


Phenotypic plasticity is a rising cancer hallmark, and lung adeno-to-squamous transition (AST) triggered by LKB1 inactivation is significantly associated with drug resistance. Mechanistic insights into AST are urgently needed to identify therapeutic vulnerability in LKB1-deficient lung cancer. Here, we find that ten-eleven translocation (TET)–mediated DNA demethylation is elevated during AST in KrasLSL-G12D/+; Lkb1L/L (KL) mice, and knockout of individual Tet genes reveals that Tet2 is required for squamous transition. TET2 promotes neutrophil infiltration through STAT3-mediated CXCL5 expression. Targeting the STAT3–CXCL5 nexus effectively inhibits squamous transition through reducing neutrophil infiltration. Interestingly, tumor-infiltrating neutrophils are laden with triglycerides and can transfer the lipid to tumor cells to promote cell proliferation and squamous transition. Pharmacological inhibition of macropinocytosis dramatically inhibits neutrophil-to–cancer cell lipid transfer and blocks squamous transition. These data uncover an epigenetic mechanism orchestrating phenotypic plasticity through regulating immune microenvironment and metabolic communication, and identify therapeutic strategies to inhibit AST.
Introduction
Phenotypic plasticity is a rising hallmark of cancer (Hanahan, 2022). For instance, adeno-to-squamous transition (AST) in lung cancer represents a typical histological transformation associated with strong cancer plasticity, the capability to switch between different lineages (Hou et al., 2016; Kanazawa et al., 2000; Qin et al., 2024). Clinical data show that squamous transition occurs in certain patients with lung adenocarcinoma (ADC) who relapse after molecularly targeted therapy using tyrosine-kinase inhibitors or KRAS inhibitor (Lin et al., 2020; Schoenfeld et al., 2020; Awad et al., 2021). Somatic loss-of-function mutations of LKB1 (also named serine-threonine kinase 11, STK11) are frequently observed in lung cancer and associated with strong cancer plasticity (Ji et al., 2007; Koivunen et al., 2008; Li et al., 2015; Tong et al., 2024). LKB1 is mutated in ∼17% of human ADC (Quintanal-Villalonga et al., 2021a). In contrast, human lung adenosquamous cell carcinoma (AdSCC), which features mixed pathologies containing both adenomatous and squamous lesions within single tumors and is associated with worse patient survival, displays an enrichment of LKB1 mutations at 39.7%, ranging from 22% to 66% according to multiple studies (Matsumoto et al., 2007; Koivunen et al., 2008; Zhang et al., 2017; Krause et al., 2020; Quintanal-Villalonga et al., 2021a; Tang et al., 2023). Quintanal-Villalonga et al. (2021a) have recently analyzed LKB1 mutations in lung tumors with squamous transition after drug resistance acquisition. This study shows an occurrence rate of STK11/LKB1 mutations at 14.3% (one out of seven) in relapsed patients with potential AST, largely due to their mutual exclusivity with epidermal growth factor receptor (EGFR) mutations (Sanchez-Cespedes et al., 2002; Mahoney et al., 2009; Ab Mutalib et al., 2014; Quintanal-Villalonga et al., 2021a). When samples with EGFR mutations were excluded from this dataset, the STK11/LKB1 mutation was observed in one out of three samples (33%) (Quintanal-Villalonga et al., 2021a). Previous studies show a prevalence of LKB1 mutations at about 39.7% in human lung AdSCC, similar to that observed in relapsed patients with potential AST (Matsumoto et al., 2007; Koivunen et al., 2008; Zhang et al., 2017; Krause et al., 2020; Quintanal-Villalonga et al., 2021a; Tang et al., 2023).
Consistent with these clinical observations, LKB1 deficiency has been proven to be able to drive transition from ADC to squamous cell carcinoma in a KrasLSL-G12D/+; Lkb1L/L (KL) mouse model (Han et al., 2014; Gao et al., 2014; Li et al., 2015; Zhang et al., 2017). We have previously shown that a decreased lysyl oxidase level and consequent extracellular matrix (ECM) remodeling serve as the important trigger for AST in the KL model as well as in KrasLSL-G12D/+; Trp53L/L mouse model, a classical ADC model (Han et al., 2014; Yao et al., 2018, Preprint). Moreover, we find that yes-associated protein (YAP) inactivation resulting from ECM remodeling precedes the appearance of squamous biomarker p63 and histological transformation in the KL model (Gao et al., 2014). The ECM remodeling seems associated with uncontrolled accumulation of excessive oxidative stress, which accelerates the process of AST (Li et al., 2015). We also find that histone methyltransferase EZH2 inhibits squamous transition by altering the expression of squamous differentiation-related genes (Zhang et al., 2017). Despite all these efforts, the epigenetic regulatory mechanisms in AST, especially associated with DNA methylation status, still remain elusive.
