


In selective autophagy of the nucleus (hereafter nucleophagy), nucleus-derived double-membrane vesicles (NDVs) are formed, sequestered within autophagosomes, and delivered to lysosomes or vacuoles for degradation. In Saccharomyces cerevisiae, the nuclear envelope (NE) protein Atg39 acts as a nucleophagy receptor, which interacts with Atg8 to target NDVs to the forming autophagosomal membranes. In this study, we revealed that Atg39 is anchored to the outer nuclear membrane via its transmembrane domain and also associated with the inner nuclear membrane via membrane-binding amphipathic helices (APHs) in its perinuclear space region, thereby linking these membranes. We also revealed that autophagosome formation-coupled Atg39 crowding causes the NE to protrude toward the cytoplasm, and the tips of the protrusions are pinched off to generate NDVs. The APHs of Atg39 are crucial for Atg39 crowding in the NE and subsequent NE protrusion. These findings suggest that the nucleophagy receptor Atg39 plays pivotal roles in NE deformation during the generation of NDVs to be degraded by nucleophagy.
Introduction
Macroautophagy (hereafter autophagy) is the mechanism whereby cellular material such as proteins and RNA—as well as larger structures such as protein aggregates, phase-separated liquid droplets, and membrane-bound organelles—are degraded, playing an important role in the maintenance and regulation of various cellular functions (Nakatogawa, 2020; Morishita and Mizushima, 2019). Autophagy initiates with the formation and expansion of the membrane cisterna known as the isolation membrane or phagophore, which bends, becomes spherical, and seals to sequester degradation targets within the resulting double-membrane vesicle, the autophagosome. The autophagosome then fuses with the lysosome in animals and the vacuole in yeast and plants for degradation of the sequestered material within these lytic organelles. Sequestration of cellular components into autophagosomes proceeds in both a selective and nonselective manner. In selective types of autophagy (hereafter selective autophagy), autophagy receptors bind to degradation targets and act to recruit core Atg proteins, which mediate autophagosome biogenesis, to the targets for their efficient sequestration into the autophagosomes (Nakatogawa, 2020; Farré and Subramani, 2016; Kirkin and Rogov, 2019). Previous studies suggested that different organelles, including mitochondria, peroxisomes, the ER, and the nucleus, are degraded by selective autophagy, and autophagy receptors responsible for degradation of these organelles have also been identified (Farré and Subramani, 2016; Kirkin and Rogov, 2019).
The nucleus contains chromosomes, and the double-membraned nuclear envelope (NE) separates specific reactions such as DNA replication and gene transcription from the cytoplasm. Quality control of nuclear proteins is important for maintaining proper nuclear functions, and the accumulation of aberrant nuclear proteins, which can be caused by alterations in the ubiquitin-proteasome system, results in the formation of intranuclear inclusion bodies, leading to neurodegenerative diseases such as Huntington's disease (Enam et al., 2018; Woulfe, 2008). Recent studies suggest that nuclear components are delivered to and degraded in lysosomes or vacuoles via autophagy (Mijaljica and Devenish, 2013; Mochida et al., 2015). In the budding yeast Saccharomyces cerevisiae, we identified the transmembrane protein Atg39 as a receptor for selective autophagy of the nucleus (hereafter nucleophagy; Mochida et al., 2015). Yeast cells lacking Atg39 exhibited abnormal nuclear morphology and an early cell death phenotype under nitrogen starvation. We and other groups also reported selective autophagy of nuclear pore complexes in yeast and of nuclear lamina and a sirtuin in mammalian cells (Tomioka et al., 2020; Lee et al., 2020; Dou et al., 2015; Li et al., 2019; Xu et al., 2020). These results suggest that selective degradation of nuclear components via autophagy also has a significant impact on nuclear homeostasis.
We previously revealed that when S. cerevisiae cells are subjected to nitrogen starvation or treated with the Tor kinase complex 1 (TORC1) inhibitor rapamycin, Atg39 is expressed and localized to the NE, and as with other autophagy receptors, it interacts with Atg11 (Zientara-Rytter and Subramani, 2020), which serves as a scaffold to recruit core Atg proteins, and Atg8, which is located on expanding isolation membranes, loading nucleus-derived double-membrane vesicles (NDVs) of ∼200 nm into the autophagosomes (Mochida et al., 2015). The outer and inner membranes of NDVs are derived from the outer nuclear membrane (ONM) and inner nuclear membrane (INM), respectively, with nucleoplasmic and nucleolar proteins existing in the vesicle lumen. To generate NDVs, the ONM and INM need to induce deformation and fission in a coordinated manner. Because NDVs do not accumulate in the cytoplasm of cells deficient for autophagosome formation (Mochida et al., 2015), these vesicles are thought to form concomitantly with autophagosome formation. We also found that Atg39 accumulates in the NE at contact with the site of autophagosome formation. However, the mechanisms underlying these processes of NDV formation during nucleophagy remain unknown.
In this study, we discovered that Atg39 binds to both the ONM and INM via its N-terminal transmembrane domain and C-terminal amphipathic helices (APHs), respectively, thereby linking these membranes. We also found that overexpression of Atg39 drives the formation of protrusions from the NE accompanied by both the ONM and INM. The APHs in the C-terminal perinuclear space (NE lumen) region of Atg39 play multiple roles in nucleophagy: Atg39 retention to the NE; the basal and enhanced assembly of Atg39 in the NE, the latter of which occurs in conjunction with autophagosome formation; and the formation of NE protrusions, the tips of which are pinched off to generate NDVs.
Results
Atg39 is an ONM protein
Although Atg39 was predicted to be an integral membrane protein with a single transmembrane domain, separating it into N-terminal (1–144) and C-terminal (165–398) regions, its membrane topology has yet to be demonstrated experimentally. To determine the membrane topology of Atg39, lysates were prepared from yeast cells expressing Atg39 N- and C-terminally tagged with HA and GFP sequences, respectively, and treated with proteinase K in the presence or absence of the detergent Triton X-100 (Fig. 1, A and B). Immunoblotting using anti-HA antibody showed that the HA tag in HA-Atg39-GFP was digested by proteinase K regardless of the presence of Triton X-100 (Fig. 1 A). In contrast, immunoblotting with anti-GFP antibody revealed that the GFP tag in Atg39-GFP was largely resistant to proteinase K in the absence of Triton X-100, although it was trimmed to a size corresponding to the protein lacking the region N-terminal to the transmembrane domain (Fig. 1 B). These results suggest that Atg39 is a single-pass membrane protein with N- and C-terminal regions exposed to the cytoplasm and the perinuclear space, respectively (Fig. 1 C), consistent with the existence of Atg8- and Atg11-binding sequences in the N-terminal region (Mochida et al., 2015).
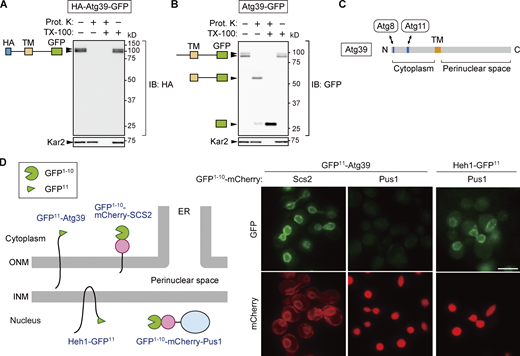
Atg39 is a transmembrane protein embedded in the ONM. (A and B) A proteinase K protection assay was performed to determine the membrane topology of Atg39. Lysates were prepared from cells overexpressing HA-Atg39-GFP (A) or Atg39-GFP (B) under the ADH1 promoter and incubated with proteinase K (Prot. K) in the absence or presence of Triton X-100 (TX-100). N-terminal HA and C-terminal GFP tags were detected by immunoblotting using anti-HA and anti-GFP antibodies, respectively. Kar2, an ER lumenal protein. (C) Schematic diagram of Atg39. TM, transmembrane domain. (D) Split GFP-based assay was performed to determine the subnuclear localization of Atg39. An N-terminal fragment of GFP (GFP1–10) and mCherry were fused to a transmembrane domain of Scs2 (GFP1–10-mCherry-Scs2) or Pus1 (GFP1–10-mCherry-Pus1), and the remaining C-terminal fragment of GFP (GFP11) was fused to the N terminus of Atg39 or the C terminus of the INM protein Heh1. Cells expressing these proteins were observed under a fluorescence microscope. GFP11-Atg39 was constitutively expressed under the HRR25 promoter. Scale bars, 5 µm. Source data are available for this figure: SourceData F1.
Atg39 is a transmembrane protein embedded in the ONM. (A and B) A proteinase K protection assay was performed to determine the membrane topology of Atg39. Lysates were prepared from cells overexpressing HA-Atg39-GFP (A) or Atg39-GFP (B) under the ADH1 promoter and incubated with proteinase K (Prot. K) in the absence or presence of Triton X-100 (TX-100). N-terminal HA and C-terminal GFP tags were detected by immunoblotting using anti-HA and anti-GFP antibodies, respectively. Kar2, an ER lumenal protein. (C) Schematic diagram of Atg39. TM, transmembrane domain. (D) Split GFP-based assay was performed to determine the subnuclear localization of Atg39. An N-terminal fragment of GFP (GFP1–10) and mCherry were fused to a transmembrane domain of Scs2 (GFP1–10-mCherry-Scs2) or Pus1 (GFP1–10-mCherry-Pus1), and the remaining C-terminal fragment of GFP (GFP11) was fused to the N terminus of Atg39 or the C terminus of the INM protein Heh1. Cells expressing these proteins were observed under a fluorescence microscope. GFP11-Atg39 was constitutively expressed under the HRR25 promoter. Scale bars, 5 µm. Source data are available for this figure: SourceData F1.
