


Maintaining long, energetically demanding axons throughout the life of an animal is a major challenge for the nervous system. Specialized glia ensheathe axons and support their function and integrity throughout life, but glial support mechanisms remain poorly defined. Here, we identified a collection of secreted and transmembrane molecules required in glia for long-term axon survival in vivo. We showed that the majority of components of the TGFβ superfamily are required in glia for sensory neuron maintenance but not glial ensheathment of axons. In the absence of glial TGFβ signaling, neurons undergo age-dependent degeneration that can be rescued either by genetic blockade of Wallerian degeneration or caspase-dependent death. Blockade of glial TGFβ signaling results in increased ATP in glia that can be mimicked by enhancing glial mitochondrial biogenesis or suppressing glial monocarboxylate transporter function. We propose that glial TGFβ signaling supports axon survival and suppresses neurodegeneration through promoting glial metabolic support of neurons.
Introduction
Information in the nervous system is carried along axons that link neuronal dendrites and somas to their synaptic targets. Maintaining axon integrity is therefore crucial to preserve neural circuit function. Axons frequently traverse great distances and have complex morphologies, such that they constitute the majority of any given neuron’s volume. For example, axons in the human sciatic nerve can be a meter long (Jessen and Mirsky, 2005), and axons of single dopaminergic neurons in the mouse substantia nigra pars compacta branch so profusely that their estimated cumulative linear length can reach nearly 800 mm (Matsuda et al., 2009). Sustaining axons for the lifetime of an animal presents a significant challenge for the neuron to overcome both because of the size of the axon and the substantial metabolic demand required for neurotransmission (Harris et al., 2012).
In most neurodegenerative diseases, it remains unclear which part of the neuron becomes sick first and ultimately leads to neurodegeneration. Does the axon degenerate and fail to support the soma, thereby resulting in neuronal cell death? Alternatively, does the cell body activate apoptotic death and axon degeneration follows as a result? Are they interdependent? The answer to these questions likely varies by cell type and context, but one can begin to explore these possibilities by examining the timing of each of these events. Dying back neuropathies, where axons are lost first, suggest that axon loss could lead to neuronal death (Fischer et al., 2004; de la Monte et al., 1988; Schaumburg et al., 1974; Sima et al., 1983). However, this is not a definitive result, and neuronal drop out in many neurodegenerative diseases is often sparse and occurs over a protracted phase such that high-resolution imaging to watch the timing of these events relative to one another is not possible. Driving expression of axon-protective proteins like WldS in neurons is an effective way to suppress axon degeneration without altering apoptotic signaling pathways (Adalbert et al., 2006; Beirowski et al., 2008; Deckwerth and Johnson, 1994) and can be used to probe whether neurodegeneration is driven primarily by the axon versus the cell body (reviewed in Coleman and Höke, 2020). For example, in the progressive motorneuronopathy (pmn) mouse, which exhibits age-dependent motor axon degeneration followed by motoneuron cell body death, supplying WldS was sufficient to block axon loss and suppress motoneuron death (Ferri et al., 2003). This observation suggested that axon degeneration was the primary driver of motoneuron loss in this model. While endogenous “axon death” genes like Sarm1 play roles primarily in Wallerian-like degeneration and could be used to further explore the role of axon loss (Adalbert et al., 2006; Beirowski et al., 2008; Fernandes et al., 2018; Ferri et al., 2003; Fischer et al., 2004; Henninger et al., 2016; Osterloh et al., 2012; Peters et al., 2018; Samsam et al., 2003; Turkiew et al., 2017), it is important to note that roles for Sarm1 have been discovered in some types of cell death, where cytoplasmic versus axonal pools of Sarm1 may play differential roles in cell destruction (Killackey et al., 2019).
Glial cells that ensheathe axons are believed to provide crucial, local, external sources of metabolic and physiological support to axons, particularly at great distances from the cell body. Human diseases such as multiple sclerosis (MS) or Charcot-Marie-Tooth (CMT), where loss of glia causes neurodegeneration, support a crucial role for glia in axon maintenance (Brennan et al., 2015; Kornek et al., 2000; Kuhlmann et al., 2002; Trapp et al., 1998). In a number of experimental models, disruption of a single glial gene can cause neurodegeneration even when glial cells appear morphologically normal (Griffiths et al., 1998; Lappe-Siefke et al., 2003). A growing body of work supports the notion that glia provide significant metabolic support to axons (Fünfschilling et al., 2012; Volkenhoff et al., 2015) and more recently, defense against iron-mediated toxicity (Mukherjee et al., 2020). However, a comprehensive understanding of the molecular mechanisms by which glia help sustain axon integrity in vivo remains elusive.
To facilitate the identification of mechanisms of glial support of axons in vivo, we conducted a screen in Drosophila to identify glial genes required for long-term axon survival in the peripheral nervous system. We identified over 200 genes that, when depleted exclusively from glia, lead to axon degeneration or animal lethality. Interestingly, we found that glial loss of the majority of known TGFβ superfamily members resulted in age-dependent axon loss and neuronal cell body death. Neurodegeneration in the absence of glial TGFβ activity could be suppressed by either expression of WldS or blockade of caspase activity, suggesting that both Wallerian degeneration and caspase-mediated pathways regulate neuronal loss in this context, and that they are interdependent to some extent. Finally, we provided evidence that TGFβ activation in glia is required to regulate glial metabolism and likely in turn metabolic support of axons. Our work identifies the TGFβ superfamily as a key regulator of in vivo glial function in supporting long-term maintenance of axons.
Results
Ablation of wrapping glia leads to axon degeneration
To explore the mechanisms by which glia support axon function and survival, we used the adult L1 wing nerve of Drosophila melanogaster, where it is possible to independently manipulate neurons and glia and examine their morphology with single cell/axon resolution in vivo (Fig. 1, A and B). This sensory nerve contains roughly 290 sensory neurons (Fig. 1 C; Hsu et al., 2021). Their cell bodies are positioned along the anterior wing margin and project their axons into the thorax; these are among the longest axons in Drosophila (Fig. 1 A; Palka et al., 1983). Each axon is individually ensheathed by wrapping glia (WG) that cover the entire nerve and interdigitate into the axon bundle separating axons from one another (Fig. 1, B and C; Hsu et al., 2021; Neukomm et al., 2014). Given their length, and their extensive ensheathment by glia, we hypothesized that the glia surrounding these neurons would be a suitable model to explore how glia provide essential support for neuronal function and maintenance.

Ablating wrapping glia results in neurodegeneration in the peripheral nerve of the wing with age. (A) Diagram of the sensory nerve in the wing of Drosophila. (B) Images from the area depicted in the box in A. A subset of glutamatergic neurons are genetically labeled with GFP, and glia are labeled with tdTomato. The orthogonal fluorescent image corresponds to the location at the asterisk. (C) Electron micrograph of a cross-section of the nerve in the wing from the same region as in A. Wrapping glial membrane (G) is psuedocolored in cyan. Ax, axons. (D–F) Representative images of control and glial-ablated wings at 28 d of age with a subset of wrapping glia labeled with tdTomato. Low magnification images showing the tdTomato (glia) channel. (1 and 2) Higher magnification images from ROIs in D–F showing neurons (green) and glia (magenta). (G) Classification of axon phenotype for each condition categorized into intact, mild, or severe. Fisher exact probability test (two-tailed, 2 × 3, n = 18–22 per condition). (H) Quantification of the number of intact neuron cell bodies at each time point. Mean ± 95% CI. Two-way ANOVA with Sidak’s multiple comparisons test, n = 61 (control), 56 (Dronc + Dark), 56 (Reaper). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Ablating wrapping glia results in neurodegeneration in the peripheral nerve of the wing with age. (A) Diagram of the sensory nerve in the wing of Drosophila. (B) Images from the area depicted in the box in A. A subset of glutamatergic neurons are genetically labeled with GFP, and glia are labeled with tdTomato. The orthogonal fluorescent image corresponds to the location at the asterisk. (C) Electron micrograph of a cross-section of the nerve in the wing from the same region as in A. Wrapping glial membrane (G) is psuedocolored in cyan. Ax, axons. (D–F) Representative images of control and glial-ablated wings at 28 d of age with a subset of wrapping glia labeled with tdTomato. Low magnification images showing the tdTomato (glia) channel. (1 and 2) Higher magnification images from ROIs in D–F showing neurons (green) and glia (magenta). (G) Classification of axon phenotype for each condition categorized into intact, mild, or severe. Fisher exact probability test (two-tailed, 2 × 3, n = 18–22 per condition). (H) Quantification of the number of intact neuron cell bodies at each time point. Mean ± 95% CI. Two-way ANOVA with Sidak’s multiple comparisons test, n = 61 (control), 56 (Dronc + Dark), 56 (Reaper). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
To assess whether glia in the L1 wing nerve were required for maintenance of axons, we selectively ablated WG and measured neuronal integrity as the animals aged. We used the Gal4/UAS binary expression system (Brand and Dormand, 1995) to overexpress the cell death molecules Dronc and Dark, or Reaper (Dorstyn et al., 1999; White et al., 1994; Zhou et al., 1999), along with tdTomato in a subset of the WG by using a Split Gal4 construct (Luan et al., 2006). This Split Gal4 was exclusively expressed in WG and is henceforth referred to as WG Split-Gal4 (Corty et al., 2022). WG Split-Gal4 labeled 87% of WG in the wing as determined by nuclear reporter expression when compared to nrv2-Gal4, which is expressed in all the WG in the wing but is also widely expressed in central nervous system glia (Fig. S1, B–E; Neukomm et al., 2014). We combined the WG split-Gal4 with the independent binary expression system QF2/QUAS (Potter et al., 2010; Riabinina et al., 2015) to fluorescently label a subset of ∼40 VGlut+ neurons in the wing in order to evaluate the effect of ablating WG on neurons in this nerve. Interestingly, ablating WG in these animals caused increased degeneration of axons in the peripheral sensory nerve of the wing as animals aged (Fig. S1 and Fig. 1, D–G). A larger proportion of nerves from 28-d-old animals whose glia were ablated exhibited mild or severe degeneration compared with the control group (control: 2/18 animals, Dronc::GFP, Dark: 11/18, P = 0.002728, Reaper: 10/18, P = 0.01763; Fig. 1 G and Fig. S1 F).

