


The lysosome is the major catabolic organelle in the cell that has been established as a key metabolic signaling center. Mutations in many lysosomal proteins have catastrophic effects and cause neurodegeneration, cancer, and age-related diseases. The vacuole is the lysosomal analog of Saccharomyces cerevisiae that harbors many evolutionary conserved proteins. Proteins reach vacuoles via the Vps10-dependent endosomal vacuolar protein sorting pathway, via the alkaline phosphatase (ALP or AP-3) pathway, and via the cytosol-to-vacuole transport (CVT) pathway. A systematic understanding of the cargo spectrum of each pathway is completely lacking. Here, we use quantitative proteomics of purified vacuoles to generate the yeast lysosomal biogenesis map. This dataset harbors information on the cargo–receptor relationship of almost all vacuolar proteins. We map binding motifs of Vps10 and the AP-3 complex and identify a novel cargo of the CVT pathway under nutrient-rich conditions. Our data show how organelle purification and quantitative proteomics can uncover fundamental insights into organelle biogenesis.
Introduction
Lysosomes and their equivalent structures (known as vacuoles in yeast cells) have long been established as the degradative end points for both intracellular and exogenous cargo. The catabolic function of the lysosome depends on two classes of proteins, soluble hydrolases and integral lysosomal membrane proteins. Lysosomal hydrolases require an acidic pH, which is established by the vacuolar H+-ATPase, an ATP-driven proton pump, in combination with other membrane-spanning ion channels (Li and Kane, 2009). Another class of lysosomal membrane proteins are permeases exporting metabolites towards the cytoplasm. Although the targeting of lysosomal/vacuolar proteins is described for some model proteins, the complete set of proteins following the different targeting pathways remains largely elusive.
Mutations in ∼50 genes encoding lysosomal hydrolases and membrane permeases cause a family of diseases known as lysosomal storage disorders (Ballabio and Gieselmann, 2009). These diseases are characterized by the accumulation of digestion products, such as lipids, amino acids, sugars, and nucleotides within the lysosomal lumen. Most of the diseases primarily affect the central nervous system. It is therefore not surprising that mutations in lysosomal sorting receptors, such as sortillin (Vps10 in yeast), are linked to Alzheimer’s disease and mutations in the AP-3 complex are causing Hermansky-Pudlak syndrome (Scherzer et al., 2004; Shotelersuk et al., 2000). To understand the role of lysosomal biogenesis in physiology and pathophysiology, it is important to know how each lysosomal protein is transported to its final destination. Identification of the transport routes of lipid metabolism enzymes is also important to understand the mechanisms of lysosomal storage disorders such as Niemann-Pick Type C (Carstea et al., 1997; Naureckiene et al., 2000), which is caused by defects in important cargoes of lysosomal sorting, such as NPC1 (Ncr1 in yeast) and NPC2 (Npc2 in yeast).
Lysosomal biogenesis requires targeting of newly synthesized lysosomal proteins from the Golgi apparatus to the lysosome. Saccharomyces cerevisiae has been a key model organism to understand the mechanisms of lysosome/vacuole biogenesis and protein targeting. Yeast possesses at least three different vacuolar protein targeting pathways. After synthesis in the ER, vacuolar proteins are transported via the secretory pathway to the Golgi apparatus. Here, vacuolar proteins are sorted either by the vacuolar protein sorting pathway (VPS pathway) via endosomes or via the adaptor protein 3 (AP-3)–dependent pathway, which is also conserved to humans (Rothman and Stevens, 1986; Simpson et al., 1996). Transmembrane proteins that follow the VPS pathway require effectors such as Golgi-associated γ-adaptin ear homology domain Arf-binding proteins (GGA; Hirst et al., 2000). The best studied soluble cargo, carboxypeptidase Y (CPY), requires the Vps10 receptor for its sorting (Marcusson et al., 1994). Many effector proteins of the VPS pathway have been identified in screens using CPY secretion as a readout (Rothman and Stevens, 1986; Bankaitis et al., 1986). VPS cargo proteins sorted into vesicles will first fuse with endosomes before they are finally delivered to vacuoles through endosome–vacuole fusion. In contrast, the AP-3 pathway transports proteins directly from the Golgi apparatus to the vacuole. The best studied cargoes of this pathway are the vacuolar alkaline phosphatase Pho8 (ALP; Cowles et al., 1997a) and the yeast casein kinase 3 Yck3 (Sun et al., 2004). A third pathway that has only been identified in yeast is the cytosol-to-vacuole transport (CVT) pathway, which requires the autophagy machinery. The CVT pathway delivers aminopeptidase 1 (Ape1) and alpha mannosidase 1 (Ams1) to the vacuole and requires the Atg19 receptor (Watanabe et al., 2010; Lynch-Day and Klionsky, 2010). All three transport pathways require fusion factors, such as tethering complexes, Sec1p/Munc-18 (S/M) proteins, such as Vps45 causing the class D vacuolar phenotype (Cowles et al., 1994), and soluble N-ethylmaleimide–sensitive factor attachment protein receptors (Sollner et al., 1993).
Aside from the few described model cargoes, little is known about which protein follows each individual pathway and its sorting requirements. Here, we developed quantitative proteomics of vacuole isolations from yeast (QPrevail) to systematically map protein targeting to the yeast vacuole. We present a complete dataset of proteins likely following the different transport routes and provide evidence on trafficking of lipid transport proteins, e.g., the Niemann-Pick Type C proteins Ncr1 and Npc2. Our data suggest that most luminal vacuolar proteins are either transported by the Vps10 sorting receptor or the CVT pathway. We also find that only few transmembrane proteins are affected in mutations in the endosomal pathway, while the majority of transmembrane cargoes are dependent on a functional AP-3 pathway. We demonstrate that QPrevail can be used to study autophagy pathways, to map cargo–receptor interactions, and to study the vacuolar protein composition in different trafficking mutants. QPrevail analyzes untagged proteins at native expression levels and highlights the importance of systematic analysis of lysosomes/vacuoles to understand their complex role in cellular metabolism.
Results
Quantitative proteomics of isolated organelles has been previously employed as a tool to determine protein transport by us and others (Eising et al., 2019; Hirst et al., 2018; Borner, 2020). However, this has never been used to systematically study protein targeting to the yeast vacuole. To assign putative cargoes to distinct vacuolar sorting pathways, we decided to disrupt key proteins of each pathway and analyze the mutant vacuole proteome. We therefore generated mutations in APL5, targeting the AP-3 complex, and in ATG19, the autophagy receptor for the CVT pathway. We generated mutations in VPS10 as the receptor for CPY and the GGA genes GGA1 and GGA2 that are necessary to generate Golgi-derived vesicles destined for the endosomes in yeast. In addition, we knocked out VPS45, an S/M protein, since its deletion affects targeting of the V-ATPase to the yeast vacuole (Sambade et al., 2005; Fig. 1 a). We first analyzed vacuolar morphology of different mutants in a strain expressing mNeon-tagged Vph1 and also stained vacuoles with FM4-64 and CMAC. This analysis revealed a largely undisturbed vacuolar morphology except for the class D mutant VPS45 (Raymond et al., 1992). In line with that, Vph1–mNeon did not reach the vacuole in vps45Δ cells (Fig. 1 b). As a quality control, we compared the purification efficiency of vacuoles from all mutants compared to WT cells. We therefore tagged Vph1 with mCherry in WT cells and with mNeon in mutant cells and mixed cells prior to lysis and vacuole isolation. For VPS45 mutants, we used GFP-tagged Yck3 as a readout. After purification, we calculated the ratio of isolated vacuoles from WT versus mutant cells. Except for VPS45 mutants, we did not detect any changes in the number or diameter of isolated vacuoles (Fig. 1 c). Although vacuoles from VPS45 mutant cells appear to be more unstable, we still decided to include this mutant in our analysis as it shows a trafficking defect for the V-ATPase (Piper et al., 1997). We thus decided to combine vacuolar purifications with quantitative proteomics. For vps45Δ vacuoles, we had to take the overall lower abundance of isolated vacuoles into account, which will be described later. We term this experimental setup QPrevail (“Quantitative Proteomics of Vacuolar Isolations”; Fig. 1 d). To confirm the crosstalk between different sorting pathways, we also tested genetic interactions of our mutant strains. Except for the double knockout of VPS45 and APL5, all tested combinations were viable (Fig. S1), suggesting that proteins can take alternative routes to reach the vacuoles as previously described for the AP-3 pathway (Reggiori et al., 2000).

Experimental setup to generate the vacuolar biogenesis map. (a) Model of vacuolar protein-targeting routes in yeast. Vacuolar proteins are delivered via the CVT pathway (orange), the CPY pathway (blue), and the AP-3 pathway (green). Deleted genes include APL5 (subunit of the AP-3 complex), the GGA1 and GGA2 adaptor protein coding genes, the CPY receptor VPS10, the S/M protein–encoding gene VPS45, and the CVT receptor ATG19. (b) Vacuolar morphology was analyzed in WT, apl5Δ, vps10Δ, gga1Δgga2Δ, vps45Δ, and atg19Δ cells using the mNeon-tagged transmembrane protein Vph1, the lipophilic dye FM4-64, and the vacuolar luminal dye CMAC. Scale bar, 5 µm. (c) Quality control of purified vacuoles. Vacuoles were purified from the same amounts of WT (Vph1-mCherry) and mutant cells (Vph1-mNeon or ADHpr-GFP-Yck3). Vacuole number and size were determined from 20 pictures (error bars = SD). All mutants, except vps45Δ, show comparable numbers of vacuoles. Scale bar, 5 µm. (d) Experimental setup for QPrevail.
Experimental setup to generate the vacuolar biogenesis map. (a) Model of vacuolar protein-targeting routes in yeast. Vacuolar proteins are delivered via the CVT pathway (orange), the CPY pathway (blue), and the AP-3 pathway (green). Deleted genes include APL5 (subunit of the AP-3 complex), the GGA1 and GGA2 adaptor protein coding genes, the CPY receptor VPS10, the S/M protein–encoding gene VPS45, and the CVT receptor ATG19. (b) Vacuolar morphology was analyzed in WT, apl5Δ, vps10Δ, gga1Δgga2Δ, vps45Δ, and atg19Δ cells using the mNeon-tagged transmembrane protein Vph1, the lipophilic dye FM4-64, and the vacuolar luminal dye CMAC. Scale bar, 5 µm. (c) Quality control of purified vacuoles. Vacuoles were purified from the same amounts of WT (Vph1-mCherry) and mutant cells (Vph1-mNeon or ADHpr-GFP-Yck3). Vacuole number and size were determined from 20 pictures (error bars = SD). All mutants, except vps45Δ, show comparable numbers of vacuoles. Scale bar, 5 µm. (d) Experimental setup for QPrevail.

