


The outer mitochondrial membrane (OMM) creates a boundary that imports most of the mitochondrial proteome while removing extraneous or damaged proteins. How the OMM senses aberrant proteins and remodels to maintain OMM integrity remains unresolved. Previously, we identified a mitochondrial remodeling mechanism called the mitochondrial-derived compartment (MDC) that removes a subset of the mitochondrial proteome. Here, we show that MDCs specifically sequester proteins localized only at the OMM, providing an explanation for how select mitochondrial proteins are incorporated into MDCs. Remarkably, selective sorting into MDCs also occurs within the OMM, as subunits of the translocase of the outer membrane (TOM) complex are excluded from MDCs unless assembly of the TOM complex is impaired. Considering that overloading the OMM with mitochondrial membrane proteins or mistargeted tail-anchored membrane proteins induces MDCs to form and sequester these proteins, we propose that one functional role of MDCs is to create an OMM-enriched trap that segregates and sequesters excess proteins from the mitochondrial surface.
Introduction
Mitochondria are double membrane–bound organelles that perform critical roles in cell physiology. While the inner mitochondrial membrane (IMM) performs essential roles in cell metabolism, the outer mitochondrial membrane (OMM) creates a key interface that supports mitochondrial biogenesis and establishes mitochondrial connections throughout the cell (Pfanner et al., 2019; Harper et al., 2020). Because 99% of the mitochondrial proteome is coded within nuclear genes, the OMM contains several protein import complexes and targeting receptors that collaborate with cytosolic chaperones to capture mitochondrial precursor proteins and direct them into mitochondria (Wiedemann and Pfanner, 2017). Failing to capture mitochondrial precursor proteins can cause dramatic consequences for the cell, as mitochondrial proteins can aberrantly target to other organelles (Vitali et al., 2018; Xiao et al., 2021; Shakya et al., 2021) or accumulate and aggregate, leading to mitoprotein-induced stress responses (Wang and Chen 2015; Wrobel et al., 2015; Boos et al., 2019). Conversely, the impaired import of mitochondrial precursor proteins induces the action of several mitochondrial quality control pathways that collaborate with the cytosolic ubiquitin-proteasome system to safeguard the integrity of the OMM. These pathways act by recruiting AAA-ATPases that use the energy from ATP hydrolysis to remove stalled, misfolded, or damaged proteins from the OMM and direct these proteins to cytosolic proteasomes for degradation (Heo et al., 2010; Weidberg and Amon, 2018; Mårtensson et al., 2019; Metzger et al., 2020).
The fidelity of mitochondrial protein targeting and import is further challenged by similarities in protein targeting mechanisms that occur at other organelles (Hegde and Keenan, 2022). The ER is a major biosynthetic organelle responsible for importing and folding ∼30% of the cellular proteome, receiving and importing precursor proteins through both co-translational and posttranslational mechanisms (Juszkiewicz and Hegde, 2018). Similar to mitochondrial protein targeting, the loss of correct protein targeting to the ER can lead to some proteins, particularly membrane proteins, mistargeting to mitochondria (Schuldiner et al., 2008; Costa et al., 2018). For example, in the absence of the guided entry of tail-anchored proteins (GET) pathway, tail-anchored (TA) membrane proteins mislocalize, generating cytosolic protein aggregates or mistarget to the OMM (Schuldiner et al., 2008; Li et al., 2019). To protect against these mistargeted TA proteins, mitochondria contain a membrane-embedded AAA-ATPase, ATAD1/Msp1, that directly removes mistargeted TA proteins and delivers these proteins for proteasomal degradation or retargeting (Chen et al., 2014; Wohlever et al., 2017; Li et al., 2019; Matsumoto et al., 2019). While both mitochondria and the ER contain mechanisms to remove aberrant proteins, the accumulation of proteins on the surface of these organelles can also induce dramatic membrane remodeling to preserve organelle homeostasis.
The accumulation of membrane proteins within the ER induces a form of ER microautophagy, whereby the ER membrane proliferates, creating stacked membrane sheets and spherical ER whorls that robustly sequester certain ER membrane proteins, and delivers them to vacuoles/lysosomes for degradation (Wright et al., 1988; Schuck et al., 2009; Schäfer et al., 2020). In mammalian cells, mitochondrial failure and impaired mitochondrial import can lead to the accumulation of PINK1 on the mitochondrial surface (Lazarou et al., 2012; Okatsu et al., 2013), initiating a signaling cascade that leads to the wholesale removal of mitochondria (Pickles et al., 2018). Conversely, rather than the removal of entire organelles, piecemeal degradative mechanisms can facilitate the removal of select portions of mitochondria in a manner that is kinetically faster and less energy-intensive. In response to some stress conditions, particularly mild oxidative damage, mitochondria form small vesicles (60–150 nm diameter) that incorporate damaged proteins and deliver them to lysosomes for degradation (Soubannier et al., 2012). These mitochondrial-derived vesicles (MDVs) have been shown to compensate for the loss of mitophagy and may be an early response pathway that attempts to protect the mitochondrial network prior to the onset of mitophagy (McClelland et al., 2014; Towers et al., 2021). Interestingly, mitochondria can also be induced to shed their outer membrane in response to infection-induced stress elicited by the targeting of pathogen proteins to the OMM (Li et al., 2022). Contrary to MDVs, these infection-induced OMM proliferations are large (several microns in diameter) and appear to contain internal membrane invaginations, somewhat reminiscent of ER whorls (Li et al., 2022).
Previously, in the budding yeast Saccharomyces cerevisiae, we identified a mitochondrial quality control pathway that also involves the remodeling of mitochondrial membranes to form a mitochondrial-derived compartment (MDC). In old-aged yeast cells and in response to acute stressors that alter cellular amino acid and lipid homeostasis, mitochondria form large, spherical compartments that robustly sequester only a minor portion of the mitochondrial proteome (Hughes et al., 2016; Schuler et al., 2021; Xiao et al., 2024). MDCs are generated from dynamic mitochondrial membrane extensions that repeatedly elongate, coalesce, and invaginate to generate distinct mitochondrial domains that robustly sequester select mitochondrial proteins (Wilson et al., 2024). Subsequently, MDCs are removed from mitochondria and delivered to yeast vacuoles for degradation, suggesting that MDCs act as a piecemeal autophagic mechanism that is induced to remodel or ensnare a portion of the mitochondrial proteome (Hughes et al., 2016). The primary cargoes identified within MDCs all include mitochondrial membrane proteins or membrane-associated proteins, including the OMM surface receptor Tom70 and members of the mitochondrial metabolite carrier superfamily. While mitochondrial metabolite carriers are incorporated into MDCs, the majority of other IMM proteins, as well as aqueous proteins from the mitochondrial matrix were all excluded from MDCs (Hughes et al., 2016). How protein cargoes are specifically sorted into MDCs, where these cargo proteins are sorted from, and the purpose for this segregation remains unclear.
Through an examination of protein selectivity in MDCs, we show here that MDCs specifically sequester proteins that are only localized at the OMM, including mitochondrial metabolite carriers mislocalized to the OMM, providing an explanation for how MDCs incorporate a minor subset of the mitochondrial proteome. Intriguingly, cargo sequestration into MDCs is also selective within the OMM, as subunits of the TOM complex are precluded from entering MDCs unless TOM complex assembly is impaired. Prompted by these results and the observation that MDCs sequester several layers of OMM (Wilson et al., 2024), we determined that MDCs form in response to the aberrant accumulation of OMM proteins. By following the mistargeting of TA proteins to the OMM, we show that MDCs can selectively sequester mistargeted TA proteins and thus may act in concordance with other mitochondrial quality control pathways to preserve OMM integrity.
Results
N-terminally tagged mitochondrial metabolite carriers are excluded from MDCs
Previously, we used the yeast GFP collection to identify the cohort of mitochondrial proteins that are sequestered into MDCs. Out of 304 detectable mitochondrial proteins that are C-terminally fused to GFP, only 26 proteins were identified to enter MDCs (Hughes et al., 2016). All of these proteins were integral membrane proteins or membrane-associated proteins from both the OMM and the IMM. A distinct class of proteins that preferentially incorporated into MDCs were mitochondrial metabolite carriers (Hughes et al., 2016). Because we observed that MDCs form in response to certain metabolic stressors (Schuler et al., 2021), we hypothesized that specific intrinsic features of mitochondrial metabolite carriers are important for their selective sorting into MDCs.
To analyze how mitochondrial metabolite carriers are incorporated into MDCs, we used the yeast oxaloacetate carrier, Oac1, as a prototype for MDC cargo sequestration. Oac1 represented an ideal cargo for examining protein selection into MDCs, as Oac1-GFP readily incorporates into MDCs (Hughes et al., 2016) and Oac1 participates in leucine biosynthesis providing a means to assess Oac1 function (Fig. 1 A; Kohlhaw 2003; Marobbio et al., 2008). A double deletion of the LEU4 and OAC1 genes created a yeast strain (leu4∆oac1∆) that is auxotrophic for leucine and growth can be rescued by ectopically expressing Oac1 from a low or high-copy plasmid (Fig. 1 B; Marobbio et al., 2008). Using this analysis, we determined that Oac1 C-terminally fused to GFP produces a non-functional protein as it cannot rescue the growth of the leu4∆oac1∆ strain when grown on a medium lacking leucine (Fig. 1 B). Alternatively, placing GFP at the N-terminus of Oac1 created a functional protein as it rescued the growth of the leu4∆oac1∆ strain (Fig. 1 B). Considering both constructs localize to mitochondria, we compared these two differentially GFP–tagged versions of Oac1 for incorporation into MDCs, which can be induced to form by treating yeast with rapamycin (Rap; Schuler et al., 2021). Intriguingly, while Oac1-GFP readily incorporates into MDCs (marked by the strong enrichment of Tom70–mCherry [Fig. 1 C, white arrows]), GFP–Oac1 is robustly excluded from MDCs (Fig. 1, C and D). The minor percentage (<20%) of GFP–Oac1 we observed at MDCs can be attributed, in part, to our scoring technique, where we first assigned MDCs based on Tom70–mCherry enrichment (which occasionally produces false MDC identifications) and where some MDCs contained a faint but detectable level of GFP–Oac1.

N-terminally tagged mitochondrial metabolite carriers are excluded from MDCs. (A) Diagram of Oac1 transport function within leucine biosynthesis in yeast. KIV, α-ketoisovalerate; α-IPM, α-isopropylmalate; and sLeu4, cytosolic version of Leu4. The enzymatic activity of Leu4 can be compensated by a second α-isopropylmalate synthase, Leu9, localized within the mitochondrial matrix, and because of Leu9 activity, a leu4∆oac1∆ double deletion is required to make leu4∆ yeast auxotrophic for leucine. (B) Cell growth spot assays of leu4∆oac1∆ yeast transformed with the indicated Oac1 constructs expressed from either low-copy (pRS416) or high-copy (pRS426) plasmids, serially diluted and plated on SD agar plates lacking either uracil (SD-Ura) or uracil and leucine (SD-Ura-Leu). (C) Super-resolution confocal fluorescence microscopy images of yeast expressing Tom70–mCherry and Oac1 endogenously tagged with GFP at the C- or N-terminus. Cells were treated with DMSO (vehicle control) or 200 nM Rap. The formation of MDCs in Rap-treated cells is indicated by white arrows. Scale bar = 1 µm. (D) Quantification of the MDC co-localization frequency of the indicated Oac1 proteins in Rap-treated cells. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (E) Quantification of the frequency N-terminally GFP-tagged mitochondrial metabolite carriers co-localize with MDCs. Error bars show mean ± SE of three or four replicates, n ≥ 100 cells per replicate.
