


Aberrant proteins located in the endoplasmic reticulum (ER) undergo rapid ubiquitination by multiple ubiquitin (Ub) E3 ligases and are retrotranslocated to the cytosol as part of the ER-associated degradation (ERAD). Despite several ERAD branches involving different Ub E3 ligases, the molecular machinery responsible for these ERAD branches in mammalian cells remains not fully understood. Through a series of multiplex knockdown/knockout experiments with real-time kinetic measurements, we demonstrate that HERC3 operates independently of the ER-embedded ubiquitin ligases RNF5 and RNF185 (RNF5/185) to mediate the retrotranslocation and ERAD of misfolded CFTR. While RNF5/185 participates in the ERAD process of both misfolded ABCB1 and CFTR, HERC3 uniquely promotes CFTR ERAD. In vitro assay revealed that HERC3 directly interacts with the exposed membrane-spanning domains (MSDs) of CFTR but not with the MSDs embedded in liposomes. Therefore, HERC3 could play a role in the quality control of MSDs in the cytoplasm and might be crucial for the ERAD pathway of select membrane proteins.
Introduction
Conformationally defective proteins in the endoplasmic reticulum (ER) resulting from genetic mutations and environmental stresses are selectively recognized by the ER quality control (ERQC) machinery and eliminated through ER-associated degradation (ERAD). Aberrant ER luminal and membrane proteins undergo retrotranslocation from the ER to the cytoplasm and ubiquitination, both of which are coordinated by the ubiquitin (Ub) ligase complexes (Christianson and Ye, 2014; Ruggiano et al., 2014). In mammalian cells, ERAD pathways involve at least 10 Ub ligases, each defining specific ERAD branches with specificity toward certain classes of substrates (Krshnan et al., 2022). Certain Ub ligases work together in concert to promote ERAD. For instance, at the ER membrane, Gp78 (AMFR) elongates the Ub chains initiated by RNF5 (RMA1) to facilitate ERAD (Morito et al., 2008). Additionally, cytoplasmic HECT Ub ligase UBE3C collaborates with the ER-embedded Ub ligase RNF185/MBRL complex to promote ERAD (van de Weijer et al., 2020). These various ERAD branches ultimately converge on the cytosolic p97/VCP complex, which extracts ubiquitinated substrates from the ER membrane for proteasomal degradation (Wu and Rapoport, 2018). Several Ub ligases, including Hrd1 (Schoebel et al., 2017; Vasic et al., 2020; Wu et al., 2020) and Doa10 (Schmidt et al., 2020), also appear to function as the retrotranslocation channel. Cytoplasmic chaperones including Bag6 (Wang et al., 2011; Xu et al., 2012) and proteasome shuttling factor UBQLNs (Lim et al., 2009) function downstream of p97-mediated substrate extraction to promote the delivery of ubiquitinated substrates to the proteasome. While the ERQC mechanism is necessary to maintain cellular proteostasis and physiological function, it is also involved in the pathogenesis of diseases such as cystic fibrosis (CF), which is caused by mutations of cystic fibrosis transmembrane conductance regulator (CFTR).
CFTR is a 1,480-residue polytopic membrane glycoprotein that is predicted to contain two membrane-spanning domains (MSD) with six transmembrane (TM) segments, two large cytosolic nucleotide-binding domains (NBD), and a cytosolic regulatory (R) domain (Riordan, 2008; Riordan et al., 1989). It belongs to the ATP-binding cassette (ABC) transporter family and functions as a cAMP-regulated Cl− channel at the apical plasma membrane (PM) of epithelial cells and its mutations cause CF, one of the most common genetic diseases in Caucasians (Boucher, 2004; Riordan, 2008; Riordan et al., 1989). The most common mutation in CF is ∆F508-CFTR in which phenylalanine at position 508 is deleted in the NBD1 located in the cytoplasmic region (Rich et al., 1990; White et al., 1990). The ∆F508 mutation destabilizes NBD1 and the interdomain assembly of CFTR (Du et al., 2005; He et al., 2013; Lewis et al., 2005; Rabeh et al., 2012). Consequently, misfolded ∆F508-CFTR is ubiquitinated and prematurely degraded by the proteasome, resulting in marginal cell surface expression (Jensen et al., 1995; Ward et al., 1995). Several Ub E3 ligases have been identified for CFTR ubiquitination at the ER. A chaperone-associated cytosolic E3 ligase CHIP (STUB1) (Meacham et al., 2001), ER-embedded E3 ligases RNF5 (Younger et al., 2006), RNF185 (El Khouri et al., 2013), and Gp78 (Morito et al., 2008) are involved in CFTR ubiquitination possibly at the distinct biosynthesis stages and at multiple sites within CFTR polypeptides (Oberdorf et al., 2006). Cytoplasmic Ub ligase CHIP appears to recognize the conformational defects of cytoplasmic regions of CFTR including NBD1 coordinated with the Hsc70/Hsp70 and Hsp90 chaperone complex (Younger et al., 2004, 2006). ER-embedded RNF5 appears to recognize the N-terminal regions of CFTR, such as MSD1 (Younger et al., 2006), and facilitates polyubiquitination in cooperation with Gp78 (Morito et al., 2008). RNF185, a paralog of RNF5, may recognize the CFTR’s MSDs in coordination with MBRL and TMUB1/2 based on a previous study (van de Weijer et al., 2020) although the exact recognition mechanism remains unknown. The ubiquitinated CFTR undergoes retrotranslocation, which may be mediated by Derlin-1 (Sun et al., 2006; Wang et al., 2008) and p97 complex (Carlson et al., 2006). Derlin-1 is thought to promote the extraction of MSD1 in the ubiquitinated CFTR (Sun et al., 2006). The extraction of the TM segments is considered generally rate-limiting in the degradation of polytopic membrane proteins. The p97 complex could specifically extract the TM segments of CFTR to accelerate ERAD (Carlson et al., 2006). The multiple Ub ligases appear to recognize various features of CFTR’s conformational defects located in multiple regions and utilize distinct downstream ERAD branches to efficiently eliminate misfolded CFTR in the early secretory pathway. However, due to a lack of comprehensive analysis, the CFTR ERAD branches have not been fully understood. Moreover, how multiple Ub ligases, including ER-embedded and cytoplasmic E3 ligases, coordinately regulate ERAD processes, including ubiquitination and retrotranslocation, still requires further investigation.
In this study, we have identified a cytoplasmic Ub ligase HERC3 that plays a crucial role in a novel ERAD branch dedicated to the selective degradation of misfolded CFTR. Through our HiBiT-based ERAD and retrotranslocation assays, we have demonstrated that both ER-embedded RNF5/185 and cytosolic HERC3 independently promote the retrotranslocation and ERAD of misfolded CFTR. Unlike RNF5/185, HERC3 displays selectivity for certain membrane proteins, such as CFTR, possibly by interacting with the MSDs. Significantly, it appears that HERC3 directly interacts with the exposed MSDs of CFTR, but not with MSDs within the lipid membrane. Our results suggest that cytoplasmic HERC3 may function as an ERQC-associated E3 ligase that interacts with the TM segments exposed on the surface of the ER membrane, thereby providing an ERAD branch specialized for a specific type of membrane proteins.
Result
HERC3 limits the cell surface expression of ∆F508-CFTR by facilitating the ERAD
Previously, we conducted a comprehensive siRNA screening in CFBE cells stably expressing ∆F508-CFTR-HRP and identified RFFL Ub ligase whose knockdown (KD) increased the PM level of rescued (r)∆F508-CFTR which was forcibly expressed at the PM by low-temperature (26°C) incubation (Okiyoneda et al., 2018). At the same time, we have identified a novel Ub ligase HERC3 whose KD also increased the r∆F508-CFTR PM level (Fig. 1 A). We ruled out the possibility of this effect being due to an off-target effect, as the increased PM CFTR was also observed in HERC3 KD using individual siRNAs (Fig. 1 A). The efficacy of these siRNAs was confirmed through reverse transcription quantitative PCR (RT-qPCR) analysis (Fig. 1 B). To determine if the increased PM CFTR was functional upon HERC3 KD, we performed a halide-sensitive YFP quenching assay in CFBE Teton cells (Okiyoneda et al., 2018). As expected, the YFP quenching induced by CFTR-mediated iodide influx was increased in the cells transfected with siHERC3, indicating that HERC3 KD significantly increased the functional r∆F508-CFTR channel at the PM (Fig. 1 C). Further analysis through Western blotting revealed that HERC3 KD increased both mature and immature r∆F508-CFTR after low-temperature rescue (Denning et al., 1992), but it increased the immature ∆F508-CFTR at 37°C, suggesting that HERC3 might regulate the CFTR level at the ER (Fig. 1 D). To investigate the impact of HERC3 KD on CFTR ERAD, we conducted a cycloheximide (CHX) chase experiment. The cell-based ELISA showed that HERC3 KD slightly but significantly decelerated the elimination of immature ∆F508-CFTR in CFBE cells (Fig. 1 E). However, HERC3 KD did not influence the PM stability of r∆F508-CFTR, in contrast to the effect observed with KD of RFFL, which is involved in the elimination of r∆F508-CFTR at the PM and endosomes (Okiyoneda et al., 2018) (Fig. 1 F). These findings suggest that the increased level of mature r∆F508-CFTR is not due to the reduced peripheral degradation of mature CFTR but could be attributed to the decelerated ERAD of immature CFTR. In line with this hypothesis, HERC3 KD resulted in reduced ubiquitination of immature ∆F508-CFTR-3HA, which is fused with an N-terminal histidine–biotin–histidine (HBH) tag (HBH-∆F508-CFTR) (Okiyoneda et al., 2018; Tagwerker et al., 2006) (Fig. 1 G). The histidine–biotin–histidine (HBH) tag, consisting of tandem His6 tags separated by a biotinylated signal, serves as a valuable tool for isolation under denaturing conditions (Okiyoneda et al., 2018; Tagwerker et al., 2006). Conversely, when HERC3 was overexpressed, it led to a decrease in immature ∆F508-CFTR levels, and this effect depended on the catalytic activity of the HECT domain as the deletion or catalytically inactive C1018A mutation of the HECT domain abolished this effect (Fig. 1, H and I). Furthermore, deleting the RCC1 Like Domain (RLD) of HERC3 also reduced its interaction with ∆F508-CFTR and the subsequent decrease in immature ∆F508-CFTR expression (Fig. 1, I and J), suggesting that the RLD is essential for HERC3’s interaction with immature ∆F508-CFTR at the ER.

HERC3 participates in the ubiquitination and ERAD of ∆F508-CFTR. (A) The PM density of r∆F508-CFTR-HRP in CFBE Teton cells transfected with 50 nM siNT or siHERC3, as indicated (n = 9–12). Each independent experiment, consisting of three to four biological replicates (n), is color-coded. (B) Quantitative PCR analysis assessed HERC3 KD efficiency in CFBE Teton ∆F508-CFTR-HRP cells (n = 3). Each biological replicate (n) is color-coded: the averages from three technical replicates are shown in triangles. (C) The channel function of r∆F508-CFTR-3HA in CFBE Teton cells transfected with 50 nM siNT or siHERC3 pool was measured by YFP quenching assay. The initial YFP quenching rate was quantified as the CFTR function (right, n = 19). Each independent experiment, consisting of four to eight biological replicates (n), is color-coded. (D) Western blotting analyzed steady-state levels of ∆F508-CFTR-3HA with (r∆F508) or without 26°C rescue (∆F508) in CFBE Teton cells transfected with 50 nM siNT or siHERC3 pool. Na+/K+ ATPase (ATPase) was used as a loading control. B, immature form; C, mature form. Western blotting also confirmed HERC3 KD in CFBE Teton ΔF508-CFTR-3HA cells. Ponceau staining was used as a loading control. A filled triangle indicates HERC3. (E) Cellular ∆F508-CFTR-3HA stability in CFBE Teton cells transfected with 50 nM siNT or siHERC3 pool was measured by cell-based ELISA using an anti-HA antibody after CHX treatment (n = 12). (F) The PM stability of r∆F508-CFTR-3HA in CFBE cells transfected with 50 nM siNT, siRFFL pool, or siHERC3 pool was measured by PM ELISA (n = 12 biological replicates). (G) Ubiquitination levels of HBH-∆F508-CFTR-3HA in CFBE Teton cells were measured by Neutravidin (NA) pull-down under denaturing conditions (NA pull-down) and Western blotting. The CFTR ubiquitination level was quantified by densitometry and normalized to CFTR in precipitates (right, n = 4). (H) A schematic diagram of the HERC3 domain composition with the residue numbers at the domain boundaries. HERC3 mutants used in this study are also shown. (I) The effects of overexpressed Myc-HERC3 variants on the steady-state level of ∆F508-CFTR-3HA were analyzed by Western blotting in co-transfected COS-7 cells. The immature ∆F508-CFTR (B band) was quantified by densitometry (right, n = 4). (J) The interaction of Myc-HERC3 variants with HBH-∆F508-CFTR-3HA in BHK cells was analyzed by NA pull-down and Western blotting. ∆F508-CFTR was rescued at 26°C incubation for 2 days, followed by a 1-h incubation at 37°C (A–D and F). Statistical significance was assessed by one-way ANOVA (A), or one-way repeated-measures (RM) ANOVA (B and I) with Dunnett’s multiple comparison tests, a two-tailed unpaired (C), or paired Student’s t test (G), or two-way ANOVA (E and F). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F1.
HERC3 participates in the ubiquitination and ERAD of ∆F508-CFTR. (A) The PM density of r∆F508-CFTR-HRP in CFBE Teton cells transfected with 50 nM siNT or siHERC3, as indicated (n = 9–12). Each independent experiment, consisting of three to four biological replicates (n), is color-coded. (B) Quantitative PCR analysis assessed HERC3 KD efficiency in CFBE Teton ∆F508-CFTR-HRP cells (n = 3). Each biological replicate (n) is color-coded: the averages from three technical replicates are shown in triangles. (C) The channel function of r∆F508-CFTR-3HA in CFBE Teton cells transfected with 50 nM siNT or siHERC3 pool was measured by YFP quenching assay. The initial YFP quenching rate was quantified as the CFTR function (right, n = 19). Each independent experiment, consisting of four to eight biological replicates (n), is color-coded. (D) Western blotting analyzed steady-state levels of ∆F508-CFTR-3HA with (r∆F508) or without 26°C rescue (∆F508) in CFBE Teton cells transfected with 50 nM siNT or siHERC3 pool. Na+/K+ ATPase (ATPase) was used as a loading control. B, immature form; C, mature form. Western blotting also confirmed HERC3 KD in CFBE Teton ΔF508-CFTR-3HA cells. Ponceau staining was used as a loading control. A filled triangle indicates HERC3. (E) Cellular ∆F508-CFTR-3HA stability in CFBE Teton cells transfected with 50 nM siNT or siHERC3 pool was measured by cell-based ELISA using an anti-HA antibody after CHX treatment (n = 12). (F) The PM stability of r∆F508-CFTR-3HA in CFBE cells transfected with 50 nM siNT, siRFFL pool, or siHERC3 pool was measured by PM ELISA (n = 12 biological replicates). (G) Ubiquitination levels of HBH-∆F508-CFTR-3HA in CFBE Teton cells were measured by Neutravidin (NA) pull-down under denaturing conditions (NA pull-down) and Western blotting. The CFTR ubiquitination level was quantified by densitometry and normalized to CFTR in precipitates (right, n = 4). (H) A schematic diagram of the HERC3 domain composition with the residue numbers at the domain boundaries. HERC3 mutants used in this study are also shown. (I) The effects of overexpressed Myc-HERC3 variants on the steady-state level of ∆F508-CFTR-3HA were analyzed by Western blotting in co-transfected COS-7 cells. The immature ∆F508-CFTR (B band) was quantified by densitometry (right, n = 4). (J) The interaction of Myc-HERC3 variants with HBH-∆F508-CFTR-3HA in BHK cells was analyzed by NA pull-down and Western blotting. ∆F508-CFTR was rescued at 26°C incubation for 2 days, followed by a 1-h incubation at 37°C (A–D and F). Statistical significance was assessed by one-way ANOVA (A), or one-way repeated-measures (RM) ANOVA (B and I) with Dunnett’s multiple comparison tests, a two-tailed unpaired (C), or paired Student’s t test (G), or two-way ANOVA (E and F). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F1.
HERC3 and RNF5/185 additively facilitate ∆F508-CFTR ERAD
Our findings suggest that HERC3 may play a role in facilitating CFTR ERAD, alongside other Ub ligases such as CHIP, RNF5, RNF185, and Gp78. To compare the impact of these Ub ligases on ∆F508-CFTR, we evaluated the effect of individually knocking down each E3 ligase in CFBE cells. Surprisingly, while HERC3 KD significantly increased cellular ∆F508-CFTR levels, KD of CHIP, RNF5, RNF185, or Gp78 had no significant effect (Fig. 2 A). Upon low-temperature rescue, the cell surface levels of r∆F508-CFTR were increased by KD of HERC3, RNF5, or RNF185, indicating their crucial role in limiting ∆F508-CFTR abundance in CFBE cells (Fig. 2 B). To examine whether HERC3 collaborates with other CFTR-related E3 Ub ligases, which may have redundant functions, we tested the effect of simultaneously knocking down HERC3 and one of these E3 ligases. While KD of either CHIP or Gp78 did not enhance the effect of HERC3 KD on the PM levels of r∆F508-CFTR, KD of RNF5 or RNF185 significantly enhanced the effect of HERC3 KD (Fig. 2 B). The absence of an impact from CHIP KD in the CFTR QC in CFBE cells was also documented previously (Okiyoneda et al., 2018), suggesting that the influence of CHIP may fluctuate depending on the cell type. This result suggests that HERC3 may regulate ∆F508-CFTR through a pathway distinct from RNF5 and RNF185.

