


IRSp53 (aka BAIAP2) is a scaffold protein that couples membranes with the cytoskeleton in actin-filled protrusions such as filopodia and lamellipodia. The protein is abundantly expressed in excitatory synapses and is essential for synapse development and synaptic plasticity, although with poorly understood mechanisms. Here we show that specific multivalent interactions between IRSp53 and its binding partners PSD-95 or Shank3 drive phase separation of the complexes in solution. IRSp53 can be enriched to the reconstituted excitatory PSD (ePSD) condensates via bridging to the core and deeper layers of ePSD. Overexpression of a mutant defective in the IRSp53/PSD-95 interaction perturbs synaptic enrichment of IRSp53 in mouse cortical neurons. The reconstituted PSD condensates promote bundled actin filament formation both in solution and on membranes, via IRSp53-mediated actin binding and bundling. Overexpression of mutants that perturb IRSp53–actin interaction leads to defects in synaptic maturation of cortical neurons. Together, our studies provide potential mechanistic insights into the physiological roles of IRSp53 in synapse formation and function.
Introduction
Dendritic spines are small protrusions that grow from dendritic shafts. Their growth initiates from thin, filopodia-like structures that form contacts with target presynaptic axon termini. Once a synaptic contact is established, a filopodium stabilizes and matures into more variable morphologies, typically characterized by a bulbous, mushroom-like head and a thin neck. Postsynaptic density (PSD) at the tip of a spine head is directly opposed to the active zone of the contacting presynaptic bouton. PSD, which is an electron-dense assembly beneath the postsynaptic membrane, is composed of thousands of proteins with a wide range of abundances (Cohen et al., 1977; Feng and Zhang, 2009; Sheng and Hoogenraad, 2007; Triller and Choquet, 2008). Examples of PSD constituents include adhesion proteins, receptors, and ion channels; scaffold proteins; signaling molecules; and actin cytoskeleton elements. In response to stimulation or inhibition, spines undergo changes in molecular composition and head morphology to tune their strengths and/or efficacy of synaptic transmission (Chen and Sabatini, 2012; Kasai et al., 2003; Matsuzaki et al., 2001; Matsuzaki et al., 2004; Nishiyama and Yasuda, 2015). Importantly, the volume of the spine head is proportional to the area of PSD and the number of glutamate receptors on PSD membranes. PSDs in excitatory synapses display layered organizations (Dosemeci et al., 2016; Harris and Weinberg, 2012; Zhu et al., 2016). The PSD core or the top layer, which refers to the layer ∼0–40 nm from the postsynaptic membranes, contains the MAGUK family scaffold proteins, such as PSD-95, that directly bind to and cluster both N-methyl-D-aspartate (NMDA) receptors and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Chen et al., 2008; MacGillavry et al., 2013; Nair et al., 2013). The PSD pallium or the bottom layer, which refers to the layer ∼40–100 nm from the postsynaptic membranes, contains scaffold proteins including Shank and Homer and their contacting actin cytoskeleton (Dosemeci et al., 2016). These two layers are connected by guanylate kinase–associated protein (GKAP; aka SAPAP or DLGAP), which shows direct interactions with the major scaffolds in both layers (Zeng et al., 2016b; Zhu et al., 2017). Our recent studies have demonstrated that formation of PSD assemblies might be driven by liquid-liquid phase separation (LLPS) of the major scaffold proteins in both layers of PSDs (Cai et al., 2021; Zeng et al., 2018; Zeng et al., 2019; Zeng et al., 2016a). The in vitro reconstituted minimal excitatory PSD (ePSD) platform provides us new toolkits to investigate the functional roles of PSD proteins and to study potential molecular mechanisms governing synapse formation and regulation.
The insulin receptor substrate protein 53 (IRSp53), also known as brain-specific angiogenesis inhibitor I–associated protein 2 (BAIAP2), is one of the most abundant proteins expressed at the ePSD (Lowenthal et al., 2015). Immuno-EM studies in cultured hippocampal neurons demonstrated that IRSp53 is distributed both in the core and pallium layers of PSDs (Burette et al., 2014; Dosemeci et al., 2017). Under excitatory conditions, an increasing amount of protein accumulates in the PSD, especially toward the pallium layer. In non-neuronal cells, the function of IRSp53 has been extensively studied. It is implicated in coupling membrane modulation with actin dynamics in the formation of filopodia and lamellipodia that underlies a wide range of cellular processes including cell motility, tumor invasion, and cell migration (Ahmed et al., 2010; Funato et al., 2004; Liu et al., 2010; Scita et al., 2008; Suetsugu et al., 2006). Human genetic studies have identified several mutations in IRSp53 that associate with neuropsychiatric disorders such as autism and schizophrenia (Kang et al., 2016). IRSp53 knockout mice show synaptic and behavioral abnormalities that resemble symptoms of patients bearing IRSp53 mutations. The IRSp53−/− mice display decreased excitatory synaptic activities and reduced spine densities in the medial prefrontal cortex, but no spine density changes were observed in the hippocampus (Chung et al., 2015; Kim et al., 2009; Sawallisch et al., 2009), indicating brain region–specific functions of the protein. These studies suggest the functional significance of IRSp53 in synaptogenesis, but it is unclear why this protein could influence spine development. It is important to understand IRSp53 function in neurons, but such studies are complicated for the following reasons: (1) the high functional redundancy in the context of synapses, whereby so many proteins in the dendrite are capable of functioning together and compensating for each other’s loss of function; (2) the severe cross-talk between different pathways, as a single domain in IRSp53 can bind to multiple synaptic proteins involved in various functions, and the specificities of these interactions are poorly characterized; and (3) the tiny size of the spine compartment and the extreme heterogeneity within spines, which have made it challenging to perform high-resolution optical imaging studies to differentiate different IRSp53-mediated interactions within a dendritic spine.
Combining biochemical reconstitution approaches with neuronal overexpression studies, we demonstrate here that IRSp53, in parallel with GKAP, serves as a connection node for scaffolding the layered PSD assemblies. Specific multivalent interactions between IRSp53 and PSD-95 or Shank3 together with the BAR domain–mediated dimerization of IRSp53 lead to formation of self-organized, condensed, and dynamic assemblies through LLPS. IRSp53 incorporation into the reconstituted PSD condensates further promotes LLPS of the PSD mixture. Here we also uncover IRSp53 as a novel and direct modulator of actin dynamics. The actin-bundling activity of IRSp53 is significantly enhanced by forming condensates with PSD proteins. Point mutations that perturb IRSp53 binding to actin strongly block the formation of filament bundles in vitro and lead to severe defects in spine head maturation when overexpressed in cultured cortical neurons. Our work identifies IRSp53 as a scaffold linker for PSD assembly and a direct regulator of actin cytoskeleton organization via LLPS in vitro, and thus suggests a model for how IRSp53 can modulate spine development in neurons.
Results
PSD-95 and IRSp53 undergo phase separation in vitro
Previous studies have demonstrated that IRSp53 binds to PDZ domains of PSD-95 via its PDZ binding motif (PBM; Choi et al., 2005; Soltau et al., 2004). IRSp53 is abundantly expressed in ePSDs with a stoichiometric ratio of about one quarter to PSD-95 (Lowenthal et al., 2015). It consists of an N-terminal inverse BAR (I-BAR) domain, a CRIB-PR domain, an SH3 domain, and a PBM (Fig. 1 A). The I-BAR is an antiparallel dimer that binds to negatively charged 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-4′,5′-bisphosphate), or PI[4,5]P2 and induces negative membrane curvatures (Lee et al., 2007; Mattila et al., 2007; Millard et al., 2005; Prevost et al., 2015; Saarikangas et al., 2009; Suetsugu et al., 2006). IRSp53 alone is in an autoinhibited conformation with the SH3 domain binding to its own CRIB-PR (Kast et al., 2014). Simultaneous bindings of Cdc42 to CRIB-PR and effectors to the SH3 domain release the autoinhibition of IRSp53.
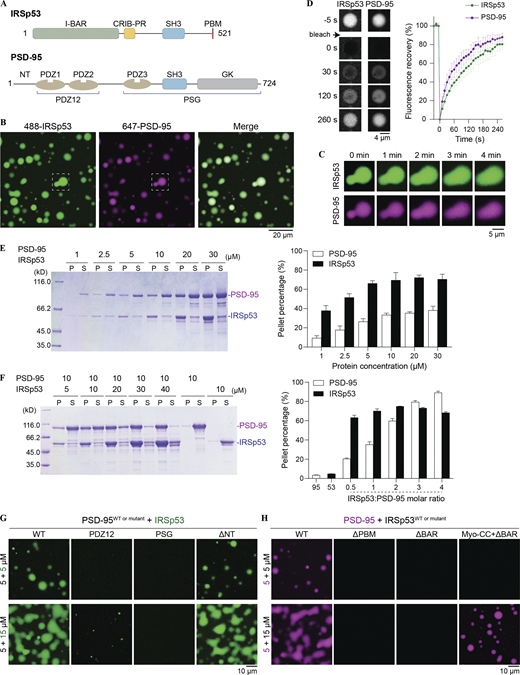
Multivalent interactions drive the condensed phase formation of the PSD-95/IRSp53 mixture. (A) Schematic diagrams showing the domain organization of IRSp53 and PSD-95. PDZ1 and PDZ2 tandem is referred to as PDZ12; PDZ3-SH3-GK supramodule is referred to as PSG. (B) Fluorescence images showing that IRSp53 and PSD-95 are colocalized and enriched in condensed droplets. The dashed box is selected for zoom-in view of the droplet fusion process in C. IRSp53 and PSD-95 were mixed at an equimolar ratio at 10 μM, and 2% of each protein was labeled by the indicated fluorophore. This fluorophore labeling ratio was used throughout the study unless otherwise stated. (C) Spherical droplets fuse into larger ones over time upon contact. (D) FRAP analysis of IRSp53 and PSD-95 in the condensed phase showing dynamic exchange of proteins between the condensates and the dilute solution. The concentration of each protein was 10 μM. The recovery curves of the fluorescence signals represent the averaged signals from three droplets, and data are presented as means ± SD. (E) Left, representative SDS-PAGE of sedimentation-based assays showing the amount of PSD-95 and IRSp53 recovered from the condensed phase or pellet (P) and the aqueous phase or supernatant (S) at indicated protein concentrations. Right, quantification of protein distribution in E. Results are from three independent batches of experiments and are presented as mean ± SD. (F) Left, sedimentation experiments showing the distribution of PSD-95 and IRSp53 in P and S at indicated protein concentrations. Right, quantification of protein distribution in F. Results are from three independent batches of experiments and are presented as mean ± SD. (G) Fluorescence images showing truncations of PSD-95 with weakened LLPS capability with IRSp53. The N-terminal 30 residues proceeding PDZ1 (NT in A) are not required for phase separation. (H) Fluorescence images showing truncations of IRSp53 with weakened LLPS capability with PSD-95. Replacement of the BAR domain with Myo-CC retained its phase separation with PSD-95. Source data are available for this figure: SourceData F1.
Multivalent interactions drive the condensed phase formation of the PSD-95/IRSp53 mixture. (A) Schematic diagrams showing the domain organization of IRSp53 and PSD-95. PDZ1 and PDZ2 tandem is referred to as PDZ12; PDZ3-SH3-GK supramodule is referred to as PSG. (B) Fluorescence images showing that IRSp53 and PSD-95 are colocalized and enriched in condensed droplets. The dashed box is selected for zoom-in view of the droplet fusion process in C. IRSp53 and PSD-95 were mixed at an equimolar ratio at 10 μM, and 2% of each protein was labeled by the indicated fluorophore. This fluorophore labeling ratio was used throughout the study unless otherwise stated. (C) Spherical droplets fuse into larger ones over time upon contact. (D) FRAP analysis of IRSp53 and PSD-95 in the condensed phase showing dynamic exchange of proteins between the condensates and the dilute solution. The concentration of each protein was 10 μM. The recovery curves of the fluorescence signals represent the averaged signals from three droplets, and data are presented as means ± SD. (E) Left, representative SDS-PAGE of sedimentation-based assays showing the amount of PSD-95 and IRSp53 recovered from the condensed phase or pellet (P) and the aqueous phase or supernatant (S) at indicated protein concentrations. Right, quantification of protein distribution in E. Results are from three independent batches of experiments and are presented as mean ± SD. (F) Left, sedimentation experiments showing the distribution of PSD-95 and IRSp53 in P and S at indicated protein concentrations. Right, quantification of protein distribution in F. Results are from three independent batches of experiments and are presented as mean ± SD. (G) Fluorescence images showing truncations of PSD-95 with weakened LLPS capability with IRSp53. The N-terminal 30 residues proceeding PDZ1 (NT in A) are not required for phase separation. (H) Fluorescence images showing truncations of IRSp53 with weakened LLPS capability with PSD-95. Replacement of the BAR domain with Myo-CC retained its phase separation with PSD-95. Source data are available for this figure: SourceData F1.