N-terminally tagged mitochondrial metabolite carriers are excluded from MDCs. (A) Diagram of Oac1 transport function within leucine biosynthesis in yeast. KIV, α-ketoisovalerate; α-IPM, α-isopropylmalate; and sLeu4, cytosolic version of Leu4. The enzymatic activity of Leu4 can be compensated by a second α-isopropylmalate synthase, Leu9, localized within the mitochondrial matrix, and because of Leu9 activity, a leu4∆oac1∆ double deletion is required to make leu4∆ yeast auxotrophic for leucine. (B) Cell growth spot assays of leu4∆oac1∆ yeast transformed with the indicated Oac1 constructs expressed from either low-copy (pRS416) or high-copy (pRS426) plasmids, serially diluted and plated on SD agar plates lacking either uracil (SD-Ura) or uracil and leucine (SD-Ura-Leu). (C) Super-resolution confocal fluorescence microscopy images of yeast expressing Tom70–mCherry and Oac1 endogenously tagged with GFP at the C- or N-terminus. Cells were treated with DMSO (vehicle control) or 200 nM Rap. The formation of MDCs in Rap-treated cells is indicated by white arrows. Scale bar = 1 µm. (D) Quantification of the MDC co-localization frequency of the indicated Oac1 proteins in Rap-treated cells. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (E) Quantification of the frequency N-terminally GFP-tagged mitochondrial metabolite carriers co-localize with MDCs. Error bars show mean ± SE of three or four replicates, n ≥ 100 cells per replicate.
To determine if other mitochondrial metabolite carriers follow a similar pattern as that observed for Oac1, we analyzed whether mitochondrial metabolite carriers are still sequestered into MDCs when fused to GFP at their N-terminus. To do so, we examined strains from the N-terminal SWAp-Tag (N-SWAT) library (Weill et al., 2018), in which each metabolite carrier is fused at the N-terminus to superfolder GFP (sfGFP) and expressed under the control of the NOP1 promoter. Indeed, the majority of N-terminally sfGFP–tagged metabolite carriers are excluded from entering MDCs unlike the C-terminally GFP–tagged carriers from the yeast GFP collection (Fig. 1 E and Fig. S1 A), which were originally analyzed in our fluorescence microscopy-based screen used to determine proteins incorporated into MDCs (Hughes et al., 2016). Importantly, sfGFP–Mcp1, an OMM protein previously identified to enrich in MDCs (Hughes et al., 2016), is still strongly incorporated into MDCs in 100% of cells analyzed (Fig. 1 E and Fig. S1 A). Furthermore, Tim50 and Ilv2, two proteins excluded from MDCs when C-terminally fused to GFP (Schuler et al., 2021), were still excluded from MDCs when analyzed from the N-SWAT library (Fig. 1 E and Fig. S1 A). Thus, in contrast to our previous analyses on C-terminally tagged mitochondrial carriers (Hughes et al., 2016; Schuler et al., 2021), mitochondrial metabolite carriers are largely excluded from MDCs when visualized using an N-terminal GFP fusion.
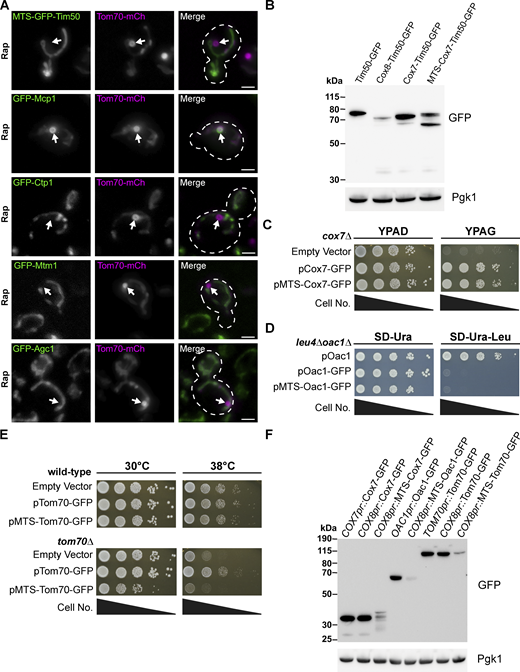
Protein level and functional analysis of MTS–containing fusion proteins. (A) Representative max projections of widefield fluorescence microscopy images of yeast expressing Tom70–mCherry and the indicated sfGFP-tagged strains examined from the N-terminal SWAp-Tag (N-SWAT) library all treated with 200 nM Rap. MDCs are indicated by the white arrows. Scale bar = 2 µm. (B) Immunoblots of whole-cell protein extracts from yeast expressing the indicated proteins. Pgk1 is shown as a loading control. (C) Cell growth spot assays of cox7∆ yeast transformed with the indicated Cox7 constructs or empty vector expressed from a low-copy (pRS416) plasmid serially diluted and plated on either rich media agar plates containing dextrose (YPAD) or glycerol (YPAG). (D) Cell growth spot assays of leu4∆oac1∆ yeast transformed with the indicated Oac1 constructs expressed from a low-copy (pRS416) plasmid serially diluted and plated on SD agar plates lacking either uracil (SD-Ura) or uracil and leucine (SD–Ura–Leu). (E) Cell growth spot assays of wild-type or tom70∆ yeast transformed with the indicated Tom70 constructs or empty vector expressed from a low-copy (pRS416) plasmid serially diluted and plated on YPAD media agar plates and grown at either 30°C or 38°C. (F) Immunoblots of whole-cell protein extracts from yeast expressing the indicated proteins from the indicated promoters. Pgk1 is shown as a loading control. Source data are available for this figure: SourceData FS1.
Protein level and functional analysis of MTS–containing fusion proteins. (A) Representative max projections of widefield fluorescence microscopy images of yeast expressing Tom70–mCherry and the indicated sfGFP-tagged strains examined from the N-terminal SWAp-Tag (N-SWAT) library all treated with 200 nM Rap. MDCs are indicated by the white arrows. Scale bar = 2 µm. (B) Immunoblots of whole-cell protein extracts from yeast expressing the indicated proteins. Pgk1 is shown as a loading control. (C) Cell growth spot assays of cox7∆ yeast transformed with the indicated Cox7 constructs or empty vector expressed from a low-copy (pRS416) plasmid serially diluted and plated on either rich media agar plates containing dextrose (YPAD) or glycerol (YPAG). (D) Cell growth spot assays of leu4∆oac1∆ yeast transformed with the indicated Oac1 constructs expressed from a low-copy (pRS416) plasmid serially diluted and plated on SD agar plates lacking either uracil (SD-Ura) or uracil and leucine (SD–Ura–Leu). (E) Cell growth spot assays of wild-type or tom70∆ yeast transformed with the indicated Tom70 constructs or empty vector expressed from a low-copy (pRS416) plasmid serially diluted and plated on YPAD media agar plates and grown at either 30°C or 38°C. (F) Immunoblots of whole-cell protein extracts from yeast expressing the indicated proteins from the indicated promoters. Pgk1 is shown as a loading control. Source data are available for this figure: SourceData FS1.
Cox7 and Cox8 differ in ability to drive cargo into MDCs
In parallel to our analysis of mitochondrial metabolite carriers, we initiated a separate set of experiments to identify parameters governing the incorporation of Cox7 into MDCs, which was the only other IMM-localized protein we previously identified in MDCs (Fig. 2 A; Hughes et al., 2016). To understand more about Cox7 sequestration into MDCs, we conducted a set of comparative analyses between Cox7 and another subunit of cytochrome c oxidase that is not incorporated into MDCs, Cox8. Both Cox7 and Cox8 are small (∼50 amino acids) proteins containing short matrix-localized segments at their N-terminus and a single C-terminal transmembrane domain (Calder and McEwan 1991; Patterson and Poyton, 1986). However, because they differ in their MDC incorporation, we assessed their ability to dominantly drive a protein into MDCs through the creation of a chimera between Cox7 or Cox8 and the intermembrane space domain of Tim50 (Fig. 2 B). Tim50 is an essential component of the presequence translocase of the inner membrane (Tim23) complex and a protein strongly excluded from MDCs (Fig. 2 A; Hughes et al., 2016). Prior work has demonstrated that the N-terminal portion (1–131) of Tim50, including its transmembrane domain, can be removed or replaced as long as the intermembrane space domain (132–476) of Tim50 is maintained, as it provides the essential function of Tim50 (Mokranjac et al., 2009). Thus, we replaced the N-terminal portion of Tim50–GFP with either full-length Cox7 or full-length Cox8 (Fig. 2 B). We first analyzed the functionality of these Tim50 chimeras by assessing if they could rescue the essentiality of Tim50 through a plasmid shuffle growth assay. A tim50∆ yeast strain expressing Tim50 from a low-copy plasmid carrying a URA3 selection marker was selected against 5-fluorooroctic acid (5-FOA), and cell growth was maintained when an additional low-copy plasmid was present that expressed a functional Tim50–GFP protein (Fig. 2 C). Using this assay, we determined that a Cox8–Tim50–GFP chimera maintains the growth of a tim50∆ strain, while the Cox7–Tim50–GFP chimera failed to rescue the essentiality of Tim50 (Fig. 2 C), demonstrating that the Cox7–Tim50–GFP chimera is non-functional. Because all of these chimeric proteins still localize to mitochondria, we assessed if any of these proteins are sequestered into MDCs. Notably, Cox7–Tim50–GFP strongly incorporated into MDCs, while both Tim50–GFP and Cox8–Tim50–GFP were excluded (Fig. 2, D and E). Importantly, a primary difference between Cox7 and Cox8 is that Cox8 contains a well-annotated, N-terminal mitochondrial targeting presequence (MTS) (Vögtle et al., 2009). When we attached the MTS from Cox8 (codons 1–27) to the N-terminus of Cox7–Tim50–GFP (creating MTS–Cox7–Tim50–GFP; Fig. 2 B), we created a chimeric protein that could rescue the growth of tim50∆ cells (Fig. 2 C), and this chimeric protein was also excluded from MDCs (Fig. 2, D and E). Notably, the MTS–Cox7–Tim50–GFP chimeric protein was processed to generate several protein species (Fig. S1 B), but based on our fluorescence microscopy, it is clear that these processed protein species were still excluded from MDCs (Fig. 2, D and E). Altogether, these results suggest that a key difference in the ability of Cox8 compared with Cox7 to support Tim50 functionality and MDC exclusion is the presence of an MTS.

Cox7 and Cox8 differ in ability to drive Tim50 into MDCs. (A) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and Tim50–GFP or Cox7–GFP. MDCs are indicated by white arrows. Scale bar = 1 µm. (B–E) Schematic of the different Tim50 chimeras analyzed in C–E. (C) Cell growth spot assays of tim50∆ yeast transformed with a low-copy (pRS416) plasmid expressing Tim50 and additionally transformed with low-copy (pRS413) plasmids that express the indicated Tim50 chimeras. Cells were serially diluted and plated on SD agar plates with a full complement of nutrients SD-Complete (SD-Comp) or SD-Complete with 1 µM 5-Fluoroorotic acid (SD-Comp + 5-FOA). (D) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated Tim50 chimeras. MDCs are indicated by the white arrows. Scale bar = 1 µm. (E) Quantification of the frequency the indicated Tim50 chimeras co-localize with MDCs. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate.
Cox7 and Cox8 differ in ability to drive Tim50 into MDCs. (A) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and Tim50–GFP or Cox7–GFP. MDCs are indicated by white arrows. Scale bar = 1 µm. (B–E) Schematic of the different Tim50 chimeras analyzed in C–E. (C) Cell growth spot assays of tim50∆ yeast transformed with a low-copy (pRS416) plasmid expressing Tim50 and additionally transformed with low-copy (pRS413) plasmids that express the indicated Tim50 chimeras. Cells were serially diluted and plated on SD agar plates with a full complement of nutrients SD-Complete (SD-Comp) or SD-Complete with 1 µM 5-Fluoroorotic acid (SD-Comp + 5-FOA). (D) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated Tim50 chimeras. MDCs are indicated by the white arrows. Scale bar = 1 µm. (E) Quantification of the frequency the indicated Tim50 chimeras co-localize with MDCs. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate.
