


TJs maintain the epithelial barrier by regulating paracellular permeability. Since TJs are under dynamically fluctuating intercellular tension, cells must continuously survey and repair any damage. However, the underlying mechanisms allowing cells to sense TJ damage and repair the barrier are not yet fully understood. Here, we showed that proteinases play an important role in the maintenance of the epithelial barrier. At TJ break sites, EpCAM–claudin-7 complexes on the basolateral membrane become accessible to apical membrane-anchored serine proteinases (MASPs) and the MASPs cleave EpCAM. Biochemical data and imaging analysis suggest that claudin-7 released from EpCAM contributes to the rapid repair of damaged TJs. Knockout (KO) of MASPs drastically reduced barrier function and live-imaging of TJ permeability showed that MASPs-KO cells exhibited increased size, duration, and frequency of leaks. Together, our results reveal a novel mechanism of TJ maintenance through the localized proteolysis of EpCAM at TJ leaks, and provide a better understanding of the dynamic regulation of epithelial permeability.

Introduction
The tight junction (TJ) is an epithelial cell–cell junction structure that controls the paracellular passage of solutes and ions to maintain barrier function (Farquhar and Palade, 1963; Turner et al., 2014; Van Itallie and Anderson, 2014; Zihni et al., 2016; Piontek et al., 2020). Transmission EM reveals that the plasma membranes of adjacent cells are apposed to each other at the TJs. Within the apposition, there are multiple membrane attachments, known as kissing points, which serve as physical barriers for the paracellular diffusion of substances (Farquhar and Palade, 1963). Using freeze-fracture replica EM (FFEM), these attachments can be visualized as a pair of grooves and ridges called TJ strands (Chalcroft and Bullivant, 1970; Wade and Karnovsky, 1974). In most epithelial cells, the TJ strands are branched and anastomosed to form a complex network. The major components of the TJ strands are claudins, which are four-pass transmembrane proteins. Claudins constitute a protein family comprising more than 20 members (Morita et al., 1999; Yamazaki et al., 2011; Mineta et al., 2011). Each epithelial cell type expresses a specific combination of multiple claudins, which determine the selectivity of paracellular permeability (Anderson and Van Itallie, 2009; Krug et al., 2014; Angelow and Yu, 2007). Pore-forming claudins, such as claudin-2, increase the ion conductivity of TJs (Furuse et al., 2001; Amasheh et al., 2002), and the gene knockout (KO) of claudin-2 drastically improves the barrier function for ions in Madin-Darby canine kidney (MDCK) II cells (Tokuda and Furuse, 2015). Two other four-pass transmembrane proteins, occludin and tricellulin, regulate the complexity of the TJ strand network and increase the tightness of the epithelial barrier (Saito et al., 2021).
Although it was believed that TJs were static structures, their components and organization are dynamically reorganized over time (Yu et al., 2010; Stephenson et al., 2019). As epithelial cells are constantly rearranged, the structure of TJs must also be reorganized to resolve tension in the cell sheet without disturbing the paracellular barrier (Madara, 1990; Jinguji and Ishikawa, 1992; Smith and Braun, 2012; Higashi et al., 2016). Recent observations using a new live imaging barrier assay indicated that TJs undergo small leakages at points where tension seems to be increased, for example, at the perimeter of dividing cells and their neighbors in developing Xenopus embryos (Stephenson et al., 2019). These leaks are followed by mechanosensitive intracellular Ca2+ flashes and transient activation of Rho GTPase, known as Rho flares (Stephenson et al., 2019; Varadarajan et al., 2022). As a result, the actomyosin cytoskeleton is rearranged, and the TJ structure is reinstated. When these processes are inhibited, repeated leaks are observed at the same sites, indicating that Rho flares play an important role in maintaining barrier function (Stephenson et al., 2019; Varadarajan et al., 2022). Notably, the magnitude of the leak begins to decrease before the Rho flare starts (Stephenson et al., 2019), suggesting the existence of another mechanism that works to stop the leaks as a first line of defense. The observation that the leaks repeatedly stop and then restart at the same sites when Ca2+ influx or the Rho pathway is inhibited (Varadarajan et al., 2022) also supports this hypothesis. The repair mechanism may involve the reorganization and/or de novo assembly of TJ strands; however, the underlying molecular mechanism has not yet been elucidated.
To date, the mechanism by which claudin is polymerized into strands remains unclear. Exogenous expression of claudins induces the formation of TJ strand–like structures in non-epithelial cells that normally do not possess TJ strands, including L fibroblasts, Rat-1 cells, HEK293 cells, and Cos-7 cells (Furuse et al., 1998; Piontek et al., 2008; Nunes et al., 2006; Van Itallie et al., 2017). However, the expression of claudins alone is not sufficient for TJ strand formation in epithelial cells. For example, keratinocytes in the mouse skin form TJs only in the second layer of the stratum granulosum although cells in the granular, spinous, and basal layers express abundant amounts of claudin-1 (Kubo et al., 2009). In a study aiming to generate epithelial cells that harbor TJ strands made of a single claudin by exogenously expressing each of the 26 claudins in the epithelial-like SF7 cells, most of the claudins failed to form a linearly arranged structure (Yamazaki et al., 2011). TJ strand formation is also regulated locally at the subcellular level. Newly synthesized claudins are first transported to the basolateral membrane and then added to the TJ strands from the basal side (Van Itallie et al., 2019). The degree of localization to the TJs varies among claudins, and the basolateral membranes of most epithelial cells contain abundant claudins (Inai et al., 2007; Kiuchi-Saishin et al., 2002; Holmes et al., 2006; Tokuda and Furuse, 2015). The basolateral claudins are likely to be maintained in an unpolymerized state. Although it have been suggested that unpolymerized claudins regulate cell migration in cancer cells (Thuma and Zöller, 2013; Wang et al., 2018), their roles remained unclear in polarized epithelial cells.
Several lines of evidence suggest that TJ strand formation is regulated by proteolytic activity. Treatment with pronase (a mixture of proteinases) induces the formation of massive TJ strands on the plasma membranes of isolated pancreatic β cells (Orci et al., 1973), which do not otherwise have TJ strands (in’t Veld et al., 1984). A series of studies by Ben-Shaul’s group showed that an adenocarcinoma cell line, HT-29, forms TJ strands when treated with serine proteinases, such as trypsin (Polak-Charcon et al., 1978; Talmon et al., 1984; Cohen et al., 1985; Faff et al., 1987, 1988; Cohen et al., 1990). In MDCK cells, the application of trypsin to the basolateral surface of the cells induced the formation of aberrant TJ strands (Lynch et al., 1995). In a canine intestinal epithelial cell line, SCBN, treatment with trypsin or TMPRSS14 (also known as suppressor of tumorigenicity [ST14], matriptase, MT-SP1, or channel-activating protein 3) improves the barrier for ions (Ronaghan et al., 2016). TMPRSS14 has been implicated in the regulation of TJ barriers (List et al., 2002, 2009; Buzza et al., 2010; Wu et al., 2017). TMPRSS14 belongs to the membrane-anchored serine proteinase (MASP) family, which contains more than 20 serine proteinases (Szabo and Bugge, 2011). PRSS8 (also known as prostasin, serine protease 8, or channel-activating protein 1), another member of the MAPS family, has also been suggested to regulate TJ formation and function in the epidermis (Leyvraz et al., 2005) and in Caco-2 cells (Buzza et al., 2013). Although various proteinases are suggested to be involved in the TJ function, it has been unclear how proteolysis regulates TJ strand assembly and epithelial barrier function.
Recent reports showed that EpCAM (epithelial cell adhesion molecule; also known as tumor-associated calcium signal transducer 1, trophoblast cell surface antigen 1, or CD326) is also important for the regulation of TJ structure and function (Ladwein et al., 2005; Sivagnanam et al., 2008; Lei et al., 2012; Guerra et al., 2012; Wu et al., 2013; Kozan et al., 2015; Salomon et al., 2017). EpCAM is a single-transmembrane protein, which is expressed exclusively in epithelial cells (Litvinov et al., 1994). Interestingly, EpCAM undergoes proteolysis in its extracellular domain, and TMPRSS14 has been shown to cleave EpCAM (Wu et al., 2017). However, the mechanism by which EpCAM regulates TJ barrier function remains unknown.
In the present study, we explored the role of EpCAM in the structure and function of TJs, using cultured MDCK II cells as a model epithelium. We also established MASP family-KO cells and investigated how proteolytic activity contributes to the maintenance of barrier function.
Results
Trypsin induces the formation of claudin-based TJ strand and cleavage of EpCAM
Application of trypsin to the basolateral surface of MDCK cells induces the formation of aberrant TJ strand–like structures on basolateral membranes (Lynch et al., 1995). We examined whether these structures were claudin-based TJ strands. We used a claudin-2-KO MDCK II cell clone (hereby referred to as “control (Ctrl)” cells throughout this manuscript) to sensitively evaluate small changes in barrier function by transepithelial electric resistance (TER) measurements. In untreated Ctrl cells, claudin-7 was diffusely localized on the basolateral membranes, and only a faint signal was observed at the TJs (Fig. 1 A) as shown previously (Hou et al., 2006; Barth et al., 2018; Otani et al., 2019). Upon treatment with a low concentration of trypsin (25 µg/ml [wt/vol]) from the basal side, claudin-7 signal at TJs became apparent (Fig. 1 A, cyan arrowheads). Claudin-1, claudin-3, and claudin-4 also exhibited similar significant increases in TJ signal upon treatment with trypsin (Fig. S1, A–D). In addition to its presence at apical TJs, claudin-7 also formed intense foci on the basolateral membranes (Fig. 1 A), which were especially evident at the basal end of the basolateral membranes (Fig. 1 A, yellow arrowheads). Claudin-1 was localized similarly to claudin-7 at the newly formed punctate structures on the basolateral membrane (Fig. S1 A). However, the TJ plaque protein ZO-1 was not incorporated into the basolateral foci (Fig. 1 A). Trypsin application from the apical side increased the claudin signals at TJs and did not induce the formation of basolateral punctate pattern signals of claudin-1 or claudin-7 (Fig. S1, E and F). FFEM analysis confirmed that in the cells treated with trypsin from the basal side, aberrant TJ strands (yellow arrowheads) were formed on the basolateral membranes in addition to the apical TJ strand network (blue arrowheads; Fig. 1 B).

Trypsin induces the TJ strand formation and cleavage of EpCAM. (A) Trypsin-induced claudin-7 puncta formation on the basolateral membranes. Untreated Ctrl cells (upper panels) and Ctrl cells treated with 25 µg/ml trypsin (lower panels) were stained with rabbit anti–claudin-7 pAb (green) and rat anti–ZO-1 mAb (red) together with DAPI (blue). Cyan and yellow arrowheads in the side-view panels indicate TJs and basolateral punctate signals, respectively. Black arrowheads on the sides of top views indicate the positions of the side view. Scale bars, 10 µm. (B) FFEM of trypsin-treated Ctrl cells. Blue and yellow arrowheads indicate the TJ strands at the TJs in the apicolateral and basolateral regions, respectively. mv, microvilli. Scale bars, 1 µm. (C) Immunoblotting of Ctrl cells treated with various concentrations of trypsin using rabbit anti-EpCAM mAb and other antibodies. (D) Trypsin-induced claudin-1–based structure in the claudin-1-GFP–expressing A431 cells. Cells were stained for F-actin with phalloidin (red) and DAPI (blue) and the fluorescence of GFP (green) was observed. Arrowheads and arrows indicate faint and intense claudin-1 signals, respectively. Scale bar, 10 µm. (E) FFEM of the trypsin-treated A431 cells. Scale bar, 1 µm. (F) Immunoblotting of A431 cells treated with various concentrations of trypsin using mouse anti-EpCAM mAb and other antibodies. Source data are available for this figure: SourceData F1.
Trypsin induces the TJ strand formation and cleavage of EpCAM. (A) Trypsin-induced claudin-7 puncta formation on the basolateral membranes. Untreated Ctrl cells (upper panels) and Ctrl cells treated with 25 µg/ml trypsin (lower panels) were stained with rabbit anti–claudin-7 pAb (green) and rat anti–ZO-1 mAb (red) together with DAPI (blue). Cyan and yellow arrowheads in the side-view panels indicate TJs and basolateral punctate signals, respectively. Black arrowheads on the sides of top views indicate the positions of the side view. Scale bars, 10 µm. (B) FFEM of trypsin-treated Ctrl cells. Blue and yellow arrowheads indicate the TJ strands at the TJs in the apicolateral and basolateral regions, respectively. mv, microvilli. Scale bars, 1 µm. (C) Immunoblotting of Ctrl cells treated with various concentrations of trypsin using rabbit anti-EpCAM mAb and other antibodies. (D) Trypsin-induced claudin-1–based structure in the claudin-1-GFP–expressing A431 cells. Cells were stained for F-actin with phalloidin (red) and DAPI (blue) and the fluorescence of GFP (green) was observed. Arrowheads and arrows indicate faint and intense claudin-1 signals, respectively. Scale bar, 10 µm. (E) FFEM of the trypsin-treated A431 cells. Scale bar, 1 µm. (F) Immunoblotting of A431 cells treated with various concentrations of trypsin using mouse anti-EpCAM mAb and other antibodies. Source data are available for this figure: SourceData F1.
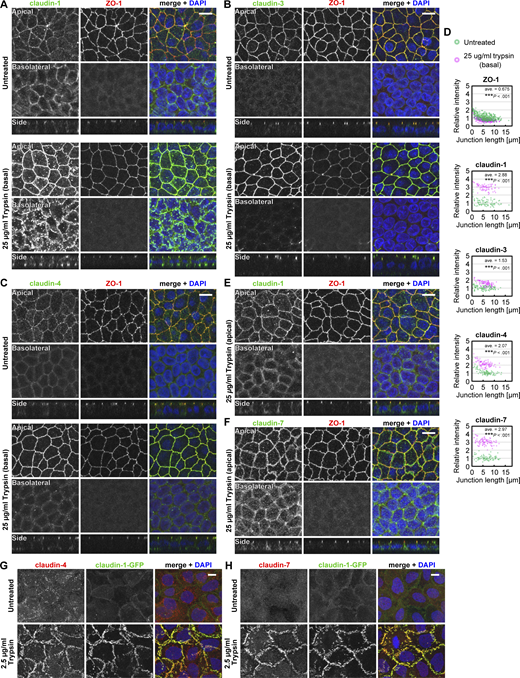
Localization patterns of claudins in the Ctrl cells and A431 cells treated with trypsin. (A–C, E, and F) Ctrl MDCK II cells grown on the filter were treated with 25 µg/ml (wt/vol) trypsin from the basal side (A–C) or the apical side (E and F) for 15 min, and were stained for claudin-1 (A and E), claudin-3 (B), claudin-4 (C), or claudin-7 (F; green) together with ZO-1 (red) and DAPI (blue). Stacked images of the apical (top) or basolateral (middle) regions and side views (bottom) are shown. Scale bars, 10 µm. (D) Quantification of fluorescence intensity of ZO-1 and claudins at cell–cell junctions. The intensity at each cell–cell junction in the trypsin-treated cells (magenta) was normalized to the averaged intensity in the untreated cells (green) and plotted against the length of the cell–cell junctions. ***, P <0.001 (weighted two-tailed Student’s t test). (G and H) Claudin-1-GFP (green)–expressing A431 cells on coverslips were treated with 2.5 µg/ml trypsin and stained for claudin-4 (G) or claudin-7 (H; red) and DAPI (blue). Stacked images are shown. Scale bars, 10 µm. Source data are available for this figure: SourceData FS1.

