


Proteins with multiple membrane-spanning segments (MS) co-translationally insert into the endoplasmic reticulum (ER) membrane of eukaryotic cells. Shr3, an ER membrane–localized chaperone in Saccharomyces cerevisiae, is required for the functional expression of a family of 18 amino acid permeases (AAP) comprised of 12 MS. We have used comprehensive scanning mutagenesis and deletion analysis of Shr3 combined with a modified split-ubiquitin approach to probe chaperone–substrate interactions in vivo. Shr3 selectively interacts with nested C-terminal AAP truncations in marked contrast to similar truncations of non-Shr3 substrate sugar transporters. Shr3–AAP interactions initiate with the first four MS of AAP and successively strengthen but weaken abruptly when all 12 MS are present. Shr3–AAP interactions are based on structural rather than sequence-specific interactions involving membrane and luminal domains of Shr3. The data align with Shr3 engaging nascent N-terminal chains of AAP, functioning as a scaffold to facilitate folding as translation completes.

Introduction
A major unsolved problem in biology is the folding of complex polytopic membrane proteins composed of multiple membrane-spanning segments (MS). In eukaryotic cells, the integration and concomitant folding of membrane proteins in the lipid bilayer of the endoplasmic reticulum (ER) are critical steps in the biogenesis of nutrient transport proteins destined to function at the plasma membrane (PM). Typically, transport proteins are co-translationally inserted into the ER membrane via the Sec61-complex, also known as the translocon. The translocon forms a protein-conducting channel that mediates protein translocation and the co-translational partitioning of MS (Rapoport et al., 2017; Seinen and Driessen, 2019; van den Berg et al., 2004). During the synthesis, each MS sequentially exits the channel and partitions into the ER membrane via the lateral gate of the translocon. Although the central channel of the translocon is too small to accommodate multiple MS, the translocon appears to have a limited capacity to promote the folding of membrane proteins with few MS; extra-channel MS-binding sites have been reported to act in a chaperone-like manner to delay the release of N-terminal MS until the translation is completed (Hou et al., 2012; Ismail et al., 2008; Sadlish et al., 2005). However, for larger and more complex membrane proteins, the challenge of preventing inappropriate interactions between MS of incompletely translated nascent chains apparently exceeds the chaperone-like activity of the translocon. Consistent with this notion, highly specialized ER-resident membrane proteins have been described in fungi that prevent translation intermediates from entering non-productive folding pathways (Kota and Ljungdahl, 2005; Lau et al., 2000; Ljungdahl et al., 1992; Luo et al., 2002; Martínez and Ljungdahl, 2000; Martínez and Ljungdahl, 2004; Sherwood and Carlson, 1999; Shurtleff et al., 2018; Trilla et al., 1999).
Shr3, the most comprehensively studied of these ER components, was identified as an integral membrane protein required for the functional expression of the conserved family of amino acid permeases (AAP) in Saccharomyces cerevisiae (Ljungdahl et al., 1992). The AAP family, belonging to the amino acid–polyamine–organocation (APC) super-family of transporters (Gilstring and Ljungdahl, 2000; Jack et al., 2000; Saier, 2000; Wong et al., 2012), is comprised of 18 genetically distinct but structurally similar proteins with 12 MS. Shr3 is composed of 210 amino acids organized into two functional domains: an N-terminal membrane domain comprised of four hydrophobic α-helices and a hydrophilic cytoplasmically oriented C-terminal domain. Initially, Shr3 was recognized as an essential factor facilitating the packaging of AAP into ER-derived secretory vesicles (Kuehn et al., 1996), an activity subsequently ascribed to facilitating the presentation of ER-exit motifs, located in the C-terminal tails of AAP, to the inner COPII coatomer subunit Sec24 (Kuehn et al., 1998; Malkus et al., 2002; Miller et al., 2002; Miller et al., 2003). The ability of Shr3 to physically interact with COPII components, primarily via its hydrophilic C-terminal tail, and the observed transient association with a newly synthesized and fully integrated AAP suggested that Shr3 facilitated ER vesicle formation in close proximity to fully integrated and folded AAP. Hence, Shr3 was designated a packaging chaperone (Gilstring et al., 1999; Gilstring and Ljungdahl, 2000).
In more recent studies, a second activity was attributed to Shr3 that is separate from and temporally preceding its packaging function. In these studies, Shr3 was found to prevent the aggregation of AAP in the ER membrane, an activity clearly associated with its N-terminal membrane domain (Kota and Ljungdahl, 2005). Critical evidence demonstrating the importance of Shr3 in facilitating the folding of AAP includes the finding that co-expressed split N- and C-terminal portions of the general amino acid permease (Gap1) assemble into a functional permease in a Shr3-dependent manner (Kota et al., 2007). The membrane domain of Shr3 is required and suffices to prevent aggregation of the first five MS of Gap1, an activity that is essential for promoting productive folding interactions with the C-terminal portions of Gap1. Similar to full-length Gap1, the N-terminal fragment displayed a propensity to aggregate in membranes isolated from cells lacking Shr3, and importantly, its aggregation appeared independent of the presence or absence of the C-terminal Gap1 fragment. In marked contrast, the aggregation status of the C-terminal fragment was dependent on the presence of both Shr3 and the N-terminal fragment. Since the N- and C-terminal fragments individually insert into the membrane, Shr3 apparently can maintain the N-terminal fragment in a conformation that enables the C-terminal fragment to interact and assemble with it.
During translocation, exclusively hydrophobic MS readily partition into the lipid phase of the membrane, whereas MS containing charged or polar residues are retained in proximity to the translocon (Heinrich and Rapoport, 2003) or to translocon-associated proteins, e.g., TRAM (translocating chain-associated membrane protein; Heinrich et al., 2000). In analogy to TRAM, Kota et al. (2007) posited that Shr3 facilitates the partitioning of MS of AAP containing charged or polar amino acid residues as they emerge from the translocon. According to this hypothesis, Shr3 physically shields charged or polar residues within more N-terminal MS, thereby preventing these thermodynamically challenging segments from engaging in non-productive interactions prior to the completion of the translation of the C-terminal MS. Similarly, the insertase/chaperone YidC (Beck et al., 2001; Dalbey and Kuhn, 2014) has been implicated as an assembly site for alpha helices of LacY in a manner that minimizes unfavorable off-pathway folding interactions during translation (Nagamori et al., 2004; Serdiuk et al., 2016; Serdiuk et al., 2019; Wagner et al., 2008; Zhu et al., 2013). Recent findings in yeast regarding the conserved ER membrane protein complex (EMC) have been interpreted in a similar manner (Miller-Vedam et al., 2020; Shurtleff et al., 2018). However, a striking difference between Shr3 and EMC function is that null alleles of SHR3 do not activate the unfolded protein response (UPR; Gilstring et al., 1999).
Despite the clear requirement of Shr3 in AAP folding, we currently lack critical information regarding the mechanisms underlying Shr3 function. Here, we have focused on the membrane domain of Shr3 and employed comprehensive scanning mutagenesis to define amino acid residues involved in recognizing AAP substrates. Further, we have exploited a modified split-ubiquitin approach to directly probe and characterize interactions with seven different AAP substrates in vivo. The data support that Shr3 acts as a membrane-localized chaperone required to help nascent chains of partially translated AAP attain and transiently maintain a structure required to follow a productive folding pathway as translation proceeds to completion. The membrane-localized chaperone–substrate interactions are highly specific but occur independent of strict sequence requirements.
Results
Systematic scanning mutagenesis of MS within the membrane domain of Shr3
We have previously shown that the membrane domain of Shr3 is required and sufficient for facilitating the folding of AAPs (Kota and Ljungdahl, 2005). To further our understanding, a systematic scanning mutagenesis approach was used to identify residues within the Shr3 membrane domain required for function. To maintain compatibility with the hydrophobic nature of membranes, the intramembrane residues were mutated to leucine; the length of consecutive substitution mutations varied, ranging from 2 to 13 residues. To minimize negative folding artifacts, the extramembrane residues within ER lumenal loops L1 and L3 and cytoplasmic oriented NT and loop L2 were mutated to alanine; the length of consecutive alanine replacements ranged from 2 to 3.
The biological activity of the 44 mutant proteins, which collectively alter all 159 aa residues comprising the N-terminal domain of Shr3, was initially assessed using growth-based assays on high-amino acid content YPD medium supplemented with metsulfuron-methyl (MM). The ability to grow in the presence of MM provides a sensitive measure of Shr3 function; MM targets and inhibits branched-chain amino acid synthesis, and growth is strictly dependent on the combined activity of multiple AAP that facilitate high-affinity isoleucine, leucine, and valine uptake (Andréasson and Ljungdahl, 2002; Jørgensen et al., 1998). Serial dilutions of cell suspensions from the strain JKY2 (shr3Δ) carrying vector control (VC), wild type (SHR3), or the mutant alleles were spotted on YPD with and without MM. Only three mutant alleles, shr3-35, shr3-50, and shr3-76, failed to support growth (Fig. 1, A and B). The steady-state levels of all three mutant proteins were found to be similar to wild-type Shr3 (Fig. 1 B), suggesting that the mutations do not grossly affect folding, and consequently, the mutant proteins are not prematurely targeted for degradation.
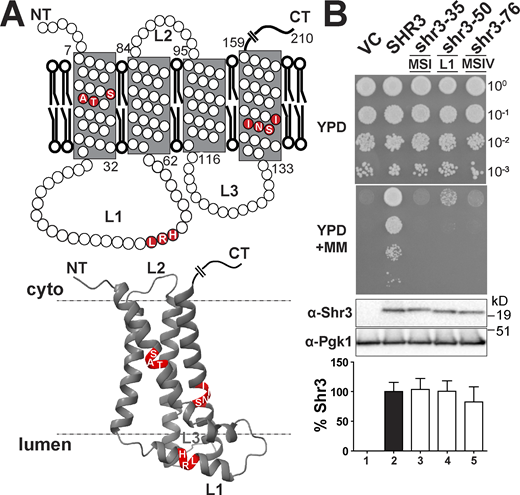
Scanning mutagenesis of the Shr3 membrane domain. (A) Graphical representation of Shr3 topology (upper) and AlphaFold-predicted structural model (lower; Jumper et al., 2021; Varadi et al., 2021). The position of residues resulting in a non-functional protein is indicated. Three-dimensional molecular graphics and analyses were performed with UCSF ChimeraX (https://www.rbvi.ucsf.edu/chimerax; Pettersen et al., 2021). (B) Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pAR4 (shr3-35), pAR18 (shr3-50), or pPL1349 (shr3-76) spotted on YPD and YPD+MM (upper panels). The plates were incubated at 30°C for 2 d and photographed. Immunoblot analysis of Shr3 proteins in extracts prepared from the strains; the levels of Pgk1 were used as loading controls (lower panels). The blots were developed using α-Shr3 and α-Pgk1 antibodies. The signal intensities of the immunoreactive forms of Shr3 and Pgk1 were quantified, and the Shr3 signals were normalized with respect to Pgk1; the mean values are plotted and error bars show standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F1.
Scanning mutagenesis of the Shr3 membrane domain. (A) Graphical representation of Shr3 topology (upper) and AlphaFold-predicted structural model (lower; Jumper et al., 2021; Varadi et al., 2021). The position of residues resulting in a non-functional protein is indicated. Three-dimensional molecular graphics and analyses were performed with UCSF ChimeraX (https://www.rbvi.ucsf.edu/chimerax; Pettersen et al., 2021). (B) Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pAR4 (shr3-35), pAR18 (shr3-50), or pPL1349 (shr3-76) spotted on YPD and YPD+MM (upper panels). The plates were incubated at 30°C for 2 d and photographed. Immunoblot analysis of Shr3 proteins in extracts prepared from the strains; the levels of Pgk1 were used as loading controls (lower panels). The blots were developed using α-Shr3 and α-Pgk1 antibodies. The signal intensities of the immunoreactive forms of Shr3 and Pgk1 were quantified, and the Shr3 signals were normalized with respect to Pgk1; the mean values are plotted and error bars show standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F1.