Although the exclusive NE localization of Atg39 is observed under a fluorescence microscope (Mochida et al., 2015), this observation does not exclude the possibility that Atg39 localizes not only to the ONM but also to the INM. To investigate whether Atg39 is embedded in the ONM only or in both the ONM and INM, we used a split-GFP–based system (Smoyer et al., 2016). In this system, an N-terminal sequence of GFP containing 10 β strands (GFP1–10) was attached to the ONM/ER membrane protein Scs2 (GFP1–10-mCherry-Scs2) or the nucleoplasmic protein Pus1 (GFP1–10-mCherry-Pus1), and the remaining C-terminal sequence containing the last β strand of GFP (GFP11) was fused to the N terminus of Atg39 (GFP11-Atg39; Fig. 1 D). When the two GFP fragments exist in the same compartment, they form a functional GFP. GFP fluorescence was observed in the NE of cells coexpressing GFP1–10-mCherry-Scs2 and GFP11-Atg39, but not those expressing GFP1–10-mCherry-Pus1 instead of GFP1–10-mCherry-Scs2. When GFP1–10-mCherry-Pus1 was coexpressed with GFP11 fused to the INM protein Heh1 (also known as Src1), GFP fluorescence was observed in the NE (INM), confirming that GFP1–10-mCherry-Pus1 can form a functional GFP if the GFP11-fused protein exists in the same compartment. These results support the conclusion that Atg39 specifically localizes to the ONM.
APHs of Atg39 bind to the INM to retain Atg39 in the NE
Because the ONM is continuous with the ER membrane and these membranes share many proteins, a mechanism must exist that retains Atg39 in the ONM. The determined topology of Atg39 suggests that the C-terminal region (165–398) is exposed to the perinuclear space; however, the importance and function of this region in nucleophagy have yet to be investigated. We therefore examined the intracellular localization of C-terminally truncated Atg39 mutants (Fig. 2, A and B). Whereas Atg391–347 and Atg391–325, as with WT Atg39, exhibited specific localization to the ONM, Atg391–296 and Atg391–194 leaked out into the ER, which spreads mainly beneath the plasma membrane in yeast cells (West et al., 2011). In contrast, deletion of the N-terminal cytoplasmic region did not affect the ONM localization of Atg39 (Fig. S1 A, Atg39136–398). These results suggest that the C-terminal region 297–325 is responsible for limiting Atg39 localization to the ONM.
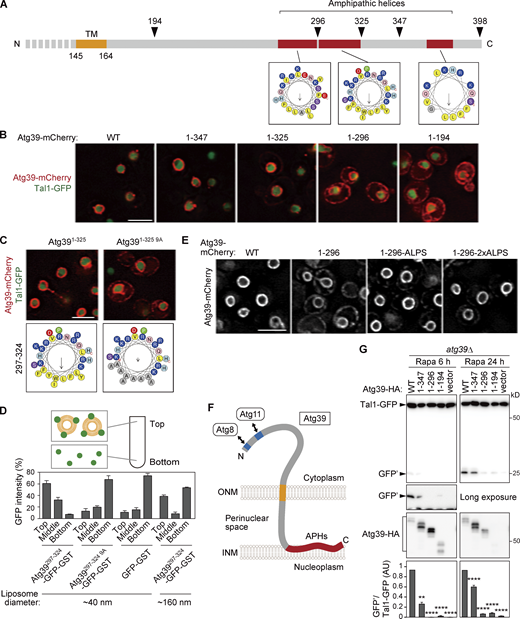
The APHs of Atg39 bind to the INM to retain Atg39 in the ONM and are important for nucleophagy. (A) Schematic diagram of the C-terminal perinuclear space region of Atg39. Helical wheels were generated using a HeliQuest server. TM, transmembrane domain. (B) mCherry-fused WT Atg39 (WT) and C-terminally truncated mutants were expressed under the constitutive HRR25 promoter and analyzed by fluorescence microscopy. Tal1, a nucleoplasmic protein. (C) The localization of Atg391–325-mCherry and Atg391–325 9A-mCherry expressed under the HRR25 promoter was analyzed by fluorescence microscopy. (D) Liposome flotation assay was performed to examine the membrane-binding ability of APH297–324. Purified GFP-GST-fused proteins were mixed with small (∼40 nm) or large (∼160 nm) liposomes, and then the liposomes and liposome-bound proteins were floated by ultracentrifugation. Data are shown as means ± SD (n = 3). (E) Atg391–296 fused with the ALPS motif was overexpressed under the CUP1 promoter. The localization of these proteins in cells not treated with rapamycin was analyzed by fluorescence microscopy. (F) The determined membrane topology of Atg39. (G)atg39Δ cells were transformed with plasmids for expressing WT Atg39 or C-terminally truncated mutants fused with the HA sequence under the ATG39 promoter. These cells were treated with rapamycin to induce nucleophagy, and degradation of Tal1-GFP was examined by immunoblotting using anti-GFP antibody. The quantification results are shown as means ± SD (n = 3). **, P < 0.01; ****, P < 0.0001 (unpaired two-tailed Student’s t test). Scale bars, 5 µm. AU, arbitrary unit. Source data are available for this figure: SourceData F2.
The APHs of Atg39 bind to the INM to retain Atg39 in the ONM and are important for nucleophagy. (A) Schematic diagram of the C-terminal perinuclear space region of Atg39. Helical wheels were generated using a HeliQuest server. TM, transmembrane domain. (B) mCherry-fused WT Atg39 (WT) and C-terminally truncated mutants were expressed under the constitutive HRR25 promoter and analyzed by fluorescence microscopy. Tal1, a nucleoplasmic protein. (C) The localization of Atg391–325-mCherry and Atg391–325 9A-mCherry expressed under the HRR25 promoter was analyzed by fluorescence microscopy. (D) Liposome flotation assay was performed to examine the membrane-binding ability of APH297–324. Purified GFP-GST-fused proteins were mixed with small (∼40 nm) or large (∼160 nm) liposomes, and then the liposomes and liposome-bound proteins were floated by ultracentrifugation. Data are shown as means ± SD (n = 3). (E) Atg391–296 fused with the ALPS motif was overexpressed under the CUP1 promoter. The localization of these proteins in cells not treated with rapamycin was analyzed by fluorescence microscopy. (F) The determined membrane topology of Atg39. (G)atg39Δ cells were transformed with plasmids for expressing WT Atg39 or C-terminally truncated mutants fused with the HA sequence under the ATG39 promoter. These cells were treated with rapamycin to induce nucleophagy, and degradation of Tal1-GFP was examined by immunoblotting using anti-GFP antibody. The quantification results are shown as means ± SD (n = 3). **, P < 0.01; ****, P < 0.0001 (unpaired two-tailed Student’s t test). Scale bars, 5 µm. AU, arbitrary unit. Source data are available for this figure: SourceData F2.

The perinuclear space region of Atg39 associates with the INM. (A) The intracellular localization of mCherry-fused WT Atg39 (WT) and the N-terminally truncated mutants expressed under the HRR25 promoter was analyzed by fluorescence microscopy. TM, transmembrane domain. (B) Cells coexpressing Atg39167–398-GFP under the HRR25 promoter and Osw5-mCherry were incubated in oleate-containing medium for 9 h to induce large lipid droplet formation and analyzed under a fluorescence microscope. (C) The five hydrophobic residues of the APH272–295 and nine hydrophobic residues of the APH297–324 were replaced with alanine (5A and 9A, respectively). These mutants were expressed under the HRR25 promoter, and their localization was examined by fluorescence microscopy. (D) The 4×HA sequence was inserted between the APH297–324 and APH365–379 of FLAG-Atg39136–398, and cells expressing this Atg39 variant under the HRR25 promoter were analyzed by immunoEM using anti-HA antibody. Immunogold signals in the vicinity of the INM and ONM (arrowheads) were counted, and the results are shown in the graph. Scale bars, 5 µm (A–C) and 200 nm (D).
The perinuclear space region of Atg39 associates with the INM. (A) The intracellular localization of mCherry-fused WT Atg39 (WT) and the N-terminally truncated mutants expressed under the HRR25 promoter was analyzed by fluorescence microscopy. TM, transmembrane domain. (B) Cells coexpressing Atg39167–398-GFP under the HRR25 promoter and Osw5-mCherry were incubated in oleate-containing medium for 9 h to induce large lipid droplet formation and analyzed under a fluorescence microscope. (C) The five hydrophobic residues of the APH272–295 and nine hydrophobic residues of the APH297–324 were replaced with alanine (5A and 9A, respectively). These mutants were expressed under the HRR25 promoter, and their localization was examined by fluorescence microscopy. (D) The 4×HA sequence was inserted between the APH297–324 and APH365–379 of FLAG-Atg39136–398, and cells expressing this Atg39 variant under the HRR25 promoter were analyzed by immunoEM using anti-HA antibody. Immunogold signals in the vicinity of the INM and ONM (arrowheads) were counted, and the results are shown in the graph. Scale bars, 5 µm (A–C) and 200 nm (D).