Localization patterns of claudins in the Ctrl cells and A431 cells treated with trypsin. (A–C, E, and F) Ctrl MDCK II cells grown on the filter were treated with 25 µg/ml (wt/vol) trypsin from the basal side (A–C) or the apical side (E and F) for 15 min, and were stained for claudin-1 (A and E), claudin-3 (B), claudin-4 (C), or claudin-7 (F; green) together with ZO-1 (red) and DAPI (blue). Stacked images of the apical (top) or basolateral (middle) regions and side views (bottom) are shown. Scale bars, 10 µm. (D) Quantification of fluorescence intensity of ZO-1 and claudins at cell–cell junctions. The intensity at each cell–cell junction in the trypsin-treated cells (magenta) was normalized to the averaged intensity in the untreated cells (green) and plotted against the length of the cell–cell junctions. ***, P <0.001 (weighted two-tailed Student’s t test). (G and H) Claudin-1-GFP (green)–expressing A431 cells on coverslips were treated with 2.5 µg/ml trypsin and stained for claudin-4 (G) or claudin-7 (H; red) and DAPI (blue). Stacked images are shown. Scale bars, 10 µm. Source data are available for this figure: SourceData FS1.
Immunoblotting analysis showed that claudins did not undergo proteolysis upon trypsin treatment (Fig. 1 C) although they exhibited altered subcellular localization. Occludin exhibited a slight decrease in the band intensity at 250 µg/ml trypsin (Fig. 1 C), which may not explain the altered localization of claudins because the localization patterns of claudin-1 and claudin-7 were unchanged in occludin-KO MDCK II cells (Saito et al., 2021). Other major TJ and adherens junction (AJ) membrane proteins, except occludin, did not change their band patterns (Fig. 1 C). Since EpCAM has been implicated in the regulation of TJ structure and function (Lei et al., 2012; Wu et al., 2013; Salomon et al., 2017; Wu et al., 2017) and it directly interacts with claudin-7 (Ladwein et al., 2005; Barth et al., 2018), we also examined whether EpCAM was cleaved by trypsin. EpCAM appeared as a 40-kD band (corresponding to the full-length protein), a faint 32-kD band, and a weak 29-kD band in untreated Ctrl MDCK II cells (Fig. 1 C). Trypsin treatment resulted in an increased intensity of the 32-kD band and a concomitant decrease in the 40-kD band in a concentration-dependent manner, suggesting that EpCAM is cleaved by trypsin (Fig. 1 C). The intensity of the 29-kD band was unaltered.
We also tested this phenomenon using the human epidermoid carcinoma cell line, A431, which expresses claudin-1, claudin-4, and claudin-7, but does not have TJs. In A431 cells stably expressing GFP-tagged claudin-1, claudin-1-GFP was diffusely distributed on the plasma membrane (Fig. 1 D). The application of trypsin induced the formation of bright concentrated claudin-1-GFP signal at the cell–cell interfaces, even at very low concentration (250 ng/ml; Fig. 1 D), which is ten-thousandth of the concentration of trypsin normally used for detaching adherent cells from the dish surface. The claudin-1-GFP signal became more evident when cells were treated with high concentrations of trypsin (Fig. 1 D). Claudin-4 and claudin-7 were also concentrated at the cell–cell interfaces and colocalized with claudin-1-GFP in trypsin-treated A431 cells (Fig. S1, G and H). FFEM analysis showed that massive TJ strands formed on the plasma membranes of trypsin-treated A431 cells (Fig. 1 E). The untreated A431 cells had only a 40-kD band of EpCAM and trypsin cleaved it to produce the 32-kD band in a concentration-dependent manner (Fig. 1 F). Occludin exhibited a decreased band intensity at 25 µg/ml and was almost completely degraded at 250 µg/ml trypsin. Angulin-1 (also known as lipolysis-stimulated lipoprotein receptor) also showed a decreased band intensity at 250 µg/ml trypsin.
These results indicate that low concentration of trypsin induces the formation of claudin-based TJ strands and the proteolysis of EpCAM in both polarized epithelial MDCK II and unpolarized A431 cells.
EpCAM is localized at the basolateral membranes of epithelial cells and involved in the regulation of the epithelial barrier
To examine the subcellular localization of EpCAM in epithelial tissues, we performed immunostaining for EpCAM in the colon and kidney of mice. EpCAM was exclusively localized at the basolateral membranes of epithelial cells in these tissues (Fig. 2 A). Cleaved EpCAM was detected in the lysates of the small and large intestines (Fig. 2 B). To examine the function(s) of EpCAM, we generated three EpCAM-KO cell clones using CRISPR/Cas9-based genome editing (Fig. 2, C–E; and Fig. S2, A–C). EpCAM was also localized at the basolateral membrane in Ctrl MDCK II cells, and the signal was abolished in EpCAM-KO cells (Fig. 2 D). Immunostaining signals for ZO-1 and claudins at tricellular contacts appeared elongated in the basal direction in EpCAM-KO cells (Fig. 2, D and E; and Fig. S2, D–F), as described previously using EpCAM-knockdown Caco-2 cells (Salomon et al., 2017). Interestingly, claudin-7 was also abolished from the basolateral membranes and became localized solely at TJs (Fig. 2 E), indicating that EpCAM is required for the basolateral localization of claudin-7, which was suggested previously using EpCAM-knockdown cells (Wu et al., 2013; Barth et al., 2018). We then evaluated the barrier function of EpCAM-KO cells using TER measurements (Fig. 2 F). On culture days 2–6, the TER values of EpCAM-KO cell clones were lower than those of the Ctrl cells, but became comparable on day 7, suggesting that EpCAM is required for the maintenance of barrier in immature cell sheets rather than in established ones. We examined the morphology of the TJ strand network on day 6 using FFEM. The TJ strand network in EpCAM-KO cells appeared to be more complex than that in Ctrl cells (Fig. 2 G). Quantification confirmed that the TJ strand network of EpCAM-KO cells contained a significantly increased number of branching points and horizontal TJ strands compared with those of the Ctrl cells (Fig. 2, H and I). We performed a computer simulation of the ionic permeability using a mathematical model of a simplified TJ strand network (Saito et al., 2021). The TJ model simulation, which is based on the branching frequency and horizontal TJ strand number, predicted that the permeability barrier of EpCAM-KO cells would actually be tighter than that of the Ctrl cells (Fig. 2 J)—the opposite of our experimental results in Fig. 2 F.

EpCAM is involved in the barrier function of epithelial cells. (A) Immunostaining of EpCAM in epithelial tissues. Mouse colon and kidney were stained with rat anti-EpCAM mAb (red) and rabbit anti–ZO-1 pAb (green) together with DAPI (blue). Enlarged image of the region in the kidney (yellow dotted rectangle) was shown (right panels). Asterisks indicate lumens. Scale bars, 10 µm. (B) Immunoblotting of epithelial tissue lysates. Lysates from mouse lung, ileum (small intestine), colon, and kidney were analyzed using rabbit anti-EpCAM and mouse anti–β-actin mAbs. Full-length (black arrowhead) and cleaved (gray arrowhead) bands of EpCAM were detected. β-actin served as an internal control to assure that similar amounts of proteins were applied. (C) Immunoblotting of the Ctrl and EpCAM-KO MDCK II cells. HEK293T cells expressing EpCAM-FLAG served as a positive control. Cell lysates were labeled with mouse anti-EpCAM and mouse anti–β-actin mAbs. (D and E) Immunostaining of the Ctrl (upper panels) and EpCAM-KO (lower panels) MDCK II cells. Cells were stained with mouse anti-EpCAM mAb (D) or rabbit anti–claudin-7 pAb (E; red) together with rat anti–ZO-1 mAb (green) and DAPI (blue). Gray arrowheads on the side of the top views indicate the location where the side-view section was made, and the black arrows and arrowheads below the side-view panels indicate the locations of the bicellular and tricellular TJs, respectively. Scale bar, 10 µm. (F) TER measurement of EpCAM-KO cells. n = 13 (Ctrl and KO#1) and 10 (KO#2 and KO#3). The values of KO cells were compared with those of Ctrl cells at each time point using two-tailed Welch’s t test with Bonferroni’s correction (ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; actual P values are shown for 0.05 < P < 0.001). (G) FFEM analysis of EpCAM-KO cells. Scale bar, 200 nm. (H) Quantification of the frequency of TJ strand branching points in the EpCAM-KO cells. Total length of TJ strands examined was 122 µm (Ctrl) and 156 µm (EpCAM-KO#1). Error bars indicate 95% confidence intervals. ***, P <0.001 (exact Poisson test). (I) Distribution of horizontal TJ strand number in the EpCAM-KO cells. Blue lines indicate mean ± SD. n = 259 (Ctrl) and 302 (EpCAM-KO#1). ***, P < 0.001 (two-tailed Welch’s t test). (J) Predicted TER values using simplified TJ strand network models based on the quantification data of TJ strand network complexity. Source data are available for this figure: SourceData F2.
EpCAM is involved in the barrier function of epithelial cells. (A) Immunostaining of EpCAM in epithelial tissues. Mouse colon and kidney were stained with rat anti-EpCAM mAb (red) and rabbit anti–ZO-1 pAb (green) together with DAPI (blue). Enlarged image of the region in the kidney (yellow dotted rectangle) was shown (right panels). Asterisks indicate lumens. Scale bars, 10 µm. (B) Immunoblotting of epithelial tissue lysates. Lysates from mouse lung, ileum (small intestine), colon, and kidney were analyzed using rabbit anti-EpCAM and mouse anti–β-actin mAbs. Full-length (black arrowhead) and cleaved (gray arrowhead) bands of EpCAM were detected. β-actin served as an internal control to assure that similar amounts of proteins were applied. (C) Immunoblotting of the Ctrl and EpCAM-KO MDCK II cells. HEK293T cells expressing EpCAM-FLAG served as a positive control. Cell lysates were labeled with mouse anti-EpCAM and mouse anti–β-actin mAbs. (D and E) Immunostaining of the Ctrl (upper panels) and EpCAM-KO (lower panels) MDCK II cells. Cells were stained with mouse anti-EpCAM mAb (D) or rabbit anti–claudin-7 pAb (E; red) together with rat anti–ZO-1 mAb (green) and DAPI (blue). Gray arrowheads on the side of the top views indicate the location where the side-view section was made, and the black arrows and arrowheads below the side-view panels indicate the locations of the bicellular and tricellular TJs, respectively. Scale bar, 10 µm. (F) TER measurement of EpCAM-KO cells. n = 13 (Ctrl and KO#1) and 10 (KO#2 and KO#3). The values of KO cells were compared with those of Ctrl cells at each time point using two-tailed Welch’s t test with Bonferroni’s correction (ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; actual P values are shown for 0.05 < P < 0.001). (G) FFEM analysis of EpCAM-KO cells. Scale bar, 200 nm. (H) Quantification of the frequency of TJ strand branching points in the EpCAM-KO cells. Total length of TJ strands examined was 122 µm (Ctrl) and 156 µm (EpCAM-KO#1). Error bars indicate 95% confidence intervals. ***, P <0.001 (exact Poisson test). (I) Distribution of horizontal TJ strand number in the EpCAM-KO cells. Blue lines indicate mean ± SD. n = 259 (Ctrl) and 302 (EpCAM-KO#1). ***, P < 0.001 (two-tailed Welch’s t test). (J) Predicted TER values using simplified TJ strand network models based on the quantification data of TJ strand network complexity. Source data are available for this figure: SourceData F2.
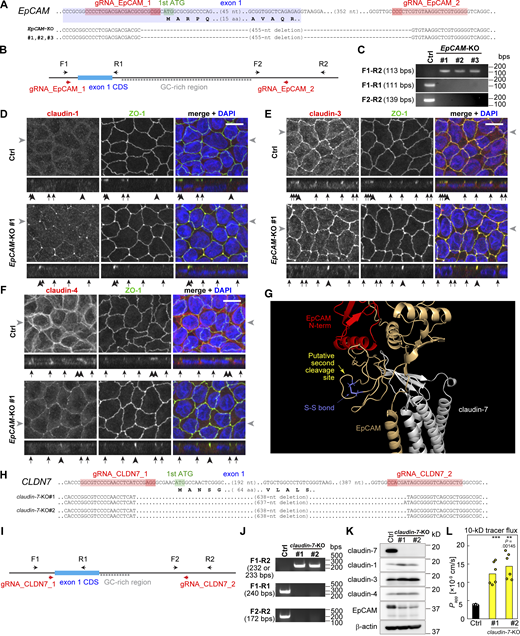
KO of EpCAM and claudin-7 genes. (A) Genomic structure and gene-KO strategies of EpCAM. gRNA sequences used for genome editing (red) are shown. The deleted region of the EpCAM gene contains the first exon with a start codon. (B) Screening strategy of EpCAM-KO cells. In the WT cells, the F1-R2 fragment is not amplified because of the high guanine-cytosine content (GC-rich) region. (C) Genomic PCR of Ctrl and EpCAM-KO clones. (D–F) Immunostaining of the Ctrl cells (top panels) and EpCAM-KO cells (bottom panels). Cells were stained with anti–claudin-1 (D), anti–claudin-3 (E), or anti–claudin-4 (F) (red) together with anti–ZO-1 (green) and DAPI (blue). Gray arrowheads on the side of the top views indicate the location where the side-view section was made, and the black arrows and arrowheads below the side-view panels indicate the locations of the bicellular and tricellular junctions, respectively. Scale bars, 10 µm. (G) Enlarged image of the predicted structure of EpCAM–claudin-7 complex. Yellow arrow indicates the putative site of second cleavage, which corresponds to the band in Fig. 4, A and B, indicated with blue arrowheads. (H) Genomic structure and gene-KO strategy of claudin-7 (CLDN7). (I) Screening strategy of claudin-7-KO cells. (J) Genomic PCR of the Ctrl and claudin-7-KO cells. (K) Immunoblotting of claudin-7-KO cells. (L) Tracer flux assay of the claudin-7-KO cells using 10-kD FITC-dextran. N = 6. Statistical significance compared with the Ctrl cells was evaluated by two-tailed Welch’s t test with Bonferroni’s correction (**, P < 0.01; ***, P < 0.001). Source data are available for this figure: SourceData FS2..