The shr3-35 allele encodes a protein with residues 17 through 19 (serine–alanine–threonine) in MS I replaced by leucine (Fig. 1 A). The shr3-76 allele carries leucine replacements at residues 139 through 142 (serine-asparagine-isoleucine-isoleucine) in MS IV (Fig. 1 A). A common feature of these non-functional proteins is that the polar residues are replaced by hydrophobic leucine residues. The third non-functional allele, shr3-50, encodes a protein with alanine substitutions at residues 51 through 53 (leucine-arginine-histidine) located within the ER lumenal loop L1 (Fig. 1 A). The importance of the individual residues affected by the shr3-35, shr3-76, and shr3-50 mutations were tested by paired leucine (shr3-35 and shr3-76) and alanine (shr3-50) substitutions, respectively (Fig. S1). With the exception of serine-139 in MS IV, various combinations of amino acid substitutions at the other residues were tolerated and could support function. Together, the data indicate that maintenance of the structure, rather than specific amino acid sequence, is of primary significance.

The importance of the individual residues affected by the shr3-35 (MSI), shr3-50 (L1), and shr3-76 (MSIV) mutations. Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pAR4 (shr3-35), pPL1330 (SHR3-36), pAR47 (shr3-37), pAR37 (SHR3-38), pAR18 (shr3-50), pAR51 (SHR3-51), pAR52 (SHR3-52), pAR50 (shr3-53), pPL1349 (shr3-76), pAR48 (shr3-77), or pAR49 (SHR3-78) spotted on YPD and YPD+MM plates. Plates were incubated at 30°C for 2 d and photographed.
The importance of the individual residues affected by the shr3-35 (MSI), shr3-50 (L1), and shr3-76 (MSIV) mutations. Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pAR4 (shr3-35), pPL1330 (SHR3-36), pAR47 (shr3-37), pAR37 (SHR3-38), pAR18 (shr3-50), pAR51 (SHR3-51), pAR52 (SHR3-52), pAR50 (shr3-53), pPL1349 (shr3-76), pAR48 (shr3-77), or pAR49 (SHR3-78) spotted on YPD and YPD+MM plates. Plates were incubated at 30°C for 2 d and photographed.
Deletion analysis of ER-lumen-oriented loops
The finding that mutations affecting residues 51–53 in the ER lumenal loop L1 abolish function raised the possibility that extramembrane sequences are important for guiding the folding of AAP sequences destined to be oriented toward the extracellular side of the PM. This prompted us to specifically test the functional significance of loops L1 and L3 (Fig. 2 A). The high-confidence AlphaFold (AF; https://alphafold.ebi.ac.uk) structural model (Fig. 2 A) and JPred4 secondary structure prediction (Fig. 2 B) of Shr3 suggest that residues 44–57 within L1 fold into an amphipathic α-helix (Fig. 2 B; Drozdetskiy et al., 2015; Jumper et al., 2021; Varadi et al., 2021). We constructed four internal deletions in L1 that affect this secondary structural motif to varying extent: shr3Δ90 (Δ34-48); shr3Δ91 (Δ39-47); shr3Δ92 (Δ44-54); and shr3Δ93 (Δ55-60). Also, an internal deletion in L3 was constructed: SHR3Δ94 (Δ121-127). The five deletion alleles directed the expression of mutant proteins at levels comparable to wild-type SHR3 (Fig. 2 C).
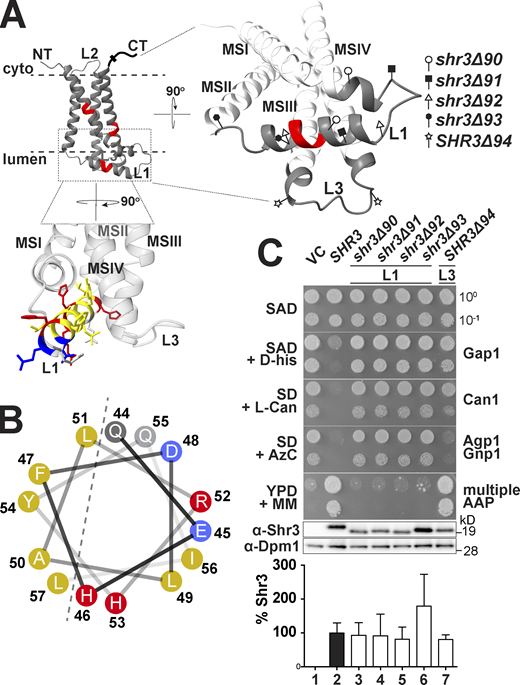
Deletion analysis of ER-lumen-oriented loops. (A) Predicted Shr3 structure (AlphaFold) and the positions of the internal deletions in loops L1 and L3. The helical segment predicted in L1 is colored with yellow for non-polar, gray for polar, blue for negatively-, and red for positively charged residues. Molecular graphics and analyses were performed with UCSF ChimeraX. (B) Helical wheel projection of the L1 α-helix with non-polar (yellow), polar (gray), negatively (blue), and positively charged (red) residues indicated. (C) Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pAR41 (shr3Δ90), pAR42 (shr3Δ91), pAR43 (shr3Δ92), pAR44 (shr3Δ93), or pAR45 (shr3Δ94) spotted on SAD containing D-histidine (D-his), SD + L-canavanine (L-Can), SD + AzC, and YPD + MM plates. Plates were incubated at 30°C for 2 d and photographed. Bottom: Immunoblot analysis of Shr3 proteins in extracts prepared from the strains; the levels of Dpm1 were used as loading controls. The blots were developed using α-Shr3 and α-Dpm1 antibodies. The signal intensities of the immunoreactive forms of Shr3 and Dpm1 were quantified and the Shr3 signals were normalized with respect to Dpm1; the mean values are plotted and the error bars show standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F2.
Deletion analysis of ER-lumen-oriented loops. (A) Predicted Shr3 structure (AlphaFold) and the positions of the internal deletions in loops L1 and L3. The helical segment predicted in L1 is colored with yellow for non-polar, gray for polar, blue for negatively-, and red for positively charged residues. Molecular graphics and analyses were performed with UCSF ChimeraX. (B) Helical wheel projection of the L1 α-helix with non-polar (yellow), polar (gray), negatively (blue), and positively charged (red) residues indicated. (C) Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pAR41 (shr3Δ90), pAR42 (shr3Δ91), pAR43 (shr3Δ92), pAR44 (shr3Δ93), or pAR45 (shr3Δ94) spotted on SAD containing D-histidine (D-his), SD + L-canavanine (L-Can), SD + AzC, and YPD + MM plates. Plates were incubated at 30°C for 2 d and photographed. Bottom: Immunoblot analysis of Shr3 proteins in extracts prepared from the strains; the levels of Dpm1 were used as loading controls. The blots were developed using α-Shr3 and α-Dpm1 antibodies. The signal intensities of the immunoreactive forms of Shr3 and Dpm1 were quantified and the Shr3 signals were normalized with respect to Dpm1; the mean values are plotted and the error bars show standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F2.
The function of these deletion alleles was assessed on YPD+MM and by more nuanced growth-based assays capable of monitoring amino acid uptake catalyzed predominantly by a single or a couple of AAP (Fig. 2 C). The latter was accomplished by examining growth on minimal media individually supplemented with toxic amino acid analogs D-histidine, L-canavanine, and azetidine-2-carboxylate (AzC), which are taken up by Gap1 (Gresham et al., 2010), Can1 (Ono et al., 1983), and Agp1/Gnp1 (Andréasson et al., 2004), respectively. The expression of functional alleles of SHR3 results in impaired growth in the presence of these toxic analogs. Serial dilutions of cell suspensions from the strain JKY2 (shr3Δ) carrying plasmids VC (vector control), SHR3, shr3Δ90, shr3Δ91, shr3Δ92, shr3Δ93, or SHR3Δ94 were spotted on SAD containing D-histidine, SD+L-canavanine, SD+AzC, and YPD+MM. The four deletion alleles affecting L1 failed to complement shr3Δ; the strains grew similar to the VC (Fig. 2 C, dilutions 1, 3, 4, 5, and 6). By contrast, the strain expressing the internal deletion in L3 showed a more complex pattern of growth. On SAD+D-histidine and SD+L-canavanine, SHR3Δ94 appeared to express a non-functional protein, and the strain grew in the presence of these toxic amino acid analogs (Fig. 2 C, compare dilution 1 with 7). However, the SHR3Δ94 allele supported growth on SD+AzC similar to wildtype, and strikingly, exhibited more robust growth on YPD+MM compared with wildtype SHR3 (Fig. 2 C, compare dilution 2 with 7); the latter phenotype was designated WT+. The finding that the deletion of seven residues in L3 results in both loss- and gain-of-function phenotypes indicates that the mutant Shr3Δ94 retains the capacity to promote the folding of some AAP but not others.
Lumenal loop L3 influences substrate specificity
The finding that SHR3Δ94 exhibited a range of phenotypes, from null to WT+, prompted us to reexamine the growth characteristics of the 44 leucine- and alanine-scanning mutant alleles using the more nuanced growth-based assays. Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying vector control (VC), SHR3, or one of the individual mutant alleles were spotted on SAD+D-histidine, SD+L-canavanine, SD+AzC, and YPD+MM (Fig. S2, A–J). The growth characteristics were evaluated, and the results, including the internal loop deletions, are summarized in an ordered heatmap (Fig. 3 A). As was found in the initial evaluation of growth on YPD+MM, most of the mutant alleles were judged to encode functional proteins; the strains grew similarly to the strain carrying wild-type SHR3. Reevaluation of the three non-functional alleles, shr3-35, shr3-50, and shr3-76, confirmed that the mutations exhibit major defects on all of the selective media (Fig. 3 A). Similar to SHR3Δ94, several mutations (SHR3-45, -63, -65, -68, -71, -74, and -75) conferred WT+ growth on YPD+MM and exhibited a null phenotype on media containing toxic amino acid analogs. In summary, growth in the presence of D-histidine was found to be the most sensitive monitor of mutations in SHR3, perhaps due to the fact that D-amino acids are taken up by a single AAP, Gap1 (Grenson et al., 1970; Rytka, 1975). We note that mutations localized to the MSIII and IV and the ER lumenal-oriented loop L3 exhibited the most pleiotropic effects, suggesting that these regions of Shr3 influence interactions with discrete AAP, and potentially contribute to apparent substrate specificity.

Growth-based assessment of Shr3 mutant protein function. Growth of serially diluted cell suspensions from strain JKY2 (shr3Δ) carrying either vector control (VC) or the indicated plasmids expressing wild-type Shr3 (SHR3) or mutated forms of Shr3 proteins (upper case indicates alleles conferring wild-type growth; lower case indicates alleles exhibiting defective growth). The cells were spotted on YPD, SD (ammonium), and SAD (allantoin) containing toxic amino analogs as indicated at the following concentrations: 200 µg/ml metsulfuron-methyl (MM); 1 mM azetidine-2-carboxylate (AzC); 1 µg/ml L-canavanine (L-can); and 0.5% wt/vol D-histidine (D-his). Plates were photographed and growth scored after 2–3 d of incubation at 30°C. (A) Mutations SHR3-30, SHR3-31, SHR3-32, and SHR3-33. (B) Mutations SHR3-34, shr3-35, SHR3-39, and SHR3-40. (C) Mutations SHR3-41, SHR3-42, SHR3-43, and SHR3-44. (D) Mutations SHR3-45, SHR3-46, SHR3-47, and SHR3-48. (E) Mutations SHR3-49, shr3-50, SHR3-54, SHR3-55, and SHR3-56. (F) Mutations SHR3-57, SHR3-58, SHR3-59, SHR3-60, and SHR3-61. (G) Mutations SHR3-62, SHR3-63, shr3-64, SHR3-65, and SHR3-66. (H) Mutations SHR3-67, SHR3-68, SHR3-69, SHR3-70, and SHR3-83. (I) Mutations SHR3-71, SHR3-72, SHR3-73, SHR3-74, and SHR3-75. (J) Mutations shr3-76, SHR3-79, SHR3-80, SHR3-81, and shr3-82.
