


Asymmetric cell division is a key tumor suppressor mechanism that prevents the uncontrolled expansion of the stem cell (SC) compartment by generating daughter cells with alternative fates: one retains SC identity and enters quiescence and the other becomes a rapidly proliferating and differentiating progenitor. A critical player in this process is Numb, which partitions asymmetrically at SC mitosis and inflicts different proliferative and differentiative fates in the two daughters. Here, we show that asymmetric Numb partitioning per se is insufficient for the proper control of mammary SC dynamics, with differential phosphorylation and functional inactivation of Numb in the two progeny also required. The asymmetric phosphorylation/inactivation of Numb in the progenitor is mediated by the atypical PKCζ isoform. This mechanism is subverted in breast cancer via aberrant activation of PKCs that phosphorylate Numb in both progenies, leading to symmetric division and expansion of the cancer SC compartment, associated with aggressive disease. Thus, Numb phosphorylation represents a target for breast cancer therapy.
Introduction
Asymmetric self-renewing division of stem cells (SCs) represents a major tumor suppressor barrier (reviewed in Knoblich [2010]; Santoro et al. [2016]). In many SC compartments, SCs generate two progenies, one of which retains the SC identity and withdraws into quiescence, while the other assumes a progenitor fate characterized by mitotic expansion and subsequent terminal differentiation (Knoblich, 2010; Santoro et al., 2016). This ensures the production of a large number of differentiated cells while limiting the size of the SC pool. The subversion of this mechanism is relevant to cancer, as skewing the replicative mode from an asymmetric (1 SC −> 1 SC + 1 progenitor) to a symmetric one (1 SC −> 2 SCs) leads to tumorigenesis (Caussinus and Gonzalez, 2005; Cicalese et al., 2009; Pece et al., 2010). Asymmetric division is determined by the unequal positioning of the two progenies relative to external cues (the “niche” concept) and/or by the asymmetric partitioning of cell fate determinants during SC mitosis (Knoblich, 2010; Santoro et al., 2016).
Numb is the best characterized cell-autonomous fate determinant. It was discovered in Drosophila, where its asymmetric partitioning—in various cellular settings—determines the destiny of the daughter cells ([Rhyu et al., 1994; Spana et al., 1995; Uemura et al., 1989], [Caussinus and Gonzalez, 2005; Knoblich, 2010; Pece et al., 2011]). Studies in developmental systems have established that Numb phosphorylation is required for its asymmetric partitioning at mitosis (Betschinger et al., 2003; Lee et al., 2006; Smith et al., 2007; Wang et al., 2006; Wirtz-Peitz et al., 2008). While events are complex and, in part, context-specific, the generally accepted view involves a series of steps in which: (i) Numb is recruited to the plasma membrane (PM) via interactions with membrane phospholipids, (ii) at mitosis, the PM-associated Numb undergoes “asymmetric” phosphorylation at one of the cell poles, executed by the asymmetrically localized polarity complex Par3/Par6/aPKC (aPKC: atypical PKC), (iii) phosphorylation determines the asymmetric release of Numb from the PM at one of the poles into the cytoplasm, while the rest remains attached to the PM at the opposite pole, and (iv) finally, when abscission occurs, one of the daughter cells inherits the majority of Numb (the PM-bound fraction plus part of the cytoplasmic fraction). Thus, at least in developmental invertebrate systems, Numb phosphorylation is thought to act predominantly as a “partitioning device” that controls its unequal inheritance. Studies in flies have also elucidated the downstream effect of Numb in cell fate determination, linking it to its ability to counteract the action of the membrane signaling receptor Notch (Babaoglan et al., 2009; Cotton et al., 2013; Frise et al., 1996; Guo et al., 1996; Spana and Doe, 1996).
Mammalian Numb shares, with the developmental model systems, the modes of regulation of asymmetric partitioning (Dho et al., 2006; Nishimura and Kaibuchi, 2007; Shen et al., 2002; Smith et al., 2007; Zhong et al., 1996) and downstream inhibition of Notch (Bultje et al., 2009; McGill et al., 2009; McGill and McGlade, 2003; Pece et al., 2004; Westhoff et al., 2009; Zhong et al., 1996; Zhong et al., 1997; also reviewed in Kandachar and Roegiers [2012]; Knoblich [2010]; Pece et al. [2011]). There are, however, important differences. The most relevant one is the presence, in mammalian Numb, of two alternatively spliced exons (exons 3 and 9) that are not present in invertebrates. Of these, exon 3 is a chordate invention (Confalonieri et al., 2019) that confers novel properties to Numb, including that of binding to Mdm2, thereby inhibiting its E3 ligase activity toward p53 and stabilizing p53 levels and tumor suppressor function (Colaluca et al., 2018; Colaluca et al., 2008; Sheng et al., 2013; Wang et al., 2015). In turn, the levels of p53 are critical to the control of proliferative fates in the SC compartment in the mammary gland (Cicalese et al., 2009; Tosoni et al., 2015), as well as other organs (Choi et al., 2020; Feldman et al., 2013; Siddique et al., 2015).
In normal murine mammary stem-like cells analyzed ex vivo (henceforth operationally defined as MaSLC for Mammary Stem-Like cells), Numb is asymmetrically partitioned in the daughter cell that retains the SC fate (Tosoni et al., 2015). It is to be pointed out that MaSLCs analyzed ex vivo do not fully represent the physiological situation. Indeed, lineage tracing studies have established that adult stem/progenitor cells in the murine mammary gland in vivo are unipotent (either basal or luminal). MaSLCs analyzed ex vivo are multipotent and should therefore be considered as de-differentiated SCs, useful as a model for investigations of cellular dynamics and biochemical events determining SC-like behavior (Davis et al., 2016; Fu et al., 2020; Koren et al., 2015; Van Keymeulen et al., 2011; Wuidart et al., 2016). In MaSLCs, high Numb levels impose high levels of p53, thereby determining quiescence, a hallmark of the SC state (Cicalese et al., 2009; Tosoni et al., 2015). Conversely, in the sibling daughter, low Numb determines low p53 levels, allowing proliferation (Tosoni et al., 2015), a characteristic of progenitor cells (Gomez-Lopez et al., 2014). Accordingly, the genetic removal of Numb causes the two daughters to acquire similar proliferative fates, a feature of cancer SC (CSC)-like cells (Tosoni et al., 2015). This was shown to be causal in human breast cancer (BC), in which loss of Numb expression or alterations in its splicing pattern, leading to loss of exon 3-containing isoforms, characterize tumors with an expanded CSC compartment and aggressive behavior (Colaluca et al., 2018). In these cancers, re-expression of Numb or restoration of p53 levels re-established the normal-like proliferative kinetics of the SC compartment and inhibited tumorigenicity and metastasis in xenotransplantation experiments performed with patient-derived xenografts (PDXs; Tosoni et al., 2017).
During the course of our studies, we made a puzzling observation (illustrated below) where many BCs display high levels of Numb phosphorylation. This observation is apparently at odds with the interpretation of Numb phosphorylation as predominantly a “partitioning device.” According to this concept, excessive Numb phosphorylation should cause a symmetric distribution of the protein, thereby imposing quiescence (via p53) on both daughters, a prediction not immediately reconcilable with the expansion of the CSC compartment characteristic of these cancers. Rather, the result suggested that phosphorylation renders Numb inactive, mimicking the situation of a Numb-defective status in cancer. The present studies were therefore undertaken to re-evaluate the role of Numb phosphorylation in the determination of SC fate in normal-like and cancer mammary SC compartments.
Results
Evolutionary conservation of the mechanisms of Numb phosphorylation and asymmetric partitioning
We dissected the mechanisms involved in Numb phosphorylation and asymmetric partitioning in mouse MaSLCs (isolated with the PKH-methodology [Pece et al., 2010; Tosoni et al., 2012]) using the blueprint of the mechanisms uncovered in developmental systems.
We monitored the partitioning of Numb, Par3, and PKCζ (one of the two atypical PKCs in mammals [Smith et al., 2007; Zhou et al., 2011]) at the first mitotic division of mouse MaSLCs using a Flag-tagged Numb and endogenous Par3 and PKCζ (Fig. 1 A). The three proteins partitioned preferentially in an asymmetric fashion, with Numb preferentially localized opposite to PKCζ and Par3 in the daughter cells (Fig. 1 A and Table S1).
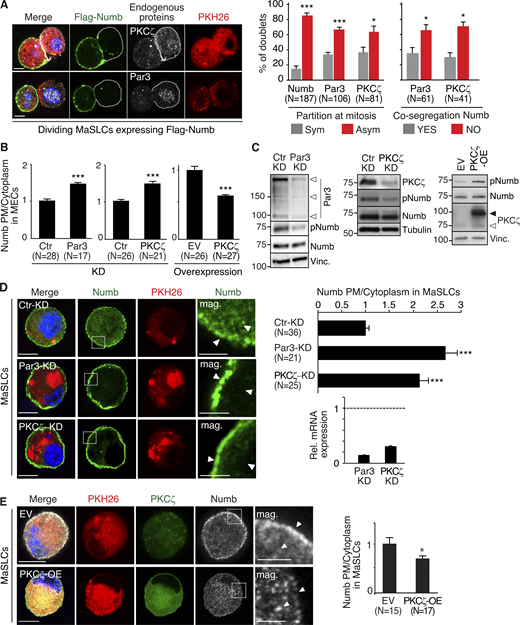
Par3- and PKCζ-KD affect Numb phosphorylation and localization. (A) Left: MaSLCs (PKHpos cells) expressing Flag-Numb were plated in suspension to allow for their division, fixed, and stained with anti-Flag (green, Flag-Numb), anti-PKCζ or anti-Par3 Ab (white), and DAPI (blue). Bar, 10 µm. Middle: Quantitative analysis of symmetric vs. asymmetric partitioning of the proteins. Right: Quantitative analysis of segregation of Par3 or PKCζ with Numb. Only doublets in which the proteins were segregated asymmetrically were counted. Data are expressed as % of doublets showing Numb/Par3 or Numb/PKCζ co-segregation (YES) or not (NO). (B) Numb (endogenous) subcellular localization in MECs stably silenced (KD) for Par3 (left) or PKCζ (middle) or overexpressing PKCζ-Venus (right; EV, empty vector). Images are in Fig. S1, A and B. Data are expressed as the ratio between plasma membrane (PM) Numb/cytoplasmic Numb, relative to controls (Ctr or EV). (C) Immunoblots (IB) of the indicated cells. pNumb, phosphorylated Numb (anti-NumbSer276 Ab in this and all subsequent figures). Left: Arrows point to three different Par3 isoforms (Dziengelewski et al., 2020; Ishiuchi and Takeichi, 2011). Right: Empty and filled arrows point to endogenous and overexpressed PKCζ-Venus, respectively. (D) Left: MaSLCs (PKHpos cells) stably silenced for Par3 (Par3-KD) and PKCζ (PKCζ-KD) were stained with anti-Numb (endogenous Numb) and DAPI. Bar, 10 μm. The boxed areas are magnified on the right (mag., arrows point to PM and cytoplasmic areas; bar, 3 μm). Right top: Quantitation of the experiment as in B. Right bottom: qPCR of Par3 and PKCζ silencing in MaSLCs, relative to control cells (=1, dashed line). (E) Left: MaSLCs (PKHpos cells) stably overexpressing PKCζ (PKCζ-OE) were stained as shown (Numb, endogenous Numb). Bar, 10 μm. The boxed areas are magnified (mag., arrows point to PM and cytoplasmic areas; bar, 3 μm). Right: Quantitation of the experiment as in B. When shown: N, number of cells analyzed. Data are reported ± SE. Statistical analysis was with the binomial test (A), or with the Student’s t test two-tailed (B, D, and E). P was calculated vs. Ctr or EV, as appropriate: *, <0.05; **, <0.01; *** <0.001, in this and all other figures. Source data are available for this figure: SourceData F1.
Par3- and PKCζ-KD affect Numb phosphorylation and localization. (A) Left: MaSLCs (PKHpos cells) expressing Flag-Numb were plated in suspension to allow for their division, fixed, and stained with anti-Flag (green, Flag-Numb), anti-PKCζ or anti-Par3 Ab (white), and DAPI (blue). Bar, 10 µm. Middle: Quantitative analysis of symmetric vs. asymmetric partitioning of the proteins. Right: Quantitative analysis of segregation of Par3 or PKCζ with Numb. Only doublets in which the proteins were segregated asymmetrically were counted. Data are expressed as % of doublets showing Numb/Par3 or Numb/PKCζ co-segregation (YES) or not (NO). (B) Numb (endogenous) subcellular localization in MECs stably silenced (KD) for Par3 (left) or PKCζ (middle) or overexpressing PKCζ-Venus (right; EV, empty vector). Images are in Fig. S1, A and B. Data are expressed as the ratio between plasma membrane (PM) Numb/cytoplasmic Numb, relative to controls (Ctr or EV). (C) Immunoblots (IB) of the indicated cells. pNumb, phosphorylated Numb (anti-NumbSer276 Ab in this and all subsequent figures). Left: Arrows point to three different Par3 isoforms (Dziengelewski et al., 2020; Ishiuchi and Takeichi, 2011). Right: Empty and filled arrows point to endogenous and overexpressed PKCζ-Venus, respectively. (D) Left: MaSLCs (PKHpos cells) stably silenced for Par3 (Par3-KD) and PKCζ (PKCζ-KD) were stained with anti-Numb (endogenous Numb) and DAPI. Bar, 10 μm. The boxed areas are magnified on the right (mag., arrows point to PM and cytoplasmic areas; bar, 3 μm). Right top: Quantitation of the experiment as in B. Right bottom: qPCR of Par3 and PKCζ silencing in MaSLCs, relative to control cells (=1, dashed line). (E) Left: MaSLCs (PKHpos cells) stably overexpressing PKCζ (PKCζ-OE) were stained as shown (Numb, endogenous Numb). Bar, 10 μm. The boxed areas are magnified (mag., arrows point to PM and cytoplasmic areas; bar, 3 μm). Right: Quantitation of the experiment as in B. When shown: N, number of cells analyzed. Data are reported ± SE. Statistical analysis was with the binomial test (A), or with the Student’s t test two-tailed (B, D, and E). P was calculated vs. Ctr or EV, as appropriate: *, <0.05; **, <0.01; *** <0.001, in this and all other figures. Source data are available for this figure: SourceData F1.