HERC3 and RNF5/185 additively reduce ∆F508-CFTR. (A and B) The cellular level of ∆F508-CFTR-3HA (A, n = 10) and PM level of r∆F508-CFTR-HRP (B, n = 8) in CFBE Teton cells transfected with 50 nM siRNA indicated was measured by cell-based ELISA using an anti-HA antibody and HRP assay, respectively. (C and D) The cellular level of ∆F508-CFTR-3HA (C, n = 15) and PM levels of r∆F508-CFTR-HRP induced by 26°C rescue (D, n = 8) in CFBE Teton cells transfected with 50 nM siRNA were measured by ELISA using an anti-HA antibody (C) and HRP assay (D), respectively. (E and F) Western blotting analyzed steady-state levels of r∆F508-CFTR-3HA in CFBE Teton cells transfected with 50 nM siRNA indicated (E). Ponceau staining was used as a loading control. B, immature form; C, mature form. The anti-RNF185 antibody detected both RNF5 and RNF185 because of the cross-reactivity. HERC3 KD was confirmed by quantitative PCR (F, n = 3). Each biological replicate (n) is color-coded: the averages from three technical replicates are shown in triangles. (G) The PM levels of r∆F508-CFTR-HRP induced by 3 µM VX-809 treatment at 37°C for 24 h in CFBE Teton cells transfected with 50 nM siRNA indicated (n = 8). (H) Representative traces (left) of the YFP fluorescence and quantification of the initial YFP quenching rate (right, n = 12) as a measure of rΔF508-CFTR function in CFBE cells transfected with 50 nM siRNA, as indicated. Each independent experiment consisting of 4 (B, D, and G), 5 (A and C), or 6 (H) biological replicates (n) is color-coded. Statistical significance was assessed by one-way RM ANOVA with Dunnett’s multiple comparison tests (F) or two-way ANOVA with Holm–Sidak multiple comparison tests, which revealed a significant main effect of HERC3 KD or RNF5/185 DKD, but no interaction between them (Pint > 0.05, C, D, and H) except for G (Pint = 0.012). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F2.
HERC3 and RNF5/185 additively reduce ∆F508-CFTR. (A and B) The cellular level of ∆F508-CFTR-3HA (A, n = 10) and PM level of r∆F508-CFTR-HRP (B, n = 8) in CFBE Teton cells transfected with 50 nM siRNA indicated was measured by cell-based ELISA using an anti-HA antibody and HRP assay, respectively. (C and D) The cellular level of ∆F508-CFTR-3HA (C, n = 15) and PM levels of r∆F508-CFTR-HRP induced by 26°C rescue (D, n = 8) in CFBE Teton cells transfected with 50 nM siRNA were measured by ELISA using an anti-HA antibody (C) and HRP assay (D), respectively. (E and F) Western blotting analyzed steady-state levels of r∆F508-CFTR-3HA in CFBE Teton cells transfected with 50 nM siRNA indicated (E). Ponceau staining was used as a loading control. B, immature form; C, mature form. The anti-RNF185 antibody detected both RNF5 and RNF185 because of the cross-reactivity. HERC3 KD was confirmed by quantitative PCR (F, n = 3). Each biological replicate (n) is color-coded: the averages from three technical replicates are shown in triangles. (G) The PM levels of r∆F508-CFTR-HRP induced by 3 µM VX-809 treatment at 37°C for 24 h in CFBE Teton cells transfected with 50 nM siRNA indicated (n = 8). (H) Representative traces (left) of the YFP fluorescence and quantification of the initial YFP quenching rate (right, n = 12) as a measure of rΔF508-CFTR function in CFBE cells transfected with 50 nM siRNA, as indicated. Each independent experiment consisting of 4 (B, D, and G), 5 (A and C), or 6 (H) biological replicates (n) is color-coded. Statistical significance was assessed by one-way RM ANOVA with Dunnett’s multiple comparison tests (F) or two-way ANOVA with Holm–Sidak multiple comparison tests, which revealed a significant main effect of HERC3 KD or RNF5/185 DKD, but no interaction between them (Pint > 0.05, C, D, and H) except for G (Pint = 0.012). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F2.
To further investigate this possibility, we assessed the effect of HERC3 KD on ∆F508-CFTR levels upon simultaneous KD of RNF5 and its paralog RNF185 (RNF5/185 DKD), both of which could be functionally redundant in CFTR ERAD (El Khouri et al., 2013). As expected, the impact of HERC3 KD on cellular ∆F508-CFTR levels remained unaffected by siRNF5/185 (Fig. 2 C). Consequently, the combined KD of HERC3 and RNF5/185 resulted in an additive increase in cellular CFTR levels (Fig. 2 C). This finding suggests that HERC3’s regulation of ∆F508-CFTR operates independently of RNF5 and RNF185. In line with these findings, the simultaneous KD of HERC3 and RNF5/185 led to an additive increase in the PM level of low-temperature rescued ∆F508-CFTR (Fig. 2 D). Similarly, comparable results were observed for the overall CFTR protein levels (Fig. 2, E and F). HERC3 KD also increased the PM levels of r∆F508-CFTR induced by the CFTR corrector VX-809 treatment in CFBE cells, and this effect was further enhanced upon RNF5/185 DKD (Fig. 2 G). VX-809 has been reported to partially correct the CFTR conformational defects such as defective MSD1/2-NBD1 interaction by binding with its MSD1, inducing partial PM expression (Farinha et al., 2013; Fiedorczuk and Chen, 2022a; Loo et al., 2013; Okiyoneda et al., 2013; Ren et al., 2013; Van Goor et al., 2011). Additionally, KD of both HERC3 and RNF5/185 additively increased the functional ∆F508-CFTR channel at the PM in the presence of VX-809 (Fig. 2 H). These findings suggest that, in addition to the RNF5/185 pathway, the HERC3 ERAD branch limits the efficacy of the CFTR corrector. Combining strategies to counteract both ERAD branches may enhance the effectiveness of CFTR correctors.
To accurately assess the role of HERC3 in CFTR ERAD, we developed a live cell CFTR degradation assay that allowed us to measure the degradation kinetics of ∆F508-CFTR fused with the HiBiT tag in the C-terminal cytosolic region (∆F508-CFTR-HiBiT[CT]) in real-time (Fig. 3 A). The HiBiT tag, comprising only 11 amino acids, exhibits a strong binding affinity to the adaptor protein LgBiT, thereby facilitating the reconstitution of the bright luminescent protein NanoBiT (Dixon et al., 2016). In this assay, we measured the luminescence signal induced by the association of the HiBiT tag with the co-expressed LgBiT in 293MSR cells at 37°C. The luminescence signal gradually attenuated during the CHX chase, and its half-life (t1/2) was ∼70 min (Fig. 3 B). The addition of a proteasome inhibitor MG-132 significantly reduced the luminescence attenuation during the CHX chase, indicating that the luminescence attenuation represents the kinetic proteasomal degradation of ∆F508-CFTR-HiBiT(CT) (Fig. 3 B). The degradation kinetics of ∆F508-CFTR in the HiBiT-based assay closely resembled those observed in traditional Western blot analyses (Fig. 3 C), validating the reliability of the new HiBiT degradation assay. However, in Western blot analyses, the ERAD rate seemed significantly faster, potentially because of its lower sensitivity in detecting CFTR. Using this innovative HiBiT assay, we examined the effect of HERC3 KD on ∆F508-CFTR ERAD in 293MSR wild-type (WT) cells and in cells with the double knockout (KO) of RNF5 and RNF185 (5/185 DKO) that we established using the CRISPR-Cas9 system (Fig. S1, A and B). Western blotting and RT-qPCR confirmed the RNF5/185 DKO and HERC3 KD in 293MSR cells, respectively (Fig. 3, D and E). The live cell degradation assay showed that consistent with the results in CFBE cells, HERC3 KD modestly decelerated the ERAD of ∆F508-CFTR-HiBiT(CT) and reduced the ERAD rate by ∼20% in 293MSR WT cells (Fig. 3 F). Moreover, the RNF5/185 DKO also decelerated CFTR ERAD and reduced the ERAD rate by ∼58% (Fig. 3 F). As expected, the combined HERC3 KD and RNF5/185 DKO resulted in an additive inhibitory effect on CFTR ERAD, reducing its ERAD rate by ∼75% (Fig. 3 F). Similar results were obtained in 293MSR, CFBE, and BEAS-2B human airway epithelial cells that stably expressed ∆F508-CFTR-Nluc (Taniguchi et al., 2022). HERC3 KD and RNF5/185 double KD (DKD) in these cells additively inhibited CFTR ERAD (Fig. 3, G–I). These results collectively indicate that HERC3, in conjunction with RNF5/185, promotes the ERAD of ∆F508-CFTR.
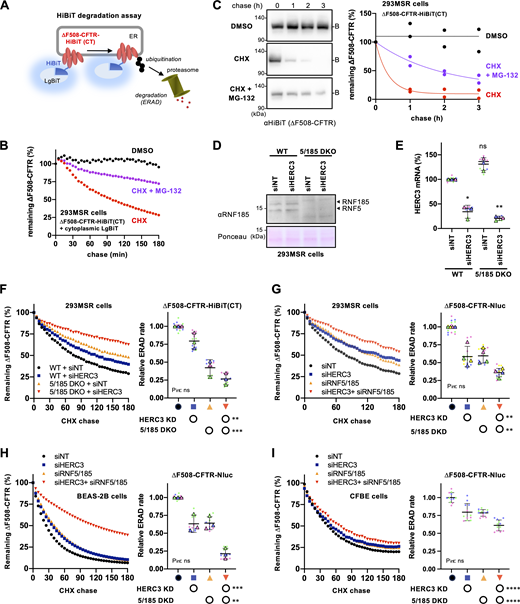
HERC3 and RNF5/185 additively facilitate ∆F508-CFTR ERAD. (A) A schematic diagram of the HiBiT degradation assay, where ∆F508-CFTR-HiBiT(CT) and cytosolic LgBiT were co-expressed. The luminescence signal generated by the interaction of the HiBiT tag and LgBiT was measured in living cells. (B) A typical measurement of ∆F508-CFTR-HiBiT(CT) ERAD in 293MSR cells. The luminescence signal during the CHX chase was measured as the remaining ∆F508-CFTR during the CHX chase, with or without 10 µM MG-132. (C) The metabolic stability of ∆F508-CFTR-HiBiT(CT) was assessed through a CHX chase at 37°C, followed by Western blotting using an anti-HiBiT antibody in 293MSR cells (n = 2). The remaining ∆F508-CFTR was expressed as a percentage of time 0, and one-phase exponential decay curves were fitted. (D) Western blotting confirmed the ablation of RNF5, and RNF185 in the WT and RNF5/185 DKO 293MSR cells transfected with siRNA indicated. Ponceau staining was used as a loading control. (E) HERC3 KD in 293MSR WT and RNF5/185 DKO cells was confirmed through quantitative PCR (n = 3). (F) Kinetic degradation of ∆F508-CFTR-HiBiT(CT) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. Luminescence was continuously monitored over 180 min in the presence of CHX and plotted normalized to the non-treated cells. The ERAD rate of ∆F508-CFTR-HiBiT(CT) was calculated by fitting the initial degradation portion of each kinetic degradation curve (right, n = 3). (G–I) Kinetic degradation of ∆F508-CFTR-Nluc(CT) in 293MSR (G, n = 4), BEAS-2B (H, n = 3), and CFBE (I, n = 12) cells transfected with 50 nM siRNA as indicated. The ERAD rate of ∆F508-CFTR-Nluc(CT) was calculated as F. Each biological replicate (n) is color-coded: the averages from three to four technical replicates are shown in triangles (E–H). Statistical significance was assessed by one-way RM ANOVA with Dunnett’s multiple comparison tests (E) or two-way RM ANOVA which revealed a significant main effect of HERC3 or RNF5/185 ablation, but no interaction between them (F–I, Pint> 0.05). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F3.
HERC3 and RNF5/185 additively facilitate ∆F508-CFTR ERAD. (A) A schematic diagram of the HiBiT degradation assay, where ∆F508-CFTR-HiBiT(CT) and cytosolic LgBiT were co-expressed. The luminescence signal generated by the interaction of the HiBiT tag and LgBiT was measured in living cells. (B) A typical measurement of ∆F508-CFTR-HiBiT(CT) ERAD in 293MSR cells. The luminescence signal during the CHX chase was measured as the remaining ∆F508-CFTR during the CHX chase, with or without 10 µM MG-132. (C) The metabolic stability of ∆F508-CFTR-HiBiT(CT) was assessed through a CHX chase at 37°C, followed by Western blotting using an anti-HiBiT antibody in 293MSR cells (n = 2). The remaining ∆F508-CFTR was expressed as a percentage of time 0, and one-phase exponential decay curves were fitted. (D) Western blotting confirmed the ablation of RNF5, and RNF185 in the WT and RNF5/185 DKO 293MSR cells transfected with siRNA indicated. Ponceau staining was used as a loading control. (E) HERC3 KD in 293MSR WT and RNF5/185 DKO cells was confirmed through quantitative PCR (n = 3). (F) Kinetic degradation of ∆F508-CFTR-HiBiT(CT) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. Luminescence was continuously monitored over 180 min in the presence of CHX and plotted normalized to the non-treated cells. The ERAD rate of ∆F508-CFTR-HiBiT(CT) was calculated by fitting the initial degradation portion of each kinetic degradation curve (right, n = 3). (G–I) Kinetic degradation of ∆F508-CFTR-Nluc(CT) in 293MSR (G, n = 4), BEAS-2B (H, n = 3), and CFBE (I, n = 12) cells transfected with 50 nM siRNA as indicated. The ERAD rate of ∆F508-CFTR-Nluc(CT) was calculated as F. Each biological replicate (n) is color-coded: the averages from three to four technical replicates are shown in triangles (E–H). Statistical significance was assessed by one-way RM ANOVA with Dunnett’s multiple comparison tests (E) or two-way RM ANOVA which revealed a significant main effect of HERC3 or RNF5/185 ablation, but no interaction between them (F–I, Pint> 0.05). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F3.

Establishment of RNF5/185 DKO 293MSR cells. (A and B) Schematic representation of the RNF5 (A) and RNF185 (B) -targeting gRNA sequences. Arrows indicate primer positions. PAM, protospacer adjacent motif. The locations of each start codon, stop codon and the catalytic cysteine residues of RNF5 (C42) and RNF185 (C39, C42) are also indicated. The sequences analyzed for each KO cell line are shown along with the deleted sequences.
Establishment of RNF5/185 DKO 293MSR cells. (A and B) Schematic representation of the RNF5 (A) and RNF185 (B) -targeting gRNA sequences. Arrows indicate primer positions. PAM, protospacer adjacent motif. The locations of each start codon, stop codon and the catalytic cysteine residues of RNF5 (C42) and RNF185 (C39, C42) are also indicated. The sequences analyzed for each KO cell line are shown along with the deleted sequences.
The involvement of HERC3 in the retrotranslocation and ubiquitination of ∆F508-CFTR
To explore how HERC3 facilitates CFTR ERAD, we measured its impact on retrotranslocation which is a crucial step for the proteasomal degradation of luminal and membrane proteins (Hampton and Sommer, 2012; Lemberg and Strisovsky, 2021; Wu and Rapoport, 2018). We developed a live cell CFTR retrotranslocation assay using ∆F508-CFTR fused with the HiBiT tag in the extracellular region (∆F508-CFTR-HiBiT[Ex]) (Taniguchi et al., 2022). The HiBiT tag initially located in the luminal side of the ER is expected to transfer to the cytoplasm where it can associate with coexpressed LgBiT during retrotranslocation (Fig. 4 A). This results in a reconstituted Nluc luminescence signal that can be measured in real-time in living cells. The luminescence signal was observed exclusively when both ∆F508-CFTR-HiBiT(Ex) and LgBiT were expressed (Fig. S2). Furthermore, in the presence of the proteasome inhibitor MG-132, the luminescence signal exhibited a continuous increase, indicating the cytoplasmic accumulation of ∆F508-CFTR-HiBiT(Ex) (Fig. 4 B and Fig. S2). Treatment with DBeQ, an inhibitor of p97/VCP, which is crucial for retrotranslocation (Chou et al., 2011), abrogated the luminescence increase induced by MG-132, suggesting that this luminescence signal can be used as an indicator of retrotranslocated CFTR from the ER to the cytoplasm (Fig. 4 B). Furthermore, we observed an elevation in the luminescent signal during MG-132 treatment, even when CHX was present. This finding suggests that the increased signal is likely attributed to the retrotranslocation of pre-existing ∆F508-CFTR-HiBiT(Ex) located within the ER (Fig. 4 C). Moreover, we observed a reduced retrotranslocation of ∆F508-CFTR-HiBiT(Ex) upon treatment with the CFTR modulator Trikafta, known for its ability to correct CFTR misfolding and prevent premature degradation (Capurro et al., 2021; Keating et al., 2018) (Fig. 4 C). The HiBiT retrotranslocation assay showed that consistent with the ERAD inhibitory effect, HERC3 KD tended to slightly reduce the retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR WT cells (Fig. 4 D). As expected, RNF5/185 DKO robustly inhibited the CFTR retrotranslocation, and this effect was additively enhanced by HERC3 KD (Fig. 4 D). These inhibitory effects on retrotranslocation were highly correlated with the effects on ERAD (Fig. S3 A).