Growth-based assessment of Shr3 mutant protein function. Growth of serially diluted cell suspensions from strain JKY2 (shr3Δ) carrying either vector control (VC) or the indicated plasmids expressing wild-type Shr3 (SHR3) or mutated forms of Shr3 proteins (upper case indicates alleles conferring wild-type growth; lower case indicates alleles exhibiting defective growth). The cells were spotted on YPD, SD (ammonium), and SAD (allantoin) containing toxic amino analogs as indicated at the following concentrations: 200 µg/ml metsulfuron-methyl (MM); 1 mM azetidine-2-carboxylate (AzC); 1 µg/ml L-canavanine (L-can); and 0.5% wt/vol D-histidine (D-his). Plates were photographed and growth scored after 2–3 d of incubation at 30°C. (A) Mutations SHR3-30, SHR3-31, SHR3-32, and SHR3-33. (B) Mutations SHR3-34, shr3-35, SHR3-39, and SHR3-40. (C) Mutations SHR3-41, SHR3-42, SHR3-43, and SHR3-44. (D) Mutations SHR3-45, SHR3-46, SHR3-47, and SHR3-48. (E) Mutations SHR3-49, shr3-50, SHR3-54, SHR3-55, and SHR3-56. (F) Mutations SHR3-57, SHR3-58, SHR3-59, SHR3-60, and SHR3-61. (G) Mutations SHR3-62, SHR3-63, shr3-64, SHR3-65, and SHR3-66. (H) Mutations SHR3-67, SHR3-68, SHR3-69, SHR3-70, and SHR3-83. (I) Mutations SHR3-71, SHR3-72, SHR3-73, SHR3-74, and SHR3-75. (J) Mutations shr3-76, SHR3-79, SHR3-80, SHR3-81, and shr3-82.
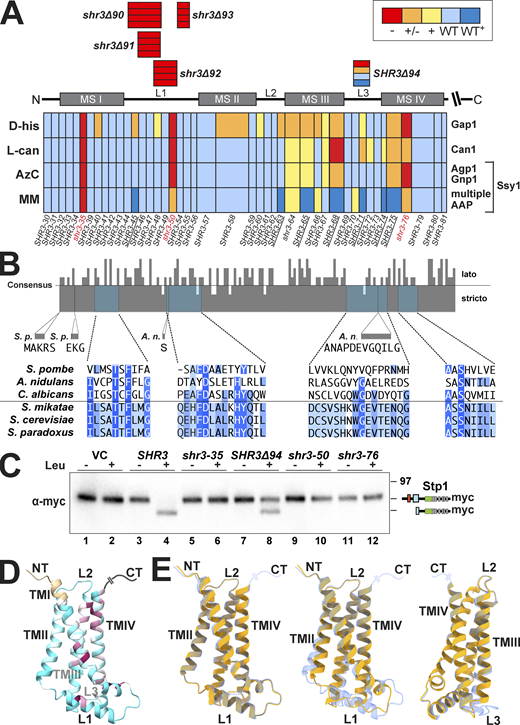
Mutational analysis of Shr3 function and substrate specificity. (A) Summary of growth characteristics of JKY2 (shr3Δ) individually expressing 44 Shr3-mutant proteins. Cells were spotted on media containing toxic amino analogs as described in Materials and methods. Growth was scored after 2–3 d of incubation at 30°C (Fig. S1). Colors reflect Shr3 function relative to wild-type activity: red, no function (−); orange, weak but detectable function (±); yellow, intermediate function but less than wild-type (+); light blue, wild-type function (WT); and dark blue, enhanced function (WT+). (B) Clustal O (Madeira et al., 2019) comparison of Shr3 sequences, corresponding to aa residues 1–159 of S. cerevisiae, and orthologs of members from the Saccharomyces sensu stricto group (S. paradoxus, S. mikatae) and orthologs from sensu lato fungi (S. pombe, A. nidulans, and C. albicans). The consensus plot (identity; Waterhouse et al., 2009) and detailed multiple sequence alignments are presented for the regions with mutations giving rise to major growth defects on selective media; identical residues in three (light blue), four (blue), and five or six homologs (dark blue) are highlighted. (C) Shr3-dependent Ssy1 folding and function assessed by Stp1 processing. Immunoblot analysis of extracts from FGY135 (shr3Δ) carrying pCA204 (STP1-13xMYC) and pRS316 (VC), pPL210 (SHR3), pAR004 (shr3-35), pAR45 (SHR3Δ94), pAR018 (shr3-50), or pPL1351 (shr3-76). Cells were grown in SD and induced for 30 min with 1.3 mM leucine (+) as indicated. (D) Predicted Shr3 structure (AlphaFold) with residues colored by sequence conservation ranging from cyan for less conserved (conservation equal or lower to a value of −0.5) to maroon for more conserved positions (conservation equal or higher to a value of 2.5). The coloring is assigned via ChimeraX, which uses the multiple sequence alignment PF08229_rp55 from Pfam (https://pfam.xfam.org/family/PF08229#tabview=tab3) and calculates conservation based on the entropy-based measure from AL2CO (Pei and Grishin, 2001). (E) Mutation-induced structural alterations predicted using AlphaFold. The individual models (yellow) of shr3-35 (left), shr3Δ92 (center), and Shr3Δ94 (right; rotated for better visualization of predicted changes) are superimposed onto the wild-type Shr3 structure (light blue). Molecular graphics and analyses were performed with UCSF ChimeraX. Source data are available for this figure: SourceData F3.
Mutational analysis of Shr3 function and substrate specificity. (A) Summary of growth characteristics of JKY2 (shr3Δ) individually expressing 44 Shr3-mutant proteins. Cells were spotted on media containing toxic amino analogs as described in Materials and methods. Growth was scored after 2–3 d of incubation at 30°C (Fig. S1). Colors reflect Shr3 function relative to wild-type activity: red, no function (−); orange, weak but detectable function (±); yellow, intermediate function but less than wild-type (+); light blue, wild-type function (WT); and dark blue, enhanced function (WT+). (B) Clustal O (Madeira et al., 2019) comparison of Shr3 sequences, corresponding to aa residues 1–159 of S. cerevisiae, and orthologs of members from the Saccharomyces sensu stricto group (S. paradoxus, S. mikatae) and orthologs from sensu lato fungi (S. pombe, A. nidulans, and C. albicans). The consensus plot (identity; Waterhouse et al., 2009) and detailed multiple sequence alignments are presented for the regions with mutations giving rise to major growth defects on selective media; identical residues in three (light blue), four (blue), and five or six homologs (dark blue) are highlighted. (C) Shr3-dependent Ssy1 folding and function assessed by Stp1 processing. Immunoblot analysis of extracts from FGY135 (shr3Δ) carrying pCA204 (STP1-13xMYC) and pRS316 (VC), pPL210 (SHR3), pAR004 (shr3-35), pAR45 (SHR3Δ94), pAR018 (shr3-50), or pPL1351 (shr3-76). Cells were grown in SD and induced for 30 min with 1.3 mM leucine (+) as indicated. (D) Predicted Shr3 structure (AlphaFold) with residues colored by sequence conservation ranging from cyan for less conserved (conservation equal or lower to a value of −0.5) to maroon for more conserved positions (conservation equal or higher to a value of 2.5). The coloring is assigned via ChimeraX, which uses the multiple sequence alignment PF08229_rp55 from Pfam (https://pfam.xfam.org/family/PF08229#tabview=tab3) and calculates conservation based on the entropy-based measure from AL2CO (Pei and Grishin, 2001). (E) Mutation-induced structural alterations predicted using AlphaFold. The individual models (yellow) of shr3-35 (left), shr3Δ92 (center), and Shr3Δ94 (right; rotated for better visualization of predicted changes) are superimposed onto the wild-type Shr3 structure (light blue). Molecular graphics and analyses were performed with UCSF ChimeraX. Source data are available for this figure: SourceData F3.
We performed multiple sequence alignments of the membrane domain of Shr3 and orthologs from two Saccharomyces sensu stricto strains, S. paradoxus and S. mikatae, and from three divergent lato fungal strains, Candida albicans (Csh3), Schizosaccharomyces pombe (Psh3), and Aspergillus nidulans (ShrA), and obtained a consensus identity plot (Fig. 3 B; Madeira et al., 2019). The Shr3 orthologs of C. albicans, S. pombe, and A. nidulans have been shown to function analogously and are required for proper amino acid uptake. Heterologous expression of CSH3 complements shr3Δ (Martínez and Ljungdahl, 2004), whereas heterologous expression of PSH3 or SHRA only partially complements and merely facilitates the functional expression of a limited subset of S. cerevisiae permeases (Erpapazoglou et al., 2006; Martínez and Ljungdahl, 2000). The Shr3 sequence is well-conserved in the Saccharomyces sensu stricto strains, exhibiting almost absolute identity. Several positions throughout the membrane domain of Shr3 are conserved between the full set of selected sequences. Interestingly, threonine 19, which is a critical amino acid residue in MSI, is conserved in all orthologs (Fig. 3 B, T19 is highlighted in dark blue). The requirement for a polar amino acid in MSIV of Shr3 is conserved as well (Fig. 3 B and Fig. S1, 39 highlighted in dark blue). A higher sequence divergence is evident in the luminal loop L3, with the extreme case of the A. nidulans ortholog that contains an extra sequence of 12 amino acid residues. The limited sequence identity in loop L3 aligns with the observation that mutations in L3 exhibit the most pleiotropic effects. Consistently, the predicted 3D AlphaFold structures of Shr3 and other fungal homologs exhibit the highest degree of variability within the L3 loop (Fig. S3).

Structural divergence between Shr3 and fungal orthologs. The predicted AlphaFold2 structure of Shr3 of S. cerevisiae (orange) and the indicated fungal orthologs (contrasting colors) were superimposed. Molecular graphics and analyses were performed with UCSF ChimeraX Graphics. The UniProt identifiers are indicated. Lumenl loop 3 (lower right corner) exhibits the greatest degree of sequence and structural divergence.
Structural divergence between Shr3 and fungal orthologs. The predicted AlphaFold2 structure of Shr3 of S. cerevisiae (orange) and the indicated fungal orthologs (contrasting colors) were superimposed. Molecular graphics and analyses were performed with UCSF ChimeraX Graphics. The UniProt identifiers are indicated. Lumenl loop 3 (lower right corner) exhibits the greatest degree of sequence and structural divergence.
The robust WT+ growth on YPD+MM exhibited by several mutant proteins clearly indicates that they retain the capacity to facilitate the folding of some AAP. In contrast to the other members of the AAP transporter family, Ssy1 does not facilitate amino acid uptake but rather functions as the primary receptor of extracellular amino acids in the context of the plasma membrane-localized SPS sensor (Didion et al., 1998; Iraqui et al., 1999; Klasson et al., 1999). Ssy1 differs from the other members of the AAP family in that it features a significantly longer cytoplasmic-oriented N-terminal domain and extended extracellular loops connecting MS V-VII and VII-VIII (Table S3). In response to extracellular amino acids, Ssy1 initiates signaling events leading to the proteolytic activation of transcription factors Stp1 and Stp2, which subsequently induce the expression of multiple AAP facilitating branched amino acid uptake, including AGP1 and GNP1. We tested the notion that Shr3Δ94 retained the ability to efficiently facilitate Ssy1 folding by examining the proteolytic cleavage of Stp1 (Fig. 3 C). Consistent with the growth assays, leucine induction led to Stp1 processing in strain FGY135 (shr3Δ) expressing SHR3 or SHR3Δ94 (Fig. 3 C, lanes 4 and 8), but not the non-functional alleles shr3-35, shr3-50, or shr3-76 (Fig. 3 C, lanes 6, 10, and 12). The data clearly demonstrate that Shr3Δ94 retains the capacity to facilitate Ssy1 folding.