We purified the full-length IRSp53 and PSD-95 to study their interaction. Individually, both proteins were soluble in solution, but the solution immediately turned turbid when IRSp53 and PSD-95 were mixed at a 1:1 molar ratio at room temperature. Fluorescence-labeled IRSp53 and PSD-95 formed spherical droplets, with enrichment of both proteins in the condensed phase (Fig. 1 B). These droplets fused into larger ones over time upon contact (Fig. 1 C). FRAP analysis of the proteins in the condensates showed that both components recovered up to ∼80% of their initial concentration, indicating that protein constituents could freely exchange between the condensate droplets and the surrounding dilute solution (Fig. 1 D). We further used sedimentation-based experiments to quantify the distribution of proteins between the condensed phase and the aqueous solution (Fig. 1 E). IRSp53/PSD-95 condensates assembled in a concentration-dependent manner. Protein condensation first appeared at 1 μM and further increased with increasing protein concentrations, reaching saturation at ≥10 μM. Manipulation of the stoichiometric ratio of IRSp53 to PSD-95 could further alter the phase separation (Fig. 1 F). When PSD-95 was kept at 10 μM, the amount of PSD-95 enrichment in the pellet fraction increased with the increasing concentrations of IRSp53 until the ratio of IRSp53 to PSD-95 reached 3:1.
Multivalent interactions drive the formation of PSD-95/IRSp53 condensates
To better understand mechanisms that govern the LLPS of the PSD-95 and IRSp53 mixture, we performed isothermal titration calorimetry (ITC)-based binding assays to measure the interaction affinity between IRSp53 and different PDZ domains of PSD-95 (Fig. S1 A). The full-length protein showed binding strengths similar to the three PDZ domains individually. The PDZ12 tandem of PSD-95 showed slightly stronger binding to IRSp53 compared with the individual PDZ domains (i.e., PDZ1 or PDZ2 alone), suggesting the presence of a weaker cooperative interaction mode. Deletion of PBM abolished IRSp53’s binding to PSD-95, further supporting that PDZ/PBM-mediated interactions underline the IRSp53/PSD-95 complex formation (Fig. S1, B and C). We then analyzed the contribution of different PDZ domains to phase separation using imaging-based assays (Fig. 1 G). The concentration of PSD-95 was fixed at 5 μM, and two concentrations of IRSp53 (5 and 15 μM) were assayed. At both concentrations, the PSG (PDZ3-SH3-GK) supramodule showed no detectable condensate formation with IRSp53. At the IRSp53-to-PSD-95 molar ratio of 3:1, the PDZ12 tandem exhibited weak phase separation with IRSp53. Deleting the N-terminal region (NT in Fig. 1 A) did not affect phase separation of PSD-95 with IRSp53. These results indicated that IRSp53/PSD-95 phase separation depends on the multiple PDZ domains of PSD-95.

IRSp53 binds to PSD-95 via PDZ/PBM interactions. (A) ITC-based measurements of interactions between the PDZ domains of PSD-95 and the full-length IRSp53. 400 μΜ of PSD-95_PDZ1, PDZ2, or PSG was titrated into 20 μM of IRSp53. 200 μΜ of PSD-95_PDZ12 was titrated into 20 μM of IRSp53. (B and C) ITC-based assay measuring the binding affinity of different PSD-95 constructs to IRSp53_ΔPBM. 200 μM PSD-95 was titrated into 20 μM IRSp53_ΔPBM.
IRSp53 binds to PSD-95 via PDZ/PBM interactions. (A) ITC-based measurements of interactions between the PDZ domains of PSD-95 and the full-length IRSp53. 400 μΜ of PSD-95_PDZ1, PDZ2, or PSG was titrated into 20 μM of IRSp53. 200 μΜ of PSD-95_PDZ12 was titrated into 20 μM of IRSp53. (B and C) ITC-based assay measuring the binding affinity of different PSD-95 constructs to IRSp53_ΔPBM. 200 μM PSD-95 was titrated into 20 μM IRSp53_ΔPBM.
We next analyzed regions on IRSp53 that may contribute to the LLPS of PSD-95 and IRSp53 (Fig. 1 H). The IRSp53_ΔPBM mutant was not capable of forming condensates with PSD-95 under both assay conditions. Deleting the dimerizing BAR domain from the full-length protein abolished IRSp53 phase separation with PSD-95. An IRSp53 mutant with its BAR domain replaced by the antiparallel coiled-coil dimer of myosin X (Myo-CC; Lu et al., 2012) retained its phase separation with PSD-95 (Figs. 1 H and S2). These results demonstrated that the self-association of IRSp53 is essential for its phase separation with PSD-95.

Oligomerization status of IRSp53 constructs in solution. (A) miniDawn analysis showing that the full-length IRSp53 exists as a dimer at 100 μM in solution. Superose 6 increase column was used for size-exclusion chromatography. Calculated MW of IRSp53 is 57.4 kD. (B) miniDawn analysis showing IRSp53 becomes monomeric after removal of the BAR domain; replacement of the BAR with a dimeric coiled coil from myosin X could restore its oligomerization. Superose 12 10/300 column was used for size-exclusion chromatography. Analysis was performed at 100 μM protein concentration. Calculated MW of IRSp53_ΔBAR is 28.5 kD, and that of Myo-CC+ΔBAR is 34.4 kD.
Oligomerization status of IRSp53 constructs in solution. (A) miniDawn analysis showing that the full-length IRSp53 exists as a dimer at 100 μM in solution. Superose 6 increase column was used for size-exclusion chromatography. Calculated MW of IRSp53 is 57.4 kD. (B) miniDawn analysis showing IRSp53 becomes monomeric after removal of the BAR domain; replacement of the BAR with a dimeric coiled coil from myosin X could restore its oligomerization. Superose 12 10/300 column was used for size-exclusion chromatography. Analysis was performed at 100 μM protein concentration. Calculated MW of IRSp53_ΔBAR is 28.5 kD, and that of Myo-CC+ΔBAR is 34.4 kD.
To summarize, interactions between multiple PDZ domains on PSD-95 and the single PBM motif on IRSp53 are important for LLPS of the mixture in vitro. Dimerization of IRSp53 via the BAR domain provides an additional level of multivalency for the protein complex to assemble into crosslinked networks that can autonomously assemble into condensed droplets via phase separation.
Shank3 binds to IRSp53, and the complex undergoes LLPS in vitro
The SH3 domain of IRSp53 was reported to bind multiple actin regulatory proteins, including Eps8 (Disanza et al., 2006; Funato et al., 2004; Kast et al., 2014), VASP (Boczkowska et al., 2013), N-WASP (Lim et al., 2008), WAVE2 (Goh et al., 2012; Miki et al., 2000), Mena (Krugmann et al., 2001), and Shank3 (Bockmann et al., 2002). As a result of its interaction with different downstream effector proteins, IRSp53 plays critical roles in the formation of filopodia and lamellipodia in a broad range of cellular activities. Shank is abundantly expressed in ePSDs and is a major scaffold protein involved in the formation and function of synapses (Naisbitt et al., 1999; Sheng and Hoogenraad, 2007). Shank family proteins are encoded by Shank1, Shank2, and Shank3. Shank3 is the best studied among the three family members. Shank3 contains an N-terminal domain, multiple ankyrin repeats (ANK), a SH3 domain, a PDZ domain, a long stretch of proline-rich sequence and a sterile α motif (SAM; Fig. 2 A). The N-terminal domain–ANK tandem was recently found to specifically interact with CaMKIIα, the most abundant protein in PSD (Cai et al., 2021). The PDZ domain specifically binds to GKAP with a high affinity (Zeng et al., 2016b). The SAM domain contributes to Shank3 oligomerization (Hayashi et al., 2009). The proline-rich region of Shank3 directly interacts with multiple SH3 domain–containing proteins, such as Homer (Tu et al., 1999), cortactin (Naisbitt et al., 1999), and IRSp53 (Bockmann et al., 2002). Previous studies have mapped the region of Shank that binds to IRSp53 (Bockmann et al., 2002; Soltau et al., 2002), and we call it proline rich motif (PRM). Multiple sequence alignment of PRMs from the Shank family members of different mammalian species revealed that this region is highly conserved (Fig. 2 A). The PRM can be further divided into two consecutive Pro-rich clusters, denoted PRM1 and PRM2, with the insertion of a spacing linker only present in Shank1.
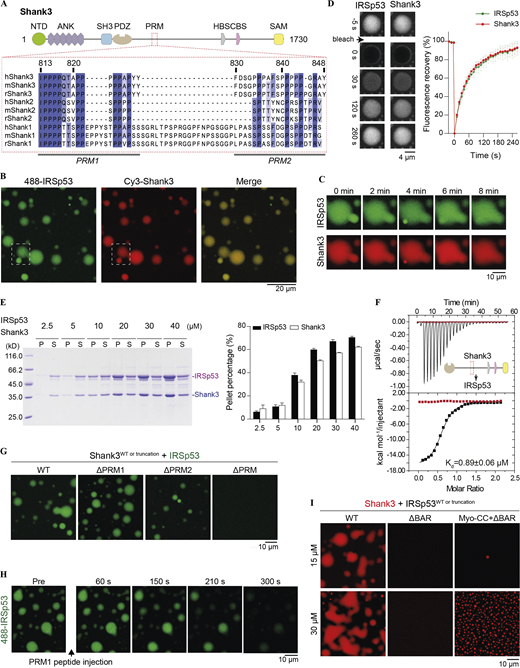
Multivalent interactions drive the condensed phase formation of the IRSp53/Shank3 mixture. (A) Schematic diagram showing the domain organization of Shank3. Multiple sequence alignment of the proline-rich motif (PRM), which is responsible for IRSp53 to interact with different isoforms of Shank from human (h), mouse (m), and rat (r). (B) Fluorescence images showing IRSp53 and Shank3 are colocalized and enriched in the condensate droplets. IRSp53 and Shank3 were mixed at an equimolar ratio at 15 μM. The dashed box is selected for zoom-in view of the droplet fusion process in C. (C) Phase droplets that are in close contact fuse into larger ones over time. (D) FRAP experiments showing fluorescence recovery of IRSp53 and Shank3 in a condensed droplet after photobleaching. Fluorescence recovery curves represent the averaged signals from three droplets, and data are presented as mean ± SD. (E) Sedimentation-based experiments showing that IRSp53 and Shank3 undergo phase separation in a concentration-dependent manner. The amounts of proteins in the condensed phase/pellet fraction (P) and in the dilute phase/supernatant fraction (S) are quantified. Data are obtained from three batches of independent experiments and are presented as mean ± SD. (F) ITC-based measurement of the binding between IRSp53 and Shank3 WT (black titration curve) or ΔPRM (red titration curve). 200 μM of Shank3 was titrated into 20 μM of IRSp53 with 200 mM NaCl in the binding buffer. (G) Fluorescence imaging analysis showing that a series of Shank3 mutants with progressively weakened phase separation with IRSp53. 15 μΜ Alexa Fluor 488–labeled IRSp53 was mixed with 15 μΜ unlabeled various truncations of Shank3. Identical imaging settings were used for all groups. (H) Fluorescence images showing that the preformed IRSp53/Shank3 condensate droplets could be gradually dispersed by the addition of the PRM1 peptide. Both IRSp53 and Shank3 were at 15 μΜ. (I) Fluorescence imaging analysis showing that the BAR domain–mediated dimerization is critical for IRSp53/Shank3 phase separation. Myo-CC restores the oligomerization status of IRSp53 (also see Fig. S2), and the replacement of BAR with Myo-CC could rescue the loss of phase separation of IRSp53 with Shank3 resulting from the BAR domain deletion. Two protein concentrations, 15 and 30 μM, were assayed, and both were mixed at an equimolar ratio. Source data are available for this figure: SourceData F2.
