


Autophagy is a lysosomal/vacuolar delivery system that degrades cytoplasmic material. During autophagy, autophagosomes deliver cellular components to the vacuole, resulting in the release of a cargo-containing autophagic body (AB) into the vacuole. AB membranes must be disrupted for degradation of cargo to occur. The lipase Atg15 and vacuolar proteases Pep4 and Prb1 are known to be necessary for this disruption and cargo degradation, but the mechanistic underpinnings remain unclear. In this study, we establish a system to detect lipase activity in the vacuole and show that Atg15 is the sole vacuolar phospholipase. Pep4 and Prb1 are required for the activation of Atg15 lipase function, which occurs following delivery of Atg15 to the vacuole by the MVB pathway. In vitro experiments reveal that Atg15 is a phospholipase B of broad substrate specificity that is likely implicated in the disruption of a range of membranes. Further, we use isolated ABs to demonstrate that Atg15 alone is able to disrupt AB membranes.
Introduction
Intracellular homeostasis is maintained by continuous synthesis and degradation of cellular components. Autophagy, a major intracellular degradation system that is highly conserved among eukaryotes, plays a key role in multiple metabolic and cellular processes (Ohsumi, 2014; Parzych and Klionsky, 2014). Disruption of this degradation results in the accumulation of cargo materials within the vacuole/lysosome and the inability to adapt to starvation conditions in yeast (Takeshige et al., 1992), or various lysosomal storage diseases in mammals (Parkinson-Lawrence et al., 2010).
Yeast provides an excellent system to elucidate the molecular mechanisms of degradation in the vacuole due to its genetic tractability and ease of use in biochemical experiments. Upon induction of autophagy in yeast, a small membrane sack is formed that elongates and subsequently closes to form a double membrane vesicle known as an autophagosome. The autophagosome sequesters a portion of cytoplasm before its outer membrane fuses with the vacuole, releasing the inner membrane of the autophagosome, which is thereafter called an autophagic body (AB), into the vacuolar lumen. ABs are immediately disintegrated, releasing their cargoes for degradation by vacuolar hydrolases into compounds that can be recycled for new synthesis. We have also previously shown that autophagy-dependent RNA degradation in the vacuole is mediated by a T2-type RNase, Rny1, and the nucleotidase Pho8 (Huang et al., 2015). All forms of degradation following delivery to the vacuole depend on the disruption of the AB membrane to expose cargoes to vacuolar enzymes.
Vacuolar proteinase A (Pep4) and proteinase B (Prb1) have been shown to be necessary for AB disruption in yeast (Takeshige et al., 1992). Pep4 is an aspartic endoprotease that triggers a protease cascade, resulting in the activation of many hydrolases that amplify the degradative capacity of the vacuole (Ammerer et al., 1986). Prb1 is a serine endoprotease that is activated by Pep4 (Mechler et al., 1988; Moehle et al., 1989). These two proteases are involved in the activation of other hydrolases including carboxypeptidase Y (CPY), aminopeptidase I (Ape1), and Pho8 (Hecht et al., 2014; Parzych and Klionsky, 2019). Treatment with a serine protease inhibitor, phenylmethylsulfonyl fluoride (PMSF), also inhibits AB disruption (Takeshige et al., 1992). Given that electron microscopy indicates that AB membranes contain few proteins (Baba et al., 1995), it is unlikely that vacuolar proteases directly disrupt AB membranes. It is therefore not clear why these vacuolar proteases are essential for the degradation of the limiting membrane of ABs.
Degradation of glycerophospholipids, which are the main components of lipid bilayers, may be essential for membrane disruption. In yeast, ATG15 (identified initially as AUT5/CVT1) encodes a vacuolar phospholipase with a single transmembrane domain (Epple et al., 2001; Teter et al., 2001). During synthesis, Atg15 undergoes glycosylation and is then transported to the vacuole via the multivesicular body (MVB) pathway (Epple et al., 2001). Absence of Atg15 causes the accumulation of ABs in the vacuole (Epple et al., 2001). Atg15 is also involved in the disruption of other transport vesicles such as those of the cytoplasm-to-vacuole targeting (Cvt) pathway, internal vesicles of the MVB pathway, and microautophagic bodies (Epple et al., 2003; Oku et al., 2017; Teter et al., 2001). However, the mechanism underlying activation of Atg15 is yet to be characterized.
Phospholipases are classified into four groups based on their ability to specifically cleave ester bonds: phospholipase A cleaves the acyl ester bond at either the sn-1 (phospholipase A1) or sn-2 (phospholipase A2) position of a phospholipid, liberating free fatty acid (FFA) and lysophospholipid (LPL); phospholipase B cleaves acyl chains at the sn-1 and sn-2 positions, releasing two FFAs; phospholipase C cleaves the glycerophosphate bond; and phospholipase D removes the polar head group (Stahelin, 2016). Ramya and Rajasekharan (2016) have reported that purified Atg15 from microsomal membrane fractions cleaves the ester bond at the sn-1 position of phospholipids. They further report that Atg15 has high activity toward phosphatidylserine (PS), moderate activity toward phosphatidylethanolamine (PE) and cardiolipin, but almost no activity toward phosphatidic acid (PA), phosphatidylinositol (PI), phosphatidylcholine (PC), phosphatidylglycerol (PG), or LPL. On the other hand, van Zutphen et al. showed that Atg15 acts as a lipase that processes the triglyceride (TG) class of neutral lipids (van Zutphen et al., 2014). It remains unclear whether these phospholipase A1 activities and reported substrate specificities are sufficient to explain the process of AB disruption. In this study, we assess Atg15 and Pep4/Prb1 activities to clarify the long-standing question of how AB membranes are disrupted.
Results
ATG15 confers vacuolar lipase activity
ATG15 encodes a protein with a single N-terminal transmembrane domain (Fig. 1 A). The residues 330–334 constitute a GXSXG lipase consensus motif, and substitution of the serine 332 with alanine (S332A) results in a defect in AB disruption (Epple et al., 2001). Since there exist several lipases in various cellular compartments (Ramya and Rajasekharan, 2016), we first set out to develop an in vitro assay system that can detect lipase activity strictly within the vacuole. Vacuoles were purified from wild type (WT) cells as previously reported (Ohsumi and Anraku, 1981) and disrupted by freeze–thaw cycles to yield a vacuolar lysate (Fig. 1 B and Fig. S1 A). To monitor lipase activity, we used NBD-PE, a PE species labeled with nitrobenzoxadiazole (NBD) on the methyl end of the FFA moiety at sn-2 (Fig. 1 B). Lipase-mediated hydrolysis of the caboxylic ester of NBD-PE at the sn-1 position (Ramya and Rajasekharan, 2016) would produce only NBD-labeled lysophosphatidylethanolamine (NBD-LPE). However, incubation of NBD-PE with vacuolar lysates from WT cells yielded both NBD-LPE and NBD-labeled FFAs (NBD-FFA; Fig. 1 C, lane 2). These represent hydrolysis of the NBD-PE acyl ester linkage at sn-1 (NBD-LPE) or the sn-2 ester bond of NBD-PE (NBD-FFA); alternatively, further hydrolysis of NBD-LPE can also yield NBD-FFA. These results clearly indicate that vacuolar lysate is able to release FFAs by hydrolysis of sn-1 and sn-2, suggesting phospholipase A1 and A2, or B-type activity.

Vacuolar lipase activity derived from Atg15 depends on Pep4/Prb1. (A) Schematic diagram of Atg15. The transmembrane domain and a lipase consensus motif are shown. (B) Schematic overview of the in vitro assay system used to measure vacuolar lipase activity. (C) Lipase activities of vacuolar lysates from WT, atg15Δ, and atg15Δ cells harboring ATG15 from expressed from either a single-copy plasmid (pRS316) or multicopy (OE) plasmid (pRS426). Lysates were incubated with NBD-PE (18:1–12:0) for 20 min. The amount of lysate used for each assay was normalized by the amount of the vacuolar membrane protein Pho8 (see Fig. S1 B). TLC plate data (upper panel) are representative results from three independent experiments. Relative intensity (lower panel) was calculated as follows: relative intensity (%) = (peak area for NBD-PE, NBD-LPE, or NBD-FFA) ×100/(peak area for NBD-PE + peak area for NBD-LPE + peak area for NBD-FFA). Error bars represent means ± SD of three independent experiments. (D) Lipase activity of vacuolar lysates from Atg15OE cells. The lysate was incubated with NBD-PE for 0, 10, 30, and 90 min. (E) Lipase activities of vacuolar lysate from Atg15OE cells. A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 5×, and 25×) and incubated with NBD-PE for 20 min. The amount of lysate used for this assay is shown in Fig. S1 D. (F) Lipase activities of vacuolar lysates from WT, prb1Δ, pep4Δ, and pep4Δ prb1Δ cells. A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 5×, and 25×) and incubated with NBD-PE for 20 min. The amount of lysates used for this assay is shown in Fig. S1 E. Source data are available for this figure: SourceData F1.
Vacuolar lipase activity derived from Atg15 depends on Pep4/Prb1. (A) Schematic diagram of Atg15. The transmembrane domain and a lipase consensus motif are shown. (B) Schematic overview of the in vitro assay system used to measure vacuolar lipase activity. (C) Lipase activities of vacuolar lysates from WT, atg15Δ, and atg15Δ cells harboring ATG15 from expressed from either a single-copy plasmid (pRS316) or multicopy (OE) plasmid (pRS426). Lysates were incubated with NBD-PE (18:1–12:0) for 20 min. The amount of lysate used for each assay was normalized by the amount of the vacuolar membrane protein Pho8 (see Fig. S1 B). TLC plate data (upper panel) are representative results from three independent experiments. Relative intensity (lower panel) was calculated as follows: relative intensity (%) = (peak area for NBD-PE, NBD-LPE, or NBD-FFA) ×100/(peak area for NBD-PE + peak area for NBD-LPE + peak area for NBD-FFA). Error bars represent means ± SD of three independent experiments. (D) Lipase activity of vacuolar lysates from Atg15OE cells. The lysate was incubated with NBD-PE for 0, 10, 30, and 90 min. (E) Lipase activities of vacuolar lysate from Atg15OE cells. A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 5×, and 25×) and incubated with NBD-PE for 20 min. The amount of lysate used for this assay is shown in Fig. S1 D. (F) Lipase activities of vacuolar lysates from WT, prb1Δ, pep4Δ, and pep4Δ prb1Δ cells. A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 5×, and 25×) and incubated with NBD-PE for 20 min. The amount of lysates used for this assay is shown in Fig. S1 E. Source data are available for this figure: SourceData F1.