Mapping mutations onto the predicted AlphaFold structure of Shr3
We mapped the conserved residues of Shr3 onto the predicted AlphaFold (https://alphafold.ebi.ac.uk) 3D structure of Shr3 (Fig. 3 D). The RP55 dataset of representative proteomes from the Pfam protein family database (https://pfam.xfam.org) was used for the multiple sequence alignment. Interestingly, although limited in number, the residues that are well-conserved localize to MSI, III, and IV and in the helical structure of L1, which are parts of the 3D structure that appear to be in proximity with each other, and thus may form the basis of interactions essential for a functional structure. In agreement with this, most of the mutations that affect function localize to the same structural elements and in the vicinity of the most conserved residues (Fig. 3, A and B). In contrast, mutations in MSII have minimal impact on Shr3 function; MSII is predicted to be more loosely associated with the other MS of Shr3 and is also the least conserved (Fig. 3 D). Although the degree of accuracy of the predictions for mutated sequences by AlphaFold is uncertain, we queried AlphaFold to predict 3D structures of the mutant Shr3 proteins and aligned them on the predicted structure of wild-type Shr3 (Fig. 3 E). Apart from certain local mismatches, the structural models of the Shr3 mutant proteins aligned remarkably well with wild-type Shr3. Strikingly, although the leucine substitutions at positions 17 through 19 (shr3-35) result in a clearly defective and non-functional protein (Fig. 3 A), no obvious structural alterations were predicted, perhaps with the exception of a minimal shift in MSI (Fig. 3 E, left). Similarly, the predicted structure of partially functional Shr3Δ94, lacking seven residues in luminal L3, aligns well with wild-type Shr3 with only a slight shift in the luminal face of MSIII and MSIV (Fig. 3 E, right). The predicted structure of the non-functional shr3Δ92, lacking the L1 residues thought to fold into an alpha-helix, exhibited a greater structural difference (Fig. 3 E, center). The absence of the α-helical element and perhaps the shorter length of L1 affected the degree of alignment with the wild-type Shr3, especially at the luminal face of all four TM helices.
Shr3-AAP substrate interactions
To critically test if the observed growth phenotypes correlate with the ability of Shr3 and shr3 mutant proteins to interact with AAP, we refined and exploited a split-ubiquitin approach to monitor interactions in vivo (Fig. 4). A sequence encoding the N-terminal fragment of ubiquitin carrying the I13A mutation (NubA), which reduces the propensity of spontaneous interactions (Johnsson, 2002; Johnsson and Varshavsky, 1994), was fused at the C-terminal end of SHR3, shr3-35, and SHR3Δ94, creating the Shr3-NubA constructs schematically depicted in Fig. 4 A. Next, we created a GAP1 allele encoding the C-terminal fragment of ubiquitin (Cub) tagged with GST-6xHA (Cub-GST; Fig. 4 A). The resulting GAP1-Cub-GST was placed under the control of the GAL1-promoter. When co-expressed, productive interactions between Shr3-NubA and Gap1-Cub-GST were expected to facilitate the assembly of the split NubA and Cub domains into a functional ubiquitin moiety recognized by ubiquitin-specific proteases, resulting in the release of the GST-6xHA reporter (Fig. 4 B). The functional attributes of the NubA and Cub fusion constructs were tested by their ability to individually complement shr3Δ and gap1Δ growth defects, respectively. Strain JKY2 (shr3Δ) carrying SHR3-NubA or SHR3Δ94-NubA grew as well as SHR3 without NubA on YPD + MM (Fig. 4 C, compare dilution 2 with 3 and 5), whereas the shr3-35-NubA allele did not (dilution 4). Strain FGY15 (gap1Δ) carrying either GAP1-Cub-GST of GAP1 grew on citrulline as a sole nitrogen source (Fig. 4 C, compare dilution 7 with 8). The functionality of Gap1-Cub-GST is dependent on its ability to exit the ER; a construct lacking the ER exit motif in the hydrophilic C-terminal domain of Gap1 is not functional (Fig. 4 C, dilution 9), presumably due to its retention in the ER. These results indicate that the presence of the Nub and Cub domains do not significantly impair the function of wild-type Shr3 and Gap1, respectively.
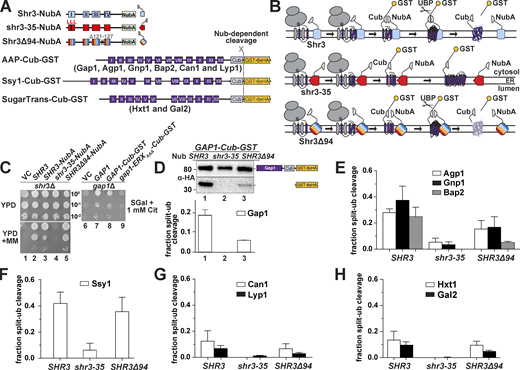
Assessing Shr3-Gap1 interactions using split ubiquitin. (A) Schematic diagram of the split ubiquitin constructs used to evaluate Shr3-AAP, Shr3-Ssy1, and Shr3-HXT interactions. (B) Overview of the split-ubiquitin assay and expected outcomes with Shr3-AAP interactions. (C) Left panels: Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pPL1262 (SHR3-NubA), pAR67 (shr3-35-NubA), or pAR76 (SHR3Δ94-NubA) spotted on YPD and YPD+MM plates. Plates were incubated at 30°C for 2 d and photographed. Right panel: Serial dilutions of cell suspensions from strain FGY15 (gap1Δ) carrying pRS317 (VC), pJK92 (GAP1), pPL1257 (GAP1-Cub-GST), or pIM28 (gap1-ERXAAA-Cub-GST) were spotted on minimal medium with 2% galactose as carbon source and 1 mM L-citrulline as sole nitrogen source. Plates were incubated for 7 d and photographed. (D) Shr3-Gap1 interactions; strain FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262), shr3-35-NubA (pAR67), or SHR3Δ94-NubA (pAR76) and carrying pPL1257 (GAP1-Cub-GST) were induced with 2% galactose for 1 h. Protein extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of full-length and cleaved Gap1 were quantified. The fraction of split-ubiquitin cleavage was determined; the mean values were plotted with error bars showing standard deviation (n = 3). (E) Shr3-Agp1, Shr3-Gnp1, and Shr3-Bap2 interactions; FGY135 strains as in D carrying pIM6 (AGP1-Cub-GST), pIM17 (GNP1-Cub-GST), or pIM7 (BAP2-Cub-GST). (F) Shr3-Ssy1 interactions; FGY135 strains as in D carrying pIM19 (SSY1-Cub-GST). (G) Shr3-Can1 and Shr3-Lyp1 interactions; FGY135 strains as in D carrying pIM8 (CAN1-Cub-GST) or pIM18 (LYP1-Cub-GST). (H) Shr3-Hxt1 and Shr3-Gal2 interactions; FGY135 strains as in D carrying pIM32 (HXT1-Cub-GST) or pIM33 (GAL2-Cub-GST). Strains in E–H were induced with 2% galactose for 1 h. Protein extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of full-length and cleaved Agp1, Gnp1, Bap2, Ssy1, Can1, Lyp1, Hxt1, and Gal2 constructs were quantified. The fraction of split-ubiquitin cleavage was determined; the mean values were plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F4.
Assessing Shr3-Gap1 interactions using split ubiquitin. (A) Schematic diagram of the split ubiquitin constructs used to evaluate Shr3-AAP, Shr3-Ssy1, and Shr3-HXT interactions. (B) Overview of the split-ubiquitin assay and expected outcomes with Shr3-AAP interactions. (C) Left panels: Serial dilutions of cell suspensions from strain JKY2 (shr3Δ) carrying pRS316 (VC), pPL210 (SHR3), pPL1262 (SHR3-NubA), pAR67 (shr3-35-NubA), or pAR76 (SHR3Δ94-NubA) spotted on YPD and YPD+MM plates. Plates were incubated at 30°C for 2 d and photographed. Right panel: Serial dilutions of cell suspensions from strain FGY15 (gap1Δ) carrying pRS317 (VC), pJK92 (GAP1), pPL1257 (GAP1-Cub-GST), or pIM28 (gap1-ERXAAA-Cub-GST) were spotted on minimal medium with 2% galactose as carbon source and 1 mM L-citrulline as sole nitrogen source. Plates were incubated for 7 d and photographed. (D) Shr3-Gap1 interactions; strain FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262), shr3-35-NubA (pAR67), or SHR3Δ94-NubA (pAR76) and carrying pPL1257 (GAP1-Cub-GST) were induced with 2% galactose for 1 h. Protein extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of full-length and cleaved Gap1 were quantified. The fraction of split-ubiquitin cleavage was determined; the mean values were plotted with error bars showing standard deviation (n = 3). (E) Shr3-Agp1, Shr3-Gnp1, and Shr3-Bap2 interactions; FGY135 strains as in D carrying pIM6 (AGP1-Cub-GST), pIM17 (GNP1-Cub-GST), or pIM7 (BAP2-Cub-GST). (F) Shr3-Ssy1 interactions; FGY135 strains as in D carrying pIM19 (SSY1-Cub-GST). (G) Shr3-Can1 and Shr3-Lyp1 interactions; FGY135 strains as in D carrying pIM8 (CAN1-Cub-GST) or pIM18 (LYP1-Cub-GST). (H) Shr3-Hxt1 and Shr3-Gal2 interactions; FGY135 strains as in D carrying pIM32 (HXT1-Cub-GST) or pIM33 (GAL2-Cub-GST). Strains in E–H were induced with 2% galactose for 1 h. Protein extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of full-length and cleaved Agp1, Gnp1, Bap2, Ssy1, Can1, Lyp1, Hxt1, and Gal2 constructs were quantified. The fraction of split-ubiquitin cleavage was determined; the mean values were plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F4.
To test in vivo interactions, we analyzed protein extracts from strain FGY135 (shr3Δ gap1Δ) carrying plasmids GAP1-Cub-GST-6xHA and SHR3-NubA, shr3-35-NubA, or SHR3Δ94-NubA by immunoblot. In cells expressing SHR3-NubA or SHR3Δ94-NubA and GAP1-Cub-GST-6xHA, two anti-HA reactive bands were detected, corresponding to full-length Gap1-Cub-GST-6xHA and the cleaved GST-6xHA (Fig. 4 D, lane 1). We calculated the fraction of split-ubiquitin cleavage in the SHR3-NubA strain to be ≈20% by dividing the intensities of the cleaved band with the intensities from full-length plus cleaved species, whereas in the SHR3Δ94-NubA strain, the cleavage was 5% (Fig. 4 D). By contrast, only a single band, full-length Gap1-Cub-GST-6xHA was detected in extracts from the strain expressing the non-functional shr3-35-NubA (Fig. 4 D, lane 2). Although expressed at similar levels, the ER retained gap1-ERXAAA-Cub-GST-6xHA protein did not exhibit an enhanced propensity to interact with Shr3 (Fig. S4), suggesting the split-ubiquitin assay primarily monitors transient interactions that presumably occur during the co-translational folding of AAP.
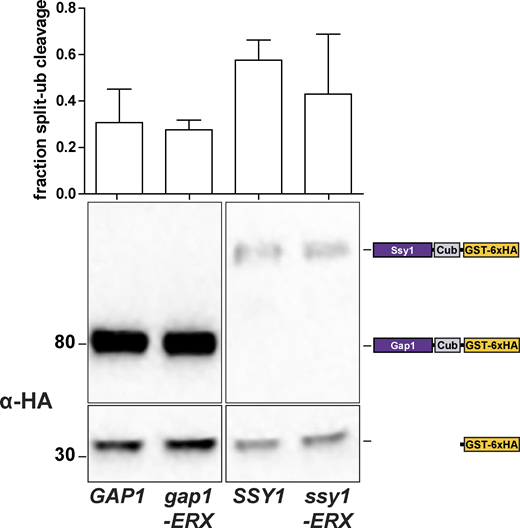
Effect of ER exit motif mutations on Shr3-AAP interactions. Strains FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262) and carrying pPL1257 (GAP1-Cub-GST), pIM19 (SSY1-Cub-GST), pIM28 (gap1-ERXAAA-Cub-GST), or pIM29 (ssy1-ERXAAA-Cub-GST) were induced with 2% galactose for 1 h. Extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of full-length and cleaved Gap1 and Ssy1 constructs were quantified. The fraction of split-ubiquitin cleavage was determined; the mean values were plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData FS4.
Effect of ER exit motif mutations on Shr3-AAP interactions. Strains FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262) and carrying pPL1257 (GAP1-Cub-GST), pIM19 (SSY1-Cub-GST), pIM28 (gap1-ERXAAA-Cub-GST), or pIM29 (ssy1-ERXAAA-Cub-GST) were induced with 2% galactose for 1 h. Extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of full-length and cleaved Gap1 and Ssy1 constructs were quantified. The fraction of split-ubiquitin cleavage was determined; the mean values were plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData FS4.