DNA methylation is reversibly and delicately regulated by multiple enzymes including DNA methyltransferases (DNMTs) and ten-eleven translocation (TET) family of DNA hydroxylases (Yin et al., 2017; Wu and Zhang, 2014). The TET family of proteins are composed of TET1, TET2, and TET3, which catalyze serial oxidation reactions: converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), then to 5-formylcytosine, and eventually to 5-carboxylcytosine (Tahiliani et al., 2009; Ito et al., 2011; He et al., 2011). Extensive studies have highlighted the tumor-suppressive role of TET family proteins in leukemia. Our recent work using a genetically engineered mouse model (GEMM) also identifies TET2 as a tumor suppressor in Kras-driven ADC (Xu et al., 2022). However, whether TET enzymes participate in AST is unclear.
Increased neutrophil infiltration is frequently observed during the AST process and is associated with squamous pathology (Xu et al., 2014; Koyama et al., 2016). Previous studies also show that temporary depletion of neutrophils could dampen squamous transition (Koyama et al., 2016; Mollaoglu et al., 2018). However, it remains unclear how neutrophil accelerates the AST process. Tumor-infiltrating neutrophils are known to promote angiogenesis and immunosuppression in the tumor microenvironment by releasing proinflammatory factors (Dumitru et al., 2013). A recent study also shows that neutrophil extracellular traps might help attract cancer cells to form distant metastases (Yang et al., 2020). A later study finds that lung neutrophils are laden with lipids and create a premetastatic niche for breast cancer (Li et al., 2020a). It remains interesting to investigate how lung neutrophil contributes to AST.
With the integrative analyses of GEMMs and clinical specimens, we here identify TET2 as an important epigenetic regulator of AST. Our results also link the TET2-mediated epigenetic mechanism to neutrophil infiltration and neutrophil–cancer cell lipid transfer during the AST process and identify multiple therapeutic strategies to inhibit lung AST.
Results
Tet2 knockout (KO) inhibits lung adeno-to-squamous transition
To study the potential link between DNA methylation and AST, we first analyzed the expression of DNA methylation regulators in normal lungs, ADC, and squamous cell carcinoma (SCC) from the KL model (Fig. 1 A). We found that KL-SCC exhibited the upregulation of Dnmt1, Dnmt3a, and Uhrf1 when compared with KL-ADC. Upregulation of Tet2 and Tet3, but not Tet1, was also observed in KL-SCC (Fig. 1 A). Using the TCGA-NSCLC datasets, we found that human lung squamous cell carcinoma (LUSC) exhibited upregulation of DNMT and TET gene expression compared with human lung adenocarcinoma (LUAD) (Fig. S1 A), indicative of a potential link of DNMT or TET with lung cancer pathologies. We focused on the roles of TETs in this process. In agreement with the upregulation of TET expression, immunohistochemistry (IHC) staining demonstrated that the levels of 5mC and 5hmC were higher in KL-SCC than KL-ADC (Fig. S1, B–D), further implying a potential role of TET and DNA demethylation in lung AST.