Wrapping glia reporter expression in the wing and degeneration classification. (A) Raw electron micrographs from Fig. 1. (B) Expression pattern of nrv2-Gal4 in the adult wing. (C) Expression pattern of the wrapping glia split-Gal4 in the adult wing. (D) Quantification of the number of wrapping glia nuclei genetically labeled by nrv2- and WG split-Gal4. (E) Estimation plot of the difference in labeling efficiency of wrapping glia nuclei for each Gal4 construct within the wing. (F) Examples of orthogonal projections of axons classified as intact, mild, or severe degeneration phenotype. All examples are from wrapping glia-ablated nerves, only the GFP channel is shown. (G) Examples of an intact (1) and neuron corpse (2) within the same wing. (1) Arrow indicates the nucleus, arrowhead indicates dendrite. Mean ± 95% CI. (D) One-tailed t test, n = 12 per condition. ****, P < 0.0001.
Wrapping glia reporter expression in the wing and degeneration classification. (A) Raw electron micrographs from Fig. 1. (B) Expression pattern of nrv2-Gal4 in the adult wing. (C) Expression pattern of the wrapping glia split-Gal4 in the adult wing. (D) Quantification of the number of wrapping glia nuclei genetically labeled by nrv2- and WG split-Gal4. (E) Estimation plot of the difference in labeling efficiency of wrapping glia nuclei for each Gal4 construct within the wing. (F) Examples of orthogonal projections of axons classified as intact, mild, or severe degeneration phenotype. All examples are from wrapping glia-ablated nerves, only the GFP channel is shown. (G) Examples of an intact (1) and neuron corpse (2) within the same wing. (1) Arrow indicates the nucleus, arrowhead indicates dendrite. Mean ± 95% CI. (D) One-tailed t test, n = 12 per condition. ****, P < 0.0001.
To determine whether the axonal degeneration we observed was also affecting overall neuronal survival, we also counted the number of intact sensory neuron cell bodies in the control and WG-ablated animals as they aged. Ablating WG caused significant loss of sensory neurons with age (Fig. 1, D–F and H; and Fig. S1 G). At baseline (4-d post eclosion [dpe] ) there were no significant differences in the average number of intact GFP+ neuron cell bodies per wing between genotypes (control: 38.0 ± 2.1 n = 21, Dronc::GFP, Dark: 36.9 ± 2.7 n = 20, Reaper: 37.2 ± 2.4 n = 20; Fig. 1 H). However, at 28 dpe, fewer neurons remained in WG-ablated animals compared to controls (control: 36.7 ± 3.1 n = 18, Dronc::GFP, Dark: 31.7 ± 8.3 n = 18, P = 0.0014, Reaper: 29.0 ± 8.1 n = 18, P < 0.0001; Fig. 1 H). Together, these data indicate that WG are required for axon and neuronal maintenance in the sensory nerve of the Drosophila wing.
WG are required for WldS to maximally prevent Wallerian degeneration after injury
The degeneration caused by eliminating WG suggested that WG play an important role in axon maintenance in the L1 nerve. To more specifically target glia→axon support mechanisms (as opposed to direct support of the cell body), we developed a system where axons would survive for weeks without their cell body. Briefly, severing wild-type axons causes the portion of the axon distal to the injury site to undergo Wallerian degeneration and dies/is eliminated (Fig. 2 A; MacDonald et al., 2006). However, this can be blocked by overexpressing WldS in neurons, which suppresses Wallerian degeneration and allows distal severed axons to remain intact for weeks after axotomy (Fig. 2 A; Glass and Griffin, 1991). It has long been known that the ability of WldS to protect axons in vivo far exceeds its ability to do so in purified neuron cultures (Adalbert et al., 2005; Buckmaster et al., 1995; Conforti et al., 2006; Lunn et al., 1989; Wang et al., 2005). We hypothesized that this greater protection was in part due to presence of glial support to axons in vivo. To directly test this, we injured nerves from animals with or without WG in a genetic background where WldS was expressed in neurons. We then measured axon survival at 10-d post axotomy (dpa; Fig. 2 B). Nerves lacking WG exhibited significantly decreased axon protection compared to controls (control = 17/24 intact, WG-ablated = 3/21 intact, P = 0.0005; Fig. 2 B). This result indicates that maximal survival of WldS-expressing axons undergoing long-term survival post-axotomy in vivo is dependent upon the presence of glia, presumably due to the support that glia provide axons.
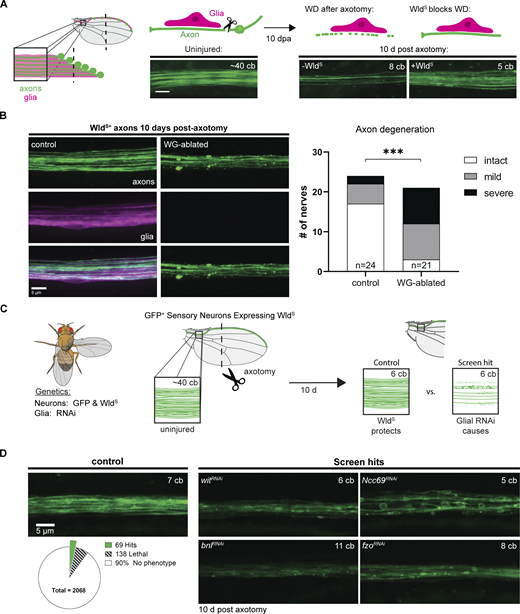
A screen for glial genes required for axon maintenance. (A) Diagram illustrating the peripheral sensory nerve in the wing (left) with neurons labeled in green and glia in magenta. Expression of WldS prevents axon degeneration 10 days post axotomy (dpa) in the wing (right). cb, cell body; scale bar, 5 µm. (B) Axon integrity in injured WldS expressing neurons in control and WG-ablated animals at 10 dpa. Chi-square test, P = 0.0005, n = 45. (C) Diagram of screen workflow. Flies expressing GFP and WldS in neurons and GAL4 in glia were crossed to UAS-RNAi flies and resulting progeny were injured and assessed for axon integrity at 10–14 dpa. (D) Summary of screen results with examples of individual screen hits: wit, Ncc69, bnl, and fzo. (See also Table S2.)
A screen for glial genes required for axon maintenance. (A) Diagram illustrating the peripheral sensory nerve in the wing (left) with neurons labeled in green and glia in magenta. Expression of WldS prevents axon degeneration 10 days post axotomy (dpa) in the wing (right). cb, cell body; scale bar, 5 µm. (B) Axon integrity in injured WldS expressing neurons in control and WG-ablated animals at 10 dpa. Chi-square test, P = 0.0005, n = 45. (C) Diagram of screen workflow. Flies expressing GFP and WldS in neurons and GAL4 in glia were crossed to UAS-RNAi flies and resulting progeny were injured and assessed for axon integrity at 10–14 dpa. (D) Summary of screen results with examples of individual screen hits: wit, Ncc69, bnl, and fzo. (See also Table S2.)
Identification of glial genes required for axon maintenance
To identify glial genes that promote long-term axon survival, we used this approach to systematically knock down genes selectively in glia in animals that express WldS in glutamatergic neurons. Following injury, we looked for those genes when knocked down in glia caused early axon loss compared to controls. Using vGlut-QF2 (Diao et al., 2015), we expressed a membrane-tethered GFP and WldS in glutamatergic neurons, allowing us to visualize axons in the L1 nerve and block Wallerian degeneration, respectively (Fig. 2 C). We removed the cell bodies from most of the neurons by cutting off the distal portion of the wing, leaving behind the Wallerian degeneration-resistant axons and glia that surround them (Fig. 2 A). We used GAL4/UAS to express RNA interference (RNAi; Perrimon et al., 2010) constructs in all glia with the pan-glial driver repo-Gal4 (Sepp et al., 2001). We then screened >2,000 publicly available UAS-RNAi lines targeting a panel of genes enriched for those encoding proteins containing predicted transmembrane domains or signal peptides (Dietzl et al., 2007; Fig. 2 D).
