


The essential Golgi protein Sly1 is a member of the Sec1/mammalian Unc-18 (SM) family of SNARE chaperones. Sly1 was originally identified through remarkable gain-of-function alleles that bypass requirements for diverse vesicle tethering factors. Employing genetic analyses and chemically defined reconstitutions of ER–Golgi fusion, we discovered that a loop conserved among Sly1 family members is not only autoinhibitory but also acts as a positive effector. An amphipathic lipid packing sensor (ALPS)-like helix within the loop directly binds high-curvature membranes. Membrane binding is required for relief of Sly1 autoinhibition and also allows Sly1 to directly tether incoming vesicles to the Qa-SNARE on the target organelle. The SLY1-20 mutation bypasses requirements for diverse tethering factors but loses this ability if the tethering activity is impaired. We propose that long-range tethers, including Golgins and multisubunit tethering complexes, hand off vesicles to Sly1, which then tethers at close range to initiate trans-SNARE complex assembly and fusion in the early secretory pathway.
Introduction
Traffic through the secretory and endocytic systems depends on accurate and timely targeting of transport vesicles to acceptor organelles. The terminal stage of targeting is membrane fusion, catalyzed by the formation of trans-SNARE complexes that zipper together, doing the mechanical work of moving two membranes into proximity and driving their merger. Although SNAREs alone can drive fusion and confer some compartmental selectivity, spontaneous SNARE assembly is slow and error prone. Consequently, an array of tethering factors and SNARE chaperones are indispensable in vivo (Baker and Hughson, 2016; Gillingham and Munro, 2019). For example, every SNARE-mediated fusion event that has been closely examined requires a cofactor of the Sec1/mammalian Unc-18 (SM) family.
For decades, the mechanisms of SM protein function were enigmatic (Carr and Rizo, 2010; Rizo and Südhof, 2012; Südhof and Rothman, 2009) but biochemical work, structural studies, and single-molecule force spectroscopy suggest that SM proteins are assembly chaperones for trans-SNARE complex formation, and that SMs act, at least in part, by templating the initial SNARE zippering reaction (Baker et al., 2015; Jiao et al., 2018) and by protecting appropriately formed prefusion complexes from kinetic proofreading by the SNARE disassembly proteins Sec17/α-SNAP and Sec18/NSF (Lobingier et al., 2014; Ma et al., 2013; Schwartz et al., 2017; Xu et al., 2010). There are four SM subfamilies. Saccharomyces cerevisiae has one representative of each. Vps33, the first SM identified genetically, controls fusion at late endosomes and lysosomes (Banta et al., 1990; Patterson, 1932; Sevrioukov et al., 1999). Vps45 controls fusion at endosomal compartments (Cowles et al., 1994; Piper et al., 1994). Sec1 and its orthologs Unc-18/Munc-18 control exocytosis (Grote et al., 2000; Novick et al., 1979; Verhage et al., 2000; Wu et al., 1998). Finally, fusion at the Golgi and endoplasmic reticulum (ER) are controlled by Sly1 (Li et al., 2005; Lupashin et al., 1996; Ossig et al., 1991; Peng and Gallwitz, 2002; Søgaard et al., 1994). The human Sly1 ortholog is SCFD1. SCFD1 variants are risk factors for amyotrophic lateral sclerosis (ALS; Cauchi, 2024).
The genetics of yeast SLY1 are intricate and revealing. Ypt1 (yeast Rab1) is an essential regulator of docking and fusion at the Golgi. SLY1 was originally identified through a dominant allele, SLY1-20, that suppresses the lethality of Ypt1 deficiency (Dascher et al., 1991; Ossig et al., 1991, 1995). Work by several groups showed that SLY1-20 suppresses deficiency not only of Ypt1 but numerous other factors that promote ER and Golgi traffic. These include the Dsl complex (Dsl1 was identified through a genetic interaction of dsl1 and SLY1-20; Reilly et al., 2001; VanRheenen et al., 2001), the COG complex (cog2, cog3; VanRheenen et al., 1998, 1999), the TRAPP complexes (bet3-1; Sacher et al., 1998), the Golgin coiled-coil tether Uso1 (yeast p115; Sapperstein et al., 1996); Ypt6 (yeast Rab6) and its nucleotide exchange complex (ric1; Bensen et al., 2001; Li et al., 2007), and the Ypt6 effector complex GARP (vps53; VanRheenen et al., 2001). In addition, SLY1-20 suppresses partial deficiencies of Golgi SNAREs (sec22; Ossig et al., 1991), COPI (sec21; Ossig et al., 1991), and the COPI Arf GAP Glo3 (VanRheenen et al., 2001).
SLY1-20 and the similar mutant SLY1-15 encode missense substitutions at adjacent positions within a loop insertion, evolutionarily conserved among Sly1 subfamily members but absent from the other three SM subfamilies (Dascher et al., 1991; Li et al., 2007). On this basis, it was hypothesized that the Sly1 loop is autoinhibitory, and that SLY1-20 and related alleles gain function by freeing the loop from the closed, autoinhibitory state (Bracher and Weissenhorn, 2002; Li et al., 2007). This idea was supported by the discovery that the Sly1 loop occludes a conserved site which, in Vps33, binds R/v-SNAREs with high affinity (Baker et al., 2015).
The mechanism by which the Sly1 loop’s putative autoinhibitory activity is released to promote SNARE complex formation is unknown but was suggested to require Ypt1, the yeast Rab1 (Bracher and Weissenhorn, 2002; Li et al., 2007). Here, we show that the loop’s inhibitory activity is released when an amphipathic helix within the loop interacts directly with an incoming vesicle membrane’s lipid bilayer. Moreover, in its open position, the loop allows Sly1 to directly tether incoming vesicles. We propose that the Sly1 N-lobe is anchored to the SNARE Sed5 on the target organelle while the Sly1 regulatory loop binds the incoming vesicle’s lipid bilayer. We further propose that the loop’s membrane binding steers Sly1 into an orientation optimal for productive R/v-SNARE association and trans-SNARE complex assembly. This schema explains how SLY1-20 bypasses the otherwise essential functions of so many different Golgi tethering factors and suggests that the Sly1 regulatory loop links Sly1 activation, identification and capture of transport vesicles addressed to organelles of the early secretory pathway, and productive fusion complex assembly.
Results
New SLY1 alleles define an autoregulatory loop
We thought it likely that early screens that identified SLY1 bypass alleles were not saturated, and that a more focused screen might yield additional informative alleles. Uso1 is a Golgin-class tether that is a direct effector of Ypt1/Rab1. Loss of Uso1 is lethal, and this lethality is suppressed by SLY1-20 (Ballew et al., 2005; Sapperstein et al., 1996). We designed a selection for dominant SLY1* alleles that could suppress the loss of USO1 (Fig. 1 A). (In this report, sets of SLY1 alleles and their protein products are referred to collectively as SLY1* and Sly1*.) Our screen retrieved many SLY1* alleles, most encoding multiple amino acid substitutions. From these, individual missense substitutions were reintroduced into wild-type SLY1 and tested for their ability to suppress Uso1 or Ypt1 deficiencies (Fig. 1 B and Table S1). Importantly, our screen independently retrieved the original SLY1-20 and SLY1-15 alleles. We also identified suppressing substitutions at nearby sites on helix α20 and on the short segment linking helices α20 and α21. Additionally, we identified suppressing substitutions at the base of the Sly1-specific loop and at positions cradling the base of the loop, but non-adjacent within the linear polypeptide sequence. Among these was T559I. Remarkably, a genome-scale survey for gene pairs with spontaneous suppressing interactions identified a substitution at the same position, T559K, that dominantly suppressed deficiencies of both the GARP subunit Vps53 and the Arf GAP Glo3 (van Leeuwen et al., 2016). Most gain-of-function single substitutions that we tested suppressed ypt1–3 but, in contrast to the multisite mutants obtained in the initial selection for uso1∆ bypass, were unable to suppress uso1∆ (Fig. S1 and Table S1). Thus, strong Sly1 gain-of-function phenotypes can arise either through individual driver substitutions or combined effects of multiple weak driver substitutions.

New gain-of-function SLY1* alleles. (A) Selection used in this study. A library of SLY1* alleles was constructed by mutagenic PCR and cloned into a single-copy plasmid. The library was then transformed into a SLY1 uso1∆ strain, with USO1 provided on a balancer plasmid bearing the counterselectable URA3 marker. Ejection of pUSO1 was forced by 5-fluoroorotic acid (5-FOA). This strategy positively selects viable cells carrying dominant mutant SLY1* alleles that bypass the otherwise essential USO1 requirement. (B) Locations within Sly1 (PDB ID 1MQS) of single missense substitutions that suppress requirements for Ypt1, or for both Ypt1 and Uso1. The loop is indicated in purple, with the dashed line denoting the portion of the loop not resolved in the crystal structure. Yellow shading indicates the domain 3a helical hairpin which, by analogy to Vps33 and Munc18-1, is hypothesized to scaffold assembly of Qa- and R-SNARE trans-complexes.
New gain-of-function SLY1* alleles. (A) Selection used in this study. A library of SLY1* alleles was constructed by mutagenic PCR and cloned into a single-copy plasmid. The library was then transformed into a SLY1 uso1∆ strain, with USO1 provided on a balancer plasmid bearing the counterselectable URA3 marker. Ejection of pUSO1 was forced by 5-fluoroorotic acid (5-FOA). This strategy positively selects viable cells carrying dominant mutant SLY1* alleles that bypass the otherwise essential USO1 requirement. (B) Locations within Sly1 (PDB ID 1MQS) of single missense substitutions that suppress requirements for Ypt1, or for both Ypt1 and Uso1. The loop is indicated in purple, with the dashed line denoting the portion of the loop not resolved in the crystal structure. Yellow shading indicates the domain 3a helical hairpin which, by analogy to Vps33 and Munc18-1, is hypothesized to scaffold assembly of Qa- and R-SNARE trans-complexes.
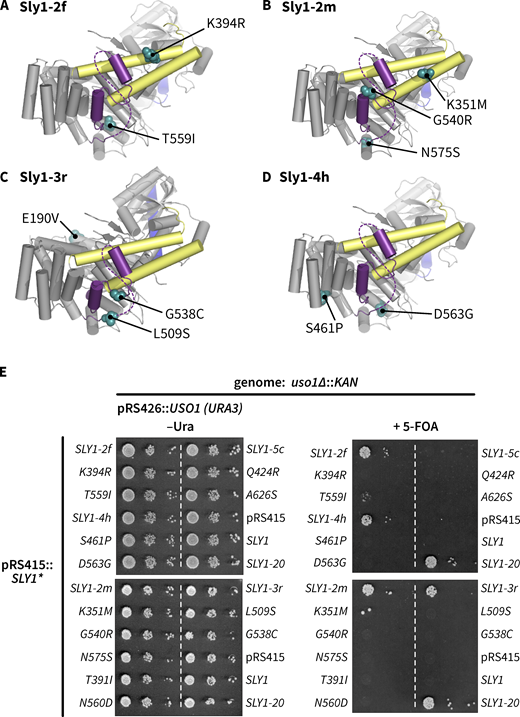
Some SLY1 alleles require multiple substitutions to suppress the lethal uso1∆ phenotype. (A–D) Locations of amino acid substitutions in four representative SLY1 alleles recovered in our screen. (E) Growth phenotypes show that most single substitutions are unable to suppress the loss of Uso1. Many of the same single mutants suppress the loss of Ypt1 (see Table S1). The multisite allele SLY1-5c, although retrieved in our primary screen, was unable to suppress the uso1∆ allele in secondary screening.
