


Guard proteins initiate defense mechanisms upon sensing pathogen-encoded virulence factors. Successful viral pathogens likely inhibit guard protein activity, but these interactions have been largely undefined. Here, we demonstrate that the human pathogen herpes simplex virus 1 (HSV-1) stimulates and inhibits an antiviral pathway initiated by NLRP1, a guard protein that induces inflammasome formation and pyroptotic cell death when activated. Notably, HSV-1 infection of human keratinocytes promotes posttranslational modifications to NLRP1, consistent with MAPK-dependent NLRP1 activation, but does not result in downstream inflammasome formation. We identify infected cell protein 0 (ICP0) as the critical HSV-1 protein that is necessary and sufficient for inhibition of the NLRP1 pathway. Mechanistically, ICP0’s cytoplasmic localization and function as an E3 ubiquitin ligase prevents proteasomal degradation of the auto-inhibitory NT-NLRP1 fragment, thereby preventing inflammasome formation. Further, we demonstrate that inhibiting this inflammasome is important for promoting HSV-1 replication. Thus, we have established a mechanism by which HSV-1 overcomes a guard-mediated antiviral defense strategy in humans.
Introduction
Host defense against microbial infection is initially mediated by the innate immune system, which includes pattern-triggered and effector-triggered immune responses (Lopes Fischer et al., 2020; Orzalli and Parameswaran, 2022; Remick et al., 2023). Pattern-triggered immune responses use germline-encoded pattern recognition receptors (PRRs) to detect and mount defenses against encounters with microbes irrespective of their pathogenicity. By contrast, effector-triggered immunity utilizes intracellular guard proteins to detect the activities of pathogen-encoded virulence factors. The importance of PRRs in immunity is well established, as PRR evasion strategies by viral pathogens are critical for their infectious potential. By contrast, while several examples exist of guard protein activation during infectious encounters, the importance of guard evasion for a successful infection is a question that remains largely unanswered.
The NACHT, leucine-rich repeat (LRR), and pyrin domain (PYD) (NLRP) containing family member NLRP1 is a guard protein that senses bacterial and viral virulence factor activities. Upon activation, NLRP1 forms a multimeric signaling platform called an inflammasome, which induces pyroptotic cell death that is important to prevent infection. NLRP1-mediated pyroptosis is characterized by the activation of the cysteine protease caspase 1 (CASP1), the formation of Gasdermin D (GSDMD)–mediated plasma membrane pores, and the extracellular accumulation of proinflammatory cytokines, such as interleukin-1β (IL-1β) and IL-18 (reviewed in Barnett et al. [2023]). NLRP1 structure and regulation are unique among the NLRP family members. The full-length (FL) protein contains N-terminal (NT) PYD, NACHT, and LRR domains, and a C-terminal (CT) caspase activation and recruitment domain (CARD). Between the NLRP1 LRR domain and CARD, there is a function-to-find domain (FIIND) consisting of ZU5 and UPA subdomains held together by a covalent bond between amino acids F1212 and S1213. A portion of FL-NLRP1 undergoes autoproteolysis between these two amino acids, producing NT and CT protein fragments that remain non-covalently associated with each other (D’Osualdo et al., 2011). This autoproteolytic event is necessary, but not sufficient, for activation of the NLRP1 inflammasome (Finger et al., 2012). The bioactive UPA-CARD containing CT-NLRP1 fragment is restrained by incorporation into a ternary inhibitory complex consisting of cellular dipeptidyl peptidases 8 or 9 (DPP8/9) and an autoproteolyzed (NT-NLRP1/CT-NLRP1) or unprocessed FL-NLRP1 (Hollingsworth et al., 2021; Huang et al., 2021; Zhong et al., 2018). When autoproteolysis is combined with the proteasome-mediated degradation of the NT-NLRP1, the CT-NLRP1 fragment is released from the inhibitory complex; it then self-oligomerizes and associates with the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) to promote CASP1 and GSDMD-dependent pyroptosis and inflammatory cytokine release.
In the context of viral infections, NLRP1 is recognized as a guard because viral proteases produced by human rhinovirus, coxsackie virus B3, and severe acute respiratory syndrome coronavirus 2 cleave NLRP1. Cleavage within the linker region between the PYD and NACHT domain unveils an NT glycine residue that is recognized as a degron by cellular Cullin-RING E3-ubiquitin ligases (Planes et al., 2022; Robinson et al., 2020; Tsu et al., 2021). In other contexts of cellular stress, ultraviolet B (UVB) irradiation and bacterial ribotoxins also activate the NLRP1 inflammasome by promoting ribotoxic stress and activating the mitogen-activated protein kinase (MAPK) ZAKα, which induces phosphorylation of the NT-NLRP1 linker region (Jenster et al., 2023; Robinson et al., 2022). Human NLRP1 has also been implicated as a sensor of nucleic acids as both long double-stranded RNA (dsRNA) and the synthetic dsDNA mimetic poly dA:dT trigger the NLRP1 inflammasome (Bauernfried et al., 2020; Zhou et al., 2023). Reductive stress and protein folding stress may also contribute to NLRP1 activation as the redox protein thioredoxin-1 has been shown to restrain NLPR1 function and aminopeptidase inhibitors can potentiate NLRP1 signaling (Ball et al., 2022; Orth-He et al., 2023; Wang et al., 2023; Zhang et al., 2023). In addition, chemical inhibitors of DPP8/9 (i.e., talabostat) destabilize the ternary inhibitory complex and subsequent turnover of FL-/NT-NLRP1 activates the inflammasome (Okondo et al., 2018; Zhong et al., 2018). Despite the different mechanisms by which these diverse stimuli initially engage the NLRP1 pathway, they all activate the inflammasome through proteasome-dependent mechanisms.
In humans, NLRP1 and downstream inflammasome components are highly expressed in keratinocytes, the major epithelial cell type that makes up cutaneous and mucocutaneous barriers (Zhong et al., 2016). These cells are the initial targets of several viruses, including herpes simplex virus 1 (HSV-1), which is a large dsDNA virus that establishes a lifelong latent infection in most of the human population and is the causative agent of recurrent cold sores at cutaneous and mucocutaneous surfaces (Knipe and Howley, 2013). Underlying the success of HSV-1 as a human pathogen is its ability to evade host innate immune defense mechanisms. Several HSV-1 immune evasive proteins target PRRs themselves or downstream signaling components (Kurt-Jones et al., 2017; Su et al., 2016). Less is understood about the recognition of HSV-1 by guard proteins and its manipulation of guard-directed host defense strategies.
In this study, we report that HSV-1 infection of human keratinocytes induces MAPK activation, posttranslational modifications to NT-NLRP1, and proteasome-dependent degradation of FL-NLRP1. However, despite these activities that should stimulate NLRP1-mediated pyroptosis, HSV-1 infection does not result in the formation of an active NLRP1 inflammasome or induce pyroptotic cell death. We found that the NLRP1 evasive activity is mediated by a single viral protein, the E3 ubiquitin ligase infected cell protein 0 (ICP0). ICP0 blocks NLPR1-induced pyroptosis by inhibiting NT-NLRP1 degradation. Mechanistically, the inhibitory activity is mediated by the E3 ubiquitin ligase activity of ICP0 and its cytoplasmic localization, and this viral protein is required for maximal replication within human keratinocytes when NLRP1 is active. These data therefore establish how HSV-1 overcomes an important guard-mediated antiviral immune pathway in human skin.
Results
HSV-1 infection activates receptor proximal events in the NLRP1 inflammasome pathway
To determine if HSV-1 engages the NLRP1 inflammasome pathway, we examined receptor proximal and downstream events in NLRP1 signaling in infected primary human keratinocytes. FL-NLRP1 (∼165 kD), NT-NLRP1 (∼135 kD), and CT-NLRP1 (∼30 kD) can be detected in keratinocyte whole-cell lysates using antibodies raised against the N-terminus and C-terminus of NLRP1, respectively (Fig. 1 A). NLRP1 antibody specificities were confirmed by loss of FL-, NT-, and CT-NLRP1 protein in cell lysates from keratinocytes transduced with lentiviruses expressing shRNAs that target NLRP1 (Fig. 1 A). Infection of keratinocytes with the WT HSV-1 strains KOS and 17syn+ resulted in a shift in NT-NLRP1 size that was observed when cell lysates were run on low-percentage (6%) SDS-PAGE gels (Fig. 1 B), consistent with the addition of a posttranslational modification(s) on this protein. The HSV-1–induced shift in NT-NLRP1 size was similar to that observed following treatment with anisomycin (Fig. 1 C), which activates the NLRP1 inflammasome by inducing MAPK-dependent phosphorylation of the NT-linker region (Jenster et al., 2023; Robinson et al., 2022). Like anisomycin treatment, HSV-1 infections in keratinocytes also induced phosphorylation of the MAPKs p38 and JNK (Fig. 1, B and C). These observations together suggest that HSV-1 infection activates a MAPK pathway in keratinocytes that may subsequently promote NT-NLRP1 phosphorylation.
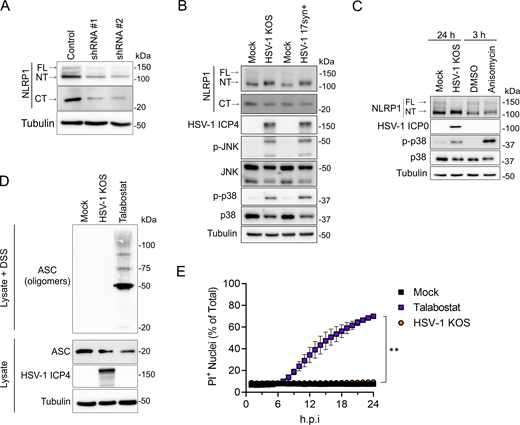
HSV-1 infection activates receptor proximal steps in the NLRP1 inflammasome pathway but does not form a functional inflammasome. (A) Immunoblots of lysates examining NLRP1 abundance in primary human keratinocytes transduced with control or shNLRP1-expressing lentiviruses. Images are representative of n = 3 biological replicates. (B) Immunoblots of lysates from mock-infected or WT HSV-1 strains KOS or 17syn+-infected primary human keratinocytes (MOI 10) at 24 h.p.i. Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from HSV-1 (MOI 10) or anisomycin (10 μM) -treated primary human keratinocytes harvested at indicated time points. Images are representative of n = 3 biological replicates. (D) Immunoblots of ASC oligomerization status in DSS-treated lysates from mock-infected, HSV-1 KOS–infected (MOI 10), or talabostat-treated (30 μM) primary human keratinocytes for 24 h. A portion of the lysates was not treated with DSS and run in parallel to examine total ASC abundance under the indicated experimental conditions. Images are representative of n = 3 biological replicates. (E) Quantification of PI+ primary human keratinocyte nuclei following infection with HSV-1 KOS (MOI 10) or treatment with talabostat (30 μM). PI+ nuclei were quantified hourly and plotted as a percentage of total nuclei detected at each time point imaged. Data are presented as the mean of three independent experiments ± SEM. The area under the curve was calculated for each condition and statistical significance was determined by Student’s t test (**P < 0.01). Source data are available for this figure: SourceData F1.