Multivalent interactions drive the condensed phase formation of the IRSp53/Shank3 mixture. (A) Schematic diagram showing the domain organization of Shank3. Multiple sequence alignment of the proline-rich motif (PRM), which is responsible for IRSp53 to interact with different isoforms of Shank from human (h), mouse (m), and rat (r). (B) Fluorescence images showing IRSp53 and Shank3 are colocalized and enriched in the condensate droplets. IRSp53 and Shank3 were mixed at an equimolar ratio at 15 μM. The dashed box is selected for zoom-in view of the droplet fusion process in C. (C) Phase droplets that are in close contact fuse into larger ones over time. (D) FRAP experiments showing fluorescence recovery of IRSp53 and Shank3 in a condensed droplet after photobleaching. Fluorescence recovery curves represent the averaged signals from three droplets, and data are presented as mean ± SD. (E) Sedimentation-based experiments showing that IRSp53 and Shank3 undergo phase separation in a concentration-dependent manner. The amounts of proteins in the condensed phase/pellet fraction (P) and in the dilute phase/supernatant fraction (S) are quantified. Data are obtained from three batches of independent experiments and are presented as mean ± SD. (F) ITC-based measurement of the binding between IRSp53 and Shank3 WT (black titration curve) or ΔPRM (red titration curve). 200 μM of Shank3 was titrated into 20 μM of IRSp53 with 200 mM NaCl in the binding buffer. (G) Fluorescence imaging analysis showing that a series of Shank3 mutants with progressively weakened phase separation with IRSp53. 15 μΜ Alexa Fluor 488–labeled IRSp53 was mixed with 15 μΜ unlabeled various truncations of Shank3. Identical imaging settings were used for all groups. (H) Fluorescence images showing that the preformed IRSp53/Shank3 condensate droplets could be gradually dispersed by the addition of the PRM1 peptide. Both IRSp53 and Shank3 were at 15 μΜ. (I) Fluorescence imaging analysis showing that the BAR domain–mediated dimerization is critical for IRSp53/Shank3 phase separation. Myo-CC restores the oligomerization status of IRSp53 (also see Fig. S2), and the replacement of BAR with Myo-CC could rescue the loss of phase separation of IRSp53 with Shank3 resulting from the BAR domain deletion. Two protein concentrations, 15 and 30 μM, were assayed, and both were mixed at an equimolar ratio. Source data are available for this figure: SourceData F2.
We first sought to verify the previously reported interaction between Shank and IRSp53 using purified recombinant proteins. To obtain soluble and well behaving Shank3, we removed part of its sequence (Zeng et al., 2018). When full-length IRSp53 was mixed with Shank3, the solution immediately turned turbid. In contrast, solutions containing each individual component were always clear. Using fluorescence microscopy, we observed formation of round droplets with significant enrichments of both proteins (Fig. 2 B). These droplets rapidly fused upon contact (Fig. 2 C). Photobleaching of the labeled proteins in droplets resulted in fast recovery of fluorescence signals to almost 100%, consistent with the liquid-like properties of the condensed phase (Fig. 2 D). Using sedimentation-based experiments, we further demonstrated that IRSp53 and Shank3 could form phase condensates at a 1:1 molar ratio at a concentration as low as 5 μM and with a saturation concentration of ≥20 μM (Fig. 2 E).
Next, we wanted to determine interactions that might drive Shank3/IRSp53 phase separation. We performed ITC-based binding assays in a buffer containing 200 mM salt to eliminate potential heat change resulting from the process of phase separation. We detected sub-micromolar binding between Shank3 and IRSp53 (Kd ∼ 0.89 ± 0.06 μM; Fig. 2 F). When proteins were combined at 15 μΜ (close to the LLPS saturation concentration) under physiological salt condition (100 mM NaCl), micrometer-sized droplets were observed. Removal of either PRM1 or PRM2 from Shank3 slightly reduced its phase separation with IRSp53, and in the absence of the entire PRM, Shank3_ΔPRM could no longer form condensates with IRSp53 (Fig. 2 G). To address the role of the SH3 domain of IRSp53 in phase separation, we attempted to introduce a point mutation in the SH3 domain or deletion of the entire domain. However, these mutants could not be analyzed owing to their instability (i.e., severe protein degradation). As an alternative, we injected an excess amount of the synthetic PRM1 peptide to the preformed Shank3/IRSp53 droplets (Fig. 2 H). We predicted that the addition of an excess amount of the PRM1 peptide to the mixture would disperse the condensed phase due to disruption of the multivalent interactions between the proteins. Indeed, the addition of the PRM1 peptide led to immediate dispersion of the droplets into homogeneous aqueous solution. Furthermore, IRSp53 dimerization was also required for phase separation to occur, since the monomeric IRSp53_ΔBAR failed to form condensates with Shank3, whereas replacement of the BAR domain with Myo-CC retained the phase separation of IRSp53 with Shank3 (Fig. 2 I). Taking the above results together, we conclude that IRSp53 is a specific binding partner of Shank3 and that the multivalent interactions between the two proteins drive LLPS of the complex in vitro.
IRSp53 enrichment into reconstituted PSD condensates
Our observations above suggested that IRSp53 might function as a scaffolding linker that can simultaneously bind to PSD-95, the top layer scaffold, and to Shank3, the bottom layer scaffold (Fig. 3 A). Previous studies have demonstrated that GKAP plays a similar role. GKAP interacts with both PSD-95 and Shank3 and is required for their synaptic localization in vivo (Zeng et al., 2016b; Zhu et al., 2017). In reconstituted PSD assemblies, removal of GKAP significantly diminished the enrichment of all other PSD components in the condensed phase (Zeng et al., 2018). Thus, we hypothesized that GKAP and IRSp53 function in parallel to drive the assembly of PSD condensates (Fig. 3 A). Consistent with previous studies, the condensation of other PSD scaffolds was nearly abolished when GKAP was removed from the 4× PSD system (PSD-95, Shank3, Homer3, and GKAP). Interestingly, the addition of IRSp53 was able to rescue the recruitment of all components into the condensed phase (Fig. 3 B).
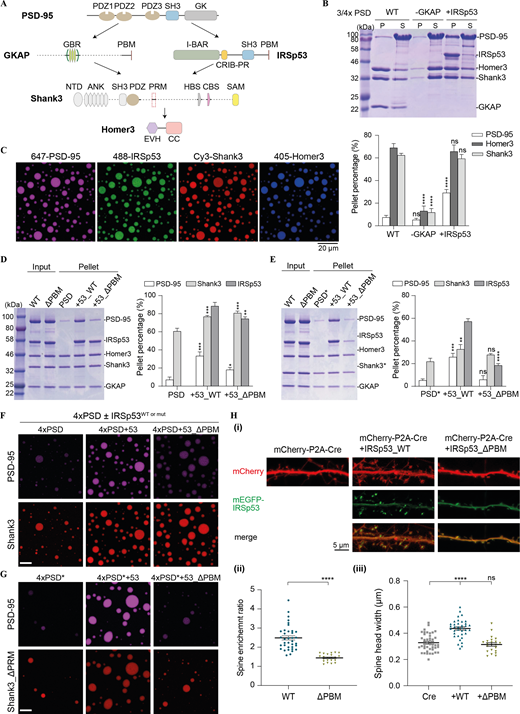
IRSp53-dependent phase separation of the PSD complex. (A) Schematic diagram showing the protein interaction network of the reconstituted 5× PSD. GKAP and IRSp53 act in parallel to connect proteins in the PSD core (PSD-95) and the pallium (Shank3 and Homer3). (B) Sedimentation experiments showing the protein distribution of different PSD components in aqueous (S) or condensed phase (P). Each component was at 5 μM in 3× PSD (PSD-95, Shank3, and Homer3) or 4× PSD (PSD-95 and GKAP or IRSp53, Shank3, and Homer3). Quantifications of the PSD-95, Homer3, and Shank3 distribution are shown underneath the SDS-PAGE. The results are from three independent batches of sedimentation experiments and are presented as mean ± SD. ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (C) Fluorescence images showing that the mixture of 5× PSD components at 5 μM undergoes LLPS at room temperature. PSD-95, IRSp53, Shank3, and Homer3 were labeled with different fluorophores as indicated and were highly enriched into the condensate droplets. GKAP was not labeled and is thus invisible. (D) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD (PSD-95, GKAP, Shank3, and Homer3) was mixed with IRSp53 (WT and mutant) at 5 μM. To help compare the LLPS level in different assay conditions, only the protein distribution in the pellet fraction is shown, with total protein input shown in the second lane. A full SDS-PAGE, including both the pellet and supernatant fractions, is included in Fig. S3 A. Quantifications of the PSD-95, Shank3, and IRSp53 distributions are shown on the right. Quantifications of all PSD components are shown in Fig. S3 A. *, P < 0.05; **, P < 0.01; ***, P < 0.001 using one-way ANOVA with Dunnett’s multiple comparison test. (E) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD* (PSD-95, GKAP, Shank3_ΔPRM, and Homer3) was mixed with IRSp53 (WT and mutant) at 5 μM. To help compare the LLPS levels in different assay conditions, only protein distribution in the pellet fraction is shown, with total protein input shown in the first lane. A full SDS-PAGE, including both the pellet and supernatant fractions, is included in Fig. S3 B. Quantifications of the PSD-95, Shank3, and IRSp53 distributions are shown on the right. Quantifications of all PSD components are shown in Fig. S3 B. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (F) Fluorescence images showing the LLPS level of PSD components when mixed with IRSp53 WT or mutant as illustrated in D. PSD-95 and Shank3 were fluorescently labeled, and their enrichment into the condensates was monitored. Identical imaging settings were used for all groups. (G) Fluorescence images showing the LLPS level of PSD components when mixed with IRSp53 WT or mutant as illustrated in E. PSD-95 and Shank3 WT or ΔPRM were fluorescently labeled, and their enrichment into the condensates was monitored. Identical imaging settings were used for all groups. (H) Representative fluorescence images of cultured IRSp53 knockout cortical neurons with the re-expression of mEGFP-tagged IRSp53 WT or ΔPBM at DIV7 (i). mCherry was cotransfected as the cell fill. Endogenous IRSp53 was deleted upon expression of Cre recombinase, and mEGFP-tagged IRSp53 WT or ΔPBM cDNA was transfected for 10 d. At DIV17, live imaging was performed without fixation. For quantification of the imaging data, spine enrichment (ii) was measured using ImageJ, and spine head width (iii) was measured using IMARIS. 22–42 neurons from three independent batches of cultures were imaged for each group in double-blinded mode (n = 42 for Cre only, n = 36 for Cre co-expression with IRSp53 WT, and n = 22 for Cre coexpression with IRSp53_ΔPBM). Note that data in Fig. 6 were collected from the same set of experiments with the same control groups (i.e., Cre only and Cre co-expression with IRSp53 WT). Error bars indicate mean ± SEM. ****, P < 0.0001 using two-tailed t test (ii) or one-way ANOVA with Dunnett’s multiple comparison test (iii). Source data are available for this figure: SourceData F3.