HERC3 and RNF5/185 facilitate ∆F508-CFTR retrotranslocation. (A) A schematic diagram of the HiBiT retrotranslocation assay, where ∆F508-CFTR-HiBiT(Ex) and cytosolic LgBiT were co-expressed. The luminescence signal generated by the interaction of LgBiT and the HiBiT tag exposed in the cytosol after retrotranslocation was measured in living cells during MG-132 treatment. (B) A typical measurement of ∆F508-CFTR-HiBiT(Ex) retrotranslocation in 293MSR cells. The luminescence signal was measured in living cells upon treatment with 10 µM MG-132, with or without 10 µM DBeQ. (C) Kinetic retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells treated with DMSO (0.3%) or Trikafta (3 µM VX-661, 3 µM VX-445, 1 µM VX-770) for 24 h at 37°C. Luminescence was continuously monitored in the presence of MG-132 with or without CHX. The signal increased by the MG-132 treatment was plotted as retrotranslocated CFTR. The retrotranslocation rate of ∆F508-CFTR-HiBiT(Ex) was calculated by linear fitting of the signal until 60 min (right, n = 4). Two-way RM ANOVA revealed a significant main effect of Trikafta or CHX, but no interaction between them (Pint > 0.05). (D) Kinetic retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. Luminescence was continuously monitored over 60 min in the presence of MG-132. The signal increased by the MG-132 treatment was plotted as retrotranslocated CFTR. The retrotranslocation rate of ∆F508-CFTR-HiBiT(Ex) was calculated by linear fitting (right, n = 3). Two-way RM ANOVA revealed a significant main effect of HERC3 KD or RNF5/185 DKO, but no interaction between them (Pint > 0.05). (E) A schematic diagram of the HiBiT ER disappearance assay, where ∆F508-CFTR-HiBiT(Ex) and ER-luminal LgBiT (ER LgBiT) were coexpressed. The luminescence signal generated by the interaction of LgBiT and the HiBiT tag in the ER was measured in living cells during the CHX chase. (F) Kinetic ER disappearance of ∆F508-CFTR-HiBiT(Ex) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. Luminescence was continuously monitored over 180 min in the presence of CHX and plotted normalized to the non-treated cells as remaining CFTR at the ER (%). The ER disappearance rate of ∆F508-CFTR-HiBiT(Ex) was calculated by fitting the kinetic ER disappearance curve (right, n = 3). Two-way RM ANOVA with Holm–Sidak multiple comparison tests revealed a significant main effect of RNF5/185 DKO and no interaction between HERC3 KD and RNF5/185 DKO (Pint > 0.05). Data distribution was assumed to be normal but was not formally tested. Each biological replicate (n) is color-coded: the averages from three or four technical replicates are shown in triangles (D and F). Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant.
HERC3 and RNF5/185 facilitate ∆F508-CFTR retrotranslocation. (A) A schematic diagram of the HiBiT retrotranslocation assay, where ∆F508-CFTR-HiBiT(Ex) and cytosolic LgBiT were co-expressed. The luminescence signal generated by the interaction of LgBiT and the HiBiT tag exposed in the cytosol after retrotranslocation was measured in living cells during MG-132 treatment. (B) A typical measurement of ∆F508-CFTR-HiBiT(Ex) retrotranslocation in 293MSR cells. The luminescence signal was measured in living cells upon treatment with 10 µM MG-132, with or without 10 µM DBeQ. (C) Kinetic retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells treated with DMSO (0.3%) or Trikafta (3 µM VX-661, 3 µM VX-445, 1 µM VX-770) for 24 h at 37°C. Luminescence was continuously monitored in the presence of MG-132 with or without CHX. The signal increased by the MG-132 treatment was plotted as retrotranslocated CFTR. The retrotranslocation rate of ∆F508-CFTR-HiBiT(Ex) was calculated by linear fitting of the signal until 60 min (right, n = 4). Two-way RM ANOVA revealed a significant main effect of Trikafta or CHX, but no interaction between them (Pint > 0.05). (D) Kinetic retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. Luminescence was continuously monitored over 60 min in the presence of MG-132. The signal increased by the MG-132 treatment was plotted as retrotranslocated CFTR. The retrotranslocation rate of ∆F508-CFTR-HiBiT(Ex) was calculated by linear fitting (right, n = 3). Two-way RM ANOVA revealed a significant main effect of HERC3 KD or RNF5/185 DKO, but no interaction between them (Pint > 0.05). (E) A schematic diagram of the HiBiT ER disappearance assay, where ∆F508-CFTR-HiBiT(Ex) and ER-luminal LgBiT (ER LgBiT) were coexpressed. The luminescence signal generated by the interaction of LgBiT and the HiBiT tag in the ER was measured in living cells during the CHX chase. (F) Kinetic ER disappearance of ∆F508-CFTR-HiBiT(Ex) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. Luminescence was continuously monitored over 180 min in the presence of CHX and plotted normalized to the non-treated cells as remaining CFTR at the ER (%). The ER disappearance rate of ∆F508-CFTR-HiBiT(Ex) was calculated by fitting the kinetic ER disappearance curve (right, n = 3). Two-way RM ANOVA with Holm–Sidak multiple comparison tests revealed a significant main effect of RNF5/185 DKO and no interaction between HERC3 KD and RNF5/185 DKO (Pint > 0.05). Data distribution was assumed to be normal but was not formally tested. Each biological replicate (n) is color-coded: the averages from three or four technical replicates are shown in triangles (D and F). Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant.

A representative luminescence trace from the ∆F508-CFTR-HiBiT(Ex) retrotranslocation assay. 293MSR cells were transiently transfected with LgBiT, with or without ∆F508-CFTR-HiBiT(Ex), followed by Endurazine loading. Luminescence was continuously monitored in live cells during treatment with or without 10 µM MG-132. This figure is associated with Fig. 4 B.
A representative luminescence trace from the ∆F508-CFTR-HiBiT(Ex) retrotranslocation assay. 293MSR cells were transiently transfected with LgBiT, with or without ∆F508-CFTR-HiBiT(Ex), followed by Endurazine loading. Luminescence was continuously monitored in live cells during treatment with or without 10 µM MG-132. This figure is associated with Fig. 4 B.

The correlation analysis. (A) The relationship between CFTR ERAD (Fig. 3 F) and the retrotranslocation rates of ∆F508-CFTR (Fig. 4 D) was analyzed for correlation. (B) Correlation analysis was performed to examine the connection between CFTR ER disappearance (Fig. 4 F) and the retrotranslocation rates of ∆F508-CFTR (Fig. 4 D). (C) The correlation between CFTR K48-linked polyubiquitination (Fig. 5 B) and K63-linked polyubiquitination (Fig. 5 C). (D) The correlation between CFTR K48-linked polyubiquitination (Fig. 5 B) and the ERAD rate (Fig. 3 F). (E) The correlation between CFTR K63-linked polyubiquitination (Fig. 5 C) and the ERAD rate (Fig. 3 F). (F) The correlation between CFTR K48-linked polyubiquitination (Fig. 5 B) and the retrotranslocation rate of ∆F508-CFTR (Fig. 4 D). (G) The correlation between CFTR K63-linked polyubiquitination (Fig. 5 C) and the retrotranslocation rate of ∆F508-CFTR (Fig. 4 D). (H) The correlation between the CFTR K48-linked polyubiquitination (Fig. 5 B) and UBQLN2 binding (Fig. 6 D). (I) The correlation between the CFTR K63-linked polyubiquitination (Fig. 5 C) and UBQLN2 binding (Fig. 6 D). (J) The correlation between the CFTR ERAD (Fig. 3 F) and UBQLN2 binding (Fig. 6 D). (K) The correlation between the retrotranslocation (Fig. 4 D) and UBQLN2 binding (Fig. 6 D).
The correlation analysis. (A) The relationship between CFTR ERAD (Fig. 3 F) and the retrotranslocation rates of ∆F508-CFTR (Fig. 4 D) was analyzed for correlation. (B) Correlation analysis was performed to examine the connection between CFTR ER disappearance (Fig. 4 F) and the retrotranslocation rates of ∆F508-CFTR (Fig. 4 D). (C) The correlation between CFTR K48-linked polyubiquitination (Fig. 5 B) and K63-linked polyubiquitination (Fig. 5 C). (D) The correlation between CFTR K48-linked polyubiquitination (Fig. 5 B) and the ERAD rate (Fig. 3 F). (E) The correlation between CFTR K63-linked polyubiquitination (Fig. 5 C) and the ERAD rate (Fig. 3 F). (F) The correlation between CFTR K48-linked polyubiquitination (Fig. 5 B) and the retrotranslocation rate of ∆F508-CFTR (Fig. 4 D). (G) The correlation between CFTR K63-linked polyubiquitination (Fig. 5 C) and the retrotranslocation rate of ∆F508-CFTR (Fig. 4 D). (H) The correlation between the CFTR K48-linked polyubiquitination (Fig. 5 B) and UBQLN2 binding (Fig. 6 D). (I) The correlation between the CFTR K63-linked polyubiquitination (Fig. 5 C) and UBQLN2 binding (Fig. 6 D). (J) The correlation between the CFTR ERAD (Fig. 3 F) and UBQLN2 binding (Fig. 6 D). (K) The correlation between the retrotranslocation (Fig. 4 D) and UBQLN2 binding (Fig. 6 D).
The ∆F508-CFTR-HiBiT(Ex) retrotranslocation was also measured using co-expressed ER luminal LgBiT (ER LgBiT), which was fused with the ER signal peptides of calnexin (CNX) at the N-terminus, and an ER retention signal (KDEL) at the C-terminus (Fig. 4 E). Luminescence was measured in real-time after adding CHX, and the decrease in luminescence corresponded to the disappearance of ∆F508-CFTR-HiBiT(Ex) from the ER. This ER disappearance assay couldn’t detect the weak effect of HERC3 KD on the CFTR retrotranslocation in the WT cells (Fig. 4 F). However, it was able to detect that RNF5/185 DKO reduced the disappearance of ∆F508-CFTR-HiBiT(Ex) from the ER (Fig. 4 F). Like the results in the HiBiT retrotranslocation assay, HERC3 KD tended to reduce the ER disappearance rate of ∆F508-CFTR in RNF5/185 DKO cells (Fig. 4 F). The results of both retrotranslocation analyses were highly correlated (Fig. S3 B). Taken together, our HiBiT assays revealed that HERC3 appears to be involved in ∆F508-CFTR retrotranslocation, albeit slightly, independently of RNF5/185.
Next, we examined whether HERC3 regulates CFTR ubiquitination independently of RNF5/185. Surprisingly, Western blotting with a pan-Ub antibody did not detect a substantial reduction in the total ubiquitination of immature HBH-∆F508-CFTR upon HERC3 KD in 293MSR WT cells and RNF5/185 DKO cells (Fig. 5 A). However, RNF5/185 DKO resulted in a significant reduction in the total CFTR ubiquitination (Fig. 5 A). To obtain more quantitative results, the CFTR ubiquitination level was assessed using Ub ELISA, a sensitive and highly quantitative method (Kamada et al., 2019; Okiyoneda et al., 2018). Ub ELISA showed that HERC3 KD reduced both K48- and K63-linked polyubiquitination of immature HBH-∆F508-CFTR by ∼45% in 293MSR WT cells (Fig. 5, B and C). As expected, RNF5/185 DKO reduced both K48- and K63-linked polyubiquitination of immature HBH-∆F508-CFTR by about 75% (Fig. 5, B and C; and Fig. S3 C). Interestingly, the effect of HERC3 KD on K48- and K63-linked polyubiquitination was somewhat antagonized in RNF5/185 DKO cells. Nonetheless, in direct comparison, the KD of HERC3 still significantly reduced both K48- and K63-linked polyubiquitination of immature ∆F508-CFTR, suggesting that HERC3, to some extent, promotes CFTR ubiquitination independently of RNF5/185 (Fig. 5 D).
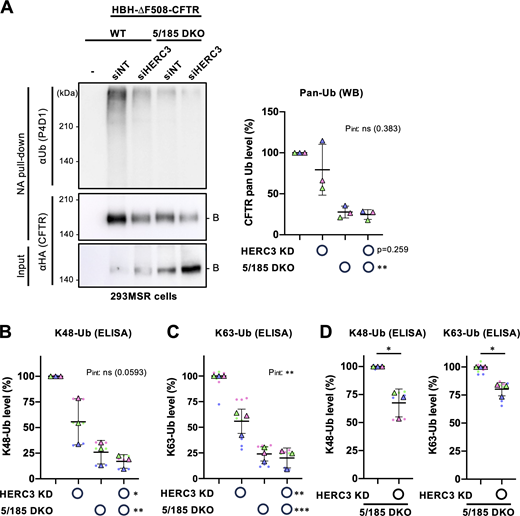
HERC3 and RNF5/185 facilitate ∆F508-CFTR ubiquitination. (A) Ubiquitination levels of HBH-∆F508-CFTR-3HA in 293MSR WT and RNF5/185 DKO cells were measured by Neutravidin pull-down under denaturing conditions (NA pull-down) and Western blotting. The CFTR ubiquitination level was quantified by densitometry and normalized to CFTR in precipitates (right, n = 3). Two-way RM ANOVA revealed a significant main effect of RNF5/185 DKO and no interaction between HERC3 KD and RNF5/185 DKO (Pint > 0.05). (B and C) K48 (B, n = 3) and K63-linked polyubiquitination (C, n = 3) of HBH-∆F508-CFTR in 293MSR WT and RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 were quantified by Ub ELISA using Ub linkage-specific antibodies. 10 µM MG-132 was treated for 3 h at 37°C. The ubiquitination level was normalized by the CFTR amount quantitated by ELISA using an anti-HA antibody. Two-way RM ANOVA revealed significant main effects of HERC3 KD or RNF5/185 DKO and a significant interaction between them in H, but not in G (Pint > 0.05). (D) The effect of HERC3 KD on K48 and K63-linked poly-ubiquitination of HBH-∆F508-CFTR in RNF5/185 DKO cells was measured by Ub ELISA using higher amounts of cell lysate. Statistical significance was assessed by a two-tailed paired t test (n = 3). Each biological replicate (n) is color-coded: the averages from three or four technical replicates are shown in triangles (B–D). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F5.
HERC3 and RNF5/185 facilitate ∆F508-CFTR ubiquitination. (A) Ubiquitination levels of HBH-∆F508-CFTR-3HA in 293MSR WT and RNF5/185 DKO cells were measured by Neutravidin pull-down under denaturing conditions (NA pull-down) and Western blotting. The CFTR ubiquitination level was quantified by densitometry and normalized to CFTR in precipitates (right, n = 3). Two-way RM ANOVA revealed a significant main effect of RNF5/185 DKO and no interaction between HERC3 KD and RNF5/185 DKO (Pint > 0.05). (B and C) K48 (B, n = 3) and K63-linked polyubiquitination (C, n = 3) of HBH-∆F508-CFTR in 293MSR WT and RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 were quantified by Ub ELISA using Ub linkage-specific antibodies. 10 µM MG-132 was treated for 3 h at 37°C. The ubiquitination level was normalized by the CFTR amount quantitated by ELISA using an anti-HA antibody. Two-way RM ANOVA revealed significant main effects of HERC3 KD or RNF5/185 DKO and a significant interaction between them in H, but not in G (Pint > 0.05). (D) The effect of HERC3 KD on K48 and K63-linked poly-ubiquitination of HBH-∆F508-CFTR in RNF5/185 DKO cells was measured by Ub ELISA using higher amounts of cell lysate. Statistical significance was assessed by a two-tailed paired t test (n = 3). Each biological replicate (n) is color-coded: the averages from three or four technical replicates are shown in triangles (B–D). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F5.
HERC3 facilitates the UBQLN2 recruitment to the misfolded CFTR during the ERAD
Given that HERC3 interacts with proteasome shuttling factors UBQLN1 and UBQLN2 (Hochrainer et al., 2008), it is plausible that UBQLN proteins play a role in the HERC3 ERAD branch. Notably, previous studies have indicated that UBQLN1 and UBQLN2 facilitate the ERAD of α1-anti-trypsin null Hong Kong mutant, a misfolded luminal protein, and CD3δ, a membrane-spanning protein (Kim et al., 2008; Lim et al., 2009). These UBQLNs are believed to serve as proteasome shuttling factors that guide ubiquitinated targets to the proteasome (Hjerpe et al., 2016; Itakura et al., 2016). However, their roles in retrotranslocation remain unclear. We particularly focused on UBQLN2 because the overexpression (OE) of UBQLN2, but not UBQLN1, reduced immature ΔF508-CFTR in a dose-dependent manner (Fig. 6 A). To examine whether HERC3 facilitates the association of ∆F508-CFTR with UBQLN2, pull-down experiments were performed using BHK cells stably expressing HBH-∆F508-CFTR-3HA. Western blotting demonstrated that HERC3 OE increased the interaction of immature HBH-ΔF508-CFTR-3HA with FLAG-UBQLN2 (Fig. 6 B). Additionally, an ELISA-based assay was used to quantify the binding of FLAG-UBQLN2 with HBH-∆F508-CFTR immobilized on NeutrAvidin (NA)-coated plates. As expected, the OE of HERC3 led to an increased interaction between CFTR and UBQLN2 (Fig. 6 C). Intriguingly, the CFTR-UBQLN2 interaction was similarly enhanced when utilizing a ∆HECT mutant devoid of E3 ligase activity (Fig. 6 C). This observation suggests that the ubiquitination activity of HERC3 is dispensable for the recruitment of UBQLN2 by HERC3. In contrast to OE, HERC3 KD modestly reduced the CFTR-UBQLN2 interaction by ∼35%, while RNF5/185 DKO robustly reduced the interaction by about 72% in 293MSR WT cells (Fig. 6 D). Like the impact on CFTR ubiquitination, the effect of HERC3 KD on the CFTR-UBQLN2 interaction was antagonized in RNF5/185 DKO cells (Fig. 6 D). However, when directly compared in RNF5/185 DKO cells transfected with an increased amount of FLAG-UBQLN2, HERC3 KD slightly but significantly reduced the CFTR-UBQLN2 interaction (Fig. 6 E). A pull-down experiment also confirmed these results, where the association of HBH-∆F508-CFTR with endogenous UBQLN2 in 293MSR cells was robustly reduced by RNF5/185 DKO and almost undetectable upon HERC3 KD in RNF5/185 DKO cells (Fig. 6 F). The reduced CFTR-UBQLN2 association upon HERC3 and RNF5/185 ablation was highly correlated with the reduction in CFTR polyubiquitination, suggesting that although the possibility that HERC3 also has ubiquitination-independent effects cannot be ruled out, HERC3 and RNF5/185 appear to primarily promote the interaction between CFTR and UBQLN2 by facilitating the ubiquitination of CFTR (Fig. S3, H and I). Furthermore, these changes in the CFTR–UBQLN2 interaction were also highly correlated with the rates of ∆F508-CFTR ERAD and retrotranslocation, suggesting that the reduced UBQLN2 association may be involved in the decelerated retrotranslocation and ERAD of misfolded CFTR (Fig. S3, J and K).