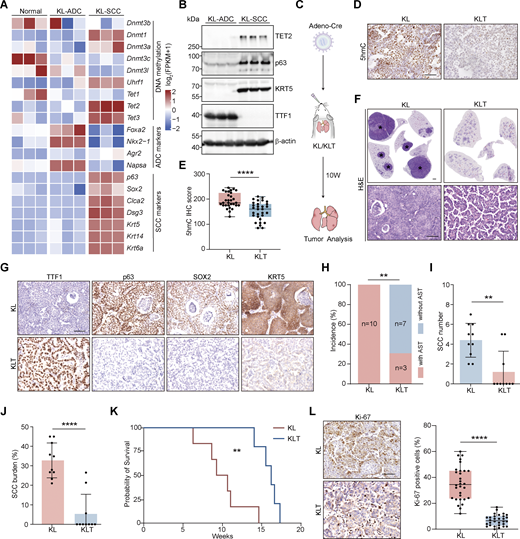
Tet2 KO inhibits squamous transition in KL model. (A) Heatmap of DNA methylation factors and adeno/squamous lineage markers in normal lung, ADC, and SCC of the KL mouse model. (B) Western blot analysis for TET2, p63, KRT5, and TTF1 in KL-ADC and KL-SCC tumors. β-Actin served as the internal loading control. Parallel blots using the same samples. (C) Schematic illustration of the KL and KLT mouse model. W, week. (D and E) (D) Representative 5hmC IHC staining of KL and KLT tumors. Scale bar: 50 μm. Statistical analysis is also shown (E). 30 representative images for each group were counted. (F) Representative H&E staining for whole lungs of KL and KLT models. The whole lungs are reused in Fig. S2 E. * indicates SCC. Scale bar: 1 mm. H&E staining with high magnification is shown. Scale bar: 50 μm. (G) Representative IHC staining for TTF1, p63, SOX2, and KRT5 in KL and KLT mice. Scale bar: 50 μm. (H–J) Statistical analyses of the AST incidence (H), the number (I), and burden (J) of SCC in KL (n = 10) and KLT (n = 10) mice. (K) Kaplan–Meier survival curves of KL (n = 6) and KLT (n = 5) mice. (L) Representative Ki-67 IHC staining and statistical analyses in KL and KLT tumors. Scale bar: 50 μm. 30 representative images for each group were counted. Data in B, D, and E represent one experiment of three independent repeats, and F–L represents one experiment of two independent repeats. **P < 0.01, ****P < 0.0001 by two-tailed unpaired Student’s t test (E, I, J, and L), Fisher’s exact test (H), log-rank (Mantel–Cox) test (K). Data are represented as mean ± SD. Source data are available for this figure: SourceData F1.
Tet2 KO inhibits squamous transition in KL model. (A) Heatmap of DNA methylation factors and adeno/squamous lineage markers in normal lung, ADC, and SCC of the KL mouse model. (B) Western blot analysis for TET2, p63, KRT5, and TTF1 in KL-ADC and KL-SCC tumors. β-Actin served as the internal loading control. Parallel blots using the same samples. (C) Schematic illustration of the KL and KLT mouse model. W, week. (D and E) (D) Representative 5hmC IHC staining of KL and KLT tumors. Scale bar: 50 μm. Statistical analysis is also shown (E). 30 representative images for each group were counted. (F) Representative H&E staining for whole lungs of KL and KLT models. The whole lungs are reused in Fig. S2 E. * indicates SCC. Scale bar: 1 mm. H&E staining with high magnification is shown. Scale bar: 50 μm. (G) Representative IHC staining for TTF1, p63, SOX2, and KRT5 in KL and KLT mice. Scale bar: 50 μm. (H–J) Statistical analyses of the AST incidence (H), the number (I), and burden (J) of SCC in KL (n = 10) and KLT (n = 10) mice. (K) Kaplan–Meier survival curves of KL (n = 6) and KLT (n = 5) mice. (L) Representative Ki-67 IHC staining and statistical analyses in KL and KLT tumors. Scale bar: 50 μm. 30 representative images for each group were counted. Data in B, D, and E represent one experiment of three independent repeats, and F–L represents one experiment of two independent repeats. **P < 0.01, ****P < 0.0001 by two-tailed unpaired Student’s t test (E, I, J, and L), Fisher’s exact test (H), log-rank (Mantel–Cox) test (K). Data are represented as mean ± SD. Source data are available for this figure: SourceData F1.