In analysis of the subcellular localization of N-terminally truncated Atg39 mutants, we found that the mutant lacking cytoplasmic and transmembrane domains (Atg39167–398) localized to the surface of lipid droplets (Fig. S1, A and B). Although this is an artifact of cytoplasmic expression of the perinuclear space region, it raised the possibility that the perinuclear space region of Atg39 has the ability to associate with lipid membranes. The C-terminal region of Atg39 is predicted to largely form a helical conformation, containing at least three APHs, including helix 297–324 (APH297–324), which is important for the ONM localization of Atg39 (Fig. 2, A and B). APHs are known to bind to membranes through their hydrophobic surface (Drin and Antonny, 2010). We therefore speculated that APH297–324 associates with the INM from the side of the perinuclear space to prevent Atg39 from leaking into the ER. To examine this possibility, nine hydrophobic residues in APH297–324 were replaced with alanine (Atg391–325 9A; Fig. 2 C). Similar to Atg391–296, this 9A mutant mislocalized to the ER, highlighting the importance of the amphipathic property of APH297–324 in Atg39 retention to the ONM. Alanine substitution of five hydrophobic residues in APH272–295 also resulted in Atg39 leakage into the ER (Fig. S1 C, Atg391–325 5A). These results suggest that, in addition to APH297–324, APH272–295 contributes to limiting Atg39 localization to the ONM.
Next, we performed a coflotation assay using liposomes and purified APH297–324 tagged with GFP-GST (Fig. 2 D). Although APH297–324-GFP-GST cofloated with small liposomes (∼40 nm in diameter) after centrifugation, the 9A mutation severely impaired liposome binding of this fusion protein, demonstrating that APH297–324 indeed has a membrane-binding ability. To verify whether imparting membrane-binding ability to Atg391–296 recovers its ONM-specific localization, the amphipathic lipid packing sensor (ALPS) motif of ArfGAP1, which binds to membranes (Bigay et al., 2005) and is irrelative to nucleophagy, was fused to the C terminus of Atg391–296. The ALPS motif is intrinsically disordered but forms an APH when this region is inserted into membranes (Bigay et al., 2005). We found that fusion of one or two copies of the ALPS motif to Atg391–296 (Atg391–296-ALPS or Atg391–296-2×ALPS) almost completely recovered its ONM localization (Fig. 2 E). Although APHs are often involved in protein folding by burying their hydrophobic residues inside of the protein, successful rescue of the ONM localization of Atg391–296 by the ALPS motif, whose amino acid sequence completely differs from Atg39 APH297–324, suggested that the mislocalization of Atg391–296 to the ER was due to loss of its membrane-binding by APH297–324 rather than misfolding of the protein. To clarify that the APHs of Atg39 associate with the INM but not the ONM in the perinuclear space, we performed immunoEM of an Atg39 variant in which the HA sequence was inserted into the region between APH297–324 and APH365–379 (Fig. S1 D). Reaction of the immunogold particles with the HA sequence was frequently detected in the vicinity of the INM. Overall, these findings suggest that, in addition to the transmembrane domain, which penetrates the ONM, Atg39 contains membrane-binding APHs and is exclusively localized to the ONM via the insertion of APHs into the perinuclear space–facing layer of the INM (Fig. 2 F). These two types of membrane-binding domains bridging the ONM and INM allowed us to speculate that Atg39 couples deformation of the ONM and INM during NDV formation in nucleophagy.
APHs of Atg39 promote Atg39 assembly at the NDV formation site
Next, we examined nucleophagy in APH-deleted Atg39 mutants. Macronucleophagy activity can be evaluated by the amounts of protease-resistant GFP fragments (GFP′) generated by vacuolar degradation of the nucleoplasmic protein Tal1 (Breker et al., 2014) fused with GFP (Fig. S2 A). The results showed that the Atg391–296 and Atg391–194 mutants were severely defective in nucleophagy and that the Atg391–347 mutant had a milder defect (Fig. 2 G), confirming that the APHs of Atg39 are important for nucleophagy. Although the expression level of Atg391–296 was lower than the WT protein when this protein was expressed under its own promoter (Fig. S2 B), cells expressing Atg391–296 at levels comparable to the WT protein using the copper-inducible CUP1 promoter and different concentrations of copper ions were still defective in degradation of Tal1-GFP (Fig. S2, C and D). Deletion of the Atg39 APHs also decreased degradation of GFP-fused Nop1 (nucleolus), Heh1 (INM), Hmg1 (ONM), and Sec63 (ER membrane; Fig. S2, E–H), suggesting that the Atg39 APHs are required for degradation of any nuclear compartments.
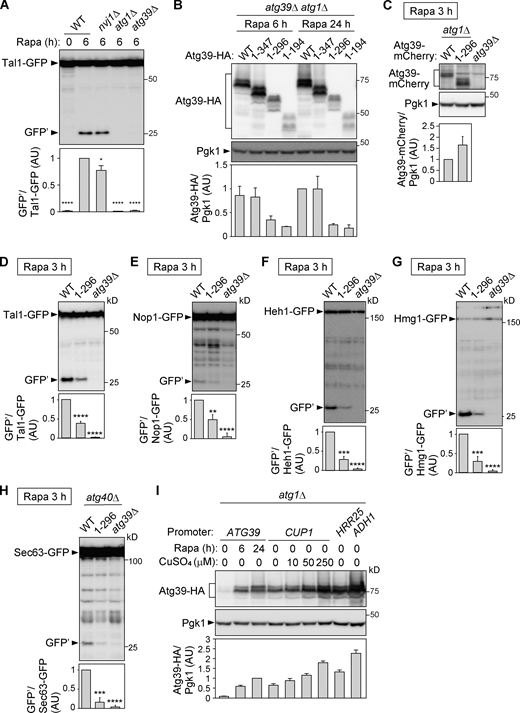
The APHs of Atg39 are required for degradation of proteins localized to different nuclear compartments. (A) Degradation of Tal1-GFP in cells lacking NVJ1 (essential for micronucleophagy), ATG1 (essential for macroautophagy), or ATG39 was analyzed by immunoblotting using anti-GFP antibody. Bars represent means ± SD (n = 3). *, P < 0.05; ****, P < 0.0001 (unpaired two-tailed Student’s t test). (B)atg39Δ atg1Δ cells carrying plasmids for expressing Atg39-HA under its own promoter were treated with rapamycin. Expression levels of Atg39-HA were analyzed by immunoblotting using anti-HA antibody. Pgk1 serves as a loading control. Bars represent means ± SD (n = 3). (C)atg1Δ cells expressing Atg39-mCherry or Atg391–296-mCherry under the CUP1 promoter were grown in the presence of 50 or 100 µM CuSO4, respectively, and treated with rapamycin for 3 h. Bars represent means ± SD (n = 3). (D–H) Degradation GFP-fused Tal1 (nucleoplasm), Nop1 (nucleolus), Heh1 (INM), Hmg1 (ONM), and Sec63 (ER) were analyzed by immunoblotting. Cells were grown and harvested under the same conditions as in C. Bars represent means ± SD (n = 3). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (unpaired two-tailed Student’s t test). (I) Expression levels of Atg39-HA under the control of different promoters used in this study and in media containing different concentrations of CuSO4 were compared. Bars represent means ± SD (n = 3). AU, arbitrary unit. Source data are available for this figure: SourceData FS2.
The APHs of Atg39 are required for degradation of proteins localized to different nuclear compartments. (A) Degradation of Tal1-GFP in cells lacking NVJ1 (essential for micronucleophagy), ATG1 (essential for macroautophagy), or ATG39 was analyzed by immunoblotting using anti-GFP antibody. Bars represent means ± SD (n = 3). *, P < 0.05; ****, P < 0.0001 (unpaired two-tailed Student’s t test). (B)atg39Δ atg1Δ cells carrying plasmids for expressing Atg39-HA under its own promoter were treated with rapamycin. Expression levels of Atg39-HA were analyzed by immunoblotting using anti-HA antibody. Pgk1 serves as a loading control. Bars represent means ± SD (n = 3). (C)atg1Δ cells expressing Atg39-mCherry or Atg391–296-mCherry under the CUP1 promoter were grown in the presence of 50 or 100 µM CuSO4, respectively, and treated with rapamycin for 3 h. Bars represent means ± SD (n = 3). (D–H) Degradation GFP-fused Tal1 (nucleoplasm), Nop1 (nucleolus), Heh1 (INM), Hmg1 (ONM), and Sec63 (ER) were analyzed by immunoblotting. Cells were grown and harvested under the same conditions as in C. Bars represent means ± SD (n = 3). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (unpaired two-tailed Student’s t test). (I) Expression levels of Atg39-HA under the control of different promoters used in this study and in media containing different concentrations of CuSO4 were compared. Bars represent means ± SD (n = 3). AU, arbitrary unit. Source data are available for this figure: SourceData FS2.
