


Danicamtiv is a second-generation myotropic sarcomere activator currently in clinical trials for treating heart failure with reduced ejection fraction. Initial clinical and preclinical studies suggest that danicamtiv improves upon the major shortcoming of the first-generation myotropic sarcomere activator, omecamtiv mecarbil (OM), which overly impaired diastolic function. However, no study has directly compared the in vivo cardiac effects of danicamtiv and OM to verify these claims. These direct comparisons are essential to understand the potential benefits of one drug over the other. Therefore, this study employed carefully controlled experiments with left ventricular pressure–volume loop and echocardiographic strain analysis to compare how danicamtiv and OM alter each phase of the cardiac cycle. Our results show that for similar increases in left ventricular stroke volume, danicamtiv reduced diastolic performance and myocardial relaxation less than OM. However, danicamtiv still significantly decreased diastolic function at higher doses, like OM. Furthermore, danicamtiv and OM elicited a qualitatively similar triphasic dose–response from the left ventricle. These similarities between danicamtiv and OM in the whole heart were surprising given recent evidence showing significant differences in the drugs’ molecular effects on myosin mechanics. We therefore conclude that danicamtiv likely has a wider therapeutic window than OM, but may be limited by the same trade-off between systolic and diastolic performance, driven by similar underlying mechanisms.
Introduction
Heart failure with reduced ejection fraction (HFrEF), a condition defined by left ventricular (LV) systolic dysfunction, is a significant public health challenge (Tsao et al., 2023). Tens of millions of HFrEF patients face a 50% 5-year mortality rate, which burdens healthcare systems with billions of dollars in annual medical expenses (Savarese et al., 2022; Cook et al., 2014). To date, the best that clinicians can offer HFrEF patients are therapies that primarily manage symptoms and delay disease progression (Mentz and Felker, 2020). These include fluid retention management, β-adrenergic blockade, antagonism of the rennin–angiotensin–aldosterone system, and, most recently, sodium–glucose cotransport inhibition (Docherty et al., 2022). While such approaches can improve clinical outcomes, they do little to address the underlying contractile deficit that drives these symptoms and disease progression (Docherty et al., 2022). Thus, developing an inotropic therapy that restores cardiac contractility has been a major goal for the field for decades (Ferrari et al., 2016).
Unfortunately, all such efforts to date have proven unsuccessful (Ahmad et al., 2019). Trying to stimulate the inotropic aspects of the β-adrenergic pathway and calcium signaling has been hindered by maladaptations to prolonged use, such as increased propensity for fatal arrhythmias (Ahmad et al., 2019). In contrast, myotropic sarcomere activation is a developing inotropic therapeutic strategy that directly modulates cardiac myosin activity to increase sarcomere force production and cardiac output (Malik et al., 2011; Voors et al., 2020; Psotka et al., 2019). Bypassing the complex upstream inotropic signaling pathways theoretically minimizes the risk of off-target effects and offers the most direct method of restoring contractility (Psotka et al., 2019). However, despite its potential benefits over previous inotropic approaches, realizing these benefits has not been easy.
The primary difficulty with developing an effective myotropic sarcomere activation therapy has been managing the trade-off between gains in systolic function and impairment of diastolic performance (Fülöp et al., 2021; Rønning et al., 2018; Mamidi et al., 2021; Mamidi et al., 2017; Mamidi et al., 2015). This is exemplified by the first-generation myotropic sarcomere activator, omecamtiv mecarbil (OM) (Malik et al., 2011). OM’s positive inotropic effects are well documented by several studies demonstrating that OM extends systolic ejection time (SET) to increase stroke volume (SV) and cardiac output (Rønning et al., 2018; Shen et al., 2010; Teerlink et al., 2016; Cleland et al., 2011). While increasing SET was initially marketed as a desirable effect, prolonging systole inevitably reduces the time available for diastolic filling. Furthermore, OM decreased measures of LV pressure relaxation and filling in pig and rodent studies, along with relaxation indices in isolated muscle fiber systems (Fülöp et al., 2021; Rønning et al., 2018; Mamidi et al., 2021; Mamidi et al., 2017; Mamidi et al., 2015). This tightly coupled trade-off between systolic and diastolic function likely contributed to OM’s narrow therapeutic window, lackluster phase III clinical trial performance, and ultimate rejection by the FDA (Teerlink et al., 2021).
More recently, the second-generation myotropic sarcomere activator, danicamtiv (DN), has entered phase II clinical trials (Voors et al., 2020). Existing data suggest that DN better preserves LV relaxation than OM. A small-scale clinical study in HFrEF patients found that a DN plasma concentration of 2,000–3,500 ng/ml significantly increased SV and SET while minimally impacting indices of diastolic function as measured by echocardiography (Voors et al., 2020). A later echocardiographic study of the acute cardiac effects of DN in healthy rats found similar changes in SV and SET (Ráduly et al., 2022). However, while the authors also reported that while DN did not change some indices of diastolic function, such as isovolumic relaxation time (IVRT), other indices, including the ratio of early to late filling velocity (E/A) and diastolic duration, were significantly decreased (Ráduly et al., 2022). Nevertheless, the authors of both the clinical and rat studies suggest that DN better maintains diastolic function when compared with OM (Voors et al., 2020; Ráduly et al., 2022).
This is consistent with a comparative study in chemically skinned human donor and failing cardiac fibers, where DN slowed crossbridge detachment less than OM (Choi et al., 2023; Choi et al., 2024). Preliminary molecular studies also indicate that DN does not impair the force dependence of crossbridge detachment, nor the canonical ATP-induced actomyosin dissociation pathway, whereas OM does (Woody et al., 2018; Scott et al., 2024, Preprint; Liu et al., 2018). Furthermore, a different study demonstrated that DN can increase systolic function in a mouse model of genetic dilated cardiomyopathy (Kooiker et al., 2023).
While the existing studies of DN offer promising results, none directly compare the in vivo cardiac effects of DN with OM. Such studies are essential to rigorously determine the potential benefits of DN over OM. Therefore, this study employed carefully controlled experiments to test whether DN better preserves diastolic function in vivo when compared with OM. To do so, we used LV pressure–volume (PV) loop and echocardiographic strain analysis in healthy mice to conduct a detailed hemodynamic and mechanical assessment of how DN and OM differentially impact each phase of the cardiac cycle. Our study is the first to make these direct in vivo comparisons between DN and OM and offers critical insights into how DN will impact cardiac function compared with OM.
Materials and methods
Study approval
All animal procedures were reviewed and approved by the Case Western Reserve University Institutional Animal Care and Use Committee in accordance with the guidelines set forth by the Institute for Laboratory Animal Research (National Research Council et al., 2011).
Animal resources
Healthy male and female mice (129SVE; Taconic Farms) aged 3–4 mo were used for all experiments. Data were analyzed with sex as an independent variable.
Experimental design
This study was designed to directly compare the acute cardiac effects of DN and OM in healthy mouse hearts. The first set of experiments assessed the heart’s cumulative dose-dependent response to the drugs. Given the complex dose responses and differences in pharmacological profiles, low and high doses for each drug (DN Low: 3 mg/kg; DN High: 6 mg/kg; OM Low: 0.75 mg/kg; OM High: 1.5 mg/kg) were chosen for more in-depth study such that the corresponding doses of each drug increased SV by similar amounts. This allowed us to assess how DN and OM differentially alter cardiac function to achieve similar increases in SV, the parameter that matters most to patients. DN High and OM High increased systolic duration by 12.5% and 20% from baseline, respectively (Tables 1 and S2). These effects match those seen in the clinically relevant doses used in clinical trials and large animal studies (Teerlink et al., 2021; Cleland et al., 2011; Shen et al., 2010; Bakkehaug et al., 2015).
Mice were divided into these four groups and a vehicle control group (DMSO) for all subsequent experiments. The volume of each dose was standardized to 0.15 ml/kg, which was used to determine the injected volume of the control group. The total bolus injection volume was <5 μl per mouse. Each mouse only received one dose to maintain statistical independence. The cardiac effects of the drugs were measured 1 and 5 min after injection in all experiments, except when measuring the cumulative dose–response.
Experimental compounds
DN (CAT# S9948, Lot 01) and OM (CAT# S2623, Lot 01) were both purchased from Selleck Chemicals as fine powders. An 80 mg/ml stock solution of each compound was prepared in DMSO and aliquoted for storage at −80°C. Dilutions in DMSO were made on the day of the experiment from these stocks. All drugs were delivered to experimental animals as solutions in 100% DMSO.
External jugular vein catheterization and drug delivery
All drugs were injected intravenously via the right external jugular vein. The vein was exposed by blunt dissection and catheterized using a 30G needle attached to 4 cm of PE-10 tubing. The needle was secured in place with surgical glue. The infusion rate and injected volume of the drug solutions were controlled by an infusion pump (Legato 100; KD Scientific) and a 50-μl glass syringe (1705TLL; Hamilton). Drug solutions delivered as bolus injections were infused over 30 s.
PV catheter placement
LV invasive hemodynamics were assessed by conductance-based PV loop analysis using the open-chest method. Anesthesia was induced with 3% (vol/vol) isoflurane in room air and an intraperitoneal injection of 10 mg/kg etomidate. Once a surgical plane of anesthesia was achieved, mice were quickly intubated with a 20G endotracheal tube using a fiber-optic illuminator and intubation stand (Kent Scientific). After confirming proper intubation, mice were transferred to a heated surgical platform, connected to a SomnoSuite unit (Kent Scientific) with the ventilation module enabled, and secured in the supine position. Ventilation parameters were calculated by the SomnoSuite based on body weight. A surgical plane of anesthesia was maintained with 2% isoflurane in room air. Body temperature was maintained at 37–38°C using a rectal temperature probe and the SomnoSuite’s homeostatic control mode in the RightTemp module. Hair was removed from the thoracic and neck regions by shaving followed by application of depilatory cream.
After external jugular catheterization, the apex of the heart was visualized by thoracotomy. First, the skin was cut longitudinally from the xiphoid process to the clavicles, and then laterally from the xiphoid process toward either flank to expose the chest wall. A small subxiphoid incision was made in the abdominal wall to expose the diaphragm. High-temperature cautery and bone scissors were then used to burn through the chest wall and cut the ribs in a longitudinal line just to the left of the sternum up to the second rib. This resulted in minimal blood loss. During the thoracotomy, a small cotton-tipped applicator soaked in 0.9% saline solution was advanced within the thoracic cavity along the path of cauterization to protect the lungs and heart from thermal injury.