IRSp53-dependent phase separation of the PSD complex. (A) Schematic diagram showing the protein interaction network of the reconstituted 5× PSD. GKAP and IRSp53 act in parallel to connect proteins in the PSD core (PSD-95) and the pallium (Shank3 and Homer3). (B) Sedimentation experiments showing the protein distribution of different PSD components in aqueous (S) or condensed phase (P). Each component was at 5 μM in 3× PSD (PSD-95, Shank3, and Homer3) or 4× PSD (PSD-95 and GKAP or IRSp53, Shank3, and Homer3). Quantifications of the PSD-95, Homer3, and Shank3 distribution are shown underneath the SDS-PAGE. The results are from three independent batches of sedimentation experiments and are presented as mean ± SD. ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (C) Fluorescence images showing that the mixture of 5× PSD components at 5 μM undergoes LLPS at room temperature. PSD-95, IRSp53, Shank3, and Homer3 were labeled with different fluorophores as indicated and were highly enriched into the condensate droplets. GKAP was not labeled and is thus invisible. (D) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD (PSD-95, GKAP, Shank3, and Homer3) was mixed with IRSp53 (WT and mutant) at 5 μM. To help compare the LLPS level in different assay conditions, only the protein distribution in the pellet fraction is shown, with total protein input shown in the second lane. A full SDS-PAGE, including both the pellet and supernatant fractions, is included in Fig. S3 A. Quantifications of the PSD-95, Shank3, and IRSp53 distributions are shown on the right. Quantifications of all PSD components are shown in Fig. S3 A. *, P < 0.05; **, P < 0.01; ***, P < 0.001 using one-way ANOVA with Dunnett’s multiple comparison test. (E) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD* (PSD-95, GKAP, Shank3_ΔPRM, and Homer3) was mixed with IRSp53 (WT and mutant) at 5 μM. To help compare the LLPS levels in different assay conditions, only protein distribution in the pellet fraction is shown, with total protein input shown in the first lane. A full SDS-PAGE, including both the pellet and supernatant fractions, is included in Fig. S3 B. Quantifications of the PSD-95, Shank3, and IRSp53 distributions are shown on the right. Quantifications of all PSD components are shown in Fig. S3 B. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (F) Fluorescence images showing the LLPS level of PSD components when mixed with IRSp53 WT or mutant as illustrated in D. PSD-95 and Shank3 were fluorescently labeled, and their enrichment into the condensates was monitored. Identical imaging settings were used for all groups. (G) Fluorescence images showing the LLPS level of PSD components when mixed with IRSp53 WT or mutant as illustrated in E. PSD-95 and Shank3 WT or ΔPRM were fluorescently labeled, and their enrichment into the condensates was monitored. Identical imaging settings were used for all groups. (H) Representative fluorescence images of cultured IRSp53 knockout cortical neurons with the re-expression of mEGFP-tagged IRSp53 WT or ΔPBM at DIV7 (i). mCherry was cotransfected as the cell fill. Endogenous IRSp53 was deleted upon expression of Cre recombinase, and mEGFP-tagged IRSp53 WT or ΔPBM cDNA was transfected for 10 d. At DIV17, live imaging was performed without fixation. For quantification of the imaging data, spine enrichment (ii) was measured using ImageJ, and spine head width (iii) was measured using IMARIS. 22–42 neurons from three independent batches of cultures were imaged for each group in double-blinded mode (n = 42 for Cre only, n = 36 for Cre co-expression with IRSp53 WT, and n = 22 for Cre coexpression with IRSp53_ΔPBM). Note that data in Fig. 6 were collected from the same set of experiments with the same control groups (i.e., Cre only and Cre co-expression with IRSp53 WT). Error bars indicate mean ± SEM. ****, P < 0.0001 using two-tailed t test (ii) or one-way ANOVA with Dunnett’s multiple comparison test (iii). Source data are available for this figure: SourceData F3.
Next, we tested whether combining IRSp53 and GKAP-driven phase separation would enhance phase separation of other scaffold proteins. When we combined each of the PSD scaffold proteins including PSD-95, GKAP, IRSp53, Shank3, and Homer3 (5× PSD) at physiological concentrations (estimated PSD concentration at ∼5 μM or less), we observed partitioning of nearly all proteins into the condensed phase (Fig. 3, C and D; and Fig. S3 A). Importantly, PSD-95 partitioned much more strongly into IRSp53-containing PSD droplets than 4× PSD droplets (Fig. 3, D and F; and Fig. S3 A). Upon deletion of the PBM motif from IRSp53, the amount of PSD-95 in the condensate droplets was dramatically reduced, but the LLPS efficiency of PSD components in the bottom layer showed only minor changes (Fig. 3, D and F; and Fig. S3 A), likely because of the remaining SH3/PRM interaction between IRSp53_ΔPBM and Shank3. To determine if the IRSp53/Shank3 phase separation also contributed to the LLPS of PSD components, we removed the PRM from Shank3 (denoted Shank3*; Fig. 3, E and G; and Fig. S3 B). Addition of IRSp53 significantly promoted the enrichment of PSD-95 into phase droplets. The overall phase separation capacities in the system also increased but were not as strong as in the 5× PSD containing WT Shank3 (Fig. 3, D and E). Further removal of the PBM motif from IRSp53 dramatically reduced the enrichment of PSD-95 and all other components into the 5× PSD droplets (Fig. 3, E and G; and Fig. S3 B). Together, these results suggest that IRSp53 and GKAP cooperatively promote phase separation of other PSD scaffold components. The simultaneous interaction of IRSp53 with scaffolds in the PSD core and pallium fits well with its distribution pattern in situ, where it is enriched in both PSD layers (Burette et al., 2014; Dosemeci et al., 2017). A possible mechanism is that IRSp53 and GKAP serve as two connection nodes to target Shank and Homer, the bottom layer scaffolds, to PSD-95, the top layer scaffold, forming a major scaffold platform to orchestrate the formation and organization of PSD condensates.
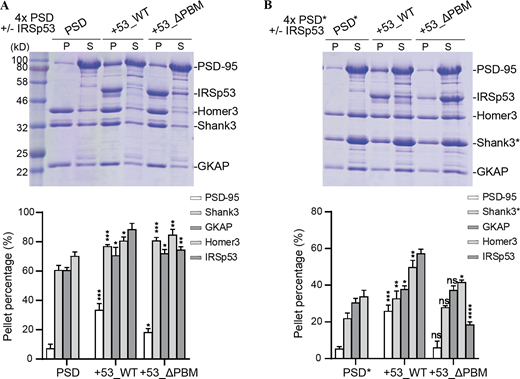
IRSp53-dependent phase separation of the PSD complex. (A) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD (PSD-95, GKAP, Shank3, and Homer3) was mixed IRSp53 (WT and mutant) at 5 μM. Distributions of each PSD component are quantified and shown as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001 using one-way ANOVA with Dunnett’s multiple comparison test. (B) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD* (PSD-95, GKAP, Shank3_ΔPRM, and Homer3) was mixed with IRSp53 (WT and mutant) at 5 μM. Distributions of each PSD component are quantified and shown as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. Source data are available for this figure: SourceData FS3.
IRSp53-dependent phase separation of the PSD complex. (A) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD (PSD-95, GKAP, Shank3, and Homer3) was mixed IRSp53 (WT and mutant) at 5 μM. Distributions of each PSD component are quantified and shown as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001 using one-way ANOVA with Dunnett’s multiple comparison test. (B) Sedimentation experiments showing the phase separation capability of PSD components when 4× PSD* (PSD-95, GKAP, Shank3_ΔPRM, and Homer3) was mixed with IRSp53 (WT and mutant) at 5 μM. Distributions of each PSD component are quantified and shown as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. Source data are available for this figure: SourceData FS3.
IRSp53/PSD condensates regulate neuronal synapse formation
The interaction between PSD-95 and IRSp53 is known to be critical for IRSp53 synaptic localization in cultured neurons (Choi et al., 2005). To overcome potential compensation by endogenous IRSp53, we expressed N-terminal mEGFP-tagged IRSp53 WT and its PSD-95 binding-deficient mutant (ΔPBM) in cortical neurons isolated from IRSp53fl/fl mice, as deletion of IRSp53 specifically impairs synaptic development of cortical neurons (Chung et al., 2015). Consistent with previous publication (Chung et al., 2015), IRSp53 knockout cortical neurons (upon Cre expression) showed significant spine reduction, which could be fully rescued by EGFP-IRSp53 WT re-expression (i.e., co-transfection of mCherry-P2A-Cre and EGFP-IRSp53 WT), but not the ΔPBM mutant (Fig. 3, Hi and Hiii). The PBM deletion also led to diminished localization of IRSp53 at synapses (Fig. 3, H ii). These results are consistent with a model in which the direct interaction and subsequently the phase separation between IRSp53 and PSD-95 are critical for the synaptic localization of IRSp53 and for its normal function in dendritic spine development.
IRSp53 in PSD condensates promotes actin bundling
IRSp53 is a well-known platform protein that couples actin cytoskeleton regulatory proteins with small GTPases for regulating actin cytoskeleton (Govind et al., 2001; Krugmann et al., 2001; Lim et al., 2008; Miki et al., 2000). We next explored whether the reconstituted PSD condensates could recruit actin-related proteins via IRSp53 and how their incorporation might affect actin polymerization in the condensates. To our surprise, when the 5× PSD mixture (with each protein at a concentration as low as 0.5 μM) was incubated with G-actin, long filamentous actin bundles were readily observed under fluorescence microscope (Fig. 4 A). Actin bundles further assembled into thick network-like structures that spanned across tens of micrometers in solution. Interestingly, the signals from the PSD constituents largely colocalized with actin bundles (Fig. 4 A; enlarged view in Fig. 4 B). It should be noted that the 5× PSD system, in the absence of G-actin, could not undergo phase separation at this concentration (Fig. S4, Bii). To investigate the dynamics of molecules in the condensates assembled on F-actin bundles, we performed FRAP analysis of the PSD-95 and Rhd-labeled actin (Fig. 4 C). We observed signal recovery for PSD-95 but there was essentially no signal recovery for the actin signal, suggesting that actin bundle-coated PSD condensates exhibit liquid-like properties as in solution and the actin filaments exist at the solid state as expected. In the absence of IRSp53, the 4× PSD system could not induce actin bundling formation under same conditions (Fig. S4, Bi). This suggested that IRSp53 can play a critical role in the promotion of actin bundle formation. To examine whether IRSp53 alone can bundle actin filaments, we incubated increasing amount of IRSp53 with 1 μM of G-actin. IRSp53 alone exhibited actin crosslinking activity only at 2 μM or above (Fig. 4 D), demonstrating that IRSp53 directly binds to and bundles actin filaments, albeit with a much lower efficiency in the absence of PSD condensates.
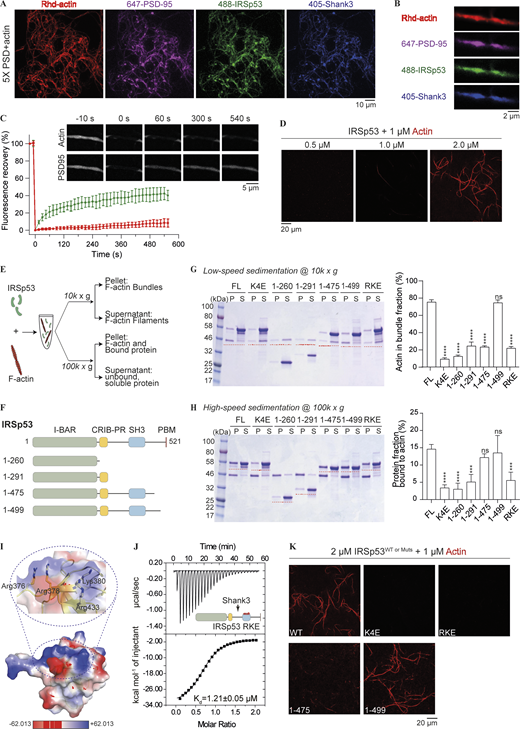
IRSp53 can bind to and bundle actin filaments. (A) Representative images showing PSD condensation on actin network when 5× PSD, each component at 0.5 μM, was mixed with 1 μM of G-actin and incubated for 20 min at room temperature. 2% of PSD-95, IRSp53, and Shank3 were labeled with different fluorophores as indicated. 10% of actin was labeled with Rhodamine dye. GKAP and Homer3 were not labeled and are thus invisible. Images were collected as a z series maximum projection. (B) Zoom-in view of PSD condensates with Rhd-actin. (C) FRAP analysis showing the fluorescence recovery of actin and PSD-95 following a bleaching event. The recovery of fluorescence signals was quantified and plotted. Data are obtained from three condensate/actin regions and are presented as mean ± SD. (D) IRSp53 can bundle actin filaments in aqueous solution. 1 μM of actin was incubated with increasing amounts of IRSp53 at indicated concentrations. 10% of actin was labeled with Rhodamine dye. (E) Experimental setup to monitor possible actin binding and bundling activity of IRSp53 via low-speed (10,000 g) and high-speed (100,000 g) centrifugations. (F) Schematic diagrams of various fragments used to map the actin binding interface in IRSp53. (G) SDS-PAGE showing the level of actin bundling induced by different fragments of IRSp53 as designated in F. 1 μM actin was mixed with 4 μM of various IRSp53 constructs. The amount of actin in the pellet fraction, which represents the bundled actin filaments, is quantified from three independent batches of experiments. Data are presented as mean ± SD. ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (H) SDS-PAGE showing binding of various IRSp53 fragments, as designated in F, to F-actin. 1 μM actin was mixed with 4 μM of various IRSp53 constructs. Note that the pellet, obtained after ultracentrifugation, was resuspended in one quarter of the initial mixture volume for concentration. The portion of protein in the pellet fraction, which represents the actin-bound protein, is quantified from three independent batches of experiments. The amount of protein sedimented without the presence of actin filaments was subtracted for correction (Fig. S4 E). The corrected pellet ratio of protein was further divided by the pellet ratio of F-actin for normalization. Data are presented as mean ± SD. ***, P < 0.001; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (I) Surface charge potential map of the IRSp53 SH3 domain (PDB accession no. 3RNJ) showing a cluster of positively charged residues (highlighted with a circle in dashed lines and enlarged to show the side chain orientations of these residues). Highlighted residues were mutated to Glu, and the mutant was referred to as IRSp53_RKE. (J) ITC-based experiment measured the binding between Shank3 and IRSp53_RKE, showing that the mutation did not affect IRSp53, via its SH3 domain, to bind to Shank3. 200 μM of Shank3 was titrated into 20 μM of IRSp53 with 200 mM NaCl in the binding buffer. (K) Representative fluorescence images showing that WT IRSp53, but not the actin binding deficient mutants (K4E, RKE), can bundle actin filaments. IRSp53 1–475 could not bundle actin filaments, and the further inclusion of 24 residues into the C-terminus (aa 1–499) rescued the actin-bundling ability to that of the full-length protein. Identical imaging settings were used for all groups. Source data are available for this figure: SourceData F4.