The relevance of the Par3/PKCζ axis to the control of Numb subcellular localization/phosphorylation was determined in primary mouse mammary epithelial cells (MECs) and mouse MaSLCs. In MECs, endogenous Numb localized at the PM and in cytoplasmic vesicular compartments (Fig. S1 A). The knockdown of Par3 (Par3-KD) or PKCζ (PKCζ-KD) increased the association of Numb with the PM and reduced Numb phosphorylation (Fig. 1, B and C; and Fig. S1 A). Conversely, overexpression (OE) of PKCζ induced the detachment of Numb from the PM and increased its phosphorylation (Fig. 1, B and C; and Fig. S1 B).
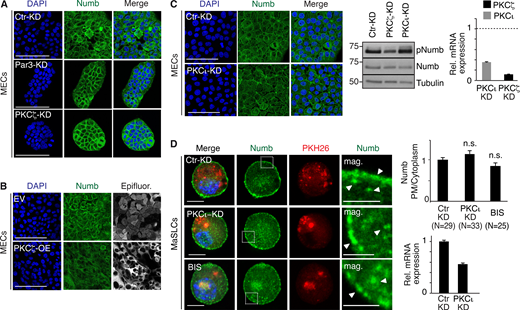
Additional data to Fig. 1. (A and B) Numb confocal immunofluorescence in mouse MECs stably silenced for Par3 (Par3-KD) and PKCζ (PKCζ -KD) or mock silenced (Ctr-KD; A) or overexpressing Venus (EV) and PKCζ-Venus (PKCζ -OE; B). Numb (green, pseudocolored from Cy3 staining), epifluorescence (gray, pseudocolored from Venus, in EV, and PKCζ-Venus, in PKCζ -OE), DAPI (blue). Bars, 100 µm. (C) Left: Numb confocal immunofluorescence in mouse MECs stably silenced for PKCι (PKCι-KD). Numb (green), DAPI (blue). Bar, 100 µm. Middle: Immunoblots of Numb and pNumb in the cell lysates indicated on top. Tubulin, loading control. Right: qPCR analysis showing the efficacy of PKCι and PKCζ silencing in MaSLCs. Data are expressed relative to control cells (dashed line). (D) Numb confocal immunofluorescence. Left: MaSLCs (PKH26POS cells) from WT mammospheres (MS) stably silenced for PKCι (PKCι-KD), were stained with anti-Numb and DAPI. Control cells were also treated with 3 μm Bisindolylmaleimide I (BIS) o/n and subjected to the same IF. The boxed areas are magnified on the right. PHK26 (red), Numb (green), DAPI (blue). Bar, 10 μm. Mag, magnification (bar, 3 μm). White arrows in magnifications point to PM or cytoplasmic areas. Right (top), quantitation of the experiment. Data are expressed as ratio between plasma membrane (PM) and cytoplasmic Numb, relative to control (N, number of cells analyzed). Right (bottom), qPCR analysis showing the efficacy of PKCι silencing in MaSLCs. Data are expressed relative to control cells ± SE; statistical analysis was with the Student’s t test two-tailed. In this and all subsequent supplementary figures: P is: *, <0.05; **, <0.01; ***, <0.001. Source data are available for this figure: SourceData FS1.
Additional data to Fig. 1. (A and B) Numb confocal immunofluorescence in mouse MECs stably silenced for Par3 (Par3-KD) and PKCζ (PKCζ -KD) or mock silenced (Ctr-KD; A) or overexpressing Venus (EV) and PKCζ-Venus (PKCζ -OE; B). Numb (green, pseudocolored from Cy3 staining), epifluorescence (gray, pseudocolored from Venus, in EV, and PKCζ-Venus, in PKCζ -OE), DAPI (blue). Bars, 100 µm. (C) Left: Numb confocal immunofluorescence in mouse MECs stably silenced for PKCι (PKCι-KD). Numb (green), DAPI (blue). Bar, 100 µm. Middle: Immunoblots of Numb and pNumb in the cell lysates indicated on top. Tubulin, loading control. Right: qPCR analysis showing the efficacy of PKCι and PKCζ silencing in MaSLCs. Data are expressed relative to control cells (dashed line). (D) Numb confocal immunofluorescence. Left: MaSLCs (PKH26POS cells) from WT mammospheres (MS) stably silenced for PKCι (PKCι-KD), were stained with anti-Numb and DAPI. Control cells were also treated with 3 μm Bisindolylmaleimide I (BIS) o/n and subjected to the same IF. The boxed areas are magnified on the right. PHK26 (red), Numb (green), DAPI (blue). Bar, 10 μm. Mag, magnification (bar, 3 μm). White arrows in magnifications point to PM or cytoplasmic areas. Right (top), quantitation of the experiment. Data are expressed as ratio between plasma membrane (PM) and cytoplasmic Numb, relative to control (N, number of cells analyzed). Right (bottom), qPCR analysis showing the efficacy of PKCι silencing in MaSLCs. Data are expressed relative to control cells ± SE; statistical analysis was with the Student’s t test two-tailed. In this and all subsequent supplementary figures: P is: *, <0.05; **, <0.01; ***, <0.001. Source data are available for this figure: SourceData FS1.
In MaSLCs, Par3-KD or PKCζ-KD (Fig. 1 D) and PKCζ-OE (Fig. 1 E) phenocopied the effects on the subcellular distribution of endogenous Numb observed in MECs. Of note, the KD of PKCι, which is the only other member of the atypical PKC subfamily, had little if any effect on Numb localization and phosphorylation, both in MECs and in MaSLCs (Fig. S1, C and D). Similarly, a chemical inhibitor of PKCs, bisindolylmaleimide I (BIS), which inhibits preferentially typical (conventional and novel) PKCs rather than atypical PKCs (Martiny-Baron et al., 1993; Toullec et al., 1991; Uberall et al., 1997), did not alter the subcellular distribution of Numb in MaSLCs (Fig. S1 D, see also Fig. S5 for specificity of BIS). These results argue that PKCζ is the major PKC isoform controlling Numb phosphorylation and distribution in MaSLCs.
To establish a causal link between Numb phosphorylation and its subcellular distribution, we engineered Numb phospho-mutants in which three PKC phosphorylation sites—Ser7, Ser276, and Ser295, conserved in evolution (Dho et al., 2006; Nishimura and Kaibuchi, 2007; Smith et al., 2007)—were mutagenized to aspartic acid (Numb-3D, phospho-mimetic) or alanine (Numb-3A, phospho-deficient; Fig. 2 A). This represents a well-established approach to dissect the contribution of PKC-mediated Numb phosphorylation to the physiology of several cellular models (Dho et al., 2006; Nishimura and Kaibuchi, 2007; Sato et al., 2011; Smith et al., 2007). A Numb mutant deprived of the phosphotyrosine-binding domain (ΔPTB) was also used as a lack-of-function control mutant (Fig. 2 A). When transduced into MaSLCs, the Numb-3A mutant showed a predominant PM localization, similar to that of wild-type full-length Numb (Numb-FL). Conversely, the Numb-3D and ΔPTB mutants were mainly localized in the cytoplasm (Fig. 2 B and Fig. S2 A).

Symmetric partitioning of Numb phosphomutants. (A) Numb mutants. The relevant Ser residues (S7, S276, and S295) and the mutations (red) are indicated. (B) MaSLCs (PKHpos cells, green) were transduced (all constructs fused to Flag-tag), stained with an anti-Numb Ab (red), and analyzed. Blue, DAPI. Bar, 10 μm. (C) Mammospheres (MS) transduced with the indicated constructs (DsRed fusion proteins) were dissociated and single cells were analyzed by time-lapse video microscopy. Epifluorescence (red). Bar, 10 μm. (D) Quantitation of the experiment in C, showing the frequency of symmetric (Sym) vs. asymmetric (Asym) partitioning. Significance was calculated vs. Numb-FL. (E) MS were transduced with Numb-DsRed (all panels) and then silenced as indicated (KD), or transduced with PKCζ (PKCζ-OE), or treated with BIS (3 μM). Cells were then analyzed as in C and D. P was calculated vs. matching control condition (Ctr-KD, EV or Ctr). When shown: N, number of doublets analyzed. Data are reported ± SD. Statistical analysis was with the nonparametric Fisher’s exact test.
Symmetric partitioning of Numb phosphomutants. (A) Numb mutants. The relevant Ser residues (S7, S276, and S295) and the mutations (red) are indicated. (B) MaSLCs (PKHpos cells, green) were transduced (all constructs fused to Flag-tag), stained with an anti-Numb Ab (red), and analyzed. Blue, DAPI. Bar, 10 μm. (C) Mammospheres (MS) transduced with the indicated constructs (DsRed fusion proteins) were dissociated and single cells were analyzed by time-lapse video microscopy. Epifluorescence (red). Bar, 10 μm. (D) Quantitation of the experiment in C, showing the frequency of symmetric (Sym) vs. asymmetric (Asym) partitioning. Significance was calculated vs. Numb-FL. (E) MS were transduced with Numb-DsRed (all panels) and then silenced as indicated (KD), or transduced with PKCζ (PKCζ-OE), or treated with BIS (3 μM). Cells were then analyzed as in C and D. P was calculated vs. matching control condition (Ctr-KD, EV or Ctr). When shown: N, number of doublets analyzed. Data are reported ± SD. Statistical analysis was with the nonparametric Fisher’s exact test.

Additional data to Fig. 2. (A) Data in Fig. 2 A were obtained by anti-Numb staining on cells transduced with various Numb mutants, in the presence of endogenous Numb. This control figure (taken from the same experiment presented in Fig. 2 B) shows that, under the conditions of image acquisition employed, only the transduced Numb is visible, while in the non-transduced cells (dashed circles) there is little endogenous Numb detected. Bar, 10 μm. (B–E) MS stably (transduced with Numb-FL-DsRed) silenced for Par3 (Par3-KD) or PKCζ (PKCζ-KD; B), or overexpressing PKCζ (PKCζ-OE; C), or silenced for PKCι (PKCι-KD; D), were dissociated and partitioning of Numb at first MaSLC mitosis was analyzed by time-lapse video microscopy. Control cells were also treated with 3 μm Bisindolylmaleimide I (BIS) (at cell plating) and subjected to the same analysis (E). Representative images at the 1st MaSLC mitotic division (1st mit.div.) are shown. Epifluor., Epifluorescence, Numb (red), PKCζ (green); b.f., bright-field. Bar, 10 μm.
Additional data to Fig. 2. (A) Data in Fig. 2 A were obtained by anti-Numb staining on cells transduced with various Numb mutants, in the presence of endogenous Numb. This control figure (taken from the same experiment presented in Fig. 2 B) shows that, under the conditions of image acquisition employed, only the transduced Numb is visible, while in the non-transduced cells (dashed circles) there is little endogenous Numb detected. Bar, 10 μm. (B–E) MS stably (transduced with Numb-FL-DsRed) silenced for Par3 (Par3-KD) or PKCζ (PKCζ-KD; B), or overexpressing PKCζ (PKCζ-OE; C), or silenced for PKCι (PKCι-KD; D), were dissociated and partitioning of Numb at first MaSLC mitosis was analyzed by time-lapse video microscopy. Control cells were also treated with 3 μm Bisindolylmaleimide I (BIS) (at cell plating) and subjected to the same analysis (E). Representative images at the 1st MaSLC mitotic division (1st mit.div.) are shown. Epifluor., Epifluorescence, Numb (red), PKCζ (green); b.f., bright-field. Bar, 10 μm.
We next analyzed the partitioning of FL and mutant Numbs at the first mitotic division of MaSLCs. Numb-FL partitioned in a predominantly asymmetric fashion, while all Numb mutants were mostly equally distributed between the two daughter cells (Fig. 2, C and D). Par3-KD or PKCζ-KD and PKCζ-OE, all caused a shift toward the symmetric partitioning of Numb (Fig. 2 E). Treatment with BIS or PKCι-KD did not alter the pattern of Numb partitioning, confirming the specific involvement of PKCζ in this process (Fig. 2 E and Fig. S2, B–E).
These results show that the molecular machinery of Numb phosphorylation and asymmetric partitioning is conserved in evolution.
Numb asymmetric partitioning is not sufficient for its action in MaSLCs
Asymmetric partitioning of Numb confers to the Numb-retaining daughter a quiescent phenotype, associated with the SC identity, while the other daughter assumes a proliferative phenotype associated with a progenitor identity (Tosoni et al., 2015; Fig. 3 A). At the biological level, this can be studied using a spheroid-based assay (mammosphere, MS), which scores for the ability of SC-like cells (MS-forming cells) to form colonies in anchorage-independent conditions (Dontu et al., 2003). In unperturbed conditions (model in Fig. 3 B, from Tosoni et al. [2015]), the MS-forming ability of MECs is gradually lost over serial passages (self-limiting behavior; Cicalese et al., 2009; Pece et al., 2010). This is compatible with the model shown in Fig. 3 A in which the number of WT stem-like cells cannot increase over time. This self-limiting behavior (Fig. 3 B) can be due to: (i) functional exhaustion of the self-renewing ability of MS-forming cells and/or (ii) their withdrawal into a prolonged quiescence state that exceeds the duration of the assay. Conversely, under Numb-KO conditions, the lack of quiescence causes the continuous expansion of MS-forming cells (Fig. 3, A and B), which is also associated with a reduced replication time.
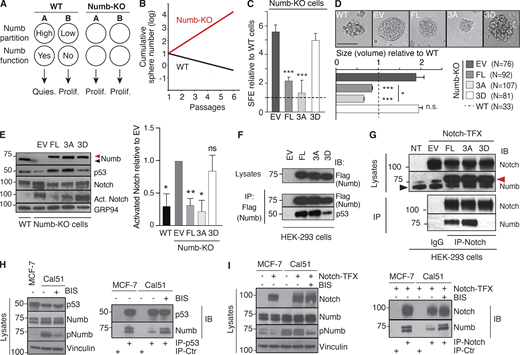
Characterization of Numb phosphomutants. (A) Scheme showing the behavior of a SC (A) and daughter progenitor cell (B) in WT vs. Numb-KO (Tosoni et al., 2015). (B) Scheme of the growth of MS from WT and Numb-KO MECs (Tosoni et al., 2015). (C and D) WT and Numb-KO cells, transduced with the indicated constructs (DsRed fusion proteins; EV, empty vector), were assessed for SFE (C, by counting only red cells or MS) and size (D, N = number of epifluorescent MS analyzed). Results are expressed relative to WT cells (see also Table S2). Significance was calculated vs. EV cells. Representative images of the MS are in D, top panel. Bar, 100 µm. (E) WT and Numb-KO MS, transduced with the indicated constructs (Flag-tagged), were analyzed by IB. Arrows, endogenous (black) or overexpressed (red) Numb (also in G). Activated Notch (Act. Notch) was detected with the anti Val1744 Ab (in this and all subsequent figures). Right: Quantitation of three independent experiments. (F) HEK-293 cells, transfected as indicated (all Numb constructs were Flag-tagged and also codify for an sh-RNA sequence against endogenous Numb; EV, empty vector), were IP and IB as shown. (G) HEK-293 cells were stably transduced with Notch-NΔE (Notch-TFX; NT, not transfected) and transfected with the indicated Numb-Flag constructs (as in F). IP and IB were as shown. (H and I) MCF-7 or Cal51 cells were either transduced with Notch-NΔE (Notch-TFX; I) or not (H). Cells were treated with BIS (or mock-treated) and IP and IB as shown. In H, IP-Ctr is anti-Flag; in I, IP-Ctr is goat IgG. Data are reported ± SD (C and E) or ± SE (D). Statistical analysis was with the Student’s t test two-tailed (C and D) or with the one-sample t test (E). Source data are available for this figure: SourceData F3.