HSV-1 infection activates receptor proximal steps in the NLRP1 inflammasome pathway but does not form a functional inflammasome. (A) Immunoblots of lysates examining NLRP1 abundance in primary human keratinocytes transduced with control or shNLRP1-expressing lentiviruses. Images are representative of n = 3 biological replicates. (B) Immunoblots of lysates from mock-infected or WT HSV-1 strains KOS or 17syn+-infected primary human keratinocytes (MOI 10) at 24 h.p.i. Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from HSV-1 (MOI 10) or anisomycin (10 μM) -treated primary human keratinocytes harvested at indicated time points. Images are representative of n = 3 biological replicates. (D) Immunoblots of ASC oligomerization status in DSS-treated lysates from mock-infected, HSV-1 KOS–infected (MOI 10), or talabostat-treated (30 μM) primary human keratinocytes for 24 h. A portion of the lysates was not treated with DSS and run in parallel to examine total ASC abundance under the indicated experimental conditions. Images are representative of n = 3 biological replicates. (E) Quantification of PI+ primary human keratinocyte nuclei following infection with HSV-1 KOS (MOI 10) or treatment with talabostat (30 μM). PI+ nuclei were quantified hourly and plotted as a percentage of total nuclei detected at each time point imaged. Data are presented as the mean of three independent experiments ± SEM. The area under the curve was calculated for each condition and statistical significance was determined by Student’s t test (**P < 0.01). Source data are available for this figure: SourceData F1.
In addition to a shift in NT-NLRP1, HSV-1 infection also resulted in a marked loss in the abundance of FL-, but not CT-, NLRP1 (Fig. 1, B and C; and Fig. S1 A). This loss occurs late in infection, between 12- and 24-h post-infection (h.p.i.) (Fig. S1 A). Proteasome-dependent turnover of FL-/NT-NLRP1 is associated with activation of the NLRP1 inflammasome (Chui et al., 2019; Robinson et al., 2020; Sandstrom et al., 2019; Tsu et al., 2021). We therefore tested whether loss of FL-NLRP1 following HSV-1 infection was proteasome dependent by examining its abundance in the presence of the proteasome inhibitor bortezomib. As a positive control for bortezomib activity, we also examined the abundance of IFI16, a cellular protein that is degraded in HSV-1–infected cells through a proteasome-dependent mechanism (Johnson et al., 2013; Orzalli et al., 2012). Both IFI16 and FL-NLRP1 protein abundance were stable in HSV-1–infected cells treated with bortezomib (Fig. S1 B), indicating the HSV-1–mediated loss of FL-NLRP1 was proteasome dependent.
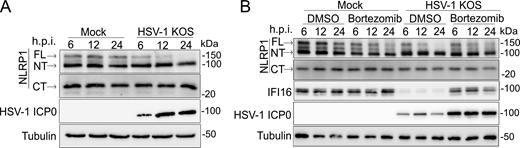
HSV-1 induces the proteasome-dependent loss of FL-NLRP1. (A) Immunoblots of lysates from mock-infected or HSV-1 KOS strain–infected primary human keratinocytes (MOI 10) at 6, 12, 24 h.p.i. Images are representative of n = 3 biological replicates. (B) Immunoblots of lysates from DMSO or bortezomib (10 μM) -treated primary human keratinocytes infected with HSV-1 strain KOS (MOI 10) and harvested at indicated time points. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData FS1.
HSV-1 induces the proteasome-dependent loss of FL-NLRP1. (A) Immunoblots of lysates from mock-infected or HSV-1 KOS strain–infected primary human keratinocytes (MOI 10) at 6, 12, 24 h.p.i. Images are representative of n = 3 biological replicates. (B) Immunoblots of lysates from DMSO or bortezomib (10 μM) -treated primary human keratinocytes infected with HSV-1 strain KOS (MOI 10) and harvested at indicated time points. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData FS1.
Based on our observations that HSV-1 infection results in MAPK (p38 and JNK) activation, posttranslational modifications to NT-NLRP1 similar to that observed with another NLRP1 activator (i.e., anisomycin), and the loss of FL-NLRP1, we expected that HSV-1–infected keratinocytes would show evidence of ASC activation and downstream pyroptosis. However, we observed no ASC oligomerization in HSV-1–infected cells when compared with those treated with talabostat, which robustly induced this phenotype (Fig. 1 D). Further, while talabostat treatment induced keratinocyte plasma membrane permeability, as measured by uptake of the membrane-impermeable dye propidium iodide (PI), this was completely absent in HSV-1–infected cells (Fig. 1 E). This latter result is consistent with reports demonstrating that HSV-1 infection does not induce human keratinocyte pyroptosis (Bauernfried et al., 2020; Orzalli et al., 2021). When combined with our finding that HSV-1 infection activates receptor proximal events in the NLRP1 inflammasome pathway, we considered a viral immune evasion activity.
HSV-1 inhibits NLRP1 inflammasome formation and keratinocyte pyroptosis
HSV-1 produces several proteins that help the virus evade cellular innate immune responses (Danastas et al., 2020; Kurt-Jones et al., 2017). Thus, the discrepancy between MAPK activation, NT-NLRP1 modification, and loss of FL-NLRP1, and the lack of downstream inflammasome activation observed in HSV-1–infected keratinocytes may have been a consequence of HSV-1 inhibiting the NLRP1 signaling pathway. To determine if HSV-1 inhibits NLRP1 signaling, we quantified PI uptake in HSV-1–infected primary keratinocytes treated with a variety of chemical agonists of NLRP1. Talabostat treatment induced robust PI uptake in keratinocytes compared with DMSO-treated control cells, but this response was reduced in cells pre-infected with HSV-1 (Fig. 2 A). PI uptake was also reduced in HSV-1–infected keratinocytes stimulated with additional NLRP1 agonists, including transfection of the dsRNA analog polyinosinic:polycytidylic acid (poly(I:C)) (Fig. 2 B) or treatment with the bacterial ribotoxin anisomycin (Fig. 2 C). By contrast, HSV-1 infection had little effect on PI uptake in keratinocytes transfected with lipopolysaccharide (LPS) (Fig. 2 D), an activator of the non-canonical caspase-4 inflammasome (Kayagaki et al., 2013, 2015). These results together suggest that HSV-1 specifically inhibits the NLRP1 signaling pathway in human keratinocytes.

HSV-1 infection inhibits the NLRP1 inflammasome. (A–D) Quantification of membrane permeability in mock- or HSV-1–infected (MOI 10) primary human keratinocytes treated with (A) talabostat (30 μM), (B) poly(I:C) (1 μg/ml), (C) anisomycin (1 μM), or (D) LPS (1 μg/ml). Lipofectamine (LTX) was used as the transfection control. PI+ nuclei were quantified hourly and results are presented as the mean of three independent experiments ± SEM. The area under the curve was calculated for each treatment condition and statistical significance was determined by Student’s t test (***P < 0.001, ****P < 0.0001). (E) Immunoblots of lysates from HSV-1–infected primary human keratinocytes (MOI 10) treated with talabostat (30 μM) for 24 h. Immunoblots of DSS-treated lysates and protein extracted from cell culture supernatants were also performed to assess ASC oligomerization and caspase-1 cleavage, respectively. Images are representative of n = 3 biological replicates. (F) ELISA for IL-1β in the supernatants of HSV-1–infected (MOI 10) keratinocytes treated with talabostat (30 μM) for 24 h. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (****P < 0.0001). (G) ASC speck detection by indirect immunofluorescence in HSV-1–infected (MOI 10) keratinocytes treated with talabostat (30 μM) for 24 h. Arrows indicate ASC specks. Images are representative of n = 3 biological replicates. Scale bar = 10 μm. (H) Quantification of ASC specks in G. Specks were quantified by counting the number of ASC+ cells as a percentage of total Hoechst+ cells. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05). (I) Immunoblots from DSS-treated lysates to assess ASC oligomerization in HSV-1–infected primary human keratinocytes (MOI 10) treated with talabostat (30 μM) or anisomycin (10 μM). Images are representative of n = 3 biological replicates. (J) Immunoblots from DSS-treated lysates to assess ASC oligomerization in HSV-1–infected primary human keratinocytes (MOI 10) treated with poly(I:C) (1 μg/ml) or anisomycin (10 μM). Images are representative of n = 3 biological replicates.
HSV-1 infection inhibits the NLRP1 inflammasome. (A–D) Quantification of membrane permeability in mock- or HSV-1–infected (MOI 10) primary human keratinocytes treated with (A) talabostat (30 μM), (B) poly(I:C) (1 μg/ml), (C) anisomycin (1 μM), or (D) LPS (1 μg/ml). Lipofectamine (LTX) was used as the transfection control. PI+ nuclei were quantified hourly and results are presented as the mean of three independent experiments ± SEM. The area under the curve was calculated for each treatment condition and statistical significance was determined by Student’s t test (***P < 0.001, ****P < 0.0001). (E) Immunoblots of lysates from HSV-1–infected primary human keratinocytes (MOI 10) treated with talabostat (30 μM) for 24 h. Immunoblots of DSS-treated lysates and protein extracted from cell culture supernatants were also performed to assess ASC oligomerization and caspase-1 cleavage, respectively. Images are representative of n = 3 biological replicates. (F) ELISA for IL-1β in the supernatants of HSV-1–infected (MOI 10) keratinocytes treated with talabostat (30 μM) for 24 h. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (****P < 0.0001). (G) ASC speck detection by indirect immunofluorescence in HSV-1–infected (MOI 10) keratinocytes treated with talabostat (30 μM) for 24 h. Arrows indicate ASC specks. Images are representative of n = 3 biological replicates. Scale bar = 10 μm. (H) Quantification of ASC specks in G. Specks were quantified by counting the number of ASC+ cells as a percentage of total Hoechst+ cells. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05). (I) Immunoblots from DSS-treated lysates to assess ASC oligomerization in HSV-1–infected primary human keratinocytes (MOI 10) treated with talabostat (30 μM) or anisomycin (10 μM). Images are representative of n = 3 biological replicates. (J) Immunoblots from DSS-treated lysates to assess ASC oligomerization in HSV-1–infected primary human keratinocytes (MOI 10) treated with poly(I:C) (1 μg/ml) or anisomycin (10 μM). Images are representative of n = 3 biological replicates.
To determine the step in the NLRP1 signaling cascade inhibited by HSV-1, we examined the activation status of critical pathway components in infected cells. Talabostat treatment of uninfected keratinocytes induced CASP1 cleavage, GSDMD cleavage, and the release of IL-1β into culture supernatants (Fig. 2, E and F). By contrast, these events were inhibited in talabostat-treated keratinocytes that were infected with HSV-1 (Fig. 2, E and F). Importantly, HSV-1 infection did not affect the abundance of GSDMD or CASP1 protein (Fig. 2 E), suggesting the virus does not inhibit NLRP1 inflammasome activity by reducing these components and that HSV-1 acts upstream of these steps in the signaling pathway. We next examined the effect of HSV-1 infection on ASC speck formation, which can be visualized by immunofluorescence microscopy. In general, the percentage of cells containing detectable ASC specks following talabostat treatment (<5%) was low compared with the number of cells that were PI positive (60–80%) (Fig. 2 H versus Fig. 2 A), suggesting that ASC speck formation may not be absolutely required for talabostat-induced pyroptosis in keratinocytes. This observation is consistent with a previous study in mouse macrophages (Dick et al., 2016). Despite the low percentage of ASC speck–positive keratinocytes following talabostat treatment, the number of ASC speck–positive cells was reduced in talabostat-treated keratinocytes infected with HSV-1 compared with uninfected cells (Fig. 2, G and H). HSV-1 infection also inhibited talabostat-induced ASC speck formation in an A549 cell line engineered to constitutively express NLRP1 and inducibly express ASC-GFP upon NF-κB activation following TNF treatment (A549-ASC(GFP)-NLRP1) (Fig. S2, A and B). Like observations in keratinocytes (Fig. 2 E), HSV-1 infection had no effect on ASC abundance in the A549-ASC(GFP)-NLRP1 cells (Fig. S2 C). However, we did observe an HSV-1–mediated increase in the abundance of FL-, NT-, and CT-NLRP1 protein in these reporter cells (Fig. S2 C). This may reflect the ability of HSV-1 to transactivate the promoter used to engineer NLRP1 expression and explain the inability of the virus to completely block talabostat-induced ASC speck formation in these cells (Fig. S2 B). Due to the low percentage of ASC speck–positive keratinocytes upon talabostat treatment, we took a complementary biochemical approach to examine the effect of HSV-1 on ASC activity by monitoring ASC oligomerization in cell lysates. Talabostat induced robust ASC oligomerization in keratinocytes, and this was reduced in HSV-1–infected cells (Fig. 2, E and I). HSV-1 infection similarly blocked ASC oligomerization induced by anisomycin (Fig. 2 I) and poly(I:C) treatment (Fig. 2 J). These results collectively establish that HSV-1 inhibits the NLRP1 signaling pathway either at the point of or upstream of ASC oligomerization.