IRSp53 can bind to and bundle actin filaments. (A) Representative images showing PSD condensation on actin network when 5× PSD, each component at 0.5 μM, was mixed with 1 μM of G-actin and incubated for 20 min at room temperature. 2% of PSD-95, IRSp53, and Shank3 were labeled with different fluorophores as indicated. 10% of actin was labeled with Rhodamine dye. GKAP and Homer3 were not labeled and are thus invisible. Images were collected as a z series maximum projection. (B) Zoom-in view of PSD condensates with Rhd-actin. (C) FRAP analysis showing the fluorescence recovery of actin and PSD-95 following a bleaching event. The recovery of fluorescence signals was quantified and plotted. Data are obtained from three condensate/actin regions and are presented as mean ± SD. (D) IRSp53 can bundle actin filaments in aqueous solution. 1 μM of actin was incubated with increasing amounts of IRSp53 at indicated concentrations. 10% of actin was labeled with Rhodamine dye. (E) Experimental setup to monitor possible actin binding and bundling activity of IRSp53 via low-speed (10,000 g) and high-speed (100,000 g) centrifugations. (F) Schematic diagrams of various fragments used to map the actin binding interface in IRSp53. (G) SDS-PAGE showing the level of actin bundling induced by different fragments of IRSp53 as designated in F. 1 μM actin was mixed with 4 μM of various IRSp53 constructs. The amount of actin in the pellet fraction, which represents the bundled actin filaments, is quantified from three independent batches of experiments. Data are presented as mean ± SD. ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (H) SDS-PAGE showing binding of various IRSp53 fragments, as designated in F, to F-actin. 1 μM actin was mixed with 4 μM of various IRSp53 constructs. Note that the pellet, obtained after ultracentrifugation, was resuspended in one quarter of the initial mixture volume for concentration. The portion of protein in the pellet fraction, which represents the actin-bound protein, is quantified from three independent batches of experiments. The amount of protein sedimented without the presence of actin filaments was subtracted for correction (Fig. S4 E). The corrected pellet ratio of protein was further divided by the pellet ratio of F-actin for normalization. Data are presented as mean ± SD. ***, P < 0.001; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (I) Surface charge potential map of the IRSp53 SH3 domain (PDB accession no. 3RNJ) showing a cluster of positively charged residues (highlighted with a circle in dashed lines and enlarged to show the side chain orientations of these residues). Highlighted residues were mutated to Glu, and the mutant was referred to as IRSp53_RKE. (J) ITC-based experiment measured the binding between Shank3 and IRSp53_RKE, showing that the mutation did not affect IRSp53, via its SH3 domain, to bind to Shank3. 200 μM of Shank3 was titrated into 20 μM of IRSp53 with 200 mM NaCl in the binding buffer. (K) Representative fluorescence images showing that WT IRSp53, but not the actin binding deficient mutants (K4E, RKE), can bundle actin filaments. IRSp53 1–475 could not bundle actin filaments, and the further inclusion of 24 residues into the C-terminus (aa 1–499) rescued the actin-bundling ability to that of the full-length protein. Identical imaging settings were used for all groups. Source data are available for this figure: SourceData F4.
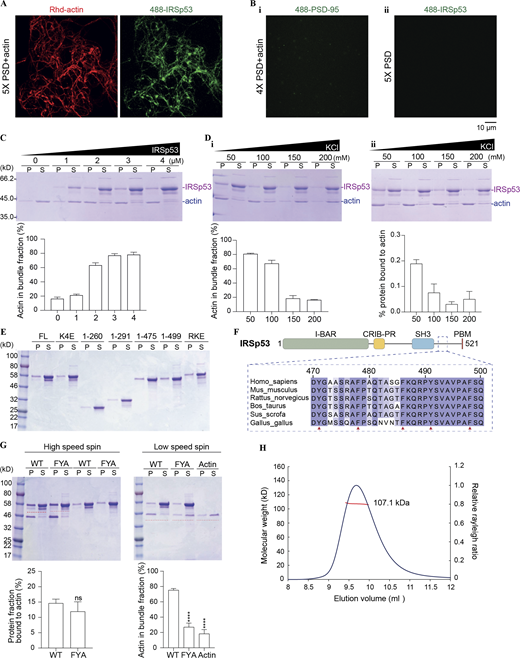
IRSp53 binds to actin via charged interactions, and its bundling activity requires hydrophobic interactions. (A and B) PSD complexes bundle actin filaments via IRSp53. Representative fluorescence images showing that 4× PSD complex with WT IRSp53 significantly promotes actin bundling (A), whereas the mixture without IRSp53 could not (Bi). The mixture of 4× PSD with IRSp53 will not undergo LLPS in the absence of G-actin (Bii). Each PSD component was at 0.5 μM, and actin was at 1 μM. (C) Effect of increasing protein concentration on the actin bundling activity of IRSp53. 1 μM G-actin was incubated with IRSp53 at indicated concentrations for 1 h at room temperature before centrifugation at 10,000 g. Amount of actin in the pellet fraction is quantified from three independent batches of experiments. Error bar indicates ± SD. (D) Effect of increasing KCl concentration on the actin bundling (i) and binding (ii) activities of IRSp53. 1 μM actin was incubated with 4 μM of IRSp53, in the presence of indicated amount of salt in the assay buffer, for 1 h at room temperature before centrifugation at 10,000 g (i) or 100,000 g (ii). Amount of actin (i) or actin-bound protein (ii) in the pellet fraction is quantified from three independent batches of experiments. Error bar indicates ± SD. (E) SDS-PAGE showing some IRSp53 protein present in the pellet fraction during the process of ultracentrifugation, in the absence of G-actin. This portion of non–actin-induced sedimentation was subtracted for correction. Note that the pellet, obtained after ultracentrifugation, was resuspended in one quarter of the initial mixture volume for concentration. (F) Multiple sequence alignment of residues 470–500 of IRSp53 from different mammalian species showing the conservation of several aromatic residues (Phe and Tyr highlighted by red arrows) throughout evolution. These residues were subjected to mutation to Ala to eliminate potential hydrophobic interactions, and the mutant was referred to as IRSp53_FYA. (G) SDS-PAGE showing that the FYA mutation in IRSp53 significantly diminished actin bundle formation (right panels) but barely interfered with its actin-binding ability (left panels). 1 μM actin was mixed with 4 μM of IRSp53 constructs. Note that the pellet, obtained after ultracentrifugation (at 100,000 g), was resuspended in one quarter of the initial mixture volume for concentration. The amount of protein sedimented independent of actin filaments (last four rows on SDS-PAGE on LHS) was subtracted for correction. The corrected pellet ratio of protein was further divided by the pellet ratio of F-actin for normalization. The amount of actin in the pellet fraction obtained after low-speed centrifugation (at 10,000 g), which represents the bundled actin filaments, was quantified. All data are obtained from three independent batches of experiments. Quantification is presented as mean ± SD. ****, P < 0.0001 using unpaired t test (protein fraction bound to actin) or one-way ANOVA with Dunnett’s multiple comparison test (actin in bundle fraction). (H) miniDawn analysis showing that the FYA mutation does not affect IRSp53 dimerization in solution. Superose 12 10/300 column was used for size-exclusion chromatography. Calculated MW of IRSp53_FYA is 57.0 kD. Analysis was performed at 100 μM protein concentration. Source data are available for this figure: SourceData FS4.
IRSp53 binds to actin via charged interactions, and its bundling activity requires hydrophobic interactions. (A and B) PSD complexes bundle actin filaments via IRSp53. Representative fluorescence images showing that 4× PSD complex with WT IRSp53 significantly promotes actin bundling (A), whereas the mixture without IRSp53 could not (Bi). The mixture of 4× PSD with IRSp53 will not undergo LLPS in the absence of G-actin (Bii). Each PSD component was at 0.5 μM, and actin was at 1 μM. (C) Effect of increasing protein concentration on the actin bundling activity of IRSp53. 1 μM G-actin was incubated with IRSp53 at indicated concentrations for 1 h at room temperature before centrifugation at 10,000 g. Amount of actin in the pellet fraction is quantified from three independent batches of experiments. Error bar indicates ± SD. (D) Effect of increasing KCl concentration on the actin bundling (i) and binding (ii) activities of IRSp53. 1 μM actin was incubated with 4 μM of IRSp53, in the presence of indicated amount of salt in the assay buffer, for 1 h at room temperature before centrifugation at 10,000 g (i) or 100,000 g (ii). Amount of actin (i) or actin-bound protein (ii) in the pellet fraction is quantified from three independent batches of experiments. Error bar indicates ± SD. (E) SDS-PAGE showing some IRSp53 protein present in the pellet fraction during the process of ultracentrifugation, in the absence of G-actin. This portion of non–actin-induced sedimentation was subtracted for correction. Note that the pellet, obtained after ultracentrifugation, was resuspended in one quarter of the initial mixture volume for concentration. (F) Multiple sequence alignment of residues 470–500 of IRSp53 from different mammalian species showing the conservation of several aromatic residues (Phe and Tyr highlighted by red arrows) throughout evolution. These residues were subjected to mutation to Ala to eliminate potential hydrophobic interactions, and the mutant was referred to as IRSp53_FYA. (G) SDS-PAGE showing that the FYA mutation in IRSp53 significantly diminished actin bundle formation (right panels) but barely interfered with its actin-binding ability (left panels). 1 μM actin was mixed with 4 μM of IRSp53 constructs. Note that the pellet, obtained after ultracentrifugation (at 100,000 g), was resuspended in one quarter of the initial mixture volume for concentration. The amount of protein sedimented independent of actin filaments (last four rows on SDS-PAGE on LHS) was subtracted for correction. The corrected pellet ratio of protein was further divided by the pellet ratio of F-actin for normalization. The amount of actin in the pellet fraction obtained after low-speed centrifugation (at 10,000 g), which represents the bundled actin filaments, was quantified. All data are obtained from three independent batches of experiments. Quantification is presented as mean ± SD. ****, P < 0.0001 using unpaired t test (protein fraction bound to actin) or one-way ANOVA with Dunnett’s multiple comparison test (actin in bundle fraction). (H) miniDawn analysis showing that the FYA mutation does not affect IRSp53 dimerization in solution. Superose 12 10/300 column was used for size-exclusion chromatography. Calculated MW of IRSp53_FYA is 57.0 kD. Analysis was performed at 100 μM protein concentration. Source data are available for this figure: SourceData FS4.