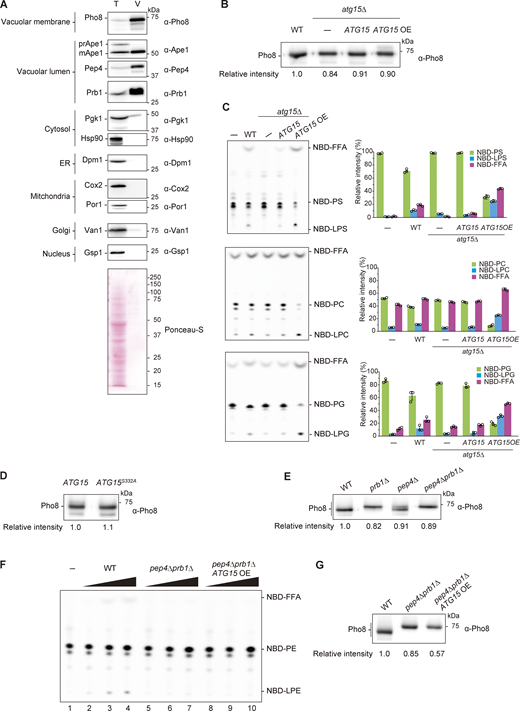
Supplemental data ofFig. 1,. (A) Purification of vacuole from WT cells. The whole-cell lysate (T) and vacuolar fraction (V) samples were subjected to western blot analysis using antibodies against Pho8 (vacuole membrane), Ape1 (autophagy cargo), Pep4, Prb1 (vacuole lumen), Pgk1, Hsp90 (cytosolic proteins), Dpm1 (ER membrane), Cox2 (mitochondrial matrix), Por1 (outer mitochondrial membrane), Van1 (Golgi), and Gsp1 (nucleus). Samples equivalent to 1/3,000 of total lysate and 1/160 of vacuolar fraction were loaded. (B) Western blotting of Pho8 in vacuolar lysates from WT, atg15Δ, and atg15Δ cells harboring ATG15 from either single plasmid (pRS316) or multicopy (OE) plasmid (pRS426). The relative band intensity of Pho8 for each strain is shown under the panel. (C) Lipase activities of vacuolar lysates from WT, atg15Δ, and atg15Δ cells harboring ATG15 from either single or multicopy (OE) plasmids. Each lysate was incubated with NBD-PS (18:1–12:0), NBD-PC (18:1–12:0), and NBD-PG (18:1–12:0) for 20 min. The amount of lysate used for the assay was the same as Fig. 1 C. TLC analysis (left panels) shows representative results from three independent experiments. The bar graphs (right panels) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (D) Western blotting of Pho8 in vacuolar lysates from atg15Δ cells expressing Atg15 or Atg15S332A from multicopy plasmids. The experimental procedure was the same as in B. (E) Western blotting of Pho8 in vacuolar lysates from WT, prb1Δ, pep4Δ, and pep4Δ prb1Δ cells. The experimental procedure was the same as in B. (F) Lipase activities of vacuolar lysates from WT, pep4Δ prb1Δ, and pep4Δ prb1Δ cells expressing Atg15 from a multicopy plasmid (pRS426). A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 5×, and 25×) and incubated with NBD-PE (18:1–12:0) for 20 min. The amount of lysates used for this assay is shown in G. (G) Western blot of Pho8 in vacuolar lysates from WT, atg15Δ, and pep4Δ prb1Δ cells expressing Atg15 from multicopy plasmids. The experimental procedure was the same as in B. Source data are available for this figure: SourceData FS1.

Supplemental data ofFig. 1,. (A) Purification of vacuole from WT cells. The whole-cell lysate (T) and vacuolar fraction (V) samples were subjected to western blot analysis using antibodies against Pho8 (vacuole membrane), Ape1 (autophagy cargo), Pep4, Prb1 (vacuole lumen), Pgk1, Hsp90 (cytosolic proteins), Dpm1 (ER membrane), Cox2 (mitochondrial matrix), Por1 (outer mitochondrial membrane), Van1 (Golgi), and Gsp1 (nucleus). Samples equivalent to 1/3,000 of total lysate and 1/160 of vacuolar fraction were loaded. (B) Western blotting of Pho8 in vacuolar lysates from WT, atg15Δ, and atg15Δ cells harboring ATG15 from either single plasmid (pRS316) or multicopy (OE) plasmid (pRS426). The relative band intensity of Pho8 for each strain is shown under the panel. (C) Lipase activities of vacuolar lysates from WT, atg15Δ, and atg15Δ cells harboring ATG15 from either single or multicopy (OE) plasmids. Each lysate was incubated with NBD-PS (18:1–12:0), NBD-PC (18:1–12:0), and NBD-PG (18:1–12:0) for 20 min. The amount of lysate used for the assay was the same as Fig. 1 C. TLC analysis (left panels) shows representative results from three independent experiments. The bar graphs (right panels) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (D) Western blotting of Pho8 in vacuolar lysates from atg15Δ cells expressing Atg15 or Atg15S332A from multicopy plasmids. The experimental procedure was the same as in B. (E) Western blotting of Pho8 in vacuolar lysates from WT, prb1Δ, pep4Δ, and pep4Δ prb1Δ cells. The experimental procedure was the same as in B. (F) Lipase activities of vacuolar lysates from WT, pep4Δ prb1Δ, and pep4Δ prb1Δ cells expressing Atg15 from a multicopy plasmid (pRS426). A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 5×, and 25×) and incubated with NBD-PE (18:1–12:0) for 20 min. The amount of lysates used for this assay is shown in G. (G) Western blot of Pho8 in vacuolar lysates from WT, atg15Δ, and pep4Δ prb1Δ cells expressing Atg15 from multicopy plasmids. The experimental procedure was the same as in B. Source data are available for this figure: SourceData FS1.
Next, we investigated the relationship between this lipase activity and Atg15. Vacuolar lysates from atg15∆ cells generated neither NBD-FFA nor NBD-LPE (Fig. 1 C, lane 3, and Fig. S1 B). These products reappeared when NBD-PE was incubated in the presence of lysates from atg15Δ cells expressing ATG15 from a plasmid (Fig. 1 C, lanes 4 and 5, and Fig. S1 B). Product formation was increased when ATG15 expression was elevated using a multicopy plasmid. Further, NBD-labeled PS, PC, and PG species all yielded NBD-labeled LPLs and NBD-FFA in a manner that correlated closely with the expression level of Atg15 (Fig. S1 C). We also found that hydrolysis progressed with incubation time (Fig. 1 D) and accelerated as more lysate was added (Fig. 1 E, lanes 2–4). Meanwhile, lysates from cells expressing Atg15S332A exhibited no hydrolytic activity at all (Fig. 1 E, lanes 5–7, and Fig. S1 D). These in vitro data indicate that Atg15 is the sole source of phospholipase activity in the vacuole, and that Atg15 is able to process a variety of phospholipid classes at the sn-1 and sn-2 positions.
Atg15 is activated by vacuolar proteases
Previously, we reported that deletion of the vacuolar proteases Pep4 and Prb1 results in accumulation of ABs in the vacuole (Takeshige et al., 1992). We next asked how vacuolar lipase activity relates to these vacuolar proteases. We performed lipase assays using vacuolar lysates from prb1Δ, pep4Δ, and pep4Δ prb1Δ cells, finding that lipase activity was completely absent in cells lacking either Prb1 or Pep4 (Fig. 1 F, lanes 5–13, and Fig. S1 E). Given that Pep4 proteolytically activates Prb1 (Mechler et al., 1988; Moehle et al., 1989), Prb1 may act directly to activate Atg15, consistent with our previous finding that disruption of ABs is inhibited by treatment with PMSF (Takeshige et al., 1992). However, it has been reported that Prb1 retains catalytic activity even in the absence of Pep4 activity (Mechler et al., 1988; Moehle et al., 1989). We therefore employed the pep4Δ prb1Δ mutant to ensure complete ablation of endoprotease activity. Even when Atg15 was overexpressed in pep4Δ prb1Δ cells, no lipase activity was detected (Fig. S1, F and G). It is therefore likely that these proteases are involved in the processing of Atg15 to an active form.
The vacuolar luminal region of Atg15 is sufficient to confer lipase activity
Atg15 is synthesized in the endoplasmic reticulum (ER) and transported to the vacuole via the MVB pathway (Fig. 2 A). Residues 13–35 of Atg15 constitute a transmembrane domain, which acts as a signal sequence for the MVB pathway (Hirata et al., 2021). To detect Atg15, we expressed Atg15 C-terminally tagged with FLAG (Atg15-FLAG). Following 1 h rapamycin treatment, vacuolar lysate samples were obtained from cells expressing Atg15-FLAG and subjected to immunoblotting. As Atg15-FLAG expressed from a single copy plasmid was only faintly detected (Fig. S2 A, lane 3), we employed a multicopy plasmid for subsequent analyses (Fig. S2 A, lane 4). To confirm that cells expressing Atg15-FLAG are able to disrupt ABs in vivo, processing of precursor Ape1 (prApe1), a cargo protein of both the autophagy and Cvt pathways, was monitored (Fig. S2 B). Following disruption of ABs and Cvt vesicles, prApe1 is processed to a mature form (mApe1) by vacuolar proteases (Baba et al., 1997; Klionsky et al., 1992). mApe1 was detected in each strain expressing Atg15-FLAG, indicating Atg15 activity. We confirmed that lipase activity is apparent in vacuolar lysates, with similar results to those shown in Fig. 1 C (Fig. S2 C).

Processing is necessary for activation of the vacuolar luminal region of Atg15. (A) Schematic illustration of the transport of Atg15 from the ER/Golgi to the vacuole via the MVB pathway. (B) Western blotting of Atg15-FLAG in the vacuole. Vacuolar fractions were obtained from atg15Δ or pep4Δ prb1Δ cells expressing Atg15-FLAG. The samples were treated with or without Endo H and analyzed using α-FLAG antibody. Red arrowhead, 75 kD band of glycosylated whole Atg15-FLAG; blue arrowhead, unglycosylated whole Atg15-FLAG. The amount of lysates used for this analysis is shown in Fig. S2 D. (C) Schematic diagram of Atg15 and CPYN50-Atg15ΔN35. TMD, transmembrane domain. (D) Fluorescence microscopy image of atg15∆ cells expressing CPYN50-Atg15ΔN35-mNG under the control of the endogenous ATG15 promoter from a multicopy plasmid (pRS426). Cells grown in SD/CA medium were treated with rapamycin for 3 h. Scale bar, 1 µm. Dashed lines indicate vacuole boundaries. DIC, differential interference contrast. (E) Analysis of the maturation of prApe1 in atg15Δ cells expressing Atg15 or CPYN50-Atg15ΔN35 under the control of the endogenous ATG15 promoter from a multicopy plasmid (pRS426). Cells grown in SD/CA medium were treated with rapamycin for 2 h. prApe1 and mApe1 were detected using α-Ape1 antibodies. (F) Localization of mCherry-Atg8 in atg15Δ cells or pep4Δ prb1Δ cells, each expressing CPYN50-Atg15ΔN35. Cells grown in YPD medium were treated with rapamycin for 3 h. Scale bar, 1 µm. See also Video 1. Dashed lines indicate vacuole boundaries. (G) Lipase activities of CPYN50-Atg15∆N35–expressing atg15Δ cells and pep4Δ prb1Δ cells. A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 2×, and 4×) and incubated with NBD-PE for 20 min. The amount of lysates used for these analyses is shown in Fig. S2 G. Source data are available for this figure: SourceData F2.