Based on the success of the split ubiquitin approach to analyze Shr3-Gap1 interactions, we created Cub-GST-6xHA tagged constructs with five additional AAP, i.e., Agp1, Gnp1, Bap2, Can1, and Lyp1, the non-transporting but Shr3-dependent AAP homolog Ssy1, and two Shr3-independent sugar transporters (HXT), i.e., the low-affinity glucose transporter Hxt1 and the galactose transporter Gal2 (Fig. 4 A). Extracts from strain FGY135 (shr3Δ gap1Δ) carrying SHR3-NubA, shr3-35-NubA, or SHR3Δ94-NubA and an individual AAP-Cub-GST-6xHA or HXT-Cub-GST-6xHA construct were prepared, and the levels of the GST-6xHA reporter were determined (Fig. 4, E–H). Consistent with the general requirement of Shr3 for AAP folding, robust interactions were detected between wild-type Shr3-NubA and Agp1-, Gnp1-, Bap2-, and Ssy1-Cub constructs (Fig. 4, E and F). Although Shr3 is required for their functional expression, Can1- and Lyp1-Cub constructs exhibited low levels of reporter cleavage (Fig. 4 G), similar to the Shr3-independent sugar transporters (Fig. 4 H).
Interactions between shr3-35-NubA and AAP-Cub were weak or absent, which precisely aligns with shr3-35 being non-functional on all selective media tested (Figs. 1 and 3). Unexpectedly, the split-Ub interactions with unrelated sugar transporters (HXT) were also significantly weaker compared with the wild-type Shr3-NubA (Fig. 4 H). This prompted us to test whether the NubA domain of Shr3-35-NubA is incorrectly oriented toward the ER lumen; however, the results indicate that NubA is correctly oriented to the cytoplasm and is presented in a context capable of supporting potential interactions (Fig. S5 A). Together, our findings indicate that the shr3-35 protein, although expressed at similar levels as Shr3 and with a correctly oriented NubA domain, is incapable of broadly engaging folding substrates.
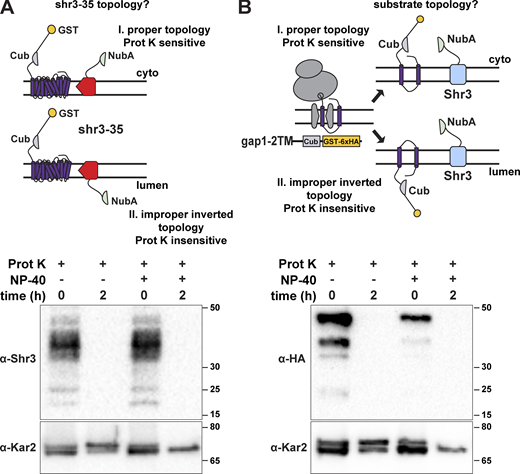
Protease cleavage assay to assess the topology of shr3-35-NubA and gap1-2TM-Cub-GST. (A) Schematic diagram of the split ubiquitin shr3-35-NubA construct (pAR67) and possible topological orientations: I. proper topology, i.e., TM in native orientation with NubA exposed to the cytoplasm and Proteinase K (Prot K) sensitive; II. improper inverted topology, i.e., TM in non-native orientation with NubA oriented toward the lumen and Prot K insensitive in the absence of detergent (NP-40). The ER-membrane topology of inserted shr3-35-NubA was examined in intact microsomes isolated from strain FGY135 (gap1Δ shr3Δ) carrying pPL1257 (GAP1-Cub-GST) and pAR67 (shr3-35-NubA). Microsomes were incubated with Prot K for 2 h with or without the addition of 0.2% NP-40 as indicated. shr3-35 was detected with the use of α-Shr3 directed to a C-terminally located epitope. Kar2 served as a control for microsome integrity. (B) Schematic diagram of the split ubiquitin gap1-2TM-Cub-GST construct (pIM1) and possible topological orientations: I. proper topology, i.e., TM in native orientation with Cub-GST exposed to the cytoplasm and Proteinase K (Prot K) sensitive; II. improper inverted topology, i.e., TM in non-native orientation with Cub-GST oriented toward the lumen and Prot K insensitive in the absence of detergent (NP-40). The ER-membrane topology of inserted gap1-2TM-Cub-GST-6xHA was examined in intact microsomes isolated from strain HKY15 (ssy1Δ) carrying (gap1-2TM-Cub-GST) and incubated with Prot K for 2 h with or without the addition of 0.2% NP-40 as indicated. Kar2 served as a control for microsome integrity. Source data are available for this figure: SourceData FS5.
Protease cleavage assay to assess the topology of shr3-35-NubA and gap1-2TM-Cub-GST. (A) Schematic diagram of the split ubiquitin shr3-35-NubA construct (pAR67) and possible topological orientations: I. proper topology, i.e., TM in native orientation with NubA exposed to the cytoplasm and Proteinase K (Prot K) sensitive; II. improper inverted topology, i.e., TM in non-native orientation with NubA oriented toward the lumen and Prot K insensitive in the absence of detergent (NP-40). The ER-membrane topology of inserted shr3-35-NubA was examined in intact microsomes isolated from strain FGY135 (gap1Δ shr3Δ) carrying pPL1257 (GAP1-Cub-GST) and pAR67 (shr3-35-NubA). Microsomes were incubated with Prot K for 2 h with or without the addition of 0.2% NP-40 as indicated. shr3-35 was detected with the use of α-Shr3 directed to a C-terminally located epitope. Kar2 served as a control for microsome integrity. (B) Schematic diagram of the split ubiquitin gap1-2TM-Cub-GST construct (pIM1) and possible topological orientations: I. proper topology, i.e., TM in native orientation with Cub-GST exposed to the cytoplasm and Proteinase K (Prot K) sensitive; II. improper inverted topology, i.e., TM in non-native orientation with Cub-GST oriented toward the lumen and Prot K insensitive in the absence of detergent (NP-40). The ER-membrane topology of inserted gap1-2TM-Cub-GST-6xHA was examined in intact microsomes isolated from strain HKY15 (ssy1Δ) carrying (gap1-2TM-Cub-GST) and incubated with Prot K for 2 h with or without the addition of 0.2% NP-40 as indicated. Kar2 served as a control for microsome integrity. Source data are available for this figure: SourceData FS5.
Interestingly, Shr3Δ94-NubA interacted robustly with Ssy1-Cub-GST-6xHA at levels comparable with wild-type Shr3-NubA (Fig. 4 F). In contrast, Shr3Δ94-NubA interacted with the other AAP-Cub constructs at significantly reduced levels. These findings suggested a mechanistic explanation for the enhanced WT+ growth phenotype conferred by Shr3Δ94-NubA. Ssy1 strictly requires Shr3 for folding (Klasson et al., 1999) and constitutes the integral membrane component of the PM-localized SPS sensor that induces the expression of AAP genes in response to extracellular amino acids (Ljungdahl and Daignan-Fornier, 2012). Consistent with the finding that the Shr3Δ94 allele supported Stp1 processing (Fig. 3 C), our data indicate that the Shr3Δ94 mutant retains the ability to assist the folding of Ssy1, restoring the transcriptional circuits abrogated by shr3Δ, resulting in enhanced branched-chain amino acid uptake. Together, the results indicate that the in vivo interactions monitored by the split ubiquitin cleavage provide a nuanced assessment of Shr3 function.
Shr3 interacts with substrates in a progressive manner
As a proxy to investigate the temporal aspects of Shr3-facilitated AAP folding, we constructed a series of truncated gap1-Cub-GST, hxt1-Cub-GST, and gal2-Cub-GST alleles capable of encoding 2, 4, 6, 8, 10, and 12 MS (Fig. 5 A). Strain FGY135 (shr3Δ gap1Δ) carrying plasmid SHR3-NubA and a truncated gap1-Cub-GST, hxt1-Cub-GST, or gal2-Cub-GST allele was employed and potential interactions were monitored by immunoblot. Shr3-NubA did not interact with gap1-2TM, even though the Cub domain is presented in the context of proper membrane topology oriented toward the cytoplasm (Fig. S5 B). The presence of two additional MS of Gap1 (gap1-4TM) supported the interaction with Shr3 (Fig. 5 B, left panel; Fig. 5 C, black bars). The intensity of the interactions increased and eventually plateaued in the strains carrying the gap1-6TM/-8TM/-10TM alleles, respectively. Strikingly, the gap1-12TM construct interacted only weakly with the functional Shr3-NubA. In marked contrast to the interaction pattern of the gap1-Cub-GST truncations with Shr3-NubA, we could not detect at appreciable levels the GST-6xHA reporter in extracts from the strain expressing SHR3-NubA and any of the truncated hxt1-Cub-GST or gal2-Cub-GST alleles (Fig. 5 B, center and right panels; Fig. 5 C, gray and white bars). We sought independent evidence regarding the clear substrate specificity exhibited by Shr3 for Gap1 constructs and applied a co-immunoprecipitation strategy to independently test the potential of substrates to co-purify with Shr3-GFP (Fig. S6); gap1-8TM and -10TM but not hxt-8TM or -10TM copurified with Shr3-GFP. The confirmatory nature of co-immunoprecipitation results strongly suggests that the split ubiquitin approach faithfully monitors specific Shr3-AAP interactions.
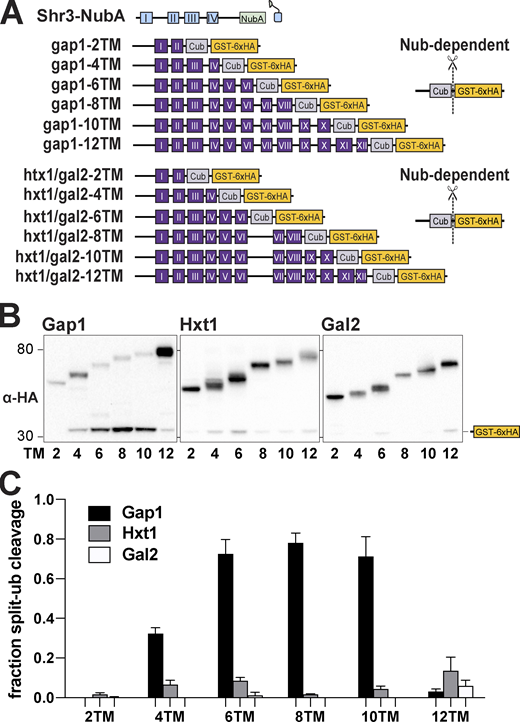
Progressivity of Shr3-Gap1 chaperone-substrate interactions. (A) Schematic diagram of split ubiquitin constructs including the gap1-Cub-GST, hxt1-Cub-GST, and gal2-Cub-GST truncation constructs. (B) Strain FGY135 (gap1Δ shr3Δ) carrying pPL1262 (SHR3-NubA) and pIM1 (gap1-2TM-Cub-GST), pIM2 (gap1-4TM-Cub-GST), pIM3 (gap1-6TM-Cub-GST), pIM4 (gap1-8TM-Cub-GST), pIM5 (gap1-10TM-Cub-GST), or pIM16 (gap1-12TM-Cub-GST; left panel), or pIM34 (hxt1-2TM-Cub-GST), pIM35 (hxt1-4TM-Cub-GST), pIM36 (hxt1-6TM-Cub-GST), pIM37 (hxt1-8TM-Cub-GST), pIM38 (hxt1-10TM-Cub-GST), or pIM39 (hxt1-12TM-Cub-GST; center panel) or pIM40 (gal2-2TM-Cub-GST), pIM41 (gal2-4TM-Cub-GST), pIM42 (gal2-6TM-Cub-GST), pIM43 (gal2-8TM-Cub-GST), pIM44 (gal2-10TM-Cub-GST), or pIM45 (gal2-12TM-Cub-GST; right panel) were induced with 2% galactose for 1 h. Extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. (C) The signal intensities of the immunoreactive forms of uncleaved Cub constructs and cleaved interaction marker (GST-6xHA) were quantified; the mean values of the fraction of split ubiquitin cleavage are plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F5.