Characterization of Numb phosphomutants. (A) Scheme showing the behavior of a SC (A) and daughter progenitor cell (B) in WT vs. Numb-KO (Tosoni et al., 2015). (B) Scheme of the growth of MS from WT and Numb-KO MECs (Tosoni et al., 2015). (C and D) WT and Numb-KO cells, transduced with the indicated constructs (DsRed fusion proteins; EV, empty vector), were assessed for SFE (C, by counting only red cells or MS) and size (D, N = number of epifluorescent MS analyzed). Results are expressed relative to WT cells (see also Table S2). Significance was calculated vs. EV cells. Representative images of the MS are in D, top panel. Bar, 100 µm. (E) WT and Numb-KO MS, transduced with the indicated constructs (Flag-tagged), were analyzed by IB. Arrows, endogenous (black) or overexpressed (red) Numb (also in G). Activated Notch (Act. Notch) was detected with the anti Val1744 Ab (in this and all subsequent figures). Right: Quantitation of three independent experiments. (F) HEK-293 cells, transfected as indicated (all Numb constructs were Flag-tagged and also codify for an sh-RNA sequence against endogenous Numb; EV, empty vector), were IP and IB as shown. (G) HEK-293 cells were stably transduced with Notch-NΔE (Notch-TFX; NT, not transfected) and transfected with the indicated Numb-Flag constructs (as in F). IP and IB were as shown. (H and I) MCF-7 or Cal51 cells were either transduced with Notch-NΔE (Notch-TFX; I) or not (H). Cells were treated with BIS (or mock-treated) and IP and IB as shown. In H, IP-Ctr is anti-Flag; in I, IP-Ctr is goat IgG. Data are reported ± SD (C and E) or ± SE (D). Statistical analysis was with the Student’s t test two-tailed (C and D) or with the one-sample t test (E). Source data are available for this figure: SourceData F3.
The described Numb mutants allowed us to dissect the effects of forced Numb symmetric distribution on the proliferative fate(s) of the MaSLC progeny. We initially tested the effects of the Numb mutants in a Numb-KO background in the MS assay. Consistent with the models shown in Fig. 3, A and B, Numb-KO MECs, transduced with a control vector (EV), displayed a ∼sixfold increased sphere-forming efficiency (SFE) vs. control MECs (Fig. 3 C, see also Table S2). Re-expression of Numb-FL in KO MECs significantly decreased the SFE vs. EV, similar to the expression of Numb-3A (Fig. 3 C). Conversely, the Numb-3D mutant did not display any significant effect (Fig. 3 C). Similar results were obtained by assessing MS size, indicative of the proliferative ability of progenitors (Fig. 3 D). As both Numb-3A and Numb-3D were symmetrically partitioned at the first MaSLC mitotic division (Fig. 2, C and D), these results argue that the presence per se of Numb in the SC and its symmetric/asymmetric partitioning is not sufficient to impart different proliferative kinetics to the progeny. Moreover, it appears that Numb-3D behaves as a null mutant, while Numb-3A is biologically competent. Together, these data suggest that phosphorylation renders Numb functionally inactive, thereby mimicking a Numb-null state.
The two major pathways controlled by Numb rely on its ability to inhibit Notch and stabilize p53. In Numb-KO MECs, we could readily observe decreased levels of p53 and increased levels of active Notch vs. WT cells (Fig. 3 E). The re-expression of Numb-FL or Numb-3A rescued these biochemical phenotypes, while Numb-3D was unable to do so (Fig. 3 E).
The ability of Numb to stabilize p53 is due to its physical interaction with Mdm2 and p53 in the context of a tri-complex, in which the E3 ligase activity of Mdm2 is inhibited (Colaluca et al., 2008). In keeping with the above biological and biochemical results, p53 co-immunoprecipitated with Numb-FL and Numb-3A, while its in vivo interaction with Numb-3D was impaired (Fig. 3 F). Similarly, the Numb-3D displayed significantly reduced co-immunoprecipitation with Notch vs. Numb-FL and Numb-3A (Fig. 3 G). In these experiments, performed in the model cell line, HEK-293, the transfected Numb constructs were engineered in a vector also containing an sh-RNA sequence against endogenous human Numb, thereby allowing the expression of the various Numb proteins (all engineered to be sh-resistant) in the absence/reduction of endogenous Numb.
To further validate the above findings, we used two Numb-proficient BC lines, MCF-7 and Cal51, displaying low and high levels of Numb phosphorylation, respectively (Fig. 3, H and I; and Fig. S3 A). Consistent with the high levels of pNumb, Cal51 cells displayed reduced levels of Numb:p53 (Fig. 3 H) and Numb:Notch (Fig. 3 I) interaction when compared with MCF-7 cells. In addition, the treatment of Cal51 cells with BIS increased the levels of co-immunoprecipitation of Numb with both p53 and Notch (Fig. 3, H and I). Note that for the experiments in Fig. 3, G and I, cells were also transduced with an activated-Notch encoding construct (Notch-TFX) to obtain sufficient levels of Notch for the co-immunoprecipitation experiments.
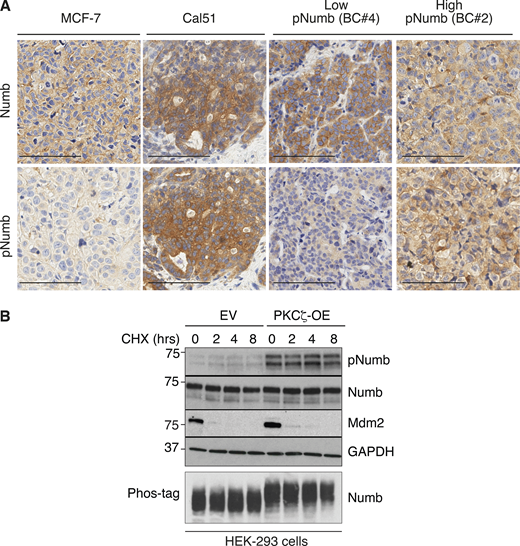
Additional data to Fig. 3. (A) The two breast cancer cell lines, MCF-7 and Cal51 used in Fig. 3, H and I, were grown as xenografts and stained for Numb and pNumb in IHC, in comparison to two PDXs from Numb-competent tumors displaying low pNumb (BC #4) and high pNumb (BC #2; the BCs in object are described in details in Fig. 8) to provide additional proof that they are representative of the situation occurring in real breast cancers. Bar, 100 μm. (B) The phosphorylation of Numb executed by PKCζ does not appreciably affect the half-life of Numb. HEK-293 cells were transduced with Venus (EV) or PKCζ-Venus (PKCζ -OE), as shown on the top, and then treated with cycloheximide (CHX, 50 μg/ml) for the indicated lengths of time. Lysates were immunoblotted as shown (top four panels). Mdm2 was used as control for a short half-lived protein. GAPDH is a loading control. In the lower panel (Phos-tag), the lysates were fractionated on an 8% Phos-tag gel (see Materials and methods) and then IB with anti-Numb. The nearly complete shift on Numb in the PKCζ-OE cells indicates that the phosphorylation of Numb executed by PKCζ is nearly stoichiometric. Source data are available for this figure: SourceData FS3.
Additional data to Fig. 3. (A) The two breast cancer cell lines, MCF-7 and Cal51 used in Fig. 3, H and I, were grown as xenografts and stained for Numb and pNumb in IHC, in comparison to two PDXs from Numb-competent tumors displaying low pNumb (BC #4) and high pNumb (BC #2; the BCs in object are described in details in Fig. 8) to provide additional proof that they are representative of the situation occurring in real breast cancers. Bar, 100 μm. (B) The phosphorylation of Numb executed by PKCζ does not appreciably affect the half-life of Numb. HEK-293 cells were transduced with Venus (EV) or PKCζ-Venus (PKCζ -OE), as shown on the top, and then treated with cycloheximide (CHX, 50 μg/ml) for the indicated lengths of time. Lysates were immunoblotted as shown (top four panels). Mdm2 was used as control for a short half-lived protein. GAPDH is a loading control. In the lower panel (Phos-tag), the lysates were fractionated on an 8% Phos-tag gel (see Materials and methods) and then IB with anti-Numb. The nearly complete shift on Numb in the PKCζ-OE cells indicates that the phosphorylation of Numb executed by PKCζ is nearly stoichiometric. Source data are available for this figure: SourceData FS3.
Finally, we demonstrated that Numb phosphorylation does not appreciably affect the stability of the protein (Fig. S3 B). The sum of the above data, therefore, argues that phosphorylation renders Numb functionally null by preventing its protein interaction ability in vivo.
Phosphorylation-mediated inactivation of Numb is required in the divisional history of MaSLCs
We directly tested the hypothesis that Numb phosphorylation leads to its loss of function. To this end, we employed Par3-KD, PKCζ-KD, and PKCζ-OE. The KD of Par3 or PKCζ caused a marked reduction in SFE (indicative of a reduction in SC number or the induction of prolonged quiescence) and in MS size (indicative of reduced progenitor proliferation; Fig. 4, A and B). These events were accompanied by a decrease in the proliferative marker Ki67 (Fig. 4 C). The opposite phenotypes were scored in PKCζ-OE cells (Fig. 4, D–G).
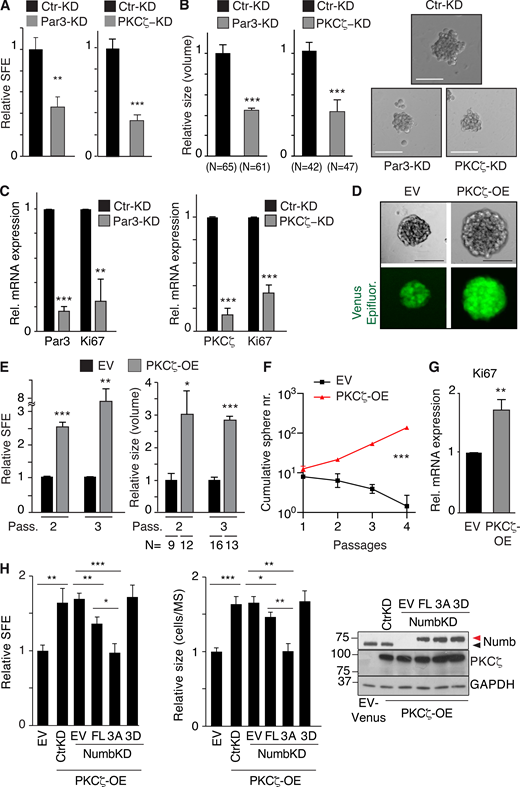
Effects of Par3/PKCζ perturbation on MaSLC. (A–C) MECs silenced for Par3 (Par3-KD) or PKCζ (PKCζ-KD) were assessed for: (A) SFE; (B) MS size (the rightmost panels show representative images; Bar, 100 µm); and (C) Par3, PKCζ KD and Ki67 expression by qPCR analysis. Data are relative to control cells (see also Table S2). (D) Cells from WT MS were transduced with control Venus (EV) or PKCζ-Venus (PKCζ-OE); Epifluorescence (green). Bar, 100 µm. (E–G) Cells, as in D, were assessed for SFE, at the indicated passages (Pass; E left, data are relative to EV) or MS size (E right, data are relative to EV, see also Table S2), or cumulative sphere number (nr) in serial replating experiments (F), or Ki67 expression by qPCR (data are relative to EV; G). (H) Cells from WT MS were stably transduced with Venus- (EV) or PKCζ-Venus (PKCζ-OE). PKCζ-OE cells were then transduced with the indicated Numb-mutants (Flag-tagged) or control vector (EV) and cells were further silenced or not for Numb expression, as shown. Cells were assessed for SFE (left, data are relative to EV, see also Table S2), MS size (middle, data are relative to EV, see also Table S2) or protein expression (right; arrows, endogenous [black] or overexpressed [red] Numb proteins). When shown: N, number of MS analyzed. Data are reported ± SE (A, B, C, E, and G) or ± SD (F and H). Statistical analysis was with the ANOVA test (F), or with the Student’s t test two-tailed (A, B, C, E, G, and H). Significance was calculated vs. Ctr-KD or EV. Source data are available for this figure: SourceData F4.
Effects of Par3/PKCζ perturbation on MaSLC. (A–C) MECs silenced for Par3 (Par3-KD) or PKCζ (PKCζ-KD) were assessed for: (A) SFE; (B) MS size (the rightmost panels show representative images; Bar, 100 µm); and (C) Par3, PKCζ KD and Ki67 expression by qPCR analysis. Data are relative to control cells (see also Table S2). (D) Cells from WT MS were transduced with control Venus (EV) or PKCζ-Venus (PKCζ-OE); Epifluorescence (green). Bar, 100 µm. (E–G) Cells, as in D, were assessed for SFE, at the indicated passages (Pass; E left, data are relative to EV) or MS size (E right, data are relative to EV, see also Table S2), or cumulative sphere number (nr) in serial replating experiments (F), or Ki67 expression by qPCR (data are relative to EV; G). (H) Cells from WT MS were stably transduced with Venus- (EV) or PKCζ-Venus (PKCζ-OE). PKCζ-OE cells were then transduced with the indicated Numb-mutants (Flag-tagged) or control vector (EV) and cells were further silenced or not for Numb expression, as shown. Cells were assessed for SFE (left, data are relative to EV, see also Table S2), MS size (middle, data are relative to EV, see also Table S2) or protein expression (right; arrows, endogenous [black] or overexpressed [red] Numb proteins). When shown: N, number of MS analyzed. Data are reported ± SE (A, B, C, E, and G) or ± SD (F and H). Statistical analysis was with the ANOVA test (F), or with the Student’s t test two-tailed (A, B, C, E, G, and H). Significance was calculated vs. Ctr-KD or EV. Source data are available for this figure: SourceData F4.