Some SLY1 alleles require multiple substitutions to suppress the lethal uso1∆ phenotype. (A–D) Locations of amino acid substitutions in four representative SLY1 alleles recovered in our screen. (E) Growth phenotypes show that most single substitutions are unable to suppress the loss of Uso1. Many of the same single mutants suppress the loss of Ypt1 (see Table S1). The multisite allele SLY1-5c, although retrieved in our primary screen, was unable to suppress the uso1∆ allele in secondary screening.
As first noted by Baker et al. (2015), Sly1 helices α20 and α21 sit atop two conserved regions that in Vps33 are of special importance for SNARE binding: domain 3a, which serves as a scaffold to nucleate the parallel, in-register assembly of the Qa- and R-SNAREs, and an aromatic pocket that serves as a high-affinity anchor point for the R-SNARE juxtamembrane linker. Baker et al. (2015) proposed that when closed, the Sly1 loop blocks R-SNARE binding to Sly1. Dominant suppressor mutants obtained in our screen and data presented below support and substantially extend that model.
Sly1 suppressor mutants are hyperactive in a minimal fusion system
In vivo genetic tests and crude in vitro transport systems (Baker et al., 1988; Ballew et al., 2005; Ruohola et al., 1988) cannot tell us whether Sly1* mutants must interact with additional proteins beyond the core SNARE fusion machinery to manifest gain of function. To overcome this limitation, we developed a chemically defined reconstituted proteoliposome (RPL) system to monitor fusion driven by ER–Golgi SNAREs (Fig. 2, A and B). This system, adapted from an assay developed to study homotypic vacuole fusion (Zucchi and Zick, 2011), employs two orthogonal pairs of Förster resonance energy transfer (FRET) probes to simultaneously monitor both lipid and content mixing in small (20 µl) reaction volumes. We mainly show content mixing because that is the reaction endpoint, but combined lipid and content mixing signals permit us to detect hemifusion intermediates or fusion accompanied by lysis.

Setup and characterization of the in vitro fusion system. (A) Reporter systems for lipid and content mixing. RPLs (reconstituted proteoliposomes) are prepared with encapsulated content mixing FRET pair, and with the membranes doped with an orthogonal FRET pair. (B) SNARE topology of the RPLs used in this study, and soluble components added to stimulate fusion. (C–E) Characterization of the system using content mixing readout. (C) Requirement for Sly1. Reactions were set up with Q- and R-SNARE RPLs, and 3% PEG. Fusion activity was monitored for 5 min, and then Sly1 was added at time = 0 (arrows) to the indicated final concentrations. Note that fusion activity is saturated at 100 nM (Sly1). (D) Requirement for tethering. Reactions were set up with Q- and R-SNARE RPLs, with the indicated final concentrations of PEG. Fusion activity was monitored for 5 min, and then Sly1 was added to a final concentration of 250 nM. Note that at 6% and 7% PEG, some Sly1-independent fusion occurs prior to Sly1 addition. (E) Effects of the SNARE disassembly machinery. Reactions were set up with Q- and R-SNARE RPLs, and with or without PEG, Sec17, Sec18, ATP, and Sly1, as indicated. Fusion was initiated by adding Sly1. For C–E, points show mean ± SEM of three independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model.
Setup and characterization of the in vitro fusion system. (A) Reporter systems for lipid and content mixing. RPLs (reconstituted proteoliposomes) are prepared with encapsulated content mixing FRET pair, and with the membranes doped with an orthogonal FRET pair. (B) SNARE topology of the RPLs used in this study, and soluble components added to stimulate fusion. (C–E) Characterization of the system using content mixing readout. (C) Requirement for Sly1. Reactions were set up with Q- and R-SNARE RPLs, and 3% PEG. Fusion activity was monitored for 5 min, and then Sly1 was added at time = 0 (arrows) to the indicated final concentrations. Note that fusion activity is saturated at 100 nM (Sly1). (D) Requirement for tethering. Reactions were set up with Q- and R-SNARE RPLs, with the indicated final concentrations of PEG. Fusion activity was monitored for 5 min, and then Sly1 was added to a final concentration of 250 nM. Note that at 6% and 7% PEG, some Sly1-independent fusion occurs prior to Sly1 addition. (E) Effects of the SNARE disassembly machinery. Reactions were set up with Q- and R-SNARE RPLs, and with or without PEG, Sec17, Sec18, ATP, and Sly1, as indicated. Fusion was initiated by adding Sly1. For C–E, points show mean ± SEM of three independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model.
In previous work, various SMs were shown to stimulate SNARE-mediated lipid mixing, but only in the presence of either tethering factors or crowding agents that functionally substitute for tethers (Furukawa and Mima, 2014; Yu et al., 2015). Consistent with these studies, content mixing in heterotypic reactions between RPLs bearing the R-SNARE Sec22, and RPLs bearing the Q-SNAREs Sed5, Bos1, and Bet1, was strongly stimulated only when both Sly1 and a crowding agent (polyethylene glycol 6000; PEG) were provided (Fig. 2, C and D). The cytoplasmic concentration of Sly1 is ∼200 nM (Table S2). In our experiments, the stimulatory effect of Sly1 saturates at 100–200 nM. Two other studies have reported in vitro stimulation of fusion by Sly1, but at concentrations 45× higher than in our standard reactions (Furukawa and Mima, 2014; Jun and Wickner, 2019). Preincubation of RPLs with Mg2+·ATP and the SNARE disassembly chaperones Sec17 and Sec18 (yeast α-SNAP and NSF) resulted in immediate and almost complete fusion upon Sly1 addition (Fig. 2 E).
With this system established, we compared the activity of wild-type Sly1 to three bypass suppressors: Sly1-20 and two of the new alleles identified in our screen. Each was tested in reactions containing 3% or 0% PEG. At 3%, all four Sly1* variants drove fusion with similar efficiency (Fig. 3 A). In marked contrast, at 0% PEG (Fig. 3 B) all three Sly1 suppressor mutants drove fusion more efficiently than the wild type. We also tested the effects of the SNARE chaperones Sec17, Sec18, and Mg2+·ATP (Fig. 3, C and D). The same overall pattern emerged. Together, these results show for the first time that Sly1 gain-of-function mutants are intrinsically hyperactive, requiring only SNAREs (or SNAREs and disassembly chaperones) to stimulate fusion, and not additional cellular factors such as Rabs, tethering factors, or crowding agents. These in vitro results mirror in vivo genetic suppression patterns observed between SLY1-20 and otherwise essential vesicle tethering regulators and effectors.

Gain-of-function Sly1 mutants alleviate the tethering requirement in vitro. (A–D) Reactions were set up as in Fig. 2, with the initial mixture containing R-SNARE and Qabc-SNARE RPLs and, as indicated for each row of panels, 0 or 3% PEG, and in the absence or presence of Sec17, Sec18 (both 100 nM), and ATP (1 mM). After a 5-min incubation, wild-type Sly1 or the indicated mutants were added (arrows) at 0, 25, 100, or 400 nM to initiate fusion. Points show the mean ± SEM from three or more independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model.
Gain-of-function Sly1 mutants alleviate the tethering requirement in vitro. (A–D) Reactions were set up as in Fig. 2, with the initial mixture containing R-SNARE and Qabc-SNARE RPLs and, as indicated for each row of panels, 0 or 3% PEG, and in the absence or presence of Sec17, Sec18 (both 100 nM), and ATP (1 mM). After a 5-min incubation, wild-type Sly1 or the indicated mutants were added (arrows) at 0, 25, 100, or 400 nM to initiate fusion. Points show the mean ± SEM from three or more independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model.
The Sly1 regulatory loop has positive as well as negative functions
If the Sly1 loop is autoinhibitory, we can predict that removal of the entire loop should hyperactivate Sly1 as much as or more than suppressing mutations characterized above. To test this hypothesis, we used the ROSETTA software environment (Leaver-Fay et al., 2011) to design a panel of 12 Sly1 variants in which the loop is replaced by short peptide linkers (Fig. 4 A and Table S3). Surprisingly, all “loopless” sly1 mutants tested in vivo exhibited either recessive lethality or slow growth when wild-type SLY1 was ejected by counterselection with 5-FOA at 30°C. The loopless mutants also exhibited temperature sensitivity and were unable to bypass Ypt1 or Uso1 deficiency. Cells carrying a single copy of sly1-0_2 grew somewhat more robustly compared with other sly1 loop deletion strains. sly1-0_2 was named sly1∆loop and subjected to further scrutiny.
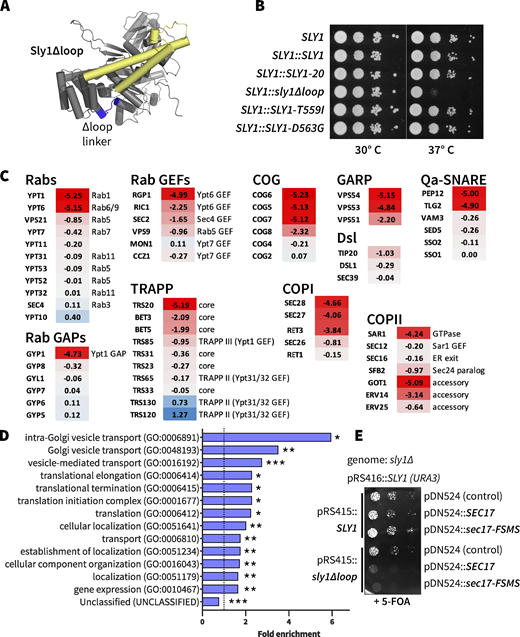
The Sly1 regulatory loop has a positive function in vivo. (A) AlphaFold2 rendering, showing the location of Sly1 loop replacement with engineered linkers (blue). Sequences of the linker insert designs, and growth phenotypes of the corresponding mutants are presented in Table S2. The domain 3a SNARE assembly template is shown in yellow. (B) The sly1∆loop mutant is temperature-sensitive for growth. Dilutions of liquid cultures were spotted as 10× serial dilutions onto YPD agar plates and incubated for 2 days at 30° or 37°C. These are knock-ins at the genomic SLY1 locus, in the Y8205 strain background used for SGA analysis. (C) Selected SGA results. Genes exhibiting synthetic interactions with sly1∆loop are shown. Scores indicate loge synthetic growth defects (red) or intergenic suppression (blue). A score of −4.6 indicates a 100× synthetic growth defect. Complete SGA results are presented in Data S1. (D) Gene Ontology Overrepresentation Test of the sly1∆loop SGA dataset. Genes with loge synthetic defect scores less than or equal to −0.5 were included in the analysis. Bars show all GO-Slim Biological Process categories with statistically significant enrichment scores (*P < 0.05; **P < 10−2; ***P < 10−6). P values were calculated using Fisher’s exact test and adjusted for multiple comparisons (Bonferroni’s correction; count = 732). Additional details are presented in Data S1. (E)SEC17 overproduction is toxic in cells expressing sly1∆loop. sly1∆ mutant cells were maintained with a counterselectable SLY1 balancer plasmid and transformed with single-copy plasmids bearing either SLY1 or sly1∆loop, as well as plasmids carrying SEC17 or sec17-FSMS (Schwartz and Merz, 2009). The balancer plasmid was ejected by plating dilutions on media with 5-FOA and growth was assayed after 2 days of growth at 30°C.