Progressivity of Shr3-Gap1 chaperone-substrate interactions. (A) Schematic diagram of split ubiquitin constructs including the gap1-Cub-GST, hxt1-Cub-GST, and gal2-Cub-GST truncation constructs. (B) Strain FGY135 (gap1Δ shr3Δ) carrying pPL1262 (SHR3-NubA) and pIM1 (gap1-2TM-Cub-GST), pIM2 (gap1-4TM-Cub-GST), pIM3 (gap1-6TM-Cub-GST), pIM4 (gap1-8TM-Cub-GST), pIM5 (gap1-10TM-Cub-GST), or pIM16 (gap1-12TM-Cub-GST; left panel), or pIM34 (hxt1-2TM-Cub-GST), pIM35 (hxt1-4TM-Cub-GST), pIM36 (hxt1-6TM-Cub-GST), pIM37 (hxt1-8TM-Cub-GST), pIM38 (hxt1-10TM-Cub-GST), or pIM39 (hxt1-12TM-Cub-GST; center panel) or pIM40 (gal2-2TM-Cub-GST), pIM41 (gal2-4TM-Cub-GST), pIM42 (gal2-6TM-Cub-GST), pIM43 (gal2-8TM-Cub-GST), pIM44 (gal2-10TM-Cub-GST), or pIM45 (gal2-12TM-Cub-GST; right panel) were induced with 2% galactose for 1 h. Extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. (C) The signal intensities of the immunoreactive forms of uncleaved Cub constructs and cleaved interaction marker (GST-6xHA) were quantified; the mean values of the fraction of split ubiquitin cleavage are plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F5.
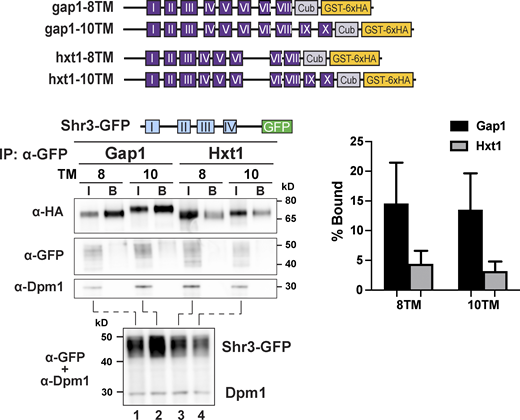
Shr3 specifically associates with Gap1 truncations. Strain FGY135 (gap1Δ shr3Δ) expressing SHR3-GFP (pAR46) and carrying plasmids gap1-8TM (pIM4), gap1-10TM (pIM5), hxt1-8TM (pIM37), or hxt1-10TM (pIM38; schematically represented) was induced with 2% galactose for 1 h. Extracts were prepared and Shr3-GFP was purified with GFP-trap agarose beads in the presence of 0.8% n-dodecyl-β-D-maltoside (DDM) as described (Methods and materials). Bound proteins, resolved by SDS-PAGE, were analyzed by immunoblot using α-HA, α-GFP, and α-Dpm1 antibodies. The input (I; lanes 1–4) correspond to 1.8% of extracts incubated with the GFP-trap beads. Extracts were separately analyzed (lower panel) to analyze levels of Shr3-GFP. The signal intensities of the immunoreactive forms of Cub-GST-6xHA constructs were quantified (right panel); the mean values of the Bound% are plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData FS6.
Shr3 specifically associates with Gap1 truncations. Strain FGY135 (gap1Δ shr3Δ) expressing SHR3-GFP (pAR46) and carrying plasmids gap1-8TM (pIM4), gap1-10TM (pIM5), hxt1-8TM (pIM37), or hxt1-10TM (pIM38; schematically represented) was induced with 2% galactose for 1 h. Extracts were prepared and Shr3-GFP was purified with GFP-trap agarose beads in the presence of 0.8% n-dodecyl-β-D-maltoside (DDM) as described (Methods and materials). Bound proteins, resolved by SDS-PAGE, were analyzed by immunoblot using α-HA, α-GFP, and α-Dpm1 antibodies. The input (I; lanes 1–4) correspond to 1.8% of extracts incubated with the GFP-trap beads. Extracts were separately analyzed (lower panel) to analyze levels of Shr3-GFP. The signal intensities of the immunoreactive forms of Cub-GST-6xHA constructs were quantified (right panel); the mean values of the Bound% are plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData FS6.
As a critical test of this notion, we examined if interactions could be detected between Shr3-NubA and two truncated Can1 constructs with 8 and 10 MS (Fig. 6). The rationale is that growth-based and biochemical assays have clearly defined Can1 as a bona fide substrate of Shr3. Despite the clear requirement for Shr3, interactions between full-length Can1 and Shr3 were found to be weak, similar to that corresponding to the non-Shr3 substrates Hxt1 and Gal2 (Fig. 4, G and H). We posited that if fully translated truncations of AAP are indeed proxies of translation intermediates, then truncations of Can1 would readily interact with Shr3. In a striking contrast to hxt1 and gal2-8TM- and -10TM-Cub, we readily detected robust interactions between the can1-8TM and -10TM-Cub and Shr3-Nub (Fig. 6, B and C). The findings are clearly consistent with Shr3 functioning specifically and at the early stages of AAP folding.
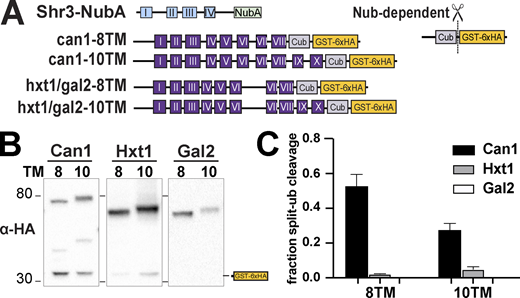
Shr3-NubA interacts with can1-Cub-GST but not hxt1-Cub-GST or gal2-Cub-GST truncations. (A) Schematic diagram of the split ubiquitin truncated constructs used to evaluate Shr3-can1 and Shr3-hxt1/gal2 interactions. (B) Strain FGY135 (gap1Δ shr3Δ) carrying pPL1262 (SHR3-NubA) and pIM46 (can1-8TM-Cub-GST), pIM47 (can1-10TM-Cub-GST), pIM37 (hxt1-8TM-Cub-GST), pIM38 (hxt1-10TM-Cub-GST), pIM43 (gal2-8TM-Cub-GST), or pIM44 (gal2-10TM-Cub-GST) was induced with 2% galactose for 1 h. Extracts were prepared, separated by SGS-PAGE, and analyzed by immunoblotting using antibodies against the 6xHA. (C) The signal intensities of the immunoreactive forms of uncleaved Cub constructs and cleaved interaction marker (GST-6xHA) were quantitated; the mean values of the fraction of split ubiquitin are plotted with error bars showing standard deviation (n = 3; biological replicates). The quantitation corresponding to hxt1-8TM-Cub-GST, hxt1-10TM-Cub-GST, gal2-8TM-Cub-GST, and gal2-10TM-Cub-GST are the same as presented in Fig. 5 C; however, the images correspond to hxt1- and gal2-Cub-constructs are from independent biological replicates. Source data are available for this figure: SourceData F6.
Shr3-NubA interacts with can1-Cub-GST but not hxt1-Cub-GST or gal2-Cub-GST truncations. (A) Schematic diagram of the split ubiquitin truncated constructs used to evaluate Shr3-can1 and Shr3-hxt1/gal2 interactions. (B) Strain FGY135 (gap1Δ shr3Δ) carrying pPL1262 (SHR3-NubA) and pIM46 (can1-8TM-Cub-GST), pIM47 (can1-10TM-Cub-GST), pIM37 (hxt1-8TM-Cub-GST), pIM38 (hxt1-10TM-Cub-GST), pIM43 (gal2-8TM-Cub-GST), or pIM44 (gal2-10TM-Cub-GST) was induced with 2% galactose for 1 h. Extracts were prepared, separated by SGS-PAGE, and analyzed by immunoblotting using antibodies against the 6xHA. (C) The signal intensities of the immunoreactive forms of uncleaved Cub constructs and cleaved interaction marker (GST-6xHA) were quantitated; the mean values of the fraction of split ubiquitin are plotted with error bars showing standard deviation (n = 3; biological replicates). The quantitation corresponding to hxt1-8TM-Cub-GST, hxt1-10TM-Cub-GST, gal2-8TM-Cub-GST, and gal2-10TM-Cub-GST are the same as presented in Fig. 5 C; however, the images correspond to hxt1- and gal2-Cub-constructs are from independent biological replicates. Source data are available for this figure: SourceData F6.
To more fully understand Shr3–substrate interactions, we created a series of truncated Agp1 and Ssy1 split-ubiquitin constructs (Fig. 7). Interestingly, in contrast to gap1-2TM, the agp1-2TM construct clearly interacted with Shr3-NubA (Fig. 7 A, black bars). Aside from this difference, the pattern of interactions with the remaining Agp1 constructs was strikingly similar to that observed with Gap1 truncations (Fig. 5); the intensity of the GST-6xHA reporter increased successively as the number of MS increased from 4 to 10 and greatly reduced when all 12 TM were present (Fig. 7 A, black bars). The interactions of the Shr3Δ94-NubA with the agp1-Cub constructs followed a similar pattern to that of the wild-type Shr3-NubA, albeit of lower intensity (Fig. 7 A, white bars). Notedly, the interaction patterns with the truncated Ssy1 constructs were quite different in that both Shr3-NubA and Shr3Δ94-NubA interacted at similar levels (Fig. 7 B). These findings are consistent with the observed interactions with full-length Ssy1-Cub (Fig. 4 F) and the ability of Shr3Δ94 to support enhanced WT+ growth (Fig. 3). Interestingly, as did agp1-2TM, ssy1-2TM engaged in readily detected interactions. The ssy1-4TM and -6TM constructs exhibited weaker interactions; however, the ssy1-8TM, -10TM, and -12TM constructs exhibited robust interactions. In summary, the data strongly suggest that Shr3 engages early during the folding of AAP.
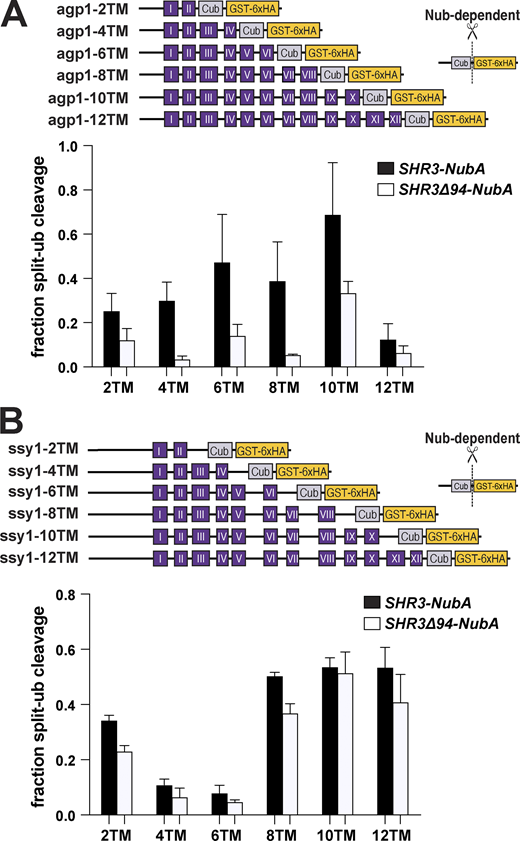
Progressive Shr3-Agp1 and Shr3-Ssy1 chaperone-substrate interactions. (A) Schematic diagram of agp1-Cub-GST truncation constructs. Strain FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262) or SHR3Δ94-NubA (pAR76) and carrying pIM9 (agp1-2TM-Cub-GST), pIM10 (agp1-4TM-Cub-GST), pIM11 (agp1-6TM-Cub-GST), pIM12 (agp1-8TM-Cub-GST), pIM13 (agp1-10TM-Cub-GST), or pIM26 (agp1-12TM-Cub-GST) were induced with 2% galactose for 1 h. Extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of uncleaved Cub constructs and cleaved interaction marker (GST-6xHA) were quantified; the mean values of the fraction of split ubiquitin cleavage are plotted with error bars showing standard deviation (n = 3; biological replicates). (B) Schematic diagram of ssy1-Cub-GST truncation constructs. Strain FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262) or SHR3Δ94-NubA (pAR76) and carrying pIM20 (ssy1-2TM-Cub-GST), pIM21 (ssy1-4TM-Cub-GST), pIM22 (ssy1-6TM-Cub-GST), pIM23 (ssy1-8TM-Cub-GST), pIM24 (ssy1-10TM-Cub-GST), or pIM25 (ssy1-12TM-Cub-GST) were induced with 2% galactose for 1 h. Extracts were prepared and analyzed as in A, and the mean values of the fraction of split ubiquitin cleavage are plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F7.
