


Mitochondrial form and function are closely interlinked in homeostasis and aging. Inhibiting mitochondrial translation is known to increase lifespan in C. elegans, and is accompanied by a fragmented mitochondrial network. However, whether this link between mitochondrial translation and morphology is causal in longevity remains uncharacterized. Here, we show in C. elegans that disrupting mitochondrial network homeostasis by blocking fission or fusion synergizes with reduced mitochondrial translation to prolong lifespan and stimulate stress response such as the mitochondrial unfolded protein response, UPRMT. Conversely, immobilizing the mitochondrial network through a simultaneous disruption of fission and fusion abrogates the lifespan increase induced by mitochondrial translation inhibition. Furthermore, we find that the synergistic effect of inhibiting both mitochondrial translation and dynamics on lifespan, despite stimulating UPRMT, does not require it. Instead, this lifespan-extending synergy is exclusively dependent on the lysosome biogenesis and autophagy transcription factor HLH-30/TFEB. Altogether, our study reveals the mechanistic crosstalk between mitochondrial translation, mitochondrial dynamics, and lysosomal signaling in regulating longevity.

Introduction
Mitochondria play an essential role in eukaryotes in maintaining cellular homeostasis through the functions of bioenergy production, macromolecules synthesis, and communicating bioenergetic status with the rest of the cell (Andreux et al., 2013; Chandel, 2015; Smith et al., 2018). A landscape of inherited genetic defects that impair mitochondrial function can result in diseases affecting the heart, brain, sensory organs, bone marrow, and muscle, and in most cases result in early mortality (Nunnari and Suomalainen, 2012). In contrast to these devastating diseases, mild mitochondrial perturbations in various animal models including mice, Drosophila melanogaster, and Caenorhabditis elegans have been shown to delay aging and age-related functional declines (Cho et al., 2011; Dell’agnello et al., 2007; Dillin et al., 2002; Houtkooper et al., 2013; Lee et al., 2003; Liu et al., 2005). Therefore, mitochondria function not only as powerhouses in cells, but also as central signaling hubs controlling the rate and quality of aging.
Mitochondrial functions are intimately intertwined with mitochondrial form (Nunnari and Suomalainen, 2012). The architecture of mitochondrial networks is governed by two opposing processes, mitochondrial fission and fusion, which coordinately regulate a flexible and adaptive mitochondrial structure to the ever-changing cellular environment (Ferree and Shirihai, 2012; Labbé et al., 2014). More specifically, fission is essentially required for the perpetual renewal of mitochondria and the segregation of impaired portions of mitochondria for elimination by autophagy (Waterham et al., 2007; Youle and van der Bliek, 2012). In contrast, fusion allows for the maximization of efficient oxidative respiration in response to starvation (Bereiter-Hahn, 2014). Therefore, these fundamental functions of mitochondrial dynamics highlight their physiological importance in aging and aging-associated health declines where mitochondrial damage and dysfunction take place.
Lysosomes are acidic membrane-bound organelles that are essential for degrading and recycling cellular components initiated by endocytosis and autophagy (Perera and Zoncu, 2016). Autophagy is required for many beneficial effects of various conserved longevity paradigms such as dietary restriction, LET-363/target of rapamycin (TOR), and germline removal (Folick et al., 2015; Hansen et al., 2018; Lapierre et al., 2011, 2013; Ramachandran et al., 2019). Importantly, lysosomes are also actively involved in quality control crosstalk with mitochondria that influences each other’s homeostasis (Soto-Heredero et al., 2017). Mitochondria-specific autophagy (or mitophagy) and the mitochondria-endosomal pathway are closely integrated with lysosomes to survey and remove mitochondrial damage (Soubannier et al., 2012; Sugiura et al., 2014; Wang and Klionsky, 2011; Youle and Narendra, 2011). However, the mechanisms that underlie the contribution of mitochondria-lysosome crosstalk to longevity are largely unclear.
We previously reported that reducing mitochondrial translation by RNAi targeting of mitochondrial ribosomal protein S5 (mrps-5) in C. elegans increases lifespan via activation of the mitochondrial unfolded protein response (UPRMT) and concurrently triggers fragmentation of the mitochondrial network (Houtkooper et al., 2013). However, the causality as well as functional and physiological roles of mitochondrial structure in this longevity paradigm remain elusive. Here, we demonstrate that altered mitochondrial dynamics caused by impaired fission or fusion cooperate with reduced mitochondrial translation to prolong lifespan and enhance stress responses such as the UPRMT. In contrast, maintaining mitochondrial network homeostasis through a simultaneous disruption of fission and fusion abrogates the lifespan increase driven by mitochondrial translation inhibition. Furthermore, evidence from proteomics reveals that shifting the mitochondrial dynamics equilibrium when mitochondrial translation is slowed down further compromises reproduction, one of the most energy-consuming processes in worm life. Last, although the UPRMT is additively provoked by the combined stress from mitochondrial fragmentation and mitochondrial translation inhibition, we find that it is not the mediators of UPRMT but the primary lysosome biogenesis and autophagy transcription factor HLH-30/transcription factor EB (TFEB) that is required for the increased lifespan. Collectively, these data indicate that mitochondrial dynamics act as downstream effectors upon mitochondrial translation stress to modulate longevity and that mitochondrial dysfunction relays stress signals to lysosomes, thereby stimulating lysosome biogenesis and consequently benefiting longevity in C. elegans.
Results
Mitochondrial network fragmentation synergizes with mitochondrial translation inhibition to promote C. elegans longevity
The genetic inhibition of mitochondrial translation by knocking down mrps-5 promotes longevity while fragmenting the mitochondrial network (Houtkooper et al., 2013). Since it remained unclear whether this fragmentation causally contributed to longevity, we wanted to investigate the association between mitochondrial dynamics and the longevity conferred by inhibiting mitochondrial translation. To explore this, we knocked down mrps-5 expression alongside one of two key mitochondrial fusion genes, eat-3 or fzo-1, using RNAi. The eat-3 gene encodes a C. elegans protein homologous to mammalian OPA1, which is required for the inner mitochondrial membrane fusion, while fzo-1 is the single C. elegans homologue of human mitofusins (MFN1 and MFN2) essential for the outer mitochondrial membrane fusion (Ichishita et al., 2008; Kanazawa et al., 2008). Depleting either eat-3 or fzo-1 blunts the fusion process, thereby driving mitochondrial dynamics toward division and fragmentation (Ichishita et al., 2008; Kanazawa et al., 2008). Using a transgenic strain that expresses mitochondrial matrix-targeted GFP in body wall muscles (pmyo-3::GFP[mit]; Benedetti et al., 2006), we found that eat-3 RNAi substantially fragmented the mitochondrial network in young (day 2) and aged (day 7) worms in the context of both wild-type and mrps-5 RNAi conditions (Fig. 1 A). Quantification of the mitochondrial network confirmed that individual or combined RNAi knockdown of mrps-5 and eat-3 significantly decreased mitochondrial connectivity (mitochondrial area:perimeter) and mitochondrial content (percentage of cytosol covered by mitochondria) in day 2 and day 7 worms compared with the empty vector (ev)-treated controls (Fig. 1 B). Moreover, the connectivity of mitochondria in mrps-5;eat-3 double RNAi animals was further reduced compared with mrps-5 single RNAi at day 7 of adulthood (Fig. 1, A and B). Similarly, depletion of fzo-1 in conjunction with mrps-5 led to a breakdown of reticular mitochondrial networks that increased in severity from day 2 to day 7 (Fig. 1, A and B).
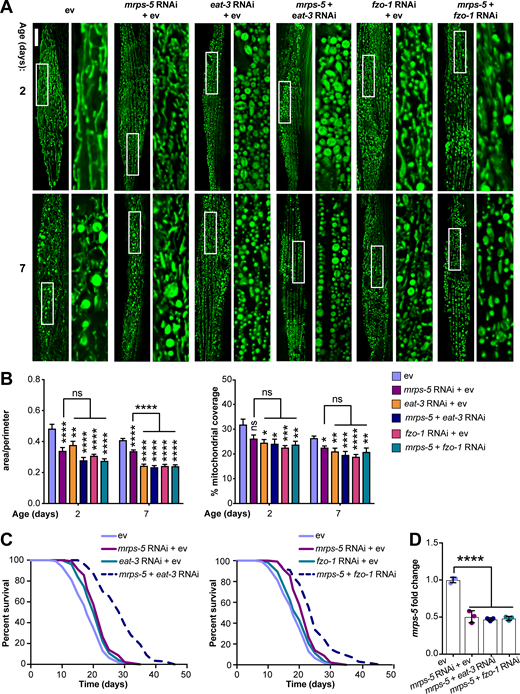
Synergistic longevity is engendered by a simultaneous suppression of mitochondrial translation and fusion.(A) Fluorescence images of mitochondrial networks in worms carrying the pmyo-3::GFP(mit) transgene fed either control bacteria expressing ev or RNAi bacteria expressing dsRNA against mrps-5, eat-3, and fzo-1, individually or in combination at day 2 and day 7 of adulthood. Scale bar in ev-treated condition on day 2 is 10 µm and valid for all the images in A. (B) Quantification of mitochondrial morphologies in A: mean ± SEM of 10–23 different animals, pooled from two independent experiments. Mitochondrial network area:perimeter ratio (left) and mitochondrial content (right, percent [%] of cytosol occupied by mitochondria). An asterisk directly above a bar refers to statistical significance compared with ev-treated controls, while an asterisk over a line or bracket indicates statistical significance compared with the condition of mrps-5 RNAi + ev. (C) Survival curves showing that double RNAi of mrps-5;eat-3 or mrps-5;fzo-1 significantly prolongs lifespan in C. elegans. Comparisons of survival curves were performed by the log-rank test. See Table S1 for lifespan statistics. (D) Transcript levels of mrps-5 in day 1 adult N2 animals. The expression of mrps-5 is comparably reduced among worms treated with mrps-5 RNAi, mrps-5;eat-3 double RNAi, or mrps-5;fzo-1 double RNAi. The expression levels of mrps-5 were normalized to reference genes Y45F10D.4 and F35G12.2 and compared with the mean value of ev-treated controls. Mean ± SD of n = 3 biological replicates. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test.
Synergistic longevity is engendered by a simultaneous suppression of mitochondrial translation and fusion.(A) Fluorescence images of mitochondrial networks in worms carrying the pmyo-3::GFP(mit) transgene fed either control bacteria expressing ev or RNAi bacteria expressing dsRNA against mrps-5, eat-3, and fzo-1, individually or in combination at day 2 and day 7 of adulthood. Scale bar in ev-treated condition on day 2 is 10 µm and valid for all the images in A. (B) Quantification of mitochondrial morphologies in A: mean ± SEM of 10–23 different animals, pooled from two independent experiments. Mitochondrial network area:perimeter ratio (left) and mitochondrial content (right, percent [%] of cytosol occupied by mitochondria). An asterisk directly above a bar refers to statistical significance compared with ev-treated controls, while an asterisk over a line or bracket indicates statistical significance compared with the condition of mrps-5 RNAi + ev. (C) Survival curves showing that double RNAi of mrps-5;eat-3 or mrps-5;fzo-1 significantly prolongs lifespan in C. elegans. Comparisons of survival curves were performed by the log-rank test. See Table S1 for lifespan statistics. (D) Transcript levels of mrps-5 in day 1 adult N2 animals. The expression of mrps-5 is comparably reduced among worms treated with mrps-5 RNAi, mrps-5;eat-3 double RNAi, or mrps-5;fzo-1 double RNAi. The expression levels of mrps-5 were normalized to reference genes Y45F10D.4 and F35G12.2 and compared with the mean value of ev-treated controls. Mean ± SD of n = 3 biological replicates. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test.
Next, we tested the effects of the disrupted mitochondrial network on longevity when mrps-5 was silenced through double RNAi knockdown of mrps-5;eat-3 or mrps-5;fzo-1. Unlike many long-lived mutants such as glp-1(e2141), eat-2(ad1116), and clk-1 RNAi that require eat-3 for their lifespan extension (Chaudhari and Kipreos, 2017), double RNAi of mrps-5;eat-3 substantially extended median lifespan compared with mrps-5 RNAi (Fig. 1 C and Table S1). Likewise, although single RNAi of fzo-1 did not affect worm lifespan (Fig. 1 C and Table S1), double RNAi of mrps-5;fzo-1 led to a more pronounced lifespan extension compared with either single RNAi control (Fig. 1 C and Table S1). Taken together, these results suggest that a simultaneous suppression of mitochondrial fusion and mitochondrial translation substantially increases lifespan in C. elegans.
To exclude the possibility that RNAi knockdown of eat-3 or fzo-1 interferes with mrps-5 expression, or vice versa, we performed quantitative real-time PCR (qPCR) analysis of the transcript level of mrps-5 on single and double RNAi–treated animals. We found that double RNAi by administering double-stranded RNA (dsRNA) in a 1:1 ratio targeting mrps-5;eat-3 or mrps-5;fzo-1 resulted in similar knockdown compared with single mrps-5 RNAi (Fig. 1 D). Similarly, RNAi of eat-3 or fzo-1 together with mrps-5 gave rise to a comparable decrease in the transcript levels of eat-3 and fzo-1 relative to their single RNAi, respectively (Fig. S1, A and B). It is worth noting that we observed a stronger knockdown upon eat-3 RNAi, when compared with fzo-1 RNAi, irrespective of cotargeting mrps-5 (Fig. S1, A and B). Collectively, these results suggest that the synergized extension of lifespan in the double RNAi experiments cannot be attributed to an additional decrease in the expression of mrps-5, eat-3, or fzo-1.