IRSp53 directly interacts with actin filaments via two surfaces
To better understand the mechanism governing actin binding and bundling activities of IRSp53, we performed cosedimentation assays (Fig. 4 E). To confirm the actin bundling activity of IRSp53, we titrated increasing amounts of protein for incubation with in vitro polymerized actin filaments for 1 h, before F-actin bundles were separated from single filaments via low-speed centrifugation at 10,000 g. Pelleted F-actin filaments were only detected above 1 μM protein concentration and reached saturation at 4 μM (Fig. S4 C). Next, we tested the influence of ionic strength on IRSp53/actin interaction. When the salt concentration in the assay buffer increased from 100 to 200 mM, IRSp53 showed limited actin bundling activity (Fig. S4, Di). We also performed ultracentrifugation of the protein/actin mixture at 100,000 g to separate F-actin filaments from free G-actin and unbound protein (Fig. S4, Dii). The amount of protein binding to the F-actin gradually decreased with increasing salt concentration, indicating that the binding between actin and IRSp53 is mainly driven by electrostatic interactions.
We next set out to map the specific actin binding sites on IRSp53. We first mutated the four positively charged residues in the BAR domain, Lys142/143/146/147, to Glu (referred to as the K4E mutant). The mutant protein could no longer bind to and bundle actin filaments in both cosedimentation assays and imaging-based analysis (Fig. 4, G, H, and K). It is notable that, even in the absence of actin filaments, some protein would sediment to the pellet fraction during ultracentrifugation (Fig. S4 E). To determine the amount of protein specifically bound to actin, non–actin-induced pellet fraction was subtracted for correction. We then wondered whether the dimeric BAR domain alone was sufficient to crosslink actin filaments. To our surprise, the purified BAR domain could neither bind to F-actin nor induce actin bundle formation (Fig. 4, G and H), suggesting that other regions in IRSp53 also contribute to the F-actin interaction. We purified various truncation constructs that cover different lengths of the protein and screened for their actin binding and bundling capacities via sedimentation assay (Fig. 4 F). Extension of the BAR domain to include the CRIB-PR region (aa 1–291) did not significantly alter the actin binding ability of IRSp53 (Fig. 4 H). Further inclusion of the SH3 domain (aa 1–475), however, could restore the actin binding efficiency to that of the full-length protein (Fig. 4 H). This indicated that the SH3 domain might present a second binding interface for F-actin. Structural analysis of the SH3 domain revealed a highly positively charged surface that positions away from its target binding site (Fig. 4, I and J). We substituted the four exposed, positively charged residues, Arg376, Arg378, Lys380, and Arg433, with Glu (referred to as the RKE mutant). This charge cluster mutation dramatically reduced the actin binding and bundling activities of the full-length IRSp53 (Fig. 4, G, H, and K). Furthermore, our mapping experiments showed that removal of the C-terminal 46 residues of IRSp53 (aa 1–475) did not alter its actin binding efficiency but nearly abolished the actin bundle formation in solution (Fig. 4, G, H, and K). We further narrowed down this critical actin crosslinking region to aa 476–499, whose inclusion restored the actin bundling activity of IRSp53 (aa 1–499) to that of the full-length protein (Fig. 4, G, H, and K). The aa 470–499 is evolutionarily conserved and contains several aromatic residues (Tyr471, Phe478, Phe486, Tyr491, and Phe498) that might participate in hydrophobic interactions (Fig. S4 F). We then investigated whether these interactions are required for crosslinking actin filaments into bundles by mutating the aromatic residues to Ala (referred to as the FYA mutant). IRSp53_FYA mutant displayed defective actin bundling, although its binding to actin was retained (Fig. S4, G and H). Overall, the above findings indicate that IRSp53 directly and specifically binds to actin via two patches of positive charges, one on the BAR domain and the other on the SH3 domain. A cluster of aromatic residues situated in the region C-terminal to the SH3 domain of IRSp53 are important for its actin bundling activity.
IRSp53 in PSD condensates bundles actin filaments on supported lipid bilayers (SLBs)
To further investigate the influence of IRSp53-mediated actin binding activity in the context of PSD condensation, we visualized the actin bundling events with various mutants under fluorescence microscope (Fig. 5 A). The actin binding–deficient mutants of IRSp53, in the 5× PSD system, failed to promote the formation of actin bundles (Fig. 5 A). It should be emphasized that both charge-cluster mutations had minimal impact on the LLPS efficiency of the PSD mixture (Fig. 5 B). On the other hand, a phase separation-deficient mutant, Shank3_ΔPRM (aka Shank3*) whose actin binding activity was retained, showed limited actin bundling activity, likely due to its reduced phase separation ability (Fig. 5, A and B).
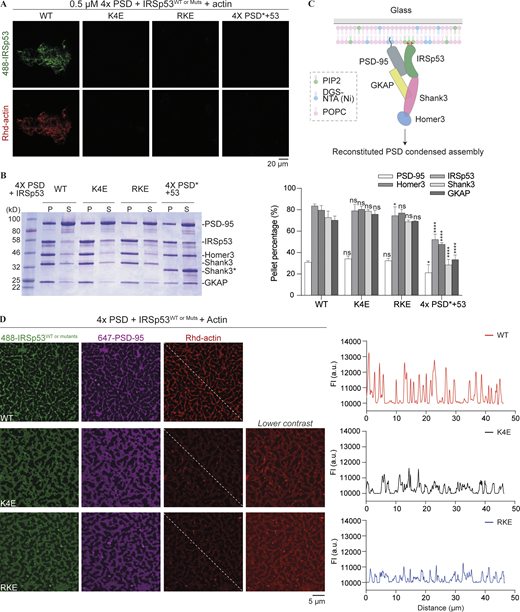
PSD condensates promote actin bundling via IRSp53-mediated actin interaction. (A) Fluorescence images showing the actin filament network assembled from PSD complexes in the presence of WT IRSp53. An actin binding–deficient mutation in IRSp53 abolished actin bundle assembly. A mutation that weakened the phase separation of PSD complexes also failed to promote actin bundling. Identical imaging settings were used for all groups. (B) Sedimentation-based experiments showing the distribution of PSD components in the dilute (S) and condensed phases (P) when 4× PSD was mixed with IRSp53 WT or actin binding deficient mutant. 5× PSD* refers to the mixture of PSD-95, IRSp53, GKAP, Shank3_ΔPRM, and Homer3. Each PSD component was at 5 μM. Quantifications of the sedimentation data are presented as mean ± SD. Data were obtained from three independent batches of experiments. *, P < 0.05; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (C) Schematic diagram showing the tethering of PSD components to the supported negatively charged lipid bilayer. (D) Representative fluorescence images showing actin filaments assembled from PSD condensates on the SLB. The actin binding–deficient mutations in IRSp53 significantly diminished actin bundling to PSD condensates on the membrane. A line plot of the Rhd-labeled actin intensities along the dashed line is presented. Identical imaging settings were used for all groups. Note that an additional panel is included for actin binding–deficient mutants of IRSp53, showing images of Rhd-actin with different contrast to the WT for better visualization. The starting concentration of the proteins in solutions (IRSp53, GKAP, Shank3, and Homer3) was 250 nM each. 10% Rhd-labeled actin was 1 μM. 10% of His8-PSD-95 and IRSp53 were labeled with the indicated fluorophores. Source data are available for this figure: SourceData F5.
PSD condensates promote actin bundling via IRSp53-mediated actin interaction. (A) Fluorescence images showing the actin filament network assembled from PSD complexes in the presence of WT IRSp53. An actin binding–deficient mutation in IRSp53 abolished actin bundle assembly. A mutation that weakened the phase separation of PSD complexes also failed to promote actin bundling. Identical imaging settings were used for all groups. (B) Sedimentation-based experiments showing the distribution of PSD components in the dilute (S) and condensed phases (P) when 4× PSD was mixed with IRSp53 WT or actin binding deficient mutant. 5× PSD* refers to the mixture of PSD-95, IRSp53, GKAP, Shank3_ΔPRM, and Homer3. Each PSD component was at 5 μM. Quantifications of the sedimentation data are presented as mean ± SD. Data were obtained from three independent batches of experiments. *, P < 0.05; ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test. (C) Schematic diagram showing the tethering of PSD components to the supported negatively charged lipid bilayer. (D) Representative fluorescence images showing actin filaments assembled from PSD condensates on the SLB. The actin binding–deficient mutations in IRSp53 significantly diminished actin bundling to PSD condensates on the membrane. A line plot of the Rhd-labeled actin intensities along the dashed line is presented. Identical imaging settings were used for all groups. Note that an additional panel is included for actin binding–deficient mutants of IRSp53, showing images of Rhd-actin with different contrast to the WT for better visualization. The starting concentration of the proteins in solutions (IRSp53, GKAP, Shank3, and Homer3) was 250 nM each. 10% Rhd-labeled actin was 1 μM. 10% of His8-PSD-95 and IRSp53 were labeled with the indicated fluorophores. Source data are available for this figure: SourceData F5.
We next asked whether IRSp53 can form LLPS-mediated clusters with PSD scaffolds on membranes (mimicking the layered PSD organization as shown in Fig. 5 C) and if such condensates can bundle actin filaments. In this assay, we first added an N-terminal His8-tag to PSD-95. We generated a system with PSD-95 on the SLB, which mimics its synaptic conformation, where its N-terminal His8-tag binding to 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt; DGS-NTA[Ni]) mimics the membrane tethering of PSD-95 via palmitoylated Cys residues. The lipid composition of our SLBs consisted of 95.9% 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 2% DGS-NTA(Ni), 2% PI(4,5)P2, and 0.1% 2-dioleoyl-sn-glycero-3-phosphoethanolamine-N (methoxy [polyethylene glycol]-5000] (PEG5000 PE). IRSp53 was bound to the PI(4,5)P2-containing membranes, and this was abolished with Lys to Glu mutations in the BAR domain but not in the mutant protein with Lys and Arg in the SH3 domain substituted with Glu (Fig. S5). This BAR domain–mediated lipid binding activity is consistent with previous reports showing that the BAR domain binds to negatively charged lipid membranes (Mattila et al., 2007; Suetsugu et al., 2006).

IRSp53 binds to negatively charged SLBs. Representative fluorescence images showing that IRSp53 WT binds to lipid membrane containing 2% PI(4,5)P2, whereas the Lys-to-Glu mutations in the BAR domain (IRSp53_K4E) abolished its lipid-binding ability. Mutation of positive charges in the SH3 domain (IRSp53_RKE) did not interfere with its lipid membrane binding. The starting concentration of IRSp53 WT or mutant proteins in solutions was at 2 μΜ. 10% of protein was labeled with Alexa Fluor 488 C5 maleimide dye.
IRSp53 binds to negatively charged SLBs. Representative fluorescence images showing that IRSp53 WT binds to lipid membrane containing 2% PI(4,5)P2, whereas the Lys-to-Glu mutations in the BAR domain (IRSp53_K4E) abolished its lipid-binding ability. Mutation of positive charges in the SH3 domain (IRSp53_RKE) did not interfere with its lipid membrane binding. The starting concentration of IRSp53 WT or mutant proteins in solutions was at 2 μΜ. 10% of protein was labeled with Alexa Fluor 488 C5 maleimide dye.
With PSD-95 coating on the membrane and upon addition of other PSD scaffolds, we observed sub-micrometer-sized clusters formed on the membrane surface (Fig. 5 D). Strikingly, actin filaments assembled on these clusters and changed the appearance of PSD condensates from round worm-like shapes to thin hair-like projections that likely resemble the underlying bundled actin structures (Fig. 5 D, top row). In contrast, PSD scaffolds assembled with actin binding deficient mutants of IRSp53 formed condensates on the membrane with wider, web-like morphologies and these condensates had dramatically reduced actin bundling activities (Fig. 5 D, middle and bottom rows). It is also noted that PSD constituents in the actin-enriched condensates showed stronger fluorescence intensities than those in the actin-depleted clusters containing mutant IRSp53. This difference likely reflects positive feedback due to the increased phase separation promoted by actin scaffolds, a result also occurring in 3D solution (Figs. 4 and S4).
Taken the above results together, we demonstrated that PSD condensates containing IRSp53, in solution and on membranes, can effectively promote actin filament bundling. The bundled actin in return promotes the condensation of PSD constituents. Thus, the scaffold proteins and the actin cytoskeleton establish a positive feedback loop that is likely connected with synapse development in neurons.
