


Vertebrate retinal photoreceptors signal light by suppressing a circulating “dark current” that maintains their relative depolarization in the dark. This dark current is composed of an inward current through CNG channels and NCKX transporters in the outer segment that is balanced by outward current exiting principally from the inner segment. It has been hypothesized that Kv2.1 channels carry a predominant fraction of the outward current in rods. We examined this hypothesis by comparing whole cell, suction electrode, and electroretinographic recordings from Kv2.1 knockout (Kv2.1−/−) and wild-type (WT) mouse rods. Single cell recordings revealed flash responses with unusual kinetics, and reduced dark currents that were quantitatively consistent with the measured depolarization of the membrane resting potential in the dark. A two-compartment (outer and inner segment) physiological model based on known ionic mechanisms revealed that the abnormal Kv2.1−/− rod photoresponses arise principally from the voltage dependencies of the known conductances and the NCKX exchanger, and a highly elevated fraction of inward current carried by Ca2+ through CNG channels due to the aberrant depolarization. Kv2.1−/− rods had shorter outer segments than WT and dysmorphic mitochondria in their inner segments. Optical coherence tomography of knockout animals demonstrated a slow photoreceptor degeneration over a period of 6 mo. Overall, these findings reveal that Kv2.1 channels carry 70–80% of the non-NKX outward dark current of the mouse rod, and that the depolarization caused by the loss of Kv2.1 results in elevated Ca2+ influx through CNG channels and elevated free intracellular Ca2+, leading to progressive degeneration.
Introduction
Vertebrate rod and cone photoreceptors signal the time-varying rate of absorbed photons by graded, hyperpolarizing responses. These photovoltage responses arise from transient suppression of a circulating “dark current,” which flows inward across the outer segment membrane and outward across the membrane of the inner segment.
The outer segment–inward limb of the rod dark current is carried primarily by CNG channels, along with a normally small (∼5%) contribution from the electrogenic Na+,Ca2+/K+ exchanger (NCKX; reviewed in Kaupp and Seifert, 2002). However, the ionic mechanisms underlying the balancing outward dark current of the inner segment have not been definitively identified. In rods with membrane potential clamped near rest, a substantial outward current is generated by voltage-gated delayed rectifier channels, historically referred to as the IKx current (Attwell and Wilson, 1980; Bader et al., 1982; Baylor and Nunn, 1986; Beech and Barnes, 1989; Barrow and Wu, 2009). The IKx current is thought to be mediated by multimeric K+ channels consisting of Kv2.1 subunits (Yazulla and Studholme, 1998) and the auxiliary subunit Kv8.2 (KCNV2). Kv2.1 channels (KCNB1) are members of the Shab-related delayed rectifier subfamily that are widely expressed in neurons, where they regulate resting membrane potential and electrical excitability (Shah and Aizenman, 2014). Both Kv2.1 and Kv8.2 are expressed in the retina (Czirják et al., 2007) and in photoreceptor inner segments in particular (Gayet-Primo et al., 2018). A previous study of Kv2.1−/− mice has shown the retina to have greatly reduced electroretinographic (ERG) a-waves (Hart et al., 2019), consistent with the idea that Kv2.1 channels carry a major portion of the outward circulating current.
Another potentially significant contributor to the outward dark current of rods is the Na+/K+ adenosine triphosphatase (NKX; α3β2 isoform), which is also strongly expressed in rod inner segments (Wetzel et al., 1999). The NKX is the only Na+ efflux mechanism balancing the Na+ influx through CNG channels and the NCKX exchanger into the rod outer segment. Because the NKX is electrogenic (stoichiometry, 3Na+ out: 2K+ in), it makes an essential contribution to outward current in the dark. Rod inner segments and cell bodies also contain hyperpolarization-activated (HCN1) channels that dynamically shape photovoltage responses during light-driven hyperpolarization (Demontis et al., 1999, 2002, 2009), but their contribution to the outward dark current at the normal resting potential is expected to be negligible because of their strong inward rectification and their reversal potential close to the resting potential. Other ionic mechanisms of rods with potential relevance to the outward dark current have been identified with transcriptomics (Siegert et al., 2012), including at least two inwardly rectifying K+ channels (KCNJ13, KCNJ14).
To clarify the role of Kv2.1 channels in rods, in this investigation we have compared the electrical and structural properties of rods lacking Kv2.1 channels (Kv2.1−/−) to those of WT littermates. The electrical properties were studied with three different methods: suction electrode (SE) recordings of outer segment photocurrents, whole cell (WC) recordings of dark electrical properties and light-evoked responses, and ERGs. Quantitative interpretation of the resultant data required developing a biophysical model that incorporates the known ionic mechanisms of the dark electrical steady state and predicts the photocurrent responses of rods of both genotypes. This analysis reveals significantly elevated intracellular Ca2+i in Kv2.1−/− rods, a result confirmed by the greatly increased NCKX current, as well as other distinctive kinetic features of their photocurrents. The chronically elevated Ca2+i in turn is consistent with features of the observed photoreceptor degeneration, producing new insights about the functional significance of Kv2.1 channels for both physiological and cell-biological homeostasis.
Materials and methods
Animals
Mice were reared and handled under the guidelines established by the National Institutes of Health using protocols approved by the Institutional Animal Care and Use Committee of the University of California at Davis. Heterozygous mice carrying the null Kv2.1 allele (RRID:IMSR MGI:3806050; Jacobson et al., 2007; Speca et al., 2014) were bred to generate Kv2.1+/+ (WT) and Kv2.1−/− littermates. Heterozygote (Kv2.1+/−) littermates were also used in certain experiments and were found to be structurally and functionally equivalent to WT littermates in all assays examined. Unless otherwise indicated, all mice used in these experiments were young adults (4–8 wk of age) housed in 12-h cyclic light (∼200 lux) and dark-adapted overnight before an experiment.
Immunohistochemistry
Mice were euthanized by CO2 narcosis and decapitated, and the eyes were removed and immersed in 4% paraformaldehyde in PBS (Thermo Fisher Scientific; AAJ75889K8) for 5 min at room temperature. The anterior segments were then cut away, the lens removed, and the eyecups fixed for 20 additional minutes before being stored in 1× PBS at 4°C until sectioning. For sectioning, eyecups were embedded in low-melting agarose (Invitrogen; 18300-012) preheated to 70°C and oriented so as to cut planar to the dorsoventral midline as described in Daniele et al. (2011) and Wagner et al, (2000). Sections 150 µm thick were cut with a vibratome (Leica Microsystems; model VT 10005) and stored in small base molds of 1× PBS at 4°C.
Sections were incubated with normal goat serum (1:20; Jackson ImmunoResearch) in 1× PBS with 0.5% BSA and 0.5% Triton X-100 (PBT) for 1 h at room temperature, then incubated in primary antibodies diluted in PBT at 4°C overnight. After three 5-min washes in PBT, secondary antibodies with 6′-diamidino-2-phenylindole (Invitrogen Life Technologies; R37606) were applied for 1.5–2 h at room temperature. The sections were washed two times for 5 min each in PBT, then one time in 1× PBS before being mounted onto slides and coverslipped with ProLong Diamond Antifade Reagent (Invitrogen Life Technologies; P36970). Primary antibodies used for immunohistochemistry include mouse anti-rhodopsin monoclonal antibody (1D4; Abcam; RRID:AB 304874), rabbit “KC” anti-Kv2.1 polyclonal antibody, in-house (Trimmer, 1991; Trimmer laboratory; RRID: AB_2315767, affinity-purified, 1:100), and mouse IgG1 anti-HCN1 monoclonal antibody N70/28.1 tissue culture supernatant (University of California, Davis/National Institutes of Health [UC Davis/NIH] NeuroMab Facility, RRID: AB_10672848) at 1:5. Alexa Fluor 488–conjugated goat anti-mouse and Alexa Fluor 633–conjugated goat anti-rabbit (Invitrogen Life Technologies) were used as secondary antibodies at 1:300 dilution in PBT. Slide-mounted sections were visualized with a Nikon Ti-E A1 multiphoton imaging system using a 40× water immersion objective and continuous wave lasers. 3-D images (30 µm in the z-direction) were sampled from the center and peripheral regions of each section using the NIS-Elements Microscope Imaging Software (Nikon). Cell numbers, morphological features, and layer thicknesses were counted and measured manually.
Paraffin-embedded sections
Paraffin embedding was done as previously described (Sun et al., 2015). Mice were euthanized by CO2 narcosis, and the eyes were removed and flash-frozen in dry ice–cooled liquid propane for 30 s before being transferred to 3% acetic acid in MeOH and stored at −80°C. After a minimum of 48 h, eyes were warmed by moving sequentially to −40°C, −20°C, then room temperature for at least 2 h at each temperature. The eyes were transferred to 100% ethanol for 1 h, replaced with fresh 100% ethanol for another hour, twice in 100% xylene, and then three 45-min changes of 60°C paraffin. Sections of 5 µm were taken with a rotary microtome and stained for hematoxylin and eosin.
Electron microscopy
Mice were perfused trans-cardially with 2% paraformaldehyde, 2% glutaraldehyde, and 0.05% CaCl2 in 50 mM 3-morpholinopropane-1-sulfonic acid, pH 7.4, as described (Ding et al., 2015). Retinas were removed and post-fixed in the same fixative for up to 2 h before being transferred to PBS for storage. Tissue was rinsed in 0.1 M sodium phosphate buffer, then stained with 1% osmium tetroxide, rinsed in distilled H2O, and dehydrated with an increasing series of ethanol. Retinas were incubated in 50% propylene oxide and 50% PolyBed resin overnight, then infiltrated with 100% PolyBed resin and polymerized in fresh resin. Thin sections (80–90 nm) were cut on an ultramicrotome, collected on copper grids, and stained with 4% uranyl acetate and 0.2% lead citrate. Images were taken using a FEI Talos L120C TEM.
Optical coherence tomography (OCT)
OCT imaging was performed with a custom-built, multi-modal scanning laser ophthalmoscopy and OCT system (Zhang et al., 2015, 2018). Anesthesia was induced with 4% isoflurane and maintained with 2–2.5%; body temperature was maintained with a heating pad at 37°C (Physitemp; TCAT-2LV). Mouse position was controlled with a stereotactic bite bar (Knopf; 923-B Mouse Head Holder) mounted on a micropositioner (Phoenix Ltd.). Pupils were dilated and cyclopleged with tropicamide and phenylephrine, and corneal surface hydration was maintained with hypromellose gel (Alcon; GenTeal Tears Severe) and a contact lens (Unicon Corporation). The scanning laser ophthalmoscopy subsystem was used for initial alignment of the mouse eye. The OCT subsystem used a broadband near-infrared superluminescent diode (Superlum; Broadlighter T-860-HP; center wavelength, 860 nm; bandwidth, 132 nm) giving an axial resolution of ∼2 µm. Widefield OCT images were acquired over 51° of visual angle (2.2 mm) and “zoomed” OCT images over ∼25° of visual angle (∼1.1 mm) with a lateral optical resolution of ∼3 µm.
OCT images were processed and flattened using standard techniques (Wojtkowski et al., 2004; Miller et al., 2019). The OCT images shown in Fig. 11 are the result of the temporal speckle averaging technique to enhance the structural information in the images (Zhang et al., 2019). In short, 30–50 zoomed OCT volumes were registered and averaged, greatly reducing speckle noise. For retinal layer thickness measurements, the flattened widefield volume was averaged to obtain an A-scan that was used to measure the lengths of the outer retina, ONL, and combined inner segment layers (ISs) and outer segment layers (OSs; Karlen et al., 2018). Layers were segmented based on the optima in the average A-scan and the gradient of the average A-scan. The inner plexiform layer thickness was defined as the neuro-fiber layer–vitreous boundary to the center of inner nuclear layer (INL). The outer nuclear layer (ONL) was from the center of the INL to the external limiting membrane (ELM). Outer and inner segment lengths were measured in OCT images using a technique recently described in Zhang et al. (2020). Briefly, the A-scan was deconvolved with the Lucy–Richardson algorithm in MATLAB (MathWorks) and the axial point-spread function of the OCT subsystem, with the result used to locate the retinal pigment epithelium (RPE) and Bruch’s membrane. Boundaries of retinal layers were then manually defined, and the location of the intensity peaks of Bruch’s membrane, the RPE, IS/OS junction, and ELM were defined by the centroid of least-squares-fitted Gaussian functions. The outer segment length was measured as the distance between the RPE and the IS/OS junction and adjusted based on known distances from the RPE to the OS tips determined in albino (BALB/c) mice (Zhang et al., 2020).
Protein analysis
Under infrared light, dark-adapted littermate pairs of WT and Kv2.1−/− mice were euthanized by cervical dislocation. The retinas were quickly dissected and sonicated in ice-chilled Ringer’s solution with protease inhibitor cocktail (Sigma-Aldrich; 4693159001). For rhodopsin quantification, an aliquot of each retinal homogenate was solubilized in PBS containing 50 mM hydroxylamine (titrated to pH 7.5 by NaOH) and 70 mM n-octylglucoside, and rhodopsin content was determined by difference spectroscopy at 500 nm before and after a full bleach, using 40,500 as the extinction coefficient. Rhodopsin quantification for each retina was performed in triplicate and averaged, and then averaged across retinas of each genotype.
For immunoblot analyses of protein expression levels, homogenates were mixed with an equal volume of 2× reducing sample buffer and boiled for 1 min. Samples of 10 µg each of retinal homogenate, 10 µg mouse brain homogenate, and 10 µg crude mouse brain membranes treated with alkaline phosphatase were separated by 7.5% SDS–PAGE and transferred to nitrocellulose membranes (Bio-Rad). Blots were blocked for 1 h with Blotto (4% wt/vol nonfat milk) in Tris-buffered saline (TBS; 50 mM Tris, pH 7.5, 150 mM NaCl) plus 0.1% vol/vol Tween-20 followed by 1 h room temperature incubation with primary antibodies. Primary antibodies were as follows: recombinant mouse anti-Kv2.1 IgG2a monoclonal K89/34R (Mandikian et al., 2014; RRID:AB_2750677; tissue culture supernatant at 1:2); mouse anti-HCN1, IgG1 monoclonal N70/28 (UC Davis/NIH NeuroMab Facility; RRID:AB_10672848; tissue culture supernatant at 1:5); and mouse anti-Grp75/mortalin, IgG1 monoclonal N52A/42 (UC Davis/NIH NeuroMab Facility, RRID:AB_2750800; tissue culture supernatant at 1:10). After three 10-min washes with TBS plus 0.1% vol/vol Tween-20, the membranes were incubated with the appropriate secondary antibodies for 1 h at room temperature in TBS. Anti-mouse secondary antibodies were anti-IgG2a Alexa 488 (Invitrogen Life Technologies; A-21131) and anti-IgG1 Alexa 647 (Life Technologies; A-21240), each at 1:1,500. After three 10-min washes with TBS, the immunoblots were visualized directly in a FluorChem Q imager (ProteinSimple), and fluorescence intensity measurements were made using associated AlphaImager software. Group differences were evaluated by independent t test. P < 0.05 was considered statistically significant.
SE recordings
Retinal tissue was dissected and maintained in complete darkness with the aid of infrared-visible converters (BE Meyers; Phantom Night Vision). Mice were euthanized by CO2 narcosis and the retinas dissected and stored in chilled Leibovitz’s L-15 medium (Invitrogen) supplemented with 10 mM glucose and 0.1 mg/ml BSA. The retina was finely chopped, and small pieces were transferred to a recording chamber perfused with bicarbonate-buffered Locke’s solution containing (in mM) 112.5 NaCl, 3.6 KCl, 2.4 MgCl2, 1.2 CaCl2, 20 NaHCO3, and 3 sodium succinate, bubbled with 95% O2/5% CO2 and warmed to 35–37°C, pH 7.4. Photocurrents were recorded from intact rods by drawing individual outer segments into a highly polished glass pipette containing (in mM) 140 NaCl, 3.6 KCl, 2.4 MgCl2, 1.2 CaCl2, 3 HEPES, and 0.2 EDTA, pH 7.4, connected to a current-to-voltage amplifier (Molecular Devices; Axopatch 200B amplifier), filtered using an analogue eight-pole Bessel filter (fc = 30 Hz), and digitized with acquisition hardware from National Instruments, Inc., and custom routines in Igor Pro (WaveMetrics).
The maximal photocurrent of the outer segment current was measured by subtracting the recorded response amplitude following a saturating flash from the baseline current level, defined as the mean level in the 0.5 s before the flash. Between 20–30 dim flash responses were averaged to calculate the average dim flash response; the time to peak was determined from the midpoint of the 10-ms flash to the dim flash response maximum, compensating for the delay imposed by the analogue filter. Light sensitivity was determined by fitting a saturating exponential function to response amplitude versus intensity data, as described (Fig. 4). The flux density of the unattenuated light source was regularly measured at the recording stage, and light stimuli were delivered through calibrated neutral density filters. The stimulus strengths are expressed in energy density units (photons μm−2). For theory, the latter values were converted to units of photoisomerizations (Φ) by multiplying by the collecting area. For WT rods, the collecting area was estimated from fluctuations in the rising phase of responses to dim flashes arising from Poisson variation in light capture. For Kv2.1−/− this was not possible, and the collecting area was estimated based on the shortened length of Kv2.1−/− rods and somewhat lowered rhodopsin expression (compare Fig. 2).
WC recordings
Adult WT and Kv2.1−/− mice (4–6 wk of age) were sacrificed under infrared light by cervical dislocation and decapitation. After enucleation, the eyecup was quickly dissected in cold, HEPES-buffered Ames and embedded in low-temperature gelling agar (Sigma-Aldrich; Agarose type VII-A; 3% wt/vol in HEPES-buffered Ames solution). Retinal slices (200-µm thick) were cut in ice-cold HEPES-buffered Ames (pH 7.4) using Leica 1000S vibratome (Leica Biosystems) stored up to 6 h inside a light-protected chamber containing bicarbonate buffered Ames (pH 7.4, 295 mOsm) maintained at 32°C and continuously bubbled with 95% O2/5% CO2.
In the dark, retinal slices were transferred to a recording chamber, rapidly perfused (3–5 ml/min) with bicarbonate buffered Ames, equilibrated with 95% O2/5% CO2, and maintained at 36°C. Cells were targeted for recording under infrared light. WC recordings were made from rod cell bodies using borosilicate glass patch pipettes (electrical resistances 12–18 MΩ) filled with (in mM) 130 K-gluconate, 10 KCl, 10 HEPES, 3 ATP-Mg, 1 GTP-Na3, and 0.1 Alexa-Fluor 488 (Invitrogen). The electrode solution was adjusted to pH 7.2 and the final osmolarity adjusted to 270–280 mOsm. WC voltage clamp recordings were made using an Axoclamp 200B amplifier (Axon Instruments). Signals were filtered digitally with an eight-pole analogue Bessel filter with 100 Hz bandwidth digitized at 1 kHz using a Digidata 1322A (Molecular Devices). Data were acquired using pClamp software (Axon Instruments) and analyzed with custom software written with Igor Pro (WaveMetrics). Membrane holding potential for rod voltage clamp experiments was −40 mV, which was not adjusted for the liquid junction potential. Subsequently, the junction potential between the external solution (ground) and the pipette was determined at 37°C, and the measured offset (−8.2 mV) was used to adjust the membrane potential values of Table 3.
For all WC experiments, the holding potential before access was set to −40 mV, as initial observations indicated this setting would be very close to the zero-current holding potential for the rods of WT mice. Upon WC access, the holding potential was immediately adjusted to achieve zero current, and the zero-current holding potential at that moment (corrected post hoc for the liquid junction potential, 8.2 ± 0.3 mV) was taken as the measurement the resting potential, Vm,rest. The saturating photocurrent was next measured. For this latter measurement, the holding potentials of both WT and Kv2.1−/− rods were preset to the same nominal value, −40 mV. After these initial measurements, response families of some rods were measured under voltage clamp or current clamp, while for others, I-V curves were measured in the dark steady state, with occasional saturating flashes delivered to gauge rundown. For the I-V curves, the holding potential was stepped from −80 to +20 for 750 ms, and the measured current averaged over a total of 100 ms after the decay of the capacitive current (capacitance compensation was not used). Only rods with a series resistance of at least 1 GΩ and at least a 5-pA maximal light-evoked response were considered for further analysis. The series resistance was checked intermittently throughout the experiment to ensure that access was stable, and a final saturating flash was delivered to ensure that the dark-adapted responses not fall below the 5-pA threshold used to gauge rundown.
In analyses of results with all methods, Kv2.1+/+ and Kv2.1−/− metrics were compared with a two-tail Student’s t test with 0.05 (95% confidence) set as the threshold for statistical significance.
ERG
Under dim red light illumination (720 nm) mice dark-adapted overnight were anesthetized with isoflurane (1.5%) and positioned on a regulated heating pad that maintained a core temperature of 37°C. After alignment of the mouse’s head in the ERG recording apparatus (Phoenix Research Labs), a mixture of phenylephrine and tropicamide (2:1) was applied to the cornea to induce pupil dilation. Methylcellulose was applied to maintain corneal moisture and electrical contact with the corneal electrode during ERG recordings. A subepidermal needle placed at the back of the head was used as a ground electrode.
Light stimuli consisted of 1-ms flashes of 510-nm light. Retinal irradiance was calculated by measuring the power output of the ERG apparatus and the stimulus duration with a calibrated photodiode. For converting retinal energy density to photoisomerizations per rod, an end-on collecting area of 0.87 µm2 was used (Lyubarsky et al., 2004). All ERG traces were acquired with Phoenix Research Labs software programmed to sample signals at a 5 kHz rate. The built-in digital filter was configured to a nominal 1 Hz to 1 kHz bandpass frequency. Recorded traces were exported to Igor Pro (WaveMetrics) for post-processing.
Theory
To address how Kv2.1 channel function and loss affects rod physiology, we used three different electrical methods: SE recording from outer segments, WC recordings from rods in retinal slices, and in vivo ERG recordings. To combine the information from these different methods into a unified picture, we developed a biophysical model of the rods of WT and Kv2.1−/− mice that includes all pertinent ionic mechanisms of the outer and inner segment membranes, as well as a model of phototransduction that incorporates voltage and calcium dependencies usually not material to characterizing WT rod function (Fig. 1).