Chest retractors were then used to expose the heart. For experiments involving preload variation, a loop of 4–0 silk suture was placed around the inferior vena cava to perform occlusions. The ends of the suture loop were threaded through a 2.5-cm segment of soft silicone tubing. The pericardium was gently removed by blunt dissection. A stab wound was made in the apex of the heart using a 27G needle, and a 1F miniature PV catheter (PVR-1045; Millar, Inc.) was quickly inserted into the LV. Prior to insertion, the catheter was equilibrated in 37°C 0.9% saline, and the pressure reading was zeroed. PV loops were recorded and visualized using LabChart 8 Pro (ADInstruments) to optimize catheter placement. After catheter placement, isoflurane was reduced to 1.5%, and cardiac function was allowed to stabilize for 10 min. During this time, a total of 100 μl of a 10% bovine serum albumin solution in sterile 0.9% saline was delivered via the external jugular vein as an initial 50 μl bolus, followed by continuous infusion at 5 μl/min.
Cumulative dose response measured by PV loops
Either DN (6 mg/kg/min at 75 μl/kg/min), OM (1 mg/kg/min at 25 μl/kg/min), or DMSO (at 75 μl/kg/min) was continuously infused IV until a lethal dose was achieved or for up to 5 min. PV loops were continuously recorded during this time. Per-mouse volume calibration was not feasible, as the mice do not survive the procedure. Therefore, average calibration parameters from other PV loop experiments in this study were used to calibrate the volume signal.
PV loop data were analyzed using LabChart 8 Pro (ADInstruments). Different phases of the dose–response were defined based on the behavior of the LV pressure waveform and its first time derivative. The transition dose between phases was determined by visual inspection for each mouse.
Bolus responses measured by PV loops
After stabilization, baseline invasive hemodynamics were recorded for 4–5 s, while the ventilator was temporarily turned off, followed by loops recorded during a transient inferior vena cava occlusion. After restarting the ventilator, cardiac function was allowed to stabilize for 5 min before repeating this procedure following infusion of either DN Low, DN High, OM Low, OM High, or DMSO as a bolus injection. PV loops were analyzed 1 min after completion of the infusion. An additional set of PV loops was analyzed 5 min after the infusion.
After the procedure, 10 μl of 15% saline solution was injected IV to measure the parallel conductance of the LV. Heparinized blood was used for cuvette volume calibration (910-1049; Millar, Inc.). Mice were euthanized by exsanguination at the end of the experiment. PV loop analysis was performed using the PV loop and blood pressure modules in LabChart 8 Pro (ADInstruments).
Echocardiography and strain mechanics
LV strain mechanics were assessed by echocardiography using a Vevo 3100 ultrasound imaging system (VisualSonics) as previously described (Holmes et al., 2023). Anesthesia was first induced in mice with 3% isoflurane in room air. The right external jugular vein was then catheterized as described above. Mice were then transferred and secured in the supine position to a heated platform with integrated ECG electrodes. Body temperature was maintained at 37°C. The parasternal long axis (PSLAX) view of the heart was obtained using an MX400 transducer (30 MHz central frequency; VisualSonics). The isoflurane concentration was reduced to 1.5%, and cardiac function was allowed to stabilize for 10 min. B-mode images of the PSLAX view at a frame rate of 230–240 frames per second were obtained at baseline and 1 min after infusion of either DN Low, DN High, OM Low, OM High, or DMSO as described above. An additional set of images was collected 5 min after infusion. Mice were euthanized by cervical dislocation after data collection and were not used in other experiments.
Echocardiographic PSLAX images were analyzed for LV strain mechanics in the VevoStrain module for VevoLab (v5.7.1; VisualSonics). The software semi-automatically defined 50 points along the endocardium and epicardium and tracked their movement over three consecutive cardiac cycles. The strain and strain rate waveforms for points within the anterior and posterior mid-wall regions of the LV were averaged together and analyzed for peak (Pk.) and time-to-peak (T2P) systolic and diastolic values. T2P values were expressed as a percentage of cardiac cycle (%CC) completion starting at the ECG R-wave.
Sarcomere phosphorylation analysis
Mice were anesthetized using 3% isoflurane in room air, then transferred to a heated surgical platform. Body temperature was maintained at 37°C. The right external jugular vein was catheterized as described above. Cardiac function was allowed to stabilize for 10 min, and the isoflurane concentration was reduced to 1.5%. The appropriate drug and dose were infused as a bolus injection into the external jugular vein, and the heart was quickly excised 1 min after infusion. Hearts were flash-frozen in liquid nitrogen and stored at −80°C until use.
Myofibril-enriched samples were made as previously described (Li et al., 2018). 20 mg of mid-wall LV tissue was thawed in 3 ml of cold relaxing solution (100 mM KCl, 20 mM imidazole, 7 mM MgCl2, 2 mM EGTA, and 4 mM MgATP, pH 7.0) and homogenized for 60 s using a polytron. The homogenates were then chemically skinned in relaxing solution containing 1% Triton X-100 (Sigma-Aldrich) for 1 h. The samples were pelleted by centrifugation at 10,000 × g for 5 min. The supernatant was discarded, and the pellets were resuspended in fresh relaxing solution containing protease inhibitors (cOmplete Ultra, 1 tablet/10 ml; Roche) and phosphatase inhibitor (PhosSTOP, 1 tablet/10 ml; Roche). The protein concentration of the samples was determined using Bradford Plus Protein Assay Kit (A55866; Thermo Fisher Scientific) according to the manufacturer’s instructions. Final samples were adjusted to a protein concentration of 1 mg/ml in relaxing solution with protease and phosphatase inhibitors and Laemmli buffer containing fresh β-mercaptoethanol.
The phosphorylation of cardiac myosin-binding protein C (cMyBPC), cardiac troponin I (cTnI), and the regulatory light chain (RLC) of myosin following treatment with DN Low, DN High, OM Low, OM High, or DMSO was assessed by ProQ Diamond staining (Thermo Fisher Scientific), followed by total protein quantification by Coomassie staining, as previously described (Li et al., 2018). 6 μg of myofibrillar protein was separated using gel electrophoresis on a 4–20% acrylamide gradient gel (Bio-Rad) at 180 V for 40 min on ice in standard Tris-glycine running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3). After electrophoresis, gels were washed in Milli-Q (MQ) water for 10 min, then fixed for 2 × 30 min in 50% methanol, 10% acetic acid, and 40% MQ water. After 3 × 10 min MQ water washes, the gels were stained for 30 min with 20 ml ProQ Diamond stain diluted in 40 ml MQ water. The gels were destained for a total of 2.75 h according to the manufacturer’s instructions using 20% acetonitrile, 5% sodium acetate, and 75% MQ water. After 2 × 5 min MQ water washes, gels were imaged on a Typhoon TRIO+ gel imager (GE Healthcare) with 50 µm pixel size, 532-nm excitation wavelength, 580-nm emission filter, normal sensitivity, and photomultiplier tube voltage of 600 V. After fluorescence imaging, gels were stained with colloidal Coomassie blue overnight, then destained in MQ water for 24 h. Coomassie-stained gels were imaged on an Azure c600 imager (Azure Biosystems). Densitometric analysis was performed using ImageJ (NIH). The level of phosphorylated cMyBPC, cTnI, and RLC was expressed as the ratio of the ProQ signal to the Coomassie signal.
Statistical analysis
The normality of the data was assessed using the Shapiro–Wilk test (Table S1). Statistical differences between groups were evaluated using t tests or ANOVA followed by Tukey’s HSD for multiple comparisons, where appropriate, as implemented in the R Core Team (2022). A type I error rate of <0.05 was considered statistically significant for all tests. All values are reported as the mean ± SEM. All error bars on graphs represent 95% confidence intervals.
Online supplemental material
Fig. S1 shows additional data related to Fig. 1. Fig. S2 presents representative changes in contractility relationships 1 min after DMSO injection. Table S1 reports the P values from Shapiro–Wilk tests for normality for the data presented in Tables 1 and 2. Table S2 lists the baseline values used to calculate the values reported in Tables 1 and 2. Table S3 reports the absolute changes in PV and strain parameters 5 min after injection. Table S4 presents an analysis of the values in Tables 1 and 2 with sex included as an independent variable.
Results
LV dose responses
We first performed a dosing study to capture the full range of DN and OM’s effects on LV function. To do so, we continuously measured LV PV loops while infusing DN (6 mg/kg/min), OM (1 mg/kg/min), or DMSO (75 μl/kg/min) intravenously, and correlated the results with the total infused dose. Control infusions with DMSO did not elicit any significant changes in LV function (Fig. S1 A).

Vehicle control and other DN continuous infusion experiments. (A and B) LV pressure response to a continuous infusion of either (A) vehicle control (DMSO) at 75 μl/kg/min or (B) DN at 3 mg/kg/min parameterized by the maximum LV pressure (Pmax), minimum LV pressure (Pmin), the maximum rate of LV pressure change (dP/dtmax), and minimum rate of LV pressure change (dP/dtmin). The x axis represents the cumulative infused dose. The two observed phases of the LV pressure response (P1 and P2) are marked with dashed lines.
Vehicle control and other DN continuous infusion experiments. (A and B) LV pressure response to a continuous infusion of either (A) vehicle control (DMSO) at 75 μl/kg/min or (B) DN at 3 mg/kg/min parameterized by the maximum LV pressure (Pmax), minimum LV pressure (Pmin), the maximum rate of LV pressure change (dP/dtmax), and minimum rate of LV pressure change (dP/dtmin). The x axis represents the cumulative infused dose. The two observed phases of the LV pressure response (P1 and P2) are marked with dashed lines.
We found that both DN and OM elicited a similar three-phase (P1, P2, P3) dose response, best characterized by their effects on LV pressure and the rate of LV pressure change (Fig. 1, A and B). We interpreted these as representing therapeutic (P1), subtoxic (P2), and toxic (P3) phases of the dose response. P1 was marked by large increases in the maximum rate of LV pressure change (dP/dtmax) with increasing dose, while the maximum LV pressure (Pmax) did not change significantly. The minimum LV pressure (Pmin) and minimum rate of pressure change (dP/dtmin) slightly decreased with DN and remained unchanged with OM. In P2, Pmax began to decline with increasing dose, and dP/dtmax either rose more slowly or began to decrease slightly. The primary feature of P2, however, was a progressive increase in Pmin and dP/dtmin. P3 was characterized by a precipitous drop in Pmax and dP/dtmax, while Pmin and dP/dtmin continued to rise until all parameters reached steady-state values. The transition doses from P1 to P2 (DN: 2.39 ± 0.12 mg/kg; OM: 1.24 ± 0.13 mg/kg) and from P2 to P3 (DN: 7.1 ± 1.1 mg/kg; OM: 2.2 ± 2.0 mg/kg) were consistently higher for DN than for OM (Fig. 1 C).