IRSp53-actin interaction modulates dendritic spine development
To explore biological roles of IRSp53-mediated actin bundling in neurons, we investigated the impact of mutating those critical actin-binding residues in IRSp53 on dendritic spine development. We used mEGFP tagged IRSp53 WT and two mutant constructs (IRSp53_K4E and RKE) and used mCherry as the cell fill and as the background transfection control (Fig. 6). Prefrontal cortical neurons obtained from IRSp53fl/fl mice were cultured, transfected with Cre recombinase alone or in combination with cDNA encoding WT IRSp53 or various mutants at 7 d in vitro (DIV7), and were collected for live imaging at DIV17. Depletion of endogenous IRSp53 led to severe reduction in the spine head width and spine density, which could be rescued by the re-expression of WT IRSp53. In contrast, re-expression of the actin binding deficient mutants failed to rescue the phenotypes, indicating that the actin bundling activity of IRSp53 is critical for normal spine development (Fig. 6, A, C, and D).

IRSp53–actin interaction regulates dendritic spine morphology and modulates synaptic enrichment of IRSp53. (A) Representative fluorescence images of cultured IRSp53 knockout cortical neurons with the re-expression of mEGFP-tagged IRSp53 WT or mutant constructs at DIV7. mCherry was cotransfected as the cell fill. Endogenous IRSp53 was deleted upon Cre expression, and mEGFP-tagged IRSp53 WT or mutant cDNA was transfected for 10 d. At DIV17, live imaging was performed without fixation. (B–D) Quantification of the imaging data showing spine enrichment (B) measured using Image J and spine head width (C) and spine density (D) measured using IMARIS. 24–43 neurons from three independent batches of cultures were imaged for each group in double-blinded mode (n = 43 for mCherry, n = 42 for Cre only, n = 36 for Cre coexpression with IRSp53 WT, n = 26 for Cre coexpression with IRSp53_K4E, and n = 24 for IRSp53_RKE). It is noted that data in Fig. 3 H were collected from the same set of experiments with the same control groups (i.e., Cre only and Cre co-expression with IRSp53 WT). Error bars indicate mean ± SEM. ***, P < 0.001, ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test.
IRSp53–actin interaction regulates dendritic spine morphology and modulates synaptic enrichment of IRSp53. (A) Representative fluorescence images of cultured IRSp53 knockout cortical neurons with the re-expression of mEGFP-tagged IRSp53 WT or mutant constructs at DIV7. mCherry was cotransfected as the cell fill. Endogenous IRSp53 was deleted upon Cre expression, and mEGFP-tagged IRSp53 WT or mutant cDNA was transfected for 10 d. At DIV17, live imaging was performed without fixation. (B–D) Quantification of the imaging data showing spine enrichment (B) measured using Image J and spine head width (C) and spine density (D) measured using IMARIS. 24–43 neurons from three independent batches of cultures were imaged for each group in double-blinded mode (n = 43 for mCherry, n = 42 for Cre only, n = 36 for Cre coexpression with IRSp53 WT, n = 26 for Cre coexpression with IRSp53_K4E, and n = 24 for IRSp53_RKE). It is noted that data in Fig. 3 H were collected from the same set of experiments with the same control groups (i.e., Cre only and Cre co-expression with IRSp53 WT). Error bars indicate mean ± SEM. ***, P < 0.001, ****, P < 0.0001 using one-way ANOVA with Dunnett’s multiple comparison test.
In addition to the increased prevalence of thin spines, the mutant proteins demonstrated significantly reduced spine localization compared with the WT protein (Fig. 6, A and B). It should be emphasized that actin binding–deficient mutations of IRSp53 did not affect direct interaction with PSD scaffolds or their LLPS efficiency in vitro (Figs. 4 J and 5 B); thus, the reduced synaptic enrichment of the mutants is likely due to their defective actin binding ability. Of course, we could not rule out the possibility that the aberrant spine development observed with actin binding deficient mutants of IRSp53 might have resulted, at least in part, from their localization deficits.
Discussion
In this study, we provided biochemical evidence showing that IRSp53 directly binds to PSD-95 via PDZ/PBM-mediated interaction and to Shank3 via SH3/PRM-mediated interaction. The BAR domain of IRSp53 provides a dimerization interface to further increase the valency in the system. These intra and intermolecular interactions together drive the LLPS of IRSp53 with PSD-95 and Shank3 in vitro. Both pairs of interactions further enhance the enrichment of IRSp53 into biochemically reconstituted PSD condensates. Mutation that abolishes IRSp53/PSD-95 binding and subsequently the phase separation in vitro failed to rescue defects in synaptic development when re-expressed in IRSp53 knockout cortical neurons and showed reduced synaptic localization when compared to WT IRSp53. Immunogold EM studies in situ have revealed that IRSp53 is distributed throughout the PSD with accumulation into both the core and the pallium. Its distribution fits well with its simultaneous binding activities with PSD-95 and Shank3. Therefore, our biochemical reconstitution demonstrated a model that GKAP and IRSp53 can act in parallel to orchestrate the assembly of other PSD scaffold proteins via phase separation and ultimately to link the upstream glutamate receptors with the downstream signaling proteins and actin cytoskeletons.
The BAR domain superfamily protein is well known to induce membrane curvatures and to function as linkers between membranes and the actin cytoskeleton (Carman and Dominguez, 2018; McMahon and Boucrot, 2015). They often possess an SH3 domain to bind actin regulatory proteins, such as WASP and WAVE complexes, which are involved in actin polymerization. The role of IRSp53 in actin dynamics has been extensively studied in filopodia and lamellipodia, but not in dendritic spines. This is largely due to the compensatory redundancy and the extreme heterogeneity within synapses. In this study, we demonstrated that the SH3 domain of IRSp53 binds to Shank3 with high specificity and affinity (Kd in the sub micromolar range). Previous in vivo studies reported that a point mutation in the SH3 domain, which abolished its binding to downstream effectors, could not rescue the spine shrinkage phenotype caused by siRNA-induced downregulation of endogenous IRSp53 expression (Choi et al., 2005). It should be noted that such mutation not only disrupts the binding of IRSp53 to actin regulatory proteins but also to the major scaffold protein, Shank. Considering the high abundance of Shank proteins in PSDs and the higher binding affinity between IRSp53 and Shank3 than the majority of the canonical SH3/PRM interactions known to date (Kd typically in the range of tens to hundreds of micromolar), the observations made with the SH3 point mutation could well be due to the defective synaptic targeting of Shank proteins and thus the aberrant dendritic spine development. The biochemically reconstituted PSD condensates can function as a valuable platform to dissect distinct functional aspects of IRSp53, and then to bridge in vitro studies to the functions of proteins in synapses.
Here, we demonstrated that IRSp53 can directly bind to actin and promote the formation of actin bundles in vitro. An interaction between the BAR domain and actin filaments was previously reported but could only occur at low salt concentration in the assay buffer (50 mM KCl) or at relatively high molar ratio to actin, suggesting a low binding affinity (Disanza et al., 2006; Mattila et al., 2007; Millard et al., 2005; Yamagishi et al., 2004). We showed that the full-length IRSp53 can bind to and bundle actin filaments under physiological conditions. The BAR domain alone is not sufficient for IRSp53 to bind to and bundle actin filaments. The SH3 domain containing an additional actin binding site is also required for the protein to bind to and bundle actin filaments. Several aromatic residues close to the C-terminus of IRSp53 further facilitate crosslinking of actin filaments into bundles. The condensed PSD assemblies can effectively promote actin filament bundling through IRSp53. Phase separation significantly lowers the threshold concentration for IRSp53-mediated actin bundling, possibly due to the local concentration of proteins bound to the actin filaments; and in return, actin filaments also promote the condensation of PSD scaffold proteins, likely via reduction in the dimensionality of molecular diffusion from 3D solution to 2D actin filament surface, a process that is analogous to surface-assisted phase separation on SLBs. A positive feedback loop is therefore established between the PSD scaffold proteins and the cytoskeleton. IRSp53 mutants, which could not bind to actin filaments but retained LLPS capability with PSD scaffolds, failed to assemble actin bundles in solution and on membranes in vitro. In IRSp53 knockout cortical neurons, expression of these mutants failed to rescue the defective maturation of dendritic spines, and their synaptic localization was affected compared with the WT protein. Our biochemical and neuronal data thus provide some evidence toward how IRSp53 deletion may lead to synaptic abnormalities in cultured neurons and mice.
The newly identified role of IRSp53 in directly modulating actin dynamics certainly raises the question of how this activity couples with its previously reported actin role (via recruitment of WASP and WAVE-dependent actin regulatory pathways). The SH3 domain not only binds to actin regulatory proteins but also directly to actin filaments, and importantly these two interfaces are not mutually exclusive as exemplified by its simultaneous binding to Shank3 and actin filaments in PSD condensates. Future work will be required to integrate the classic actin cytoskeletal pathway into reconstituted PSD condensates and analyze potential cooperation between the two pathways.
The direct binding of IRSp53 to actin also raises the question of how this interaction is regulated. We speculate that the regulation might be fulfilled by changes in the distribution pattern of IRSp53 molecules as well as modulation of phase separation in PSDs. The BAR domain, in addition to binding to actin, can bind to negatively charged membranes, and hence there might be a competition between plasma membrane and F-actin for binding to the BAR domain. Immuno-EM studies in situ revealed that IRSp53 molecules showed increased accumulation into the PSD pallium under excitatory conditions. This fraction of IRSp53 is likely to be too far away to reach the membrane and thus is free to interact with the actin cytoskeleton. Furthermore, our biochemical data suggest that LLPS efficiency strongly affects the actin bundling ability of IRSp53, and the actin bundles further promote PSD condensate formation. Thus, one might envision that the size of the PSD assembly and the level of bundled actin are mutually coupled to regulate dendritic spine morphology and synaptic activity.
Materials and methods
Protein expression and purification
Various protein constructs were generated using standard PCR-based methods and cloned into vectors containing an N-terminal Trx/His6-affinity tag flanked by an HRV-3C protease cleavage site. All constructs were confirmed by DNA sequencing.
All proteins were recombinantly expressed using Escherichia coli BL21 cells in LB medium at 16°C overnight. Bacterial cells were grown at 37°C until reaching OD600 0.6–0.8 before induction with IPTG at 0.5 mM at 16°C. Full-length IRSp53 and its point mutants (K4E, RKE, and ΔPBM) were expressed at 37°C for 3 h before cell harvest.
In general, proteins were purified using a Ni2+-NTA affinity column followed by size-exclusion chromatography (Superdex 200 or Superdex 75) with a buffer containing 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 2 mM DTT. For IRSp53 WT and mutant proteins, the column buffer contained 50 mM Tris, pH 8.0, 300 mM NaCl, 1 mM EDTA, and 2 mM DTT. The affinity tag of each protein was cleaved by HRV-3C protease at 4°C overnight, and the untagged protein was further purified by another step of size-exclusion chromatography using a buffer containing 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 2 mM DTT. For IRSp53 WT and mutant proteins, the column buffer contained 50 mM Tris, pH 8.0, 200 mM NaCl, 1 mM EDTA, and 2 mM DTT. For full-length PSD-95, ion exchange chromatography was performed to purify untagged protein following 3C cleavage. The purified PSD-95 protein was then buffer exchanged into 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 2 mM DTT using a desalting column. Shank3_PRM1 peptide (sequence: IPPPPQTAPPPPPAPYYFDSG) was commercially synthesized by ChinaPeptides with purity >97%.
Protein labeling with fluorophore
For amide labeling
The fluorophores, including iFluor 405/488/Cy3 NHS ester (AAT Bioquest) and Alexa Fluor 647 NHS ester (Thermo Fisher Scientific), were dissolved in DMSO. Purified protein was exchanged into a buffer containing 20 mM Hepes, pH 7.6, 100 mM NaCl, 1 mM EDTA, and 1 mM DTT using a desalting column. Protein was concentrated to ≥2 mg/ml before incubation with the fluorophore at 1:1 molar ratio at room temperature for 2 h. The labeling reaction was quenched by 200 mM Tris, pH 8.2, and the labeled protein was buffer exchanged into 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 2 mM DTT using a desalting column. Fluorescence labeling efficiency was detected using Nanodrop 2000 (Thermo Fisher Scientific).