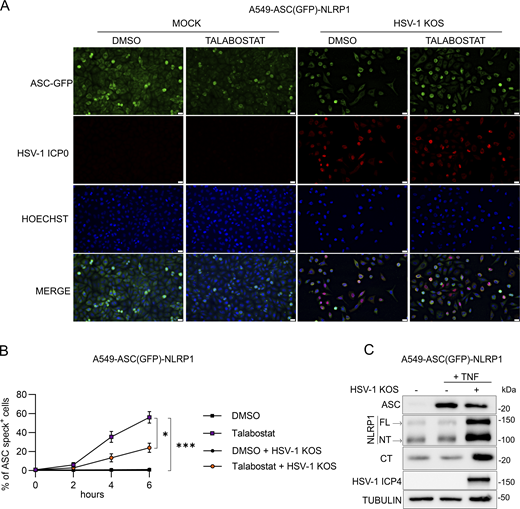
HSV-1 inhibits talabostat-induced ASC speck formation in A549-ASC(GFP)-NLRP1 cells. (A) Micrographs visualizing ASC speck formation in HSV-1–infected (MOI 10) A549-ASC(GFP)-NLRP1 cells followed by treatment with talabostat (30 μM) or DMSO. Images are representative of n = 3 biological replicates. Scale bars = 20 μm. (B) Quantification of specks in A549-ASC(GFP)-NLRP1 cells infected with HSV-1 (MOI 10) followed by treatment with DMSO or talabostat (30 μM). Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (*P < 0.05, ***P < 0.001). (C) Immunoblot of lysates from TNFα-treated (2.5 μg/ml) A549-ASC(GFP)-NLRP1 cells infected with HSV-1 (MOI 10) for 6 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData FS2.
HSV-1 inhibits talabostat-induced ASC speck formation in A549-ASC(GFP)-NLRP1 cells. (A) Micrographs visualizing ASC speck formation in HSV-1–infected (MOI 10) A549-ASC(GFP)-NLRP1 cells followed by treatment with talabostat (30 μM) or DMSO. Images are representative of n = 3 biological replicates. Scale bars = 20 μm. (B) Quantification of specks in A549-ASC(GFP)-NLRP1 cells infected with HSV-1 (MOI 10) followed by treatment with DMSO or talabostat (30 μM). Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (*P < 0.05, ***P < 0.001). (C) Immunoblot of lysates from TNFα-treated (2.5 μg/ml) A549-ASC(GFP)-NLRP1 cells infected with HSV-1 (MOI 10) for 6 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData FS2.
HSV-1 infection blocks proteasome-dependent degradation of NT-NLRP1
We next focused on receptor proximal events in the NLRP1 signaling pathway upstream of ASC. Proteasome-dependent degradation of the auto-inhibitory NT-NLRP1 fragment is critical for activation of the NLRP1 inflammasome (Chui et al., 2019; Sandstrom et al., 2019). We did not observe a noticeable loss of NT-NLRP1 in talabostat-treated primary keratinocytes (Fig. 2 E), although talabostat-induced membrane permeability is inhibited in these cells following treatment with bortezomib (Fig. S3). By contrast, anisomycin treatment in keratinocytes resulted in an observable loss of NT-NLRP1 that was inhibited by bortezomib treatment (Fig. 3 A). Importantly, bortezomib treatment had no effect on anisomycin-induced p38 phosphorylation, suggesting that proteosome inhibition did not affect the upstream MAPK pathway activated by this ribotoxin (Fig. 3 A). To determine if HSV-1 infection inhibits NT-NLRP1 degradation, we examined the abundance of NT-NLRP1 in HSV-1–infected cells treated with anisomycin. While anisomycin treatment reduced the abundance of NT-NLRP1 in uninfected cells, this was not observed in cells preinfected with HSV-1 (Fig. 3 B). A similar inhibition of NT-NLRP1 degradation was observed following poly(I:C) transfection in HSV-1–infected keratinocytes (Fig. 3 C). Importantly, HSV-1 infection did not reduce anisomycin- (Fig. 3 B) or poly(I:C)-induced p38 phosphorylation (Fig. 3 C), indicating that HSV-1 does not inhibit ribotoxic stress–induced MAPK signaling and instead acts at the level of proteasome-dependent turnover of NT-NLRP1.

Talabostat-induced PI uptake in primary human keratinocytes is proteasome and neddylation dependent. Quantification of membrane permeability of primary human keratinocytes treated with talabostat (30 μM) in the presence or absence of bortezomib (10 μM) or MLN4924 (2.5 μM). Data are presented as the mean ± SEM of three independent experiments.
Talabostat-induced PI uptake in primary human keratinocytes is proteasome and neddylation dependent. Quantification of membrane permeability of primary human keratinocytes treated with talabostat (30 μM) in the presence or absence of bortezomib (10 μM) or MLN4924 (2.5 μM). Data are presented as the mean ± SEM of three independent experiments.
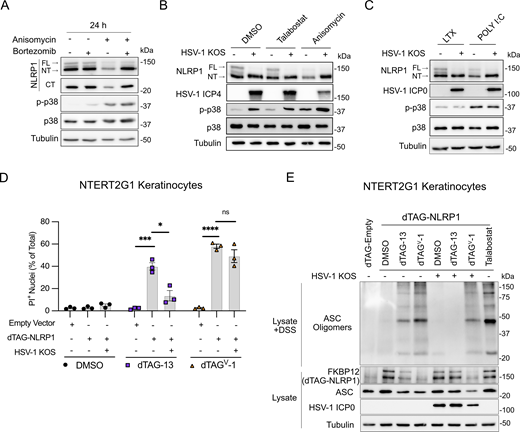
HSV-1 inhibits proteasome-dependent NT-NLRP1 degradation. (A) Immunoblots of lysates from primary human keratinocytes treated with anisomycin (1 μM) and/or bortezomib (10 μM) for 24 h. Images are representative of n = 3 biological replicates. (B) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 KOS (MOI 10) for 4 h followed by treatment with talabostat (30 μM) or anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from primary keratinocytes infected with HSV-1 KOS (MOI 10) for 4 h followed by transfection with poly(I:C) (1 μg/ml) for 24 h. Images are representative of n = 3 biological replicates. (D) NTERT2G1 keratinocytes expressing an empty vector or dTAG-NLRP1 were infected with HSV-1 KOS for 4 h followed by treatment with dTAG-13 or dTAGV-1. Membrane permeability was assessed at 8 h after treatment. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, ***P < 0.001, ****P < 0.0001). (E) Immunoblot of ASC oligomers and lysates from NTERT2G1 keratinocytes expressing dTAG-NLRP1 infected with HSV-1 for 4 h followed by treatment with DMSO, dTAG-13, or dTAGV-1 for 4 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData F3.
HSV-1 inhibits proteasome-dependent NT-NLRP1 degradation. (A) Immunoblots of lysates from primary human keratinocytes treated with anisomycin (1 μM) and/or bortezomib (10 μM) for 24 h. Images are representative of n = 3 biological replicates. (B) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 KOS (MOI 10) for 4 h followed by treatment with talabostat (30 μM) or anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from primary keratinocytes infected with HSV-1 KOS (MOI 10) for 4 h followed by transfection with poly(I:C) (1 μg/ml) for 24 h. Images are representative of n = 3 biological replicates. (D) NTERT2G1 keratinocytes expressing an empty vector or dTAG-NLRP1 were infected with HSV-1 KOS for 4 h followed by treatment with dTAG-13 or dTAGV-1. Membrane permeability was assessed at 8 h after treatment. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, ***P < 0.001, ****P < 0.0001). (E) Immunoblot of ASC oligomers and lysates from NTERT2G1 keratinocytes expressing dTAG-NLRP1 infected with HSV-1 for 4 h followed by treatment with DMSO, dTAG-13, or dTAGV-1 for 4 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData F3.
The exact mechanism by which NT-NLRP1 is targeted for proteasomal degradation in response to ribotoxic stress and DPP9 inhibition is unclear. Members of the Cullin-RING E3 ubiquitin ligase (CRL) family have been implicated in targeting NT-NLRP1 for degradation based on observations that protein neddylation (which positively regulates CRL activity) and a functional ubiquitin-activating enzyme (E1) are required for NLRP1 inflammasome signaling in response to these stimuli (Jenster et al., 2023; Robinson et al., 2020, 2022). We have similarly observed that treatment of primary keratinocytes with the neddylation-activating enzyme inhibitor MLN4924 blocks talabostat-induced membrane permeability in primary keratinocytes (Fig. S3). We therefore hypothesized that HSV-1 infection might inhibit NT-NLRP1 degradation by blocking the activity of a specific CRL complex. To investigate the effect of HSV-1 on CRL-mediated degradation of NT-NLRP1, we made use of a recently described construct where a degradation tag (dTAG), FKBP12F36V, was fused to the N-terminus of NLRP1 (dTAG-NLRP1) (Hollingsworth et al., 2021). The addition of this tag allows for rapid Cullin 4 or Cullin 2 (CUL4/CUL2)-mediated proteasomal degradation of the fusion protein in the presence of the chemicals dTAG-13 or dTAGV-1, respectively (Nabet et al., 2018, 2020). Stable transduction of an hTERT (telomerase reverse transcriptase)-immortalized normal keratinocyte cell line (NTERT2G1) with a lentivirus expressing dTAG-NLRP1, but not an empty vector, resulted in PI uptake following treatment with dTAG-13 or dTAGV-1 (Fig. 3 D). Consistent with our observations with other activators of the NLRP1 inflammasome, HSV-1 KOS infection inhibited dTAG-13–induced PI uptake (Fig. 3 D), ASC oligomerization (Fig. 3 E), and loss of dTAG-NLRP1 protein (Fig. 3 E). However, these HSV-1 inhibitory activities were not observed in cells treated with dTAGV-1, suggesting that HSV-1 specifically inhibits the NLRP1 inflammasome when it is activated by a CUL4-containing CRL complex. The ability of HSV-1 to inhibit ASC oligomerization in response to dTAG-13, but not dTAGV-1, further underscores that HSV-1 inhibits the pathway upstream of ASC. Together, these data strongly argue that HSV-1 inhibits the proteasome-dependent loss of NT-NLRP1 and may act by inhibiting the functions of specific CRL complexes involved in endogenous NLRP1 activation.