The Sly1 regulatory loop has a positive function in vivo. (A) AlphaFold2 rendering, showing the location of Sly1 loop replacement with engineered linkers (blue). Sequences of the linker insert designs, and growth phenotypes of the corresponding mutants are presented in Table S2. The domain 3a SNARE assembly template is shown in yellow. (B) The sly1∆loop mutant is temperature-sensitive for growth. Dilutions of liquid cultures were spotted as 10× serial dilutions onto YPD agar plates and incubated for 2 days at 30° or 37°C. These are knock-ins at the genomic SLY1 locus, in the Y8205 strain background used for SGA analysis. (C) Selected SGA results. Genes exhibiting synthetic interactions with sly1∆loop are shown. Scores indicate loge synthetic growth defects (red) or intergenic suppression (blue). A score of −4.6 indicates a 100× synthetic growth defect. Complete SGA results are presented in Data S1. (D) Gene Ontology Overrepresentation Test of the sly1∆loop SGA dataset. Genes with loge synthetic defect scores less than or equal to −0.5 were included in the analysis. Bars show all GO-Slim Biological Process categories with statistically significant enrichment scores (*P < 0.05; **P < 10−2; ***P < 10−6). P values were calculated using Fisher’s exact test and adjusted for multiple comparisons (Bonferroni’s correction; count = 732). Additional details are presented in Data S1. (E)SEC17 overproduction is toxic in cells expressing sly1∆loop. sly1∆ mutant cells were maintained with a counterselectable SLY1 balancer plasmid and transformed with single-copy plasmids bearing either SLY1 or sly1∆loop, as well as plasmids carrying SEC17 or sec17-FSMS (Schwartz and Merz, 2009). The balancer plasmid was ejected by plating dilutions on media with 5-FOA and growth was assayed after 2 days of growth at 30°C.
To gain genome-scale insight into the sly1∆loop mutant’s loss of function, we used synthetic genome array (SGA) analysis. SGA measures the synthetic sickness or rescue (suppression) of a query allele versus a genome-scale collection of loss-of-function alleles (Tong and Boone, 2006). In an SGA query strain, we exchanged the genomic SLY1 gene with sly1∆loop. The resulting strain grew normally on rich YPD medium containing 5-FOA at 30°C but slowly compared with strains containing wild-type or hyperactive SLY1* at 37°C (Fig. 4 B). When subjected to SGA analysis, sly1∆loop exhibited synthetic-sick or synthetic-lethal interactions with 10 of the 12 genes previously reported to exhibit positive suppressing interactions with SLY1-20, as well as dozens of additional genes that function in organelle biogenesis and membrane traffic—particularly traffic into and through the cis and medial Golgi, and retrograde traffic from Golgi to ER (Fig. 4 C and Data S1). Gene ontology analysis (Mi et al., 2019; The Gene Ontology Consortium, 2019) verified significant enrichment for these functions (Fig. 4 D). The synthetic sick and synthetic lethal interactions of sly1∆loop are a mirror inversion of suppressing interactions seen with SLY1-20 and similar alleles.
Wild-type SLY1 activity is required for resistance to the toxic effects of SEC17 overproduction because SMs are needed to prevent premature disassembly of trans-SNARE complexes (Lobingier et al., 2014; Ma et al., 2013; Schwartz et al., 2017; Xu et al., 2010). SEC17 overproduction caused a severe growth defect in sly1∆loop cells, consistent with deficient SM function (Fig. 4 E). Overexpression of a Sec17 mutant defective for membrane interaction, Sec17-FSMS (Schwartz et al., 2017; Song et al., 2017; Winter et al., 2009), caused an even more severe growth defect in sly1∆loop mutants. Together, the genetic and functional genomic results show that sly1∆loop is a recessive loss-of-function allele and not, as predicted, a dominant suppressor.
To assess the molecular basis of the sly1∆loop mutant’s defect, we returned to the chemically defined fusion system. As shown in Fig. 5, Sly1∆loop drove slower fusion compared with wild-type Sly1. Moreover, Sly1∆loop was unable to bypass a tethering requirement in vitro (Figs. 5, A and C), consistent with its inability to suppress Ypt1 and Uso1 deficiency in vivo. In dose-response experiments, both Sly1∆loop and wild-type Sly1 exhibited saturating fusion activity at ∼100 nM (compare Sly1∆loop in Fig. 5 to wild-type Sly1 in Fig. 3). Moreover, the Sly1∆loop protein was properly folded as indicated by circular dichroism (Fig. 5 E). We conclude that Sly1∆loop is, on a per-molecule basis, a less efficient promoter of SNARE-mediated fusion compared with the wild-type. Together the data indicate that the Sly1 regulatory loop is not merely autoinhibitory, but also harbors a positive fusion-stimulating activity.

The Sly1 regulatory loop has a positive function in vitro. (A–D) Fusion activity of Sly1∆loop versus wild-type Sly1. Master mixes were assembled as in Fig. 3 and incubated for 5 min at 30°C. Fusion was initiated by adding (arrows) Sly1 or Sly1∆loop at the concentrations indicated in the legend adjacent to panel B. Reactions were run in the absence (A and C) or presence (B and D) of 3% PEG; and in the absence (A and B) or presence (C and D) of Sec17, Sec18 (both 100 nM), and ATP. Points show mean ± SEM from three or more independent experiments; in some cases, the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model. (E) Purified Sly1∆loop protein is folded. Circular dichroism spectra of wild-type Sly1 and Sly1∆loop. The spectra are normalized to account for small differences in molecular mass and concentration. A comparison of lipid and content mixing signals for the experiments in panels B and D is presented in Fig. S3.
The Sly1 regulatory loop has a positive function in vitro. (A–D) Fusion activity of Sly1∆loop versus wild-type Sly1. Master mixes were assembled as in Fig. 3 and incubated for 5 min at 30°C. Fusion was initiated by adding (arrows) Sly1 or Sly1∆loop at the concentrations indicated in the legend adjacent to panel B. Reactions were run in the absence (A and C) or presence (B and D) of 3% PEG; and in the absence (A and B) or presence (C and D) of Sec17, Sec18 (both 100 nM), and ATP. Points show mean ± SEM from three or more independent experiments; in some cases, the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model. (E) Purified Sly1∆loop protein is folded. Circular dichroism spectra of wild-type Sly1 and Sly1∆loop. The spectra are normalized to account for small differences in molecular mass and concentration. A comparison of lipid and content mixing signals for the experiments in panels B and D is presented in Fig. S3.
The loop’s positive function resides within ALPS-like helix α21
The regulatory loop’s most conserved region is helix α21 (Fig. 6, A and B). Interestingly, none of the activating gain-of-function mutations isolated to date map to α21. On closer inspection, we noticed that α21 is amphipathic (Fig. 6 C). We therefore designed a mutant, Sly1-pα21, in which helix α21 is mutated to make it polar rather than amphipathic. Unexpectedly, sly1-pα21 caused recessive lethality (Fig. 6 D)—a phenotype more severe than that of sly1∆loop.

Amphipathic helix α21 is indispensable for normal Sly1 function. (A) CONSURF analysis of evolutionary conservation within the Sly1 loop. Helix α21 is the most highly conserved portion of the loop. Locations of gain-of-function mutations, and hydrophobic residues within the loop are indicated, as are the five substitutions in the Sly1-pα21 mutant. (B) Position of helix α21 within Sly1. Note that no gain-of-function mutations within α21 have been identified. The loop is purple; the domain 3a templating domain is yellow. (C) Helix α21 and residues immediately upstream have the potential to fold into a strongly amphipathic α-helix. The helical wheel renderings comprise the region underlined in black and were produced using HELIQUEST; hydrophobic moment (µH) is indicated. (D) Growth phenotypes of cells carrying sly1-pα21, SLY1-20-pα21, and other alleles were assayed in a sly1∆ strain with a SLY1 balancer plasmid, which is ejected in the presence of 5-FOA. (E–J) RPL fusion with (E–G) Sly1-pα21 and (H–J) the compound mutant Sly1-20-pα21. For reference, fusion is also plotted for Sly1 and Sly1∆loop. Reactions were set up with (E and H) 0% PEG, Sec17 and Sec18 (100 nM each), and ATP (1 mM); (F and I) 3% PEG and no Sec17, Sec18 (100 nM each), or ATP; or (G, J, and F) 0% PEG, Sec17 and Sec18 (100 nM each), and ATP (1 mM). Fusion was initiated at time = 0 by adding Sly1 or its mutants, at the concentrations indicated in the legends at the right side of the figure. Points show mean ± SEM from three or more independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model. Lipid mixing traces for panels G and J are presented in Fig. S4.
Amphipathic helix α21 is indispensable for normal Sly1 function. (A) CONSURF analysis of evolutionary conservation within the Sly1 loop. Helix α21 is the most highly conserved portion of the loop. Locations of gain-of-function mutations, and hydrophobic residues within the loop are indicated, as are the five substitutions in the Sly1-pα21 mutant. (B) Position of helix α21 within Sly1. Note that no gain-of-function mutations within α21 have been identified. The loop is purple; the domain 3a templating domain is yellow. (C) Helix α21 and residues immediately upstream have the potential to fold into a strongly amphipathic α-helix. The helical wheel renderings comprise the region underlined in black and were produced using HELIQUEST; hydrophobic moment (µH) is indicated. (D) Growth phenotypes of cells carrying sly1-pα21, SLY1-20-pα21, and other alleles were assayed in a sly1∆ strain with a SLY1 balancer plasmid, which is ejected in the presence of 5-FOA. (E–J) RPL fusion with (E–G) Sly1-pα21 and (H–J) the compound mutant Sly1-20-pα21. For reference, fusion is also plotted for Sly1 and Sly1∆loop. Reactions were set up with (E and H) 0% PEG, Sec17 and Sec18 (100 nM each), and ATP (1 mM); (F and I) 3% PEG and no Sec17, Sec18 (100 nM each), or ATP; or (G, J, and F) 0% PEG, Sec17 and Sec18 (100 nM each), and ATP (1 mM). Fusion was initiated at time = 0 by adding Sly1 or its mutants, at the concentrations indicated in the legends at the right side of the figure. Points show mean ± SEM from three or more independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model. Lipid mixing traces for panels G and J are presented in Fig. S4.
Amphipathic helices operate as membrane recognition modules across a wide range of proteins, particularly within the early secretory pathway (Bigay and Antonny, 2012). In silico analyses using PMIpred (Fig. S2) predict that α21 binds membranes more strongly than the well-characterized amphipathic lipid packing sensor (ALPS) domain in GMAP-210, while the pα21 mutant should neither bind membranes nor sense curvature (Magdeleine et al., 2016; van Hilten et al., 2023a, 2023b, Preprint).

In silico estimation of membrane binding by Sly1 helix α21 and its polar mutant pα21. (A and B) PMIpred results for helices α21 (A) and pα21 (B) were generated by the server at https://pmipred.fkt.physik.tu-dortmund.de/curvature-sensing/ (van Hilten et al., 2023a, 2023b, Preprint). Similar results for α21 and pα21 were obtained when the model was initialized with either negatively charged or neutral membranes. These estimated binding parameters for α21 predict stronger binding than for the well-characterized GMAP-210 ALPS domain, and similar binding to the engineered high-affinity ALPScond mutant (Magdeleine et al., 2016; van Hilten et al., 2023a).
In silico estimation of membrane binding by Sly1 helix α21 and its polar mutant pα21. (A and B) PMIpred results for helices α21 (A) and pα21 (B) were generated by the server at https://pmipred.fkt.physik.tu-dortmund.de/curvature-sensing/ (van Hilten et al., 2023a, 2023b, Preprint). Similar results for α21 and pα21 were obtained when the model was initialized with either negatively charged or neutral membranes. These estimated binding parameters for α21 predict stronger binding than for the well-characterized GMAP-210 ALPS domain, and similar binding to the engineered high-affinity ALPScond mutant (Magdeleine et al., 2016; van Hilten et al., 2023a).