Reducing mitochondrial translation and fusion suppresses animals’ growth and exclusively provokes mitochondrial stress response. (A) Transcript level of eat-3 upon eat-3 RNAi or mrps-5;eat-3 double RNAi was measured by qPCR in day 1 adult N2 animals. RNAi of eat-3 alone or in conjunction with mrps-5 RNAi results in an around 66.3% reduction in the mRNA level. (B) Transcript level of fzo-1 upon fzo-1 RNAi or mrps-5;fzo-1 double RNAi was measured in day 1 adult N2 animals. Around 43.7% decrease in the mRNA level of fzo-1 was observed in worms treated with RNAi against fzo-1 alone or mrps-5; fzo-1 in combination. The expression levels of eat-3 or fzo-1 were normalized to reference genes y45f10d.4 and f35g12.2 and compared with the mean value of ev-treated controls. Mean ± SD of n = 3 biological replicates. ns, not significant; *, P < 0.5; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparison tests. (C) Representative micrographs of worms showing that double RNAi of mrps-5;eat-3 exhibits the strongest suppression on age-dependent growth in body length. Scale bar in ev-treated day 3 condition is 200 µm and valid for all the images in C. The length of worms was normalized to the mean value of ev-treated day 3 controls. Mean ± SD of n = 17–24 images. Significance was calculated using one-way ANOVA with Tukey’s multiple comparison tests; ***, P < 0.001; ****, P < 0.0001; ns, not significant. (D) Fragmenting mitochondrial network through RNAi against eat-3 or fzo-1 triggers the UPRMT, visualized in hsp-6::GFP reporter strain on day 3 and day 7 of adulthood. Scale bar in ev-treated hsp-6::GFP animals represents 200 µm and is valid for all the images in D. (E) Quantification of hsp-6::GFP expression in D. The GFP fluorescence intensity was normalized to the mean value of ev-treated controls. Mean ± SD of n = 17 images. Significance was calculated using one-way ANOVA with Tukey’s multiple comparisons test; ****, P < 0.0001. (F) RNAi of mrps-5, eat-3, and fzo-1, individually or in combination, does not influence ER proteostasis, visualized in hsp-4::GFP reporter strain at day 7 of adulthood. Scale bar in ev-treated hsp-4::GFP animals represents 1 mm and is valid for all the images in F. (G) Measurement of hsp-4::GFP fluorescence intensity in F using a TECAN plate reader. Mean ± SD of n = 5 or 6 replicates. ns, not significant; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (H) Basal and maximum OCR upon the RNAi knockdown of eat-3 and fzo-1. Basal OCR is not influenced by RNAi of eat-3 or fzo-1, whereas maximum OCR is strongly diminished. Mean ± SD of 11–16 replicates; Significance was calculated using one-way ANOVA with Tukey’s multiple comparisons test; ****, P < 0.0001; ns, not significant. (I) Raw averaged traces of oxygen consumption from wild type N2 worms treated with RNAi against eat-3 and fzo-1, respectively. Mean ± SEM (n = 16); FCCP and sodium azide were added at the indicated time.
Reducing mitochondrial translation and fusion suppresses animals’ growth and exclusively provokes mitochondrial stress response. (A) Transcript level of eat-3 upon eat-3 RNAi or mrps-5;eat-3 double RNAi was measured by qPCR in day 1 adult N2 animals. RNAi of eat-3 alone or in conjunction with mrps-5 RNAi results in an around 66.3% reduction in the mRNA level. (B) Transcript level of fzo-1 upon fzo-1 RNAi or mrps-5;fzo-1 double RNAi was measured in day 1 adult N2 animals. Around 43.7% decrease in the mRNA level of fzo-1 was observed in worms treated with RNAi against fzo-1 alone or mrps-5; fzo-1 in combination. The expression levels of eat-3 or fzo-1 were normalized to reference genes y45f10d.4 and f35g12.2 and compared with the mean value of ev-treated controls. Mean ± SD of n = 3 biological replicates. ns, not significant; *, P < 0.5; ***, P < 0.001 by one-way ANOVA with Tukey’s multiple comparison tests. (C) Representative micrographs of worms showing that double RNAi of mrps-5;eat-3 exhibits the strongest suppression on age-dependent growth in body length. Scale bar in ev-treated day 3 condition is 200 µm and valid for all the images in C. The length of worms was normalized to the mean value of ev-treated day 3 controls. Mean ± SD of n = 17–24 images. Significance was calculated using one-way ANOVA with Tukey’s multiple comparison tests; ***, P < 0.001; ****, P < 0.0001; ns, not significant. (D) Fragmenting mitochondrial network through RNAi against eat-3 or fzo-1 triggers the UPRMT, visualized in hsp-6::GFP reporter strain on day 3 and day 7 of adulthood. Scale bar in ev-treated hsp-6::GFP animals represents 200 µm and is valid for all the images in D. (E) Quantification of hsp-6::GFP expression in D. The GFP fluorescence intensity was normalized to the mean value of ev-treated controls. Mean ± SD of n = 17 images. Significance was calculated using one-way ANOVA with Tukey’s multiple comparisons test; ****, P < 0.0001. (F) RNAi of mrps-5, eat-3, and fzo-1, individually or in combination, does not influence ER proteostasis, visualized in hsp-4::GFP reporter strain at day 7 of adulthood. Scale bar in ev-treated hsp-4::GFP animals represents 1 mm and is valid for all the images in F. (G) Measurement of hsp-4::GFP fluorescence intensity in F using a TECAN plate reader. Mean ± SD of n = 5 or 6 replicates. ns, not significant; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (H) Basal and maximum OCR upon the RNAi knockdown of eat-3 and fzo-1. Basal OCR is not influenced by RNAi of eat-3 or fzo-1, whereas maximum OCR is strongly diminished. Mean ± SD of 11–16 replicates; Significance was calculated using one-way ANOVA with Tukey’s multiple comparisons test; ****, P < 0.0001; ns, not significant. (I) Raw averaged traces of oxygen consumption from wild type N2 worms treated with RNAi against eat-3 and fzo-1, respectively. Mean ± SEM (n = 16); FCCP and sodium azide were added at the indicated time.
The lifespan extension of mitochondrial electron transport chain worm mutants is associated with a reduction of their body size (Houtkooper et al., 2013; Rea et al., 2007). We therefore asked if mitochondrial network fragmentation synergized with mitochondrial translation inhibition to increase lifespan by compromising growth capacity. A slight but significant decrease in body length occurred at the young adult stage (day 3) upon mrps-5;eat-3 double RNAi compared with mrps-5 RNAi alone, which was further manifested upon age-related growth as observed at day 7 of adulthood (Fig. S1 C). Co-inactivation of mrps-5;fzo-1 also led to a significant reduction in body length at day 7 of adulthood compared with mrps-5 RNAi alone (Fig. S1 C). Taken together, these data lend support to the previously reported notion that in mitochondrial dysfunction–modulated longevity, there might be a common regulatory circuit to control a shortening of size and a lengthening of lifespan.
Mitochondrial translation inhibition and network fragmentation jointly activate the UPRMT
Having determined the effects of a concurrent inhibition of mitochondrial fusion and mitochondrial translation on lifespan, we then asked whether fragmenting the mitochondrial network influenced longevity by increasing stress responses. Specifically, the activation of the UPRMT has been causally associated with the increased lifespan induced by mitochondrial translation reduction in C. elegans (Houtkooper et al., 2013). Therefore we speculated that double RNAi of mrps-5;eat-3 or mrps-5;fzo-1 might additively contribute to the UPRMT activation to prolong lifespan. To test this hypothesis, we monitored the level of UPRMT for RNAi-treated worms using the GFP reporter strain for hsp-6 (mitochondrial HSP70), a prominent downstream target of the UPRMT (Yoneda et al., 2004). Fragmenting mitochondrial structure by RNAi ablation of either eat-3 or fzo-1 stimulated the UPRMT (Fig. S1, D and E), in line with recent findings (Zhang et al., 2018). Furthermore, mrps-5;eat-3 double RNAi triggered the strongest UPRMT response, consistent with its most prominent lifespan increase (Fig. 1 C; and Fig. 2, A and B). Double RNAi of mrps-5;fzo-1 also resulted in an additive stimulation of the UPRMT, though to a lesser extent when compared with mrps-5;eat-3 double RNAi (Fig. 2, A and B). Furthermore, while the activation pattern of the UPRMT in aged animals (day 7) followed the same trend in all these conditions, the level of activation was reduced upon aging (Fig. 2, A and B). Taken together, these data show that the activation of UPRMT correlates well with longevity, implying its important role in the life-extending synergy between fragmented mitochondrial morphology and reduced mitochondrial translation.

Simultaneous suppression of mitochondrial translation and fusion amplifies mitochondrial stress. (A) Double RNAi of mrps-5;eat-3 or mrps-5;fzo-1 synergistically activates the UPRMT, visualized by hsp-6::GFP reporter strain at day 3 and day 7 of adulthood. Scale bar in ev-treated condition on day 3 represents 200 µm and is valid for all the images in A. (B) Quantification of GFP fluorescence intensity in A, expressed as fold change relative to wild type N2 fed on ev. Mean ± SD of n = 17 images. (C) Mitochondrial OCR after mrps-5 RNAi, mrps-5;eat-3 double RNAi, or mrps-5;fzo-1 double RNAi. The strongest reduction of basal OCR occurs in N2 worms treated with mrps-5;eat-3 double RNAi, while maximum OCR is reduced to an equal extent by three types of RNAi treatments. The OCR was measured in day 7 adult worms. Mean ± SD of n = 11–16 biological replicates. (D) Raw averaged traces of oxygen consumption from day 7 adult N2 worms treated with RNAi against mrps-5 alone or RNAi against mrps-5;eat-3 or mrps-5;fzo-1. Mean ± SEM (n = 16); FCCP, an uncoupler reagent, was added at the indicated time to achieve the maximum mitochondrial respiration, while sodium azide, a complex IV inhibitor, was added to fully block mitochondrial respiration, thereby measuring nonmitochondrial oxygen consumption. ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test.
Simultaneous suppression of mitochondrial translation and fusion amplifies mitochondrial stress. (A) Double RNAi of mrps-5;eat-3 or mrps-5;fzo-1 synergistically activates the UPRMT, visualized by hsp-6::GFP reporter strain at day 3 and day 7 of adulthood. Scale bar in ev-treated condition on day 3 represents 200 µm and is valid for all the images in A. (B) Quantification of GFP fluorescence intensity in A, expressed as fold change relative to wild type N2 fed on ev. Mean ± SD of n = 17 images. (C) Mitochondrial OCR after mrps-5 RNAi, mrps-5;eat-3 double RNAi, or mrps-5;fzo-1 double RNAi. The strongest reduction of basal OCR occurs in N2 worms treated with mrps-5;eat-3 double RNAi, while maximum OCR is reduced to an equal extent by three types of RNAi treatments. The OCR was measured in day 7 adult worms. Mean ± SD of n = 11–16 biological replicates. (D) Raw averaged traces of oxygen consumption from day 7 adult N2 worms treated with RNAi against mrps-5 alone or RNAi against mrps-5;eat-3 or mrps-5;fzo-1. Mean ± SEM (n = 16); FCCP, an uncoupler reagent, was added at the indicated time to achieve the maximum mitochondrial respiration, while sodium azide, a complex IV inhibitor, was added to fully block mitochondrial respiration, thereby measuring nonmitochondrial oxygen consumption. ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test.
The mitochondrial network is physically and functionally connected with the ER via mitochondrial-associated membranes to regulate lipid exchange, calcium homeostasis, mitochondrial fission, and autophagosome formation (Murley and Nunnari, 2016). Therefore, to examine the potential influences of the disrupted mitochondrial network on ER homeostasis, we analyzed the ER unfolded protein response (UPRER). However, neither the combined RNAi of mrps-5;eat-3 and mrps-5;fzo-1 nor the single RNAi of mrps-5, eat-3, or fzo-1 activated the UPRER (Fig. S1, F and G), examined by a GFP reporter for the ER chaperone immunoglobulin heavy chain–binding protein (hsp-4; Calfon et al., 2002). Combined, these results suggest that fragmenting mitochondria specifically interferes with mitochondrial protein homeostasis.
Translation of oxidative phosphorylation complexes involves the coordination of cytosolic and mitochondrial translation apparatus (Couvillion et al., 2016; Houtkooper et al., 2013). Therefore, inhibiting mitochondrial translation through mrps-5 RNAi leads to a substantial decline in mitochondrial respiration (Houtkooper et al., 2013). To assess whether fragmenting the mitochondrial network further impairs mitochondrial respiration, we measured oxygen consumption in worms treated with RNAi against mrps-5 and eat-3 (or mrps-5 and fzo-1), individually or in combination. Consistent with previous findings (Houtkooper et al., 2013), mrps-5 RNAi profoundly decreased the basal respiration and disabled the increase normally observed upon the treatment of mitochondrial uncoupler carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP; Fig. 2, C and D). In addition, RNAi against eat-3 or fzo-1 alone strongly attenuated the maximum respiratory capacity without significantly affecting the basal respiration (Fig. S1, H and I). An additive reduction of basal respiration was observed in mrps-5;eat-3 double RNAi-treated animals compared with those in mrps-5 RNAi-treated group (Fig. 2, C and D), whereas this phenotype was absent upon double RNAi of mrps-5;fzo-1 (Fig. 2, C and D). In addition, no further decrease was observed in the maximum respiration upon double RNAi of mrps-5;eat-3 or mrps-5;fzo-1 compared with that upon mrps-5 RNAi alone, suggesting a nadir of maximum respiration after mrps-5 RNAi (Fig. 2, C and D). Taken together, these data demonstrate that double RNAi of mrps-5;eat-3 exhibits not only the most potent prolonging effect on lifespan, but also the strongest suppression on mitochondrial respiration, thereby intensifying the stresses induced by mrps-5 RNAi.
Simultaneously abrogating mitochondrial fission and fusion prevents mitochondrial translation inhibition-mediated lifespan extension
Given our results that a simultaneous ablation of mitochondrial fusion and mitochondrial translation has a strong synergistic effect on longevity in C. elegans, we next investigated the effects on lifespan when we blocked the opposite process, mitochondrial fission, in conjunction with reduced mitochondrial translation. Mutation of drp-1 in C. elegans triggers strong fission defects, resulting in a hyper-fused mitochondrial network (Labrousse et al., 1999; Weir et al., 2017). Similar to knockdown of fusion genes, mutation of drp-1 amplified the effects of mrps-5 RNAi on longevity (Fig. 3 A and Table S1). A direct role for mitochondrial dynamics in the longevity in C. elegans has been previously suggested (Weir et al., 2017). Simultaneous depletion of drp-1 and fzo-1 promotes longevity as it induces a homeostatic equilibrium between fission and fusion that is resistant to aging-driven mitochondrial fragmentation (Weir et al., 2017). To aid in understanding the synergy and crosstalk between the mitochondrial network and mitochondrial translation in lifespan regulation, we further tested the influences of such an immobilized mitochondrial network on mrps-5 RNAi-induced longevity using the drp-1;fzo-1 double mutant (Weir et al., 2017). Double mutation of drp-1;fzo-1 abrogated the mrps-5 RNAi induction of longer lifespan without affecting its inhibitory role in growth (Fig. 3, B and C; Fig. S2, A and B; and Table S1). In contrast, mrps-5 RNAi in the fzo-1(tm1133) single mutant still robustly extended the lifespan (Fig. S2 C and Table S1). This resembles our previous lifespan data obtained in N2 worms treated with mrps-5;fzo-1 RNAi (Fig. 1 C and Table S1).