De novo expression of HSV-1 ICP0 is necessary and sufficient to inhibit NT-NLRP1 degradation and inflammasome activation
To determine how HSV-1 evades NLRP1 signaling by blocking NT-NLRP1 degradation, specific viral proteins were examined. HSV-1 gene expression occurs in a temporal cascade beginning with immediate-early (IE), followed by early (E), and then late (L) gene expression. Expression of an HSV-1 gene class (i.e., IE, E, or L) can be inhibited using specific viral mutants within the preceding gene class or chemical inhibitors that block progression of the cascade. We first determined whether DNA replication and thus L gene expression were required for HSV-1 to block NLRP1 signaling by measuring PI uptake in talabostat-treated keratinocytes infected with HSV-1 in the presence of phosphonoacetic acid (PAA), a viral DNA polymerase inhibitor (Honess and Watson, 1977). PAA treatment reduced HSV-1 yields in keratinocytes (Fig. S4 A) as expected but had no effect on the ability of HSV-1 to inhibit talabostat-induced PI uptake (Fig. S4 B), suggesting that late events in the virus replication cycle (i.e., the production of L gene products) were not required to inhibit NLRP1 signaling. We then tested multiple HSV-1 strains carrying null mutations in specific IE and E viral genes for their effects on talabostat and anisomycin-induced PI uptake. Notably, the HSV-1 mutant 7134 was unable to prevent talabostat- or anisomycin-induced PI uptake (Fig. 4 A and Fig. S4 C). By contrast, infection with the rescue virus HSV-1 7134R inhibited PI uptake by both NLRP1 stimuli (Fig. 4 A and Fig. S4 C). The gene absent in HSV-1 7134 was therefore required to inhibit NLRP1 signaling. HSV-1 7134 is deleted for ICP0, a RING E3 ubiquitin ligase that has not previously been implicated in inflammasome regulation in keratinocytes. Consistent with HSV-1 7134 being unable to block talabostat-induced PI uptake, we observed mostly normal ASC oligomerization, GSDMD cleavage, and mature IL-1β (p17) secretion in HSV-1 7134–infected cells treated with talabostat (Fig. 4 B). The ICP0 complemented strain 7134R, akin to WT HSV-1, prevented all these talabostat-induced activities (Fig. 4 B). In addition, HSV-1 7134 was unable to prevent talabostat-induced ASC speck formation in A549-ASC(GFP)-NLRP1 cells as compared with HSV-1 7134R–infected cells (Fig. S4 D). These results together indicate the ICP0 expression following HSV-1 infection is required to inhibit the formation of a functional NLRP1 inflammasome.
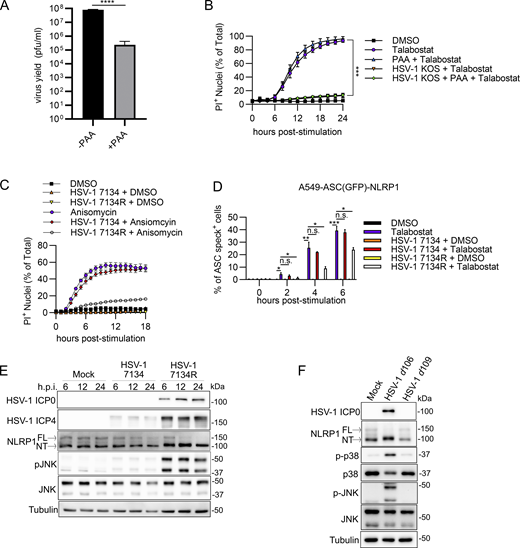
HSV-1 ICP0 is required to inhibit the NLRP1 inflammasome. (A) Viral yields at 24 h.p.i. from PAA-treated (125 μg/ml) primary human keratinocytes infected with HSV-1 (MOI 10). Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated by Student’s t test (****P < 0.0001). (B) Kinetics of membrane permeability in HSV-1–infected primary human keratinocytes treated with talabostat (30 μM) in the presence or absence of PAA. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated by Student’s t test using the area under the curve for each treatment condition (***P < 0.001). (C) Kinetics of membrane permeability in primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 and the corresponding rescue 7134R at MOI 10 followed by treatment with anisomycin (10 μM). Data are representative of two independent experiments and are presented as the mean ± SEM of three technical replicates. (D) Quantification of ASC specks in A549-ASC(GFP)-NLRP1 cells. Cells were infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10 followed by treatment with talabostat (30 μM). Data represent the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001). (E) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10. Lysates were harvested at 6, 12, and 24 h.p.i. Images are representative of n = 3 biological replicates. (F) Immunoblots of lysates from primary human keratinocytes infected with non-replicative viruses HSV-1 d106 and d109 (MOI 10) for 24 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData FS4.
HSV-1 ICP0 is required to inhibit the NLRP1 inflammasome. (A) Viral yields at 24 h.p.i. from PAA-treated (125 μg/ml) primary human keratinocytes infected with HSV-1 (MOI 10). Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated by Student’s t test (****P < 0.0001). (B) Kinetics of membrane permeability in HSV-1–infected primary human keratinocytes treated with talabostat (30 μM) in the presence or absence of PAA. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated by Student’s t test using the area under the curve for each treatment condition (***P < 0.001). (C) Kinetics of membrane permeability in primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 and the corresponding rescue 7134R at MOI 10 followed by treatment with anisomycin (10 μM). Data are representative of two independent experiments and are presented as the mean ± SEM of three technical replicates. (D) Quantification of ASC specks in A549-ASC(GFP)-NLRP1 cells. Cells were infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10 followed by treatment with talabostat (30 μM). Data represent the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001). (E) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10. Lysates were harvested at 6, 12, and 24 h.p.i. Images are representative of n = 3 biological replicates. (F) Immunoblots of lysates from primary human keratinocytes infected with non-replicative viruses HSV-1 d106 and d109 (MOI 10) for 24 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData FS4.
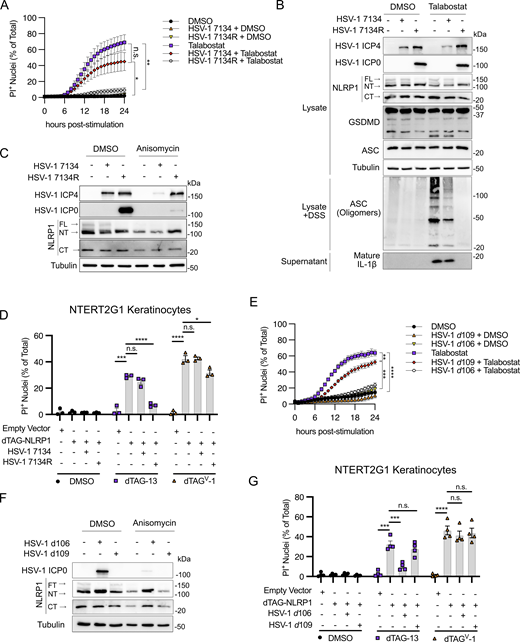
HSV-1 ICP0 is required to inhibit the NLRP1 inflammasome. (A) Kinetics of membrane permeability in primary keratinocytes infected with HSV-1 ∆ICP0 virus 7134 and the corresponding rescue 7134R at MOI 10 followed by treatment with talabostat (30 μM). Data are presented as the mean of three independent experiments ± SEM. Area under the curves was calculated for each treatment and statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01). (B) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10 for 4 h followed by treatment with talabostat (30 μM) for 24 h. Immunoblots were also run from DSS-treated lysates to assess ASC oligomerization and from protein extracted from supernatants to test for caspase-1 cleavage. Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10 for 4 h followed by treatment with anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (D) Quantification of membrane permeability from control NTERT2G1 keratinocytes (empty vector) or cells expressing dTAG-NLRP1 infected with HSV-1 7134 or 7134R (MOI 10) for 4 h followed by treatment with dTAG-13 or dTAGV-1 for 8 h. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, ***P < 0.001, ****P < 0.0001). (E) Kinetics of membrane permeability in primary human keratinocytes infected with non-replicative viruses HSV-1 d106 and d109 (MOI 10) for 4 h followed by treatment with talabostat (30 μM). Data are presented as the mean of three independent experiments ± SEM. Area under the curve was calculated for each treatment and statistical significance was calculated by Student’s t test (**P < 0.01, ***P < 0.001, ****P < 0.0001). (F) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 d106 or d109 at MOI 10 for 4 h followed by treatment with anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (G) Quantification of membrane permeability from control NTERT2G1 keratinocytes (empty vector) or cells expressing dTAG-NLRP1 infected with HSV-1 d106 or d109 for 4 h followed by treatment with dTAG-13 or dTAGV-1 for 8 h. Data are presented as the mean of four independent experiments ± SEM. Statistical significance was calculated by Student’s t test (***P < 0.001, ****P < 0.0001) Source data are available for this figure: SourceData F4.
HSV-1 ICP0 is required to inhibit the NLRP1 inflammasome. (A) Kinetics of membrane permeability in primary keratinocytes infected with HSV-1 ∆ICP0 virus 7134 and the corresponding rescue 7134R at MOI 10 followed by treatment with talabostat (30 μM). Data are presented as the mean of three independent experiments ± SEM. Area under the curves was calculated for each treatment and statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01). (B) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10 for 4 h followed by treatment with talabostat (30 μM) for 24 h. Immunoblots were also run from DSS-treated lysates to assess ASC oligomerization and from protein extracted from supernatants to test for caspase-1 cleavage. Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 ∆ICP0 virus 7134 or the 7134R rescue virus at MOI 10 for 4 h followed by treatment with anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (D) Quantification of membrane permeability from control NTERT2G1 keratinocytes (empty vector) or cells expressing dTAG-NLRP1 infected with HSV-1 7134 or 7134R (MOI 10) for 4 h followed by treatment with dTAG-13 or dTAGV-1 for 8 h. Data are presented as the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, ***P < 0.001, ****P < 0.0001). (E) Kinetics of membrane permeability in primary human keratinocytes infected with non-replicative viruses HSV-1 d106 and d109 (MOI 10) for 4 h followed by treatment with talabostat (30 μM). Data are presented as the mean of three independent experiments ± SEM. Area under the curve was calculated for each treatment and statistical significance was calculated by Student’s t test (**P < 0.01, ***P < 0.001, ****P < 0.0001). (F) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 d106 or d109 at MOI 10 for 4 h followed by treatment with anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (G) Quantification of membrane permeability from control NTERT2G1 keratinocytes (empty vector) or cells expressing dTAG-NLRP1 infected with HSV-1 d106 or d109 for 4 h followed by treatment with dTAG-13 or dTAGV-1 for 8 h. Data are presented as the mean of four independent experiments ± SEM. Statistical significance was calculated by Student’s t test (***P < 0.001, ****P < 0.0001) Source data are available for this figure: SourceData F4.
To determine if ICP0 was required to inhibit agonist-induced degradation of NT-NLRP1, we compared NT-NLRP1 abundance in anisomycin-treated primary keratinocytes infected with HSV-1 7134 or 7134R. Anisomycin treatment substantially reduced the abundance of ICP0 but not another IE protein, ICP4, in HSV-1 7134R–infected cells (Fig. 4 C). This reduction in ICP0 was not observed with other NLRP1 agonists (i.e., talabostat, Fig. 4 B) and suggests that ICP0 might be more susceptible than ICP4 to anisomycin-mediated protein synthesis inhibition. However, despite the reduced abundance of ICP0, infections with HSV-1 7134R, but not 7134, inhibited the anisomycin-mediated reduction in NT-NLRP1 protein abundance in these cells (Fig. 4 C). Consistent with these observations, HSV-1 7134 infection was unable to inhibit dTAG-13–induced membrane permeability in dTAG-NLRP1 expressing NTERT2G1 keratinocytes when compared with cells infected with HSV-1 7134R (Fig. 4 D). In contrast to observations with HSV-1 KOS (Fig. 3 D), HSV-1 7134R also significantly reduced dTAG-V1–induced PI uptake, but its effects were modest compared with its inhibition of dTAG-13–mediated PI uptake (Fig. 4 D). These findings together suggest that ICP0 is required to inhibit NT-NLRP1 degradation mediated by NLRP1 agonists.
ICP0 may act alone or in concert with other viral proteins to inhibit the NLRP1 signaling pathway. We reasoned that if ICP0 is sufficient for evasion of NLRP1, then expression of ICP0 alone in the absence of other viral proteins should inhibit the signaling pathway. To initially assess this possibility, we utilized two mutant viruses, HSV-1 d109 and d106. HSV-1 d109 has mutations in each of the IE genes or their promoters resulting in no substantial de novo viral gene expression upon infection (Samaniego et al., 1998). As such, the d109 strain does not express ICP0. By contrast, ICP0 is the only IE protein produced following infections with the d106 strain of HSV-1 (Samaniego et al., 1998). Consistent with our findings using strains that genetically lack only ICP0, we found that HSV-1 d109 (which does not express any viral genes) was unable to maximally prevent talabostat-induced PI uptake by infected primary keratinocytes (Fig. 4 E), block anisomycin-induced NT-NLRP1 degradation in primary keratinocytes (Fig. 4 F), or reduce dTAG-13–induced membrane permeability in dTAG-NLRP1 expressing NTERT2G1 keratinocytes (Fig. 4 G). Also consistent with our emerging model, HSV-1 d106 (which expresses ICP0) retained the ability to perform all three of these activities (Fig. 4, E–G). These results together strongly argue that ICP0 is the critical viral protein required for robust inhibition of the NLRP1 signaling pathway and does not require additional viral gene expression to mediate evasion.
