


Nucleotide-binding oligomerization domain (NBD), leucine-rich repeat (LRR) containing protein family (NLRs) are intracellular pattern recognition receptors that mediate innate immunity against infections. The endothelium is the first line of defense against blood-borne pathogens, but it is unclear which NLRs control endothelial cell (EC) intrinsic immunity. Here, we demonstrate that human ECs simultaneously activate NLRP1 and CARD8 inflammasomes in response to DPP8/9 inhibitor Val-boro-Pro (VbP). Enterovirus Coxsackie virus B3 (CVB3)—the most common cause of viral myocarditis—predominantly activates CARD8 in ECs in a manner that requires viral 2A and 3C protease cleavage at CARD8 p.G38 and proteasome function. Genetic deletion of CARD8 in ECs and human embryonic stem cell–derived cardiomyocytes (HCMs) attenuates CVB3-induced pyroptosis, inflammation, and viral propagation. Furthermore, using a stratified endothelial–cardiomyocyte co-culture system, we demonstrate that deleting CARD8 in ECs reduces CVB3 infection of the underlying cardiomyocytes. Our study uncovers the unique role of CARD8 inflammasome in endothelium-intrinsic anti-viral immunity.
Introduction
A healthy endothelium maintains hemodynamic homeostasis and serves as the body’s first line of defense against blood-borne pathogens and their pathogen associated molecular patterns (PAMPs). Pattern recognition receptors (PRRs) detect these PAMPs to trigger innate immune responses aimed at eliminating the threat (Xiao et al., 2014; Shao et al., 2020). NLRPs (nucleotide binding and oligomerization, leucine-rich repeat containing protein super family) are a category of PRRs that sense PAMPs and initiate the formation of multiprotein complexes known as inflammasomes (Lamkanfi and Dixit, 2014). At least 22 different kinds of NLRs are present in humans (Martinon and Tschopp, 2005; Ting et al., 2008), which vary in terms of tissue distribution. These sensors have a general domain organization of an NACHT/NBD domain, an LRR domain (leucine-rich repeat domain), and a Pyrin domain (PYD; Lamkanfi and Dixit, 2014). Human NLRP1 contains an additional CARD (caspase-1 recruitment domain; Broz and Dixit, 2016; Ting et al., 2008). PYD or CARD mediate homotypic interactions with the adaptor protein apoptosis-associated speck-like protein (ASC; Broz and Dixit, 2016; Hayward et al., 2018). NLR activation leads to proximity-induced auto-proteolytic activation of caspase 1 (CASP1) and the maturation of gasdermin (GSDM) proteins and IL-1 family of pro-inflammatory cytokines such as IL-1β and IL-18, enabling their secretion through GSDM pores (Heilig et al., 2018; Evavold et al., 2018). When the source of cellular stress cannot be removed, GSDM pores in the cell membrane result in pro-inflammatory cell death known as pyroptosis (Shi et al., 2015; Liu et al., 2016b). Although the endothelium as the internal barrier forms the first line of defense against blood-borne pathogens, the role of endothelial-intrinsic inflammasomes has not been well studied.
Amongst the NLR proteins, NLRP3 is the most widely studied (Afonina et al., 2017; Chen and Chen, 2018; Swanson et al., 2019) and has been implicated in a wide variety of common diseases with inflammatory etiology (Van Der Heijden et al., 2017; Cruz-Tapias et al., 2013; Dong et al., 2020; Jin and Fu, 2019; Jia et al., 2019; Zhai et al., 2018). However, it is increasingly recognized that different tissues and cell types employ distinct repertoires of NLR sensors. In particular, recent studies have highlighted a unique role of the human NLRP1 inflammasome in barrier tissues, such as the skin and the lung (Zwicker et al., 2017; Jin et al., 2007; Drutman et al., 2019; Mamaï et al., 2015; Zhong et al., 2016; Grandemange et al., 2017). NLRP1, together with a related sensor CARD8 (also known as CARDINAL/TUCAN/NDDP1), is restrained by dipeptidyl peptidase 8/9 (DPP8/9) through its enzymatic activity and/or physical interaction (Johnson et al., 2020; Okondo et al., 2017; Zhong et al., 2018; Johnson et al., 2018). In contrast to other sensors, the involvement of CARD8 in human disease is still less understood (Paramel et al., 2015; Ito et al., 2014; Yang et al., 2011; Zhang et al., 2017; Hoffman et al., 2001; Mao et al., 2018). Recent studies confirmed CARD8 as a bona fide inflammasome sensor with a unique mechanism of action (Johnson et al., 2018). CARD8 cannot induce ASC oligomerization and therefore functions in an ASC-independent manner (Gong et al., 2021). Instead, CARD8 directly engages CASP1 upon activation, while human NLRP1 requires ASC to bridge interactions with CASP1 (Ball et al., 2020; Qin et al., 2020; Johnson et al., 2018). It is worth noting that rodents do not encode a discernible CARD8 gene, which suggests limited utility of rodent models for the functional characterization of human CARD8.
Although the ligands for rodent Nlrp1 have been characterized (Boyden and Dietrich, 2006; Griswold et al., 2019; Gai et al., 2019; Sandstrom et al., 2019; Broz and Dixit, 2016; Ball et al., 2021), the natural ligands of human NLRP1 and CARD8 have remained elusive. Recently, enteroviral 3C proteases, long dsRNA, cytosolic peptide accumulation, and reductive stress were identified as the natural triggers for the human NLRP1 inflammasome (Orth-He et al., 2022; Wang et al., 2022; Tsu et al., 2021; Robinson et al., 2020; Bauernfried et al., 2020). This occurs through a process of “functional degradation” in a proteasome-dependent manner (Robinson et al., 2020). In a similar fashion, HIV-1 protease has also been found to activate the CARD8 inflammasome through N-terminal cleavage of CARD8 (Wang et al., 2021).
Myocarditis is the inflammation of the heart, most commonly caused by viral infections. Myocarditis can resolve fully without lasting damage or progress to inflammatory cardiomyopathy and eventual heart failure (Tschöpe et al., 2021). The latter is characterized by chronic and excessive lymphocytic infiltration that causes pathogenic remodeling and is associated with viral persistence (Bouin et al., 2019). Coxsackie virus B3 (CVB3) is an enterovirus that accounts for 15–30% of viral myocarditis cases (Garmaroudi et al., 2015; Buskiewicz et al., 2016; Tschöpe et al., 2021). CVB3 contains a 7.4 kb positive, single-stranded RNA genome that translates a single polyprotein which relies on two virally encoded proteases 2A (2Apro) and 3C (3Cpro) for polyprotein cleavage. 2A protease and 3C protease also take part in evasion of host immune responses by cleaving host proteins (Maghsoudi et al., 2010; Kuyumcu-Martinez et al., 2002). In murine models, CVB3-induced myocardial damage is Casp-1-dependent (Wang et al., 2014). The NOD2-NLRP3 inflammasome has also previously been implicated in both human endomyocardial biopsies (EMB) and animal models as a key mediator of viral uptake and inflammation in CVB3 myocarditis (Tschöpe et al., 2017). Furthermore, inhibition of Casp1 (Yu et al., 2020) improves CVB3-induced myocarditis in mice.
Here, we demonstrate that NLRP1 and CARD8 are the sole mediators of inflammasome activation in primary human endothelial cells (ECs). Furthermore, ECs concurrently activate both NLRP1 and CARD8 as functional inflammasomes in response to DPP8/9 inhibitor Val-boroPro (VbP). To the best of our knowledge, ECs are the only primary human cell types that are capable of doing so. We further show that CARD8 is the principal inflammasome sensor triggered by CVB3 infection of aortic ECs. Mechanistically, CVB3-dependent, CARD8-driven EC pyroptosis requires viral proteases, 2Apro and 3Cpro, which cooperate to degrade the CARD8 N-terminal fragment. Genetic removal of CARD8 alleviates CVB3-induced cell death and cytokine release, leading to reduced viral replication in both ECs and human embryonic stem cell–derived human cardiomyocytes. Furthermore, deletion of CARD8 in ECs protects underlying cardiomyocytes from CVB3 infection. Our work elucidates CVB3 as a trigger of the endothelial CARD8 inflammasome and highlights the potential therapeutic benefit of inhibiting CARD8 in CVB3-driven myocarditis.
Results and discussion
NLRP1 and CARD8 are the predominant inflammasome sensors in ECs
We first profiled the expression of NLRP sensors and known inflammasome components in primary human aortic ECs (HAECs; Fig. S1 A). This analysis revealed that HAECs express all known components required for a functional inflammasome. The predominant NLR sensors in HAECs as determined by mRNA levels are CARD8 and NLRP1 (Fig.1 A and Fig. S1 A). We confirmed that NLRP1, CARD8, and ASC protein are expressed in HAECs (Fig. S1 B). NLRP3 was not expressed either in unstimulated HAECs or after lipopolysaccharide (LPS) stimulation (Fig. S1 C), a common NLRP3 priming signal. In HAECs, CARD8 is expressed at higher levels than NLRP1 (Fig. 1 A and Fig. S1 A). We validated the expression of NLRP1 and CARD8 in three different endothelial subtypes—umbilical venous (HUVECs), microvascular (HMVECs), and lymphatic ECs (HMLECs; Fig. S1 D). Single-cell RNAseq of human atherosclerotic plaque tissue (Wirka et al., 2019) confirmed that NLRP1 and CARD8 are expressed at high levels in ECs in vivo, but not NLRP3, which is almost exclusively expressed by macrophages (Fig. S1 E). These results suggest that primary human HAECs express NLRP1 and CARD8 as the primary inflammasome sensors.
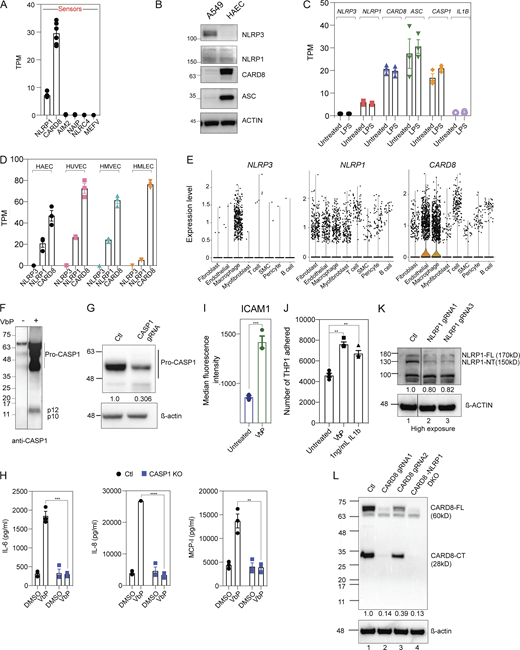
NLRP1 and CARD8 are simultaneously triggered by VbP in primary human endothelial cells lines. Related to Fig. 1. (A) RNA sequencing of HAECs from two healthy donors showing mRNA levels of known inflammasome sensors. (B) Immunoblots of untreated A549 cell lysates and HAECs lysates showing levels of inflammasome proteins, with β-actin loading control. (C) mRNA levels of indicated inflammasome sensors in control and 16 h LPS-treated HAECs as measured by RNA sequencing. (D) mRNA levels of NLRP1, NLRP3, and CARD8 in three different primary endothelial cell lines (HUVEC, HMVEC, and HAEC) as measured by RNA sequencing (Lim et al., 2019). (E) Single-cell RNA sequencing indicating expression levels of NLRP1, CARD8, and NLRP3 in atherosclerotic lesions of a human coronary artery (Wirka et al., 2019). Each dot indicates a single cell under each cell type. (F) Immunoblots of VbP conditioned EC cell-culture supernatant showing levels of caspase-1 (CASP-1) cleavage, with β-actin loading control. (G) Immunoblots of EC cell lysates showing procaspase-1 levels. (H) Levels of IL-6, IL-8, and MCP-1 secreted by VbP-treated control (Ctl) and CASP-1 KO ECs. (I) Levels of ICAM-1 in the presence or absence of VbP measured by flow cytometry. (J) Number of THP-1 cells adhered to ECs treated with and without VbP. IL-1β treatment was used as a positive control. (K) Immunoblot of EC cell lysates showing levels of full-length (FL) and N-terminal (NT) NLRP1 with β-actin loading control. (L) Immunoblot of EC cell lysates showing levels of CARD8 with β-actin loading control. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test unless otherwise indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. Each symbol represents one biological replicate, unless otherwise stated. All experiments were repeated thrice. Source data are available for this figure: SourceData FS1.