Genetic interactions of the vacuole trafficking mutants analyzed in this study. (a) Tetrad analysis of vps10Δ (blue) mutants crossed with atg19Δ (yellow). (b) Tetrad analysis of apl5Δ (blue) mutants crossed with vps45Δ (yellow). (c) Tetrad analysis of vps10Δ (blue) mutants crossed with gga1Δ (green) gga2Δ (yellow). (d) Tetrad analysis of apl5Δ (blue) mutants crossed with atg19Δ (yellow). (e) Tetrad analysis of apl5Δ (blue) mutants crossed with gga1Δ (green) gga2Δ (yellow). (f) Tetrad analysis of vps10Δ (blue) mutants crossed with vps45Δ (yellow). (g) Tetrad analysis of apl5Δ (blue) mutants crossed with vps10Δ (yellow).
Genetic interactions of the vacuole trafficking mutants analyzed in this study. (a) Tetrad analysis of vps10Δ (blue) mutants crossed with atg19Δ (yellow). (b) Tetrad analysis of apl5Δ (blue) mutants crossed with vps45Δ (yellow). (c) Tetrad analysis of vps10Δ (blue) mutants crossed with gga1Δ (green) gga2Δ (yellow). (d) Tetrad analysis of apl5Δ (blue) mutants crossed with atg19Δ (yellow). (e) Tetrad analysis of apl5Δ (blue) mutants crossed with gga1Δ (green) gga2Δ (yellow). (f) Tetrad analysis of vps10Δ (blue) mutants crossed with vps45Δ (yellow). (g) Tetrad analysis of apl5Δ (blue) mutants crossed with vps10Δ (yellow).
A systematic proteomic approach for generation of the vacuolar biogenesis map
To generate a systematic vacuole biogenesis map, we analyzed the proteomes of multiple replicates of isolated vacuoles of knockouts of APL5, ATG19, VPS10, and VPS45 and a double deletion of GGA1 and GGA2 labeled with “heavy” lysine and compared them with ”light”-labeled WT vacuoles (Eising et al., 2019; Ong et al., 2002). We included replicates of isolated vacuoles of heavy- and light-labeled WT cells. We also measured replicates of total proteomes of mutants to control for changes in total protein amount. The resulting data for vacuolar and total proteomes are shown in Table S1 and Fig. S2. To visualize changes in vacuolar protein abundances, we first generated a list of proteins that have been annotated as vacuolar before (Table S2). An overview of the logarithmic ratios for each vacuolar protein is shown in hierarchical clusters in Fig. 2 as the vacuolar biogenesis map. This analysis shows clustering of the different knockouts and highlights clusters of proteins that are potentially transported to the vacuole via different trafficking routes. Importantly, known model cargoes of each pathway were identified in the corresponding clusters. For example, a potential AP-3–dependent cluster (inset 1) contained the known AP-3 cargoes Pho8, Nyv1, and Yck3. Knockout of ATG19 affected the known CVT cargoes Ape1 and Ape4 (inset 4). CPY was affected in vps10Δ cells, in the GGA double deletion as well as VPS45 knockout cells (inset 2). The vacuole biogenesis map also highlights proteins that are more abundant in mutant vacuoles, e.g., the cluster of plasma membrane proteins in gga1Δgga2Δ cells (insert 3). As expected, all identified subunits of the V1 domain of the V-ATPase were only affected in vps45Δ cells (inset 5). Several proteins only show very small changes in their vacuolar abundance in all analyzed mutants (inset 6 and Table S1). These were mainly peripheral membrane proteins such as subunits of the homotypic fusion and protein sorting (HOPS) complex (Rieder and Emr, 1997) and the target of rapamycin (TOR) complex 1. Besides minimal changes in the SILAC ratios of these proteins, subunits of each complex formed distinct clusters. Together, this suggests that for HOPS and TORC1 all subunits are affected similarly, even though changes in the ratios are only minimal.

Comparison of total proteome levels and vacuolar proteome levels in the different trafficking mutants. (a) SILAC ratios from heavy-labeled vps10Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (b) SILAC ratios from heavy-labeled gga1Δgga2Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (c) SILAC ratios from heavy-labeled vps45Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (d) SILAC ratios from heavy-labeled atg19Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (e) SILAC ratios from heavy-labeled apl5Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue.
Comparison of total proteome levels and vacuolar proteome levels in the different trafficking mutants. (a) SILAC ratios from heavy-labeled vps10Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (b) SILAC ratios from heavy-labeled gga1Δgga2Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (c) SILAC ratios from heavy-labeled vps45Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (d) SILAC ratios from heavy-labeled atg19Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue. (e) SILAC ratios from heavy-labeled apl5Δ cells and light-labeled WT cells of total proteomes are plotted on the x-axis against the SILAC ratios from enriched vacuoles. Vacuolar annotated proteins are plotted in yellow and other proteins in blue.

Overview of the vacuolar biogenesis map. Hierarchical clustering of vacuolar trafficking mutants (left to right) and vacuolar proteins (top to bottom). Proteins in the different mutants are color coded by a gradient from green (low heavy/light ratio) to red (high heavy/light ratio). The legend for the color code is in the bottom left of the figure. Gray fields represent proteins that were not reliably quantified with more than two unique peptides. Clusters for different trafficking pathways are shown: (1) AP-3, (2) endolysosomal/CPY, (3) GGA1/GGA2-dependent accumulation, (4) CVT pathway, (5) sorting defect of V-ATPase subunits in vps45∆ cells, and (6) subunits of protein complexes TORC1 and HOPS.
Overview of the vacuolar biogenesis map. Hierarchical clustering of vacuolar trafficking mutants (left to right) and vacuolar proteins (top to bottom). Proteins in the different mutants are color coded by a gradient from green (low heavy/light ratio) to red (high heavy/light ratio). The legend for the color code is in the bottom left of the figure. Gray fields represent proteins that were not reliably quantified with more than two unique peptides. Clusters for different trafficking pathways are shown: (1) AP-3, (2) endolysosomal/CPY, (3) GGA1/GGA2-dependent accumulation, (4) CVT pathway, (5) sorting defect of V-ATPase subunits in vps45∆ cells, and (6) subunits of protein complexes TORC1 and HOPS.
Several proteins are not reliably quantified in some mutants and appear as grey labeled boxes. Some proteins appear in clusters of pathways even though they do not show any ratios (e.g., Pib1 in the AP-3 cluster). This protein would therefore be falsely annotated in the AP-3 cluster. To further validate our dataset, we analyzed proteins affected by the different pathways.
Most soluble proteins of the vacuolar lumen are sorted by Vps10
Encouraged by the specificity of our vacuolar proteomics, we began to dissect the requirements for specific sorting pathways. We started our analysis with the CPY receptor Vps10 (Fig. 3 a). To determine statistically significant cargoes of Vps10, we compared the SILAC ratios of vps10Δ (heavy) versus WT (light) vacuoles against the SILAC ratios of heavy- and light-labeled WT vacuoles. We performed a t test between the two groups and visualized the results in form of a volcano plot (Fig. 3 b). We used the Perseus software package (Tyanova et al., 2016) to calculate a significance curve that separates the cargo proteins from the background. The most depleted proteins in the vacuoles of vps10Δ cells were, as expected, Vps10 itself and the known cargo CPY (Prc1; Fig. 3 b). Detection of Vps10 is possible because of the re-quant option of MaxQuant (for details see materials and methods). Besides the luminal CPY, other proteins annotated as potential soluble peptidases of the vacuole were significantly depleted (Yhr202W, Ydr415C, Pep4, Atg42, and Ape3), suggesting that they are also targets of Vps10 (a list of all soluble proteins is shown in Fig S3 a and Table S3). Importantly, the soluble proteases are not depleted in general in the cells (Fig. S3 b). One of the most depleted proteins from the vacuoles of vps10Δ cells was the Niemann-Pick Type C protein homolog, Npc2 (Fig. 3 b; Berger et al., 2005b). Npc2 has been described as the cargo of the AP-3 pathway and GGA proteins (Berger et al., 2007). Our data suggest that Npc2 is exclusively sorted by Vps10. We also identified several proteins enriched in the vacuoles of vps10Δ cells. Amongst them are components of the CVT pathway (Ape1 and Ams1), suggesting an upregulation of this pathway to compensate for the loss of Vps10 and maintain the lytic capacity of the vacuoles (Fig. 3 b). All results of vps10Δ analysis can be found in Table S4. To confirm our results, we next asked if the soluble proteases and Npc2 are secreted in a vps10Δ background by comparing the supernatant of light-labeled WT cells with heavy-labeled vps10Δ cells (Fig. 3 c). As expected for cargoes of Vps10, we identified Yhr202W, Ydr415C, Pep4, Ape3, Atg42, and Npc2 together with the positive control CPY in the supernatant (Fig. 3 d and Table S5). In addition, we confirmed the secretion of HA-tagged Npc2 and CPY in vps10Δ cells by Western blot (Fig. 3 e).
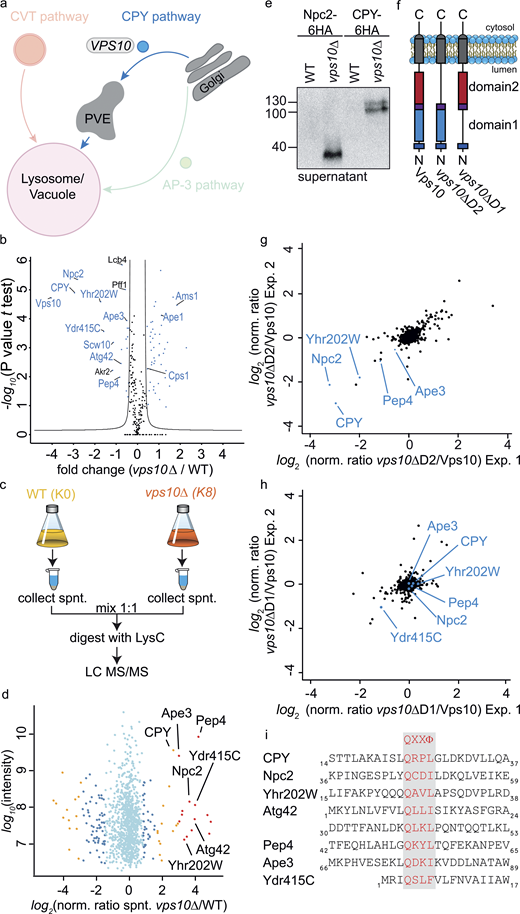
Identification of cargo proteins of the Vps10 sorting receptor. (a) Model highlighting the analysis of the CPY trafficking pathway. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of vps10Δ cells. Fold changes calculated from the independent experiments comparing SILAC ratios from (vps10Δ K8 versus WT K0) with (WT K8 versus WT K0) on the x-axis were plotted against negative logarithmic P values of the t test performed from replicates. The hyperbolic curve separates depleted proteins (left side, blue dots) and enriched proteins (right side, blue dots) from unaffected proteins (black dots). (c) Experimental setup to compare secreted proteins in vps10Δ and WT cells. (d) Identified proteins in the supernatant of vps10∆ compared with WT. Averaged peptide intensities are plotted against average heavy/light SILAC ratios from two different experiments. Significant outliers (P < 1e−14) are color coded in red (P < 0.0001), orange, or blue (P < 0.05); other identified proteins are shown in light blue. (e) Validation of MS results via Western blot. Npc2 and CPY were tagged with a 6HA tag and supernatants of WT and mutant cells decorated with anti-HA antibody. (f) Structure of Vps10 variants including full-length Vps10, Vps10-lacking domain 2 (vps10ΔD2), and Vps10 lacking domain 1 (vps10ΔD1). (g and h) Proteomic comparison of vacuoles isolated from vps10ΔD2 cells and WT cells (g) and of vps10ΔD1 cells and WT cells (h). SILAC ratios of two independent experiments are plotted against each other. Previously identified Vps10 cargo proteins are labeled. (i) Proteins which bind to Vps10 domain 2 show leucin or isoleucine as hydrophobic amino acid. Only the domain 1–dependent Ydr415C harbors a phenylalanine. Source data are available for this figure: SourceData F3.