The E3 ubiquitin ligase activity and cytoplasmic localization of HSV-1 ICP0 mediate NLRP1 signaling evasion
ICP0 has functions during infection that depend on its E3 ubiquitin ligase activity and functions that do not (Rodriguez et al., 2020). To determine the role of ICP0 enzymatic activities in NLRP1 signaling disruption, we utilized a mutant virus (KOS RING finger mutant [RFm]) that carries mutations in the RING finger domain that disrupt ubiquitin ligase functions (i.e., C116G/C156A). We also examined the rescue virus known as KOS RING finger rescue (RFr), which restores ICP0 enzymatic activity (Orzalli et al., 2012). Similar to infections with ICP0-deficient HSV-1 7134 (Fig. 4, A–C), cells infected with HSV-1 KOS RFm were unable to inhibit talabostat-induced PI uptake (Fig. 5 A), ASC oligomerization (Fig. 5 B), or anisomycin-induced NT-NLRP1 degradation (Fig. 5 C) in primary human keratinocytes. In addition, KOS RFm was unable to reduce dTAG-13–induced PI uptake in dTAG-NLRP1 expressing NTERT2G1 keratinocytes (Fig. 5 D) or talabostat-induced ASC speck formation in A549-ASC(GFP)-NLRP1 cells as compared to infections with enzymatically competent KOS RFr (Fig. S5). To further examine the role of the RING finger domain in ICP0-mediated inhibition of NLRP1 signaling, we examined ASC speck formation in talabostat-treated A549-ASC(GFP)-NLRP1 cells transfected with WT ICP0 or a mutant plasmid containing the C116G RING finger domain mutation (modeled in Fig. 5 E), which disrupts E3 ubiquitin ligase activity (Lium and Silverstein, 1997). We quantified ASC speck formation only in cells where ICP0 protein could be detected by indirect immunofluorescence. Exogenous expression of WT ICP0 or the C116G mutant did not induce ASC speck formation in A549-ASC(GFP)-NLRP1 cells in the absence of talabostat (Fig. 5 G). We found that talabostat was unable to efficiently induce ASC speck formation in cells expressing WT ICP0 (Fig. 5, F and G). By contrast, ICP0 C116G was unable to prevent talabostat-induced ASC speck formation (Fig. 5, F and G). These collective data indicate that ICP0 and its ubiquitin ligase activity are necessary and sufficient to inhibit NLRP1 inflammasome formation.
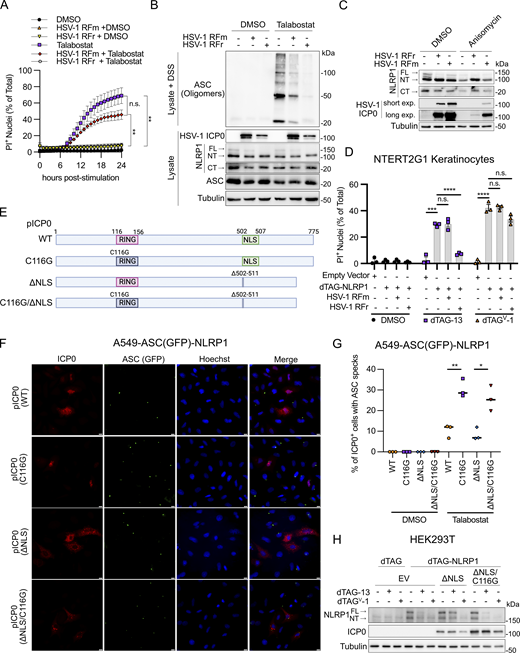
Cytoplasmic ICP0’s E3 ubiquitin ligase activity is required to inhibit the NLRP1 inflammasome. (A) Kinetics of membrane permeability in primary human keratinocytes infected with HSV-1 ICP0 RING finger mutant (RFm) and RING finger rescue (RFr) at a MOI of 10 for 4 h followed by treatment with talabostat (30 μM). Data are presented as the mean ± SEM of three independent experiments. Area under the curves were calculated for each treatment and statistical significance was calculated by Student’s t test (**P < 0.01). (B) Immunoblots of ASC oligomers in DSS-treated primary human keratinocytes infected with HSV-1 RFm or RFr at a MOI of 10 for 4 h followed by treatment with talabostat (30 μM). Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 RFm or RFr rescue virus at a MOI of 10 for 4 h followed by treatment with Anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (D) Quantification of membrane permeability from control or dTAG-NLRP1 NTERT2G1 keratinocytes infected with HSV-1 RFm or RFr (MOI 10) for 4 h followed by treatment with dTAG-13 or dTAGV-1 for 8 h. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated by Student’s t test (***P < 0.001, ****P < 0.0001). (E) Diagram of WT and mutant ICP0 constructs generated for this study and used in panels F–H. (F) Micrographs of A549-ASC(GFP)-NLRP1 cells transfected with indicated ICP0 constructs followed by TNF and talabostat (30 μM) treatment (scale bar = 10 μm). Images are representative of n = 3 biological replicates. (G) Quantification of ASC speck formation in micrographs from F. Data are presented as the mean ± SEM of three experiments. Statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01). (H) Immunoblots from HEK293T cells transfected with dTAG-NLRP1 and/or pICP0ΔNLS and pICP0ΔNLS C116G followed by treatment with dTAG-13 or dTAGV-1 for 24 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData F5.
Cytoplasmic ICP0’s E3 ubiquitin ligase activity is required to inhibit the NLRP1 inflammasome. (A) Kinetics of membrane permeability in primary human keratinocytes infected with HSV-1 ICP0 RING finger mutant (RFm) and RING finger rescue (RFr) at a MOI of 10 for 4 h followed by treatment with talabostat (30 μM). Data are presented as the mean ± SEM of three independent experiments. Area under the curves were calculated for each treatment and statistical significance was calculated by Student’s t test (**P < 0.01). (B) Immunoblots of ASC oligomers in DSS-treated primary human keratinocytes infected with HSV-1 RFm or RFr at a MOI of 10 for 4 h followed by treatment with talabostat (30 μM). Images are representative of n = 3 biological replicates. (C) Immunoblots of lysates from primary human keratinocytes infected with HSV-1 RFm or RFr rescue virus at a MOI of 10 for 4 h followed by treatment with Anisomycin (1 μM) for 24 h. Images are representative of n = 3 biological replicates. (D) Quantification of membrane permeability from control or dTAG-NLRP1 NTERT2G1 keratinocytes infected with HSV-1 RFm or RFr (MOI 10) for 4 h followed by treatment with dTAG-13 or dTAGV-1 for 8 h. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated by Student’s t test (***P < 0.001, ****P < 0.0001). (E) Diagram of WT and mutant ICP0 constructs generated for this study and used in panels F–H. (F) Micrographs of A549-ASC(GFP)-NLRP1 cells transfected with indicated ICP0 constructs followed by TNF and talabostat (30 μM) treatment (scale bar = 10 μm). Images are representative of n = 3 biological replicates. (G) Quantification of ASC speck formation in micrographs from F. Data are presented as the mean ± SEM of three experiments. Statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01). (H) Immunoblots from HEK293T cells transfected with dTAG-NLRP1 and/or pICP0ΔNLS and pICP0ΔNLS C116G followed by treatment with dTAG-13 or dTAGV-1 for 24 h. Images are representative of n = 3 biological replicates. Source data are available for this figure: SourceData F5.

HSV-1 ICP0 E3 ubiquitin ligase activity is required to inhibit the NLRP1 inflammasome in A549-ASC(GFP)-NLRP1 cells. Quantification of ASC specks in A549-ASC(GFP)-NLRP1 cells. Cells were infected with HSV-1 RFm or the RFr rescue virus at MOI 10 followed by treatment with talabostat (30 μM). Data represent the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001).
HSV-1 ICP0 E3 ubiquitin ligase activity is required to inhibit the NLRP1 inflammasome in A549-ASC(GFP)-NLRP1 cells. Quantification of ASC specks in A549-ASC(GFP)-NLRP1 cells. Cells were infected with HSV-1 RFm or the RFr rescue virus at MOI 10 followed by treatment with talabostat (30 μM). Data represent the mean of three independent experiments ± SEM. Statistical significance was calculated by Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001).
Following HSV-1 infection, ICP0 initially localizes to the nucleus but accumulates in the cytoplasm as infection progresses (Lopez et al., 2001). Given that NLRP1 inflammasome components are cytoplasmic (Yang et al., 2022) and inflammasome formation occurs in the cytoplasm of keratinocytes (Fig. 2 G), we sought to determine whether the inflammasome evasive activity of ICP0 involves its cytoplasmic localization. To test this possibility, we constructed ICP0 expression plasmids with a small internal deletion (a.a. 502–511) containing the nuclear localization signal (pICP0ΔNLS) with or without a functional RING finger domain (pICP0ΔNLS/C116G) and examined their ability to block NLRP1-dependent inflammasome formation (Fig. 5 E). As expected, ICP0ΔNLS and ICP0ΔNLS/C116G were solely localized to the cytoplasm in transfected A549-ASC(GFP)-NLRP1 cells when compared to WT ICP0, which could be detected in both the nucleus and cytoplasm (Fig. 5 F). Similar to observations with WT ICP0 transfection, we found that talabostat was unable to efficiently induce ASC speck formation in cells expressing the ΔNLS mutant, and this was dependent on ICP0 RING finger activity (Fig. 5, F and G). We further examined the ability of these ICP0 constructs to modulate dTAG-NLRP1 degradation in cotransfected HEK293T cells. Like our observations in NTERT2G1 keratinocytes infected with HSV-1 KOS (Fig. 3 E), ICP0ΔNLS expression inhibited dTAG-13, but not dTAGv-1, induced loss of dTAG-NLRP1, and this loss was restored in the presence of ICP0ΔNLS/C116G (Fig. 5 H). These data together indicate that cytoplasmic ICP0 blocks NT-NLRP1 degradation and inhibits the NLRP1 inflammasome.
ICP0 expression is required for the activation of upstream events in NLRP1 signaling in HSV-1–infected keratinocytes
If ICP0 were the sole viral protein preventing NLRP1 inflammasome formation in HSV-1–infected keratinocytes, then removal of ICP0 should allow HSV-1 to promote NLRP1-dependent pyroptosis in this cell type. However, contrary to this expectation, we did not observe hallmarks of NLRP1 inflammasome activation, including ASC oligomerization or membrane permeability, in keratinocytes infected with ICP0-deficient HSV-1 7134 (Fig. 4, A and B; and Fig. S4 C) or the ICP0 RING finger mutant HSV-1 RFm (Fig. 5, A and B). This may be explained if, in addition to inhibiting the NLRP1 pathway, ICP0 contributes to the activation of NLRP1. To test this hypothesis, we closely examined upstream signals associated with NLRP1 activation following infections with HSV-1 7134 (ICP0-deficient) and HSV-1 d106 (expresses only ICP0). MAPK activation (p-JNK) (Fig. S4 E), the shift in NT-NLRP1 size (Fig. 4, B and C; and Fig. S4 E), and loss of FL-NLRP1 (Fig. 4, B and C; and Fig. S4 E) were substantially reduced in keratinocytes infected with HSV-1 7134 when compared to infections with the ICP0 expressing rescue virus, HSV-1 7134R, suggesting that ICP0 expression was required for these phenotypes during HSV-1 infection. Further, infections with HSV-1 d106, but not d109, were sufficient to activate p38 and JNK (Fig. S4 F), promote the shift in NT-NLRP1 (Fig. 4 F and Fig. S4 F), and promote loss of FL-NLRP1 (Fig. 4 F and Fig. S4 F), indicating that ICP0 expression alone during HSV-1 infection induces these responses. These observations are consistent with ICP0 expression being critical for engaging the NLRP1 pathway and may explain the lack of inflammasome activation in cells infected with ICP0-deficient or enzymatically inactive viruses.