In control animals, WldS prevented Wallerian degeneration and axons remained completely intact at 10 dpa (Fig. 2 D). Our screen sought to identify genes that, when knocked down in glia, resulted in significant axon degeneration by day 10 (Fig. 2 C). We identified 69 candidate genes whose loss in glia resulted in axon degeneration or defects in axon morphology (Table S2). These included a TGFβ receptor (Wit), a fibroblast growth factor (FGF) Branchless (Bnl), and a mitofusin Fuzzy onions (Fzo; Fig. 2 D). Other axonal phenotypes were also observed in our screen, including a severe axon blebbing phenotype after glial loss of the sodium-chloride co-transporter Ncc69 (Fig. 2 D). This phenotype is remarkably similar to the neuronal activity-dependent axon disruption observed in zebrafish slc12a2b (NKCC1b) mutants (Marshall-Phelps et al., 2020). In addition, and consistent with previous work (Mukherjee et al., 2020), we identified 138 genes that caused animal lethality—defined by absence of viable adult progeny—when selectively knocked down in glia (Fig. 2 D).
Glial TGFβ signaling is required for WldS-mediated long-term axon survival
The TGFβ receptor Wit was one of several members of the TGFβ superfamily identified in our screen. The TGFβ superfamily is made up of two major branches (TGFβ and BMP; Fig. 3 A; Upadhyay et al., 2017). We obtained RNAi lines to test the majority of known genes in this pathway in our axon survival screen (Fig. 3 B). We found that most RNAi constructs targeting components of this superfamily caused axon degeneration or lethality when expressed in glia (Fig. 3, B and C). These data strongly suggest a role for TGFβ signaling in glial support of long-term axon survival. Surprisingly, knockdown of both TGFβ ligands and their receptors selectively in glia (with repo-Gal4) caused premature axon degeneration (Fig. 3, B and C). This observation supports the notion that autocrine TGFβ signaling may be important for long-term axon survival in our assay; however, below, we also explored the roles for TGFβ signaling in neurons.

Glial-specific knockdown of TGFβ and BMP pathway components causes axon degeneration in the sensitized sensory nerve of the wing. (A) Diagram of the TGFβ superfamily members in the Drosophila genome. (B) Table of the RNAis targeting the TGFβ superfamily genes shown in C. VDRC—“v#”, Bloominton—“BL#”. (C) Images of WldS+ axons in the sensory nerve of the wing 10 dpa from control and TGFβ superfamily knockdown animals. Scale bar, 5 µm. Pan-glial knockdown of the ligand myo or the receptor tkv were lethal (not shown).
Glial-specific knockdown of TGFβ and BMP pathway components causes axon degeneration in the sensitized sensory nerve of the wing. (A) Diagram of the TGFβ superfamily members in the Drosophila genome. (B) Table of the RNAis targeting the TGFβ superfamily genes shown in C. VDRC—“v#”, Bloominton—“BL#”. (C) Images of WldS+ axons in the sensory nerve of the wing 10 dpa from control and TGFβ superfamily knockdown animals. Scale bar, 5 µm. Pan-glial knockdown of the ligand myo or the receptor tkv were lethal (not shown).
TGFβ signaling in glia is required for neuron maintenance as animals age
An underlying goal of the above long-term axon survival screen was to identify glial molecules required to sustain neuronal health under normal conditions. We therefore targeted each of the TGFβ superfamily genes using RNAi (driven by repo-Gal4 in all glia) and evaluated axon integrity and neuron survival in uninjured nerves as animals aged at three timepoints: 4, 14, and 28 dpe. Interestingly, we found robust degeneration of axons and neuron cell bodies in aged animals in several TGFβ knockdown conditions compared to controls, with the strongest phenotype elicited by knockdown of the TGFβ receptor babo (Fig. 4, A–C; and Fig. S2, A and B). TGFβ signaling was also inhibited by overexpressing a dominant negative form of the receptor (baboDN) that lacked the kinase domain (Brummel et al., 1999). Glial expression of baboDN phenocopied the RNAi-mediated knockdown (Fig. S2 E), further supporting the conclusion that disruption of Babo signaling in glia is sufficient to induce neuron degeneration. We also confirmed the specificity of the RNAi targeting babo and its downstream target Smox with additional non-overlapping RNAi constructs (Fig. 4 D).

Glial-specific knockdown of TGFβ superfamily members results in age-dependent neurodegeneration in the sensory nerve in the adult wing. (A) Comparison of the number of intact neuron cell bodies per wing at 28 d for control and TGFβ superfamily glial-knockdown conditions, n = 196 (control) 12–55 (RNAi). (B) Quantification of the number of intact neuron cell bodies per wing at each time point in control and babo-knockdown animals n = 127 (control), 80 (baboRNAi). Note: Same data as shown in A and S2. (C) Images from control and babo-knockdown animals of the axon bundle in the wing at each time point (left) and the proportion of wings in each classification (right) for all conditions. Fisher exact probability test, 2 × 3, 4 d, P = 1, 14 d, P = 0.0004638, 28 d, P=0.0001142, n = 127 (control), 80 (baboRNAi). (D) Quantification of the number of intact neuron cell bodies in control, babo, and Smox knockdown animals using two non-overlapping RNAis each, n = 41 (control), 45 (baboRNAi1), 39 (baboRNA2), 37 (SmoxRNAi1), 33 (SmoxRNAi2). (E) Quantification of the number of intact neuron cell bodies in adult-specific knockdown animals using Gal80ts, n = 34 (control), 32 (RNAi). Mean ± 95% CI. (A) One-way ANOVA with Dunnett’s T3 multiple comparisons test. (B, D, and E) Two-way ANOVA with Sidak’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Glial-specific knockdown of TGFβ superfamily members results in age-dependent neurodegeneration in the sensory nerve in the adult wing. (A) Comparison of the number of intact neuron cell bodies per wing at 28 d for control and TGFβ superfamily glial-knockdown conditions, n = 196 (control) 12–55 (RNAi). (B) Quantification of the number of intact neuron cell bodies per wing at each time point in control and babo-knockdown animals n = 127 (control), 80 (baboRNAi). Note: Same data as shown in A and S2. (C) Images from control and babo-knockdown animals of the axon bundle in the wing at each time point (left) and the proportion of wings in each classification (right) for all conditions. Fisher exact probability test, 2 × 3, 4 d, P = 1, 14 d, P = 0.0004638, 28 d, P=0.0001142, n = 127 (control), 80 (baboRNAi). (D) Quantification of the number of intact neuron cell bodies in control, babo, and Smox knockdown animals using two non-overlapping RNAis each, n = 41 (control), 45 (baboRNAi1), 39 (baboRNA2), 37 (SmoxRNAi1), 33 (SmoxRNAi2). (E) Quantification of the number of intact neuron cell bodies in adult-specific knockdown animals using Gal80ts, n = 34 (control), 32 (RNAi). Mean ± 95% CI. (A) One-way ANOVA with Dunnett’s T3 multiple comparisons test. (B, D, and E) Two-way ANOVA with Sidak’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.

Age-dependent neurodegeneration and decreased survival in glial-TGFβ knockdown animals. (A and B) Quantification of the number of intact VGlut+ neuron cell bodies present per wing of 4 (A) and 14 (B) dpe nerves in control and pan-glial RNAi conditions (A) n = 181 (control), 10–25 (RNAi) and (B) n = 208 (control), 13–37 (RNAi). (C) Quantification of the number of intact VGlut+ neuron cell bodies present in the L1 nerve at 28 d in control and wrapping glia-specific knockdown conditions, n = 90 (control), 18–30 (RNAi). (D) Quantification of the number of intact VGlut+ neuron cell bodies present in the L1 nerve at 28 d in control and neuronal knockdown conditions, n = 37 (control), 5–30 (RNAi). (E) Quantification of the number of intact VGlut+ cell bodies in the L1 nerve in aging animals from control animals and animals overexpressing a dominant negative version of Babo (baboDN), n = 69 (control), 43 (baboDN). (F) The percent of flies that died by 28 d of age for control and pan-glial RNAi knockdown conditions. Graphs show mean ± 95% CI. (A–D) Brown-Forsythe and Welch ANOVA with Dunnett’s T3 multiple comparisons test. (E) Two-way ANOVA with Sidak’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Age-dependent neurodegeneration and decreased survival in glial-TGFβ knockdown animals. (A and B) Quantification of the number of intact VGlut+ neuron cell bodies present per wing of 4 (A) and 14 (B) dpe nerves in control and pan-glial RNAi conditions (A) n = 181 (control), 10–25 (RNAi) and (B) n = 208 (control), 13–37 (RNAi). (C) Quantification of the number of intact VGlut+ neuron cell bodies present in the L1 nerve at 28 d in control and wrapping glia-specific knockdown conditions, n = 90 (control), 18–30 (RNAi). (D) Quantification of the number of intact VGlut+ neuron cell bodies present in the L1 nerve at 28 d in control and neuronal knockdown conditions, n = 37 (control), 5–30 (RNAi). (E) Quantification of the number of intact VGlut+ cell bodies in the L1 nerve in aging animals from control animals and animals overexpressing a dominant negative version of Babo (baboDN), n = 69 (control), 43 (baboDN). (F) The percent of flies that died by 28 d of age for control and pan-glial RNAi knockdown conditions. Graphs show mean ± 95% CI. (A–D) Brown-Forsythe and Welch ANOVA with Dunnett’s T3 multiple comparisons test. (E) Two-way ANOVA with Sidak’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
To examine a role for these genes in WG, we repeated our aging experiments using nrv2-Gal4, which in peripheral nerves is specific to WG. We observed in most cases similar age-dependent neuron loss with WG-specific elimination when comparing to pan-glial knockdown (Fig. S2 C). In particular, WG-specific knockdown of babo, mav, gbb, dpp, and Smox all resulted in degeneration of vGlut+ neurons by 28 dpe. We note that knockdown of some genes selectively in WG did not cause significant loss of sensory neurons or resulted in a less severe phenotype compared to when they were depleted from all glia (Fig. S2 C). This could suggest additional roles for these genes in other glial subtypes, glia-glia signaling, or could result from technical differences (such as differences in driver strength affecting RNAi efficiency). Finally, to determine whether TGFβ signaling was required in neurons for survival, we knocked down each of these genes in glutamatergic neurons. We found that knockdown of Smox led to significant age-dependent neuron loss, approaching loss of 50% of all vGlut+ neurons (Fig. S2 D). From these data, we conclude that the TGFβ pathway is required in glia, and to some extent neurons, for long-term neuron survival.