NLRP1 and CARD8 are simultaneously triggered by VbP in primary human endothelial cells lines. Related to Fig. 1. (A) RNA sequencing of HAECs from two healthy donors showing mRNA levels of known inflammasome sensors. (B) Immunoblots of untreated A549 cell lysates and HAECs lysates showing levels of inflammasome proteins, with β-actin loading control. (C) mRNA levels of indicated inflammasome sensors in control and 16 h LPS-treated HAECs as measured by RNA sequencing. (D) mRNA levels of NLRP1, NLRP3, and CARD8 in three different primary endothelial cell lines (HUVEC, HMVEC, and HAEC) as measured by RNA sequencing (Lim et al., 2019). (E) Single-cell RNA sequencing indicating expression levels of NLRP1, CARD8, and NLRP3 in atherosclerotic lesions of a human coronary artery (Wirka et al., 2019). Each dot indicates a single cell under each cell type. (F) Immunoblots of VbP conditioned EC cell-culture supernatant showing levels of caspase-1 (CASP-1) cleavage, with β-actin loading control. (G) Immunoblots of EC cell lysates showing procaspase-1 levels. (H) Levels of IL-6, IL-8, and MCP-1 secreted by VbP-treated control (Ctl) and CASP-1 KO ECs. (I) Levels of ICAM-1 in the presence or absence of VbP measured by flow cytometry. (J) Number of THP-1 cells adhered to ECs treated with and without VbP. IL-1β treatment was used as a positive control. (K) Immunoblot of EC cell lysates showing levels of full-length (FL) and N-terminal (NT) NLRP1 with β-actin loading control. (L) Immunoblot of EC cell lysates showing levels of CARD8 with β-actin loading control. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test unless otherwise indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. Each symbol represents one biological replicate, unless otherwise stated. All experiments were repeated thrice. Source data are available for this figure: SourceData FS1.
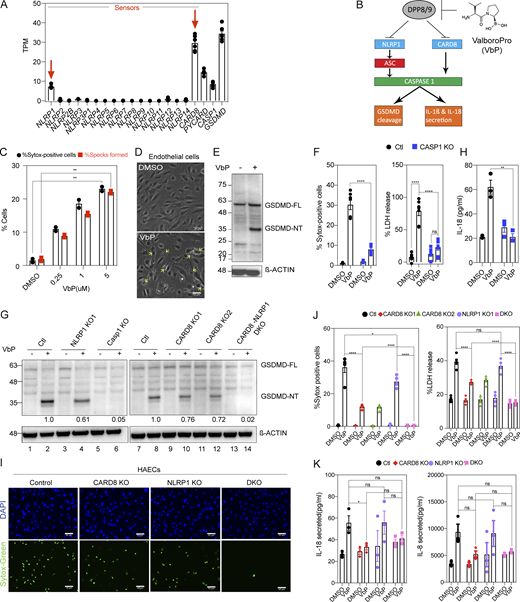
NLRP1 and CARD8 are simultaneously triggered by VbP in primary human endothelial cell lines. (A) RNA sequencing of HAECs from two healthy donors showing mRNA levels of inflammasome components. Each dot represents one biological replicate. (B) Schematic of NLRP1 and CARD8 inflammasome activation. VbP (also known as Talabostat) inhibits DPP8/9 to initiate inflammasome activation. (C) Number of ASC Specks formed and SYTOX-positive cells (i.e., percentage cell death) for WT ECs treated with VbP expressed as a percentage of all cells imaged. (D) Phase contrast image of WT ECs treated with the media control (Ctl) or VbP for 16 h. Images taken at 20×. Yellow arrows indicate a few of the cells having pyroptotic morphology. Scale bars, 50 µm. (E) Immunoblot of HAECs 16-h after treatment with VbP, showing levels of GSDMD cleavage from full-length (FL) into activated (NT) form, with a β-actin loading control. (F) Number of SYTOX-positive cells (left) and LDH release (right; expressed as a percentage of LDH released in the lysis control sample) of Ctl and CASP-1 KO ECs treated with 5 µM VbP or DMSO. (G) Immunoblots of VbP or DMSO-treated Ctl, NLRP1 KO, CARD8 KO 1 and 2, DKO, and CASP-1 KO EC cell lysates showing levels of GSDMD cleavage with densitometry ratios of N-terminal fragment normalized to β-actin control. (H) Levels of IL-18 secreted by VbP-treated Ctl and CASP-1 KO ECs. (I) HAECs were treated with VbP or DMSO followed by SYTOX green cell viability stain with Hoechst as a counterstain. Images were taken at 20×. (J) Percentage SYTOX-positive cells (left) and percentage LDH release (right) of Ctl and HAECs of indicated genotypes treated with VbP for 16 h. Media supernatant for the LDH assay was taken from the same samples used in the cell death assay. (K) Levels of IL-18 (left) and IL-8 (right) secreted by HAECs treated with VbP or DMSO. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test unless otherwise indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. Each symbol represents a distinct biological replicate. Each symbol in the SYTOX green cell death assay represents cell death as a percentage of all cells imaged across 12 fields of one well/sample. All Western blots are a representative image after conducting a minimum of three biological replicates (n = 3) for each of the independent experiments with WT, KO, DKO cells, etc. All experiments were repeated thrice. Source data are available for this figure: SourceData F1.
NLRP1 and CARD8 are simultaneously triggered by VbP in primary human endothelial cell lines. (A) RNA sequencing of HAECs from two healthy donors showing mRNA levels of inflammasome components. Each dot represents one biological replicate. (B) Schematic of NLRP1 and CARD8 inflammasome activation. VbP (also known as Talabostat) inhibits DPP8/9 to initiate inflammasome activation. (C) Number of ASC Specks formed and SYTOX-positive cells (i.e., percentage cell death) for WT ECs treated with VbP expressed as a percentage of all cells imaged. (D) Phase contrast image of WT ECs treated with the media control (Ctl) or VbP for 16 h. Images taken at 20×. Yellow arrows indicate a few of the cells having pyroptotic morphology. Scale bars, 50 µm. (E) Immunoblot of HAECs 16-h after treatment with VbP, showing levels of GSDMD cleavage from full-length (FL) into activated (NT) form, with a β-actin loading control. (F) Number of SYTOX-positive cells (left) and LDH release (right; expressed as a percentage of LDH released in the lysis control sample) of Ctl and CASP-1 KO ECs treated with 5 µM VbP or DMSO. (G) Immunoblots of VbP or DMSO-treated Ctl, NLRP1 KO, CARD8 KO 1 and 2, DKO, and CASP-1 KO EC cell lysates showing levels of GSDMD cleavage with densitometry ratios of N-terminal fragment normalized to β-actin control. (H) Levels of IL-18 secreted by VbP-treated Ctl and CASP-1 KO ECs. (I) HAECs were treated with VbP or DMSO followed by SYTOX green cell viability stain with Hoechst as a counterstain. Images were taken at 20×. (J) Percentage SYTOX-positive cells (left) and percentage LDH release (right) of Ctl and HAECs of indicated genotypes treated with VbP for 16 h. Media supernatant for the LDH assay was taken from the same samples used in the cell death assay. (K) Levels of IL-18 (left) and IL-8 (right) secreted by HAECs treated with VbP or DMSO. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test unless otherwise indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. Each symbol represents a distinct biological replicate. Each symbol in the SYTOX green cell death assay represents cell death as a percentage of all cells imaged across 12 fields of one well/sample. All Western blots are a representative image after conducting a minimum of three biological replicates (n = 3) for each of the independent experiments with WT, KO, DKO cells, etc. All experiments were repeated thrice. Source data are available for this figure: SourceData F1.
ECs undergo VbP-dependent pyroptosis
We next tested whether CARD8 and/or NLRP1 are capable of inducing pyroptosis in HAECs in response to VbP (also known as Talabostat mesylate), the universal activator of NLRP1 and CARD8 (Fig. 1 B; Okondo et al., 2018; Gai et al., 2019; Johnson et al., 2018, 2020). Using reporter HAECs expressing ASC-mCherry, we confirmed that VbP induced ASC-mCherry speck formation—a readout of NLRP1 inflammasome activation—in a dose-dependent manner (Fig. 1 C). In unmodified HAECs, VbP induced marked cell death characterized by ruptured cell membranes (Fig. 1 D, yellow arrows), which are indicative of lytic cell death. Consistently, VbP induced the cleavage of GSDMD into the p30 form (Fig. 1 E) and the release of CASP1 p10/12 fragment into the culture supernatant (Fig. S1 F). Furthermore, CRISPR/Cas9-mediated knockout of CASP1 in primary HAECs (Fig. S1 G) abrogated VbP-induced cell death measured by SYTOX green cell staining, lactate dehydrogenase (LDH) release (Fig. 1 F), and GSDMD cleavage (Fig. 1 G, lanes 5–6), confirming that the cell death observed is indeed pyroptosis. VbP treatment also resulted in the secretion of IL-18—another readout of inflammasome activity (Fig. 1 H)— which was abrogated in CASP1 KO HAECs. Altogether, these results validate that primary HAECs are capable of undergoing inflammasome-driven pyroptosis. Aside from IL-18, CASP1 deletion also blunted VbP-dependent secretion of pro-inflammatory cytokines IL-6, IL-8, and MCP-1 in a CASP1-dependent manner (Fig. S1 H), and upregulation of neutrophil adhesion glycoprotein ICAM-1 (Fig. S1 I) and increased adhesion of THP-1 monocytes (Fig. S1 J). These results suggest that EC pyroptosis leads to a general inflammatory response in ECs.
VbP simultaneously trigger CARD8 and NLRP1 in endothelial cells
As ECs express NLRP1 and CARD8, we asked if both sensors are simultaneously activated in response to VbP. For unknown reasons, in all human cell types studied to date, VbP-induced inflammasome activation goes through either NLRP1 or CARD8, but not both at the same time, even when both are expressed in a particular cell type. For example, in keratinocytes, VbP-induced pyroptosis and IL-18 secretion depend only on NLRP1, but not CARD8, even though both are present (Zhong et al., 2018), while in blood cells such as THP-1 and multiple leukemic cell lines like OCI-AML2 and MV4;11, only CARD8 is triggered by VbP (Johnson et al., 2018; Okondo et al., 2017). To address if either or both engaged in response to VbP in ECs, we generated NLRP1 KO, CARD8 KO, and double KO (DKO) HAECs. NLRP1 single KO HAECs (Fig. S1 K) reduced GSDMD cleavage by 40% (Fig. 1 G, lanes 3–4) but only modestly reduced cell death as measured by both SYTOX positivity (Fig. 1 I) and LDH release (Fig. 1 J) and did not significantly reduce IL-18 and IL-8 pro-inflammatory cytokine secretion in response to VbP (Fig. 1 K). These results suggest that in HAECs, VbP-mediated inflammasome activation is only partially dependent on NLRP1. On the other hand, CARD8 KO HAECs (Fig. S1 L) reduced GSDMD cleavage by about 30% (Fig.1 G, lanes 9–12), but reduced cell death and LDH release to a greater extent than the NLRP1 KO HAECs (Fig.1, I and J). This effect was particularly evident for IL-18 and IL-8 secretion, which was largely eliminated in CARD8 KO HAECs (Fig. 1 K). This indicates that CARD8 is also activated by VbP in HAECs and might play even a more major role in VbP-mediated inflammasome activation compared to NLRP1 (Fig.1 B). To confirm the simultaneous engagement of NLRP1 and CARD8, we measured VbP-mediated pyroptosis and cytokine secretion in NLRP1 and CARD8 DKO HAECs (Fig. S1 L, lane 4). DKO HAECs completely abrogated GSDMD cleavage (Fig.1 G), cell death (Fig. 1, I and J), and cytokine secretion (Fig. 1 K) in response to VbP. These results (summarized in Table S1) demonstrate that VbP activates both NLRP1 and CARD8 simultaneously in HAECs.
CVB3 activates the endothelial CARD8 inflammasome
We next sought to find potential pathogen/disease-related triggers of endothelial NLRP1 and CARD8 inflammasomes. Hornung and colleagues recently reported that dsRNA intermediates of the Semliki Forrest virus activate human NLRP1 (Bauernfried et al., 2020). Since the endothelium serves as an internal line of defense against blood-borne pathogens, we hypothesized that viruses with dsRNA replication intermediates might also activate NLRP1 or CARD8 in HAECs. Members of the Flaviviridae family like Dengue virus (DENV) and Zika virus (ZIKV) are positive sense, single-stranded RNA viruses with tropism to ECs. DENV induces endothelial dysfunction after prolonged infection and causes vascular leakage during Dengue hemorrhagic fever (DHF)/Dengue shock syndrome (DSS; Dalrymple and Mackow, 2012). ZIKV is also known to infect ECs in blood-brain and placental barriers (Liu et al., 2016a). To test if DENV or ZIKV can induce NLRP1/CARD8 activation, we assayed for GSDMD cleavage in DENV- and ZIKV-infected HAECs. While DENV and ZIKV effectively replicated in HAECs (Fig. S2 A), neither induced GSDMD cleavage (Fig. S2 B) nor IL-18 secretion (Fig. S2 C), even at a high multiplicity of infection (MOI) of 1. These results suggest that positive sense single-stranded RNA viruses are not all activators of inflammasome-driven pyroptosis in HAECs.