Schematic of a rod with its outer segment in an SE. (A) Infrared image of a Kv2.1+/− rod outer segment in a suction pipette with overlay of key elements of the electrical recording system, and outline of the outer and inner segments (dashed line). A physical model of the pipette with neck constriction length of 4–5 µm accounts for the measured resistance of the pipette without (2.5 MΩ) and with (5.0 MΩ) the rod drawn into the pipette, constraining the seal and series resistances to Rseal ∼3.3 MΩ and Rseries ∼1.7 MΩ, giving an overall collection efficiency for the current-divider circuit of approximately two thirds. The inset shows an image of a rod filled with fluorescein during a WC recording experiment from a retinal slice; the electrode (hazy fluorescence shadow at left) was sealed to the cell body. (B) Two-compartment electrical schematic of the rod identifying the respective currents of the inner and outer segments. All current components, including those in the outer segment, have material voltage dependence in the highly depolarized Kv2.1−/– rod (Fig. 5). Resistance values used: Rtissue = 10 KΩ; Rin = 158 MΩ and 113 MΩ for WT and KO rods, respectively.
Schematic of a rod with its outer segment in an SE. (A) Infrared image of a Kv2.1+/− rod outer segment in a suction pipette with overlay of key elements of the electrical recording system, and outline of the outer and inner segments (dashed line). A physical model of the pipette with neck constriction length of 4–5 µm accounts for the measured resistance of the pipette without (2.5 MΩ) and with (5.0 MΩ) the rod drawn into the pipette, constraining the seal and series resistances to Rseal ∼3.3 MΩ and Rseries ∼1.7 MΩ, giving an overall collection efficiency for the current-divider circuit of approximately two thirds. The inset shows an image of a rod filled with fluorescein during a WC recording experiment from a retinal slice; the electrode (hazy fluorescence shadow at left) was sealed to the cell body. (B) Two-compartment electrical schematic of the rod identifying the respective currents of the inner and outer segments. All current components, including those in the outer segment, have material voltage dependence in the highly depolarized Kv2.1−/– rod (Fig. 5). Resistance values used: Rtissue = 10 KΩ; Rin = 158 MΩ and 113 MΩ for WT and KO rods, respectively.
A two-compartment electrophysiological model of the rod
Electrical balance and ion species homeostasis in the dark steady state
Two general conditions must be satisfied for a rod to be in steady state in darkness: (1) electrical balance or electroneutrality, which requires that the sum of all ionic currents over the membrane must be zero; and (2) ion species homeostasis, which requires that the inward and outward fluxes of each permeant ion species (in particular, Na+, K+, Ca2+) must be equal. For characterizing the electrical balance, the measured dark resting potential was assumed to correspond to that of the inner segment (Vm,IS,rest) in the model, as the WC electrode was patched to the cell body (Fig. 1, inset). The magnitude of the total outer segment membrane current in the dark was determined from analysis of the saturating photocurrent, with both CNG (ICNG) and NCXK (INCKX) contributing net inward current suppressed by strong stimulation. A key empirical parameter characterizing the saturating photocurrent is the fraction arising from the NCKX, estimated from SE photocurrents to be 0.06 for WT rods (Makino et al., 2004) and herein from 0.15 to 0.45 for Kv2.1−/− rods (Fig. S1 and Fig. S2). Because INCKX is voltage-dependent (Lagnado et al., 1988; Lagnado and McNaughton, 1990; Eq. 4), the magnitude of INCKX extracted from the photocurrent after a bright flash must be understood to be that at the hyperpolarized potential of the saturating response, not at the resting potential, and thus the magnitude of the NCKX current at the resting potential must be inferred from the voltage dependence of the NCKX current (Fig. 8). Analysis of the saturating photocurrent also allowed the magnitude of ICNG in darkness before the flash to be determined (Fig. 7 and Fig. S2), which in combination with INCKX predicts fCa, the CNG current carried by Ca2+, and the complementary fraction carried by Na+ (fNa = 1 – fCa), as described below (Eq. 5). With the dark current partitioned into ICNG and INCKX, the inward flux of Na+ was readily calculated from these currents, fCa, and the stoichiometry of the NCKX (4Na+ in: 1 Ca2+, 1 K+ out). Because the only path for Na+ efflux from the rod is via Na+/K+ exchange (NKX) in the inner segment, the contribution of the electrogenic NKX to the outward dark current was thus readily determined, and was found to be ∼35% for WT and ∼40% for knockout (KO) rods (these values depend somewhat on the absolute magnitudes of the dark current of the individual rods as well as the partitioning between ICNG and INCKX; Fig. 7, Fig. S1, and Fig. S2). The residual non-NKX outward dark current was distributed over IKv2.1 (WT), IHCN1 (WT and KO), and a leak K+ current (WT and KO), as described in Results (Fig. 6).

Extraction of the shoulder amplitudes of Kv2.1−/− rod photocurrents. (A) Saturating photocurrent responses of a Kv2.1−/− rod to flashes of four intensities: 7.3 × 104, 3.9 × 104, 1.9 × 104, and 1.1 × 104 photons μm−2 (identified by the nominal neutral density filters labels f0, f3, f6, and f9). Each trace is the average of 2–4 responses. The raw traces were first scaled by 1.5× to adjust for SE collection efficiency (two thirds), and then were interpolated on a 100-µs time grid using the MATLAB “interp1” routine with the piecewise cubic Hermite interpolating polynomial (pchip) option. (B) Same traces as in A, but slid laterally for concurrence in the initial portion of the rising phase. (C) Average of traces in B (thick black line), plotted with running error bar (gray region) corresponding to ±2 SDs (C is replotted as Fig. S2 F). (D) Amplitudes of the shoulder levels of the traces in B, taken as the average of each trace over the interval 37–43 ms, and plotted as a function of the nominal ND filter density. (E) Saturated amplitudes of the responses in B, taken as the average amplitude over the interval 120–400 ms. (F) Scatterplot of the results in D and E. The tight bunching of the points implies that the shoulder amplitude was invariant with flash strength over this range of intensities. (G) Scatterplot of the amplitudes of the shoulders versus those of the saturating photocurrent for a population of Kv2.1−/− rods recorded with SEs and analyzed as in A–F (Pearson r = 0.84, P < 10−6); each symbol is the result from an analysis such as shown in A–F, and is thus the average of the x and y values in a panel like F. The black line is an unconstrained straight line fitted to the data by least squares, has a slope of 0.72, and accounts for 71% of the variance, i.e., r2 = 0.71. Based on the kinetics and light dependence (compare Fig. 4 and Fig. 7), the component of the photocurrent below the shoulder can be assigned unambiguously to suppression of CNG current, and so the slope of the line indicates that on average, CNG current suppression comprises 72% of the Kv2.1−/− photocurrent regardless of the underlying cause of cell-to-cell variation (which may arise, for example, from outer segment length variation). The blue line is the upper limit set by the hypothesis fCa = 1 (Eq. 5): four of the data points fall above the limit, and many others are situated close to the limit. (H) The magnitudes of the residual (non-CNG channel) saturating Kv2.1−/− rod photocurrents are plotted against the total photocurrent amplitude. In this plot, the hypothesis fCa = 1 is represented by the red line; theory (Eq. 5) predicts that no points can lie below this line if the magnitudes of the two components correspond to the dark levels of the CNG and NCKX currents; many of the points lie very near or below the limit. The closer the points to the line, the higher resting Ca2+ would be expected to be (the cyan symbols in G and H are from the rod whose photocurrents are analyzed in A–F). Avg., average; Sat, saturation.
Extraction of the shoulder amplitudes of Kv2.1−/− rod photocurrents. (A) Saturating photocurrent responses of a Kv2.1−/− rod to flashes of four intensities: 7.3 × 104, 3.9 × 104, 1.9 × 104, and 1.1 × 104 photons μm−2 (identified by the nominal neutral density filters labels f0, f3, f6, and f9). Each trace is the average of 2–4 responses. The raw traces were first scaled by 1.5× to adjust for SE collection efficiency (two thirds), and then were interpolated on a 100-µs time grid using the MATLAB “interp1” routine with the piecewise cubic Hermite interpolating polynomial (pchip) option. (B) Same traces as in A, but slid laterally for concurrence in the initial portion of the rising phase. (C) Average of traces in B (thick black line), plotted with running error bar (gray region) corresponding to ±2 SDs (C is replotted as Fig. S2 F). (D) Amplitudes of the shoulder levels of the traces in B, taken as the average of each trace over the interval 37–43 ms, and plotted as a function of the nominal ND filter density. (E) Saturated amplitudes of the responses in B, taken as the average amplitude over the interval 120–400 ms. (F) Scatterplot of the results in D and E. The tight bunching of the points implies that the shoulder amplitude was invariant with flash strength over this range of intensities. (G) Scatterplot of the amplitudes of the shoulders versus those of the saturating photocurrent for a population of Kv2.1−/− rods recorded with SEs and analyzed as in A–F (Pearson r = 0.84, P < 10−6); each symbol is the result from an analysis such as shown in A–F, and is thus the average of the x and y values in a panel like F. The black line is an unconstrained straight line fitted to the data by least squares, has a slope of 0.72, and accounts for 71% of the variance, i.e., r2 = 0.71. Based on the kinetics and light dependence (compare Fig. 4 and Fig. 7), the component of the photocurrent below the shoulder can be assigned unambiguously to suppression of CNG current, and so the slope of the line indicates that on average, CNG current suppression comprises 72% of the Kv2.1−/− photocurrent regardless of the underlying cause of cell-to-cell variation (which may arise, for example, from outer segment length variation). The blue line is the upper limit set by the hypothesis fCa = 1 (Eq. 5): four of the data points fall above the limit, and many others are situated close to the limit. (H) The magnitudes of the residual (non-CNG channel) saturating Kv2.1−/− rod photocurrents are plotted against the total photocurrent amplitude. In this plot, the hypothesis fCa = 1 is represented by the red line; theory (Eq. 5) predicts that no points can lie below this line if the magnitudes of the two components correspond to the dark levels of the CNG and NCKX currents; many of the points lie very near or below the limit. The closer the points to the line, the higher resting Ca2+ would be expected to be (the cyan symbols in G and H are from the rod whose photocurrents are analyzed in A–F). Avg., average; Sat, saturation.