We, then, tested the ability of the Numb mutants to rescue the biological effects of PKCζ overexpression. Under these conditions, one would predict that the 3A mutant should revert the effects of PKCζ-OE, while the 3D mutant should be unable to do so. Indeed, these predictions were experimentally confirmed upon reconstitution of MaSLCs depleted of endogenous Numb (Numb-KD) with the various mutants (Fig. 4 H). Of note, the reconstitution with Numb-FL was less effective vs. the 3A mutant, as also expected based on the sum of all previous results (Fig. 4 H).
These results were mirrored by consistent effects on Numb-regulated pathways. In particular, in Par3-KD and PKCζ-KD cells, the decrease in phospho-Numb (pNumb) was accompanied by increased levels of p53 and p53 target genes, and by decreased levels of active Notch and Notch target genes (Fig. 5, A and B). The opposite effects were detected in PKCζ-OE cells (Fig. 5 C).
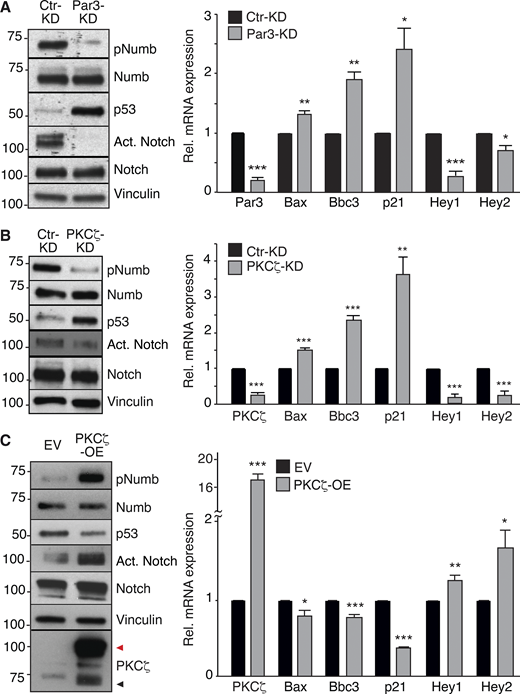
Effects of Par3/PKCζ perturbation on Notch and p53 signaling. (A–C) Left: IB in Par3-KD, PKCζ-KD, and PKCζ-OE MECs. Black and red arrows point to endogenous and overexpressed PKCζ (Venus-tagged), respectively. Right: qPCR analysis of the indicated genes (p53 targets: Bax, Bbc3, p21; Notch targets: Hey1, Hey2). Data are relative to mRNA levels in control cells (=1) and are reported ± SE. Statistical analysis was with with the Student’s t test two-tailed. In all panels, significance was calculated vs. Ctr-KD or EV. Source data are available for this figure: SourceData F5.
Effects of Par3/PKCζ perturbation on Notch and p53 signaling. (A–C) Left: IB in Par3-KD, PKCζ-KD, and PKCζ-OE MECs. Black and red arrows point to endogenous and overexpressed PKCζ (Venus-tagged), respectively. Right: qPCR analysis of the indicated genes (p53 targets: Bax, Bbc3, p21; Notch targets: Hey1, Hey2). Data are relative to mRNA levels in control cells (=1) and are reported ± SE. Statistical analysis was with with the Student’s t test two-tailed. In all panels, significance was calculated vs. Ctr-KD or EV. Source data are available for this figure: SourceData F5.
The sum of our results is interpretable according to the model shown in Fig. 6. In this model, the stereotypical all-or-none partitioning of Numb at the division of a MaSLC is substituted by a closer-to-reality scenario in which Numb is predominantly, but not exclusively, partitioned in one of the daughter cells. This is consistent with the fact that a fraction of Numb (WT panel, Fig. 6), which remains associated with one of the cellular poles, is asymmetrically partitioned, while another fraction—released into the cytoplasm as a consequence of phosphorylation—would be equally divided between the two daughter cells. This cytoplasmic fraction, however, would be inactive, as a consequence of phosphorylation, thereby yielding a complete asymmetric partitioning of “functional Numb.” The results presented so far support this model in which phosphorylation is central not only to allow unequal partitioning of Numb but also to determine its functional status and thus the differential regulation of Numb-controlled pathways (p53 and Notch) in the daughter cells (Fig. 6).

A model for the effects of Numb perturbations. The cartoon shows various conditions of Numb perturbations. (A) The localization of Numb immediately before the mitosis of the MaSLC is shown (solid line, Numb associated with the PM; dots, Numb released in the cytoplasm). After mitosis, the two daughters are indicated as A and B. (B) The partition of Numb in the two daughters is shown. (C) The functionality of Numb (as the result of its phosphorylation status) is shown. (D) The effects of on the p53 and Notch pathways is shown. H, high activity; L, low activity. (E) The predicted state (Quies., quiescent; Prol., proliferating) of the two daughters is shown. Note that only in the WT condition, an asymmetric fate is obtained. (F) Prediction of the effects on MS growth.
A model for the effects of Numb perturbations. The cartoon shows various conditions of Numb perturbations. (A) The localization of Numb immediately before the mitosis of the MaSLC is shown (solid line, Numb associated with the PM; dots, Numb released in the cytoplasm). After mitosis, the two daughters are indicated as A and B. (B) The partition of Numb in the two daughters is shown. (C) The functionality of Numb (as the result of its phosphorylation status) is shown. (D) The effects of on the p53 and Notch pathways is shown. H, high activity; L, low activity. (E) The predicted state (Quies., quiescent; Prol., proliferating) of the two daughters is shown. Note that only in the WT condition, an asymmetric fate is obtained. (F) Prediction of the effects on MS growth.
Subversion of Numb phosphorylation is frequent in BC
Loss of Numb expression is causal in BC and is selected for in the natural history of the tumor by conferring metastatic ability, a property associated with the expansion of the CSC compartment (Tosoni et al., 2015). We reasoned that if Numb phosphorylation phenocopies loss of Numb, then it might also be selected as an advantage-conferring event in mammary tumorigenesis. To test this hypothesis, we evaluated the phosphorylation status of Numb in a large consecutive cohort of BCs (N = 2,453 [Pece et al., 2019]) by immunohistochemistry (IHC) on tissue microarrays (TMA). We obtained IHC staining for Numb and pNumb in 1,675 cases (Fig. 7 A and Table S3; note that the loss of samples was mostly due to detachment of TMA cores during the IHC procedure or to the insufficient presence of tumor in the analyzed slice of the TMA in some cores). In a significant number of cases, the tumor core arrayed on the TMA also contained normal breast tissue, also permitting the measurement of physiological pNumb levels. The intensity of pNumb staining in the majority of normal breast glands was 0-1 on our scoring scale (Fig. 7 A). In BCs, the majority of tumors displayed normal-like pNumb staining, while a sizable and significant fraction showed high levels of pNumb (>1).
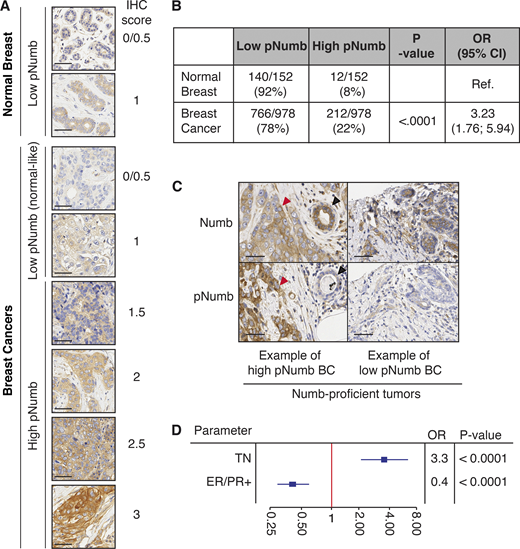
Clinical relevance of pNumb in human BCs. (A) IHC of pNumb in normal and tumor breast tissues, with IHC scores. Bars, 50 µm. (B) Analysis of pNumb (low pNumb, IHC ≤ 1; high pNumb, IHC > 1). OR, odd ratio, CI, confidence interval. (C) Representative IHC images of Numb and pNumb status in a high pNumb and a low pNumb BC. Both tumors are Numb-proficient (Numb IHC ≥ 2, top panels). In the high pNumb BC, the tumor (red arrow) is pNumb high, while a normal gland (black arrow) is pNumb low. Bar. 50 µm. (D) Forest Plot of association between high pNumb and some clinical and pathological parameters. TN, triple-negative BCs, ER/PR+, estrogen and progesterone receptor positive BCs. OR, odds ratio. The complete set of data is in Table S4. In B and D, statistical analysis was with the nonparametric Fisher’s exact test.
Clinical relevance of pNumb in human BCs. (A) IHC of pNumb in normal and tumor breast tissues, with IHC scores. Bars, 50 µm. (B) Analysis of pNumb (low pNumb, IHC ≤ 1; high pNumb, IHC > 1). OR, odd ratio, CI, confidence interval. (C) Representative IHC images of Numb and pNumb status in a high pNumb and a low pNumb BC. Both tumors are Numb-proficient (Numb IHC ≥ 2, top panels). In the high pNumb BC, the tumor (red arrow) is pNumb high, while a normal gland (black arrow) is pNumb low. Bar. 50 µm. (D) Forest Plot of association between high pNumb and some clinical and pathological parameters. TN, triple-negative BCs, ER/PR+, estrogen and progesterone receptor positive BCs. OR, odds ratio. The complete set of data is in Table S4. In B and D, statistical analysis was with the nonparametric Fisher’s exact test.
To obtain quantitative results, we had to further select cases. We have previously shown that a sizable fraction of BCs is Numb-null (i.e., its expression is lost or severely attenuated, either at the transcriptional or the post-translational level; Colaluca et al., 2018; Pece et al., 2004). In these tumors, the levels of pNumb would obviously be low, simply because of the lack of Numb protein. Thus, we selected, within the cohort, only the Numb-proficient tumors, i.e., those tumors that displayed readily detectable Numb in IHC assays. This yielded a total of 978 tumor samples and 152 normal tissues present on the same cores (Table S3). Within this subcohort, 22% of BCs displayed high pNumb (OR vs. normal 3.2; P < 0.0001; Fig. 7, B and C); overall high pNumb was present in ∼12% of all analyzed BCs (212/1675). Importantly, a high pNumb status in BC correlated with some clinical and pathological parameters of aggressive disease course; in particular with negative hormone receptor status and triple-negative BC (TNBC; Fig. 7 D and Table S4).
Thus, aberrant Numb phosphorylation is frequent in BC and correlates with some parameters of aggressive disease. This behavior is similar to that previously described for BCs in which Numb expression is lost (Colaluca et al., 2008; Pece et al., 2004; Tosoni et al., 2017). Together, this evidence supports the concept that pNumb is functionally null, and argues that in BC, the loss of Numb function can be due to either loss of expression or its hyperphosphorylation.
Molecular and biological impact of altered Numb phosphorylation in BC
We sought for functional evidence that the pNumb status in BC affects the self-renewal kinetics of the SC compartment. As the two major signaling pathways controlled by Numb, Notch, and p53 were similarly affected by Numb hyperphosphorylation or by loss of expression, we dissected their relative contribution to the division of MaSLCs and their daughters in BC. We used the following pharmacological tools: (i) a γ-presenilin inhibitor (GSI-IX), which prevents the activating proteolytic cleavage of Notch, thus acting as a Notch inhibitor (Dovey et al., 2001; Lu et al., 2014) and (ii) Nutlin-3, which by inhibiting Mdm2 restores p53 levels, thereby phenocopying the action of Numb on p53 (Vassilev et al., 2004). We tested these drugs in serial MS assays in various conditions.
To establish a molecularly defined framework of reference, we initially tested these drugs on the growth of MS derived from the mouse mammary gland. The conditions, frequently detected in BC, of Numb loss-of-expression or Numb hyperphosphorylation were mimicked by Numb-KO and by PKCζ-OE, respectively. In both cases, treatment with Nutlin-3 caused a shift from a logarithmically expanding phenotype to a self-limiting one in the serial MS assay (Fig. 8, A and B), demonstrating that restoration of p53 function leads to the reversion of the symmetric pattern of division of the MS-forming cells toward an asymmetric one. The effects of GSI-IX were less pronounced, albeit significant. In particular, under conditions of GSI-IX treatment, both Numb-KO and PKCζ-OE MECs still displayed amplification of the MS-forming compartment over serial passages (Fig. 8, A and B). However, under the same conditions, GSI-IX had a pronounced effect on MS size, consistent with its predominant role in the divisional history of the progenitors (Fig. 8, C and D).
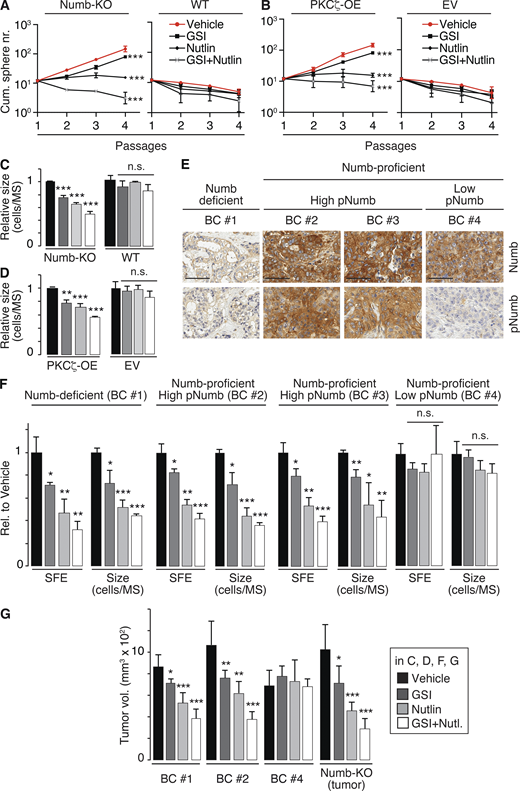
Numb phosphorylation in the SC compartment in BCs. (A and B) MS serial propagation assay under the shown drug treatments in: (A) Numb-KO MS vs. WT MS; (B) PKCζ-OE MS vs. EV MS (empty vector). (C and D) Cells, as in A and B, were evaluated for SFE and MS size (expressed relative to vehicle, see also Table S2). (E) IHC images of Numb and pNumb in the primary BCs used to generate the PDX-derived primary cultures for the experiments in F and G. Bar, 70 µm. (F) SFE and MS size of the indicated primary cultures, under various pharmacological treatments, expressed relative (Rel.) to vehicle. (G) Tumor cells from the indicated BC primary cultures or from a tumor that formed in a Numb-KO mouse, were treated ex vivo with the indicated drugs and orthotopically transplanted (50,000 cells per injection) in immunocompromised NOD/SCID mice (6/group). In all panels: GSI-IX (GSI), 10 μM; Nutlin-3 (Nutlin) 10 μM; GSI + Nutlin (10 μM each) for the duration of the experiments, except for G, in which cells were treated for 72 h before being injected. In all panels data are reported ± SD. Statistical analysis was with the ANOVA test (A and B) or with the Student’s t test two-tailed (C, D, F, and G).