The mitochondrial targeting sequence precludes cargo from entering MDCs
Considering that the addition of an MTS to Cox7–Tim50–GFP excluded this protein from entering MDCs, we examined if the addition of an MTS generally precludes proteins from MDC sequestration. We attached the MTS of Cox8 to Cox7–GFP, Oac1–GFP, and Tom70–GFP and assessed the colocalization of these proteins with MDCs. Indeed, while Cox7–GFP, Oac1–GFP, and Tom70–GFP are all strongly enriched within MDCs, the addition of an MTS to each of these proteins precluded them from entering MDCs (Fig. 3, A and B). Adding an MTS to Cox7–GFP still produces a functional protein because it can rescue the growth of a cox7∆ yeast strain in a medium containing a non-fermentable carbon source (Fig. S1 C). However, adding an MTS to Oac1-GFP or Tom70–GFP creates non-functional proteins (Fig. S1, D and E), which would be expected as the addition of an MTS to Oac1 would disrupt the orientation of Oac1 membrane insertion, and MTS addition to Tom70 would be expected to drive the inward import of Tom70–GFP into mitochondria. Of note, each MTS–containing protein is expressed from the COX8 promoter and is present at lower steady-state levels than the non-MTS–containing proteins. However, this lower protein level is not due to expression from the COX8 promoter because both Cox7–GFP and Tom70–GFP maintain similar steady-state levels regardless of whether they are expressed from their endogenous promoter or the COX8 promoter (Fig. S1 F). Thus, the presence of an MTS, which facilitates the import of proteins into the mitochondrial IMM and matrix, prevents protein incorporation into MDCs.

A mitochondrial targeting sequence precludes cargo from entering MDCs. (A) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and either Cox7–GFP, Oac1–GFP, or Tom70–GFP without or with an N-terminal mitochondrial targeting sequence (MTS). MDCs are indicated by the white arrows. Scale bar = 1 µm. (B) Quantification of the frequency the indicated proteins colocalize with MDCs. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate.
A mitochondrial targeting sequence precludes cargo from entering MDCs. (A) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and either Cox7–GFP, Oac1–GFP, or Tom70–GFP without or with an N-terminal mitochondrial targeting sequence (MTS). MDCs are indicated by the white arrows. Scale bar = 1 µm. (B) Quantification of the frequency the indicated proteins colocalize with MDCs. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate.
MDC cargo are localized to the mitochondrial outer membrane
At this point, our data show that protein functionality does not correlate with MDC incorporation and that the presence of an N-terminal MTS, which normally drives proteins into the IMM or matrix, prevents MDC targeting. Considering our data in an accompanying manuscript shows that MDCs are derived from a distinct remodeling of the OMM (Wilson et al., 2024), we hypothesized that the subdomain localization of mitochondrial proteins is a key determinant for incorporation into MDCs and that Oac1–GFP and Cox7–GFP are sequestered into MDCs because they are mislocalized to the OMM. Thus, to assess the mitochondrial subdomain localization of proteins differentially incorporated into MDCs, we initially used a protease protection assay. While our results supported the interpretation that Cox7–GFP is more restricted to the OMM than MTS–Cox7–GFP and not clearly localized in the IMM, we also observed that some of the membrane proteins we investigated formed protease-resistant populations, including Cox7–GFP, complicating our interpretations of the results from this protease-based assay (Fig. S2 A, see below for MTS–Oac1-GFP). Instead, we adopted a different strategy based on the observation that we could directly visualize isolated mitochondria using super-resolution confocal fluorescence microscopy to assess protein localization within mitochondria. Using this strategy, we could resolve that Tim50–GFP, as well as Tim18–GFP and Tim21–GFP (two other IMM proteins that do not localize to MDCs), were all internally localized within mitochondria compared with Tom70–mCherry, an OMM protein (Fig. 4 A and Fig. S2 B). Similarly, both Cox15–GFP (IMM protein) and Lat1–GFP (matrix protein) showed a diffuse internal fluorescence inside mitochondria compared with Tom70–mCherry (Fig. 4 A). Alternatively, mitochondria isolated from cells expressing Tom20–GFP and Tom70–mCherry showed that these proteins co-localized, which would be expected as both are OMM proteins (Fig. 4 A). These results are consistent with prior observations on the mitochondrial subdomain localizations for each of these proteins (Vögtle et al., 2017) and demonstrate that direct visualization of isolated mitochondria in vitro can be utilized to identify proteins localized to the OMM from those localized to the interior of mitochondria.
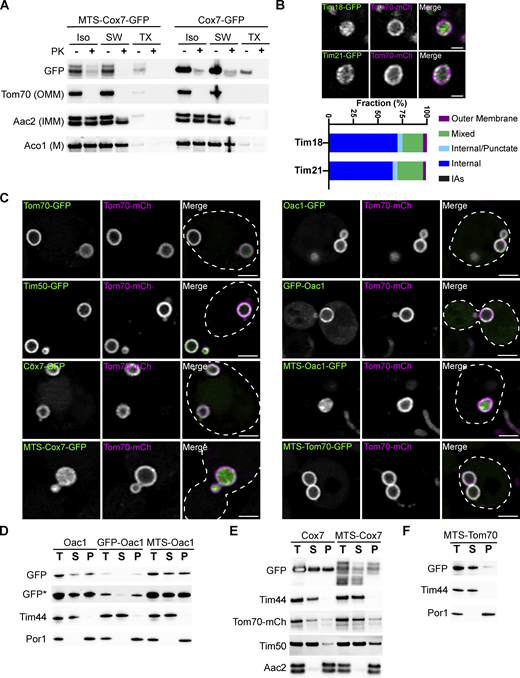
MDC cargo are localized to the outer mitochondrial membrane. (A) Immunoblot of a protease protection assay on isolated mitochondria from yeast cells expressing Tom70–mCherry and either Cox7–GFP or MTS–Cox7–GFP. The extent of proteinase K (PK) protection was analyzed through immunoblot with antibodies specific to either GFP, mCherry for Tom70–mCherry (OMM), Aac2 (IMM), or Aco1 (matrix, M). ISO = iso-osmolar buffer, SW = hypo-osmolar swelling, TX = 1% Triton X-100. (B) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and either Tim18-GFP or Tim21-GFP. Scale bar = 1 µm. Quantifications of the localization of each indicated protein are shown below as a percent of the total mitochondria observed, n ≥ 100. (C) Super-resolution confocal fluorescence microscopy images of swollen mitochondria in yeast cells expressing Tom70–mCherry, Mdm12-6xFLAG-AID and OsTir1 treated with 1 mM auxin for 3 h. These yeast cells were also transformed with a low-copy plasmid (pRS416) expressing the indicated GFP–tagged proteins. Scale bar = 2 µm. (D–F) Mitochondrial membrane integration was analyzed through a carbonate extraction assay. Mitochondria isolated from yeast cells expressing either Oac1-GFP (D), GFP–Oac1 (D), MTS–Oac1-GFP (D), Cox7–GFP (E), MTS–Cox7–GFP (E), or MTS–Tom70–GFP (F) were treated with 100 mM Na2CO3 pH 11 to transform mitochondrial membranes into membrane sheets. Membranes were precipitated through ultracentrifugation and the extent that proteins remained in the membrane pellet fraction (P) compared to the supernatant (S) was determined through immunoblot with antibodies specific to either GFP, mCherry for Tom70–mCherry (OMM protein), Tim50 (IMM protein), Aac2 (polytopic IMM protein), Por1 (OMM protein), or Tim44 (membrane-associated matrix protein). A total (T) untreated sample of isolated mitochondria was also analyzed as a control for input. * indicates a longer exposure. Source data are available for this figure: SourceData FS2.
MDC cargo are localized to the outer mitochondrial membrane. (A) Immunoblot of a protease protection assay on isolated mitochondria from yeast cells expressing Tom70–mCherry and either Cox7–GFP or MTS–Cox7–GFP. The extent of proteinase K (PK) protection was analyzed through immunoblot with antibodies specific to either GFP, mCherry for Tom70–mCherry (OMM), Aac2 (IMM), or Aco1 (matrix, M). ISO = iso-osmolar buffer, SW = hypo-osmolar swelling, TX = 1% Triton X-100. (B) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and either Tim18-GFP or Tim21-GFP. Scale bar = 1 µm. Quantifications of the localization of each indicated protein are shown below as a percent of the total mitochondria observed, n ≥ 100. (C) Super-resolution confocal fluorescence microscopy images of swollen mitochondria in yeast cells expressing Tom70–mCherry, Mdm12-6xFLAG-AID and OsTir1 treated with 1 mM auxin for 3 h. These yeast cells were also transformed with a low-copy plasmid (pRS416) expressing the indicated GFP–tagged proteins. Scale bar = 2 µm. (D–F) Mitochondrial membrane integration was analyzed through a carbonate extraction assay. Mitochondria isolated from yeast cells expressing either Oac1-GFP (D), GFP–Oac1 (D), MTS–Oac1-GFP (D), Cox7–GFP (E), MTS–Cox7–GFP (E), or MTS–Tom70–GFP (F) were treated with 100 mM Na2CO3 pH 11 to transform mitochondrial membranes into membrane sheets. Membranes were precipitated through ultracentrifugation and the extent that proteins remained in the membrane pellet fraction (P) compared to the supernatant (S) was determined through immunoblot with antibodies specific to either GFP, mCherry for Tom70–mCherry (OMM protein), Tim50 (IMM protein), Aac2 (polytopic IMM protein), Por1 (OMM protein), or Tim44 (membrane-associated matrix protein). A total (T) untreated sample of isolated mitochondria was also analyzed as a control for input. * indicates a longer exposure. Source data are available for this figure: SourceData FS2.

MDC cargo are localized to the outer mitochondrial membrane. (A) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and either Tim50–GFP, Cox15-GFP, Lat1-GFP, or Tom20–GFP. Scale bar = 1 µm. (B) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and either Cox7–GFP, MTS–Cox7–GFP, MTS–Tom70–GFP, Oac1–GFP, GFP–Oac1, or MTS–Oac1–GFP. Scale bar = 1 µm. (C–E) Examples of the diverse protein localizations observed in purified mitochondria compared to Tom70–mCherry used for the quantifications in D and E. (D) Quantification of the localization of each indicated protein imaged in A within purified mitochondria shown as a percent of the total mitochondria observed, n ≥ 100. (E) Quantification of the localization of each indicated protein imaged in B within purified mitochondria shown as a percent of the total mitochondria observed, n ≥ 100.
MDC cargo are localized to the outer mitochondrial membrane. (A) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and either Tim50–GFP, Cox15-GFP, Lat1-GFP, or Tom20–GFP. Scale bar = 1 µm. (B) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and either Cox7–GFP, MTS–Cox7–GFP, MTS–Tom70–GFP, Oac1–GFP, GFP–Oac1, or MTS–Oac1–GFP. Scale bar = 1 µm. (C–E) Examples of the diverse protein localizations observed in purified mitochondria compared to Tom70–mCherry used for the quantifications in D and E. (D) Quantification of the localization of each indicated protein imaged in A within purified mitochondria shown as a percent of the total mitochondria observed, n ≥ 100. (E) Quantification of the localization of each indicated protein imaged in B within purified mitochondria shown as a percent of the total mitochondria observed, n ≥ 100.
Occasionally, we found that protein localizations were more complex than either overlapping with Tom70–mCherry or residing inside the Tom70–mCherry marked outer membrane ring (Fig. 4 B). To quantify our observations, we assigned protein localizations to the following categories: overlapping with Tom70–mCherry (outer membrane), overlapping with Tom70–mCherry and internal luminal fluorescence (mixed), internal luminal fluorescence with discrete puncta that overlapped with Tom70–mCherry (internal/punctate), internal luminal fluorescence only (internal), and bright internal puncta (internal aggregates, IAs) (Fig. 4 C). Using these categories to quantify protein localizations for several of our control proteins, we observed that Tim50–GFP, Tim18–GFP, Tim21–GFP, and Cox15–GFP are internally localized in >70% of mitochondria, Lat1–GFP was always internally localized, while Tom20–GFP strongly overlapped with Tom70–mCherry on the outer membrane of isolated mitochondria (Fig. 4 D and Fig. S2 B). These results are consistent with observations performed in vivo using swollen mitochondria to determine the subdomain localizations of mitochondrial proteins (Wurm and Jakobs, 2006), except they can additionally resolve intramitochondrial proteins that localize to the inner boundary membrane.