Identification of cargo proteins of the Vps10 sorting receptor. (a) Model highlighting the analysis of the CPY trafficking pathway. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of vps10Δ cells. Fold changes calculated from the independent experiments comparing SILAC ratios from (vps10Δ K8 versus WT K0) with (WT K8 versus WT K0) on the x-axis were plotted against negative logarithmic P values of the t test performed from replicates. The hyperbolic curve separates depleted proteins (left side, blue dots) and enriched proteins (right side, blue dots) from unaffected proteins (black dots). (c) Experimental setup to compare secreted proteins in vps10Δ and WT cells. (d) Identified proteins in the supernatant of vps10∆ compared with WT. Averaged peptide intensities are plotted against average heavy/light SILAC ratios from two different experiments. Significant outliers (P < 1e−14) are color coded in red (P < 0.0001), orange, or blue (P < 0.05); other identified proteins are shown in light blue. (e) Validation of MS results via Western blot. Npc2 and CPY were tagged with a 6HA tag and supernatants of WT and mutant cells decorated with anti-HA antibody. (f) Structure of Vps10 variants including full-length Vps10, Vps10-lacking domain 2 (vps10ΔD2), and Vps10 lacking domain 1 (vps10ΔD1). (g and h) Proteomic comparison of vacuoles isolated from vps10ΔD2 cells and WT cells (g) and of vps10ΔD1 cells and WT cells (h). SILAC ratios of two independent experiments are plotted against each other. Previously identified Vps10 cargo proteins are labeled. (i) Proteins which bind to Vps10 domain 2 show leucin or isoleucine as hydrophobic amino acid. Only the domain 1–dependent Ydr415C harbors a phenylalanine. Source data are available for this figure: SourceData F3.

Comparison of soluble vacuolar proteins. (a) Normalized heavy/light ratios for all soluble vacuolar proteins in isolated vacuoles across the different mutants were analyzed (error bars show the SD from 3 to 4 measurements). (b) Normalized heavy/light ratios for all soluble vacuolar proteins in whole cell extracts across the different mutants analyzed. (c) Sorting of AP-3 cargoes according to their binding motifs. Transmembrane proteins were divided into groups with the tyrosine motif, dileucine motif, or both according to their amino acid sequence. Only cytosolic parts were counted (according to Uniprot database). (d) Comparison of protein ratios from vacuole samples of two experiments, APM3 D217A G469K versus APM3. Proteins with a dileucine motif are colored in red, proteins bearing a tyrosine motif in blue, and proteins with both motifs in purple. The asterisks indicate non–TM proteins. (e) Comparison of protein ratios from vacuole samples of two experiments, APS3 V106D L121S versus APS3. Known AP-3 cargoes are colored in red and cargoes identified in our screen colored in blue. The asterisks indicate non–TM proteins. (f) Vacuolar proteomics of the AP-3 cargo Atg27. atg27Δ cells show a significant decrease in CVT cargo proteins and Atg8 in isolated vacuoles in comparison to WT cells.
Comparison of soluble vacuolar proteins. (a) Normalized heavy/light ratios for all soluble vacuolar proteins in isolated vacuoles across the different mutants were analyzed (error bars show the SD from 3 to 4 measurements). (b) Normalized heavy/light ratios for all soluble vacuolar proteins in whole cell extracts across the different mutants analyzed. (c) Sorting of AP-3 cargoes according to their binding motifs. Transmembrane proteins were divided into groups with the tyrosine motif, dileucine motif, or both according to their amino acid sequence. Only cytosolic parts were counted (according to Uniprot database). (d) Comparison of protein ratios from vacuole samples of two experiments, APM3 D217A G469K versus APM3. Proteins with a dileucine motif are colored in red, proteins bearing a tyrosine motif in blue, and proteins with both motifs in purple. The asterisks indicate non–TM proteins. (e) Comparison of protein ratios from vacuole samples of two experiments, APS3 V106D L121S versus APS3. Known AP-3 cargoes are colored in red and cargoes identified in our screen colored in blue. The asterisks indicate non–TM proteins. (f) Vacuolar proteomics of the AP-3 cargo Atg27. atg27Δ cells show a significant decrease in CVT cargo proteins and Atg8 in isolated vacuoles in comparison to WT cells.
We next asked how Vps10 binds to its different cargoes. Vps10 belongs to the family of sortilins and has two homologous sortilin-like domains, domain 1 and domain 2 (Fig. 3 f; Jørgensen et al., 1999). Vps10 binds CPY via a QXXΦ motif (where Φ is a hydrophobic amino acid) via its domain 2 (Jørgensen et al., 1999). To determine which of the two domains is required for Vps10 mediated sorting, we compared the proteomes of light-labeled cells expressing full length Vps10 with heavy-labeled cells expressing Vps10 with either domain 1 or domain 2 deleted (vps10ΔD1, vps10ΔD2; Fig. 3 f and Table S6). Plotting the SILAC ratios of two replicates of Vps10 full length (K0) and vps10ΔD2 (K8) against each other revealed depletion of most previously identified cargoes from the vacuoles of cells lacking domain 2 of Vps10 (Npc2, Yhr202W, Pep4, CPY, and Ape3; Fig. 3 g). In contrast, deletion of domain 1 in Vps10 resulted only in the loss of Ydr415C (Fig. 3 h). We were able to identify a QXXΦ sorting motif in all cargoes. The only difference we detected between Ydr415C and the other cargoes is the hydrophobic amino acid in the 4th position which is a phenylalanine in Ydr415C in comparison to a leucine or isoleucine in the other cargoes (Fig. 3 i). In summary, QPrevail allowed us to identify soluble cargoes of the Vps10 and their potential Vps10 binding domains.
Gga proteins are necessary for Vps10 sorting and affect the localization of plasma membrane permeases
To understand the role of the sorting machinery at the Golgi, we next focused on the two vesicle coat proteins Gga1 and Gga2 (Fig. 4 a). Gga proteins have been implicated in the sorting of proteins from the Golgi apparatus to the endosome as well as of ubiquitinated proteins (Black and Pelham, 2000; Scott et al., 2004). As before, we used QPrevail to analyze vacuoles from gga1∆gga2∆ cells and identified the known cargo Vps10 to be depleted in mutant vacuoles. In line with our data on Vps10 itself (Fig. 3 b), also cargoes of Vps10 were depleted (green labels; Fig. 4 b). In addition, multiple plasma membrane permeases (Mup1, Dip5, and Tna1, orange labels) and the Rsp5 adaptor Ear1 are found less abundant in gga1Δgga2Δ vacuoles, suggesting a deficiency in sorting of ubiquitinated plasma membrane proteins to the vacuole (Fig. 4 b). Interestingly, we also found the palmitoyltransferase Akr2 depleted in GGA mutants as well as the known palmitoylated lipid kinases Lsb6 and Lcb4 (Fig. 4 b, pink dots; Roth et al., 2006). In contrast, we identified several proteins that were enriched in the vacuoles of GGA mutants (blue labels). Amongst them are the soluble N-ethylmaleimide–sensitive factor attachment protein receptor Tlg2 and Sso1 as well as Ptm1 which has an unknown function but was previously co-purified with Tlg2-containing vesicles (Inadome et al., 2005). To our surprise, besides the endosomal type-I transmembrane receptor Vps10, only very few other vacuolar transmembrane proteins appear to be dependent on sorting by the Gga proteins. The complete dataset for the gga1Δgga2Δ mutant can be found in Table S7.

Identification of cargo proteins depending on the Gga1 Gga2 adaptor proteins. (a) Model highlighting the analysis of the GGA mutants within the CPY trafficking pathway. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of gga1Δgga2Δ cells. Analysis was performed as in Fig. 3 b. Vps10-dependent cargoes are labeled in green, depleted proteins in orange, and enriched plasma membrane proteins in blue. (c) Gga proteins affect the uptake of Mup1 from the plasma membrane. WT cells (upper panels) or gga1Δgga2Δ cells expressing Mup1-GFP were grown to the mid-log phase in SDC medium lacking methionine to induce Mup1 expression and retention at the plasma membrane. Cells were costained with FM4-64 to mark vacuoles and imaged 0 min, 30 min, and 60 min after the addition of methionine. Scale bar, 5 µm. (d) Localization of N-terminally GFP-tagged Sso1 and FM4-64–stained vacuoles was analyzed in WT cells (upper panel) and gga1Δgga2Δ cells (lower panel). Scale bar, 5 µm.
Identification of cargo proteins depending on the Gga1 Gga2 adaptor proteins. (a) Model highlighting the analysis of the GGA mutants within the CPY trafficking pathway. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of gga1Δgga2Δ cells. Analysis was performed as in Fig. 3 b. Vps10-dependent cargoes are labeled in green, depleted proteins in orange, and enriched plasma membrane proteins in blue. (c) Gga proteins affect the uptake of Mup1 from the plasma membrane. WT cells (upper panels) or gga1Δgga2Δ cells expressing Mup1-GFP were grown to the mid-log phase in SDC medium lacking methionine to induce Mup1 expression and retention at the plasma membrane. Cells were costained with FM4-64 to mark vacuoles and imaged 0 min, 30 min, and 60 min after the addition of methionine. Scale bar, 5 µm. (d) Localization of N-terminally GFP-tagged Sso1 and FM4-64–stained vacuoles was analyzed in WT cells (upper panel) and gga1Δgga2Δ cells (lower panel). Scale bar, 5 µm.
To confirm our results, we analyzed Mup1 localization in WT and gga1Δgga2Δ cells. We grew cells without methionine to induce full expression of Mup1 at the plasma membrane and then added methionine to induce internalization of the transporter (Fig. 4 c; Lin et al., 2008). Most of the GFP-tagged Mup1 reached the FM4-64–labelled vacuole of WT cells after 30 min of methionine chase. In contrast, even after 60 min of methionine chase, most of Mup1 remained at the plasma membrane in gga1Δgga2Δ cells (Fig. 4 c), indicating that permease trafficking is strongly affected in this mutant.
To confirm the trafficking defect of Sso1, we analyzed the localization of N-terminally tagged GFP-Sso1. Although GFP-Sso1 is completely localized at the cell periphery of WT cells, we detected a considerable fraction of GFP-Sso1 at the vacuolar membrane of gga1Δgga2Δ cells, supporting our proteomics experiments (Fig. 4 d).
Taken together, QPrevail revealed Vps10 as one of very few transmembrane proteins that require the Gga proteins for sorting. Consequently, sorting of all identified Vps10 cargoes is dependent on Gga proteins. In addition, we show Gga proteins to be necessary for other transport processes based on the enrichment of Sso1 at the vacuole of GGA mutants as well as the depletion of Mup1.