Numb phosphorylation in the SC compartment in BCs. (A and B) MS serial propagation assay under the shown drug treatments in: (A) Numb-KO MS vs. WT MS; (B) PKCζ-OE MS vs. EV MS (empty vector). (C and D) Cells, as in A and B, were evaluated for SFE and MS size (expressed relative to vehicle, see also Table S2). (E) IHC images of Numb and pNumb in the primary BCs used to generate the PDX-derived primary cultures for the experiments in F and G. Bar, 70 µm. (F) SFE and MS size of the indicated primary cultures, under various pharmacological treatments, expressed relative (Rel.) to vehicle. (G) Tumor cells from the indicated BC primary cultures or from a tumor that formed in a Numb-KO mouse, were treated ex vivo with the indicated drugs and orthotopically transplanted (50,000 cells per injection) in immunocompromised NOD/SCID mice (6/group). In all panels: GSI-IX (GSI), 10 μM; Nutlin-3 (Nutlin) 10 μM; GSI + Nutlin (10 μM each) for the duration of the experiments, except for G, in which cells were treated for 72 h before being injected. In all panels data are reported ± SD. Statistical analysis was with the ANOVA test (A and B) or with the Student’s t test two-tailed (C, D, F, and G).
Next, we established PDXs from BCs and derived primary cultures from them for biological experiments. We selected (Fig. 8 E): (i) a Numb-deficient BC (Numb IHC score <0.5, BC #1); (ii) two Numb-proficient BCs (Numb IHC score ≥2, BC #2 and BC #3) displaying high pNumb (pNumb IHC ≥ 1.5); (iii) one Numb-proficient BC (BC #4) displaying low pNumb (pNumb IHC < 1.5).
We selected BCs showing a similar logarithmic expansion of the CSC compartment (Fig. S4 A) to ensure fair comparison among the samples. In addition, all selected BCs were p53-competent (i.e., not harboring mutations/deletions of the p53 gene, Fig. S4 B). Furthermore, BC #1, 2, and 3 (either Numb-deficient or displaying high pNumb) showed activation of the Notch pathway, as witnessed by increased levels of Notch targets when compared with BC #4 (Numb-proficient, low pNumb; Fig. S4 C).

Additional data to Fig. 8. (A) Serial MS assay of the cells from the BC PDXs employed in the experiments shown in Fig. 8, F and G. Results are reported ± SD. A normal counterpart of BC #1 (N#1) is also reported (red line). P, *** = < 0.001 by ANOVA test. (B) Top panels: Representative images of p53 status in FFPE samples of the BCs analyzed in Fig. 8 E. Bottom panels: The scale of p53 IHC staining used in representative BC tumors. BCs were classified into three groups according to the p53 nuclear staining (see also Materials and methods and Alsner et al., 2008; Colaluca et al., 2018; Yemelyanova et al., 2011): 0–1, 0–1% positive nuclei (indicative of complete loss of p53 protein/nonsense mutations); 2–79, 2–79% positive nuclei (indicative of WT levels of p53); 80+, positive p53 nuclei ≥80% (indicative of missense mutations of p53). Bar, 100 μm. (C) The indicated BCs (see Fig. 8 for detailed descriptions) were analyzed by qPCR for the indicated Notch pathway targets. Data are expressed (±SE) relative to mRNA levels in BC #4 (=1; BC #4 is a Numb-proficient, low-pNumb BC). In C, significance was calculated with the Student’s t test two-tailed, vs. BC #4.
Additional data to Fig. 8. (A) Serial MS assay of the cells from the BC PDXs employed in the experiments shown in Fig. 8, F and G. Results are reported ± SD. A normal counterpart of BC #1 (N#1) is also reported (red line). P, *** = < 0.001 by ANOVA test. (B) Top panels: Representative images of p53 status in FFPE samples of the BCs analyzed in Fig. 8 E. Bottom panels: The scale of p53 IHC staining used in representative BC tumors. BCs were classified into three groups according to the p53 nuclear staining (see also Materials and methods and Alsner et al., 2008; Colaluca et al., 2018; Yemelyanova et al., 2011): 0–1, 0–1% positive nuclei (indicative of complete loss of p53 protein/nonsense mutations); 2–79, 2–79% positive nuclei (indicative of WT levels of p53); 80+, positive p53 nuclei ≥80% (indicative of missense mutations of p53). Bar, 100 μm. (C) The indicated BCs (see Fig. 8 for detailed descriptions) were analyzed by qPCR for the indicated Notch pathway targets. Data are expressed (±SE) relative to mRNA levels in BC #4 (=1; BC #4 is a Numb-proficient, low-pNumb BC). In C, significance was calculated with the Student’s t test two-tailed, vs. BC #4.
Based on the previous set of results, the prediction is that in the pharmacological assays, the Numb-deficient (BC #1) and the high-pNumb tumors (BC #2 and #3) should behave similarly and phenocopy the results obtained with Numb-KO MECs and PKCζ-OE MECs (Fig. 8, A–D), while the Numb-proficient/low-pNumb BC (BC #4) should be insensitive to the treatments. These predictions were confirmed in MS assays (Fig. 8 F) and in xenotransplantation assays in mice (Fig. 8 G). We note that in the Numb-proficient tumor with low pNumb (BC #4), we do not know the alteration(s) leading to the logarithmic expansion of the CSC compartment. However, this tumor served as a suitable control for the specificity of the effects of Nutlin-3 or GSI-IX.
We concluded that Numb hyperphosphorylation in BC phenocopies Numb loss and is causal in the expansion of the CSC-like compartment that occurs in these malignancies.
Mechanisms of Numb phosphorylation in human BCs
We sought to gain insights into the mechanisms upstream of Numb phosphorylation in human BC. Initially, we surveyed the METABRIC and TGCA BC databases. The most obvious candidate, PKCζ, was not altered at a high frequency (∼2%; Fig. S5 A). However, the overall rate of overexpression of all PKCs was significant (∼20–25%). In particular, typical PKCs (conventional and novel) were overexpressed in ∼18–20% of BC cases with frequent co-occurrence of upregulation events (Fig. S5, A and B).
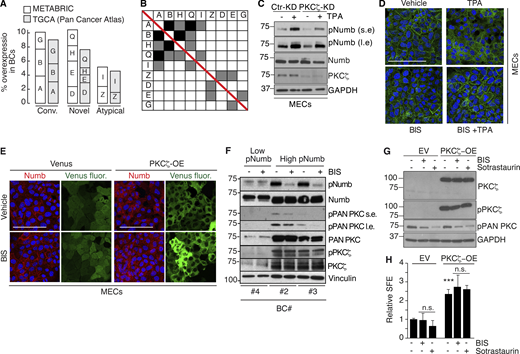
Additional data to Fig. 9. (A and B) Overexpression of PKC isoforms in human breast cancers. (A) The overexpression of PKC isoforms was evaluated in the METABRIC and TGCA (Pan Cancer Atlas) BC datasets, as described in Materials and methods. Parameters used were: mRNA expression z-scores relative to all samples (log microarray); z-score threshold + 2.0. The percentage of alterations in the two datasets (METABRIC, N = 1904; TGCA, N = 994) is reported for the three classes of PKCs and further subdivided for each gene (A, PRKCA or α; B, PRKCB or β; G, PRKCG or γ; D, PRKCD or δ; E, PRKCE or ε; H, PRKCH or η; Q, PRKCQ or θ; Z, PRKCZ or ζ; I, PRKCI or ι). (B) Matrix showing the co-occurrence of overexpression of PKC isoforms. The matrix shows the co-occurrence (P < 0.05 by one-tailed Fisher’s exact test). Color code: black, co-occurrence in both datasets (METABRIC and TGCA); gray, co-occurrence in only one dataset. (C–H) Various controls for the specificity of the effects of TPA and of PKC inhibitors. (C) The indicated MEC transfectants were stimulated with TPA (1 μM for 12′) followed by IB with the indicated Ab. GAPDH is a loading control. S.e, short exposure; l.e, long exposure. The blot shows that also in the absence of PKCζ, TPA can efficiently phosphorylate Numb. (D) Numb confocal immunofluorescence in mouse MECs treated with TPA (1 μM TPA, 12 min), BIS (3 μM, 6 h) or a combination of TPA + BIS. In the combination treatment, cells were pretreated with BIS for 6 hr and then induced with TPA for 1 μM TPA for 12 min. Numb (green), DAPI (blue). Bar, 100 µm. (E) Numb confocal immunofluorescence in mouse MECs stably overexpressing Venus (Venus) or PKCζ-Venus (PKCζ -OE) treated with BIS (3 μM, 6 h). Numb (red), epifluorescence (Venus), DAPI (blue). Bars, 100 µm. (F) The indicated BC cultures were treated or not with BIS (3 μM o/n) and immunoblotted as shown on the right. Vinculin, loading control; l.e., long exposure. The figure shows that in BC displaying high pNumb, BIS significantly reduces the level of pNumb. (G) MS from WT mice were dispersed and infected with Venus (EV) or PKCζ-Venus (PKCζ -OE), and treated with BIS (3 μM o/n) or Sotrastaurin (0.5 μM o/n), another PKC inhibitor (Albert et al., 2022) and subjected to IB as indicated. (H) Cells as in G were treated with BIS (3 μM for the duration of the experiment) or Sotrastaurin (0.5 μM for the duration of the experiment), and subjected to a MS assay, as indicated. Data are expressed relative to control (EV untreated) cells ± SD (see also Table S2). Statistical analysis was with the Student’s t test two-tailed. The combined results of G and H show that treatment with two different PKC inhibitors cannot revert the biochemical and biological effects of PKCζ -OE. Source data are available for this figure: SourceData FS5.
Additional data to Fig. 9. (A and B) Overexpression of PKC isoforms in human breast cancers. (A) The overexpression of PKC isoforms was evaluated in the METABRIC and TGCA (Pan Cancer Atlas) BC datasets, as described in Materials and methods. Parameters used were: mRNA expression z-scores relative to all samples (log microarray); z-score threshold + 2.0. The percentage of alterations in the two datasets (METABRIC, N = 1904; TGCA, N = 994) is reported for the three classes of PKCs and further subdivided for each gene (A, PRKCA or α; B, PRKCB or β; G, PRKCG or γ; D, PRKCD or δ; E, PRKCE or ε; H, PRKCH or η; Q, PRKCQ or θ; Z, PRKCZ or ζ; I, PRKCI or ι). (B) Matrix showing the co-occurrence of overexpression of PKC isoforms. The matrix shows the co-occurrence (P < 0.05 by one-tailed Fisher’s exact test). Color code: black, co-occurrence in both datasets (METABRIC and TGCA); gray, co-occurrence in only one dataset. (C–H) Various controls for the specificity of the effects of TPA and of PKC inhibitors. (C) The indicated MEC transfectants were stimulated with TPA (1 μM for 12′) followed by IB with the indicated Ab. GAPDH is a loading control. S.e, short exposure; l.e, long exposure. The blot shows that also in the absence of PKCζ, TPA can efficiently phosphorylate Numb. (D) Numb confocal immunofluorescence in mouse MECs treated with TPA (1 μM TPA, 12 min), BIS (3 μM, 6 h) or a combination of TPA + BIS. In the combination treatment, cells were pretreated with BIS for 6 hr and then induced with TPA for 1 μM TPA for 12 min. Numb (green), DAPI (blue). Bar, 100 µm. (E) Numb confocal immunofluorescence in mouse MECs stably overexpressing Venus (Venus) or PKCζ-Venus (PKCζ -OE) treated with BIS (3 μM, 6 h). Numb (red), epifluorescence (Venus), DAPI (blue). Bars, 100 µm. (F) The indicated BC cultures were treated or not with BIS (3 μM o/n) and immunoblotted as shown on the right. Vinculin, loading control; l.e., long exposure. The figure shows that in BC displaying high pNumb, BIS significantly reduces the level of pNumb. (G) MS from WT mice were dispersed and infected with Venus (EV) or PKCζ-Venus (PKCζ -OE), and treated with BIS (3 μM o/n) or Sotrastaurin (0.5 μM o/n), another PKC inhibitor (Albert et al., 2022) and subjected to IB as indicated. (H) Cells as in G were treated with BIS (3 μM for the duration of the experiment) or Sotrastaurin (0.5 μM for the duration of the experiment), and subjected to a MS assay, as indicated. Data are expressed relative to control (EV untreated) cells ± SD (see also Table S2). Statistical analysis was with the Student’s t test two-tailed. The combined results of G and H show that treatment with two different PKC inhibitors cannot revert the biochemical and biological effects of PKCζ -OE. Source data are available for this figure: SourceData FS5.
Based on these observations, we hypothesized that Numb hyperphosphorylation in human BCs is predominantly (albeit not necessarily exclusively) driven by activation of typical PKCs, rather than atypical PKCs. To test this possibility, we initially evaluated the effects of activation of typical PKCs on Numb and the divisional history of MaSLCs. We employed TPA, a widely used activator of typical (but not atypical) PKCs (Ono et al., 1989; Ron and Kazanietz, 1999; Ways et al., 1992). As expected, in MECs, TPA induced the phosphorylation of Numb in control and PKCζ-KD cells (Fig. S5 C), supporting the notion that its effects are exerted through the activation of typical PKCs. In MaSLCs, TPA induced the prompt release of Numb from the PM (Fig. 9 A) and Numb symmetric partitioning at the first mitotic division of the MaSLC (Fig. 9 B). This was mirrored by a TPA-induced expansion of the SC-like compartment, witnessed by increased SFE (Fig. 9 C), which was reversed by Nutlin-3, and to a lesser extent by GSI-IX treatment, similar to what was detected under conditions of Numb-KO or PKCζ-OE. In MECs, TPA treatment induced redistribution of Numb from the PM to the cytoplasm (Fig. 9 D, left), accompanied by high levels of pNumb and activated Notch, and low levels of p53 (Fig. 9 D, right). Thus, activation of typical PKCs by TPA phenocopies the effects of the atypical PKC and PKCζ.