Next, we determined which nucleophagy step is defective in Atg39 mutants lacking the APHs. To this end, we first determined the intracellular dynamics of Atg39 during the process of nucleophagy. Because the expression of Atg39 is repressed under normal conditions (Mochida et al., 2015), in these experiments, we expressed Atg39 using the constitutive HRR25 promoter (Fig. S2 I) and observed changes in Atg39 localization upon rapamycin treatment (nucleophagy induction). Before adding rapamycin, Atg39-mCherry was almost uniformly distributed in the NE, although a significant proportion of cells had faint Atg39-mCherry puncta that did not colocalize with the autophagosomal membrane marker Atg8 tagged with mNeonGreen (mNG; Fig. 3 A). In contrast, 30 min after rapamycin addition, Atg39-mCherry assembled and formed bright puncta that colocalized with mNG-Atg8, and after 3 h, these puncta disappeared, and mCherry and mNG fluorescence was observed within the vacuole, showing the progression of nucleophagy (Fig. 3 A). Moreover, after 30 min of rapamycin treatment, the NE frequently formed protrusions (or buds), with notable accumulation of Atg39-mCherry and mNG-Atg8 (Fig. 3 A). Colocalization of the INM protein Heh1 suggested that the INM protrudes together with the ONM (Figs. 3 B and S3 A). These results further suggest that NE protrusions associated with bright Atg39-mCherry puncta represent forming NDVs. In contrast, the bright puncta of Atg39-mCherry did not form in cells lacking the core Atg protein Atg1 or Atg2, and this was also the case in cells lacking the adapter protein Atg11, which recruits the core Atg proteins (Fig. 4 A). Meanwhile, the formation of Atg8-negative, faint Atg39 puncta, which did not increase following rapamycin treatment (Fig. 3 A), was largely unaffected by deletion of genes encoding these Atg proteins, including Atg8 (Figs. 4 A and S3 B; Atg1 was used to discriminate Atg39 puncta associated with autophagosome formation in atg8Δ cells). These results suggest that Atg39 partially assembles in the NE, and that this assembly is strongly enhanced in line with the formation of the autophagosomal membrane. One would expect that the latter mode of Atg39 assembly is mediated by the interaction with Atg8 or Atg11. Indeed, introducing mutations into either the Atg8-interacting motif (AIM) or the Atg11-binding region (11BR) of Atg39 largely blocked the formation of bright Atg39-mCherry puncta that colocalize with Atg8, while Atg8-negative Atg39-mCherry puncta were still observed in these cells (Fig. 4 B). Since autophagy receptors bind to Atg11 to recruit core Atg proteins for autophagosome formation, decreased numbers of Atg8-positive Atg39 puncta in cells expressing the 11BR mutant would be caused by the defective initiation of autophagosome formation. These results suggest that the interaction of Atg39 with Atg8 on forming autophagosomal membranes mediates higher-order assembly of Atg39.

Atg39 accumulates at NE protrusions upon nucleophagy induction. (A) Cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin and observed under a fluorescence microscope. The number of mNG-Atg8–positive and –negative puncta of Atg39-mCherry (bottom left) and the fluorescence intensity of these puncta (bottom right) were measured and are shown graphically. Bars represent means ± SD (n = 3; bottom left). ***, P < 0.001 (unpaired two-tailed Student’s t test; bottom left). ****, P < 0.0001 (unpaired two-tailed Mann–Whitney U test; bottom right). Arrowheads, mNG-Atg8–positive Atg39-mCherry puncta; arrows, mNG-Atg8–negative Atg39-mCherry puncta. Scale bars, 5 µm. (B) Cells constitutively expressing Atg39-mCherry under the HRR25 promoter were treated with rapamycin for 0.5 h and analyzed by fluorescence microscopy. Fluorescence intensity along the dashed line was measured and is graphically shown. Arrowheads, Atg39-mCherry–positive NE protrusions with mNG-Atg8 puncta. Scale bars, 2 µm. AU, arbitrary unit.
Atg39 accumulates at NE protrusions upon nucleophagy induction. (A) Cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin and observed under a fluorescence microscope. The number of mNG-Atg8–positive and –negative puncta of Atg39-mCherry (bottom left) and the fluorescence intensity of these puncta (bottom right) were measured and are shown graphically. Bars represent means ± SD (n = 3; bottom left). ***, P < 0.001 (unpaired two-tailed Student’s t test; bottom left). ****, P < 0.0001 (unpaired two-tailed Mann–Whitney U test; bottom right). Arrowheads, mNG-Atg8–positive Atg39-mCherry puncta; arrows, mNG-Atg8–negative Atg39-mCherry puncta. Scale bars, 5 µm. (B) Cells constitutively expressing Atg39-mCherry under the HRR25 promoter were treated with rapamycin for 0.5 h and analyzed by fluorescence microscopy. Fluorescence intensity along the dashed line was measured and is graphically shown. Arrowheads, Atg39-mCherry–positive NE protrusions with mNG-Atg8 puncta. Scale bars, 2 µm. AU, arbitrary unit.

Analysis of Atg39 assembly at protruded nuclear regions. (A) Cells constitutively expressing Atg39-mCherry under the HRR25 promoter were treated with rapamycin for 0.5 h and analyzed by fluorescence microscopy. Fluorescence intensity along the dashed line was measured, and the results are shown in the graph. Arrowheads, Atg39-mCherry–positive NE protrusions with mNG-Atg8 puncta. Scale bars, 2 µm. (B) Cells coexpressing Atg39-GFP under the HRR25 promoter and Atg1-mCherry were analyzed by fluorescence microscopy. The number of Atg39-GFP puncta per cell is shown. Arrows, Atg1-mCherry–negative Atg39 puncta. Bars represent means ± SD (n = 3). Scale bars, 5 µm. AU, arbitrary unit.
Analysis of Atg39 assembly at protruded nuclear regions. (A) Cells constitutively expressing Atg39-mCherry under the HRR25 promoter were treated with rapamycin for 0.5 h and analyzed by fluorescence microscopy. Fluorescence intensity along the dashed line was measured, and the results are shown in the graph. Arrowheads, Atg39-mCherry–positive NE protrusions with mNG-Atg8 puncta. Scale bars, 2 µm. (B) Cells coexpressing Atg39-GFP under the HRR25 promoter and Atg1-mCherry were analyzed by fluorescence microscopy. The number of Atg39-GFP puncta per cell is shown. Arrows, Atg1-mCherry–negative Atg39 puncta. Bars represent means ± SD (n = 3). Scale bars, 5 µm. AU, arbitrary unit.

Atg39 clusters in the NE through the APHs and binds to Atg8 on forming autophagosomal membranes. (A and B) Cells expressing Atg39-mCherry under the HRR25 promoter (A) or mCherry-fused WT Atg39 or mutants defective in binding to Atg8 and/or Atg11 under the CUP1 promoter (B) were treated with rapamycin for 0.5 h and analyzed by fluorescence microscopy. (C) Puncta formation of WT Atg39 and Atg391–296 expressed under the CUP1 promoter was examined by fluorescence microscopy. Bars represent means ± SD (n = 3). P values were determined by unpaired two-tailed Student’s t test (bar graphs) or unpaired two-tailed Mann–Whitney U test (boxplots). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Arrowheads, mNG/GFP-Atg8–positive Atg39-mCherry puncta; arrows, mNG/GFP-Atg8–negative Atg39 puncta. Scale bars, 5 µm. AU, arbitrary unit.
Atg39 clusters in the NE through the APHs and binds to Atg8 on forming autophagosomal membranes. (A and B) Cells expressing Atg39-mCherry under the HRR25 promoter (A) or mCherry-fused WT Atg39 or mutants defective in binding to Atg8 and/or Atg11 under the CUP1 promoter (B) were treated with rapamycin for 0.5 h and analyzed by fluorescence microscopy. (C) Puncta formation of WT Atg39 and Atg391–296 expressed under the CUP1 promoter was examined by fluorescence microscopy. Bars represent means ± SD (n = 3). P values were determined by unpaired two-tailed Student’s t test (bar graphs) or unpaired two-tailed Mann–Whitney U test (boxplots). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Arrowheads, mNG/GFP-Atg8–positive Atg39-mCherry puncta; arrows, mNG/GFP-Atg8–negative Atg39 puncta. Scale bars, 5 µm. AU, arbitrary unit.
As with the membrane-binding APHs analyzed previously (Drin and Antonny, 2010), APH297–324 of Atg39 preferentially bound to smaller liposomes (membranes with high positive curvature; Fig. 2 D). Because protruding membrane domains have higher curvature than other parts of the NE, it is possible that the APHs of Atg39 sense the local curvature in the INM during assembly. Indeed, deletion of the APHs decreased the formation of both Atg8-negative (faint, rapamycin-independent) and Atg8-positive (bright, rapamycin-dependent) Atg39-mCherry puncta (Fig. 4 C). These results suggest that the APHs of Atg39 are involved in both basal and enhanced Atg39 assembly in the NE. As mentioned above, enhanced Atg39 assembly depends on autophagosome formation (Fig. 4 A) and binding to Atg8 (Fig. 4 B), and thus it is likely that autophagosome formation in the cytoplasm and APHs bound to the INM act together to assemble Atg39 at the site of NDV formation.
APHs of Atg39 are involved in deformation of the NE during NDV formation
The insertion of APHs into one side of the lipid bilayer can bend the membrane (Drin and Antonny, 2010). We therefore examined whether Atg39 itself can induce local NE remodeling for the generation of NDVs. Atg39-mCherry was overexpressed using the CUP1 promoter in atg1Δ cells to block nucleophagy and accumulate Atg39-mCherry in the NE. We found that the accumulation of Atg39 in the NE caused the extension of tubules from the NE (Fig. 5 A), consistent with a previous report (Vevea et al., 2015). Rapamycin treatment increased the formation of these NE tubules in terms of both number and length in atg1Δ cells (Fig. 5 A). In addition, additional deletion of neither ATG8 nor ATG11 in atg1Δ cells affected formation of the NE tubules upon overexpression of Atg39 (Fig. S4 A). These results suggest that rapamycin treatment promotes NE tubule formation independently of autophagosome formation and the interactions of Atg39 with Atg8 and Atg11. EM further confirmed that both the ONM and INM join to form these tubules (Fig. 5 B). The tips of the tubules were often swollen and contained components similar to the nucleoplasm, consistent with the idea that these structures are related to NDV formation.