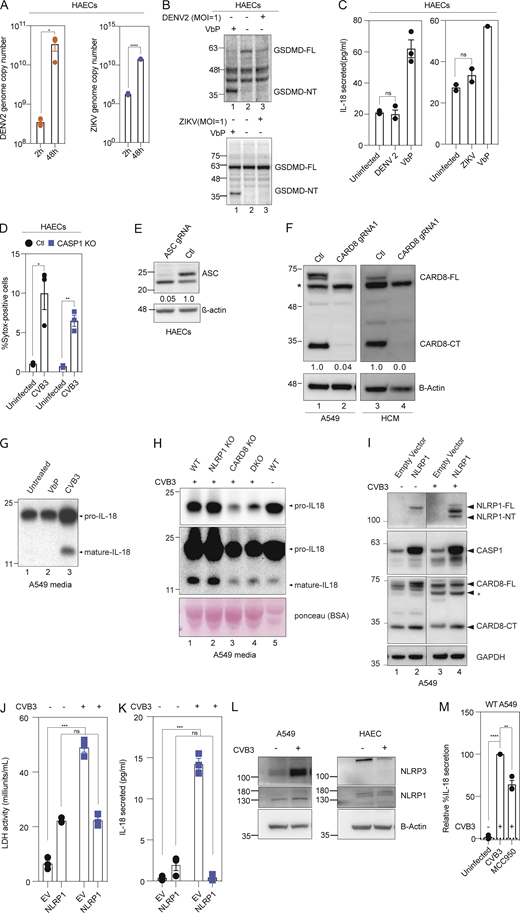
CVB3 activates the CARD8 inflammasome in endothelial cells and human cardiomyocytes. Related to Fig. 2. (A) Viral genome copy numbers of DENV2 (left) or ZIKV (right) in ECs infected with DENV2 or ZIKV, respectively, at an MOI = 1, 2 and 48 h after infection, measured by qPCR. (B) Immunoblots of WT EC cell lysates for GSDMD N-terminal fragment (GSDMD-NT) after treatment with DENV2 (top) or ZIKV (bottom) for 24 h, MOI = 1, with β-actin loading control. (C) Levels of IL-18 secreted by WT ECs after 24 h DENV2, ZIKV treatment at an MOI = 1 or 16 h VbP treatment as a positive control. (D) Percentage SYTOX-positive cells of HAECs 16-h post-CVB3 infection. (E) Immunoblot of ECs showing ASC levels, with β-actin loading control. (F) Immunoblots of A549 cell lysates and hPSC-derived HCMs indicating levels of CARD8, with β-actin loading control. (G) Immunoblots of VbP- and CVB3-treated A549 cell conditioned media showing levels of pro and mature forms of IL-18 secreted. (H) Immunoblots of CVB3 treated WT, NLRP1 KO, CARD8 KO, and DKO A549 cell-conditioned media showing levels of pro and mature forms of IL-18 secreted, with Ponceau staining used as a loading control. (I) Immunoblots of uninfected or CVB3 infected A549 cells over expressing NLRP1 or an empty vector (EV) harvested after 16 h, showing levels of NLRP1, CASP-1, and CARD8, with GAPDH loading control. (J) LDH activity in media collected from uninfected and CVB3 infected A549 cells over expressing NLRP1 or an EV. (K) Levels of IL-18 secreted by uninfected and CVB3 infected A549 cells over expressing NLRP1 or an empty vector EV. (L) Immunoblots of uninfected and CVB3 infected (for 16 h) WT A549 cell lysates and WT EC lysates indicating levels of NLRP3 and NLRP1, with β-actin loading control. (M) Relative levels of IL-18 secreted by CVB3 infected A549 cells in the presence or absence of NLRP3 inhibitor MCC950. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI is 0.1 unless indicated otherwise. Each symbol represents one biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData FS2.
CVB3 activates the CARD8 inflammasome in endothelial cells and human cardiomyocytes. Related to Fig. 2. (A) Viral genome copy numbers of DENV2 (left) or ZIKV (right) in ECs infected with DENV2 or ZIKV, respectively, at an MOI = 1, 2 and 48 h after infection, measured by qPCR. (B) Immunoblots of WT EC cell lysates for GSDMD N-terminal fragment (GSDMD-NT) after treatment with DENV2 (top) or ZIKV (bottom) for 24 h, MOI = 1, with β-actin loading control. (C) Levels of IL-18 secreted by WT ECs after 24 h DENV2, ZIKV treatment at an MOI = 1 or 16 h VbP treatment as a positive control. (D) Percentage SYTOX-positive cells of HAECs 16-h post-CVB3 infection. (E) Immunoblot of ECs showing ASC levels, with β-actin loading control. (F) Immunoblots of A549 cell lysates and hPSC-derived HCMs indicating levels of CARD8, with β-actin loading control. (G) Immunoblots of VbP- and CVB3-treated A549 cell conditioned media showing levels of pro and mature forms of IL-18 secreted. (H) Immunoblots of CVB3 treated WT, NLRP1 KO, CARD8 KO, and DKO A549 cell-conditioned media showing levels of pro and mature forms of IL-18 secreted, with Ponceau staining used as a loading control. (I) Immunoblots of uninfected or CVB3 infected A549 cells over expressing NLRP1 or an empty vector (EV) harvested after 16 h, showing levels of NLRP1, CASP-1, and CARD8, with GAPDH loading control. (J) LDH activity in media collected from uninfected and CVB3 infected A549 cells over expressing NLRP1 or an EV. (K) Levels of IL-18 secreted by uninfected and CVB3 infected A549 cells over expressing NLRP1 or an empty vector EV. (L) Immunoblots of uninfected and CVB3 infected (for 16 h) WT A549 cell lysates and WT EC lysates indicating levels of NLRP3 and NLRP1, with β-actin loading control. (M) Relative levels of IL-18 secreted by CVB3 infected A549 cells in the presence or absence of NLRP3 inhibitor MCC950. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI is 0.1 unless indicated otherwise. Each symbol represents one biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData FS2.
Recently, the HIV-1 protease was shown to cleave CARD8 at its N-terminus, leading to its activation and pyroptotic cell death in HIV-infected T lymphocytes (Wang et al., 2021). We, therefore, tested the alternative possibility that endothelial NLRP1/CARD8 can be activated by proteases encoded by EC-tropic viruses. CVB3 infects human ECs and causes myocarditis (Huber et al., 1990; Buskiewicz et al., 2016). We and others recently showed that CVB3-3Cpro can directly cleave and activate NLRP1 in heterologous systems (Robinson et al., 2020; Tsu et al., 2021); however, the effect of CVB3 infections in human primary ECs has not been characterized. This question is especially relevant, as the ability of ECs to simultaneously engage CARD8 and NLRP1 is not shared by other human cell types including skin keratinocytes. To address this question, we carried out live CVB3 infection in primary HAECs (Nancy strain, MOI = 0.1; Fig. 2 A). In infected cells, we observed morphological evidence of pyroptosis (Fig. 2 A, yellow arrowheads), similar to that seen in VbP-treated cells. Consistent with this finding, CVB3-infected cells caused rapid uptake of SYTOX green (Fig. 2 B) and GSDMD cleavage (Fig. 2 C, lane 2) in a CASP-1 dependent manner (Fig. S2 D and Fig. 2 C, lane 7). To our surprise, NLRP1 KO only modestly reduced GSDMD cleavage (Fig. 2 C, lane 4) and even enhanced CVB3-induced cell death (Fig. 2 B). Since human NLRP1 requires ASC to activate caspase-1, we further tested ASC KO HAECs (Fig. S2 E). Similar to NLRP1 KO, ASC KO failed to abolish CVB3-induced cell death or GSDMD cleavage (Fig. 2, B and C, lane 3), confirming that NLRP1 is dispensable for CVB3-induced inflammasome activation. In contrast, CARD8 KO completely abrogated GSDMD cleavage following CVB3 infection, as did the NLRP1 and CARD8 DKO ECs (Fig. 2 C, lanes 5–6). Consequently, CARD8 KO and DKO EC cells showed less pyroptotic morphology compared with control or NLRP1 KO HAECs (Fig. 2, A and B). In an orthogonal system, we infected A549 lung epithelial carcinoma cells, which express endogenous CARD8, with CVB3. CVB3 induced robust IL-18 secretion from A549, which was suppressed by knocking out CARD8 (Fig. 2 D and Fig. S2 F). As CARD8 was shown to induce little to no pro–IL-1β processing and release in hematopoietic cells in response to VbP (Johnson et al., 2018, 2020; Okondo et al., 2017), we checked if the IL-18 secreted from A549 was of the pro or mature form through immunoblotting (Fig. S2, G and H). Indeed, CVB3 but not VbP resulted in the secretion of mature, cleaved IL-18 (Fig. S2 G), which was reduced in CARD8 KO and DKO A549 cells (Fig. S2 H). This suggests that the ability of CARD8 to induce cytokine processing and release might be ligand- or cell type–dependent. The findings NLRP1 KO HAECs and A549 had higher cell death and IL-18 release in response to CVB3 infection, respectively (Fig. 2, B and D; and Fig. S2, G and H), prompted us to examine the relationship between NLRP1 and CARD8, namely if NLRP1 exerted an antagonistic effect on CARD8 in a cell type where both are present and functional. Indeed, overexpression of NLRP1 in A549 cells completely abrogated CVB3-mediated cell death and IL-18 secretion (Fig. S2, I–K). However, this was not due to an increase in CARD8 expression or autoproteolysis, which were also unchanged (Fig. S2 I). Hence, while NLRP1 appears to antagonize CVB3-dependent CARD8 activation, it does not appear to directly regulate the activity of CARD8 through its levels, autoproteolytic or viral protease-induced cleavage (see Fig. 3). It might regulate the downstream effects of CARD8 activation in yet unknown ways. Unlike HAECs, A549 upregulates NLRP3 (Fig. S2 L) upon CVB3 infection. Pharmacological inhibition of NLRP3 by MCC950 indeed reduced CVB3-induced IL-18 secretion (Fig. S2 M), confirming previous findings that CVB3 can also activate NLRP3 in cells that are NLRP3-competent (Wang et al., 2019).
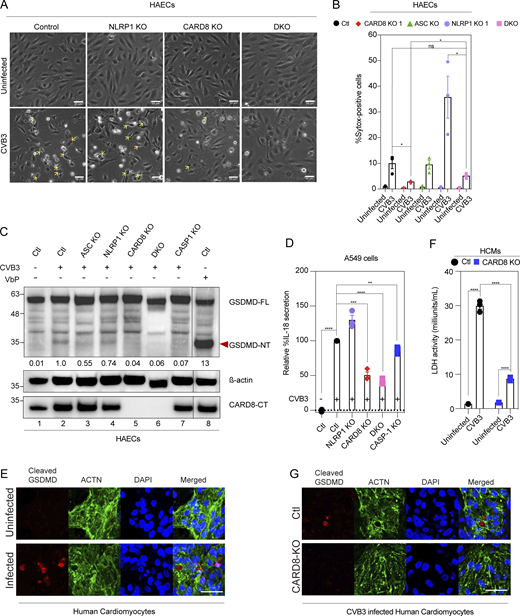
CVB3 activates the CARD8 inflammasome in endothelial cells and human cardiomyocytes. (A) Phase contrast images of HAECs of indicated genotypes 16-h post-CVB3 infection (20×). Yellow arrows highlight pyroptotic morphology. Scale bars, 50 µm. (B) Percentage SYTOX-positive HAECs of indicated genotypes 16-h post-CVB3 infection. (C) Immunoblot of HAECs infected with CVB3 for 16 h, showing levels of cleaved GSDMD with normalized densitometry values of N-terminal fragment (GSDMD-NT) with β-actin loading control. (D) Relative levels of IL-18 secreted by CVB3-infected A549 cell of indicated genotype, normalized to scrambled control (ctl). (E) Immunofluorescence of human cardiomyocytes (HCM) infected with CVB3 for 16 h, stained for cleaved GSDMD and cardiomyocyte marker ACTN. Scale bars, 50 µm. (F) Cell death as measured by LDH activity in supernatant of HCMs 16-h post-CVB3 infection. (G) Immunofluorescence of control and CARD8 KO HCM infected with CVB3 for 16 h for cleaved GSDMD and cardiomyocyte marker ACTN. Scale bars, 50 µm. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. The CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData F2.
CVB3 activates the CARD8 inflammasome in endothelial cells and human cardiomyocytes. (A) Phase contrast images of HAECs of indicated genotypes 16-h post-CVB3 infection (20×). Yellow arrows highlight pyroptotic morphology. Scale bars, 50 µm. (B) Percentage SYTOX-positive HAECs of indicated genotypes 16-h post-CVB3 infection. (C) Immunoblot of HAECs infected with CVB3 for 16 h, showing levels of cleaved GSDMD with normalized densitometry values of N-terminal fragment (GSDMD-NT) with β-actin loading control. (D) Relative levels of IL-18 secreted by CVB3-infected A549 cell of indicated genotype, normalized to scrambled control (ctl). (E) Immunofluorescence of human cardiomyocytes (HCM) infected with CVB3 for 16 h, stained for cleaved GSDMD and cardiomyocyte marker ACTN. Scale bars, 50 µm. (F) Cell death as measured by LDH activity in supernatant of HCMs 16-h post-CVB3 infection. (G) Immunofluorescence of control and CARD8 KO HCM infected with CVB3 for 16 h for cleaved GSDMD and cardiomyocyte marker ACTN. Scale bars, 50 µm. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. The CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData F2.
In myocarditis, tissue damage in the myocardium ultimately accounts for the pathogenicity of CVB3 infection. Cardiomyocytes have been shown to be directly infected by CVB3 in both human endomyocardial biopsies (Tschöpe et al., 2017) and in mouse models of CVB3 infection (Tschöpe et al., 2017; Wang et al., 2014; Yu et al., 2020). Hence, we asked if CARD8 also plays a role in human cardiomyocyte infection. To this end, we utilized human pluripotent stem cell (hPSC)-derived cardiomyocytes (HCM; Yap et al., 2019), which express high levels of CARD8 (Fig. S2 F, lane 3). Cleaved GSDMD immunoreactivity was detected in HCM following CVB3 infection (Fig. 2 E), demonstrating inflammasome activation. CARD8 KO HCM (Fig. S2 F) had lower cell death compared to control HCMs following CVB3 infection as evidenced by reduced LDH release (Fig. 2 F), demonstrating that pyroptotic cell death is attenuated in the absence of CARD8 in cardiomyocytes in addition to ECs. This is also supported by reduced cleaved GSDMD in the CARD8 KO cardiomyocytes (Fig. 2 G). Altogether, our findings (summarized in Table S2) indicate that CVB3 predominantly activates the CARD8 inflammasome in multiple cardiovascular cell types such as ECs and cardiomyocytes.