Using this fluorescence microscopy strategy to analyze protein localizations on isolated mitochondria, we observed that both Cox7–GFP and Oac1–GFP, two proteins that incorporate into MDCs, strongly colocalized with Tom70–mCherry on the outer membrane of isolated mitochondria (Fig. 4, B and E). Conversely, MTS–Cox7–GFP and MTS–Oac1–GFP, two protein constructs that are excluded from MDCs, were strongly localized within the interior of isolated mitochondria (Fig. 4, B and E). Note that MTS–Oac1–GFP frequently formed internal aggregates (Fig. 4, B and E), which was not surprising considering that the addition of an MTS to Oac1 would be expected to disrupt the topology of Oac1 within the IMM. Both GFP–Oac1 and MTS–Tom70–GFP, two other constructs that were excluded from MDCs, showed more of an internal localization within isolated mitochondria (Fig. 4, B and E), albeit not as robustly as MTS–Cox7–GFP or MTS–Oac1–GFP.
Supporting our observations from isolated mitochondria, we also observed an intramitochondrial localization for MTS–Cox7–GFP and MTS–Oac1–GFP by acutely swelling mitochondria in vivo (Fig. S2 C). To do so, we used an auxin-induced degron system to acutely dissolve the ER–mitochondria encounter structure (ERMES), which resulted in the appearance of swollen mitochondrial spheres as previously reported (John Peter et al., 2022). While this experiment allowed us to observe MTS–Cox7–GFP and MTS–Oac1–GFP within the interior of swollen mitochondria, we could not distinguish some proteins (Tim50–GFP and Oac1–GFP) expected to localize to the inner boundary membrane from proteins (Tom70–mCherry) that localize to the OMM similar to previous observations by others (Fig. S2 C; Wurm and Jakobs, 2006). One additional possibility for the difference in protein localization of the MTS–containing membrane proteins is that they were aberrantly imported into the mitochondrial matrix. However, upon using a carbonate extraction assay to examine the integration of these proteins within mitochondrial membranes, we observed that MTS–Oac1–GFP, MTS–Cox7–GFP, and MTS–Tom70–GFP all integrate into membranes to a similar extent as the non-MTS containing versions (Fig. S2, D–F).
Based on observations with Oac1, we also compared the subdomain localization of other metabolite carriers that can be fused to GFP at the N- or C-termini and still target mitochondria. Similar to Oac1, both Dic1–GFP and Yhm2–GFP localized more frequently to the OMM compared with the N-terminally GFP–tagged versions, GFP–Dic1 and GFP–Yhm2 (Fig. S3, A and B). Consistent with this observation, Dic1–GFP and Yhm2–GFP also colocalized with ∼60% of Tom70–mCherry marked MDCs, while GFP–Dic1 and GFP–Yhm2 were excluded from the majority of MDCs (Fig. S3, C and D). Conversely, both Mtm1 and Ymc2 localized internally within isolated mitochondria regardless of whether they were N- or C-terminally tagged with GFP (Fig. S3, A and B). While we had previously shown that Mtm1–GFP and Ymc2–GFP can become enriched within MDCs (Schuler et al., 2021), our quantification on the percent of MDCs that contained these metabolite carriers revealed that Ymc2–GFP, GFP–Ymc2, and GFP–Mtm1 are strongly excluded from MDCs in >80% of cells, while Mtm1–GFP is excluded from MDCs in ∼65% of cells (Fig. S3, C and D). In total, the combination of our results using MTS–containing chimeric proteins and these mitochondrial subdomain localization studies all suggest that a key determinant for proteins sequestered into MDCs is their localization to the OMM, which could include proteins aberrantly localized to the OMM, such as Oac1–GFP and Cox7–GFP.

GFP–tagged mitochondrial metabolite carriers that localize within mitochondria are excluded from MDCs. (A) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and the indicated GFP-tagged mitochondrial metabolite carriers. Scale bar = 1 µm. (B) Quantifications of the localization of each indicated protein are shown in A as a percent of the total mitochondria observed, n ≥ 100. (C) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated mitochondrial metabolite carrier C-terminally tagged to GFP. MDCs are indicated by the white arrows. Scale bar = 1 μm. (D) Quantification of the frequency of the indicated GFP-tagged mitochondrial metabolite carriers co-localize with MDCs. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (E) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated intermembrane space proteins C-terminally tagged to GFP. MDCs are indicated by the white arrows. Scale bar = 1 µm. Images from Ccp1-GFP cells are max projections, the other images are from a single z-slice.
GFP–tagged mitochondrial metabolite carriers that localize within mitochondria are excluded from MDCs. (A) Super-resolution confocal fluorescence microscopy images of purified mitochondria from cells expressing Tom70–mCherry and the indicated GFP-tagged mitochondrial metabolite carriers. Scale bar = 1 µm. (B) Quantifications of the localization of each indicated protein are shown in A as a percent of the total mitochondria observed, n ≥ 100. (C) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated mitochondrial metabolite carrier C-terminally tagged to GFP. MDCs are indicated by the white arrows. Scale bar = 1 μm. (D) Quantification of the frequency of the indicated GFP-tagged mitochondrial metabolite carriers co-localize with MDCs. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (E) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated intermembrane space proteins C-terminally tagged to GFP. MDCs are indicated by the white arrows. Scale bar = 1 µm. Images from Ccp1-GFP cells are max projections, the other images are from a single z-slice.
Previously, we determined that all detectable mitochondrial matrix proteins and IMM proteins (which now include mitochondrial metabolite carriers and Cox7) were excluded from MDCs, and originally annotated IMS proteins were also excluded from MDCs (Hughes et al., 2016). We considered that IMS proteins may still enter MDCs but potentially could not be detected in our initial widefield fluorescence microscopy-based screen. However, using super-resolution confocal fluorescence microscopy, we still observed that several IMS proteins are strongly excluded from MDCs (Fig. S3 E). Thus, our results suggest that MDCs incorporate membrane cargo proteins exclusively from the OMM and restrict proteins from intramitochondrial subdomains.
Subunits of the TOM complex are excluded from MDCs unless complex assembly is impaired
While MDC cargo is incorporated from the OMM, we also observed that not all OMM proteins are sequestered into MDCs to the same extent. For example, the OMM receptor for MTS–containing mitochondrial proteins, Tom20, is incorporated into MDCs, but unlike Tom70, it is not enriched within MDCs (Fig. 5, A and B). A line-scan analysis showed that the amount of Tom20–GFP incorporated into MDCs is similar to the concentration of Tom20–GFP found throughout the mitochondrial tubule, highlighted by a mean MDC to mitochondrial tubule fluorescence intensity ratio close to 1 (1.22; Fig. 5, B and C). This observation is in contrast to Tom70–mCherry, which is strongly enriched within MDCs with a mean MDC to mitochondrial tubule fluorescence intensity ratio of 8.20 (Fig. 5 C). To begin to assess how Tom20 is limited from MDC sequestration, we truncated Tom20 at regions flanking the tetratricopeptide motif previously shown to bind MTS–containing precursor proteins (Abe et al., 2000), creating a truncation that only maintained the N-terminal transmembrane domain of Tom20 (Tom201–50-GFP) and a truncation that removed the final 38 C-terminal amino acids of Tom20 (Tom201–145-GFP; Fig. 5 A). Intriguingly, while Tom201–145-GFP acted similarly to full-length Tom20–GFP and was not enriched within MDCs (Fig. 5, B and C), Tom201–50-GFP was robustly sequestered within MDCs (Fig. 5 B), highlighted by a threefold enrichment into MDCs (Fig. 5 C), demonstrating that the Tom20 C-terminal precursor-binding domain is required to limit Tom20 incorporation into MDCs. Notably, we also observed that removing the precursor-binding domain of Tom70 (Tom701–98-GFP) did not disrupt the robust accumulation of this protein into MDCs (Fig. 5, B and C), but appeared to increase enrichment within MDCs, potentially indicating that an OMM-targeting transmembrane domain, by itself, is sufficient for MDC sequestration.
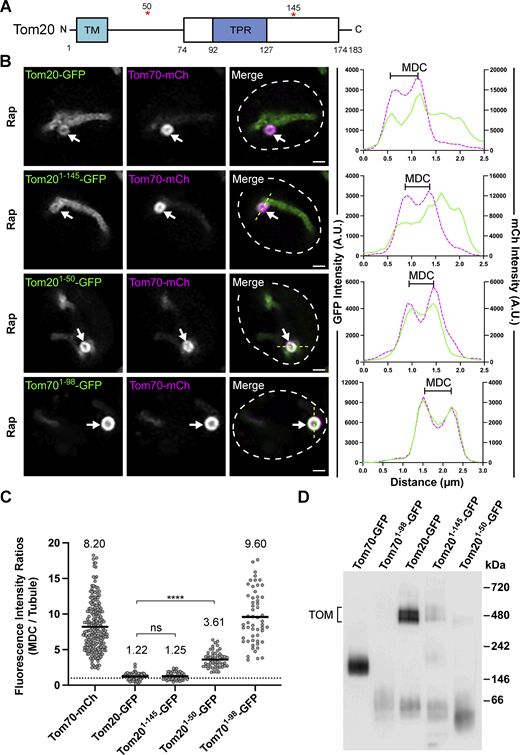
Tom20 becomes enriched in MDCs when truncated to its N-terminal transmembrane domain. (A) Schematic of the Tom20 protein. The white box indicates the cytoplasmic core domain of Tom20 that contains a single tetratricopeptide (TPR) motif. The red asterisks indicate the residues where Tom20 was truncated in B–D. TM = Transmembrane domain. (B) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated Tom20–GFP proteins or a truncated version of Tom70, Tom701–98-GFP. MDCs are indicated by the white arrows. Scale bar = 1 µm. The yellow line marks the position of the line-scan fluorescence intensity profile. The left and right Y axis corresponds to the GFP and mCherry fluorescence intensity, respectively. Bracket marks MDC. (C) Quantification of protein enrichment within MDCs from the yeast strains analyzed in A. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared with the fluorescence intensity in the mitochondrial tubule. The dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. Unpaired t tests with Welch’s correction were used to determine statistical significance; ns, not significant; ****, P < 0.0001. (D) Blue native electrophoresis on isolated mitochondria from yeast strains expressing the indicated proteins. Proteins were detected with an antibody against GFP and the bracket indicates the TOM complex. Source data are available for this figure: SourceData F5.
Tom20 becomes enriched in MDCs when truncated to its N-terminal transmembrane domain. (A) Schematic of the Tom20 protein. The white box indicates the cytoplasmic core domain of Tom20 that contains a single tetratricopeptide (TPR) motif. The red asterisks indicate the residues where Tom20 was truncated in B–D. TM = Transmembrane domain. (B) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and the indicated Tom20–GFP proteins or a truncated version of Tom70, Tom701–98-GFP. MDCs are indicated by the white arrows. Scale bar = 1 µm. The yellow line marks the position of the line-scan fluorescence intensity profile. The left and right Y axis corresponds to the GFP and mCherry fluorescence intensity, respectively. Bracket marks MDC. (C) Quantification of protein enrichment within MDCs from the yeast strains analyzed in A. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared with the fluorescence intensity in the mitochondrial tubule. The dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. Unpaired t tests with Welch’s correction were used to determine statistical significance; ns, not significant; ****, P < 0.0001. (D) Blue native electrophoresis on isolated mitochondria from yeast strains expressing the indicated proteins. Proteins were detected with an antibody against GFP and the bracket indicates the TOM complex. Source data are available for this figure: SourceData F5.