Decomposition of the amplitude and rate-saturated photocurrents of Kv2.1−/− rods into three membrane current components: CNG, NCKX, and capacitive. Related to Fig. 7. The traces (heavy black line) are averages of several responses to flashes of at least three intense flash strengths up to 20,000 photons μm−2. The responses to different flash strengths were interpolated on a 100-µs grid and shifted for maximal congruence with the early rising phase (CNG channel closure) of the most intense flash. The running error (gray shaded region) is ±1 SD. A–H provide three numbers characterizing the theory traces: the saturated photocurrent (PC) amplitude; fNCKX, the fraction of the photocurrent ascribed to NCKX current; and Carest, the estimated Ca2+i in the dark-adapted rod immediately before the flash. The panels are arranged in order of increasing CaD, which ranges from approximately two times normal (A, 560 nM) to >4 µM (G); in the latter case, Carest greatly exceeds the internal binding constant Kex (1.1 µM) of the NCKX, which serves as a gauge (Fig. 7). The “average” value of Carest is 10-fold higher than the normal WT level. The values of fNCKX colored red exceed the naive theoretical limit of 0.33, which neglects the effect of hyperpolarization on the NCKX (the value of the half-saturation constant Kex of the NCKX current limits the reliability of estimating Carest, as it is highly nonlinearly dependent on fNCKX as the latter approaches its limit; thus, Carest estimates >4 µM were not differentiated). The preprocessing of the photocurrent data of each of the rods was performed with the procedure illustrated in Fig. S1.
Decomposition of the amplitude and rate-saturated photocurrents of Kv2.1−/− rods into three membrane current components: CNG, NCKX, and capacitive. Related to Fig. 7. The traces (heavy black line) are averages of several responses to flashes of at least three intense flash strengths up to 20,000 photons μm−2. The responses to different flash strengths were interpolated on a 100-µs grid and shifted for maximal congruence with the early rising phase (CNG channel closure) of the most intense flash. The running error (gray shaded region) is ±1 SD. A–H provide three numbers characterizing the theory traces: the saturated photocurrent (PC) amplitude; fNCKX, the fraction of the photocurrent ascribed to NCKX current; and Carest, the estimated Ca2+i in the dark-adapted rod immediately before the flash. The panels are arranged in order of increasing CaD, which ranges from approximately two times normal (A, 560 nM) to >4 µM (G); in the latter case, Carest greatly exceeds the internal binding constant Kex (1.1 µM) of the NCKX, which serves as a gauge (Fig. 7). The “average” value of Carest is 10-fold higher than the normal WT level. The values of fNCKX colored red exceed the naive theoretical limit of 0.33, which neglects the effect of hyperpolarization on the NCKX (the value of the half-saturation constant Kex of the NCKX current limits the reliability of estimating Carest, as it is highly nonlinearly dependent on fNCKX as the latter approaches its limit; thus, Carest estimates >4 µM were not differentiated). The preprocessing of the photocurrent data of each of the rods was performed with the procedure illustrated in Fig. S1.
Calcium ion homeostasis in the rod outer segment
Rate equations of the primary reactions of phototransduction
Rate equations describing phototransduction in mouse rods, including calcium modulation of guanylate cyclase, have been described previously (Gross et al., 2012a). Modifications to these equations, including extension of several calcium-dependent functions to higher than normal Ca2+i levels, were made in our investigation and are described in the Supplemental text (see end of the PDF). To accommodate the depolarization of Kv2.1−/− rods, the rate equation governing changes in Ca2+i in the outer segment was generalized (Eq. S15) to include the voltage-dependent relations Eq. 4 and Eq. 5. In addition, a continuous saturating Boltzmann description of fCa(Vm) was developed, constrained by values extracted from WT and Kv2.1−/− rod photocurrents (Fig. 8 C).
Rate equations for voltage-gated inner segment ion channels

Kinetic features of the two-state Boltzmann models of Kv2.1 and HCN1 channels used in the theoretical model. (A) Steady state activation functions for Kv2.1 (green) and HCN1 (magenta) channels. Dashed lines show description of the IKx current of rabbit rods (Beech and Barnes, 1989) and of Ih current (Demontis et al., 2002). (B) Voltage-dependence of the rate constants of closed → open (green curve) and open → closed (red curve) state transitions of the Kv2.1 model compared with the description (dashed curve) of Beech and Barnes (1989) of IKx kinetics. (C) Voltage dependence of rate constants of closed → open (green curve) and open → closed (red curve) state transitions of the HCN1 model compared with the characterization of Ih kinetics (dashed curve) by Demontis et al. (2002). In B and C, the unbroken black curve is the equilibrium rate constant, the sum of the rates described by the red and green curves (Demontis et al. (2002) did not characterize the deactivation of HCN1 channels in their experiments, and provided no information about their behavior for Vm greater than −60 mV. A two-state Boltzmann model that perfectly recapitulates their results and includes voltage-dependent deactivation predicts that rods reach a saturating hyperpolarization more negative than −80 mV, which has never been reported in rod photovoltage recordings; Baylor and Nunn, 1986; Cobbs and Pugh, 1987; Schneeweis and Schnapf, 1995; Cangiano et al., 2012). In addition, using an HCN1 two-state model that recaps the activation curve and kinetics of Demontis et al. (2002) requires the resting conductance parameter GHCN1 to be 1.75–2 nS to account for the WT I-V data of Fig. 6, because of the leftward voltage shift in pO. Further, adoption of such a two-state model predicts that the photovoltage response to flashes that strongly saturate, i.e., close, the CNG channel current will have a plateau of −60 mV, more negative than ever observed. In contrast, the two-state Boltzmann HCN1 model described herein predicts a photovoltage plateau of −53 mV.
Kinetic features of the two-state Boltzmann models of Kv2.1 and HCN1 channels used in the theoretical model. (A) Steady state activation functions for Kv2.1 (green) and HCN1 (magenta) channels. Dashed lines show description of the IKx current of rabbit rods (Beech and Barnes, 1989) and of Ih current (Demontis et al., 2002). (B) Voltage-dependence of the rate constants of closed → open (green curve) and open → closed (red curve) state transitions of the Kv2.1 model compared with the description (dashed curve) of Beech and Barnes (1989) of IKx kinetics. (C) Voltage dependence of rate constants of closed → open (green curve) and open → closed (red curve) state transitions of the HCN1 model compared with the characterization of Ih kinetics (dashed curve) by Demontis et al. (2002). In B and C, the unbroken black curve is the equilibrium rate constant, the sum of the rates described by the red and green curves (Demontis et al. (2002) did not characterize the deactivation of HCN1 channels in their experiments, and provided no information about their behavior for Vm greater than −60 mV. A two-state Boltzmann model that perfectly recapitulates their results and includes voltage-dependent deactivation predicts that rods reach a saturating hyperpolarization more negative than −80 mV, which has never been reported in rod photovoltage recordings; Baylor and Nunn, 1986; Cobbs and Pugh, 1987; Schneeweis and Schnapf, 1995; Cangiano et al., 2012). In addition, using an HCN1 two-state model that recaps the activation curve and kinetics of Demontis et al. (2002) requires the resting conductance parameter GHCN1 to be 1.75–2 nS to account for the WT I-V data of Fig. 6, because of the leftward voltage shift in pO. Further, adoption of such a two-state model predicts that the photovoltage response to flashes that strongly saturate, i.e., close, the CNG channel current will have a plateau of −60 mV, more negative than ever observed. In contrast, the two-state Boltzmann HCN1 model described herein predicts a photovoltage plateau of −53 mV.
Currents of the inner segment ionic mechanisms

Voltage dependence of NKX electrogenic current. (A) Normalized currents of different NKX isoforms from Stanley et al. (2015) (symbols), and identification of the retinal cell types in which they are expressed, including the rod isoform α3β2 (black circles). Data of each isoform were fitted using the MATLAB least squares fitting tool (cftool) with a Boltzmann function of the form of Eq. 12. For the curve describing the rod isoform data γ = 0.33, Vm,0.5 = −57.1 mV, and sNKX = 27.6 mV. (B) The voltage dependence of the NKX current in a WT mouse rod with 22 pA dark current; the smooth curve plots the function fitted to the rod isoform in panel scaled to have the magnitude (7.5 pA) of the NKX current at the resting potential (−32 mV) required to extrude all the Na+ that enters the rod outer segment through CNG channels and the NCKX exchanger. The saturating level of the WT mouse rod NKX current is thus predicted to be 9.3 pA. Analysis of the dependence of the turnover number of the NKX on K+o and Na+i (Stanley et al., 2015) along with the information that K+o = 6 mM in the SRS (Steinberg et al., 1980) predict the turnover number at the Vm,rest to be ∼45 s−1. Consequently, the rod inner segment would have a total of 1.3 × 106 NKX units, and given a membrane surface area 80 µm2 (length, 15 µm; diameter, 1.7 µm), an NKX expression density of ∼16,000 µm−2.
Voltage dependence of NKX electrogenic current. (A) Normalized currents of different NKX isoforms from Stanley et al. (2015) (symbols), and identification of the retinal cell types in which they are expressed, including the rod isoform α3β2 (black circles). Data of each isoform were fitted using the MATLAB least squares fitting tool (cftool) with a Boltzmann function of the form of Eq. 12. For the curve describing the rod isoform data γ = 0.33, Vm,0.5 = −57.1 mV, and sNKX = 27.6 mV. (B) The voltage dependence of the NKX current in a WT mouse rod with 22 pA dark current; the smooth curve plots the function fitted to the rod isoform in panel scaled to have the magnitude (7.5 pA) of the NKX current at the resting potential (−32 mV) required to extrude all the Na+ that enters the rod outer segment through CNG channels and the NCKX exchanger. The saturating level of the WT mouse rod NKX current is thus predicted to be 9.3 pA. Analysis of the dependence of the turnover number of the NKX on K+o and Na+i (Stanley et al., 2015) along with the information that K+o = 6 mM in the SRS (Steinberg et al., 1980) predict the turnover number at the Vm,rest to be ∼45 s−1. Consequently, the rod inner segment would have a total of 1.3 × 106 NKX units, and given a membrane surface area 80 µm2 (length, 15 µm; diameter, 1.7 µm), an NKX expression density of ∼16,000 µm−2.
This completes the description of the ionic mechanisms underlying the two-compartment model of the rod (Eq. 1 and Eq. 2), with the exception of IK,leak, which had to be postulated to electrically balance with INKX the dark current of the Kv2.1−/− rod. We considered several different types of IK channels, including channels with an ideal linear I-V curve through EK (Eq. 10a, with pO = 1), channels that follow the GHK current formula (Eq. 10b, with pO = 1), channels with strongly inwardly rectifying I-V curves, and voltage-gated K+ channels with “Kv2.1-like” voltage dependence (Eq. 10a).
Implementation of the dynamic model
The two-compartment model rod is not isopotential. In the dark state, for example, the outer segment is depolarized relative the inner segment to an extent dependent on the magnitudes of the ionic currents and the internal resistance between the segments (Rin in Fig. 1). Moreover, the local transmembrane potentials of the inner and outer segments are affected by the resistance through the tissue to the chamber ground (Rtissue), the resistance of the suction pipette to the SE virtual ground (RSE,series), and the SE seal resistance (Rseal), and the capacitances of inner and outer segments have separable but linked effects. As a consequence, Eq. 1 and Eq. 2 are not independent, but “cross-coupled” (Eq. S1, Eq. S2, and Eq. S3). We found that Eq. 1 and Eq. 2 could be reexpressed in terms of the internal potentials Vin,IS and Vin,OS in a manner that allowed them to be more readily solved for the dark steady state, and by numerical integration with a predictor-corrector method for light-driven changes (see Additional details of the theoretical model in the Supplemental text).
The driving force for light responses was light-activated spatially homogeneous phosphodiesterase (PDE) activity, expressed as in previous papers in terms of the number activated PDE catalytic subunits in the outer segment generated by photoactivated rhodopsin (R*) with a specified lifetime (Lamb and Pugh, 1992; Nikonov et al., 1998; Gross et al., 2012a). To better predict families of Kv2.1−/− rod responses, it was useful to assume that R* deactivated with a shorter time constant (25–45 ms) for subsaturating flashes, segueing to a slower one (70–100 ms) that prevailed for saturating flashes (a rationale for this assumption is provided in the Supplement). For both WT and Kv2.1−/− rods, the lifetime τE of the Gtα-PDE (“G*-E*”) complex was assigned to the “dominant time constant” extracted from the temporal spacing of the saturating responses and the logarithm of flash strengths with a “Pepperberg plot” analysis (Nikonov et al., 1998; Krispel et al., 2006; Burns and Pugh, 2009).
A set of ordinary differential equations (ODEs) governing the 11 time-dependent variables was developed and integrated with the MATLAB integrator ODE35s, and the solutions used with the current relations (Eq. 3, Eq. 4, Eq. 5, Eq. 10, Eq. 11, and Eq. 12) to predict the dark currents of the inner and outer segments, and the time course of the photoresponses for different strength flashes (compare Table S1). The variables are as follows: E*(t), number of activated PDE catalytic subunits in the outer segment; cG(t), cytoplasmic free cGMP; Ca(t), cytoplasmic free Ca2+i; Gc1a(t), active guanylate cyclase, complexed with guanylate cyclase binding proteins (GCAPs)-1–3Mg2+ (Fig. S5 A); Gc1it(t), inactive guanylate cyclase, complexed with GCAPs1-3Ca2+ (Fig. S5 A); Gc2a(t), active guanylate cyclase, complexed with GCAPs2-3Mg2+ (Fig. S5 A); Gc2it(t), inactive guanylate cyclase, complexed with GCAPs2-3Ca2+ (Fig. S5 A); pO,Kv2.1(t), open probability of Kv2.1 channels; pO,HCN1(t), open probability of HCN1 channels; Vin,OS(t), potential (reexterior ground) in the outer segment (Fig. 1 A); and Vin,IS(t), potential (reexterior ground) in the inner segment (Fig. 1 A). The initial values of the variables in the rod before the flash were specified by a genotype- and cell-specific determination of the values of all variables for the dark steady state and included adherence to both electrical balance and ion species homeostasis, as described above. Three-state models of GCAPs-1 and GCAPs-2 regulation of rod guanylate cyclase (Fig. S5 A; Makino et al., 2012) were introduced for two reasons: (1) to provide an explicit molecular description of Ca2+ buffering, and (2) to provide a means of “damping” the activation and deactivation of guanylate cyclase by rapid changes in Ca2+i predicted to occur in the Kv2.1−/− rod arising from rapid changes in Ca2+ fluxes in or out of the outer segment attendant membrane hyperpolarization and return to its normally depolarized state. Further details are provided in the Supplemental text, along with a list of the parameters (Table S2).