A subset of transmembrane proteins is sorted to the vacuole via endosomes
To identify all cargo delivered via the endo-lysosomal pathway, we analyzed the class D mutant vps45Δ that blocks all fusion events in the endo-lysosomal and the CVT pathway (Fig. 5 a and Table S8; Cowles et al., 1994; Abeliovich et al., 1999). As described previously, vacuoles from vps45Δ cells appear to be more instable and less in abundance in our purifications. However, working with normalized ratios from the MaxQuant output allowed us to determine proteins strongly affected in vps45Δ cells. As expected, we were able to detect depletion of all previously identified Vps10 cargoes (green labels; Fig. 5 b). Interestingly, the depletion of Vps10 itself is not statistically significant (Fig. 5 b). However, based on the depletion of all its identified cargoes, we consider the level of Vps10 depletion in vps45Δ cells biologically relevant. In addition, the known CVT cargoes Ape1, Ape4, and Ams1 were depleted in vacuoles isolated from vps45Δ cells (orange labels; Fig. 5 b). This is in line with previous results showing the requirement of Vps45 for the fusion of CVT carriers with vacuoles (Abeliovich et al., 1999). We also detected multiple transmembrane and membrane-associated proteins that were only depleted in vps45Δ cells but not in vps10Δ or gga1Δgga2Δ cells. Amongst those are the V-ATPase V0 subunit Vph1 as well as multiple subunits of the V1 domain (Vma13, Vma4, Vma1, Vma5, Vma7, and Vma2) as expected (purple labels; Fig. 5 b). We also found the Niemann-Pick Type C 1 homolog Ncr1, the lipase Atg15 as well as the proteinase B, Prb1, depleted from vacuoles of vps45Δ cells (blue labels; Fig. 5 b). To determine the trafficking defect of Ncr1, we tagged the protein with mNeonGreen and observed it in multiple dots in vps45Δ cells, thus confirming our proteomics results (Fig. 5 c). Interestingly, Ncr1 has a short cytoplasmic C-terminus (Fig. 5 d), while Atg15 harbors a very short N-terminal cytoplasmic tail (Fig. 5 f) and Prb1 is a luminal protein. We therefore hypothesize that the sorting motif of these proteins is not located in the cytoplasm and that they are sorted by an unknown mechanism. Deletion of the cytosolic C-terminus of Ncr1 did not affect vacuolar targeting of the protein, suggesting that it does not harbor a sorting signal (Fig. 5, d and e). Replacing the short cytosolic N-terminus of Atg15 by GFP did also not affect the localization of the protein (Fig. 5, f and g). However, N-terminal tagging retained large amounts of the protein in the ER. We therefore compared vacuolar proteomes of Atg15ΔN versus full-length Atg15 and atg15Δ versus WT cells to check if the N-terminal depleted version is still functional. Atg15 deletion shows an increase in plasma membrane–localized proteins in the vacuole, suggesting that Atg15 is also required for degradation of multivesicular bodies. In contrast, Atg15ΔN does not show this phenotype, suggesting that it is localized at the vacuole and functional (Fig. 5 h and Table S9). Together, this supports our hypothesis that Atg15 does not require any cytosolic sorting motif.

Identification of vacuolar proteins that require Vps45 mediated membrane fusion for their localization. (a) Model highlighting the analysis of VPS45 mutants within the CPY and CVT trafficking pathways. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of vps45Δ cells. Analysis was performed as in Fig. 3 b. Vps10-dependent cargoes are labeled in green, CVT cargo proteins are labeled in red, V-ATPase subunits are labeled in purple, and other proteins are labeled in blue. (c) Localization of C-terminal mNeon-tagged Ncr1 and FM4-64–stained vacuoles was analyzed in WT cells (upper panel) and vps45Δ cells (lower panel). Scale bar, 5 µm. (d) Model of the Ncr1 domain architecture of the full-length protein (left) and the C-terminal truncation (right). (e) Localization of C-terminal mNeon-tagged Ncr1 (upper panel) and Ncr1ΔC (lower panel) with Vph1-mCherry as the vacuole marker was analyzed. Scale bar, 5 µm. (f) Model of the Atg15 full-length protein (left) and the N-terminal truncation (right). (g) Localization of N-terminal GFP-tagged Atg15 (upper panel) and Atg15ΔN (lower panel) with Vph1-mCherry as the vacuole marker was analyzed. Scale bar, 5 µm. (h) N-terminal truncation of Atg15 shows no effect on the vacuolar proteome. Proteomic comparison of vacuoles from NOPpr-Atg15ΔN/NOPpr-Atg15 and atg15Δ/WT cells.
Identification of vacuolar proteins that require Vps45 mediated membrane fusion for their localization. (a) Model highlighting the analysis of VPS45 mutants within the CPY and CVT trafficking pathways. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of vps45Δ cells. Analysis was performed as in Fig. 3 b. Vps10-dependent cargoes are labeled in green, CVT cargo proteins are labeled in red, V-ATPase subunits are labeled in purple, and other proteins are labeled in blue. (c) Localization of C-terminal mNeon-tagged Ncr1 and FM4-64–stained vacuoles was analyzed in WT cells (upper panel) and vps45Δ cells (lower panel). Scale bar, 5 µm. (d) Model of the Ncr1 domain architecture of the full-length protein (left) and the C-terminal truncation (right). (e) Localization of C-terminal mNeon-tagged Ncr1 (upper panel) and Ncr1ΔC (lower panel) with Vph1-mCherry as the vacuole marker was analyzed. Scale bar, 5 µm. (f) Model of the Atg15 full-length protein (left) and the N-terminal truncation (right). (g) Localization of N-terminal GFP-tagged Atg15 (upper panel) and Atg15ΔN (lower panel) with Vph1-mCherry as the vacuole marker was analyzed. Scale bar, 5 µm. (h) N-terminal truncation of Atg15 shows no effect on the vacuolar proteome. Proteomic comparison of vacuoles from NOPpr-Atg15ΔN/NOPpr-Atg15 and atg15Δ/WT cells.
QPrevail allows for the analysis of autophagy-related processes under nonstarvation conditions
As a second pathway, we analyzed the constitutive autophagy-related CVT pathway (Fig. 6 a). We compared proteomes of vacuoles from cells lacking Atg19, the CVT autophagy receptor (Scott et al., 2001), with those of WT cells. Similar to vps45Δ cells, we identified the known CVT cargoes Ape1, Ape4, and Ams1 to be depleted from atg19Δ vacuoles (Fig. 6 b and Table S10). In addition, we identified Lap3, a soluble cytosolic cysteine protease that was previously shown to be associated with Ape1 and characterized as selectively transported to the vacuole during nitrogen starvation (Kageyama et al., 2009). The sensitivity of our setup identified Lap3 also as a target of the CVT pathway under regular growth conditions. We also detected colocalization of overexpressed GFP-Lap3 and BFP-Ape1 as previously shown (Kageyama et al., 2009). Interestingly, colocalization of the two overexpressed proteins is lost in cells lacking the CVT receptor gene ATG19 (Fig. 6, c and d). In addition, overexpressed Lap3-GFP formed large cytosolic foci even in the absence of Atg19, Ape1, or both, suggesting that it has an inherent property to self-assemble (Fig. 6 e). To test the sensitivity of the assay for studying the CVT pathway, we compared proteomes of vacuoles isolated from WT (K0-labeled) and atg4Δ (K8-labeled) cells. Atg4 cleaves Atg8 and thus makes it accessible for lipidation with phosphatidylethanolamine (Kirisako et al., 2000). As expected, Atg8 was depleted from vacuoles along with the known CVT cargoes Ape1 and Ape4 (Fig. 6 f and Table S11). Together, our analysis revealed the constitutive targeting of Lap3 to the vacuole by the CVT pathway.

Identification of vacuolar proteins delivered by the Atg19-dependent CVT pathway. (a) Model highlighting the analysis of ATG19 mutants within the CVT trafficking pathway. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of atg19Δ cells. Analysis was performed as in Fig. 3 b. (c) Localization of overproduced GFP-Lap3 and overproduced BFP-Ape1 in WT (upper panel) and atg19∆ (lower panel) under nutrient-rich growth conditions. Overproduction from the CUP1 promoter was induced with 2 mM CuSO4 for 90 min. Vacuoles were stained with FM4-64. Scale bar, 5 µm. (c and d) Quantification of images in c from three different experiments. The amount of Lap3 co-localizing, in proximity or not colocalizing with Ape1 was calculated. Bars show average of three experiments (red dots, error bars = SD). (e) Analysis of GFP-Lap3 overproduced from the GPD promoter in respect to FM4-64–stained vacuoles is shown in WT cells (top row), atg19Δ cells (top middle row), ape1Δ cells (bottom middle row), and ape1Δatg19Δ cells (bottom row). Scale bar, 5 µm. (f) Proteomic comparison of vacuoles from atg4Δ cells and WT cells. Averaged peptide intensities are plotted against heavy/light SILAC ratios. Significant outliers (P < 1e−14) are color coded in red (P < 0.0001), orange, or blue (P < 0.05); other identified proteins are shown in light blue.
Identification of vacuolar proteins delivered by the Atg19-dependent CVT pathway. (a) Model highlighting the analysis of ATG19 mutants within the CVT trafficking pathway. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of atg19Δ cells. Analysis was performed as in Fig. 3 b. (c) Localization of overproduced GFP-Lap3 and overproduced BFP-Ape1 in WT (upper panel) and atg19∆ (lower panel) under nutrient-rich growth conditions. Overproduction from the CUP1 promoter was induced with 2 mM CuSO4 for 90 min. Vacuoles were stained with FM4-64. Scale bar, 5 µm. (c and d) Quantification of images in c from three different experiments. The amount of Lap3 co-localizing, in proximity or not colocalizing with Ape1 was calculated. Bars show average of three experiments (red dots, error bars = SD). (e) Analysis of GFP-Lap3 overproduced from the GPD promoter in respect to FM4-64–stained vacuoles is shown in WT cells (top row), atg19Δ cells (top middle row), ape1Δ cells (bottom middle row), and ape1Δatg19Δ cells (bottom row). Scale bar, 5 µm. (f) Proteomic comparison of vacuoles from atg4Δ cells and WT cells. Averaged peptide intensities are plotted against heavy/light SILAC ratios. Significant outliers (P < 1e−14) are color coded in red (P < 0.0001), orange, or blue (P < 0.05); other identified proteins are shown in light blue.