Tri-phasic LV-pressure dose response to DN and OM. (A and B) LV pressure response to a continuous infusion of either (A) DN at 6 mg/kg/min or (B) OM at 1 mg/kg/min parameterized by the maximum LV pressure (Pmax), minimum LV pressure (Pmin), the maximum rate of LV pressure change (dP/dtmax), and minimum rate of LV pressure change (dP/dtmin). The x axis represents the cumulative infused dose. The three different phases of the LV pressure response (P1, P2, and P3) are marked with dashed lines. (C) Doses at which the pressure response transitions from phase 1 to phase 2 (P1 → P2) and phase 2 to phase 3 (P2 → P3) for DN and OM. N = 4 mice/group.
Tri-phasic LV-pressure dose response to DN and OM. (A and B) LV pressure response to a continuous infusion of either (A) DN at 6 mg/kg/min or (B) OM at 1 mg/kg/min parameterized by the maximum LV pressure (Pmax), minimum LV pressure (Pmin), the maximum rate of LV pressure change (dP/dtmax), and minimum rate of LV pressure change (dP/dtmin). The x axis represents the cumulative infused dose. The three different phases of the LV pressure response (P1, P2, and P3) are marked with dashed lines. (C) Doses at which the pressure response transitions from phase 1 to phase 2 (P1 → P2) and phase 2 to phase 3 (P2 → P3) for DN and OM. N = 4 mice/group.
Fig. 2 shows representative pressure waveforms and PV loops during each phase. The pressure waveforms reflect the trends observed in Pmax, Pmin, dP/dtmax, and dP/dtmin. PV loops initially widened during P1, then progressively contracted toward the lower left corner as developed pressure and volumes decreased during P2 and P3. Further insights from the volume signal obtained during this protocol were limited due to frequent breathing artifacts. These prevented accurate parameterization. Additionally, the elevated contractile state of the heart at higher drug doses may have shifted the position of the conductance electrodes within the LV. Due to these limitations, the volume data shown in Fig. 2 are only suitable for qualitative analysis.

LV-pressure waveforms and PV-loops during tri-phasic dose response to DN and OM. (A and B) Representative LV pressure waveforms (top panels) and PV loops (bottom panels) at baseline (Pre) and at each phase (P1, P2, and P3) of the response to continual infusion of either (A) DN at 6 mg/kg/min or (B) OM at 1 mg/kg/min. The three phases represent the therapeutic, subtoxic, and toxic phases, respectively.
LV-pressure waveforms and PV-loops during tri-phasic dose response to DN and OM. (A and B) Representative LV pressure waveforms (top panels) and PV loops (bottom panels) at baseline (Pre) and at each phase (P1, P2, and P3) of the response to continual infusion of either (A) DN at 6 mg/kg/min or (B) OM at 1 mg/kg/min. The three phases represent the therapeutic, subtoxic, and toxic phases, respectively.
These experiments highlight the complex manner in which the LV responds to different doses of myotropic sarcomere activators. Notably, the exact location of the transition doses depends on both the infusion rate and the pharmacokinetics of the drugs. For example, when DN was infused at half the rate, only P1 and P2 were observed (Fig. S1 B), despite delivering twice the total dose used in Figs. 1 and 2. As we discuss below, this is likely due to DN’s high rate of systemic clearance and low half-life in mice (Grillo et al., 2021). Therefore, we highly caution against using these curves as a direct map between injected dose and dose–response phase. Instead, the most important observation from these experiments is that DN and OM elicited qualitatively similar dose responses from the LV.
SV-matched doses
The differences in DN and OM’s transition doses and their complex dose–response profiles make rigorous quantitative comparisons between the two challenging. We therefore selected two doses for each drug (DN Low: 3 mg/kg; DN High: 6 mg/kg; OM Low: 0.75 mg/kg; OM High: 1.5 mg/kg) that produced similar increases in SV 1 min after infusion (Fig. 3). We chose to standardize drug effects to SV, as this is the primary parameter that needs to be restored in HFrEF patients. At the low doses, DN and OM increased SV by 4.39 ± 0.35 and 4.1 ± 0.4 μl, respectively (Fig. 3 B; P = 0.99). At the high doses, DN and OM increased SV by 7.7 ± 0.8 and 7.6 ± 0.9 μl, respectively (Fig. 3 B; P = 0.99). These changes were accompanied by similar dose-dependent decreases in end-diastolic volume (EDV, Fig. 3 C) and end-systolic volume (ESV, Table 1) across dose brackets. These effect-matched doses are critical for enabling direct comparisons of how DN and OM alter the cardiac cycle to achieve the same increase in SV.

SV matched DN and OM doses. Similar effects of DN and OM on LV SV 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A) Representative PV loops at baseline (Pre) and after (Post) drug infusion. Arrows indicate the locations for measuring SV and EDV. (B and C) Quantification of the absolute change (∆) in (B) SV and (C) EDV. †P < 0.05 compared with the DMSO control group, **P < 0.01, ***P < 0.001, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (B) DN Low–DN High, P = 0.0044; OM Low–OM High, P = 0.0029; DN Low–OM Low, P = 1.0; DN High–OM High, P = 1.0; DMSO–DN Low, p 0.0048; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.012; DMSO–OM High, P < 1e-7. (C) DN Low–DN High, P = 0.00072; OM Low–OM High, P = 0.0058; DN Low–OM Low, P = 0.95; DN High–OM High, P = 1.0; DMSO–DN Low, P = 0.086; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.016; DMSO–OM High, P < 1e-7. Group sizes are as follows: (B) DMSO, N = 9; DN Low, N = 10; DN High, N = 10; OM Low, N = 9, OM High, N = 10. (C) DMSO, N = 9; DN Low, N = 10; DN High, N = 9; OM Low, N = 9, OM High, N = 8.
SV matched DN and OM doses. Similar effects of DN and OM on LV SV 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A) Representative PV loops at baseline (Pre) and after (Post) drug infusion. Arrows indicate the locations for measuring SV and EDV. (B and C) Quantification of the absolute change (∆) in (B) SV and (C) EDV. †P < 0.05 compared with the DMSO control group, **P < 0.01, ***P < 0.001, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (B) DN Low–DN High, P = 0.0044; OM Low–OM High, P = 0.0029; DN Low–OM Low, P = 1.0; DN High–OM High, P = 1.0; DMSO–DN Low, p 0.0048; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.012; DMSO–OM High, P < 1e-7. (C) DN Low–DN High, P = 0.00072; OM Low–OM High, P = 0.0058; DN Low–OM Low, P = 0.95; DN High–OM High, P = 1.0; DMSO–DN Low, P = 0.086; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.016; DMSO–OM High, P < 1e-7. Group sizes are as follows: (B) DMSO, N = 9; DN Low, N = 10; DN High, N = 10; OM Low, N = 9, OM High, N = 10. (C) DMSO, N = 9; DN Low, N = 10; DN High, N = 9; OM Low, N = 9, OM High, N = 8.
PV loop parameters
| DMSO | DN Low | DN High | OM Low | OM High | |
|---|---|---|---|---|---|
| Δ HR (bpm) | 7 ± 5 | 4 ± 5 | −6 ± 6 | 1.1 ± 2.6 | −10.9 ± 3.4 |
| Δ ESV (µl) | −1.4 ± 0.6 | −6.6 ± 0.5a | −12.6 ± 1.2a,b | −7.2 ± 0.5a | −11.0 ± 1.1a,b |
| Δ EDV (µl) | −0.08 ± 0.23 | −1.86 ± 0.28 | −4.8 ± 0.7a,b | −2.3 ± 0.4a | −4.9 ± 0.7a,b |
| Δ SV (µl) | 1.0 ± 0.4 | 4.39 ± 0.35a | 7.7 ± 0.8a,b | 4.1 ± 0.4a | 7.6 ± 0.9a,b |
| Δ EF (%) | 3.5 ± 1.5 | 17.2 ± 2.1a | 36.2 ± 2.9a,b | 16.2 ± 1.0a | 30.7 ± 3.3a,b |
| Δ SW (mmHg * μl) | 146 ± 31 | 440 ± 50a | 780 ± 90a,b | 400 ± 50 | 720 ± 80a,b |
| Δ CO (µl/min) | 550 ± 210 | 1,950 ± 270a | 4,000 ± 500a,b | 2,010 ± 190a | 3,500 ± 400a,b |
| Δ PFR (µl/s) | 15 ± 13 | 157 ± 25 | 390 ± 80a,b | 131 ± 28 | 300 ± 50a |
| Δ PER (µl/s) | 38 ± 10 | 126 ± 14 | 280 ± 50a,b | 63 ± 15 | 83 ± 31c |
| Δ Pmax (mmHg) | 5.5 ± 1.4 | 5.2 ± 1.2 | 4.0 ± 2.1 | 1.0 ± 1.6 | 4.5 ± 1.6 |
| Δ Pmin (mmHg) | −0.15 ± 0.29 | 0.21 ± 0.27 | 0.15 ± 0.26 | −0.12 ± 0.19 | 1.08 ± 0.26a,b |
| Δ ESP (mmHg) | 5.6 ± 1.4 | 2.8 ± 1.9 | −1.4 ± 2.7 | −1.5 ± 2.4 | 2.0 ± 1.9 |
| Δ EDP (mmHg) | −0.19 ± 0.33 | −0.2 ± 0.4 | −0.44 ± 0.24 | −0.41 ± 0.27 | −0.09 ± 0.26 |
| Δ dP/dtmax (mmHg/s) | 520 ± 170 | 1,780 ± 90a | 3,400 ± 500a,b | 1,830 ± 160a | 3,120 ± 320a,b |
| Δ dP/dtmin (mmHg/s) | −1,080 ± 290 | −1,280 ± 200 | −200 ± 400 | −540 ± 270 | 30 ± 230 |
| Δ Tau (ms) | −0.39 ± 0.21 | 0.10 ± 0.08 | 1.48 ± 0.12a,b | 0.43 ± 0.10a | 2.72 ± 0.25a,b,c |
| Δ Systolic duration (ms) | −1.0 ± 0.7 | 1.0 ± 0.5 | 6.5 ± 1.3a,b | 3.4 ± 0.5a | 11.1 ± 1.0a,b,c |
| Δ Systolic duration (% CC) | −0.09 ± 0.27 | 1.06 ± 0.22 | 4.1 ± 0.7a,b | 2.8 ± 0.4a,c | 7.3 ± 0.4a,b,c |
| Δ Diastolic duration (ms) | −1.1 ± 1.0 | −1.8 ± 0.9 | −4.0 ± 1.1 | −3.5 ± 0.6 | −8.0 ± 0.8a,b,c |
| Δ Diastolic duration (% CC) | 0.09 ± 0.27 | −1.06 ± 0.22 | −4.1 ± 0.7a,b | −2.8 ± 0.4a,c | −7.3 ± 0.4a,b,c |
| Δ S/D | −0.003 ± 0.009 | 0.035 ± 0.007 | 0.144 ± 0.025a,b | 0.099 ± 0.015a,c | 0.264 ± 0.015a,b,c |
| Δ ESPVR slope (mmHg/μl) | 0.34 ± 0.14 | −1.4 ± 0.5a | −1.40 ± 0.28a | −2.25 ± 0.32a | −1.7 ± 0.6a |
| Δ ESPVR intercept (mmHg) | 5.7 ± 2.2 | 49 ± 9a | 66 ± 10a | 60 ± 10a | 58 ± 17a |
| Δ dP/dtmax-EDV slope (mmHg/s/μl) | 0 ± 7 | −9 ± 10 | −83 ± 17a,b | −6 ± 12 | −83 ± 29a,b |
| Δ dP/dtmax-EDV intercept (mmHg/s) | 590 ± 250 | 1,900 ± 500 | 6,300 ± 400a,b | 2,930 ± 340a | 5,400 ± 700a,b |
| Δ EDPVR (1/μl) | 0.027 ± 0.022 | 0.019 ± 0.020 | −0.036 ± 0.015 | −0.030 ± 0.014 | −0.041 ± 0.018 |
Absolute change (∆) in LV PV loop parameters 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). N = 7–11 mice/group.