These observations suggested a working model: helix α21 probes for the presence of an incoming vesicle and binds to the vesicle membrane, holding the loop open and exposing the R-SNARE binding site. This allows Sly1 to bind the R-SNARE and initiate the assembly of trans-SNARE complexes. In this model, Sly1-pα21 is nonfunctional because α21 cannot recognize incoming vesicle membranes and the loop is trapped in its closed, autoinhibited state. To test this hypothesis, we engineered a compound mutant, SLY1-20-pα21. This mutant has both the activating Sly1-20 mutation (E532K) in α20 and the five polar substitutions in α21 (Fig. 6 C). Remarkably, SLY1-20-pα21 cells exhibited wild-type growth (Fig. 6 D). Unlike SLY1-20, however, SLY1-20-pα21 was unable to suppress the lethality of ypt1-3 or uso1∆ deficiencies (Table S1). The amphipathic character of helix α21 is therefore essential for normal Sly1 function, and for gain-of-function phenotypes that are conferred by SLY1-20.
Because α21 is predicted to embed itself in the vesicle membrane, we hypothesized that fusion deficiencies seen with Sly1 loop mutants were attributable to failure to transit from a lipid mixing (hemifusion) state to opening of a stable fusion pore, and content mixing. This should result in accumulation of hemifused reaction intermediates. However, excess lipid mixing relative to content mixing was not observed (Figs. S3 and S4). We therefore conclude that the Sly1 loop and its amphipathic helix α21 promote the onset of lipid mixing and hemifusion but not the subsequent formation of a stable fusion pore.

Lipid as well as content mixing is defective with Sly1∆loop. Parallel lipid (top) and content mixing (bottom) results are shown from the same sets of reactions. The content mixing traces are identical to those shown in Fig. 5, B and D, and are shown here to facilitate comparison with the lipid mixing data. All reactions contained 3% PEG. (A) The reactions in A omitted Sec17 and Sec18. (B) The reactions in B contained 100 nM each of Sec17 and Sec18, and ATP. Lipid mixing is reported as raw fluorescence counts in arbitrary units. As the membranes mix, FRET from Marina Blue DHPE to NBD-DHPE (initially in separate liposomes) quenches Marina Blue emission at 465 nm. Points and bars in all traces show means ± SEM of data from three separate experiments.
Lipid as well as content mixing is defective with Sly1∆loop. Parallel lipid (top) and content mixing (bottom) results are shown from the same sets of reactions. The content mixing traces are identical to those shown in Fig. 5, B and D, and are shown here to facilitate comparison with the lipid mixing data. All reactions contained 3% PEG. (A) The reactions in A omitted Sec17 and Sec18. (B) The reactions in B contained 100 nM each of Sec17 and Sec18, and ATP. Lipid mixing is reported as raw fluorescence counts in arbitrary units. As the membranes mix, FRET from Marina Blue DHPE to NBD-DHPE (initially in separate liposomes) quenches Marina Blue emission at 465 nm. Points and bars in all traces show means ± SEM of data from three separate experiments.

Lipid, as well as content mixing, is defective with Sly1∆ polar loop mutants. Parallel lipid (top) and content mixing (bottom) results are shown from the same sets of reactions. (A and B) The content mixing data in A and B are identical to Fig. 6, G and J, and are shown here to facilitate comparison with the lipid mixing data. All reactions contained 3% PEG and 100 nM each of Sec17 and Sec18, and ATP. Lipid mixing is reported as raw fluorescence counts in arbitrary units. As the membranes mix, FRET from Marina Blue DHPE to NBD-DHPE (initially in separate liposomes) quenches Marina Blue emission at 465 nm. Points and bars in all traces show means ± SEM of data from three separate experiments.
Lipid, as well as content mixing, is defective with Sly1∆ polar loop mutants. Parallel lipid (top) and content mixing (bottom) results are shown from the same sets of reactions. (A and B) The content mixing data in A and B are identical to Fig. 6, G and J, and are shown here to facilitate comparison with the lipid mixing data. All reactions contained 3% PEG and 100 nM each of Sec17 and Sec18, and ATP. Lipid mixing is reported as raw fluorescence counts in arbitrary units. As the membranes mix, FRET from Marina Blue DHPE to NBD-DHPE (initially in separate liposomes) quenches Marina Blue emission at 465 nm. Points and bars in all traces show means ± SEM of data from three separate experiments.
Genetic results for the Sly1 amphipathic helix and loop mutants were closely mirrored in fusion experiments with RPLs (Fig. 6, E–G). Under every condition tested, Sly1-pα21 was less efficient at stimulating fusion than Sly1∆loop. Fusion in the presence of Sly1-pα21 was reduced in the absence or presence of PEG, as well as in the absence or presence of Sec17, Sec18, and ATP. Compared to Sly1-pα21, the compound mutant Sly1-20-pα21 (Fig. 6, H–J) exhibited a greater ability to stimulate fusion under every tested condition. Sly1∆loop and the Sly1-20-pα21 compound mutant had similar properties. Hence, the amphipathic character of helix α21 is required for the loop’s positive functions: activation and normal function of wild-type Sly1, as well as hyperactivity of Sly1-20, both in vivo and in vitro.
To further test the hypothesis that the regulatory loop has a positive function, we prepared chimeras with fragments of the loop appended to the amino terminus of Sly1∆loop (Fig. 7 A and Table S4). In vivo, chimeras bearing the entire loop, or α20-21 or α21 alone, restored normal growth to sly1∆loop (Fig. 7 B). Mutation of five hydrophobic residues within α21 eliminated rescue by loop-SLY1∆loop or by α20-21-SLY1∆loop. However, at 30°C, the polar mutant pα21-SLY1∆loop grew almost as well as the α21-SLY1∆loop. The mechanism of rescue by this mutant construct is unclear.
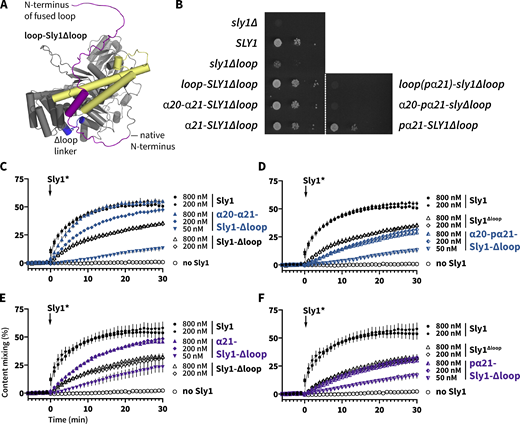
Appending the Sly1 loop to the amino terminus of Sly1∆loop partially restores function. (A) Chimeric constructs were prepared with different fragments of the Sly1 loop appended to the N-terminus of Sly1 via a short, flexible linker (see Table S3 for details). Mutants designated pα21 had the five polar substitutions in the appended loop as described in Fig. 6 C. (B) The loop-Sly1 mutants were expressed from the native SLY1 promoter on single-copy plasmids. Growth of a sly1∆ strain was assessed in the presence of the indicated constructs following ejection of a SLY1 balancer plasmid by plating on media containing 5-FOA. (C–F) Fusion driven by mutants with fragments of the loop (C and E) or polar derivatives of the same fragments (D and F). Points show mean ± SEM of three independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model.
Appending the Sly1 loop to the amino terminus of Sly1∆loop partially restores function. (A) Chimeric constructs were prepared with different fragments of the Sly1 loop appended to the N-terminus of Sly1 via a short, flexible linker (see Table S3 for details). Mutants designated pα21 had the five polar substitutions in the appended loop as described in Fig. 6 C. (B) The loop-Sly1 mutants were expressed from the native SLY1 promoter on single-copy plasmids. Growth of a sly1∆ strain was assessed in the presence of the indicated constructs following ejection of a SLY1 balancer plasmid by plating on media containing 5-FOA. (C–F) Fusion driven by mutants with fragments of the loop (C and E) or polar derivatives of the same fragments (D and F). Points show mean ± SEM of three independent experiments; in many cases the error bars are smaller than the symbols. Gray lines show least-squares nonlinear fits of a second-order kinetic model.
In vitro, the α20-21-Sly1∆loop chimera drove almost wild-type fusion when added at 800 nM, whereas its polar mutant (α20-pα21-Sly1∆loop) exhibited stronger defects similar to the Sly1∆loop. α21-Sly1∆loop exhibited gain of function relative to Sly1∆loop, while the polar mutant, pα21-Sly1∆loop, had no gain-of-function relative to Sly1∆loop. Overall (with the interesting exception of the pα21-SLY1∆loop allele’s in vivo phenotype), our results indicate that that the evolutionarily conserved portion of the Sly1 loop can partially replace the loop’s positive function, even when attached to Sly1 at a non-native location.
Helix α21 binds lipid bilayers directly with a preference for high curvature
The above data suggest the hypothesis that Sly1 helix α21 binds to membranes and this allows Sly1 to tether vesicles. To test whether α21 binds membranes directly we used a FRET assay. A peptide was synthesized comprising α21 and flanking residues, with an N-terminal tetramethylrhodamine fluorophore (TMR-α21). A control peptide, TMR-pα21, contained the same five substitutions as the Sly1-pα21 mutant (see Fig. 6 A). Protein-free liposomes were prepared by extrusion with 0.8% Texas Red-phosphatidylethanolamine (TRPE) to serve as a FRET acceptor for TMR. Representative emission spectra for the peptides and liposomes are shown in Fig. 8 A. Liposomes with either 6.7% or 30% ergosterol were extruded to two nominal diameters (30 and 200 nm). When mixed with TRPE-doped liposomes, the α21-TMR peptide generated a reproducible FRET signal, evident mainly as donor quenching (Fig. 8, B and C). Under the same conditions, the polar TMR-pα21 peptide exhibited smaller FRET signals. Moreover, the TMR-α21 peptide yielded a larger FRET signal with smaller liposomes at both sterol concentrations (Fig. 8 C). In contrast, the TMR-pα21 FRET signals did not depend on liposome diameter. We conclude that helix α21 binds membranes directly through a mechanism involving the apolar residues within α21, and it more avidly binds membranes with higher curvature. This is reminiscent of the behavior of ALPS domains, proposed to operate as membrane selectivity filters in the early secretory pathway.

Sly1 helix α21 binds membranes, with a preference for higher curvature. TMR-α21 and TMR-pα21 peptides were added to liposomes of nominal diameter 30 and 200 nm, which contained 1% TRPE as a fluorescence acceptor. (A) Emission spectra of peptides or liposomes (30 nm diameter, 6.7% ergosterol) measured separately, and the sums of the peptide and liposome spectra. The sums represent the no-FRET condition. Both the TMR-α21 and TMR-pα21 spectra are plotted; they overlap almost exactly. Vertical dashed lines at 585 and 610 nm indicate emission peaks for labeled peptides and liposomes, respectively. (B) Example of FRET data. Spectra from binding reactions containing liposomes (30 nm diameter, 6.7% ergosterol, 500 µM total lipid) and 25 µM TMR-α21 or TMR-pα21 are shown. The no-FRET condition is shown for reference. (C) Normalized FRET ratios for binding reactions containing the indicated combinations of liposomes and peptides, as in panel B. Traces and bars in A–C show means and ±95% confidence bands from four independent experiments.