The majority of vacuolar transmembrane proteins are sorted via the AP-3 pathway
Finally, we used QPrevail to analyze the AP-3 pathway that directly transports cargo from the Golgi apparatus to the vacuole (Fig. 7 a; Cowles et al., 1997b). We identified 52 proteins depleted from apl5Δ vacuoles compared with WT vacuoles (Fig. 7 b). Amongst these proteins were the known AP-3 cargoes Pho8, Nyv1, Atg27, Yck3, and Ego3 and the recently described Tag1 and Vac7 (Fig. 7 b; green dots; Kira et al., 2021; Schoppe et al., 2020). Most proteins identified in this experiment are described as transmembrane or membrane-associated proteins, and the results are shown in (Table S12). Some AP-3 cargoes localize to the plasma membrane in apl5∆ cells (Reggiori et al., 2000; Sun et al., 2004, GNS, Yck3). However, only lipidated cargoes such as Ego1 showed this phenotype, and Ego3 reflects this (Fig. 7 c). Since the TORC1 activity–controlling EGO complex appears to be less abundant at the vacuole of apl5Δ cells, we tested if mutants of all AP-3 complex subunits become sensitive to rapamycin. Indeed, serial dilutions of apl5Δ, apl6Δ, apm3Δ, and aps3Δ cells show a higher sensitivity to rapamycin compared with WT cells and similar to that of ego3Δ cells (Fig. 7 d).

Identification of vacuolar proteins delivered by the AP-3 pathway. (a) Model highlighting the analysis of the AP-3 pathway by deletion of the AP-3 subunit APL5. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of apl5Δ cells. Analysis was performed as in Fig. 3 b. Known AP-3 cargo proteins are color coded in green. The two insets represent magnifications of the dense areas in close proximity to the hyperbolic curve. (c) Localization of Ego3-GFP in Vph1-mCherry–stained WT cells (upper panel) and apl5Δ cells (lower panel). Scale bar, 5 µm. (d) AP-3 mutants are hypersensitive to rapamycin treatment. WT, apl5Δ, apl6Δ, apm3Δ, aps3Δ, and ego3Δ cells were spotted on control plates or plates containing 4 ng/ml rapamycin. (e) Localization of N-terminal GFP-tagged Vac7 in FM4-64 WT cells (upper panel) and apl5Δ cells (lower panel). Scale bar, 5 µm. (f) Localization of N-terminal mCherry-tagged Ypk9 in WT (left panel) and apl5∆ (right panel) cells. The vacuolar lumen was stained with CMAC. Scale bar, 5 µm. (f and g) Superplot showing the quantification of images in f from three different experiments (error bars = SD). Single experiments are color coded and the average is shown as the black line. (h) Experimental setup for the visual genetics screen. Vph1-mKate–expressing WT cells, apl6Δ cells, and vps45Δ cells were crossed against a library of mutants expressing N-terminally GFP-tagged proteins annotated to localize to the vacuole, sporulated, selected for haploids, and imaged with an automated microscope. (h and i) N-terminally GFP-tagged Syg1- and Vph1-mKate–expressing WT cells (upper panel), vps45Δ cells (middle panel), and apl6Δ cells (lower panel) are shown as an example from the screen performed in h. Scale bar, 5 µM.
Identification of vacuolar proteins delivered by the AP-3 pathway. (a) Model highlighting the analysis of the AP-3 pathway by deletion of the AP-3 subunit APL5. (b) Volcano plot identifying cargoes that are enriched or depleted at vacuoles of apl5Δ cells. Analysis was performed as in Fig. 3 b. Known AP-3 cargo proteins are color coded in green. The two insets represent magnifications of the dense areas in close proximity to the hyperbolic curve. (c) Localization of Ego3-GFP in Vph1-mCherry–stained WT cells (upper panel) and apl5Δ cells (lower panel). Scale bar, 5 µm. (d) AP-3 mutants are hypersensitive to rapamycin treatment. WT, apl5Δ, apl6Δ, apm3Δ, aps3Δ, and ego3Δ cells were spotted on control plates or plates containing 4 ng/ml rapamycin. (e) Localization of N-terminal GFP-tagged Vac7 in FM4-64 WT cells (upper panel) and apl5Δ cells (lower panel). Scale bar, 5 µm. (f) Localization of N-terminal mCherry-tagged Ypk9 in WT (left panel) and apl5∆ (right panel) cells. The vacuolar lumen was stained with CMAC. Scale bar, 5 µm. (f and g) Superplot showing the quantification of images in f from three different experiments (error bars = SD). Single experiments are color coded and the average is shown as the black line. (h) Experimental setup for the visual genetics screen. Vph1-mKate–expressing WT cells, apl6Δ cells, and vps45Δ cells were crossed against a library of mutants expressing N-terminally GFP-tagged proteins annotated to localize to the vacuole, sporulated, selected for haploids, and imaged with an automated microscope. (h and i) N-terminally GFP-tagged Syg1- and Vph1-mKate–expressing WT cells (upper panel), vps45Δ cells (middle panel), and apl6Δ cells (lower panel) are shown as an example from the screen performed in h. Scale bar, 5 µM.
In contrast to palmitoylated proteins such as Ego1 and Yck3, analysis of the appearance of proteins at the plasma membrane fails for most transmembrane cargoes of the AP-3 pathway, possibly because the length of their transmembrane domain may not match the thickness of the plasma membrane (Sharpe et al., 2010). In line with this, Vac7 as a subunit of the Fab1 complex and an identified cargo of the AP-3 pathway (Schoppe et al., 2020) does not localize to the cortex of apl5Δ cells. Instead, it appears in several intracellular dots in addition to the vacuole staining (Fig. 7 e). Other cargoes such as the potential AP-3 cargo Ypk9 localize to the vacuole in both WT and apl5Δ cells. However, we detected a strong decrease in the signal at the vacuole of Ypk9 in apl5Δ cells (Fig. 7, f and g). Together, this shows that the conventional approach of monitoring GFP-tagged cargoes in AP-3 mutants may not uncover their trafficking pathway, whereas our unbiased proteomic approach suggests them as AP-3 cargoes. We also tested this via a visual genetics screen. We assembled a collection of 362 strains expressing N-terminally GFP-tagged proteins (Table S13). We introduced either the vacuolar marker Vph1-mKate alone or together with either a vps45Δ or an apl6Δ mutation (Fig. 7 h). Our analysis revealed the localization of only four proteins to the plasma membrane in apl6Δ cells (Yck3, Syg1, Tag1, and Slm4). Syg1 localization in WT, vps45Δ, and apl6Δ cells is represented as an example from the screen in Fig. 7 i. However, during our analysis, we realized that one readout for AP-3 cargo proteins is the localization to the class D vacuoles in vps45Δ cells. Although cargo proteins of the endosomal pathway do not reach the class D vacuole, AP-3 cargoes are still present at this structure. 33 out of the 52 potential AP-3 cargoes identified by QPrevail localized to the vacuole in vps45Δ cells (Fig. S4). We also used this readout to identify 14 additional proteins that we were unable to detect by proteomics (9 proteins Avt6, Yjl163c, Mch1, Ydl199c, Ydl206w, Vtc5, Yjr124c, Yml018c, and Zrc1) or did not appear as AP-3 cargoes based on their fold change values (5 proteins Avt7, Mnr2, Vam3, Vcx1, and Ydl180w). Among those is only Vam3 as a known Ap-3 cargo which we cannot account for using the QPrevail method. Together, we identified 64 proteins as potential AP-3 cargoes (Table S14). A total of 18 proteins did not clearly localize to the vacuole (Table S14, orange). For several of these 18 proteins, this is most likely a problem of N-terminal tagging such as Zrt3, Fet5, Ybt1, Vnx1, Mam3, and Vtc2 as they localized to the ER. Among the potential hits is Atg8, which is a protein targeted to the autophagosome via conjugation to phosphatidylethanolamine and thus unlikely a target of the AP-3 pathway. However, the autophagy-related gene Atg27 is a clear target of the AP-3 pathway. QPrevail of atg27Δ cells shows a decrease in Atg8 abundance at the vacuole along with cargoes of the CVT pathway. Therefore, we consider the lower abundance of Atg8 on vacuoles in AP-3 mutants a consequence of the Atg27 trafficking defect (Fig. S3 f and Table S15). To further test which of the 64 proteins are potential AP-3 cargoes, we analyzed their localization in the class E mutant vps27Δ (Piper et al., 1995). We systematically crossed Vph1-mKate vps27Δ cells with the N-terminal–tagged vacuolar GFP library and analyzed the localization of the 64 potential AP-3 cargoes (Fig. S5). As expected, the V-ATPase subunit Vph1 accumulated in the class E compartment. Of the 48 proteins that we had previously localized to the vacuole, only three were exclusively found in the class E compartment (Rmd1, Hsv2, and Fmp42; yellow). Five proteins accumulated in class E compartments but were still found at the vacuole (Ego3, Cps1, Vba1, Atg22, and Cwp1; green). Interestingly, Ego3 was previously shown to be transported by both, the AP-3 pathway as well as an endosomal pathway (Hatakeyama et al., 2019). For 30 proteins, we did not find colocalization with the class E-compartment marked by Vph1-mKate in vps27Δ cells, suggesting that these are most likely cargo proteins of the AP-3 pathway (Fig. S5 and Table S14).

Analysis of the 64 potential AP-3 cargoes in the visual genetics screen. Each N-terminally GFP-tagged protein and Vph1-mKate signal is shown for WT cells, apl6Δ cells, and vps45Δ cells.
Analysis of the 64 potential AP-3 cargoes in the visual genetics screen. Each N-terminally GFP-tagged protein and Vph1-mKate signal is shown for WT cells, apl6Δ cells, and vps45Δ cells.

Analysis of potential AP-3 cargoes in the visual genetics screen compared to the ESCRT mutant. Each N-terminally GFP-tagged protein and Vph1-mKate signal is shown for WT cells and vps27Δ (ESCRT-0 subunit) cells.
Analysis of potential AP-3 cargoes in the visual genetics screen compared to the ESCRT mutant. Each N-terminally GFP-tagged protein and Vph1-mKate signal is shown for WT cells and vps27Δ (ESCRT-0 subunit) cells.
52 of the 64 depleted proteins at vacuoles of apl5Δ vacuoles harbor transmembrane domains. Analysis of the 52 transmembrane proteins revealed that 43 harbored at least one known sorting motif for AP-3, a YXXΦ, or a dileucine motif (E/DXXXLL; Vowels and Payne, 1998; Darsow et al., 1998; Ohno et al., 1998), in regions exposed to the cytosol (Fig. S3 c). To test if we can map AP-3 cargo relationships based on their sorting motifs, we generated mutations in the µ and the σ subunit of AP-3. We generated Apm3 D217A G469K mutants to affect the YXXΦ binding motif (Mardones et al., 2013). In addition, we generated Aps3 V106D L121S mutations based on the sequence homology of AP-3 and AP-2 (Kelly et al., 2008) to affect the dileucine sorting motif of the AP-3 complex. We compared the vacuolar proteome of both mutants compared to WT vacuoles in duplicates and plotted the heavy-to-light ratios of the duplicates against each other. This analysis revealed for the Apm3 mutant that the YXXΦ-containing AP-3 cargo protein Atg27 (Segarra et al., 2015) was strongly depleted from the vacuoles of the mutant cells (Fig. S3 d). We also identified several cargoes depleted from vacuoles of Aps3 (V106D; L121S) cells. Among them are proteins we have identified as AP-3 cargoes. However, most of the identified potential AP-3 cargoes (Fig. S3 e) were unaffected in both mutants, suggesting that the mutations do not have a full functional impact or that sorting of AP-3 cargoes requires additional protein features (Table S16). However, our data reveal that QPrevail is a powerful method to uncover almost all cargoes that traffic to the vacuole via the AP-3 pathway.