NLRP1 inflammasome activation inhibits the replication of ICP0-deficient HSV-1
Our findings described above suggest a working model whereby HSV-1 infection triggers the NLRP1 inflammasome, which leads to pyroptosis and restriction of viral replication. ICP0 may then have evolved a mechanism to prevent NLRP1 signaling to ensure viral replication. Central to this idea is the premise that NLRP1 inflammasome activation prevents efficient HSV-1 replication. To test this concept, we examined the effect of talabostat on the replication of the ICP0 deletion virus HSV-1 7134, which cannot evade the signaling pathway. Consistent with other studies demonstrating that ICP0 is required for robust HSV-1 replication in other human cell types (Cai and Schaffer, 1989, 1992; Everett, 1989; Stow and Stow, 1986), HSV-1 7134 replication was restricted (∼3–4 logs) in DMSO-treated human keratinocytes compared with HSV-1 7134R (Fig. 6 A). Talabostat treatment further reduced the replication of HSV-1 7134 with the greatest restriction observed at 24 and 36 h.p.i. (Fig. 6 A). By contrast, HSV-1 7134R replication was not affected by talabostat treatment, consistent with its ability to inhibit NLRP1 signaling. Because talabostat is an inhibitor of DPP8/9, it was possible that DDP8/9 activity may play a pro-viral role in HSV-1 7134–infected cells independent of its ability to restrain NLRP1. To address this possibility, we first determined whether talabostat-dependent restriction of HSV-1 7134 was dependent on caspase activity by examining viral yields in talabostat-treated keratinocytes in the presence of the pan-caspase inhibitor z-VAD-FMK. Blocking all caspase activity in keratinocytes restored HSV-1 7134 replication in the presence of talabostat (Fig. 6 B), suggesting that this restriction was caspase dependent. We next examined the requirement for NLRP1 in talabostat-mediated restriction by depleting NLRP1 from primary keratinocytes by transduction with shRNA expressing lentiviruses (Fig. 1 A). Depletion of NLRP1 by this method completely inhibited talabostat-induced PI uptake in these cells when compared with cells transduced with a non-targeting (shNT) control lentivirus (Fig. 6 C). In addition, talabostat was unable to restrict the replication of HSV-1 7134 in NLRP1-deficient cells (Fig. 6 D). Importantly, neither caspase inhibition nor NLRP1 depletion had any effect on the replication of the ICP0 expressing rescue virus HSV-1 7134R (Fig. 6, B and D). Together, these results argue that activation of the NLRP1 inflammasome can restrict HSV-1 replication and that HSV-1 ICP0 inhibits NLRP1 inflammasome signaling to overcome this antiviral activity.

NLRP1 inflammasome activation inhibits replication of ICP0-deficient HSV-1. (A) Viral yields from primary human keratinocytes infected with HSV-1 7134 or 7134R (MOI 1) followed by treatment with DMSO or talabostat (30 μM). Samples were collected for plaque assays at 6, 12, 24, and 36 h.p.i. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (**P < 0.01, ***P < 0.001). (B) Viral yields from primary human keratinocytes infected with HSV-1 7134 or 7134R (MOI 1) followed by treatment with DMSO or talabostat (30 μM) in the presence or absence of the pan-caspase inhibitor zVAD-FMK (20 μM). Samples were collected for plaque assays at 36 h.p.i. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (*P < 0.05). (C) Kinetics of membrane permeability in response to talabostat in primary human keratinocytes transduced with non-targeting (shNT) control or shNLRP1 expressing lentiviruses. Data are presented as the mean ± SEM of three independent experiments. Area under the curves were calculated for each treatment condition and statistical significance was calculated by Student's t test (***P < 0.001). (D) Viral yields from non-targeting (shNT) control or shNLRP1 expressing primary human keratinocytes infected with HSV-1 7134 or 7134R (MOI 1) followed by treatment with DMSO or talabostat (30 μM). Samples were collected for plaque assays at 36 h.p.i. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (**P < 0.01).
NLRP1 inflammasome activation inhibits replication of ICP0-deficient HSV-1. (A) Viral yields from primary human keratinocytes infected with HSV-1 7134 or 7134R (MOI 1) followed by treatment with DMSO or talabostat (30 μM). Samples were collected for plaque assays at 6, 12, 24, and 36 h.p.i. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (**P < 0.01, ***P < 0.001). (B) Viral yields from primary human keratinocytes infected with HSV-1 7134 or 7134R (MOI 1) followed by treatment with DMSO or talabostat (30 μM) in the presence or absence of the pan-caspase inhibitor zVAD-FMK (20 μM). Samples were collected for plaque assays at 36 h.p.i. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (*P < 0.05). (C) Kinetics of membrane permeability in response to talabostat in primary human keratinocytes transduced with non-targeting (shNT) control or shNLRP1 expressing lentiviruses. Data are presented as the mean ± SEM of three independent experiments. Area under the curves were calculated for each treatment condition and statistical significance was calculated by Student's t test (***P < 0.001). (D) Viral yields from non-targeting (shNT) control or shNLRP1 expressing primary human keratinocytes infected with HSV-1 7134 or 7134R (MOI 1) followed by treatment with DMSO or talabostat (30 μM). Samples were collected for plaque assays at 36 h.p.i. Data are presented as the mean ± SEM of three independent experiments. Statistical significance was calculated for individual time points by Student’s t test (**P < 0.01).
Discussion
Viral pathogens manipulate PRR-mediated innate immune defense strategies to replicate successfully in their hosts; however, whether viruses employ mechanisms to overcome guard-mediated immune responses has remained unclear. The identification of multiple viral agonists (Bauernfried et al., 2020; Robinson et al., 2020; Tsu et al., 2021; Planès et al., 2022; Yang et al., 2022; Jenster et al., 2023) and inhibitors of the NLRP1 inflammasome in this study and others (Gerlic et al., 2013; Gregory et al., 2011) suggest that this pathway plays an important role in host defense following virus infection. In this study, we provide evidence that HSV-1 stimulates and inhibits the activities of the guard protein NLRP1 (Fig. 7), and we propose that inhibiting the consequences of NLRP1 activation contributes to its success as a human pathogen.

Model figure. HSV-1 infections in primary human keratinocytes induce ICP0-dependent phosphorylation of the MAPKs p38 and JNK and the addition of posttranslational modifications to the N-terminus of NLRP1 indicative of NLRP1 activation. Instead of the formation of a functional inflammasome in these infected cells that will limit HSV-1 replication, the virus blocks the NLRP1 inflammasome pathway by inhibiting Cullin RING E3 ubiquitin ligase-dependent degradation of NT-NLRP1. This inhibitory function is also mediated by ICP0, suggesting a single virulence factor produced by HSV-1 activates and inhibits the NLRP1 inflammasome pathway. Created with https://BioRender.com.
Model figure. HSV-1 infections in primary human keratinocytes induce ICP0-dependent phosphorylation of the MAPKs p38 and JNK and the addition of posttranslational modifications to the N-terminus of NLRP1 indicative of NLRP1 activation. Instead of the formation of a functional inflammasome in these infected cells that will limit HSV-1 replication, the virus blocks the NLRP1 inflammasome pathway by inhibiting Cullin RING E3 ubiquitin ligase-dependent degradation of NT-NLRP1. This inhibitory function is also mediated by ICP0, suggesting a single virulence factor produced by HSV-1 activates and inhibits the NLRP1 inflammasome pathway. Created with https://BioRender.com.
How HSV-1 infection initially activates receptor proximal steps in the NLRP1 pathway has not been fully established. Following HSV-1 infection of human keratinocytes, we observed a shift in NT-NLRP1 size that correlated with HSV-1–mediated activation of the MAPKs p38 and JNK. Recently, MAPK family members were demonstrated to play critical roles in the activation of the NLRP1 pathway in response to pathogens and environmental inducers of ribotoxic stress (Jenster et al., 2023; Pinilla et al., 2023; Robinson et al., 2022, 2023). Specifically, ribotoxic stress activates a ZAKα-dependent MAPK signaling cascade that promotes NT-NLRP1 phosphorylation and proteasomal degradation. Thus, the observed shift in NT-NLRP1 size following HSV-1 infection may reflect MAPK-dependent NT-NLRP1 phosphorylation. HSV-1 lytic infection is not known to induce ribotoxic stress but has been reported to activate p38 and JNK in other cell types through unclear mechanisms involving the viral protein ICP27 (Hargett et al., 2005; McLean and Bachenheimer, 1999; Zachos et al., 1999). In contrast to these previous reports, we found that ICP0 was the major driver of p38/JNK activation and the shift in NT-NLRP1 size in HSV-1–infected keratinocytes, suggesting a unique interaction between ICP0 and MAPK signaling pathways in this cell type. Future studies will be necessary to determine whether ICP0 expression in HSV-1–infected keratinocytes activates ZAKα or another MAPK pathway that engages NLRP1.
In addition to changes in NT-NLRP1 size, we also observed a specific and potent proteasome-dependent loss of FL-NLRP1 in HSV-1–infected keratinocytes. The exact role that FL-NLRP1 plays in the NLRP1 inflammasome pathway has not been thoroughly explored, but previous studies suggest it can be a component of the ternary inhibitory complex and thus may act as an inhibitor of the signaling pathway. Specifically, DPP9 coimmunoprecipitates with FL-NLRP1 and the NT- and CT-NLRP1 fragments (Zhong et al., 2018), and overexpression of an uncleavable NLRP1 protein reduces inflammasome activation by free CT-NLRP1 when NT-NLRP1 is not present (Hollingsworth et al., 2021). In addition, mutations at the interface between DPP9 and the ZU5 subdomain present in both FL- and NT-NLRP1 are sufficient to activate the NLRP1 inflammasome (Hollingsworth et al., 2021). Thus, HSV-1–induced proteasome-dependent degradation of FL-NLRP1 may result in destabilization of a subset of ternary inhibitory complexes containing FL-NLRP1 in cells. Notably, we found that loss of FL-NLRP1 in HSV-1–infected cells (like MAPK activation) was dependent on ICP0 expression. Future work will therefore be necessary to differentiate between the contributions of ICP0-dependent MAPK signaling and FL-NLRP1 degradation as potential mechanisms of NLRP1 inflammasome activation in HSV-1–infected keratinocytes.