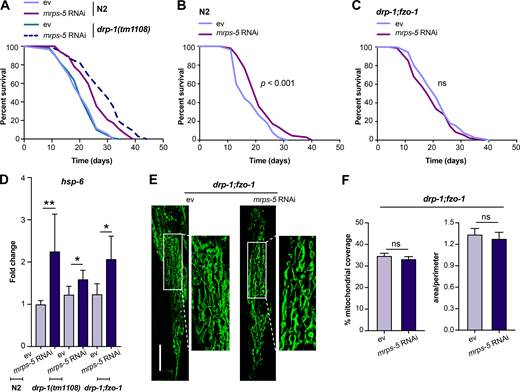
An immobilized mitochondrial network abrogates mrps-5 RNAi-mediated lifespan extension.(A) Survival curves performed in N2 and drp-1(tm1108) demonstrating that mutation of drp-1 significantly enhances mrps-5 RNAi-mediated longevity. See Table S1 for lifespan statistics. (B) Lifespan analysis of N2 revealing the pro-longevity effects of mrps-5 RNAi. Survival curves were compared using the log-rank test. See Table S1 for lifespan statistics. (C) Lifespan analysis of drp-1;fzo-1 double mutant showing that combined mutation of fzo-1 and drp-1 abrogates mrps-5 RNAi-mediated lifespan extension. Survival curves were compared using the log-rank test. See Table S1 for lifespan statistics. (D) The transcript level of hsp-6 in day 1 adult N2, drp-1(tm1108), and drp-1;fzo-1 mutant upon mrps-5 RNAi. The expression levels of hsp-6 were normalized to reference genes ama-1, cdc-42, and F35G12.2, and compared with the mean value of ev-treated N2 worms. The results were derived from two independent experiments. Differences were determined by Student’s t test; *, P < 0.5; **, P < 0.01. (E) Mitochondrial networks in muscle cells of drp-1;fzo-1 double mutant upon mrps-5 RNAi. Animals were examined at day 2 of adulthood. Scale bar in ev-treated condition is 10 µm and valid for all the images in E. (F) Quantification of mitochondrial morphologies in E: mean ± SEM of 33–42 different animals, pooled from two independent experiments. ns, not significant by Student’s t test.
An immobilized mitochondrial network abrogates mrps-5 RNAi-mediated lifespan extension.(A) Survival curves performed in N2 and drp-1(tm1108) demonstrating that mutation of drp-1 significantly enhances mrps-5 RNAi-mediated longevity. See Table S1 for lifespan statistics. (B) Lifespan analysis of N2 revealing the pro-longevity effects of mrps-5 RNAi. Survival curves were compared using the log-rank test. See Table S1 for lifespan statistics. (C) Lifespan analysis of drp-1;fzo-1 double mutant showing that combined mutation of fzo-1 and drp-1 abrogates mrps-5 RNAi-mediated lifespan extension. Survival curves were compared using the log-rank test. See Table S1 for lifespan statistics. (D) The transcript level of hsp-6 in day 1 adult N2, drp-1(tm1108), and drp-1;fzo-1 mutant upon mrps-5 RNAi. The expression levels of hsp-6 were normalized to reference genes ama-1, cdc-42, and F35G12.2, and compared with the mean value of ev-treated N2 worms. The results were derived from two independent experiments. Differences were determined by Student’s t test; *, P < 0.5; **, P < 0.01. (E) Mitochondrial networks in muscle cells of drp-1;fzo-1 double mutant upon mrps-5 RNAi. Animals were examined at day 2 of adulthood. Scale bar in ev-treated condition is 10 µm and valid for all the images in E. (F) Quantification of mitochondrial morphologies in E: mean ± SEM of 33–42 different animals, pooled from two independent experiments. ns, not significant by Student’s t test.
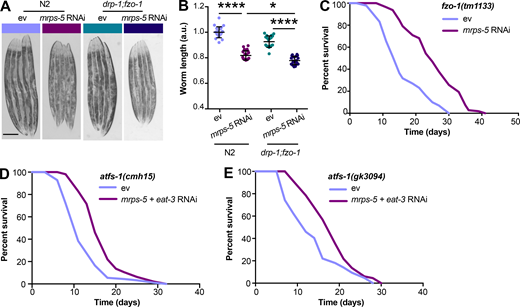
Double mutation of fzo-1;drp-1 does not suppress mrps-5 RNAi-mediated inhibitory effects on growth, and deletion of atfs-1 does not prevent mrps-5;eat-3 double RNAi from prolonging lifespan.(A) Representative light microscopy images of N2 and drp-1;fzo-1 worms at day 3 of adulthood. Scale bar in ev-treated N2 animals is 200 µm and valid for all the images in A. (B) Body length quantification of N2 and drp-1;fzo-1 mutant treated with RNAi against mrps-5. The length of worms was quantified and normalized to the mean value of ev-treated N2 worms. Mean ± SD of n = 20–22 images. *, P < 0.5; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (C) Lifespan analyses of fzo-1(tm1133) showing that mrps-5 RNAi promotes lifespan. See Table S1 for lifespan statistics. (D) Lifespan analyses of atfs-1(cmh15) showing that double RNAi of mrps-5;eat-3 extends lifespan upon loss of afts-1. See Table S1 for lifespan statistics. (E) Lifespan analyses of atfs-1(gk3094) confirm the findings obtained in D on mrps-5;eat-3 double RNAi-induced lifespan increase upon loss of atfs-1. See Table S1 for lifespan statistics.
Double mutation of fzo-1;drp-1 does not suppress mrps-5 RNAi-mediated inhibitory effects on growth, and deletion of atfs-1 does not prevent mrps-5;eat-3 double RNAi from prolonging lifespan.(A) Representative light microscopy images of N2 and drp-1;fzo-1 worms at day 3 of adulthood. Scale bar in ev-treated N2 animals is 200 µm and valid for all the images in A. (B) Body length quantification of N2 and drp-1;fzo-1 mutant treated with RNAi against mrps-5. The length of worms was quantified and normalized to the mean value of ev-treated N2 worms. Mean ± SD of n = 20–22 images. *, P < 0.5; ****, P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (C) Lifespan analyses of fzo-1(tm1133) showing that mrps-5 RNAi promotes lifespan. See Table S1 for lifespan statistics. (D) Lifespan analyses of atfs-1(cmh15) showing that double RNAi of mrps-5;eat-3 extends lifespan upon loss of afts-1. See Table S1 for lifespan statistics. (E) Lifespan analyses of atfs-1(gk3094) confirm the findings obtained in D on mrps-5;eat-3 double RNAi-induced lifespan increase upon loss of atfs-1. See Table S1 for lifespan statistics.
We next investigated the activation of the UPRMT in the drp-1:fzo-1 double mutant upon depletion of mrps-5. We quantified the transcript level of hsp-6 and first determined that the knockdown of mrps-5 in N2 worms significantly increased its expression (Fig. 3 D), consistent with our previous observations using a hsp-6::GFP reporter strain (Fig. 2, A and B). We next measured a rise in hsp-6 expression occurring in both the drp-1(tm1108) and the drp-1;fzo-1 mutants upon mrps-5 RNAi compared with their corresponding ev-treated controls (Fig. 3 D). Together, these results suggest that the UPRMT is likely a universal stress response upon mitochondrial translation inhibition regardless of the mitochondrial network environment.
Finally, we examined the impact of mrps-5 RNAi on the mitochondrial network of the drp-1;fzo-1 mutant. No detectable change in mitochondrial connectivity or mitochondrial content occurred upon mrps-5 RNAi in the drp-1;fzo-1 mutant (Fig. 3, E and F). These results suggest that the double mutation of drp-1 and fzo-1 leaves the mitochondrial network resistant to mrps-5 RNAi-mediated fragmentation, likely explaining the absence of any observed lifespan increase after mrps-5 is depleted from the drp-1;fzo-1 double mutant.
Lifespan extension upon simultaneous inhibition of mitochondrial translation and fusion is independent of the UPRMT
Given that disrupting the mitochondrial network through eat-3 RNAi synergizes with mrps-5 RNAi to extend lifespan and activate the UPRMT (Fig. 1 C; and Fig. 2, A and B), we sought to determine the requirement of the UPRMT for the lifespan phenotype. HAF-1, a mitochondrial peptide transporter, is a key mediator of the UPRMT and is required for mrps-5 RNAi-induced lifespan increase, UPRMT activation, and respiration loss (Haynes et al., 2010; Houtkooper et al., 2013). We therefore tested the requirement of HAF-1 in the prolonged lifespan conferred by mrps-5;eat-3 double RNAi. As expected, mrps-5 RNAi failed to extend lifespan in the haf-1(ok705) mutant (Fig. 4 A and Table S1). Surprisingly, mutation of haf-1 did not abrogate the lifespan increase in mrps-5;eat-3 double RNAi-treated animals (Fig. 4 A and Table S1). To confirm this observation, we checked the requirement of another key player of the UPRMT, the transcription factor ATFS-1 (Nargund et al., 2012). Following mitochondrial dysfunction, import of ATFS-1 into the mitochondria is impaired, allowing it instead to translocate to the nucleus and activate a number of stress response genes (Nargund et al., 2012). We observed that RNAi of mrps-5 robustly prolonged the lifespan in the eat-3(tm1107) mutant akin to the lifespan extension evoked by double RNAi of mrps-5;eat-3 in wild-type N2 (Fig. 1 C, Fig. 4 B, and Table S1). Although atfs-1 RNAi partly reversed the mrps-5 RNAi-mediated lifespan increase in eat-3(tm1107) (Fig. 4 B and Table S1), it also resulted in a comparable lifespan reduction in eat-3(tm1107) treated with ev (Fig. 4 B and Table S1). Therefore, these data suggest that afts-1 is nonspecifically required for the lifespan of eat-3(tm1107), independent of mrps-5 RNAi.
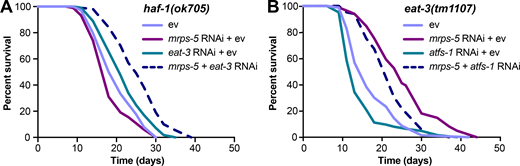
mrps-5;eat-3 RNAi-mediated lifespan extension is independent of the UPRMT.(A) Lifespan analysis performed in haf-1(ok705) showing that deletion of haf-1 does not block mrps-5;eat-3 double RNAi-mediated lifespan extension. See Table S1 for lifespan statistics. (B) Lifespan analysis of eat-3(tm1107) showing that atfs-1 RNAi fails to block mrps-5 RNAi-mediated increase in longevity. See Table S1 for lifespan statistics.
mrps-5;eat-3 RNAi-mediated lifespan extension is independent of the UPRMT.(A) Lifespan analysis performed in haf-1(ok705) showing that deletion of haf-1 does not block mrps-5;eat-3 double RNAi-mediated lifespan extension. See Table S1 for lifespan statistics. (B) Lifespan analysis of eat-3(tm1107) showing that atfs-1 RNAi fails to block mrps-5 RNAi-mediated increase in longevity. See Table S1 for lifespan statistics.
We then tested the effects of mrps-5;eat-3 double RNAi on longevity upon a complete loss of atfs-1. Two previously described atfs-1 deletion mutants, atfs-1(cmh15) and atfs-1(gk3094) (Deng et al., 2019), were used for these experiments. Consistent with our prior findings (Fig. 4, A and B; and Table S1), significant lifespan extension occurred in both atfs-1(cmh15) and atfs-1(gk3094) after mrps-5;eat-3 double RNAi (Fig. S2, D and E; and Table S1). Taken together, these results suggest that the UPRMT is not required for the observed lifespan increase mediated by double RNAi of mrps-5;eat-3 and that simultaneous disruption of mitochondrial translation and fusion employs signaling pathways beyond the UPRMT to delay aging.
Proteomics demonstrates involvement of reduced reproductive capacity, though not causal for lifespan extension
To resolve in greater detail how the mitochondrial network coordinates signals derived from mitochondrial translation inhibition to regulate longevity, we performed proteomics on worms treated with RNAi against mrps-5 or double RNAi against mrps-5;eat-3, mrps-5;fzo-1, and mrps-5;drp-1. Partial least squares-discriminant analysis (PLS-DA) showed a clear separation between mrps-5 RNAi- and ev-treated worms (Fig. 5 A). In addition, gene ontology (GO) term enrichment analyses using the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resource (Huang et al., 2009) revealed that the down-regulated proteins were overrepresented in cellular processes including “respiratory electron transport chain,” “positive regulation of growth rate,” “larval development,” “embryonic development,” and “ribosome biogenesis” (Fig. 5 B). Among them, the most down-regulated GO term of “respiratory electron transport chain” likely explains the repressed oxygen consumption rates (OCRs) that were observed in mrps-5 RNAi-treated animals (Fig. 2 D and Fig. 5 B). In addition, confirming the remodeled mitochondrial network upon mrps-5 RNAi (Fig. 1 A), “mitochondrion organization,” was found among the significantly up-regulated GO terms (Fig. 5 B).