Processing is necessary for activation of the vacuolar luminal region of Atg15. (A) Schematic illustration of the transport of Atg15 from the ER/Golgi to the vacuole via the MVB pathway. (B) Western blotting of Atg15-FLAG in the vacuole. Vacuolar fractions were obtained from atg15Δ or pep4Δ prb1Δ cells expressing Atg15-FLAG. The samples were treated with or without Endo H and analyzed using α-FLAG antibody. Red arrowhead, 75 kD band of glycosylated whole Atg15-FLAG; blue arrowhead, unglycosylated whole Atg15-FLAG. The amount of lysates used for this analysis is shown in Fig. S2 D. (C) Schematic diagram of Atg15 and CPYN50-Atg15ΔN35. TMD, transmembrane domain. (D) Fluorescence microscopy image of atg15∆ cells expressing CPYN50-Atg15ΔN35-mNG under the control of the endogenous ATG15 promoter from a multicopy plasmid (pRS426). Cells grown in SD/CA medium were treated with rapamycin for 3 h. Scale bar, 1 µm. Dashed lines indicate vacuole boundaries. DIC, differential interference contrast. (E) Analysis of the maturation of prApe1 in atg15Δ cells expressing Atg15 or CPYN50-Atg15ΔN35 under the control of the endogenous ATG15 promoter from a multicopy plasmid (pRS426). Cells grown in SD/CA medium were treated with rapamycin for 2 h. prApe1 and mApe1 were detected using α-Ape1 antibodies. (F) Localization of mCherry-Atg8 in atg15Δ cells or pep4Δ prb1Δ cells, each expressing CPYN50-Atg15ΔN35. Cells grown in YPD medium were treated with rapamycin for 3 h. Scale bar, 1 µm. See also Video 1. Dashed lines indicate vacuole boundaries. (G) Lipase activities of CPYN50-Atg15∆N35–expressing atg15Δ cells and pep4Δ prb1Δ cells. A vacuolar lysate preparation was added at three concentrations relative to standard assay conditions (1×, 2×, and 4×) and incubated with NBD-PE for 20 min. The amount of lysates used for these analyses is shown in Fig. S2 G. Source data are available for this figure: SourceData F2.
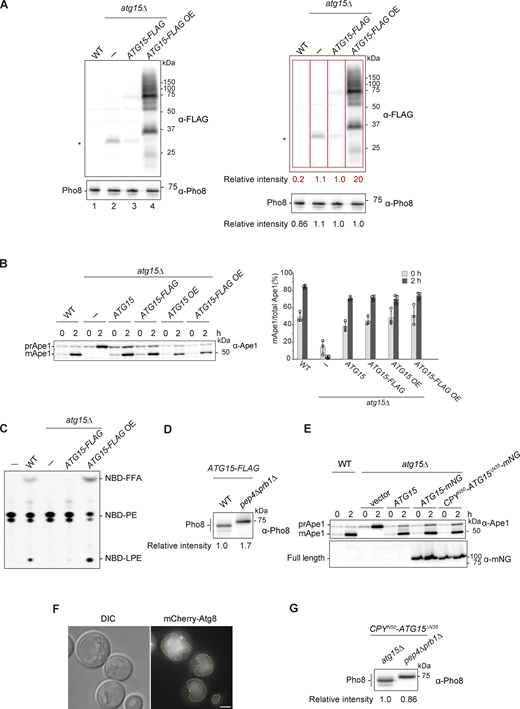
Supplemental data ofFig. 2,. (A) Western blotting of vacuolar fractions from WT, atg15Δ, and atg15Δ cells expressing C-terminally FLAG-tagged Atg15 (Atg15-FLAG) from either a single plasmid (pRS316) or a multicopy plasmid (pRS426). The ratio of the band intensities detected by α-FLAG (red squares) and α-Pho8 antibodies for each strain are shown in the right panels. *, nonspecific band. (B) Maturation of prApe1 in cells expressing Atg15 tagged with FLAG in C-terminus. Cells were grown in SD/CA medium and treated with rapamycin for 2 h. prApe1 and mApe1 were detected with α-Ape1 antibodies. Western blotting (left panel) shows representative results from three independent experiments. The band intensities of prApe1 and mApe1 were measured, and the percentage of mApe1 to the sum of prApe1 and mApe1 were determined (right panel). Error bars represent means ± SD of three independent experiments. (C) Lipase activity of vacuolar lysates from the strains in A. Each lysate was incubated with NBD-PE for 20 min. (D) Western blotting of vacuolar fractions from atg15Δ and pep4Δ prb1Δ cells each expressing Atg15-FLAG from a multicopy plasmid. The experimental procedure was the same as in Fig. S1 B. (E) Maturation of prApe1 in mNG-tagged Atg15 expressing cells. Culture condition was the same as in B. prApe1 and mApe1 were detected with α-Ape1 antibodies. Full length of C-terminally mNG-tagged Atg15 and CPYN50-Atg15ΔN35 were detected with α-mNG antibodies. (F) Fluorescence microscopy image of mCherry-Atg8 in WT cells. Cells were grown in SD/CA medium and then treated with rapamycin for 3 h. Scale bar, 1 µm. Dashed lines, vacuole boundaries. DIC, differential interference contrast. (G) Western blotting of vacuolar fractions from atg15Δ and pep4Δ prb1Δ cells each expressing CPYN50-Atg15∆N35. The experimental procedures were the same as in Fig. S1 B. Source data are available for this figure: SourceData FS2.

Supplemental data ofFig. 2,. (A) Western blotting of vacuolar fractions from WT, atg15Δ, and atg15Δ cells expressing C-terminally FLAG-tagged Atg15 (Atg15-FLAG) from either a single plasmid (pRS316) or a multicopy plasmid (pRS426). The ratio of the band intensities detected by α-FLAG (red squares) and α-Pho8 antibodies for each strain are shown in the right panels. *, nonspecific band. (B) Maturation of prApe1 in cells expressing Atg15 tagged with FLAG in C-terminus. Cells were grown in SD/CA medium and treated with rapamycin for 2 h. prApe1 and mApe1 were detected with α-Ape1 antibodies. Western blotting (left panel) shows representative results from three independent experiments. The band intensities of prApe1 and mApe1 were measured, and the percentage of mApe1 to the sum of prApe1 and mApe1 were determined (right panel). Error bars represent means ± SD of three independent experiments. (C) Lipase activity of vacuolar lysates from the strains in A. Each lysate was incubated with NBD-PE for 20 min. (D) Western blotting of vacuolar fractions from atg15Δ and pep4Δ prb1Δ cells each expressing Atg15-FLAG from a multicopy plasmid. The experimental procedure was the same as in Fig. S1 B. (E) Maturation of prApe1 in mNG-tagged Atg15 expressing cells. Culture condition was the same as in B. prApe1 and mApe1 were detected with α-Ape1 antibodies. Full length of C-terminally mNG-tagged Atg15 and CPYN50-Atg15ΔN35 were detected with α-mNG antibodies. (F) Fluorescence microscopy image of mCherry-Atg8 in WT cells. Cells were grown in SD/CA medium and then treated with rapamycin for 3 h. Scale bar, 1 µm. Dashed lines, vacuole boundaries. DIC, differential interference contrast. (G) Western blotting of vacuolar fractions from atg15Δ and pep4Δ prb1Δ cells each expressing CPYN50-Atg15∆N35. The experimental procedures were the same as in Fig. S1 B. Source data are available for this figure: SourceData FS2.
In pep4Δ prb1Δ cells, a glycosylated full-length Atg15-FLAG band at 75 kD and heavily glycosylated smear bands were observed, as previously reported (Fig. 2 B, lane 2, and Fig. S2 D; Epple et al., 2001). In WT cells, a single band slightly smaller than the 75 kD band was also apparent, the amount of which was reduced due to processing at the C-terminus (Fig. 2 B, lane 1, and Fig. S2 D). When lysates from pep4Δ prb1Δ cells were treated with endoglycosidase H, the 75 kD band and smeared bands converged into one lower band (Fig. 2 B, lane 4). Further, compared with this band, a single band of apparently smaller molecular mass appeared in WT lysates (Fig. 2 B, lane 3). These observed differences in mobility of Atg15-FLAG suggest that all Atg15 molecules were cleaved at the N-terminus by Pep4 and Prb1 as all observed bands have an intact C-terminus. This most likely occurs rapidly at an N-terminal region adjacent to the transmembrane domain immediately following delivery of Atg15 to the vacuole.
We next expressed Atg15 lacking this transmembrane domain to gain more insights into the processing of Atg15 in the vacuole. Expression of Atg15 lacking residues 1–35 (“vacuolar luminal region,” Atg15ΔN35) in fusion with 50 N-terminal residues (1–50) of CPY (CPYN50) allows for the transport of the resulting CPYN50-Atg15ΔN35 (Fig. 2 C) to the vacuole via the CPY pathway (Johnson et al., 1987). By tagging CPYN50-Atg15ΔN35 with mNeonGreen (mNG) at its C-terminus, we confirmed that this fusion protein localizes to the vacuole under growing and autophagy-inducing conditions (Fig. 2 D). To determine whether CPYN50-Atg15ΔN35–expressing cells are able to disrupt ABs in the absence of endogenous Atg15, processing of prApe1 was monitored. Ape1 maturation occurred normally in atg15Δ cells expressing CPYN50-Atg15ΔN35 under both growing and autophagy-inducing conditions (Fig. 2 E and Fig. S2 E). Therefore, we conclude that the vacuolar luminal region is sufficient to generate activated Atg15 that is capable of AB and Cvt vesicle disruption.