CARD8 activation is dependent on CVB3-induced degradation of CARD8 NT
Viral proteases have been implicated in the N-terminal cleavage-induced activation of NLRP1 and CARD8 (Wang et al., 2019; Tsu et al., 2021; Robinson et al., 2020). HRV-3C protease cleaves NLRP1 between Q130 and G131, generating an N-glycine degron that leads to the proteasomal degradation of the N-terminal auto-inhibitory domain (Robinson et al., 2020). Similarly, HIV-1 protease cleaves CARD8 between F58 and F59 to activate it in a proteasome-dependent manner (Wang et al., 2021). We first observed that full-length endogenous CARD8 but not NLRP1 was reduced in both HAECs and A549 following CVB3 infection (Fig. 3 A), suggesting a mechanism of infection-induced degradation. In HEK293T cells stably expressing CARD8, CVB3 infection caused a loss of reactivity against a specific antibody recognizing the N-terminus of CARD8, while the C-terminal fragment remained at comparable levels to uninfected cells (Fig. 3 B). CVB3 infection therefore increases the ratio of C-terminal/N-terminal fragments, which is predicted to disrupt ternary complex formation leading to CARD8 inflammasome activation (Sharif et al., 2021). CVB3-induced GSDMD cleavage in HAECs was sensitive to proteasome inhibitor Bortezomib (BTZ), as well as to cullin-RING E3 ubiquitin ligase inhibitor MLN4924 (Robinson et al., 2020; Fig. 3 C). These results suggest that upon CVB3 infection, the N-terminal fragment of CARD8 is degraded in a proteasome-dependent manner.
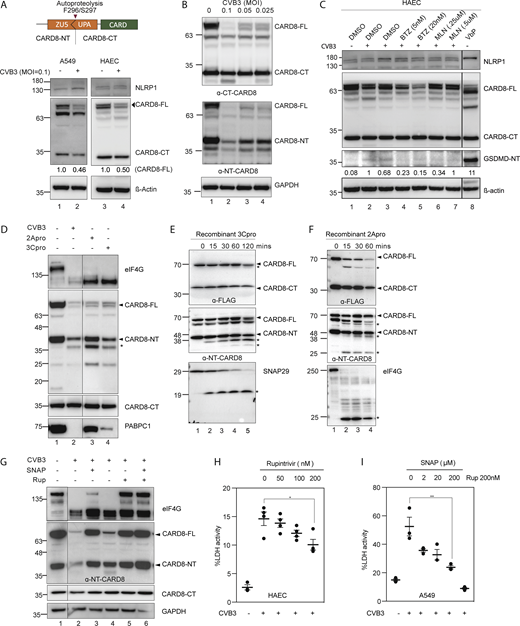
CVB3 2A and 3C protease mediate CARD8 activation. (A) Top: Domain organization of CARD8, which undergoes autoproteolysis to generate an N-terminal (NT) and C-terminal (CT) fragment. Bottom: CVB3-infected A549 and HAECs immunoblotted with C-terminal CARD8 antibody with normalized densitometry levels of full length CARD8 (CARD8-FL), with β-actin loading control. (B) CARD8 expressing HEK293T cells (HEK293T-CARD8) infected with indicated MOI of CVB3 and immunoblotted with ɑ-C- (top) and N-terminal (bottom) CARD8 antibody, with GAPDH loading control. (C) HAEC pretreated with the indicated drugs followed by CVB3 infection for 16 h. Immunoblotting and normalized densitometry values of GSDMD-NT indicate reduced inflammasome activation with BTZ and MLN pretreatments (at concentrations as indicated), with β-actin loading control. (D) HEK293T-CARD8 cells were transfected with CVB3-2A protease or 3C-protease and immunoblotted for ɑ-N-terminal (NT) and ɑ-C-terminal (CT) CARD8. eIF4G and PABPC1 are endogenous substrates of 2Apro and 3Cpro, respectively. * indicates cleavage product. (E) HEK293T-CARD8 cell lysates were incubated with purified CVB3-3C protease and immunoblotted for CARD8 using FLAG tag and ɑ-N-terminal (NT) CARD8 antibody. SNAP29 serves as a positive control for 3C-protease cleavage. (F) HEK293T-CARD8 cell lysates were incubated with purified CVB3-2A protease and immunoblotted for CARD8 using FLAG tag and ɑ-N-terminal (NT) CARD8 antibody. eIF4G serves as a positive control for 2A-protease cleavage. (G) HEK293T-CARD8 cells were infected with CVB3 in the presence or absence of increasing concentrations of 2A and 3C-protease inhibitors, SNAP and Rupintrivir, respectively, and immunoblotted with ɑ-NT-CARD8 and ɑ-CT-CARD8 antibodies, with GAPDH loading control. (H) Percentage cell death (measured by LDH activity) of HAECs infected with CVB3 in the presence of increasing concentrations of 3C-protease inhibitor, Rupintrivir. (I) Percentage cell death (measured by LDH activity) of A549 infected with CVB3 in the presence of increasing concentrations of 2A-protease inhibitor, SNAP. Unless indicated otherwise, all experiments have been carried out in HEK293T-CARD8 cells. Data are represented by mean ± SEM, P values indicated are from one-way ANOVA test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData F3.
CVB3 2A and 3C protease mediate CARD8 activation. (A) Top: Domain organization of CARD8, which undergoes autoproteolysis to generate an N-terminal (NT) and C-terminal (CT) fragment. Bottom: CVB3-infected A549 and HAECs immunoblotted with C-terminal CARD8 antibody with normalized densitometry levels of full length CARD8 (CARD8-FL), with β-actin loading control. (B) CARD8 expressing HEK293T cells (HEK293T-CARD8) infected with indicated MOI of CVB3 and immunoblotted with ɑ-C- (top) and N-terminal (bottom) CARD8 antibody, with GAPDH loading control. (C) HAEC pretreated with the indicated drugs followed by CVB3 infection for 16 h. Immunoblotting and normalized densitometry values of GSDMD-NT indicate reduced inflammasome activation with BTZ and MLN pretreatments (at concentrations as indicated), with β-actin loading control. (D) HEK293T-CARD8 cells were transfected with CVB3-2A protease or 3C-protease and immunoblotted for ɑ-N-terminal (NT) and ɑ-C-terminal (CT) CARD8. eIF4G and PABPC1 are endogenous substrates of 2Apro and 3Cpro, respectively. * indicates cleavage product. (E) HEK293T-CARD8 cell lysates were incubated with purified CVB3-3C protease and immunoblotted for CARD8 using FLAG tag and ɑ-N-terminal (NT) CARD8 antibody. SNAP29 serves as a positive control for 3C-protease cleavage. (F) HEK293T-CARD8 cell lysates were incubated with purified CVB3-2A protease and immunoblotted for CARD8 using FLAG tag and ɑ-N-terminal (NT) CARD8 antibody. eIF4G serves as a positive control for 2A-protease cleavage. (G) HEK293T-CARD8 cells were infected with CVB3 in the presence or absence of increasing concentrations of 2A and 3C-protease inhibitors, SNAP and Rupintrivir, respectively, and immunoblotted with ɑ-NT-CARD8 and ɑ-CT-CARD8 antibodies, with GAPDH loading control. (H) Percentage cell death (measured by LDH activity) of HAECs infected with CVB3 in the presence of increasing concentrations of 3C-protease inhibitor, Rupintrivir. (I) Percentage cell death (measured by LDH activity) of A549 infected with CVB3 in the presence of increasing concentrations of 2A-protease inhibitor, SNAP. Unless indicated otherwise, all experiments have been carried out in HEK293T-CARD8 cells. Data are represented by mean ± SEM, P values indicated are from one-way ANOVA test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData F3.
By reducing the dose or duration of CVB3 infection, we were able to detect the appearance of distinct CARD8 N-terminal cleavage products (Fig. S3 A). We therefore sought to identify the proteases responsible. We first ruled out that endogenous apoptotic caspases were responsible, since CARD8 cleavage was not affected by a pan-caspase inhibitor z-VAD-fmk (Fig. S3 B). The CVB3 genome encodes two proteases, known as 3Cpro and 2Apro (Fung et al., 2016; Maghsoudi et al., 2010). Transient overexpression of 3Cpro and 2Apro in HEK293T-CARD8 cells led to degradation of full-length CARD8 and the appearance of N-terminal cleavage products (Fig. 3 D) while preserving C-terminal fragments. Cleavage of known 2Apro substrate, eIF4G and 3Cpro substrate, PABPC1 (Maghsoudi et al., 2010; Kuyumcu-Martinez et al., 2002; Garmaroudi et al., 2015) confirmed the activity of transfected proteases. The observed N-terminal cleavage of CARD8 by 2A and 3Cpro correlated with inflammasome activation as measured by CASP1 cleavage and IL-18 release (Fig. S3 C), both of which were reduced in CARD8 KO cells. These data suggest that 2A/3Cpro-mediated N-terminal cleavage of CARD8 is sufficient to activate CARD8. Next, we performed in vitro cleavage experiments using purified CVB3 3Cpro (Fig. 3 E) and 2Apro (Fig. 3 F) incubated with HEK293T-CARD8 lysates. Both proteases cleaved CARD8 to yield truncated N-terminal fragments revealed by blotting for the C-terminal FLAG tag on the overexpressed CARD8, concurrently with the emergence of shorter N-terminal fragments similar to those seen with live CVB3 infection (Fig. 3 D). NLRP1, on the other hand, was cleaved only by 3Cpro (Fig. S3, D and E), as previously reported (Robinson et al., 2020), but not by 2Apro (Fig. S3 F). Of note, although in vivo cleavage of CARD8 by 2Apro and 3Cpro yielded similar cleavage bands (Fig. 3 D), the two proteases do not appear to cleave at the same sites, since the immediate cleavage products of the in vitro cleavage assays did not have the same sizes (Fig. 3, E and F). This discrepancy might reflect the inherent instability of cleavage products in vivo, such that only the most stable intermediates persist (Fig. 3 D). As such, the loss of N-terminal CARD8 reactivity is a more reliable readout of CARD8 degradation rather than the appearance of specific cleavage products. We next asked if 2Apro inhibitor, S-nitroso-N-acetyl-penicillamine (SNAP; Zell et al., 2004) or 3Cpro inhibitor, Rupintrivir (Dragovich et al., 1999) could prevent CARD8 cleavage. SNAP and Rupintrivir both suppressed CVB3-induced CARD8 degradation in HEK293T-CARD8 cells (Fig. 3 G and Fig. S3 G) and the activation of transfected CASP1 (Fig. S3 G). Consistently, SNAP and Rupintrivir treatment also rescued the levels of full-length and N-terminal CARD8 in 2Apro and 3Cpro-transfected HEK293T-CARD8 cells (Fig. S3 H). In HAECs, Rupintrivir treatment suppressed CVB3-induced cell death measured by LDH released (Fig. 3 H) and reduced GSDMD cleavage (Fig. S3 I) while SNAP also reduced CVB3-induced cell death in A549 (Fig. 3 I). However, the observed reduction in cell death in HAECs following live CVB3 infection cannot be solely attributed to inhibition of CARD8-mediated pyroptosis, since Rupintrivir—by inhibiting 3Cpro—also suppresses viral replication in both control and CARD8 KO HAECs (Fig. S3 J). The effects of various inhibitors on the outcomes of CVB3 infection are summarized in Table S3. Altogether, these results suggest that CVB3 activates the CARD8 inflammasome in human HAECs in a manner that requires both 2Apro and 3Cpro. These proteases likely cleave CARD8 leading to reduced N:C-terminal CARD8 ratio in a proteasome-dependent manner. As 2Apro is encoded 5′ to the 3Cpro in the CVB3 genome, our findings also imply that 2Apro would trigger CARD8-dependent pyroptosis before 3Cpro (Toyoda et al., 1986; Maghsoudi et al., 2010). This order of events might help explain why CARD8 plays a more important role than NLRP1 during CVB3 infection.