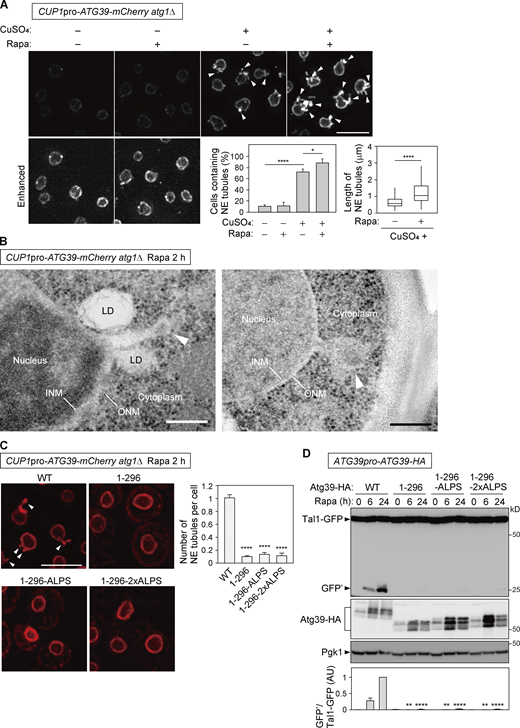
Atg39 accumulation in the NE causes NE tubulation depending on the APHs. (A)atg1Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were grown in the presence or absence of 250 µM CuSO4 overnight and treated with rapamycin for 2 h. The images are maximum-intensity projections of Z stacks. The percentage of cells containing NE tubules (left) and length of NE tubules (right) are shown. Bars represent means ± SD (n = 3). *, P < 0.05; ****, P < 0.0001 (Tukey’s multiple comparisons test; left). ****, P < 0.0001 (unpaired two-tailed Mann–Whitney U test; right). (B)atg1Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 2 h, and the nuclear morphology of these cells was examined by EM. Arrowheads, NE tubules with swollen tips. LD, lipid droplet. (C) mCherry-fused Atg39, Atg391–296, Atg391–296-ALPS, and Atg391–296-2×ALPS were overexpressed under the CUP1 promoter in atg1Δ cells. These cells were treated with rapamycin for 2 h, and the formation of NE tubules was analyzed by fluorescence microscopy. Arrowheads, NE tubules. Bars represent means ± SD (n = 3). ****, P < 0.0001 (unpaired two-tailed Student’s t test). (D) Plasmids for expressing HA-fused Atg39, Atg391–296, Atg391–296-ALPS, and Atg391–296-2×ALPS from their own promoter were integrated into the genome of atg39Δ cells. Degradation of Tal1-GFP in these cells was analyzed by immunoblotting. Pgk1 serves as a loading control. Bars represent means ± SD (n = 3). **, P < 0.01; ****, P < 0.0001 (unpaired two-tailed Student’s t test). Scale bars, 5 µm (A and C) and 200 nm (B). AU, arbitrary unit. Source data are available for this figure: SourceData F5.
Atg39 accumulation in the NE causes NE tubulation depending on the APHs. (A)atg1Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were grown in the presence or absence of 250 µM CuSO4 overnight and treated with rapamycin for 2 h. The images are maximum-intensity projections of Z stacks. The percentage of cells containing NE tubules (left) and length of NE tubules (right) are shown. Bars represent means ± SD (n = 3). *, P < 0.05; ****, P < 0.0001 (Tukey’s multiple comparisons test; left). ****, P < 0.0001 (unpaired two-tailed Mann–Whitney U test; right). (B)atg1Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 2 h, and the nuclear morphology of these cells was examined by EM. Arrowheads, NE tubules with swollen tips. LD, lipid droplet. (C) mCherry-fused Atg39, Atg391–296, Atg391–296-ALPS, and Atg391–296-2×ALPS were overexpressed under the CUP1 promoter in atg1Δ cells. These cells were treated with rapamycin for 2 h, and the formation of NE tubules was analyzed by fluorescence microscopy. Arrowheads, NE tubules. Bars represent means ± SD (n = 3). ****, P < 0.0001 (unpaired two-tailed Student’s t test). (D) Plasmids for expressing HA-fused Atg39, Atg391–296, Atg391–296-ALPS, and Atg391–296-2×ALPS from their own promoter were integrated into the genome of atg39Δ cells. Degradation of Tal1-GFP in these cells was analyzed by immunoblotting. Pgk1 serves as a loading control. Bars represent means ± SD (n = 3). **, P < 0.01; ****, P < 0.0001 (unpaired two-tailed Student’s t test). Scale bars, 5 µm (A and C) and 200 nm (B). AU, arbitrary unit. Source data are available for this figure: SourceData F5.

Analysis of NE tubules induced by Atg39 overexpression. (A) NE tubule formation was induced by the overexpression of Atg39 under the CUP1 promoter and 2-h rapamycin treatment in atg1Δ cells, and the effects of additional deletion of ATG8 or ATG11 on NE tubule formation were examined by fluorescence microscopy. The images are maximum-intensity projections of Z stacks. The percentage of cells containing NE tubules (top right) and the length of NE tubules (bottom right) are shown. Bars represent means ± SD (n = 3). Statistical significance was determined using Tukey’s multiple comparisons test (top right) and the Kruskal–Wallis test (bottom right), respectively. Arrowheads, NE tubules with GFP signals. (B)ypt7Δ cells and ypt7Δ atg1Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 2 h to induce the formation of NDVs and NE tubules. The number of Atg39-mCherry puncta (bottom left) and the length of NE tubules (bottom right) are shown. Bars represent means ± SD (n = 3). P values were determined by unpaired two-tailed Student’s t test (bottom left) or unpaired two-tailed Mann–Whitney U test (bottom right). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Scale bars, 5 µm.
Analysis of NE tubules induced by Atg39 overexpression. (A) NE tubule formation was induced by the overexpression of Atg39 under the CUP1 promoter and 2-h rapamycin treatment in atg1Δ cells, and the effects of additional deletion of ATG8 or ATG11 on NE tubule formation were examined by fluorescence microscopy. The images are maximum-intensity projections of Z stacks. The percentage of cells containing NE tubules (top right) and the length of NE tubules (bottom right) are shown. Bars represent means ± SD (n = 3). Statistical significance was determined using Tukey’s multiple comparisons test (top right) and the Kruskal–Wallis test (bottom right), respectively. Arrowheads, NE tubules with GFP signals. (B)ypt7Δ cells and ypt7Δ atg1Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 2 h to induce the formation of NDVs and NE tubules. The number of Atg39-mCherry puncta (bottom left) and the length of NE tubules (bottom right) are shown. Bars represent means ± SD (n = 3). P values were determined by unpaired two-tailed Student’s t test (bottom left) or unpaired two-tailed Mann–Whitney U test (bottom right). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Scale bars, 5 µm.
To further examine the relationship between NE protrusions and NDV formation, we analyzed both the length of NE tubules and the number of NDVs in cells lacking Ypt7, a Rab GTPase essential for autophagosome–vacuole fusion (Kirisako et al., 1999). While rapamycin treatment in ypt7Δ cells overexpressing Atg39-mCherry caused the accumulation of NDVs (enclosed within autophagosomes) in the cytoplasm, it did not significantly affect the length of NE tubules (Fig. S4 B; Mochida et al., 2015). On the other hands, ypt7Δ atg1Δ cells treated with rapamycin, in which NDVs are not formed, contained longer NE tubules than ypt7Δ cells (Fig. S4 B). These results are consistent with the idea that NE protrusions represent intermediates in NDV formation.
Although NE tubulation is an exaggerated phenomenon caused by Atg39 overexpression, puncta of endogenous Atg39 were often observed at the tip of the NE protrusions (Fig. S5 A). Furthermore, time-lapse microscopy showed that budding and fission of Atg39-mCherry–enriched structures can occur at the tip of NE tubules (Fig. S5 B). In addition, NE tubules were observed in atg1Δ cells expressing Atg39 under its own promoter after prolonged rapamycin treatment (Fig. S5 C). Overall, these results suggest that endogenous Atg39 also deforms the NE and generates short protrusions, the tip of which provides a site for NDV formation.

NE protrusion during nucleophagy. (A) Cells expressing Atg39-mCherry under their own promoter at the original chromosomal locus were treated with rapamycin for 4 h, and Atg39-mCherry puncta formed at the NE protrusion tip were analyzed by fluorescence microscopy. Fluorescence intensity along the dashed line was measured and is shown graphically. Arrowhead, NE protrusion. (B) Cells expressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 20 min, and then fluorescence images were taken at 15-s intervals. Arrowhead, a GFP-Atg8–positive Atg39-mCherry punctum at the tip of an NE tubule; arrow, an NDV. (C)atg1Δ cells expressing Atg39-mCherry under their own promoter at the original chromosomal locus were treated with rapamycin for 24 h. Representative EM images of NE protrusions are shown. Arrowhead, NE protrusion. (D) A representative EM image of an NE tubule without the INM (arrowhead) generated in atg1Δ cells overexpressing Atg39 under the CUP1 promoter and treated with rapamycin for 2 h. Scale bars, 5 µm (A), 2 µm (B), and 200 nm (C and D).