Sly1 helix α21 binds membranes, with a preference for higher curvature. TMR-α21 and TMR-pα21 peptides were added to liposomes of nominal diameter 30 and 200 nm, which contained 1% TRPE as a fluorescence acceptor. (A) Emission spectra of peptides or liposomes (30 nm diameter, 6.7% ergosterol) measured separately, and the sums of the peptide and liposome spectra. The sums represent the no-FRET condition. Both the TMR-α21 and TMR-pα21 spectra are plotted; they overlap almost exactly. Vertical dashed lines at 585 and 610 nm indicate emission peaks for labeled peptides and liposomes, respectively. (B) Example of FRET data. Spectra from binding reactions containing liposomes (30 nm diameter, 6.7% ergosterol, 500 µM total lipid) and 25 µM TMR-α21 or TMR-pα21 are shown. The no-FRET condition is shown for reference. (C) Normalized FRET ratios for binding reactions containing the indicated combinations of liposomes and peptides, as in panel B. Traces and bars in A–C show means and ±95% confidence bands from four independent experiments.
Hyperactive Sly1* tethers high-curvature vesicles to the Qa-SNARE
Sly1 binds the Sed5 SNARE’s N-peptide-Habc domain (residues 1–210) with sub-nM affinity (Demircioglu et al., 2014; Grabowski and Gallwitz, 1997; Yamaguchi et al., 2002). Thus, we hypothesized that Sly1 may tether heterotypically, with one side of Sly1 binding to the N-terminal domain of Sed5 on the target membrane while the other side of Sly1, via helix α21, binds directly to the membrane of an incoming vesicle. To test this hypothesis, we adapted a bead-based assay (Fig. 9 A) previously used to study Rab-mediated tethering (Lo et al., 2011). First, GST-Sed5 cytoplasmic domain (GST-Sed5cyt) or control GST protein were adsorbed onto glutathione–agarose beads. Then wild-type or mutant Sly1* was allowed to bind to the immobilized GST-Sed5cyt (Fig. S5). Finally, fluorescent liposomes or RPLs were added and imaged by confocal microscopy. If Sly1 or its mutants mediate tethering between Sed5 and the membranes, we should see a corona of fluorescent vesicles surrounding the beads. Qualitative results with wild-type and mutant forms of Sly1 are shown in Fig. 9 A. To quantify this tethering, a bead spin-down assay was used (Fig. 9, B–D). When Sly1-20, Sly1-T559I, or Sly1-D563G were added to the beads, robust tethering of SNARE-free liposomes was observed (Fig. 9, A and B). Tethering was eliminated if either Sly1 or Sed5 (“GST control”) was omitted. Tethering was attenuated with wild-type Sly1, Sly1∆loop, and Sly1-pα21. An intermediate tethering signal was observed with Sly1-20-pα21. The partial tethering observed with this compound mutant might be due to eight hydrophobic residues on the loop that are still present in our Sly1-pα21 and Sly1-20-pα21 mutants (see Fig. 6 C). Robust tethering therefore requires that the loop be present, the loop be open, and helix α21 be amphipathic. Moreover, as in the peptide binding assays, Sly1-20 mediated tethering was most efficient with small-diameter vesicles and was insensitive to sterol concentration (compare Figs. 8 C and 9 C). Together, these findings indicate that both helix α21 in isolation and the Sly1 loop, in the context of Sly1-20, sense membrane curvature.

Hyperactive forms of Sly1 tether high-curvature vesicles to immobilized Sed5. (A) The ability of Sed5-bound Sly1 to directly tether vesicles was tested using a bead-based assay system. GST-Sed5 was adsorbed to glutathione-sepharose (GSH) beads, and wild-type or mutant forms of Sly1 were added to the reaction mixture. After 5 min, Texas Red-DHPE labeled liposomes were added to the mixtures, incubated for 15–20 min, and imaged by confocal microscopy (10× objective). A false-color scale was chosen to emphasize small differences in contrast under conditions with less tethering. The micrographs are representative of at least two independent assays per condition. (B–D) To quantify tethering efficiency, we used a spin-down assay. Binding reactions set up as for microscopy were subjected to low-speed centrifugation to sediment the GSH beads and associated proteins and vesicles. The supernatant was discarded and detergent was added to the pellet to liberate bound fluorescent lipids; the resulting signal was quantified by fluorometry. In C, Sly1-20 was present for each condition. In D, Sly1* was preincubated with a 6:1 excess of soluble Sed5-Habc or Sed5-N-Habc, as indicated. Y-axes show bead-associated fluorescence (au, arbitrary units) after subtracting background from blanks containing only buffer. Bars indicate means ±95% confidence intervals for 4–10 independent experiments. Binding of the Sly1 variants to immobilized Sed5 was efficient and nearly stoichiometric (Fig. S5).
Hyperactive forms of Sly1 tether high-curvature vesicles to immobilized Sed5. (A) The ability of Sed5-bound Sly1 to directly tether vesicles was tested using a bead-based assay system. GST-Sed5 was adsorbed to glutathione-sepharose (GSH) beads, and wild-type or mutant forms of Sly1 were added to the reaction mixture. After 5 min, Texas Red-DHPE labeled liposomes were added to the mixtures, incubated for 15–20 min, and imaged by confocal microscopy (10× objective). A false-color scale was chosen to emphasize small differences in contrast under conditions with less tethering. The micrographs are representative of at least two independent assays per condition. (B–D) To quantify tethering efficiency, we used a spin-down assay. Binding reactions set up as for microscopy were subjected to low-speed centrifugation to sediment the GSH beads and associated proteins and vesicles. The supernatant was discarded and detergent was added to the pellet to liberate bound fluorescent lipids; the resulting signal was quantified by fluorometry. In C, Sly1-20 was present for each condition. In D, Sly1* was preincubated with a 6:1 excess of soluble Sed5-Habc or Sed5-N-Habc, as indicated. Y-axes show bead-associated fluorescence (au, arbitrary units) after subtracting background from blanks containing only buffer. Bars indicate means ±95% confidence intervals for 4–10 independent experiments. Binding of the Sly1 variants to immobilized Sed5 was efficient and nearly stoichiometric (Fig. S5).
Association of Sly1 and its variants with immobilized GST-Sed5. Binding reactions were set up as in Fig. 9 with the indicated Sly1 variants (80 pmol per 500 µl reaction) and GST-Sed5 cytoplasmic domain immobilized on glutathione-agarose (GST-Sed5Cyt; 150 pmol added to beads per reaction). The binding and wash buffer contained 10 mg/ml BSA. After sedimenting and washing the beads, proteins in the pellet were eluted with SDS-PAGE sample buffer, separated on 10% polyacrylamide gels, and stained with Coomassie blue. Source data are available for this figure: SourceData FS5.
Association of Sly1 and its variants with immobilized GST-Sed5. Binding reactions were set up as in Fig. 9 with the indicated Sly1 variants (80 pmol per 500 µl reaction) and GST-Sed5 cytoplasmic domain immobilized on glutathione-agarose (GST-Sed5Cyt; 150 pmol added to beads per reaction). The binding and wash buffer contained 10 mg/ml BSA. After sedimenting and washing the beads, proteins in the pellet were eluted with SDS-PAGE sample buffer, separated on 10% polyacrylamide gels, and stained with Coomassie blue. Source data are available for this figure: SourceData FS5.
Sly1 binds the Sed5 N-terminal domain with sub-nM affinity (Bracher and Weissenhorn, 2002; Demircioglu et al., 2014; Yamaguchi et al., 2002). To test the importance of this binding interaction in tethering, Sly1-20 was preincubated with a 6:1 molar excess of Sed5-N-Habc (aa 1–210; Fig. 9 D). This abolished tethering. In contrast, tethering was not blocked by Sed5-Habc lacking the N-peptide required for high-affinity Sly1 binding (aa 22–210). This shows that Sly1 cannot tether incoming vesicles unless it is anchored to the Qa-SNARE through a high-affinity interaction. Taken together, the present and previously reported genetic data, and our assays of in vitro fusion, peptide binding, and tethering, all support the conclusion that the amphipathic helix α21 is necessary and sufficient for direct Sly1 binding to the incoming vesicle’s lipid bilayer. This binding both activates Sly1 and allows it to tether incoming vesicles to the target membrane Qa-SNARE.
Discussion
SLY1 was identified through isolation of SLY1-20 as a dominant single-copy suppressor of deficiency in Ypt1, the yeast Rab1 protein. Previous studies suggested that SLY1 gain-of-function alleles become hyperactive through the loss of autoinhibition by the Sly1 loop. The present experiments directly support that hypothesis but further show that the loop has a positive function. Both functions are essential for bypass of tethering requirements by the SLY1-20 mutation, and both require the presence of conserved apolar residues within α21. Sly1 mutants with a constitutively open loop that has reduced membrane affinity, as well as mutants that lack the loop entirely, exhibit loss of function relative to the wild-type.
In a working model (Fig. 10 A), long-range tethers mediate the initial capture of the vesicle by the target membrane, operating at ranges from 30 to >200 nm. Multisubunit tethering complexes (MTCs) including GARP, Dsl, and COG have long appendages (Chou et al., 2016; Ha et al., 2016; Ren et al., 2009). Golgins have rod-like coiled-coil domains interspersed with hinge-like domains (Cheung and Pfeffer, 2016; Gillingham, 2018). In three cases (Uso1, Golgin-210, and the Golgin-like endosomal tether EEA1), there is evidence that the hinges cause the tether to buckle or collapse, allowing the vesicle to approach the target membrane (Yamakawa et al., 1996; Cheung et al., 2015; Murray et al., 2016). We propose that long-range tethering factors hand vesicles off to Sly1, which then tethers vesicles at a range of ∼15 nm from the target membrane to promote trans-SNARE complex assembly (Fig. 10 B). The Sly1 loop’s preference for small-diameter vesicles is similar to ALPS helices, which operate as selectivity filters that recognize bulk physical properties of membranes in the early secretory pathway (Bigay et al., 2005; Bigay and Antonny, 2012; Drin et al., 2008; Magdeleine et al., 2016). We propose that Sly1’s gated, close-range tethering function adds an additional membrane selectivity filter to this system.

Working model. (A) Long-range tethering is mediated by coiled-coil Golgin family tethers and multisubunit tethering complexes (MTC’s). Flexibility or buckling of long-range tethers allows the vesicle to dwell in the region near Sly1 so that handoff can occur. (B) Mechanism of close-range tethering. Sly1 is anchored to the N-terminal domain of the Qa-SNARE on the target membrane. Note that in the closed ground state, the loop and helix α21 (magenta) occlude the sec22 binding site on the Sly1 SNARE templating domain (yellow). Binding of α21 to an incoming vesicle’s membrane pulls open the autoinhibitory loop and tethers the vesicle to Sly1, likely in a spatial orientation optimal for Sec22 binding to Sly1’s SNARE templating domain (yellow). In panels ii and iii, helix α21 is shown but the unstructured portion of the loop is omitted for clarity. (C) The Sly1 loop is conformationally heterogeneous in the crystal structure (PDB ID 1MQS). Temperature (B) factors in the Sly1 crystal are shown by color and backbone trace thickness. The highest disorder is in α20–α21, the thick red peptide segment.
Working model. (A) Long-range tethering is mediated by coiled-coil Golgin family tethers and multisubunit tethering complexes (MTC’s). Flexibility or buckling of long-range tethers allows the vesicle to dwell in the region near Sly1 so that handoff can occur. (B) Mechanism of close-range tethering. Sly1 is anchored to the N-terminal domain of the Qa-SNARE on the target membrane. Note that in the closed ground state, the loop and helix α21 (magenta) occlude the sec22 binding site on the Sly1 SNARE templating domain (yellow). Binding of α21 to an incoming vesicle’s membrane pulls open the autoinhibitory loop and tethers the vesicle to Sly1, likely in a spatial orientation optimal for Sec22 binding to Sly1’s SNARE templating domain (yellow). In panels ii and iii, helix α21 is shown but the unstructured portion of the loop is omitted for clarity. (C) The Sly1 loop is conformationally heterogeneous in the crystal structure (PDB ID 1MQS). Temperature (B) factors in the Sly1 crystal are shown by color and backbone trace thickness. The highest disorder is in α20–α21, the thick red peptide segment.