Significance is defined as P < 0.05 for all comparisons as determined by ANOVA and Tukey’s HSD. HR, heart rate; ESV, end-systolic volume; EDV, end-diastolic volume; SV, stroke volume; EF, ejection fraction; SW, stroke work; CO, cardiac output; PFR, peak filling rate; PER, peak ejection rate; Pmax, maximum LV pressure; Pmin, minimum LV pressure; ESP, end-systolic pressure; EDP, end-diastolic pressure; dP/dtmax, maximum rate of LV pressure change; dP/dtmin, minimum rate of LV pressure change; Tau, the time constant of LV pressure relaxation; % CC, percent cardiac cycle; S/D, the ratio of systolic to diastolic duration; ESPVR, end-systolic pressure–volume relationship; EDPVR, exponent of the end-diastolic pressure–volume relationship.
Significant difference when compared to the DMSO group.
Significant difference when compared to the low dose of the same drug.
Significant difference when compared to the corresponding dose of DN.
The low doses fall within the therapeutic phase, while the high doses lie near the boundary between the therapeutic and subtoxic regimes. Notably, the DN doses had to be significantly higher than the P1 to P2 transition dose identified in Fig. 1 C due to DN’s fast systemic clearance and short half-life in mice (Grillo et al., 2021).
PV loop analysis
In-depth PV loop analysis was used to assess the acute effects of DN and OM on pressure generation, ejection, pressure relaxation, and load-independent measures of contractility 1 min after drug infusion (Fig. 4 and Table 1). All baseline PV parameters were similar between groups, aside from slight differences in heart rate (P = 0.04), which remained within 8% across groups (Table S2). Table 1 reports the absolute change (Δ) in all PV parameters from baseline 1 min after drug infusion for each group, along with statistical significance values.

Bolus responses to DN and OM measured by steady-state LV–PV loops. Analysis of LV pressure and volume signals 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A and B) Representative LV pressure and (B) volume waveforms at baseline (Pre) and after (Post) drug infusion. Panels are annotated with where the systolic duration, time constant of pressure relaxation (Tau), maximum rate of pressure development (dP/dtmax), and PER measurements are taken. (D–F) Quantifications of the absolute change (∆) in (C) dP/dtmax, (D) systolic duration, (E) PER, and (F) Tau. †P < 0.05 compared with the DMSO control group, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (C) DN Low–DN High, P = 0.0032; OM Low–OM High, P = 0.049; DN Low–OM Low, P = 1.0; DN High–OM High, P = 0.95; DMSO–DN Low, P = 0.049; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.046; DMSO–OM High, P = 1.2e-5. (D) DN Low–DN High, P = 0.0001; OM Low–OM High, p < 1e-7; DN Low–OM Low, P = 0.043; DN High–OM High, P = 2.5e-5; DMSO–DN Low, P = 0.35; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.00024; DMSO–OM High, P < 1e-7. (E) DN Low–DN High, P = 0.0034; OM Low–OM High, P = 0.99; DN Low–OM Low, P = 0.51; DN High–OM High, P = 0.00015; DMSO–DN Low, P = 0.23; DMSO–DN High, P = 1e-5; DMSO–OM Low, P = 0.98; DMSO–OM High, P = 0.81. (F) DN Low–DN High, P = 2.6e-5; OM Low–OM High, P < 1e-7; DN Low–OM Low, P = 0.65; DN High–OM High, P = 9.6e-6; DMSO–DN Low, P = 0.29; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.012; DMSO–OM High, P < 1e-7. Group sizes are as follows: (C) DMSO, N = 8; DN Low, N = 10; DN High, N = 9; OM Low, N = 8, OM High, N = 8. (D) DMSO, N = 9; DN Low, N = 11; DN High, N = 10; OM Low, N = 10, OM High, N = 10. (E) DMSO, N = 7; DN Low, N = 10; DN High, N = 8; OM Low, N = 10, OM High, N = 10. (F) DMSO, N = 9; DN Low, N = 10; DN High, N = 8; OM Low, N = 10, OM High, N = 10.
Bolus responses to DN and OM measured by steady-state LV–PV loops. Analysis of LV pressure and volume signals 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A and B) Representative LV pressure and (B) volume waveforms at baseline (Pre) and after (Post) drug infusion. Panels are annotated with where the systolic duration, time constant of pressure relaxation (Tau), maximum rate of pressure development (dP/dtmax), and PER measurements are taken. (D–F) Quantifications of the absolute change (∆) in (C) dP/dtmax, (D) systolic duration, (E) PER, and (F) Tau. †P < 0.05 compared with the DMSO control group, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (C) DN Low–DN High, P = 0.0032; OM Low–OM High, P = 0.049; DN Low–OM Low, P = 1.0; DN High–OM High, P = 0.95; DMSO–DN Low, P = 0.049; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.046; DMSO–OM High, P = 1.2e-5. (D) DN Low–DN High, P = 0.0001; OM Low–OM High, p < 1e-7; DN Low–OM Low, P = 0.043; DN High–OM High, P = 2.5e-5; DMSO–DN Low, P = 0.35; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.00024; DMSO–OM High, P < 1e-7. (E) DN Low–DN High, P = 0.0034; OM Low–OM High, P = 0.99; DN Low–OM Low, P = 0.51; DN High–OM High, P = 0.00015; DMSO–DN Low, P = 0.23; DMSO–DN High, P = 1e-5; DMSO–OM Low, P = 0.98; DMSO–OM High, P = 0.81. (F) DN Low–DN High, P = 2.6e-5; OM Low–OM High, P < 1e-7; DN Low–OM Low, P = 0.65; DN High–OM High, P = 9.6e-6; DMSO–DN Low, P = 0.29; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.012; DMSO–OM High, P < 1e-7. Group sizes are as follows: (C) DMSO, N = 8; DN Low, N = 10; DN High, N = 9; OM Low, N = 8, OM High, N = 8. (D) DMSO, N = 9; DN Low, N = 11; DN High, N = 10; OM Low, N = 10, OM High, N = 10. (E) DMSO, N = 7; DN Low, N = 10; DN High, N = 8; OM Low, N = 10, OM High, N = 10. (F) DMSO, N = 9; DN Low, N = 10; DN High, N = 8; OM Low, N = 10, OM High, N = 10.
Neither drug significantly impacted heart rate (Table 1). Both DN and OM significantly increased dP/dtmax in a dose-dependent manner (∆ dP/dtmax: DMSO, 520 ± 170 mmHg/s; DN Low, 1,780 ± 90 mmHg/s; DN High, 3,400 ± 500 mmHg/s; OM Low, 1,830 ± 160 mmHg/s; OM High, 3,120 ± 320 mmHg/s; Fig. 4 C). However, this was accompanied by only a slight increase in Pmax, which was not significantly different from the DMSO control group (Table 1). End-systolic pressure (ESP) was also unchanged by either drug compared with DMSO (Table 1).
Both drugs significantly increased systolic duration, expressed as a %CC, in a dose-dependent fashion (Δ systolic duration: DMSO, −0.09 ± 0.27 %CC; DN Low, 1.06 ± 0.22 %CC; DN High, 4.1 ± 0.7 %CC; OM Low, 2.8 ± 0.4 %CC; OM High, 7.3 ± 0.4 %CC; Fig. 4 D). However, DN consistently increased systolic duration less than OM, and the increase with DN Low was not significantly different from DMSO. Consequently, DN also decreased diastolic duration less than OM (Table 1).
Normalization to cardiac cycle duration was appropriate since heart rate did not significantly change following drug infusion (Table 1). Furthermore, these results remained consistent when systolic and diastolic durations were expressed in absolute time (ms), except for the comparison between DN Low and OM Low, which was no longer statistically significant (P = 0.3).
To maintain the same change in SV, DN increased the peak ejection rate (PER) in a dose-dependent manner (Δ PER: DMSO, 38 ± 10 μl/s; DN Low, 126 ± 14 μl/s; DN High, 280 ± 50 μl/s; Fig. 4 E). However, PER was only significantly increased compared with the DMSO group at the DN High dose. In contrast, OM did not significantly increase PER at either dose compared with DMSO (Δ PER: OM Low, 63 ± 15 μl/s; OM High, 83 ± 31 μl/s).
Finally, both drugs affected pressure relaxation. DN High and OM High significantly increased the time constant of pressure relaxation (Tau), with DN producing a smaller effect (Δ Tau: DMSO, −0.39 ± 0.21 ms; DN High, 1.48 ± 0.12 ms; OM High, 2.72 ± 0.25 ms; Fig. 4 F). The changes in Tau with DN Low and OM Low were much smaller and not significantly different from each other (Δ Tau: DN Low, 0.10 ± 0.08 ms; OM Low, 0.43 ± 0.10 ms). However, only OM Low showed a statistically significant increase in Tau compared with DMSO, whereas DN Low did not.
The change in dP/dtmin was not statistically different from the DMSO control for any drug or dose (Table 1). Similarly, the change in Pmin and the end-diastolic pressures were either not statistically or physiologically significantly altered by either drug at the tested doses (Table 1).
The slopes of conventional load-independent contractility indices generally decreased following administration of DN and OM. DMSO did not significantly impact any of the load-independent contractility indices (Fig. S2). Both drugs at both doses reduced the slope of the end-systolic pressure–volume relationship (ESPVR) (Δ ESPVR: DMSO, 0.34 ± 0.14 mmHg/μl; DN Low, −1.4 ± 0.5 mmHg/μl; DN High, −1.40 ± 0.28 mmHg/μl; OM Low, −2.25 ± 0.32 mmHg/μl; OM High, −1.7 ± 0.6 mmHg/μl; Fig. 5, A and B). However, the effect was not dose-dependent within the range tested.

LV contractility response to bolus injection of vehicle control. (A and B) Representative (A) ESP and ESV data and (B) maximum rate of pressure development (dP/dtmax) and EDV data before (Pre) and 1 min after (Post) injection with DMSO at 0.15 ml/kg. Dashed lines are linear fits to the data. ESV, end-systolic volume.