NE protrusion during nucleophagy. (A) Cells expressing Atg39-mCherry under their own promoter at the original chromosomal locus were treated with rapamycin for 4 h, and Atg39-mCherry puncta formed at the NE protrusion tip were analyzed by fluorescence microscopy. Fluorescence intensity along the dashed line was measured and is shown graphically. Arrowhead, NE protrusion. (B) Cells expressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 20 min, and then fluorescence images were taken at 15-s intervals. Arrowhead, a GFP-Atg8–positive Atg39-mCherry punctum at the tip of an NE tubule; arrow, an NDV. (C)atg1Δ cells expressing Atg39-mCherry under their own promoter at the original chromosomal locus were treated with rapamycin for 24 h. Representative EM images of NE protrusions are shown. Arrowhead, NE protrusion. (D) A representative EM image of an NE tubule without the INM (arrowhead) generated in atg1Δ cells overexpressing Atg39 under the CUP1 promoter and treated with rapamycin for 2 h. Scale bars, 5 µm (A), 2 µm (B), and 200 nm (C and D).
Next, we investigated the importance of the APHs in NE tubulation. The formation of NE tubules was much less efficient in cells overexpressing Atg391–296 compared with the WT Atg39 (Fig. 5 C), suggesting that the APHs of Atg39 are involved in NE tubulation. We also found that overexpression of the ALPS-motif–fused Atg391–296 mutants (Atg391–296-ALPS and Atg391–296-2×ALPS), which almost normally localized to the ONM in untreated cells (Fig. 2 E), failed to induce the formation of NE tubules (Fig. 5 C). (It should be noted that Atg391–296-ALPS partially leaked out into the ER in rapamycin-treated atg1Δ cells, whereas Atg391–296-2×ALPS was still retained in the ONM.) Moreover, Atg391–296-ALPS and Atg391–296-2×ALPS were defective in degradation of Tal1-GFP (Fig. 5 D). These results suggest that the ALPS motif of ArfGAP1 is sufficient for Atg39 anchoring to the ONM but not for the formation of NE tubules and nucleophagy. Thus, we concluded that the APHs of Atg39 play important roles not only in Atg39 anchoring to the NE but also in NE deformation. The APHs of Atg39 are thought to have a feature(s) different from the ArfGAP1 ALPS motif, which is required for NE tubulation.
Chromosomes are excluded from NE tubules and NDVs
Lastly, we investigated the incorporation of nuclear components into NE tubules under Atg39 overexpression. Consistent with the EM observations showing that these tubules are composed of both the ONM and INM (Fig. 6 A), fluorescence microscopy showed signals of the INM protein Heh1 in most tubules positive for Atg39 (Fig. 6 A). Moreover, although lower but significant proportions of NE tubules also contained the nucleoplasmic protein Tal1 and the nucleolar protein Nop56, the histone H2A Hta2 seldom colocalized with these tubules. Consistent with these results, GFP fragments derived from Hta2-GFP were scarcely detected and were only partially blocked by deletion of ATG39, while Atg39 was indispensable for degradation of Heh1-GFP, Tal1-GFP, and Nop56-GFP (Fig. 6, B–E). We also examined the incorporation of histone proteins into NDVs using ypt7Δ cells. Whereas puncta of Tal1-GFP were observed outside the nucleus, which colocalized with cytoplasmic Atg39-mCherry puncta representing NDVs sequestered within the autophagosomes, the GFP-fused histones Hta2 and Htz1 did not form puncta (Fig. 6 F). Moreover, Hoechst-stained DNA was not detected in Atg39-mCherry puncta in these cells (Fig. S6), suggesting that chromosomes do not enter NE protrusions and are excluded from sequestration into NDVs.
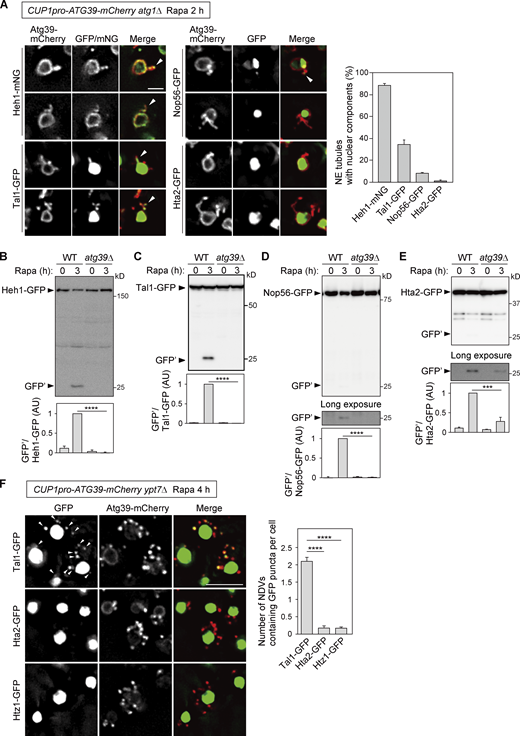
NE tubules and NDVs exclude chromosomes. (A) NE tubule formation was induced by the overexpression of Atg39-mCherry under the CUP1 promoter in atg1Δ cells, followed by rapamycin treatment for 2 h. The colocalization of Atg39-mCherry–positive NE tubules with mNG or GFP-fused Heh (INM), Tal1 (nucleoplasm), Nop56 (nucleolus), and Hta2 (histone) was examined by fluorescence microscopy. Arrowheads, NE tubules containing mNG/GFP signals. The percentage of NE tubules containing nuclear components is shown in the graph. Bars represent means ± SD (n = 3). (B–E) Degradation of GFP-fused Heh1, Tal1, Nop56, and Hta2 was examined in cells overexpressing Atg39-mCherry under the CUP1 promoter. Bars represent means ± SD (n = 3). ***, P < 0.001; ****, P < 0.0001 (unpaired two-tailed Student’s t test). (F)ypt7Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 4 h to induce NDV accumulation. Arrowheads, puncta of Tal1-GFP that colocalized with those of Atg39-mCherry. Bars represent means ± SD (n = 3). ****, P < 0.0001 (unpaired two-tailed Student’s t test). Scale bars, 2 µm (A) and 5 µm (F). AU, arbitrary unit. Source data are available for this figure: SourceData F6.
NE tubules and NDVs exclude chromosomes. (A) NE tubule formation was induced by the overexpression of Atg39-mCherry under the CUP1 promoter in atg1Δ cells, followed by rapamycin treatment for 2 h. The colocalization of Atg39-mCherry–positive NE tubules with mNG or GFP-fused Heh (INM), Tal1 (nucleoplasm), Nop56 (nucleolus), and Hta2 (histone) was examined by fluorescence microscopy. Arrowheads, NE tubules containing mNG/GFP signals. The percentage of NE tubules containing nuclear components is shown in the graph. Bars represent means ± SD (n = 3). (B–E) Degradation of GFP-fused Heh1, Tal1, Nop56, and Hta2 was examined in cells overexpressing Atg39-mCherry under the CUP1 promoter. Bars represent means ± SD (n = 3). ***, P < 0.001; ****, P < 0.0001 (unpaired two-tailed Student’s t test). (F)ypt7Δ cells overexpressing Atg39-mCherry under the CUP1 promoter were treated with rapamycin for 4 h to induce NDV accumulation. Arrowheads, puncta of Tal1-GFP that colocalized with those of Atg39-mCherry. Bars represent means ± SD (n = 3). ****, P < 0.0001 (unpaired two-tailed Student’s t test). Scale bars, 2 µm (A) and 5 µm (F). AU, arbitrary unit. Source data are available for this figure: SourceData F6.

NDVs do not contain DNA. ypt7Δ cells overexpressing Atg39-mCherry were treated with rapamycin for 8 h, and the DNA was stained with Hoechst 33258. Arrowhead, a nucleus. Small puncta marked by asterisks and that did not colocalize with Atg39-mCherry puncta represent mitochondrial DNA. Fluorescence intensity along the dashed line was measured and is shown in the graph. Scale bars, 5 µm. AU, arbitrary unit.
NDVs do not contain DNA. ypt7Δ cells overexpressing Atg39-mCherry were treated with rapamycin for 8 h, and the DNA was stained with Hoechst 33258. Arrowhead, a nucleus. Small puncta marked by asterisks and that did not colocalize with Atg39-mCherry puncta represent mitochondrial DNA. Fluorescence intensity along the dashed line was measured and is shown in the graph. Scale bars, 5 µm. AU, arbitrary unit.
Discussion
In this study, we showed that the nucleophagy receptor Atg39 is a single membrane-spanning ONM protein whose N-terminal cytoplasmic region interacts with Atg8 and Atg11 (Mochida et al., 2015) and whose C-terminal perinuclear space region contains APHs that associate with the INM. Deletion of these APHs resulted in Atg39 mislocalization to the ER, with impaired basal (autophagosome formation– and Atg8 binding–independent) and enhanced (autophagosome formation– and Atg8 binding–dependent) assembly in the NE as well as reduced nucleophagy activity. Overexpression of Atg39 induced the extension of double-membrane tubules from the NE depending on the APHs. Based on these results, we propose the following model for the mechanism of NDV formation during Atg39-mediated nucleophagy (Fig. 7).