Contrary to Tom70, Tom20 is more stably associated with the TOM complex as shown by blue native electrophoresis (Fig. 5 D and Fig. S4 A; Dekker et al., 1998), which could partially explain why Tom20–GFP is not enriched within MDCs. Supporting this idea, the Tom201–50-GFP mutant no longer associates with the TOM complex while the Tom201–145–GFP still associates with the TOM complex, albeit to a lesser extent than full-length Tom20–GFP (Fig. 5 D). Prompted by these results analyzing Tom20, we examined if core subunits of the TOM complex also showed a limited enrichment within MDCs. Strikingly, GFP–Tom22, GFP–Tom5, and GFP–Tom7 all showed a strong de-enrichment within MDCs, as each of these TOM complex subunits was observed to have mean MDC to mitochondrial tubule fluorescence intensity ratios below 1 (Fig. 6, A–D). Because TOM complex assembly is not disrupted during MDC-inducing conditions (Fig. S4 B), we examined if assembly into TOM complexes is a key determinant that restricts TOM complex subunits from entering MDCs. To do so, we analyzed MDC enrichment of TOM complex subunits in cells lacking TOM6 (tom6∆), which encodes a key but non-essential subunit of the TOM complex that supports complex assembly (Fig. 6, E–G and Fig. S4 A; Dekker et al., 1998). Intriguingly, within tom6∆ cells, GFP–Tom22, GFP–Tom5, and GFP–Tom7 all robustly sequestered within MDCs with a comparable threefold enrichment (Fig. 6, A–D), demonstrating that core subunits of the TOM complex are typically precluded from MDC sequestration unless TOM complex assembly is impaired. Supporting our observations analyzing subunits of the TOM complex, we also observed that a functional GFP–Sam37 protein (assessed by growth with a tom70∆ strain, Fig. S4 C; Gratzer et al., 1995), which is a subunit of the sorting and assembly machinery (SAM) complex, was precluded from MDC sequestration (Fig. S4, D and E). Additionally, by using indirect immunofluorescence, we observed that the yeast voltage-dependent anion channel, Por1, is de-enriched from MDCs (Fig. S4, F and G). Thus, multiple OMM proteins that are subunits of large protein complexes are all restricted from MDC sequestration. While the current mechanism for how these protein complexes are precluded from MDCs remains unclear, these results demonstrate that additional sorting mechanisms exist within the OMM to direct certain protein cargoes into MDCs.
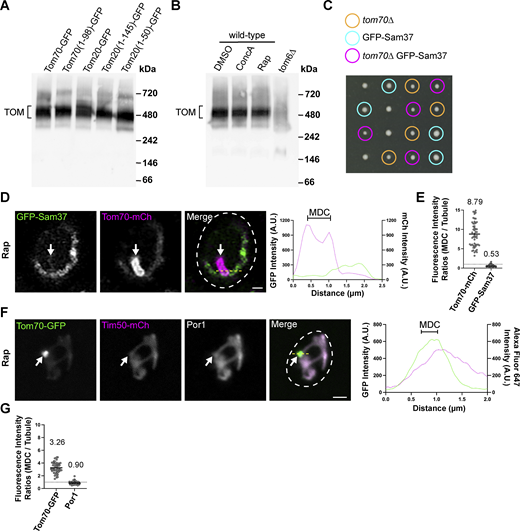
Por1 and GFP–Sam37 are excluded from MDCs. (A) Blue native electrophoresis on isolated mitochondria from yeast strains expressing the indicated proteins. Protein complexes were detected with an antibody against Tom40 and the bracket indicates the TOM complex. (B) Blue native electrophoresis on isolated mitochondria from wild-type or tom6∆ yeast strains. Wild-type yeast was treated with either DMSO, 500 nM ConcA, or 200 nM Rap for 2 h prior to mitochondria isolation. Protein complexes were detected with an antibody against Tom40 and the bracket indicates the TOM complex. (C) Growth of individual haploid spores from a tetrad dissection of spores generated from a heterozygous diploid yeast strain (TOM70/tom70∆ GFP–SAM37/SAM37). The genotype of the spores are indicated. (D) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and GFP–Sam37. MDCs are indicated by the white arrows. Scale bar = 1 µm. Yellow line marks the position of the line-scan fluorescence intensity profile. Left and right Y axis corresponds to the GFP and mCherry fluorescence intensity, respectively. Bracket marks MDC. (E) Quantification of protein enrichment within MDCs from the yeast strain analyzed in D. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared to the fluorescence intensity in the mitochondrial tubule. The dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. (F) Indirect immunofluorescence on yeast expressing Tom70–yeGFP and Tim50–mCherry after Rap-induced MDC formation. Por1 localization was detected with monoclonal primary antibodies and secondary antibodies conjugated to Alexa Fluor 647. MDCs are indicated by the white arrows. Scale bar = 2 µm. Yellow line marks the position of the line-scan fluorescence intensity profile. Left and right Y axis corresponds to the GFP and Alexa Fluor 647 fluorescence intensity, respectively. Bracket marks MDC. (G) Quantification of protein enrichment within MDCs from the yeast strain analyzed in F. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared to the fluorescence intensity in the mitochondrial tubule. Dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. Source data are available for this figure: SourceData FS4.
Por1 and GFP–Sam37 are excluded from MDCs. (A) Blue native electrophoresis on isolated mitochondria from yeast strains expressing the indicated proteins. Protein complexes were detected with an antibody against Tom40 and the bracket indicates the TOM complex. (B) Blue native electrophoresis on isolated mitochondria from wild-type or tom6∆ yeast strains. Wild-type yeast was treated with either DMSO, 500 nM ConcA, or 200 nM Rap for 2 h prior to mitochondria isolation. Protein complexes were detected with an antibody against Tom40 and the bracket indicates the TOM complex. (C) Growth of individual haploid spores from a tetrad dissection of spores generated from a heterozygous diploid yeast strain (TOM70/tom70∆ GFP–SAM37/SAM37). The genotype of the spores are indicated. (D) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in yeast expressing Tom70–mCherry and GFP–Sam37. MDCs are indicated by the white arrows. Scale bar = 1 µm. Yellow line marks the position of the line-scan fluorescence intensity profile. Left and right Y axis corresponds to the GFP and mCherry fluorescence intensity, respectively. Bracket marks MDC. (E) Quantification of protein enrichment within MDCs from the yeast strain analyzed in D. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared to the fluorescence intensity in the mitochondrial tubule. The dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. (F) Indirect immunofluorescence on yeast expressing Tom70–yeGFP and Tim50–mCherry after Rap-induced MDC formation. Por1 localization was detected with monoclonal primary antibodies and secondary antibodies conjugated to Alexa Fluor 647. MDCs are indicated by the white arrows. Scale bar = 2 µm. Yellow line marks the position of the line-scan fluorescence intensity profile. Left and right Y axis corresponds to the GFP and Alexa Fluor 647 fluorescence intensity, respectively. Bracket marks MDC. (G) Quantification of protein enrichment within MDCs from the yeast strain analyzed in F. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared to the fluorescence intensity in the mitochondrial tubule. Dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. Source data are available for this figure: SourceData FS4.
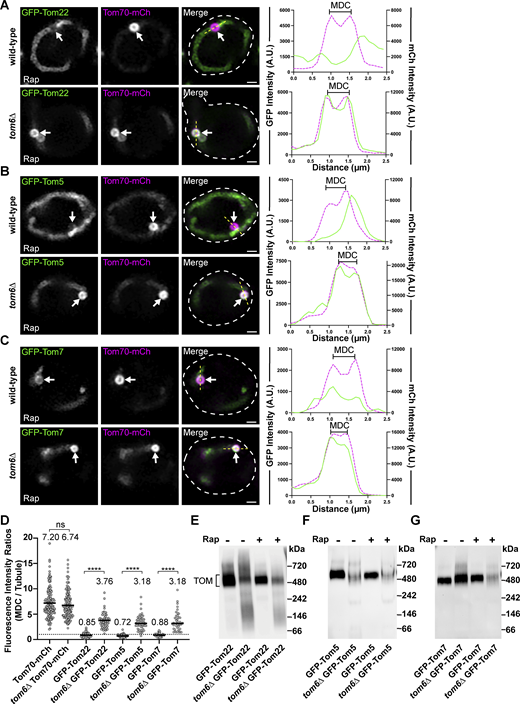
Subunits of the TOM complex are excluded from MDCs unless complex assembly is impaired. (A–C) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in wild-type or tom6∆ yeast expressing Tom70–mCherry and the indicated TOM complex subunits N-terminally tagged with GFP. MDCs are indicated by the white arrows. Scale bar = 1 µm. The yellow line marks the position of the line-scan fluorescence intensity profile. Left and right Y axis corresponds to the GFP and mCherry fluorescence intensity, respectively. Bracket marks MDC. (D) Quantification of protein enrichment within MDCs from wild-type or tom6∆ yeast strains analyzed in A–C. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared to the fluorescence intensity in the mitochondrial tubule. Dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. Unpaired t tests with Welch’s correction were used to determine statistical significance; ns, not significant; ****, P < 0.0001. (E–G) Blue native electrophoresis on isolated mitochondria from wild-type or tom6∆ yeast strains expressing the indicated proteins and treated with either DMSO (−) or 200 nM Rap (+) for 2 h prior to mitochondria isolation. Proteins were detected with an antibody against GFP and the bracket indicates the TOM complex. Source data are available for this figure: SourceData F6.
Subunits of the TOM complex are excluded from MDCs unless complex assembly is impaired. (A–C) Super-resolution confocal fluorescence microscopy images of Rap-induced MDC formation in wild-type or tom6∆ yeast expressing Tom70–mCherry and the indicated TOM complex subunits N-terminally tagged with GFP. MDCs are indicated by the white arrows. Scale bar = 1 µm. The yellow line marks the position of the line-scan fluorescence intensity profile. Left and right Y axis corresponds to the GFP and mCherry fluorescence intensity, respectively. Bracket marks MDC. (D) Quantification of protein enrichment within MDCs from wild-type or tom6∆ yeast strains analyzed in A–C. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared to the fluorescence intensity in the mitochondrial tubule. Dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. Unpaired t tests with Welch’s correction were used to determine statistical significance; ns, not significant; ****, P < 0.0001. (E–G) Blue native electrophoresis on isolated mitochondria from wild-type or tom6∆ yeast strains expressing the indicated proteins and treated with either DMSO (−) or 200 nM Rap (+) for 2 h prior to mitochondria isolation. Proteins were detected with an antibody against GFP and the bracket indicates the TOM complex. Source data are available for this figure: SourceData F6.
Overloading the OMM with membrane proteins induces MDC formation
An analysis of MDC formation demonstrated that OMM-derived extensions repeatedly elongate, coalesce, and invaginate to generate compartments that capture OMM cargo and cytosol within a protected domain (Wilson et al., 2024). These structures are similar to the membrane-enriched domains generated by the ER in response to the overexpression and aberrant accumulation of some ER membrane proteins, which has been described as a form of ER microautophagy (Wright et al., 1988; Schuck et al., 2014). Considering the striking similarity between MDCs and ER microautophagy, we examined if the overexpression of OMM proteins induces the generation of MDCs from mitochondria. Indeed, a vast array of OMM proteins were all capable of inducing constitutive MDC formation when individually overexpressed (Fig. 7 A and Fig. S5 A). Using blue native electrophoresis, we observed that TOM complex assembly was not strongly impaired in cells overexpressing Scm4, Tom7, and Ysc83, all strong inducers of MDC formation, demonstrating that the overexpression of these proteins was not inducing MDC formation through disruption of TOM complex assembly (Fig. S5 B). Notably, many of the proteins that led to robust MDC formation are all OMM proteins that are strongly sequestered in MDCs (Fig. S5 C); however, these proteins are not strongly sequestered into MDCs just because they are abundant OMM proteins (Fig. S5 D). Rather some abundant proteins, like Tom20 and TOM complex subunits, are excluded from MDCs, while less abundant proteins, like Fzo1 and Tcd2, can become enriched within MDCs (Fig. S5 D), suggesting that while protein overexpression can induce MDC formation, protein abundance within the OMM cannot fully explain why some proteins become enriched while other OMM proteins are excluded.

Overloading the OMM with membrane proteins induces MDC formation. (A) Quantification of the percent of cells forming Tom70–GFP–enriched MDCs in cells constitutively overexpressing the indicated proteins. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (B) Quantification of the percent of cells forming Tom70–GFP–enriched MDCs in the indicated yeast strains treated with DMSO (vehicle control), 500 nM Concanamycin A (ConcA), or 200 nM Rap. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (C) Super-resolution confocal fluorescence microscopy images of wild-type or get1∆get2∆ yeast strains expressing Tom70–mCherry and overexpressing GFP–Pex15 treated with DMSO or 200 nM Rap. Yellow arrows indicate mitochondrial tubules, white arrows indicate MDCs and red arrows indicate peroxisomes. Scale bar = 1 µm. (D) Super-resolution confocal fluorescence microscopy images of wild-type or get1∆get2∆ yeast strains expressing Tom70–mCherry and overexpressing GFP–Ubc6 treated with DMSO or 200 nM Rap. Yellow arrows indicate mitochondrial tubules and white arrows indicate MDCs. Scale bar = 1 µm. (E) Quantification of the percent of cells containing mitochondrial localized GFP–Pex15 or GFP–Ubc6 in the indicated yeast strains. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (F) Quantification of protein enrichment within MDCs from the yeast strains analyzed in C and D. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared with the fluorescence intensity in the mitochondrial tubule. Dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. (G) Model of MDC sequestration of surplus proteins from the OMM.