It is possible that the phenotypes in neuronal survival that we observe after glial depletion of TGFβ signaling could result from perturbation of a developmental requirement for these genes. To determine whether TGFβ signaling is required in the adult, we combined glial-specific knockdown with a temperature-sensitive Gal80 (Gal80ts) construct to temporally control RNAi expression (McGuire et al., 2003). We inhibited RNAi expression during development by rearing animals at 18°C. We then transferred adult animals to 31°C to allow glia-specific RNAi expression to knock down babo or Smox. At 4-day post-temperature shift, there was no significant difference in the number of neuron cell bodies between control and TGFβ knockdown animals (control: 44.0 ± 1.65, babo: 43.9 ± 1.65, P = 0.9807, Smox: 44.4 ± 1.88, P = 0.8130; Fig. 4 E). However, after 14 days of RNAi-mediated knockdown, both babo and Smox knockdown animals had fewer intact neuron cell bodies (control: 44.0 ± 1.71, babo: 41.8 ± 1.94, P = 0.0090, Smox: 40.9 ± 3.31, P = 0.0001; Fig. 4 E). These data indicate that babo and Smox are required in glia in the mature nerve to promote neuron survival and suppress neuron degeneration.
babo is expressed in WG in the adult wing
We utilized a transgenic fly line where the Gal4 coding sequence was inserted into the endogenous babo coding region with a splice acceptor resulting in truncation of the babo transcript and expression of Gal4 in its place (Lee et al., 2018). Using this genetic driver we examined co-localization of a nuclear reporter (UAS-lamin::GFP) driven by babo-Gal4 expression with an antibody that specifically labels WG nuclei within the larval peripheral nerves (Oaz; Corty et al., 2022) as well as the pan-glial nuclear protein Repo (Fig. S3 A). All Oaz+ nuclei within larval nerves were GFP+/Repo+ (n = 18 Oaz+ nuclei from n = 3 larvae, Fig. S3 A) indicating that babo was expressed in WG in peripheral nerves during development. Additionally, all Repo+/Oaz− nuclei were also GFP+, indicating that babo was also expressed in other nerve glia as well as WG (n = 123 Repo+/Oaz− nuclei from n = 3 larvae, Fig. S3 A [arrow]). To further test whether babo was also expressed in adult WG, we crossed the babo-Gal4 animals to a nuclear reporter (UAS-mCherry.NLS) in a genetic background where WG were labeled independently with GFP (a nrv2-GFP protein trap which labels all WG; Stork et al., 2008). As expected, our positive control (nrv2-Gal4) had nuclear reporter expression in WG nuclei (arrows) surrounded by GFP+ WG at all timepoints tested (n = 12 wings, Fig. 5, A and B). The experimental babo-Gal4 wings also labeled nuclei (arrow) within the GFP+ WG at 4 (n = 4 wings), 14 (n = 4 wings), and 28 (n = 4 wings) dpe (Fig. 5, A and B; and Fig. S3, B and D). Importantly, the only nuclei present within the nerve in this region are glial nuclei (Neukomm et al., 2014), indicating that babo is expressed in mature WG within the peripheral sensory nerve in the wing. We additionally stained wings from animals expressing nuclear-mCherry under control of babo-Gal4 with the pan-glial nuclear marker Repo (Fig. 5, C and D). We found many Repo+ nuclei (arrow) that were positive for nuclear-mCherry within the GFP+ axon bundle (Fig. 5 D). We also observed that babo-Gal4 labeled neuronal nuclei in the wing in addition to glia at all timepoints tested, including in the glutamatergic neurons (Fig. S3, B and C), which is consistent with our finding above of a role for TGFβ signaling in neurons.

babo is expressed in peripheral nerves during development and in the adult wing. (A) Nerves from wandering L3 animals expressing a nuclear membrane tethered GFP in babo+ cells. Nerves were co-stained for oaz (WG nuclei), HRP (neurons), and Repo (all glial nuclei). The region of interest indicated by the box is shown at higher magnification (bottom right panel). Yellow arrow indicates a GFP+/oaz−/Repo + nucleus. (B) Overview of L1 nerve expression from animals with glutamatergic neurons expressing membrane GFP via QF/QUAS and UAS-driven nuclear mCherry, nrv2-Gal4 (WG) (left) and babo-Gal4 (right) animals are shown. (C) Higher magnification images from boxes in B showing a glutamatergic neuron (arrow) with a mCherry- nucleus when nrv2-Gal4 is present (left) and one that is mCherry+ (arrow) when babo-Gal4 (right) is present. (D) Higher magnification images from boxes in B showing glutamatergic axons with mCherry+ nuclei (arrows) within the axon bundle in both conditions.
babo is expressed in peripheral nerves during development and in the adult wing. (A) Nerves from wandering L3 animals expressing a nuclear membrane tethered GFP in babo+ cells. Nerves were co-stained for oaz (WG nuclei), HRP (neurons), and Repo (all glial nuclei). The region of interest indicated by the box is shown at higher magnification (bottom right panel). Yellow arrow indicates a GFP+/oaz−/Repo + nucleus. (B) Overview of L1 nerve expression from animals with glutamatergic neurons expressing membrane GFP via QF/QUAS and UAS-driven nuclear mCherry, nrv2-Gal4 (WG) (left) and babo-Gal4 (right) animals are shown. (C) Higher magnification images from boxes in B showing a glutamatergic neuron (arrow) with a mCherry- nucleus when nrv2-Gal4 is present (left) and one that is mCherry+ (arrow) when babo-Gal4 (right) is present. (D) Higher magnification images from boxes in B showing glutamatergic axons with mCherry+ nuclei (arrows) within the axon bundle in both conditions.

TGFβ receptor B abo is expressed in adult WG. (A) Nuclear mCherry reporter expression under nrv2 (left) or babo (right) transcriptional control in a background where membrane tethered GFP is expressed in WG at 28 d. Scale bar, 200 µm. (B) Higher magnification images from boxes in A showing mCherry+ nuclei enclosed in GFP+ WG membrane (yellow arrows). Scale bar, 5 µm. (C) Low magnification image of proximal stained wing. Scale bar, 20 µm. (D) Higher magnification images from region of interest in C. Glutamatergic neurons expressing GFP were stained with an anti-GFP antibody (green). Nuclear mCherry (magenta) was driven by babo-Gal4 expression and endogenous signal was detected. An antibody against the pan-glial nuclear marker Repo was also stained (cyan). A mCherry+/Repo+ nucleus is indicated (arrow) within the axon bundle. Orthogonal views are shown from z-stack images. Scale bar, 20 µm.
TGFβ receptor B abo is expressed in adult WG. (A) Nuclear mCherry reporter expression under nrv2 (left) or babo (right) transcriptional control in a background where membrane tethered GFP is expressed in WG at 28 d. Scale bar, 200 µm. (B) Higher magnification images from boxes in A showing mCherry+ nuclei enclosed in GFP+ WG membrane (yellow arrows). Scale bar, 5 µm. (C) Low magnification image of proximal stained wing. Scale bar, 20 µm. (D) Higher magnification images from region of interest in C. Glutamatergic neurons expressing GFP were stained with an anti-GFP antibody (green). Nuclear mCherry (magenta) was driven by babo-Gal4 expression and endogenous signal was detected. An antibody against the pan-glial nuclear marker Repo was also stained (cyan). A mCherry+/Repo+ nucleus is indicated (arrow) within the axon bundle. Orthogonal views are shown from z-stack images. Scale bar, 20 µm.
Loss of TGFβ does not alter ensheathment of axons
We next examined whether loss of TGFβ signaling in glia would affect glial development and/or morphology. We evaluated the overall coverage of the L1 nerve by labeling the glia using a genetically encoded fluorescent reporter (tdTomato) and imaged the nerve at 4 and 28 dpe. When we examined nerves where TGFβ genes were knocked down in all glia, there were no obvious defects in glial morphology or coverage of the nerve in knockdown animals compared to controls at either timepoint (4 dpe: control: n = 24 animals, babo: n = 23, Smox: n = 21; 28 dpe: control: n = 21, babo: n = 22, Smox: n = 16; Fig. 6 A). Even in aged nerves that had axonal debris present and fewer neuron cell bodies, glial ensheathment appeared normal (Fig. 6 A and Fig. S4 A). We also specifically assessed WG morphology by examining reporter expression in WG-specific knockdown conditions compared to controls. Again, we did not find obvious gaps or changes in morphology of WG despite the presence of neurodegeneration (4 dpe: control: n = 23 animals, babo: n = 23, Smox: n = 23; 28 dpe: control: n = 22, babo: n = 24, Smox: n = 23; Fig. 6 B and Fig. S4 B).