Model mechanism of NDV formation in Atg39-mediated nucleophagy. Atg39 forms a small cluster depending on the APHs in the NE region, where the INM would partially protrude via an unknown mechanism. Then, Atg39 in the cluster recruits the core Atg proteins via Atg11 on the cytoplasmic surface of the NE, initiating autophagosome formation, which triggers the further assembly of Atg39 via the interaction with Atg8 on the forming autophagosomal membrane. The condensation of Atg39 locally enhances its APH-dependent membrane-deforming activity, protruding the NE toward the cytoplasm at the site. An unknown mechanism mediates fission of the tip of the protrusion to release an NDV, which is sequestered into the autophagosome via the interaction between Atg39 and Atg8, a canonical function of autophagy receptors.
Model mechanism of NDV formation in Atg39-mediated nucleophagy. Atg39 forms a small cluster depending on the APHs in the NE region, where the INM would partially protrude via an unknown mechanism. Then, Atg39 in the cluster recruits the core Atg proteins via Atg11 on the cytoplasmic surface of the NE, initiating autophagosome formation, which triggers the further assembly of Atg39 via the interaction with Atg8 on the forming autophagosomal membrane. The condensation of Atg39 locally enhances its APH-dependent membrane-deforming activity, protruding the NE toward the cytoplasm at the site. An unknown mechanism mediates fission of the tip of the protrusion to release an NDV, which is sequestered into the autophagosome via the interaction between Atg39 and Atg8, a canonical function of autophagy receptors.
First, a certain amount of Atg39 assembles in the NE in a manner dependent on its APHs and independent of autophagosome formation (Atg8-negative Atg39 puncta). Dependence of this basal Atg39 assembly on the APHs suggests that at least the INM protrudes into the perinuclear space at this stage. The APHs of Atg39 and/or other proteins (see below) may also be involved in this initial deformation of the NE/INM. The cytoplasmic region of Atg39 in the assemblage subsequently recruits the core Atg proteins via Atg11, thereby initiating autophagosome formation on the NE. Atg39 then assembles further via interaction with Atg8 on expanding isolation membranes, thereby inducing protrusion of the INM into the perinuclear space through the APHs while remaining anchored to the ONM in the transmembrane domain, resulting in the formation of double-membrane protrusions out of the NE. Both the ONM and INM cause fission at the tip of the protrusions, resulting in the formation of NDVs containing intranuclear components, which are then sequestered into the autophagosomes and delivered to the vacuole for degradation.
We found that fusion of the ALPS motif derived from ArfGAP1 to Atg391–296 recovered its NE-specific localization but failed to generate NE tubules when this protein was overexpressed and could not rescue nucleophagy activity in atg39Δ cells. These results allowed us to conclude that Atg39 APHs-dependent NE protrusion is crucial for nucleophagy. The ALPS motif is a sort of APH but has features different from many other APHs. For example, the ALPS motif is intrinsically unfolded but forms an APH upon association with membranes, and a polar face of the helix is highly enriched in serine and threonine residues (Drin and Antonny, 2010). Although a determinant(s) for NE deformation that the APHs of Atg39 have but the ALPS motif lacks remains unknown, it may be related to these characteristics specific to the ALPS or the difference in some properties such as length, insertion depth into membranes, and amino acid compositions between these helices.
Although membrane bending via the wedging effect of APH insertion has been well documented, the perinuclear space region of Atg39 may also generate membrane curvature through an additional mechanism that can cooperate with the wedging mechanism. For example, a number of BAR proteins contain APHs and form crescent-shaped dimers, which serve as scaffolds and act together with the APHs to tubulate membranes (Baumgart et al., 2011). Steric pressure generated by local crowding of peripheral membrane proteins can also cause the tubulation of flat membranes (Stachowiak et al., 2012). Structural and in vitro studies of the perinuclear space region of Atg39 should further clarify the detailed mechanisms by which this region drives NE deformation. In addition, it is also possible that Atg39 recruits other membrane-deforming proteins in the cytoplasm and perinuclear space to generate NDVs. It should be noted that tubules extending without the INM were also observed in Atg39-overexpressing cells (Figs. 6 A and S5 D). These ONM tubules were not formed when Atg39 lacking the APHs was overexpressed. This observation may be merely an artifact of Atg39 overexpression but implies that the APHs of Atg39 are also involved in ONM deformation. However, because the perinuclear space surface of the protruding ONM has negative curvature, the perinuclear space region other than the APHs might contribute to deformation of the ONM.
Although the mechanism of NDV fission from the NE remains to be elucidated, the endosomal sorting complex required for transport (ESCRT)-III complex is a potential candidate for fission of the INM. ESCRT-III proteins form spiral filaments and mediate the invagination and fission of intralumenal vesicles in late endosomes, and these proteins are also known to exist on the nucleoplasmic surface of the INM, where they cause NE deformation similar to that observed in nucleophagy (McCullough et al., 2018; Lee et al., 2012; Webster et al., 2014). During the nucleus-to-cytoplasm exit of some herpesviruses, the INM, in association with nucleocapsids, invaginates and causes fission, depending on ESCRT-III to release nucleocapsid-containing vesicles into the perinuclear space (Arii et al., 2018; Lee et al., 2012). Similarly, ESCRT-III may contribute to initial deformation, protrusion, and fission of the INM during NDV formation in nucleophagy. The molecular mechanism of ONM fission also remains unclear but is thought to involve membrane-deforming proteins such as dynamin-related proteins and reticulon-family proteins, which are reported to mediate organelle fission during selective autophagy of mitochondria and the ER, respectively (Mao et al., 2013; Khaminets et al., 2015; Mochida et al., 2020).
In this study, INM insertion of the APHs was found to be important for the NE retention of Atg39. However, continuity between the ONM and the ER membrane poses the further question of why the APHs of Atg39 associate with the INM in the NE, but not with the opposite membrane in the ER. The difference in thickness (the distance between the two opposing membranes) might be one answer to this question, given that the ER is thicker than the NE in S. cerevisiae (West et al., 2011), which might prevent the APHs of Atg39 from reaching the opposite membrane in the ER. In this case, Atg39 that has been synthesized in the ER is diffused to the NE and anchored to the INM via the APHs or degraded in the ER due to unstable APHs without membrane insertion. It is also conceivable that Atg39 APHs disfavor the lumen-facing leaflet of the ER due to its flatness or negative curvature, especially with the tubular ER. The perinuclear space region of Atg39 may also contain a sequence interacting with an INM protein, which cooperates with the APHs in Atg39 anchoring to the INM.
Although nucleophagy is thought to be advantageous for cells in that it can serve as a powerful system for quality and quantity control of nuclear components, it must avoid degrading components such as chromosomes that are essential for cell viability. In this study, we showed that NE protrusions induced by overexpression of Atg39, as well as NDVs, do not contain histones and DNA. We speculate that NE protrusions are thin enough to block the entry of chromosomes into NDVs, guaranteeing the safety in degradation of the nucleus.
NDV formation during nucleophagy is associated with complicated membrane dynamics in the NE. This study focused on the APHs of Atg39, providing a deeper understanding of the underlying mechanism. However, unresolved issues remain, including how the ONM deforms and how membrane fission occurs to release NDVs from NE protrusions. Further analysis of Atg39 and identification of other proteins involved in nucleophagy will be important in addressing these issues.
Materials and methods
Yeast strains and plasmids
Yeast strains used in this study are listed in Table S1. Gene deletion and tagging were based on a standard PCR-based method (Janke et al., 2004). pRS303-derived plasmids were integrated into the genome at the HIS3 locus after linearization using the restriction enzyme NsiI (Sikorski and Hieter, 1989; Janke et al., 2004). Plasmids used in this study are listed in Table S2. They were constructed by amplifying appropriate DNA fragments by PCR and assembling them using the Gibson Assembly method (New England Biolabs). pRS315-NOP1pro-GFP1–10-mCherry-PUS1 and pRS315-NOP1pro-GFP1–10-mCherry-SCS2TM were generated using PCR products amplified from Addgene plasmids #86413 and #86419 and Addgene plasmids #86416 and #86419, respectively.
Yeast cell growth conditions
Yeast cells were grown at 30°C in YPD medium (1% yeast extract, 2% peptone, and 2% glucose) or SD+CA medium (0.17% yeast nitrogen base without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 0.5% casamino acids, and 2% glucose) appropriately supplemented with 0.002% adenine, 0.002% uracil, and 0.002% tryptophan. For nucleophagy induction, cells were grown to mid-log phase and treated with 200 ng/ml rapamycin. For Atg39 overexpression, cells in which ATG39 was put under the control of the CUP1 promoter were grown in SD+CA medium containing 250 µM CuSO4 overnight, except Figs. 2 E, 4 C, and 5 C and Fig. S2, C–H. In Fig. 4 C, 10 µM (for WT Atg39 cells) or 250 µM (for Atg391–296 cells) CuSO4 was added to the medium to express Atg39 variants at comparable levels. In Figs. 2 E and 5 C, 150 µM (for WT Atg39 cells), 250 µM (for Atg391–296 cells), 10 µM (for Atg391–296-ALPS cells), or 200 µM (for Atg391–296-2×ALPS cells) CuSO4 was added to the medium to express Atg39 variants at similar levels. To develop large lipid droplets (Fig. S1 B), cells were grown in SO+CA medium (0.17% yeast nitrogen base without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 0.12% oleate, 0.2% Tween 40, 0.1% glucose, 1% casamino acids, 0.1% yeast extract, 0.002% adenine sulfate, 0.002% tryptophan, and 0.002% uracil) for 9 h.