Three-state guanylate cyclase (GC)–GCAPs model and Ca2+ buffering by GCAPs and recoverin. (A) Schematic of regulation of rod guanylate cyclase activity through Ca2+ and Mg2+ binding and unbinding to tightly associated GCAP1 protein (after Dizhoor et al., 2010). Each GC holomer is a dimer with three tightly associated guanylate cyclase type 1 activating proteins (GCAP1), and each GCAP1 has three functional EF hand calcium binding sites, for a total of six binding sites per GC. The calcium-bound GCAP1s inhibit the GC (inactive). When Ca2+i declines in the outer segment during the light response, Ca2+ dissociates from GCAP1 and is replaced by Mg2+, stimulating increased GC synthesis of cGMP. The schematic postulates the existence of a transient intermediate state in which the GCAPs are free of divalent cation. A parallel three-state scheme (not shown) is assumed to govern the interaction of GCAP2 with GC. (B) Decomposition of total steady state rod GC activity into a component regulated by GCAP1 and a second component regulated by GCAP2 (after Makino et al., 2008). The function defining total GC activity (black curve) is essentially that of Gross et al. (2012a), but manipulated for Ca2+i greater than the WT level (open circle at 320 nM) so that in a Kv2.1−/− rod at rest (filled circle), cGMP synthesis in the dark gives the appropriate level of CNG channel current (compare Fig. 8 B). The green and blue curves represent the activity of GC-GCAP2 and GC-GCAP1, respectively, and their summed activity is given by the red curve. The blue and green curves are determined Eq. S1 and Eq. S2 with parameters Kcyc,2 = 48 nM, ncyc,2 = 2.0, Kcyc,1 = 103 nM, and ncyc,1 = 2.3, respectively. The absolute scaling of the two curves was set by the least square fitting to the overall activity function (black curve), which saturates at 150 µM s−1; given a total GC concentration of 5 µM and the same turnover for each GC-GCAPj enzyme complex, the fitted concentrations of the two components are GC-GCAP1 = 1.77 µM and GC-GCAP2 = 3.23 µM. (C) Rod Ca2+ buffer power can be described in terms of Ca2+ binding by the EF hand proteins, GCAP1 and GCAP2, with three functional Ca2+ binding sites each; and recoverin, with two functional Ca2+ binding sites. Given the known stoichiometry of Ca2+ binding and the concentrations of GC (taken to be 5 µM referred to the outer segment cytoplasm) and recoverin (taken to be 25 µM), overall buffer power can be computed. The results of the calculation (thickened black curve) are compared with description of calcium buffering of salamander rods by Lagnado et al. (1992) (black dashed curve); also shown is the latter curve scaled by 65% (lighter dashed curve). The symbols plot the calculated Ca2+ buffer power at the resting Ca2+ levels of the WT (open circle) and Kv2.1−/− rod (filled circle; a buffer component with calcium-independent buffer power of 5 is not plotted; this component helps stabilize Ca2+i at nanomolar concentrations where the buffering power of GCAP2 declines).
Three-state guanylate cyclase (GC)–GCAPs model and Ca2+ buffering by GCAPs and recoverin. (A) Schematic of regulation of rod guanylate cyclase activity through Ca2+ and Mg2+ binding and unbinding to tightly associated GCAP1 protein (after Dizhoor et al., 2010). Each GC holomer is a dimer with three tightly associated guanylate cyclase type 1 activating proteins (GCAP1), and each GCAP1 has three functional EF hand calcium binding sites, for a total of six binding sites per GC. The calcium-bound GCAP1s inhibit the GC (inactive). When Ca2+i declines in the outer segment during the light response, Ca2+ dissociates from GCAP1 and is replaced by Mg2+, stimulating increased GC synthesis of cGMP. The schematic postulates the existence of a transient intermediate state in which the GCAPs are free of divalent cation. A parallel three-state scheme (not shown) is assumed to govern the interaction of GCAP2 with GC. (B) Decomposition of total steady state rod GC activity into a component regulated by GCAP1 and a second component regulated by GCAP2 (after Makino et al., 2008). The function defining total GC activity (black curve) is essentially that of Gross et al. (2012a), but manipulated for Ca2+i greater than the WT level (open circle at 320 nM) so that in a Kv2.1−/− rod at rest (filled circle), cGMP synthesis in the dark gives the appropriate level of CNG channel current (compare Fig. 8 B). The green and blue curves represent the activity of GC-GCAP2 and GC-GCAP1, respectively, and their summed activity is given by the red curve. The blue and green curves are determined Eq. S1 and Eq. S2 with parameters Kcyc,2 = 48 nM, ncyc,2 = 2.0, Kcyc,1 = 103 nM, and ncyc,1 = 2.3, respectively. The absolute scaling of the two curves was set by the least square fitting to the overall activity function (black curve), which saturates at 150 µM s−1; given a total GC concentration of 5 µM and the same turnover for each GC-GCAPj enzyme complex, the fitted concentrations of the two components are GC-GCAP1 = 1.77 µM and GC-GCAP2 = 3.23 µM. (C) Rod Ca2+ buffer power can be described in terms of Ca2+ binding by the EF hand proteins, GCAP1 and GCAP2, with three functional Ca2+ binding sites each; and recoverin, with two functional Ca2+ binding sites. Given the known stoichiometry of Ca2+ binding and the concentrations of GC (taken to be 5 µM referred to the outer segment cytoplasm) and recoverin (taken to be 25 µM), overall buffer power can be computed. The results of the calculation (thickened black curve) are compared with description of calcium buffering of salamander rods by Lagnado et al. (1992) (black dashed curve); also shown is the latter curve scaled by 65% (lighter dashed curve). The symbols plot the calculated Ca2+ buffer power at the resting Ca2+ levels of the WT (open circle) and Kv2.1−/− rod (filled circle; a buffer component with calcium-independent buffer power of 5 is not plotted; this component helps stabilize Ca2+i at nanomolar concentrations where the buffering power of GCAP2 declines).
Online supplemental material
Fig. S1 gives a detailed description of the analysis used to extract the amplitude of the unusual “shoulder” component from saturating outer segment photocurrents of Kv2.1−/− rods, and provides statistics of this analysis from a population of rods. Fig. S2 applies the theory to saturating photocurrent responses to extract the CNG channel, NCKX, and capacitive components of a population of Kv2.1−/− rods, supplementing the same analysis as applied in Fig. 7 of the main text. Fig. S3 graphically presents the two-state Boltzmann models used to describe the voltage-dependent activation and deactivation of Kv2.1 and HCN1 channels. Fig. S4 presents a reanalysis of the voltage dependence of electrogenic currents of various NKX isoforms from the study of Stanley et al. (2015), parametrically characterizing them as modified Boltzmann functions. The I-V curve of the rod isoform, α3β2, is a key determinant of the outward dark current of the inner segment. Fig. S5 presents a graphic analysis of the dependence of the guanylate cyclase activity of the mouse rod, extending that dependence into the “high calcium” regimen required for understanding the Kv2.1−/− rod phenotype. It also presents a novel analysis of the contribution of GCAPs to the calcium buffer power of the rod. These analyses play key roles in the theory. Scheme 1 presents a schematic of a four-state CNG channel model developed in the effort to understand the unusual kinetics of the recovery phases of subsaturating Kv2.1−/− rod photocurrent responses in terms of voltage-dependent divalent cation block and rapid relief of block. Table S1 shows variables and rate equations of the two-compartment rod. Table S2 shows parameters of the two-compartment rod model and values employed.

Four-state CNG channel model incorporating control by both cGMP and membrane potential (Vm), with the latter determining whether the channel is blocked by divalent cations or unblocked. The four boxes identify the concentrations of channels in the open-blocked (OB), open-unblocked (OU), closed-blocked (CB), and closed-unblocked states. The simplest dynamic version consistent with Eq. 2 has voltage-dependent rates that are independent of cGMP and cGMP-dependent rates independent of membrane potential. The dynamic model potentially allows transitions blocked → unblocked and unblocked → blocked to have different dependence on membrane potential, which could serve to stabilize CNG current against rapid increases in Ca2+.
Four-state CNG channel model incorporating control by both cGMP and membrane potential (Vm), with the latter determining whether the channel is blocked by divalent cations or unblocked. The four boxes identify the concentrations of channels in the open-blocked (OB), open-unblocked (OU), closed-blocked (CB), and closed-unblocked states. The simplest dynamic version consistent with Eq. 2 has voltage-dependent rates that are independent of cGMP and cGMP-dependent rates independent of membrane potential. The dynamic model potentially allows transitions blocked → unblocked and unblocked → blocked to have different dependence on membrane potential, which could serve to stabilize CNG current against rapid increases in Ca2+.
Results
Kv2.1 is expressed in the distal inner segments of rod photoreceptor cells
To assess the structural status of Kv2.1−/− photoreceptors in vivo, we used OCT to image the retinas of littermate pairs of young mice (Fig. 2 A). Compared with those of Kv2.1+/− littermate controls, the ONL thickness of the Kv2.1−/− retina was reduced by ∼33% at 8 wk of age, indicating a comparable loss of rods. OCT also revealed the outer segments of Kv2.1−/− rods of young mice to be ∼30% shorter than those of Kv2.1+/− littermates (Fig. 2, A and B; cf. also Fig. 11). All else being equal, the combination of rod loss and outer segment shortening predicts an ∼50% reduction in total rhodopsin in Kv2.1−/− retina, close to the 60% reduction in rhodopsin content that we measured spectrophotometrically from dark-adapted littermate retinas (Fig. 2 C).

OCT reveals thinning of photoreceptor layers of the Kv2.1−/− retina, and immunostaining distinctive patterns of Kv2.1 and HCN1 expression in the rod IS. (A) OCT B-scans of central retina of live heterozygous and Kv2.1−/− mice. CC, choriocapillaris; OPL, outer plexiform layer; GCL, ganglion cell fiber layer. The OCT B-scans have been aligned at the ELM; a red dashed line through the ELM is extended across the figure to the histochemical sections in D to show alignment of the two kinds of section. (B) Axial profile plots of the scattering intensity of the B-scans in A, aligned at the ELM (red dashed line) to help identify highly scattering retinal features. (C) Measurement of rhodopsin content of WT and Kv2.1−/− (KO) retinas (n = 5 retinas from three mice of each genotype). Error bars are SD. (D) Retinal sections stained for Kv2.1 and HCN1 reveal Kv2.1 expression to be narrowly confined to the apical inner segment compartment of photoreceptors (orange), while HCN1 resides more broadly in the basal inner segment as well as the synaptic layers (bottom, green). Scale bars, 20 µm. (E) Higher magnified views of representative sections of WT retina shown in D, with red, green, and merged channels separately shown to highlight the differences in Kv2.1 and HCN1 channel staining. Scale bars, 5 μm. (F) Representative immunoblot of homogenates of WT and Kv2.1−/− mice labeled with antibodies for Kv2.1 (blue) and HCN1 (red, upper band), with Grp75 served as loading control (red, lower band). Numbers at right indicate positions of molecular weight standards (kD). Individual regions/channels from the blot are also shown below in black and white. (G) Quantification of Kv2.1 and HCN1 protein levels by Western blot (mean ± SEM, n = 10 mice/group). Fluorescence intensities were background-subtracted, normalized by the loading control (Grp75), and expressed relative to WT. HCN1 expression was not statistically different between WT and KO samples (P > 0.05).
OCT reveals thinning of photoreceptor layers of the Kv2.1−/− retina, and immunostaining distinctive patterns of Kv2.1 and HCN1 expression in the rod IS. (A) OCT B-scans of central retina of live heterozygous and Kv2.1−/− mice. CC, choriocapillaris; OPL, outer plexiform layer; GCL, ganglion cell fiber layer. The OCT B-scans have been aligned at the ELM; a red dashed line through the ELM is extended across the figure to the histochemical sections in D to show alignment of the two kinds of section. (B) Axial profile plots of the scattering intensity of the B-scans in A, aligned at the ELM (red dashed line) to help identify highly scattering retinal features. (C) Measurement of rhodopsin content of WT and Kv2.1−/− (KO) retinas (n = 5 retinas from three mice of each genotype). Error bars are SD. (D) Retinal sections stained for Kv2.1 and HCN1 reveal Kv2.1 expression to be narrowly confined to the apical inner segment compartment of photoreceptors (orange), while HCN1 resides more broadly in the basal inner segment as well as the synaptic layers (bottom, green). Scale bars, 20 µm. (E) Higher magnified views of representative sections of WT retina shown in D, with red, green, and merged channels separately shown to highlight the differences in Kv2.1 and HCN1 channel staining. Scale bars, 5 μm. (F) Representative immunoblot of homogenates of WT and Kv2.1−/− mice labeled with antibodies for Kv2.1 (blue) and HCN1 (red, upper band), with Grp75 served as loading control (red, lower band). Numbers at right indicate positions of molecular weight standards (kD). Individual regions/channels from the blot are also shown below in black and white. (G) Quantification of Kv2.1 and HCN1 protein levels by Western blot (mean ± SEM, n = 10 mice/group). Fluorescence intensities were background-subtracted, normalized by the loading control (Grp75), and expressed relative to WT. HCN1 expression was not statistically different between WT and KO samples (P > 0.05).
To localize the expression of Kv2.1 in the mouse retina, we imaged fixed retinal sections of WT and Kv2.1−/− littermates immunolabeled for Kv2.1. In WT retinas, Kv2.1 labeling was found in rod inner segments, and appeared more intense in the distal inner segment near the inner/outer segment border (Fig. 2, D and E). In contrast, the hyperpolarization-activated channel HCN1 appeared evenly distributed throughout the inner segment. Notably, Kv2.1 labeling was undetectable in all other photoreceptor compartments, and far lower in all other cell types of the WT retina under our immunolabeling and imaging conditions (Fig. 2 D, left). Kv2.1 labeling was completely absent in Kv2.1−/− sections processed and imaged in parallel (Fig. 2 D, right), demonstrating the specificity of the Kv2.1 antibody. To our knowledge, this is the first time that the distal and proximal regions of the inner segment plasma membrane have been shown to have distinct ion channel expression patterns. To test whether the expression of HCN1 was also affected by the loss of Kv2.1, we used quantitative Western blotting (Fig. 1, F and G). Samples from Kv2.1−/− and WT littermate controls normalized by the loading control (Grp75) showed comparable levels of HCN1 in the presence of absence of Kv2.1. This suggests that Kv2.1 does not play an essential role in the proper protein trafficking of other inner segment ion channels.
In summary, Kv2.1 is highly expressed in the rod inner segment, but expression in the other compartments of mouse photoreceptors is below detection. This expression pattern is consistent with it playing a major role in the outward component of the rod circulating current, which exits predominantly from the inner segment region in rodent rods (Hagins et al., 1970).
The rod circulating current of Kv2.1−/− mice is substantially reduced from its normal level
The ERG provides a noninvasive in vivo functional assessment of the retina at multiple levels of processing. The ERGs of WT (Fig. 3, A and B) and KO (Fig. 3, C and D) littermates were strikingly different (Table 1). The magnitude of the a-waves, the initial, negative-going field potential generated by the suppression of the rod circulating current, was reduced in the Kv2.1−/− eye to ∼50% of its WT level. The activation kinetics (slope of the a-wave) of the normalized a-wave differed negligibly between WT and Kv2.1−/− mice (Fig. 3 E), suggesting that the biochemistry of phototransduction is similarly activated by light in the two genotypes (Pugh and Lamb, 1993). The saturated amplitude of the b-wave, which is generated by inner retinal neurons, was reduced by ∼20%, and its initiation was notably delayed. The slow and positive-going c-wave, which is generated by the RPE layer in response to the light-evoked decrease in extracellular K+ in the subretinal space (SRS; Oakley and Green, 1976; Steinberg et al., 1980), was definitely present in the Kv2.1−/− ERG, but was reduced to half the WT amplitude, and displayed a substantially shortened time to peak (1,680 ± 30 ms in WT versus 740 ± 80 ms in Kv2.1−/−; Fig. 3, B and D). To obtain a more detailed description of the rod photocurrent, we turned to recordings from outer segments with SEs.