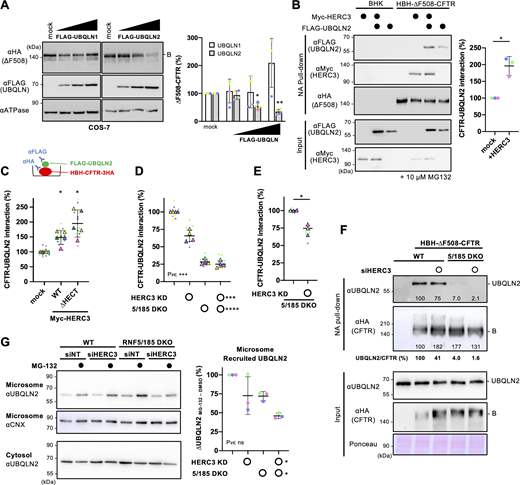
HERC3 facilitates ∆F508-CFTR interaction with UBQLN2. (A) Western blotting showed the steady-state level of ΔF508-CFTR-3HA under OE of FLAG-UBQLN1 or FLAG-UBQLN2 in transiently coexpressed COS-7 cells. The CFTR level was quantified by densitometry (right, n = 4). Na+/K+ ATPase (ATPase) was used as a loading control. B, immature form. (B) The interaction between FLAG-UBQLN2 and HBH-∆F508-CFTR-3HA in BHK cells transfected with or without Myc-HERC3 was assessed using NA pull-down and Western blotting. The amount of UBQLN2 bound to HBH-∆F508-CFTR-3HA was quantified by densitometry and normalized to CFTR levels in the precipitates (right, n = 3). (C) The effect of Myc-HERC3 OE on the FLAG-UBQLN2 and HBH-∆F508-CFTR-3HA interaction in 293MSR WT cells was measured by ELISA using an anti-FLAG antibody. The level of FLAG-UBQLN2 binding was normalized to the CFTR level, which was measured by ELISA using an anti-HA antibody (n = 5). (D and E) The interaction between FLAG-UBQLN2 and HBH-∆F508-CFTR-3HA in 293MSR WT and RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 was measured by ELISA as C (D, n = 4). Additionally, under conditions of increased FLAG-UBQLN2 expression, the UBQLN2 binding to HBH-∆F508-CFTR-3HA in RNF5/185 DKO cells was quantified by ELISA (E, n = 3). (F) The association of HBH-∆F508-CFTR with endogenous UBQLN2 in 293MSR WT or RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 was analyzed by NA pull-down after DSP cross-linking. The quantities of UBQLN2 and ∆F508-CFTR in the precipitates were measured using densitometry and expressed as a percentage of the control. The quantities of CFTR-bound UBQLN2 were normalized to CFTR levels as UBQLN2/CFTR and expressed as a percentage of the control. (G) The level of endogenous UBQLN2 in the microsomes of 293MSR WT and RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 was measured. Cells were treated with or without 10 µM MG-132 for 3 h before subcellular fractionation. Microsomes enriched with ER membranes were confirmed using an anti-calnexin (CNX) antibody. The quantities of the ER-recruited UBQLN2 were quantified by subtracting the amount of UBQLN2 before MG-132 treatment from the amount after MG-132 treatment and were expressed as a percentage of the control (n = 4, right). Each biological replicate (n) is color-coded: the averages from three technical replicates are shown in triangles (C–E). Statistical significance was assessed by one-way RM ANOVA with Dunnett’s multiple comparison tests (A and C), a two-tailed paired t test (B and E), or two-way RM ANOVA (D and G). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F6.
HERC3 facilitates ∆F508-CFTR interaction with UBQLN2. (A) Western blotting showed the steady-state level of ΔF508-CFTR-3HA under OE of FLAG-UBQLN1 or FLAG-UBQLN2 in transiently coexpressed COS-7 cells. The CFTR level was quantified by densitometry (right, n = 4). Na+/K+ ATPase (ATPase) was used as a loading control. B, immature form. (B) The interaction between FLAG-UBQLN2 and HBH-∆F508-CFTR-3HA in BHK cells transfected with or without Myc-HERC3 was assessed using NA pull-down and Western blotting. The amount of UBQLN2 bound to HBH-∆F508-CFTR-3HA was quantified by densitometry and normalized to CFTR levels in the precipitates (right, n = 3). (C) The effect of Myc-HERC3 OE on the FLAG-UBQLN2 and HBH-∆F508-CFTR-3HA interaction in 293MSR WT cells was measured by ELISA using an anti-FLAG antibody. The level of FLAG-UBQLN2 binding was normalized to the CFTR level, which was measured by ELISA using an anti-HA antibody (n = 5). (D and E) The interaction between FLAG-UBQLN2 and HBH-∆F508-CFTR-3HA in 293MSR WT and RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 was measured by ELISA as C (D, n = 4). Additionally, under conditions of increased FLAG-UBQLN2 expression, the UBQLN2 binding to HBH-∆F508-CFTR-3HA in RNF5/185 DKO cells was quantified by ELISA (E, n = 3). (F) The association of HBH-∆F508-CFTR with endogenous UBQLN2 in 293MSR WT or RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 was analyzed by NA pull-down after DSP cross-linking. The quantities of UBQLN2 and ∆F508-CFTR in the precipitates were measured using densitometry and expressed as a percentage of the control. The quantities of CFTR-bound UBQLN2 were normalized to CFTR levels as UBQLN2/CFTR and expressed as a percentage of the control. (G) The level of endogenous UBQLN2 in the microsomes of 293MSR WT and RNF5/185 DKO cells transfected with 50 nM siNT or siHERC3 was measured. Cells were treated with or without 10 µM MG-132 for 3 h before subcellular fractionation. Microsomes enriched with ER membranes were confirmed using an anti-calnexin (CNX) antibody. The quantities of the ER-recruited UBQLN2 were quantified by subtracting the amount of UBQLN2 before MG-132 treatment from the amount after MG-132 treatment and were expressed as a percentage of the control (n = 4, right). Each biological replicate (n) is color-coded: the averages from three technical replicates are shown in triangles (C–E). Statistical significance was assessed by one-way RM ANOVA with Dunnett’s multiple comparison tests (A and C), a two-tailed paired t test (B and E), or two-way RM ANOVA (D and G). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, ***P < 0.001, ****P < 0.0001, ns, not significant. Source data are available for this figure: SourceData F6.
During ERAD, cytoplasmic UBQLN proteins are recruited to the ER membrane to aid in the proteasomal degradation of ubiquitinated proteins (Lim et al., 2009). To investigate the role of HERC3 in UBQLN2 recruitment to the ER membrane, we measured the endogenous UBQLN2 abundance in microsomes. Consistent with a previous study (Lim et al., 2009), proteasome inhibitor MG-132 treatment increased the endogenous UBQLN2 in microsomes, indicating the recruitment of UBQLN2 from the cytoplasm to the ER membrane during ERAD (Fig. 6 G). We quantified the ER-recruited UBQLN2 by measuring the increase in UBQLN2 abundance in the microsome after MG-132 treatment. The results of the quantification showed that the KD of HERC3 and the DKO of RNF5/185 both independently reduced the recruitment of UBQLN2 to the ER membrane. Interestingly, when HERC3 KD and RNF5/185 DKO were combined, there was an additive reduction in the recruitment of UBQLN2 to the ER membrane (Fig. 6 G). This suggests that HERC3 and RNF5/185 cooperate to promote UBQLN2 recruitment during ERAD.
UBQLN proteins facilitate the retrotranslocation of misfolded CFTR
Next, we determined whether UBQLN2 plays a role in facilitating the retrotranslocation and ERAD of ∆F508-CFTR, as its function in retrotranslocation has not been clearly understood despite its role as proteasome shuttling factors (Hochrainer et al., 2008). Initially, a single KD of UBQLN1, UBQLN2, or UBQLN4 did not significantly reduce ∆F508-CFTR ERAD, possibly due to functional redundancy among UBQLN proteins, which are widely expressed in all tissues (Marín, 2014) (Fig. S4 A). Therefore, a triple KD of UBQLN1/2/4 was performed and confirmed by Western blotting (Fig. 7 A). The triple KD of UBQLNs reduced ∆F508-CFTR ERAD by ∼23% and retrotranslocation by ∼42% in 293MSR cells (Fig. 7, B and C). Likewise, we observed reduced retrotranslocation upon UBQLN triple KD even in experiments using MG-132 and CHX chase, indicating that UBQLNs partially facilitate retrotranslocation of pre-existing ∆F508-CFTR-HiBiT within the ER (Fig. S4 B). Furthermore, the triple KD of UBQLNs resulted in a reduction of ∆F508-CFTR disappearance from the ER lumen (Fig. 7 D). These findings suggest that UBQLNs not only promote ERAD but also participate in facilitating the retrotranslocation of misfolded CFTR from the ER to the cytoplasm. The detergent solubility analyses revealed that UBQLN triple KD showed a tendency to increase the insolubility of ∆F508-CFTR-HiBiT(CT) in 293MSR cells, a phenotype similar to that observed with MG-132 treatment (Fig. 7 E). This suggests that UBQLNs may play a crucial role in maintaining the solubility of ∆F508-CFTR, thereby partially facilitating retrotranslocation and ERAD. Alternatively, the reduced retrotranslocation and ERAD observed upon UBQLNs KD could lead to increased CFTR aggregation.

Effects of UBQLN single KD on the CFTR ERAD, triple KD on CFTR retrotranslocation, and establishment of TCRα-HiBiT and Insig-1-HiBiT ERAD assay. (A) The kinetic degradation of ∆F508-CFTR-HiBiT(CT) in 293MSR WT cells transfected with 50 nM siNT or siUNQLN1, 2, or 4. The ERAD rate was calculated by fitting the initial degradation portion of each kinetic degradation curve (right, n = 2). Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles. Data represent mean. (B) The retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells upon UBQLN1/2/4 triple KD was measured during the MG-132 and CHX chase (n = 3, unpaired t test). (C and D) The HiBiT degradation assay confirmed the proteasomal degradation of TCRα-HiBiT (C, n = 4) and Insig-1-HiBiT (D, n = 3) in 293MSR cells. Data represent mean ± SD. **P < 0.01.
Effects of UBQLN single KD on the CFTR ERAD, triple KD on CFTR retrotranslocation, and establishment of TCRα-HiBiT and Insig-1-HiBiT ERAD assay. (A) The kinetic degradation of ∆F508-CFTR-HiBiT(CT) in 293MSR WT cells transfected with 50 nM siNT or siUNQLN1, 2, or 4. The ERAD rate was calculated by fitting the initial degradation portion of each kinetic degradation curve (right, n = 2). Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles. Data represent mean. (B) The retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells upon UBQLN1/2/4 triple KD was measured during the MG-132 and CHX chase (n = 3, unpaired t test). (C and D) The HiBiT degradation assay confirmed the proteasomal degradation of TCRα-HiBiT (C, n = 4) and Insig-1-HiBiT (D, n = 3) in 293MSR cells. Data represent mean ± SD. **P < 0.01.
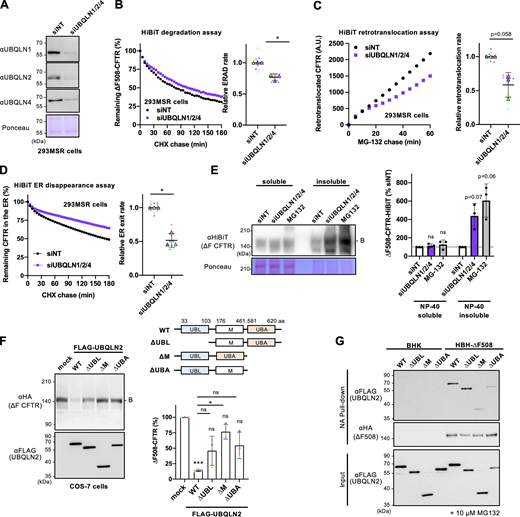
UBQLN proteins facilitate ∆F508-CFTR retrotranslocation and ERAD. (A) Western blotting confirmed the triple KD of UBQLN1, 2, and 4 in 293MSR cells transfected with 50 nM siRNA as indicated. Ponceau staining was used as a loading control. (B) Kinetic degradation of ∆F508-CFTR-HiBiT(CT) in 293MSR WT cells transfected with 50 nM siNT or siUBQLN1/2/4. The ERAD rate was calculated by fitting the initial degradation portion of each kinetic degradation curve (right, n = 3). (C) Kinetic retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells upon UBQLN triple KD. The retrotranslocation was calculated by linear fitting (right, n = 3). (D) Kinetic ER disappearance of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells upon UBQLN triple KD. The ER disappearance rate was calculated by fitting the kinetic ER disappearance curve (right, n = 3). (E) The detergent NP-40 solubility of ∆F508-CFTR-HiBiT(CT) in 293MSR cells was assessed following UBQLN1/2/4 triple KD or MG-132 treatment (10 µM, 3 h) using Western blotting with an anti-HiBiT antibody (n = 3). The soluble (100 µg) and insoluble (40 µg) fractions were analyzed. (F) The effects of overexpressed FLAG-UBQLN2 variants on the steady-state level of ∆F508-CFTR-3HA were analyzed by Western blotting in co-transfected COS-7 cells. The immature ∆F508-CFTR (B band) was quantified by densitometry (right, n = 3). A schematic diagram of the UBQLN2 domain composition with the residue numbers at the domain boundaries. UBQLN2 mutants used in this study are also shown. (G) The interaction of FLAG-UBQLN2 variants with HBH-∆F508-CFTR-3HA in BHK cells was analyzed by NA pull-down and Western blotting. Cells were treated with 10 µM MG-132 for 3 h before cell lysis. Statistical significance was assessed by a two-tailed paired t test (B–D), or one-way RM ANOVA with Dunnett’s multiple comparison tests (E and F). Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles (B–D). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, ns, not significant. Source data are available for this figure: SourceData F7.
UBQLN proteins facilitate ∆F508-CFTR retrotranslocation and ERAD. (A) Western blotting confirmed the triple KD of UBQLN1, 2, and 4 in 293MSR cells transfected with 50 nM siRNA as indicated. Ponceau staining was used as a loading control. (B) Kinetic degradation of ∆F508-CFTR-HiBiT(CT) in 293MSR WT cells transfected with 50 nM siNT or siUBQLN1/2/4. The ERAD rate was calculated by fitting the initial degradation portion of each kinetic degradation curve (right, n = 3). (C) Kinetic retrotranslocation of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells upon UBQLN triple KD. The retrotranslocation was calculated by linear fitting (right, n = 3). (D) Kinetic ER disappearance of ∆F508-CFTR-HiBiT(Ex) in 293MSR cells upon UBQLN triple KD. The ER disappearance rate was calculated by fitting the kinetic ER disappearance curve (right, n = 3). (E) The detergent NP-40 solubility of ∆F508-CFTR-HiBiT(CT) in 293MSR cells was assessed following UBQLN1/2/4 triple KD or MG-132 treatment (10 µM, 3 h) using Western blotting with an anti-HiBiT antibody (n = 3). The soluble (100 µg) and insoluble (40 µg) fractions were analyzed. (F) The effects of overexpressed FLAG-UBQLN2 variants on the steady-state level of ∆F508-CFTR-3HA were analyzed by Western blotting in co-transfected COS-7 cells. The immature ∆F508-CFTR (B band) was quantified by densitometry (right, n = 3). A schematic diagram of the UBQLN2 domain composition with the residue numbers at the domain boundaries. UBQLN2 mutants used in this study are also shown. (G) The interaction of FLAG-UBQLN2 variants with HBH-∆F508-CFTR-3HA in BHK cells was analyzed by NA pull-down and Western blotting. Cells were treated with 10 µM MG-132 for 3 h before cell lysis. Statistical significance was assessed by a two-tailed paired t test (B–D), or one-way RM ANOVA with Dunnett’s multiple comparison tests (E and F). Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles (B–D). Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, ns, not significant. Source data are available for this figure: SourceData F7.
To gain further insight into the mechanism of UBQLNs' action in CFTR ERAD, we tested the effects of UBQLN2 mutants with the Ub-like domain (UBL), central M domain (M), or Ub-associated domain (UBA) deleted (Fig. 7 F). The UBL and UBA domains are known to be involved in proteasome binding (Chen et al., 2016; Ko et al., 2004) and poly-Ub chain binding (Zhang et al., 2008), respectively, while the central M domain containing stress-inducible 1 (STI1) domain is believed to bind to exposed TM segments in the cytosol to prevent aggregation (Itakura et al., 2016). Western blotting showed that FLAG-UBQLN2 OE reduced immature ∆F508-CFTR, likely due to facilitating ERAD (Fig. 7 F). In contrast, the deletion of UBL or UBA slightly reduced the effect of UBQLN2, suggesting that UBQLN2 likely recognizes poly-Ub chains on ∆F508-CFTR through its UBA domain and transfers to the proteasome via its UBL domain during ERAD (Fig. 7 F). Interestingly, the deletion of the central M domain reduced the UBQLN2 effect, indicating the possibility that the M domain might be crucial in shielding the exposed CFTR TM segments in the cytosol to partially promote ∆F508-CFTR retrotranslocation and ERAD (Fig. 7 F). To investigate the interaction between UBQLN2 and ∆F508-CFTR during ERAD, we assessed the CFTR-UBQLN2 association in BHK cells following treatment with the proteasome inhibitor MG-132. Pull-down experiments revealed that the deletion of the M domain or UBA domain reduced the association of HBH-∆F508-CFTR with FLAG-UBQLN2, suggesting the possibility that during ERAD, the M domain, and UBA domain might be involved in the interaction with the exposed TM segments and poly-Ub chains in ∆F508-CFTR, respectively (Fig. 7 G). On the other hand, the UBL domain appeared to be dispensable for the CFTR interaction (Fig. 7 G) and was likely involved in CFTR ERAD by binding to proteasomes.