Overloading the OMM with membrane proteins induces MDC formation. (A) Quantification of the percent of cells forming Tom70–GFP–enriched MDCs in cells constitutively overexpressing the indicated proteins. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (B) Quantification of the percent of cells forming Tom70–GFP–enriched MDCs in the indicated yeast strains treated with DMSO (vehicle control), 500 nM Concanamycin A (ConcA), or 200 nM Rap. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (C) Super-resolution confocal fluorescence microscopy images of wild-type or get1∆get2∆ yeast strains expressing Tom70–mCherry and overexpressing GFP–Pex15 treated with DMSO or 200 nM Rap. Yellow arrows indicate mitochondrial tubules, white arrows indicate MDCs and red arrows indicate peroxisomes. Scale bar = 1 µm. (D) Super-resolution confocal fluorescence microscopy images of wild-type or get1∆get2∆ yeast strains expressing Tom70–mCherry and overexpressing GFP–Ubc6 treated with DMSO or 200 nM Rap. Yellow arrows indicate mitochondrial tubules and white arrows indicate MDCs. Scale bar = 1 µm. (E) Quantification of the percent of cells containing mitochondrial localized GFP–Pex15 or GFP–Ubc6 in the indicated yeast strains. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (F) Quantification of protein enrichment within MDCs from the yeast strains analyzed in C and D. Each dot represents the ratio of the fluorescence intensity measured within MDCs compared with the fluorescence intensity in the mitochondrial tubule. Dark line indicates the mean with the mean value shown above. The dotted line demarcates a ratio of 1. (G) Model of MDC sequestration of surplus proteins from the OMM.
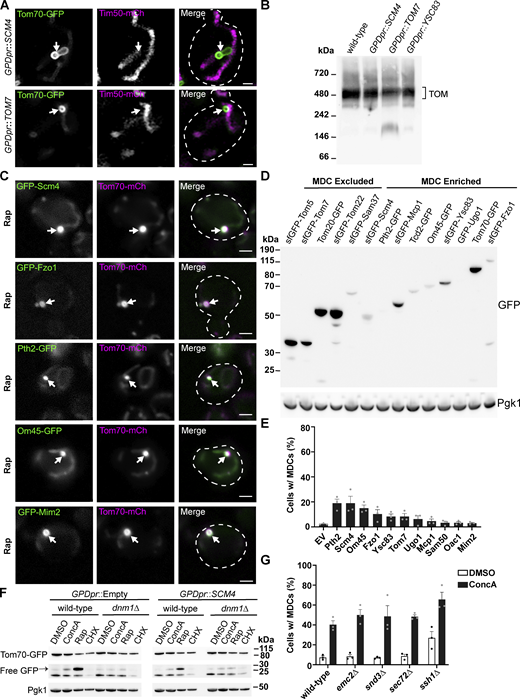
Overloading the OMM with membrane proteins induces MDC formation. (A) Super-resolution confocal fluorescence microscopy images of cells expressing Tom70–GFP and Tim50–mCherry and constitutively overexpressing Scm4 or Tom7. MDCs are indicated by the white arrows. Scale bar = 1 µm. (B) Blue native electrophoresis on isolated mitochondria from yeast strains overexpressing the indicated proteins. Protein complexes were detected with an antibody against Tom40 and the bracket indicates the TOM complex. (C) Representative max projections of widefield fluorescence microscopy images of MDC formation of yeast expressing Tom70–mCherry and the indicated OMM proteins tagged with GFP. MDCs are indicated by the white arrows. Scale bar = 2 µm. (D) Immunoblots of whole-cell protein extracts from yeast expressing the indicated GFP-tagged proteins. GFP-tagged proteins shown to be excluded or enriched in MDCs are indicated. Pgk1 is shown as a loading control. (E) Quantification of the percent of cells forming MDCs in tom70∆ cells constitutively overexpressing the indicated proteins. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (F) Immunoblots of whole-cell protein extracts from wild-type or dnm1∆ yeast expressing Tom70–yeGFP in control strains (GPDpr::Empty) or strains constitutively overexpressing Scm4 (GPDpr::SCM4). Yeast was treated with the indicated compounds for 6 h prior to obtaining protein extracts. Tom70–GFP and free-GFP were detected with antibodies for GFP. Pgk1 is shown as a loading control. (G) Quantification of the percent of cells forming MDCs in the indicated yeast strains treated with DMSO (vehicle control) or 500 nM ConcA. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. Source data are available for this figure: SourceData FS5.
Overloading the OMM with membrane proteins induces MDC formation. (A) Super-resolution confocal fluorescence microscopy images of cells expressing Tom70–GFP and Tim50–mCherry and constitutively overexpressing Scm4 or Tom7. MDCs are indicated by the white arrows. Scale bar = 1 µm. (B) Blue native electrophoresis on isolated mitochondria from yeast strains overexpressing the indicated proteins. Protein complexes were detected with an antibody against Tom40 and the bracket indicates the TOM complex. (C) Representative max projections of widefield fluorescence microscopy images of MDC formation of yeast expressing Tom70–mCherry and the indicated OMM proteins tagged with GFP. MDCs are indicated by the white arrows. Scale bar = 2 µm. (D) Immunoblots of whole-cell protein extracts from yeast expressing the indicated GFP-tagged proteins. GFP-tagged proteins shown to be excluded or enriched in MDCs are indicated. Pgk1 is shown as a loading control. (E) Quantification of the percent of cells forming MDCs in tom70∆ cells constitutively overexpressing the indicated proteins. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. (F) Immunoblots of whole-cell protein extracts from wild-type or dnm1∆ yeast expressing Tom70–yeGFP in control strains (GPDpr::Empty) or strains constitutively overexpressing Scm4 (GPDpr::SCM4). Yeast was treated with the indicated compounds for 6 h prior to obtaining protein extracts. Tom70–GFP and free-GFP were detected with antibodies for GFP. Pgk1 is shown as a loading control. (G) Quantification of the percent of cells forming MDCs in the indicated yeast strains treated with DMSO (vehicle control) or 500 nM ConcA. Error bars show mean ± SE of three replicates, n ≥ 100 cells per replicate. Source data are available for this figure: SourceData FS5.
Because Tom70 facilitates the targeting and import of OMM proteins and performs an unknown supportive role in MDC formation (Hughes et al., 2016), we assessed if Tom70 is required for MDCs to form in response to the overexpression of OMM proteins. In tom70∆ cells, we analyzed MDC formation upon the constitutive overexpression of ten OMM proteins or the metabolite carrier, Oac1, all of which had produced the strongest MDC formation in wild-type cells. In most cases, we observed that MDC formation was strongly blunted in tom70∆ cells, with the exception of Scm4 and Pth2, both of which still induced MDCs to form in ∼20% of cells (Fig. S5 D). Previously, we followed the degradation of MDCs (which were induced to form by treatment with Concanamycin A [ConcA]) by observing Tom70–GFP marked foci within the yeast vacuole, which can also be observed by immunoblot through the release of free GFP (Fig. S5 F; Hughes et al., 2016). Using this assay, we observed the release of free GFP from Tom70–GFP after treatment with ConcA or Rap, which can be blocked by impairing the mitochondrial fission machinery (dnm1∆ cells; Fig. S5 F; Hughes et al., 2016). Conversely, we did not observe that Tom70–GFP was constitutively degraded upon protein overexpression (GPDpr::SCM4) or efficiently degraded after cycloheximide (CHX) treatment, suggesting that MDC formation may primarily act by sequestering cargo from the OMM, while the subsequent degradation of MDCs requires separable steps that include the mitochondrial fission machinery and the core autophagy machinery (Hughes et al., 2016).
As our results suggest that MDCs can sequester excess proteins from the OMM, we hypothesized that the MDC pathway functions in concordance with other mitochondrial quality control pathways known to remove excess OMM proteins. To investigate this idea, we analyzed MDC formation in msp1∆ cells, which lack the mitochondrial membrane-embedded AAA-ATPase shown to extract mistargeted tail-anchored (TA) OMM proteins (Chen et al., 2014). We observed that msp1∆ cells had both an increase in steady-state MDC formation and enhanced MDC formation after treatment with ConcA and a modest increase upon Rap treatment (Fig. 7 B). Consistent with these results, we also observed enhanced MDC formation in get1∆, get2∆, get3∆, and get1∆get2∆ cells, which lack key proteins required for TA protein targeting and insertion at the ER, leading to enhanced mistargeting of TA proteins to the OMM (Fig. 7 B; Schuldiner et al., 2008). Prompted by these results, we examined if disrupting other ER-protein import pathways also led to enhanced MDC formation. To do so, we generated genetic deletions to remove non-essential subunits of the Sec63 complex (sec72∆), Ssh1 translocon complex (ssh1∆), SRP-independent protein targeting (snd3∆), and the ER transmembrane complex (emc2∆), and analyzed MDC formation (Green et al., 1992; Finke et al., 1996; Aviram et al., 2016; Jonikas et al., 2009). While ssh1∆ cells demonstrated a dramatic increase in constitutive MDC formation and enhanced MDC formation upon ConcA treatment, disruption of the other ER targeting and import pathways did not significantly affect MDC formation (Fig. S5 G).
Because MDCs strongly sequester some but not all OMM proteins, we were curious if MDCs were capable of sequestering mistargeted TA proteins. When we ectopically overexpressed GFP–Ubc6 and GFP–Pex15, two TA proteins that mistarget to the OMM in cells with an impaired GET pathway (Schuldiner et al., 2008; Chen et al., 2014), we observed that both GFP–Ubc6 and GFP–Pex15 localized more frequently to mitochondria in get1∆get2∆ yeast compared with wild-type yeast (yellow arrows, Fig. 7, C and D; quantification Fig. 7 E). Consistent with our results that MDCs form more frequently in get1∆get2∆ cells, we also observed the enrichment of GFP–Pex15 and GFP–Ubc6 within MDCs in vehicle-treated cells (white arrows, Fig. 7 D). Upon Rap treatment, mitochondrial-localized GFP–Pex15 and GFP–Ubc6 were always observed to colocalize with MDCs (Fig. 7, C and D), and these proteins were also enriched within MDCs (Fig. 7 F), demonstrating that MDCs are capable of sequestering mistargeted proteins. Collectively, we observed that MDCs preferentially ensnare membrane protein cargo from a single membrane, the OMM, that MDCs form in response to disturbances in OMM protein concentration, and that through the formation of an MDC both membrane and membrane protein cargoes are sequestered into a distinct, protected domain (Fig. 7 G).
Discussion
The OMM creates a critical interface between mitochondria and the rest of the cell. While the OMM supports mitochondrial biogenesis and establishes contacts with other organelles, it can also be a target for both cellular proteins mistargeted to the mitochondrial surface and pathogen proteins (Schuldiner et al., 2008; Costa et al., 2018; Li et al., 2022). In response, mitochondria have several mechanisms to safeguard the OMM, including the remodeling of the OMM to package aberrant proteins into distinct domains, which can subsequently be removed and degraded by lysosomes (Soubannier et al., 2012; Hughes et al., 2016). Previously, we identified a mitochondrial remodeling mechanism, called mitochondrial-derived compartments (MDCs), that robustly sequesters some mitochondrial membrane proteins (Hughes et al., 2016). Through an examination of protein selectivity in MDCs, we have shown here that MDCs concentrate cargo proteins from a single membrane, the OMM, and exclude proteins from internal mitochondrial domains. These results provide an explanation for how only a minor subset of mitochondrial proteins become robustly incorporated into MDCs. Based on these results and observations from an accompanying manuscript (Wilson et al., 2024), we considered that MDCs may act as a membrane-enriched trap to sequester surplus proteins from the OMM. In support, we observed that the overexpression of OMM proteins, as well as the mistargeting of TA proteins to the OMM all induced constitutive MDC formation. Furthermore, these aberrant proteins were sequestered into MDCs, which is not the case for all proteins residing in the OMM, as subunits of the TOM complex are excluded from MDCs unless their assembly or association with the TOM complex becomes impaired. In total, these results suggest that MDCs may facilitate the removal of membrane and protein from mitochondria while maintaining critical functional aspects of the organelle.