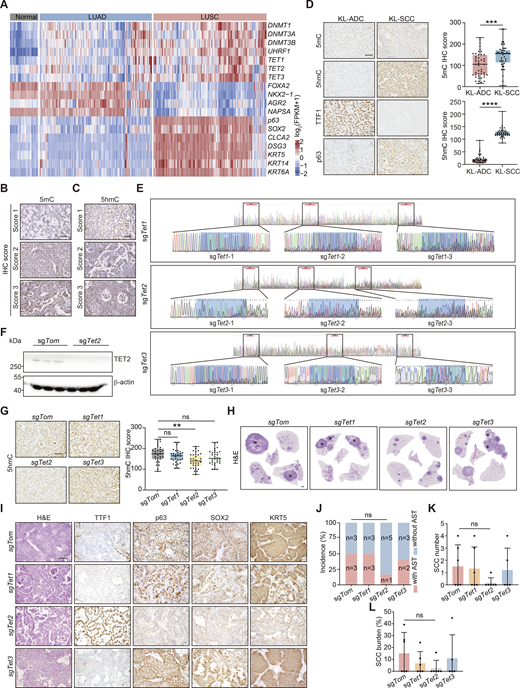
CRISPR-mediated Tet2 KO inhibits squamous transition in KL model. (A) Heatmap of gene expression of DNA methylation regulators and lineage-defined biomarkers in human normal lung, LUAD, and LUSC (from TCGA dataset). (B) Representative IHC staining of various levels of 5mC (relative scores: 1, 2, 3). Scale bar: 50 μm. (C) Representative IHC staining of various intensity of 5hmC (relative scores: 1, 2, 3). Scale bar: 50 μm. (D) Representative IHC staining of 5mC, 5hmC, TTF1, and p63 in KL-ADC and KL-SCC. Scale bar: 50 μm. Statistical analyses are shown on the right. 45 representative images for each group were counted. (E) KO efficiency detection of sgTet1/2/3 through Sanger sequence. The targeting site is marked by box. (F) KO efficiency detection of sgTet2 through western blot in tumors with Tet2 KO using lentivirus in KL mice. β-Actin served as the internal loading control. (G) Representative IHC staining of 5hmC in KL lung tumors with Tet1/2/3 individual KOs. Scale bar: 50 μm. Statistical analyses are shown on the right. 30 representative images for each group were counted. (H) Representative H&E staining of whole mouse lungs with Tet1/2/3 individual KOs. * indicates SCC. Scale bar: 1 mm. (I) Representative H&E and IHC staining for TTF1, p63, SOX2, and KRT5 in mouse lung tumors with Tet1/2/3 individual KOs. Scale bar: 50 μm. (J–L) Statistical analyses of the AST incidence (J), the number (K), and burden (L) of SCC in KL mice with Tet1/2/3 individual KOs (sgTom: n = 6; sgTet1: n = 6; sgTet2: n = 6; sgTet3: n = 5). Data in B–F represent one experiment of three independent repeats, and G–L represent one experiment of two independent repeats. **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (D), one-way ANOVA test (G, K and L), Fisher’s exact test (J). ns: not significant. Data are represented as mean ± SD. Source data are available for this figure: SourceData FS1.
CRISPR-mediated Tet2 KO inhibits squamous transition in KL model. (A) Heatmap of gene expression of DNA methylation regulators and lineage-defined biomarkers in human normal lung, LUAD, and LUSC (from TCGA dataset). (B) Representative IHC staining of various levels of 5mC (relative scores: 1, 2, 3). Scale bar: 50 μm. (C) Representative IHC staining of various intensity of 5hmC (relative scores: 1, 2, 3). Scale bar: 50 μm. (D) Representative IHC staining of 5mC, 5hmC, TTF1, and p63 in KL-ADC and KL-SCC. Scale bar: 50 μm. Statistical analyses are shown on the right. 45 representative images for each group were counted. (E) KO efficiency detection of sgTet1/2/3 through Sanger sequence. The targeting site is marked by box. (F) KO efficiency detection of sgTet2 through western blot in tumors with Tet2 KO using lentivirus in KL mice. β-Actin served as the internal loading control. (G) Representative IHC staining of 5hmC in KL lung tumors with Tet1/2/3 individual KOs. Scale bar: 50 μm. Statistical analyses are shown on the right. 30 representative images for each group were counted. (H) Representative H&E staining of whole mouse lungs with Tet1/2/3 individual KOs. * indicates SCC. Scale bar: 1 mm. (I) Representative H&E and IHC staining for TTF1, p63, SOX2, and KRT5 in mouse lung tumors with Tet1/2/3 individual KOs. Scale bar: 50 μm. (J–L) Statistical analyses of the AST incidence (J), the number (K), and burden (L) of SCC in KL mice with Tet1/2/3 individual KOs (sgTom: n = 6; sgTet1: n = 6; sgTet2: n = 6; sgTet3: n = 5). Data in B–F represent one experiment of three independent repeats, and G–L represent one experiment of two independent repeats. **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (D), one-way ANOVA test (G, K and L), Fisher’s exact test (J). ns: not significant. Data are represented as mean ± SD. Source data are available for this figure: SourceData FS1.