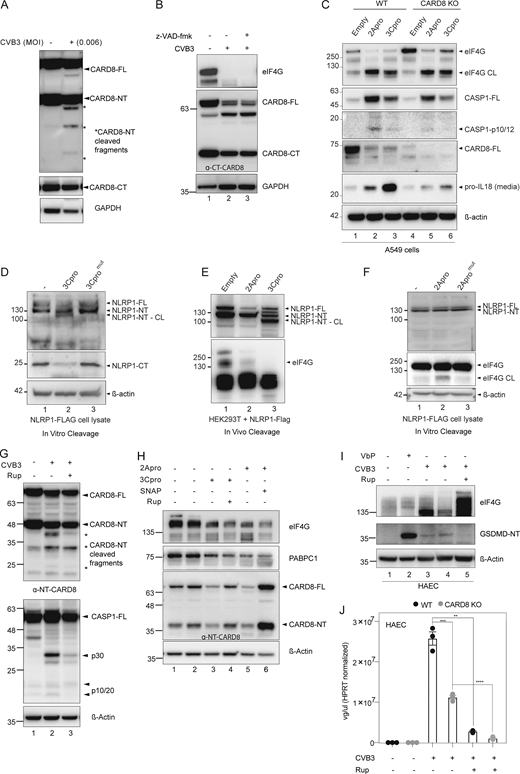
CVB3 2A and 3C protease mediate CARD8 activation. Related to Fig. 3. (A) HEK293T CARD8 infected with low dose (MOI = 0.006) of CVB3 and immunoblotted for NT-CARD8 and GAPDH loading control. (B) HEK293T CARD8 infected with CVB3 in the presence of pan-caspase inhibitor z-VAD-fmk. eIF4G served as a positive control indicating CVB3 infection, with GAPDH loading control. (C) Immunoblots of WT and CARD8 KO A549 cells transfected with 2Apro and 3Cpro showing levels of CASP-1, CARD8, and pro–IL-18, with β-actin loading control. eIF4G serves as a positive control for 2Apro-induced cleavage. (D) Cell lysates from NLRP1 overexpressing HEK293T cells were incubated with purified WT and catalytically inactive CVB3-3C protease, and immunoblotted for NLRP1 using FLAG tag and α-C-terminal NLRP1 antibody, with β-actin loading control. (E) HEK293T cells overexpressing NLRP1 were transfected with pIRES-2Apro or 3Cpro and immunoblotted 24 h later for NLRP1-NT. eIF4G serves as a positive control for 2A protease mediated cleavage. (F) Cell lysates from NLRP1 overexpressing HEK293T cells were incubated with purified WT and catalytically inactive CVB3-2A protease and immunoblotted for NLRP1 using FLAG tag, with β-actin loading control. eIF4G is a positive control for CVB3-2A protease-induced cleavage. (G) HEK293T CARD8 were transfected with CASP1 and infected with CVB3 in the presence of Rupintrivir (200 nM) and immunoblotted with α-NT-CARD8 and α-CASP1 antibodies. eIF4G degradation serves as a positive control indicating CVB3 infection. * indicates cleavage product. (H) HEK293T CARD8 were transfected with 2A protease and 3C-protease in the presence or absence of SNAP (200 μM) or Rupintrivir (200 nM) and immunoblotted with α-NT-CARD8, α-eIF4G, and α-PABPC1. eIF4G and PABPC1 are endogenous substrates of 2Apro and 3Cpro, respectively. (I) HAECs were infected with CVB3 in the presence of Rupintrivir (200 nM) and immunoblotted with α-GSDMD. eIF4G degradation served as a positive control indicating CVB3 infection. (J) WT and CARD8-KO HAECs were infected with CVB3 in the presence or absence of Rupintrivir (200 nM). Intracellular viral genome copy number was quantitated with qPCR. Each symbol represents one biological replicate. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI is 0.1 unless indicated otherwise. All experiments were repeated twice. Source data are available for this figure: SourceData FS3.
CVB3 2A and 3C protease mediate CARD8 activation. Related to Fig. 3. (A) HEK293T CARD8 infected with low dose (MOI = 0.006) of CVB3 and immunoblotted for NT-CARD8 and GAPDH loading control. (B) HEK293T CARD8 infected with CVB3 in the presence of pan-caspase inhibitor z-VAD-fmk. eIF4G served as a positive control indicating CVB3 infection, with GAPDH loading control. (C) Immunoblots of WT and CARD8 KO A549 cells transfected with 2Apro and 3Cpro showing levels of CASP-1, CARD8, and pro–IL-18, with β-actin loading control. eIF4G serves as a positive control for 2Apro-induced cleavage. (D) Cell lysates from NLRP1 overexpressing HEK293T cells were incubated with purified WT and catalytically inactive CVB3-3C protease, and immunoblotted for NLRP1 using FLAG tag and α-C-terminal NLRP1 antibody, with β-actin loading control. (E) HEK293T cells overexpressing NLRP1 were transfected with pIRES-2Apro or 3Cpro and immunoblotted 24 h later for NLRP1-NT. eIF4G serves as a positive control for 2A protease mediated cleavage. (F) Cell lysates from NLRP1 overexpressing HEK293T cells were incubated with purified WT and catalytically inactive CVB3-2A protease and immunoblotted for NLRP1 using FLAG tag, with β-actin loading control. eIF4G is a positive control for CVB3-2A protease-induced cleavage. (G) HEK293T CARD8 were transfected with CASP1 and infected with CVB3 in the presence of Rupintrivir (200 nM) and immunoblotted with α-NT-CARD8 and α-CASP1 antibodies. eIF4G degradation serves as a positive control indicating CVB3 infection. * indicates cleavage product. (H) HEK293T CARD8 were transfected with 2A protease and 3C-protease in the presence or absence of SNAP (200 μM) or Rupintrivir (200 nM) and immunoblotted with α-NT-CARD8, α-eIF4G, and α-PABPC1. eIF4G and PABPC1 are endogenous substrates of 2Apro and 3Cpro, respectively. (I) HAECs were infected with CVB3 in the presence of Rupintrivir (200 nM) and immunoblotted with α-GSDMD. eIF4G degradation served as a positive control indicating CVB3 infection. (J) WT and CARD8-KO HAECs were infected with CVB3 in the presence or absence of Rupintrivir (200 nM). Intracellular viral genome copy number was quantitated with qPCR. Each symbol represents one biological replicate. Data are represented by mean ± SEM, P values indicated are from parametric, unpaired, two-tailed student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI is 0.1 unless indicated otherwise. All experiments were repeated twice. Source data are available for this figure: SourceData FS3.
CVB3 3Cpro cleaves CARD8 at p.G38
We sought to define the 3Cpro cleavage sites in the CARD8 N-terminal region. As enteroviral 3Cpro typically requires a glycine at the P1 position, we mutated all glycine residues before the FIIND autoproteolytic cleavage site. CARD8 mutant 1 encompasses the first six N-terminal glycine residues (G38A, G57A, G112_113A, G127A, G131A; Fig. 4 A) and was insensitive to in vitro 3Cpro-mediated cleavage (Fig. 4 B). CARD8 mutants 2 and 3, each containing five glycine mutations within the FIIND domain (Fig. 4 A), abrogated autoproteolytic cleavage in addition to 3C cleavage (Fig. 4 B) and were hence not further examined. We further isolated the putative cleavage site within CARD8 mutant 1 to p.G38, since mutating G38 alone (along with Q37 i.e., p.Q37L_G38P; Fig. 4 C) but not the other glycine residues in mutant 1 (Fig. 4 D) abolished 3Cpro-mediated N-terminal cleavage. This cleavage site is consistent with the predicted size of the N-terminal cleavage fragment (<10 kD). Consistent with G38 being the CVB3 3Cpro cleavage recognition site, CARD8Q37L_G38P was not cleaved following CVB3 infection in HEK293T cells, using CARD8G166E as a negative control that underwent cleavage (Fig. 4 E). Furthermore, CARD8Q37L_G38P, unlike WT CARD8, failed to increase CVB3-induced pyroptosis as measured by LDH release in when overexpressed in A549 cells (Fig. 4 F).
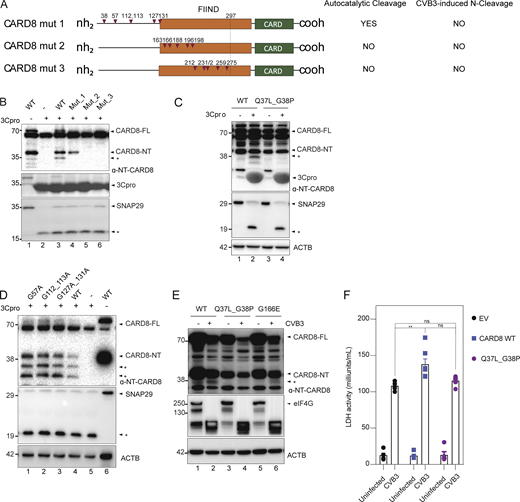
CVB3 3C-protease cleaves CARD8 at p.G38. (A) Domain organization of CARD8 mutants generated by site-directed mutagenesis. Autoproteolytic function and CVB3-induced CARD8-NT cleavage ability of each mutant are indicated on the right. Each red arrowhead represents a glycine residue. (B) Cell lysates of WT and mutant CARD8 transfected into HEK293T cells were incubated with CVB3 3C-protease, and immunoblotted for CARD8 NT. (C) Cell lysates of WT and mutant CARD8 (p.Q37L_G38P) transfected into HEK293T cells were incubated with CVB3 3C-protease, and immunoblotted for CARD8. SNAP29 is used as a positive control for 3C protease-induced cleavage, with β-actin loading control. (D) Cell lysates of WT and mutant CARD8 (G57A, G112_113A, G127A_131A) transfected into HEK293T cells were incubated with CVB3 3C-protease, and immunoblotted for CARD8. SNAP29 is used as a positive control for 3C protease-induced cleavage, with β-actin loading control. (E) HEK293T cells stably expressing WT and mutant CARD8 (p.Q37L_G38P and G166E) were infected with CVB3 (MOI = 0.006) and immunoblotted 16 h later for CARD8 NT, with β-actin loading control. EIF4G is used as a positive control of CVB3-induced cleavage products. (F) Cell death as measured by LDH release of A549 cells stably expressing empty vector (EV), WT CARD8 or cleavage mutant CARD8Q37L_G38P 16-h post-CVB3 infection. Data are represented by mean ± SEM, P values indicated are from one-way ANOVA test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData F4.
CVB3 3C-protease cleaves CARD8 at p.G38. (A) Domain organization of CARD8 mutants generated by site-directed mutagenesis. Autoproteolytic function and CVB3-induced CARD8-NT cleavage ability of each mutant are indicated on the right. Each red arrowhead represents a glycine residue. (B) Cell lysates of WT and mutant CARD8 transfected into HEK293T cells were incubated with CVB3 3C-protease, and immunoblotted for CARD8 NT. (C) Cell lysates of WT and mutant CARD8 (p.Q37L_G38P) transfected into HEK293T cells were incubated with CVB3 3C-protease, and immunoblotted for CARD8. SNAP29 is used as a positive control for 3C protease-induced cleavage, with β-actin loading control. (D) Cell lysates of WT and mutant CARD8 (G57A, G112_113A, G127A_131A) transfected into HEK293T cells were incubated with CVB3 3C-protease, and immunoblotted for CARD8. SNAP29 is used as a positive control for 3C protease-induced cleavage, with β-actin loading control. (E) HEK293T cells stably expressing WT and mutant CARD8 (p.Q37L_G38P and G166E) were infected with CVB3 (MOI = 0.006) and immunoblotted 16 h later for CARD8 NT, with β-actin loading control. EIF4G is used as a positive control of CVB3-induced cleavage products. (F) Cell death as measured by LDH release of A549 cells stably expressing empty vector (EV), WT CARD8 or cleavage mutant CARD8Q37L_G38P 16-h post-CVB3 infection. Data are represented by mean ± SEM, P values indicated are from one-way ANOVA test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice. Source data are available for this figure: SourceData F4.
CARD8 deletion enhances EC-intrinsic immunity against CVB3 infection
We next asked if CARD8-dependent pyroptosis during CVB3 infection affects viral replication and dissemination. HAECs and HCMs are permissive for CVB3 replication (Fig. 5 A). We first tested the effect of CARD8 inflammasome activation to CVB3 propagation by measuring intracellular genome copy number. CARD8 KO reduced intracellular CVB3 genome replication in both HAECs and HCMs (Fig. 5 A). This was accompanied by reduced viral propagation, as evidenced by the reduced functional viral titer (measured using plaque assays) in the supernatant of infected cells (Fig. 5 B). We confirmed these results by infecting HAECs with Timer-CVB3 (Robinson et al., 2014), a recombinant virus harboring a fluorescent timer protein that undergoes slow conversion from green to red over time during viral maturation. Timer-CVB3 allowed us to track viral infection and dissemination in the same controlled experiment. This analysis revealed that CARD8 KO HAECs had reduced GFP-positivity following infection (Fig. 5 C), indicating that early stages of viral infection and replication were inhibited by the loss of CARD8. Alternatively, reduced pyroptosis following CVB3 infection could also impair viral dissemination and propagation, leading to an overall reduction in viral reproduction in a bulk culture. This hypothesis predicts that inhibiting pyroptosis downstream of CARD8 would phenocopy CARD8 KO cells vis-à-vis viral titer. Indeed, suppressing pyroptosis using CASP1 KO in HAECs, or pretreatment with CASP1 inhibitor Belnacasan, or GSMD inhibitor Disulfuram (Hu et al., 2020) effectively reduced CVB3 viral titer in the supernatant following CVB3 infection (Fig. 5, D and E). Hence, depletion of CARD8 attenuates viral propagation in HAECs and HCMs, likely by impairing shedding of functional viruses from infected cells.