KO of EpCAM and claudin-7 genes. (A) Genomic structure and gene-KO strategies of EpCAM. gRNA sequences used for genome editing (red) are shown. The deleted region of the EpCAM gene contains the first exon with a start codon. (B) Screening strategy of EpCAM-KO cells. In the WT cells, the F1-R2 fragment is not amplified because of the high guanine-cytosine content (GC-rich) region. (C) Genomic PCR of Ctrl and EpCAM-KO clones. (D–F) Immunostaining of the Ctrl cells (top panels) and EpCAM-KO cells (bottom panels). Cells were stained with anti–claudin-1 (D), anti–claudin-3 (E), or anti–claudin-4 (F) (red) together with anti–ZO-1 (green) and DAPI (blue). Gray arrowheads on the side of the top views indicate the location where the side-view section was made, and the black arrows and arrowheads below the side-view panels indicate the locations of the bicellular and tricellular junctions, respectively. Scale bars, 10 µm. (G) Enlarged image of the predicted structure of EpCAM–claudin-7 complex. Yellow arrow indicates the putative site of second cleavage, which corresponds to the band in Fig. 4, A and B, indicated with blue arrowheads. (H) Genomic structure and gene-KO strategy of claudin-7 (CLDN7). (I) Screening strategy of claudin-7-KO cells. (J) Genomic PCR of the Ctrl and claudin-7-KO cells. (K) Immunoblotting of claudin-7-KO cells. (L) Tracer flux assay of the claudin-7-KO cells using 10-kD FITC-dextran. N = 6. Statistical significance compared with the Ctrl cells was evaluated by two-tailed Welch’s t test with Bonferroni’s correction (**, P < 0.01; ***, P < 0.001). Source data are available for this figure: SourceData FS2..
These data and analyses indicate that EpCAM-KO cells have delayed development of barrier function and require more complex morphology of the TJ strand network than Ctrl cells for full barrier development, suggesting that EpCAM is involved in the process of either barrier establishment or maintenance.
Claudin-7 is complexed with EpCAM and released upon proteolytic cleavage of EpCAM
To identify EpCAM-interacting protein(s) and gain insights into its functions, we established a cell clone stably expressing carboxy-terminally FLAG-tagged EpCAM (EpCAM-FLAG) using EpCAM-KO cells as a parental clone and performed immunoprecipitation using an anti-FLAG antibody. Silver staining showed that only an 18-kD band (arrow) was specifically co-immunoprecipitated with EpCAM-FLAG (arrowhead; Fig. 3 A, left panel). Since claudin-7 has been reported to interact with EpCAM (Ladwein et al., 2005) and most classical claudins migrate at 18-kD in our electrophoresis setting, we speculated that this band is claudin-7. Immunoblotting analysis confirmed that claudin-7 co-precipitated with EpCAM-FLAG (Fig. 3 A, right panel). Claudin-1 and claudin-3 were also detected, although the EpCAM-bound population of claudin-7 was much more abundant than those of claudin-1 and claudin-3. Claudin-4 was not detected in the immunoprecipitated product. The total amount of claudins was decreased in EpCAM-KO cells, while claudin-3, claudin-4, and claudin-7 exhibited an increased amount of a presumed degradation product at 13 kD (asterisks; Fig. 3 A, right panels), suggesting that EpCAM contributes to the stability of claudins. We noted that the bands for claudin-1 and claudin-3 that co-immunoprecipitated with EpCAM-FLAG appeared to be slightly lower than those in the input for unknown reasons. To confirm the interaction between claudins and EpCAM, we co-transfected HEK293T cells with EpCAM-FLAG and GFP-tagged claudin-1, -2, -3, -4, and -7, and performed immunoprecipitation using NP-40. Among the five claudins, only claudin-7-GFP was specifically co-immunoprecipitated with EpCAM-FLAG (Fig. 3 B). Previous reports have shown that coimmunoprecipitation of membrane proteins and their partners are dependent on the detergents in the lysis buffer (Izumi et al., 2016; Charrin et al., 2009; Haining et al., 2012; Hemler, 2005; Yáñez-Mó et al., 2009). Thus, we lysed the cells with a buffer containing Brij97 and performed immunoprecipitation. In Brij97 lysate, claudin-1 as well as claudin-7 was co-immunoprecipitated with EpCAM-FLAG (Fig. 3 B). Claudin-2, claudin-3, and claudin-4 were also weakly detected (Fig. 3 B), suggesting that EpCAM can interact with these claudins. When claudin-7-mCherry was co-expressed with these claudins, the interaction with EpCAM was abolished (Fig. 3 C), suggesting that these claudins compete for the binding to EpCAM with claudin-7. Next, to assess whether claudin-7 is released from EpCAM upon trypsin treatment, we treated EpCAM-FLAG–expressing MDCK II cells with trypsin from the basolateral side. Trypsin induced the cleavage of EpCAM-FLAG (Fig. 3 D). Then, the cell lysates were separated into Brij97-soluble and Brij97-insoluble fractions. Claudin-7 was co-immunoprecipitated with EpCAM-FLAG from the Brij97-soluble fraction. Trypsin treatment increased the amount of Brij97-insoluble claudin-7 in a concentration-dependent manner, whereas EpCAM-FLAG remained completely Brij97-soluble regardless of its cleavage status (Fig. 3 D), indicating that claudin-7 was dissociated from the cleaved EpCAM. It is generally considered that adhesion molecules become detergent-insoluble when incorporated into junctional structures (Hirano et al., 1987; Sakakibara et al., 1997; Takahashi et al., 2009). Thus, Brij97-insoluble claudin-7, which was released from EpCAM, is likely to be polymerized and incorporated into TJ strands. Claudin-1 and claudin-3, but not claudin-4, were also co-immunoprecipitated with EpCAM from the untreated cells. These claudins became insoluble to Brij97 upon trypsin treatment. These data suggest that cleaved EpCAM releases complexed claudin-7 and then released claudin-7 is incorporated into TJ strands.

EpCAM restrains claudin-7 on the basolateral membranes. (A) Pull-down assay of EpCAM-FLAG. Immunoprecipitates (IPs) from EpCAM-KO MDCK II cells expressing EpCAM-FLAG were compared with those from EpCAM-KO cells by silver staining (left) and immunoblotting using HRP-linked anti-FLAG (M2), mouse anti-EpCAM mAb, and other antibodies (right). Arrowhead and arrow in the left panel indicate EpCAM-FLAG and claudin(s), respectively. Asterisks in the right panels indicate degradation products of claudins. (B) Immunoprecipitation of EpCAM-FLAG from HEK293T cells expressing EpCAM-FLAG and claudin-GFP using 1% NP-40 (upper panels) or 1% Brij97 (lower panels). (C) Immunoprecipitation of EpCAM-FLAG from HEK293T cells expressing EpCAM-FLAG and claudin-GFP with or without claudin-7-mCherry using 1% Brij97. Asterisk indicates the heavy chain of IgG. (D) Fractionation of EpCAM-FLAG–expressing EpCAM-KO MDCK II cells treated with various concentrations of trypsin into Brij97-soluble and insoluble fractions and immunoprecipitation of EpCAM-FLAG from the soluble fraction. (E) Predicted structure of the EpCAM–claudin-7 complex. Side views of the complex from three directions are shown. Red and orange indicate the regions before and after the putative cleavage site of EpCAM, respectively. Light blue, salmon, yellow, and pale green indicate the first extracellular loop, second extracellular loop, transmembrane region, and intracellular region of claudin-7, respectively. Out, extracellular space. In, cytoplasmic space. Mem, plasma membrane. (F) Predicted structure of cleaved EpCAM and claudin-7. Black arrow indicates the exposed first extracellular loop of claudin-7. (G) The model of paired claudin-15 molecules (white and teal), superimposed with full-length (red and orange) and cleaved (orange) EpCAM. Black arrow indicates that orange EpCAM molecule and teal claudin molecule cause steric hindrance. Source data are available for this figure: SourceData F3.
EpCAM restrains claudin-7 on the basolateral membranes. (A) Pull-down assay of EpCAM-FLAG. Immunoprecipitates (IPs) from EpCAM-KO MDCK II cells expressing EpCAM-FLAG were compared with those from EpCAM-KO cells by silver staining (left) and immunoblotting using HRP-linked anti-FLAG (M2), mouse anti-EpCAM mAb, and other antibodies (right). Arrowhead and arrow in the left panel indicate EpCAM-FLAG and claudin(s), respectively. Asterisks in the right panels indicate degradation products of claudins. (B) Immunoprecipitation of EpCAM-FLAG from HEK293T cells expressing EpCAM-FLAG and claudin-GFP using 1% NP-40 (upper panels) or 1% Brij97 (lower panels). (C) Immunoprecipitation of EpCAM-FLAG from HEK293T cells expressing EpCAM-FLAG and claudin-GFP with or without claudin-7-mCherry using 1% Brij97. Asterisk indicates the heavy chain of IgG. (D) Fractionation of EpCAM-FLAG–expressing EpCAM-KO MDCK II cells treated with various concentrations of trypsin into Brij97-soluble and insoluble fractions and immunoprecipitation of EpCAM-FLAG from the soluble fraction. (E) Predicted structure of the EpCAM–claudin-7 complex. Side views of the complex from three directions are shown. Red and orange indicate the regions before and after the putative cleavage site of EpCAM, respectively. Light blue, salmon, yellow, and pale green indicate the first extracellular loop, second extracellular loop, transmembrane region, and intracellular region of claudin-7, respectively. Out, extracellular space. In, cytoplasmic space. Mem, plasma membrane. (F) Predicted structure of cleaved EpCAM and claudin-7. Black arrow indicates the exposed first extracellular loop of claudin-7. (G) The model of paired claudin-15 molecules (white and teal), superimposed with full-length (red and orange) and cleaved (orange) EpCAM. Black arrow indicates that orange EpCAM molecule and teal claudin molecule cause steric hindrance. Source data are available for this figure: SourceData F3.
To understand how the cleavage of EpCAM results in the release of claudin-7, we predicted the structure of the EpCAM–claudin-7 complex using AlphaFold2 (Jumper et al., 2021; Fig. 3 E). The extracellular domain of EpCAM (orange and red) interacted with and restrained the first extracellular loop of claudin-7 (light blue) in the predicted complex structure (Fig. 3 E), although the predicted local distance difference test score of this region was not high (44.42–74.58). A recent study determined the cleavage site of EpCAM at Arg80-Arg81 (Wu et al., 2017). Based on this information, we created a predicted model of the cleaved EpCAM–claudin-7 complex (Fig. 3 F). In the cleaved EpCAM model, the extracellular domain of EpCAM appeared raised by 15° compared with the full-length model, and the first extracellular loop of claudin-7 was no longer restrained (Fig. 3 F, arrow). Although the cleaved region (red) does not directly interact with claudin-7 in the predicted structure of the full-length EpCAM–claudin-7 complex (Fig. 3 E), it may affect the structure of the rest of the extracellular domain of EpCAM and stabilize the binding of EpCAM to the first extracellular loop of claudin-7. The orientation model of claudin molecules in polymerized TJ strands has been proposed using the crystal structure of claudin-15 (Suzuki model; Suzuki et al., 2015). Using this model, we examined whether EpCAM interferes with the transcellular interactions of claudins (Fig. 3 G). Although full-length EpCAM (orange and red) was predicted to cause steric hindrance with the trans-interacting claudin molecule (green; Fig. 3 G, arrow), cleaved EpCAM did not interfere with this interaction, suggesting that the cleavage of EpCAM releases claudin-7 and makes it available for polymerization.
Together, our results and the predicted structures suggest that EpCAM sequesters claudin-7 at the basolateral membrane, maintaining claudin-7 in a polymerization-competent pool, so that it can incorporate into apical TJ strands upon release from EpCAM.
Membrane-anchored serine proteinases cleave EpCAM
Our data suggested that the EpCAM–claudin-7 complex is maintained at the basolateral membrane and the cleavage of EpCAM by trypsin releases claudin-7, which then is incorporated into TJ structure. We hypothesized that some endogenous proteinase in the extracellular fluids or on the cell membrane is responsible for the cleavage of EpCAM and may contribute to the regulation of epithelial barrier.
To identify the proteinase(s) responsible for the proteolysis of EpCAM, we screened for proteinase inhibitors. Treatment of Ctrl MDCK II cells with serine proteinase inhibitors (aprotinin, benzamide, and leupeptin) decreased the intensity of the cleaved band of EpCAM at 32 kD, whereas treatment with an aspartate proteinase inhibitor (pepstatin A) had no effect (Fig. 4 A). The cysteine proteinase inhibitor E-64 increased the intensity of the 32-kD band, probably because it delayed the degradation of the cleaved band in lysosomes by inhibiting cysteine cathepsins, such as cathepsin B and cathepsin L (Turk et al., 2012). Camostat, a serine proteinase inhibitor specific for the MASP family, also decreased the intensity of the cleaved band (gray arrowheads in Fig. 4 A), suggesting that MASPs are responsible for the cleavage of EpCAM (Fig. 4 A). In cell lysates treated with leupeptin or camostat, the band at 29 kD (blue arrowheads in Fig. 4 A) became evident. We deduced that the cleavage site of this 29-kD band was located between Cys99 and Cys110. Since these cysteines form an S-S bond, cleavage between them does not appear to affect the overall structure of the EpCAM–claudin complex in the AlphaFold2-predicted model (Fig. S2 G). Thus, we focused on the 32-kD band in this study. Camostat decreased the 32-kD band intensity in a concentration-dependent manner (Fig. 4 B).