HERC3 selectively facilitates ERAD of misfolded CFTR
To investigate the substrate selectivity of HERC3, we tested the effect of HERC3 KD on several ERAD models including TCRα (an ERAD-Lm substrate [Horimoto et al., 2013]), Insig-1 (an ERAD-M substrate [Lee et al., 2006; Leto et al., 2019]), and D18G-TTR (an ERAD-L substrate [Sato et al., 2012; Sato et al., 2007]) (Fig. 8 A). Kinetic ERAD of TCRα-HiBiT and Insig-1-HiBiT were successfully measured in 293MSR cells co-transfected with cytoplasmic LgBiT as a proteasome inhibitor blocked their degradation (Fig. S4, C and D). The HiBiT degradation assay showed that HERC3 KD and/or RNF5/185 DKO did not lead to a reduction in the ERAD rates of TCRα and Insig-1, indicating that neither HERC3 nor RNF5/185 is involved in their ERAD processes (Fig. 8, B and C). Similarly, the KD of HERC3 and/or the DKO of RNF5/185 did not result in a decrease in D18G-TTR ERAD (Fig. 8 D). In line with the KD phenotypes, the OE of HERC3 had no impact on the steady-state levels of TCRα-HA and FLAG-D18G-TTR in COS-7 cells. However, HERC3 OE resulted in a dose-dependent reduction in the levels of ∆F508-CFTR (Fig. 8 E). Notably, unlike WT-CFTR, HERC3 OE did not affect the levels of normally folded WT-CFTR, suggesting that HERC3 may specifically facilitate the degradation of misfolded or structurally abnormal CFTR (Fig. 8 E). We additionally investigated the influence of HERC3 KD on the ERAD of an ABC transporter ABCB1 (MDR1/P-glycoprotein) with a ∆Y490 mutation (∆Y490-ABCB1), which is analogous to the ∆F508 mutation in CFTR (Hoof et al., 1994). Since both ∆F508-CFTR and ∆Y490-ABCB1 exhibit folding defects in the cytosolic NBD1 region, the ERAD-C pathway might be involved in their degradation based on previous studies in yeast (Gnann et al., 2004; Nakatsukasa et al., 2008). The HiBiT degradation assay showed that like ∆F508-CFTR, the ERAD of ∆Y490-ABCB1 was decelerated in RNF5/185 DKO cells compared with the WT cells (Fig. 8 F). However, HERC3 KD did not result in a deceleration of ∆Y490-ABCB1 ERAD in both WT and RNF5/185 DKO cells (Fig. 8 F). These findings indicate that RNF5/185 is responsible for the degradation of both ∆F508-CFTR and ∆Y490-ABCB1. In contrast, HERC3 may selectively recognize specific molecular determinants that are present only in misfolded CFTR, but not in the ABCB1 mutant.

The substrate selectivity of HERC3 in ERAD. (A) A schematic diagram of the ERAD substrate models used in this study. The misfolded region is indicated by a star. The HiBiT tag was fused in the cytoplasmic region except for D18G-TTR. (B and C) The HiBiT degradation assay measured the ERAD of TCRα-HiBiT (B, n = 4) and Insig-1-HiBiT (C, n = 3) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3, as indicated. (D) The metabolic stability of D18G-TTR was measured by CHX chase at 37°C and Western blotting with an anti-TTR antibody in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3 as indicated. The remaining TTR was quantified by densitometry and expressed as a percentage of the initial amount (right, n = 3). (E) Western blotting analyzed the effects of Myc-HERC3 OE on co-transfected ∆F508-CFTR-3HA, TCRα-HA, D18G-TTR-FLAG, or WT-CFTR-3HA in COS-7 cells. The immature ∆F508-CFTR (B band), TCRα, D18G-TTR, and total WT-CFTR (B and C bands) were quantified by densitometry (n = 3). (F) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1-HiBiT (E, n = 3) in 293MSR WT and RNF5/185 KO cells as B and C. Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles (B, C, and F). Statistical significance was assessed by a one-way RM ANOVA with Dunnett’s multiple comparison tests (E) or two-way RM ANOVA revealed no significant main effect of HERC3 KD or RNF5/185 DKO, and no interaction between them (Pint > 0.05), except for a significant main effect of RNF5/185 DKO in F. Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ns, not significant. Source data are available for this figure: SourceData F8.
The substrate selectivity of HERC3 in ERAD. (A) A schematic diagram of the ERAD substrate models used in this study. The misfolded region is indicated by a star. The HiBiT tag was fused in the cytoplasmic region except for D18G-TTR. (B and C) The HiBiT degradation assay measured the ERAD of TCRα-HiBiT (B, n = 4) and Insig-1-HiBiT (C, n = 3) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3, as indicated. (D) The metabolic stability of D18G-TTR was measured by CHX chase at 37°C and Western blotting with an anti-TTR antibody in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3 as indicated. The remaining TTR was quantified by densitometry and expressed as a percentage of the initial amount (right, n = 3). (E) Western blotting analyzed the effects of Myc-HERC3 OE on co-transfected ∆F508-CFTR-3HA, TCRα-HA, D18G-TTR-FLAG, or WT-CFTR-3HA in COS-7 cells. The immature ∆F508-CFTR (B band), TCRα, D18G-TTR, and total WT-CFTR (B and C bands) were quantified by densitometry (n = 3). (F) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1-HiBiT (E, n = 3) in 293MSR WT and RNF5/185 KO cells as B and C. Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles (B, C, and F). Statistical significance was assessed by a one-way RM ANOVA with Dunnett’s multiple comparison tests (E) or two-way RM ANOVA revealed no significant main effect of HERC3 KD or RNF5/185 DKO, and no interaction between them (Pint > 0.05), except for a significant main effect of RNF5/185 DKO in F. Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ns, not significant. Source data are available for this figure: SourceData F8.
To investigate the molecular determinants crucial for HERC3 interaction, we generated CFTR fragments fused with the HiBiT tag at the C-terminus, located in the cytoplasm (Fig. 9 A). By utilizing the HiBiT degradation assay, we measured the contributions of HERC3 and RNF5/185 to the ERAD of these CFTR fragments. Consistent with previous findings (Du and Lukacs, 2009), MSD1 (M1), NBD1 with the ∆F508 mutation (∆F508-NBD1), MSD1-NBD1 with ∆F508 mutation (M1-N1[∆F]), and MSD2 (M2) fragments were rapidly eliminated, indicating that individually expressed CFTR domains are recognized as nonnative polypeptides by the ERQC mechanism (Fig. 9, B–D and Fig. S5 A). Like the full-length ∆F508-CFTR, HERC3 KD and RNF5/185 DKO additively reduced the ERAD rates of M1, M1-N1(∆F), and M2 fragments in 293MSR cells (Fig. 9, B–D). In contrast, the KD of HERC3 and/or RNF5/185 DKO did not have any effect on the ERAD of the cytoplasmic ∆F508-NBD1 (Fig. S5 A). These findings suggest that HERC3 and RNF5/185 could play a role in identifying structural abnormalities in the MSDs of CFTR, thereby aiding in the ERAD of misfolded CFTR. This interpretation gains support from the results showing that HERC3 KD and RNF5/185 DKO had an additional impact on reducing the ERAD rate of N1303K-CFTR (Fig. S5 B). N1303K-CFTR is known for its NBD2 mutation, which leads to the unfolding of MSD1 and MSD2, as evidenced by limited protease susceptibility (Du and Lukacs, 2009). Furthermore, correlation analyses revealed that the impact of HERC3 and/or RNF5/185 ablation on ERAD was almost equivalent between ∆F508-CFTR and M1 (Fig. S5 C, slope 0.81) or M1-N1 (Fig. S5 D, slope 1.06), indicating that HERC3 and RNF5/185 might primarily sense conformational defects in the N-terminal region of CFTR, which is crucial for ∆F508-CFTR ERAD. In contrast, the effect on the ERAD of M2 was weaker compared to that of ∆F508-CFTR (Fig. S5 E, slope 0.532). These results suggest that in addition to RNF5/185 and HERC3, other E3 ligases, such as CHIP, may also participate in the ERQC checkpoints of M2 at the late stages of CFTR biogenesis as proposed previously (Younger et al., 2006).

HERC3 selectively facilitates ERAD by interacting with the CFTR-MSDs. (A) A schematic diagram of the CFTR fragment models used in this study. The misfolded region is indicated by a star. M1; MSD1, M1-N1(∆F); MSD1 and NBD1 with ∆F508 mutation, M2; MSD2, N1(∆F); NBD1 with ∆F508 mutation. ∆Y490-ABCB1-MSD1CFTR and ∆Y490-ABCB1-MSD2CFTR are the chimeras in which the MSD1 and MSD2 of ABCB1 were replaced with respective MSDs of CFTR. The HiBiT tag was fused in the C-terminal region located in the cytoplasm. (B–D) The HiBiT degradation assay measured the ERAD of M1-HiBiT (B, n = 3), M1-N1(∆F)-HiBiT (C, n = 3), and M2-HiBiT (D, n = 3) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3, as indicated. (E and F) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1, ∆Y490-ABCB1-∆M1, ∆Y490-ABCB1-∆M2, ∆Y490-ABCB1-M1CFTR, and ∆Y490-ABCB1-M2CFTR in 293MSR WT cells (F, n = 3). The ABCB1-HiBiT constructs analyzed were illustrated in E. (G and H) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1-M1CFTR (G, n = 4) and ∆Y490-ABCB1-M2CFTR (H, n = 4) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3, as indicated. Statistical significance was assessed using a two-tailed paired t test (F) or two-way RM ANOVA (B–D, G, and H) which revealed a significant main effect of HERC3 KD or RNF5/185 DKO, but no significant interaction between them (Pint > 0.05), except for C. Each biological replicate (n) is color-coded, and the averages from four technical replicates are represented by triangles. Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant.
HERC3 selectively facilitates ERAD by interacting with the CFTR-MSDs. (A) A schematic diagram of the CFTR fragment models used in this study. The misfolded region is indicated by a star. M1; MSD1, M1-N1(∆F); MSD1 and NBD1 with ∆F508 mutation, M2; MSD2, N1(∆F); NBD1 with ∆F508 mutation. ∆Y490-ABCB1-MSD1CFTR and ∆Y490-ABCB1-MSD2CFTR are the chimeras in which the MSD1 and MSD2 of ABCB1 were replaced with respective MSDs of CFTR. The HiBiT tag was fused in the C-terminal region located in the cytoplasm. (B–D) The HiBiT degradation assay measured the ERAD of M1-HiBiT (B, n = 3), M1-N1(∆F)-HiBiT (C, n = 3), and M2-HiBiT (D, n = 3) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3, as indicated. (E and F) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1, ∆Y490-ABCB1-∆M1, ∆Y490-ABCB1-∆M2, ∆Y490-ABCB1-M1CFTR, and ∆Y490-ABCB1-M2CFTR in 293MSR WT cells (F, n = 3). The ABCB1-HiBiT constructs analyzed were illustrated in E. (G and H) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1-M1CFTR (G, n = 4) and ∆Y490-ABCB1-M2CFTR (H, n = 4) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3, as indicated. Statistical significance was assessed using a two-tailed paired t test (F) or two-way RM ANOVA (B–D, G, and H) which revealed a significant main effect of HERC3 KD or RNF5/185 DKO, but no significant interaction between them (Pint > 0.05), except for C. Each biological replicate (n) is color-coded, and the averages from four technical replicates are represented by triangles. Data distribution was assumed to be normal but was not formally tested. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant.

Effects of ablation of HERC3 and/or RNF5/185 on the ERAD of ∆F508-NBD1, N1303K-CFTR, and ∆Y490-ABCB1-∆M1, correlation of the ERAD between ∆F508-CFTR and CFTR fragments. (A and B) The HiBiT degradation assay measured the ERAD of ∆F508-NBD1-HiBiT (A, n = 4) and N1303K-CFTR-HiBiT (B, n = 3) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3 as indicated. Two-way RM ANOVA revealed a significant main effect of HERC3 KD (A and B) or RNF5/185 DKO (B), but no interaction between them (Pint > 0.05, in A and B). (C–E) The correlation of ERAD rates between ∆F508-CFTR (Fig. 3 F) and M1 (C, Fig. 9 B), M1-N1(∆F) (D, Fig. 9 C), or M2 (E, Fig. 9 D) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. (F) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1-∆M1-HiBiT in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3 as indicated (n = 3). Two-way RM ANOVA revealed a significant main effect of RNF5/185 DKO, but not that of HERC3 KD, and no interaction between them. Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles (A, B, and F). Data represent mean ± SD. *P < 0.05, **P < 0.01, ****P < 0.0001, ns, not significant.
Effects of ablation of HERC3 and/or RNF5/185 on the ERAD of ∆F508-NBD1, N1303K-CFTR, and ∆Y490-ABCB1-∆M1, correlation of the ERAD between ∆F508-CFTR and CFTR fragments. (A and B) The HiBiT degradation assay measured the ERAD of ∆F508-NBD1-HiBiT (A, n = 4) and N1303K-CFTR-HiBiT (B, n = 3) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3 as indicated. Two-way RM ANOVA revealed a significant main effect of HERC3 KD (A and B) or RNF5/185 DKO (B), but no interaction between them (Pint > 0.05, in A and B). (C–E) The correlation of ERAD rates between ∆F508-CFTR (Fig. 3 F) and M1 (C, Fig. 9 B), M1-N1(∆F) (D, Fig. 9 C), or M2 (E, Fig. 9 D) in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3. (F) The HiBiT degradation assay measured the ERAD of ∆Y490-ABCB1-∆M1-HiBiT in 293MSR WT and RNF5/185 KO cells transfected with 50 nM siNT or siHERC3 as indicated (n = 3). Two-way RM ANOVA revealed a significant main effect of RNF5/185 DKO, but not that of HERC3 KD, and no interaction between them. Each biological replicate (n) is color-coded: the averages from four technical replicates are shown in triangles (A, B, and F). Data represent mean ± SD. *P < 0.05, **P < 0.01, ****P < 0.0001, ns, not significant.
To investigate whether the CFTR MSDs play a crucial role in HERC3- and RNF5/185-mediated ERAD, we conducted domain-swapped experiments. We generated ∆Y490-ABCB1-M1CFTR and ∆Y490-ABCB1-M2CFTR, where the M1 or M2 of ABCB1 was replaced with the respective CFTR’s MSD (Fig. 9 E). Additionally, as controls, ∆Y490-ABCB1-∆M1 and ∆Y490-ABCB1-∆M2 were constructed (Fig. 9 E). The HiBiT degradation assay revealed that the deletion of M1 from ∆Y490-ABCB1 facilitated the ERAD of ∆Y490-ABCB1 (Fig. 9 F). In contrast, the deletion of M2 almost completely inhibited the ERAD (Fig. 9 F). Introducing CFTR-M1 or CFTR-M2 instead of the corresponding ABCB1 MSDs enhanced ERAD, suggesting that CFTR-M1 and -M2 facilitated the ERAD (Fig. 9 F). Notably, HERC3 KD and RNF5/185 DKO additively attenuated the ERAD of both ∆Y490-ABCB1-M1CFTR and ∆Y490-ABCB1-M2CFTR chimeras in contrast to the observed effect on ∆Y490-ABCB1 (Fig. 9, G and H). Furthermore, the introduction of CFTR-M1 to ∆Y490-ABCB1-∆M1 reinstated the contribution of HERC3 to ERAD (Fig. 9 G and Fig. S5 F). These findings suggest the possibility that HERC3 selectively identifies specific features within CFTR’s MSDs, thereby facilitating the ERAD of specific membrane proteins.
HERC3 directly interacts with the exposed CFTR-MSDs
To investigate whether HERC3 directly interacts with the CFTR-MSDs, we produced recombinant FLAG-HERC3, biotinylated (Bio)-∆F508-CFTR, Bio-CFTR-M1, and Bio-CFTR-M2 proteins using the wheat cell-free system. Western blotting confirmed their expression (Fig. 10 A). As a negative control, FLAG-dihydrofolate reductase (DHFR) was utilized. CFTR proteins were also synthesized in the wheat cell-free system in the presence of liposomes to embed their membrane-spanning regions. AlphaScreen analysis using crude translation products containing these recombinant proteins demonstrated that FLAG-HERC3 binds to Bio-∆F508-CFTR but shows no binding to CFTR incorporated into liposomes (Fig. 10 B). Furthermore, FLAG-HERC3 bound to Bio-CFTR-M1 and Bio-CFTR-M2, but not to versions incorporated into liposomes (Fig. 10 C). These findings strongly suggest that HERC3 may not recognize CFTR-MSDs when embedded in the membrane but could potentially directly interact with MSDs exposed to the membrane.