CARD8 deletion in ECs and HCMs improves cell-intrinsic immunity against CVB3. (A) Viral genome (vg) copy numbers of CVB3 in control (Ctl) and CARD-KO HAECs and HCMs determined by qPCR. (B) Functional titers of CVB3 in Ctl and CARD8-KO HAECs and HCMs measured by plaque assays. P values indicated are from parametric, unpaired, two-tailed student’s t test. (C) Epifluorescence images of Ctl and CARD8-KO HAECs infected with Timer-CVB3 in the GFP channel, indicating early infection. Scale bars, 50 µm. (D) vg copy numbers of CVB3 in WT HAECs treated with specific inhibitors Belnacasan at 10 μM (caspase-1 inhibitor) and Disulfuram at 10 μM (GSDMD inhibitor) or both, determined by qPCR. CVB3 infection MOI = 0.006. (E) vg copy numbers of CVB3 in WT and CASP-1 KO HAECs, determined by qPCR. CVB3 infection MOI = 0.006. (F) Expression of ICAM-1 in CVB3-infected HAECs measured by flow cytometry. (G) Number of THP1 cells adhered to Ctl and CARD8 KO HAECs infected with CVB3 at the indicated time points. (H) mRNA levels of representative pro-inflammatory genes induced by CVB3 infection in HAECs, determined by qPCR. (I) mRNA of indicated interferon-response genes in Ctl and CARD8-KO HCMs measured by qPCR. (J) mRNA levels of representative pro-inflammatory and interferon response genes induced by CVB3 infection in HAECs in the presence and absence of specific inhibitors, Belnacasan at 10 μM (caspase-1 inhibitor), Disulfuram at 10 μM (GSDMD inhibitor), or both, determined by qPCR. CVB3 infection MOI = 0.006. (K) Schematic of co-culture between Ctl and CARD8-KO HAECs infected with the Timer-CVB3 and HCMs. (L) Epifluorescence images of HCMs co-cultured with Ctl and CARD8-KO HAECs and infected from the apical side of the endothelium with Timer-CVB3 in the DsRed channel indicating a later stage of infection. Scale bars, 50 µm. (M) CVB3 vg copy number in HCMs co-cultured with Ctl and CARD8 KO HAECs infected with the Timer-CVB3 measured by qPCR. P values indicated are from parametric, unpaired, two-tailed student’s t test. Data are represented by mean ± SEM, P values indicated are from two-way ANOVA test unless otherwise stated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice.
CARD8 deletion in ECs and HCMs improves cell-intrinsic immunity against CVB3. (A) Viral genome (vg) copy numbers of CVB3 in control (Ctl) and CARD-KO HAECs and HCMs determined by qPCR. (B) Functional titers of CVB3 in Ctl and CARD8-KO HAECs and HCMs measured by plaque assays. P values indicated are from parametric, unpaired, two-tailed student’s t test. (C) Epifluorescence images of Ctl and CARD8-KO HAECs infected with Timer-CVB3 in the GFP channel, indicating early infection. Scale bars, 50 µm. (D) vg copy numbers of CVB3 in WT HAECs treated with specific inhibitors Belnacasan at 10 μM (caspase-1 inhibitor) and Disulfuram at 10 μM (GSDMD inhibitor) or both, determined by qPCR. CVB3 infection MOI = 0.006. (E) vg copy numbers of CVB3 in WT and CASP-1 KO HAECs, determined by qPCR. CVB3 infection MOI = 0.006. (F) Expression of ICAM-1 in CVB3-infected HAECs measured by flow cytometry. (G) Number of THP1 cells adhered to Ctl and CARD8 KO HAECs infected with CVB3 at the indicated time points. (H) mRNA levels of representative pro-inflammatory genes induced by CVB3 infection in HAECs, determined by qPCR. (I) mRNA of indicated interferon-response genes in Ctl and CARD8-KO HCMs measured by qPCR. (J) mRNA levels of representative pro-inflammatory and interferon response genes induced by CVB3 infection in HAECs in the presence and absence of specific inhibitors, Belnacasan at 10 μM (caspase-1 inhibitor), Disulfuram at 10 μM (GSDMD inhibitor), or both, determined by qPCR. CVB3 infection MOI = 0.006. (K) Schematic of co-culture between Ctl and CARD8-KO HAECs infected with the Timer-CVB3 and HCMs. (L) Epifluorescence images of HCMs co-cultured with Ctl and CARD8-KO HAECs and infected from the apical side of the endothelium with Timer-CVB3 in the DsRed channel indicating a later stage of infection. Scale bars, 50 µm. (M) CVB3 vg copy number in HCMs co-cultured with Ctl and CARD8 KO HAECs infected with the Timer-CVB3 measured by qPCR. P values indicated are from parametric, unpaired, two-tailed student’s t test. Data are represented by mean ± SEM, P values indicated are from two-way ANOVA test unless otherwise stated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. CVB3 infection MOI used is 0.1 unless indicated otherwise. Unless otherwise stated, each dot represents a biological replicate. All experiments were repeated twice.
Myocarditis is characterized by unrestrained recruitment of leukocytes to the myocardium, which inflicts tissue damage leading to myocarditis (Gao et al., 2008; Tschöpe et al., 2021). Since the endothelium is the major gatekeeper of tissue infiltrating leukocytes (Huber et al., 1990), we asked if CARD8 affected endothelial activation and leukocyte adhesion. Indeed, CARD8 KO HAECs had significantly reduced expression of the prototypical activation marker ICAM-1, which is robustly induced following CVB3 infection (Fig. 5 F). ICAM-1 is a ligand for LFA-1, an integrin found on the surface of leukocytes. ICAM-1/LFA stabilizes the interactions between leukocytes and the endothelium, allowing them to transmigrate (Yang et al., 2005). Leukocyte recruitment is also considered as a hallmark of endothelial inflammation (Kumar et al., 2020). In line with reduced ICAM-1 expression, CARD8 KO HAECs recruited fewer THP-1 monocytes indicating reduced leukocyte adhesion (Fig. 5 G), and expressed lower levels of pro-inflammatory genes (Fig. 5 H) following CVB3 infection. Likewise, CARD8 KO in HCM suppressed the expression of pro-inflammatory and interferon-responsive genes such as IFITM, MX2, OAS1, IL-1β, and TNFα (Fig. 5 I). We wondered if this anti-inflammatory phenotype of CARD8 KO cells was simply due to reduced viral propagation. Belnacasan- and Disulfuram-mediated inhibition of pyroptosis in CVB3-infected HAECs both reduced viral release (Fig. 5 E; Hu et al., 2020). However, this was not sufficient to reduce pro-inflammatory and interferon-responsive gene expression (Fig. 5 J). These results indicate that in addition to facilitating viral release through pyroptosis, CARD8 also promotes induction of pro-inflammatory pathways through mechanisms independent of pyroptosis.
Finally, we asked if CARD8 attenuation protects underlying cardiomyocytes from infection. To this end, we performed co-culture experiments between HAECs and HCM separated by a 0.4 µM transwell membrane (Fig. 5 K), which allows the passage of virions and cytokines but not cells. We infected WT or CARD8 KO HAECs on the apical side with Timer-CVB3 (Robinson et al., 2014), and examined the development of red fluorescence in the underlying HCMs as an indication of established infection. CARD8 KO HAECs indeed protected the underlying HCM from apical CVB3 infection as judged by reduced frequency and intensity of HCMs with red fluorescence (Fig. 5 L). This was corroborated by reduced qPCR-quantified CVB3 genome copy number in the HCMs co-cultured with CARD8 KO HAECs (Fig. 5 M). Altogether, these results indicate that CARD8 potentiates CVB3 propagation and endothelial activation following CVB3 infection. In the absence of CARD8, CVB3 infection of cardiovascular target tissues such as ECs and cardiomyocytes leads to less cell death, viral propagation, and reduced endothelial barrier disruption. As such, inhibiting the CARD8 inflammasome in the context of CVB3 infection might provide protection against CVB3-induced myocarditis.
The endothelium plays a major role in the innate immune machinery of the human body (Shao et al., 2020). Here, we provide evidence that the NLRP1 and CARD8 inflammasomes mediate endothelial pyroptosis and inflammatory activation. To the best of our knowledge, human ECs are the first cell type reported to activate both NLRP1 and CARD8 in response to VbP. Another aspect in which ECs differ from other inflammasome competent cell types is that they do not secrete large amounts of IL-1B or IL-18 despite prominent GSDMD cleavage. This is in agreement with previous reports demonstrating that CARD8 activation can lead to GSDMD-mediated cell death in the absence of IL-1 cleavage and secretion (Johnson et al., 2020). Primary human ECs also do not express NLRP3, even upon LPS priming. This suggests that NLRP3 inhibitors may not be as effective in preventing EC-intrinsic inflammasome activation. Furthermore, deletion of murine Nlrp3 (Wang et al., 2019) exacerbates CVB3-induced cardiomyopathy in mice.
We also provide evidence that CVB3 predominantly activates the CARD8 inflammasome within human ECs likely through 2Apro- and 3Cpro-dependent CARD8 cleavage. For 3Cpro, this cleavage occurs at p.Glycine_38 in the N-terminus, which leads to functional degradation and activation of CARD8. Unlike 3Cpro, 2A cleavage motifs are less understood. We have not been able to map the exact CARD8 cleavage site by 2Apro using direct mutagenesis or through mass spectrometry due to the difficulty in obtaining sufficient active recombinant 2Apro (Fig. 3 D). Nonetheless, 2Apro likely recognizes a distinct site of cleavage as 3Cpro, since the cleavage products of both enzymes are distinct (Fig. 3, E and F). Future work is required to fully elucidate the molecular mechanism on how 2Apro recognizes and cuts the CARD8 N-terminus.
At first glance, our results are incongruent with the discovery that CVB3-3Cpro activates human NLRP1 (Tsu et al., 2021), as reported recently by us and other groups. However, it is important to highlight that these earlier studies did not use cell types that could simultaneously activate both NLRP1 and CARD8—a property that is unique to human ECs. Since 2Apro is expressed earlier than 3Cpro during the enteroviral life cycle, 2Apro-triggered CARD8 activation very likely precedes and dominates over 3Cpro-driven NLRP1 activation during CVB3 infection of ECs. In addition, CARD8 does not require ASC oligomerization to activate NLRP1, we hypothesize that once started, CARD8 activation leads to pyroptotic cell death that occurs more rapidly than NLRP1 even though both can be cleaved by the viral proteases. As a result, NLRP1 activation might not lead to any major increase in pyroptotic readouts. Our functional experiments also revealed that CARD8 potentiates CVB3 propagation in ECs and cardiomyocytes. This could occur as a direct result of CARD8-mediated pyroptosis aiding viral spread. An alternate possibility is that CARD8 suppresses anti-viral response through non-interferon-dependent pathways and is co-opted by viruses to evade host immune responses. Future studies are required to distinguish between these two possibilities.
In summary, this work demonstrates that ECs express both CARD8 and NLRP1 inflammasomes and that both are functional in response to the DPP9 inhibitor VbP. These findings also highlight CARD8 as a major sensor for CVB3 in multiple cell types in the human cardiovascular system. While NLRP1 is dispensable for CVB3-induced inflammasome activation, we postulated that pharmacologic inhibition of the CARD8 inflammasome might be useful in limiting pathogenic inflammation in certain cardiovascular conditions. However, it remains to be seen whether a pathogen may engage in both inflammasome sensors within a single cell to orchestrate host protection or whether a potential interplay between CARD8 and NLRP1 in ECs exists.
Materials and methods
Cell culture and reagents used
Cryopreserved primary HAECs were purchased from PromoCell (C12271; PromoCell). HAECs were grown in endothelial growth medium MV2 (EGMV2) media containing 5% EGMV supplement from PromoCell (C22022), 1% penicillin/streptomycin mixture (15140122; Gibco), and filter sterilized through a 0.2-µm filter. To subculture, HAECs were washed once with PBS, trypsinized with 0.25% trypsin (25200072; Life Technologies) for 90 s, and then neutralized with an equal volume of EGMV2 complete media. They were then counted and centrifuged at 500 rcf, resuspended and seeded at density between 3,000 and 5,000 cells/cm2. For maintenance, plates were coated with 7 µg/ml fibronectin in PBS (C43050; Promocell) for 45 min and aspirated before seeding the cells.
HeLa (CCL2; ATCC), A549 (CCL-185; ATCC), and HEK293T (CRL-3216; ATCC) were cultured in DMEM (SH30022.01; Cytiva) supplemented with 10% FBS (SV30160.03HI; Cytiva) and 1% penicillin/streptomycin mixture (15140122; Gibco). HEK293T-pCDH-puro-CARD8-FLAG cells were generated as previously described (Robinson et al., 2020).
Media was changed every 48 h until the cells grew to confluence. To subculture, cells were washed once with PBS (C43050; Promocell), trypsinized (0.25% trypsin; 25200072; Life Technologies) for 2 min, neutralized with an equal volume of 10% FBS/DMEM media and centrifuged at 500 rcf for 5 min, resuspended, and seeded at a density of 8,000 cells /cm2.
All human pluripotent stem cell studies were carried in accordance with National University of Singapore’s Institutional Review Board (IRB-12-451). Cardiomyocytes were generated as described previously (Yap et al., 2019). Human embryonic stem cells (H1, WiCell Research Institute) were routinely cultured on 10 µg/ml of laminin-521 in PBS (Biolamina) and daily Nutristem media change to maintain pluripotency. For differentiation, 6 million H1 cells were seeded onto 10 cm2 dish (Thermo Fisher Scientific) precoated with LN-521+221. Cells were maintained from day 0 to 3 on Nutristem media. On day 4, differentiation RPMI media with B27 supplement without insulin (Thermo Fisher Scientific) with 10 μM of CHIR 99021 (Tocris) were added into the wells. Medium was changed the next day. On day 7, differentiation media with 5 μM of IWP2 (Tocris) was added into the wells, followed by media changed on day 9. Cells were then dissociated from the dish with TrypLE (Thermo Fisher Scientific) at 8 min, 37°C and replated at 1 million cells onto 24-well plate.