Rod-driven ERG a-waves are reduced 50% in amplitude in the Kv2.1−/− retina . (A and C) ERG response families from WT (black) and Kv2.1−/− mice (red). The initial corneal-negative component is the a-wave, whose underlying generator is the suppression of the rod circulating current. The immediately following corneal-positive component is the b-wave, whose generators are largely the ON (depolarizing) bipolar cells post-synaptic to the rods and cones (see labels). Range of flash strengths is shown in F. (Lighter bands surrounding the traces are running SEMs of recordings to the same stimuli from multiple mice (n = 4 for Kv2.1−/−; n = 5 for WT, age 42 d). (B and D) The same ERGs as in A and C presented on a much slower time base to display the corneal-positive going c-waves. (E) Representation of a selection of the a-waves from A and C after normalization to −1 reveals the rising phase of the a-waves of the WT and Kv2.1−/− mice to have similar activation kinetics and dependence on flash strength. (F) Dependence of the a-wave amplitude on flash intensity for WT (black symbols) and Kv2.1−/− (red symbols) mice.
Rod-driven ERG a-waves are reduced 50% in amplitude in the Kv2.1−/− retina . (A and C) ERG response families from WT (black) and Kv2.1−/− mice (red). The initial corneal-negative component is the a-wave, whose underlying generator is the suppression of the rod circulating current. The immediately following corneal-positive component is the b-wave, whose generators are largely the ON (depolarizing) bipolar cells post-synaptic to the rods and cones (see labels). Range of flash strengths is shown in F. (Lighter bands surrounding the traces are running SEMs of recordings to the same stimuli from multiple mice (n = 4 for Kv2.1−/−; n = 5 for WT, age 42 d). (B and D) The same ERGs as in A and C presented on a much slower time base to display the corneal-positive going c-waves. (E) Representation of a selection of the a-waves from A and C after normalization to −1 reveals the rising phase of the a-waves of the WT and Kv2.1−/− mice to have similar activation kinetics and dependence on flash strength. (F) Dependence of the a-wave amplitude on flash intensity for WT (black symbols) and Kv2.1−/− (red symbols) mice.
Properties of WT and Kv2.1–/– mouse ERGs
| Maximal a-wave, μVa | Maximal b-wave, μV | Maximal c-wave, μVa | |
|---|---|---|---|
| WT | 1,075 ± 15 (4) | 2,103 ± 16 (4) | 1,828 ± 25 (4) |
| Kv2.1–/– | 497 ± 13 (4) | 1,710 ± 23 (4) | 977 ± 13 (4) |
All values are mean ± SEM; values in parentheses give the number of eyes. See Fig. 3 for graphical identification of the ERG component waves.
P < 0.01.
Photocurrents of Kv2.1−/− rod outer segments have reduced saturating amplitude, decreased light sensitivity, biphasic rising phases, and recovery undershoots
The saturated amplitudes of photocurrents of Kv2.1−/− rods recorded from outer segments with SEs were substantially reduced relative to those of WT littermates (Fig. 4 and Table 2). Kv2.1−/− rods were less sensitive to light, with peak response versus intensity curves shifted to ∼2.5-fold higher intensities (Fig. 4 F and Table 2, Io values). A natural interpretation of these results consistent with the ERG data would be that in the absence of the outward current normally supplied by Kv2.1 channels, the CNG channels of Kv2.1−/− rods carry roughly half their normal inward dark current. However, the SE data revealed the Kv2.1−/− rod outer segment’s physiology to be altered in a more drastic manner than just reduction of inward current. The first clue to such alteration was provided by highly unusual kinetic features of the photocurrent responses. Specifically, most KO rods exhibited shoulders on the rising phase of the responses to saturating (and sometimes subsaturating) flashes (Fig. 4, D and E; Fig. S1; and Fig. S2), and the recoveries from both dim and bright flashes invariably exhibited prominent undershoots below baseline (Fig. 4, C and E). As explained below, both these features point to substantial alteration in mechanisms governing Ca2+ balance in the Kv2.1−/− outer segment. To obtain further insight into the physiological processes underlying these features, we performed WC recordings (Fig. 5 and Table 3), and developed a theoretical model of the rod’s dark- and photo-currents, separating current contributions from the inner and outer segments (see Theory and Fig. 1).

In the absence of Kv2.1 channels, rods generate a reduced photocurrent response whose rising phase has a shoulder, and whose recovery phase undershoots the dark-adapted baseline. (A–C) Families of photocurrent responses of a WT rod, a rod of a Kv2.1−/− mouse (−/−), and one from a heterozygote (+/−). Flash strengths ranged from 7 to 4,000 photons μm−2. (D) A comparison of averaged saturating photocurrent responses of WT and Kv2.1−/− rods; the dashed line presents the WT response scaled to the amplitude of the Kv2.1−/− response. (E) Selected responses from C separated and replotted on an expanded time scale (arbitrarily shifted vertically with the response to the dimmest flash at the bottom). (F) Response versus intensity relations extracted from the response families in A–C (traces for some strongly saturating responses are not shown in A–C). Data were fitted with exponential saturation functions, 1 − exp(−I/Io) with Io values of 108, 112, and 262 photons μm−2 for the WT, +/−, and −/− rod responses, respectively. (G) Pepperberg plot of time in saturation as a function of the logarithm of the flash strength. The abscissa values (I’) were scaled by the ratio of the Io value of each group to Io for the WT group. The slope of the lower component estimates the dominant recovery time constant for flashes producing less than ∼1 photoisomerization per disc face; see Table 2 for average values.
In the absence of Kv2.1 channels, rods generate a reduced photocurrent response whose rising phase has a shoulder, and whose recovery phase undershoots the dark-adapted baseline. (A–C) Families of photocurrent responses of a WT rod, a rod of a Kv2.1−/− mouse (−/−), and one from a heterozygote (+/−). Flash strengths ranged from 7 to 4,000 photons μm−2. (D) A comparison of averaged saturating photocurrent responses of WT and Kv2.1−/− rods; the dashed line presents the WT response scaled to the amplitude of the Kv2.1−/− response. (E) Selected responses from C separated and replotted on an expanded time scale (arbitrarily shifted vertically with the response to the dimmest flash at the bottom). (F) Response versus intensity relations extracted from the response families in A–C (traces for some strongly saturating responses are not shown in A–C). Data were fitted with exponential saturation functions, 1 − exp(−I/Io) with Io values of 108, 112, and 262 photons μm−2 for the WT, +/−, and −/− rod responses, respectively. (G) Pepperberg plot of time in saturation as a function of the logarithm of the flash strength. The abscissa values (I’) were scaled by the ratio of the Io value of each group to Io for the WT group. The slope of the lower component estimates the dominant recovery time constant for flashes producing less than ∼1 photoisomerization per disc face; see Table 2 for average values.
Properties of photocurrents of WT and Kv2.1–/– rods recorded with SEs
| Maximal photocurrent, pAa | Dim flash time to peak, msa | Dominant time constant of recovery, ms | Io, photons µm−2a | |
|---|---|---|---|---|
| WT | 12.8 ± 0.7 (22) | 110 ± 5 (22) | 168 ± 9 (22) | 41 ± 3 (22) |
| Kv2.1–/– | 5.1 ± 0.3 (15) | 71 ± 3 (7) | 140 ± 6 (15) | 106 ± 8 (14) |
Io, flash strength that elicited a half-maximal photocurrent response. All values are mean ± SEM; values in parentheses give the number of rods.
P < 0.05.

Current clamp photovoltage responses of Kv2.1−/− rods have greatly slowed rising phases. (A) Photovoltage family of a WT rod. (B) Photovoltage family of a Kv2.1−/− rod. Flash strengths ranged from 8 to 800 photons μm−2. (C) Average response versus intensity functions for photovoltage responses; smooth curves are exponential saturation functions (the flash strengths available in the slice recording apparatus were limited in range to ∼10-fold less than those used in the SE experiments; theory and experiments in other species show that the nose seen in the WT response to the strongest flash [A] is expected to reach approximately −75 mV, with relatively rapid relaxation to the approximately −55 mV plateau; Baylor and Nunn, 1986; Cobbs and Pugh, 1987; Schneeweis and Schnapf, 1995; Cangiano et al., 2012).
Current clamp photovoltage responses of Kv2.1−/− rods have greatly slowed rising phases. (A) Photovoltage family of a WT rod. (B) Photovoltage family of a Kv2.1−/− rod. Flash strengths ranged from 8 to 800 photons μm−2. (C) Average response versus intensity functions for photovoltage responses; smooth curves are exponential saturation functions (the flash strengths available in the slice recording apparatus were limited in range to ∼10-fold less than those used in the SE experiments; theory and experiments in other species show that the nose seen in the WT response to the strongest flash [A] is expected to reach approximately −75 mV, with relatively rapid relaxation to the approximately −55 mV plateau; Baylor and Nunn, 1986; Cobbs and Pugh, 1987; Schneeweis and Schnapf, 1995; Cangiano et al., 2012).
Kv2.1−/− rods are depolarized in the dark-adapted state, and exhibit higher Rm and slowed photovoltages
The dark-adapted state is a special set point for the normal rod, a state in which inward and outward currents are balanced, the fluxes of all permeant ion species are in homeostatic equilibrium, and the steady state biochemistry of the normal outer segment is optimized for reproducibly signaling single photons. Loss of the hyperpolarizing Kv2.1 current would be expected to lead to depolarization, and indeed the resting membrane potential of Kv2.1−/− rods was substantially depolarized relative to WT rods: Vm,rest = −12.1 ± 3.0 mV versus −31.5 ± 1.1 mV, respectively (Table 3, dark Vm, P < 0.001). Photovoltage responses of WT rods to the maximally intense stimulus (800 photons μm−2) usually exhibited a small “nose” followed by relaxation to a plateau between −50 mV and −55 mV (Fig. 5 A). Comparison with previous recordings from mammalian rods (Schneeweis and Schnapf, 1995; Cangiano et al., 2012) suggests that had it been possible to deliver more intense flashes, the peak nose amplitude would likely have reached −70 to −75 mV, while the plateau level would be unchanged. In addition to being depolarized, Kv2.1−/− rods generated photovoltages (current clamp responses) with notably slowed rising phases (Fig. 5). This slowing may arise in part from an increase of membrane resistance of Kv2.1−/− rods (Table 3, dark Rm, P < 0.05).
Properties of WT and Kv2.1−/− rods recorded with WC electrodes
| Dark Vm, mV | Light Vm, mV | Dark Rm, GΩ | Rmax, pA | tpeak, ms | |
|---|---|---|---|---|---|
| WT | −31.5a ± 1.1 (10) | −45.4 ± 5.2 (5) | 1.77b ± 0.12 (7) | 17.9 ± 1.6 (15) | 77b ± 8 (6) |
| Kv2.1−/− | −12.1a ± 3.0 (4) | −50.8 ± 4.8 (5) | 2.31b ± 0.24 (4) | 14.6 ± 1.8 (9) | 110b ± 17 (7) |
Numerical quantities are the mean ± SEM, with the number of biological replicates in parentheses. Membrane potentials (dark Vm) were corrected for the liquid junction potential of the K gluconate internal solution relative to ground (8.2 ± 0.3 mV, n = 3). Vm was measured immediately upon access as the zero current holding potential, then Rm as the total series resistance to ground, followed by measurement of the amplitude of responses recorded under voltage clamp (Rmax), and current clamp (light Vm) that produced saturated photocurrents. The time to peak (tpeak) was taken as that of the photovoltage response producing a half-saturated amplitude.
P < 0.001.
P < 0.05.
WC voltage clamp to the WT Vm,rest restores Kv2.1−/− rod outer segments’ dark current
The saturating amplitude of WC photocurrents of WT rods voltage clamped to their resting potential and that of unclamped WT rods recorded with SEs are statistically indistinguishable (P = 0.47): Rmax = 17.9 pA (Table 3) versus Rmax = 19.2 pA (Table 2, adjusted for two thirds SE collection efficiency). This equivalency between Rmax obtained from WT rods with the two recording modes contrasts strikingly with the highly reliable (P < 10−4) difference between the saturating amplitudes of Kv2.1−/− rods under clamped and unclamped conditions: Rmax = 14.6 pA (Table 3) versus Rmax = 7.7 pA (Table 2, adjusted for SE efficiency). A further striking result is that, when adjusted for the measured 30% shorter length of the Kv2.1−/− rod outer segment (Fig. 2), under voltage clamp to the WT resting potential, the saturating photocurrents of WT and Kv2.1−/− rods are indistinguishable: Rmax = 17.9 pA versus Rmax = 18.9 pA (P = 0.7). The simplest interpretation of these results is that clamping its membrane potential to the resting potential of the WT rod restores the CNG and NCKX currents of the Kv2.1−/− rod to an essentially normal level. In contrast, the depolarization of the unclamped Kv2.1−/− rod can be expected to reduce NCKX activity (Eq. 4), reduce the magnitude of the CNG channel current (Eq. 3), and increase the fraction of the CNG channel current carried by Ca2+ (Eq. 4), leading to increased intracellular calcium and reduced synthesis of cGMP, further contributing to decreased CNG current. In the following sections, we make the case that these predictable changes in fact occur.
Evaluation of ionic mechanisms contributing to the dark current of WT and Kv2.1−/− rods
WC voltage clamp current/voltage (I-V) measurements, combined with saturating photocurrents to measure the magnitude of outer segment membrane current, provides a means of assessing how different components of membrane current contribute to the dark steady state (Fig. 6). The hypothesis to be tested by application of the theory to I-V data is that the only difference between rods of the two genotypes is the absence of Kv2.1 current. The known membrane currents to be summed to test this hypothesis are those illustrated in Fig. 1, viz., in the outer segment the CNG channel and electrogenic NCKX currents, and in the inner segment the NKX, Kv2.1 and HCN1 currents, and an unidentified K+ leak current. In testing this hypothesis, the sum of the CNG and NCKX currents is forced to go through the respective measured −Rmax values (Fig. 6, B and D, filled black symbols). Other constraints on the theory are that the efflux of Na+ via the NKX of the inner segment must match the influx of Na+ in the outer segment through CNG channels and the NCKX at WT Vm,rest (red symbol), the potential to which both genotype rods were clamped when not undergoing voltage steps; and that the HCN1 currents for both genotypes should be the same in all respects. For both WT and Kv2.1−/− rods held at membrane potentials below −50 mV the I-V, curves are dominated by HCN1 current (compare pink and red curves between Fig. 6, B and D) with conductance parameters (GHCN1,rest; Table S2) differing by 15% for the two genotypes, a difference undetectable with histology or Western analysis (Fig. 2). The voltage dependence of activation of HCN1 channels used here (Fig. 6, B and D; and Fig. S3) was shifted to more depolarized potentials by −25 mV from that reported by Demontis et al. (2002). Without this shift, the HCN1 conductance parameter (GHCN1,rest) required to account for the inward current below −40 mV in both genotypes would have to be 1.75–2 nS, and even with such values, the description of the I-V data is qualitatively poorer (compare Fig. S3 and attendant text).