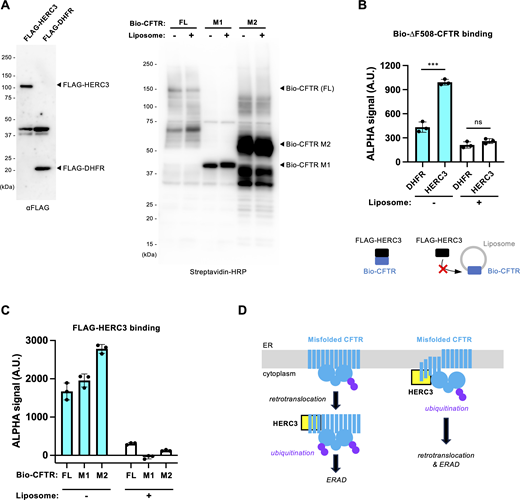
HERC3 directly interacts with the exposed CFTR-MSDs in vitro. (A) Western blotting confirmed the synthesis of FLAG-HERC3 (left) and biotinylated CFTR full-length (FL), M1, and M2 (right) using a wheat cell-free synthesis system in the presence or absence of asolectin liposomes. (B and C) AlphaScreen was employed to evaluate the direct binding of FLAG-HERC3 and biotinylated CFTR synthesized in the presence or absence of asolectin liposomes. FLAG- DHFR served as a negative control. The specific binding signal of FLAG-HERC3, subtracted by the DHFR binding, was measured in C. (D) The proposed model illustrates the function of HERC3 in the CFTR ERQC. HERC3 appears to selectively interact with specific regions of MSDs, typically embedded in the ER membrane. It is speculated that HERC3 monitors the MSDs of select membrane proteins at the ER membrane’s surface and facilitates the ERAD when the TM segments become exposed to the cytosol. Source data are available for this figure: SourceData F10.
HERC3 directly interacts with the exposed CFTR-MSDs in vitro. (A) Western blotting confirmed the synthesis of FLAG-HERC3 (left) and biotinylated CFTR full-length (FL), M1, and M2 (right) using a wheat cell-free synthesis system in the presence or absence of asolectin liposomes. (B and C) AlphaScreen was employed to evaluate the direct binding of FLAG-HERC3 and biotinylated CFTR synthesized in the presence or absence of asolectin liposomes. FLAG- DHFR served as a negative control. The specific binding signal of FLAG-HERC3, subtracted by the DHFR binding, was measured in C. (D) The proposed model illustrates the function of HERC3 in the CFTR ERQC. HERC3 appears to selectively interact with specific regions of MSDs, typically embedded in the ER membrane. It is speculated that HERC3 monitors the MSDs of select membrane proteins at the ER membrane’s surface and facilitates the ERAD when the TM segments become exposed to the cytosol. Source data are available for this figure: SourceData F10.
Discussion
In this study, we have identified HERC3 as an auxiliary E3 ligase that operates alongside RNF5/185 in the ERQC of membrane proteins. Additionally, we have developed HiBiT-based assays to monitor the kinetic ERAD and retrotranslocation of various ERAD substrates. While previous investigations employed split Venus (Grotzke et al., 2013) or GFP (Zhong et al., 2015) for retrotranslocation measurement during ERAD, our approach presents several advantages. We utilize luminescence, a method that offers greater sensitivity and is particularly suitable for screening chemicals susceptible to autofluorescence. Additionally, our assay allows for real-time analysis of live cells in a 96-well plate using a plate reader. This feature not only provides high temporal resolution but also facilitates the concurrent analysis of multiple samples, establishing it as a superior choice for this purpose. By utilizing the HiBiT and Nluc technologies for quantitative retrotranslocation and ERAD measurements, we have discovered that HERC3, in parallel with the RNF5/185 pathway, accelerates the retrotranslocation and ERAD of misfolded CFTR by promoting polyubiquitination. Both HERC3 and RNF5/185 collaborate in recruiting UBQLNs, which appear to partially contribute to facilitating the retrotranslocation and proteasomal delivery of ubiquitinated CFTR. While RNF5/185 contributes to both retrotranslocation and the proteasomal degradation of ∆F508-CFTR, the role of HERC3 in retrotranslocation is relatively minor. HERC3 likely plays a more significant role in facilitating the proteasomal degradation of CFTR following retrotranslocation. Considering that HERC3 is primarily localized in the cytoplasm (Cruz et al., 2001), it is reasonable that HERC3 primarily acts on the misfolded CFTR dislocated to the cytoplasm. Moreover, this presumed function is in line with previous findings that HERC3 promotes the proteasomal degradation of cytosolic proteins such as RPL23A (Zhang et al., 2022b), EIF5A (Zhang et al., 2022a), and SMAD7 (Li et al., 2019). On the other hand, HERC3 appears to be at least partially involved in retrotranslocation, and its contribution may be particularly important when the function of RNF5/185 is insufficient. We speculate that in the absence of RNF5/185, other ER-based Ub ligases might contribute to facilitating CFTR retrotranslocation, especially preceding the HERC3 ERAD pathway. Further research in the future will be necessary to confirm this hypothesis.
Our findings indicate that HERC3 plays a role in facilitating both K48- and K63-linked polyubiquitination of ∆F508-CFTR (Fig. S3 C). The significant correlation between the reduced polyubiquitination and ERAD upon HERC3 KD suggests that HERC3 promotes the CFTR ERAD by enhancing polyubiquitination (Fig. S3, D and E). In contrast, HERC3 KD led to a ∼45% decrease in the CFTR polyubiquitination, while only marginally affecting the CFTR retrotranslocation (Fig. S3, F and G). This may also support our model that HERC3-mediated ubiquitination primarily influences the downstream ERAD process rather than retrotranslocation (Fig. 10 D). However, HERC3 seems to at least play a role in the regulation of CFTR ubiquitination at the ER before retrotranslocation (Fig. 10 D), as evidenced by the fact that its KD led to an increase in the abundance of the foldable ∆F508-CFTR biogenic intermediate, which can reach the PM, especially at low temperatures or in the presence of a CFTR corrector. In contrast to HERC3, RNF5/185 appears to primarily facilitate retrotranslocation by promoting K48- and K63-linked polyubiquitination (Fig. S3, C–G). Surprisingly, while HERC3 KD showed an additive effect on reducing ERAD and retrotranslocation when combined with RNF5/185 DKO, the effect of HERC3 KD on CFTR K48- and K63-linked polyubiquitination was counteracted by RNF5/185 DKO. Thus, it appears that the HERC3-mediated CFTR ubiquitination is at least partially dependent on RNF5/185. Similar to Gp78 (Morito et al., 2008), HERC3 may function as an E4 enzyme that elongates the Ub chains initiated by RNF5/185, as it directly binds to Ub (Cruz et al., 2001). Alternatively, HERC3 may modify the Ub chains initiated by RNF5/185 by adding non-canonical Ub chains, such as K27 chains, which promote the p97-proteasome pathway (Shearer et al., 2022), as HERC3 has been shown to promote K27-linked polyubiquitination (Zhang et al., 2022a).
Based on our findings, it appears that HERC3 and RNF5/185 cooperatively facilitate the recruitment of UBQLN2 to the misfolded CFTR at the ER membrane, likely by enhancing CFTR polyubiquitination. In addition to their role as proteasome shuttling factors (Hjerpe et al., 2016; Itakura et al., 2016), our HiBiT-based retrotranslocation assays revealed the partial involvement of UBQLNs in promoting retrotranslocation. This contribution in retrotranslocation aligns with previous findings that the depletion of UBQLNs leads to ER stress, which arises from the accumulation of abnormal proteins in the ER (Lim et al., 2009). While UBQLNs appear to bind ubiquitinated CFTR through their UBA domain, the central M domain of UBQLNs may shield the exposed CFTR TM segments of the dislocation intermediate as proposed (Itakura et al., 2016). Structural models also suggest that the M domain forms a hydrophobic groove that could potentially recognize the TM segments (Fry et al., 2021). Considering that the hydrophobicity of TM helices presents an energetic barrier during the retrotranslocation of integral membrane ERAD substrates (Guerriero et al., 2017), UBQLNs may serve to protect the exposed TM segments of the dislocation intermediate from undesired interactions or aggregation on the surface of the ER membrane, thereby facilitating the retrotranslocation. This model is supported by our observation that UBQLNs’ triple KD increased the insolubility of ∆F508-CFTR. UBQLNs may also contribute to facilitating retrotranslocation through a polyubiquitin-mediated ratcheting mechanism, as proposed previously (Baldridge and Rapoport, 2016).
Our kinetic ERAD assays, utilizing a variety of substrates, have revealed the impressive substrate specificity of HERC3. It appears that neither the HERC3 nor RNF5/185 ERAD pathways are involved in the degradation of TCRα, Insig-1, and D18G-TTR. These substrates are primarily targeted by the Hrd1 (Kikkert et al., 2004; Sato et al., 2012) and/or Gp78 ERAD branches (Chen et al., 2006; Song et al., 2005). Unlike a cytosolic E3 ligase CHIP involved in the ubiquitination of conformationally defective cytoplasmic NBD1 (Meacham et al., 2001; Rabeh et al., 2012; Younger et al., 2004), HERC3 does not play a crucial role in the QC checkpoint of the CFTR’s NBD1. Consistent with previous studies (van de Weijer et al., 2020; Zhong et al., 2009), the RNF5/185 ERAD branch appears to specifically eliminate certain types of membrane proteins, such as ∆F508-CFTR and ∆Y490-ABCB1. Both CFTR and ABCB1 belong to the ABC transporter family, and conserved residues in these ABC transporters are primarily located in the cytosolic region, while sequences in the TM region show high variability (Liu et al., 2017). Like the ∆F508 mutation in CFTR, the ∆Y490 mutation causes misfolding of ABCB1, disrupting the packing of the TM segments (Loo et al., 2002). Therefore, RNF5/185 appears to play a role primarily in the ERQC checkpoint of the MSDs in mutants of both ABCB1 and CFTR, as proposed previously (Younger et al., 2006). Based on our results using the ABCB1-CFTR chimera, it seems that the CFTR’s M1 and M2 may contain potential degrons that facilitate the ERAD of polytopic membrane protein. This finding aligns with recent research showing that a type I CFTR corrector, tezacaftor (VX-661), and a type III corrector, elexacaftor (VX-445), bind to M1 and M2, respectively, stabilizing these domains and reducing the susceptibility of mutant CFTR to degradation by the ERQC machinery (Fiedorczuk and Chen, 2022b). Since introducing the CFTR’s M1 or M2 induced the HERC3-dependent ERAD of ∆Y490-ABCB1, HERC3 likely identifies specific features of conformationally defective CFTR’s MSDs. Consistent with this hypothesis, our in vitro study provides evidence supporting the direct binding of HERC3 to CFTR’s MSDs. Intriguingly, HERC3 displayed no binding to MSDs embedded in liposomes, suggesting that HERC3 may selectively recognize the exposed TM regions typically present within the ER membranes. In contrast to ABCB1 TM segments which lack charged residues (Wang et al., 2007), the insertion and orientation of CFTR TM segments could be more error-prone due to the presence of a substantial number of charged residues in these segments (Sadlish and Skach, 2004). Specifically, previous studies have shown that TM segment 6 of CFTR, housing three positively charged residues, exhibits high instability within the lipid bilayer (Tector and Hartl, 1999). Consequently, the CFTR’s MSDs are predicted to be unstable in the membrane (Fiedorczuk and Chen, 2022a). We propose that alongside the ER-embedded RNF5/185, the cytosolic E3 ligase HERC3 monitors the exposed TM regions of membrane proteins on the cytoplasmic surface of the ER membrane, providing an ERAD branch for select membrane proteins (Fig. 10 D).
Materials and methods
The key resource table is listed in Table 1.
Key resources table
| Reagents or resource | ||
|---|---|---|
| Experimental models: Cell lines | Source | Identifier |
| CFBE teton ΔF508-CFTR-HRP | PMID: 29503157 | N/A |
| CFBE teton ΔF508-CFTR-3HA, YFP-H148Q/I152L/F46L | PMID: 29503157 | N/A |
| CFBE ΔF508-CFTR-3HA | PMID: 29503157 | N/A |
| CFBE teton HBH-ΔF508-CFTR | PMID: 29503157 | N/A |
| CFBE teton ∆F508-CFTR-3HA-Nluc(CT) | This paper | N/A |
| COS-7 | JCRB cell bank | Cat#JCRB9127 |
| BHK | PMID: 29503157 | N/A |
| BHK HBH-ΔF508-CFTR-3HA | PMID: 29503157 | N/A |
| 293MSR | Thermo Fisher Scientific | Cat#R79507 |
| 293MSR RNF5/185 DKO | This paper | N/A |
| 293MSR HBH-∆F508-CFTR-3HA | This paper | N/A |
| 293MSR RNF5/185 DKO HBH-∆F508-CFTR-3HA | This paper | N/A |
| 293MSR teton ∆F508-CFTR-3HA-Nluc(CT) | This paper | N/A |
| BEAS-2B | ECACC | Cat#95102433 |
| BEAS-2B teton ∆F508-CFTR-3HA-Nluc(CT) | This paper | N/A |
| Recombinant DNA | Source | Identifier |
| pNUT ∆F508-CFTR-3HA | PMID: 7691813 | N/A |
| pcDNA 6xmyc-HERC3 | This paper | N/A |
| pcDNA 6xmyc-HERC3 ∆HECT | This paper | N/A |
| pcDNA 6xmyc-HERC3 ∆RLD | This paper | N/A |
| pcDNA 6xmyc-HERC3-C1018A | This paper | N/A |
| pNUT ∆F508-CFTR-3HA-HiBiT(CT) | This paper | N/A |
| pBiT1.1-N [TK/LgBiT] | Promega | Cat#N198A |
| pBiT1.1-N [TK/CNXss-LgBiT-KDEL] | This paper | N/A |
| pcDNA6 ∆F508-CFTR-HiBiT (Ex) | This paper | N/A |
| pCS2+ FLAG-hPLIC-1(UBQLN1) | Addgene | Cat#8663 |
| pCS2+ FLAG-hPLIC-2(UBQLN2) | Addgene | Cat#8661 |
| pCS2+ FLAG-UBQLN2 ∆UBL | This paper | N/A |
| pCS2+ FLAG-UBQLN2 ∆M | This paper | N/A |
| pCS2+ FLAG-UBQLN2 ∆UBA | This paper | N/A |
| pDONR221 | Addgene | Cat#3388 |
| pLX304 | Addgene | Cat#25890 |
| pLX303 HBH-∆F508 CFTR-3HA | This paper | N/A |
| pLIX402 | Addgene | Cat#41394 |
| pLIX402 blast | This paper | N/A |
| pLIX402 Nluc(CT) | This paper | N/A |
| pLIX402 ∆F508-CFTR-3HA-Nluc(CT) | This paper | N/A |
| pNUT-HBH-∆F508-CFTR-3HA | PMID: 29503157 | N/A |
| pCMV-TCRα-HA | PMID: 10601236 | N/A |
| pBiT2.1 TCRα-HiBiT | This paper | N/A |
| pBiT2.1 Insig-1-HiBiT | This paper | N/A |
| pBiT2.1 ∆Y490-ABCB1-HiBiT | This paper | N/A |
| pNUT CFTR M1 (1–413 aa)-HiBiT | This paper | N/A |
| pNUT ∆F508-CFTR M1N1 (1–635 aa)-HiBiT | This paper | N/A |
| pNUT CFTR M2 (733–1218 aa)-3HA-HiBiT | This paper | N/A |
| pBiT2.1 ∆F508-CFTR N1 (389–678 aa)-HiBiT | This paper | N/A |
| pNUT N1303K-CFTR-3HA-HiBiT | This paper | N/A |
| pEF6 D18G-TTR | PMID: 17431395 | N/A |
| pEF6 D18G-TTR-FLAG | PMID: 17431395 | N/A |
| pBiT2.1 ∆Y490-ABCB1 ∆M1 (1–379aa)-HiBiT | This paper | N/A |
| pBiT2.1 ∆Y490-ABCB1 ∆M2 (681–1025aa)-HiBiT | This paper | N/A |
| pBiT2.1 ∆Y490-ABCB1 (∆1-379)-CFTR-MSD1 (1–413 aa)-HiBiT (∆Y490-ABCB1-MSD1CFTR) | This paper | N/A |
| pBiT2.1 ∆Y490-ABCB1 (∆681-1025)-CFTR-MSD2 (837–1156 aa)-HiBiT (∆Y490-ABCB1-MSD2CFTR) | This paper | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Addgene | Cat#62988 |
| pSpCas9(BB)-2A-Puro-RNF5 gRNA #1 | This paper | N/A |
| pSpCas9(BB)-2A-Puro-RNF5 gRNA #2 | This paper | N/A |
| pSpCas9(BB)-2A-Puro-RNF185 gRNA #1 | This paper | N/A |
| pSpCas9(BB)-2A-Puro-RNF185 gRNA #2 | This paper | N/A |
| pEU-E01-nbls-∆F508-CFTR (Biotin-∆F508-CFTR) | This paper | N/A |
| pEU-E01-nbls-CFTR M1 (1-413) (Biotin-CFTR M1) | This paper | N/A |
| pEU-E01-nbls-CFTR M2 (733-1218) (Biotin-CFTR M2) | This paper | N/A |
| pEU-E01-nFLAG-DHFR (FLAG-DHFR) | PMID: 27249653 | N/A |
| pEU-E01-nFLAG-HERC3 (FLAG-HERC3) | This paper | N/A |
| pCold-His-SUMO-RNF185 ΔTM | This paper | N/A |
| Antibodies | Source | Identifier |