We further examined whether CPYN50-Atg15ΔN35 is able to disrupt ABs in the absence of Pep4 and Prb1. ABs were labeled with mCherry-tagged Atg8 (Kirisako et al., 1999). Upon induction of autophagy, mCherry-Atg8 signal was homogenously diffused in the vacuole of WT cells (Fig. S2 F). atg15Δ cells expressing CPYN50-Atg15ΔN35 exhibited a similar pattern (Fig. 2 F and Video 1), indicating that autophagy proceeded normally in these cells. On the other hand, in pep4Δ prb1Δ cells expressing CPYN50-Atg15ΔN35, intact ABs were observed in vacuoles (Fig. 2 F and Video 1). We also observed that vacuolar lysates from CPYN50-Atg15ΔN35–expressing cells do not exhibit lipase activity in the absence of Pep4 and Prb1 (Fig. 2 G and Fig. S2 G). Together, these data suggest that release of Atg15 from MVB vesicles is not sufficient for acquisition of lipase activity. Rather, we find that processing within the vacuolar luminal region is required.
Fluorescence microscopy real-time imaging of mCherry-Atg8–expressing atg15Δ cells (left) and pep4Δ prb1Δ cells (right), all expressing CPYN50-Atg15ΔN35. Cells grown in YPD medium were treated with rapamycin for 3 h. The frame rate of the video is 10 fps. See also Fig. 2 F. Scale bar, 1 µm.
Fluorescence microscopy real-time imaging of mCherry-Atg8–expressing atg15Δ cells (left) and pep4Δ prb1Δ cells (right), all expressing CPYN50-Atg15ΔN35. Cells grown in YPD medium were treated with rapamycin for 3 h. The frame rate of the video is 10 fps. See also Fig. 2 F. Scale bar, 1 µm.
The vacuolar luminal region of Atg15 binds to the AB membrane
We sought to understand the behavior of the vacuolar luminal region of Atg15. Atg15 and CPYN50-Atg15ΔN35 were tagged with mNG at their C-termini and expressed in pep4Δ prb1Δ cells to avoid processing. Spinning-disk confocal fluorescence microscopy revealed that Atg15-mNG signal was detected as circular structures, which suggests a nuclear ER localization as previously reported (Fig. 3 and Video 2; Epple et al., 2001). We also observed rapid movement of Atg15-mNG puncta within vacuoles. These most likely represent MVB vesicles, as previously reported (Hirata et al., 2021). Occasionally, distinct bright foci were observed that likely represent aggregate forms of Atg15. On the other hand, CPYN50-Atg15ΔN35-mNG were observed as ring structures surrounding mCherry-Atg8 foci (Fig. 3 and Video 3), indicating that the vacuolar luminal domain is bound to the AB membrane. These data suggest that following delivery to the vacuole, Atg15 might bind to AB membranes upon its release from MVBs.

The vacuolar luminal region of Atg15 binds to the AB membrane. Spinning-disk confocal microscopy image of pep4Δ prb1Δ cells expressing Atg15-mNG or CPYN50-Atg15ΔN35-mNG. Cells were grown in YPD medium and treated with rapamycin for 3 h. ABs are indicated by mCherry-Atg8 fluorescence. The green and pink arrowheads indicate ER and aggregated Atg15-mNG, respectively. Dashed lines indicate vacuole (yellow) and cell (cyan) boundaries. Scale bar, 1 µm. See also Videos 2 and 3.
The vacuolar luminal region of Atg15 binds to the AB membrane. Spinning-disk confocal microscopy image of pep4Δ prb1Δ cells expressing Atg15-mNG or CPYN50-Atg15ΔN35-mNG. Cells were grown in YPD medium and treated with rapamycin for 3 h. ABs are indicated by mCherry-Atg8 fluorescence. The green and pink arrowheads indicate ER and aggregated Atg15-mNG, respectively. Dashed lines indicate vacuole (yellow) and cell (cyan) boundaries. Scale bar, 1 µm. See also Videos 2 and 3.
Spinning-disk confocal microscopy real-time imaging of pep4Δ prb1Δ cells expressing Atg15-mNG. Cells were grown in YPD medium and treated with rapamycin for 3 h. ABs are indicated by mCherry-Atg8 fluorescence. The frame rate of the video is 10 fps. See also Fig. 3. Scale bar, 1 µm.
Spinning-disk confocal microscopy real-time imaging of pep4Δ prb1Δ cells expressing Atg15-mNG. Cells were grown in YPD medium and treated with rapamycin for 3 h. ABs are indicated by mCherry-Atg8 fluorescence. The frame rate of the video is 10 fps. See also Fig. 3. Scale bar, 1 µm.
Spinning-disk confocal microscopy real-time imaging of pep4Δ prb1Δ cells expressing CPYN50-Atg15ΔN35. Cells were grown in YPD medium and treated with rapamycin for 3 h. ABs are indicated by mCherry-Atg8 fluorescence. The frame rate of the video is 10 fps. See also Fig. 3. Scale bar, 1 µm.
Spinning-disk confocal microscopy real-time imaging of pep4Δ prb1Δ cells expressing CPYN50-Atg15ΔN35. Cells were grown in YPD medium and treated with rapamycin for 3 h. ABs are indicated by mCherry-Atg8 fluorescence. The frame rate of the video is 10 fps. See also Fig. 3. Scale bar, 1 µm.
Isolation of active Atg15 from vacuolar pellet fractions
Due to the cleavage of the terminal tag (Fig. 2 B), we constructed an internally FLAG-tagged Atg15 variant to further study processing of Atg15 in the vacuole. We first set out to identify the region necessary to disrupt ABs by making a series of N- and C-terminal truncation mutants of Atg15 that we fused with CPYN50. By expressing these truncation mutants in atg15Δ cells, we found that the deletion of residues 1–58 had little effect on the maturation of prApe1, but further deletion of Arg59 severely impaired maturation (Fig. S3 A). When C-terminal residues 467–520 were deleted, prApe1 was still processed into mApe1, whereas the deletion of Trp466 caused an almost complete maturation defect (Fig. S3 B). These results suggest that residues 59–466 of Atg15 are required to disrupt ABs, which is in close agreement with a previous study (Hirata et al., 2021). Further optimization revealed that the FLAG tag can be inserted after Asp168 (Atg15iFLAG) without affecting prApe1 maturation (Fig. 4 A).
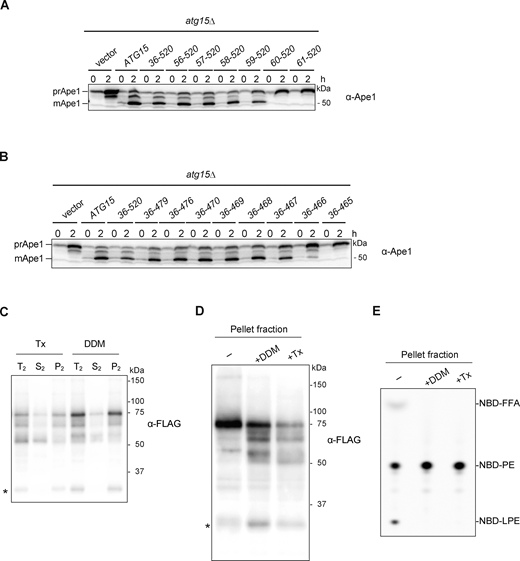
Supplemental data ofFig. 4,. (A and B) Western blotting of the maturation of prApe1 in cells expressing N-terminally (A) or C-terminally (B) truncated Atg15 mutants. atg15Δ cells expressing WT Atg15 or fusion of truncated Atg15 with CPYN50 under the control of ATG15 promoter from a multicopy-plasmid (pRS426) were cultured in the same conditions as in Fig. 2 D. A series of N- and C-terminally truncated forms of Atg15 were constructed by PCR from pRS426-CPY(1–50)-ATG15(Δ1–35)-mNeonGreen (YPL073) (see Materials and methods). (C) Vacuolar pellet fraction (Fig. 4 C, lane 3) was treated with 2% Tx or 0.5% DDM (T2) and incubated at 4°C for 30 min. Then samples were centrifuged at 135,000 g for 30 min, followed by separation into pellet (P2) and supernatant (S2) fractions before being subjected to western blot analysis with α-FLAG antibodies. *, nonspecific band. (D) Vacuolar pellet fraction (Fig. 4 C, lane 3) was treated with 0.5% DDM or 2% Tx. Samples were then subjected to western blotting with α-FLAG antibodies. *, nonspecific band. (E) Lipase activities of vacuolar pellet fraction treated with detergents. NBD-PE was incubated with each fraction in D for 20 min. Final DDM and Tx concentrations in the reaction mixture were 0.05% and 0.2%, respectively. Source data are available for this figure: SourceData FS3.

Supplemental data ofFig. 4,. (A and B) Western blotting of the maturation of prApe1 in cells expressing N-terminally (A) or C-terminally (B) truncated Atg15 mutants. atg15Δ cells expressing WT Atg15 or fusion of truncated Atg15 with CPYN50 under the control of ATG15 promoter from a multicopy-plasmid (pRS426) were cultured in the same conditions as in Fig. 2 D. A series of N- and C-terminally truncated forms of Atg15 were constructed by PCR from pRS426-CPY(1–50)-ATG15(Δ1–35)-mNeonGreen (YPL073) (see Materials and methods). (C) Vacuolar pellet fraction (Fig. 4 C, lane 3) was treated with 2% Tx or 0.5% DDM (T2) and incubated at 4°C for 30 min. Then samples were centrifuged at 135,000 g for 30 min, followed by separation into pellet (P2) and supernatant (S2) fractions before being subjected to western blot analysis with α-FLAG antibodies. *, nonspecific band. (D) Vacuolar pellet fraction (Fig. 4 C, lane 3) was treated with 0.5% DDM or 2% Tx. Samples were then subjected to western blotting with α-FLAG antibodies. *, nonspecific band. (E) Lipase activities of vacuolar pellet fraction treated with detergents. NBD-PE was incubated with each fraction in D for 20 min. Final DDM and Tx concentrations in the reaction mixture were 0.05% and 0.2%, respectively. Source data are available for this figure: SourceData FS3.