Contributions of outer segment and inner segment ionic mechanisms to the voltage clamp currents of dark-adapted WT and Kv2.1−/− rods. (A and C) WC voltage clamp current as a function of holding potential for a population of dark-adapted WT (A; n = 5) and Kv2.1−/− (B; n = 5) mouse rods; error bars are 95% confidence intervals, and the shaded gray areas reflect the observed physiological range of membrane potentials measured from the WC recordings. Zero-current holding potentials (filled red symbols with lateral error bars; Table 2) were measured immediately upon WC access. The solid black curves are generated as the sum of the red and black curves in the respective lower panels. In A and C, the dashed curves present the I-V curve used to describe the data of the other genotype; thus, for example, the dashed curve in A replots the unbroken black curve in C. (B) Component analysis of the voltage clamp current of the dark-adapted WT rod. The two outer segment inward components are CNG current (dark blue) and NCKX current (cyan); their sum is given by the black curve; this curve was required to go through the filled black symbols, which plot the negative of the photocurrents (−Rmax, Table 3) measured at the effective outer segment holding potential; the error bars are 95% confidence intervals. The CNG current curve is the Boltzmann function of Eq. 3 with reversal potential VCNG = +8.5 mV and steepness factor sCNG = 14 mV (Baylor and Nunn, 1986). The NCKX current curve is given by Eq. 4, with VNCKX = −14 mV, sNCKX = 70 mV, KNCKX = 1,100 nM (Schnetkamp et al., 1991), and INCKX,sat(VNCKX) = −2.0 pA and −1.6 pA for WT and Kv2.1−/−, respectively (Lagnado and McNaughton, 1991). The inner segment components are the NKX electrogenic current (α3β2 isoform; Eq. 12; Supplemental text), Kv2.1 and HCN1 currents, and an unidentified K+ leak current; the sum of the inner segment components is given by the red curve. The Kv2.1 curve was generated with Eq. 10b with PK adjusted so that the current at Vm,rest is 9.1 pA, corresponding (with EK = −91 mV) to GKv2.1 = 0.18 nS (Eq. 10a). The leak current magnitude is assumed to be identical in both genotypes, and has the magnitude 2.7 pA at −32.0 mV. The HCN1 I-V curve was generated with Eq. 11 with GHCN1,rest = 1.45 nS and EHCN1 = −31 mV. The sum of the red and black curves in B is plotted as the black curve in A. The inner segment zero-current holding potential is plotted as a red circle at the average Vm, rest for WT rods (−32 mV). The filled black circle, which has the same magnitude, is plotted at the resting potential of the outer segment, which is situated slightly positively relative to the red symbol because the circulating current flows through the internal resistance of the rod (Fig. 1). At the WT resting potential, the currents are not only in electrical equilibrium but also produce homeostasis of the permeant ions Na+, K+, and Ca2+, based on the stoichiometry and other properties of the NCXK and NKX exchangers (see Theory). (D) Component analysis of the Kv2.1−/− rod dark current. The components are the same as those of the WT rod, but in the absence of Kv2.1 channels, an unidentified K+ leak current of 5.0 pA at the resting potential (−12 mV) of the inner segment is required for electrical balance. Extrapolated to Vm,rest of the WT rod, this current would have the amplitude 2.7 pA, which is equal to the magnitude of the leak current assigned to the WT rod at Vm,rest in B. For the HCN1 current, GHCN1,rest = 1.6 nS, and EHCN1 = −31 mV as in the WT rod. At the resting potential, the Kv2.1 current accounts for 80% of the total non-NKX, non-HCN1 outward current (the ordinate scale in D has been expanded relative to that in B to facilitate inspection of the component curves).
Contributions of outer segment and inner segment ionic mechanisms to the voltage clamp currents of dark-adapted WT and Kv2.1−/− rods. (A and C) WC voltage clamp current as a function of holding potential for a population of dark-adapted WT (A; n = 5) and Kv2.1−/− (B; n = 5) mouse rods; error bars are 95% confidence intervals, and the shaded gray areas reflect the observed physiological range of membrane potentials measured from the WC recordings. Zero-current holding potentials (filled red symbols with lateral error bars; Table 2) were measured immediately upon WC access. The solid black curves are generated as the sum of the red and black curves in the respective lower panels. In A and C, the dashed curves present the I-V curve used to describe the data of the other genotype; thus, for example, the dashed curve in A replots the unbroken black curve in C. (B) Component analysis of the voltage clamp current of the dark-adapted WT rod. The two outer segment inward components are CNG current (dark blue) and NCKX current (cyan); their sum is given by the black curve; this curve was required to go through the filled black symbols, which plot the negative of the photocurrents (−Rmax, Table 3) measured at the effective outer segment holding potential; the error bars are 95% confidence intervals. The CNG current curve is the Boltzmann function of Eq. 3 with reversal potential VCNG = +8.5 mV and steepness factor sCNG = 14 mV (Baylor and Nunn, 1986). The NCKX current curve is given by Eq. 4, with VNCKX = −14 mV, sNCKX = 70 mV, KNCKX = 1,100 nM (Schnetkamp et al., 1991), and INCKX,sat(VNCKX) = −2.0 pA and −1.6 pA for WT and Kv2.1−/−, respectively (Lagnado and McNaughton, 1991). The inner segment components are the NKX electrogenic current (α3β2 isoform; Eq. 12; Supplemental text), Kv2.1 and HCN1 currents, and an unidentified K+ leak current; the sum of the inner segment components is given by the red curve. The Kv2.1 curve was generated with Eq. 10b with PK adjusted so that the current at Vm,rest is 9.1 pA, corresponding (with EK = −91 mV) to GKv2.1 = 0.18 nS (Eq. 10a). The leak current magnitude is assumed to be identical in both genotypes, and has the magnitude 2.7 pA at −32.0 mV. The HCN1 I-V curve was generated with Eq. 11 with GHCN1,rest = 1.45 nS and EHCN1 = −31 mV. The sum of the red and black curves in B is plotted as the black curve in A. The inner segment zero-current holding potential is plotted as a red circle at the average Vm, rest for WT rods (−32 mV). The filled black circle, which has the same magnitude, is plotted at the resting potential of the outer segment, which is situated slightly positively relative to the red symbol because the circulating current flows through the internal resistance of the rod (Fig. 1). At the WT resting potential, the currents are not only in electrical equilibrium but also produce homeostasis of the permeant ions Na+, K+, and Ca2+, based on the stoichiometry and other properties of the NCXK and NKX exchangers (see Theory). (D) Component analysis of the Kv2.1−/− rod dark current. The components are the same as those of the WT rod, but in the absence of Kv2.1 channels, an unidentified K+ leak current of 5.0 pA at the resting potential (−12 mV) of the inner segment is required for electrical balance. Extrapolated to Vm,rest of the WT rod, this current would have the amplitude 2.7 pA, which is equal to the magnitude of the leak current assigned to the WT rod at Vm,rest in B. For the HCN1 current, GHCN1,rest = 1.6 nS, and EHCN1 = −31 mV as in the WT rod. At the resting potential, the Kv2.1 current accounts for 80% of the total non-NKX, non-HCN1 outward current (the ordinate scale in D has been expanded relative to that in B to facilitate inspection of the component curves).
For Kv2.1−/− rods, the NKX electrogenic current, acting solely to balance Na+ influx at Vm,rest, cannot supply the requisite outward current to electrically balance the inward current of the outer segment, requiring the postulation of another outward current, identified as K+ leak. Given its I-V characteristic, the leak current is fully determined by the constraint that, when summed with all the other curves, the resultant I-V prediction (Fig. 6 C, solid black curve) has a zero crossing at −12 mV, the resting potential of the unclamped Kv2.1−/− rod (Fig. 6 C, red symbol). Determined thus, the leak current has a magnitude of 7 pA at −12 mV and 3.6 pA at −32 mV. On the assumption (Occam’s razor) that the leak current is identical in WT and Kv2.1−/− rods, the Kv2.1 current of the WT rod is then completely determined by the requirement that the sum of all currents (Fig. 6 A, solid black curve) has its zero crossing at the WT resting potential, –32 mV (Fig. 6 A, red symbol).
From the theoretical decomposition, the relative contributions of the different ionic mechanisms to the outward dark current of the WT rod at rest are found to be 34% NKX, 20% leak, and 46% Kv2.1 (the HCN1 current at −32 mV is negligibly small, −0.2 pA). Thus, Kv2.1 is estimated to contribute 70% of the non-NKX outward current of the WT rod. The prediction of a small K+ leak current does not rest solely on the WC data. The average unclamped rod recorded with SEs has a photocurrent of 7.7 pA (see above; cf. Fig. 7). Depending somewhat on the magnitude and partitioning of the preflash dark current into CNG and NCKX current, the electrogenic NKX current that balances the influx of Na+ into the outer segment of such a rod is estimated to be 3.0 pA, requiring a leak current of 4.7 pA to maintain electroneutrality at the Kv2.1−/− rod resting potential of –12 mV. Projected along its I-V curve to –32 mV, the leak current would be 2.7 pA. From similar analysis of the photocurrents of a population of Kv2.1−/− rods (Fig. S1), the leak current was predicted to range from 2 to 4 pA at –32 mV, providing a second estimate consistent with that (3.6 pA) from the WC voltage-clamp data. Assuming all the non-NKX, nonleak outward current of the WT rod is carried by Kv2.1 channels, these considerations of unclamped Kv2.1−/− rods lead to the conclusion that these channels carry 70–80% of the normal outward dark current not carried by the NKX.

Decomposition of the bright-flash photocurrents of WT and Kv2.1−/− rods into components from CNG channel, NCKX, and capacitive currents. (A) Solid black line presents the average photocurrent of an individual WT rod, adjusted for a suction pipette collection efficiency of two thirds; the gray band presents the running SD of the traces averaged. The position of the data trace on the time axis was not adjusted for the flash duration (10 ms) or the analogue filtering (30 Hz, eight-pole Bessel) in this plot. (B) The three components comprising the theoretical photocurrent response are separately plotted as membrane current (see legend on figure). The theoretical currents were digitally filtered by a 30 Hz, eight-pole Bessel filter whose characteristics matched those used in the suction pipette recordings, and otherwise were adjusted only for a several-millisecond delay arising from the early stages of phototransduction (Lamb and Pugh, 1992), and 5 ms for the flash duration. The sum of the three components (red dashed line) is identical to the dashed red trace shown in A, except for the baseline offset, which converts the trace in A to “photocurrent.” (C and D) The same analysis as in A and B of the photocurrent of a Kv2.1−/− rod. Here the NCKX and capacitive currents contribute almost equally in determining the shoulder amplitude. The fraction of the saturated photocurrent attributable to the NCKX is 0.2, 3.3-fold greater than that (0.06) estimated for the WT mouse rod (Makino et al., 2004), and the estimated Ca2+i 1,030 nM. Similar results were obtained from analysis of the saturating photocurrents of an additional population of Kv2.1−/− rods (Fig. S2).
Decomposition of the bright-flash photocurrents of WT and Kv2.1−/− rods into components from CNG channel, NCKX, and capacitive currents. (A) Solid black line presents the average photocurrent of an individual WT rod, adjusted for a suction pipette collection efficiency of two thirds; the gray band presents the running SD of the traces averaged. The position of the data trace on the time axis was not adjusted for the flash duration (10 ms) or the analogue filtering (30 Hz, eight-pole Bessel) in this plot. (B) The three components comprising the theoretical photocurrent response are separately plotted as membrane current (see legend on figure). The theoretical currents were digitally filtered by a 30 Hz, eight-pole Bessel filter whose characteristics matched those used in the suction pipette recordings, and otherwise were adjusted only for a several-millisecond delay arising from the early stages of phototransduction (Lamb and Pugh, 1992), and 5 ms for the flash duration. The sum of the three components (red dashed line) is identical to the dashed red trace shown in A, except for the baseline offset, which converts the trace in A to “photocurrent.” (C and D) The same analysis as in A and B of the photocurrent of a Kv2.1−/− rod. Here the NCKX and capacitive currents contribute almost equally in determining the shoulder amplitude. The fraction of the saturated photocurrent attributable to the NCKX is 0.2, 3.3-fold greater than that (0.06) estimated for the WT mouse rod (Makino et al., 2004), and the estimated Ca2+i 1,030 nM. Similar results were obtained from analysis of the saturating photocurrents of an additional population of Kv2.1−/− rods (Fig. S2).
Overall, the depolarization of the Kv2.1−/− rod in the dark steady state is a hallmark feature. Near Vm,rest = −12 mV, both outer segment ionic mechanisms, the CNG channels and the NCKX, are voltage-dependent (Eq. 3 and Eq. 4), and these dependencies, as we now explain, are critical for understanding the distinctive shoulders of the photocurrent rising phase (Fig. 4).
The high fraction of photocurrent from the NCKX implies that Ca2+i is elevated in Kv2.1−/− rods
Suction pipette and WC recordings have long established that the outer segment plasma membrane has only three components of membrane current: (1) CNG channel current, (2) NCKX current, (3) capacitive current (e.g., Baylor and Nunn, 1986; Cobbs and Pugh, 1987; Hodgkin et al., 1987; Hodgkin and Nunn, 1988; Lagnado et al., 1992). The shoulders of the rising phase of Kv2.1−/− rod responses are reminiscent of shoulders in the responses of rods loaded with Ca2+ in the dark and stimulated with strong flashes that rapidly close all CNG channels (Lagnado et al., 1992). The unequivocal interpretation by Lagnado et al. (1992) of the shoulders of the rod photoresponses in their experiments is that they represent the saturated level of electrogenic transport by the rod outer segment NCKX, a level reached when Ca2+i exceeds its internal binding constant of ∼1.6 µM (Lagnado et al., 1992). The shoulders in the rising phase of Kv2.1−/− rod photocurrents averaged 72% of the saturating photocurrent amplitude (Fig. S1 G). Interpretation of the shoulder as the current level set by rapid suppression of CNG channel current and subsequent rate-limited NCKX extrusion of Ca2+ implies that fCa in the preflash dark-adapted state of the Kv2.1−/− rod is near unity (Eq. 5). However, this interpretation neglects the increase in the NCKX current owing to hyperpolarization (Eq. 4 and Fig. 6) and any contribution of outer segment capacitive current to the photocurrent. To investigate the relative contributions of CNG, NCKX, and capacitive currents, we used the two-compartment model to describe the saturated photocurrents of WT and Kv2.1−/− rods (Fig. 7 and Fig. S2). The model revealed that capacitive current likely contributes materially to the Kv2.1−/− photocurrent response to strong flashes, effectively lowering the level of the observed shoulder.
Prediction of the bright-flash photocurrents (Fig. 7 D and Fig. S2) of Kv2.1−/− rods also provides a means of estimating Ca2+i in the dark-adapted Kv2.1−/− rod outer segment. Specifically, the NCKX current magnitude in darkness before the saturating flash is determined by the level of Ca2+ in the outer segment, with the effective binding constant Kex of the exchanger serving as the gauge (Fig. 8). Thus, assuming that WT and Kv2.1−/− rods hyperpolarize to approximately the same level from their different resting potentials (Fig. 5 and Table 3), Eq. 5 predicts Ca2+i in this particular dark-adapted Kv2.1−/− rod to have been 1,030 nM, more than three times the normal resting level. The theory also predicts the time course of the decline of NCKX current (Fig. 7). This latter time course is affected not only by the NCKX but also by the total extrudable Ca2+, which depends on the total calcium buffering power over the entire range of Ca2+i excursion from the dark-adapted level to the lowest level achieved in the absence of influx through the CNG channels (Lagnado et al., 1992).