MASPs cleave EpCAM in MDCK II cells and increase claudin-1 at cell–cell boundaries in A431 cells. (A) Effects of protease inhibitors on the cleavage of EpCAM. Immunoblotting of Ctrl cell lysates treated with 200 nM of aprotinin, 3 mM of benzamide, 100 µM of leupeptin (serine protease inhibitors; Ser-PI), 20 µM of E-64 (cysteine protease inhibitor; Cys-PI), 20 µM of pepstatin A (aspartate protease inhibitor; Asp-PI), 10 µM of camostat (MASP inhibitor; MASP inh.), or corresponding vehicles with rabbit anti-EpCAM mAb. Blue arrowhead indicates the 29-kD band of the new degradation product. (B) Effects of camostat treatment for 16 h. Band intensities were quantified and relative mean intensities (black) and individual measurements (n = 4; gray) are shown. ***, P < 0.001 (two-tailed Welch’s t test). (C) Effects of camostat on TER of Ctrl cells. ns, P > 0.05; **, P < 0.01 (two-tailed paired t test). (D) Effects of camostat on TER of EpCAM-KO cells. **, P < 0.01; ***, P < 0.001 (two-tailed paired t test). (E) RT-PCR of MASP genes using the cDNA library of Ctrl cells. Genome DNA and reaction without reverse transcriptase (RT−) served as positive and negative controls, respectively. (F) Claudin-1-GFP (green) accumulation at cell–cell boundaries of A431 cells transiently expressing MASPs labeled with nuclear mCherry (red; asterisks). Cells were stained with DAPI (blue). Scale bar, 10 µm.
MASPs cleave EpCAM in MDCK II cells and increase claudin-1 at cell–cell boundaries in A431 cells. (A) Effects of protease inhibitors on the cleavage of EpCAM. Immunoblotting of Ctrl cell lysates treated with 200 nM of aprotinin, 3 mM of benzamide, 100 µM of leupeptin (serine protease inhibitors; Ser-PI), 20 µM of E-64 (cysteine protease inhibitor; Cys-PI), 20 µM of pepstatin A (aspartate protease inhibitor; Asp-PI), 10 µM of camostat (MASP inhibitor; MASP inh.), or corresponding vehicles with rabbit anti-EpCAM mAb. Blue arrowhead indicates the 29-kD band of the new degradation product. (B) Effects of camostat treatment for 16 h. Band intensities were quantified and relative mean intensities (black) and individual measurements (n = 4; gray) are shown. ***, P < 0.001 (two-tailed Welch’s t test). (C) Effects of camostat on TER of Ctrl cells. ns, P > 0.05; **, P < 0.01 (two-tailed paired t test). (D) Effects of camostat on TER of EpCAM-KO cells. **, P < 0.01; ***, P < 0.001 (two-tailed paired t test). (E) RT-PCR of MASP genes using the cDNA library of Ctrl cells. Genome DNA and reaction without reverse transcriptase (RT−) served as positive and negative controls, respectively. (F) Claudin-1-GFP (green) accumulation at cell–cell boundaries of A431 cells transiently expressing MASPs labeled with nuclear mCherry (red; asterisks). Cells were stained with DAPI (blue). Scale bar, 10 µm.
At the same concentration range, camostat decreased the TER values of established Ctrl MDCK II cell sheets, suggesting that MASPs are also involved in the maintenance of the epithelial barrier (Fig. 4 C). To test whether the effect of MASPs on barrier function was dependent on EpCAM, we treated EpCAM-KO cells with camostat. The reduction in TER values of EpCAM-KO cells by camostat treatment was slight, albeit significant (Fig. 4 D), indicating that the effect of MASPs on barrier function was largely mediated by EpCAM.
MASPs are a large family of proteins (Szabo and Bugge, 2011). To identify the MASP responsible for EpCAM cleavage and barrier function, we evaluated the expression levels of MASPs in MDCK II cells (Fig. 4 E). Semi-quantitative RT-PCR revealed that TMPRSS1 (TP1; also known as hepsin and Hpn), TMPRSS4 (TP4; also known as MT-SP2 and channel-activating protein 2), TMRPSS14 (TP14), and PRSS8 (P8) were the most abundantly expressed MASPs in MDCK II cells (Fig. 4 E). Exogenous expression of TP1, TP14, and P8 in claudin-1-GFP–expressing A431 cells induced bright foci of claudin-1-GFP at cell–cell interfaces, suggesting that these proteinases can induce TJ strand formation (Fig. 4 F). Induction of this structure was not evident in TP4-expressing cells (Fig. 4 F).
These data suggest that MASP family members can cleave EpCAM and may be involved in the regulation of epithelial barrier function.
MASPs cleave EpCAM and contribute to barrier development in epithelial cells
To further examine whether MASPs regulate epithelial barrier function, we established TP1/TP4/TP14/P8-quadruple-KO (MASP-qKO) cell clones by sequentially knocking out each of the four MASPs using the CRISPR/Cas9 system (Fig. 5, A and B; Fig. S3; and Fig. S4 A). In MASP-qKO cells, EpCAM cleavage was drastically reduced (Fig. 5 B), consistent with the results of MASP inhibitor treatment (Fig. 4, A and B). The cleaved band at 32 kD was still evident in the MASP-single KO (sKO) cells and some of the MASP-double KO (dKO) cells, but was almost diminished in MASP-triple KO (tKO) cells (Fig. S4, B–D).

MASPs regulate the epithelial barrier function. (A) RT-PCR of MASP-qKO cells. Reaction without reverse transcriptase (RT−) served as a negative control. (B) Immunoblotting of MASP-qKO cells using rabbit anti-EpCAM mAb and other antibodies. Black and gray arrowheads indicate full-length and cleaved EpCAM, respectively. Arrows and asterisks indicate specific and non-specific bands, respectively. (C and D) Staining of MASP-qKO cells using mouse anti-EPCAM mAb (C) or rabbit anti–claudin-7 pAb (D; red) together with rat anti–ZO-1 mAb (green) and DAPI (blue). Scale bars, 10 µm. (E) FFEM of MASP-qKO cells. Scale bar, 200 nm. (F) Quantification of the frequency of TJ strand branching points in the MASP-qKO cells. Ctrl data is the same one used in Fig. 2 H. Total length of TJ strands examined was 111 µm. Error bars indicate 95% confidence intervals. *, P < 0.05 (exact Poisson test). (G) Distribution of horizontal TJ strand number in the MASP-qKO cells. Blue lines indicate mean ± SD. Ctrl data is the same one used in Fig. 2 I. n = 400 (MASP-qKO#1). ***, P < 0.001 (two-tailed Welch’s t test). (H) Predicted TER values using simplified TJ strand network models based on the quantification data of TJ strand network complexity in the MASP-qKO cells. (I) TER measurements of MASP-qKO cells. n = 5. ***, P < 0.001 (two-tailed Welch’s t test with Bonferroni’s correction). (J) Effects of trypsin treatment form the apical side of the MASP-qKO cells and EpCAM-KO cells. n = 5. *, P < 0.05; ***, P < 0.001 (two-tailed paired t test). (K) FFEM images of the Ctrl cells (top) and MASP-qKO cells (bottom) treated with trypsin from the apical side. Scale bars, 200 nm. Source data are available for this figure: SourceData F5.
MASPs regulate the epithelial barrier function. (A) RT-PCR of MASP-qKO cells. Reaction without reverse transcriptase (RT−) served as a negative control. (B) Immunoblotting of MASP-qKO cells using rabbit anti-EpCAM mAb and other antibodies. Black and gray arrowheads indicate full-length and cleaved EpCAM, respectively. Arrows and asterisks indicate specific and non-specific bands, respectively. (C and D) Staining of MASP-qKO cells using mouse anti-EPCAM mAb (C) or rabbit anti–claudin-7 pAb (D; red) together with rat anti–ZO-1 mAb (green) and DAPI (blue). Scale bars, 10 µm. (E) FFEM of MASP-qKO cells. Scale bar, 200 nm. (F) Quantification of the frequency of TJ strand branching points in the MASP-qKO cells. Ctrl data is the same one used in Fig. 2 H. Total length of TJ strands examined was 111 µm. Error bars indicate 95% confidence intervals. *, P < 0.05 (exact Poisson test). (G) Distribution of horizontal TJ strand number in the MASP-qKO cells. Blue lines indicate mean ± SD. Ctrl data is the same one used in Fig. 2 I. n = 400 (MASP-qKO#1). ***, P < 0.001 (two-tailed Welch’s t test). (H) Predicted TER values using simplified TJ strand network models based on the quantification data of TJ strand network complexity in the MASP-qKO cells. (I) TER measurements of MASP-qKO cells. n = 5. ***, P < 0.001 (two-tailed Welch’s t test with Bonferroni’s correction). (J) Effects of trypsin treatment form the apical side of the MASP-qKO cells and EpCAM-KO cells. n = 5. *, P < 0.05; ***, P < 0.001 (two-tailed paired t test). (K) FFEM images of the Ctrl cells (top) and MASP-qKO cells (bottom) treated with trypsin from the apical side. Scale bars, 200 nm. Source data are available for this figure: SourceData F5.

Gene-KO strategies and genomic structure of MASP genes. (A) Single, double, triple, and quadruple KOs of MASPs were isolated from the claudin-2 (CLDN2)-KO MDCK II (Ctrl) cell clone. (B) Genomic structure of KO cells. Two gRNAs were simultaneously introduced to the cells to induce large deletions. The sequences of gRNAs (red) and genome sequences of the KO clones are shown. The deleted regions of TMPRSS1 (TP1) and TMPRSS4 (TP4) contain the sequences encoding transmembrane region, which is supposed to be critical for proper localization and function of these genes. For TMPRSS14 (TP14) and PRSS8(P8), the first exon with a start codon (green) was removed.

Gene-KO strategies and genomic structure of MASP genes. (A) Single, double, triple, and quadruple KOs of MASPs were isolated from the claudin-2 (CLDN2)-KO MDCK II (Ctrl) cell clone. (B) Genomic structure of KO cells. Two gRNAs were simultaneously introduced to the cells to induce large deletions. The sequences of gRNAs (red) and genome sequences of the KO clones are shown. The deleted regions of TMPRSS1 (TP1) and TMPRSS4 (TP4) contain the sequences encoding transmembrane region, which is supposed to be critical for proper localization and function of these genes. For TMPRSS14 (TP14) and PRSS8(P8), the first exon with a start codon (green) was removed.
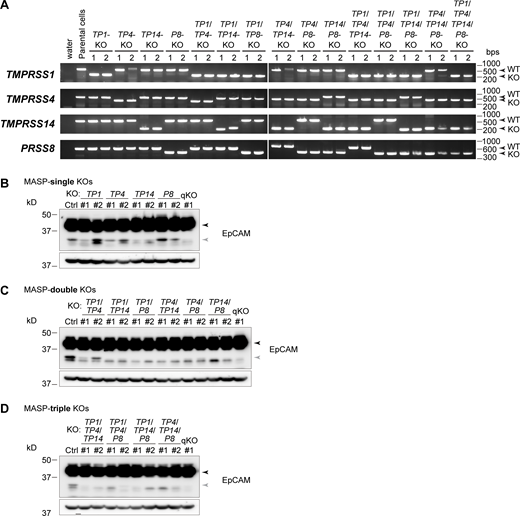
RT-PCR and immunoblotting of the MASP-KO cells. (A) Transcripts of each MASP-KO cell clone were analyzed by RT-PCR. The primers were designed at the upstream and downstream regions of the deletion. (B–D) Lysates of the MASP-sKO (B), -dKO (C), and -tKO (D) cells were analyzed using rabbit anti-EpCAM mAb. Black and gray arrowheads indicate full-length and cleaved EpCAM, respectively. β-actin served as loading controls. Source data are available for this figure: SourceData FS4.

RT-PCR and immunoblotting of the MASP-KO cells. (A) Transcripts of each MASP-KO cell clone were analyzed by RT-PCR. The primers were designed at the upstream and downstream regions of the deletion. (B–D) Lysates of the MASP-sKO (B), -dKO (C), and -tKO (D) cells were analyzed using rabbit anti-EpCAM mAb. Black and gray arrowheads indicate full-length and cleaved EpCAM, respectively. β-actin served as loading controls. Source data are available for this figure: SourceData FS4.
The localizations of EpCAM, claudin-7, and ZO-1 were not apparently altered in MASP-qKO cells (Fig. 5, C and D). In FFEM, the TJ strand network appeared slightly simpler in MASP-qKO cells than in the Ctrl cells (Fig. 5 E). Quantitative analyses confirmed that the branching points of TJ strands were slightly, albeit significantly, reduced, and the horizontal strand number was markedly reduced in the MASP-qKO cells (Fig. 5, F and G). TJ model simulation based on the quantification data of TJ strand morphology predicted that TER would be reduced by 36% (Fig. 5 H). In fact, the measured TER value of the MASP-qKO cells was reduced even more—by 70% in both clones (Fig. 5 I), suggesting that MASPs may regulate not only the structure of TJs but also their dynamics. The TER values of MASP-sKO, MASP-dKO, and MASP-tKO cells were reduced by 0-43%, 9–63%, and 52–74%, respectively (Fig. S5, A–C), suggesting that the barrier does not rely on any single MASP, but rather on all four MASPs. Similar to the results of TER, tracer flux measurement of 10-kD fluorescent dextran showed that the permeation of macromolecules was also dependent on all four MASPs (Fig. S5, D–G). To test whether the reduction in TER was caused by the loss of proteolytic activity in MASP-qKO cells, we applied low-concentration trypsin to the apical side of the cells. Trypsin application drastically increased the TER of MASP-qKO cells after 6 h, whereas the same treatment only slightly increased the TER of Ctrl cells and even decreased that of EpCAM-KO cells (Fig. 5 J), suggesting that the barrier function of MASP-qKO cells was restored through proteinase activity on EpCAM. FFEM analysis showed that there were no significant consistent changes in MASP-qKO cells treated with trypsin from the apical side (Fig. 5 K).

Barrier function of MASP-KO cells. (A–G) TER measurement (A–C) and tracer flux assay (D–G) of MASP-sKO (A and D), -dKO (B and E), -tKO (C and F), and -qKO (G) cells. Two independent clones were analyzed (n = 5; circles) for each genotype and the averaged values are shown (bars). Statistical significances compared with the Ctrl cells were evaluated by two-tailed Welch’s t test with Bonferroni’s correction (ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001).