Glial morphology appears intact in TGFβ knockdown animals. (A) Axons (green) and glia (magenta) in control and pan-glial TGFβ knockdown animals from 4- (top) and 28-(bottom)-d-old L1 nerves. (B) Nerves from WG-specific TGFβ knockdown animals from 4- (top) and 28-(bottom)-d-old L1 nerves. (C–E) Electron micrographs of a cross section of the L1 nerve at 28 d from control (C), babo (D), and Smox (E) knockdown animals. Wrapping glia are highlighted in cyan. (F) Quantification of the wrapping index ([individually wrapped axons + bundles of axons]/total axons). Mean ± 95% CI. One-way ANOVA with Sidak’s multiple comparisons test, n = 4–5 per condition. ns, not significant.
Glial morphology appears intact in TGFβ knockdown animals. (A) Axons (green) and glia (magenta) in control and pan-glial TGFβ knockdown animals from 4- (top) and 28-(bottom)-d-old L1 nerves. (B) Nerves from WG-specific TGFβ knockdown animals from 4- (top) and 28-(bottom)-d-old L1 nerves. (C–E) Electron micrographs of a cross section of the L1 nerve at 28 d from control (C), babo (D), and Smox (E) knockdown animals. Wrapping glia are highlighted in cyan. (F) Quantification of the wrapping index ([individually wrapped axons + bundles of axons]/total axons). Mean ± 95% CI. One-way ANOVA with Sidak’s multiple comparisons test, n = 4–5 per condition. ns, not significant.

Quantification of neuron loss for nerves shown in Fig. 6, A and B. Electron micrographs corresponding to Fig. 6, C–E. (A) Quantification of neuron loss in pan-glial knockdown animals represented in Fig. 6 A, n = 45 (control), 45 (baboRNAi), 37 (SmoxRNAi). (B) Quantification of neuron loss in WG-specific knockdown animals represented in Fig. 6 B, n = 45 (control), 47 (baboRNAi), 46 (SmoxRNAi). (C–E) Electron micrographs corresponding to Fig. 6, C–E (C) Control (D) baboRNAi (E) SmoxRNAi. Scale bar 1 μm. Mean ± 95%CI. Two-way ANOVA with Sidak’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Quantification of neuron loss for nerves shown in Fig. 6, A and B. Electron micrographs corresponding to Fig. 6, C–E. (A) Quantification of neuron loss in pan-glial knockdown animals represented in Fig. 6 A, n = 45 (control), 45 (baboRNAi), 37 (SmoxRNAi). (B) Quantification of neuron loss in WG-specific knockdown animals represented in Fig. 6 B, n = 45 (control), 47 (baboRNAi), 46 (SmoxRNAi). (C–E) Electron micrographs corresponding to Fig. 6, C–E (C) Control (D) baboRNAi (E) SmoxRNAi. Scale bar 1 μm. Mean ± 95%CI. Two-way ANOVA with Sidak’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
To examine finer detail of axonal ensheathment we assessed the ultrastructure of this nerve using transmission electron microscopy. We examined nerves from control, babo, and Smox pan-glial knockdown animals and found no consistent evidence at the ultrastructural level of defects in glial ensheathment at 28 dpe (control: n = 4 animals, babo: n = 5, Smox: n = 4; Fig. 6, C–F and Fig. S4, C–E). To quantify this form of multi-axonal ensheathment, we measured the wrapping index for each nerve from the electron micrographs (Matzat et al., 2015). When we calculated the wrapping index for each condition, there was no significant difference between the conditions, although one nerve from the Smox knockdown condition did appear to have reduced ensheathment (control: 0.88 ± 0.020 n = 4, babo: 0.84 ± 0.083 n = 5, Smox: 0.81 ± 0.21 n = 4; Fig. 6 F). Together, our data indicate that inhibiting TGFβ in glia does not cause consistent defects in glial ensheathment and therefore disrupted glial ensheathment of axons does not explain the degeneration observed in TGFβ knockdown animals.
Finally, we quantified the number of WG present in the nerve in control and knockdown animals by using a genetically encoded nuclear reporter (UAS-lamin::GFP). Knock down of babo or Smox in WG caused a modest increase in the number of WG in the L1 nerve at 4 dpe (nrv2>: 24.9 ± 3.52, nrv2 > lacZ: 26.9 ± 3.37, nrv2 > baboRNAi: 30.1 ± 3.54, nrv2 > SmoxRNAi: 29.0 ± 3.76; Fig. S5, A and B). At 28 dpe, the number of WG nuclei in babo knockdown animals remained elevated compared to controls, but the number of WG nuclei in Smox knockdown animals was not significantly different from controls (nrv2>: 24.5 ± 3.40, nrv2 > lacZ: 26.2 ± 3.39, nrv2 > baboRNAi: 29.5 ± 3.38, nrv2 > SmoxRNAi: 25.4 ± 6.76; Fig. S5, A and B). We hypothesized that this increase in WG nuclei could be due to inhibition of a role for TGFβ signaling in promoting apoptosis (Schuster and Krieglstein, 2002). We tested this by overexpressing the caspase inhibitor p35 in WG. Blocking caspase activity phenocopied the increase WG nuclei observed in babo knockdown conditions, suggesting a role for this receptor in inducing apoptosis in a subset of WG (Fig. S5, C and D). Importantly however, inhibition of apoptosis alone by p35 overexpression in glia did not cause neuron loss in aged animals (Fig. S5 E). We concluded that increased glial nuclei on its own was not responsible for neuron loss in aged animals.

Greater number of wrapping glia nuclei in TGFβ knockdown animals. (A) Images from 4 dpe wings expressing nuclear GFP and membrane-tethered tdTomato in wrapping glia in control (n/a & lacZ; top) and TGFβ knockdown animals (bottom). Higher magnification images from the boxed area are shown below each wing. (B) Quantification of the number of GFP+ WG nuclei per wing, n = 46 (nrv2>), 48 (nrv2 > lacZ), 48 (baboRNAi), 46 (SmoxRNAi). (C) Images of wings at 4 dpe from control (top), WG babo-KD (bottom left), and WG p35-OE (bottom right) animals. (D) Quantification of the number of wrapping glia nuclei in 4-d control, glial babo-KD, and glial p35-OE animals, n = 24 (control), 24 (nrv2 > lacZ), 24 (baboRNAi), 24 (p35). (E) Quantification of the number of intact neuron cell bodies in nerves from control animals, glial-baboRNAi, and glial-p35 expressing animals, n = 32 (control), 19 (baboRNAi), 26 (p35). Mean ± 95% CI. (B) Two-way ANOVA with Tukey’s multiple comparisons test. (D) One-way ANOVA with Tukey’s multiple comparisons test. (E) Two-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Greater number of wrapping glia nuclei in TGFβ knockdown animals. (A) Images from 4 dpe wings expressing nuclear GFP and membrane-tethered tdTomato in wrapping glia in control (n/a & lacZ; top) and TGFβ knockdown animals (bottom). Higher magnification images from the boxed area are shown below each wing. (B) Quantification of the number of GFP+ WG nuclei per wing, n = 46 (nrv2>), 48 (nrv2 > lacZ), 48 (baboRNAi), 46 (SmoxRNAi). (C) Images of wings at 4 dpe from control (top), WG babo-KD (bottom left), and WG p35-OE (bottom right) animals. (D) Quantification of the number of wrapping glia nuclei in 4-d control, glial babo-KD, and glial p35-OE animals, n = 24 (control), 24 (nrv2 > lacZ), 24 (baboRNAi), 24 (p35). (E) Quantification of the number of intact neuron cell bodies in nerves from control animals, glial-baboRNAi, and glial-p35 expressing animals, n = 32 (control), 19 (baboRNAi), 26 (p35). Mean ± 95% CI. (B) Two-way ANOVA with Tukey’s multiple comparisons test. (D) One-way ANOVA with Tukey’s multiple comparisons test. (E) Two-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Neuronal WldS expression or caspase inhibition rescues neurodegeneration induced by glial babo knockdown
In aged animals, both the axons and cell body of the neurons were affected by babo knockdown in glia. This could result from a lack of glial support of neuronal cell bodies, axons, or both. The neuroprotective effects of WldS are selective to Wallerian degeneration and do not block apoptosis (Beirowski et al., 2008; Deckwerth and Johnson, 1994; Hoopfer et al., 2006). We overexpressed WldS in the glutamatergic neurons to suppress Wallerian degeneration pathways in aged control and babo knockdown animals and found that blocking Wallerian degeneration rescued axon degeneration observed in babo knockdown animals at 28 dpe (control -WldSn = 17, control +WldSn = 19, baboRNAi -WldSn = 27, baboRNAi +WldSn = 20; Fig. 7, A and B). These data suggest that loss of babo in glia leads to activation of a WldS-sensitive axon degeneration pathway in axons. Interestingly, we found that suppressing axon degeneration with WldS also completely rescued neuron cell death (Fig. 7, A and C). These findings suggest that glial loss of Babo leads to activation of an axon degeneration pathway in neurons that can ultimately result in neuronal cell death.