Voltage dependence of NCKX current, CNG channel current, and the fraction of CNG current carried by Ca2+ for voltage clamped and unclamped conditions. (A) Dependence of the NCKX current on membrane potential for the average WT and Kv2.1−/− (KO) rod under WC voltage-clamp to the WT resting potential, −32 mV. The black (WT) and red (KO) curves plot the I-V curve of the rod NCKX (Lagnado and McNaughton, 1990; Eq. 4) scaled to pass through the values (open circles) based on the assumption that the rods have the same normal resting Ca2+i, Cadark = 320 nM. The thicker black curve plots the calcium-saturated NCKX activity function of the WT rod, obtained by scaling the curve for the resting condition by (320 + Kex)/320, with Kex = 1,100 nM (Schnetkamp et al., 1991). The dashed red curve plots the calcium-saturated NCKX activity function of the Kv2.1−/− rod, adjusted for the shorter average length of its outer segment. (B) Dependence of the CNG channel current on membrane potential for WT and Kv2.1−/− rods (Eq. 3); the symbols plot the negative of the Rmax values in Table 3 (the lateral offset of the symbols arises from differential depolarization of the outer segments when the inner segment is clamped to the same potential; cf. Fig. 1). (C) Fraction of the CNG current carried by Ca2+ (the smooth curve is the same at that in F). (D) Dependence of NCKX current on membrane potential of average WT and Kv2.1−/− (KO) rod under unclamped conditions; open symbols plot the values at respective resting potentials, while filled symbols give measured values upon maximal hyper-polarization. (E) CNG channel current dependence on membrane potential; the filled symbols represent the saturated magnitude of recorded currents, while the open symbols represent the values projected along the curves to the unclamped resting potentials. For the WT rod, the saturated photocurrent (CNG + NCKX) is 20 pA; for the KO rod, 9.5 pA. The red curve not drawn through the points represents the CNG current that would have been recorded if the KO rod had the same length as the WT rod. Depolarization alone cannot fully account for the ratio of the KO to WT currents, so based on Eq. 3, an ∼12% reduction in resting cGMP level by elevated Ca2+i is implicated. (F) Dependence of fCa, the fraction of CNG current carried by Ca2+ on membrane potential. The values of fCa for the WT rod in darkness (open black symbol, fCa = 0.072) and the Kv2.1−/− rod (open red symbol, fCa = 0.29) are derived from the homeostasis relation (Eq. 5), given the respective curves in D and E. A “saturating Boltzmann” function (smooth black trace) was fitted through the data points to provide a continuous function fCa(Vm), needed for analysis of dynamic changes in Ca2+ fluxes during phototransduction as the outer segment membrane potential varies. CNGC, CNG channels.
Voltage dependence of NCKX current, CNG channel current, and the fraction of CNG current carried by Ca2+ for voltage clamped and unclamped conditions. (A) Dependence of the NCKX current on membrane potential for the average WT and Kv2.1−/− (KO) rod under WC voltage-clamp to the WT resting potential, −32 mV. The black (WT) and red (KO) curves plot the I-V curve of the rod NCKX (Lagnado and McNaughton, 1990; Eq. 4) scaled to pass through the values (open circles) based on the assumption that the rods have the same normal resting Ca2+i, Cadark = 320 nM. The thicker black curve plots the calcium-saturated NCKX activity function of the WT rod, obtained by scaling the curve for the resting condition by (320 + Kex)/320, with Kex = 1,100 nM (Schnetkamp et al., 1991). The dashed red curve plots the calcium-saturated NCKX activity function of the Kv2.1−/− rod, adjusted for the shorter average length of its outer segment. (B) Dependence of the CNG channel current on membrane potential for WT and Kv2.1−/− rods (Eq. 3); the symbols plot the negative of the Rmax values in Table 3 (the lateral offset of the symbols arises from differential depolarization of the outer segments when the inner segment is clamped to the same potential; cf. Fig. 1). (C) Fraction of the CNG current carried by Ca2+ (the smooth curve is the same at that in F). (D) Dependence of NCKX current on membrane potential of average WT and Kv2.1−/− (KO) rod under unclamped conditions; open symbols plot the values at respective resting potentials, while filled symbols give measured values upon maximal hyper-polarization. (E) CNG channel current dependence on membrane potential; the filled symbols represent the saturated magnitude of recorded currents, while the open symbols represent the values projected along the curves to the unclamped resting potentials. For the WT rod, the saturated photocurrent (CNG + NCKX) is 20 pA; for the KO rod, 9.5 pA. The red curve not drawn through the points represents the CNG current that would have been recorded if the KO rod had the same length as the WT rod. Depolarization alone cannot fully account for the ratio of the KO to WT currents, so based on Eq. 3, an ∼12% reduction in resting cGMP level by elevated Ca2+i is implicated. (F) Dependence of fCa, the fraction of CNG current carried by Ca2+ on membrane potential. The values of fCa for the WT rod in darkness (open black symbol, fCa = 0.072) and the Kv2.1−/− rod (open red symbol, fCa = 0.29) are derived from the homeostasis relation (Eq. 5), given the respective curves in D and E. A “saturating Boltzmann” function (smooth black trace) was fitted through the data points to provide a continuous function fCa(Vm), needed for analysis of dynamic changes in Ca2+ fluxes during phototransduction as the outer segment membrane potential varies. CNGC, CNG channels.
Because the same-shaped NCKX and CNG channel I-V curves must account for both WT and Kv2.1−/− data, one can use Eq. 3, Eq. 4, and Eq. 5 to deduce that in addition to being depolarized, cGMP in the Kv2.1−/− must be somewhat reduced (Fig. 8). To understand the principle, it is helpful to compare the expectations for the condition in which rods of both genotypes are clamped to the WT resting potential (Fig. 8, A–C) with the unclamped condition that applies to the SE recording (Fig. 8, D–F). Under WC voltage clamp to −32 mV, the photocurrents (Rmax) and dark (CNG channel + NCKX) currents have the same magnitude and opposite sign, and the only difference between the WT and Kv2.1−/− rod is the shortened length of the latter’s outer segment; thus, in this condition, both CNG channel and NCKX currents are proportionately reduced in scale, so that fCa is estimated with Eq. 5 to be the same for both genotypes (Fig. 8 C). In contrast, in the unclamped condition, both NCXK (Fig. 8 D) and CNG channel (Fig. 8 E) curves “pull apart” in opposite directions for the two genotype rods; the extracted magnitude of the NCKX component of the photocurrent (red filled symbol in Fig. 8 D) is larger in the Kv2.1−/− than in the WT rod, and the CNG channel component is reduced (black filled symbol). Extrapolation along the theory curves to the respective unclamped resting potentials predicts the CNG current in darkness (Fig. 8 E, open symbols). Since mere “sliding” along the CNG channel curve of the WT rod to –12 mV does not account for the vertical shift of the open circle, in addition to the depolarization, there must be a reduction in cGMP in the Kv2.1−/− relative to that in the WT outer segment. The shorter-length outer segment of the Kv2.1−/− is material to estimation of the reduction in cGMP; thus, were the Kv2.1−/− outer segment normal length, the CNG channel current would be that given by the second red curve; using this latter curve, the cGMP is found with Eq. 3 to be reduced by 12%. Finally, application of Eq. 4 to the results in panels D and E leads to determination of fCa, which is found to be threefold higher for the Kv2.1−/− rod (0.20) than for the WT rod (0.066; Fig. 8 F), implying a substantially increased influx of Ca2+. A parallel application of the theory to a population of Kv2.1−/− rods reveals that on average, resting Ca2+i in Kv2.1−/− rods exceeds the normal resting level by 10-fold or more (Fig. S2).
When a Kv2.1−/− rod is stimulated with a strongly saturating flash, the CNG channel current is very rapidly suppressed, quantitatively accounting for the initial phase of the photocurrent up to the shoulder (Fig. 7 and Fig. S2). The shoulder amplitude, however, is affected both by NCKX saturation due to the elevated Ca2+i in the prior dark-adapted state (Lagnado et al., 1988, 1992) and by the capacitive transient arising from hyperpolarization. This latter conclusion follows first from the fact that the “super-shoulder” component exceeds the one-third limit set by Eq. 5 (Fig. S1), and is strengthened by the prediction of the time course with a model that includes the outer segment capacitive transients (Fig. 7, B and D; and Fig. S2). The time course of the NCKX decline is less perfectly predicted than the decline of the CNG channel current, depending as it does not only on free Ca2+i but also on the quantity of Ca2+ exchangeable from intracellular buffers over the entire range of Ca2+i excursion. Nonetheless, our explanation of the shoulder of the Kv2.1−/− rod photocurrent is consistent with the fact that the rod outer segment plasma membrane has only two ionic mechanisms, CNG channels and the NCKX.
The undershoots are caused by rapid activation of guanylate cycle above its resting level
Some previous studies have reported undershoots or inverted responses in recordings from outer segments with SEs, particularly when the outer segment is incompletely drawn into the recording pipette and the portion of the outer segment external to the pipette constriction is stimulated (Baylor and Lamb, 1982; Jarvinen and Lamb, 2005). Thus, one potential explanation for the undershoot is that the rod had been drawn into the suction pipette to the extent that some of the inner segment is past the constriction (Fig. 1), leading to detection of inner segment voltage-sensitive currents, in particular hyperpolarization-activated inward cation current (Ih) arising from the HCN1 channels that are strongly expressed in the inner segment (Fig. 2). This latter hypothesis predicts that the undershoot would not be responsive to light. To test this hypothesis, we performed paired-flash experiments, whereby the first saturating flash elicited a response with an undershoot and the second saturating flash was timed to occur during the undershoot (Fig. 9). In all paired-flash experiments, the flash delivered during the undershoot produced a larger amplitude photocurrent, and the amplitude of the response to the second flash response was indistinguishable from the sum of the magnitudes of the first response and its undershoot (Fig. 9). These results are inconsistent with the undershoot arising from a voltage-sensitive current like Ih in the inner segment, and rather imply that in Kv2.1−/− rod responses the undershoot corresponds to an increase in light-suppressible outer segment CNG channel current arising from transiently increased cGMP.

Light suppression of the undershoot of Kv2.1−/− rod photoresponses reveals it to arise from CNG channel current. (A) Paired-flash paradigm: the response of the rod to a single saturating flash (black trace); response of the same rod to a pair of identical flashes (red trace), with the second delivered during the undershoot of the response to the first flash. The ordinate scale is normalized by the saturating amplitude of the photocurrent response to the first flash. (B) Bar chart summarizing results from 10 experiments. The red filled region gives the magnitudes of saturating photocurrents to the first and second flashes in the paired-flash experiment; the gray-filled region stacked on the first flash plots the magnitude of the undershoot. All magnitudes are scaled by the amplitude of the response to the first flash, as in A. The magnitude of the photocurrent response to the second flash is indistinguishable from the sum of the magnitudes of the initial photocurrent and its undershoot.
Light suppression of the undershoot of Kv2.1−/− rod photoresponses reveals it to arise from CNG channel current. (A) Paired-flash paradigm: the response of the rod to a single saturating flash (black trace); response of the same rod to a pair of identical flashes (red trace), with the second delivered during the undershoot of the response to the first flash. The ordinate scale is normalized by the saturating amplitude of the photocurrent response to the first flash. (B) Bar chart summarizing results from 10 experiments. The red filled region gives the magnitudes of saturating photocurrents to the first and second flashes in the paired-flash experiment; the gray-filled region stacked on the first flash plots the magnitude of the undershoot. All magnitudes are scaled by the amplitude of the response to the first flash, as in A. The magnitude of the photocurrent response to the second flash is indistinguishable from the sum of the magnitudes of the initial photocurrent and its undershoot.
The rising phase of the second response in the paired-flash experiments did not exhibit the shoulder seen in the initial response, and moreover, the duration of the second response was shorter (Fig. 9 A). These results indicate that Ca2+i has not yet returned to its elevated resting level at the time of the second flash, and further suggest that the elevated resting Ca2+i present at the time of the first flash inhibits the deactivation of an early step in phototransduction (Murnick and Lamb, 1996).
Predictions of photocurrent response families of WT and Kv2.1−/− rods
The theory was next used to generate photocurrent response families of WT and Kv2.1−/− rods (Fig. 10). The model was able to recapitulate key qualitative features of Kv2.1−/− rod response families, including the rapid downturns of subsaturating responses, undershoots below baseline, and the spacing of the recoveries from saturating flashes (Fig. 10 E). The rapid downturn in the subsaturating responses of the Kv2.1−/− rod is predicted by the model as a correspondingly rapid activation of guanylate cyclase. This fast activation of cyclase arises from an extremely rapid decline in Ca2+i caused by the step-like dependence of fCa on membrane potential; thus, hyperpolarization of only 10–15 mV can greatly decrease Ca2+ influx through CNG channels (Fig. 8 C), while NCKX extrusion continues at near basal level. In this way, the Kv2.1−/− rod is very different from WT rods, in which fCa is effectively voltage-independent over the normal range of Vm (Fig. 8 C), so that the time course of Ca2+i decline in WT rods is dictated mainly by the NCKX and the Ca2+-dependent Ca2+ buffer power of the rod (Fig. S5). In effect, the gain of the Ca2+ feedback to guanylate cyclase in the Kv2.1−/− rod during subsaturating responses is greatly increased by the steep voltage-dependence of fCa, the fraction of CNG channel current carried by Ca2+.
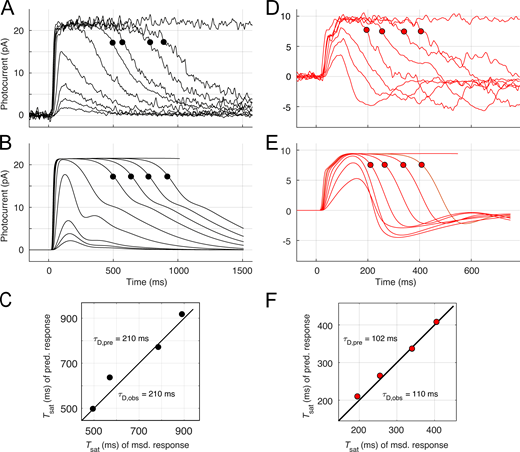
Key features of the photocurrent responses of Kv2.1−/− rods can be explained with an extension of phototransduction theory to include the voltage dependence of CNG channels and the NCKX, and the voltage-dependent and voltage-activated conductances of the inner segment. Related to Fig. 1. (A) Response family of a WT rod: with the exception of that to the most intense, the traces were restricted to subsaturating responses and saturated responses that exhibit recovery translation invariance (Nikonov et al., 1998). The measured flash strengths were 4.6, 8.7, 18, 108, 427, 1,422, 2,774, 5,263, 10,570, and 73,000 photons μm−2. The filled symbols are plotted on the traces at the times (Tsat) when the dark currents had recovered 20% from saturation. (B) Predictions of the two-compartment model; filled symbols identify times 20% recovery from saturation, as in A. (C) Comparison of the times in saturation (Tsat) extracted from the empirical traces (msd.) and the predicted traces (pred.). The symbols τD, Obs and τD, pre give the values of the dominant time constants estimated by least-squares fitting of straight lines of Tsat versus the natural log of the flash strength for the observed and predicted traces, respectively (Nikonov et al., 1998). In the theoretical model, the lifetime τE of the Gtα-PDE (G*-E*) complex was assumed equal to τD, Obs (210 ms), and the lifetime of photoactivated rhodopsin (R*) was set to 45 ms (Eq. S9). Theory predicts the points to follow a line of unit slope (unbroken line). (D and E) Response family of a Kv2.1–/– rod and model predictions (the same rods whose response to the most intense flash were predicted and plotted in Fig. 7 on a much faster time scale); the circular symbols plot the times of 20% recovery from saturation (Tsat), as in A and B. The measured flash strengths were 108, 224, 427, 747, 1,423, 2,774, 5,263, and 73,000 photons μm −2. In the model calculations the lifetime τE of the Gtα-PDE (G*-E*) complex was assumed equal to τD, Obs (110 ms), while R* was assumed to deactivate with a time constant of 30 ms for subsaturating flashes, segueing to 70 ms for saturating flashes (Eq. S11). Most other parameters were set to the same value for WT and Kv2.1–/– rods (Table S2). (F) Comparison of the dominant time constants extracted from analysis of the empirically measured and theoretically predicted traces (the predicted responses to the most intense flashes in A and C are provided in Fig. 7; predicting the time course of the recoveries to this intensity flash, estimated to produce more than 75 R* per disc face, is beyond the scope of the model).
Key features of the photocurrent responses of Kv2.1−/− rods can be explained with an extension of phototransduction theory to include the voltage dependence of CNG channels and the NCKX, and the voltage-dependent and voltage-activated conductances of the inner segment. Related to Fig. 1. (A) Response family of a WT rod: with the exception of that to the most intense, the traces were restricted to subsaturating responses and saturated responses that exhibit recovery translation invariance (Nikonov et al., 1998). The measured flash strengths were 4.6, 8.7, 18, 108, 427, 1,422, 2,774, 5,263, 10,570, and 73,000 photons μm−2. The filled symbols are plotted on the traces at the times (Tsat) when the dark currents had recovered 20% from saturation. (B) Predictions of the two-compartment model; filled symbols identify times 20% recovery from saturation, as in A. (C) Comparison of the times in saturation (Tsat) extracted from the empirical traces (msd.) and the predicted traces (pred.). The symbols τD, Obs and τD, pre give the values of the dominant time constants estimated by least-squares fitting of straight lines of Tsat versus the natural log of the flash strength for the observed and predicted traces, respectively (Nikonov et al., 1998). In the theoretical model, the lifetime τE of the Gtα-PDE (G*-E*) complex was assumed equal to τD, Obs (210 ms), and the lifetime of photoactivated rhodopsin (R*) was set to 45 ms (Eq. S9). Theory predicts the points to follow a line of unit slope (unbroken line). (D and E) Response family of a Kv2.1–/– rod and model predictions (the same rods whose response to the most intense flash were predicted and plotted in Fig. 7 on a much faster time scale); the circular symbols plot the times of 20% recovery from saturation (Tsat), as in A and B. The measured flash strengths were 108, 224, 427, 747, 1,423, 2,774, 5,263, and 73,000 photons μm −2. In the model calculations the lifetime τE of the Gtα-PDE (G*-E*) complex was assumed equal to τD, Obs (110 ms), while R* was assumed to deactivate with a time constant of 30 ms for subsaturating flashes, segueing to 70 ms for saturating flashes (Eq. S11). Most other parameters were set to the same value for WT and Kv2.1–/– rods (Table S2). (F) Comparison of the dominant time constants extracted from analysis of the empirically measured and theoretically predicted traces (the predicted responses to the most intense flashes in A and C are provided in Fig. 7; predicting the time course of the recoveries to this intensity flash, estimated to produce more than 75 R* per disc face, is beyond the scope of the model).
While the model was able to predict response undershoots below baseline (Fig. 10 E), it did not recapitulate the detailed form of the undershoots, and moreover often predicted oscillations that were not observed in the data. The early portion of the observed recoveries from saturating responses of rods of both genotypes obeyed “recovery translation invariance” (Nikonov et al., 1998), and the dominant time constant extracted from these traces was interpretable in the model as the lifetime of the τE of the Gtα-PDE (G*-E*) complex (Krispel et al., 2006; Burns and Pugh, 2009; Fig. 10 F). However, predictions of subsaturating responses and the spacing of the saturating responses were improved if it was assumed that photoactivated rhodopsin (R*) inactivated somewhat faster during responses to subsaturating flashes than during responses to saturating flashes (Fig. 10 E, Eq. S11, and Table S2). This formulation of a variable R* lifetime is admittedly ad hoc and presumed to be a placeholder for an effect of the high Ca2+i on the dark-adapted Kv2.1–/– rod. However, the only known mechanism of regulation of R* lifetime by calcium, recoverin-mediated inhibition of Grk1, is not known to shorten the R* lifetime in high Ca2+i (Nikonov et al., 2000; Makino et al., 2004).
Photoreceptor degeneration in Kv2.1−/− rods
Elevated intracellular Ca2+ such as occurs during hypoxia is well known to produce detrimental effects on mitochondrial and ER structure and function (Stirling and Stys, 2010). Consistent with such expectations, electron micrographs show severe distortions of mitochondria in the Kv2.1−/− rod inner segment relative to those in WT samples (Fig. 11 D). Moreover, OCT data and histology show thinning of the ONL and decreased outer segment length (Fig. 2 and Fig. 11). The rod degeneration and loss are progressive with age (Fig. 11 C). The relatively static shortening of Kv2.1−/− rod outer segments by ∼30% relative to those of WT mice (Fig. 2 B and Fig. 11 C) may result in part from reduced production in the ER of proteins and lipids required for the daily renewal of 10% of the outer segment.