Barrier function of MASP-KO cells. (A–G) TER measurement (A–C) and tracer flux assay (D–G) of MASP-sKO (A and D), -dKO (B and E), -tKO (C and F), and -qKO (G) cells. Two independent clones were analyzed (n = 5; circles) for each genotype and the averaged values are shown (bars). Statistical significances compared with the Ctrl cells were evaluated by two-tailed Welch’s t test with Bonferroni’s correction (ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
These data indicate that MASPs are required for the maintenance of the epithelial barrier.
MASPs regulate TJ repair
Next, we examined the dynamics of TJ permeability using the zinc-based ultrasensitive microscopic barrier assay (ZnUMBA; Stephenson et al., 2019), which can detect local barrier defects during live cell imaging using Zn2+ ions and the zinc ion indicator, FluoZin-3. We added Zn2+ and FluoZin-3 into the apical and basal compartments, respectively, and imaged the fluorescence of FluoZin-3, which indicates the position of TJ break sites. Our first attempt to image the cell sheet made of MASP-qKO cells alone failed because MASP-qKO cells were so leaky that the fluorescence of the FluoZin-3 signal increased too rapidly all over the cell sheet. Thus, we mixed a small amount of leaky MASP-qKO cells with tight Ctrl cells labeled with nuclear localization signal (nls)–conjugated mCherry and observed the cell sheet made up of both cell clones (Fig. 6 A). Although sporadic increases in the FluoZin-3 signal were observed in both the Ctrl and MASP-qKO cell regions, the size, length, and frequency of the FluoZin-3 signal were increased in the MASP-qKO cell region than in the Ctrl cell region (Fig. 6 B and Video 1). Quantification of the number of cell–cell junctions that were contained in a leak showed that the leak size was significantly greater in or next to the MASP-qKO cell area than in the Ctrl cell area (Fig. 6 C). We divided cell–cell junctions into four categories: (1) MASP-qKO cell–MASP-qKO cell junctions, (2) MASP-qKO cell–Ctrl cell junctions, (3) Ctrl cell–Ctrl cell junctions, in which the Ctrl cell is located next to MASP-qKO cells, and (4) Ctrl cell–Ctrl cell junctions, in which none of the Ctrl cells are neighboring MASP-qKO cells. Quantification of leak duration showed that the leaks in categories 1 and 2 persisted for a longer period compared with those in category 4 (Fig. 6 D). The frequency of leak occurrence was also greater in categories 1, 2, and 3 than in category 4 (Fig. 6 E), indicating that the cell–cell junctions of MASP-qKO cells are more prone to leakage and take more time to stop it compared with those of the Ctrl cells.

MASPs stabilize dynamics in TJ permeability. (A) Snapshots of the ZnUMBA assay using MASP-qKO cells mixed with Ctrl cells labeled with nuclear mCherry (pseudocolor blue). FluoZin-3 signals in the qKO cell region (yellow arrows) and Ctrl cell region (white arrows) are shown with a Gem lookup table. Scale bar, 10 µm. (B) FluoZin-3 signals at each time point are shown in orange. Cell–cell junctions are indicated with gray lines. Blue areas indicate the qKO cells. All signals observed during imaging are shown in the bottom-right panel. (C) Quantification of the leak sizes. A set of interconnected cell–cell junctions with FluoZin-3 signal are regarded as a single leak. The number of cell–cell junctions in each leak was counted and plotted. The leaks were divided into two categories: leaks located within or next to the MASP-qKO cells and leaks in the Ctrl cells. 25, 50, and 75 percentiles are shown (magenta; n = 517 [within or next to MASP-qKO] and 385 [Ctrl]). ***, P < 0.001 (two-tailed Welch’s t test). (D) Quantification of the duration of the leaks. Cell–cell junctions were categorized into four groups: qKO-qKO (n = 144), qKO-Ctrl (n = 218), Ctrl-Ctrl next to qKO (n = 130), and Ctrl-Ctrl not neighboring to qKO (n = 400). *, P < 0.05 (two-tailed Welch’s t test with Bonferroni’s correction). (E) Frequency of leaks. *, P < 0.05; ***, P < 0.001 (Steel-Dwass test). Source data are available for this figure: SourceData F6.
MASPs stabilize dynamics in TJ permeability. (A) Snapshots of the ZnUMBA assay using MASP-qKO cells mixed with Ctrl cells labeled with nuclear mCherry (pseudocolor blue). FluoZin-3 signals in the qKO cell region (yellow arrows) and Ctrl cell region (white arrows) are shown with a Gem lookup table. Scale bar, 10 µm. (B) FluoZin-3 signals at each time point are shown in orange. Cell–cell junctions are indicated with gray lines. Blue areas indicate the qKO cells. All signals observed during imaging are shown in the bottom-right panel. (C) Quantification of the leak sizes. A set of interconnected cell–cell junctions with FluoZin-3 signal are regarded as a single leak. The number of cell–cell junctions in each leak was counted and plotted. The leaks were divided into two categories: leaks located within or next to the MASP-qKO cells and leaks in the Ctrl cells. 25, 50, and 75 percentiles are shown (magenta; n = 517 [within or next to MASP-qKO] and 385 [Ctrl]). ***, P < 0.001 (two-tailed Welch’s t test). (D) Quantification of the duration of the leaks. Cell–cell junctions were categorized into four groups: qKO-qKO (n = 144), qKO-Ctrl (n = 218), Ctrl-Ctrl next to qKO (n = 130), and Ctrl-Ctrl not neighboring to qKO (n = 400). *, P < 0.05 (two-tailed Welch’s t test with Bonferroni’s correction). (E) Frequency of leaks. *, P < 0.05; ***, P < 0.001 (Steel-Dwass test). Source data are available for this figure: SourceData F6.
Live imaging of permeability in MASP-qKO cells by ZnUMBA assay. MASP-qKO cells mixed with Ctrl cells (labeled with nuclear mCherry) were imaged. FluoZin-3 signals are shown with a Gem lookup table (left). Schematic illustration of MASP-qKO (blue) and Ctrl (green) cells are shown (right). Cell boundaries with or without FluoZin-3 signals at each time point are shown in light green and gray, respectively. Time is indicated in min:s. Frame rate is 30 fps.
Live imaging of permeability in MASP-qKO cells by ZnUMBA assay. MASP-qKO cells mixed with Ctrl cells (labeled with nuclear mCherry) were imaged. FluoZin-3 signals are shown with a Gem lookup table (left). Schematic illustration of MASP-qKO (blue) and Ctrl (green) cells are shown (right). Cell boundaries with or without FluoZin-3 signals at each time point are shown in light green and gray, respectively. Time is indicated in min:s. Frame rate is 30 fps.
Together, these data suggest that MASPs are required for the repair of dynamic TJ leakage.
Claudin-7 is required for the establishment of tight epithelial barrier
Since our results suggest a model where EpCAM sequesters claudin-7 at the basolateral membrane, and claudin-7 can be incorporated into apical TJ strands upon release from EpCAM following MASP-mediated proteolysis, we also knocked out claudin-7 using CRISPR/Cas9 system (Fig. S2, H–J) and evaluated the structure and function of TJs. In the claudin-7-KO cells, the localization of EpCAM at the basolateral membranes was drastically reduced (Fig. 7 A), suggesting that EpCAM and claudin-7 are mutually dependent on each other for their stability and localization at the basolateral membrane. Consistent with this, the amount of EpCAM protein decreased in claudin-7-KO cells (Fig. S2 K). The basolateral localization of claudin-1 and claudin-3 was decreased and that of claudin-4 was slightly increased (Fig. 7 A). The amounts of claudin-1, claudin-3, and claudin-4 proteins slightly increased (Fig. S2 K), suggesting the existence of a compensatory mechanism to maintain the total amount of claudins. The TJ-strand network of claudin-7-KO cells revealed by FFEM was less branched and contained a smaller number of horizontal strands compared with that of Ctrl cells (Fig. 7, B–D). The TJ model simulation using FFEM data predicted that the TER value would be reduced to less than one-third in claudin-7-KO cells (Fig. 7 E). Indeed, the measured TER values were in a similar range (Fig. 7 F), suggesting that the structural changes in the claudin-7-KO cells caused the barrier loss. The permeability of the 10-kD tracer was also increased in the claudin-7-KO cells (Fig. S2 L). Since the claudin-7-KO cells lacked the EpCAM–claudin-7 complex, we expected that these cells would no longer respond to treatment with proteinases. Indeed, treatment of claudin-7-KO cells with trypsin from the apical side did not restore the barrier function (Fig. 7 G), which contrasts with MASP-qKO cells (Fig. 5 J).

Claudin-7 is required for the formation of complex TJ strand network and barrier function. (A) Staining of the claudin-7-KO cells mixed with Ctrl cells using mouse anti-EpCAM, mouse anti–claudin-1, or mouse anti–claudin-4 mAbs (red) together with rabbit anti–claudin-7 pAb (green) and rat anti–ZO-1 mAb (blue; top three rows), or mouse anti-EpCAM mAb (green), rabbit anti–claudin-3 pAb (red), and rat anti–ZO-1 mAb (blue; bottom row). Side view positions are indicated with black arrowheads. Scale bars, 10 µm. (B) FFEM of the claudin-7-KO cells. Scale bar, 200 nm. (C) Quantification of the frequency of TJ strand branching points in the claudin-7-KO cells. Ctrl data are the same as those used in Fig. 2 H. Total length of TJ strands examined was 111 µm. Error bars indicate 95% confidence intervals. ***, P < 0.001 (exact Poisson test). (D) Distribution of horizontal TJ strand number in the claudin-7-KO cells. Blue lines indicate mean ± SD. Ctrl data are the same as those used in Fig. 2 I. n = 450 (claudin-7-KO#1). ***, P < 0.001 (two-tailed Welch’s t test). (E) Predicted TER values using simplified TJ strand network models based on the quantification data of TJ strand network complexity in the claudin-7-KO cells. (F) TER measurements of claudin-7-KO cells. n = 5. ***, P < 0.001 (two-tailed Welch’s t test with Bonferroni’s correction). (G) Effects of trypsin treatment form the apical side of claudin-7-KO cells. n = 5. ***, P < 0.001 (two-tailed paired t test). Source data are available for this figure: SourceData F7.
Claudin-7 is required for the formation of complex TJ strand network and barrier function. (A) Staining of the claudin-7-KO cells mixed with Ctrl cells using mouse anti-EpCAM, mouse anti–claudin-1, or mouse anti–claudin-4 mAbs (red) together with rabbit anti–claudin-7 pAb (green) and rat anti–ZO-1 mAb (blue; top three rows), or mouse anti-EpCAM mAb (green), rabbit anti–claudin-3 pAb (red), and rat anti–ZO-1 mAb (blue; bottom row). Side view positions are indicated with black arrowheads. Scale bars, 10 µm. (B) FFEM of the claudin-7-KO cells. Scale bar, 200 nm. (C) Quantification of the frequency of TJ strand branching points in the claudin-7-KO cells. Ctrl data are the same as those used in Fig. 2 H. Total length of TJ strands examined was 111 µm. Error bars indicate 95% confidence intervals. ***, P < 0.001 (exact Poisson test). (D) Distribution of horizontal TJ strand number in the claudin-7-KO cells. Blue lines indicate mean ± SD. Ctrl data are the same as those used in Fig. 2 I. n = 450 (claudin-7-KO#1). ***, P < 0.001 (two-tailed Welch’s t test). (E) Predicted TER values using simplified TJ strand network models based on the quantification data of TJ strand network complexity in the claudin-7-KO cells. (F) TER measurements of claudin-7-KO cells. n = 5. ***, P < 0.001 (two-tailed Welch’s t test with Bonferroni’s correction). (G) Effects of trypsin treatment form the apical side of claudin-7-KO cells. n = 5. ***, P < 0.001 (two-tailed paired t test). Source data are available for this figure: SourceData F7.
These data indicate that claudin-7 is required for the formation of a tight TJ strand network and that the EpCAM–claudin-7 complex is responsible for proteinase-induced reinforcement of the TJ barrier.
EpCAM is cleaved at leak sites and helps repair TJ leaks
To examine whether EpCAM cleavage and claudin-7 incorporation into the TJ structure occur at TJ break sites, we utilized a cell surface–labeling technique using sulfo-NHS-biotin (Fig. 8 A, left). We cultured Ctrl cells on a transwell filter and applied sulfo-NHS-biotin from either the apical or basal side, and then biotinylated proteins were isolated and analyzed by immunoblotting. The full-length EpCAM band was preferentially labeled from the basal side, although the band was also detected in the apically labeled sample (Fig. 8 A, right). The basolateral membrane marker E-cadherin exhibited a similar pattern (Fig. 8 A, right), suggesting that a small amount of sulfo-NHS-biotin might be transported from the apical compartment to the basal side in our experimental setup. In contrast, cleaved EpCAM was only detected in the apically labeled cell sample (Fig. 8 A, right), similar to the apical marker glycoprotein 135 (GP135; also known as podocalyxin). As EpCAM was localized on the basolateral membranes in immunostaining (Fig. 2 D), it is likely that EpCAM cleavage occurs only at the sites of TJ breaks, which are typically observed in ZnUMBA imaging, rather than at the apical surface. The MASP P8 was also detected in the apically labeled sample and claudin-7 exhibited the distribution similar to basolateral proteins (Fig. 8 A, right).