Discussion
The analysis described here is, to the best of our knowledge, the first comprehensive comparison of protein transport pathways to the vacuole, the lysosome equivalent of yeast cells. We find evidence that most luminal proteins, except for the proteinase B, Prb1, are generally transported by the sorting receptor Vps10 or CVT pathway. Sorting of the Vps10 receptor itself requires the Gga proteins and Vps45, which is necessary for fusion events with endosomes along the CPY pathway. This endosomal sorting pathway transports only few transmembrane and membrane-associated proteins such as Ncr1 and Atg15. Surprisingly, sorting of Prb1 is not affected by deletion of Vps10 or the GGA proteins and is only affected in VPS45 mutant cells. Our analysis confirms cargoes of the CVT pathway and identifies Lap3 as a CVT cargo under nitrogen starvation conditions, as previously reported (Kageyama et al., 2009), and also under standard growth conditions. Finally, we show that the majority of vacuolar transmembrane and membrane-associated proteins are transported via the AP-3 pathway.
Systematic analysis of the vacuolar proteome using QPrevail allowed us to dissect sorting pathways in cells with unprecedented sensitivity. This approach has multiple advantages over fluorescent microscopy–based methods. First, all proteins analyzed by are untagged, thus guaranteeing functionality and native expression of the analyzed proteins. This bypasses the heterogeneity of phenotypes observed for fluorescently tagged cargoes of the AP-3 pathway. Our analysis also revealed high flexibility of different transport routes. Cargo proteins of pathways and receptors are not completely absent from the vacuoles of the respective knockout mutants as noticed before (Sun et al., 2004; Darsow et al., 1998). However, this is advantageous for our quantitative method. Finally, our analysis is sufficiently sensitive to measure CVT transport of endogenous proteins under nonstarvation conditions. Therefore, this approach might also be useful to analyze nutrient availability–independent autophagy pathways (Wilfling et al., 2020; Schäfer et al., 2020).
Our analysis revealed that only few vacuolar proteins are sorted via the endosomal pathway. Among them are the V-ATPase subunits, the homologous ABC transporters Bpt1 and Ycf1 (Sharma et al., 2002; Li et al., 1996), the vacuolar lipase Atg15 (Teter et al., 2001), the proteinase B, Prb1 (Moehle et al., 1987), the two Niemann-Pick Type C yeast homologs Ncr1 and Npc2 (Berger et al., 2005b; a), and Vps10 along with its cargo proteins. In contrast, our data suggest that the bulk of vacuolar membrane proteins is transported via the AP-3 pathway. Why are these transporters following different routes to the vacuole? Likely, complexes such as the V-ATPase and the NPC homology proteins already function in endosomes. The V-ATPase starts to acidify the endosomal compartments (Lafourcade et al., 2008). The mammalian homolog of Vps10, sortillin, undergoes conformational changes at low pH, resulting in cargo release (Leloup et al., 2017). A similar mechanism can be expected for Vps10. Mammalian Npc proteins have been shown to start the export of cholesterol at the endosomal level (Higgins et al., 1999). However, Npc1 appears to be most efficient at the low pH of lysosomes (Deffieu and Pfeffer, 2011). Our data support findings that both Npc proteins of yeast are sorted via the MVB pathway (Du et al., 2013) and are independent of the AP-3 pathway which had also been previously suggested (Berger et al., 2007).
In agreement with previous studies, we do not identify cytosolic sorting motifs on membrane proteins transported to the vacuole via endosomes (Conibear and Stevens, 1998). The only strict dependence is apparent for the sorting of Vps10 that requires the function of Gga adaptor proteins, similar to clathrin-coated vesicles (Hirst et al., 2012). However, deletion of the cytosolic part of Vps10 does not affect its trafficking to the vacuole (Arlt et al., 2015). Similar to the cytosolic part of Vps10, the cytosolic C-termini of Ncr1 and Atg15 are also not negligible for sorting. This either suggests the presence of a luminal sorting motif or clustering of proteins depending on the structure and or composition of the membrane itself.
In contrast, our analysis suggests that AP-3–dependent sorting requires cytosolic sorting motifs. However, mutations previously shown to affect the interaction of the AP-3 µ-subunit with the YXXΦ sorting motif (Mardones et al., 2013) did not abolish trafficking of all YXXΦ containing proteins. Similarly, mutations predicted on the interaction of the AP-2 σ-subunit with a dileucine-containing motif (Kelly et al., 2008) only affected a fraction of potential AP-3 cargoes. This suggests that AP-3 cargoes harbor multiple signals to ensure trafficking via the AP-3 pathway.
We anticipate this dataset to fuel numerous mechanistic studies. In the future, it will be of interest to understand how the cargo is bound by receptors such as Vps10/sortillin or adaptors such as the AP-3 complex. Knowing multiple cargoes of a given pathway will tremendously promote identification of the general underlying biochemical mechanisms of these interactions. In addition, a large fraction of the vacuolar proteins in yeast has functional homologs in mammalian cells (Table S17). Thus, our data suggest that many of the transport processes are conserved throughout evolution. Our analysis highlights that mutations in the sorting pathways have pleiotropic effects on the vacuolar proteome. Since many lipid metabolism enzymes are also sorted to the vacuole and the lysosome in mammalian cells, we also expect the vacuolar lipid composition to be affected. Since mutations in lysosomal proteins and endo-lysosomal sorting proteins cause a variety of diseases, the understanding of such mutations in the context of cellular metabolism will strongly profit from systematic methods. The vacuolar biogenesis map presented is an entry point into such analyses.
Materials and methods
Yeast strains and plasmids
Yeast strains used in this study are described in Table S18. Plasmids used in this study are shown in Table S19.
Gene sequences were cloned into plasmid vectors via Fast cloning (Li et al., 2011) and point mutations inserted using Q5 mutagenesis.
Growth conditions and media
Yeast strains were grown under standard conditions. For rapamycin spotting assays, the cells were grown in YPD (2% glucose, 2% peptone, and 1% yeast extract) and the drug added in a final concentration of 4 ng/ml. Plates were incubated for 48 h at 30°C.
For SILAC labeling, yeast cells were grown in SDC–lysine medium (2% glucose, 6.7 g/liter yeast nitrogen base without amino acids and 1.92 g/liter yeast synthetic dropout without lysine; Sigma Aldrich). Precultures were grown over day at 30°C in the presence of 30 mg/liter normal lysine or heavy lysine (L-lysine 13C615N2; Cambridge Isotope Laboratories), diluted in the evening and cells grown to the log phase in the next morning before harvesting.
For microscopy experiments, cells were grown in SDC–lysine medium with the addition of 30 mg/liter normal lysine. The Mup1 uptake assay was performed in SDC–methionine medium, inducing Mup1 endocytosis by the addition of 1 mM methionine.
For tetrad dissections, diploid cells generated from mating of haploid deletion mutants in YPD were sporulated on sporulation plates (3% agar, 1% KOAc) for 5 d at 30°C.
Vacuole isolation
For SILAC experiments, vacuoles were purified from logarithmic-phase cells in 500 ml of SDC–lysine media with the addition of 30 mg/liter (final) lysine (control culture with light lysine, K0, and mutant culture with heavy lysine, K8). After harvesting the same OD units of light and heavy grown cultures, the cells were mixed and the pellet treated with Tris-buffer (0.1 M Tris, pH 9.4; 10 mM DTT) for 10 min at 30°C. After centrifugation (4,000 rpm, 4 min at RT), cells were incubated in spheroblasting buffer (0.6 M sorbitol, 50 mM KPi, pH 7.4, in 0.2× YPD) with lyticase for 20 min at 30°C. The cells were centrifuged again (3,500 rpm, 3 min, 4°C) and the pellet taken up in 2.5 ml of 15% Ficoll solution (in 10 mM PIPES/KOH, pH 6.8, 0.2 M sorbitol) on ice. 200 μl of DEAE dextran (0.4 mg/ml) was added and the cells incubated on ice for 3 min and at 30°C for 1.5 min. The cells were transferred to a centrifugation tube (Seton Scientific, for Beckman SW40 rotor) and the gradient poured on top by adding 3 ml of 8% Ficoll, 3 ml of 4% Ficoll, and 0% Ficoll solution. The tubes were centrifuged for 1.5 h (30,000 rpm, 4°C) and the vacuoles collected as the interphase between 0% and 4% Ficoll solution (for details, see (Cabrera and Ungermann, 2008). Protein concentration was determined with Bradford assay and the vacuole samples directly used for the proteomics protocol. For quality control, WT cell vacuoles were labeled with Vph1-mCherry and mutant vacuoles with Vph1-mNeon or GFP-Yck3. The same amounts of WT and mutant cells were mixed before vacuole isolation and purified vacuoles examined via fluorescence microscopy by determining the size and amount of WT and mutant vacuoles.
Fluorescence microscopy
For microscopy experiments, cells were grown in SDC media at 30°C to the logarithmic phase if not other mentioned. Drugs were added at indicated concentrations. Cells were imaged at RT on an Olympus IX-71 inverted microscope equipped with 100× NA 1.49 and 60× NA 1.40 objectives, a sCMOS camera (PCO), an InsightSSI illumination system, 4′,6-diamidino-2-phenylindole, GFP, and mCherry filters, and SoftWoRx software (Applied Precision). We used constrained-iterative deconvolution (SoftWoRx). All microscopy image processing and quantification was performed using ImageJ (RRID:SCR_003070; National Institutes of Health). If not stated otherwise, pictures were taken with same settings and processed in the same way. For quantification of microscopy data, error bars were defined based on the standard deviation. As the statistical test, the t test was performed (two-tailed homoscedastic) to calculate the P value.
Western blot
For Western blot of the secreted proteins, cells were grown in YPD for 24 h, and the supernatant was concentrated in a centrifugal filter tube (Amicon Ultra 10K, 0.5 ml, Merck Millipore), adjusting the volume according to OD units. After washing with PBS twice, the proteins were precipitated with TCA and resuspended in SDS loading buffer. HA-tagged proteins were detected with a 1:2,000 diluted mouse anti-HA antibody 12CA5 (Roche; RRID:AB_514505) and HRP-coupled mouse IgG κ–binding protein (RRID:AB_2687626; Santa Cruz Biotechnology), diluted to 1:10,000. For visualization, a chemiluminescent substrate solution (SuperSignal West Pico PLUS; Thermo Fisher Scientific) was used.