Wld S overexpression in neurons rescues neurodegeneration in babo knockdown animals. (A) Representative images of neuron cell bodies and axons within the wing in control and babo knockdown animals at 4 (top) and 28 (bottom) days of age with and without WldS expressed in neurons. Scale bar 5 μm. (B) Classification of axon integrity in control and babo knockdown animals at 28 d with and without WldS. Pairwise Fisher Exact Probability test (two-tailed, 2 × 3, n = 17 [control], 19 [control +WldS], 27 [baboRNAi], 20 [baboRNAi +WldS]). (C) Quantification of the number of VGlut+ neuron cell bodies in the wing from control and babo knockdown animals with and without WldS (n = 40 [control], 46 [control +WldS], 53 [baboRNAi], 48 [baboRNAi +WldS]). Mean ± 95% CI. Two-way ANOVA with Tukey’s multiple comparisons test, n = 187. *, P < 0.05; ****, P < 0.0001; ns, not significant.
Wld S overexpression in neurons rescues neurodegeneration in babo knockdown animals. (A) Representative images of neuron cell bodies and axons within the wing in control and babo knockdown animals at 4 (top) and 28 (bottom) days of age with and without WldS expressed in neurons. Scale bar 5 μm. (B) Classification of axon integrity in control and babo knockdown animals at 28 d with and without WldS. Pairwise Fisher Exact Probability test (two-tailed, 2 × 3, n = 17 [control], 19 [control +WldS], 27 [baboRNAi], 20 [baboRNAi +WldS]). (C) Quantification of the number of VGlut+ neuron cell bodies in the wing from control and babo knockdown animals with and without WldS (n = 40 [control], 46 [control +WldS], 53 [baboRNAi], 48 [baboRNAi +WldS]). Mean ± 95% CI. Two-way ANOVA with Tukey’s multiple comparisons test, n = 187. *, P < 0.05; ****, P < 0.0001; ns, not significant.
We next explored potential roles for caspases in neuronal loss after manipulation of glial TGFβ signaling. Using a potent caspase inhibitor p35, we genetically blocked caspase-mediated death in glutamatergic neurons while simultaneously knocking down babo in glia. Surprisingly, this resulted in complete rescue of axon degeneration (control -P35 n = 18, control +P35 n = 18, baboRNAi -P35 n = 12, baboRNAi +P35 n = 15; Fig. 8, A and B) and rescued neuron cell loss (Fig. 8, A and C). These data indicate that caspase signaling is activated in neurons in response to blockade of glial TGFβ signaling. Moreover, coupled with our observation that WldS can also rescue this phenotype, this data indicates that both Wallerian degeneration-like and caspase-dependent signaling pathways are activated in neurons to drive neurodegeneration after glial depletion of TGFβ activity. Suppression of either of these prodegenerative pathways is sufficient to provide strong rescue of neurodegenerative phenotypes.

p35 overexpression in neurons rescues neurodegeneration in babo knockdown animals. (A) Representative images of neuron cell bodies and axons within the wing in control and babo knockdown animals at 4 (top) and 28 (bottom) days of age with and without p35 expressed in neurons. Scale bar 5 µm. (B) Classification of axon integrity in control and babo knockdown animals at 28 d with and without p35. Pairwise Fisher Exact Probability Test (two-tailed, 2 × 3, n = 18 [control], 18 [control +P35], 12 [baboRNAi], 15 [baboRNAi +P35]). (C) Quantification of the number of VGlut+ neuron cell bodies in the wing from control and babo knockdown animals with and without p35. Mean ± 95% CI. Two-way ANOVA with Tukey’s multiple comparisons test (n = 36 [control], 36 [control +P35], 30 [baboRNAi], 33 [baboRNAi +P35]). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
p35 overexpression in neurons rescues neurodegeneration in babo knockdown animals. (A) Representative images of neuron cell bodies and axons within the wing in control and babo knockdown animals at 4 (top) and 28 (bottom) days of age with and without p35 expressed in neurons. Scale bar 5 µm. (B) Classification of axon integrity in control and babo knockdown animals at 28 d with and without p35. Pairwise Fisher Exact Probability Test (two-tailed, 2 × 3, n = 18 [control], 18 [control +P35], 12 [baboRNAi], 15 [baboRNAi +P35]). (C) Quantification of the number of VGlut+ neuron cell bodies in the wing from control and babo knockdown animals with and without p35. Mean ± 95% CI. Two-way ANOVA with Tukey’s multiple comparisons test (n = 36 [control], 36 [control +P35], 30 [baboRNAi], 33 [baboRNAi +P35]). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Glial TGFβ signaling regulates glial metabolism and support of neurons
How does manipulation of TGFβ signaling in glia alter glial support of axons? One mechanism by which glia support axons is by providing metabolites to fuel energy demands of the axon (Fünfschilling et al., 2012; Volkenhoff et al., 2015). Work in Drosophila fat body has shown that TGFβ signaling regulates key metabolic genes, and its loss leads to increased TCA cycle function, ATP levels, and mitochondrial biogenesis (Ghosh and O’Connor, 2014). First, we examined whether metabolism was altered in WG when TGFβ signaling was perturbed. Using a genetically encoded ATP sensor, we measured the relative ATP concentrations in WG in control animals (UAS-lacZ) and animals where babo or Smox were knocked down in WG (Lobas et al., 2019). In both knockdown conditions, ATP levels were increased around 46 and 32%, respectively, compared to controls (Fig. 9, A and B). We next artificially increased mitochondrial biogenesis in WG by overexpressing the transcriptional co-activator srl (PGC-1α in mammals; Dominy and Puigserver, 2013), to test whether this would phenocopy blockade of TGFβ signaling. srlOE resulted in a ∼36% increase in ATP levels, similar to baboRNAi and SmoxRNAi conditions (Fig. 9, A and B). Monocarboxylate transporters (MCTs) are well-known mediators of glial support of axons through delivery of lactate (Fünfschilling et al., 2012), so we next tested whether reduction of MCTs in WG would impact overall ATP levels by knocking down Bsg, the adaptor protein required for plasma membrane localization of all MCTs (Besse et al., 2007; Halestrap and Wilson, 2012; Kirk et al., 2000). We found that WG depletion of Bsg also led to an increase in WG ATP, similar to suppression of TGFβ signaling in WG (Fig. 9, A and B). Finally, we examined whether enhancing mitochondrial biogenesis in WG, or blockade of the MCT chaperone Bsg, would also cause age-dependent loss of VGlut+ neurons. We aged control animals and animals overexpressing srl in glia, or BsgRNAi and counted the number of intact glutamatergic neurons per nerve at 28 dpe. srl overexpression was sufficient to cause neuron loss, as was blockade of MCT function through knockdown of Bsg (Fig. 9 C). These results are consistent with a model in which loss of TGFβ signaling in glia leads to changes in glial metabolic function and thus support of neurons that in turn causes neurodegeneration.

Blocking TGFβ signaling alters wrapping glial metabolism and support of axons. (A) ATP sensor signal in wrapping glia (WG) in the L1 nerve. Asterisks indicate WG nuclei. (B) Quantification of the cytosolic ATP sensor intensity (n = 64 [control], 24 [baboRNAi], 18 [SmoxRNAi], 32 [srlOE], 18 [BsgRNAi]). (C) Quantification of intact neuron cell bodies from aged, 28-day animals from control, srlOE, or BsgRNAi expressed in WG n = 23 (control), 27 (srlOE), 29 (BsgRNAi). Genotypes are as indicated. Mean ± 95% CI. One-way ANOVA with Tukey’s multiple comparisons test, n = 78. **, P < 0.01; ****, P < 0.0001.
Blocking TGFβ signaling alters wrapping glial metabolism and support of axons. (A) ATP sensor signal in wrapping glia (WG) in the L1 nerve. Asterisks indicate WG nuclei. (B) Quantification of the cytosolic ATP sensor intensity (n = 64 [control], 24 [baboRNAi], 18 [SmoxRNAi], 32 [srlOE], 18 [BsgRNAi]). (C) Quantification of intact neuron cell bodies from aged, 28-day animals from control, srlOE, or BsgRNAi expressed in WG n = 23 (control), 27 (srlOE), 29 (BsgRNAi). Genotypes are as indicated. Mean ± 95% CI. One-way ANOVA with Tukey’s multiple comparisons test, n = 78. **, P < 0.01; ****, P < 0.0001.
Discussion
Axons can be remarkably complex and enormous structures and maintaining their integrity and synaptic connections is essential for long-term nervous system function. Glial cells are thought to provide crucial support to axons to enable and sustain their energetically demanding functions. In this study, we used Drosophila as a screening platform to identify a collection of glial genes required for axon survival in vivo. We found that wrapping glia were essential for long-term survival of neurons in peripheral nerves, and that the TGFβ signaling pathway plays a key role in supporting long-term axon survival. Similar to its role in the fat body, TGFβ signaling in glia appears to regulate glial metabolism and in turn metabolic support of neurons. In the absence of TGFβ signaling in glia, both WldS-sensitive axon degeneration pathways and caspase-mediated cell death programs become active. Together our work reveals TGFβ activity as an important new pathway required for glial support of neurons and axons and argues for an interdependence between the axon and soma for long-term maintenance of neuronal integrity in the Drosophila L1 nerve.
Identifying genes involved in glial support of axons in vivo
Through a genetic screen to identify genes with roles in promoting axon survival, we assayed a library consisting of most of the secreted and transmembrane proteins encoded in the Drosophila genome. We identified an array of molecules that, when depleted selectively from glia, lead to axon degeneration. Components of the TGFβ superfamily were over-represented in our candidates from this screen, and we showed that loss of glial TGFβ signaling—ligands, receptors, or downstream signaling molecules—led to age-dependent axon degeneration and neuronal loss. Surprisingly, we found that providing WldS or the caspase inhibitor P35 to neurons was sufficient to overcome neurodegeneration caused by reduced TGFβ signaling in glia. We proposed that TGFβ signaling in glia normally promotes axon maintenance, and this support is required to sustain neuronal survival over the animal’s lifespan.