Reagents and antibodies
The small molecule inhibitor of DPP8/9, Talabostat mesylate (Val-boroPro mesylate), was purchased from MedChem Express (HY-13233A). LPS (InvivoGen tlrl-3pelps) was used at 1,000 EU/ml made in EGMV2 media. Nigericin was purchased from MedChem Express (HY-100381).
For cell-culture experiments, Talabostat was prepared in DMSO solution containing 0.1% TFA to prevent cyclization. TFA was purchased from Sigma-Aldrich (T6508-5Ml). Triton-X 100 was purchased from Merck (T8787-250 ml). The inhibitors Bortezomib (HY-10227), Rupintrivir (HY-106161), SNAP (82250-25), and Z-VAD-FMK (HY-16658B) were purchased from Cayman Chemical. Specific inhibitor Disulfuram (#PHR1690; Sigma-Aldrich) was purchased from Sigma, and Belcanasan (HY-13205) was purchased from MedChem Express.
Antibodies used were anti-NLRP1 (Cat.no AG-25B-0005-C100; Adipogen), anti-C-terminal–CARD8 (ab24186; Abcam), anti-N-terminal–CARD8 (ab194585; Abcam), anti-ASC (AG-25B-0006-C100; Adipogen), anti-pro-caspase-1+ p10 +p12 (ab719515; Abcam), anti-N-terminal–Gasdermin-D (ab215203; Abcam), cleaved GSDMD-NT (Asp275; 36425; Cell Signaling Technology), GSDMDC1 (NBP2-33422; Novus Biologicals), anti-eIF4G(C45A4) (2469; Cell Signaling Technology), anti-PABP (10910-1-AP; Proteintech), anti–IL-18 (ab201324; Abcam), anti–IL-1β (AB201; R&D systems), and anti–β-actin (A2228-100 µl; Sigma-Aldrich).
Preparation of the RNAseq libraries and downstream analysis
mRNA was enriched from 1 µg total RNA using NEBNext poly(A) mRNA Magnetic Isolation Module (E7490L; New England Biolabs). The library was prepared from the mRNA-enriched fraction using NEBNext UltraTM II Directional RNA Library Prep (E7765L; New England Biolabs) according to the manufacturer’s instructions. The library was sequenced on the Illumina HiSeq 2500 platform, generating 150 bp paired end reads. The reads were trimmed for adaptors using Trimmomatic and aligned to GRCh38 genome assembly using the STAR aligner and gene-wise read coverage was quantified using Feature Counts from the Subread package. All downstream analysis was performed using R. Genes with TPM below 1 in every sample were discarded. DESeq2 was used to identify differentially expressed genes.
Cell death–SYTOX green cell staining assay
HAECs were seeded at 20,000 cells per well, in a 96-well ultra-cell carrier plate from PerkinElmer (6055302), with a final media volume of 100 µl per well. 24 h later, the cells were treated with 5 µM of VbP or DMSO control or CVB3 (ATCC VR-30) for 16–18 h. The media was then used for the LDH cytotoxicity assay. At the same time, the cells were stained with Hoechst 33342, 1:10,000 (sc-200908; Santa Cruz) and with the SYTOX green dead cell stain, 1:2,000 (S34860; Thermo Fisher Scientific) in media for 45 min and then imaged using the Operetta High Content Imager (PerkinElmer) at 37°C and 5% CO2 supply. 12 fields were randomly selected and imaged per well. The images were quantified on the Columbus Image Data Storage and Analysis system (PerkinElmer). The total number of Hoechst positive cells was considered as the total number of cells (both live and dead) in one well. The total number of SYTOX + Hoechst positive cells was considered as dead cells. Percentage of SYTOX-positive cells i.e., cell death was calculated and plotted using PRISM 8. Each experiment was performed in triplicate (n = 3).
LDH cytotoxicity assay
HAECs, A549 cells, or HCMs, were seeded at 20,000 cells per well, in a 96-well ultra-cell carrier plate (6055302; Perkin-Elmer), with a final media volume of 100 µl per well. 24 h later, the cells were treated with either 5 µM VbP or DMSO or CVB3 (VR-30; ATCC) for 16–18 h. The media was spun at 4°C at 500 rcf for 5 min in order to sediment the cellular debris. The media supernatant was analyzed for LDH activity using LDH activity assay kit (MAK066; Sigma-Aldrich). An extra well of untreated cells was also lysed with 0.2% Triton X-100 as positive control for total cell death. Concentrations of 0, 2.5, 5, 7.5, and 12.5 nM/well of NADH standards were prepared in duplicate as suggested by the kit. After addition of 30–40 µl of LDH substrate into each well, the plate was shaken for 60–90 s and then read using Tecan Infinite M200 at 37°C using the manufacturer’s instructions. The concentration of LDH was calculated based on the manufacturer’s protocol, and LDH cytotoxicity was plotted as a percentage of the LDH activity of the lysis control well.
ASC speck formation assay
ASC was overexpressed in our HAECs using ASC-mCherry pCDH plasmid construct which consists of the ASC ORF cloned into pCDH plasmid under the control of a CMV-7 promoter (Zhong et al., 2018). ASC-mCherry lentiviral particles were made as described in Materials and methods and transduced into HAECs at an MOI of 1. For speck formation assay, ASC-mCherry expressing cells were seeded at 20,000 cells per well in a 96-well ultra-cell carrier plate from PerkinElmer (6055302), with a final media volume of 100 µl per well. After 24 h later, the cells were treated with either 0.25, 1, 5 µM VbP, or DMSO for 16–18 h. The media was discarded, and cells were stained with Hoechst and SYTOX green cell stain for 45 min according to the previous specifications. The cells were imaged using Operetta High Content Imager (PerkinElmer) at 37°C and 5% CO2. Cell death was measured as based on SYTOX intensity as described in Materials and methods. ASC specks were also quantified on the Columbus Image Data Storage and Analysis system (PerkinElmer). The results were plotted as a percentage of the cells showing the presence of a red (mCherry) speck compared to the total number of cells seeded in the well.
Plasmid transfection, lentivirus production, and lentiviral transduction
Four kinds of expression plasmids were used for the overexpression and knockout experiments: pCDH vector, pBABE, pIRES, and LentiCRISPR v2 (plasmid 52961; Addgene; a gift from Feng Zhang, Massachusetts Institute of Technology, Cambridge, MA; Sanjana et al., 2014).
For lentivirus production, 4.4 × 106 HEK293T cells (4.4 × 106) were seeded on poly-L-lysine (P4707; Sigma-Aldrich)-coated 10-cm dishes. 3 µg of the transfer plasmid were co-transfected with three third-generation packaging plasmids: 0.8 µg pREV (encodes viral Rev gene), 1.2 µg pVSG (encodes vesicular stomatitis virus – G, glycoprotein for the viral envelope), and 2 µg pMDL (contains Gag and Pol required for viral packaging) using Lipo-fectamine 2000 (11668019; Thermo Fisher Scientific) according to the manufacturer’s instructions. All lentiviruses were prepared in 10-cm petri dishes. Media was changed 8 h later. Virus containing media was collected at 24 and 48 h after transfection. The virus collected was spun at 1,000 rcf for 5 min, followed by filtration through a 0.45-µm filter (16555K; Sartorius) and syringe. It was then concentrated using Amicon Ultra-15 Centrifugal Filter Unit (UFC910024; Sigma-Aldrich) and kept frozen at −80°C. Viral MOI was calculated and the appropriate titer was used for lentiviral transduction onto the HAECs, A549 cells, or HCMs along with 8 µg /ml of polybrene (9268; Sigma-Aldrich). Media was replaced with fresh media the next day. After 72 h, cells containing the vector were selected in either 500 µg /ml G418 (sc-29065A; Santa Cruz) or 600 µg/ml puromycin (InvivoGen ant-pr-1) depending on the selection marker for 72 h.
HEK293T cells stably expressing CARD8 were transfected at 4 × 104 cells in suspension in a 6-well dish with 2 µg of various expression plasmids (pIRES-CVB3-2A, pIRES-CVB3-3C, pBABE-Casp1-Myc) using Lipofectamine 3000 (L3000001; Thermo Fisher Scientific) according to the manufacturer’s instructions. pIRES-CVB3-2A, pIRES-CVB3-3C, and pBABE-Casp1-Myc transfected HEK293T-CARD8 cells were harvested 48 h post-transfection.
CRISPR/Cas9 gene editing
ASC, Caspase-1, NLRP1, and CARD8 were knocked out using CRISPR/Cas9 gene-editing tool.
The guide RNA for CARD8 was constructed based on Johnson et al., 2018. The lentiCRISPRv2 transfer plasmid (Addgene plasmid 52961) was used as the plasmid backbone (a gift from Feng Zhang).
The gRNA sequences are as follows: ASC gRNA 1:5′-CGCTAACGTGCTGCGCGACA-3′; ASC gRNA 2: 5′-GCTAACGTGCTGCGCGACAT-3′; ASC gRNA 3: 5′-CATGTCGCGCAGCACGTTAG-3′; Caspase 1 gRNA: 5′-TGACTCCGTTATTCCGAAAG-3′; NLRP1 gRNA 1: 5′-GATAGCCCGAGTGACATCGG-3′; NLRP1 gRNA 2: 5′-AGCCCGAGTGACATCGGTGG-3′; CARD8 gRNA 1: 5′-AGCGTTTGGTTCCCCACTGC-3′; CARD8 gRNA 2: 5′-TGACGATTGCGTTTGGTTCC-3′.
Generation of stable cell lines
All the guide RNAs were cloned into LentiCRISPRv2 expression plasmids. LentiCRISPRv2 backbone alone was transduced in ECs to generate the control cell line. ECs, A549 cells, or HCMs were transduced with lentivirus containing gRNAs at an MOI of roughly 14. After 16 h, media was changed out. 72 h later, cells were selected for stable expression of gRNAs with 0.6 µg/ml puromycin for another 72 h. Therefore, after a week, cells were passaged and used for experimentation.
Immunoblotting
Cells were harvested by trypsinization and washed once with PBS. Cell pellets were lysed using RIPA buffer containing complete protease inhibitor cocktail (04693159001; Roche). Protein concentration was measured by bicinchoninic acid (BCA) assay (23225; Thermo Fisher Scientific). Equal amount of lysates were boiled for 5 min in 1× laemmli sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 12.5 mM EDTA, 0.02% bromophenol blue, and 50 mM DTT) at 95°C. Proteins were then loaded onto SDS-PAGE 4–12% gradient gels (NW04122BOX; Thermo Fisher Scientific) and ran in NuPAGE MES SDS running buffer (Thermo Fisher Scientific) between 120 and 130 V. The proteins were then transferred to a 0.2 μM PVDF membrane (22860; Thermo Fisher Scientific) before blocking in 5% non-fat milk TBS-Tween 20 0.1% (v/v) for 1 h at room temperature. Membranes were probed with primary antibodies in TBST/milk overnight rolling at 4°C. Membranes were extensively washed four times at intervals of 30-30-15 min TBST. The membrane was then probed with HRP-conjugated secondary antibodies against mouse or rabbit IgG (Jackson ImmunoResearch) at 1:5,000 and washed again following the same regiment. Chemiluminescence was captured using the Chemidoc imaging system (Bio-Rad).
Cytokine array and analysis
Media that was conditioned by ECs, A549 cells, or HCMs treated with Val-boroPro or virus was collected after 16–18 h and centrifuged at 500 g and 4°C to remove cellular debris. Media was frozen at −80°C for storage or used immediately for the cytokine array. Cytokines from the supernatant were quantified using LEGENDplex (740809; BioLegend) according to the manufacturer’s instructions. Media was diluted 10× with the assay buffer before they were added to the LEGENDplex beads in order to overcome the matrix effect. Fluorescence intensity of the beads was acquired by LSR Fortessa (BD Biosciences). The levels of cytokines were analyzed using the LEGENDplex software according to the manufacturer’s instructions.
IL-18 determination
Concentrations of IL-18 in cell culture supernatants were determined by Human IL-18 ELISA kit (RAB0543; Sigma-Aldrich) according to the manufacturer’s instructions.
Dengue and Zika virus infection of ECs
200,000 human aortic ECs were seeded per well of a 6-well plate. After 48 h, they were incubated at 37°C with DENV (strain Eden-2) or ZIKV (STRAIN h/pf/2013) for 2 h at an MOI of 1, in serum-free EGMV2 media and then washed twice with PBS. They were then incubated in 5% serum supplemented EGMV2 media with 1% penicillin/streptomycin (complete EGMV2 media) for 24 h. Media and cells were then harvested for experimentation purposes.