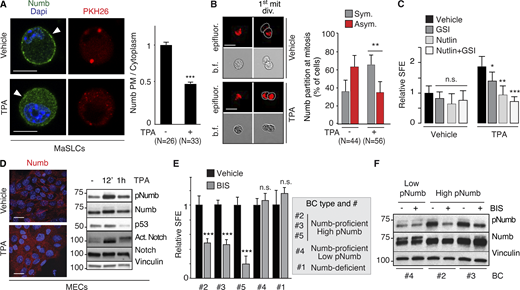
PKCs and Numb phosphorylation in human BCs. (A) Left: MaSLCs (PKHpos cells) were treated with 1 μM TPA (12 min) and stained with anti-Numb and DAPI. PHK26 (red), Numb (green), DAPI (blue). Arrowheads point to PM. Bar, 10 μm. Right: Quantitation of experiment, data are expressed as the ratio between plasma membrane (PM) Numb and cytoplasmic Numb, relative to vehicle. (B) Left: Numb partitioning at mitosis of MaSLCs (1st mit. div.), treated as indicated (TPA, 1 μM for the duration of the experiment). Bar, 10 μm. b.f., bright-field.; epifluorescence (red). Right: Quantitation of the experiment, showing the frequency of symmetric (Sym) vs. asymmetric (Asym) partitioning of Numb. (C) SFE of mouse WT MECs treated as indicated (TPA treatment as in B, other drugs as in Fig. 8); data are relative to Vehicle control (see also Table S2). (D) Left: Numb confocal immunofluorescence in MECs treated with TPA as in A. Numb (red), DAPI (blue); Bar, 20 μm. Right: IB of MECs treated with TPA (1 μM) for the indicated time. (E) SFE, in the presence of BIS of the indicated BC primary cultures. (F) The indicated BC primary cultures were treated with BIS (3 μM o/n) and IB as shown. When shown: N, number of cells or doublets analyzed, as appropriate. Data are reported ± SD (B, C, and E) or ± SE (A). Statistical analysis was with the nonparametric Fisher’s exact test (B) or with the Student’s t test two-tailed (A, C, and E). Source data are available for this figure: SourceData F9.
PKCs and Numb phosphorylation in human BCs. (A) Left: MaSLCs (PKHpos cells) were treated with 1 μM TPA (12 min) and stained with anti-Numb and DAPI. PHK26 (red), Numb (green), DAPI (blue). Arrowheads point to PM. Bar, 10 μm. Right: Quantitation of experiment, data are expressed as the ratio between plasma membrane (PM) Numb and cytoplasmic Numb, relative to vehicle. (B) Left: Numb partitioning at mitosis of MaSLCs (1st mit. div.), treated as indicated (TPA, 1 μM for the duration of the experiment). Bar, 10 μm. b.f., bright-field.; epifluorescence (red). Right: Quantitation of the experiment, showing the frequency of symmetric (Sym) vs. asymmetric (Asym) partitioning of Numb. (C) SFE of mouse WT MECs treated as indicated (TPA treatment as in B, other drugs as in Fig. 8); data are relative to Vehicle control (see also Table S2). (D) Left: Numb confocal immunofluorescence in MECs treated with TPA as in A. Numb (red), DAPI (blue); Bar, 20 μm. Right: IB of MECs treated with TPA (1 μM) for the indicated time. (E) SFE, in the presence of BIS of the indicated BC primary cultures. (F) The indicated BC primary cultures were treated with BIS (3 μM o/n) and IB as shown. When shown: N, number of cells or doublets analyzed, as appropriate. Data are reported ± SD (B, C, and E) or ± SE (A). Statistical analysis was with the nonparametric Fisher’s exact test (B) or with the Student’s t test two-tailed (A, C, and E). Source data are available for this figure: SourceData F9.
These results argue that Numb phosphorylation (and its biological effects) could be equally sustained by atypical PKCs (in particular PKCζ, which is the “physiological” kinase) or by typical PKCs. This latter condition might apply to BC. We directly tested this hypothesis in BCs using BIS. We used this inhibitor at a concentration of 3 μM, which—based on extant literature—has negligible effects on the activities of atypical PKCs (PKCζ and PKCι), while potently inhibiting conventional and novel PKCs (Martiny-Baron et al., 1993; Toullec et al., 1991; Uberall et al., 1997; Fig. S5, D–H for controls on the effects and specificity of BIS). We used the inhibitor on the same BC primary cultures used in Fig. 8 F (plus an additional Numb-proficient, high-pNumb tumor, BC #5) in an MS assay. The three high-pNumb tumors (BC #2, #3, and #5) were significantly affected by BIS and displayed reduced SFE. Conversely, the Numb-deficient and the low-pNumb tumors (BC #1 and #4) were not affected by the treatment (Fig. 9 E). In addition, BIS significantly decreased the levels of pNumb in representative high-pNumb tumors (BC #2 and #3), but not in a low-pNumb tumors (BC #4; Fig. 9 F).
We concluded that in a significant fraction of BCs, the activation of PKCs, and preponderantly of typical (canonical and novel) PKCs, induces aberrant phosphorylation of Numb, leading to its inhibition and to the loss of its tumor suppression function, which results in expansion of the CSC-like compartment.
Discussion
Herein, we report an investigation of the consequences of Numb phosphorylation in the regulation of mammary SC compartments. We demonstrated that Numb asymmetric partitioning, under normal-like ex vivo conditions, is necessary but not sufficient for the establishment of alternative cell fates in the daughters of a MaSLC, as Numb inactivation—in the daughter cell that assumes the progenitor fate—is also required. Interestingly, both the asymmetric partitioning of Numb and its functional inactivation are controlled by its phosphorylation status.
These results resolve a potential incongruity of the asymmetric partitioning model. In that model, the supposedly Numb-negative daughter (the progenitor cell) would still inherit a significant amount of Numb (Fig. 6). Indeed, in a recent study in hematopoietic SCs, careful quantitative analysis showed that the ratio of inherited Numb, between sister cells rarely exceeds twofold (Loeffler et al., 2019). However, based on our results, the complement of Numb inherited by the progenitor would be phosphorylated and therefore functionally null; this determines a true “all-or-none” scenario for Numb activity between the two daughter cells, leading to a clear-cut and opposite activity of the two major pathways regulated by Numb, namely Notch and p53. In the daughter that retains the SC fate, quiescence would be imposed by high levels of p53 in the absence of Notch. Vice versa, in the progenitor cell, Notch-dependent signaling could occur in the absence of p53-mediated inhibition, leading to expansion of the transit-amplifying compartment.
Biochemically, Numb acts as an inhibitor of pathways by binding and functionally sequestering its interactors, e.g., it binds to and inhibits Notch (Bultje et al., 2009; McGill et al., 2009; McGill and McGlade, 2003; Pece et al., 2004; Westhoff et al., 2009; Zhong et al., 1996; Zhong et al., 1997) and also Mdm2 (Colaluca et al., 2018; Colaluca et al., 2008; Sheng et al., 2013; Wang et al., 2015). Phosphorylation prevents Numb from interacting with its targets in vivo, leading to the simultaneous activation of an oncogenic pathway (Notch) and the inhibition of a tumor suppressor (p53). Therefore, Numb phosphorylation critically controls the balance of the two pathways in the progeny of the MaSLC undergoing fate determination. This mechanism could extend to other SC compartments as it has been shown that Numb phosphorylation destabilizes p53 and promotes self-renewal in liver cancer tumor-initiating cells (Siddique et al., 2015). Presently, we do not know whether the lack of interaction of pNumb with its partners in vivo is due to the “mislocalization” of pNumb or to alterations in the intrinsic protein:protein interaction abilities of the protein.
In the mammary gland, the centrality of Numb in the maintenance of SC homeostasis is underscored by its “oncogenic fragility.” In itself, the Numb gene does not meet the definition of a cancer-associated gene, as its genetic alterations (deletions, amplifications, and mutations) are rare. However, it appears to be a convergence point for many oncogenic insults that all lead to its loss (or attenuation) of function. In the course of our studies, we have uncovered three possible mechanisms of Numb subversion in BC: (i) exaggerated ubiquitination/degradation of the protein (Pece et al., 2004), (ii) alterations in the splicing pattern leading to loss of exon 3-bearing isoforms (Colaluca et al., 2018), and (iii) hyperphosphorylation (this study). In all cases, these alterations are associated with indicators of aggressive disease course and unfavorable prognosis (Colaluca et al., 2018; Colaluca et al., 2008; Pece et al., 2004; and this paper). In addition, the reproduction of these alterations in ex vivo systems leads to the emergence of CSC-like cells and the expansion of a CSC-like compartment (Tosoni et al., 2017; Tosoni et al., 2015; and this paper). The sum of these results argues that Numb subversion is a major determinant in BC pathogenesis.
In dissecting the mechanisms responsible for Numb hyperphosphorylation in BC, we discovered that the major culprit is represented by typical PKCs. This is interesting since under normal conditions the major regulator of Numb phosphorylation is the atypical PKC, PKCζ. From our data, it seems that, in principle, all PKCs are capable of efficiently phosphorylating Numb. However, PKCζ, thanks to its activation by polarity complexes (Betschinger et al., 2003; Henrique and Schweisguth, 2003; Macara, 2004; Suzuki and Ohno, 2006; Wirtz-Peitz et al., 2008), is the sole one capable of executing Numb phosphorylation in a topologically restricted fashion: an occurrence of critical importance for the determination of asymmetric fates in the normal SC compartment (Bellaiche et al., 2001; Besson et al., 2015; Dho et al., 2006; Smith et al., 2007). While this might account for the specificity of PKCζ in the normal setting, it also raises several questions. For instance, our data clearly show that unchecked, ubiquitous phosphorylation of Numb can lead to loss of its tumor suppressor function. So, how is Numb “shielded,” under normal conditions, from the activity of typical PKCs that are not topologically restricted and/or activated (Hong, 2018)? While we can offer no experimental explanation, at this stage, one possible answer is that only under conditions of hyperactivation of PKCs (occurring in tumors but not in normal settings), the levels of Numb phosphorylation might become stoichiometrically significant.
This hypothesis begs the next question: are PKCs activated in BC? And if so, which ones? In this contention, it is worth noting that several studies point to the relevance of altered PKC activation to BC (Urtreger et al., 2012). For instance, PKCs have been found to be elevated in malignant vs. normal breast tissues (Jarzabek et al., 2002; O'Brian et al., 1989) and associated with aggressive biological behavior in BC (Lonne et al., 2010; Pan et al., 2005). Moreover, in BCs there is a correlation between PKC activity/abundance and estrogen-receptor-negative status (Assender et al., 2007; Gundimeda et al., 1996; Platet et al., 1998). In particular, overexpression of PRKCA has been associated with anti-estrogen resistance and tumor aggressiveness (Frankel et al., 2007; Nabha et al., 2005; Tonetti et al., 2003) and shown to be relevant as a therapeutic target for TNBCs (Pham, 2016). PRKCA expression seems to be connected to the activation of epithelial-to-mesenchymal transition in BC cell lines (Llorens et al., 2019), a phenomenon linked to the emergence of CSCs (Mani et al., 2008), and metastasis and therapy resistance (Pham et al., 2017). The pharmacological inhibition of PRKCA selectively targets BC CSCs vs. non-CSCs in vitro (Tam et al., 2013). Moreover, data mining of BC databases revealed an interesting pattern of co-occurrence of overexpression of the different PKC isoforms, arguing that their transcriptional regulation might be altered en bloc (or partially so) by a common upstream event in BC, a speculative scenario that warrants further investigation.
The results herein presented converge on the possibility that altered Numb phosphorylation, executed by aberrantly regulated PKCs, is a key mechanism to derange the tumor suppressive function of Numb. A note of caution is due since some of our conclusions are based on the use of pharmacological PKC inhibitors. While we provided several controls for the specificity of BIS, off-target effects of the drug cannot be excluded. Similarly, active PKCs might modulate other signaling pathways, in addition to Numb phosphorylation. We note, however, that—if further confirmed—our findings have potential therapeutic implications in the perspective of restoring the functionality of the Numb network in BC. This in principle could be achieved by Mdm2 inhibiting drugs (e.g., Nutlin-3), by Notch inhibitors (e.g., GSI-IX), PKC inhibitors (e.g., BIS), or by combinations of the above. While some of these strategies (Mdm2 inhibition) might find application in p53-competent BCs, others (Notch inhibition or PKC inhibition) might be useful also in p53-mutated BCs, especially in TNBC. The status of Numb, pNumb, and p53 in BCs would provide the necessary stratification tools to select the most appropriate treatment.
Materials and methods
Antibodies and chemicals
Primary antibodies (Ab) for immunofluorescence (IF) were directed against Flag (Cat. F3165; mouse monoclonal from SIGMA), Numb (C29G11; rabbit monoclonal from Cell Signaling Technologies, Cat. 4140), PKCζ (C-20 [Cat. Sc-216] or H-1 [Cat. Sc-17781], Santa Cruz Biotechnology, used in Fig. 1, A and E, respectively), and Par3 (Cat. 07-330; Millipore). Fluorochrome-conjugated secondary Ab were obtained from Jackson ImmunoResearch Laboratories (Alexa Fluor 488 Donkey Anti-Mouse IgG, Cat. 715-545-150, used in Fig. 1, A and E; Alexa Fluor 488 Donkey Anti-Rabbit IgG, Cat. 715-545-152, used in Fig. 1 D and Fig. 9 A and in Fig. S1, A, C, and D and Fig. S5 D; Cy3 AffiniPure Donkey Anti-Rabbit IgG, Cat. 715-165-152, used in Fig. 2 B and 9 D and Figs. S1 B, S2 A, and S5 E; Alexa Fluor 647 AffiniPure Donkey Anti-Rabbit IgG, Cat. 715-605-152, used in Fig. 1, A and E).