EpCAM is likely to be cleaved at TJ break sites. (A) Surface biotin labeling experiment. Sulfo-NHS-biotin was applied to the apical or basal side of Ctrl cells and biotinylated proteins were captured with streptavidin (SA) beads and analyzed by immunoblotting using rabbit anti-EpCAM mAb and other antibodies. Arrows and asterisks indicate the specific and non-specific bands, respectively. (B) Immunostaining of apically biotinylated cells extracted with detergent. Sulfo-NHS-biotin was applied to the apical side of Ctrl (upper panels) or MASP-qKO (lower panels) cells. NP-40–soluble proteins were extracted before fixation and cells were stained with anti–claudin-7 pAb (green), streptavidin (red), and DAPI (blue). Top views of TJ and basolateral levels and side view are shown. Black arrowheads on the side of the top views indicate the location where the side-view section was made, and the small yellow arrowheads on the side-view panels indicate the locations of the tricellular contacts. Note that NP-40–insoluble claudin-7 was detected at the TJ break sites (white arrows). Scale bar, 10 µm. Source data are available for this figure: SourceData F8.
EpCAM is likely to be cleaved at TJ break sites. (A) Surface biotin labeling experiment. Sulfo-NHS-biotin was applied to the apical or basal side of Ctrl cells and biotinylated proteins were captured with streptavidin (SA) beads and analyzed by immunoblotting using rabbit anti-EpCAM mAb and other antibodies. Arrows and asterisks indicate the specific and non-specific bands, respectively. (B) Immunostaining of apically biotinylated cells extracted with detergent. Sulfo-NHS-biotin was applied to the apical side of Ctrl (upper panels) or MASP-qKO (lower panels) cells. NP-40–soluble proteins were extracted before fixation and cells were stained with anti–claudin-7 pAb (green), streptavidin (red), and DAPI (blue). Top views of TJ and basolateral levels and side view are shown. Black arrowheads on the side of the top views indicate the location where the side-view section was made, and the small yellow arrowheads on the side-view panels indicate the locations of the tricellular contacts. Note that NP-40–insoluble claudin-7 was detected at the TJ break sites (white arrows). Scale bar, 10 µm. Source data are available for this figure: SourceData F8.
If EpCAM is cleaved at TJ break sites, it is expected that claudin-7 is released from EpCAM and incorporated into the TJ strand network to repair the break at these sites. Since claudin-7 is abundantly expressed on the basolateral membrane, it was difficult to assess the polymerization state of claudin-7 by conventional immunofluorescence staining method. Thus, we took advantage of detergent extraction technique. In apically biotinylated and NP-40–treated Ctrl cells, the biotin label was sporadically detected on the basolateral membrane in a pattern similar to that observed in ZnUMBA imaging (Fig. 8 B). Claudin-7 signal was clearly detected around the biotin signal, suggesting that claudin-7 is polymerized around the break sites. The detergent-insoluble claudin-7 was also detected at the tricellular regions, which might have a different mechanism to incorporate claudins into a detergent-insoluble structure. In MASP-qKO cells, the size and frequency of biotin-labeled TJ break sites were much more increased than Ctrl cells (Fig. 8 B), which is consistent with the ZnUMBA imaging. At these TJ break sites in MASP-qKO cells, there was no evident accumulation of detergent-insoluble claudin-7 although the claudin-7 signal was detected at the tricellular contacts (Fig. 8 B).
These results suggest that EpCAM might be cleaved and release claudin-7 at TJ breaks, where MASPs, including P8, on the apical surface can access the EpCAM–claudin-7 complex on the basolateral surface.
EpCAM- and claudin-7–dependent barrier maintenance in other systems
We explored whether the EpCAM-based mechanism functions in other epithelial models. In parental MDCK II cells, which express claudin-2, EpCAM appears as a major 40-kD full-length band and faint lower bands (32 kD and 29 kD; Fig. 9 A). The 32-kD band was increased by trypsin treatment in a concentration-dependent manner (Fig. 9 A), indicating that EpCAM is cleaved by trypsin. Camostat treatment diminished the 32-kD band, suggesting that the cleavage is mediated by MASPs (Fig. 9 B). Trypsin treatment also increased the intensity of claudin-7 at TJs and induced basolateral punctate structures (Fig. 9 C). The localization of claudin-2 at TJs was also increased by trypsin (Fig. 9 C). To examine whether MASPs contribute to the maintenance of barrier function of parental MDCK II cells, the effects of camostat on TER and trace flux were evaluated (Fig. 9 D). Although the baseline TER of parental MDCK II cells is low (∼65 Ω cm2), camostat treatment significantly lowered the TER value, and increased the tracer flux by ∼100% (Fig. 9 D). These results suggest that MASPs are responsible for the maintenance of barrier in parental MDCK II cells.

EpCAM/claudin-7–dependent barrier maintenance in other epithelial models. (A and E) Immunoblotting of parental MDCK II cells (A) and Caco-2 cells (E) treated with various concentrations of trypsin using rabbit anti-EpCAM mAb and other antibodies. Black, gray, and blue arrowheads indicate full-length, cleaved, and 29-kD bands, respectively. (B and F) Effects of camostat treatment for 16 h on parental MDCK II cells (B) and Caco-2 cells (F). (C and G) Effects of trypsin on the localization of claudin-7 (red) in parental MDCK II cells (C) and Caco-2 cells (G). Cells were co-stained with claudin-2 (green) and ZO-1 (blue; C) or ZO-1 (green) and DAPI (blue; G). Scale bars, 10 µm. (D and H) Effects of camostat on TER and permeability of 10-kD FITC-dextran in parental MDCK II cells (D) and Caco-2 cells (H). ns, P > 0.05; *, P < 0.05; ***, P < 0.001 (two-tailed paired t test). (I and J) Immunostaining of frozen sections of the small intestine (I) and colon (J). Tissue sections were either untreated (upper panels) or treated with NP-40 before fixation (lower panels) and stained for claudin-7 (green), ZO-1 (red), and DAPI (blue). White arrows indicate the tips of the villi. The tip region of villi in the left panels (white rectangle in I) was enlarged and shown in the right panels. l, lumen; v, villus; c, crypt. Scale bars, 50 µm. Source data are available for this figure: SourceData F9.
EpCAM/claudin-7–dependent barrier maintenance in other epithelial models. (A and E) Immunoblotting of parental MDCK II cells (A) and Caco-2 cells (E) treated with various concentrations of trypsin using rabbit anti-EpCAM mAb and other antibodies. Black, gray, and blue arrowheads indicate full-length, cleaved, and 29-kD bands, respectively. (B and F) Effects of camostat treatment for 16 h on parental MDCK II cells (B) and Caco-2 cells (F). (C and G) Effects of trypsin on the localization of claudin-7 (red) in parental MDCK II cells (C) and Caco-2 cells (G). Cells were co-stained with claudin-2 (green) and ZO-1 (blue; C) or ZO-1 (green) and DAPI (blue; G). Scale bars, 10 µm. (D and H) Effects of camostat on TER and permeability of 10-kD FITC-dextran in parental MDCK II cells (D) and Caco-2 cells (H). ns, P > 0.05; *, P < 0.05; ***, P < 0.001 (two-tailed paired t test). (I and J) Immunostaining of frozen sections of the small intestine (I) and colon (J). Tissue sections were either untreated (upper panels) or treated with NP-40 before fixation (lower panels) and stained for claudin-7 (green), ZO-1 (red), and DAPI (blue). White arrows indicate the tips of the villi. The tip region of villi in the left panels (white rectangle in I) was enlarged and shown in the right panels. l, lumen; v, villus; c, crypt. Scale bars, 50 µm. Source data are available for this figure: SourceData F9.
We also used human colorectal adenocarcinoma cell line Caco-2. EpCAM appears as 40- and 32-kD bands, which have almost the same intensity (Fig. 9 E). Trypsin treatment reduces the 40-kD band intensity and simultaneously increased the 32-kD band (Fig. 9 E), suggesting that EpCAM is cleaved by trypsin. Camostat treatment did not change the intensity of either band (Fig. 9 F), suggesting that the majority of 32-kD band in untreated Caco-2 cells is not produced by MASPs. Trypsin treatment from the basal side induced claudin-7 puncta on the basolateral membrane and increased the intensity of claudin-7 at TJs (Fig. 9 G). Camostat treatment lowered the TER value by ∼20% and increased the tracer flux by ∼40% (Fig. 9 H), indicating that MASPs play a role in maintaining barrier function in Caco-2 cells.
Finally, we examined whether claudin-7 is incorporated into TJ structures at the sites where TJs are reorganized in vivo. In the small intestine, new cells are provided from the region around the crypts and old cells are extruded at the tips of the villi. At the extrusion zones, TJs maintain barrier while undergoing dynamic rearrangement. New TJ strands were observed on the basolateral membranes at these sites (Madara, 1990). We examined whether these TJ strands are composed of claudin-7. Immunostaining of mouse jejunum revealed that claudin-7 was broadly expressed in the entire epithelium and was distributed on the basolateral membranes (Fig. 9 I). At the tips of the villi, claudin-7 exhibited slightly intense signal. We took advantage of the detergent extraction method to visualize claudin-7 incorporated into junctional structures by removing unpolymerized claudin-7. In the NP-40–treated section, only the tips of the villi retained strong signal of claudin-7 and most of the other regions lost the staining (Fig. 9 I), indicating that claudin-7 is assembled into the junctional structure at the extrusion zones. In the colon, claudin-7 signal was observed at the focused area of luminal surface, the region where old cells are extruded (Fig. 9 J).
Discussion
In this study, we investigated the mechanism by which EpCAM and MASPs regulate the TJ barrier using an MDCK II epithelial cell model, and we propose a novel model mechanism of TJ maintenance (Fig. 10). In this model, MASPs are localized on the apical surface, and the EpCAM–claudin-7 complex is located at the basolateral membrane when the TJ is intact. Claudin-1 and part of claudin-3 might also be associated with and sequestered by EpCAM. At sites where the TJ is breached, MASPs can access the EpCAM–claudin-7 complex and cleave EpCAM. Claudin-7 and other claudins released from cleaved EpCAM are competent for polymerization and participate in TJ repair. In EpCAM-KO cells, the EpCAM–claudin-7 complex is missing, and the cells do not have the proteolysis-based repair mechanism. However, the TJ barrier is maintained by the constitutive incorporation of free claudin-7 into the TJ strands, which build up a TJ strand network with increased number of bifurcations and horizontal strands. In contrast, claudin-7-KO cells exhibit impaired barrier function, which is probably due to two reasons. First, the claudin-7–EpCAM-based repair mechanism is missing in these cells. Second, the TJ strand network becomes simpler than that of the Ctrl cells. In MASP-qKO and camostat-treated cells, the repair of TJ breaks is impaired or delayed, which results in increased permeability of ions and macromolecules. This model explains how the epithelial barrier is maintained, even during the dynamic remodeling processes of epithelial sheets. TJs also possess fence function, which maintains segregation of apical and basolateral membrane proteins. EpCAM- and claudin-7–dependent surveillance and repair mechanism of TJs might also contribute to the maintenance of fence function and apicobasal polarity, which should be examined in the future study.