Despite observing hallmarks of NLRP1 activation in HSV-1–infected keratinocytes, we did not observe the formation of a functional NLRP1 inflammasome in these cells. Instead, we found that HSV-1 actively inhibits NLRP1 signaling. This statement is supported by biochemical evidence demonstrating decreased NT-NLRP1 degradation, ASC oligomerization, caspase-1 cleavage, and GSDMD-mediated plasma membrane permeability in HSV-1–infected cells stimulated with diverse chemical agonists (i.e., talabostat, poly(I:C), and anisomycin) of the NLRP1 inflammasome pathway. By screening viral mutants with individual gene deletions, we identified HSV-1 ICP0 as the critical viral protein responsible for this inhibition. Viral proteins from other viruses, including vaccinia virus F1L and Kaposi sarcoma-associated herpesvirus ORF63, are reported to inhibit reconstituted NLRP1 inflammasomes by directly interacting with NLRP1 (Gerlic et al., 2013; Gregory et al., 2011). We did not observe a physical interaction between ICP0 and endogenous NLRP1 in keratinocytes. Instead, ICP0 inhibits proteasomal degradation of the inhibitory NT-NLRP1 fragment that, along with DPP9, restrains the bioactive CT-NLRP1 fragment that seeds the inflammasome. The CRL family of ubiquitin ligases (CRL1-7) are implicated in NT-NLRP1 degradation in response to diverse NLRP1 stimuli, and two CRL2 complexes (CUL2ZYG11B/ZER1) have been identified as essential for viral protease-dependent NLRP1 inflammasome formation (Robinson et al., 2020). Notably, CUL2ZYG11B/ZER1 has not been implicated in inflammasome activation in response to other NLRP1 stimuli and the identity of the CRL complexes that promote NLRP1 activation under these conditions has remained elusive. Using a protein degradation system (dTAG) that recruits individual CRL complexes to target proteins, we demonstrated that ICP0 can specifically inhibit CRL4-mediated degradation of NLRP1. This observation, coupled with ICP0-mediated inhibition of endogenous NT-NLRP1 degradation, lead us to hypothesize that a CRL4 complex(es) is an important component of the NLRP1 inflammasome pathway. The requirement for CRL4 complexes in NT-NLRP1 degradation in keratinocytes and how ICP0 regulates their functions are currently being explored. Additionally, the role and identity of the CRL complexes involved in inflammasome formation initiated by mouse Nlrp1b (Chui et al., 2019; Sandstrom et al., 2019), gain-of-function mutations in human NLRP1 (Ball et al., 2019, Preprint; Zhong et al., 2016, 2018), or the closely related inflammasome sensor CARD8 (Chui et al., 2020; Hsiao et al., 2022) are either unknown or currently undefined. Studying the effect of ICP0 on the inflammasome pathways engaged by these proteins could provide additional insight into their regulation.
ICP0 is an E3 ubiquitin ligase that transactivates viral gene expression by overcoming intrinsic innate defense mechanisms that are present in the cell nucleus (reviewed in Rodriguez et al. [2020]). The ubiquitin ligase activity of ICP0 is critical for these activities in HSV-1–infected cells, and here, we demonstrate that this function is similarly required for NLRP1 inflammasome evasion. Why the E3 ubiquitin ligase activity of ICP0 is required to inhibit NLRP1 signaling is not clear since we observed no reduction in the abundance of cellular proteins known to be involved in the formation of this inflammatory complex that could account for this inhibition. This suggests the existence of additional unknown cellular proteins (e.g., CRL complexes) important for the formation of the NLRP1 inflammasome that might be targeted for degradation by ICP0. While proteomic studies in other cell types (e.g., human fibroblasts, HEK293T cells, and Neuro 2A cells) have identified several cellular targets of ICP0’s E3 ubiquitin ligase activity (Conwell et al., 2015; Hou et al., 2022; Kim et al., 2021), these studies have not yet been performed in primary human keratinocytes where the NLRP1 inflammasome pathway functions. Future studies will therefore be necessary to further define the role of the RING finger domain and novel ICP0 target proteins in inhibiting the NLRP1 signaling pathway in this cell type.
In addition to demonstrating a role for the E3 ubiquitin ligase function of ICP0 in inhibiting the NLRP1 inflammasome pathway, we also found that the subcellular localization of ICP0 plays an important role in this evasion process. ICP0 is found both in the nucleus and the cytoplasm of infected cells, and here we demonstrate that cytoplasmic ICP0 is sufficient to inhibit the NLRP1 inflammasome. The role of cytoplasmic ICP0 in HSV-1 infection is not well understood, and few functions have been attributed to its cytoplasmic localization. Studies have found that cytoplasmic ICP0 is required for its ability to block PRR-induced antiviral transcriptional responses in human embryonic lung fibroblasts (Paladino et al., 2010; Taylor et al., 2014), disrupt microtubule networks (Liu et al., 2010), and regulate autophagy adaptor protein abundance (Waisner and Kalamvoki, 2019). This contrasts with the large number of studies defining nuclear roles for ICP0 during HSV-1 infection (reviewed in Rodríguez et al. [2020]). Our study therefore expands the functions of cytoplasmic ICP0 following HSV-1 infection to include evasion of a guard-mediated host defense pathway that is present in human keratinocytes.
The consequences of NLRP1 activation in antiviral defense and viral pathogenesis have remained undefined, partly due to the lack of a small animal model that recapitulates the expression pattern and sensing activities of human NLRP1. Unlike their human counterparts, murine keratinocytes do not express homologs of NLRP1 (Sand et al., 2018) and are therefore poor models to investigate the importance of epithelial NLRP1 in host defense against HSV-1 in vivo. In this study, we, therefore, chose to investigate the antiviral consequences of NLRP1 activation in human keratinocytes, which are the initial target cell type for HSV-1 and thus represent a biologically relevant system to study its antiviral activity. Activation of NLRP1 in this cell type by talabostat restricted HSV-1 replication only when the viral inhibitor of this pathway, ICP0, was absent. The restriction of ICP0-deficient HSV-1 in keratinocytes was modest, likely a consequence of the substantial replication defect of ICP0-deficient viruses in these cells, which has been noted for other primary human cell types (Everett et al., 2004; Orzalli et al., 2013). NLRP1-mediated keratinocyte pyroptosis may have additional consequences for HSV-1 infection in cutaneous and mucocutaneous tissues where multiple cell types exist whose responses to viral infection can be shaped by the consequences of inflammasome activation. We have previously demonstrated that IL-1 cytokines released from keratinocytes promote an antiviral state in neighboring dermal fibroblasts, and this response plays an important role in cutaneous antiviral defense in human skin (Orzalli et al., 2018, 2021). Thus, NLRP1 activation following acute infection may restrict HSV-1 replication in this organ. By contrast, NLRP1 may also promote virus replication in the context of HSV-1 reactivation following establishment of latency in sensory neurons that innervate the skin. Reactivation of HSV-1 is associated with prolonged exposure to UVB light, an activator of the NLRP1 pathway (Robinson et al., 2022), and IL-1 cytokines promote the reactivation of HSV-1 from latently infected neuronal cultures in vitro (Cuddy et al., 2020). Thus, it is tempting to speculate that UVB-induced NLRP1 inflammasome activation may contribute to reactivation of HSV-1 in humans following extensive sun exposure. Future studies will be necessary to fully understand the interactions between HSV-1 and NLRP1 in human skin.
Overall, our study highlights the complex interactions between the effector-triggered NLRP1 inflammasome pathway and HSV-1 in human skin. We demonstrate that the HSV-1 virulence factor ICP0 is likely a novel agonist of the NLRP1 inflammasome in this barrier tissue, but consequently, this same viral protein inhibits this antiviral pathway to promote its replication in its human host.
Materials and methods
Cell culture
Normal human epidermal keratinocytes were derived from discarded human foreskin in accordance with University of Massachusetts (UMass) Chan Medical School Institutional Review Board #H00021295. Keratinocytes were initially cultured on mitomycin-treated 3T3 cells as described previously (Orzalli et al., 2021). Prior to experimentation, keratinocytes were transferred to keratinocyte serum-free media (KSFM) supplemented with epidermal growth factor (EGF) and bovine pituitary extract (BPE) from Gibco (#17005042). NTERT2G1 keratinocytes were also cultured in KSFM with EGF and BPE. For experiments, both primary and immortalized keratinocytes were plated at a concentration of 1.5 × 105 cells/well in a 24-well plate and incubated overnight at 37°C. Cell plating concentrations for other plate sizes were scaled up or down as a ratio of the plate well area. HEK293T and U2OS cells were cultured in high-glucose (4.5 g/liter) Dulbecco’s Modified Eagle Medium (DMEM) (#11965092; Gibco) supplemented with 5% fetal bovine serum (FBS) (#16000-044; Gibco) and 5% newborn calf serum (#11965092; Gibco) and Pen/Strep (100 U/ml) (#15140122; Gibco). A549 cells expressing NLRP1 and ASC-GFP (#a549-ascg-nlrp1; Invivogen) were cultured in high-glucose DMEM supplemented with 10% FBS, 25 mM HEPES, Pen/Strep, and 1 μg/ml blasticidin (#anti-bl-05; Invivogen).
Viruses
HSV-1 WT strains KOS (Schaffer et al., 1970) and 17syn+ were grown and titrated on Vero cells following standard procedures. HSV-1 7134, 7134R, KOS RFm, and RFr have been described previously (Cai and Schaffer, 1989; Orzalli et al., 2012) and were grown and titrated on U2OS cells. HSV-1 d106 and d109 (Samaniego et al., 1998) were grown and titrated on U2OS cells expressing ICP4 and ICP27 (Miyagawa et al., 2015).
Lentivirus production and transduction of primary keratinocytes
HEK293T cells were plated at a density of 4.5 × 106 cells/10-cm dish and incubated overnight at 37°C. The next day, the media was replaced with high-glucose DMEM supplemented with 10% FBS and Pen/Strep. Transfection reactions were set up in 500 μl serum-free high-glucose DMEM, with 6 μg of pGIPZ non-silencing control vector (UMass Chan RNAi Core, Clone ID: RHS4346) or pGIPZ shNLRP1 vectors (UMass Chan RNAi Core, Clone ID: V2LHS_386953, V2LHS_386956), 3 μg of psPAX2 (Eckard et al., 2014), and 1.5 μg pVSV-G (Eckard et al., 2014) with 40 μl polyethylenimine (1 μg/μl; Stock concentration) (#408727; Sigma-Aldrich). To generate dTAG-NLRP1 expressing keratinocytes, HEK293T cells were transfected with 6 μg dTAG-NLRP1 (plasmid # 166825; Addgene) or dTAG-empty (plasmid # 91797; Addgene) in addition to psPAX2 and pVSV-G. After 24 h, the transfection complex containing supernatants was replaced with 5 ml of fresh high-glucose DMEM supplemented with 10% FBS and Pen/Strep. Primary keratinocytes or NTERT2G1 were plated at a density of 4.5 × 106 cells per well in a 6-well plate and incubated overnight prior to transduction. Supernatants from transfected HEK293T cells were collected at 48 h after transfection and lentivirus was concentrated using Lenti-X Concentrator (#631231; Takara Bio). The lentivirus pellet was resuspended in PBS, diluted in KSFM, and added to the keratinocytes. Cells were then subjected to centrifugation at 2,000 rpm for 30 min. 24 h after transduction, primary keratinocytes were cultured in KSFM containing 1 μg/ml puromycin (#ant-pr-1; Invivogen), while NTERT2G1 cells were cultured in KSFM with 20 μg/ml puromycin for selection until all control cells were dead (∼72–96 h).