We then performed CRISPR/Cas9-mediated Tet (Tet1/2/3) gene KO in the KL model through lentivirus nasal inhalation as previously described (Sánchez-Rivera et al., 2014; Wu et al., 2018). DNA sequencing confirmed the proper edits in the targeted alleles (Fig. S1 E). We further confirmed the KO efficiency of Tet2 by western blot analysis (Fig. S1 F). We observed a remarkable decrease in global 5hmC levels in Tet2 KO tumors, but not Tet1 KO or Tet3 KO tumors (Fig. S1 G). Interestingly, only Tet2 KO dramatically suppressed squamous transition (Fig. S1 H), as evidenced by the fact that most tumors showed ADC pathology with high TTF1 expression and no p63, SOX2, and KRT5 expression (Fig. S1 I). Tet2 KO also reduced the AST incidence, the number, and burden of SCC in the KL model (Fig. S1, J–L). In agreement with the transcriptome data (Fig. 1 A), western blot data further confirmed the upregulation of TET2 protein in KL-SCC (Fig. 1 B). These results indicate that Tet2, but not Tet1 or Tet3, may be important for AST in the KL model.
We next generated a conditional KrasLSL-G12D/+; Lkb1L/L; Tet2L/L (KLT) mouse strain for AST analyses (Fig. 1 C). Tet2 KO efficiency was confirmed by western blot analysis (Fig. S2 A). Further PCR analysis verified Tet2 KO in cancer cells (CD45−/EpCAM+) but not in immune cells (CD45+/EpCAM−) isolated from KLT lung tumors (Fig. S2 B). In agreement with our earlier finding (Fig. S1 G), Tet2 KO resulted in a dramatic reduction of 5hmC level (Fig. 1, D and E). We found that Tet2 KO increased the growth of ADC (Fig. S2, C and D), consistent with our previous study about the suppressive role of Tet2 in KrasG12D-driven ADC (Xu et al., 2022). Interestingly, Tet2 KO dramatically suppressed squamous transition (Fig. 1, F and G; and Fig. S2 E). Statistical analyses also showed a significant decrease of AST incidence, the number, and burden of SCC in KLT mice (Fig. 1, H–J). Moreover, the KLT mouse survival was prolonged compared with KL mice (Fig. 1 K). IHC staining of Ki-67 confirmed a remarkable decrease in cell proliferation in KLT total lung tumors (Fig. 1 L). These data together identify TET2 as an important regulator of AST in the KL model.
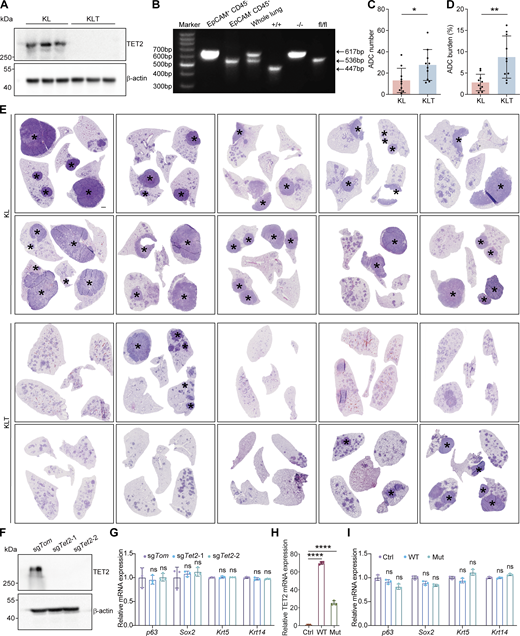
Tet2 KO inhibits lung AST in KL model. (A) Western blot analysis for TET2 in KL and KLT tumors. β-Actin served as the internal loading control. (B) KO efficiency detection of Tet2 in cancer cells (CD45− EpCAM+), tumor-associated immune cells (CD45+ EpCAM−), and whole lung isolated from KLT tumors. “+/+” means the lung tissue isolated from wild-type mice. “−/−” means the homozygous deletion of Tet2 gene alleles. “fl/fl” means the floxed Tet2 gene alleles with intact gene expression before Cre administration. (C and D) Statistical analyses of number (C) and burden (D) of ADC in KL (n = 10) and KLT (n = 10) mice after 10 wk of Adeno-Cre treatment. (E) Representative H&E staining of whole lungs from KL and KLT mice. * indicates SCC. Scale bar: 1 mm. (F) Detection of TET2 protein in the KL squamous organoids with CRISPR-mediated Tet2 KO by western blotting. β-Actin serves as an internal loading control. (G) Relative mRNA expression of p63, Sox2, Krt5, and Krt14 in the KL squamous organoids upon Tet2 KO by RT-qPCR. (H) Relative mRNA expression of TET2 in the KL squamous organoids after overexpressing wild-type (WT) and inactive mutant (Mut) TET2 by RT-qPCR. (I) Relative mRNA expression of p63, Sox2, Krt5, and Krt14 in the KL squamous organoids after overexpressing wild-type (WT) and inactive mutant (Mut) TET2 by RT-qPCR. Data in A, B, and F–I represent one experiment of three independent repeats, and C–E represent one experiment of two independent repeats. *P < 0.05, **P < 0.01 by two-tailed unpaired Student’s t test (C and D), ****P < 0.0001 by one-way ANOVA test (G–I). ns: not significant. Data are represented as mean ± SD. Source data are available for this figure: SourceData FS2.