In the anterograde ER–Golgi pathway, both Sly1 and the Qa-SNARE Sed5 must be present on the Golgi acceptor membrane—they cannot fulfill their functions if located only on COPII-derived transport vesicles (Cao and Barlowe, 2000). Sly1 is anchored to Sed5 through direct, sub-nanomolar interaction with the Sed5 N-terminal domain (Bracher and Weissenhorn, 2002; Demircioglu et al., 2014; Yamaguchi et al., 2002). As our experiments show, Sly1 binding to the Sed5 N-peptide is indispensable for Sly1-mediated tethering (Fig. 10 B). However, a previous report argued that the Sed5–Sly1 interaction is of relatively minor importance (Peng and Gallwitz, 2004). In parallel work (Duan et al., 2024; Gao and Banfield, 2020), we show that Sly1 binding to the Sed5 N-terminal domain is important for fusion in vitro and indispensable in vivo.
Sitting opposite Sly1’s N-peptide-binding cleft is the Sly1 loop. The loop is mobile. In the Sly1 crystal structure (PDB ID 1MQS), the poorly conserved N-terminal half of the loop is unresolved, while the better-conserved C-terminal half of the loop is partially resolved but has a large temperature (B) factor, indicating conformational polymorphism (Fig. 10 C). We speculate that when Sly1 is in its autoinhibited ground state, helix α21 undergoes “logrolling” excursions about its long axis, intermittently exposing apolar side chains to probe for the presence of incoming vesicle membranes. Helix α21 binding to the vesicle’s bilayer then has two consequences. First, the loop is pulled open, exposing the R-SNARE binding surface on Sly1. Second, the open loop operates as a close-range tether, stabilizing the vesicle and target membrane within a distance sufficient to favor R-SNARE binding to Sly1. When the R-SNARE is bound to Sly1, the loop cannot close. Sly1 binding to the R-SNARE and vesicle membrane then promotes formation of the trans-SNARE template complex on Sly1 domain 3a (Fig. 10 B).
The Sly1 loop probably constrains the rotational motion of Sly1 so that Sly1 and the Qa-SNARE Sed5 are optimally oriented for productive R-SNARE engagement. AlphaFold2 modeling of Sly1-SNARE complex structures exactly predicts that geometry, with the deployed helix α21 precisely flanking the membrane entry point of Sec22’s transmembrane domain. In the companion manuscript (Duan et al., 2024), we present experimental evidence that α21 promotes selective trans- versus cis-SNARE complex assembly.
In our in vitro tethering assays, Sly1-20 and other gain-of-function mutants allow efficient tethering, consistent with the ability of these mutants to suppress requirements for other tethering factors. In the same in vitro assays, however, wild-type Sly1 tethers less efficiently. This raises the question of whether close-range tethering is important for wild-type Sly1. The Sly1∆loop mutant cannot be autoinhibited, yet it exhibits substantial tethering and fusion defects in vitro. In vivo, our SGA analyses reveal that the sly1∆loop allele exhibits synthetic sickness or lethality with dozens of genes involved in ER and Golgi traffic, including many genes that encode tethering factors or their regulators. In other words, when close-range tethering is prevented, even partial impairment of long-range tethering results in catastrophe and death. We suggest that a key function of Golgi long-range tethers is to allow incoming vesicles to dwell in the vicinity of Sly1 for long enough to allow inspection of vesicle membrane properties by α21, leading to loop opening, close-range tethering, R-SNARE engagement, and assembly of the fusogenic trans-SNARE complex.
Additional mechanisms might contribute to Sly1 loop function. First, it is possible that as-yet unidentified proteins bind Sly1, contributing to loop opening and tethering. Second, when open (as in Sly1-20), the loop may be intrinsically disordered, generating a “steric cushion” that exerts a bending force on the adjacent docked membranes (Busch et al., 2015; D’Agostino et al., 2017). Our evidence for a steric cushion mechanism is mixed. In vitro, the behavior of Sly1∆loop, which completely lacks the loop, and of Sly1-20-pα20, which has a full-length loop that is constitutively open but partially defective in membrane binding, exhibit similar defects in most assays. This argues against the steric cushion hypothesis. In vivo, however, SLY1-20-pα20 allows almost wild-type growth, while the sly1∆loop mutant grows slowly. In a different model, helix α21 penetration of the vesicle perturbs local membrane structure, lowering the energy barrier for the initiation of lipid mixing. Our data neither support nor refute the membrane disruption hypothesis but do suggest that the loop’s major functions occur at or before lipid mixing, not at the subsequent transition from lipid to content mixing.
Sly1 has been proposed to promote vesicle fusion in several ways. (1) The Golgi Qa SNARE Sed5 can adopt a tightly closed conformation. Sly1 can open closed Sed5, allowing SNARE complexes to form more readily, at least in aqueous solution (Demircioglu et al., 2014; Kosodo et al., 1998). (2) As we show here, helix α21 binding to membranes both de-represses and directly promotes Sly1 activity through a mechanism involving close-range vesicle tethering. (3) Sly1 has conserved structural features that in Munc18-1 and Vps33 have been shown to catalyze trans-SNARE complex assembly through a Qa–R-SNARE templating mechanism. (4) We have shown (again in aqueous solution, but corroborated by genetic experiments) that Sly1 and another SM, Vps33, can decrease the rate of SNARE complex disassembly by Sec17 and Sec18 (Lobingier et al., 2014; Sheffield et al., 1999). In the accompanying study (Duan et al., 2024), we show that each of these mechanisms contributes to Sly1 function and that all are required for full Sly1 activity.
The Sly1 loop is conserved among Sly1 homologs from yeast to human but absent from representatives of other SM sub-families: Sec1/Munc18, Vps45, and Vps33. Why is the loop unique to Sly1? We suggest that accessory proteins provide similar functions for these SMs. For example, the endosomal SM Vps45 associates with a scaffold protein, Vac1 (in mammals, Rabenosyn-5). Vac1 binds both Rab5 and phosphatidylinositol-3-phosphate, and could mediate close-range tethering in a manner analogous to the Sly1 loop (Burd et al., 1997; Peterson et al., 1999; Rahajeng et al., 2010; Tall et al., 1999). Sec1 physically and functionally interacts with the exocyst tethering complex. Vps33 is stably associated with Vps-C tethering complexes (HOPS and CORVET), which subsume both tethering and SNARE assembly functions (Morgera et al., 2012; Rieder and Emr, 1997). In HOPS, an ALPS-like domain within the Vps41 subunit is proposed to select high-curvature endocytic vesicles for docking and fusion (Cabrera et al., 2010). Munc18-1, despite lacking the Sly1-specific regulatory loop, is reported to tether vesicles in a reaction that requires at least the Qa-SNARE N-peptide and the R-SNARE on the opposing membrane (Arnold et al., 2017; Tareste et al., 2008). There is no evidence that this tethering occurs through a direct interaction between Munc18-1 and the vesicle bilayer, but the parallels suggest that various forms of close-range tethering will facilitate SM activity in many or all SNARE-mediated fusion systems.
Which long-range tethers hand vesicles off to Sly1? Persuasive experiments show that Sly1 operates in concert with Ypt1 and Uso1 (yeast Rab1 and p115, respectively) on the anterograde ER–Golgi pathway (Cao and Barlowe, 2000). However, direct interactions between Sly1 and Ypt1 or Uso1 have not been detected, and the mechanisms of Uso1/p115 tethering are controversial. Binding interactions are reported between human Sly1 and COG, and perhaps between yeast Sly1 and Dsl (Kraynack et al., 2005; Laufman et al., 2009). The positive suppressing interactions of SLY1-20, the negative synthetic sickness or lethal interactions of sly1∆loop, and the known SNARE interactions of Sly1, all point to Sly1 working as a common “receiving agent” for vesicle fusion at ER and cis, medial, and perhaps trans Golgi target membranes. Several ER and Golgi tethers bind Sly1 client SNAREs directly. Sly1 probably facilitates the delivery of cargo containers initially captured by COG, Dsl, various Golgins, and possibly the TRAPP and GARP complexes. An outstanding challenge for the future is to identify which combinations of long-range tethers and SNAREs operate in concert with Sly1, and mechanisms that coordinate handoffs from long-range tethers to the core SM-SNARE fusion machinery.
Materials and methods
Yeast strains and SLY1 gain-of-function screen
We use standard Saccharomyces genetic nomenclature (Dunham et al., 2015). Dominant alleles, whether wild-type or mutant, are named in uppercase type (e.g., SLY1-20); recessive alleles are named in lowercase (e.g., sly1∆loop). Strains and plasmids used in this study are described in Table S5. To obtain new suppressors of uso1∆, a library of SLY1* mutant alleles was constructed using the GeneMorph II Random Mutagenesis Kit (#200550; Agilent). The SLY1 open reading frame was amplified using the “medium mutation rate” PCR protocol. Four mutagenic PCR pools were separately purified and cloned into a derivative of the yeast vector pRS415, which contained 431 bp of the SLY1 promoter and 249 bp of the SLY1 terminator, using traditional restriction–ligation methods. Aliquots of the pRS415::SLY1 mutant library ligation products were transformed into TOP10F´chemically competent E. coli cells and 10 individual clones were Sanger-sequenced to assess cloning fidelity and mutation frequency. Each clone sequenced contained the SLY1 open reading frame with 0–4 mutations, with about 50% of the clones containing mutations. After the SLY1 mutant library pools were verified, aliquots of the mutant library ligation products were transformed into Bioline Alpha-Select Gold Efficiency Competent E. coli cells. Transformant colonies were scraped from the LB + Amp transformation plates (maintaining four separate mutant pools) and allowed to grow for about two doublings. Plasmid DNA was extracted and purified from each of the pooled cultures using Qiaquick columns. 1 µg of plasmid DNA from each SLY1 mutagenic pool was transformed into S. cerevisiae strain AMY2144 (CBY1297: uso1∆ pRS426::USO1). Transformant colonies were grown under selection for leucine auxotrophy, then replica plated to a synthetic complete medium containing 5-FOA and incubated for 2 days. Yeast colonies that grew on 5-FOA (thus “kicking out” the WT copy of USO1) were struck out on –Leu plates, and plasmid DNA was purified from 10 or more clones from each pooled library, using the Smash and Grab procedure. Plasmids were Sanger-sequenced. On the basis of these results, pRS415::SLY1* single mutant alleles were constructed using PCR and Gibson assembly, and are described in Tables S1, S3, S4, and S5. The second half of sly1-pα21 gene and its derivative SLY1-20-pα21 (see Table S2) were ordered as a gBlock (IDT) and cloned into the BamHI and NcoI sites on the wild-type SLY1 plasmid.
Design of Sly1∆loop variants and AlphaFold2 modeling
A crystal structure for Sly1 (PDB ID 1MQS) was used as the input structure for in silico design (Leaver-Fay et al., 2011). First, loop residues in the crystal structure were removed: either just the loop residues (Ser503 to Thr555) or a larger region including some residues from the adjacent α helices (Ser500 to Ile558). Next, varying-length loop backbones were modeled into the loop gap as previously described (Silva et al., 2019). We used Rosetta to assign amino acid sequences to the new loop backbones. We used the Rosetta foldability filter to assess the folding compatibility of the assigned sequences and designed backbones. Finally, we visually evaluated the final design outputs and constructed 12 mutants to test in the lab (Table S3). AlphaFold2 modeling was done in the ColabFold environment (Jumper et al., 2021; Mirdita et al., 2022). Template selection was set to pdb100, msa_mode was set to mmseqs2_uniref_env, pair_mode was set to unpaired_paired, and relax_max_iterations was set to 200 with greedy pairing.