Form is often indicative of function. In an accompanying manuscript, we show that MDCs form through mitochondria creating OMM extensions that repeatedly elongate, coalesce, and invaginate to create a domain that not only concentrates OMM cargo, but can also segregate OMM cargo into a protected compartment (Wilson et al., 2024). Similar membrane proliferations form multilamellar whorls generated from ER membranes in response to the accumulation of certain membrane proteins and in response to ER stress (Wright et al., 1988; Schuck et al., 2009; Schäfer et al., 2020). These ER membrane proliferations have been described as a type of ER microautophagy, as they are eventually directly engulfed by vacuoles/lysosomes (Schuck et al., 2014). Prompted by the similarities between ER microautophagy and MDC formation, we examined and observed that MDCs similarly arise from the accumulation of proteins at the OMM. Considering that MDCs also concentrate membrane, it seems likely that MDC formation may occur in response to a variety of mechanisms that challenge OMM integrity and thus could sequester diverse hydrophobic cargo.
Previously, we showed that MDCs form in response to conditions that all increase intracellular amino acid levels and we originally proposed that MDCs may protect against this stress by removing metabolite carriers of the SLC25A family from mitochondria (Schuler et al., 2021). Here, we demonstrate that proteins specifically enter MDCs from the OMM, that some metabolite carriers previously observed to enter MDCs were mislocalized to the OMM, and that many N-terminally GFP–tagged metabolite carriers are excluded from MDCs. These results argue against our initial model that MDCs sequester metabolite carriers from the IMM to limit metabolite import into mitochondria in response to elevated amino acid levels. Furthermore, we have subsequently observed that the cellular response to the MDC inducers, Rap, CHX, and ConcA, is complex and additionally leads to alterations in cellular and mitochondrial lipid profiles (Xiao et al., 2024). Currently, we consider that MDCs act as a content sequestration structure from the OMM, which could form in response to various distinct stresses that may not share overlapping mechanisms. In support, bacteria also remodel their outer membrane and release outer membrane vesicles in response to distinct stressors that include, the presence of excess or aberrant proteins, alterations in lipid composition, excess membrane production, structural alterations, or as a stress-activated pathway (Schwechheimer and Kuehn, 2013; Roier et al., 2016). Thus, because studies on the MDC pathway and other mitochondrial remodeling pathways are incipient, it is likely that MDCs form in response to diverse mechanisms, many of which may not share common mechanisms for activating MDC formation.
How cargo proteins are selectively targeted and enriched within MDCs still needs to be determined. In the formation of an MDC, the OMM would need to proliferate without a connection to the IMM, and thus proteins that can more readily diffuse through the OMM may become preferentially captured by MDCs. Such a mechanism might provide a convenient method to capture aberrant proteins that do not have strong, restrictive interactions with other mitochondrial proteins. This possibility may explain why Tom70, a protein known to be diffusely spread throughout the OMM, incorporates into MDCs, while subunits of the TOM complex are de-enriched from MDCs, as TOM complexes localize to intramitochondrial contact sites to mediate protein import (Chacinska et al., 2010; Schulte et al., 2023). Furthermore, a diffusion mechanism may also explain why truncating both Tom20 and Tom70 to only their OMM-targeting N-terminal transmembrane domains was sufficient for the robust incorporation of these proteins into MDCs and could explain how disrupting TOM complex assembly in the tom6∆ strain led to TOM complex subunits becoming sequestered into MDCs.
While a diffusion mechanism could explain how certain OMM proteins preferentially enter MDCs, there are likely other features of MDCs that lead to the strong enrichment of proteins within these domains. One possibility is that the membrane composition of MDCs is distinct allowing for the enrichment of certain OMM proteins (Xiao et al., 2024). Another possibility is that there are specific cargo-selecting proteins that facilitate the enrichment of proteins within MDCs. For example, we consistently observe a robust concentration of Tom70 within MDCs, and MDC formation is impaired in the absence of Tom70 (Hughes et al., 2016; Schuler et al., 2021). While Tom70 is a multifunctional protein that may perform various roles in supporting MDC formation, it is notable that a premier function of Tom70 is as a co-chaperone and receptor for hydrophobic precursor proteins and other aggregation-prone proteins (Backes et al., 2018, 2021), many of which are MDC cargo proteins. Additionally, Tom70 supports other quality control pathways that selectively remove proteins from the OMM (Weidburg and Amon, 2018), indicating that Tom70 could act as a protein chaperone that maneuvers cargo proteins into MDCs. Determining the molecular mechanisms that support cargo selection and enrichment within MDCs will be a primary focus of future studies.
Several quality control mechanisms have been identified that monitor the integrity of the OMM by removing precursor proteins from stalled protein import complexes or by removing mistargeted, misfolded, or damaged proteins from the OMM (Heo et al., 2010; Weidberg and Amon, 2018; Mårtensson et al., 2019; Metzger et al., 2020). Our results suggest that MDCs function in concordance with these pathways, but rather than directly extracting aberrant proteins from the OMM, the OMM is instead remodeled into a sequestering domain. Protein and membrane sequestration into MDCs may function as a distinct quality control mechanism that is induced when other OMM monitoring pathways become overwhelmed, which is supported by the enhanced MDC formation that occurs in cells lacking Msp1. Intriguingly, we have also previously observed that Msp1 is another protein strongly sequestered into MDCs, along with Tom70, both of which function in several protein quality control pathways (Chen et al., 2014; Hughes et al., 2016; Weidburg and Amon, 2018; Backes et al., 2021). Based on these observations, it also seems possible that MDCs could provide a distinct membrane platform that concentrates aberrant proteins or precursor proteins and functions with other quality control mechanisms to efficiently remove proteins from mitochondria.
Materials and methods
Yeast strains
All yeast strains are derivatives of S. cerevisiae S288C (BY) (Brachmann et al., 1998) and are listed in Table S1. Deletion strains were created by one-step PCR-mediated gene replacement using the previously described pRS series of vectors (Brachmann et al., 1998; Sikorski and Hieter, 1989) and oligo pairs listed in Table S2. Correct gene deletions were confirmed by colony PCR across the chromosomal insertion site. Strains expressing proteins with attached C-terminal fluorescent proteins were created by one-step PCR-mediated C-terminal endogenous epitope tagging using standard techniques and oligo pairs listed in Table S2. Plasmid templates for fluorescent epitope tagging were from the pKT series of vectors (Sheff and Thorn, 2004). Correct integrations were confirmed by a combination of colony PCR across the chromosomal insertion site and correctly localized expression of the fluorophore by microscopy. An integrated yEGFP–Oac1 fusion expressed from its endogenous promoter was generated by integrating a PCR construct containing the URA3 gene flanked by two halves of yEGFP directly upstream of the OAC1 ORF. URA3 was subsequently selected against 5F-OA, and the in-frame integration of yEGFP at the N-terminus of Oac1 was determined by sequencing. Where indicated, strains expressing proteins with superfolder GFP fused to the N-terminus are from the SWAp-Tag library described in Weill et al. (2018) and were a gift from Maya Schuldiner (Weizmann Institute of Science, Rehovot, Israel).
Yeast cell culture and growth assays
Yeast cells were grown exponentially for 15–16 h at 30°C to a final optical (wavelength 600 nm) density of 0.5–1 before the start of all experiments. This period of overnight log-phase growth was carried out to ensure vacuolar and mitochondrial uniformity across the cell population and is essential for consistent MDC formation. Unless otherwise indicated, cells were cultured in YPAD medium (1% yeast extract, 2% peptone, 0.005% adenine, 2% glucose). Otherwise, cells were cultured in a synthetic defined (SD) medium that contained the following unless specific nutrients were removed to select for growth or plasmid retention: 0.67% yeast nitrogen base without amino acids, 2% glucose, supplemented nutrients 0.072 g/L each adenine, alanine, arginine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, histidine, myo-inositol, isoleucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, uracil, valine, 0.369 g/L leucine, and 0.007 g/L para-aminobenzoic acid. Unless otherwise indicated, rapamycin, concanamycin A, cycloheximide, and indole-3-acetic acid (auxin) were added to cultures at final concentrations of 200 nM, 500 nM, 10 µg/ml, and 1 mM, respectively. For serial-dilution growth assays, exponentially growing yeast cells were all set to a density of 1 OD600/ml in water and five 10-fold serial dilutions were created. 3 μl of each dilution was spotted onto the indicated nutritional media plus agar plates. Unless otherwise indicated, the growth assays were performed at 30°C, and the images were taken 2 or 3 days later.
Plasmids
The majority of plasmids generated for this study were created through a similar process that involved insertion of PCR-amplified genes of interest (GOI) into the yeast expression pRS series plasmids (Sikorski and Hieter, 1989) via Gibson assembly (Gibson et al., 2009), and expression was driven from the inclusion of the GOI endogenous promoter or a low expression promoter (CPY promoter for N-terminal GFP constructs or COX8 promoter for MTS–containing constructs). All oligo pairs used in the amplification of GOIs are listed in Table S2, and all plasmids used in this study are listed in Table S3. All plasmids were checked for sequence errors through the ORFs of GOIs, checked for protein expression, and often the function of expressed proteins was analyzed. A brief description of the construction of all OAC1-containing plasmids is provided for detail. The OAC1 ORF plus ∼500 base pairs (bp) upstream (5′UTR) and downstream (3′UTR) were PCR-amplified from BY4741 genomic DNA and contained additional 20–25 bp flanking regions that were homologous to the DNA regions directly flanking the EcoR1 recognition site found in the MCS of pRS416 and pRS426 plasmids. This OAC1 amplicon was inserted into both pRS416 and pRS426 plasmids linearized by EcoR1 digestion and the plasmids were reassembled containing the OAC1 locus via Gibson assembly. Similarly, OAC1 was fused to GFP in frame at the N-terminus by inserting via Gibson assembly the OAC1 ORF plus 500 bp of its 3′UTR into pGO36 and pGO35, both of which were linearized by digestion with EcoR1. Both pGO36 and pGO35 express GFP from the CPY promoter in pRS416 and pRS426, respectively, and were gifts from Greg Odorizzi (University of Colorado Boulder, Boulder, CO, USA). The OAC1-yeGFP ORF was PCR amplified from AHY8509 genomic DNA and contained ∼500 bp upstream (5′UTR) and the ADH1 terminator sequence and was also inserted into the EcoR1 cut-site of pRS416 and pRS426 plasmids. Alternatively, only the OAC1-yeGFP ORF (minus the first start codon) and the ADH1 terminator sequence were PCR-amplified from AHY8509 genomic DNA and fused behind a PCR-amplicon that contained ∼500 bp of the COX8 5′UTR and the Cox8 MTS (codons 1–27), both of which were inserted into the EcoR1 cut-site of pRS416 by tandem Gibson assembly. Plasmids for GPD-driven overexpression of OMM proteins, OAC1, AAC1, or GFP–TA proteins were generated by gateway-mediated transfer of the corresponding ORF (Harvard Institute of Proteomics) from pDONR201/221 into pAG306GPD-ccdB chromosome I or pAG413GFP–ccdb (Hughes and Gottschling, 2012) using Gateway LR Clonase II Enzyme mix (Thermo Fisher Scientific) according to the manufacturer’s instructions. To integrate the resulting expression plasmid into yeast chromosome I (199456-199457), pAG306GPD-ORF chromosome I plasmids were digested with NotI.