The intense ensheathment of axons by WG in the adult L1 wing nerve suggests that isolating individual axons in this tissue is critical for neuronal function, maintenance, or both. Reduced glial ensheathment does not appear to explain the phenotypes reported here in babo and Smox knockdown animals. Our live imaging and ultrastructural analyses both indicate that glia remain tightly associated with axons in these knockdown animals. This ensheathment creates a physical barrier that likely prevents axons from directly accessing metabolites outside the nerve to support their activity. This anatomical arrangement supports the notion that in Drosophila, as in mammals, glia act as the go-between and provide support to the axons they enwrap. Using a new, highly specific Split Gal4 line (Corty et al., 2022), we showed that ablating most of the WG in the adult peripheral nerve in the wing caused robust age-dependent neurodegeneration. These results are remarkably similar to what has been seen in vertebrate models of CMT where loss of myelinating glia results in neurodegeneration in peripheral nerves (Adlkofer et al., 1995; Verhamme et al., 2011). Whether the loss of WG in the fly leads to axon degeneration because of a loss of general support mechanisms, as is observed in mammals (Fünfschilling et al., 2012; Lee et al., 2012), or is a direct result of a lack of survival cues through TGFβ (or other) signaling remains to be determined.
TGFβ signaling is context-dependent and can impact multiple cellular processes at once
Inhibiting members of both branches of the TGFβ superfamily (TGFβ and BMP) in glia caused neurodegeneration in aged animals. The TGFβ receptor Babo phosphorylates and activates the Smad transcription factor Smox, allowing it to enter the nucleus (Upadhyay et al., 2017). Smox has the potential to modify many genetic pathways simultaneously, and given that loss of Smox phenocopies Babo depletion, our data suggest that at least part of this pathway’s role in supporting axon maintenance involves gene regulation. Identifying the key transcriptional targets for Smox that promote this pro-survival function in axons will be an exciting next step in understanding the full array of molecules involved in glial support of axon maintenance.
Using a genetically encoded reporter, we found that babo is expressed in both glia and neurons in the adult wing, supporting a role for this receptor in the mature nerve in both cell types. More importantly, we showed that conditional knockdown of either babo or Smox specifically in mature glia was sufficient to induce neurodegeneration. The adult-specific knockdown was modest compared to the constitutive knockdown conditions and could indicate that TGFβ signaling is required both during development and in the mature nerve. Alternatively, this could result from technical differences between experiments. We were only able to examine adult-specific knockdown animals out to 14 dpe due to increased lethality in these animals at 31°C. Nevertheless, the adult expression of babo combined with the requirement for TGFβ components in adult glia support a role for the TGFβ pathway in the mature nerve to promote neuronal survival.
TGFβ signaling is important for regulation of a battery of metabolic genes in the fat body, and loss of TGFβ signaling through Babo and Smox leads to increased TCA cycle function and changes in nuclear-encoded mitochondrial gene expression (Ghosh and O’Connor, 2014). Interestingly, we found that glial depletion of Babo or Smox led to increased ATP as would be predicted from increased mitochondrial respiration. Indeed, enhancing mitochondrial biogenesis (by overexpressing srl) increased ATP and phenocopied neuronal loss observed after glial depletion of TGFβ signaling. Finally, we found that glial depletion of Bsg, which is a key chaperone required for the localization of lactate-transporting MCTs, also led to increased glial ATP and phenocopy of age-dependent neuron loss in the peripheral nerve. We therefore proposed that glial TGFβ signaling plays an important role regulating glial metabolic function and in turn support of neurons in the peripheral nerve, and that loss of this support is sufficient to induce neurodegeneration.
Which degenerates first: the axon or the cell body?
Axon degeneration and neuronal cell body loss in our experimental system was coordinately timed in both our glial-ablated and TGFβ knockdown animals. As such, we are unable to determine which compartment of the cell began to degenerate first. We found that suppressing Wallerian degeneration by expressing WldS in uninjured, normally aging axons was sufficient to rescue neurodegeneration in animals lacking the TGFβ receptor in glia. We concluded from this that loss of glial TGFβ signaling leads to the activation of WldS-sensitive axon degeneration pathways and ultimately cell body degeneration. While it remains possible that WldS directly protects the neuron cell bodies, this would depart from the substantial body of data supporting an axon-specific role for WldS in suppressing neurodegeneration. At the same time, we found that genetic blockade of caspase activity also rescued cell body loss and axon degeneration. Together these results suggest that blockade of glial TGFβ activativity induces a degenerative program in neurons involving WldS- and caspase-sensitive mechanisms. One interpretation of this result is that Wallerian degeneration pathways are activated in the axon, while caspase-dependent pathways are activated in the soma, and both are required to drive neuronal loss. P35 is a potent inhibitor that broadly blocks caspases (Bertin et al., 1996; Bump et al., 1995; LaCount et al., 2000; Xue and Robert Horvitz, 1995; Zhou et al., 1998). In some cases, caspases have been implicated in neurite degeneration and so we cannot formally rule out such a non-cell death associated role based on our data (Kuo et al., 2006; Nikolaev et al., 2009; Simon et al., 2012; Williams et al., 2006). It remains possible that P35 could also be preventing axon degeneration directly rather than secondarily through protection of the cell body.
Glia provide the necessary support for WldS to maximally protect axons in vivo
A yet-to-be explained phenomenon in the study of axon degeneration is the observation that WldS is a much more potent suppressor of Wallerian degeneration in vivo as compared to in vitro. Severed WldS+ axons survive for many weeks in vivo (Adalbert et al., 2005; Lunn et al., 1989), yet in purified neuron cultures, protection persists only for days (Buckmaster et al., 1995; Wang et al., 2005). While there are many differences between these environments, one striking difference is the presence or absence of a glial sheath around axons. Our findings that WldS protection of injured axons in vivo is significantly impaired when glia are absent indicates that when axons lack a neuronal cell body their survival is highly dependent upon glial support.
In summary, our work provides multiple lines of direct in vivo evidence that non-myelinating glia are crucial for supporting the survival of long axons. When they are eliminated, axons show increased age-dependent degeneration and even WldS is unable to protect axons without this glial support. We have identified several candidate glial genes required for this glial support of axon integrity and neuronal survival in vivo and show that TGFβ signaling plays a crucial role in glia to promote long-term axon maintenance, potentially through regulation of glial metabolic support of axons.
Materials and methods
Fly husbandry
Flies were grown on standard molasses cornmeal agar with extra yeast and maintained at 25°C. The following fly (Drosophila melanogaster) stocks used in this study were obtained from the following sources. Bloomington: OK371-QF2 (66473), 10xQUAS-6xGFP, UAS-mtdTomato-3xHA (66479), Repo-GAL4 (7415), UAS-Reaper14 (5824), UAS-dronc::GFP (56759), UAS-lacZ.NZ312 (3956), UAS-lacZ.NZ20b (3955), VGlut-QF2 (60315), QUAS-mCD8::GFP (30002), UAS-lamin::GFP (BL7376), nrv2-GAL4 (6799), UAS-baboDN (64423), baboRNAi2 (40866), SmoxRNAi2 (41670), tkvRNAi (40937), mavRNAi (34650), putRNAi (39025), UAS-mCherry.NLS3 (38424), babo-Gal4CRIMIC00274 (83164), tubP-Gal80ts-20 (7019). Vienna Drosophila Resource Center RNAi lines are listed in the supplementary excel file (Table S2). Additional RNAi lines including tkvRNAi, putRNAi, and saxRNAi were generously provided by Dr. Michael O’Connor. UAS-dark was kindly provided by Dr. John M. Abrams (Akdemir et al., 2006). The protein trap nrv2-GFP published in Stork et al. (2008). UAS-iATPsnFR (Lobas et al., 2019). The WG split-Gal4 (Corty et al., 2022) line was established using the nrv2-DNA-binding domain construct previously reported in Coutinho-Budd et al. (2017) combined with a VP16 activation domain converted from the IT.0117-Gal4 (BL62647) using methods described in Gohl et al. (2011). Additional lines that we generated to complete this work were QUAS-Wlds(III).
Sensitized RNAi screen
RNAi lines were crossed to the w*; VGlut-QF2, QUAS-mCD8::GFP/CyO; QUAS-WldS, Repo-Gal4/TM3 driver line. After 7 d, parents were discarded and progeny returned to 25°C. Progeny were then anesthetized on CO2 fly pads, sorted for genotype using visible markers, aged 4 d at 25°C, anesthetized on CO2 and one wing was cut between the two cross-veins of the wing using spring scissors (#15002-08; F.S.T), while the other wing served as an uninjured control. Injured flies were transferred to fresh vials every 3–7 d and then imaged 10- or 14-d post axotomy (see imaging). For each RNAi line, at least five wings were evaluated, and results are reported in the supplemental excel file (Table S2). RNAi lines were scored as lethal if no viable adult flies of the correct genotype emerged or if adults died before the imaging timepoint. Both female and male progeny were used except where genetics prohibited use of males. RNAi lines used in the screen are publicly available from Vienna Drosophila Resource Center and their sequences are available on the webpage https://stockcenter.vdrc.at/control/main.