LV contractility response to bolus injection of vehicle control. (A and B) Representative (A) ESP and ESV data and (B) maximum rate of pressure development (dP/dtmax) and EDV data before (Pre) and 1 min after (Post) injection with DMSO at 0.15 ml/kg. Dashed lines are linear fits to the data. ESV, end-systolic volume.

Bolus response to DN and OM measured by contractility indices. Analysis of LV load–independent contractility indices 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A) Representative effects on the relationship between ESP and ESV at baseline (Pre) and after (Post) drug infusion. The dashed line is the linear fit of the data. (B) Quantification of the absolute change (∆) in the slope of the ESPVR. (C) Representative effects on the relationship between the maximum rate of LV pressure change (dP/dtmax) and EDV at baseline (Pre) and after (Post) drug infusion. The dashed line is the linear fit of the data. (D) Quantification of the absolute change in the slope of the dP/dtmax-EDV relationship. †P < 0.05 compared with the DMSO control group, *P < 0.05, **P < 0.01, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (B) DN Low–DN High, P = 1.0; OM Low–OM High, P = 0.88; DN Low–OM Low, P = 0.59; DN High–OM High, P = 0.96; DMSO–DN Low, P = 0.022; DMSO–DN High, P = 0.045; DMSO–OM Low, P = 0.00071; DMSO–OM High, P = 0.013. (D) DN Low–DN High, P = 0.0074; OM Low–OM High, P = 0.028; DN Low–OM Low, P = 1.0; DN High–OM High, P = 0.93; DMSO–DN Low, P = 0.99; DMSO–DN High, P = 0.0019; DMSO–OM Low, P = 1.0; DMSO–OM High, P = 0.011. Group sizes are as follows: (B) DMSO, N = 7; DN Low, N = 10; DN High, N = 9; OM Low, N = 8, OM High, N = 10. (D) DMSO, N = 8; DN Low, N = 9; DN High, N = 7; OM Low, N = 8, OM High, N = 10. ESV, end-systolic volume.
Bolus response to DN and OM measured by contractility indices. Analysis of LV load–independent contractility indices 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A) Representative effects on the relationship between ESP and ESV at baseline (Pre) and after (Post) drug infusion. The dashed line is the linear fit of the data. (B) Quantification of the absolute change (∆) in the slope of the ESPVR. (C) Representative effects on the relationship between the maximum rate of LV pressure change (dP/dtmax) and EDV at baseline (Pre) and after (Post) drug infusion. The dashed line is the linear fit of the data. (D) Quantification of the absolute change in the slope of the dP/dtmax-EDV relationship. †P < 0.05 compared with the DMSO control group, *P < 0.05, **P < 0.01, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (B) DN Low–DN High, P = 1.0; OM Low–OM High, P = 0.88; DN Low–OM Low, P = 0.59; DN High–OM High, P = 0.96; DMSO–DN Low, P = 0.022; DMSO–DN High, P = 0.045; DMSO–OM Low, P = 0.00071; DMSO–OM High, P = 0.013. (D) DN Low–DN High, P = 0.0074; OM Low–OM High, P = 0.028; DN Low–OM Low, P = 1.0; DN High–OM High, P = 0.93; DMSO–DN Low, P = 0.99; DMSO–DN High, P = 0.0019; DMSO–OM Low, P = 1.0; DMSO–OM High, P = 0.011. Group sizes are as follows: (B) DMSO, N = 7; DN Low, N = 10; DN High, N = 9; OM Low, N = 8, OM High, N = 10. (D) DMSO, N = 8; DN Low, N = 9; DN High, N = 7; OM Low, N = 8, OM High, N = 10. ESV, end-systolic volume.
At low doses, neither drug significantly altered the slope of the dP/dt max end-diastolic volume relationship (dP/dt max-EDV) compared with DMSO (Δ dP/dtmax-EDV slope: DMSO, 6 ± 5 mmHg/s/μl; DN Low, −9 ± 10 mmHg/s/μl; OM Low, −6 ± 12 mmHg/s/μl; Fig. 5, C and D). In contrast, both high doses caused significant reductions in the dP/dtmax-EDV slope (DN High, −83 ± 17 mmHg/s/μl; OM High, −83 ± 29 mmHg/s/μl).
Despite the reductions in slope, the overall ESPVR and dP/dtmax-EDV relationships were shifted upward relative to baseline, as indicated by increases in their intercept terms (Fig. 5, A and C; and Table 1). This suggests that both drugs enabled the heart to maintain higher ESP and dP/dtmax across a broader range of preloads. The end-diastolic pressure–volume relationship (EDPVR) was unchanged across all treatment groups compared with DMSO (Table 1).
Strain mechanics
Radial strain mechanics of the LV mid-wall were assessed using speckle-tracking echocardiography (Fig. 6 and Table 2). This approach directly measures the relative thickening and thinning of the LV wall during ejection and filling. All baseline values were similar between groups (Table S2). Table 2 reports the absolute change (Δ) in all strain parameters from baseline 1 min after drug infusion for each group, along with corresponding statistical significance values. Doppler-based measures of diastolic function were not collected due to DN’s exceptionally high clearance rate in mice and the time required to reposition the ultrasound transducer for Doppler imaging (Grillo et al., 2021). Moreover, the effects of DN and OM on Doppler-based indices of diastolic function have already been described in human clinical trials (Voors et al., 2020; Cleland et al., 2011). We discuss how our results align with these reported Doppler findings in the Discussion section below.

Bolus response to DN and OM measured by LV strain mechanics. Analysis of LV radial strain and SR mechanics 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A and B) Group average ± 95% confidence interval radial strain and (B) SR waveform at baseline (Pre) and after (Post) drug infusion. Panels are annotated with where Pk. SRS, T2P SRS, Pk. SRSR, and T2P SRSR are measured from. (C–F) Absolute change (∆) in (C) Pk. SRS, (D) T2P SRS, (E) Pk. DRSR, and (F) T2P DRSR. †P < 0.05 compared with the DMSO control group, *P < 0.05, **P <0.01, ***P < 0.001, ****P < 0.0001, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (C) DN Low–DN High, P = 0.0043; OM Low–OM High, P = 0.023; DN Low–OM Low, P = 0.057; DN High–OM High, P = 0.16; DMSO–DN Low, P = 9e-6; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.00062; DMSO–OM High, P < 1e-7. (D) DN Low–DN High, P = 0.0052; OM Low–OM High, P < 1e-7; DN Low–OM Low, P = 1.0; DN High–OM High, P = 0.024; DMSO–DN Low, P = 1.0; DMSO–DN High, P = 0.002; DMSO–OM Low, P = 1.0; DMSO–OM High, P < 1e-7. (E) DN Low–DN High, P = 0.043; OM Low–OM High, P = 0.00029; DN Low–OM Low, P = 0.52; DN High–OM High, P = 1.0; DMSO–DN Low, P = 0.13; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.88; DMSO–OM High, P = 2e-5. (F) DN Low–DN High, P = 0.00063; OM Low–OM High, P < 1e-7; DN Low–OM Low, P = 0.61; DN High–OM High, P = 0.024; DMSO–DN Low, P = 0.086; DMSO–DN High, P = 2e-7; DMSO–OM Low, P = 0.78; DMSO–OM High, P < 1e-7. Group sizes are as follows: (C) DMSO, N = 9; DN Low, N = 8; DN High, N = 8; OM Low, N = 9, OM High, N = 9. (D) DMSO, N = 10; DN Low, N = 9; DN High, N = 9; OM Low, N = 9, OM High, N = 9. (E) DMSO, N = 9; DN Low, N = 8; DN High, N = 9; OM Low, N = 10, OM High, N = 10. (F) DMSO, N = 9; DN Low, N = 10; DN High, N = 10; OM Low, N = 9, OM High, N = 10. Pk. SRSR, peak systolic radial strain rate; SR, strain rate; T2P SRS, time-to-peak radial systolic strain; T2P SRSR, time-to-peak diastolic radial strain rate.
Bolus response to DN and OM measured by LV strain mechanics. Analysis of LV radial strain and SR mechanics 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A and B) Group average ± 95% confidence interval radial strain and (B) SR waveform at baseline (Pre) and after (Post) drug infusion. Panels are annotated with where Pk. SRS, T2P SRS, Pk. SRSR, and T2P SRSR are measured from. (C–F) Absolute change (∆) in (C) Pk. SRS, (D) T2P SRS, (E) Pk. DRSR, and (F) T2P DRSR. †P < 0.05 compared with the DMSO control group, *P < 0.05, **P <0.01, ***P < 0.001, ****P < 0.0001, ns P > 0.05 as determined by ANOVA and Tukey’s HSD. Exact P values are as follows: (C) DN Low–DN High, P = 0.0043; OM Low–OM High, P = 0.023; DN Low–OM Low, P = 0.057; DN High–OM High, P = 0.16; DMSO–DN Low, P = 9e-6; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.00062; DMSO–OM High, P < 1e-7. (D) DN Low–DN High, P = 0.0052; OM Low–OM High, P < 1e-7; DN Low–OM Low, P = 1.0; DN High–OM High, P = 0.024; DMSO–DN Low, P = 1.0; DMSO–DN High, P = 0.002; DMSO–OM Low, P = 1.0; DMSO–OM High, P < 1e-7. (E) DN Low–DN High, P = 0.043; OM Low–OM High, P = 0.00029; DN Low–OM Low, P = 0.52; DN High–OM High, P = 1.0; DMSO–DN Low, P = 0.13; DMSO–DN High, P < 1e-7; DMSO–OM Low, P = 0.88; DMSO–OM High, P = 2e-5. (F) DN Low–DN High, P = 0.00063; OM Low–OM High, P < 1e-7; DN Low–OM Low, P = 0.61; DN High–OM High, P = 0.024; DMSO–DN Low, P = 0.086; DMSO–DN High, P = 2e-7; DMSO–OM Low, P = 0.78; DMSO–OM High, P < 1e-7. Group sizes are as follows: (C) DMSO, N = 9; DN Low, N = 8; DN High, N = 8; OM Low, N = 9, OM High, N = 9. (D) DMSO, N = 10; DN Low, N = 9; DN High, N = 9; OM Low, N = 9, OM High, N = 9. (E) DMSO, N = 9; DN Low, N = 8; DN High, N = 9; OM Low, N = 10, OM High, N = 10. (F) DMSO, N = 9; DN Low, N = 10; DN High, N = 10; OM Low, N = 9, OM High, N = 10. Pk. SRSR, peak systolic radial strain rate; SR, strain rate; T2P SRS, time-to-peak radial systolic strain; T2P SRSR, time-to-peak diastolic radial strain rate.