Tet2 KO inhibits lung AST in KL model. (A) Western blot analysis for TET2 in KL and KLT tumors. β-Actin served as the internal loading control. (B) KO efficiency detection of Tet2 in cancer cells (CD45− EpCAM+), tumor-associated immune cells (CD45+ EpCAM−), and whole lung isolated from KLT tumors. “+/+” means the lung tissue isolated from wild-type mice. “−/−” means the homozygous deletion of Tet2 gene alleles. “fl/fl” means the floxed Tet2 gene alleles with intact gene expression before Cre administration. (C and D) Statistical analyses of number (C) and burden (D) of ADC in KL (n = 10) and KLT (n = 10) mice after 10 wk of Adeno-Cre treatment. (E) Representative H&E staining of whole lungs from KL and KLT mice. * indicates SCC. Scale bar: 1 mm. (F) Detection of TET2 protein in the KL squamous organoids with CRISPR-mediated Tet2 KO by western blotting. β-Actin serves as an internal loading control. (G) Relative mRNA expression of p63, Sox2, Krt5, and Krt14 in the KL squamous organoids upon Tet2 KO by RT-qPCR. (H) Relative mRNA expression of TET2 in the KL squamous organoids after overexpressing wild-type (WT) and inactive mutant (Mut) TET2 by RT-qPCR. (I) Relative mRNA expression of p63, Sox2, Krt5, and Krt14 in the KL squamous organoids after overexpressing wild-type (WT) and inactive mutant (Mut) TET2 by RT-qPCR. Data in A, B, and F–I represent one experiment of three independent repeats, and C–E represent one experiment of two independent repeats. *P < 0.05, **P < 0.01 by two-tailed unpaired Student’s t test (C and D), ****P < 0.0001 by one-way ANOVA test (G–I). ns: not significant. Data are represented as mean ± SD. Source data are available for this figure: SourceData FS2.
Tet2 KO inhibits neutrophil infiltration into KL tumor microenvironment
We further checked if TET2 regulated the gene expression of squamous lineage markers. We found that Tet2 KO in KL squamous organoids didn’t affect the expression of p63, Sox2, Krt5, and Krt14 (Fig. S2, F and G). Moreover, ectopic expression of wild-type TET2 or its catalytically inactive mutant (Xu et al., 2022) failed to alter the expression of squamous markers (Fig. S2, H and I). These results indicate that Tet2 might not directly regulate squamous transition.
Tumor immune microenvironment is frequently observed with an alteration during lung AST, e.g., fewer T lymphocytes with higher expression of immune checkpoint receptor and increased numbers of neutrophils were detected in KL model when compared with KrasLSL−G12D/+; Trp53L/L(KP) model, a typical ADC mouse model (Koyama et al., 2016). Through transcriptomic analyses of KL-ADC and KL-SCC using gene set variation analysis (GSVA), we found that inflammatory signaling pathways, especially the interferon (IFN) pathway, were significantly enriched in KL-SCC (Fig. S3 A). Consistently, immunofluorescence (IF) staining for myeloperoxidase (MPO) showed that neutrophil infiltration was remarkably increased in KL-SCC (Fig. S3, B and C).