Cell lysate extraction and immunoblotting
Frozen yeast cells were resuspended in 200 mM NaOH and left on ice for 5 min. After centrifugation at 3,000 g and removal of the supernatants, the cell pellets were resuspended in urea SDS sample buffer (100 mM MOPS-KOH, pH 6.8, 4% SDS, 100 mM DTT, and 8 M urea) and incubated at 65°C for 10 min. Sample preparation for GFP cleavage assay of Heh1-GFP, Hta2-GFP, Nop1-GFP, Hmg1-GFP, and Sec63-GFP was performed as follows. Frozen yeast cells were incubated with 200 mM NaOH containing 100 mM DTT and 1 mM phenylmethylsulfonyl fluoride (PMSF) and then incubated with excess amounts of 20% TCA. After centrifugation, pellets were washed with acetone, dried, and incubated with urea SDS sample buffer. Sample preparation for GFP cleavage assay of Nop56-GFP was performed as described previously (Mochida et al., 2015). Briefly, cells were treated with 20% TCA, washed with acetone, resuspended in urea SDS sample buffer, and disrupted using zirconia beads and FastPrep-24 (MP Biomedicals). Proteins were resolved by SDS-PAGE, transferred to PVDF membranes, and incubated with primary antibodies against GFP (632381; Clontech), HA (11867431001; Roche), mRFP (a gift from Dr. Toshiya Endo, Kyoto Sangyo University, Kamigamo-motoyama, Kyoto, Japan), and Kar2 (a gift from Dr. Toshiya Endo). After incubation with HRP-conjugated secondary antibodies against rabbit IgG (111-035-144; Jackson Immuno Research Laboratories), mouse IgG (315-035-003; Jackson Immuno Research Laboratories), and rat IgG (112-035-003; Jackson Immuno Research Laboratories), the blots were visualized using ImageQuant LAS 4000 (GE Healthcare). All immunoblot data were quantified in Fiji software (Schindelin et al., 2012) and analyzed in Excel (Microsoft). Data distribution was assumed to be normal, but this was not formally tested.
Fluorescence microscopy
For fluorescence microscopy, yeast cells were grown in SD+CA medium containing appropriate supplements unless otherwise indicated. Cells were analyzed using two different fluorescence microscopy systems, as described previously (Mochida et al., 2020). The images in Figs. 1 D, S1 A, S5 B, and S6 were acquired using an inverted microscope (IX81; Olympus) equipped with an electron-multiplying charge-coupled device camera (ImagEM C9100-13; Hamamatsu Photonics), a 150× objective lens (UAPON 150XOTIRF, NA/1.45; Olympus), a Z drift compensator (IX3-ZDC2; Olympus), appropriate lasers and filters, and MetaMorph software (Molecular Devices). For time-lapse imaging, cells were grown in the glass-bottom dish and kept at 30°C using a stage-top incubator (TOKAI HIT). The images in Fig. S5 B were deconvoluted by AutoQuant X3 software (Media Cybernetics). All other fluorescence microscopy images were acquired using a Delta Vision Elite microscope system (GE Healthcare) equipped with a scientific complementary metal-oxide-semiconductor camera (pco.edge 5.5; PCO AG), a 60× objective lens (PLAPON, NA/1.42; Olympus), a 100× objective lens (UPlanSApo, NA/1.40; Olympus), and SoftWoRx software. Images acquired by a Delta Vision were deconvoluted using SoftWoRx software. All acquired images were analyzed using Fiji.
Quantification of fluorescence microscopy images
Atg39-mCherry puncta and mNG-Atg8 puncta were detected using the Find Maxima function of Fiji as described previously (Mochida et al., 2020). When the maxima of Atg39-mCherry puncta existed within 272 nm of those of GFP- or mNG-Atg8 puncta, they were classified as GFP/mNG-Atg8–positive. The number of NDVs containing nuclear components (Fig. 6 F) was counted similarly, but Atg39-mCherry puncta existing inside or in the vicinity of the nucleus were excluded. The number of Atg39-mCherry-positive tubules extending from the NE was manually counted. The length of the NE tubules was measured using the segmented line selection tool of Fiji. In branched tubules, the length of the longest tubule was measured. Statistical tests for microscopy data were performed in Excel, R Studio, and GraphPad Prism. Parametric tests were used when data distribution was assumed to be normal, but this was not formally tested.
Proteinase K protection assay
Yeast cells grown to mid-log phase were washed with 100 mM Tris-HCl, pH 8.0, containing 10 mM DTT and then converted to spheroplasts by incubation in 0.5× YPD containing 1 M sorbitol and 200 µg/ml zymolyase 100T (Nacalai Tesque) at 30°C for 30 min. Cells were washed with 20 mM HEPES-KOH, pH 7.2, containing 1.2 M sorbitol and incubated in 0.5× YPD containing 1 M sorbitol and 200 ng/ml rapamycin for 3 h. After centrifugation, pelleted cells were resuspended in HSE buffer (20 mM HEPES-KOH, pH 7.2, 1 M sorbitol, and 1 mM EDTA) containing 0.5× Complete protease inhibitor cocktail (Roche) and passed through a polycarbonate membrane filter with a 3-µm pore size (Merck Millipore). The supernatants (lysates) were obtained by removing cell debris by centrifugation and then were treated with 1% Triton X-100 and 100 µg/ml proteinase K on ice for 30 min. Proteolysis was stopped by the addition of 10 mM PMSF, and after trichloroacetic acid precipitation, the proteins were solubilized in urea SDS sample buffer and analyzed by immunoblotting.
Protein purification
BJ3505 cells overexpressing Atg39297–324-GFP-GST, Atg39297–324 9A-GFP-GST, or GFP-GST were grown to late-log phase and harvested. Frozen cells were resuspended in buffer A (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1 mM EDTA, and 10% glycerol) containing 2× protease inhibitor cocktail, 2 mM PMSF, and 1 mM DTT, disrupted using a Multi-beads Shocker (Yasui Kikai) and 0.5-mm YZB zirconia beads, and solubilized with 1% Triton X-100. The lysates were cleared by centrifugation at 100,000 g at 4°C for 30 min and then mixed with Glutathione Sepharose 4B (GE Healthcare). The Sepharose beads were washed with buffer B (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM EDTA, and 10% glycerol) containing 0.01% Triton X-100, and then the bound proteins were eluted with buffer B containing 0.01% Triton X-100 and 10 mM reduced glutathione.
Liposome flotation assay
Liposomes were prepared as follows. Lipids in chloroform were mixed (55:25:20:0.005 mol% POPC:DOPE:bovine liver PI:Rhodamine-DOPE) and dried in a glass tube. The lipid film was hydrated with buffer C (20 mM HEPES-KOH, pH 7.2, and 150 mM NaCl) at a lipid concentration of 1 mM before the suspension was repeatedly frozen and thawed. To prepare larger liposomes, the liposome suspension was extruded through a polycarbonate membrane filter with a 200-nm pore size (Merck Millipore). Smaller liposomes were prepared by sonicating the liposome suspension. The size of the liposomes was measured using a Zetasizer Nano S (Malvern Instruments).
40 µl of the prepared liposomes was mixed with 5 µl of 250 nM protein solution and 80 µl of buffer C and incubated for 1 h at 30°C. 125 µl of 100% OptiPrep (Abbott Diagnostics Technologies) was added to the mixture, which was then overlaid with 450 µl of buffer C containing 40% OptiPrep and 200 µl of OptiPrep-free buffer C. After centrifugation at 200,000 g at 4°C for 105 min, the top, middle, and bottom fractions (300 µl each) were collected, and GFP fluorescence in each fraction was measured using a Varioskan Flash (Thermo Scientific).
EM
EM was performed by Tokai-EMA Inc. Yeast cells were sandwiched with copper disks, rapidly frozen at −175°C, and freeze-substituted with ethanol containing 2% glutaraldehyde and 0.5% tannic acid. After dehydration, the samples were embedded in Quetol-651 resin and ultra-thin sectioned for observation under a transmission electron microscope (JEM-1400Plus; JEOL). For immunoEM, the samples were prepared in a similar manner, except that cells were freeze-substituted with ethanol containing 1% tannic acid and 2% water and embedded in LR white resin. The sections were incubated with an antibody against HA (11867431001; Roche) and then with a secondary antibody conjugated to 10-nm gold particles.
Online supplemental material
Fig. S1 shows the results related to INM binding of the Atg39 APHs. Fig. S2 shows the results of the analysis of Atg39 mutants deleted for the APHs. Figs. S3, S4, and S5 show the results related to Atg39-mediated NE protrusion. Fig. S6 shows the absence of DNA in Atg39-positive autophagosomes. Table S1 and Table S2 list the yeast strains and plasmids used in this study, respectively.
Acknowledgments
We thank the members of our laboratory for materials, discussions, and technical and secretarial support; and the Biomaterials Analysis Division of the Open Facility Center at the Tokyo Institute of Technology for DNA sequencing.
This work was supported in part by KAKENHI Grants-in-Aid for Scientific Research JP17H01430 and JP19H05708 (to H. Nakatogawa) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Japan Agency for Medical Research and Development grant JP21gm1410004 (to H. Nakatogawa); and a STAR Grant funded by the Tokyo Tech Fund (to H. Nakatogawa).
The authors declare no competing financial interests.
Author contributions: K. Mochida, T. Kotani, and H. Nakatogawa designed the project. K. Mochida, T. Otani, and Y. Katsumata performed the experiments with the help of H. Kirisako and C. Kakuta. K. Mochida and H. Nakatogawa wrote the manuscript. All authors analyzed and discussed the results and commented on the manuscript.