| HA (16B12) | BioLegend | Cat#901515 |
| Na+/K+-ATPase | Santa Cruz Biotechnology | Cat#sc-48345 |
| HERC3 (2C1) | Abnova | Cat#H00008916-M01A |
| Ub (P4D1) | Santa Cruz Biotechnology | Cat#sc-8017 |
| Myc | Wako | Cat#017-21871 |
| RNF185 antiserum | This paper | N/A |
| K48-Ub (Apu2) | Merck Millipore | Cat#05-1307 |
| K63-Ub (Apu3) | Merck Millipore | Cat#05-1308 |
| FLAG (1E6) | Wako | Cat#014-22383 |
| UBQLN2 (D7R2Z) | Cell Signaling Technology | Cat#85509 |
| CNX | MBL | Cat#PM060 |
| UBQLN1 (D3T7F) | Cell Signaling Technology | Cat#14526 |
| UBQLN4 | Bethyl Laboratories | Cat#A305-237A |
| Prealbumine (TTR, E-1) | Santa Cruz Biotechnology | Cat#sc-377517 |
| HERC3 (2C1) | Abnova | Cat#H00008916-M01A |
| HiBiT | Promega | Cat#N7200 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson Immuno Research | Cat#115-035-166 |
| Peroxidase AffiniPure Donkey Anti- Rabbit IgG (H+L) | Jackson Immuno Research | Cat#711-035-152 |
| Chemicals, peptides, and recombinant proteins | Source | Identifier |
| Lipofectamine RNAiMAX Transfection reagent | Invitrogen | Cat#13778150 |
| Doxycycline Hydrochloride n-Hydrate (Dox) | FUJIFILM Wako | Cat#049-31121 |
| 8-cpt-cAMP | Santa Cruz Biotechnology | Cat#sc-201569A |
| Forskolin | FUJIFILM Wako | Cat#067-02191 |
| 3-isobutyl-1-methyl-xanthine (IBMX) | FUJIFILM Wako | Cat#095-03413 |
| Genistein | FUJIFILM Wako | Cat#073-05531 |
| Cycloheximide (CHX) | FUJIFILM Wako | Cat#037-20991 |
| MG-132 | Cayman | Cat#10012628 |
| N-Ethylmaleimide (NEM) | FUJIFILM Wako | Cat#054-02063 |
| Pepstatin A | Peptide institute | Cat#4397 |
| Leupeptin Hemisulfate monohydrate | FUJIFILM Wako | Cat#122-03751 |
| Benzylsulfonyl Fluoride (PMSF) | FUJIFILM Wako | Cat#164-12181 |
| Ponceau-S | Sigma-Aldrich | Cat#24-3875-5 |
| Ponceau-S | Beacle, Inc | Cat#BCL-PSS-01 |
| NeutrAvidin agarose | Thermo Fisher Scientific | Cat#29200 |
| VX-809 | Cayman | Cat#22196 |
| VX-661 | Selleck Chemicals | Cat#S7059 |
| VX-445 | Selleck Chemicals | Cat#S8851 |
| VX-770 | Chemscene LLC | Cat#CS-0497 |
| Nunc FluoroNunc/LumiNunc 96-Well Plates | Thermo Fisher Scientific | Cat#437796 |
| Avidin, NeutrAvidin Biotin-binding protein | Invitrogen | Cat#A2666 |
| Nunc MicroWell 96-Well, Nunclon Delta-Treated, Flat-Bottom Microplate | Thermo Fisher Scientific | Cat#136101 |
| Nano-Glo Endurazine Live Cell Substrate | Promega | Cat#N2571 |
| Albumin, from Bovine Serum, Fraction V pH7.0 (BSA) | FUJIFILM Wako | Cat#015-27053 |
| (+)-Biotin | FUJIFILM Wako | Cat#029-08713 |
| Urea | FUJIFILM Wako | Cat#217-00615 |
| DbeQ | Selleck | Cat#S7199 |
| CO2 independent medium | Thermo Fisher Scientific | Cat#18045088 |
| Polyethylenimine (PEI) Max | Polyscience, Inc | Cat#24765 |
| G418 | FUJIFILM Wako | Cat#074-05963 |
| Puromycin | Sigma-Aldrich | Cat#P9620-10ML |
| Blasticidin S, Hydrochloride | Funakoshi | Cat#KK-400 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34580 |
| ImmunoStar Zeta | FUJIFILM Wako | Cat#295-72404 |
| ImmunoStar LD | FUJIFILM Wako | Cat#292-69903 |
| Oligonucleotides | Source | Identifier |
| RNF5 gRNA#1: 5′-GCCCAACGATCGTGGGCAGG-3′ | This paper | N/A |
| RNF5 gRNA#2: 5′-GACTCTTACCTGTACCCCGG-3′ | This paper | N/A |
| RNF185 gRNA#1: 5′-TGTAGTGGTACGGTGCAAAC-3′ | This paper | N/A |
| RNF185 gRNA#2: 5′-AATGGCGCTGGCGAGAGCGG-3′ | This paper | N/A |
| AllStars Neg. Control siRNA (siNT) | Qiagen | Cat#1027281 |
| HS_HERC3_4 siRNA: 5′-CAGCCCGAGCTGATCGCTTTA-3′ | Qiagen | Cat#SI00107233 |
| HS_HERC3_5 siRNA: 5′-CAGCGATCGGATTCCCATCTA-3′ | Qiagen | Cat#SI03066959 |
| HS_HERC3_8 siRNA: 5′-TTGAGTCTCTATATTGTCCAA-3′ | Qiagen | Cat#SI05092101 |
| Hs_RNF5_11 siRNA: 5′-CAGCCAGAGAAGAATCAGTAT-3′ | Qiagen | Cat#SI05063114 |
| Hs_RNF5_6 siRNA: 5′-GGGCGCGACCTTCGAATGTAA-3′ | Qiagen | Cat#SI03221484 |
| Hs_RNF5_7 siRNA: 5′-AACGGCAAGAGTGTCCAGTAT-3′ | Qiagen | Cat#SI04204116 |
| Hs_RNF185_2 siRNA: 5′-CACCTTCTGGAAATCCGCAAA-3′ | Qiagen | Cat#SI03059385 |
| Hs_RNF185_3 siRNA: 5′-CTCCGAGGCAGTAATAACTGA-3′ | Qiagen | Cat#SI03089982 |
| Hs_RNF185_8 siRNA: 5′-TCCCATCAGCTTTCTCCCTAA-3′ | Qiagen | Cat#SI04959598 |
| Hs_RFFL_1 siRNA 5′-ATCGGTTTCTTCAGTGCCTTA-3′ | Qiagen | Cat#SI00148232 |
| Hs_RFFL_3 siRNA 5′-TCGCAACTTTGTCAACTACAA-3′ | Qiagen | Cat#SI00148246 |
| Hs_RFFL_5 siRNA 5′-CTCCATGACATCTCTACCGAA-3′ | Qiagen | Cat#SI03089653 |
| RNF5 genome PCR primer pair: 5′-CCAGCCGAATGAGTTGGGTTAACAC-3′ (FW) 5′-GGCGAGAAACAGGAAGAGGGAATCC-3′ (Rev) | This paper | N/A |
| RNF185 genome PCR primer pair: 5′-ACCTAGCAAGCTAGAGGCTG-3′ (FW) 5′-AGGGCTCCTGTAACCGTCAA-3′ (Rev) | This paper | N/A |
| M13F sequence primer: 5′-GTAAAACGACGGCCAGT-3′ | This paper | N/A |
| M13R pUC sequence primer: 5′-CAGGAAACAGCTATGAC-3′ | This paper | N/A |
| HERC3 qPCR primer pair: 5′-GATCTGCCGAGAAAGCTATGG-3′ (FW) 5′-TTTGCCACCACACACCTTTAG-3′ (Rev) | This paper | N/A |
| GAPDH qPCR primer pair: 5′-CATGAGAAGTATGACAACAGCCT-3′ (FW) 5′-AGTCCTTCCACGATACCAAAGT-3′ (Rev) | This paper | N/A |
| Software and algorithms | Source | Identifier |
| GraphPad Prism 8 | GraphPad Software, Inc | RRID:SCR_002798 |
| Evolution Capt | Vilber-Lourmat | N/A |
| ImageQuant | Cytiva | RRID:SCR_014246 |
Resource availability
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Tsukasa Okiyoneda ([email protected]).
Materials availability
Reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Cell lines and cell culture
CFBE41o-, BHK cells and corresponding transfectants, and COS-7 cells have been previously described (Okiyoneda et al., 2018). Briefly, CFBE41o- and their respective transfectants were cultured in E-MEM medium (FUJIFILM Wako Pure Chemical Corporation) supplemented with 10% FBS. BHK and COS-7 cells were cultured in D-MEM/F12 medium (FUJIFILM Wako Pure Chemical Corporation) supplemented with 5% FBS and D-MEM medium (FUJIFILM Wako Pure Chemical Corporation) supplemented with 10% FBS, respectively. 293MSR and RNF5/185 DKO cells were cultured in D-MEM medium (FUJIFILM Wako Pure Chemical Corporation) supplemented with 10% FBS, 0.5 mg/ml G418. 293MSR and RNF5/185 DKO cells stably expressing HBH-∆F508-CFTR-3HA were established by lentivirus transduction as previously (Okiyoneda et al., 2018) and were cultured in D-MEM medium supplemented with 10% FBS, 0.5 mg/ml G418, and 5 µg/ml blasticidin S. BEAS-2B, 293MSR, and CFBE cells stably expressing inducible ∆F508-CFTR-3HA-Nluc were established by lentivirus transduction as previously (Taniguchi et al., 2022). The CFTR expressions in CFBE and BEAS-2B cells were induced by treating them with 1 µg/ml of doxycycline (Dox) for 4 and 2 days, respectively, unless otherwise specified. All cell culture media were supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (FUJIFILM Wako Pure Chemical Corporation), and cells were cultured at 37°C under 5% CO2.
Plasmids and antibodies
Plasmids and primers used in this study are listed in Table 1. Truncated, deletion, or mutated constructs of HERC3, UBQLN2, and CFTR were created by PCR-based cloning methods. The HiBiT tag on CFTR variants, Insig-1 and ABCB1 was inserted by PCR-based cloning methods. In most instances, the HiBiT tag was attached to the C-terminus of proteins situated within the cytosol. For the CFTR-HiBiT(Ex) construct, the extracellular HiBiT(Ex) tag was inserted into the fourth extracellular loop of CFTR, replacing the 3xHA tag on the CFTR-3HA (Okiyoneda et al., 2010). TCRα-HiBiT was created by inserting TCRα from pCMV-TCRα-HA (a kind gift from Dr. R. Kopito, Stanford University, Stanford, CA, USA) into the pBiT2.1-HiBiT vector by In-fusion cloning. To produce lentivirus vectors, CFTR variants or UBQLN2 variants were first cloned into pDONR221 (#3388; Addgene) using BP reaction with BP clonase II Enzyme mix (Thermo Fisher Scientific). Subsequently, they were transferred to pLX304 (#25890; Addgene) or pLIX402 Nluc DEST vector via LR reaction with LR clonase II Enzyme mix (Thermo Fisher Scientific). The pLIX402 Nluc DEST vector was created by replacing the HA tag in pLIX402 DEST vector (#41394; Addgene) with Nluc cDNA in-frame using In-fusion cloning. The pLIX402 ∆F508-CFTR-Nluc (CT) was constructed by LR reaction. CNXss-LgBiT-KDEL was constructed by introducing the N-terminal signal sequence of CNX and the C-terminal KDEL ER retention signal into LgBiT in pBiT1.1-N [TK-LgBiT] (Promega) through PCR-based mutagenesis. Other mutant and chimera constructs were generated through PCR-based mutagenesis or in-fusion cloning.
CRISPR–Cas9 mediated gene knockout of RNF5 or RNF185 was performed using the pSpCas9(BB)-2A-Puro (PX459) V2.0 (#62988; Addgene) containing RNF5 gRNA#1 (5′-GCCCAACGATCGTGGGCAGG-3′), RNF5 gRNA#2 (5′-GACTCTTACCTGTACCCCGG-3′), RNF185 gRNA#1 (5′-TGTAGTGGTACGGTGCAAAC-3′), and RNF185 gRNA#2 (5′-AATGGCGCTGGCGAGAGCGG-3′). Pairs of oligonucleotides with BpiI (BbsI, Cat#ER1012; Thermo Fisher Scientific) overhangs were annealed and ligated into the BpiI-digested vector. All constructs generated were verified by DNA sequencing.
For wheat germ cell-free protein synthesis, Biotin-∆F508-CFTR, Biotin-CFTR-M1 (1-413), Biotin-CFTR-M2 (733-1218), FLAG-DHFR, and FLAG-HERC3 in a pEU vector were utilized as previously (Takahashi et al., 2016). For the biotinylated proteins, a biotin ligase recognition sequence (bls) was fused to the N-terminus.
The RNF185 antiserum was produced by immunizing a rabbit with recombinant human RNF185 ∆TM (1-130 amino acid) protein purified from E. coli, and its specificity was confirmed by ELISA. The RNF185 ∆TM protein fused with an N-terminal His6-sumo tag was expressed in BL21 rosetta2 E. coli strain (Merck Millipore) using the pCold vector (Takara Bio) and purified using Ni-NTA agarose (Qiagen) as described previously (Taniguchi et al., 2023). The His6-sumo tag was subsequently removed using Ulp1 protease and Ni-NTA agarose to prepare non-tagged RNF185 ∆TM proteins. Other antibodies used in this study are listed in Table 1.
Mammalian cell transfection
Transient transfection in COS-7, BHK, 293MSR, and RNF5/185 DKO cells was achieved using polyethylenimine (PEI) max (Polysciences, Inc.), and experiments were performed 2 days after transfection. siRNA transfections (50 nM each) were performed using Lipofectamine RNAiMax transfection reagent (Invitrogen) according to the manufacturer’s instructions. Cells transfected with siRNA were generally used for the experiments 4–5 days after transfection unless specified otherwise. The target sequences of siRNA are listed in Table 1. When not specifically mentioned, pooled siRNA was used for KD. The pooled siRNA was prepared by mixing an equal amount of individual siRNA for HERC3 (siHERC3 #4, #5, #8), RNF5 (siRNF5 #11, #6, #7), RNF185 (siRNF185 #2, #3, #8), and RFFL (siRFFL #1, #3, #5). AllStars Negative Control siRNA from Qiagen was utilized as a negative control non-targeting siRNA (siNT).
Generation of CRISPR/Cas9-mediated knockout cells
For CRISPR/Cas9-mediated knockouts, 293MSR cells were transfected with specific sgRNAs targeting RNF5 (gRNA#1 and gRNA#2) and RNF185 (gRNA#1 and gRNA#2) using PEI Max (Polyscience, Inc.). On the next day, the cells were treated with puromycin (3 µg/ml; Sigma-Aldrich) for 24 h to enrich the transfected cells. The enriched cells were then diluted and seeded into 96-well plates to allow for the growth of single colonies. Colonies were screened by genome PCR using KOD polymerase (Toyobo), and specific primers are listed in Table 1. The targeted genome deletions were confirmed by genomic DNA sequencing (Fig. S1, A and B) and at the protein level by Western blotting. Confirmation of RNF5 and RNF185 KO was performed through Western blotting and DNA sequencing. For sequencing of RNF5 and RNF185, the genomic locus was amplified by PCR using specific RNF5 and RNF185 genome PCR primer pairs listed in Table 1. The PCR product was cloned into pMD20-T vector using a Mighty TA-cloning Kit (Takara Bio). The DNA sequence of eight colonies was determined by DNA sequencing using M13F and M13R pUC sequence primers, which resulted in the same result (Fig. S1, A and B).
Measurement of PM level and stability of CFTR
Cell-surface expression of ∆F508-CFTR-HRP in CFBE Teton cells on 96-well plates was measured after the addition of the HRP substrate (SuperSignal West Pico PLUS; Thermo Fisher Scientific), as previously described (Okiyoneda et al., 2018; Taniguchi et al., 2022). Briefly, the subconfluent CFBE cells were transfected with siRNA (50 nM each) in 96 wells, and the CFTR expression was induced by 1 µg/ml Dox treatment for 4 days. The ∆F508-CFTR-HRP was rescued through corrector treatment, either at 37°C as specified in the figure legend or at 26°C for 2 days, followed by a 1-h incubation at 37°C before the experiment. Subsequently, cells underwent six washes with PBS, and HRP activity was assessed by adding 100 μl/well of SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific). After a 5-min incubation, chemiluminescence was quantified using a Valioskan microplate reader (Thermo Fisher Scientific) or the EnSpire Alpha plate reader (Perkin Elmer).
The cell surface stability of ∆F508-CFTR-3HA in the CFBE cells on 96-well plates was measured by cell surface ELISA using an anti-HA antibody (16B12; BioLegend) as previously described (Okiyoneda et al., 2013). Cell surface expression of ∆F508-CFTR was induced by 26°C incubation for 2 days, followed by a 1-h incubation at 37°C to induce the thermal unfolding. The cellular level and stability of ∆F508-CFTR-3HA in CFBE cells on 96-well plates were measured by ELISA using an anti-HA antibody (16B12; BioLegend) after 4% paraformaldehyde (PFA) fixation and cell permeabilization using 0.1% Triton-X100. After blocking with 0.5% bovine serum albumin (BSA)–PBS, cells were incubated with HRP-conjugated anti-mouse IgG, and the luminescent signal was detected using the HRP substrate (SuperSignal West Pico PLUS; Thermo Fisher Scientific). The luminescent signal was measured using either the Luminoskan (Thermo Fisher Scientific), Varioskan Flash microplate reader (Thermo Fisher Scientific), or the EnSpire Alpha plate reader (Perkin Elmer).
Quantitative real-time PCR
Total RNA was extracted from cells using TRIzol (Thermo Fisher Scientific) according to the manufacturer’s protocols. An amount of 500 ng of total RNA was then used for the reverse transcription reaction using ReverTra Ace qPCR RT Master Mix. Quantitative RT-PCR was performed in the LightCycler 480 System (Roche Diagnostics), and the gene expression was examined by SYBR Advantage qPCR Premix. The relative quantity of the target gene mRNA was normalized using human GAPDH as the internal control. PCR amplification was performed in two steps, and the reaction protocol included preincubation at 95°C for 3 min; then the amplification of 40 cycles was set for 5 s at 95°C and 30 s at 60°C, and the melting curve of 5 min at 95°C, 60 s at 60°C, and 97°C. The sequences of primers used for quantitative RT-PCR are listed in Table 1.