Strain mechanics parameters
| DMSO | DN Low | DN High | OM Low | OM High | |
|---|---|---|---|---|---|
| Δ HR (bpm) | 1.6 ± 3.3 | −6 ± 5 | −18 ± 9 | −5 ± 7 | −11 ± 5 |
| Δ Pk. SRS (%) | −3.2 ± 1.5 | 9.7 ± 1.4a | 18.4 ± 1.7a,b | 6.4 ± 1.9a | 13.2 ± 1.2a,b |
| Δ T2P SRS (% CC) | 0.8 ± 1.1 | 1.2 ± 1.1 | 6.9 ± 1.1a,b | 0.6 ± 0.8 | 11.6 ± 1.2a,b,c |
| Δ Pk. SRSR (1/s) | 0.3 ± 0.5 | 0.73 ± 0.25 | −0.5 ± 0.6 | 0.25 ± 0.30 | −1.57 ± 0.32a,b |
| Δ T2P SRSR (% CC) | −0.9 ± 1.5 | −1.4 ± 1.5 | 1.1 ± 1.1 | −2.8 ± 1.7 | −0.8 ± 1.7 |
| Δ Pk. DRSR (1/s) | 0.8 ± 0.4 | −1.7 ± 0.6 | −4.6 ± 0.5a,b | −0.1 ± 0.7 | −4.4 ± 0.9a,b |
| Δ T2P DRSR (% CC) | 0.7 ± 1.0 | 5.3 ± 1.1 | 12.8 ± 1.1a,b | 2.7 ± 0.7 | 18.1 ± 1.9a,b,c |
Absolute change (∆) in LV strain parameters 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). N = 8–10 mice/group.
Significance is defined as P < 0.05 for all comparisons as determined by ANOVA and Tukey’s HSD. HR, heart rate; Pk. SRS, peak systolic radial strain; T2P SRS, time-to-peak systolic radial strain; % CC, percent cardiac cycle; Pk. SRSR, peak systolic radial strain rate; T2P SRSR, time-to-peak systolic radial strain rate; Pk. DRSR, peak diastolic radial strain rate; T2P DRSR, time-to-peak diastolic radial strain rate.
Significant difference when compared to the DMSO group.
Significant difference when compared to the low dose of the same drug.
Significant difference when compared to the corresponding dose of DN.
The drugs caused a slightly greater reduction in heart rate during echocardiography compared with the PV loop experiments (Table 2); however, none of these differences were statistically significant relative to the DMSO control group. At the low doses, both drugs similarly increased peak systolic radial strain (Pk. SRS), reflecting enhanced systolic wall thickening (Δ Pk. SRS: DMSO, −3.2 ± 1.5%; DN Low, 9.7 ± 1.4%; OM Low, 6.4 ± 1.9%, Fig. 6, A and C). These changes occurred without significantly affecting T2P SRS (Δ T2P SRS: DMSO, 0.8 ± 1.1 %CC; DN Low, 1.2 ± 1.1 %CC; OM Low, 0.6 ± 0.8 %CC, Fig. 6 D). At the high doses, both drugs further increased Pk. SRS (Δ Pk. SRS: DN High, 18.4 ± 1.7%; OM High, 13.2 ± 1.2%) and significantly delayed T2P SRS (Δ T2P SRS: DN High, 6.9 ± 1.1 %CC; OM High, 11.6 ± 1.2 %CC). This rightward shift in T2P SRS indicates a prolonged systolic duration. Notably, DN High increased T2P SRS to a lesser extent than OM High.
The primary effects on radial strain rate, the time derivative of radial strain, were observed during the diastolic phase of the curve. At low doses, neither drug significantly changed the peak diastolic radial strain rate (Pk. DRSR) or its time to peak (T2P DRSR) compared with DMSO (Δ Pk. DRSR: DMSO, 0.8 ± 0.4 1/s; DN Low, −1.7 ± 0.6 1/s; OM Low, −0.1 ± 0.7 1/s; Δ T2P DRSR: DMSO, 0.7 ± 1.0 %CC; DN Low, 5.3 ± 1.1 %CC; OM Low, 2.7 ± 0.7 %CC, Fig. 6, B, E, and F).
At high doses, both drugs made Pk. DRSR significantly more negative (Δ Pk. DRSR: DN High, −4.6 ± 0.5 1/s; OM High, −4.4 ± 0.9 1/s) and significantly delayed T2P DRSR (Δ T2P DRSR: DN High, 12.8 ± 1.1 %CC; OM High, 18.1 ± 1.9 %CC). As with T2P SRS, DN High increased T2P DRSR to a lesser extent than OM High. However, the delay in the diastolic strain rate peaks at high doses was sufficient to eliminate the first peak typically seen during early diastolic filling.
Sarcomere phosphorylation analysis
The phosphorylation of cardiac sarcomere proteins was quantified using ProQ Diamond staining to determine whether the drugs altered sarcomere phosphorylation status 1 min after administration. We found that acute treatment with DN or OM did not significantly change the phosphorylation levels of cMyBPC, cTnI, or myosin RLC (Fig. 7). These results suggest that the observed functional changes at 1 min are independent of sarcomere phosphorylation and can be primarily attributed to the drugs' mechanical effects.
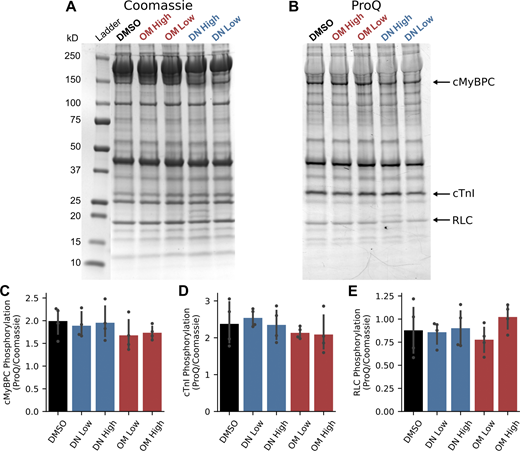
Sarcomere phosphorylation response to bolus injection of DN and OM. Analysis of sarcomere phosphorylation 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A–E) Representative (A) Coomassie blue– and (B) ProQ Diamond–stained gels with annotations indicating the molecular weights of the ladder and the locations of cMyBPC, cTnI, and myosin’s RLC. ProQ Diamond signals normalized to Coomassie blue signal for (C) cMyBPC, (D) cTnI, and (E) RLC. N = 4 hearts/group. No comparison was statistically significant and all P > 0.05 as determined by ANOVA. Exact P values are as follows: (C) P = 0.52; (D) P = 0.54; (E) P = 0.53. Source data are available for this figure: SourceData F7.
Sarcomere phosphorylation response to bolus injection of DN and OM. Analysis of sarcomere phosphorylation 1 min after infusion of either vehicle control (DMSO), DN at 3 mg/kg (DN Low), DN at 6 mg/kg (DN High), OM at 0.75 mg/kg (OM Low), or OM at 1.5 mg/kg (OM High). (A–E) Representative (A) Coomassie blue– and (B) ProQ Diamond–stained gels with annotations indicating the molecular weights of the ladder and the locations of cMyBPC, cTnI, and myosin’s RLC. ProQ Diamond signals normalized to Coomassie blue signal for (C) cMyBPC, (D) cTnI, and (E) RLC. N = 4 hearts/group. No comparison was statistically significant and all P > 0.05 as determined by ANOVA. Exact P values are as follows: (C) P = 0.52; (D) P = 0.54; (E) P = 0.53. Source data are available for this figure: SourceData F7.
5-min PV loop and strain mechanics analysis
PV loops and LV strain mechanics were also analyzed 5 min after drug infusion (Table S3). None of the parameters for DN Low were significantly different from the DMSO group after 5 min. Many parameters in the DN High group, such as SV, dP/dtmax, Tau, systolic duration, and T2P DRSR, were either significantly reduced from the 1-min time point or not significantly different from the DMSO group. In contrast, the OM Low group had several parameters that remained significantly different from the DMSO group after 5 min, including SV, dP/dtmax and systolic duration. Most of the OM Low 5-min parameters were not significantly different from the 1-min time point. Finally, in the OM High group, only Pk. DRSR was significantly reduced at 5 min compared with the 1-min time point and the vast majority of parameters remained significantly different from the DMSO group. These data show that the effect of OM persisted much longer than that of DN and justify our focus on the 1-min time point data.
Sex differences
The 1-min PV loop and strain mechanics data were analyzed for sex-based differences (Table S4). The only statistically significant interaction between sex and drug was observed for non-normalized systolic and diastolic durations. However, this difference was no longer significant when durations were normalized to the cardiac cycle duration (Table S4). These results suggest that both DN and OM acutely affect cardiac function similarly in male and female mice.
Discussion
In this study, we directly compared the in vivo cardiac effects of DN, a second-generation myotropic sarcomere activator, and OM, the first-generation compound. We specifically investigated prior claims that DN impairs diastolic function less than OM. This is the first study to make direct in vivo comparisons between the two drugs to address this question. To do so, we evaluated the LV dose–response to DN and OM and examined how each drug modified the cardiac cycle to achieve a similar increase in SV. Overall, our findings support the idea that DN better preserves diastolic function compared with OM. However, DN still fundamentally alters the cardiac cycle in a manner similar to OM, which may ultimately limit its therapeutic window.
DN better preserves diastolic function with a similar in vivo mechanism to OM
While DN preserved diastolic function better than OM, both myotropic sarcomere activators had similar overall effects on cardiac function in healthy mice. During continuous intravenous infusion, both drugs elicited a qualitatively similar three-phase dose–response (Figs. 1 and 2). This response began with a therapeutic phase (P1), followed by an intermediate subtoxic phase (P2), and concluded with a toxic phase (P3). Although a previous study documented the toxic response to OM (Fülöp et al., 2021), our study is the first to fully characterize all three phases.
We specifically highlight the significance of the shared toxic response. At sufficiently high concentrations, both DN and OM drove the LV into a state of sustained contraction that became uncoupled from normal pacing and relaxation mechanisms. During this phase, left atrial contraction was the only source of LV filling. As a result, the LV was unable to fill adequately or generate substantial pressure. The fact that both drugs produced this same toxic response provides strong evidence that their sarcomere effects converge at high doses. A later subsection will explore how these shared in vivo effects relate to the drugs’ molecular mechanisms. The following subsections will compare and contrast how DN and OM affect each phase of the cardiac cycle within their therapeutic ranges.