Ab for immunoblot (IB) were directed against Numb (AB21, a mouse monoclonal Ab against amino acids 537–551 of hNumb [Colaluca et al., 2008]), and for the experiment in Fig. S3 B, the anti-Numb C29G11, rabbit monoclonal from Cell Signaling Technologies (Cat. 4140) was used; Vinculin (mouse monoclonal; Sigma-Aldrich, Cat. V9131); GRP94 (9G10, rat monoclonal, Cat. ADI-SPA-851; Enzo Life Sciences); Tubulin (11H10, rabbit monoclonal, Cat. 2125; Cell Signaling Technologies); p53 (1C12, mouse monoclonal, Cat. 2524; Cell Signaling Technologies, Figs. 3 E, 5, A–C, and 9 D; FL393, Santa Cruz Biotechnology, goat polyclonal, Cat. sc-6243-G, Fig. 3 F; goat polyclonal, Bio-techne, Cat. AF1355 Fig. 3 H); PKCζ (C24E6, rabbit monoclonal, Cat. 9368; Cell Signaling Technologies); p-PKCζ (H-2, mouse monoclonal, Cat. sc-271962; Santa Cruz Biotechnology); PAN-PKC (A-9, mouse monoclonal, Cat. sc-17804; Santa Cruz Biotechnology); pPAN PKC (rabbit polyclonal, Cat. 9371; Cell Signaling Technologies); Notch (5B5, rat monoclonal, Cat. 3447; Cell Signaling Technologies); Cleaved Notch1 Val1744 (D3B8, rabbit monoclonal, Cat. 4147; Cell Signaling Technologies); Flag (D6W5B, rabbit monoclonal, Cat. 2368; Cell Signaling Technologies); Par3 (Cat. 07-330; Millipore); Mdm2 (Mouse Monoclonal, Cat. Op46; Calbiochem); anti GAPDH (rabbit monoclonal, Cat. 5174; Cell Signaling Technologies); and pNumb-Ser276 (rabbit monoclonal, Cat. 4140; Cell Signaling Technologies). In all, IB, vinculin, tubulin, GRP94, or GAPDH were used as loading controls.
Ab for IHC were directed against Numb or pNumb-Ser276 (same as for IB) and p53 (DO-1, mouse monoclonal, Cat. sc-126; Santa Cruz Biotechnology).
Ab for immunoprecipitation (IP) were directed against Flag (Cat. A2220; M2-agarose affinity gel from Sigma-Aldrich); p53 (DO-1, mouse conjugated to agarose, Cat. sc-126 AC; Santa Cruz Biotechnology); and Notch (C-20, goat polyclonalCat. sc-6014; Santa Cruz Biotechnology).
Chemicals used were as follows: Nutlin-3 (Cayman Chemical or supplied by S. Minucci and M. Varasi); GSI-IX (DAPT; Sigma-Aldrich); BIS (Bisindolylmaleimide I; Santa Cruz Biotechnology); TPA (Sigma-Aldrich); methylcellulose (Sigma for mammosphere assays or StemCell Technologies for time-lapse experiments); and Sotrastaurin (Aurogene). BIS was used at 3 μm. TPA was used at 1 µM, Nutlin-3 and GSI-IX were used at 10 µM in in vitro and ex vivo experiments, and Sotrastaurin was used at 0.5 μM. PKH26-GL or PKH67-GL dyes were from Sigma-Aldrich.
Engineering of vectors, quantitative PCR, and shRNA experiments
All Numb constructs employed in this study were engineered in the isoform I of human Numb. The lentiviral construct harboring the Numb-DsRed (pLVX-PURO-Numb-DsRed) was previously described (Tosoni et al., 2017; Tosoni et al., 2015).
For all other experiments involving ectopic expression of Numb or Numb mutants, a vector containing a Flag-tagged full-length wild-type human Numb (FL in all figures) together with a human sh-Numb sequence (directed against 5′-GGTTAAGTACCTTGGCCATGT-3′) was engineered to express ectopic Numb, while simultaneously silencing the endogenous human protein (see also Table S1). This vector, called pLL3.7-Numb-Flag, was engineered by subcloning the Flag-Numb fragment from the pcDNA3-Flag-Numb construct (Colaluca et al., 2008) into the NheI/EcoRI sites of the pLLox3.7-shNumb vector (Colaluca et al., 2008). The Numb cDNA coding sequence was mutagenized by site-directed mutagenesis in the sequence gttaaAtaTTtAggAcaCgt to render Numb expression resistant to its sh-RNA. The construct was sequence verified. For the experiments in mouse cells in which the endogenous Numb protein was silenced, a mouse-specific shNumb vector was co-transfected (see below and Table S1).
Point mutations of Numb at Ser7, 276, and 295 were introduced either in the pLVX-PURO-Numb-DsRed or pLL3.7-Numb-Flag constructs by PCR-based site-directed mutagenesis. pLV-Venus and pLV-PKCζ-Venus lentiviral vectors were a kind gift from K. Kaibuchi (Nagoya University Graduate School of Medicine, Nagoya, Japan; Nishimura and Kaibuchi, 2007). pLL3.7-Notch1-ΔE (Notch-TFX, Fig. 3, G and I), encoding the C-terminal Flag-tagged version of the human activated form of Notch-ΔE mutant, was engineered subcloning the Notch-ΔE fragment from the vector pcDNA3-Flag-Notch1-ΔE (Westhoff et al., 2009) into the NheI/EcoRI sites of the pLLox3.7 vector, followed by sequence verification.
Total RNA was extracted with QIAzol (Qiagen) and purified using the RNeasy kit (Qiagen). Reverse transcription was performed starting from 0.02 to 2 μg of total RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Each sample was tested at least in duplicate. The ΔCt method was used to calculate the mRNA levels of each target gene normalized against two different housekeeping genes (from those listed below). The 2^−ΔΔCt method was used to compare the mRNA levels of each target gene, normalized to the housekeeping genes, relative to an external standard. Taqman Gene Expression Assays (Thermo Fisher Scientific) were as follows: PKCζ (hs00177051_m1 and mm00776345_g1), PKCι (mm00435769_m1), Par3 (mm00473929_m1), Ki67 (mm01278616_m1), p21 (mm00432448_m1), Bax (mm00432050_m1), Bbc3 (mm00519268_m1), Hey1 (mm00468865_m1 and hs00232618_m1), Hey2 (mm00469280_m1 and hs00232622_m1), Hes1 (hs00172878_m1), Hes5 (hs01387463_g1), and HeyL (hs00232718_m1). Housekeeping genes used for normalization were as follows: Gapdh (mm99999915_g1), B2m (mm00437762_m1), Tbp (mm00446973_m1 and hs99999910_m1), and Gusb (mm01197698_m1 and hs99999908_m1).
All silencing experiments were performed with shRNA lentiviral vectors obtained from Dharmacon; in particular, the silencing of Par3, PKCζ, and PKCι expressions was achieved using pLKO.1-shPar3 (Clone ID: TRCN0000094401), pLKO.1-shPKCζ (Clone IDs: TRCN0000022869 and TRCN0000022871), and pLKO.1-shPKCι (Clone ID: TRCN0000022754), respectively. In some experiments (corresponding to those of Figs. 1 B, 4 A, 5 B, and S1 A), a second shRNA vector for PKCζ was also used (Clone ID: TRCN0000022871) with results comparable with those obtained with TRCN0000022869 (not shown). The silencing of mouse Numb (Fig. 4 H) was obtained with Clone ID: TRCN0000105739. Cells from dissociated MS were lentivirally transduced in suspension and allowed to form next-generation MS for 7 d under puromycin selection. In all KD experiments, Ctr populations were transduced with a Ctr shRNA-Luciferase vector (pLKO-Luc, TRCN0000072243).
Protein studies
Immunoblot (IB)
Cells were lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with a protease inhibitor cocktail (CALBIOCHEM) and phosphatase inhibitors (50 mM NaF, 10 mM Na3VO4 pH 7.5) for 30 min on ice, and the lysates were clarified by centrifugation at 16,000 g for 30 min at 4°C. For the experiments in Fig. S3 B, EDTA was omitted from the lysis buffer to allow for Phos-tag gel electrophoresis. Protein concentration was measured by the Bradford assay (Bio-Rad) following the manufacturer’s instructions. After SDS-PAGE, proteins were transferred onto nitrocellulose filters. Filters were blocked for 1 h (or o/n) in 5% milk in TBS (50 mM Tris-HCl, pH 7.4, 150 mM NaCl) supplemented with 0.1% Tween (TBS-T), and then incubated with the primary Ab, diluted in TBS-T plus 5% milk, for 1 h at RT or o/n, followed by three washes of 5 min each in TBS-T. Filters were then incubated with the appropriate horseradish peroxidase-conjugated secondary Ab (anti-mouse IgG HRP-linked 7076 or anti-rabbit IgG HRP-linked 7074 or 7077 anti-rat IgG HRP-linked, Cell Signaling) diluted 1:2,000 in TBS-T for 30 min. After three washes in TBS-T (5 min each), the bound secondary Ab was revealed using the ECL method (Biorad). The Phos-tag SDS-PAGE in Fig. S3 B was prepared according to the manufacturer’s instructions for the Zn2+-Phos-tag SDS-PAGE (WAKO) at an 8% concentration polyacrylamide and with 25 μM Zn2+-Phos-tag.
For several IB experiments, samples were loaded on different gels (since many proteins have similar molecular weight), which were subsequently cut in the region of interest to allow detection with multiple Ab. Reblotting was also performed in some cases. All IB were performed at least twice independently, with the exception of the control IB of Fig. 4 H right and Fig. S5 H, which were performed once.
Co-immunoprecipitation (co-IP)
For co-IP experiments (Fig. 3, F–I), cells were plated on 150-mm plates. Cells (at a confluence between 80 and 95%) were lysed in 10% Glycerol, 50 mM Hepes pH 7.5, 100 mM NaCl, 50 mM EDTA pH 8, 1% Triton. At least 4 mg of cellular lysate was incubated in the presence of specific Ab o/n at 4°C with rotation. Protein G was added and incubated for an additional 1 h. The supernatant was then removed and beads were washed with lysis buffer. Immunoprecipitated proteins were eluted in Laemmli buffer 2× and analyzed by IB.
Immunofluorescence (IF)
For IF, cells (from various procedures as specified in “Imaging studies”) were fixed with 4% formaldehyde (20 min at RT) and then stained with the appropriate Ab. Briefly, cells were permeabilized in PBS 0.1% Triton X-100 for 5 min at RT. To prevent non-specific binding of the Ab, cells were incubated with PBS in the presence of 3% BSA for 30 min and then with primary Ab diluted in PBS (+3% BSA) for 1 h at RT. Coverslips were washed three times with PBS and incubated for 45 min at RT with secondary Ab. After three washes with PBS, nuclei were DAPI-stained for 30 min at RT, and after three washes in PBS, the coverslips were mounted in Mowiol-Dabco or glycerol mounting medium (Sigma-Aldrich). Images were obtained using a Leica TCS SP8 FSU AOBS or SP5 AOBS confocal microscope equipped with a 63X/1.4NA oil-immersion objective or 40X/1.3NA, PMT and HyD detectors and LasX 3.5.5 acquisition software (Leica Microsystems GmbH). Images were processed using Fiji/ImageJ software (v1.52, National Institutes of Health). Images shown in Figs. 1, A, D, and E, 2 B, 9, A and D, S1, A–D, S2 A; and Fig. S5 D and E represent a single confocal plane where brightness and contrast were optimized.
Immunohistochemistry (IHC)
A total of 3-μm sections were prepared from formalin-fixed paraffin-embedded (FFPE) tissue and dried at 37°C o/n. The sections were processed with Bond-RX fully Automated stainer system (Leica Biosystems) according to the following protocol. First, tissues were deparaffinized and pretreated with the Epitope Retrieval Solution 1 (pH 6) at 100°C for 20 min for Numb and pNumb staining, 40 min for p53 staining. After the washing steps, peroxidase blocking was performed for 10 min using the Bond Polymer Refine Detection Kit (#DC9800; Leica Biosystems). Tissues were incubated for 30 min with the appropriate Ab diluted in Bond Primary Ab Diluent (#AR9352). Subsequently, tissues were incubated with postprimary and polymer for 16 min, developed with DAB-chromogen for 10 min, and counterstained with hematoxylin for 5 min.
Cell biology procedures and flow cytometry
Isolation of primary cells and MS growth
Primary MECs isolated from murine tissues and primary tumor cells from human BC PDXs were cultivated in suspension to generate MS, as described previously (Pece et al., 2010; Tosoni et al., 2012; Tosoni et al., 2015). Briefly, murine mammary glands from WT or Numb-KO mice or transplants of Numb-KO tumor cells or PDXs were mechanically dissociated and digested in DMEM/F12 medium supplemented with 1 mM glutamine (Euroclone 200 U/ml collagenase (Sigma-Aldrich) and 100 U/ml hyaluronidase (Sigma-Aldrich) at 37°C for 4 h. Cell suspensions were then centrifuged (80 g, 5 min), resuspended in 0.2% NaCl to lyse red blood cells, and sequentially filtered through membrane syringe filters of decreasing pore sizes (100-, 70-, 40-, and 20-µm meshes). The resulting single cells were plated onto ultralow attachment plates (Falcon) at a density of 100,000 viable cells/ml (to obtain primary MS) in a stem cell medium (SCM) composed of serum-free mammary epithelial basal medium (Clonetics), 5 µg/ml insulin (Sigma-Aldrich), 0.5 µg/ml hydrocortisone (Sigma-Aldrich), B27 (Invitrogen), 20 ng/ml EGF and bFGF (BD Biosciences), and 4 µg/ml heparin (Sigma-Aldrich). MS were collected after 7–10 d and mechanically dissociated by pipetting up and down several times with a fire-polished pipette to yield a single-cell suspension. For serial MS propagation experiments, 5,000 cells derived from the dissociation of primary murine or human MS, respectively, were plated in SCM (liquid suspension culture) using 24-multiwell plates coated with a Poly HEMA (Sigma-Aldrich) solution (1.2% in 95% ethanol) or in SCM containing 1% methyl-cellulose (Sigma-Aldrich). After 7–10 d, secondary MS were counted and then dissociated to calculate the SFE (total number of MS/total number of plated cells × 100). The same procedure was repeated to obtain subsequent MS generations.
For the experiments described in Fig. 3, C and D, cells obtained by the dissociation of Numb-KO MS were transduced with the indicated DsRed-tagged constructs, counted (annotating the number of red cells), and plated in 1% methylcellulose for 7 d to measure SFE (panel C) or size (panel D). As the efficiency of transduction was different for the various constructs, the SFE was calculated using only the number of red cells or MS, according to the formula: number of red MS/number of single red cells at plating. For the measurement of MS size (in panel D) only red MS were counted.
MS size was calculated either as volume (μm3) based on the diameter of the MS (Fig. 3 D; and Fig. 4, B and E, right) or it was calculated as the number of cells obtained from the dissociation of MS/number of total MS (all other experiments; Tosoni et al., 2015).