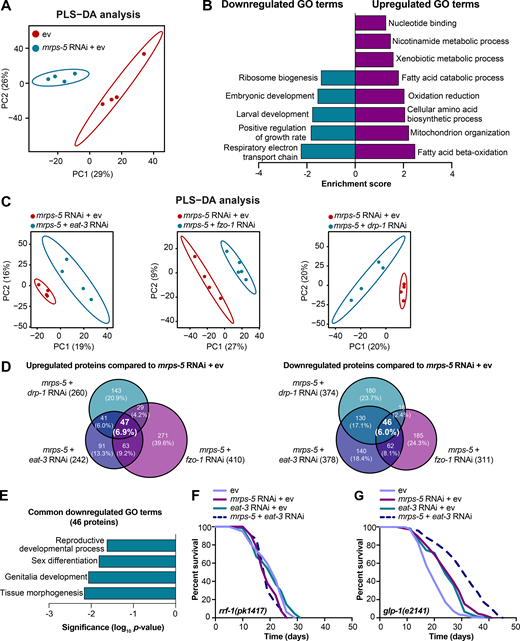
Proteomic analysis reveals reduced reproductive capacity as a consequence rather than a cause of lifespan extension.(A) PLS-DA showing group separations based on differentially expressed proteins in worms treated with mrps-5 RNAi compared with ev control. (B) GO term enrichment analyses (biological processes) of the significantly up- and down-regulated proteins performed using DAVID Bioinformatics Database with an EASE score <0.05. (C) PLS-DA score plot showing a clear group separation between double RNAi-treated groups (mrps-5;eat-3, mrps-5;fzo-1, and mrps-5;drp-1) and mrps-5 RNAi-treated group. (D) Venn diagram depicting overlap of differentially expressed proteins in groups treated with double RNAi against mrps-5;eat-3, mrps-5;fzo-1, and mrps-5;drp-1 compared with mrps-5 RNAi + ev. These differentially expressed proteins were determined by a VIP score >1. In total, 242, 410, and 260 proteins are up-regulated and 378, 311, and 374 proteins are down-regulated when comparing mrps-5;eat-3 RNAi, mrps-5;fzo-1 RNAi, and mrps-5;drp-1 RNAi to mrps-5 RNAi + ev, respectively. 47 up- and 46 down-regulated proteins are shared by three double RNAi-treated groups. (E) GO term enrichment analyses of 46 significantly down-regulated proteins performed using DAVID Bioinformatics Database with an EASE score <0.05. Reproduction-related GO terms were found to be significantly enriched. (F) Survival curves of rrf-1(pk1417) subjected to RNAi against mrps-5 and eat-3, individually or in combination, to achieve germline-specific RNAi. Germline-specific knockdown of mrps-5 does not benefit longevity, and neither does germline-specific double RNAi of mrps-5;eat-3. See Table S1 for lifespan statistics. (G) Survival curves of glp-1(e2141) treated with RNAi against mrps-5 and eat-3, individually or in combination. Double RNAi of mrps-5;eat-3 synergistically prolongs the lifespan of glp-1(e2141) mutant worms. See Table S1 for lifespan statistics.
Proteomic analysis reveals reduced reproductive capacity as a consequence rather than a cause of lifespan extension.(A) PLS-DA showing group separations based on differentially expressed proteins in worms treated with mrps-5 RNAi compared with ev control. (B) GO term enrichment analyses (biological processes) of the significantly up- and down-regulated proteins performed using DAVID Bioinformatics Database with an EASE score <0.05. (C) PLS-DA score plot showing a clear group separation between double RNAi-treated groups (mrps-5;eat-3, mrps-5;fzo-1, and mrps-5;drp-1) and mrps-5 RNAi-treated group. (D) Venn diagram depicting overlap of differentially expressed proteins in groups treated with double RNAi against mrps-5;eat-3, mrps-5;fzo-1, and mrps-5;drp-1 compared with mrps-5 RNAi + ev. These differentially expressed proteins were determined by a VIP score >1. In total, 242, 410, and 260 proteins are up-regulated and 378, 311, and 374 proteins are down-regulated when comparing mrps-5;eat-3 RNAi, mrps-5;fzo-1 RNAi, and mrps-5;drp-1 RNAi to mrps-5 RNAi + ev, respectively. 47 up- and 46 down-regulated proteins are shared by three double RNAi-treated groups. (E) GO term enrichment analyses of 46 significantly down-regulated proteins performed using DAVID Bioinformatics Database with an EASE score <0.05. Reproduction-related GO terms were found to be significantly enriched. (F) Survival curves of rrf-1(pk1417) subjected to RNAi against mrps-5 and eat-3, individually or in combination, to achieve germline-specific RNAi. Germline-specific knockdown of mrps-5 does not benefit longevity, and neither does germline-specific double RNAi of mrps-5;eat-3. See Table S1 for lifespan statistics. (G) Survival curves of glp-1(e2141) treated with RNAi against mrps-5 and eat-3, individually or in combination. Double RNAi of mrps-5;eat-3 synergistically prolongs the lifespan of glp-1(e2141) mutant worms. See Table S1 for lifespan statistics.
To probe how remodeling mitochondrial structure affects mrps-5 RNAi-mediated changes in cellular processes, we first compared double RNAi-treated worms (mrps-5;eat-3, mrps-5;fzo-1, and mrps-5;drp-1) and mrps-5 RNAi-treated worms to ev-treated control worms. PLS-DA analysis showed a strong similarity between mrps-5 RNAi-treated and all double RNAi-treated groups (Fig. S3 A). As such, a strong overlap of the differentially regulated proteins between mrps-5 RNAi-treated and double RNAi-treated animals was observed when comparing them to the control group (Fig. S3 B). The GO terms that were enriched in the 174 commonly up-regulated and 103 commonly down-regulated proteins recapitulated those that were influenced by mrps-5 RNAi alone including the down-regulated GO terms “respiratory electron transport chain” and “embryonic development,” and the up-regulated GO term “mitochondrial organization” (Fig. 5 B and Fig. S3 C). These data suggest that these biological processes constitute the core cellular responses to the genetic interventions in mitochondrial translation, which at least in part explain some of the physiological manifestations in worms, such as the deficiency in growth and mitochondrial respiration.

Similar biological processes are altered among worms with impaired mitochondrial translation and dynamics. (A) PLS-DA showing a great similarity among groups of mrps-5 RNAi, mrps-5;eat-3 RNAi, mrps-5;fzo-1 RNAi, and mrps-5;drp-1 RNAi when compared with ev-treated worms on day 1 of adulthood. (B) Venn diagram showing overlap of differentially expressed proteins between animal groups treated with mrps-5 RNAi, mrps-5;eat-3 RNAi, mrps-5;fzo-1 RNAi, and mrps-5;drp-1 RNAi when compared with ev. These differentially expressed proteins were determined by a VIP score >1. In total, 382, 414, 427, and 352 proteins are up-regulated and 285, 257, 220, and 230 proteins are down-regulated when comparing mrps-5 RNAi, mrps-5;drp-1 RNAi, mrps-5;eat-3 RNAi, and mrps-5;fzo-1 RNAi to the ev-treated group, respectively. 174 up- and 103 down-regulated proteins are shared by mrps-5 RNAi and double RNAi-treated groups compared with ev-treated controls. (C) GO term enrichment analyses of the 103 down- and 174 up-regulated proteins performed using DAVID Bioinformatics Database with an EASE score <0.05. (D) Double RNAi of mrps-5;eat-3 leads to infertility. Shown are light microscopy shots of four populations of worms having reached day 1 of adulthood. mrps-5;eat-3 RNAi leads to infertility in worms. Scale bar in ev-treated condition is 1 mm and applied to all the images in D.
Similar biological processes are altered among worms with impaired mitochondrial translation and dynamics. (A) PLS-DA showing a great similarity among groups of mrps-5 RNAi, mrps-5;eat-3 RNAi, mrps-5;fzo-1 RNAi, and mrps-5;drp-1 RNAi when compared with ev-treated worms on day 1 of adulthood. (B) Venn diagram showing overlap of differentially expressed proteins between animal groups treated with mrps-5 RNAi, mrps-5;eat-3 RNAi, mrps-5;fzo-1 RNAi, and mrps-5;drp-1 RNAi when compared with ev. These differentially expressed proteins were determined by a VIP score >1. In total, 382, 414, 427, and 352 proteins are up-regulated and 285, 257, 220, and 230 proteins are down-regulated when comparing mrps-5 RNAi, mrps-5;drp-1 RNAi, mrps-5;eat-3 RNAi, and mrps-5;fzo-1 RNAi to the ev-treated group, respectively. 174 up- and 103 down-regulated proteins are shared by mrps-5 RNAi and double RNAi-treated groups compared with ev-treated controls. (C) GO term enrichment analyses of the 103 down- and 174 up-regulated proteins performed using DAVID Bioinformatics Database with an EASE score <0.05. (D) Double RNAi of mrps-5;eat-3 leads to infertility. Shown are light microscopy shots of four populations of worms having reached day 1 of adulthood. mrps-5;eat-3 RNAi leads to infertility in worms. Scale bar in ev-treated condition is 1 mm and applied to all the images in D.
To distinguish the cellular processes that contribute to the synergistic effects between the mitochondrial network and mitochondrial translation on longevity, we further compared the proteome datasets of double RNAi-treated worms to that of mrps-5 RNAi-treated worms. Even though abrogating fission and fusion does not intrinsically shift the GO term features present in mrps-5 RNAi-treated worms (Fig. S3, A-C), it triggered additional modifications in protein profiles that unambiguously discriminate mrps-5;eat-3, mrps-5;fzo-1, and mrps-5;drp-1 from mrps-5 RNAi-treated animals, analyzed by PLS-DA analyses (Fig. 5 C). To pinpoint the shared mechanisms underlying the synergized lifespan observed repeatedly in three types of double RNAi conditions, we focused on the differentially regulated proteins concurrently present in three types of double RNAi-treated animals compared with mrps-5 RNAi. There are 47 up- and 46 down-regulated proteins that meet this criterion (Fig. 5 D). GO term enrichment analyses detected reproduction-associated GO terms including “reproductive development,” “sex differentiation,” and “genitalia development” significantly overrepresented for the 46 commonly down-regulated proteins (Fig. 5 E), while no significant GO terms were found in the 47 up-regulated proteins. To confirm this result, we examined the reproduction capacity of the most long-lived double RNAi worms (mrps-5;eat-3). Indeed, mrps-5;eat-3 double RNAi gave rise to a complete loss of fecundity of N2 worms (Fig. S3 D).
The reproductive system is known to intertwine with longevity in C. elegans, particularly the germline, which is involved in canonical longevity signaling pathways such as the mechanistic TOR (mTOR) and DAF-2/insulin-like signaling pathways (Maklakov and Immler, 2016). We therefore asked whether inhibiting mitochondrial translation and fusion extended lifespan through germline-derived signals and fecundity-longevity trade-off mechanisms. We first performed lifespan in the rrf-1(pk1417) mutant where RNAi of any introduced double-stranded RNA is restricted to the germline (Sijen et al., 2001). Upon RNAi against mrps-5 and eat-3, we did not observe any lifespan increase of rrf-1(pk1417) (Fig. 5 F and Table S1). This suggests that germline-specific RNAi of those genes is not sufficient to reproduce the lifespan extension that occurred upon the RNAi knockdown at the whole organism level. We also examined the effects of mrps-5;eat-3 double RNAi on the lifespan of the germline-deficient glp-1(e2141) mutant, which is sterile when raised at the restrictive temperature (Priess et al., 1987). We found that double RNAi of mrps-5;eat-3 synergistically prolonged the lifespan of glp-1(e2141) (Fig. 5 G and Table S1), which resembles the longevity of N2 upon RNAi of these genes (Fig. 1 C). Taken together, these results suggest that although reproduction-related GO terms are overrepresented as being further down-regulated when the stoichiometry of mitochondrial dynamics is altered together with mrps-5 depletion, the germline is likely not the functional tissue producing longevity signals, and the loss of fecundity is not causal for the lifespan extension.
HLH-30/TFEB regulates mitochondrial translation inhibition-mediated longevity in mitochondrial fission or fusion mutants
To elucidate the mechanism underlying the longevity induced by the combined stress imposed by mitochondrial network fragmentation and mitochondrial translation reduction, we examined the involvement of pathways beyond the UPRMT. Nine mediators of diverse cellular signaling pathways are reported linked to the lifespan extension induced by mitochondrial dysfunction. These include the genes hif-1, skn-1, cep-1, pink-1, dct-1, taf-4, atf-5, daf-16, and hlh-30 (Dillin et al., 2002; Khan et al., 2013; Lapierre et al., 2013; Lee et al., 2010; Munkácsy and Rea, 2014; Palikaras et al., 2015; Quirós et al., 2017; Schiavi et al., 2013; Ventura et al., 2009). We performed knockdown experiments for each of these genes (Fig. S4 A) and determined the effect on lifespan in combination with mrps-5 RNAi in the eat-3(tm1107) mutant (Fig. 6 A and Fig. S4, B–I; and Table S1). RNAi against seven of these nine genes (hif-1, skn-1, atf-5, cep-1, pink-1, dct-1, and taf-4) did not shorten the mrps-5 RNAi-induced lifespan in eat-3(tm1107) (Fig. S4, B–H; and Table S1). In contrast, RNAi against hlh-30 or daf-16 reduced the lifespan under these conditions, albeit to different extents (Fig. 6 A, Fig. S4 I, and Table S1). The conserved forkhead transcription factor DAF-16 is a central player in the coordination of many stress responses such as detoxification, antimicrobial response, and protein folding stress, and the helix-loop-helix transcription factor HLH-30 is required for long lifespan in multiple longevity paradigms such as germline removal, inhibition of insulin/insulin-like growth factor signaling, and impaired mitochondrial respiration (Lapierre et al., 2013; Murphy et al., 2003; O’Rourke and Ruvkun, 2013). We observed that hlh-30 RNAi specifically abolished mrps-5 RNAi-induced lifespan increase in eat-3(tm1107) without affecting its basal lifespan, whereas daf-16 RNAi shortened the lifespan of eat-3(tm1107), irrespective of mrps-5 RNAi (Fig. 6 A, Fig. S4 I, and Table S1). Combined, these results suggest that while daf-16 is generally required for sustaining a normal lifespan in eat-3(tm1107), hlh-30 is dispensable under normal conditions but becomes crucial under stress.
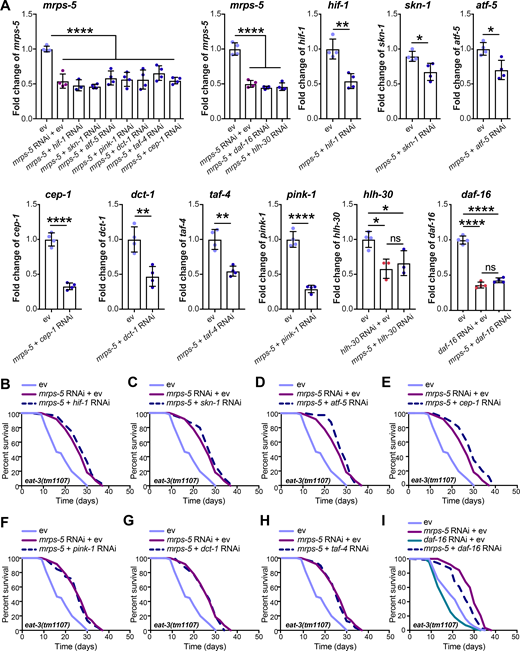
Lifespan screen for downstream mediators of mrps-5 RNAi-induced lifespan increase in eat-3(tm1107).(A) RNAi knockdown efficiencies were measured in day 1 adult eat-3(tm1107) for the genes examined in the lifespan screen. Transcript levels of genes upon RNAi knockdown were normalized to the reference genes cdc-42 and f35g12.2 and compared with the mean value of ev-treated controls. Mean ± SD of at least three biological replicates. Significance was calculated using two-tailed unpaired Student’s t test for two conditions and one-way ANOVA with Tukey’s multiple comparisons tests for three or more conditions. ns, not significant; *, P < 0.5; **, P < 0.01; ****, P < 0.0001. (B–I) Lifespan screen performed in eat-3(tm1107). Lifespans were measured in the eat-3(tm1107) mutant worms upon RNAi knockdown of mrps-5 or double RNAi of mrps-5;hif-1 (B), mrps-5;skn-1 (C), mrps-5;atf-5 (D), mrps-5;cep-1 (E), mrps-5;pink-1 (F), mrps-5;dct-1 (G), mrps-5;taf-4 (H), and mrps-5;daf-16 (I). daf-16 RNAi reduces lifespan nonspecifically in the eat-3(tm1107) mutant with and without mrps-5 RNAi treatment. See Table S1 for lifespan statistics.