Isolation of active Atg15 from pellet fraction of the vacuole. (A) Maturation of prApe1 and expression of Atg15iFLAG, Atg15S332A,iFLAG, CPYN50-Atg15ΔN35,iFLAG, or CPYN50-Atg15ΔN35,S332A,iFLAG. Cells were grown in SD/CA medium and treated with rapamycin for 2 h. *, nonspecific band. (B) Overview of the strategy employed for the fractionation of prcAtg15s. Total vacuolar lysate (T) was fractionated to supernatant (S) and pellet (P) fractions by centrifugation. The pellet (P = T2) was then resuspended in buffer containing 2% Tx, 0.1 M Na2CO3, 2.5 M urea, or 1 M KCl and subjected to a second round of centrifugation to yield a pellet fraction (P2) and supernatant (S2) fraction. (C) Western blotting of prcAtg15 and vacuolar membrane proteins. Vacuolar lysates from Atg15iFLAG-expressing cells were subjected to fractionation as in B. Equivalent amounts (relative to starting materials) of each fraction were analyzed. (D) Lipase activities of T, S, and P fractions (panel C, lane 1–3). Each fraction was incubated with NBD-PE for 20 min. TLC analysis (upper panel) shows representative data from three independent experiments. The bar graph (lower panel) represents the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (E) Western blotting of the processed forms of CPYN50-Atg15ΔN35,iFLAG (prcAtg15ΔN35s) and vacuolar membrane proteins (as in C). (F) Western blotting of prcAtg15s obtained by immunoprecipitation. Untreated vacuolar pellet fraction (P), vacuolar pellet fraction treated with 0.5% DDM (P+DDM), prcAtg15s pulled down by α-FLAG antibody-conjugated agarose beads (Bound), and prcAtg15s eluted from bound by an excess amount of 3xFLAG peptides (Eluate) were analyzed. Samples equivalent to 0.2% of P and P+DDM, and 5% of Bound and Eluate were subjected to Ponceau S staining (left) and western blotting using α-FLAG antibodies (right). *, nonspecific band; **, mouse-IgG heavy chain; ⁂, mouse-IgG light chain. Source data are available for this figure: SourceData F4.
Isolation of active Atg15 from pellet fraction of the vacuole. (A) Maturation of prApe1 and expression of Atg15iFLAG, Atg15S332A,iFLAG, CPYN50-Atg15ΔN35,iFLAG, or CPYN50-Atg15ΔN35,S332A,iFLAG. Cells were grown in SD/CA medium and treated with rapamycin for 2 h. *, nonspecific band. (B) Overview of the strategy employed for the fractionation of prcAtg15s. Total vacuolar lysate (T) was fractionated to supernatant (S) and pellet (P) fractions by centrifugation. The pellet (P = T2) was then resuspended in buffer containing 2% Tx, 0.1 M Na2CO3, 2.5 M urea, or 1 M KCl and subjected to a second round of centrifugation to yield a pellet fraction (P2) and supernatant (S2) fraction. (C) Western blotting of prcAtg15 and vacuolar membrane proteins. Vacuolar lysates from Atg15iFLAG-expressing cells were subjected to fractionation as in B. Equivalent amounts (relative to starting materials) of each fraction were analyzed. (D) Lipase activities of T, S, and P fractions (panel C, lane 1–3). Each fraction was incubated with NBD-PE for 20 min. TLC analysis (upper panel) shows representative data from three independent experiments. The bar graph (lower panel) represents the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (E) Western blotting of the processed forms of CPYN50-Atg15ΔN35,iFLAG (prcAtg15ΔN35s) and vacuolar membrane proteins (as in C). (F) Western blotting of prcAtg15s obtained by immunoprecipitation. Untreated vacuolar pellet fraction (P), vacuolar pellet fraction treated with 0.5% DDM (P+DDM), prcAtg15s pulled down by α-FLAG antibody-conjugated agarose beads (Bound), and prcAtg15s eluted from bound by an excess amount of 3xFLAG peptides (Eluate) were analyzed. Samples equivalent to 0.2% of P and P+DDM, and 5% of Bound and Eluate were subjected to Ponceau S staining (left) and western blotting using α-FLAG antibodies (right). *, nonspecific band; **, mouse-IgG heavy chain; ⁂, mouse-IgG light chain. Source data are available for this figure: SourceData F4.
We next undertook biochemical characterization of the processed forms of Atg15iFLAG (hereafter referred to as prcAtg15s). Vacuolar lysates from cells expressing Atg15iFLAG were subjected to centrifugation at 135,000 g (Fig. 4 B). Almost all prcAtg15s were detected in the pellet fraction, with only a very faint signal in the supernatant (Fig. 4 C, lanes 1–3). In addition, lipase activity was also detected mainly in the pellet fraction (Fig. 4 D). To extract processed Atg15 from pellets, we next subjected the pellet fraction to a range of conditions, including high pH, urea, high salt, or incubation in the presence of a detergent. High pH, urea, and high salt are known to strip peripheral proteins from membranes, whereas detergent treatment results in solubilization of both integral and peripheral membrane proteins. prcAtg15s were not extracted following high pH, urea, or high salt treatments (Fig. 4 C, lanes 10–18), but were partially solubilized with the detergent Triton X-100 (Tx; Fig. 4 C, lanes 7–9). Vph1 and Pho8, which are integral vacuolar membrane proteins, were also extracted by Tx. Other detergents, such as n-dodecyl-β-D-maltoside (DDM), solubilized the prcAtg15s to a nearly equal extent as Tx (Fig. S3 C). We further examined the membrane-binding properties of CPYN50-Atg15ΔN35, finding that the vacuolar luminal region is tightly bound to the membrane (Fig. 4 E), and also determined that prcAtg15s were partially degraded when subjected to detergent treatments (Fig. 4 C, lanes 7–9, Fig. 4 E, lanes 7–9, and Fig. S3 C). This likely reflects an increased sensitivity of prcAtg15s to proteases following solubilization.
We also investigated the lipase activity of prcAtg15s, but failed to detect any such activity in the presence of detergents (Fig. S3, D and E). To obtain detergent-free prcAtg15s, homogenates of vacuolar pellets were first solubilized with 0.5% DDM in the presence of α-FLAG antibody-conjugated agarose beads, following which the beads were collected and subsequently washed with a buffer solution to remove unbound materials and the detergent. The beads were then incubated with an excess amount of 3xFLAG peptide to elute prcAtg15s. Although the amount of the final eluted fraction was too small to be detected by Ponceau-S staining, several processed forms of Atg15iFLAG were detected by western blotting with α-FLAG antibodies (Fig. 4 F).
Activated Atg15 functions as a phospholipase B
Following the above procedures, we prepared eluates from both prcAtg15 and prcAtg15S332A samples (Fig. 5 A) to characterize their lipase activities. Upon incubation in the presence of prcAtg15 eluates, NBD-PE levels as determined by TLC decreased in a time-dependent manner to an almost undetectable level at 6 h. Meanwhile, NBD-LPE increased up to 1 h before exhibiting a gradual decrease. NBD-FFA increased throughout the incubation, indicating that NBD-FFA is generated from both NBD-PE and NBD-LPE (Fig. 5 B). In contrast, the prcAtg15S332A eluate did not exhibit any lipase activity at all (Fig. 5 B).

Activated Atg15 is a phospholipase B of broad head-group specificity. (A) Eluates of prcAtg15s and prcAtg15S332As were obtained as in Fig. 4 F. Samples equivalent to 0.3% of P and P+DDM, and 22% of Eluate were subjected to western blotting with α-FLAG antibodies. *, nonspecific band. (B) Time course of lipase activity in prcAtg15s and prcAtg15S332As. At each point, 3 μl of eluates in A were incubated with NBD-PE. Representative results from three independent experiments are shown (upper panel). The line graphs (lower panel) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (C and D) Lipase activity toward NBD-PE, NBD-PS, NBD-PC, and NBD-PG, each labeled at sn-2 (C–E), and NBD-LPC (12:0) (D). Each phospholipid was incubated with 1 μl of Eluate (A) for 1 h. (E) Lipase activity toward TF-PI (18:1–6:0) labeled at sn-2. TF-PI was incubated with 4 μl of Eluate (A) for 2 h. (F) Lipase activity toward TF-TG and BODIPY-CE. Each non-polar lipid was incubated with 4 μl of Eluate (A) for 2 h. Source data are available for this figure: SourceData F5.
Activated Atg15 is a phospholipase B of broad head-group specificity. (A) Eluates of prcAtg15s and prcAtg15S332As were obtained as in Fig. 4 F. Samples equivalent to 0.3% of P and P+DDM, and 22% of Eluate were subjected to western blotting with α-FLAG antibodies. *, nonspecific band. (B) Time course of lipase activity in prcAtg15s and prcAtg15S332As. At each point, 3 μl of eluates in A were incubated with NBD-PE. Representative results from three independent experiments are shown (upper panel). The line graphs (lower panel) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (C and D) Lipase activity toward NBD-PE, NBD-PS, NBD-PC, and NBD-PG, each labeled at sn-2 (C–E), and NBD-LPC (12:0) (D). Each phospholipid was incubated with 1 μl of Eluate (A) for 1 h. (E) Lipase activity toward TF-PI (18:1–6:0) labeled at sn-2. TF-PI was incubated with 4 μl of Eluate (A) for 2 h. (F) Lipase activity toward TF-TG and BODIPY-CE. Each non-polar lipid was incubated with 4 μl of Eluate (A) for 2 h. Source data are available for this figure: SourceData F5.
Next, we examined substrate specificity. NBD-PS was hydrolyzed as NBD-PE (Fig. 5 C). Further, while commercially available NBD-PC and NBD-PG were somewhat degraded, these species were clearly hydrolyzed by prcAtg15 eluates (Fig. 5 C). Meanwhile, NBD-labeled lysophosphatidylcholine (LPC) was cleaved to NBD-FFA (Fig. 5 D), and we additionally observed hydrolysis of topflour-labeled PI (TF-PI) to TF-lysophosphatidylinositol (TF-LPI) and TF-FFA (Fig. 5 E). On the other hand, we were unable to detect any activity toward non-polar lipids such as TG and cholesterol ester (CE) in our assay conditions (Fig. 5 F). These results correspond well with those from vacuolar lysates (Fig. S4). Taken together, these data indicate that active Atg15 is a phospholipase B of broad head-group specificity.
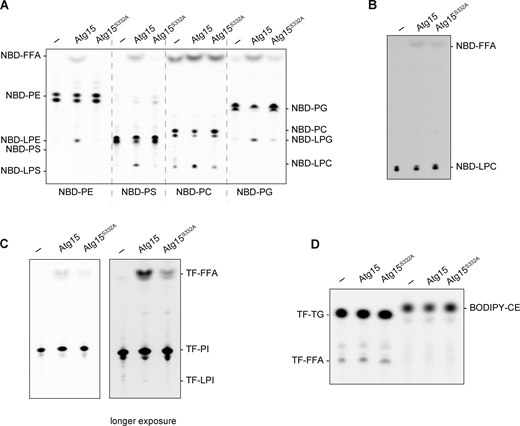
Vacuolar lysate has lipase activity derived from Atg15 of broad specificity. (A and B) Lipase activity of vacuolar lysates from atg15Δ cells expressing Atg15iFLAG and Atg15iFLAG,S332A. The lysates were incubated with NBD-PE, NBD-PS, NBD-PC, and NBD-PG, each labeled at sn-2 (A), and NBD-LPC (B) for 1 h. (C) Lipase activity of vacuolar lysates toward TF-PI labeled at sn-2. The incubation time was 1 h. (D) Lipase activity of vacuolar lysates toward TF-TG and BODIPY-CE. The incubation time was 2 h. Source data are available for this figure: SourceData FS4.