Progressive Shr3-Agp1 and Shr3-Ssy1 chaperone-substrate interactions. (A) Schematic diagram of agp1-Cub-GST truncation constructs. Strain FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262) or SHR3Δ94-NubA (pAR76) and carrying pIM9 (agp1-2TM-Cub-GST), pIM10 (agp1-4TM-Cub-GST), pIM11 (agp1-6TM-Cub-GST), pIM12 (agp1-8TM-Cub-GST), pIM13 (agp1-10TM-Cub-GST), or pIM26 (agp1-12TM-Cub-GST) were induced with 2% galactose for 1 h. Extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting using α-HA antibody. The signal intensities of the immunoreactive forms of uncleaved Cub constructs and cleaved interaction marker (GST-6xHA) were quantified; the mean values of the fraction of split ubiquitin cleavage are plotted with error bars showing standard deviation (n = 3; biological replicates). (B) Schematic diagram of ssy1-Cub-GST truncation constructs. Strain FGY135 (gap1Δ shr3Δ) expressing SHR3-NubA (pPL1262) or SHR3Δ94-NubA (pAR76) and carrying pIM20 (ssy1-2TM-Cub-GST), pIM21 (ssy1-4TM-Cub-GST), pIM22 (ssy1-6TM-Cub-GST), pIM23 (ssy1-8TM-Cub-GST), pIM24 (ssy1-10TM-Cub-GST), or pIM25 (ssy1-12TM-Cub-GST) were induced with 2% galactose for 1 h. Extracts were prepared and analyzed as in A, and the mean values of the fraction of split ubiquitin cleavage are plotted with error bars showing standard deviation (n = 3; biological replicates). Source data are available for this figure: SourceData F7.
Discussion
Since its discovery 30 years ago (Ljungdahl et al., 1992), the precise function of Shr3 and its high degree of selectivity for AAP has remained a mystery. What is known is that in the absence of Shr3 (shr3Δ), the MS of AAP insert correctly oriented into the ER membrane but fail to fold into native structures, and consequently aggregate forming high molecular weight complexes that do not exit the ER (Kota et al., 2007; Kota and Ljungdahl, 2005). Shr3 productively engages with the first five MS (I-V) of split Gap1, an interaction required to enable the subsequent assembly of an independently expressed C-terminal split-Gap1 fragment comprising MS VI-XII (Kota et al., 2007). These previous findings suggest that Shr3 engages with nascent N-terminal MS of AAP to promote folding of the holoprotein as translation completes. Together with the ability of Shr3 to interact with COPII components via its C-terminal cytoplasmic tail (Gilstring et al., 1999; Kuehn et al., 1998), Shr3 apparently operates at the nexus between folding and packaging of AAP into COPII-coated vesicles.
Here, we addressed the role of Shr3 in facilitating AAP folding within the two-dimensional constraints of the ER membrane. Hydrophobic MS of AAP exit the translocon as α-helices, secondary structures that form concomitantly with translation. During de novo synthesis of membrane proteins, the process of properly incorporating N-terminal MS into a native structure can only be achieved as more C-terminal MS partition into the membrane. In the absence of Shr3, partially unfolded forms of AAPs apparently exhibit greater stability than unfolded states and thus become kinetically trapped, either in off-pathway intraprotein folding events or to interfering interprotein interactions with hydrophobic MS of other proteins present in the ER membrane. Our current findings are consistent with a model in which Shr3 facilitates the folding of AAP by selectively engaging as a structural scaffold initially involving interactions with N-terminal MS of AAP as they emerge from the translocon (Fig. 8). Shr3 exerts its essential chaperone functions in a co-translational and transient manner. As more C-terminal MS of the growing AAP sequence sequentially partition into the ER membrane and folding progresses, the requirement of the scaffold function of Shr3 is expected to lessen. Accordingly, the Shr3–AAP association is strongest as AAP fold and becomes less evident as AAP attain their native structures. The members of evolutionarily conserved protein families are likely to experience similar constraints during synthesis, and thus, it is not surprising that their folding exhibits a shared requirement for a highly selective chaperone.

Model of Shr3 - facilitated AAP folding. Shr3 interacts transiently with AAP as they co-translationally insert into the ER membrane. Shr3-AAP interactions start early when the first 2–4 N-terminal MS of an AAP have partitioned into the lipid bilayer. As translation progresses and C-terminal MS partition into the membrane, AAP can fold to attain native conformations, displacing Shr3. The chaperone activity of Shr3 is based on structural rather than sequence-specific interactions. Fully folded AAP enter the secretory pathway and are targeted to the PM where they function to facilitate amino acid uptake. In the absence of Shr3 (shr3Δ), AAP misfold and are retained in the ER, forming high-molecular weight aggregates that are recognized as ERAD substrates.
Model of Shr3 - facilitated AAP folding. Shr3 interacts transiently with AAP as they co-translationally insert into the ER membrane. Shr3-AAP interactions start early when the first 2–4 N-terminal MS of an AAP have partitioned into the lipid bilayer. As translation progresses and C-terminal MS partition into the membrane, AAP can fold to attain native conformations, displacing Shr3. The chaperone activity of Shr3 is based on structural rather than sequence-specific interactions. Fully folded AAP enter the secretory pathway and are targeted to the PM where they function to facilitate amino acid uptake. In the absence of Shr3 (shr3Δ), AAP misfold and are retained in the ER, forming high-molecular weight aggregates that are recognized as ERAD substrates.
The foundation of our model of Shr3 function is based on saturation scanning mutagenesis of the N-terminal membrane domain of Shr3 that collectively assessed the requirement of each of the 159 amino acids that comprise the domain. Strikingly, very few substitutions at three discrete sites resulted in a complete loss of function, suggesting that Shr3 generally recognizes its folding substrates independently of strict and localized sequence-specific interactions (Fig. S2, A–J and Fig. 3 A). Rather, our data are consistent with Shr3 specifically interacting with the emerging MS of the nascent chains of AAP, and thus it seems that recognition of substrates is based on the structural characteristics of Shr3 and common structural constraints shared by AAP. The notion that structure, rather than sequence-based recognition, underlies interactions between Shr3 and AAP folding substrates aligns well with the high-confidence AlphaFold structure of Shr3. Mapping the shr3-35, -50, and -76 loss-of-function mutations onto this structure shows that the affected residues, some of them featuring polar sidechains, are largely predicted to face the interior of the Shr3. The fact that we did not recover mutations affecting obvious external surface residues is clearly consistent with Shr3 acting as a structural scaffold for AAP folding. Additional support for this includes our finding that the lumen-oriented loop L1 (aa 33–61), predicted to contain an α-helix with amphipathic characteristics (aa 44–57; Fig. 2), is a critical structural element. All tested internal deletions in L1 alter the length of this helix and result in loss of function. Accordingly, and consistent with the AlphaFold structure of Shr3, L1 mutations deform the Shr3 scaffold, preventing the structural recognition of AAP.
By contrast to mutations affecting L1, the deletion within L3 (SHR3Δ94) exhibited a variable effect on amino acid uptake (Fig. 2), a phenotype that we traced to distinct AAP substrates. Hence, the data implicate L3 as an important determinant that influences substrate interactions. This region appears to be critical for interactions with Gap1 and Can1 but less important for interactions with Agp1 and Gnp1, and clearly dispensable for interactions with Ssy1 (Figs. 2, 3, 4, and 7). The observation that Ssy1, the only non-transporting AAP, exhibits a more lax requirement for the lumen-oriented L3 of Shr3 suggests that transporting AAP may have an enhanced requirement for the Shr3 scaffold due to relatively short extramembrane sequences and to fold into conformations that allows cycling between outward and inward facing states. The loop regions of AAP that face the extracellular milieu are lumen-oriented during biogenesis. Our data are consistent with the longer lumenal loops of the non-transporting Ssy1, connecting MSV-VI and VII-VIII (Table S3), providing greater flexibility, and enabling Ssy1 to maintain productive interactions with Shr3Δ94. Interestingly, the clear phenotypes of Shr3 mutations within lumenal domains are consistent with recent evidence that extracellular loops of AAP have important roles affecting intracellular trafficking and transport function (van’t Klooster et al., 2020). Interactions between the lumen-oriented loops of Shr3 may be direct, maintaining the extracellular loops in a more flexible state required for subsequent folding events, e.g., involving MS that fold in a context with more distal C-terminal MS. In analogy, proper trafficking and functional expression of the closest AAP homologs in mammals, the L-type amino acid transporters (LAT; SLC7 family), depend on extramembrane region–mediated recognition by the 4F2hc or rBAT, members of the SLC3 protein family (Fotiadis et al., 2013; Rosell et al., 2014).
The scaffold-like function of Shr3 may be exquisitely sensitive to minor shifts in intraprotein helix–helix interactions, a notion consistent with the subtle mutant-induced changes predicted by AlphaFold (Fig. 3 E). Although the accuracy of AlphaFold to predict structural effects of missense mutations has recently been questioned (Buel and Walters, 2022; Pak et al., 2023), we note that the Shr3 mutant variants examined, including the most severe shr3-35 mutant, were expressed at similar levels as wild-type Shr3 (Fig. 1 C and Fig. 2 B), suggesting that the mutations did not result in gross misfolding. Obviously, the lack of larger more pronounced predicted structural changes may simply reflect the limitations of AlphaFold and the present status of the algorithms used in making predictions, which are based on the structures of largely functional proteins in the protein databases. However, as a scaffold, the highly specific function of Shr3 in facilitating AAP folding and its ability to select AAP substrates among the multitude of potential substrates in the ER membrane may depend on alternating sets of distant residues that sequentially emerge on the substrates as their MS partition into the membrane. This could account for major defects resulting from minimal shifts predicted in the relative angles between MS of Shr3. Clearly, structural features of the Shr3 mutant proteins need to be experimentally addressed, and hopefully, future findings may provide input to advance the predictive power of machine-based and artificial intelligence methodologies.
Our findings regarding the patterns of specific interactions between Shr3 and truncated Gap1, Agp1, Can1, and Ssy1 are striking and consistent with Shr3 acting in a transient manner as the MS of AAP partition into the ER membrane (Figs. 5, 6, and 7). The observed interactions persist until all MS are available, at which point the intramolecular interactions inherent to the native 3D structure of AAP likely contribute to displacing Shr3 (Fig. 8). As previously postulated (Kota et al., 2007; Kota and Ljungdahl, 2005), Shr3 may prevent AAP translation intermediates from engaging in non-productive interactions, perhaps shielding polar residues, which, as has been more recently shown, can potentially be recognized by a hydrophilic pocket in the structure of the ERAD-associated E3 ubiquitin ligase Hrd1 (Schoebel et al., 2017). Indeed, folding and degradation are tightly coupled processes during AAP biogenesis (Kota et al., 2007). Our model for the Shr3 chaperone function in the biogenesis of AAP exhibits certain similarities to that of the bacterial insertase/chaperone YidC (Beck et al., 2001; Dalbey and Kuhn, 2014). YidC apparently acts as an assembly site for alpha helices, relying on hydrophobic interactions with LacY to minimize energetically unfavorable off-pathway intramolecular interactions involving LacY segments in the non-native structure during translation (Nagamori et al., 2004; Serdiuk et al., 2016; Serdiuk et al., 2019; Wagner et al., 2008; Zhu et al., 2013). Similar transient membrane-localized chaperone substrate interactions have been reported in the case of the mammalian PAT intramembrane chaperone complex comprising four MS, three contributed by Asterix and one by CCDC47 (Chitwood and Hegde, 2020). Although in these studies an in vitro translation system was employed, in contrast to our in vivo split-ubiquitin approach, the PAT complex interaction was found to be selective for truncated, immature β1-adrenergic receptor constructs compared with the full-length substrate, which presumably is capable of helix packing and polar residue shielding.