Isovolumic contraction
First, both drugs had a pronounced effect on isovolumic contraction by significantly increasing dP/dtmax at the two doses studied (Fig. 1 and 4). Similar increases in the rate of pressure development have been reported in previous studies of OM using healthy rat and pig in vivo models (Fülöp et al., 2021; Rønning et al., 2018). The effect of DN on dP/dtmax is also consistent with findings in intact trabeculae, where 1 µM DN significantly increased the rate of tension development (Kooiker et al., 2023). Importantly, we did not detect significant changes in heart rate or sarcomere phosphorylation 1 min after administration of either drug. This suggests that the observed enhancement in pressure development results directly from cardiac myosin modulation, rather than from activation of upstream signaling pathways (Brodde and Michel, 1999; Blanton, 2020). However, our findings do not rule out changes in phosphorylation or other posttranslational modifications following chronic treatment.
Other studies did find that high concentrations of OM can alter cardiac excitation–contraction (EC) coupling (Nánási et al., 2017; Szentandrassy et al., 2016). It is unknown whether DN has a similar effect on cardiac EC coupling. This effect may contribute slightly to our observed results, but the majority of the observed results are likely due to the direct impact of DN and OM on cardiac myosin.
Notably, our findings for OM appear to contradict early claims that OM does not accelerate pressure development (Malik et al., 2011). However, OM’s effect on dP/dtmax may depend on the disease state. While studies in healthy rodents and pigs have reported increased dP/dtmax following OM administration (Fülöp et al., 2021; Rønning et al., 2018), the original preclinical studies in dogs with systolic heart failure and two independent studies in pig models of ischemic heart failure did not observe significant increases in dP/dtmax (Malik et al., 2011; Rønning et al., 2018; Shen et al., 2010). Interestingly, only the first-party preclinical data in healthy dogs failed to detect an increase in dP/dtmax under normal physiological conditions (Malik et al., 2011). This may reflect unrecognized species differences in drug response.
To date, no study has reported the in vivo effect of DN on dP/dtmax in a diseased heart. However, experiments in intact trabeculae from a mouse model of genetic dilated cardiomyopathy demonstrated that DN significantly increased the rate of tension development (Kooiker et al., 2023). These findings suggest that DN and OM may differentially affect pressure development depending on the pathological context. Regardless, future studies are needed to evaluate DN’s effects in diverse in vivo models of heart failure.
Although DN and OM increased dP/dtmax, both drugs decreased the slope of the linear relationship between dP/dtmax and EDV at higher doses (Fig. 5). To our knowledge, our study is the first to describe this effect. For comparison, positive inotropy mediated by endogenous β-adrenergic signaling also increases dP/dtmax but increases the dP/dtmax-EDV slope as well (Little, 1985). β-adrenergic signaling achieves this by coordinated phosphorylation of several sarcomere proteins, including cMyBPC, cTnI, and titin, which enhances the dependence of crossbridge recruitment and pressure development on preload (Brodde and Michel, 1999; Little, 1985; Gresham and Stelzer, 2016).
Decreases in the dP/dtmax-EDV slope are commonly associated with reduced contractility in diseased states, often resulting from β-adrenergic desensitization and impaired preload-dependent regulation of pressure development (Little et al., 1987; Lohse et al., 2003). However, since DN and OM did not alter sarcomere phosphorylation and the experiments were conducted in healthy hearts, these mechanisms are unlikely to explain the observed effects. Instead, DN and OM likely act by partially uncoupling the sarcomere from endogenous preload-dependent regulatory mechanisms, forcing greater crossbridge recruitment over a broader range of preloads. Notably, the dP/dtmax-EDV relationship was preserved at lower doses of both drugs, suggesting that the extent of uncoupling depends on the proportion of drug-bound myosin heads.
Ejection
DN and OM also significantly affected multiple aspects of systolic ejection. Both drugs notably prolonged systolic duration in a dose-dependent manner (Fig. 4, A and D), consistent with findings from prior preclinical and clinical studies (Malik et al., 2011; Voors et al., 2020; Fülöp et al., 2021; Rønning et al., 2018; Shen et al., 2010; Cleland et al., 2011). T2P SRS closely mirrored these changes in systolic duration (Fig. 6, A and D), suggesting that T2P SRS is a reliable surrogate for systolic timing.
Notably, DN consistently increased both systolic duration and T2P SRS less than OM, despite producing similar increases in SV. DN achieved this, at least in part, by increasing PER more than OM—a key mechanistic difference between the two drugs (Fig. 4, B and C). As previously discussed, this may be a favorable property of DN, as preserving systolic duration allows for greater diastolic duration and filling time. Regardless of mechanism, both drugs significantly increased Pk. SRS, indicating enhanced wall thickening due to increased myocardial contraction (Fig. 6, A and C). These in vivo findings are consistent with in vitro studies in isolated unloaded cardiomyocytes, where both DN and OM increased the magnitude of cell shortening (Ráduly et al., 2022; Horváth et al., 2017).
As with the dP/dtmax-EDV relationship, both DN and OM decreased the slope of ESPVR (Fig. 5). The decreases in ESPVR that we observed were not dose-dependent, suggesting that the effect may saturate at lower doses than those tested here. Additionally, DN and OM treatments left-shifted the overall ESPVR curve, as indicated by the change in intercept (Table 1). As others have noted, the combination of decreased ESPVR slope and left-shift following inotropic treatment likely indicates a change in the nonlinear behavior of the ESPVR rather than a decrease in contractility (Cingolani and Kass, 2011; Kass et al., 1989). We interpret our results similarly.
These alterations to the ESPVR could reflect changes in sarcomere length–dependent activation (LDA), the basis of many LV load–dependent phenomena (Kosta and Dauby, 2021; Konhilas et al., 2002). For instance, fiber-level studies have shown that OM abolished LDA by saturating crossbridge-mediated thin-filament activation effects at shorter sarcomere lengths (Gollapudi et al., 2017). While the effect of DN on fiber-level LDA has yet to be studied, our previous work demonstrated that DN and OM alter cardiac fiber mechanism in very similar ways (Choi et al., 2023, 2024). Therefore, similar reductions in LDA may also account for DN’s impact on the ESPVR.
Notably, our results conflict with some previous studies that assessed the effect of OM on ESPVR in other species. First-party preclinical studies in dogs reported nearly a 60% increase in normalized end-systolic elastance (a measure synonymous with ESPVR) 10 min after a 1 mg/kg bolus injection of OM (Malik et al., 2011). An independent study in healthy rats observed a more modest but still significant increase in ESPVR following dose titration with 0.6 and 1.2 mg/kg of OM (Fülöp et al., 2021). These discrepancies may be attributed to species-specific physiological differences, such as lower heart rates and greater LV volume reserves in dogs and rats compared with mice (Milani-Nejad and Janssen, 2014).
However, our findings are not without precedent. An ex vivo study using cross-circulated healthy and isoproterenol-induced heart failure rat hearts reported no change in ESPVR following treatment with 200–500 ng/ml OM (Obata et al., 2019). Similarly, a study in postischemic pigs observed minimal or even slightly negative changes in ESPVR following OM administration, although the authors did not discuss the significance of these results (Rønning et al., 2018). Additionally, interpretation of OM’s effects on time-varying elastance in the first-party dog study is limited by the lack of clarity on whether healthy or heart failure animals were used, as well as the absence of group summary statistics and variance measures (Malik et al., 2011). These omissions complicate direct comparison with our results.
Some internal inconsistencies in the rat study also complicate the interpretation of their results (Fülöp et al., 2021). The authors report that 1.2 mg/kg OM caused both systolic and diastolic blood pressures to significantly drop from a reported average of 149 and 130 mm Hg to 49 and 37 mm Hg, respectively (Fülöp et al., 2021). Such a severe decline in blood pressure would be consistent with OM-induced toxicity, as described in our study. However, these hemodynamic changes are not reflected in their representative PV loops or their reported increase in ESPVR (Fülöp et al., 2021). Regardless, our data suggest that the effect of myotropic sarcomere activators on contractility measures may be more complex than initially reported.
Isovolumic relaxation
Following ejection, DN and OM similarly affected isovolumic relaxation. While Pmin and dP/dtmin were not dramatically altered by the bolus injections compared with the DMSO group (Table 1), DN did cause a slight drop in these parameters during the P1 phase of the continuous infusions (Fig. 1 A). Low doses of DN may reduce Pmin and dP/dtmin due to increased deformation of elastic elements within the LV during systole (Sengupta et al., 2008). As a result, the LV walls may spring back with greater force during early diastole, producing greater suction within the chamber. The negative diastolic effects of higher DN concentrations likely override this mechanism. OM may not have reduced Pmin and dP/dtmin as much due to its more negative impact on diastolic function compared with DN.
Both drugs profoundly impacted Tau (Fig. 4 F). DN and OM significantly increased Tau at higher doses, consistent with slowed crossbridge detachment kinetics observed in permeabilized muscle fibers and with findings from other in vivo studies of OM (Fülöp et al., 2021; Mamidi et al., 2017; Mamidi et al., 2015; Ráduly et al., 2022; Choi et al., 2023). In contrast, lower doses of both drugs resulted in only minor or nonsignificant increases in Tau, suggesting a nonlinear dose–response relationship. These findings align with clinical data from DN trials, which showed a similar nonlinear increase in IVRT with drug–plasma concentration in patients with HFrEF (Voors et al., 2020). IVRT was not reported in the corresponding clinical trial for OM (Cleland et al., 2011), but a similar relationship likely exists.
As with its effect on systolic duration, DN consistently increased Tau less than OM, supporting the claim that DN impairs diastolic function to a lesser extent (Fig. 4 F). This observation aligns with fiber-level studies showing that DN slows crossbridge detachment less than OM (Choi et al., 2023). The tight correlation between the effects of both drugs on Tau and systolic duration suggests that these parameters are likely governed by the same underlying molecular mechanism.
However, our recent studies found that isolated human heart failure tissue is more sensitive to the slowing effects of both DN and OM compared with nonfailing donor tissue (Mamidi et al., 2017; Choi et al., 2024). Although DN still slowed crossbridge detachment less than OM in heart failure fibers (Choi et al., 2024), it may produce a greater increase in Tau in the context of HFrEF pathophysiology than what we observed in the healthy heart. Future experiments are needed to determine how HFrEF pathophysiology modulates DN’s effects on pressure relaxation and other aspects of diastolic function in vivo.
Filling
Finally, both DN and OM impaired ventricular filling to some extent. At high doses, both drugs significantly shortened diastolic duration, as measured by the LV pressure signal, and reduced filling time, as indicated by the time remaining after T2P SRS in the strain waveform (Table 1; and Fig. 6, A and D). These pressure- and strain-based findings are consistent with Doppler-derived measurements of filling time in healthy rats treated with DN (Ráduly et al., 2022). Lower doses produced only mild effects on filling. These impairments are likely direct consequences of prolonged systolic duration and slowed pressure relaxation. As with other diastolic parameters, DN preserved diastolic filling better than OM at both doses, further supporting the conclusion that DN impairs diastolic function less than OM.