Detection and repair of TJ breaks by EpCAM
We and others have developed assays to detect TJ break sites by adding a pair of compounds, one into the apical compartment and the other into the basal compartment, and detecting their interaction at TJ break sites by microscopy (Richter et al., 2016; Reiche et al., 2018; Stephenson et al., 2019). Our data show that living epithelial cells use the same concept to survey the integrity of the epithelial barrier and detect TJ breaks. The sensor protein, EpCAM, resides at the basolateral surface of the plasma membrane and is separated from MASPs or other proteolytic activities in the apical region by intact TJs. At sites where the TJ is damaged, opening of a route between the apical and basal compartments is detected by the proteolytic activity on EpCAM. Upon cleavage, EpCAM initiates the repair process by releasing claudin-7, a TJ strand building block. Thus, EpCAM plays a pivotal role in TJ-break surveillance and repair.
Retention of EpCAM and claudins at basolateral membrane
It is still unclear how EpCAM complexed with claudin-7 (or claudin-1/3) is maintained on the surface of basolateral membrane. It was suggested that palmitoylated claudin-7 is preferentially incorporated into glycolipid-enriched membranes (Heiler et al., 2015). In contrast to other claudins, claudin-4 was not associated with EpCAM in epithelial cells, although claudin-4 was abundantly localized on the basolateral membranes. Recent studies revealed that claudin-4 does not polymerize into strands and exhibits uniform distribution when expressed in U2OS or Cos-7 cells (Shashikanth et al., 2022; Gonschior et al., 2022). Interestingly, when co-expressed with other claudins (claudin-3 or claudin-7), claudin-4 becomes integrated with the TJ strands (Shashikanth et al., 2022; Gonschior et al., 2022). Based on these reports, it is likely that most part of claudin-4 is diffusely located on the basolateral surface. If other claudins are excessively assembled into TJ structures, claudin-4 might be integrated with the assembled TJ strands and becomes resistant to Brij97 solubilization.
Intestinal barrier maintenance by EpCAM and claudin-7
The dynamic repair mechanism of barrier function by EpCAM may be important in organs such as the intestinal tract, where cell–cell junctions are constantly affected by peristaltic movements, cell proliferation, differentiation, translocation within the same epithelial sheet, and cell extrusion. This hypothesis was supported by the observation of detergent-resistant claudin-7 staining at the cell extrusion sites of small intestine and colon in mice. Mutations in the EpCAM gene cause congenital tufting enteropathy (CTE) with intestinal malabsorption (Sivagnanam et al., 2008; Guerra et al., 2012; Kozan et al., 2015). The malabsorption phenotype of EpCAM-KO mice might be caused by the loss of the TJ-break surveillance and repair mechanism by EpCAM and claudin-7. Double KO mice of EpCAM and its close homolog TROP2 (trophoblast cell surface antigen 2) exhibited more severe phenotypes compared with EpCAM-KO mice, suggesting that TROP2 also contributes to the TJ maintenance (Szabo et al., 2022). Claudin-7-KO mice exhibit electrolyte wasting and severe dehydration and die within 12 d after birth (Tatum et al., 2010). This is attributed to a barrier defect in renal epithelial cells. In addition to the general barrier phenotype of the kidney, the claudin-7-KO mice also exhibit intestinal villous atrophy, crypt hyperplasia (Ding et al., 2012), impaired barrier function, and reduced expression of EpCAM protein (Tanaka et al., 2015), as observed in CTE. Conditional claudin-7-KO in the intestine also resulted in severe intestinal defects and death in mice (Li et al., 2018). Based on these reports, it is likely that EpCAM and claudin-7 are involved in the pathogenesis of CTE through the same pathway. Alternatively, the intestinal phenotypes of claudin-7-KO mice could be explained by the increased production of cytokines and matrix metalloproteinases and altered localization of integrins (Ding et al., 2012).
MASPs mediate TJ formation and maintenance
In this study, we focused on four MASPs expressed in MDCK II cells. It is still unclear whether MASPs are required for the maintenance of TJs in other epithelial cell types. Among these four MASPs, P8 (prostasin) has been implicated in the regulation of the epithelial barrier function and TJ development. Epidermis-specific P8-KO mice exhibited impaired barrier function of the skin and could not survive for >60 h after birth due to water loss from the skin (Leyvraz et al., 2005). Dotted staining of occludin in the second layer of the stratum granulosum was lost in P8-KO mice, suggesting that no mature TJ was formed in these mice. This phenotype resembles that of claudin-1-KO mice (Furuse et al., 2002), strongly suggesting that P8 regulates TJ formation in the skin. TP14 (matriptase) is also associated with epithelial barrier function (List et al., 2002; List et al., 2009; Buzza et al., 2010; Wu et al., 2017). KO or suppression of TP14 expression in the intestine results in altered localization of TJ proteins and increased permeability of ions and macromolecules (List et al., 2009; Buzza et al., 2010). These phenotypes are well explained by our model showing that MASP-mediated proteolysis induces de novo TJ strand formation. The functions of TP1 (hepsin), TP4, and other MASPs in TJs have not yet been extensively explored. Mutations in TMPRSS3, one of the MASPs, cause hearing loss due to hair cell degeneration (Scott et al., 2001; Fasquelle et al., 2011). As loss of claudins in the inner ear causes the degenerative death of cochlear hair cells, resulting in congenital hearing loss in humans and mice (Wilcox et al., 2001; Ben-Yosef et al., 2003; Kitajiri et al., 2004; Nakano et al., 2009; Ramzan et al., 2021), TMPRSS3 may also be involved in the formation or maintenance of TJs in the cochlear sensory epithelium.
Camostat, the MASP inhibitor, has been known to improve the absorption of peptides, such as insulin, vasopressin, and calcitonin, through the epithelial sheets of the nasal mucosa, skin, and intestinal mucosa (Morimoto et al., 1991, 1992; Yamamoto et al., 1994; Tozaki et al., 1998). The improved absorption has been understood to be due to the prevention of peptide degradation by endogenous proteases, but inhibition of the dynamic repair mechanism of barrier function by MASP-mediated proteolysis may also contribute to the improved permeability and absorption of peptides. From this perspective, elucidation of the mechanism of barrier maintenance by proteolysis may lead to the development of novel drug delivery systems focusing on MASPs.
De novo TJ formation through proteolysis
It has been long known that proteinases can induce TJ strand formation in isolated pancreatic islet cells (Orci et al., 1973; in’t Veld et al., 1984), the oral epithelium (Shimono and Clementi, 1977), and the cultured carcinoma cell line, HT29 (Polak-Charcon et al., 1978; Talmon et al., 1984; Cohen et al., 1985; Faff et al., 1987, 1988; Cohen et al., 1990). Since pancreatic endocrine cells are non-epithelial cells and do not have TJs (in’t Veld et al., 1984), they do not require a mechanism to maintain TJs. Rather, it is plausible that proteinase-induced rapid TJ formation can prevent the invasion of toxic fluids containing proteinases, such as digestive pancreatic juice, into the interstitial space and protect the humoral environment of the endocrine system against perturbations. It would be interesting to assess whether EpCAM also functions in islet cells, the oral epithelium, and HT29 cells.
Maintenance of TJ barrier by local repair
Transient and localized activation of Rho GTPase (Rho flares) and Rho flare–associated accumulation of the actomyosin cytoskeleton have also been proposed to repair TJ breaks in Xenopus embryos (Stephenson et al., 2019). Based on our findings, we suggest that epithelial cells have two distinct repair mechanisms: (1) rapid closure of the TJ strand break by proteolysis of EpCAM and supply of polymerization-competent claudin-7, and (2) reinstatement of TJ structure by Rho flare–mediated actomyosin contraction and crowding of TJ components and prevention of the repeated loss of integrity. Similar multiple cellular mechanisms are known to operate in the repair of plasma membrane damage: (1) rapid resealing of the breaks by the formation of a membrane “patch” derived from intracellular compartments or membrane budding and (2) reinstatement of cortical membrane and cytoskeleton by Rho family GTPase-mediated formation and closure of actomyosin contractile ring encircling the wound (Terasaki et al., 1997; Benink and Bement, 2005; Davenport et al., 2016; Nakamura et al., 2018; Zhen et al., 2021).
The permeability of epithelial sheets is determined by the total sum of the transcellular and paracellular fluxes. Hence, in an epithelium with tight barrier function, the weakest points of the TJs have the greatest contribution to the permeability of the whole sheet (Krug, 2017). To maintain barrier function effectively, epithelial cells should be equipped with mechanisms to deal with local damage in TJs. From this perspective, both mechanisms are ideal for maintaining the robustness of the barrier.
Currently, it is unclear how widely these two mechanisms are conserved among the different cell types and species. Although Rho flares have only been described in the surface epithelium of Xenopus gastrula-stage embryos, similar local accumulation of actin filaments has been observed in MDCK II cells (Tokuda et al., 2014), suggesting that this mechanism exists in a broad range of epithelial cells across species. Recent research has indicated that EpCAM regulates the cortical RhoA zone in cultured epithelial cells (Gaston et al., 2021), suggesting that there is a crosstalk between these pathways. Further studies examining the spatiotemporal regulation of both pathways and their mutual dependency using the same experimental setup are required to understand how these distinct mechanisms cooperatively maintain the epithelial barrier.
Regulation of MASP activation
We demonstrated that the MASP P8 is localized to the apical membranes (Fig. 8 A), and our model is based on this observation (Fig. 10). However, we could not confirm whether the other three MASPs we characterized were on the apical membranes, as we did not have antibodies applicable for immunostaining or immunoblotting of biotin-labeled proteins. Other studies have shown that TP1 (hepsin) as well as P8 are localized on apical surfaces (Brunati et al., 2015). In contrast, TP14 (matriptase) was localized on the basolateral membranes in immunohistochemistry of the mouse skin and intestine (List et al., 2009), immunostaining of Caco-2 cells (Buzza et al., 2010; Wu et al., 2017), T84 cells, and human colon (Wu et al., 2017). Together with our results that the expression of a single MASP was not sufficient to support the barrier (Fig. S5), these reports suggest that MASPs are not solely regulated by their localization but might require activation by other MASPs. It has been proposed that TP14 activates P8 in the skin (Netzel-Arnett et al., 2006). Others have suggested that P8 is required for the activation of TP14 in Caco-2 cells (Buzza et al., 2013). Therefore, it is important to clarify the hierarchy of MASPs and their activation or inhibition mechanisms.
Concluding remarks
Here, we demonstrate a novel EpCAM-based mechanism for the surveillance and repair of TJs. This mechanism can explain past observations of proteinase-induced TJ formation and enhances our understanding of the dynamic regulation of TJ barriers and congenital tufting enteropathy pathogenesis.
Materials and methods
Cell culture
MDCK II, HEK293T, and SP2 cells were cultured in Dulbecco’s DMEM (D5796; Sigma-Aldrich) supplemented with 5% FBS (F7524; Sigma-Aldrich). A431 and Caco-2 cells were cultured in DMEM with 10 and 20% FBS, respectively. Cells were maintained in a 5% CO2 incubator at 37°C. All KO clones of MDCK II cells were established from a parental claudin-2-KO MDCK II cell clone (Saito et al., 2021). Thus, the claudin-2-KO cell line is referred to as the “Ctrl” cell line throughout this manuscript.
Antibodies
Mouse anti-EpCAM mAb (clone OTI1H9; SC322331) was purchased from Origene Technologies. Rabbit anti-EpCAM mAb (clone E6V8Y; #93790) and rabbit anti–α-catenin mAb (clone 23B2; #3240) were purchased from Cell Signaling Technology. Rabbit anti-TMPRSS1 (HPN) polyclonal antibody (pAb; HPA006804), mouse anti–β-actin mAb (clone AC-15; #A1978), and mouse anti-FLAG M2 HRP-linked mAb (#A8592) were purchased from Sigma-Aldrich. Rabbit anti-PRSS8 (Prostasin) pAb (GTX103976) was purchased from GeneTex. Rabbit anti–ZO-1 pAb (#61-7300), rabbit anti–claudin-2 pAb (#51-6100), rabbit anti–claudin-3 pAb (#34-1700), mouse anti–ZO-1 mAb (#ZO1-1A12), and mouse anti–claudin-4 mAb (#32-9400) were from Thermo Fisher Scientific. Rat anti-EpCAM mAb (clone EBA-1; sc-66020), rat anti–ZO-1 (alpha+) mAb (clone R40.76; sc-33725), mouse anti–claudin-1 mAb (clone XX7; sc-81796), and mouse anti-occludin mAb (clone E-5; sc-13325) were obtained from Santa Cruz Biotechnology. Rabbit anti–claudin-7 pAb (#18875) was obtained from Immuno-Biological Laboratories. Rabbit anti-GFP pAb (#598) was purchased from Medical and Biological Laboratories. Mouse anti-GFP tag mAb (66002-1-Ig) mAb and rabbit anti-mCherry pAb (26765-1-AP) were obtained from Proteintech. Mouse anti–E-cadherin mAb (clone 36; #610182) was purchased from BD Biosciences. Mouse anti-Podocalyxin (GP135) mAb (clone 3F2:D8; MABS1327) was purchased from EMD Millipore. Rabbit anti–angulin-1 pAb (#00301) was obtained from BiCell Scientific. Mouse anti-DYKDDDDK (FLAG)-tag mAb (clone 1E6; #018-22386) was purchased from Fujifilm-WAKO. The rat anti–claudin-2 mAb (clone 2D7) was described previously (Saito et al., 2021). The rat anti-TP14 mAb (clone 3A1) was produced in-house using the rat medial iliac lymph node method (Kishiro et al., 1995) using a peptide (NH2-Cys-MSGVEEGVEFLPVNN-COOH; synthesized by Genscript) corresponding to the N-terminus of the cytoplasmic domain of canine TP14 as an immunogen, as described previously (Saito et al., 2021). The peptide was conjugated with Keyhole limpet hemocyanin (#77605; Thermo Fisher Scientific), dialyzed against PBS, and mixed with Freund’s complete adjuvant (#263810; BD Biosciences) to make an emulsion. Then, the emulsion was intracutaneously injected into the footpads of 8-wk-old Wistar rats. Animal experiments strictly adhered to the Japanese Guidelines for Proper Conduct of Animals Experiments. The protocols for the experiments using animals (#30112, #201923, #2020023, and #2021034) were reviewed by the Animal Care and Use Committee of Fukushima Medical University and approved by the university’s president. The rats were euthanized by cervical dislocation under deep anesthesia using sevoflurane (Fujifilm WAKO) after 2 wk. The iliac lymph nodes were isolated and minced. The lymphocytes were isolated from the lymph nodes, filtrated with a nylon cell strainer (#352350; Corning), and mixed with mouse myeloma cell line SP2. The cells were washed with DMEM and fused by slowly adding 1 ml of 50% PEG4000 (#1.09727.0100; Merck Millipore) in DMEM containing 5% DMSO (D2650; Sigma-Aldrich). The cell suspension was slowly diluted with 9 ml of DMEM, and washed with DMEM. Hybridoma cells were selected using GIT medium (#637-25715; Fujifilm WAKO) containing hypoxanthin-aminopterin-thymidine (#21060017; HAT; Thermo Fisher Scientific) supplemented with 10% BM-Condimed H1 (11088947001; Roche) for 7–10 d. Positive clones were screened with a solid-phase ELISA using the immunogen peptide-coated 96-well microplate (#655101; Greiner bio-one) and evaluated with immunoblotting.
Alexa Fluor 488–conjugated donkey anti-mouse IgG pAb (#715-545-151), Alexa Fluor 488–conjugated donkey anti-rabbit IgG pAb (#711-545-152), Alexa Fluor 488–conjugated donkey anti-rat IgG pAb (#712-545-153), Cy3-conjugated donkey anti-mouse IgG pAb (#715-165-151), Cy3-conjugated donkey anti-rabbit IgG pAb (#711-165-152), Cy3-conjugated donkey anti-rat IgG pAb (#712-165-153), and Alexa Fluor 647–conjugated donkey anti-rat IgG pAb (#712-605-153) were purchased from Jackson ImmunoResearch Laboratories. HRP-linked sheep anti-mouse IgG pAb (#NA931V; GE Healthcare), HRP-linked goat anti-rabbit IgG pAb (#7074P; Cell Signaling Technology), and HRP-linked goat anti-rat IgG pAb (#NA935V; GE Healthcare) were also used in this study.