Vacuolar lysate has lipase activity derived from Atg15 of broad specificity. (A and B) Lipase activity of vacuolar lysates from atg15Δ cells expressing Atg15iFLAG and Atg15iFLAG,S332A. The lysates were incubated with NBD-PE, NBD-PS, NBD-PC, and NBD-PG, each labeled at sn-2 (A), and NBD-LPC (B) for 1 h. (C) Lipase activity of vacuolar lysates toward TF-PI labeled at sn-2. The incubation time was 1 h. (D) Lipase activity of vacuolar lysates toward TF-TG and BODIPY-CE. The incubation time was 2 h. Source data are available for this figure: SourceData FS4.
The active form of Atg15 is sufficient to disrupt ABs
To determine whether phospholipids are degraded in membranes as well as in solution, we next synthesized NBD-PE containing liposomes. We observed degradation of NBD-PE by prcAtg15s in both 400- and 100-nm-diameter liposomes (Fig. S5). We also developed an in vitro assay system to evaluate AB disruption (Fig. 6 A). To this end, we employed our recently established method for the isolation of ABs that retain autophagosome cargo (Kawamata et al., 2022), which allowed us to use purified ABs as a native substrate of Atg15. We isolated ABs from atg15Δ cells expressing GST-GFP, which localizes to the cytosol and is delivered to the vacuole via autophagy. prApe1 and GST-GFP in the AB were resistant to Proteinase K (Pro K) treatment (Fig. 6 B, lanes 1 and 2). Upon treatment of ABs with detergent, prApe1 and GFP-GST were cleaved to produce degraded Ape1 (dApe1) and free GFP (Fig. 6 B, lane 3), respectively. Previous work has shown that the GFP moiety is resistant to Pro K treatment and that Pro K treatment results in the formation of dApe1 (Nair et al., 2011). Therefore, the amount of free GFP or dApe1 reflects the degree of AB membrane disruption. When ABs were treated with prcAtg15 eluates, dApe1 and free GFP appeared (Fig. 6 B, lane 4 and 6), indicating that prcAtg15 eluates are capable of AB membrane disruption. Furthermore, the extent of this disruption was dependent on the amount of prcAtg15 eluate added (Fig. 6 B, lanes 4 and 6). On the other hand, treatment of ABs with prcAtg15S332A did not result in membrane disruption (Fig. 6 B, lanes 5 and 7). These results indicate that active Atg15 is sufficient to disrupt AB membranes in vitro.
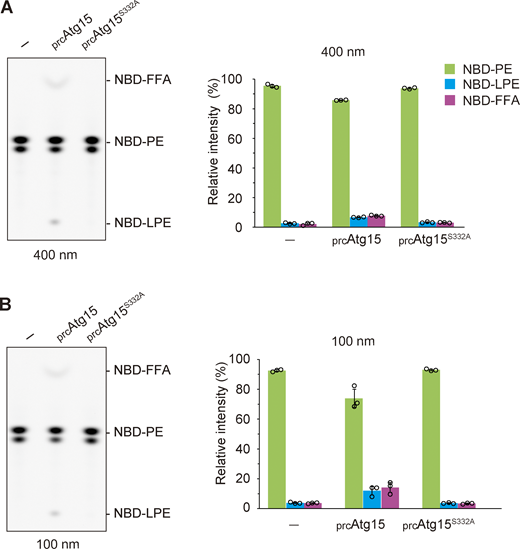
Active Atg15 degrades phospholipids in membrane. (A) Lipase activity of vacuolar lysates toward 400-nm-diameter liposomes containing NBD-PE. The incubation time was 2 h. TLC analysis (left panels) shows representative results from three independent experiments. The bar graphs (right panels) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (B) Lipase activity of vacuolar lysates toward 100-nm-diameter liposomes containing NBD-PE. The incubation time was 2 h. TLC analysis (left panels) shows representative results from three independent experiments. The bar graphs (right panels) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. Source data are available for this figure: SourceData FS5.

Active Atg15 degrades phospholipids in membrane. (A) Lipase activity of vacuolar lysates toward 400-nm-diameter liposomes containing NBD-PE. The incubation time was 2 h. TLC analysis (left panels) shows representative results from three independent experiments. The bar graphs (right panels) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. (B) Lipase activity of vacuolar lysates toward 100-nm-diameter liposomes containing NBD-PE. The incubation time was 2 h. TLC analysis (left panels) shows representative results from three independent experiments. The bar graphs (right panels) represent the relative intensity calculated as described in Fig. 1 C. Error bars represent means ± SD of three independent experiments. Source data are available for this figure: SourceData FS5.

Activated Atg15 is sufficient to disrupt ABs. (A) Schematic view of the in vitro assay employed to evaluate AB disruption. (B) Disruption of ABs by activated Atg15. Isolated ABs were incubated with 0.2% Tx, prcAtg15, or prcAtg15S332A eluates at 30°C for 0 or 3 h. Eluate was added at two concentrations relative to standard assay conditions (1× and 3×). The reaction mixture was subjected to western blotting using α-Ape1 and α-GFP antibodies, respectively. (C) A model of AB disruption by active Atg15 in the vacuole. (i) Upon delivery to the vacuole, Atg15 is cleaved from MVB vesicles by Pep4/Prb1. (ii) Processed forms of Atg15 are tightly bound to the AB membrane. Atg15 is activated by Pep4/Prb1 during or following localization to the AB membrane. (iii) The active form of Atg15 hydrolyzes acyl ester linkages of phospholipids of AB membranes, releasing two acyl groups and disrupting the membrane. (iv) AB contents are released into the vacuole, where they are degraded by vacuolar hydrolases including Pep4 and Prb1. Source data are available for this figure: SourceData F6.
Activated Atg15 is sufficient to disrupt ABs. (A) Schematic view of the in vitro assay employed to evaluate AB disruption. (B) Disruption of ABs by activated Atg15. Isolated ABs were incubated with 0.2% Tx, prcAtg15, or prcAtg15S332A eluates at 30°C for 0 or 3 h. Eluate was added at two concentrations relative to standard assay conditions (1× and 3×). The reaction mixture was subjected to western blotting using α-Ape1 and α-GFP antibodies, respectively. (C) A model of AB disruption by active Atg15 in the vacuole. (i) Upon delivery to the vacuole, Atg15 is cleaved from MVB vesicles by Pep4/Prb1. (ii) Processed forms of Atg15 are tightly bound to the AB membrane. Atg15 is activated by Pep4/Prb1 during or following localization to the AB membrane. (iii) The active form of Atg15 hydrolyzes acyl ester linkages of phospholipids of AB membranes, releasing two acyl groups and disrupting the membrane. (iv) AB contents are released into the vacuole, where they are degraded by vacuolar hydrolases including Pep4 and Prb1. Source data are available for this figure: SourceData F6.
Overall, we propose a model for the processing of Atg15 to an active form and the subsequent lysis of AB membranes in the vacuole by active Atg15 (Fig. 6 C). First, Atg15 is delivered to the vacuole by the MVB pathway, where it is immediately cleaved from MVB vesicles by Pep4/Prb1. The processed forms are tightly bound to the AB membrane and activated by Pep4/Prb1 either during or following the localization of Atg15 to AB membrane. Following activation, Atg15 is able to hydrolyze ester bonds at the sn-1 and sn-2 positions of AB membrane phospholipids. Finally, released AB contents are degraded by vacuolar hydrolases including Pep4 and Prb1, completing the degradation of autophagy cargos.
Discussion
Both Atg15 and the proteases Pep4/Prb1 are necessary for AB disruption, but how these proteins are involved in this process has remained unclear. In this study, we established an in vitro lipase assay to investigate Atg15, which revealed that Pep4 and Prb1 process Atg15 to an activated form with lipase activity. Furthermore, we used isolated ABs to develop an in vitro system to evaluate AB membrane disruption, which allowed us to show that activated Atg15, which has phospholipase B activity, is sufficient to disrupt AB membranes.
Activation of Atg15 by Pep4/Prb1-mediated processing
Our in vitro lipase assay helped clarify the relationship between Atg15 lipase activity and other vacuolar proteins that have previously been implicated in AB disruption. We showed that the appearance of Atg15 lipase activity completely depends on Pep4 and Prb1 (Fig. 1 F). The fact that cells overexpressing Atg15 show no lipase activity in the absence of Pep4 or Prb1 (Fig. S1 F) strongly suggests that Pep4 and Prb1 activate Atg15 by direct processing of the protein rather than indirectly via the degradation of an inhibitor of Atg15.
Our analyses suggest that Atg15 is cleaved at the region adjacent to the transmembrane domain by Pep4 and Prb1 to liberate it from MVB vesicles, after which it binds AB membranes (Fig. 2 B and Fig. 3). We also report that the processing of the vacuolar luminal region of Atg15 is necessary for Atg15 to acquire lipase activity (Fig. 2 G). Creation of an internally FLAG-tagged Atg15 enabled us to analyze the processing of Atg15 by immunopurification (Fig. 4). Using this strategy, we successfully obtained active form(s) of Atg15 with lipase activity (Fig. 4 F). The 3D structure of Atg15, which is currently unsolved, will provide important clues that allow for a more nuanced understanding of the activation mechanism.
Enzymatic features of Atg15 following activation
Ramya and Rajasekharan previously reported that Atg15 purified from microsomal membrane fractions of yeast preferentially hydrolyzes PS rather than other phospholipids (Ramya and Rajasekharan, 2016). In our study, active Atg15 did not show such a preference for PS: PC, PG, and PE were also hydrolyzed (Fig. 5 C). This discrepancy may be related to technical differences in assay conditions: our lipase assay was performed at physiological pH (pH 6.9), whereas Ramya and Rajasekharan measured at pH 8.0.
Importantly, our results reveal for the first time that active Atg15 exhibits phospholipase B activity (Fig. 5 B). The ability of phospholipase B enzymes to cleave both acyl groups of phospholipids is likely important for efficient membrane disruption, and the low substrate specificity of Atg15 may also be advantageous for the degradation of diverse membrane lipids of Cvt bodies, MVB, and microautophagic vesicles, as well as organellar membranes such as those of mitochondria, the ER, and peroxisomes following delivery to the vacuole. For non-polar lipids (TG and CE), we were unable to observe hydrolysis by activated Atg15 using our lipase assay (Fig. 5 F). Further analyses are needed to determine if Atg15 has such activity under different conditions or if there are alternative lipases in the vacuole that are able to hydrolyze such non-polar lipids.