MDC assays
For MDC assays, overnight log-phase cell cultures were grown in the presence of dimethyl sulfoxide (DMSO) or the indicated drug for 2 h. For MDC assays with cells containing plasmids, overnight log-phase yeast cultures grown in selective SD medium were back-diluted to an OD600 = 0.1–0.2 in YPAD medium and allowed to grow for at least 4 h prior to MDC induction. Prior to visualization, cells were harvested by centrifugation, washed once, and resuspended in 100 mM HEPES containing 5% glucose. Subsequently, yeast were directly plated onto a slide at small volumes to allow the formation of a monolayer, and optical z-sections of live yeast cells were acquired with a ZEISS Axio Imager M2 or for super-resolution confocal fluorescence microscopy images a ZEISS LSM800 with Airyscan was used. The percent cells with MDCs were quantified in each experiment at the 2-h time point. All quantifications show the mean ± SE from three biological replicates with n = 100 cells per experiment. MDCs were identified as Tom70–positive, Tim50–negative structures that were enriched for Tom70 versus the mitochondrial tubule. In MDC colocalization assays, MDCs were identified as large, Tom70–enriched, spherical structures prior to assessing the colocalization of different proteins of interest.
Fluorescence microscopy
Fluorescence microscopy was performed as described by English et al. (2020). In brief, optical z-sections of live yeast cells were acquired with a ZEISS Axio Imager M2 equipped with a ZEISS Axiocam 506 monochromatic camera, 100× oil-immersion objective (plan apochromat, NA 1.4) or 63× oil-immersion objective (plan apochromat, NA 1.4) or a ZEISS LSM800 equipped with an Airyscan detector, 63× oil-immersion objective (plan apochromat, NA 1.4) at room temperature (RT). Crudely purified mitochondria were immobilized on glass slides coated with poly-L-lysine and optical z-sections were acquired with a ZEISS LSM800 equipped with an Airyscan detector, 63× oil-immersion objective (plan apochromat, NA 1.4) at RT. For yeast indirect immunofluorescence, the fixation and antibody staining were performed as described in Schuler et al. (2021). Except prior to blocking and antibody staining, cells were permeabilized with methanol for 6 min at −20°C and briefly incubated for 30 sec in acetone at −20°C. To detect Por1, samples were incubated with primary antibody (monoclonal Por1 antibody produced in mouse, diluted 1:100 or 1:200 in wash buffer containing 1% BSA) for 90 min at RT and subsequently incubated with secondary antibody (goat anti-mouse IgG [H+L] cross absorbed secondary antibody, Alexa Fluor 647, 1:100 diluted in wash buffer containing 1% BSA) for 45 min at RT. Subsequently, optical z-sections of the fixed cells were acquired with a Zeiss AxioObserver 7 equipped with a PCO Edge 4.2LT Monochrome, Air Cooled, USB 3 CCD camera with a Solid-State Colibri 7 LED illuminator and 63× oil-immersion objective (Plan Apochromat, NA 1.4) at RT. All widefield images were acquired with ZEN (Carl Zeiss) and processed with Fiji (Schindelin et al., 2012). Super-resolution images were acquired with ZEN (Carl Zeiss) and processed using the automated Airyscan processing algorithm in ZEN (Carl Zeiss) and further processed in Fiji. Fluorochromes are indicated in figure legends. Individual channels of all images were minimally adjusted in Fiji to match the fluorescence intensities between channels for better visualization. Line-scan analysis was performed on non-adjusted, single z-sections in Fiji.
Protein preparation and immunoblotting
For Western blot analysis of protein levels, yeast cultures were grown to log-phase (OD600 = 0.5–1) and 2 OD600 cell equivalents were isolated by centrifugation, washed with dH2O, and incubated in 0.1 M NaOH for 5 min at RT. Subsequently, cells were reisolated by centrifugation at 16,000 × g for 10 min at 4°C and lysed for 5 min at 95°C in lysis buffer (10 mM Tris pH 6.8, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% SDS and containing cOMPLETE protease inhibitor cocktail [Millipore Sigma]). Upon lysis, samples were denatured in Laemmli buffer (63 mM Tris pH 6.8, 2% SDS, 10% glycerol, 1 mg/ml bromophenol blue, 1% β-mercaptoethanol) for 5 min at 95°C. To separate proteins based on molecular weight, equal amounts of protein were subjected to SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Millipore Sigma) by semi-dry transfer. Nonspecific antibody binding was blocked by incubation with Tris-buffered saline + 0.05% Tween-20 (TBST) containing 10% dry milk for 1 h at RT. After incubation with the primary antibodies at 4°C overnight, membranes were washed four times with TBST and incubated with secondary antibody (goat-anti-rabbit or donkey-anti-mouse HRP-conjugated, 1:5,000 in TBST + 10% dry milk; Sigma-Aldrich) for 1 h at RT. Subsequently, membranes were washed twice with TBST and twice with TBS, and enhanced chemiluminescence solution (Thermo Fisher Scientific) was applied and the antibody signal was detected with a BioRad Chemidoc MP system. All blots were exported as TIFFs and cropped in Adobe Photoshop CC.
Protein localization within swollen mitochondria
All yeast strains examined for protein localization within swollen mitochondria contained Mdm12 C-terminally fused to AID-6xFLAG from the constructs described in Morawska and Ulrich (2013). Subsequently, GPD1–OsTIR1 was integrated into the LEU2 locus using the plasmid pNH605-pGPD1-osTIR1 digested with Swa1 as described in Chan et al. (2018). Auxin-induced protein degradation was performed essentially as described in John Peter et al. (2022), except 3-indole acetic acid (auxin) was added to a final concentration of 1 mM. After a 3-h incubation, cells were prepped for fluorescence microscopy analysis and the auxin-induced degradation of Mdm12 and swelling of mitochondria were visually confirmed as shown in John Peters et al. (2022).
Isolation of yeast mitochondria
Crudely purified mitochondria were isolated from yeast cells as described in Schuler et al. (2021). Briefly, yeast were grown overnight in log-phase to an OD600 = 0.5–1, then isolated by centrifugation, washed with dH2O, and the pellet weight was determined. Cells were then resuspended in 2 ml/g pellet dithiothreitol (DTT) buffer (0.1 M Tris, 10 mM DTT, pH 9.4) and incubated for 20 min at 30°C under constant shaking. After reisolation by centrifugation, DTT-treated cells were washed once with zymolyase buffer (1.2 M sorbitol, 20 mM K2HPO4, pH 7.4 with HCl), and the cell walls were digested for 30 min at 30°C under constant shaking in 7 ml zymolyase buffer/g cell pellet containing 1 mg zymolyase 100T/g cell pellet. After zymolyase digestion, cells were reisolated by centrifugation, washed with zymolyase buffer, and lysed by mechanical disruption in 6.5 ml/g pellet homogenization buffer (0.6 M sorbitol, 10 mM Tris pH 7.4, 1 mM ethylenediaminetetraacetate [EDTA] pH 8.0 with KOH, 0.2% BSA, 1 mM phenylmethylsulfonylfluoride) at 4°C. Cell debris was removed from the homogenate twice by centrifugation at 5,000 × g for 5 min at 4°C and mitochondria were pelleted at 14,000 × g for 15 min at 4°C. The mitochondrial pellet was resuspended in SEM buffer (250 mM sucrose, 1 mM EDTA pH 8.0 with KOH, 10 mM 3-(N-morpholino)-propansulfonic acid pH 7.2), reisolated by differential centrifugation as described above, resuspended in SEM buffer, and the mitochondria were shock-frozen in liquid nitrogen and stored at −80°C.
Protease protection and carbonate extraction
For protease protection assays, 50 µg of crudely purified mitochondria were incubated on ice with or without proteinase K (PK) under different conditions: mitochondria were stabilized in an iso-osmolar buffer (SEM buffer), the OMM was ruptured by hypo-osmolar swelling (EM buffer: 1 mM EDTA, 10 mM MOPS/KOH, pH 7.2), or mitochondria were lysed by addition of 1% Triton X-100 (TX) to SEM buffer. Subsequently, PK activity was inhibited by the addition of 2 mM PMSF, mitochondria were pelleted by centrifuging at 20,000 × g at 4°C and analyzed by SDS-PAGE and immunoblotting as described above. For carbonate extraction experiments, 100 µg of mitochondria were incubated on ice with 0.1 M Na2CO3, pH 11 for 30 min. Subsequently, mitochondrial membranes were pelleted by centrifuging at 126,000 × g for 45 min at 4°C, and analyzed by SDS-PAGE and immunoblotting as described above.
Blue native electrophoresis
Mitochondria were isolated as described above and protein complexes were solubilized on ice for 15 min in 1X NativePAGE sample buffer (Thermo Fisher Scientific) with 1% digitonin. Non-solubilized membrane fractions were removed by centrifugation at 20,000 × g for 30 min at 4°C and the protein content of the solubilized fraction was determined and normalized using a bicinchoninic assay. 0.25% of Coomassie G-250 was added to the solubilized fraction and protein complexes were separated by electrophoresis on a NativePAGE 4–16% Bis-Tris Gel (Thermo Fisher Scientific). Subsequently, separated protein complexes were transferred to a PVDF membrane (MilliporeSigma) overnight at 4°C using NuPAGE Transfer Buffer (Thermo Fisher Scientific). Membranes were then incubated in 8% acetic acid at RT for 20 min and washed with methanol to remove background Coomassie G-250. To detect protein complexes, the PVDF membrane was first blocked from nonspecific antibody binding by incubation with PBS + 0.05% Tween-20 (PBST) containing 5% dry milk for 1 h at RT. Membranes were then incubated with primary antibodies for either Tom40 or GFP diluted 1:1,000 in PBST containing 5% dry milk for 1 h at RT. Subsequently, membranes were washed four times with PBST and incubated with a secondary antibody (goat-anti-rabbit or donkey-anti-mouse HRP-conjugated,1:5,000 in PBST + 5% dry milk) for 1 h at RT. After washing twice with PBST and twice with PBS, an enhanced chemiluminescence solution (Thermo Fisher Scientific) was applied and the antibody signal was detected with a BioRad Chemidoc MP system. All blots were exported as TIFFs and cropped in Adobe Photoshop CC.
Quantification and statistical analysis
The number of replicates, what n represents, and dispersion and precision measures are indicated in the figure legends. Sample sizes were as large as possible to be representative but of a manageable size for quantifications. For most experiments, quantifications show the mean ± SE from three biological replicates with n = 100 cells per experiment. In experiments with data depicted from a single biological replicate, the experiment was repeated with the same results. All statistical analysis was performed in Prism (GraphPad) and the statistical test used is indicated in the corresponding figure legend.
Online supplemental material
Fig. S1 shows representative widefield images of N-terminally GFP-tagged metabolite carriers excluded from MDCs, as well as the protein levels and functionality assays for chimeric proteins fused to an MTS. Fig. S2 provides additional analysis of MDC cargo proteins localizing to the OMM. Fig. S3 shows that GFP-tagged metabolite carriers that localize within mitochondria are excluded from MDCs. Fig. S4 provides evidence that both Por1 and GFP–Sam37 are also excluded from MDCs. Fig. S5 provides additional analysis on the induction of MDC formation by excess OMM proteins. Table S1 lists the yeast strains used in this study. Table S2 lists the oligonucleotides used in this study. Table S3 lists bacterial strains, chemicals, antibodies, plasmids, and software used in this study.
Data availability
All reagents used in this study are available upon request. All other data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank members of the A.L. Hughes group for discussion and manuscript comments. We thank members of Janet. M. Shaw laboratory for providing reagents and support early on in the project. We would like to also thank Greg Odorizzi for plasmid gifts and Maya Schuldiner for gifting the yeast SWAp-Tag library described in Weill et al. (2018).
Z.N. Wilson, Ph.D., was supported by an American Heart Association Postdoctoral Fellowship 20POST35200110 and an American Cancer Society–Give Mas, Live Mas Southern Multifoods Postdoctoral Fellowship PF-20-018-01-CCG. Support for this research was also generously provided by grant number PF-19-0209 from the United Mitochondrial Disease Foundation (Z.N. Wilson) and National Institutes of Health grants 5T32HL007576-36 (S. Wong), GM119694 (A.L. Hughes), and AG061376 (A.L. Hughes).
Author contributions: Z.N. Wilson: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing—original draft, Writing—review & editing, S.S. Balasubramaniam: Investigation, Validation, S. Wong: Investigation, Validation, M.-H. Schuler: Conceptualization, Investigation, Methodology, M.J. Wopat: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing—review & editing, A.L. Hughes: Conceptualization, Funding acquisition, Project administration, Supervision, Writing—review & editing.