Both drugs delayed T2P DRSR (Fig. 6 F). Under normal conditions, T2P DRSR occurs during to early diastolic filling, when the ventricular wall rapidly thins as the ventricle relaxes and fills with blood (Dokainish et al., 2008; Dahl et al., 2016). A smaller second peak typically follows, representing late filling driven by atrial contraction. At higher doses, both DN and OM eliminated the early filling peak and shifted T2P DRSR to the late filling phase (Fig. 6 B), suggesting that pressure relaxation was delayed to the point that left atrial systole occurred before LV pressure had fallen below left atrial pressure.
While lower doses preserved some of the double-peak morphology, the effect remained appreciable. This pattern is expected to reduce the E/A ratio as measured by Doppler imaging, consistent with previous studies in healthy rats treated with DN and OM (Fülöp et al., 2021; Ráduly et al., 2022). Species with longer diastolic filling times, such as humans, may be less sensitive to these delays (Kass et al., 2004). However, clinical trials of both DN and OM show dose-dependent reductions in E-wave velocity and E/A ratio (Voors et al., 2020; Cleland et al., 2011), indicating that this mechanism likely contributes to altered diastolic function in humans as well.
As a secondary consequence of delayed T2P DRSR, both DN and OM increased the magnitude of Pk. DRSR (Fig. 6 E). This is consistent with the modest increases in peak filling rate observed following high-dose treatment with both drugs (Table 1) and may reflect enhanced left atrial contraction. Isolated cardiac muscle fiber studies have shown that DN and OM affect left atrial muscle, albeit to a lesser extent than LV tissue (Voors et al., 2020; Nakanishi et al., 2022).
In vivo studies have also reported increases in late filling velocity attributed to left atrial contraction in both humans and rats treated with DN or OM (Voors et al., 2020; Fülöp et al., 2021; Cleland et al., 2011; Ráduly et al., 2022). Although left atrial function was not assessed for OM, DN has been shown to increase both left atrial emptying fraction and the left atrial function index in dogs with systolic heart failure and in HFrEF patients (Voors et al., 2020). These findings suggest that left atrial function may play a meaningful role in modulating whole-heart hemodynamics following DN or OM treatment.
Additionally, the shortening of diastolic duration may lead to partial overlap between early and late filling phases. The combined effect of these overlapping phases would contribute to an increased peak filling velocity (Appleton, 1991).
Finally, neither drug at either dose significantly altered the EDPVR (Table 1). Initially, this might suggest that DN and OM do not alter LV end-diastolic compliance at the doses tested. However, the effect of DN and OM on EDPVR may be partly masked. The reduction in EDV following drug treatment could shift the PV loops leftward onto the flatter part of the EDPVR, thereby obscuring any significant curvature of the relationship during preload reduction. Furthermore, fitting to flat EDPVR data can substantially reduce confidence in the fitted parameters, so we caution against overinterpretation (Cingolani and Kass, 2011). However, we expect that EDPVR and end-diastolic compliance would increase at higher (subtoxic and toxic) DN and OM doses, given the increases in Pmin (Figs. 1 and 2).
Both DN and OM likely enhance cooperative thin-filament activation
DN and OM likely affect sarcomere function similarly given that both drugs alter whole-heart hemodynamics in mostly the same way. Our results are consistent with a model where DN enhances cooperative thin-filament activation like OM (Woody et al., 2018; McKillop and Geeves, 1993). In the case of OM, drug binding near the converter domain of myosin stabilizes strongly bound crossbridges in the prepower stroke conformation, increasing the crossbridge duty ratio (Woody et al., 2018; Planelles-Herrero et al., 2017). As a consequence, OM-bound crossbridges prop open tropomyosin, activating neighboring thin-filament regulatory units independent of calcium. Studies have also shown that OM directly activates the thick filament, allowing more myosin heads to form crossbridges (Kampourakis et al., 2018).
Like OM, previous studies suggest that DN stabilizes strongly bound crossbridges and increases the duty ratio (Woody et al., 2018; Kooiker et al., 2023). DN may also directly activate the thick filament, as observed in muscle X-ray diffraction studies (Auguin et al., 2024; Kooiker et al., 2023; Governali et al., 2020). Thus, these DN-bound crossbridges would prime neighboring thin-filament regulatory units similar to OM. Under therapeutic concentrations, both drugs would allow for more crossbridge formation earlier in the Ca2+ transient and explain the increase in dP/dtmax. These drug-bound crossbridges would also extend ejection and slow pressure relaxation by prolonging thin-filament activation and inhibiting cooperative thin-filament deactivation.
At toxic concentrations, the high number of DN- or OM-bound crossbridges likely maintains thin-filament activation even after intracellular Ca2+ levels decline. This phenomenon is supported by solution-based experiments showing that the thin filament can remain fully activated when only one third of its binding sites are occupied by myosin, independent of Ca2+ (McKillop and Geeves, 1993; Stehle and Iorga, 2010). This mechanism likely underlies the sustained contractile state observed during DN and OM toxicity and establishes a hard upper limit on the therapeutic window of myotropic sarcomere activators. In the whole heart, decreased filling, and thus decreased preload, during DN and OM toxicity may also contribute to the decline in Pmax and dP/dtmax via Frank–Starling effects (Konhilas et al., 2002; Kosta and Dauby, 2021).
Ultimately, at higher concentrations, the effects of enhanced cooperative thin-filament activation appear to dominate the whole-heart response, regardless of any differences in molecular mechanism between DN and OM. These findings suggest that any myosin modulator that stabilizes strongly bound crossbridges may face similar limitations.
DN likely preserves diastolic function better than OM due to differences in molecular mechanisms
Although cooperative thin-filament activation likely accounts for the general cardiac response to DN and OM at high concentrations, differences in their specific molecular mechanisms are nevertheless critically important. Variations in how DN and OM modulate the actomyosin ATPase cycle and myosin mechanics likely explain why DN can achieve similar increases in SV while better preserving diastolic duration and pressure relaxation.
For example, an early report of single-molecule and stopped-flow studies indicates that DN preserves the force dependence of myosin detachment and maintains the canonical pathway of ATP-induced actomyosin dissociation (Scott et al., 2024, Preprint). Stopped-flow experiments also suggest that DN accelerates actomyosin association kinetics, resulting in increased actin-activated myosin S1 ATPase cycling (Voors et al., 2020; Scott et al., 2024, Preprint). In contrast, OM prolongs crossbridge attachment by eliminating the force dependence of myosin detachment and inhibiting the canonical ATP-induced dissociation pathway (Woody et al., 2018; Liu et al., 2018). As a result, OM reduces actin-activated myosin S1 ATPase cycling (Malik et al., 2011; Liu et al., 2018).
These mechanistic differences may allow DN to preserve more normal crossbridge detachment kinetics at lower concentrations when cooperative thin-filament activation is less dominant. This could explain why DN did not prolong systolic duration as much as OM, despite producing similar increases in SV (Fig. 4 D).
Additionally, optical trap experiments have shown that DN may impede the power stroke less than OM (Woody et al., 2018; Scott et al., 2024, Preprint). As a result, DN-bound myosins likely retain the ability to contribute to force generation and shortening, whereas OM-bound myosins are largely nonfunctional and instead increase inter-filament drag. This mechanistic distinction may explain why DN increased PER while OM did not (Fig. 4 E), a finding consistent with fiber-level data (Choi et al., 2023). Therefore, although DN’s favorable molecular properties do not eliminate the effects of cooperative thin-filament activation at high concentrations, they may widen its therapeutic window and improve tolerability in patients with HFrEF.
Limitations
There are several important limitations to this study that warrant consideration. First, all experiments were conducted in healthy mice. This approach was necessary to isolate and interpret the fundamental differences in drug effects without the added complexity of HFrEF pathophysiology (Diwan and Hill, 2020; Nabeebaccus et al., 2020). However, since both DN and OM are intended for use in patients with HFrEF, it will be essential for future studies to investigate how specific components of HFrEF pathophysiology may differentially modify their cardiac effects.
Second, several species-specific differences between mouse and human cardiac physiology may complicate the direct translation of our findings. These include differences in sarcomere protein isoforms, baseline heart rate, and diastolic reserve (Milani-Nejad and Janssen, 2014). In addition, the pharmacokinetic clearance of DN is substantially higher in mice than in humans (Grillo et al., 2021). This is reflected in our 5-min data, where OM’s cardiac effects persist significantly longer than DN’s (Table S3). Nevertheless, comparative studies using skinned myocardial fibers demonstrate that both DN and OM exert similar effects on crossbridge function across species (Mamidi et al., 2017; Mamidi et al., 2015; Choi et al., 2023; Kooiker et al., 2023). Therefore, despite these physiological and pharmacokinetic differences, we expect our results to retain strong translational relevance.
Third, our PV loop data were collected using an open-chest approach, while our ultrasound data were collected using a closed-chest configuration. While the data collected by the two methods were highly consistent with each other, the open-chest approach is highly invasive. However, these trade-offs are justifiable given the superior control over PV catheter position within the LV compared with the closed-chest PV loop approach, as well as direct access to the inferior vena cava (Cingolani and Kass, 2011). Nevertheless, these differences should be taken into account when comparing between our PV data and other closed-chest data sets (Voors et al., 2020; Teerlink et al., 2016; Cleland et al., 2011). Finally, all of our observations were made following acute administration of DN or OM, which may not predict differences in long-term efficacy.
Conclusion
To summarize, we used carefully controlled in vivo experiments to determine whether the second-generation myotropic sarcomere activator DN better preserves diastolic function compared with the first-generation myotropic sarcomere activator OM. Overall, our data support the notion that DN improves upon OM’s primary weakness. We found that DN achieved the same increase in SV while impairing diastolic function less. This may benefit DN in clinical trials by contributing to a wider therapeutic window, assuming that DN can achieve a greater increase in SV for the same tolerated decrease in diastolic function as OM. However, DN still reduced diastolic function at higher doses and resulted in the same toxic response as OM. Thus, the same underlying mechanism that limited OM’s therapeutic window will likely limit DN’s. Other pharmacokinetic and pharmacodynamics factors may also impact DN’s therapeutic window. As stated by others, further clinical trials will be needed to assess the appropriate therapeutic window of DN in HFrEF patients (Voors et al., 2020). Future iterations of myotropic sarcomere activating therapies and other sarcomere activating approaches should continue to maximize increases in systolic performance while minimizing reductions in diastolic function.
Data availability
All data are available from the corresponding author upon reasonable request.
Acknowledgments
Henk L. Granzier served as editor.
This study was supported by the following American Heart Association grant awarded to J.B. Holmes: 25PRE1374126; and by the following NIH grants from the National Heart Lung and Blood Institute awarded to J.E. Stelzer: R01HL114770, R01HL153236, R01HL146676, and R01HL173989.
Author contributions: J.B. Holmes: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, validation, visualization, and writing—original draft, review, and editing. J.E. Stelzer: conceptualization, funding acquisition, project administration, resources, supervision, validation, visualization, and writing—review and editing.