TET2 promotes neutrophil infiltration potentially through the upregulation of CXCL5 expression. (A) Heatmap of inflammatory and IFN signaling gene expression in KL tumors at serial time points. W, week. Both ADC and SCC were isolated from 8-wk time point, e.g., 8W-A means ADC and 8W-S means SCC. (B) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in indicated KL tumors at serial time points. Neutrophil is indicated by MPO-positive cells. Red: MPO; blue: DAPI. Scale bar: 100 μm. (C) Statistical analyses of neutrophil (MPO-positive cells) infiltration in indicated KL tumors at a serial time points. 7W and 8W-A tumors analyzed were ADC and 8W-S, 9W, and 10W tumors analyzed were SCC. 30 representative images for each group were counted. (D) Representative H&E staining and IHC staining for TTF1, p63, SOX2, and KRT5 in lung tumors from KL or KLT mice after 7 wk (7W) of Adeno-Cre treatment. Scale bar: 50 μm. (E) Statistical analyses of tumor burden of KL-7W (n = 5) and KLT-7W (n = 5) mice. (F) Schematic illustration of immune infiltration analysis by FACS in KL and KLT mice. (G) Gating strategy for analyzing immune infiltration subtypes in KL and KLT mice. (H) The number of indicated immune subpopulations per mg lung were shown in KL (n = 6) and KLT (n = 5) models. Each dot represents one mouse. (I) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KLT-7W and KLT-9W tumors; red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analyses are shown on the right. (J) Relative expression of CXCL subfamily genes in KL tumors. (K) Relative mRNA expression of Cxcl1, 2, 3, and 5 in KL and KLT tumors by RT-qPCR. (L) Representative whole lungs of KL mice treated with saline and αCXCL5. * indicates SCC. Scale bar: 1 mm. (M) Representative IHC staining for CXCL5 in KL model with sgTom or sgCxcl5. Scale bar: 50 µm. (N) Representative H&E staining and IHC staining in KL model with sgTom or sgCxcl5. Scale bar: 50 µm. (O and P) Statistical analyses of the number (O) and burden (P) of SCC in KL model with sgTom (n = 5) or sgCxcl5 (n = 7). (Q) Representative whole lungs of KL model with sgTom or sgCxcl5. * indicates SCC. Scale bar: 1 mm. Data in B, C, H, and K represent one experiment of three independent repeats, and D, E, I, and L–Q represent one experiment of two independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (E, H, I, O, and P), one-way ANOVA test (C). ns: not significant. Data are represented as mean ± SD.
TET2 promotes neutrophil infiltration potentially through the upregulation of CXCL5 expression. (A) Heatmap of inflammatory and IFN signaling gene expression in KL tumors at serial time points. W, week. Both ADC and SCC were isolated from 8-wk time point, e.g., 8W-A means ADC and 8W-S means SCC. (B) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in indicated KL tumors at serial time points. Neutrophil is indicated by MPO-positive cells. Red: MPO; blue: DAPI. Scale bar: 100 μm. (C) Statistical analyses of neutrophil (MPO-positive cells) infiltration in indicated KL tumors at a serial time points. 7W and 8W-A tumors analyzed were ADC and 8W-S, 9W, and 10W tumors analyzed were SCC. 30 representative images for each group were counted. (D) Representative H&E staining and IHC staining for TTF1, p63, SOX2, and KRT5 in lung tumors from KL or KLT mice after 7 wk (7W) of Adeno-Cre treatment. Scale bar: 50 μm. (E) Statistical analyses of tumor burden of KL-7W (n = 5) and KLT-7W (n = 5) mice. (F) Schematic illustration of immune infiltration analysis by FACS in KL and KLT mice. (G) Gating strategy for analyzing immune infiltration subtypes in KL and KLT mice. (H) The number of indicated immune subpopulations per mg lung were shown in KL (n = 6) and KLT (n = 5) models. Each dot represents one mouse. (I) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KLT-7W and KLT-9W tumors; red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analyses are shown on the right. (J) Relative expression of CXCL subfamily genes in KL tumors. (K) Relative mRNA expression of Cxcl1, 2, 3, and 5 in KL and KLT tumors by RT-qPCR. (L) Representative whole lungs of KL mice treated with saline and αCXCL5. * indicates SCC. Scale bar: 1 mm. (M) Representative IHC staining for CXCL5 in KL model with sgTom or sgCxcl5. Scale bar: 50 µm. (N) Representative H&E staining and IHC staining in KL model with sgTom or sgCxcl5. Scale bar: 50 µm. (O and P) Statistical analyses of the number (O) and burden (P) of SCC in KL model with sgTom (n = 5) or sgCxcl5 (n = 7). (Q) Representative whole lungs of KL model with sgTom or sgCxcl5. * indicates SCC. Scale bar: 1 mm. Data in B, C, H, and K represent one experiment of three independent repeats, and D, E, I, and L–Q represent one experiment of two independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (E, H, I, O, and P), one-way ANOVA test (C). ns: not significant. Data are represented as mean ± SD.