Finally, the conserved eukaryotic ER membrane protein complex (EMC) insertase (Bai et al., 2020; O’Donnell et al., 2020; Pleiner et al., 2020; Volkmar and Christianson, 2020) has, like Shr3 (Kota and Ljungdahl, 2005), also been implicated in promoting the folding of nascent polytopic membrane proteins typically enriched for MS containing polar or charged residues (Miller-Vedam et al., 2020; Shurtleff et al., 2018). Importantly, the EMC has been shown to associate with a number of ER-integral substrate-specific chaperones such as Sop4, Gsf2, and Ilm1. Notedly, Shr3, although an abundant ER membrane protein, has not been identified as an interacting partner of EMC components (Shurtleff et al., 2018). Thus, it is likely that Shr3 and EMC act independently and in parallel with Sec61 translocons engaged with distinct ribosome populations and thereby help facilitate the efficient folding of more challenging versus canonical membrane protein substrates (O’Keefe and High, 2020). The potential network of dynamic interactions in the ER remains to be explored, particularly for an integral substrate-specific chaperone like Shr3. Also, the structural determinants in the AAP substrates of Shr3 that dictate the remarkable degree of substrate specificity need to be defined. To this end, our broad collection of mutations mapped on 3D model structures, in combination with extended growth-based phenotypic analysis and in vivo interaction studies, provides a strong foundation for future progress.
Materials and methods
Yeast strains and plasmids
Media
Standard media, YPD (yeast extract, peptone, and dextrose), and SD (synthetic defined with ammonium as nitrogen source and glucose as carbon source) were prepared as previously described (Burke et al., 2000). Ammonia-based synthetic complete dextrose (SC) drop-out medium wase prepared as described (Andréasson and Ljungdahl, 2002) and SAD (synthetic minimal dextrose, with allantoin as sole nitrogen source) was prepared as previously described. Media were made solid with 2% (wt/vol) bacto Agar (Difco), 2% (wt/vol) washed bacto Agar (Difco), or 2% (wt/vol) washed pure Agar where indicated. Sensitivity to 200 µg/ml MM (2-{[({[(4-methoxy-6-methyl)-1,3,5-triazin-2-yl]-amino}carbonyl) amino-]-sulfonyl}-benzoic acid) was tested on YPD as described previously (Jørgensen et al., 1998). Sensitivity to 1 mM AzC (azetidine-2-carboxylate), 10 µg/ml DL-ethionine, 50 µg/ml p-Fluoro-DL-phenylalanine, and 1 µg/ml L-canavanine was tested on SD. Sensitivity to 0.5% (wt/vol) D-histidine was tested on SAD media made solid with washed pure Agar. Cells were grown overnight in SC-uracil medium, then resuspended in water to OD = 1, and 10-fold dilutions were prepared in water and then spotted on the indicated medium. Plates were then incubated at 30°C for 2–3 d and photographed. Gap1-dependent citrulline uptake was monitored on a minimal medium containing 2% galactose as the carbon source, 1 mM L-citrulline as the sole nitrogen source, and uracil. Media were made solid with washed bacto Agar. Plates were incubated at 30°C for 7 d and photographed.
Immunoblot analysis
Whole-cell extracts were prepared under denaturing conditions using NaOH and trichloroacetic acid as described previously (Silve et al., 1991). Proteins were separated using SDS-PAGE and blotted onto Amersham Protran 0.45 µm nitrocellulose membrane (GE Healthcare). The primary antibodies and dilutions were mouse anti-Dpm1 5C5A7 (5C5A7; Abcam), 1:2,500; rat anti-HA-HRP 3F10 (Cat#12013819001; Roche Applied Science), 1:2,500–1:5,000; mouse anti-Pgk1 22C5D8 (Cat #459250; Thermo Fisher Scientific), 1:10,000; and rabbit anti-Shr3 (Kota and Ljungdahl, 2005), 1:9,000, respectively. Secondary antibodies and dilutions used were goat anti-mouse-poly-HRP (Cat#31430; Invitrogen; Thermo Fisher Scientific), 1:5,000 and goat anti-rabbit-poly-HRP (Cat#31460; Thermo Fisher Scientific; Invitrogen), 1:5,000, respectively. Immunoreactive bands were visualized by chemiluminescence using SuperSignal West Dura Extended-Duration Substrate (Thermo Fisher Scientific) as substrate in either a ChemiDoc (Bio-Rad) or Azure 280 (AH Diagnostics) imaging system.
Split-ubiquitin assay
Cells were pregrown to a logarithmic phase in SD+Raff (2% raffinose and 0.1% glucose as carbon sources). 10 OD600 of cells, induced in 5 ml of SD+Gal (2% galactose as sole carbon source) for 1 h, were collected, washed once in ddH2O, and resuspended in 150 μl lysis buffer (0.8 M sorbitol; 10 mM MOPS, pH 7.2; 2 mM EDTA; 1 mM PMSF; 1X complete EDTA-free protease inhibitor cocktail [Roche]). Cells were lysed by bead beating, i.e., 0.5-mm glass beads (Cat #11079105; BioSpec Products) and 3 × 20 s pulses at 6.5 m/s (Fastprep-24; MP Biomedical). The lysates were cleared by centrifugation at 500 × g for 10 min, and 25 μl of the resulting supernatant was diluted 1:1 with 2× sample buffer. Proteins were separated using SDS-PAGE and analyzed by immunoblot.
Co-immunopreciptation
Cells were pregrown to the logarithmic phase in SD+Raff (2% raffinose and 0.1% glucose as carbon sources). 40 OD600 of cells, induced in 15 ml of SD+Gal (2% galactose as sole carbon source) for 1 h, were collected, washed once in ddH20, and resuspended in 300 μl IP-lysis buffer (40 mM Hepes-KOH, pH 7.4; 150 mM KCl; 5 mM. MgCl2; 1 mM PMSF; 1X complete EDTA-free protease inhibitor cocktail [Roche]; 5% [vol/vol] glycerol). Cells were lysed by bead beating, i.e., 0.5-mm glass beads (Cat #11079105; BioSpec Products), 3 × 20 s pulses at 6.5 m/s (Fastprep-24, MP Biomedical). The lysates were cleared by centrifugation at 4,000 × g for 5 min at 4°C and 200 μl of the resulting supernatant was diluted 1:1 with lysis buffer supplemented with n-dodecyl-β-D-maltoside (Sigma-Aldrich) to reach a final concentration of 0.8%. The extracts were incubated for 1 h under continuous inversion at 4°C, after which the solubilized extracts were clarified by centrifugation at 15,000 × g for 10 min at 4°C. The supernatant fractions were diluted to a protein concentration of 1–3 mg/ml with lysis buffer and incubated for 1 h with GFP-trap agarose beads (ChromoTek) under continuous inversion at 4°C. The beads were pelleted at 2,700 × g for 2 min at 4°C and washed four times with lysis buffer containing 0.1% n-dodecyl-β-D-maltoside. Proteins were denatured with the addition of 50 μl 5× sample buffer and heated at 45°C for 10 min. An aliquot representing 1.8% of the total protein incubated with the beads was analyzed as Input. Proteins were separated by SDS-PAGE and analyzed by immunoblot, The primary antibodies and dilutions used were rat anti-HA-HRP 3F10 (Cat #12013819001; Roche Applied Science), 1:5,000; mouse anti-GFP (Cat #1814460; Roche Applied Science) 1:1,000; and mouse anti-Dpm1 5C5A7 (5C5A7; Abcam), 1:2,500, respectively.
Protease protection assay
Cells were pregrown in SD+R (synthetic defined with ammonium as nitrogen source and 2% raffinose and 0.1% glucose as carbon source) to the logarithmic phase. Approximately, 5 OD of logarithmic cells were induced in 5 ml of SD+G (synthetic defined with ammonium as nitrogen source and 2% galactose as carbon source) for 1 h. Cells were collected and washed once in ddH2O. Cells were resuspended in 150 μl lysis buffer (0.8 M sorbitol; 10 mM MOPS, pH 7.2; 2 mM EDTA; 1 mM PMSF; and 1X complete, mini, EDTA-free protease inhibitor cocktail; Roche). Cells were lysed by bead beating with 0.5 mm glass beads for 3 × 20 s at 6.5 m/s in a benchtop homogenizer (Fastprep-24; MP Biomedical). The cell lysates were centrifuged at 500 g for 10 min and 100 μl of the resulting supernatant was centrifuged at 100,000 g for 30 min. The membrane pellet was resuspended in 50 μl lysis buffer (0.8 M sorbitol; 10 mM MOPS, pH 7.2; 2 mM EDTA; 5 mM CaCl2). The resulting membrane preparations were digested with 20 µg Proteinase K (Thermo Fisher Scientific) on ice with 0.2% NP-40 as indicated. Time points were taken at 0 and 2 h. Proteins were precipitated using trichloroacetic acid as described previously (Silve et al., 1991). Proteins were separated using SDS-PAGE and blotted onto Amersham Protran 0.45 µm nitrocellulose membrane (GE Healthcare). The primary antibodies and dilutions used were rat anti-HA-HRP 3F10 (Cat #12013819001; Roche Applied Science), 1:2,500–1:5,000 and rabbit anti-Kar2, 1:5,000 (obtained from Mark Rose, Princeton University), respectively. The secondary antibody and dilution used were goat anti-rabbit-poly-HRP (Cat #31460; Invitrogen; Thermo Fisher Scientific), 1:5,000, respectively. Immunoreactive bands were visualized by chemiluminescence using SuperSignal West Dura Extended-Duration Substrate (Thermo Fisher Scientific) as substrate in a ChemiDoc imaging system (Bio-Rad).
AlphaFold predictions
The Python notebook AlphaFold2_advanced.ipynb (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/AlphaFold2_advanced.ipynb) from ColabFold (Mirdita et al., 2022) was modified to install and run on a local computer independent of Google Colab. All models were automatically batch-built using AlphaFold2_advanced with default settings using msa_method, homooligomer, and used_amber_relax set as jackhammer, 1, and True, respectively. Multiple sequence alignments (MSAs) were created by searching Uniref90, MGnify v.2018_12, and a small BIG Fantastic Database (bfd-first_non_consensus_sequences) with the jackhammer.
Online supplemental material
Fig. S1 contains additional data regarding the growth phenotypes associated with the shr3-35, shr3-50, and shr3-76 mutations. Fig. S2 documents the growth-based assessment of the Shr3 mutant protein function that was used to compile the summary presented in Fig. 3 A. Fig. S3 provides a comparison between AlphaFold predicted structures of Shr3 and nine fungal orthologs; the images reveal a striking degree of structural similarity within membrane-spanning segments and extensive variation in lumenal-oriented L3. Fig. S4 includes data indicating that Shr3–AAP interactions are transient and not influenced by mutations that block the exit of AAP from the ER. Fig. S5 documents necessary control experiments demonstrating the correct topology of shr3-35-NubA and gap1-2TM-Cub-GST constructs. Fig. S6 documents that truncated gap1-8TM and -10TM, but not truncated hxt1-8TM or -10TM, co-purify with Shr3. Table S1 lists the yeast strains constructed and used in this study. Table S2 lists the plasmids engineered and used in this study. Table S3 provides a summary of the number of amino acid residues in the loops and termini of representative AAP.
Data availability
The data underlying all figures are available in the published article and its online supplemental material. Primers used in the construction of plasmids are available on request.
Acknowledgments
We thank the members of the Ljungdahl laboratory and Claes Andréasson (Stockholm University, Department of Molecular Biosciences, The Wenner-Gren Institute) for constructive comments throughout the course of this work. In particular, we acknowledge Nina Horwege and Carlos Sacristán for creating originating plasmid constructs. We thank Mark Rose, Princeton University, for providing the polyclonal anti-Kar2 antibody. We gratefully acknowledge Alexey Amunts (Stockholm University, Department of Biochemistry and Biophysics) for early guidance with structural modeling and Arne Elofsson (Stockholm University, Department of Biochemistry and Biophysics) for critical discussions and comments. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
This research was supported by funding from Swedish Research Council (P.O. Ljungdahl), grant/award numbers: 2015-04202, 2019-01547, and 2022-01190.
Author contribution: Conceptualization: I. Myronidi, A. Ring, and P.O. Ljungdahl. Formal analysis: I. Myronidi, A. Ring, F. Wu, and P.O. Ljungdahl. Investigation: I. Myronidi, A. Ring, and F. Wu. Methodology: I. Myronidi, A. Ring, and F. Wu. Resources: I. Myronidi, A. Ring, and F. Wu. Software: F. Wu. Validation and Visualization: I. Myronidi, A. Ring, and F. Wu. Formal analysis: I. Myronidi, A. Ring, F. Wu, and P.O. Ljungdahl. Writing original draft: A. Ring, I. Myronidi, and P.O. Ljungdahl. Writing review and editing: all authors. Supervision and funding acquisition: P.O. Ljungdahl.