SGA analysis
A query strain (AMY2443) was constructed in the Y9205 genetic background (Tong and Boone, 2006), with sly1∆loop and a linked nourseothricin (NAT) marker integrated through allelic replacement at the native SLY1 locus. This query strain was crossed to the MAT a haploid deletion and DAmP libraries, where each individual genetic perturbation is marked with a KAN resistance marker (Breslow et al., 2008; Tong and Boone, 2006). Diploids were selected by robotic pinning (Singer RoToR) onto YPD + 100 mg/liter clonNAT + 200 mg/liter G418, then induced to sporulate by pinning to sporulation medium (20 g/liter agar, 10 g/liter potassium acetate, 1 g/liter yeast extract, 0.5 g/liter glucose, 0.1 g/liter amino acid supplement [2 g histidine, 10 g leucine, 2 g lysine, 2 g uracil]), and grown at room temperature for 5 days. Spores were subsequently pinned to haploid selection medium (SD -His/Arg/Lys + 50 mg/liter canavanine + 50 mg/liter thialysine) and MAT, a meiotic progeny grown for 2 days at 25°C. This haploid selection step was repeated, and the resulting colonies were imaged using a Phenobooth (Singer) imaging system. These colonies encompass all potential meiotic progeny and serve as the control strains for phenotypic normalization. Haploid double mutants carrying both the KAN deletion allele and the sly1∆loop::NAT allele were selected by pinning meiotic progeny to a double selection medium (SD/MSG -His/Arg/Lys + 50 mg/liter canavanine + 50 mg/liter thialysine +100 mg/liter clonNAT + 200 mg/liter G418). After 2 days of growth at 25°C, this selection step was repeated and duplicate plates incubated at either 30°C or 37°C. Plates were imaged using the Phenobooth system, and colony size differences were calculated using PhenoSuite software and web app (https://singerinstruments.shinyapps.io/phenobooth/).
Protein purification
Full-length SNARE proteins were produced as previously described (Furukawa and Mima, 2014) with modifications. E. coli Rosetta2 (DE3) pLysS cells (Novagen), harboring each of the SNARE expression plasmids with 3C protease-cleavable N-terminal tags (pET-41/GST-His6 for SEC22 and pET-30/His6 for SED5, BOS1, and BET1), were inoculated from a 1:1,000 dilution of the starter culture grown in MDAG-135 medium (Studier, 2005) into 1 liter of Terrific Broth supplemented with 100 µg/mliter Kanamycin and 34 µg/ml Chloramphenicol and grown at 37°C, 275 rpm until OD600 was reached ∼1. Cultures were then induced with 1 mM IPTG for 3 h at 37°C. Cultures were harvested at 5,000 × g and cell pellets were snap-frozen with liquid nitrogen. Each liter yielded ∼10 g of wet cells, which were stored at −70°C. For purification, the frozen pellets were warmed to −10°C and broken into small pieces with a metal spatula, then resuspended at a ratio of 5 ml of buffer per gram of cell paste in 1× SNARE buffer (20 mM Na·PO4, 500 mM NaCl, 10% [m/vol] glycerol, 1 mM DTT, pH 7) supplemented with 30–40 mM imidazole, 0.25 mg/ml chicken egg lysozyme, 125 U benzonase per g of cells, and 1× Sigmafast Protease inhibitor cocktail. 4 ml (1/10 volume) of 1 M n-octyl-β-D-glucopyranoside in (β-OG, Anatrace; dissolved in water) was added to 100 mM final concentration; the suspension was rotated at room temperature for 25 min to allow detergent-aided enzymatic lysis. Lysates were clarified at 16,500 × g, 4°C for 10 min, transferred to clean centrifuge tubes, and centrifuged again for 20 min. Clarified lysates were batch-bound with 2 ml of Ni-Sepharose HP equilibrated in 1× SNARE buffer with β-OG for 30 min. The SNARE-bound resin was washed in plastic disposable columns with 25 ml of SNARE buffer supplemented with β-OG and 60–100 mM imidazole. SNARE proteins were eluted with SNARE buffer supplemented with β-OG and 200–300 mM imidazole, and snap-frozen in liquid nitrogen. Purified protein was quantified by using absorbance at 280 nm, and purity was assessed with SDS-PAGE with Coomassie blue staining. Protein aliquots were stored at −70°C until reconstitution. We note that 3C protease caused substantial unintended cleavage of Bos1 in its N-terminal linker domain due to a cryptic 3C site (148-GLPLYQ/GL-155). Mutation of the poorly conserved residue Q153 to aspartic acid eliminated unintended proteolysis.
Soluble domains of Sed5 were expressed from the pET-30 vector (for H6-Habc and H6-N21-Habc) or pET-49 vector (for GST-H6-SED5∆TM) and purified in the same way as the full-length protein, except that the temperature was lowered to 35°C prior to induction. The buffers did not contain β-OG, and lysis was performed using Emulsiflex-C5 high-pressure homogenizer (Avestin). Eluted protein was exchanged into FB160M1 (20 mM HEPES·KOH, 160 mM KOAc, 10% [m/vol] Glycerol, 1 mM MgOAc2, pH 7) using a PD-10 desalting column. Precipitated material was removed by centrifugation at 10,000 × g for 10 min, and soluble protein aliquots were snap-frozen in liquid nitrogen in 250 µl PCR tubes and stored at −80°C. Sec22(SNARE)-GFP-His8 was expressed from the pST50Trc1 vector in Rosetta2(DE3) cells grown in ZYM-5052 autoinduction media (Studier, 2005) supplemented with carbenicillin (100 µg/ml) and chloramphenicol (34 µg/ml) overnight (>16 h) at 30°C from a 1:1,000 dilution of starter culture. Cells were harvested and protein was purified as for soluble domains of Sed5. Sec17 was purified as described (Schwartz and Merz, 2009) except that the culture was grown in ZYM-5052 autoinduction media (Studier, 2005) at 37°C until OD600nm was ∼0.8; the temperature was then lowered to 18°C and the culture was incubated for ∼24 h. Sec18 was purified as described (Haas and Wickner, 1996).
Sly1 and its mutants were expressed in Rosetta2(DE3) cells from pHIS-Parallel1 vectors (Lobingier et al., 2014; Sheffield et al., 1999). Frozen glycerol stocks were used to inoculate overnight starter cultures at 37°C in MDAG-135 containing 100 mg/liter carbenicillin and 50 mg/liter chloramphenicol (Studier, 2005). Each starter culture was diluted 1/1,000 to seed 1–2 liter of Terrific Broth containing 100 mg/liter carbenicillin and 34 mg/liter chloramphenicol. These cultures were grown in an orbital shaker (37°C, 275 rpm) to OD600nm ∼1. Cultures were then transferred to a prechilled shaker at 16°C for 1 h before induction with 0.1–1 mM IPTG for 18 h. Cells were sedimented and resuspended in cold Sly1 buffer (20 mM Na·PO4, 500 mM NaCl, 10% [m/vol] glycerol, and 1 mM DTT, pH 7) supplemented with 30 mM imidazole, 0.25 mg/ml chicken egg lysozyme, and 1× Sigmafast Protease inhibitor cocktail at a ratio of 5 ml of buffer per gram of cell paste. The cells were lysed by passing through Emulsiflex-C5 high pressure homogenizer (Avestin) two to four times, and the lysate was clarified by centrifugation at (16,500 × g, 25 min, 4°C). Clarified lysate from 1 liter of culture (2 liters for Sly1∆loop and Sly1-20) was bound in a batch with 1 ml equilibrated Ni2+-Sepharose HP resin (GE Healthcare) for 30 min at 4°C. Sly1-bound resin was collected in a 25 ml-disposable Econo-Pac column (Bio-Rad) by gravity and washed with 25 ml of SLY1 buffer supplemented with 50 mM imidazole at pH 7. Sly1 was eluted with Sly1 buffer supplemented with 300 mM imidazole pH 7 in 0.5 ml fractions. Most of the protein eluted in fractions 3–7. Sly1 was exchanged into FB160M1 (20 mM HEPES·KOH, 160 mM KOAc, 10% m/vol Glycerol, 1 mM MgOAc2, pH 7) using a PD-10 column (GE Healthcare). Precipitated material was removed by centrifugation at 10,000 × g for 10 min and soluble proteins were diluted or concentrated to ∼2.4 mg/ml. Aliquots were snap-frozen in liquid nitrogen in thin-wall PCR tubes and stored at −70°C.
Recombinant HRV3C protease was prepared either as an N-terminal His8-tag fusion (AMP2019) or an N-terminal GST-His6-(Thrombin) fusion (AMP2016). 1 liter of 1/1,000 dilution of an overnight culture of Rosetta2(DE3) cells harboring the expression plasmid was grown overnight at 37°C in ZYM-5052 autoinduction media with 100 µg/ml kanamycin and 34 µg/ml chloramphenicol. Cells were centrifuged, resuspended in four times the volume of Lysis buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM DTT, and no protease inhibitors) supplemented with 15 mM imidazole and 0.5 mg/ml lysozyme, and lysed using Emulsiflex-C5 high-pressure homogenizer (Avestin). Clarified lysate was incubated with 3 ml Ni2+-Sepharose HP (GE Healthcare) for ∼30 min and strained in a disposable column. Resin was washed thoroughly with Lysis buffer supplemented with 40–60 mM imidazole, and the protein was eluted with 200 mM imidazole in about 7.5 ml. Concentrated fractions were combined and EDTA was added to 1 mM. The yield was ∼100 mg of purified protease per 1 liter of culture. Purified protease was diluted to 10 mg/ml and exchanged into storage buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, and 20% glycerol), frozen in liquid N2, and stored at −80°C. Protease activity of the preparations was assayed using a homemade assay based on a linked FRET pair of fluorescent proteins (Evers et al., 2006), modified with an HRV 3C-cleavable linker. Reduction in FRET due to proteolysis was monitored in real time using a SpectraMax Gemini microplate reader (Molecular Devices). GST-His6 was expressed and purified using conventional Ni2+ IMAC chromatography methods. Protein was exchanged into FB160M1 before freezing in liquid N2 and stored at −80°C.
Circular dichroism spectroscopy
Purified SLY1wt or SLY1∆loop was exchanged into CD buffer (20 mM Na-Pi and 100 mM NaCl, pH 7.2), diluted to 0.2 mg/ml, and loaded into a 0.1-cm path length cuvette. Spectroscopy was performed using a J-1500 CD Spectrophotometer (JASCO) at 25°C. CD and absorbance were measured from λ = 195–260 nm in steps of 0.1 nm. The protein concentration during each read was calculated from the absorbance at 205 nm (Anthis and Clore, 2013). Molar ellipticity for each protein was calculated by dividing the CD at each wavelength by the cuvette pathlength and protein concentration. The mean residue ellipticity for each protein was calculated by dividing the molar ellipticity by the number of amino acids per protein.