Purification of MaSLCs by the PKH methodology
For the isolation of MaSLCs with the PKH method, we followed the procedure described in Pece et al. (2010) and Tosoni et al. (2015). Briefly, when cultured in nonadherent conditions, the majority of cells in single-cell suspensions obtained from mammary glands do not proliferate and undergo anoikis, whereas MaSLCs gave rise to MS. These MS can be isolated and dissociated, infected with appropriate expression vectors (if required), and stained with PKH dyes, either PKH26 (red) or PKH67 (green). During the growth of subsequent MS generations, the PKH dye dilutes at each cell division, resulting in an MS population composed mostly of dull cells except for rare intensely stained PKHpos cells, which retained the dye since they do not divide (a hallmark of stem-like cells). PKHpos cells can be isolated by FACS sorting. Briefly, single-cell suspensions from dissociated PKH-labeled MS were FACS sorted using a BD FACS Aria Fusion (Becton Dickinson) equipped with a 488 nm laser and a band-pass 585/42 optical filter. A mean sorting rate of 800 events per second at a sorting pressure of 10 PSI nozzle 130 μm was maintained.
Imaging studies
Analysis of Numb partitioning at mitosis (Figs. 2, C–E, 9 B, and S2, B–E)
For these experiments, MaSLCs were identified with the “retrospective methodology,” described in Tosoni et al. (2015). In brief, cells expressing the appropriate fluorochrome-tagged constructs (or subjected to the indicated genetic manipulations or pharmacological treatments) were resuspended (5,000 cells) in 400 μl of methylcellulose in a complete medium and plated onto glass-bottom six-well plates followed by time-lapse video microscopy using the Scan^R screening station (Olympus-SIS) equipped with an Olympus IX 81 inverted microscope with a Hamamatsu Orca R2 Cooled CCD camera, an Olympus MT20E illumination system and an incubation chamber (Evotec/Okolab). Alternatively, a Nikon time-lapse station equipped with a Nikon Eclipse Ti microscope, a Lumencor SpectraX-6 light source, an Andor Zyla 4.2 (Oxford Instruments) sCMOS camera, and an incubation chamber (Okolab s.r.l.) was used. All images were acquired at 37°C and 5% CO2 with a 10X/0.3NA objective lens. Differential interference contrast, GFP, and DsRed-epifluorescence images were collected with autofocusing procedures and different focal planes were recorded to prevent loss of image contrast caused by axial cell movement. Images were captured every hour for at least 4 d starting 8–16 h after plating and reconstructed using ImageJ or Matlab software (MathWorks).
Under the culture conditions utilized, only MaSLCs give raise to MS. Thus, at the end of the image acquisition, each single MaSLC expressing Numb-DsRed, which gave rise to a MS (∼40 μm diameter), was analyzed retrospectively at the first mitosis (two-cell stage level) for Numb partitioning and classified as “symmetric” if the Numb-DsRed epifluorescence was equally partitioned in both daughter cells, and “asymmetric” when the epifluorescence was present predominantly in one daughter cell.
Analysis of Numb distribution in cellular compartments (Figs. 1, B, D, and E, 9 A, and S1 D)
To evaluate the relative amounts of Numb at the PM vs. cytoplasm, the sum of Numb pixel values in a region of interest (ROI; 1 µm thickness) at the level of the PM and the cytoplasm was calculated and the raw integrative density (RID), normalized for the PM and cytoplasmic area, was evaluated at the individual cell level using ImageJ software. Confocal acquisitions were performed as described in the section “Immunofluorescence” with a Leica TCS SP5 and SP8 AOBS microscope system equipped with Leica HyD (high-quantum-efficiency hybrid detector) and PMT detectors. Images were acquired using 40X 1.3 and 63X 1.4 NA oil-immersion objectives under the control of LAS AF or LasX Software (Leica). For the analysis performed in MaSLCs, PKHpos cells were first sorted and purified, as described in “Purification of MaSLCs by the PKH methodology,” from MS previously transduced with the appropriate lentiviral fluorochrome-tagged constructs or KD vectors. PKHpos cells were then plated on poly-lysine coated coverslips, left to adhere for 10 min, fixed with 4% formaldehyde (20 min at RT), followed by IF, and finally subjected to the imaging analysis to evaluate the Numb subcellular localization as described.
Analysis of co-segregation of PKCζ and Par3 with asymmetrically partitioned Numb (Fig. 1 A)
MaSLCs were purified by the PKH method, as described in “Purification of MaSLCs by the PKH methodology,” from MS already transduced with a lentiviral vector encoding Numb-Flag. PKHpos cells were then plated in suspension conditions for ∼36 h to allow for the first mitotic division to occur and then transferred onto poly-lysine coated coverslips, left to adhere for 10 min, and then fixed with 4% formaldehyde (20 min at RT), followed by IF. The segregation of either PKCζ or Par3 with Numb was evaluated by staining with the appropriate Ab on PKHpos cell doublets. To avoid the possibility of analyzing cell aggregates, instead of doublets of clonal origin, only cell doublets displaying a similar intensity of the PKH-dye in both cells of the doublet were considered.
Analysis of IHC slides
Slides were digitally scanned with the Aperio ScanScope XT (Leica Microsystem) and analyzed by a pathologist. For the quantitation of p53, slides were automatically analyzed with the Aperio ImageScope IHC algorithm (Aperio Technologies, Inc.). Digital images were processed with Adobe Photoshop CS3. It has been reported that p53 expression by IHC analysis can be used as a surrogate for mutational analysis (Alsner et al., 2008; Colaluca et al., 2018). A strong IHC staining is regarded as indicative of a missense mutation of the p53 gene, whereas the absence of expression may indicate a nonsense mutation, leading to the formation of a truncated protein (Alsner et al., 2008; Yemelyanova et al., 2011). Patients were classified according to the p53 nuclear staining into three groups: 0–1, 0–1% positive nuclei (indicative of complete loss of p53 protein/nonsense mutations); 2–79, 2–79% positive nuclei (indicative of WT levels of p53); 80+, positive p53 nuclei ≥80% (indicative of missense mutations of p53) as described in Colaluca et al. (2018).
In vivo studies
Numb-KO mice were previously described (Tosoni et al., 2015). Briefly, mice were generated by crossing Numblox/lox mice (Wilson et al., 2007; Zilian et al., 2001) with CK5-Cre mice (Ramirez et al., 2004). Targeted deletion of the Numb allele was confirmed by PCR genotyping, as described previously (Ramirez et al., 2004; Zilian et al., 2001). NOD/SCID-IL-2R gamma chain-null mice (The Jackson Laboratory) were used as a recipient for transplantation experiments (Cicalese et al., 2009; Pece et al., 2010).
To generate PDX models, fresh specimens from Numb-deficient and Numb-proficient human BCs were cut into 4 × 2 mm pieces using a razor blade, removing necrotic tissue, if present. The resulting fragments were embedded in ice-cold Matrigel (BD Matrigel, BD Biosciences) and immediately injected into the fourth inguinal mammary glands of 8-wk-old female NOD/SCID/IL2Rγ−/− (NSG) mice (#5557; Jackson Laboratories stock) anesthetized by i.p. injection of 150 mg/kg tribromoethanol (Avertin). Animals were euthanized when tumors were 1–1.2 cm in the largest diameter. Tumor volume (Fig. 8 G) was determined by measuring the tumor in two dimensions and using the mathematical formula for a prolate ellipsoid, which is tumor volume=(L × W2)/2, where L = length of the longest diameter and W = length of the shortest diameter. Mice were monitored twice weekly by an investigator blinded to the assigned treatment. No animals were excluded from the analysis. The size of the animal cohort was established with the aim to minimize the number of animals used considering that inter-animal variations in the mammary fat pad engraftment are generally low starting from fixed numbers of primary cells under standardized transplantation conditions. For transplantation studies, 50,000 cells from PDXs or Numb-KO tumors, treated in vitro as indicated, were used.
All in vivo experiments were approved by the Italian Ministry of Health and performed in accordance with the Italian laws (D.L.vo 116/92 and following additions), which enforce the EU 86/609 directive, and under the control of the institutional organism for animal welfare and ethical approach to animals in experimental procedures (Cogentech OPBA).
Clinical samples and analyses
Fresh, frozen, or archival FFPE mammary tissue specimens were collected at the European Institute of Oncology (IEO, Milan, Italy) using standard operating procedures approved by the Institutional Ethical Board. Informed consent was obtained for all tissue specimens linked with clinical data. Research on human samples was approved by the IEO Institutional Review Board.
For the analyses reported in Fig. 7 and Tables S3 and S4, we used a large retrospective consecutive cohort of 2453 BC patients operated at the IEO between 1997 and 2000 (the IEO BC 97-00 cohort). Detailed descriptions of the selection criteria and the clinical and pathological characteristics of this cohort are reported elsewhere (Pece et al., 2019). The Numb status was attributed to the tumors and normal mammary glands by measuring the levels of Numb expression by IHC. Normal mammary glands displayed homogenous and intense Numb staining in the luminal layers (IHC score 2-3; see also [Colaluca et al., 2008; Pece et al., 2004]). Tumors were classified on a Numb IHC scale from 0 to 3 (0 = undetectable expression; score ≥2, expression comparable to that of normal luminal mammary cells). Tumors were labeled as Numb-deficient if >70% of cells displayed a score <2 or as Numb-proficient if >60% of the cells displayed a score of ≥2. Tumors were also classified on a pNumb scale (by IHC with anti-Numb-pSer276) from 0 to 3 (0–1 = undetectable expression or expression comparable to that of normal luminal mammary cells; score ≥1.5, high expression). Tumors were labeled as high pNumb if >70% of the cells displayed a score ≥1.5 or as low pNumb if >60% of the cells displayed a score <1.5.
For the experiments involving primary BC cultures (Figs. 8, E–G, 9, E and F, and S3 A (right part), Fig. S4 and Fig. S5 F), the selected BCs were all WT p53, as assessed by IHC ([Dumay et al., 2013] and Fig. S4 B) and classified as high/low-Numb and high/low-pNumb, as described above (see Fig. 8 E).
The analyses of the METABRIC (Curtis et al., 2012; Pereira et al., 2016) and TGCA (PanCancer Atlas, [Hoadley et al., 2018]) BC datasets were performed on the cBioPortal (Cerami et al., 2012; Gao et al., 2013).
Statistical analyses
In all figures, N indicates the number of cells or MS analyzed (as appropriate). All other biological experiments were performed on the same preparations of transduced cells or mouse-derived cells (as appropriate), independently plated at least in triplicate. PCR experiments were performed in biological triplicates with the exception of the control PCR experiments of Fig. 1 D (experimental duplicates), and Fig. S1, C and D (experimental triplicates). Results are reported as mean values, and error bars indicate standard errors, except for experiments in which bars indicate standard deviation (Figs. 2, D and E, 3, C, 4, F and H, 8, A–D, F, and G, 9, B, C, and E, S4 A, and S5 H).
Statistical analyses were performed using the binomial test (Fig. 1 A), the nonparametric Fisher’s exact test (Figs. 2, D and E, 7, B and D, and 9 B), the ANOVA test (Figs. 4 F, 8, A and B, and S4 A), the one-sample t test (Fig. 3 E), or with the Student’s t test two-tailed (all other experiments). Data distribution was assumed to be normal but this was not formally tested. In each panel, P is: *, <0.05; **, <0.01; ***, <0.001; n.s. (not significant), >0.05.
Online supplemental material
Fig. S1 shows additional data and controls to Fig. 1 on the effects of various KDs and OE on Numb phosphorylation and localization. Fig. S2 shows additional data and controls to Fig. 2 on the symmetric partitioning of Numb phosphomutants. Fig. S3 shows additional data to Fig. 3, and in particular, it shows the status of Numb and pNumb in BC cell lines and BCs ex vivo (A) and the fact that Numb phosphorylation does not alter the half-life of Numb (B). Fig. S4 shows additional data to Fig. 8 on BCs ex vivo, in particular the proliferation kinetics in suspension culture of the employed BCs (A), their p53 status (B), and the level of expression of Notch target genes (C). Fig. S4 shows additional data to Fig. 9 on the impact of the overexpression of PKCs and their inhibition, in actual BCs and in BCs ex vivo. Table S1 shows details of Numb transduction/detection in the various experiments of the main text. Table S2 shows details of the measurements of SFE and sphere size in various experiments on MaSLCs. Table S3 shows the clinical and pathological characteristics of the 1675- and the 978-BC cohorts. Table S4 shows the correlation of pNumb with clinical and pathological parameters in the 978-BC cohort.
Data availability
Mice strains, established cell lines, and reagents described or generated in this study are available upon request. The availability of primary cultures (murine or human) and patient specimens (FFPE sections) might be restricted by their limited lifespan (primary cultures) by present legislation on privacy (FFPE sections and human primary cultures).
Acknowledgments
We thank S. Minucci (Istituto Europeo di Oncologia IRCCS, Milan, Italy) and M. Varasi (Istituto FIRC di Oncologia Molecolare, Milan, Italy) for Nutlin-3; M. Coazzoli for technical assistance; the Veterinary Facility, the IEO Biobank, the IEO Imaging Unit, the IEO Flow Cytometry Unit, the Real Time PCR and DNA Sequencing Service of Cogentech (Cogentech Srl, Milan), and the Molecular Pathology of the IEO; R. Gunby for critically editing the manuscript.
This study was funded by the Associazione Italiana per la Ricerca sul Cancro (AIRC—IG 18988, IG 23060, IG 11904, IG 15538, and MultiUnit-5x1000 MCO 10.000); MIUR, the Italian Ministry of University and Scientific Research (Prot. 2015XS92CC, 20177E9EPY_002); Italian Ministry of Health (Ricerca Corrente; 5×1000 funds, RF-2016-02361540, and RF-2013-02358446). M.G. Filippone was supported by a fellowship from the Fondazione Umberto Veronesi.
The authors declare no competing financial interests.
Author contributions: Conceptualization: S. Pece, D. Tosoni, and P.P. Di Fiore; data curation and analysis: M.G. Filippone, S. Freddi, G. Bertalot, and D. Tosoni; funding acquisition: S. Pece and P.P. Di Fiore; investigation: M.G. Filippone, S. Zecchini, S. Restelli, I.N. Colaluca, and G. Bertalot; methodology: M.G. Filippone, D. Tosoni, and I.N. Colaluca; project administration: D. Tosoni and P.P. Di Fiore; supervision: S. Pece, D. Tosoni, and P.P. Di Fiore; writing (original draft): M.G. Filippone, D. Tosoni, and P.P. Di Fiore; writing (review and editing): all authors. D. Tosoni is the custodian of all the original documentations.