Proteomics
Mass spectrometry was done with purified vacuole samples and total cell lysate. Peptide digestion and purification were performed using a commercial proteomics kit (iST Kit; PreOmics). 200 µg of purified vacuoles was precipitated with TCA and the pellet resuspended in the lysis buffer of the kit. For cell lysate samples, 2 OD units of yeast cells were centrifuged and the cell pellet resuspended in the lysis buffer. For supernatant samples, supernatants of heavy- and light-labeled cultures were mixed before concentration and TCA precipitation and the protein pellet resuspended in lysis buffer. Protein digestion was performed with LysC according to the standard protocol of the kit. Reversed-phase chromatography was performed on a Thermo Ultimate 3000 RSLCnano system connected to a Q ExactivePlus mass spectrometer (Thermo Fisher Scientific) through a nanoelectrospray ion source as described before with following adjustments (Eising et al., 2019). Peptides were eluted from the column with a linear gradient of acetonitrile from 10 to 35% in 0.1% formic acid for 118 min, from 35 to 60% for 20 min, and from 60 to 80% for 10 min at a constant flow rate of 200 nl/min. The 10 most intense multiply charged ions (z = 2) from the survey scan were selected with an isolation width of 1.4 m/z. The dynamic exclusion of sequenced peptides was set at 20 s. The resulting spectra were analyzed with MaxQuant (version 1.6.14.0, www.maxquant.org; Cox and Mann, 2008; Cox et al., 2011) as described previously. The re-quant option of MaxQuant was enabled to identify also proteins that only yield one SILAC pair (Fröhlich et al., 2013).
Proteomics data analysis and generation of the vacuole biogenesis map
MaxQuant output files (proteinGroups) for all QPrevail experiments were analyzed using the Perseus software package (Perseus version 1.6.15.0; Tyanova et al., 2016). First, SILAC ratios for all proteins annotated as vacuolar (Table S2) were extracted. Ratios were log2 transformed, and afterward, the dataset was filtered for a minimum of three valid ratios. Hierarchical clustering was performed for Euclidean distance, with average linkage and no constraints according to standard values in Perseus. The result of this clustering is represented as the vacuole biogenesis map in Fig. 2.
Significant cargoes of each condition were determined by a volcano plot–based strategy, combining t test P values with ratio information. A standard equal group variance t test was applied based on the logarithmic SILAC ratios of the replicates of the control experiments (heavy-labeled WT vacuoles compared with light-labeled WT vacuoles, annotated as WT in the datasets) and the logarithmic SILAC ratios of the replicates for each trafficking mutant (e.g., heavy-labeled vps10Δ vacuoles compared with light-labeled WT vacuoles, annotated as VPS10). Significance lines in the volcano plot corresponding to a given FDR were determined by a permutation-based method (Tusher et al., 2001). Threshold values (= FDR) were selected as 0.5 and SO values (= curve bend) between 0.1 and 1.0 (vps10Δ = 1; gga1Δgga2Δ = 1; vps45Δ = 1; atg19Δ = 0.1; and apl5Δ = 0.18). The output tables of the calculations are supplemented for each trafficking mutant as Tables S3–S7. The SPS file of the Perseus software package is available as Data S1, where all calculations are annotated.
For SILAC plots (ratio versus intensity), the P value was defined as significance A as described before (Cox and Mann, 2008). Significant outliers are colored in red (P < 1e−14), orange (P < 0.0001), or steel blue (P < 0.05); other proteins are shown in light blue.
Genetic interaction screen
Sporulated cells were incubated with zymolyase to degrade cell walls and single spores dissected with the help of a micromanipulator. Spores were grown on YPD plates for 2 d and replica plated on selection plates followed by incubation at 30°C for 1 d.
Visual genetics screens
An array of 362 haploid MATa strains expressing N-terminally GFP-tagged variants of vacuolar proteins under control of a NOP1 promoter was assembled from the genome-wide SWAp-TAG mutant collection (Weill et al., 2018). The desired mutation combinations Vph1-mKate, Vph1-mKate vps45Δ, and Vph1-mKate apl6Δ were constructed in a synthetic genetic array–ready haploid MATα background and introduced into the collection of GFP–vacuole mutants by the synthetic genetic array method (Tong and Boone, 2006; Cohen and Schuldiner, 2011). Haploid strains were mated, diploid cells were selected, and sporulation was induced via a 7-d nitrogen starvation. Haploid cells were selected using thialysine and canavanine (Sigma-Aldrich), and mutants harboring the complete desired combinations of mutations were selected. Throughout the process, mutant collections were handled with a RoToR bench-top colony array instrument (Singer Instruments). Representative strains were validated by PCR and manual microscopy. For automated imaging, cells were grown to the mid-logarithmic phase in SDC media at 30°C and imaged on an inverted fluorescence microscopic ScanR system (Olympus) using a 40× air lens (NA 0.9). Images were manually reviewed using ImageJ. For the class E compartment phenotype screen, the N-terminally GFP-tagged variants of vacuolar proteins under control of a NOP1 promoter carrying strains were crossed to a Vph1-mKate vps27Δ Matα strain. Haploid strains were mated, diploid cells were selected, and sporulation was induced via a 7-d nitrogen starvation. Haploid cells were selected using thialysine and canavanine (Sigma-Aldrich), and mutants harboring the complete desired combinations of mutations were selected.
Primer list
See Table 1.
Primer list
| Primer | Sequence |
| NPC2 S3 | 5ʹ-AGGATGATCTGATCACTTGCTTAACCGGAGAAGTCATCTTCCCACCAAGGCGTACGCTGCAGGTCGAC-3ʹ |
| NPC2 S2 | 5ʹ-GAACGAGAAGGGAAATAAACACGGATCAATGAGTTGTATGAATCAGATCAATCGATGAATTCGAGCTCG-3ʹ |
| VPS10 S1 | 5ʹ-CTGTATATCTGGAAAAGCCCTGAAGTGTCCAGTAGTCATCACACGTTATGCGTACGCTGCAGGTCGAC-3ʹ |
| VPS10 S2 | 5ʹ-GTTTATGAAAAGTATATGGAATTATCTACTCTATGTAAAGTAATCTCTCTAATCGATGAATTCGAGCTCG-3ʹ |
| CPY S2 | 5ʹ-TAAAGCGTGTATGTGTAGGCATACCGTTTTTATTATCAGCTACGATCGAAATCGATGAATTCGAGCTCG-3ʹ |
| CPY S3 | 5ʹ-AAAACGCCTTAAGTATGGTTAACGAATGGATCCACGGTGGTTTCTCCTTACGTACGCTGCAGGTCGAC-3ʹ |
| APM3 S1 | 5ʹ-GACTTGGGCAACAACAGAGGCTGTAAACCTTACCAACCCAACCAAAATAGATGCGTACGCTGCAGGTCGAC-3ʹ |
| APM3 S2 | 5ʹ-CTCATTATATTCTATTTAGTTTCGCATGGAATTTCAAGTACACATATAACTAATCGATGAATTCGAGCTCG-3ʹ |
| APM3 for | 5ʹ-GATATCGAATTCCTGGAGGGGTAGAAGTCGCTG-3ʹ |
| APM3 rev | 5ʹ-GGATCCCCCGGGCTGTGCGGAAGTCTTCCCTAAG-3ʹ |
| pRS APM3 for | 5ʹ-AGGGAAGACTTCCGCACAGCCCGGGGGATCCAC-3ʹ |
| pRS APM3 rev | 5ʹ-GCGACTTCTACCCCTCCAGGAATTCGATATCAAGCTTATCG-3ʹ |
| APM3 D217A for | 5ʹ-GCTTTATGTGGCTTTGTTAGAAAC-3ʹ |
| APM3 D217A rev | 5ʹ-TCATTATTCTCATGTTTGCTAG-3ʹ |
| APM3 G469K for | 5ʹ-GCTGTACAAGAAGGCAAAGTATAAGAC-3ʹ |
| APM3 G469K rev | 5ʹ-TTTGTATTTTTGCCAATTGTTAATC-3ʹ |
| APS3 S1 | 5ʹ-CTATACCAGATCTGGGTTACAGAGGCCAGAACACATATAACGTTACACAATGCGTACGCTGCAGGTCGAC-3ʹ |
| APS3 S2 | 5ʹ-CAGATCTTGACGAACGAAAATAACCATCATCCGATTAATTTGTTTGTTTAATCGATGAATTCGAGCTCG-3ʹ |
| APS3 for | 5ʹ-TGATATCGAATTCCTGCTTTTGAGTATGAGGAGTTCCAC-3ʹ |
| APS3 rev | 5ʹ-TGGATCCCCCGGGCTGGTGGATTTTGCCATTTCTAAAGAG-3ʹ |
| pRS APS3 for | 5ʹ-AAATGGCAAAATCCACCAGCCCGGGGGATCCAC-3ʹ |
| pRS APS3 rev | 5ʹ-TCCTCATACTCAAAAGCAGGAATTCGATATCAAGCTTATCG-3ʹ |
| APS3 V106D for | 5ʹ-CCAAACTTTTGACGAATCATTGGAC-3ʹ |
| APS3 V106D rev | 5ʹ-ATCAGATCTAATATGGCC-3ʹ |
| APS3 L121S for | 5ʹ-TGAACTTGATTCGATTTTTAACTGG-3ʹ |
| APS3 L121S rev | 5ʹ-TTGACTTCAGTAAAACAACG-3ʹ |
| GGA1 S1 | 5ʹ-GGGACAAGTCACTACTTCAAGTATAACCCAGACAAGAGTCTTTTAAAATGCGTACGCTGCAGGTCGAC-3ʹ |
