D.H. Murray is supported by the Wellcome Trust grant 211193/Z/18/Z and Royal Society RGS\R2\180284. A. Gueho and T. Soldati were supported by the Swiss National Science Foundation research grants 310030_149390, 310030_169386, and 310030_188813 to T. Soldati. J.H. Vines is funded by Royal Society grant RGF\EA\180126 and J.S. King is funded by a Royal Society University Research Fellowship URF\R\201036. Open Access funding provided by University of Sheffield.
Author contributions: Conceptualization: J.S. King and J.H. Vines; investigation: J.H. Vines, H. Maib, C.M. Buckley, A. Gueho, Z. Zhou, and J.S. King; writing—original draft: J.H. Vines and J.S. King; writing - edit and review: all authors; funding acquisition: J.S. King, D.H. Murray, and T. Soldati; supervision: J.S. King, D.H. Murray, and T. Soldati.