Determination of DENV2 and ZIKV copy number in the genome after infection
Cells were washed once with PBS before they were lysed in lysis buffer for RNA extraction. RNA was extracted using the MN Nucleospin RNA kit (740955.50; Macherey-Nagel) according to the manufacturer’s instructions. cDNAs were synthesized using iScript Select cDNA synthesis kit (Bio-Rad) using 5 pmol of primer C69B for DENV and 1162C for ZIKV. DENV and ZIKV genome copy numbers were determined by quantitative real-time PCR using primer pair C14A and C69B with probe VICD2C38B for DENV102 and primer pair 1086 and 1162C with probe 1107 for ZIKV103 with SsoAdvanced Universal Probes Supermix (1725281; Bio-Rad) in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Copy numbers were then calculated using standard curves generated from serial dilutions of DNA plasmids containing either DENV or ZIKV genome sequence. Fold increase was calculated by dividing the total genomic copy numbers by the input virus amount. The primer sequences are: C14A 5′-AATATGCTGAAACGCGAGAGAAACCGCG-3′; C69B: 5′-CCCATCTCITCAIIATCCCTGCTGTTGG-3′; VICD2C38B: 5′-AGCATTCCAAGTGAGAATCTCTTTGTCAGCTGT-3′; 1086: 5′-CCGCTGCCCAACACAAG-3′; 1162c: 5′-CCACTAACGTTCTTTTGCAGACAT-3′; 1107: 5′-AGCCTACCTTGACAAGCAGTCAGACACTCAA-3′.
Primers and probes were purchased from Integrated DNA Technologies, Inc. (IDT).
Preparation of CVB3
2 million HeLa cells were seeded on the day preceding infection so that the cells were confluent in 24 h. CVB3 from ATCC (VR-30; ATCC) was thawed on ice for 20 min. 80 µl of CVB3 was added to 2.3 ml of serum-free DMEM. After washing the cells once with PDS, the virus preparation was added to the cells for 1 h at 37°C, with gentle rocking of the plate every 10 min to ensure even spreading of the virus. An additional 2.3 ml of serum-free DMEM with 1% penicillin/streptomycin was added to the cells without removing the previous DMEM-virus preparation. After 24 h incubation, the media containing the virus was collected and centrifuged at 2,000 rpm for 5 min in a swinging bucket centrifuge at 4°C to sediment the cell debris. Virus supernatant was aliquoted into eppendorf tubes, para-filmed, and stored at −80°C.
Titering of CVB3
Viral titers were determined by plaque assay. Briefly, A549 cells were plated at ∼90% confluency and then washed with serum-free media and infected with serial dilution of CVB3 for 1 h. The cells were then washed once with serum-free media and overlaid with 0.8% aquacide II (CFAH-17851; Chromadex) in DMEM containing 2% FBS for 3 d. The cells were fixed with 4% formaldehyde solution (F8775; Sigma-Aldrich) for 2 h and stained with 1% crystal violet solution (V5265; Sigma-Aldrich). The plaques were counted, and the numbers of viral PFU/ml were calculated.
CVB3 infection
Cells at 90% confluency were incubated with various inhibitors (Bortezomib, Rupintrivir, MLN4924) for 30 min. CVB3 virus B3 virus prepared was thawed on ice for 20 min. The cells were washed twice with serum-free media and infected with CVB3 at a MOI as indicated for 2 h in serum-free media. The cells were then washed with serum-free media once and replenished with growth media with 2% FBS containing fresh inhibitors until they were harvested. The supernatants and cell lysates were collected after 16 h of infection. The supernatants were then used for LDH, cytokine secretion, and plaque assay. The lysates were used for Western blot or RNA purification.
Preparation of CVB3
2 million HeLa cells were seeded on the day preceding infection so that the cells were confluent in 24 h. CVB3 from ATCC (VR-30; ATCC) was thawed on ice for 20 min. 80 µl of CVB3 was added to 2.3 ml of serum-free DMEM. After washing the cells once with PDS, the virus preparation was added to the cells for 1 h at 37°C, with gentle rocking of the plate every 10 min to ensure even spreading of the virus. An additional 2.3 ml of serum-free DMEM with 1% penicillin/streptomycin was added to the cells without removing the previous DMEM-virus preparation. After 24 h incubation, the media containing the virus was collected and centrifuged at 2,000 rpm for 5 min in a swinging bucket centrifuge at 4°C to sediment the cell debris. Virus supernatant was aliquoted into eppendorf tubes, parafilmed, and stored at −80°C.
Determination of CVB3 copy number in the genome after infection
After RNA extraction with the Quick-RNA MicroPrep Kit (#R1051; Zymo Research), CVB3 RNA were quantified by the Luna Universal Probe One-Step RT-qPCR Kit (#E3006X; New England Biolabs). The sequences of the CVB3 oligos used were: forward primer 5′-GCATATGGTGATGATGTGATC-3′, reverse primer 5′-GGGGTACTGTTCATCTGCTC-3′, and probe 5′-FAM-AGGGTTACGGGCTGATCAT-3IABkFQ-3'. Reactions were performed in the Bio-Rad CFX96 Real-Time PCR Detection system as follows: 45°C for 15 min (reverse transcription), 95°C for 2 min, followed by 40 cycles at 95°C for 15 s, and 60°C for 1 min. The corresponding CVB3 sequences were inserted in the pIDTSMART-AMP and used as templates for the amplification to generate equations of standard curve for the absolute quantification of CVB3 copy number. The calculated CVB3 copy numbers were then normalized to hHPRT housekeeping gene.
Detection of cleaved GSDMD by immunofluorescence
CVB3-infected Ctl and CARD8 KO HCMs were fixed and permeabilized with FIX & PERM Cell Permeabilization Kit (GAS004; Thermo Fisher Scientific), before they were blocked in 2% BSA/PBS for 30 min. They were then incubated in primary antibodies anti-Actinin (1:100; A7811; Sigma-Aldrich) diluted in 2% BSA/PBS overnight at 4°C. Cells were washed with PBS before incubating with secondary antibodies overnight at 4°C (1:1,000 Alexa Fluor 568, 647; A11004, A21244; Invitrogen) and counter-stained with DAPI (1:1,000, D1306; Invitrogen). Samples were imaged on LSM710 Carl Zeiss Confocal Microscope.
In vitro cleavage assay
In vitro cleavage assay was performed as previously described (Mohamud et al., 2020). Briefly, lysates from stable HEK293T cells expressing CARD8-Flag (20 µg) were incubated with purified 3Cpro or 2Apro in cleavage assay buffer (20 mM Hepes, pH 7.4, 150 mM KOAc, and 1 mM DTT) for the indicated times at 37°C. Reactions were terminated with 6× sample buffer and subjected to Western blot analysis.
CARD8 site-directed mutagenesis
The mutations were generated using the Q5 site-directed mutagenesis kit (#E0552S; New England Biolabs) according to the manufacturer’s protocol with pCDH-CARD8 as the PCR template. The resulting constructs were verified by sequencing (Table 1).
Constructs
| Primer | Sequence 5′ to 3′ |
|---|---|
| QG37LA forward primer | TAGACTACTAGCATCACGGAAAC |
| QG37LA reverse primer | ATGAGTTTGGATGCATCTATG |
| TG57SA forward primer | CTGCAATACACAAAAAGTGCAATTTTTTTTC |
| TG57SA reverse primer | TTCCCGTATGCTATTGTCAACCA |
| G112A G113A forward primer | TATCTGCGGCGGACATTCCCAG |
| G112A G113A reverse primer | GTTGACACTCAGGAACAGCA |
| SG131TA forward primer | GATACAGCAGACATTTGCTCAGA |
| SG131TA reverse primer | TTGTGCCTCTGAAGATTCCTGC |
| G166E forward primer | GGCCTGAAGAAAATGTGGATG |
| G166E reverse primer | CCAGAAACTGACGATTTTTATAATC |
Belcanasan- and disulfuram-specific inhibition assays
100,000 cells were seeded on 12-well plate overnight and pretreated with 10 μM Belnacasan or 10 μM Disulfiram (#PHR1690; Sigma-Aldrich) for 1 h then infected with CVB3 in serum-free media for 1 h in the presence of the respective inhibitors. Media was then replaced with freshly made inhibitors in 2% supplemented EGMV2. The supernatants were collected after 16 h and used for viral quantification by qPCR.
Timer-CVB3 infection
Timer-CVB3 was a gift from Ralph Feuer lab (Robinson et al., 2014). 20,000 ECs/well were seeded in CellCarrier-96 Ultra Microplates (6055300; PerkinElmer) and infected with Timer CVB3. After 16 h, the cells were fixed and imaged with a fluo-rescent light microscope in the GFP channel.
Timer-CVB3 infected HAECs-HCM co-culture
1 million cardiomyocytes were seeded onto the underside of a LN521 precoated transwell (CLS3401; Sigma-Aldrich). Transwell were kept upside down in the incubator for 5 h before flipping upright and placed into a cell-culture plate containing differentiation media. After 16 h, 60,000 ECs were seeded in the upper chamber and cultured in 300 µl EGMV2 media. The ECs were infected with CVB3 or Timer-CVB3 at MOI 0.1 for 1 h, after which the cells were washed with PBS and incubated in 2% EGMV2 media. After 16 h, CM were harvested from the transwell for viral genome quantification or fixed for confocal imaging.
THP1 adhesion assay
20,000 ECs/well were seeded in CellCarrier-96 Ultra Microplates (6055300; PerkinElmer). 4 million THP1 cells were stained with 10 µg/ml Hoechst 33342 (sc-200908; Santa Cruz) for 2 h. They were then washed three times with PBS, after which they were resuspended at 1,600 cells/µl in EGMV2. The media on the ECs were removed and 25 µl of THP1 cells were added to each well. After 10 min, unadhered THP1 cells were gently aspirated and washed twice with PBS. 50 µl of EGMV2 was added into each well, before the plate was imaged and quantified by the Operetta CLS High-Content Analysis System (PerkinElmer).
RT-qPCR
Purified RNA was reverse-transcribed with iScript RT Supermix (1708841; Bio-Rad Laboratories). Quantitative PCR using gene-specific primers was performed with iTaq Universal SYBR Green Supermix (1725124; Bio-Rad Laboratories). The sequences of the primers: IFITM 5′-GGTCCCTGTTCAACACCCTC-3′ and 5′-CCTGTCCCTAGACTTCACGG-3′; MX2 5′-AACGTGCAGCGAGCTTGTC-3′ and 5′-TGGCTGTTGCTGGAAGGAAT-3′; OAS1 5′-TCTGCTGGCTGAAAGCAACA-3′ and 5′-CAGTCCTCTTCTGCCTGTGG-3′; IL1b 5′-ATGATGGCTTATTACAGTGGCAA-3′ and 5′-GTCGGAGATTCGTAGCTGGA-3′ TNFa 5′-CCCATGTTGTAGCAAACCCTC-3′; and 5′-TATCTCTCAGCTCCACGCCA-3′. Primers were purchased from Integrated DNA Technologies, Inc. (IDT).
Flow cytometry
HAECs or HCMs treated with DMSO, VbP, or CVB3 were trypsinsed, pelleted at 500 g for 5 min, and blocked in 1% BSA/PBS for 15 min. The cells were then incubated with Near-IR Live/Dead dye (L10119; Life Technologies), anti–ICAM1-PE 1:100 (sc-107 PE; Santa Cruz Biotechnology), and anti–VCAM1-PE-Vio770 1:100 (130-104-127; Miltenyi Biotec) for 30 min on ice. After washing with 1% BSA/PBS, the cells were fixed in 2% PFA for 30 min. Fluorescence intensity of the cells was acquired by LSRFortessa (BD Biosciences) using the BD FACSDiva 8 software and analyzed by FlowJo version 10.6.1.
Statistical analysis
Statistical analysis performed was parametric, unpaired, two-tailed student’s t test, and one-way ANOVA test as indicated. PRISM 8 was used for the analysis. Error bars show the mean values with SEM. A one-way ANOVA test was also carried out. All significant values were calculated based on the parametric, unpaired, two tailed student’s t test or the one-way ANOVA (as indicated in the individual figure legends) from a minimum of three biological replicates (n = 3). Data are represented by mean ± SEM, P values where * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001, and **** indicates P < 0.0001.
Online supplemental material
Fig. S1 shows that NLRP1 and CARD8 are simultaneously triggered by VbP in primary human ECs lines. Fig. S2 shows that CVB3 activates the CARD8 inflammasome in ECs and human cardiomyocytes. Fig. S3 shows that CVB3 2A and 3C protease mediate CARD8 activation. Table S1 summarizes the effects of VbP on HAECs. Table S2 summarizes the effects of CVB3 infection across different cell lines used in this study. Table S3 summarizes the effect of specific inhibitors on CVB3 infection across different cell lines used in this study.
Acknowledgments
Timer-CVB3 was a kind gift of Dr. Ralph Feuer (SDSU). We thank Swarnaseetha Adusumalli and Baptiste Kerouanton for bioinformatics assistance. We thank Clarissa Tan Ling Ling from Duke-NUS for technical assistance with cardiomyocyte derivation.
This work is supported by NRF-NRFF2017-05 from the National Research Foundation and MOH-000439 from the National Medical Research Council/Ministry of Health awarded to L. Ho, and NRF-NRFF11-2019-0006 from the National Research Foundation and Nanyang Assistant Professorship (Nanyang Technological University) awarded to F. Zhong.
Author contributions: R. Nadkarni performed all experiments in Fig. 1 and wrote the manuscript, W.C. Chu performed all experiments in Figs. 2, 3, and 4 and C.Q.E. Lee performed all experiments in Fig. 5. L. Ho conceptualized the project with F. Zhong, provided supervision, and wrote the manuscript. Y. Mohamud, L. Yap, G.A. Toh, S. Beh, K. Robinson, K. Tryggvason, and H. Luo contributed to specific experiments.