Preparation of RPLs
The FB160 buffer system and lipid mixtures used here are derived from B88 buffer, used extensively in COPII vesicle budding assays (Baker et al., 1988), and from lipidomic studies. The ER lipid mix is based on “Major-Minor” mixtures used for COPII budding (Antonny et al., 2001; Matsuoka et al., 1998). The Golgi mix is based on lipidomic surveys (Klemm et al., 2009; Schneiter et al., 1999). In particular, the study of Schneiter et al. (1999) used a highly enriched Golgi fraction known to be competent for docking and fusion of COPII carrier vesicles (Lupashin et al., 1996). Relatively high concentrations of ergosterol were used based on prior work on COPII budding, which demonstrated that higher sterol levels yielded more morphologically homogenous COPII vesicles (Matsuoka et al., 1998). In pilot studies, however, RPLs prepared with lower ergosterol concentrations exhibited similar fusion characteristics, including Sly1 and PEG dependence, as the high-sterol RPLs used in the experiments presented here. Lipids were obtained from Avanti Polar Lipids as chloroform stocks except for ergosterol, which was from Sigma-Aldrich. Table S6 lists the proportions, working stocks, and volumes of lipids and detergent used to prepare ER-mix and Golgi-mix RPLs. Lipid stocks were prepared or purchased in chloroform, except for ergosterol and phosphatidylinositol-4-phosphate, which were dissolved in 1:1 chloroform:methanol. β-OG stock solutions were prepared in methanol. Lipid–detergent films were prepared by transferring lipid and β-OG stocks to a glass vial (typically, 8 µmol total lipids and 70 µmol β-OG). The mixture was dried under a nitrogen stream; residual solvent was removed using a Speedvac evaporator. The lipid–detergent film was hydrated and solubilized with 400 µl 5× FB160M1 by three cycles of bath sonication and shaking. To the lipid–β-OG mixture, content mixing FRET reporters were then added (500 µl of 4 mg/ml solution of R-phycoerythrin-biotin conjugate, or 296 µl of 2 mg/ml Alexa-Streptavidin; both reagents from Thermo Fisher Scientific/Molecular Probes). SNARE stocks in SNARE elution buffer with β-OG were then added to a final molar ratio of 1:600 (each Q-SNARE) or 1:300 (Sec22) to total phospholipids. Water was used to fill the headspace necessary to dilute 5× FB160M1 buffer to 1× concentration (2 ml final volume). Mixtures were nutated for 30 min before recombinant 3C protease was added (in 1:10 ratio to total SNAREs) to cleave affinity tags from the SNARE proteins during dialysis. The resulting mixtures were dialyzed (20 kD cutoff) for ∼18 h at 4°C in the dark against 250 volumes of FB160M1 containing 2 g BioBeads SM2 (Bio-Rad) per 2 ml of RPL mixture. The RPL mixture was then separated from the unencapsulated content mixing probe by floating the RPLs up a step gradient of iso-osmotic Histodenz (35/25/0%) in FB160M1 (SW60Ti rotor at 55,000 rpm for 90 min), harvested, and diluted to 2 mM phospholipid. Phospholipid was quantified by measuring the fluorescence of the membrane fluorophore, initially verified by inorganic phosphate analysis (Chen et al., 1956). 32 µl aliquots of RPLs were transferred to thin-wall PCR tubes and frozen by immersion in liquid N2. RPLs prepared by this method and stored at −80°C were stable and fusion-competent, with minimal leakage of encapsulated FRET probes, for at least 1 year.
RPL fusion assays
Unless noted otherwise, a standard order-of-addition was always used to initiate RPL assays. 250 µM (final phospholipid) of each RPL was premixed with PEG6K and other fusion components such as Sec17, Sec18, and ATP. Fusion assays were performed in 20 µl sample volumes in 384-well plates (#4514; Corning). The reactions were monitored in a plate-based fluorimeter (Molecular Devices Gemini XPS or EM) for 5 min to establish a baseline, and then Sly1 was added to initiate fusion. Except as noted, the moment of Sly1 addition was defined as time = 0. Lipid mixing was monitored at Ex370nm and Em465nm. Content mixing was monitored at Ex565nm and Em670nm. The content mixing assays were normally performed in the presence of a large excess of unlabeled streptavidin so that any biotinylated FRET probe escaping due to RPL leakage cannot form a FRET pair with fluorescent streptavidin. To calibrate the assay, RPLs were lysed with detergent in the presence or absence of unlabeled streptavidin, respectively, yielding values for assay FRET background (defined as 0% fusion) and complete probe mixing (defined as 100% fusion). For content mixing, the typical signal for a complete reaction over a fusion background (e.g., no Sly1) exceeded 50:1. Graphs show mean ± SEM of n ≥ 3 independent assays. Curves on the graphs show a second-order kinetic model fit to each dataset using a weighted least-squares algorithm in GraphPad Prism. Note that positive and negative control traces are in some cases repeated between figure panels. These are shared controls from larger experiments with multiple treatments, executed in parallel.
Yeast growth assays
Yeast strains containing pRS426::USO1 or pRS416::YPT1 and pRS415::SLY1 mutant plasmids were grown in –Leu liquid media, then diluted using a 48-pin manifold or a multichannel pipettor onto 5-FOA plates. The 5-FOA plates were grown at restrictive or non-restrictive temperatures, as indicated. Growth was scored relative to positive and negative control strains after 2–3 days.
Peptide–liposome binding assay
Bead-based tethering assays
Beads were prepared in 100 µl (10 reactions) or 1 ml (100 reactions) batches. In small disposable spin columns, 100 µl of beads were washed in FB160M1 supplemented with 1% (ml/vol) bovine serum albumin (FB160M1BSA) and were loaded with 100 µg of GST-Sed5 cytoplasmic domain (1/5 of the resin’s nominal binding capacity; 150 pmol protein per 10 µl resin in a 1× reaction) in a volume of 500 µl FB160BSA; this mixture was incubated, with slow agitation, for 30 min at room temperature. Unbound material was removed by gentle centrifugation (∼70 × g, 10 s), the beads were washed once with FB160BSA, and the beads were then blocked by adding excess recombinant GST-His6 protein (1.25 mg; 2.5 times the resin’s nominal binding capacity) in FB160BSA in 500 µl final volume. Unbound GST-H6 was not removed. The bead-SED5-GST suspension was stored at 4°C for up to a week. For tethering assays, 1× reaction aliquots of the bead-SED5-GST suspension (50 µl, containing ∼10 µl packed beads) were transferred to 250 µl PCR tubes, then Sly1* (75 pmol; a 1:2 M ratio to Sed5cyt) was added to each reaction tube in 50 µl volume, allowing the Sly1* to bind to the immobilized GST-Sed5cyt in 100 µl final volume. For competition experiments, the Sly1* was preincubated with a sixfold molar excess (450 pmol) of Sed5 Habc or N-Habc domain for 10 min at room temperature before adding the Sly1*-competitor mixture to the beads. Tethering was initiated by adding Texas-red-DHPE labeled liposomes to each 1× tethering reaction (1–6 µl depending on stock concentration). The tethering reactions were incubated for 15–20 min at room temperature and then transferred to wells of chambered coverslips that had been preincubated with FB160BSA for at least 20 min. These preparations were observed at ambient temperature (23 ± 2°C) using a Nikon Ti2 microscope equipped with a Yokogawa CSU-X1 spinning disk confocal unit, a Toptica iChrome MLE laser combiner and launch; 405, 488, 561, and 647 nm diode lasers (Coherent); a Finger Lakes high-speed emission filter wheel; and a Mad City piezoelectric Z-stage. The microscope was controlled by Nikon Elements software, and data analysis and figure preparation were done with the Fiji package of ImageJ software and plug-ins. Tethering reactions were observed using a 10× 0.30 NA Plan Fluor objective and an Andor 888 EMCCD camera operated at an EM gain of 300 with 200-ms exposure per frame.
Tethering was also quantified using bead spin-down assays. Binding reactions were initiated as in the microscopy-based tethering experiments. To quantify liposomes tethered to the beads, the beads were washed once in 1.3 ml of FB160BSA and then sedimented for 1 min at 500 × g in a swinging-bucket rotor. The supernatant was carefully removed and resin-bound lipids were eluted from the beads with 50 µl of BugBuster protein extraction reagent (Millipore). The beads were again sedimented. To quantify the amount of eluted TRPE lipid, 20 µl of the final supernatant was analyzed in a plate-reading fluorimeter (Molecular Devices Gemini XPS or Gemini EM; excitation 595 nm; cutoff 610 nm; and emission 615 nm).
Online supplemental material
Fig. S1 shows the locations and growth phenotypes of select SLY1 alleles in which more than one missense substitution is required to suppress uso1∆. Fig. S2 shows in silico PMIPred estimates of Sly1 α21 and Sly1 pα21 membrane binding parameters. Fig. S3 shows that fusion reactions driven by the Sly1∆loop mutant do not accumulate intermediate reaction products that exhibit lipid but not content mixing. Fig. S4 shows that fusion reactions driven by Sly1 polar loop mutants do not accumulate intermediates exhibiting lipid but not content mixing. Fig. S5 presents GST pulldown assays showing that a set of Sly1 variants do not exhibit major defects in direct binding to Sed5. Table S1 lists SLY1 mutants and summarizes their growth phenotypes. Table S2 shows estimated in vivo protein concentrations and compares them to in vitro concentrations used in fusion reconstitution assays. Table S3 describes the set of engineered loopless Sly1 mutants and summarizes their growth phenotypes. Table S4 describes Sly1 chimeric proteins with the loop fused to the Sly1 N-terminus and describes growth phenotypes of corresponding mutants. Table S5 describes the plasmids and yeast strains used in this study. Table S6 shows the lipid compositions of reconstituted SNARE proteoliposomes used in this study. Data S1 shows full SGA results for sly1∆loop. The same dataset is archived at the DRYAD repository (Plemel et al., 2024; doi: http://doi.org/10.5061/dryad.dr7sqvb5b).
Data availability
The data, as well as plasmids and yeast strains constructed for this study, are available from the corresponding author upon reasonable request. We may ask that requestors pay shipment costs for plasmids or strains. The SGA data and analyses summarized in Fig. 4 and presented in the Data S1 are also archived at the DRYAD repository (Plemel et al., 2024; doi: http://doi.org/10.5061/dryad.dr7sqvb5b).
Acknowledgments
We are grateful to Drs. M. Ailion, J. Bai, R. Baker, J. Cattin, S. Hoppins, I. Topalidou, and M. Zick for helpful advice and critical comments on the manuscript; C. Barlowe (Geisel School of Medicine at Dartmouth, Hanover, NH), C. Boone (University of Toronto, Ontario, Canada), and D. Waugh (National Cancer Institute, Frederick, MD) for antibodies and strains, D. Baker and the University of Washington Institute for Protein Design (Seattle, WA) for computational resources, and D. Beacham (Molecular Probes/Thermo Fisher Scientific, Eugene, Oregon) for gifts of fluorescent reagents.
Our studies are supported by National Institutes of Health (NIH)/National Institute of General Medical Sciences (NIGMS) R01 grants GM077349 and GM130644, and the University of Washington (A.J. Merz), NIH/NIGMS T32 GM007270 (U. Nattermann), NIH MARC T34 GM083883 (B.M. Delgado), and Medical Research Council MC_UP_1201/10 (E.A. Miller).
Author contributions: Conceptualization: A.J. Merz, R.L. Plemel, M. Duan, T. Takenaka, and J. Mima; data curation: R.L. Plemel and A.J. Merz; Formal analysis: A.J. Merz, M. Duan, R.L. Plemel, U. Nattermann, and E.A. Miller; Funding acquisition: A.J. Merz, E. Miller, and D.P. Nickerson; Investigation: M. Duan, R.L. Plemel, T. Takenaka, A. Lin, B.M. Delgado, U. Nattermann, D.P. Nickerson, E.A. Miller, and A.J. Merz; Methodology: M. Duan, R.L. Plemel, T. Takenaka, A. Lin, U. Nattermann, J. Mima, and E.A. Miller; Project administration: A.J. Merz and R.L. Plemel; Resources: J. Mima; Software: U. Nattermann; Supervision: A.J. Merz; Visualization: A.J. Merz and U. Nattermann; Writing—original draft: A.J. Merz; Writing - review and editing: All authors.