| GGA2 S2 | 5ʹ-ATATAATATGGCATCTACTTTTTTTTCAACTTCTCTACCGAATTTGATTAATCGATGAATTCGAGCTCG-3ʹ |
| GGA2 S1 | 5ʹ-AAGAGCAACATAACTCATAGCTGAATTGCTAATCGTGATACTGCATCATGCGTACGCTGCAGGTCGAC-3ʹ |
| GGA2 S2 | 5ʹ-AAAATATACATAGAGAAGAGAAAGGATTGATAAGAAACGCCAGAGGATTAATCGATGAATTCGAGCTCG-3ʹ |
| VPS45 S1 | 5ʹ-GACTTGGTTTTGAGTTAAGGCCATCTTTTACTGTATAGAACAAAGAAATGCGTACGCTGCAGGTCGAC-3ʹ |
| VPS45 S2 | 5ʹ-GATTTATGCCTCATATATAAAATAGAATTTTAGAATAAGATAATCCTTATTTAATCGATGAATTCGAGCTCG-3ʹ |
| APL5 S1 | 5ʹ-GAAGTGGGAAAGGCAGTTGCCACAACCAGAAGAGCATAACATATTGTCATGCGTACGCTGCAGGTCGAC-3ʹ |
| APL5 S2 | 5ʹ-CGTAATCGCTGTCATTGAGAATTCACGACTTCGATTTTTTTATCGATCGATGAATTCGAGCTCG-3ʹ |
| ATG19 S1 | 5ʹ-TTGCGGCGGCACTTGCTTCAGTAACGCCCAAAGGAGAGTTCTGGTAAATGCGTACGCTGCAGGTCGAC-3ʹ |
| ATG19 S2 | 5ʹ-TGTGTATGTGAAAAGGTACTCATTGCTGTATAAAAATAGAGTTTGACCTAATCGATGAATTCGAGCTCG-3ʹ |
| LAP3 S1 | 5ʹ-CGTTTAGAAGCCACCTTTTACGAGCACCACAAATTGTTTTAAAAAGAATGCGTACGCTGCAGGTCGAC-3ʹ |
| LAP3 S4 | 5ʹ-TGAAACTCTTTGTTCCAAGAGTTGATCTTACTGATATCGATGGAAGAGGACATCGATGAATTCTCTGTCG-3ʹ |
| VPH1 S2 | 5ʹ-TATTTAATGAAGTACTTAAATGTTTCGCTTTTTTTAAAAGTCCTCAAAATATCGATGAATTCGAGCTCG-3ʹ |
| VPH1 S3 | 5ʹ-GTATAAAGACATGGAAGTCGCTGTTGCTAGTGCAAGCTCTTCCGCTTCAAGCCGTACGCTGCAGGTCGAC-3ʹ |
| APL6 S1 | 5ʹ-GAAAGGTAAACCAAAGACAGCAAACAATCGAAAAGTGGACAACCAGCAATGCGTACGCTGCAGGTCGAC-3ʹ |
| APL6 S2 | 5ʹ-CCTATATCTATATCTATAAATATACATATTTATATAGTATTATCTCCGAGTTCTAATCGATGAATTCGAGCTCG-3ʹ |
| SLM4 S1 | 5ʹ-AAAAGGCAGTTATCAGCAGCAAACGGTATCCAAAATATTGAAGCAATATGCGTACGCTGCAGGTCGAC-3ʹ |
| SLM4 S2 | 5ʹ-CGGAGACGCATGAAAAGGTGTGGCCTCGATACATATTGTTAAACCCATCAATCGATGAATTCGAGCTCG-3ʹ |
| NCR1 S2 | 5ʹ-TTTTACCTATTTTTTCACTACGTAAAATATAGTATAATCTGCTATGGCTAATCGATGAATTCGAGCTCG-3ʹ |
| NCR1 S3 | 5ʹ-CATTGTTTGGTGGTGAAAGCTATAGGGACGATTCCATCGAAGCAGAAGATCGTACGCTGCAGGTCGAC-3ʹ |
| APE1 S1 | 5ʹ-TTAGTGCAATTGTAGAAACCTGCACAACCAACAAAATTAAGAAAAAAAGAATGCGTACGCTGCAGGTCGAC-3ʹ |
| APE1 S2 | 5ʹ-AAAAGGATAAAGAAACAGAAATCAAAAGAAATAAAAAGAGTGTGGCAAAATCAATCGATGAATTCGAGCTCG-3ʹ |
| YPK9 S1 | 5ʹ-TAAAAAGGAGCCCAGACTTACTGATAGATCTTGCATATACTCCGGTAATGCGTACGCTGCAGGTCGAC-3ʹ |
| YPK9 S4 | 5ʹ-TTCCTTTCACTCCTTTGGCCATGTTGAATTTGGTTTGAAGAGGGAATATCCATCGATGAATTCTCTGTCG-3ʹ |
| pRS405 for | 5ʹ-TATTTATAGTACTTCATTCAGCCCGGGGGATCCAC-3ʹ |
| pRS405 rev | 5ʹ-CGGATACTTTTTAGTTTCAGGAATTCGATATCAAGCTTATCG-3ʹ |
| VPS10 for | 5ʹ-TTGATATCGAATTCCTGAAACTAAAAAGTATCCGCCTG-3ʹ |
| VPS10 rev | 5ʹ-GTGGATCCCCCGGGCTGAATGAAGTACTATAAATATTAAAGTACG-3ʹ |
| VPS10 D1 del for | 5ʹ-AAGGGAACGAATGATAAAGAAATA-3ʹ |
| VPS10 D1 del rev | 5ʹ-TGCTCCAGCACCAGCACC-3ʹ |
| VPS10 D2 del for | 5ʹ-CGCCATCTAACGAAAACCAG-3ʹ |
| VPS10 D2 del rev | 5ʹ-CTTGAAATCAAATTTGTTTTCTGTAACC-3ʹ |
| NPC2 S1 | 5ʹ-CATAACCATATTAAATCTTCTCCTTCAAAGCTAGCACGCCTTCCAAAATGCGTACGCTGCAGGTCGAC-3ʹ |
| VAC7 S1 | 5ʹ-CCTTATCGTTTCATCTCAGGCAAGTTAAAGCATTTGGGAAACGTGCTAGATGCGTACGCTGCAGGTCGAC-3ʹ |
| VAC7 S4 | 5ʹ-CGGGTGCCTCAACTGTTTCTGTCTCTACAGTGAGCTTTCTATCTTCTTCTGTCATCGATGAATTCTCTGTCG-3ʹ |
| SLM4 S3 | 5ʹ-AAAGAGCTATGAGAGAGTTGACTGATTTGTTTGGCTACAAGCTAGGTCGTACGCTGCAGGTCGAC-3ʹ |
| MUP1 S2 | 5ʹ-GTTCATACGTGATTATAAGAATCGAGATGAGATGGTAAGTACCTTTTTGGTTAATCGATGAATTCGAGCTCG-3ʹ |
| MUP1 S3 | 5ʹ-CGTTATTGAAACGAATATAATCGAACATTACAAAAGTGAACAAGAAAAATCGCTGCGTACGCTGCAGGTCGAC-3ʹ |
| YCK3 S1 | 5ʹ-GTGGTATCTCATTCTGAAGAAAAAGTGTAAAAGGACGATAAGGAAAGATGCGTACGCTGCAGGTCGAC-3ʹ |
| YCK3 S4 | 5ʹ-GGTCCTACAGCATAATGAATACCTACAATGTGTTGTGAAGATCGTTGGGACATCGATGAATTCTCTGTC-3ʹ |
| Ncr1 S2 Cterm cytosol del | 5ʹ-ATCTTCTGCTTCGATGGAATCGTCCCTATAGCTTTCACCACCAAACAATCGATGAATTCGAGCTCG-3ʹ |
| Ncr1 S3 Cterm cytosl del | 5ʹ-CATTGTAGCAGCATTGCATGCTCTCCTATTTTTACCTGCTTTACTTTCATCGTACGCTGCAGGTCGAC-3ʹ |
| ATG15 S1 | 5ʹ-AACTGATCTAGGCATTACAATTAAAGGAAACAAGGGAAATATTCTATTGAATGCGTACGCTGCAGGTCGAC-3ʹ |
| ATG15 S4 | 5ʹ-CCTAGATGCAAAGGAGAAGCAAATCTCTTTCTTGAAGGGCTTTTATGCAACATCGATGAATTCTCTGTCG-3ʹ |
| Atg15 S1 Nterm cytosl del | 5ʹ-TTGCATAAAAGCCCTTCAAGAAAGAGATTTGCTTCTCCTTTGCGTACGCTGCAGGTCGAC-3ʹ |
| Atg15 S4 Nterm cytosl del | 5ʹ-GCAATAAGGCAGAGCACTGTAAGCGTTAGAATGCATCCTAGATGCATCGATGAATTCTCTGTCG-3ʹ |
| VPS27 S1 | 5ʹ-TTGCTAAGGTGAATGAGTAGTGAGTAAAGAACTAAGAACAGTATGCGTACGCTGCAGGTCGAC-3ʹ |
| VPS27 S2 | 5ʹ-GCGCTAGGTTTCTTTTTACAAATACATAGAAAAGGCTACAATATTAAATCGATGAATTCGAGCTCG-3ʹ |
| ATG15 S2 | 5ʹ-CGCATAGGCCCTAAAACAACACTAGGGTCATAATAGATGTATGGGTCTTAATCGATGAATTCGAGCTCG-3ʹ |
| ATG27 S1 | 5ʹ-GAGATCTTCAATCGATGCGATAGATAAAGGTAAGGAAAGCTTTCACGATGCGTACGCTGCAGGTCGAC-3ʹ |
| ATG27 S2 | 5ʹ-TAGCACTGCTGTTGCAAAAATATCGAATTGTAAGCCAGTAAACTTATTTAATCGATGAATTCGAGCTCG-3ʹ |
| ATG4 S1 | 5ʹ-GTAGATGAAGAATGGACGACTTCTTATCACGTATAGGAGTGATATACATGCGTACGCTGCAGGTCGAC-3ʹ |
| ATG4 S2 | 5ʹ-GGGAATATATTAAAACAAGTATATATGCTTATGAACTAGTGAATTCCTTACACTAATCGATGAATTCGAGCTCG-3ʹ |
Online supplemental material
Fig. S1 shows genetic interactions of the vacuole trafficking mutants analyzed in this study. Fig. S2 shows comparison of total proteome levels and vacuolar proteome levels in the different trafficking mutants. Fig. S3 shows comparison of soluble vacuolar proteins. Fig. S4 shows analysis of the 64 potential AP-3 cargoes in the visual genetics screen. Fig. S5 shows analysis of potential AP-3 cargoes in the visual genetics screen compared to ESCRT mutant. Table S1 is a summary of all identified proteins in the SILAC experiment for total proteomes and vacuolar isolations. Table S2 lists of all proteins that have been annotated as vacuolar/endosmal based on GO terms. Table S3 lists of SILAC ratios of all vacuolar luminal annotated proteins in isolated vacuoles. Table S4 lists the Perseus output for the analysis of all Vps10-dependent proteins. Table S5 lists proteomics results for the analysis of secreted proteins in vps10 cells compared to WT cells. Table S6 lists proteomics results for the analysis of the proteome of isolated vacuoles from vps10ΔD1 and vps10ΔD2 cells compared to WT cells. Table S7 lists the Perseus output for the analysis of all Gga1 Gga2-dependent proteins. Table S8 lists the Perseus output for the analysis of all Vps45-dependent proteins. Table S9 lists of proteomics results for the analysis of the proteome of isolated vacuoles from atg15ΔN and atg15Δ cells compared to WT cells. Table S10 lists of the Perseus output for the analysis of all Atg19-dependent proteins. Table S11 lists proteomics results for the analysis of the proteome of isolated vacuoles from atg4Δ cells compared to WT cells. Table S12 lists the Perseus output for the analysis of all Apl5-dependent proteins. Table S13 lists N-terminally GFP-tagged genes used for the visual genetics screen. Table S14 lists potential AP-3 cargoes. Table S15 lists proteomics results for the analysis of the proteome of isolated vacuoles from atg27Δ cells compared to with WT cells. Table S16 lists of proteomics results for the analysis of the proteome of isolated vacuoles from AP-3 mutant cells compared to with WT cells corresponding to Fig. S3, d and e. Table S17 lists vacuolar yeast genes and their corresponding human homologs. Table S18 lists of all yeast strains used in this study. Table S19 lists of all plasmids used in this study. Data S1 provides Perseus output file recapitulating all steps of data analysis.
Acknowledgments
We thank members of the Fröhlich lab for critical comments.
This work was supported by the SFB 944 (German Research Society [DFG] Project P20 to F. Fröhlich and Project P11 to C. Ungermann). M. Bohnert is supported the Gerty Cori Programme, Medical Faculty, University of Münster, Germany, and by the SFB 1348.
The authors declare no further competing financial interests.
Author contributions: S. Eising and F. Fröhlich conceptualized the project and prepared figures. S. Eising, B. Esch, P. Vargas Duarte, and M. Wälte conducted the investigation. S. Walter ran mass spectrometry samples. M. Bohnert and C. Ungermann conceptualized the project. F. Fröhlich wrote the manuscript.