Hydrophobic features of Atg15
Many vacuolar hydrolytic enzymes undergo processing by proteases (Hecht et al., 2014). We assume that Atg15 is maintained in an inactive state during trafficking from the ER to Golgi, instead becoming active only after delivery to the vacuole. Atg15 is very sensitive to proteases, especially when detached from membranes in the vacuole (Fig. 4, C, E, and F). Free Atg15 is therefore rapidly degraded in the vacuole, as previously reported (Epple et al., 2001; Teter et al., 2001). These properties may be critical for the spaciotemporal regulation of potentially deleterious lipase activity.
Our analysis of the vacuolar luminal region raises two intriguing implications. First, this region binds to the AB membrane, but not the vacuolar membrane (Fig. 3). This finding likely explains why Atg15 is unable to digest the vacuolar membrane. We also found that this region binds the AB membrane even when the protein is not activated (Fig. 2, F and G; and Fig. 3), suggesting that activation of Atg15 might occur on AB membranes, which provides an excellent way to restrict Atg15 lipase activity to specific membranes. The apparent preference of Atg15 for AB membranes may be explained by the high membrane curvature of these membranes (vacuolar membranes are generally characterized by very low or negative curvature). Alternatively, glycoproteins abundant on the inner surface of the vacuolar membrane may not allow Atg15 to bind to the vacuolar membrane.
The second implication of our study is that the processed forms of the vacuolar luminal region of Atg15 are tightly bound to the membrane in a manner reminiscent of a transmembrane protein (Fig. 4, C and E). Although a search of the domain database (Letunic et al., 2021) revealed no identified transmembrane domains within the vacuolar luminal region (Fig. 1 A), Atg15 probably contains membrane-binding sites besides the active center, as observed in many other lipases (Cao et al., 2013). Indeed, the structure of Atg15, as predicted by AlphaFold2 (Tunyasuvunakool et al., 2021), shows that Atg15 has several clusters of hydrophobic residues facing toward the exterior of the protein, some of which may become embedded in AB membranes. Our finding that Atg15 appears to run at a slightly higher molecular weight than expected (15–20% larger than predicted), even in the absence of glycosylation (Fig. 2 B), also likely reflects the hydrophobic nature of the Atg15 protein.
Physiological functions of the vacuole/lysosome lipases
Recently, lysosomal phospholipase A2 (PLA2G15/LPLA-2) was identified as a key lipase responsible for degrading membranes in lysosomes. Deletion of PLA2G15/LPLA-2 results in the formation of enlarged lysosomes that accumulate abundant membranous structures (Li et al., 2021). However, no studies have yet assessed how PLAG15/LPLA-2 in lysosomes degrade membrane lipids delivered by autophagy or other transport routes. The complexity of lysosomal hydrolases may make it difficult to analyze the protein network responsible for membrane disruption within lysosomes. The relative simplicity of yeast vacuolar enzymes was particularly advantageous for our study as it allows us to clarify the relationship between proteases and lipase activity in the vacuole.
Characterization of lipase activity in the vacuole/lysosome is essential to understand how lipids are recycled. Our identification of Atg15 as a phospholipase B with broad substrate specificity indicates that various species of FFAs, LPLs, and water-soluble glycerophosphodiesters (GPDs) are generated from membranes implicated in autophagy. Very recently, Spns1 was identified as a transporter that effluxes LPC and LPE in the lysosome for their recycling into cellular phospholipid pools (He et al., 2022). Further, Laqtom et al. showed in a recent study that CLN3, which is a lysosomal transmembrane protein involved in Batten disease, is required for the efflux of GPDs from lysosomes in mice (Laqtom et al., 2022). This protein shares the same function with its yeast homolog, Btn1 (Mirza et al., 2019). Considering these reports, LPLs and GPDs in the vacuole might be reused as lipids or other components. On the other hand, excessive accumulation of FFAs in the vacuole/lysosome could lead to lipotoxicity and vacuole/lysosome dysfunction. Further studies on the fate of these degradation products will uncover the impact of lipid degradation by autophagy on cellular metabolism.
Materials and methods
Lipids used in this study
NBD-PE (1-oleoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-sn-glycero-3-phosphoethanolamine), NBD-PS (1-oleoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-sn-glycero-3-phosphoserine), NBD-PC (1-oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-glycero-3-phosphocholine), NBD-PG (1-oleoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-sn-glycero-3-[phospho-rac-(1-glycerol)]), TF-PI (1-oleoyl-2-{6-[4-(dipyrrometheneboron difluoride)butanoyl]aminhexanoyl-sn-glycero-3-phosphoinositol), NBD-LPC (1-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-2-hydroxy-sn-glycero-3-phosphocholine), DOPE (dioleoylphosphatidylethanolamine), POPC (1-palmitoyl-2-oleoylphosphatidylcholine), and TF-TG (1,2-dioleoyl-3-[11-(dipyrrometheneboron difluoride)undecanoyl]-sn-glycerol) were obtained from Avanti Polar Lipids. BODIPY-CE (cholesteryl 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoate) was obtained from Life Technologies. Lipids (with the exception of NBD-LPC) were dissolved in 5 mM CHAPS at a concentration of 125 ng/μl and sonicated for 1–5 min. NBD-LPC was dissolved in water.
Buffers used in this study
Buffer A (10 mM 2- (N-morpholino)ethanesulfonic acid-tris (hydroxymethyl)aminomethane [MES-Tris], pH 6.9, 0.2 M sorbitol, 12% wt/vol Ficoll 400, and 0.1 mM MgCl2), buffer B (10 mM MES-Tris, pH 6.9, 0.2 M sorbitol, 8% wt/vol Ficoll 400, and 0.1 mM MgCl2), buffer B’ (10 mM MES-Tris, pH 6.9, 0.2 M sorbitol, 4% wt/vol Ficoll 400, and 0.1 mM MgCl2), buffer C (10 mM MES-Tris, pH 6.9, 0.2 M sorbitol, and 0.1 mM MgCl2), buffer D (10 mM MES-Tris, pH 6.9, 5 mM MgCl2, and 150 mM KCl), buffer E (30 mM MES-Tris, pH 6.9 and, 200 mM KCl), buffer F (30 mM MES-Tris, pH 6.9, 100 µM CaCl2, and 200 mM KCl), and spheroplast buffer (50 mM Tris-HCl, pH 7.5, and 1.2 M sorbitol) were used.
Yeast strains and media
The yeast strains used in this study are listed in Table S1. Gene deletion and tagging were performed using standard PCR-based methods, as described previously (Janke et al., 2004; Knop et al., 1999), and validated by PCR. Cells were cultured at 30°C in YPD medium (1% yeast extract [Gibco], 2% Bacto peptone [Gibco], 2% glucose) or in a synthetic defined medium comprising 0.17% yeast nitrogen base without amino acids and ammonium sulfate (BD), 0.5% ammonium sulfate, 0.5% casamino acids (BD), 2% glucose, 20 µg/ml adenine sulfate, and 20 µg/ml tryptophan (SD/CA medium). To induce autophagy, cells were treated with rapamycin (R-5000; LC Laboratories) at a concentration of 0.2 µM.
Plasmid construction
Plasmids and primers used in this study are listed in Tables S2 and S3, respectively. To generate pRS316-ATG15 (TPL000) and pRS426-ATG15 (TPL001), a DNA fragment containing the ATG15 gene from 1,000 nt upstream of the initiation codon was amplified by PCR from genomic DNA. The resulting DNA fragment was inserted into pRS316 or pRS426 using BamHI and HindⅢ sites. To generate pRS426-ATG15 (S332A; TPL002), the Atg15 active site S332 (serine at amino-acid position 332) was replaced with alanine in pRS426/ATG15 (TPL001) by inverse PCR. To generate pRS426-ATG15-mNeonGreen (YPL046), the recognition sequence of NheⅠ was inserted into the region upstream of the stop codon of ATG15 in pRS426-ATG15 (TPL001). The resulting plasmid was cut using NheⅠ, into which a PCR-amplified DNA fragment containing the mNeonGreen gene (Shaner et al., 2013) was inserted using the in-Fusion reaction (Clontech). To generate pRS426-CPY(1–50)-ATG15(Δ1–35)-mNeonGreen (YPL073), a DNA fragment containing the PRC1 gene was first amplified by PCR from genomic DNA and inserted into pBluescript SK(+) using HindⅢ and EcoRⅠ sites. This was subsequently used as a PCR template to amplify a DNA fragment encoding the first 50 amino acids of the CPY protein, while a DNA fragment encoding the 36–520 amino acids of Atg15 fused with mNG at the C-terminus was amplified by PCR from pRS426-ATG15-mNeonGreen (YPL046). These DNA fragments were then fused by in-fusion reaction. To generate pRS426-CPY(1–50)-ATG15(Δ1–35; YPL103), both pRS426-CPY(1–50)-ATG15(Δ1–35)-mNeonGreen (YPL073) and pRS426-ATG15 (TPL001) were cut using EcoRⅠ and HindⅢ, and a fusion construct containing nucleotides of CPY (1–50) and Atg15∆N35 was generated by ligation (Clontech). To generate pRS426-ATG15-3xFLAG (TPL003), a DNA fragment encoding a glycine linker followed by a 3xFLAG sequence was inserted between the ATG15 gene and its stop codon by inverse PCR.
To generate the plasmids pRS426-ATG15(168-3xFLAG-169; YPL183), pRS426-ATG15(S332A, 168-3xFLAG-169; YPL185), pRS426-CPY(1–50)-ATG15(Δ1–35, 168-3xFLAG-169; YPL187), and pRS426-CPY(1–50)-ATG15(S332A, Δ1–35, 168-3xFLAG-169; YPL189), a DNA fragment encoding a glycine linker followed by a 3xFLAG sequence was inserted at aspartic acid 168 of Atg15 by inverse PCR.
Isolation of vacuoles
Vacuoles were obtained from cells as previously described with some modifications (Kawamata et al., 2022; Ohsumi and Anraku, 1981; Takeshige et al., 1992). Cells grown in 10 L YPD to OD600 = 1.0–2.0 were treated with 0.2 µM rapamycin for 1 h. Cells were then collected and washed once with water. Spheroplasts were prepared by incubation of cells with Zymolyase 100T (Nacalai Tesque) at 10 µg/OD600 unit in 600 ml spheroplast buffer for 30 min. Spheroplasts were then collected by centrifugation, washed with spheroplast buffer, resuspended in 130 ml of ice-cold buffer A, and homogenized using a Dounce homogenizer on ice. The lysate was then divided into six volumes that were each transferred to a new ultracentrifuge tube and layered with 10 ml of buffer B. These layered samples were centrifuged at 72,000 g in a P28S swing rotor (Hitachi Koki) for 30 min at 4°C. The upper white layer of each tube was collected and combined into a new ultracentrifuge tube. This crude vacuole isolate was then mixed with 9 ml of buffer B, divided into two volumes that were each transferred to a new centrifuge tube, overlaid with 13 ml of buffer B’ and 13 ml of buffer C, and centrifuged at 72,000 g in a P28S swing rotor (Hitachi Koki) for 30 min at 4°C. The band at the 0–4% Ficoll interface was collected using a Piston Gradient Fractionator (BioComp Instruments, Inc). The vacuolar fraction was stored at −75°C. The yield of the vacuole was estimated by measuring the enzymatic activity of the vacuolar enzyme Pho8, as previously described (Makino et al., 2021), or by calculating the band intensity of Pho8 detected by western blotting. Protease inhibitors and reducing agents were not used during the isolation of vacuoles.