Lifespan screen for downstream mediators of mrps-5 RNAi-induced lifespan increase in eat-3(tm1107).(A) RNAi knockdown efficiencies were measured in day 1 adult eat-3(tm1107) for the genes examined in the lifespan screen. Transcript levels of genes upon RNAi knockdown were normalized to the reference genes cdc-42 and f35g12.2 and compared with the mean value of ev-treated controls. Mean ± SD of at least three biological replicates. Significance was calculated using two-tailed unpaired Student’s t test for two conditions and one-way ANOVA with Tukey’s multiple comparisons tests for three or more conditions. ns, not significant; *, P < 0.5; **, P < 0.01; ****, P < 0.0001. (B–I) Lifespan screen performed in eat-3(tm1107). Lifespans were measured in the eat-3(tm1107) mutant worms upon RNAi knockdown of mrps-5 or double RNAi of mrps-5;hif-1 (B), mrps-5;skn-1 (C), mrps-5;atf-5 (D), mrps-5;cep-1 (E), mrps-5;pink-1 (F), mrps-5;dct-1 (G), mrps-5;taf-4 (H), and mrps-5;daf-16 (I). daf-16 RNAi reduces lifespan nonspecifically in the eat-3(tm1107) mutant with and without mrps-5 RNAi treatment. See Table S1 for lifespan statistics.
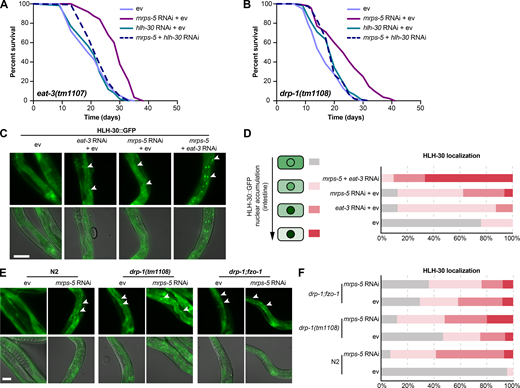
HLH-30/TFEB drives longevity from combined inhibition of mitochondrial translation and dynamics.(A and B) Lifespan analysis performed in eat-3(tm1107) (A) and drp-1(tm1108) (B) subjected to RNAi against mrps-5 and hlh-30, individually or in combination. hlh-30 RNAi abolishes mrps-5 RNAi-mediated lifespan increase in both the eat-3(tm1107) and the drp-1(tm1108) mutant worms. See Table S1 for lifespan statistics. (C) Representative micrographs of worms expressing HLH-30::GFP grown on ev, mrps-5 RNAi, eat-3 RNAi, or mrps-5;eat-3 double RNAi bacteria (as indicated). Images were taken at day 2 of adulthood. χ2 tests were performed to assess the significance of differences in the levels of HLH-30::GFP nuclear enrichment among RNAi-treated N2 worms (P = 0.0001, P = 0.0005, and P = 0 when comparing mrps-5 RNAi + ev, eat-3 RNAi + ev, and mrps-5 + eat-3 RNAi to ev-treated controls, respectively). The upper panels show fluorescent images of HLH-30::GFP nuclear localization, while the lower panels display overlay of HLH-30::GFP fluorescent images with bright field images of worms. Scale bar in ev-treated condition is 100 µm and applies to all the images in C. White arrowheads indicate intestinal nuclei. (D) Quantification of HLH-30 nuclear localization in C: n = 16–29 different animals per RNAi treatment, pooled from two independent experiments. (E) HLH-30::GFP nuclear localization in intestinal cells of wild-type, drp-1(tm1108) mutant, and drp-1;fzo-1 double mutant worms at day 2 of adulthood. Scale bar in ev-treated N2 is 100 µm and applies to all the images in E. White arrowheads indicate intestinal nuclei. (F) Quantification of HLH-30 nuclear enrichment in E: n = 21–32 different animals per RNAi treatment, pooled from two independent experiments. χ2 tests were performed to assess the significance of differences in the levels of HLH-30::GFP nuclear enrichment in N2, drp-1(tm1108) mutant, and drp-1;fzo-1 mutant upon mrps-5 RNAi (P = 0, P = 0.0314, and P = 0.5533 when comparing mrps-5 RNAi to ev-treated controls in N2, drp-1(tm1108), and drp-1;fzo-1, respectively).
HLH-30/TFEB drives longevity from combined inhibition of mitochondrial translation and dynamics.(A and B) Lifespan analysis performed in eat-3(tm1107) (A) and drp-1(tm1108) (B) subjected to RNAi against mrps-5 and hlh-30, individually or in combination. hlh-30 RNAi abolishes mrps-5 RNAi-mediated lifespan increase in both the eat-3(tm1107) and the drp-1(tm1108) mutant worms. See Table S1 for lifespan statistics. (C) Representative micrographs of worms expressing HLH-30::GFP grown on ev, mrps-5 RNAi, eat-3 RNAi, or mrps-5;eat-3 double RNAi bacteria (as indicated). Images were taken at day 2 of adulthood. χ2 tests were performed to assess the significance of differences in the levels of HLH-30::GFP nuclear enrichment among RNAi-treated N2 worms (P = 0.0001, P = 0.0005, and P = 0 when comparing mrps-5 RNAi + ev, eat-3 RNAi + ev, and mrps-5 + eat-3 RNAi to ev-treated controls, respectively). The upper panels show fluorescent images of HLH-30::GFP nuclear localization, while the lower panels display overlay of HLH-30::GFP fluorescent images with bright field images of worms. Scale bar in ev-treated condition is 100 µm and applies to all the images in C. White arrowheads indicate intestinal nuclei. (D) Quantification of HLH-30 nuclear localization in C: n = 16–29 different animals per RNAi treatment, pooled from two independent experiments. (E) HLH-30::GFP nuclear localization in intestinal cells of wild-type, drp-1(tm1108) mutant, and drp-1;fzo-1 double mutant worms at day 2 of adulthood. Scale bar in ev-treated N2 is 100 µm and applies to all the images in E. White arrowheads indicate intestinal nuclei. (F) Quantification of HLH-30 nuclear enrichment in E: n = 21–32 different animals per RNAi treatment, pooled from two independent experiments. χ2 tests were performed to assess the significance of differences in the levels of HLH-30::GFP nuclear enrichment in N2, drp-1(tm1108) mutant, and drp-1;fzo-1 mutant upon mrps-5 RNAi (P = 0, P = 0.0314, and P = 0.5533 when comparing mrps-5 RNAi to ev-treated controls in N2, drp-1(tm1108), and drp-1;fzo-1, respectively).
Next, we examined the role of hlh-30 in mrps-5 RNAi-mediated longevity in the drp-1(tm1108) mutant. Upon hlh-30 RNAi, the long lifespan conferred by mrps-5 RNAi in the drp-1(tm1108) mutant was substantially abrogated (Fig. 6 B and Table S1). This further establishes a central role of HLH-30 in mediating the synergistic longevity effects engendered by simultaneously suppressing mitochondrial translation and disrupting mitochondrial dynamics.
In C. elegans, HLH-30 localizes to the nucleus in response to a variety of stresses such as heat stress and starvation (Fig. S5 A) to ensure the transcriptional induction of its downstream protective genes (Lapierre et al., 2013; Lin et al., 2018; O’Rourke and Ruvkun, 2013). To investigate the localization of HLH-30, we employed a transgenic C. elegans line that expresses the protein tagged with GFP (Lapierre et al., 2013). We found that although individual RNAi of eat-3 and mrps-5 stimulated HLH-30 nuclear localization, combined knockdown of mrps-5;eat-3 resulted in a stronger nuclear enrichment of HLH-30 (Fig. 6, C and D). Depletion of drp-1 also increased the HLH-30 nuclear localization, which was further enhanced upon mrps-5 RNAi (Fig. 6, E and F). We also analyzed the HLH-30::GFP nuclear enrichment upon the double mutation of drp-1 and fzo-1, in which we found an increased nuclear localization of HLH-30::GFP compared with wild-type N2 animals (Fig. 6, E and F). However, no further increase in this nuclear localization was observed by mrps-5 RNAi in the drp-1;fzo-1 mutant (Fig. 6, E and F). Taken together, these results suggest that HLH-30 functions as the downstream effector to mediate longevity in response to inhibition of mitochondrial dynamics and mitochondrial translation.

External stress promotes HLH-30 nuclear localization and mrps-5;eat-3 double RNAi elevates MVBs levels without altering energy charge. (A) Representative micrographs of worms expressing HLH-30::GFP upon starvation for 8 h or upon heat stress at 35°C for 3 h. Images were taken on day 2 of adulthood. Scale bar in untreated condition is 100 µm and applied to all the images in A. (B) Worms with impaired mitochondrial translation and mitochondrial dynamics do not exhibit changes in energy charge, an index reflecting energy status based on ([ATP]+1⁄2[ADP])/([ATP]+[ADP]+[AMP]). Worms at L4 were collected for polar metabolites analyses. ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (C) Examples of the different types of MVBs and lysosome-like structures. Electron microscopy was conducted in day 1 adult N2 worms. Scale bar in each image is 200 nm. (D) Table representing the density of electron-lucent (e-lucent) and electron-dense (e-dense) MVBs per 100 µm2. E-lucent and e-dense MVBs were quantified from 21, 20, and 33 random fields of hypodermis and intestine for worms treated with ev, mrps-5 RNAi, and mrps-5;eat-3 double RNAi, respectively. Three animals were used for each group.
External stress promotes HLH-30 nuclear localization and mrps-5;eat-3 double RNAi elevates MVBs levels without altering energy charge. (A) Representative micrographs of worms expressing HLH-30::GFP upon starvation for 8 h or upon heat stress at 35°C for 3 h. Images were taken on day 2 of adulthood. Scale bar in untreated condition is 100 µm and applied to all the images in A. (B) Worms with impaired mitochondrial translation and mitochondrial dynamics do not exhibit changes in energy charge, an index reflecting energy status based on ([ATP]+1⁄2[ADP])/([ATP]+[ADP]+[AMP]). Worms at L4 were collected for polar metabolites analyses. ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (C) Examples of the different types of MVBs and lysosome-like structures. Electron microscopy was conducted in day 1 adult N2 worms. Scale bar in each image is 200 nm. (D) Table representing the density of electron-lucent (e-lucent) and electron-dense (e-dense) MVBs per 100 µm2. E-lucent and e-dense MVBs were quantified from 21, 20, and 33 random fields of hypodermis and intestine for worms treated with ev, mrps-5 RNAi, and mrps-5;eat-3 double RNAi, respectively. Three animals were used for each group.
TFEB/HLH-30, identified as a global regulator of energy metabolism, maintains energy homeostasis in response to environmental cues such as nutrient availability through regulation of genes in the autophagy-lysosomal pathway (Settembre et al., 2013). As inhibiting mitochondrial translation and dynamics severely compromises the cell’s major energy-producing process, namely mitochondrial respiration, we asked whether a change in energy status upon these mitochondrial stresses may have triggered the HLH-30 nuclear localization. We performed semi-targeted metabolomics and specifically determined the energy charge, which is an index based on the following calculation: ([ATP]+1⁄2[ADP])/([ATP]+[ADP]+[AMP]). Upon depletion of mrps-5 with eat-3 or drp-1, the energy charge was not significantly altered (Fig. S5 B), suggesting that there is no overall cellular energy crisis. Combined, our results suggest that the mitochondrial stress induced by blocking mitochondrial translation and impairing mitochondrial dynamics extends lifespan by promoting the nuclear enrichment of HLH-30.
Combined inhibition of mitochondrial translation and fusion stimulates lysosome biogenesis
HLH-30 and its mammalian orthologue TFEB are identified as master regulators of lysosome biogenesis and autophagy processes (Lapierre et al., 2013; O’Rourke and Ruvkun, 2013; Sardiello et al., 2009). Therefore, we expected an increase in the number of lysosomes upon simultaneous inhibition of mitochondrial translation and fusion. To test this, we visualized lysosomes in coelomocytes using a transgenic strain expressing GFP-labeled lysosomal marker LMP-1 (Treusch et al., 2004). The coelomocytes in C. elegans are scavenger cells that actively require endosome-lysosome pathways, in which LMP-1::GFP localizes to bona fide lysosomal structures (Fares and Greenwald, 2001; Treusch et al., 2004). When mrps-5 and eat-3 were silenced together, a significant increase in the fluorescence intensity of LMP-1::GFP was observed (Fig. 7, A and B). Additionally, double RNAi of mrps-5;eat-3 significantly reduced the number of enlarged lysosomal compartments (Fig. 7, A and B) that are likely a form of abnormal lysosomes occurring upon lysosomal dysfunction (Eguchi et al., 2018; Li et al., 2018; Sambri et al., 2017).