Aging assay
Animals of the appropriate genotypes were crossed, as described above, selected for markers at eclosion, and adults were aged for the indicated time windows at 25°C. Aging flies were transferred into fresh vials every 3–7 d. The number of dead flies in each vial was recorded during each transfer and these tallies can be found in Fig. S2 F. Subsets of wings from each cohort were imaged at 4, 14, and 28 d after progeny were originally collected. All wings were inspected at 63× for injuries and were not evaluated if they had any visible tears or scars in the L1 wing vein containing the nerve.
Adult-specific knockdown
Crosses were performed at 18°C, and the progeny were allowed to develop at 18°C. Adults of the correct genotype were collected into fresh cornmeal agar vials and transferred to 31°C. Flies were maintained at 31°C and transferred to fresh vials every 3–5 d until imaging.
Imaging
Imaging of the wing nerve was done as previously described in Neukomm et al. (2014). Briefly, flies were anesthetized using CO2 and their wings were removed using spring scissors, mounted on a slide in Halocarbon oil 27 (#H8773; Sigma-Aldrich), covered with #1.5 cover glass, and imaged within 15 min of mounting. Z-stack images were taken of the nerve on a Zeiss Axio Examiner with a Yokogawa spinning disk and Hamamatsu camera using a 63×1.4NA oil-immersion objective at room temperature. Zeiss Zen Blue 3.4 software was used to acquire and Zen Blue and Adobe photoshop were used to process images. Processing included orthogonal projections using maximum intensity, stitching of tiled images, rotation, and cropping. The same acquisition settings were used across samples for each of the experiments and control samples were imaged in the same imaging session as experimental samples. VGlut+ neuron cell bodies in the L1 vein were counted under 63× magnification. Cells were counted as intact if they had a clear nucleus and dendrite or were considered dead if they were shrunken and the dendrite or nucleus were not clearly visible (examples shown in Fig. S1 G).
Quantification of axon degeneration
Images were classified into phenotypic categories (intact, mild, or severe degeneration) with the conditions blinded to the scorer (Fig. S1 F). Images were given randomized numerical names and all genotypes and ages for a given experiment were scored together in one session and later decoded. For experiments in which the wrapping glia were ablated, the channel containing the axons was first extracted from the two-color images before blinding and scoring so that the scorer remained blind to the presence or absence of glia.
Quantification of iATPsnFR
To quantify iATPsnFR intensity, images were acquired as z-stacks and maximum intensity projections were derived for each nerve. Using ZEN 3.4 (blue edition) software, each nerve was then traced, excluding nuclei to control for variability due to differences in nuclei numbers. The average intensity for the traced region for each nerve was then used for statistical analysis.
Immunofluorescence
Wandering third instar larvae were dissected and pinned open as filets in cold PBS and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. Larvae were then permeabilized in 0.3% PBST (PBS + Triton X-100) for 15 min at room temperature with agitation, and remaining wash and antibody solutions were made in 0.3% PBST with 1% BSA. Samples were incubated in primary antibody solution overnight at 4°C followed by one quick wash then 5 × 15 min washes using 0.3% PBST before incubating in secondary antibody solution overnight at 4°C. Samples were then washed again as before, then incubated in Vectashield (#H-1000; Vector Labs) for 1 h at room temperature before mounting. Antibodies used were: (1°) anti-Repo (Mouse anti-Repo, #8D12; DSHB), Alexa Fluor 647 anti-HRP (Goat anti-HRP, #123-605-021; Jackson Labs), anti-oaz (Rabbit anti-oaz, (Corty et al., 2022), anti-GFP (Chicken anti-GFP #ab13970; Abcam); (2°) DyLight 405 Donkey anti-Mouse (#715-475-150; Jackson Labs), Alexa Fluor 488 Donkey anti-Chicken (#703-545-155; Jackson Labs), and Rhodamine Red-X Donkey anti-Rabbit (#711-295-152; Jackson Labs). After staining, larva filets were mounted in Vectashield and covered with #1.5 cover glass (#1404-15; Globe scientific) and stored at 4°C. The same protocol was used for adult wings; however, both primary and secondary incubation steps were done for 5 d at 4°C.
Electron microscopy
Aged flies were maintained as described above. For EM procedures, we used a modified microwave protocol from Cunningham and Monk (2018); Czopka and Lyons (2011). Flies were anesthetized with CO2, and their wings were removed with spring scissors and immediately put into freshly made fix solution (2% glutaraldehyde, 4% paraformaldehyde, 0.1 M sodium cacodylate buffer). Forceps were used to gently submerge the tissue in a microcentrifuge tube and microwaved using the following settings: 2× (100W for 1 min, OFF for 1 min), then immediately followed by 5× (450W for 20 s, OFF for 20 s) before storing the tissue at 4°C overnight in fix solution. The following day samples were washed in 0.1 M sodium cacodylate buffer followed by secondary fixation in 2% osmium tetroxide, 0.1 M sodium cacodylate buffer and 0.1 M imidazole pH 7.5 and microwaved 2× (100W for 1 min, OFF for 1 min), 5× (450W for 20 s, OFF for 20 s). Following osmium fixation, samples were rinsed in distilled water for 3 × 10 min washes. Next, samples were stained in saturated uranyl acetate (UA) ∼8% in water and microwaved 2× (450W for 1 min, OFF for 1 min). This was followed by dehydration steps with an escalating ethanol series with each step microwaved at 250W for 45 s. The final 100% EtOH step was repeated three times and each step was microwaved for 2× (250W for 1 min, OFF for 1 min). Following EtOH dehydration, samples were dehydrated in 100% acetone and microwaved 2× (250W for 1 min, OFF for 1 min) and repeated three times. Next, samples were transferred to a 50:50 resin/acetone solution and agitated overnight at room temperature. Final resin infiltration was done in 100% resin and agitated at room temperature for at least 1 h. Tissues were embedded in Embed 812 resin (#14120; EMS) and cured in a 60°C oven overnight. Ultrathin 70-nm sections were cut on a Leica ultramicrotome and transferred to 100 mesh Formvar grids (#FCF100-Cu; EMS). Grids were counter-stained for 20 min in 5% uranyl acetate followed by 7 min in Reynold’s lead citrate. Micrographs were acquired on a FEI Tecnai T12 interfaced to Advanced Microscopy Techniques (AMT) CCD camera.
Statistical analysis
Statistical analyses were performed in GraphPad Prism 8. Fisher Exact test or χ-square test were used to compare experimental to control conditions for axon degeneration data. For quantification of neuron cell bodies, when analyzing the effect of two variables (genotype and age) two-way ANOVA was used with Sidak’s or Dunnett’s multiple comparisons test to analyze the effect of genotype at each age compared to control. When comparing all groups to each other, Tukey’s multiple comparisons test was used. To compare multiple experimental groups to the same control group from a single timepoint, Brown–Forsythe and Welch’s ANOVA was used with Dunnett’s T3 or Tukey’s multiple comparisons test. When comparing one experimental group to a control, a one-tailed Welch’s t test was used. For parametric tests, data distribution was assumed to be normal but this was not formally tested. Significance was determined using an α of 0.05. P values are represented as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Online supplemental material
Fig. S1 shows wrapping glia reporter expression in the wing and degeneration classification. Fig. S2 shows age-dependent neurodegeneration and decreased survival in glial-TGFβ knockdown animals. Fig. S3 shows babo is expressed in peripheral nerves during development and in the adult wing. Fig. S4, A and B, show quantification of neuron loss for nerves shown in Fig. 6, A and B. Fig. S4, C–E, show electron micrographs corresponding to Fig. 6, C–E. Fig. S5 shows greater number of wrapping glia nuclei in TGFβ knockdown animals. Table S1 shows genotypes corresponding to figures. Table S2 shows RNAi screen results.
Acknowledgments
We thank all the Freeman Lab members for their discussion and feedback. We would like to acknowledge the technical support and expertise of the staff in the electron microscopy core facility, particularly Dr. Robert Kayton as well as Dr. Deborah Hegarty from the Aicher Lab. We also thank Dr. Kelly Monk and members of the Monk Lab for sharing their expertise and equipment for electron microscopy, and Drs. Kevin Wright, Ben Emery, and Rachel Dresbeck for critical feedback on the manuscript. We acknowledge the fly community for generous sharing of reagents and the VDRC for providing the RNAi collection used in the screen.
The study was supported by National Institute of Neurological Disorders and Stroke P30 NS061800 (to S.A. Aicher) and National Institutes of Health grants NS053538 and NS112215 (to M.R. Freeman).
Author contributions: Conceptualization, A.P. Lassetter and M.R. Freeman; Formal Analysis, A.P. Lassetter; Funding Acquisition, S.A. Aicher and M.R. Freeman; Investigation, A.P. Lassetter, M.M. Corty, R. Barria., A.E. Sheehan, J.Q. Hill, and A.N. Fox; Methodology, A.P. Lassetter, M.M. Corty, J.Q. Hill, A.N. Fox, and M.R. Freeman; Resources, S.A. Aicher and M.R. Freeman; Supervision, M.R. Freeman; Visualization, A.P. Lassetter; Writing—Original Draft, A.P. Lassetter and M.R. Freeman. Writing—Review & Editing, A.P. Lassetter, M.M. Corty, S.A. Aicher, and M.R. Freeman.