Kv2.1 −/− rods undergo slow degeneration. (A) Paraffin-embedded ultrathin sections of heterozygous and Kv2.1−/− retinas of 6-mo-old mice stained for hematoxylin and eosin. Sections have been aligned at the ELM, showing that the OS, IS, and ONL layers are smaller in the Kv2.1−/− mouse. Scale bar, 10 µm. (B) OCT B-scans of central retina of Kv2.1−/− mice at 6 wk and 24 wk of age. There is a notable decrease in the OS, IS, and ONL layer thickness over time in Kv2.1−/− mice. Scale bar, 100 µm. (C) Quantification of the OS, IS, ONL, and IPL layers in the same mice from 6 wk to 6 mo of age (mean ± SEM, n = 4 per genotype). The OS layer is shorter in Kv2.1−/− mice at all ages, and there is a progressive decline in IS and ONL thickness over time. (D) TEM sections showing individual rod photoreceptors in WT and Kv2.1−/− mice. Mitochondria in the Kv2.1−/− rod inner segments are more abundant and look distorted as compared with WT. Dark adaptation before euthanasia did not change this effect. Scale bar, 1 µm. IPL, inner plexiform layer; OPL, outer plexiform layer; GCL, ganglion cell fiber layer; TEM, transmission EM.
Kv2.1 −/− rods undergo slow degeneration. (A) Paraffin-embedded ultrathin sections of heterozygous and Kv2.1−/− retinas of 6-mo-old mice stained for hematoxylin and eosin. Sections have been aligned at the ELM, showing that the OS, IS, and ONL layers are smaller in the Kv2.1−/− mouse. Scale bar, 10 µm. (B) OCT B-scans of central retina of Kv2.1−/− mice at 6 wk and 24 wk of age. There is a notable decrease in the OS, IS, and ONL layer thickness over time in Kv2.1−/− mice. Scale bar, 100 µm. (C) Quantification of the OS, IS, ONL, and IPL layers in the same mice from 6 wk to 6 mo of age (mean ± SEM, n = 4 per genotype). The OS layer is shorter in Kv2.1−/− mice at all ages, and there is a progressive decline in IS and ONL thickness over time. (D) TEM sections showing individual rod photoreceptors in WT and Kv2.1−/− mice. Mitochondria in the Kv2.1−/− rod inner segments are more abundant and look distorted as compared with WT. Dark adaptation before euthanasia did not change this effect. Scale bar, 1 µm. IPL, inner plexiform layer; OPL, outer plexiform layer; GCL, ganglion cell fiber layer; TEM, transmission EM.
Discussion
The outward dark current of the normal mouse rod is generated predominantly by Kv2.1 channels and the electrogenic NKX
The electrogenic NKX current of the mouse rod inner segment must carry approximately one third of the normal outward dark current (Fig. 6). This conclusion follows directly from the homeostatic requirement that the net flux of each permeant ion must be zero in the dark steady state, and the fact that the NKX is the only efflux route for Na+ that enters the outer segment through CNG channels and the Na+/ Ca2+, K+ exchanger (NCKX1). Genetic elimination of the hyperpolarizing Kv2.1 current strongly depolarizes the rod (Table 3 and Fig. 5), implying that Kv2.1 channels contribute critically to the normal hyperpolarizing outward dark current. The theoretical analysis shows that to maintain Na+ homeostasis, Kv2.1−/− rods must have a non-NKX outward current at their resting potential of –12 mV. Assuming that this latter leak current is carried by an unidentified class of K+ channels that have the same expression level in WT channels leads to the conclusion that Kv2.1 channels carry 70–80% of the normal outward dark current (Fig. 6 and attendant text). Further support for the conclusion that Kv2.1 current dominates the non-NKX outward dark current of the rod comes from its localization to, and very strong expression in, the inner segment (Fig. 2), which was shown in the original current source density analysis that discovered the dark current to be the predominant membrane source of outward current (Hagins et al., 1970; Penn and Hagins, 1972).
The primary mechanisms of the physiological phenotype of Kv2.1−/− rods: depolarization and elevated Ca2+ influx
The primary physiological defect in rods lacking Kv2.1 channels is the strong depolarization resulting from the loss of the potent hyperpolarizing current that these channels normally provide (Fig. 6). Fortunately for the experimenter, a good fraction of the depolarized Kv2.1−/− rods is able to survive this loss for several months (Fig. 2) and achieve a distinct dark steady state; however, the static depolarization has dire consequences. For a dark-adapted Kv2.1−/− rod whose NCKX is responsible for ∼20% of the total photocurrent (Fig. 7, C and D), depolarization to –12 mV causes fCa, the fraction of inward current carried by Ca2+ through the CNG channels, to increase more than threefold, from ∼6% to 20% (Fig. 7 and Fig. 8). This effect is a direct consequence of the I-V relations of the CNG channels and NCKX (Fig. 8, A and B; Eq. 3; and Eq. 4), and the homeostatic requirement of balanced Ca2+ flux in the dark steady state (Eq. 5). Free Ca2+ in the dark-adapted Kv2.1−/− rod outer segment is also predicted to be much higher than normal, at least 750 nM and likely 2–4 µM on average (Fig. S2), as compared with a normal level of ∼300 nM (Woodruff et al., 2002; Gross et al., 2012a). The abnormally elevated fCa of the CNG channels at rest in Kv2.1−/− rods no doubt arises from relief of divalent cation block of rod CNG channels, which are much more permeable to Ca2+ than to Na+ when the block is relieved by depolarization (Zimmerman and Baylor, 1992), highlighting the essential role of this block in maintaining Ca2+i at a level proper for normal rod function.
The steep voltage dependence of the fraction of CNG channel current carried by Ca2+ underlies the strange photocurrent kinetics of Kv2.1−/− rod
In addition to reduction in photocurrent magnitude and shoulders on the rising phase of the response to bright flashes, Kv2.1−/− rods exhibit unusually rapid, “truncating” downturns of their subsaturating responses, followed by undershoots below baseline (Fig. 4, C and E). To explain these phenomena, the domains of a number of dynamic variables of the model rod, e.g., fCa of the CNG channels (Fig. 8), guanylate cyclase activity (Fig. S5 B), and Ca2+ buffering (Fig. S5 C), had to be extended into membrane potential and Ca2+i regimens not usually encompassed by phototransduction theory. With reasonable extensions, moderately successful predictions could be made of the characteristic kinetic features of Kv2.1−/− rods, including the rapid downturns of subsaturating responses and undershoots (Fig. 10 E), as well as the spacing of recoveries from saturating flashes (Fig. 10 F). The theory predicts that even small hyperpolarizations “jump” fCa from a high to low value (Fig. 8 C), causing Ca2+i to decline very quickly and strongly activate cGMP synthesis of guanylate cyclase. While successful in qualitatively explaining the strange features of Kv2.1−/− rod photocurrent kinetics, the present theory should be taken as an installment on future work that not only incorporates all the rod membrane’s ionic mechanisms but also includes additional molecular information about the consequences of chronic exposure to high Ca2+i on these mechanisms and on the components of phototransduction.
Considerations about mouse rod WC recordings
The WT photovoltage responses obtained under WC current clamp recording displayed kinetics that at first glance appear to be unusually slow. However, closer inspection reveals that the substaturating responses were quite similar to those previously published, especially in recordings from mouse rods (Cangiano et al., 2012; Jin et al., 2015; Pahlberg et al., 2017). Insufficient flash strength in our apparatus is likely responsible for the absence from our recordings of the classic nose in WT rods, whereby intense flashes drive the rod photovoltage to approximately −75 mV, followed by relaxation to a plateau of approximately −55 mV while CNG channels remain completely closed (Baylor and Nunn, 1986; Cobbs and Pugh, 1987; Schneeweis and Schnapf, 1995). Uncompensated series resistance and capacitance may have contributed to the slowing of photovoltage kinetics, but the greater slowing in photovoltages of Kv2.1−/− relative to WT rods, and the fact that this was observed in all cells (Fig. 5 and Table 3), suggest that the increased slowing of photovoltage responses of Kv2.1−/−arise from their altered physiology.
The total volume of the mouse rod cytoplasm is only 150–170 fl, so dialysis of the inner and outer segments by the relatively low resistance electrode patched onto the cell body should be relatively fast under our recording conditions. It bears emphasis in this context that photovoltage response families take considerably more time to collect than the other WC measurements, inasmuch as repeated stimulation with each flash strength is required to obtain reliable average responses, and time must be allotted for complete recovery between flashes. That the time to peak of observed subsaturating photovoltage responses tends to increase rather than decrease with flash strength as in the recordings of much larger amphibian rods (Baylor and Nunn, 1986; Cobbs and Pugh, 1987) suggests that dialysis could have lowered the concentration of agents that normally act to shorten the time to peak of the subsaturating photoresponse. Cyclic nucleotides and their precursors are examples of such agents; thus, for example, reduced cAMP or cGMP concentration in the inner segment could lower the resting HCN1 conductance by an allosteric mechanism (e.g., Eq. S23), diminishing the magnitude of the HCN1 current. Alternatively, and not mutually exclusively, reduced GTP concentration in the outer segment could diminish the calcium feedback activation of guanylate cyclase, an established mechanism of shortening the time to peak of subsaturating photocurrent responses (Burns et al., 2002). Finally, alteration of Na+i, K+i, or Ca2+i by dialysis could weaken NCKX function and thereby diminish the calcium feedback activation of guanylate cyclase.
Photovoltage response families of mouse rods have to date been published by only two other groups (Cangiano et al., 2012; Pahlberg et al., 2017). The response family in Cangiano et al. (2012) was recorded at room temperature, and the subsaturating responses are much slower in reaching peak amplitude than our WT responses (Fig. 5). The response family in Pahlberg et al. (2017) is very similar to ours, and also exhibits a slowing in the time to peak of subsaturating responses as a function of increasing flash strength. Both experimental technique and theory will no doubt undergo further modification as additional mouse rod WC experiments are undertaken. We stress that a great value of theory is its ability to generate testable predictions about unexplained features of the data. It can be anticipated that photoresponses of mouse rods having genetically targeted manipulations not just of outer segment proteins but also of channels (such as Kv2.1), and modulators responsible for the outward limb of the circulating current and other features of the overall electrical response will be important in better understanding the rod photovoltage.
Presence of the c-wave in the Kv2.1−/− eye
A recent study (Hart et al., 2019) reported that mice lacking Kv2.1 channels do not generate an ERG c-wave, whereas the c-wave was found to be present in the Kv2.1−/− mice of this study (Fig. 3 D). The c-wave is a transepithelial potential response of the RPE to the decrease in K+o in the SRS surrounding the photoreceptors that occurs as a consequence of the rod light response (Steinberg et al., 1980; Steinberg, 1985). The decrease in SRS K+o is driven both by reduced efflux of K+ through inner segment K+ channels as the rod hyperpolarizes, and continued influx of K+ into the rod through the NKX. As expected from the reduced a-wave amplitude, and confirmed with WC and SE recordings, in the dark-adapted Kv2.1−/− rod, there is a reduction of inward dark current into the outer segment. As a consequence, there must be reduced efflux of K+ from the Kv2.1−/− rod relative to that of the WT via both ionic mechanisms. Nonetheless, as the Kv2.1−/− rod is able to maintain electrical balance in the dark via its NKX and an unidentified K+ leak current, the NKX must continue to pump K+ from the SRS into the Kv2.1−/− rod, and so it would be expected that a c-wave would still be present in ERG responses, though reduced in amplitude, as we have observed (Fig. 3 D).
Kv2.1 channels in retinal disease conditions
Cone dystrophy with supernormal rod response (CDSRR) is a recessive retinal disorder characterized by color vision deficits, reduced acuity, and larger than normal rod-driven ERG responses (Gouras et al., 1983; Alexander and Fishman, 1984; Kato et al., 1993; Hood et al., 1996; Michaelides et al., 2005; Wu et al., 2006), which arises from mutations in KCNV2 (Kv8.2; Wissinger et al., 2008, 2011; Zelinger et al., 2013). This disorder has been proposed to arise because “Kv2.1/Kv8.2 channels contribute to the delayed depolarizing phase of the photovoltage response; with reduced or absent Kv current deactivation, the light-induced photoreceptor hyperpolarization becomes more sustained” (Gayet-Primo et al., 2018). Our results suggest an alternative explanation of CDSRR and related photoreceptor disorders arising from mutations in Kv2.1 and Kv8.2 genes, namely that loss of the current normally responsible for most of the outward dark current causes chronic and severe depolarization in the dark, with the consequence of greatly increased Ca2+ influx through the CNG channels (Fig. 7, Fig. 8, and Fig. S2), increased Ca2+i in the dark, and supernormal light-driven hyperpolarization (Table 3). Eventually, the highly elevated Ca2+i will cause inexorable stress on mitochondria (Fig. 11), ER, and other basic cellular functions (Stirling and Stys, 2010), leading to photoreceptor loss. Applying our results and analysis to other CDSRR models may provide further insights into the role of mutant Kv2.1 and associated subunits in rod function and disease-associated dysfunction.
A phenotype of severe Kv2.1 dysfunction of potential clinical interest is presented by the ERG b-wave of the Kv2.1−/− mice (Fig. 3 C), which is strikingly delayed in onset by ∼20 ms. (Fig. 3 C), and reduced in amplitude by ∼20% (Table 2). The delay in b-wave generation could arise from slowed photoreceptor hyperpolarization, which we observed in the WC recordings under current clamp mode. It also could reflect slowed removal of elevated glutamate from the photoreceptor synapses (e.g., if the glutamate concentration were strongly elevated by the rod’s depolarization). The decreased b-wave amplitude could arise from a partial degeneration of ON-bipolar cells, but the absence of any obvious change in the INL (bipolar cell body layer) thickness (Fig. 2) makes substantial loss of bipolar cells unlikely. The smaller decrease in b-wave relative to the reduction in a-wave amplitude Kv2.1−/− mice appears to be another instance of the general resilience of retinal bipolar cells during photoreceptor degeneration (Hood et al., 1996; Aleman et al., 2001). The extracellular dipole current underlying the saturating a-wave of the Kv2.1−/− retina, however, comprises a mixture of the three components of rod photocurrent—suppression of CNG channel current, decline of NCKX current, and decline of capacitive current—that is quite distinct from that underlying the a-wave in the WT retina (Fig. 7).
Overall, the slow degeneration of Kv2.1−/− rods is likely the consequence of their fragile life on a tightwire of high Ca2+i. From this perspective, the Kv2.1−/− rod may provide a valuable experimental model for investigating Ca2+-dependent photoreceptor degeneration, as well regulatory mechanisms that help cells cope with chronically elevated Ca2+i.
Acknowledgments
Jeanne M. Nerbonne served as editor.
We thank Christopher Kessler for performing early SE experiments and analysis, Emily Levine for initial immunohistochemistry and morphological quantitation, Syed Abbas for rhodopsin quantification and early PCR genotyping, and Pengfei Zhang for help with OCT analysis. We also thank the University of California, Davis, EyePod laboratory for use of their OCT imaging facilities.
Supported by National Institutes of Health grants R01-EY14047 (M.E. Burns) and R01-NS114210 (J.S. Trimmer) and National Eye Institute core grant P30-EY012576.
The authors declare no competing financial interests.
Author contributions: C. Fortenbach performed WC recordings and analysis; G. Peinado Allina performed ERGs and analysis; C.M. Shores performed SE recordings and analysis; S.J. Karlen performed electron microscopy; S.J. Karlen and E.B. Miller performed OCT and analysis; H. Bishop and J.S. Trimmer performed quantitative initial breeding of the Kv2.1−/− mouse line and quantitative immunoblotting; J.S. Trimmer and M.E. Burns conceived of the project; E.N. Pugh, Jr. and M.E. Burns directed experimental design, theoretical analysis, and data interpretation; E.N. Pugh, Jr. performed the computational analysis; and M.E. Burns and E.N. Pugh, Jr. wrote the paper, with help from coauthors.