Combined inhibition of mitochondrial translation and fusion promotes lysosome biogenesis. (A) Representative micrographs of LMP-1::GFP-labeled lysosomes in coelomocytes. Animals were imaged at day 6 of adulthood. Scale bar in ev-treated condition is 10 µm and applies to all the images in A. (B) Quantification of LMP-1::GFP fluorescence intensity (left) and lysosome size (right) in A. Fluorescence intensity of LMP-1::GFP per coelomocyte was quantified and normalized to the area of the coelomocyte using ImageJ. 20 different animals per RNAi treatment were included for quantification. Two independent experiments have been performed. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (C) Ultrastructural characteristics of lysosome-like structures and MVBs upon RNAi against mrps-5 and eat-3. The images display the longitudinal cross section of day 1 adult worms. Black and red arrows indicate MVBs and lysosome-like structures, respectively. The images were captured at the magnification of 11,000×, 18,500×, and 13,000× for ev, mrps-5 RNAi, and mrps-5;eat-3 RNAi. Cut, cuticle; Hyp, hypodermis. The scale bar in each image and in the inset denotes 1 µm and 200 nm. (D) Quantification of lysosome-like structures and MVBs (the sum of both electron dense [e-dense] and electron lucent [e-lucent] MVBs) in C. RNAi knockdown of mrps-5;eat-3 profoundly promotes the formation of both lysosome-like structures and MVBs when compared with mrps-5 RNAi- or ev-treated animals. χ2 tests were performed to assess the significance of differences in relation to the RNAi treatments and the levels of lysosome-associated structures in N2 worms (mrps-5 RNAi vs. ev, P = 0.0001; mrps-5;eat-3 double RNAi vs. ev, P = 0.0042; mrps-5;eat-3 double RNAi vs. mrps-5 RNAi, P = 0.0045). Lysosome-like structures and MVBs were quantified in a blind manner from 21, 20, and 33 random fields of hypodermis and intestine for ev, mrps-5 RNAi, and mrps-5;eat-3. Data were derived from three animals for each group. (E) Ultrastructural analysis by electron microscopy reveals a suppressive effect of hlh-30 RNAi on mrps-5 RNAi-mediated increase in lysosome-associated structures in eat-3(tm1107). The images display the longitudinal cross-section of young adult eat-3(tm1107) worms. Black and red arrows indicate MVBs and lysosome-like structures, respectively. The images were captured at the magnification of 18,500× for both mrps-5 RNAi and mrps-5;hlh-30 RNAi conditions. The insets represent (1) e-dense MVBs, (2) e-lucent MVBs, and (3) lysosome-like structures in eat-3(tm1107) upon mrps-5 RNAi. The fourth inset represents e-dense MVBs in eat-3(tm1107) upon mrps-5;hlh-30 double RNAi. The scale bar in each image denotes 1 µm, and the scale bar in the inset 1 is 200 nm and applied to all the insets in E. (F) Quantification of lysosome-like structures and MVBs in E. The number of MVBs is the sum of both e-dense and e-lucent compartments. χ2 tests were performed to assess the significance of differences in relation to the RNAi treatments and the levels of lysosome-associated structures in the eat-3(tm1107) mutant (mrps-5 RNAi vs. mrps-5;hlh-30 double RNAi, P = 0). Lysosome-like structures and MVBs were quantified in a blind manner from 79 and 87 random fields of hypodermis and intestine of eat-3(tm1107) upon RNAi against mrps-5 and hlh-30, individually or in combination. Data were derived from at least three animals for each group.
Combined inhibition of mitochondrial translation and fusion promotes lysosome biogenesis. (A) Representative micrographs of LMP-1::GFP-labeled lysosomes in coelomocytes. Animals were imaged at day 6 of adulthood. Scale bar in ev-treated condition is 10 µm and applies to all the images in A. (B) Quantification of LMP-1::GFP fluorescence intensity (left) and lysosome size (right) in A. Fluorescence intensity of LMP-1::GFP per coelomocyte was quantified and normalized to the area of the coelomocyte using ImageJ. 20 different animals per RNAi treatment were included for quantification. Two independent experiments have been performed. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (C) Ultrastructural characteristics of lysosome-like structures and MVBs upon RNAi against mrps-5 and eat-3. The images display the longitudinal cross section of day 1 adult worms. Black and red arrows indicate MVBs and lysosome-like structures, respectively. The images were captured at the magnification of 11,000×, 18,500×, and 13,000× for ev, mrps-5 RNAi, and mrps-5;eat-3 RNAi. Cut, cuticle; Hyp, hypodermis. The scale bar in each image and in the inset denotes 1 µm and 200 nm. (D) Quantification of lysosome-like structures and MVBs (the sum of both electron dense [e-dense] and electron lucent [e-lucent] MVBs) in C. RNAi knockdown of mrps-5;eat-3 profoundly promotes the formation of both lysosome-like structures and MVBs when compared with mrps-5 RNAi- or ev-treated animals. χ2 tests were performed to assess the significance of differences in relation to the RNAi treatments and the levels of lysosome-associated structures in N2 worms (mrps-5 RNAi vs. ev, P = 0.0001; mrps-5;eat-3 double RNAi vs. ev, P = 0.0042; mrps-5;eat-3 double RNAi vs. mrps-5 RNAi, P = 0.0045). Lysosome-like structures and MVBs were quantified in a blind manner from 21, 20, and 33 random fields of hypodermis and intestine for ev, mrps-5 RNAi, and mrps-5;eat-3. Data were derived from three animals for each group. (E) Ultrastructural analysis by electron microscopy reveals a suppressive effect of hlh-30 RNAi on mrps-5 RNAi-mediated increase in lysosome-associated structures in eat-3(tm1107). The images display the longitudinal cross-section of young adult eat-3(tm1107) worms. Black and red arrows indicate MVBs and lysosome-like structures, respectively. The images were captured at the magnification of 18,500× for both mrps-5 RNAi and mrps-5;hlh-30 RNAi conditions. The insets represent (1) e-dense MVBs, (2) e-lucent MVBs, and (3) lysosome-like structures in eat-3(tm1107) upon mrps-5 RNAi. The fourth inset represents e-dense MVBs in eat-3(tm1107) upon mrps-5;hlh-30 double RNAi. The scale bar in each image denotes 1 µm, and the scale bar in the inset 1 is 200 nm and applied to all the insets in E. (F) Quantification of lysosome-like structures and MVBs in E. The number of MVBs is the sum of both e-dense and e-lucent compartments. χ2 tests were performed to assess the significance of differences in relation to the RNAi treatments and the levels of lysosome-associated structures in the eat-3(tm1107) mutant (mrps-5 RNAi vs. mrps-5;hlh-30 double RNAi, P = 0). Lysosome-like structures and MVBs were quantified in a blind manner from 79 and 87 random fields of hypodermis and intestine of eat-3(tm1107) upon RNAi against mrps-5 and hlh-30, individually or in combination. Data were derived from at least three animals for each group.
We next conducted electron microscopic analyses to examine the presence of lysosomes and lysosome-related organelles in order to further investigate if lysosome biogenesis is differentially regulated in these RNAi-treated worms. We observed three types of cellular compartments in worm samples that are related to lysosome biogenesis and macromolecule degradation: (1) electron-lucent multivesicular bodies (MVBs), (2) electron-dense MVBs, and (3) lysosome-like structures (Fig. S5 C). Studies in C. elegans suggest a distinct difference between electron-lucent and electron-dense MVBs, where the electron-lucent MVBs are associated with exosome excretion, and the electron-dense MVBs are the intermediates toward lysosomes to participate in lysosome biogenesis or intracellular digestion (Hyenne et al., 2015; Liégeois et al., 2006). We quantified each of these lysosome-associated structures in the hypodermal and intestinal regions of worms. A profound increase in the number of lysosome-like structures occurred in the double RNAi of mrps-5;eat-3, about six times higher than that of mrps-5 RNAi alone (Fig. 7, C and D). In contrast, these structures were not observed in ev-treated worms (Fig. 7, C and D). In addition, upon double RNAi of mrps-5;eat-3, we found a marked increase in the overall number of MVBs, including both electron-lucent and electron-dense MVBs, compared with those in mrps-5 RNAi-treated and ev-treated worms (Fig. 7, C and D). The increase of the electron-dense MVBs was even more pronounced than that of the electron-lucent MVBs (Fig. S5 D).
We then asked whether the increase in the lysosome-associated compartments upon the combined inhibition of mrps-5;eat-3 depends on HLH-30. We conducted an additional electron microscopy analysis on the eat-3(tm1107) mutant treated with mrps-5 RNAi or mrps-5;hlh-30 double RNAi. This revealed that the cosilencing of hlh-30 with mrps-5 significantly reduced the number of lysosome-like structures and MVBs in the hypodermal and intestinal regions of eat-3(tm1107), compared with mrps-5 RNAi alone (Fig. 7, E and F). These results confirmed that slowing down mitochondrial translation in conjunction with reducing fusion stimulates HLH-30–mediated lysosome biogenesis.
Combined inhibition of mitochondrial translation and fusion promotes autophagic activity
Finally, we aimed to determine whether the increase in lysosome number after inhibition of mitochondrial translation and fusion led to an increase in autophagy. We used a transgenic strain expressing an mCherry::GFP::LGG-1 tandem reporter that allows us to assess autophagic flux (Chang et al., 2017). In this system, autophagosomes are visualized as yellow punctae due to colocalization of GFP and mCherry fluorescence, while the acidic environment of lysosomes quenches the GFP fluorescence and leaves only the red signal of mCherry representing autolysosomes (ALs; Chang et al., 2017). We found that double RNAi of mrps-5;eat-3 elevated AL formation without significantly affecting autophagosome levels in both the pharynx and the hypodermal seam cells in day 2 adult worms (Fig. 8, A–D). This suggests an increased autophagic flux toward ALs by decreasing mitochondrial translation and fusion, which is likely due to the increased lysosome biogenesis.

Combined RNAi of mrps-5;eat-3 promotes autophagic activity. (A) The number of AP and AL was measured in the pharynx of day 2 adult worms expressing tandem autophagy reporter mCherry::GFP::LGG-1 and treated with RNAi against mrps-5 alone or against mrps-5 and eat-3 simultaneously. Scale bar, 20 µm. (B) Quantification of AP and AL in A. mCherry::GFP::LGG-1- and mCherry::LGG-1–positive punctae were counted in 30 worms per genotype at day 2 of adulthood. Data were derived from two independent experiments. Mean ± SEM, *, P < 0.05; **, P < 0.01; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (C) The number of AP and AL was measured in the seam cell of day 2 adult mCherry::GFP::LGG-1 reporter worms fed on bacteria expressing ev, mrps-5, or mrps-5;eat-3 in combination. Scale bar, 20 µm. (D) Quantification of AP and AL in C. mCherry::GFP::LGG-1- and mCherry::LGG-1–positive punctae were counted in 24–26 worms per genotype at day 2 of adulthood. Data were pooled from two independent experiments. Mean ± SEM; ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (E) Model for mitochondrial dynamics and mitochondrial translation-mediated longevity signaling pathway. (a) Upon absence of external stress in mitochondrial translation and dynamics, HLH-30 resides in the cytosol and animals live a normal lifespan. (b and c) Inhibition of either fusion (b) or fission (c) in conjunction with impaired mitochondrial translation synergistically promotes HLH-30 trafficking to the nucleus, whereupon it prolongs lifespan by up-regulating lysosome biogenesis and autophagy process. (d) Simultaneous suppression of fission and fusion abrogates the effect of mrps-5 RNAi on the HLH-30 nuclear localization, which in turn compromises its downstream longevity benefits.
Combined RNAi of mrps-5;eat-3 promotes autophagic activity. (A) The number of AP and AL was measured in the pharynx of day 2 adult worms expressing tandem autophagy reporter mCherry::GFP::LGG-1 and treated with RNAi against mrps-5 alone or against mrps-5 and eat-3 simultaneously. Scale bar, 20 µm. (B) Quantification of AP and AL in A. mCherry::GFP::LGG-1- and mCherry::LGG-1–positive punctae were counted in 30 worms per genotype at day 2 of adulthood. Data were derived from two independent experiments. Mean ± SEM, *, P < 0.05; **, P < 0.01; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (C) The number of AP and AL was measured in the seam cell of day 2 adult mCherry::GFP::LGG-1 reporter worms fed on bacteria expressing ev, mrps-5, or mrps-5;eat-3 in combination. Scale bar, 20 µm. (D) Quantification of AP and AL in C. mCherry::GFP::LGG-1- and mCherry::LGG-1–positive punctae were counted in 24–26 worms per genotype at day 2 of adulthood. Data were pooled from two independent experiments. Mean ± SEM; ***, P < 0.001; ****, P < 0.0001; ns, not significant by one-way ANOVA with Tukey’s multiple comparisons test. (E) Model for mitochondrial dynamics and mitochondrial translation-mediated longevity signaling pathway. (a) Upon absence of external stress in mitochondrial translation and dynamics, HLH-30 resides in the cytosol and animals live a normal lifespan. (b and c) Inhibition of either fusion (b) or fission (c) in conjunction with impaired mitochondrial translation synergistically promotes HLH-30 trafficking to the nucleus, whereupon it prolongs lifespan by up-regulating lysosome biogenesis and autophagy process. (d) Simultaneous suppression of fission and fusion abrogates the effect of mrps-5 RNAi on the HLH-30 nuclear localization, which in turn compromises its downstream longevity benefits.
Altogether, we suggest a model whereupon the impairment of mitochondrial translation, disrupting the equilibrium cycle of mitochondrial fission and fusion, by suppressing either process results in HLH-30/TFEB localization to nuclei. This enables HLH-30/TFEB to initiate lysosome biogenesis and lysosome-based degradation processes, which in turn promotes the lifespan extension in C. elegans. This model is illustrated in Fig. 8 E.
Discussion
We have taken advantage of a C. elegans model with inhibited mitochondrial translation to identify the genetic requirements for mitochondrial dynamics in promoting longevity. Enhancing a disequilibrium of mitochondrial fission and fusion by depleting either one of these processes reinforces the effects of mitochondrial translation inhibition on the UPRMT activation and longevity. Conversely, immobilizing the mitochondrial network through a combined mutation of drp-1 and fzo-1 abolishes mitochondrial translation inhibition-mediated lifespan extension, despite the induction of UPRMT. Blocking UPRMT by depleting its two major mediators including ATFS-1 and HAF-1, respectively, fails to prevent the synergistic lifespan-extending effects from the combined inhibition of mitochondrial translation and fusion. Instead, this lifespan-extending synergy is dependent on the lysosome biogenesis transcription factor, HLH-30/TFEB.