For cysteine labeling
The fluorophore, Alexa Fluor 488 C5 maleimide (Thermo Fisher Scientific), was dissolved in DMSO. Purified protein was exchanged into a buffer containing 20 mM Hepes, pH 7.6, 200 mM NaCl, and 1 mM EDTA using a desalting column. Protein was concentrated to ≥2 mg/ml before incubation with the fluorophore at 1:1 molar ratio at 4°C for 16 h. The labeling reaction was quenched by 5 mM β-mercaptoethanol, and the labeled protein was buffer exchanged into 50 mM Tris, pH 8.0, 200 mM NaCl, 1 mM EDTA, and 2 mM DTT using a desalting column. Fluorescence labeling efficiency was detected using Nanodrop 2000 (Thermo Fisher Scientific).
Phase separation sedimentation and imaging assay
For phase separation assays, proteins were directly mixed to reach specified concentrations. The final buffer of LLPS reactions was 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 2 mM DTT.
For sedimentation-based assays, the total volume of each mixture was 50 μl. The mixture was incubated at room temperature for 10 min before centrifugation at 16,873 g and 25°C for 10 min. The supernatant was collected, and the pellet was resuspended in 50 μl of the assay buffer; both fractions were then mixed with 50 μl of 2× SDS loading dye and boiled at 90°C for 5 min. Samples from both fractions were analyzed by SDS-PAGE with Coomassie blue R250 staining. The intensity of each band on SDS-PAGE was quantified by ImageJ.
For imaging-based assays, each mixture was injected into a homemade chamber composed of a coverslip and a glass slide assembled with one layer of double-sided tape. Fluorescence images were captured using a Zeiss LSM 800 microscope.
FRAP assay
FRAP experiments were performed on a Zeiss LSM 800 microscope at room temperature, using a 63× oil lens objective. For each FRAP experiment, fluorescence intensities of a neighboring droplet with similar size to the one subjected to photobleaching were recorded for fluorescence intensity correction, and a third region in the background with the same size was also recorded for background signal subtraction. Alexa Fluor 488 signal was photobleached by 488-nm laser beam, Cy3 signal by 561-nm laser beam and Alexa Fluor 647 signal by 633-nm laser beam at 100% power, respectively. All experiments were completed within 2 h after initiation of phase separation. For data analysis, the intensity at the prebleach point was normalized to 100%, and the intensity right after the bleaching event was set to 0%.
ITC assay
ITC experiments were performed using MicroCal VP-ITC calorimeter (Malvern) at 25°C. For ITC measurements of interactions between PSD-95 and IRSp53 constructs, proteins were exchanged into the binding buffer containing 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 2 mM DTT. For ITC measurements of interactions between Shank3 and IRSp53 constructs, proteins were exchanged into the binding buffer containing 50 mM Tris, pH 8.0, 200 mM NaCl, 1 mM EDTA, and 2 mM DTT. Each titration point was performed by injecting 10 μl aliquot of one protein in the syringe into its binding protein in the cell at a time interval of 120 s to ensure that the titration peak returned to the baseline. Titration data were fitted with the one-site binding model using Origin 7.0.
Actin binding and bundling sedimentation assay
Rabbit skeletal muscle actin (ALK99; Cytoskeleton) was diluted in G-buffer (5 mM Tris, pH 8.0, 0.2 mM CaCl2, and 0.2 mM ATP) and allowed to depolymerize on ice for 1 h before centrifugation at 16,873 g and 4°C for 0.5 h to remove residual polymerized material. Actin was polymerized at 20 μM for 1 h in polymerization buffer (10 mM Tris, pH 8.0, 50 mM KCl, 2 mM MgCl2, 1 mM ATP, and 2 mM DTT). In vitro polymerized actin was then mixed with protein constructs at indicated concentrations and incubated for 1 h at room temperature. All protein samples were centrifuged at 16,873 g for 10 min to remove potential precipitations before assay setup.
For actin binding sedimentation assay, the final reaction buffer contained 50 mM Tris, pH 8.0, 50 mM KCl, 2 mM MgCl2, 1 mM ATP, and 2 mM DTT; the reaction volume was 100 μl. Actin/protein mixture was centrifuged at 100,000 g at 4°C for 0.5 h. The supernatant was collected, and the pellet was resuspended in 25 μl of assay buffer. Samples from both fractions were analyzed by SDS-PAGE with Coomassie blue R250 staining. The intensity of each protein band on SDS-PAGE was quantified by ImageJ.
For actin bundling sedimentation assay, the final reaction buffer contained 50 mM Tris, pH 8.0, 100 mM KCl, 2 mM MgCl2, 1 mM ATP, and 2 mM DTT; the reaction volume was at 50 μl. Actin/protein mixture was centrifuged at 10,000 g and 4°C for 0.5 h. The supernatant was collected, and the pellet was resuspended in 50 μl of assay buffer. Samples from both fractions were analyzed by SDS-PAGE with Coomassie blue R250 staining. The intensity of each actin band on SDS-PAGE was quantified by ImageJ.
Imaging of actin bundling
For visualization of actin bundle formation, 10% rhodamine-labeled G-actin (Cytoskeleton) at 1 μM was mixed with desired protein components at indicated concentrations. The reaction buffer contained 50 mM Tris, pH 8.0, 100 mM NaCl, 2 mM MgCl2, 1 mM ATP, and 2 mM DTT. The actin/protein mixture was then added into a homemade chamber and incubated for 20 min at room temperature before imaging. Fluorescence images were captured using a Zeiss LSM 800 microscope.
SLB preparation and phase separation assay
Lipids comprising 95.9% POPC, 2% DGS-NTA(Ni), 2% PI(4,5)P2, and 0.1% PEG5000 PE (all lipids purchased from Avanti Polar Lipids and dissolved in chloroform) were mixed in a 2-ml glass vial with glass pipette and dried under a stream of nitrogen gas, followed by ≥1-h vacuum to ensure removal of residual chloroform. The lipid mixture was resuspended in PBS buffer supplemented with 1% sodium cholate. Lipids completely dissolved in this detergent environment should be transparent. The dissolved lipid mixture was subjected to a HiTrap desalting column (GE Healthcare) with reaction buffer that contained 50 mM Tris, pH 8.0, 100 mM NaCl, and 1 mM TCEP. Small, unilamellar vesicles (SUVs) were formed during the detergent removal process.
Chambered cover glass (Lab-tek) was initially washed with Hellmanex II (Helma Analytics) overnight and thoroughly rinsed with MilliQ water. The cover glass was then washed with 5 M NaOH for 1 h at 50°C and thoroughly rinsed with MilliQ water, and this process was repeated three times. The cleaned cover glass was then equilibrated with PBS buffer, followed by addition of 150 μl SUVs and incubation at 42°C for 1 h to generate SLB. SLBs were washed with the reaction buffer three times (sixfold dilution each time). It should be noted that SLBs should not be exposed to the atmosphere, to prevent oxidation. Then SLBs were blocked with 1 mg/ml BSA in the reaction buffer at room temperature for 30 min. 2 μM 10% Alexa Fluor 647–labeled, His8-tagged PSD-95 was added and incubated with SLBs for 1 h at room temperature, followed by washing with the reaction buffer three times to remove unbound His-PSD-95. Other PSD components (each at 250 nM) together with 10% Rhodamine-labeled G-actin (1 μM), were premixed in the reaction buffer supplemented with 1 mM ATP and 2 mM MgCl2 and were then added to the His8-PSD-95-bound SLBs, waiting for phase separation and actin polymerization to happen on the lipid bilayers. All data were collected within 8 h after lipid coating started. Experiments were carried out at room temperature.
Cortical neuron culture and transfection
Cultured cortical neurons were prepared from embryonic day 17 fetal IRSp53 floxed mice carrying exons 4–6 of the Baiap2 gene, encoding IRSp53, floxed by LoxP (IRSp53fl/fl) in a C57BL/6J background, designed and generated by Biocytogen (Kim et al., 2020). Briefly, dissected cortex were dissociated with papain and plated on poly-D-lysine–coated 18-mm glass coverslips in a 60-mm Petri dish with plating medium (Neurobasal-A medium supplemented with 2% B-27, 10% FBS, 1% GlutaMax, and 1 mm sodium pyruvate; all from Thermo Fisher Scientific). 4 h after plating, plating medium was replaced with FBS free culture medium (Neurobasal-A medium supplemented with 2% B-27, 1% GlutaMax, and 1 mM sodium pyruvate) and then 50% replacement every 7 d.
Cortical neurons were transfected by the calcium-phosphate transfection method at DIV7. Briefly, 7 μg of plasmid DNA and 9 μl of 2 M CaCl2 were mixed in distilled water to a total volume of 75 μl, added to an equal volume of 2× BBS (in mM: 50 BES, 280 NaCl, and 1.5 Na2HPO4, pH 7.1). The DNA mixture was incubated for 15 min at room temperature and added to neurons in transfection medium (MEM, 1% GlutaMax, 1 mM sodium pyruvate, 0.6% glucose, and 10 mM Hepes, pH 7.65). The DNA mixture–treated neurons were incubated for 90 min in a 5% CO2 incubator, and washed twice for 20 min with washing medium (MEM, 1% GlutaMax, 1 mM sodium pyruvate, 0.6% glucose, and 10 mM Hepes, pH 7.35). The washing medium was then replaced with original culture medium.
Image acquisition of live neurons and analysis
DIV17–18 transfected neurons were mounted in a magnetic chamber (CM-B18-1; Live Cell Instrument) on the stage of a Zeiss LSM 780 Confocal Microscope. Images ware acquired in Tyrode’s solution (136 mM NaCl, 2 mM CaCl2, 2.5 mM KCl, 2 mM MgCl2, 10 mM glucose, and 10 mM Hepes, pH 7.4, 285–290 mOsm) using a 63× objective lens.
Spine density and head width were analyzed by an automated method using Imaris software (Bitplane; RRID:SCR_007370) using maximum-intensity projected 2D images. In Imaris Surpass mode, a new filament was created using the AutoPath mode of the FilamentTracer to define the dendrite, with minor modifications from previous studies (Lee et al., 2019). All filaments counted were devoid of crossing neurites or additional dendritic branch points between defined start and end points. For the automatic spine detection, the minimum spine end diameter and maximum spine length were set at 0.2 and 4 μm. Automatic thresholds were used for generating spine seed points and surface rendering. After generating the trace, a filter was applied to ensure that all dendritic protrusions <2 μm2 were defined as spines. For each condition, three dendrites were measured for each neuron. Spine enrichment was measured with ImageJ. Transfected neurons were chosen randomly for quantification from three independent batches of cultures.
Size-exclusion chromatography coupled with multiangle light scattering assay
Experiments were performed on an AKTA purifier system (GE Healthcare) coupled with a static light scattering detector (miniDawn; Wyatt) and a differential refractive index detector (Optilab; Wyatt). Protein samples at 100 μM were filtered and loaded into a Superose 6 Increase column or a Superose 12 10/300 GL column pre- equilibrated by a column buffer composed of 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 1 mM DTT. Data were analyzed using ASTRA6 (Wyatt).
Online supplemental material
Fig. S1 shows that IRSp53 binds to PSD-95 via PDZ/PBM interactions. Fig. S2 shows the oligomerization status of IRSp53 constructs in solution. Fig. S3 shows IRSp53-dependent phase separation of the PSD complex. Fig. S4 shows that IRSp53 binds to actin via charged interactions and that its bundling activity requires hydrophobic interactions. Fig. S5 shows that IRSp53 binds to negatively charged SLBs.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of China (2019YFA0508402), the National Science Foundation of China (82188101), Research Grants Council (RGC) of Hong Kong (AoE-M09-12, 16104518 and 16101419), a Human Frontier Science Program research grant (RGP0020/2019) to M. Zhang, the Institute of Basic Science (IBS-R002-D1) to E. Kim, and an RGC General Research Fund (GRF) grant (16102120) to Z. Feng. M. Zhang was a Kerry Holdings Professor of Science at HKUST.
The authors declare no competing financial interests.
Author contributions: Z. Feng, S. Lee, B. Jia, and T. Jian performed experiments; Z. Feng, S. Lee, and M. Zhang analyzed data; Z. Feng, E. Kim, and M. Zhang designed the research; Z. Feng and M. Zhang drafted the manuscript; and all authors commented on the paper. M. Zhang coordinated the project.