Generation of KO cell lines
Claudin-2-KO cells were described previously (Saito et al., 2021). To generate EpCAM-KO, MASP-KO, and claudin-7-KO cells, DNA oligonucleotides encoding single-guide RNAs (sgRNAs) were synthesized (Macrogen), and the annealed DNA oligonucleotides were cloned into pSpCas9 (BB)-2A-Puro (PX459) plasmids (plasmid #62988; Addgene; Ran et al., 2013) at the Bbs I cleavage sites. Two gRNAs were designed for each gene to induce an excision of exons encoding the initiation codon or important domain. Parental cells were transiently transfected with a pair of plasmids encoding sgRNAs using PEI-max (Polysciences) and selected using 3 µg/ml of puromycin (Sigma-Aldrich) for 1 d. The cells were then sparsely seeded onto 10-cm dishes and cell clones were obtained by scraping off the colonies. Genomic DNA was extracted from cell clones and screened by PCR using GoTaq DNA polymerase (Promega) and specific primers. PCR products of KO clones were subcloned into pGEM-T-easy (Promega) and sequenced (Macrogen) to determine the precise deletion sites. After the establishment of cell clones, we confirmed that the cells were no longer resistant to puromycin.
Cloning
Total RNA of MDCK II cells was isolated using TRIzol (#15596026; Thermo Fisher Scientific) according to the manufacturer’s instruction. A cDNA library of MDCK II cells was synthesized using the PrimeScript II 1st strand cDNA synthesis kit (Takara Bio). cDNAs encoding canine HPN (TP1), TMPRSS4 (TP4), ST14 (TP14), PRSS8 (P8), EPCAM, CLDN1, CLDN2, CLDN3, CLDN4, and CLDN7 were amplified using PrimeSTAR GXL DNA polymerase (R050A; Takara-Bio, Japan) from the cDNA library of MDCK II cells. Primer sets used are 5′-ATAGATCTCGTGACATGGCGGAGAAGGAGG-3′ and 5′-ATGAATTCAGAGCTGGGTCACCATGCCGC-3′ (TP1), 5′-ATAGATCTGCCGGCATGGATCCGGACAGC-3′ and 5′-ATGAATTCATGGTGTAGACTTTCGGACATTG-3′ (TP4), 5′-ATAGATCTCAGAGCATGAGTGGCGTCGAGG-3′ and 5′-ATGAATTCACACCCCCGTCTCCTCTCTGATC-3′ (TP14), 5′-ATAGATCTTGGGCCATGGCCCACAGGGCAG-3′ and 5′-ATGAATTCAGCAGAGGAGGCTGAGGGTCAG-3′ (P8), 5′-ATAGATCTCGCGGCATGGCGCGGCCCCAGG-3′ and 5′-ATGAATTCTGCATTGAGTTCCCTATGCATCTCAC-3′ (EPCAM); 5′-ATGGATCCCGAGCCATGGCCAACGCGGGGC-3′ and 5′-ATGAATTCCACGTAGTCTTTCCCACTGGAAG-3′ (CLDN1); 5′-ATGGATCCTCCGCCATGGCCTCTCTCGGCC-3′ and 5′-ATGAATTCCACATACCCTGTCAGGCTGTAG-3′ (CLDN2); 5′-ATGGATCCGCAGCCATGTCCATGGGCCTGG-3′ and 5′-ATGAATTCCACGTAGTCCTTGCGGTCGTAG-3′ (CLDN3); 5′-ATGGATCCCGAGCCATGGCCTCCATGGGGC-3′ and 5′-ATGAATTCCACGTAGTTGCTGGCTGGGGCG-3′ (CLDN4); 5′-ATGGATCCGCGAACATGGCCAACTCGGGCC-3′ and 5′-ATGAATTCCACGTACTCCTTGGCAGAGTTG-3′ (CLDN7). Restriction sites (Bgl II and EcoR I for TP1, TP4, TP14, P8, and EPCAM and BamH I and EcoR I for CLDN1, CLDN2, CLDN3, CLDN4, and CLDN7) are italicized and the start and termination codons are underlined. The restriction sites in the cDNA sequences were disrupted by introducing a silent mutation using specific primer sets (5′-GTGGACGGCAAGATTTGCACGGTGACCGGC-3′ and 5′-GCCGGTCACCGTGCAAATCTTGCCGTCCAC-3′ [TP1]; 5′-CAGCCCAGACCAAGACCTGGATGTTGTTGG-3′ and 5′-CCAACAACATCCAGGTCTTGGTCTGGGCTG-3′ [TP4]; 5′-CGGGTTCAGAAGATTTTCAATGGCTACCTG-3′ and 5′-CAGGTAGCCATTGAAAATCTTCTGAACCCG-3′ [TP14]; 5′-GGAACCTTCACGGCATCCTGCGGGACTTC-3′ and 5′-GAAGTCCCGCAGGATGCCGTGAAGGTTCC-3′ [CLDN2]). The fragments were digested with restriction enzymes, purified using NucleoSpin Gel and PCR Clean-up kit (U0609C; Takara-Bio), and cloned into the pCAG, pCAG-IRES-mCherry-nls, pCAG-cGFP, or pCAG-cmCherry vectors (Saito et al., 2021; Saito et al., 2022) at Bgl II and EcoR I restriction sites using T4 DNA ligase (#2011B, Takara-Bio). The DNA sequences were verified (Macrogen).
RT-PCR
To identify MASPs expressed in MDCK II cells, fragments within an exon of each MASP gene were amplified from the cDNA library of MDCK II cells using the GoTaq DNA polymerase. Genomic DNA was used as a positive control. Reaction mix without reverse transcriptase was used as a negative control. For the evaluation of MASP-KO cells, cDNA libraries were synthesized from each clone and examined with specific primers, that could amplify a fragment including the deleted region.
Sulfo-NHS-biotin labeling of apical and basal proteins
Cells were seeded onto 6-well polyethylene terephthalate Transwell filters with 0.4-µm pore size (#353090; Corning) at 2.0 × 105 cells/ml and cultured for 4 d. Both the apical and basal sides of the cells were washed thrice with HBSS (#14025-092; Thermo Fisher Scientific) at 37°C three times. Then, 0.25 mg/ml of EZ-link Sulfo-NHS-SS-biotin (#A39258; Thermo Fisher Scientific) in HBSS was applied to either the apical or basal sides of the cells and incubated at RT for 30 min. To analyze the biotinylated proteins by immunoblotting, cells were washed with cold TBS three times and lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1% Triton X-100, cOmplete protease inhibitors cocktail [#04693116001; Roche]) at 4°C. Lysates were briefly sonicated (#UR-21P; Tomy digital biology) and centrifuged at 15,000×g for 20 min at 4°C. The supernatants were incubated with Streptavidin Sepharose (#17-5113-01; GE Healthcare) with rotation for 2 h at 4°C, washed five times with the lysis buffer containing 0.5% Triton X-100, and boiled in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.005% bromophenol blue) for 5 min. Bound proteins were subjected to immunoblotting. To stain the biotinylated proteins, cells were washed with cold TBS thrice and the detergent-soluble proteins were extracted with 1% Nonidet P-40 (NP-40; #25223-04, Nacalai) in PBS supplemented with cOmplete proteinase inhibitors cocktail on ice for 10 min. After washing with PBS, cells were fixed with 100% methanol at −20°C for 15 min and subjected to the staining using CF568-conjugated streptavidin (#29035; Biotium).
Co-immunoprecipitation
To isolate EpCAM-FLAG-binding proteins, confluent EpCAM-FLAG–expressing cell sheets from four 10-cm dishes were lysed with IP lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% or 1% NP-40 or 1% Brij97 [P6136; Sigma-Aldrich], 1 mM DTT, and 2 mM EDTA) and centrifuged at 15,000×g for 20 min at 4°C. The supernatant was incubated with Protein G-sepharose (#17061801; Cytiva) with rotation for 2 h at 4°C, washed with the IP lysis buffer containing 0.1% NP-40 or 0.1% Brij97 five times, and the bound proteins were eluted with SDS sample buffer. To examine the interaction between EpCAM-FLAG and claudin-GFP, HEK293T cells were co-transfected with the expression vectors in a 6-well plate. 2 d later, the cells were lysed and immunoprecipitation was performed as described above.
Immunofluorescence microscopy and image acquisition
For fluorescence microscopy of MDCK II cells, 2 × 105 cells/ml of cells were seeded onto polycarbonate Transwells with 0.4-µm pore size (#3401; Corning). After 6 d, the cells were fixed with 99.9% methanol (Fujifilm-Wako) for 15 min at −20°C. For staining of mouse epithelial tissues, dissected tissue blocks were freshly embedded in OCT compound (Sakura Finetek) and quickly frozen in liquid nitrogen. The frozen tissues were cut into ∼8-µm-thick sections in a cryostat (Cryostar NX70; Thermo Fisher Scientific) at −20°C. The sections were mounted on coverslips, air-dried for 30 min at RT. For NP-40 extraction, sections were treated with 1% NP-40 in PBS containing proteinase inhibitors for 10 min on ice. Sections were fixed with 95% ethanol at −20°C for 20 min. For staining A431 cells, the cells were seeded onto coverslips and fixed with 99.9% methanol for 15 min at −20°C. For staining mCherry-nls–expressing A431 cells, the cells were fixed with 1% formaldehyde (Fujifilm-Wako) for 30 min at RT and permeabilized with 0.2% Triton X-100 in PBS for 5 min at RT. The filter membranes or coverslips were then washed with PBS thrice. After blocking with 2% BSA in PBS, the samples were incubated with primary antibodies diluted in PBS containing 0.2% BSA for 1 h at RT. After washing with PBS, the samples were incubated with fluorescently labeled secondary antibodies diluted in PBS containing 0.2% BSA for 1 h at RT. After washing, the cells were embedded in FLUORO-GEL II with DAPI (Electron Microscopy Sciences). The samples were observed at RT using a laser scanning confocal microscope (FV1000; Olympus) with a 60x oil-immersion objective lens (UPlanSApo 60×; Olympus) at laser wavelengths of 405, 488, and 559 nm, or a fluorescence microscope (BX61; Olympus) with a 40× objective lens (UPlanSApo 40×; Olympus) equipped with a mercury lamp, dichroic filter sets (NIBA, WIG, and WU) and a cooled CCD camera (DP71; Olympus). Images were acquired using the FluoView ver. 4.2b (Olympus) or CellSens ver. 1.14 (Olympus) and processed using ImageJ and Photoshop (Adobe).
Immunoblotting
Cells or bead-bound proteins were boiled in the SDS sample buffer for 5 min. Proteins were separated by SDS-PAGE using 5–20% gradient gels (Fujifilm Wako), and transferred to a polyvinylidene fluoride membrane (Immobilon, Merck). The membranes were blocked with 5% non-fat dried milk in TBS containing 0.1% Tween-20 (TBS-T) for 30 min at RT, followed by incubation with a primary antibody diluted in TBS-T overnight at 4°C. After washing with TBS-T, the membranes were incubated with an HRP-conjugated secondary antibody in TBS-T for 1 h at RT. After washing with TBS-T, the membranes were incubated with ECL Prime (GE Healthcare) and developed using LAS4000 (GE Healthcare).
FFEM
Freeze fracture replicas were produced using a previously described method (Tarusawa et al., 2009) with some modifications. Cells (2 × 105 cells/ml) were seeded onto polyethylene terephthalate filters with 0.4-µm pore size (#353090; Corning) and cultured for 6 d. The cells were washed with 0.1 M phosphate buffer (PB) and fixed with 2% glutaraldehyde in 0.1 M PB at 4°C overnight. After washing with 0.1 M PB, the samples were cryoprotected with 30% glycerol in 0.1 M PB at 4°C overnight, and then rapidly frozen in between two copper carriers using a high-pressure freezing machine (HPM010; BAL-TEC). The cells were then fractured by separation of the two carriers at −120°C and replicated by platinum (45° unidirectional from horizontal level, 2 nm thick) and carbon (20 nm thick) in a freeze-fracture replica machine (BAF060; BAL-TEC). The replicated materials were transferred to a solution containing kitchen bleach (50%) and incubated with shaking until cell debris was removed from the replicas. The replicas were washed twice with distilled water and picked up onto grids coated with Pioloform (Agar Scientific). The samples were observed with a JEM1010 transmission EM (JEOL) at 100 kV accelerating voltage. Images were captured with a Veleta CCD camera using iTEM software (Olympus Soft Imaging Solutions). All freeze-fracture images are presented apical-side up in the figures.
Treatment of cells with trypsin
Cells were cultured on culture dishes (for immunoprecipitation), coverslips (for immunostaining of A431 cells), or 12-well polycarbonate Transwell filters with 0.4-µm pore size (#3401; Corning) at 1.0 × 105 cells/ml (for immunostaining of MDCK II cells). Cells were washed with FBS-free DMEM thrice at 37°C, treated with Trypsin (0.25% [wt/vol] Trypsin-1 mM EDTA•4Na solution; #209-16941; Fujifilm-WAKO) diluted with FBS-free DMEM, and incubated at 37°C for 15 min in a 5% CO2 incubator. As EDTA was diluted more than 10 times with DMEM, which contains 1.8 mM CaCl2 and 1 mM MgCl2, it did not affect the cell adhesion. The cells were then washed and processed for subsequent applications.
TER measurement
Cells were seeded onto 12-well polycarbonate Transwell filters with 0.4-µm pore size at 1.0 × 105 cells/ml and cultured for 7 d. The TER of the cell sheet was measured using a volt-ohm meter (Millicell ERS-2; EMD Millipore). All measurements were subtracted by a blank measurement of a Transwell filter with medium alone and then multiplied by the culture area of the Transwell filter to calculate the unit area resistance.
Tracer flux measurement
Simulation of ion permeability using simplified TJ-strand network models
To obtain the TER value for the model, the values for 106 s are averaged. The simulation was performed using Matlab software (Release 2021a; The MathWorks).
AlphaFold2 prediction of the structure of EpCAM–claudin-7 complex
The prediction was performed using AlphaFold Colab on Google Scholar (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb; Jumper et al., 2021) and visualized using Open-Source PyMOL (The PyMOL Molecular Graphics System, Version 2.5.0, Schrödinger, LLC).
ZnUMBA
A detailed protocol for ZnUMBA using MDCK II cells will be published elsewhere (Higashi et al., 2022 Preprint). Briefly, the cells were seeded onto the bottom surfaces of 12-well polycarbonate Transwell filters with 0.4-µm pore size (#3401; Corning) at a density of 1.0 × 105 cells/cm2 and cultured until TER reached a plateau. Before imaging, both the apical and basal sides of the cells were washed twice with Z medium (30% HBSS, 65% phenol red-free DMEM [D1145; Sigma-Aldrich], and 5% FBS). The filter was placed on a glass-bottom dish containing 2 mM of ZnCl2 in Z medium, which faced the apical side of the cells. 10 µM of FluoZin-3 and 1 µM of Ca-EDTA in Z medium were placed in the filter, which faced the basal side of the cells. The cells were imaged using a confocal microscope, as described above. At the end of the imaging, the addition of excess ZnCl2 to the basal medium helped in the identification of the cell borders.
Statistical analysis
The results are presented as the mean ± SD, unless otherwise indicated. Statistical significance of the differences was evaluated using the two-tailed Welch’s t test (Figs. 2, F and I, 4 B, 5, G and I, 6, C and D, 7, D and G, S2 L, and S5, A–G), the two-tailed paired t test (Figs. 4, C and D, 5 J, 7 G, and 9, D and H), the exact Poisson test (Figs. 2 H, 5 F, and 7 C) and Steel-Dwass test (Fig. 6 E). Bonferroni correction was used to address the multiple comparison problem in Figs. 5 I, 6 D, 7 F, S2 L, and S5, A–G. All tests were performed using Microsoft Excel, except for the calculation of P value in the Steel-Dwass test, which was performed using the R software.
Online supplemental material
Fig. S1 shows localization patterns of claudins in the Ctrl cells and A431 cells treated with trypsin. Fig. S2 shows KO of EpCAM and claudin-7 genes. Fig. S3 shows gene-KO strategies and genomic structure of MASP genes. Fig. S4 shows RT-PCR and immunoblotting of the MASP-KO cells. Fig. S5 shows barrier function of MASP-KO cells. Video 1 shows live imaging of permeability in MASP-qKO cells by ZnUMBA assay.
Acknowledgments
We would like to express our gratitude to Seiko Watanabe, Ayumi Hozumi, Keiko Watari, Misa Sato, Takako Maegawa, and Hitoshi Takagi for their technical assistance; the members of the Laboratory Animal Research Center of Fukushima Medical University for the maintenance of animals; and Dr. Ann L. Miller (University of Michigan), Dr. Ryutaro Shirakawa (Tohoku University), and Dr. Tetsuhisa Otani (National Institute for Physiological Sciences, Japan) for the critical review of our manuscript.
This work was supported by the Cooperative Study Program (19-136 and 22NIPS107) of the National Institute for Physiological Sciences, and by the Japan Society for the Promotion of Science KAKENHI (Grant Number JP16H06280), Grant-in-Aid for Scientific Research on Innovative Areas - Platforms for Advanced Technologies and Research Resources “Advanced Bioimaging Support (ABiS)”to Y. Fukazawa. This work was funded by the Japan Society for the Promotion of Science KAKENHI (Grant Number 21K06156) to T. Higashi, and partly supported by the Grant Program for Biomedical Engineering Research from the Nakatani Foundation to T. Higashi.
The authors declare no competing financial interests.
Author contributions: This project was initiated by M. Furuse and designed by T. Higashi. A.C. Saito and Y. Fukazawa performed the FFEM microscopy. A.C. Saito performed the prediction of permeability using simplified TJ-strand network models. T. Higashi and A. Higashi produced a monoclonal antibody against TMPRSS14. A. Higashi performed immunostaining of frozen sections of mouse epithelial tissues. M. Ono performed part of the immunoprecipitation experiments with input from A.C. Saito. T. Higashi designed and carried out all other experiments and conducted the analyses, with inputs from M. Furuse and H. Chiba. T. Higashi wrote the manuscript with inputs from other authors, and all authors reviewed and revised the manuscript.