Knockdown of haf-1 prevented the lifespan extension observed upon mrps-5 RNAi (Houtkooper et al., 2013). However, we found here that neither haf-1 nor atfs-1 is necessary for the lifespan increase induced by mrps-5;eat-3 RNAi. This seemingly incompatible observation can be reconciled by the fact that in response to different degrees of cellular damage, mitochondria execute a hierarchical surveillance network encompassing UPRMT, mitochondrial fission and fusion, and lysosome-based degradation pathway such as autophagy to control mitochondrial quality (Andreux et al., 2013; Fischer et al., 2012; Ni et al., 2015; Youle and van der Bliek, 2012). We speculate that upon the intensive mitochondrial stress caused by simultaneous inhibition of mitochondrial dynamics and mitochondrial translation, HLH-30/TFEB acts primarily to maintain cellular homeostasis and to promote longevity, likely through the activation of lysosome biogenesis and autophagy pathway. Mitophagy is a specific mode of autophagy by which impaired mitochondria are eliminated (Wang and Klionsky, 2011). In our lifespan screen, knocking down neither pink-1 nor dct-1—two key mitophagy genes—prevents mrps-5 RNAi from prolonging the lifespan of eat-3(tm1107). However, we do not rule out the possibility of an important role of mitophagy in regulating this longevity, as RNAi knockdown of a single mitophagy gene might not entirely block the mitophagy process. Therefore, future studies using null mutants in the mitophagy pathway are merited to determine the impact of mitophagy in this longevity paradigm.
In this work, combined inactivation of fission and fusion in the drp-1;fzo-1 null mutant abrogates mitochondrial translation inhibition-mediated lifespan increase. Recent studies in mouse and yeast demonstrate that a simultaneous impairment of mitochondrial fission and fusion compromises the mitophagy/autophagy process (Bernhardt et al., 2015; Song et al., 2017). Importantly, we also found that double mutation of drp-1;fzo-1 prevents mrps-5 RNAi from increasing the HLH-30 nuclear enrichment. Therefore, we postulate that the immobilized mitochondrial network in the drp-1;fzo-1 null mutant prevents the influences of mitochondrial translation inhibition on lifespan via a similar mechanism involving the suppression of HLH-30–mediated autophagy and lysosome function.
Our work highlights an important role for HLH-30/TFEB nuclear localization and its downstream effects on lysosome biogenesis and autophagy upon stresses from mitochondrial translation and mitochondrial dynamics. We did not find up-regulation of HLH-30 target genes, such as those described in a previous study in which nuclear localization of HLH-30 in the glp-1(e2141) mutant up-regulated a set of 11 genes that are involved in autophagy and lysosome pathways (Lapierre et al., 2013). Similar to our findings, the majority of these HLH-30 target genes were not up-regulated upon heat stress or in the long-lived daf-2(e1370) mutant, both of which increase the HLH-30 nuclear enrichment (Lapierre et al., 2013; Lin et al., 2018). We hence conclude that HLH-30 activates specific gene sets depending on the type of stress that in the context of this study remain to be uncovered. Similarly, we did not find obvious up-regulation of lysosome-related pathways in our proteomics screen. This might be due to the limited resolution of our proteomics that covers only about a quarter of the proteins in lysosome-related pathways in C. elegans. Importantly, we confirmed that hlh-30 is required for the longevity upon combined inhibition of mitochondrial translation and dynamics, and through various imaging techniques we established that downstream HLH-30 pathways such as lysosome biogenesis and autophagic activity are activated.
Our work identifies the crucial role of HLH-30/TFEB in the lifespan increase and reveals its enhanced nuclear localization mediated by the combined repression of mitochondrial network homeostasis and mitochondrial translation. In the current mechanisms underlying nutrient depletion-induced TFEB activation in mammalian cells, inactivated rapamycin complex 1 leads to TFEB dephosphorylation on Ser211, which in turn frees TFEB from 14–3-3 protein sequestration in the cytosol, thereby promoting its localization to the nucleus (Martina et al., 2012; Roczniak-Ferguson et al., 2012; Settembre et al., 2012). Other cell-based studies suggest that mitochondrial stresses such as TFAM depletion and mitochondrial respiratory inhibition induce TFEB nuclear localization through mechanisms associated with Parkin activity and Ca2+ efflux from lysosomes (Baixauli et al., 2015; Nezich et al., 2015; Zhang et al., 2016). The E3 ligase Parkin activates TFEB likely by degrading a transcriptional repressor of PGC-1α, thereby activating PGC-1α–promoted TFEB expression, while the lysosome-localized Ca2+ release promotes TFEB dephosphorylation by activating calcineurin (Medina et al., 2015; Nezich et al., 2015; Siddiqui et al., 2015). Future work will have to elucidate the involvement of these pathways as mechanistic players in the mitochondrial dysfunction-mediated HLH-30/TFEB nuclear localization.
Our study clarifies the connections between the UPRMT, mitochondrial dynamics, and lysosome biogenesis in the context of C. elegans longevity. We showed that the synergistic effect of inhibiting both mitochondrial function and dynamics on lifespan, despite inducing the UPRMT, is not dependent on it. Instead, the connection of this synergy to longevity is exclusively the result of the induction of lysosome biogenesis and lysosome-based degradation pathway, specifically through the nuclear localization of HLH-30. The results highlight how various environments may activate a multitude of signaling pathways that have known roles in lifespan extension, and yet the extension itself may stem from a single pathway. In addition, our work illustrates a functional interaction between mitochondria and lysosomes and their relative implications in longevity. Upon stresses from mitochondrial translation and the mitochondrial network, mitochondria communicate with lysosomes to stimulate lysosome biogenesis and autophagic activity through HLH-30/TFEB nuclear localization. Our results provide a better understanding of how mitochondrial dysfunction drives longevity through crosstalk with lysosome biogenesis signals and suggest that the strategic manipulation of mitochondrial-lysosomal communication may present new opportunities for targeted therapeutic interventions in aging.
Materials and methods
Worm strains and maintenance
C. elegans strains N2 Bristol, drp-1(tm1108)IV, fzo-1(tm1133)II, haf-1(ok705)IV, SJ4100: zcls13[hsp-6p::GFP], SJ4005: zcls4[hsp-4p::GFP], SJ4103: zcls14[myo-3p::GFP(mit)], MAH235: sqIs19 [hlh-30p::hlh-30::gfp + rol-6(su1006)], RT258: unc-119(ed3)III; pwls50[lmp-1p::lmp-1::gfp, Cbr-unc-119(+)], ZC376.7(gk3094)V, rrf-1(pk1417)I, glp-1(e2141)III were obtained from the Caenorhabditis Genetics Center (University of Minnesota). C. elegans strain eat-3(tm1107) was obtained from S. Mitani (National Bioresource Project of Japan, Tokyo Women’s Medical University School of Medicine, Tokyo, Japan). C. elegans strains MAH215: sqls11[lgg-1p::mcherry::gfp::lgg-1 + rol-6(su1106)] and atfs-1(cmh15) were kind gifts from L.R. Lapierre (Brown University, Providence, RI) and C.M. Haynes (University of Massachusetts Medical School, Worcester, MA), respectively. C. elegans strains fzo-1(tm1133)II;drp-1(tm1108)IV, WBM671: wbmEx289[myo-3p::tomm20(aa1-49)::gfp::unc54 3′UTR], WBM752: fzo-1(tm1133)II;drp-1(tm1108)IV; wbmEx289[myo-3p::tomm20(aa1-49)::gfp::unc54 3′UTR], fzo-1(tm1133)II;drp-1(tm1108)IV; sqIs19 [hlh-30p::hlh-30::gfp + rol-6(su1006)], and drp-1(tm1108)IV; sqIs19 [hlh-30p::hlh-30::gfp + rol-6(su1006)] were kind gifts from W.B. Mair (Harvard T.H. Chan School of Public Health, Harvard University, Boston, MA). All nematode strains were grown and maintained on nematode growth media (NGM) agar plates at 20°C as previously described (Brenner, 1974) with the exception of glp-1(e2141)III, which was maintained at 15°C.
Bacterial feeding strains and RNAi experiments
Escherichia coli HT115 (DE3) with the ev L4440 was obtained from the Caenorhabditis Genetics Center. Bacterial feeding RNAi experiments were performed as described (Kamath et al., 2001). RNAi E. coli feeding clones used were mrps-5(E02A10.1), eat-3(D2013.5), fzo-1(ZK1248.14), atfs-1(ZC376.7), hif-1(F38A6.3), skn-1(T19E7.2), cep-1(F52B5.5), atf-5(T04C10.4), pink-1(EEED8.9), dct-1(C14F5.1), taf-4(R119.6), daf-16(R13H8.1), and hlh-30(W02C12.3) derived from the Ahringer RNAi library (Kamath and Ahringer, 2003). All these clones were confirmed by sequencing. In all RNAi experiments described in this study, worms were subjected to RNAi bacteria from the time of hatching. In the case of double RNAi experiments, two bacterial cultures were mixed in equal proportion based on OD measurements.
Lifespan analysis
Age-synchronized nematodes were plated as embryos on NGM plates supplemented with 2 mM IPTG (Promega, cat. no. v3951) and 25 µg/ml carbenicillin (Sigma-Aldrich, cat. no. c1389) that had been seeded with HT115 (DE3) bacteria transformed with either pL4440 ev or the indicated RNAi construct, as described before (Liu et al., 2019). All assays were performed at 20°C except for the lifespan of glp-1(e2141), in which glp-1(e2141) worms were first grown at the restrictive temperature 25°C to eliminate germ cells, then switched to 20°C at larval stage 4 (L4) for the rest of the assay. 5-Fluorouracil (Sigma-Aldrich, cat. no. f6627) was not used in the glp-1(e2141) lifespan assay. For all the other strains, worms were transferred to 10 µM 5-fluorouracil (Sigma-Aldrich, cat. no. f6627) supplemented plates at the L4 larval stage. L4 in all lifespan assays was counted as day 0 of life. Approximately 100–180 worms were used per condition in every lifespan experiment. Survival was analyzed every other day, and worms were considered dead when they did not respond to repeated prodding. Worms that were missing, displaying internal egg hatching, losing vulva integrity, and burrowing into NGM agar were censored. SPSS software was used for statistical analysis, and significance was calculated using the log-rank (Mantel–Cox) method. All the key lifespan assays were repeated at least three times by two independent observers and one-time blinded. The lifespan statistics are shown in Table S1.
Extraction of mRNA and qPCR
Worms were subjected to RNAi bacteria upon hatching and grown at 20°C until day 1 adulthood, as described before (Liu et al., 2019). Approximately 500 synchronized worms were collected for total mRNA extraction at day 1 of adulthood. At least three biological replicates were prepared per condition. Total RNA was isolated according to the manufacturer’s protocol. Briefly, samples were homogenized in TRIzol (Invitrogen) by shaking using a TissueLyser II (QIAGEN) for 5 min at a frequency of 30 times/s. RNA was quantified with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized using the QuantiTect Reverse Transcription kit (QIAGEN) after the elimination of genomic DNA. The qPCR reaction was performed in 8 µl with a primer concentration of 1 µM and SYBR Green Master mix (Roche) in a Roche LightCycler 480 system. About qPCR experiments related to Fig. 1 D and Fig. S1, A and B, the geometric mean of Y45F10D.4 and F35G12.2 was used for normalization; about qPCR experiments related to Fig. S4 A, the geometric mean of cdc-42 and F35G12.2 was used for normalization; about qPCR experiments related to Fig. 3 D, the geometric mean of ama-1, cdc-42, and F35G12.2 was used for normalization. The oligonucleotides used for PCR are listed in Table S2.
Worm respiration assays
Oxygen consumption of whole worms was measured using the Seahorse XF24 (Seahorse Bioscience) at room temperature (25°C) as previously described (Koopman et al., 2016). Briefly, around 250 worms were washed off NGM plates at the desired stage with M9 solution and washed three times to remove residual bacteria, and then resuspended in 500 µl M9 solution (∼10 worms per 20 µl). Worms were transferred in 96-well microplates (∼10 worms per well), and oxygen consumption was measured. FCCP (Abcam, cat. no. ab120081) and sodium azide (Sigma-Aldrich, cat. no. s8032) treatments were used at 10 µM and 40 mM, respectively. Each experiment was repeated at least two times.
Microscopy, GFP expression, and quantification
To examine the UPRMT and the UPRER, respectively, SJ4100 hsp-6p::GFP and SJ4005 hsp-4p::GFP were bleached, synchronized, and plated on NGM plates seeded with RNAi bacteria against the genes of interest. For the UPRMT, worms at day 3 and day 7 of adulthood were anesthetized with 40 mM levamisole (Santa Cruz Biotech, cat. no. sc205730) and imaged at room temperature using a Leica M205 FA fluorescent microscope with 1× PlanAPO objective and Leica DFC 365 FX camera. Images were captured using Leica Application Suite X software. 100–120 worms were imaged for each condition. Each experiment was repeated three times. To quantify GFP fluorescence, the whole area of worm was chosen and quantified using ImageJ software (http://rsbweb.nih.gov/ij/). Fluorescence was normalized to the area of worms to compare between genotypes. For the UPRER, day 7 adult worms treated with 25 µg/ml solution of tunicamycin (Sigma-Aldrich, cat. no. t7765) on NGM plates for 6 h were used as positive controls. Control animals were treated in an equivalent solution of DMSO (Sigma-Aldrich, cat. no. d8418) on plates. Worms were washed off plates with M9 buffer three times to remove residual bacteria, and then transferred into 96-well microplates containing 20 µl of 40 mM levamisole (Santa Cruz Biotech, cat. no. sc205730; 15 worms per well). Subsequently, GFP fluorescence was measured at excitation 479 nm and emission 515 nm in an TECAN infinite M200 Pro plate reader. To measure length of worms, worms were immobilized with 50 mM sodium azide and imaged using Leica M205 FA microscope with Lecia DFC 365 FX camera in bright field mode. The length of worms was quantified using ImageJ software.
Fluorescent microscopy
For mitochondrial network imaging, animals at day 2 and day 7 were immobilized for 2 min in 40 mM levamisole (Santa Cruz Biotech, cat. no. sc205730) in M9 buffer and mounted on 2% agarose pads. The mitochondrial network was visualized in transgenic strains expressing GFP-tagged mitochondrial matrix protein pmyo-3::GFP(mit) or GFP-tagged mitochondrial outer membrane protein TOMM-20 at room temperature with a Leica fluorescent microscope DM6 B containing a 100×, 1.4-NA oil-immersion objective lens and Lecia DFC 9000 GT camera. The region of worms between the pharynx and vulva was selected for muscle mitochondria. Subsequently, images were deconvoluted with the Huygens professional deconvolution software (Scientific Volume Imaging) using a theoretical point spread function for GFP channel and the classic maximum likelihood estimation algorithm. The signal to noise ratios and background intensities were automatically determined and taken into account during processing. Two independent experiments were performed for mitochondrial network quantification. Quantification of mitochondrial content and network connectivity was performed with ImageJ using a previously developed macro (Dagda et al., 2009).