Fractionation of vacuolar lysate into soluble and pellet fractions
The vacuolar lysate was mixed with an equal volume of 2× buffer D and centrifugated at 135,000 g in a S55A2 angle rotor (Hitachi Koki) at 4°C for 30 min to obtain supernatant (S) and pellet (P) fractions. The P fraction was resuspended in buffer D containing either 2% Tx (Sigma-Aldrich), 0.5% DDM (Sigma-Aldrich), 0.1 M Na2CO3, 2.5 M urea, or 1 M KCl and incubated for 30 min at 4°C with shaking at 1,400 rpm. Finally, samples were centrifuged again at 135,000 g for 30 min at 4°C to obtain a second supernatant (S2) fraction and pellet (P2) fraction.
Immunoprecipitation and elution of processed forms of Atg15
Vacuolar pellet fractions obtained from a 5-L yeast culture were suspended in 5 ml of buffer D containing 0.5% DDM by vortexing and sonication and then incubated with 60 μl (slurry 50% vol/vol) of α-FLAG antibody-conjugated agarose beads (Millipore) for 1 h at 4°C. The beads were washed three times with buffer E and then mixed with 60 µg of 3xFLAG peptide (Sigma-Aldrich) in 60 μl of buffer E for 1 h with shaking at 1,400 rpm at 4°C. The samples were transferred to a Biospin column (BIORAD) and eluted by centrifugation. The volume of the eluate was ∼60 μl from 5 L culture.
Liposome preparation
To prepare liposomes, POPC:DOPE:NBD-PE were mixed at a volumetric ratio of 88:6:6 in glass tubes. Dried films were prepared by evaporating chloroform. Films were hydrated in buffer E to obtain 1 mM lipid solutions. The lipid suspension was incubated at room temperature for 1 h and mixed by vortex. Homogenously sized unilamellar vesicles were obtained using a mini-extruder set in combination with polycarbonate filters with a pore size of 400 and 100 nm (Avanti). To separate liposomes from NBD-PE, the liposome solution was centrifuged at 125,000 g for 1 h at 4°C and the supernatant was removed carefully. Then, buffer E was added and liposomes were analyzed using a Zetasizer Nano S (Malvern Instruments) as described previously (Kawamata et al., 2022).
In vitro lipase activity assay
To measure the lipase activity of vacuolar lysates, 125 ng of NBD-PE was added to each vacuolar lysate fraction in a total volume of 20 μl of buffer B’ and incubated at 30°C for the indicated time periods. The amount of vacuolar lysate was normalized using either the amount of Pho8, as determined by the band intensity of Pho8 detected by western blotting, or the enzymatic activity of Pho8. Lipase assays of prcAtg15 eluates were performed by incubating samples with 125 ng of each fluorescent lipid (NBD-PE, NBD-PS, NBD-PC, NBD-PG, NBD-LPC, TF-PI, TF-TG, or BODIPY-CE) in a total volume of 20 μl buffer E. Where concentrations of vacuolar lipase were adjusted, volumes of vacuolar lipase were increased as indicated in the figure legend with corresponding reductions in buffer E volume. For in vitro lipase activity assays using liposomes, lipase assays of prcAtg15 eluates were performed by incubating samples with 1 μl of liposome (1 mM) containing NBD-PE in a total volume of 20 μl buffer. Total lipids of the reaction mixture were extracted according to the BUME method (Löfgren et al., 2012). The lipids were dissolved in chloroform/methanol (2:1, vol/vol) and applied to a TLC plate (HPTLC SILICA GEL 60 F254 50 GLASS PL 1.05642.0001; Millipore), which was developed with a solution containing chloroform/methanol/water at a ratio of 65:35:8 for NBD-PE, NBD-PS, NBD-PC, NBD-PG, NBD-LPC, TF-PI, or hexane/diethyl ether/acetic acid at a ratio of 50/50/1 for TF-TG and BODIPY-CE, as described previously (Ishibashi et al., 2019). Fluorescence on the TLC plate was detected using a FUSION-FX7 imaging system (Vilber-Lourmat). The intensity of each fluorescent band was quantified using FUSION-FX7 analysis software.
Isolation of ABs
ABs were isolated as described previously (Kawamata et al., 2022). In brief, atg15∆ cells (SMY345) were grown in 2.5 L YPD cultures to a density of OD600 = 1.0 and then treated with 0.2 µM rapamycin for 6 h. Vacuoles were isolated as described above and then passed through a 0.8-μm PC membrane filter (ATTP01300; Millipore) to liberate ABs. Filtrates were subjected to 0–30% OPTIPREP density gradient centrifugation at 72,000 g for 90 min using a P40ST swing rotor and AB-enriched fractions (∼3–4 ml) were collected.
Evaluation of AB disruption in vitro
20 μl of AB fraction was mixed with 10 or 30 μl of prcAtg15 eluates or 0.2% Tx in a total volume of 50 μl of buffer F and incubated at 30°C for 3 h. Samples were then treated with 160 µg/ml Pro K on ice for 30 min and the reaction was terminated by adding PMSF. Samples were precipitated using TCA and analyzed by western blotting.
Western blotting
Immunoblot analyses of Ape1 were performed as previously described (Urban et al., 2007). To analyze the processing of Atg15, the samples were precipitated with 10% of TCA and 1.8 µg/ml tRNA and resuspended in the sample buffer (50 mM Tris-HCl, pH 7.5, 70 mM SDS, 8% Glycerol, 20 mM DTT, Bromophenol Blue), followed by incubation at 65°C for 15 min. For deglycosylation of Atg15, the samples were treated with Endo H (NEB) and GlycoBuffer (NEB) at 30°C for 2 h. Samples were then separated by SDS-PAGE and subjected to western blotting. α-FLAG (F3165, 1:1,000; Sigma-Aldrich), α-Pho8 (ab113688, 1:1,000; Abcam), α-Ape1 (1:5,000; Hamasaki et al., 2003), α-Prb1 (1:5,000; laboratory stock), and α-GFP (11814460001, 1:1,000; Roche) were used as primary antibodies. Femtoglow HRP Substrate (21008; Michigan Diagnostics) was used to initiate chemiluminescence and blots were visualized and quantified using a FUSION-FX7 imaging system.
Microscopy
Fluorescence microscopy was performed using an inverted fluorescence microscope (IX81; Olympus) equipped with an electron-multiplying CCD camera (ImagEM C9100-13; Hamamatsu Photonics) and 150× objective lens (UAPON 150× OTIRF, NA/1.45; Olympus). mNG and mCherry were excited using 488 and 561 nm lasers, respectively, both with a maximum output of 50 mW (Coherent). Emitted fluorescence was filtered using a Di01-R488/561-25 dichroic mirror (Semrock) and an Em01-R488/568-25 bandpass filter (Semrock) and separated into two channels using a U-SIP splitter (Olympus) equipped with a DM565HQ dichroic mirror (Olympus). Fluorescence was further filtered using an FF02-525/50-25 bandpass filter (Semrock) for the mNG channel and an FF01-624/40-25 bandpass filter (Semrock) for the mCherry channel.
For spinning-disk confocal microscopy, an Olympus IXplore SpinSR10 equipped with an sCMOS camera (ORCA-Flash4.0; Hamamatsu) and a 100× objective lens (UPLAPO 100×OHR, NA/1.50; Olympus) was employed. mNG was excited using a 488 nm laser and fluorescence was passed through a mirror unit U-FGFP equipped with a DM490GFP dichroic mirror (Semrock), a BP460–480GFP excitation filter, and a BA495–540GFP absorption filter. mCherry was excited using a 561 nm laser and fluorescence passed through a U-FMCHE mirror unit (Olympus) equipped with a DM595 dichroic mirror, a BP565–585 excitation filter, and a BA600–690 absorption filter. Images were acquired as individual slices through a central plane of the cell using super-resolution mode.
Both microscopes were controlled using Olympus CellSens software and acquired images were subsequently processed using ImageJ software (Fiji Ver. 1.53f51; Schindelin et al., 2012).
Online supplemental material
Fig. S1, related to Fig. 1, shows western blotting and lipase activity assay of vacuolar fractions. Fig. S2, related to Fig. 2, shows western blotting of vacuolar and cellular lysates, lipase activity of vacuolar lysates from Atg15-FLAG–expressing cells, and microscopy images of mCherry-Atg8 in WT cells. Fig. S3, related to Fig. 4, shows maturation of prApe1 in cells expressing truncated Atg15 mutants and western blotting of prcAtg15 and lipase activity of vacuolar lysates from Atg15iFLAG-expressing cells. Fig. S4, related to Fig. 5, shows lipase activity of vacuolar lysates from Atg15 and Atg15S332A toward various lipid species. Fig. S5, related to Fig. 6, shows lipase assay of active Atg15 using liposomes. Table S1 lists the yeast strains used in this study. Table S2 lists the plasmids used in this study. Table S3 lists the primers used in this study. Video 1 shows dynamics of ABs labeled mCherry-Atg8 in CPYN50-Atg15∆N35–expressing cells. Video 2 shows localization of Atg15-mNG and ABs in pep4∆ prb1∆ cells. Video 3 shows localization of CPYN50-Atg15∆N35 and ABs in pep4∆ prb1∆ cells.
Acknowledgments
We are grateful to members of the Ohsumi laboratory, Dr. Nobuo Noda, Dr. Yohei Ishibashi, and Dr. Masato Umeda for discussions and critical comments, and the Biomaterials Analysis Division, Open Facility Center, Tokyo Institute of Technology for DNA sequencing.
This work was supported in part by Grants-in-Aid for Scientific Research 16H06375 and 19H05708 (to Y. Ohsumi) and 18H02399 (to T. Kawamata) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. T. Kawamata was also supported by the Japan Science and Technology Agency FOREST program (JPMJFR214X).
Author contributions: Conceptualization and experimental design: Y. Kagohashi, T. Kawamata, and Y. Ohsumi; data collection: Y. Kagohashi, M. Sasaki, A.I. May, and T. Kawamata; manuscript preparation: Y. Kagohashi, A.I. May, T. Kawamata, and Y. Ohsumi.