Plasmids and transfections of A549-ASC(GFP)-NLRP1 cells
A codon-optimized ICP0 cDNA based on the HSV-1 KOS ICP0 sequence (GenBank accession #KT899744) (Colgrove et al., 2016) was synthesized by TWIST Bioscience and cloned into the pTWIST EF1α vector to make pICP0. The pICP0 C116G construct was generated by site-directed mutagenesis (5′-GGATGTAGGTGCAGTCTGTACTGACGAAATTGCC-3′) of a BamH1/Kpn1 fragment of pICP0 containing the RING finger domain followed by subcloning of this mutated fragment back into the pICP0 backbone. The pICP0ΔNLS construct was generated by inverse PCR (5′-GAGAATCCTAGCCCGCAATCAACA-3′, 5′-GAGCGTGGAACAAGAAGCCGCAGTA-3′). The pICP0ΔNLS/C116G constructs were generated by subcloning the BamH1/Kpn1 fragment of pICP0 C116G containing the RING finger domain into pICP0ΔNLS. Plasmids were fully sequenced (Plasmidsaurus) to confirm identity prior to use. For transfections, A549-ASC(GFP)-NLRP1 cells were plated on coverslips in 24-well dishes at a concentration of 75,000 cells/well and incubated overnight at 37°C. Transfection complexes were generated using 2.5 μl Lipofectamine LTX (#A12621; Thermo Fisher Scientific), 0.5 μl Plus reagent, and 0.5 μg plasmid DNA diluted in 100 μl Opti-MEM Reduced Serum Media (#31985062; Gibco) per reaction. Fresh culture media was added to the cells and the DNA/Lipofectamine LTX complexes were added dropwise to each well and incubated for 24 h prior to infection/treatments.
dTAG-NLRP1 transfections of HEK293T cells
HEK293T cells were plated at a concentration of 200,000 cells/well in a 12-well plate and incubated overnight at 37°C. The next day, the media was replaced with high-glucose DMEM supplemented with 10% FBS and Pen/Strep and transfected with 975 ng of indicated ICP0 constructs and 25 ng of dTAG-NLRP1 (Addgene #166825) (Hollingsworth et al., 2021). After 24 h, the transfection complexes were removed and the cells treated with DMSO, dTAG-13 (500 nM), or dTAGV-1 (500 nM) for 24 h before collecting lysates for immunoblotting.
Viral infections and chemical treatments
Infections of keratinocytes and A549-NLRP1-ASC-GFP cells were performed in KSFM or DMEM supplemented with 1% FBS and Pen/Strep (DMEV), respectively, at the specified multiplicity of infection (MOI). Viruses were diluted in media, added to cells, and incubated for an hour at 37°C with gentle shaking. After virus adsorption, the viral inoculum was aspirated and replaced with fresh media containing 1% FBS (A549-ASC(GFP)-NLRP1 cells) or KSFM (keratinocytes). At 4 h after infection, cells were treated with 30 μM talabostat (#HY-13233; MedChemExpress) or DMSO, transfected with poly(I:C) (1 μg/ml) (#tlr-pic; Invivogen) or LPS (1 μg/ml) (#tlrl-peklps; Invivogen) using Lipofectamine LTX, or treated with 10 μM anisomycin (#HY-18982; MedChemExpress) and incubated for the indicated time points at 37°C. Bortezomib (#HY-10227; MedChemExpress) was resuspended in DMSO, added to cells 1 h after infection, and used at a final concentration of 10 μM. PAA (#S1885; Sigma-Aldrich) treatments were done at 125 μg/ml. Cells were treated with dTAG-13 (#SML2601; Sigma-Aldrich) or dTAGV-1 (#HY-145514; MedChemExpress) at a final concentration of 500 nM. Treatments with zVAD-FMK (#tlrl-VAD; Invivogen) were done at 20 μM.
Plaque assays
Lysate samples for plaque assays were collected by adding an equal volume of autoclaved 10% instant nonfat dry milk (Nestle Carnation, Amazon) (wt/vol) in water to each infected well. The samples were frozen at −80°C and thawed on ice prior to use. Thawed samples were serially diluted in DMEV, added to a monolayer of Vero or U2OS cells in a 6-well dish, and incubated for 1 h at 37°C with gentle shaking. The virus was then aspirated and overlaid with DMEV supplemented with purified human immunoglobulin (IgG) (#I4506; Sigma-Aldrich) and incubated at 37°C for 72 h. Each lot of human IgG was empirically tested to define the concentration required to inactivate extracellular virus. Assays were terminated by fixing the cells with ice-cold methanol followed by staining with crystal violet. The plates were washed and dried, and plaques were counted to calculate viral titers.
SDS-PAGE and immunoblots
Cells were lysed in 1× Laemmli Sample buffer (#1610747; BioRad) supplemented with β-mercaptoethanol (BME) and boiled for 10 min. Samples were run on 6, 10, or 15% Tris-Glycine gels, transferred onto polyvinylidene fluoride membranes overnight at 4°C, and blocked for 30 min in TBST (Tris Buffered Saline with 0.05% Tween) containing 5% milk followed by overnight incubation with primary antibodies (1:1,000) in TBST containing 2.5% bovine serum albumin (TBST-BSA). The blots were washed 3× in TBST for 10 min, incubated with goat anti-rabbit or anti-mouse HRP-tagged secondary antibodies (#111-035-003, #115-035-003, 1:5,000; Jackson Labs) at room temperature for 1 h in TBST-BSA followed by 2× washes in TBST and 1× TBS wash for 10 min each. The blots were then developed using Pico or Femto SuperSignal western blot substrate (Thermo Fisher Scientific) on a Chemidoc MP imaging system (BioRad). Primary antibodies used for immunoblots were anti-NALP1 (#56719; Cell Signaling), anti-NLRP1 (#679802; BioLegend), anti-HSV-1 ICP0 (#H1A027; East Coast Bio), anti-HSV-1 ICP4 (H1A021-100; Virusys), anti-ASC (#sc514414; Santa Cruz), anti-GSDMD (#97558; Cell Signaling), anti-CASP1 (#2225; Cell Signaling), anti-IL1β (#12703; Cell Signaling), anti-FKBP12 (#AB24373; Abcam), anti-p38 (#9212; Cell Signaling), anti-pp38 (#4511T; Cell Signaling), anti-JNK (#9252; Cell Signaling), anti-p-JNK (#9251; Cell Signaling), and anti-β-Tubulin (#86298S; Cell Signaling).
Disuccinimidyl suberate (DSS) oligomerization
DSS oligomerization experiments were performed on keratinocytes from 12-well plates that were initially seeded at a density of 300,000 cells/well. Cells were collected by trypsinization and washed 3× with PBS. Cell pellets were then lysed with 200 μl of 1% IGEPAL in PBS on ice for 10 min. The samples were centrifuged at 20,000 g for 10 min and the pellet was then resuspended in 100 μl of PBS containing 1 mM DSS (#S1885; Sigma-Aldrich), followed by incubation with rotation at 37°C for 15 min. Following centrifugation at 20,000 g, the insoluble pellet was resuspended in 1× Laemmli Buffer containing 2.5% BME. Samples were boiled for 10 min before proceeding with SDS-PAGE and immunoblot.
Membrane permeability assays
All PI uptake assays were conducted on cells in 96-well plates. Cells were overlaid with media supplemented with 1:30,000 Hoechst 33342 (#P3566; Invitrogen) and 1:10,000 PI (#H3570; Invitrogen) and imaged hourly for 24 h using a Lionheart FX automated microscope (Biotek). Image analysis was performed using Gen5 software, and ratios of PI+ to Hoechst+ cells were plotted as a measurement of the percentage of membrane-permeable cells.
Protein extraction from cell culture supernatants
Cell culture supernatants were collected from experiments in 12-well plates. The supernatant samples were centrifuged at 300 g for 5 min to remove cell debris. The supernatants were then transferred to a microcentrifuge tube containing an equal volume of methanol and one-fourth the volume of chloroform. This mixture was vortexed for 20 s and centrifuged at 20,000 g for 10 min at room temperature. The upper phase was removed and 500 μl of methanol was added to the intermediate phase. The samples were vortexed followed by centrifugation at 20,000 g. The supernatant was removed, and the resulting pellet was dried at 55°C for 10 min and then resuspended in 1× Laemmli Buffer supplemented with BME.
Immunofluorescence microscopy
Cells were plated on glass coverslips on a 24-well plate and incubated overnight. At indicated time points after infections and treatments, the cells were washed with PBS and fixed in 4% paraformaldehyde at room temperature for 10 min. The cells were then washed with PBS containing 0.1% Tween20 (PBST), followed by permeabilization with 0.5% NP-40 for 10 min. The cells were washed again and blocked with 5% Normal Goat Serum (#005-000-121; Jackson ImmunoResearch) in PBS overnight at 4°C. Blocked cells were washed and incubated with primary antibody (1:100) at 37°C for 2 h followed by incubation with secondary antibody (1:500) and Hoechst 33342 (1:20,000) for 1 h in the dark. The cells were washed 2× with PBST, 1× with PBS, and the coverslips were mounted on slides using ProLong Diamond Antifade mounting reagent (#P36965; Jackson ImmunoResearch). Images of the cells were taken on a Leica DMi8S Inverted Microscope using 63× oil immersion magnification and processed with the Thunder Imaging System. Primary antibodies used for immunofluorescence were anti-HSV-1 ICP0 (#sc53070; SantaCruz), anti-ASC (#sc22514R; SantaCruz), and anti-GFP (#2956S; Cell Signaling). Alexa Fluor 488 goat anti-rabbit IgG H+L (#A11008) and Alexa Fluor 568 goat anti-mouse IgG H+L (#A11031) secondary antibodies were from Thermo Fisher Scientific.
ASC speck quantification
Keratinocytes and A549-ASC(GFP)-NLRP1 cells were plated on coverslips at a density of 150,000 cells/well and 75,000 cells/well, respectively, and incubated for 24 h at 37°C prior to experimentation. Keratinocytes were infected with HSV-1 for 1 h as described above, followed by treatment with DMSO or 30 μM talabostat for 24 h. Cells were fixed and stained for ASC according to the immunofluorescence microscopy protocol described above. 10 representative images from each condition were taken at 10× magnification and ASC speck+ cells were counted and plotted as a percentage of the total number of cells imaged. A549-ASC(GFP)-NLRP1 cells were treated with 2.5 ng/ml TNF (#5701012; BioLegend) for 6 h, after which the cells were either mock-infected or infected with HSV-1. At 1 h.p.i., the cells were treated with Hoechst (1:30,000) to stain nuclei and either DMSO or 30 µM talabostat and imaged every 2 h on a Lionheart FX automated microscope. ASC-GFP speck+ cells were quantified using Gen5 software (BioTek). For transfection experiments, DMSO or talabostat-treated cells were fixed and stained with anti-GFP and anti-ICP0 primary antibodies and fluorescently conjugated secondary antibodies following the immunofluorescence microscopy protocol described above. 1,000 ICP0+ cells were examined per experiment for the presence of ASC-GFP specks.
ELISA
IL1β ELISAs were performed using the ELISA MAX Deluxe Set (Cat#437004; BioLegend) per the kit’s protocol. Cell supernatants from experiments were first centrifuged at 300 g for 5 min to remove cell debris or floating cells.
Online supplemental material
Fig. S1 contains supporting data demonstrating HSV-1–mediated degradation of FL-NLRP1. Fig. S2 demonstrates that HSV-1 infection inhibits NLRP1 inflammasome activation in an A549-ASC(GFP)-NLRP1 reporter cell line. Fig. S3 shows confirmation that talabostat-induced inflammasome activation in primary human keratinocytes is proteasome and neddylation dependent. Fig. S4 contains supporting data demonstrating that HSV-1 ICP0 expression is required for activation and inhibition of the NLRP1 inflammasome pathway. Fig. S5 demonstrates that ICP0’s E3 ubiquitin ligase activity inhibits NLRP1 inflammasome activation in an A549-ASC(GFP)-NLRP1 reporter cell line.
Data availability
All data underlying the findings of this study are available as supplementary files included in this publication or from the corresponding author upon request.
Acknowledgments
We thank the members of the Fitzgerald lab (UMass Chan) for their insight during the development of this study. We thank Dr. Jonathan Kagan, Dr. Read Pukkila-Worley, and Dr. Melanie Trombly for their comments on the manuscript.
Work for this study was supported by National Institutes of Health grant R00AI130258 (to M.H. Orzalli), the Smith Family Award for Excellence in Biomedical Research from the Richard and Susan Smith Family Foundation (to M.H. Orzalli), and startup funds from UMass Chan Medical School.
Author contributions: P. Parameswaran: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing - original draft, Writing - review & editing, L. Payne: Investigation, J. Powers: Investigation, Writing - review & editing, M. Rashighi: Conceptualization, Methodology, Resources, Writing - review & editing, M.H. Orzalli: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing.