For HLH-30::GFP imaging in N2 worms upon RNAi treatments, the nuclear localization of HLH-30::GFP in day 2 animals was visualized with a Leica fluorescent microscope DM6 B containing a 40×, 1.25-NA oil-immersion objective lens, and a Leica DFC 9000 GT camera. Images were captured using Leica Application Suite X software. All samples were imaged at room temperature. To avoid subtle localization caused by starvation, mounting, and imaging conditions, all photomicrographs were taken within 5 min of mounting. For heat stress, animals were exposed to 35°C for 3 h; for starvation, animals were transferred onto regular NGM plates without food for 8 h. Two independent experiments were performed with consistent results.
For HLH::30 GFP imaging in N2, drp-1(tm1108) mutant, drp-1;fzo-1 mutant upon mrps-5 RNAi, widefield fluorescence imaging was done using a Nikon 20X Plan Apochromat dry lens/NA 0.8. Day 2 adult C. elegans were anesthetized using tetramisole (2 mg/ml) and mounted on 2% agarose pads between a glass slide and coverslip. Images were taken immediately following slide preparation, less than 5 min following removal from food.
Confocal microscopy
For lysosome imaging, day 6 LMP-1::GFP transgenic animals were immobilized in 50 mM sodium azide and mounted on 2% agarose pads. LMP-1::GFP-labeled lysosomes were imaged in the coelomocytes of animals at room temperature. Confocal analysis was performed using a Leica TCS SP8 X confocal microscope equipped with 63×, 1.4-NA oil-immersion objective lens, a 488-nm laser for GFP, and a fast sCMOS camera. Confocal images were captured in a 4.48-µm range with 0.3-µm step size using Leica Application Suite X software, and the pinhole used was 1 AU Deconvolution was performed in the Huygens professional deconvolution with classic maximum likelihood estimation algorithm. Quantification was performed in ImageJ to measure the integrated intensity/area and lysosomes diameter. Images were taken from 20 animals per condition with two independent experiments performed.
For autophagy analysis, transgenic worms expressing the tandem reporter mCherry::GFP::LGG-1 (Chang et al., 2017) were treated with RNAi against mrps-5 or mrps-5;eat-3 from the time of hatching and imaged at day 2 of adulthood using a Leica TCS SP8 X confocal microscope equipped with 63×, 1.4-NA oil-immersion objective lens, and a fast sCMOS camera, as previously described (Chang et al., 2017). In brief, day 2 adult worms were anesthetized using 0.1% sodium azide in M9 medium, and images of 10–15 different worms for each genotype were acquired from a single optical section. To visualize autophagosome (AP) and AL in pharynx, confocal images were taken where the lumen of the pharyngeal bulb was in focus, while for seam cell, the Z-position was chosen when the nuclei were in focus. Quantification of the number of mCherry-only and mCherry::GFP-positive punctae used ImageJ, and data were derived from two independent experiments.
Polar metabolite analysis
Synchronized worms were collected at L4 with five biological replicates per condition. A 75 µl mixture of the following internal standards in water was added to each sample: adenosine-15N5-monophosphate (100 µM), adenosine-15N5-triphosphate (1 mM), D4-alanine (100 µM), D7-arginine (100 µM), D3-aspartic acid (100 µM), D4-citric acid (100 µM), 13C1-citrulline (100 µM), 13C6-fructose-1,6-diphosphate (100 µM), guanosine-15N5-monophosphate (100 µM), guanosine-15N5-triphosphate (1 mM), 13C6-glucose (1 mM), 13C6-glucose-6-phosphate (100 µM), D3-glutamic acid (100 µM), D5-glutamine (100 µM), 13C6-isoleucine (100 µM), D3-leucine (100 µM), D4-lysine (100 µM), D3-methionine (100 µM), D6-ornithine (100 µM), D5-phenylalanine (100 µM), D7-proline (100 µM), 13C3-pyruvate (100 µM), D3-serine (100 µM), D5-tryptophan (100 µM), D4-tyrosine (100 µM), and D8-valine (100 µM). Subsequently, 425 µl water, 500 µl methanol, and 1 ml chloroform were added to the same 2-ml tube before thorough mixing and centrifugation for 10 min at 14,000 rpm. The top layer, containing the polar phase, was transferred to a new 1.5-ml tube and dried using a vacuum concentrator at 60°C. Dried samples were reconstituted in 100 µl methanol/water (6/4 vol/vol). Metabolites were analyzed using a Waters Acquity ultra-high performance liquid chromatography system coupled to a Bruker Impact II Ultra-High Resolution Qq-Time-Of-Flight mass spectrometer. Samples were kept at 12°C during analysis, and 5 µl of each sample was injected. Chromatographic separation was achieved using a Merck Millipore SeQuant ZIC-cHILIC column (polyetheretherketone 100 × 2.1 mm, 3 µm particle size). Column temperature was held at 30°C. The mobile phase consisted of (A) 1:9 acetonitrile:water and (B) 9:1 acetonitrile:water, both containing 5 mM ammonium acetate. Using a flow rate of 0.25 ml/min, the liquid chromatography gradient consisted of 100% B for 0–2 min, ramp to 0% B at 28 min, 0% B for 28–30 min, ramp to 100% B at 31 min, and 100% B for 31–35 min. Mass spectrometry data were acquired using negative and ionization in full scan mode over the range of m/z 50–1,200. Data were analyzed using Bruker TASQ software version 2.1.22.3. All reported metabolites were corrected to internal standards and then normalized to the total amount of metabolites in each sample. Metabolite identification has been based on a combination of accurate mass, (relative) retention times, and fragmentation spectra, compared with the analysis of a library of standards. The metabolomics analysis was performed as described previously (Baardman et al., 2018; Koenis et al., 2018).
Worm electron microscopy and data analysis
Wild type N2 and eat-3(tm1107) were bleached and plated on NGM plates seeded with RNAi bacteria, which express dsRNA against the genes of interest. At day 1 for N2 and young adult for eat-3(tm1107), three biological replicates with each containing multiple worms per condition were fixed using high-pressure freezing (Leica EM PACT2, Leica Microsystems) in 20% BSA (Sigma-Aldrich), then freeze-substituted in 0.5% osmium tetroxide (EMS) and 0.5% uranyl acetate (Agar Scientific) in acetone (EMS) at −90°C. Free substitution was performed using an automated free-substitution machine (AFS2 + freeze substitution processor, Leica Microsystems). Specifically, worm samples were maintained at −90°C for 110 h, and then slowly warmed up to −20°C (5°C/h) where worm samples were held for another 16 h, and subsequently warmed up slowly to 0°C (5°C/h). Next, worm samples were rinsed in pure acetone (EMS) three times (20 min each time) at 0°C and then removed from specimen carrier at room temperature (20°C), followed by three times rinsing in pure acetone (EMS; 20 min each time). Thereafter, worm samples were placed in a Pella Microporous Specimen Capsule and submerged in increasing concentrations of epon resin (TAAB Laboratories Equipment Ltd.) at room temperature (starting with 1:3 resin:acetone mixture for 3 h, then 1:1 resin:acetone mixture for 3 h, and 3:1 resin:acetone mixture for 16 h, finishing with 100% resin for 24 h). Last, worms were transferred to embedding mold and incubated at 60°C for 2 d. Ultrathin sections (≈70 nm) from the imaged animals were cut and stained with uranyl acetate/lead citrate (Agar Scientific) and imaged in a Tecnai T12 transmission electron microscope (Thermo Fisher Scientific) operating at 100 kilovolts equipped with a digital camera (Veleta). The regions of hypodermis and intestine of animals were analyzed and the quantification analyses for multivesicular bodies and multilamellar lysosomes were performed blind by two independent examiners after randomization of the images.
SWATH proteomics—library acquisition
Synchronized worms were collected at the stage of young adult for proteomics analysis with five biological replicates per condition. For the acquisition of a spectral library, 150 µg of digested peptide from three sets of pooled N2 wildtype adult C. elegans were pH fractionated (protocol no. 84868, Thermo Fisher Scientific) into 10 fractions. The resulting 30 fractions were separated on an Eksigent liquid chromatography machine coupled with a 20 cm PicoFrit emitter injected on an AbSciex 5600+ TripleTOF mass spectrometer using a 120-min gradient going from 2% to 35% acetonitrile at 300 nanoliter/min. At the MS1 level, the 20 most intense precursors were selected in the range of 350–1,460 m/z with a 500-ms survey scan. At the MS2 level, spectra were acquired at 150-ms survey scans between 50 and 2,000 m/z. These samples were used for preparing a spectral library to support the data-independent acquisition (DIA)/SWATH data (Röst et al., 2014). iRT peptides (Biognosys) were added as external calibrants to all samples, for both DDA injections (library acquisition) and DIA injections (for quantification).
SWATH proteomics—sample acquisition
For all samples measured for quantitative DIA (SWATH) acquisition, 1 µg of nonfractionated samples were injected in the same AbSciex 5600+ TripleTOF using the same parameters as above, except that a 60-min gradient was used and the instrument was operated in data-independent acquisition mode using 64 m/z windows (Gillet et al., 2012). The output .wiff files from both DIA and data-dependent acquisition (DDA) acquisition mode were converted to centroided mzXML files using FileConverter v2.2.0. For library generation from the DDA mzXML files, samples were searched against the canonical UniProtKB C. elegans proteome database containing 27,481 proteins and searched with Comet v2016.01 r3. Reverse decoy proteins were generated, and up to two tryptic missed cleavages were allowed with a precursor mass error of 50 ppm and fragment error of 0.1 D. Cysteine carbamidomethylation was used as the fixed modification and methionine oxidation as the variable modification. PeptideProphet was used to search the data and scored with iProphet. A 1% protein false discovery rate was used for significance cutoff. The in-house results from the DDA runs were combined with extensive published C. elegans DDA data (PXD004584; Narayan et al., 2016). The light channel from these SILAC-labeled samples was also searched with Comet as above, using the same parameters, using CiRT peptides instead of iRT peptides (Parker et al., 2015). The results from the in-house and downloaded DDA files were combined, and peptides with retention time variance of ≥150 were removed. For peptides identified in-house and in the downloaded dataset, the in-house peptide was retained and the other peptide discarded. The final assembled library contains 67,612 peptides (of which 18,072 are from the in-house DDA runs and 49,540 from the downloaded DDA runs), corresponding to 9,438 unique proteins. This library was then used as the reference library for OpenSWATH v2.1.0. OpenSWATH was run on all DIA acquisitions, followed by mProphet scoring using the msproteomicstools package available on GitHub (September 2017). 2,124 proteins were quantified exclusively from proteotypic peptides. Protein data were generated from the peptide matrix using MSstats v3.12.0. Differential expression was determined using either a Student's t test (with P values corrected for multiple testing using the Benjamini–Hochberg false discovery rate, presented for reference) or PLS-DA with mixomics (Rohart et al., 2017) setting a variable of importance (VIP) score of >1 as significant. The DIA data and the assembled library file are available on Proteomics Identifications Database (PRIDE) under the ID PXD009256.
Functional annotation
GO analyses were conducted using the DAVID bioinformatics resource (Huang et al., 2009). Proteins that were differentially regulated with a VIP score >1 were subjected to functional annotation clustering. To retrieve significantly enriched GO terms, enrichment threshold (expression analysis systematic explorer score) was set as 0.05 for all analyses.
Statistical analysis
Data were analyzed by two-tailed unpaired Student’s t test to compare one independent variable between two conditions. One-way ANOVA with Tukey’s multiple comparisons test was used to compare one independent variable between three or more conditions. Categorical data were analyzed using χ2 tests. Survival curves were calculated using the log-rank (Mantel–Cox) method. For all experiments, data are shown as mean ± SD unless indicated otherwise, and a P value <0.05 was considered significant.
Online supplemental material
Fig. S1 shows suppressive effects on animals’ growth and mitochondrial respiration and stimulative effects on the UPRMT upon inhibition of mitochondrial translation and fusion. Fig. S2 shows that double mutation of fzo-1 and drp-1 does not suppress the reduction in body length after mrps-5 RNAi and that mrps-5;eat-3 double RNAi promotes longevity upon loss of atfs-1. Fig. S3 displays proteomics analyses of worms with impaired mitochondrial translation and mitochondrial dynamics. Fig. S4 shows RNAi efficiency tests and lifespan screen for downstream mediators of mrps-5 RNAi-induced lifespan extension in eat-3(tm1107). Fig. S5 shows HLH-30 localization to the nucleus upon starvation and heat stress, and a robust increase in MVBs with no change in energy charge upon mrps-5;eat-3 double RNAi. Table S1 contains statistical analyses of lifespan data in this study. Table S2 shows a list of primer sets used for qPCR analyses in this study.
Acknowledgments
The authors thank the Caenorhabditis Genetics Center at the University of Minnesota and National Bioresource Project of Japan for providing C. elegans strains. The authors also thank Louis R. Lapierre and Cole M. Haynes for proving transgenic C. elegans strains, and Jan van Weering for helping with high-pressure freezing of samples.
Work in the Houtkooper group is financially supported by an European Research Council Starting grant (no. 638290), a VIDI grant from The Netherlands Organization for Health Research and Development (ZonMw; no. 91715305), and the Velux Stiftung (no. 1063). A.W. MacInnes is supported by E-Rare-2, the ERA-Net for Research on Rare Diseases (ZonMw no. 40-44000-98-1008). G.E. Janssens is supported by a Federation of European Biochemical Society long-term fellowship and a VENI grant from ZonMw (no. 09150161810014). E.G. Williams is supported by an National Institutes of Health F32 Ruth Kirchstein Fellowship (F32GM119190). R. Aebersold is supported by ERC grant Proteomics 4D ERC-20140AdG 670821.
R.H. Houtkooper is co-inventor on a patent related to mitochondrial ribosomal proteins as aging regulators. The other authors declare no competing financial interests.
Author contributions: Y.J. Liu and R.H. Houtkooper conceived and designed the project. Y.J. Liu, R.L. McIntyre, and P. Yao performed experiments. Y.J. Liu and R.L. McIntyre analyzed the data. G.E. Janssens, E.G. Williams, J. Lan, and R. Aebersold performed proteomic and bioinformatic analyses. N.N. van der Wel and H. van der Veen performed electron microscopy analyses. B. Schomakers and M. van Weeghel performed and analyzed metabolomics. W.B. Mair provided worm strains and advice. Y.J. Liu, R.L. McIntyre, A.W. MacInnes, and R.H. Houtkooper interpreted data. Y.J. Liu, A.W. MacInnes, and R.H. Houtkooper wrote the manuscript with contributions from all other authors.