Halide-sensitive YFP quenching assay
∆F508-CFTR function assay by halide-sensitive YFP fluorescence quenching was performed as described (Okiyoneda et al., 2018; Veit et al., 2014). Briefly, CFBE cells expressing both inducible ∆F508-CFTR-3HA and halide sensor YFP-F46L/H148Q/I152L were seeded onto black 96-well microplates and transfected with siRNA (50 nM each). The CFTR expression was induced by 1 µg/ml Dox treatment for 4 days. Cell surface expression of low temperature-rescued ∆F508-CFTR was induced for 2 days at 26°C, followed by a 1-h incubation at 37°C before analysis. At the time of assay, cells were washed four times with 400 μl of phosphate-buffered saline (PBS)-chloride (140 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4, 1.1 mM MgCl2, 0.7 mM CaCl2, and 5 mM glucose) and incubated with PBS-chloride (50 μl per well), followed by well wise-injection of an activator solution (50 μl per well) containing 20 µM forskolin (FUJIFILM Wako Pure Chemical Corporation), 0.5 mM 3-isobutyl-1-methyl-xanthine (IBMX; FUJIFILM Wako Pure Chemical Corporation), 0.5 mM 8-(4-chlorophenylthio)-adenosine-3′,5′-cyclic monophosphate (CPT-cAMP; Santa Cruz Biotechnology), and 0.1 mM genistein (FUJIFILM Wako Pure Chemical Corporation). The cells were then incubated for 57 s. The fluorescence was continuously recorded (200 ms per point) for 3 s (baseline) and for 32 s after the rapid addition of 100 μl PBS-iodide, where NaCl was replaced with NaI. The fluorescence measurements were carried out using a VICTOR Nivo multimode microplate reader (Perkin Elmer) with a dual syringe pump (excitation/emission 500/535 nm). After normalizing the YFP signals before PBS-iodide injection, the I− influx rate was calculated by curve-fitting the YFP fluorescence decay using GraphPad Prism 8 software (GraphPad Software).
Western blotting
Cells were solubilized in a RIPA buffer supplemented with 1 mM PMSF (FUJIFILM Wako Pure Chemical Corporation), 5 µg/ml leupeptin (FUJIFILM Wako Pure Chemical Corporation), and 5 µg/ml pepstatin A (Peptide Institute, Inc.). The cell lysates were analyzed by a Western blot as previously described (Okiyoneda et al., 2010). Briefly, an equal amount of proteins from cell lysates underwent SDS-PAGE, and the gels were transferred onto nitrocellulose membranes using the Mini Trans-blot cell system (Bio-Rad). Following this, the membranes were blocked with 5% skim milk and incubated with primary and HRP-conjugated secondary antibodies. Subsequently, the membranes underwent three washes with 0.1% Tween-PBS. Western blots were visualized using a SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific), ImmunoStar Zeta (FUJIFILM Wako Pure Chemical Corporation), or ImmunoStar LD (FUJIFILM Wako Pure Chemical Corporation) and analyzed by LAS 4000 mini (Cytiva Life Sciences) or FUSION Chemiluminescence Imaging System (Vilber Bio Imaging). In some cases, the staining of Ponceau S was used as a loading control. Densitometric analysis for Western blot quantification was performed using ImageQuant (LAS 4000 mini software; Cytiva) or Evolution-Capt (Fusion software; Vilber Bio Imaging).
CFTR ubiquitination measurement by Western blotting
CFTR ubiquitination was measured as previously described (Okiyoneda et al., 2010, 2018). Briefly, CFBE Teton HBH-∆F508-CFTR-3HA cells, 293MSR, and RNF5/185 DKO cells stably expressing HBH-∆F508-CFTR-3HA were lysed in RIPA buffer containing 5 µg/ml Leupeptin, 5 µg/ml Pepstatin A, 1 mM PMSF, 10 µM MG-132, and 5 mM N-ethylmaleimide (NEM) after 10 µM MG-132 treatment at 37°C for 1 h (CFBE) or 3 h (293MSR, RNF5/185 DKO). The CFTR expression was induced by 1 µg/ml Dox treatment for 4 days. Immature HBH-∆F508-CFTR was purified using Neutravidin agarose (Thermo Fisher Scientific) under denaturing conditions and analyzed by Western blotting with anti-Ub (P4D1) or anti-K48 (Apu2) and anti-HA antibodies.
Ub ELISA
CFTR ubiquitination levels in CFBE cells, 293MSR, and RNF5/185 DKO cells were performed as previously described (Kamada et al., 2019; Okiyoneda et al., 2018). Briefly, cells were lysed in RIPA buffer supplemented with 5 µg/ml Leupeptin, 5 µg/ml Pepstatin A, 1 mM PMSF, 10 µM MG-132, and 5 mM NEM after 10 µM MG-132 treatment at 37°C for 1 h (CFBE) or 3 h (293MSR, RNF5/185 DKO). The HBH-∆F508-CFTR in the cell lysate was immobilized on NA-coated 96 well-white plates and denatured in 8 M urea at room temperature for 5 min. After the denaturation, the plate was washed five times with 2 M urea in RIPA buffer. Following four washes with 0.1% NP-40-PBS, the plate was blocked with 0.1% BSA. CFTR ubiquitination was detected using anti-K48 Ub (Apu2; Merck Millipore) and anti-K63 Ub (Apu3; Merck Millipore) antibodies. The ubiquitination levels were quantified with an HRP-conjugated secondary antibody. Linkage-specific CFTR ubiquitination levels were normalized for the CFTR level quantified using the anti-HA antibody (16B12; BioLegend).
Pull-down experiments
To detect the interaction of HBH-∆F508-CFTR-3HA and Myc-HERC3 and/or FLAG-UBQLN2, BHK cells stably expressing HBH-∆F508-CFTR-3HA were transfected with Myc-HERC3 and/or FLAG-UBQLN2. Two days after transfection, the cells were solubilized in mild lysis buffer (150 mM NaCl, 20 mM Tris, 0.1% NP-40, pH 8.0) supplemented with 1 mM PMSF, 5 mg/ml leupeptin, and pepstatin. The cell lysates were then incubated with NA agarose (Thermo Fisher Scientific) for 2 h at 4°C. After being washed four times with mild lysis buffer, the complex was eluted in urea elution buffer (8 M urea, 2% SDS, 3 mM biotin) at room temperature for 30 min and analyzed by Western blotting. In Fig. 6 B and Fig. 7 G, cells were treated with 10 µM MG-132 for 3 h before cell lysis.
HiBiT and Nluc degradation assay
For the HiBiT degradation assay, subconfluent 293MSR and RNF5/185 DKO cells in six-well plates were transfected with siRNA (50 nM each). After 1 day of culture, the cells were trypsinized, reseeded in six-well plates, and cultured for another day. Then, the cells were transfected with different HiBiT fusion constructs, including CFTR variants-HiBiT (CT), TCRα-HiBiT, Insig-1-HiBiT, or ∆Y490-ABCB1-HiBiT, along with cytosolic LgBiT (pBiT1.1-N [TK/LgBiT], Promega). The following day, the cells were trypsinized again and seeded in Nunc MicroWell 96-Well Nunclon Delta-Treated Flat-Bottom Microplates (Thermo Fisher Scientific) and cultured for 18–24 h.
HiBiT retrotranslocation assay
Subconfluent cells were transfected with ∆F508-CFTR-HiBiT (Ex) and cytosolic LgBiT (pBiT1.1-N [TK/LgBiT]; Promega). The next day, cells were trypsinized and seeded in 96-well plates (2 × 104 cells/well) and cultured for 18–24 h. Cells were washed with 100 μl of the CO2-independent medium. Then, 50 μl of 0.5× Nano-Glo Endurazine (Promega) in the CO2-independent medium was added to each well. The cells were incubated for 2.5 h at 37°C and 5% CO2. To detect the retrotranslocated ∆F508-CFTR signal, 10 μl of 60 µM MG-132 (final 10 µM) with or without 600 µg/ml CHX (final 100 µg/ml) in the CO2-independent medium was added to each well. Luminescence was continuously measured at 5-min intervals for 3 h using a Luminoskan plate reader (Thermo Fisher Scientific). The amount of retrotranslocated CFTR was calculated by subtracting the luminescence signal before MG-132 treatment from the signal after MG-132 treatment. The retrotranslocation rate was then calculated by linear fitting the increased retrotranslocation during MG-132 treatment. In the Trikafta experiments, cells were incubated with Trikafta (consisting of 3 µM VX-661, 3 µM VX-445, and 1 µM VX-770) for 24 h at 37°C. Trikafta treatment was also maintained during Endurazine loading and the MG-132 chase phase when luminescent measurements were conducted.
HiBiT ER disappearance assay
Subconfluent cells were transfected with ∆F508-CFTR-HiBiT (Ex) and ER luminal LgBiT (CNXss-LgBiT-KDEL). The CNXss-LgBiT-KDEL construct contains a signal sequence of calnexin (CNX) fused with LgBiT at the N-terminus and an ER retention signal (KDEL) fused with LgBiT at the C-terminus. The next day, the transfected cells were trypsinized and seeded in 96-well plates (2 × 104 cells/well) and cultured for 18–24 h. After washing cells with 100 μl of the CO2-independent medium, 50 μl of 0.1× Nano-Glo Endurazine (Promega) in the CO2-independent medium was added to each well. The cells were then incubated for 2.5 h at 37°C, 5% CO2. To measure the ER disappearance of CFTR-HiBiT(Ex), 10 μl of 600 µg/ml CHX in the CO2-independent medium was added to each well (final 100 µg/ml). Luminescence was continuously measured at 5-min intervals for 3 h using a Luminoskan plate reader (Thermo Fisher Scientific). The luminescence signal (Lum) of CHX-treated cells (LumCHX) was normalized to the signal of untreated cells (Lumuntreat) to calculate the decrease in luminescence signal due to CHX treatment as Eq. 1 like the HiBiT and Nluc degradation assay. The ERAD rate was calculated by fitting the luminescence decay with a one-phase exponential decay function using GraphPad Prism 8.
ELISA to detect CFTR-UBQLN2 interaction
293MSR cells cotransfected with HBH-∆F508-CFTR-3HA and FLAG-UBQLN2 were treated with 10 µM MG-132 for 1 h at 37°C and incubated with 0.1% PFA in PBS (+) for 15 min for crosslinking nearby proteins. After quenching by adding 125 mM glycine, cells were washed with PBS (+) twice and lysed in RIPA buffer containing 5 µg/ml leupeptin, 5 µg/ml pepstatin A, and 1 mM PMSF. HBH-∆F508-CFTR-3HA in cell lysate was immobilized on an NA-coated 96-plate at 4°C for 2 h. After five times washing with 0.1% NP-40 in PBS (−) and blocking with 0.5% BSA in 0.1% NP-40 PBS (−), HBH-∆F508-CFTR and FLAG-UBQLN2 were probed with anti-HA (16B12) and anti-FLAG (1E6) antibodies, respectively, and quantified with an HRP-conjugated secondary antibody. After incubating with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific), the luminescent signals were measured using a Variokan plate reader (Thermo Fisher Scientific). The specific signals of FLAG-UBQLN2 and HBH-∆F508-CFTR-3HA were quantified by subtracting the background signals obtained from the lysates of cells transfected with FLAG-UBQLN2 alone, without HBH-∆F508-CFTR-3HA. FLAG-UBQLN2 binding on the HBH-∆F508-CFTR-3HA was normalized for the CFTR level quantified by an anti-HA antibody (16B12).
Subcellular fractionation
DSP crosslink pull-down assay
293MSR WT and RNF5/185 DKO cells stably expressing HBH-∆F508-CFTR-3HA were transfected with siRNA (50 nM each) and treated with 10 µM MG-132 for 1 h at 37°C. After washing with PBS(+), cells were incubated with 0.1 mM dithiobis[succinimidyl propionate] (DSP; Thermo Fisher Scientific) in PBS(+) for 15 min at room temperature for cross-linking. After washing with PBS(+) and 0.5% BSA in PBS(+) several times, cells were solubilized in mild lysis buffer (150 mM NaCl, 20 mM Tris, 0.1% NP-40, pH 8.0, supplemented with 1 mM PMSF, 5 mg/ml leupeptin and pepstatin). Cell lysates were incubated with Neutravidin agarose (Thermo Fisher Scientific) for 2 h at 4°C. After being washed five times with mild lysis buffer, the complex was eluted in urea elution buffer (8 M urea, 2% SDS, 3 mM biotin) at 30°C for 30 min and analyzed by Western blotting.
Wheat cell-free protein synthesis
The recombinant FLAG-HERC3 and a series of biotinylated CFTRs were synthesized using a wheat cell-free synthesis system (CellFree Sciences), following the previously described protocol (Takahashi et al., 2016) with slight modifications. In some instances of biotinylated CFTR synthesis, 10 mg/ml (final concentration) of asolectin liposomes, prepared as previously reported (Takeda et al., 2015), were introduced into the reaction. To assess the expression of each recombinant protein, 5 μl of the crude translation mixtures were subjected to SDS-PAGE, followed by Western blot analysis using anti-FLAG antibody (M2; Sigma-Aldrich) and HRP-conjugated streptavidin (Abcam), respectively.
AlphaScreen
The binding interactions between FLAG-HERC3 and biotinylated CFTRs were evaluated using AlphaScreen technology (PerkinElmer). 1 µl of each crude translation product containing biotinylated CFTR or FLAG-HERC3 was combined in a 384-well OptiPlate (PerkinElmer) with a total volume of 15 μl of binding reaction buffer (50 mM Tris-HCl, pH 7.5, 1 mg/ml bovine serum albumin, 0.01% Tween 20, 100 mM NaCl). Following 1 h of incubation at 26°C, 10 μl of binding reaction buffer containing 0.3 ng/μl Anti-FLAG antibody (1E6, FUJIFILM Wako Pure Chemical Corporation), 0.08 μl streptavidin-coated donor beads, and 0.08 μl protein A-conjugated acceptor beads were added to each well of the OptiPlate. The plate was subsequently incubated at 26°C for an additional hour. Luminescence was detected using the EnVision multi-plate reader (PerkinElmer) and analyzed with the AlphaScreen detection program.
Statistical analysis
For quantification, data from at least three independent experiments were used where the data are expressed as means ± standard deviation (SD). Statistical significance was assessed from at least three biological replicates (n) by one-way or two-way ANOVA or a two-tailed Student’s t test, as specified in figure legends using GraphPad Prism 8. A P value <0.05 was defined as statistically significant. Data distribution was assumed to be normal, but this was not formally tested.
Online supplemental material
Fig. S1 provides a schematic representation of the gRNA targeting RNF5 and RNF185 for the creation of RNF5/185 DKO cells. Fig. S2 displays a representative luminescence trace from the ∆F508-CFTR-HiBiT(Ex) retrotranslocation assay. Fig. S3 illustrates the correlation of the effects of HERC3 KD and RNF5/185 DKO on ∆F508-CFTR ERAD, retrotranslocation, ubiquitination, and CFTR-UBQLN2 interaction. Fig. S4 depicts the effects of UBQLN single KD on ∆F508-CFTR ERAD, triple KD on ∆F508-CFTR retrotranslocation, and the establishment of TCRα-HiBiT and Insig-1-HiBiT ERAD assays. Fig. S5 shows the effects of HERC3 and/or RNF5/185 ablation on the ERAD of ∆F508-NBD1, N1303K-CFTR, and ∆Y490-ABCB1-∆M1, along with the correlation of the effects of HERC3 KD and RNF5/185 DKO on ERAD of ∆F508-CFTR and CFTR fragments.
Data availability
The raw data required to reproduce these findings are available from the corresponding authors upon reasonable request. All data generated and analyzed during this study are included in this manuscript and the supplemental material. This paper does not report the original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank R. Kopito (Stanford University, Stanford, CA, USA) and Y. Ye (National Institutes of Health, Bethesda, MD, USA; TCRα-HA), M. Gottesman (National Institutes of Health, Bethesda, MD, USA; Addgene #10957 MDRwt /ABCB1), P. Howley (Harvard Medical School, Boston, MA, USA; Addgene #8661 FLAG-hPLIC-2/UBQLN2, Addgene #8663 FLAG-hPLIC-1/UBQLN1), H. Kai (Kumamoto University, Kumamoto, Japan; D18G-TTR), D. Root (Broad Institute of MIT and Harvard, Cambridge, MA, USA; Addgene #41394 pLIX402, #25890 pLX304), and F. Zhang (Broad Institute of MIT and Harvard, Cambridge, MA, USA; Addgene #62988 pSpCas9(BB)-2A-Puro (PX459) V2.0) for providing plasmids, D. Gruenert (University of California San Francisco, San Francisco, CA, USA) for the CFBE41o- cells, H. Takeda (Ehime University, Matsuyama, Japan) for asolectin liposomes, and C. Furukawa (Ehime University, Matsuyama, Japan) for technical support.
This work was supported by JSPS/MEXT KAKENHI (21H00294, 22H02576 to T. Okiyoneda, 21H00287 to H. Takahashi), the Japan Agency for Medical Research and Development (AMED, JP23fk0210086h0003 to H. Takahashi), a grant for research on Japan Program for Infectious Diseases Research and Infrastructure from AMED (21fm0208101j0005, 22fm0208101j0006 to H. Takahashi), Takeda Science Foundation (to T. Okiyoneda), and Individual Special Research Subsidy with grants from Kwansei Gakuin University (to T. Okiyoneda).
Author contributions: Y. Kamada: Investigation, Methodology, and Writing—original draft; Y. Ohnishi: Investigation, Methodology, C. Nakashima: Investigation, Methodology, A. Fujii: Investigation, Methodology, M. Terakawa: Investigation and Methodology; I. Hamano: Investigation and Methodology, U. Nakayamada: Investigation and Methodology, S. Kato: Investigation and Methodology, N. Hirata: Investigation and Methodology, H. Tateishi: Investigation and Methodology, R. Fukuda: Investigation and Project administration, H. Takahashi: Investigation, Methodology, and Resources, G.L. Lukacs: Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, and Writing—review & editing, T. Okiyoneda: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing—original draft, and Writing—review & editing.