To test the early change of tumor immune microenvironment, we performed flow cytometry analysis of the dissociated lung tumor from KL and KLT mice at 7 wk after Adeno-Cre infection, a time point when all tumors uniformly maintained ADC pathology (Fig. S3, D–G) (Han et al., 2014). Despite comparable tumor burdens (Fig. S3, D and E), we observed a dramatic decrease of neutrophils (CD11b+/Ly6G+/CD45+) in KLT tumors (Fig. 2 A and Fig. S3 H), which was further confirmed by MPO staining (Fig. 2, B and C). We compared neutrophil infiltration between the 7- and 9-wk tumors from the KLT model and found no significant change over this period (Fig. S3 I). Besides neutrophils, tumor-associated macrophages (TAM) were found to be elevated in KLT tumors, with dominant M1-like TAM (F4/80+/CD11clow(lo)/CD11bhigh(hi)/CD45+) (Fig. 2 A and Fig. S3 H). No substantial change of CD4+ T cells (CD4+/CD3+/CD45+), CD8+ T cells (CD8+/CD3+/CD45+), regulatory T (Treg) cells (Foxp3+/CD4+/CD3+/CD45+), natural killer cells (NK, CD49b+/CD3−/CD45+), or natural killer T cells (NKT, CD49b+/CD3+/CD45+) was observed (Fig. 2 A and Fig. S3 H).

TET2 promotes neutrophil infiltration and AST potentially through CXCL5 production. (A) The percentage of indicated immune subpopulations in KL (n = 6) and KLT (n = 5) mouse lungs. Each dot represents one mouse. (B and C) (B) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL and KLT tumors; red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analyses (C). 30 representative images for each group were counted. (D) ELISA assay detection of CXCL5 protein levels in KL (n = 8) and KLT (n = 8) lung tumors. (E) Schematic illustration of αCXCL5 treatment experiment. W, week. (F) Representative CXCL5 IHC staining in KL lung tumors. Scale bar: 50 μm. (G) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL mice treated with saline and αCXCL5. red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analysis is shown on the right. The statistical fields include both ADC and SCC pathologies. 30 representative images for each group were counted. (H) Representative H&E staining and IHC staining in KL mice treated with saline or αCXCL5. The whole lungs are reused in Fig. S3 L. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (I–K) Statistical analyses of the AST incidence (I), the number (J), and burden (K) of SCC in KL mice treated with saline (n = 5) or αCXCL5 (n = 5). (L) Representative Ki-67 IHC staining and statistical analyses in KL mice treated with saline or αCXCL5. Scale bar: 50 μm. 30 representative images for each group were counted. (M) Representative fluorescence staining of neutrophils (MPO-positive cells) in KL tumors with sgTom or sgCxcl5. red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analyses are shown on the right. The statistical fields include both ADC and SCC pathologies. 30 representative images for each group were counted. (N) Representative whole lungs of KL model with sgTom or sgCxcl5. The whole lungs are reused in Fig. S3 Q. * indicates SCC. Scale bar: 1 mm. (O) Statistical analyses of the AST incidence in KL model with sgTom (n = 5) or sgCxcl5 (n = 7). Data in A–D represent one experiment of three independent repeats, and F–O represent one experiment of two independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (A, C, D, G, and J–M), Fisher’s exact test (I and O). ns: not significant. Data are represented as mean ± SD.
TET2 promotes neutrophil infiltration and AST potentially through CXCL5 production. (A) The percentage of indicated immune subpopulations in KL (n = 6) and KLT (n = 5) mouse lungs. Each dot represents one mouse. (B and C) (B) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL and KLT tumors; red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analyses (C). 30 representative images for each group were counted. (D) ELISA assay detection of CXCL5 protein levels in KL (n = 8) and KLT (n = 8) lung tumors. (E) Schematic illustration of αCXCL5 treatment experiment. W, week. (F) Representative CXCL5 IHC staining in KL lung tumors. Scale bar: 50 μm. (G) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL mice treated with saline and αCXCL5. red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analysis is shown on the right. The statistical fields include both ADC and SCC pathologies. 30 representative images for each group were counted. (H) Representative H&E staining and IHC staining in KL mice treated with saline or αCXCL5. The whole lungs are reused in Fig. S3 L. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (I–K) Statistical analyses of the AST incidence (I), the number (J), and burden (K) of SCC in KL mice treated with saline (n = 5) or αCXCL5 (n = 5). (L) Representative Ki-67 IHC staining and statistical analyses in KL mice treated with saline or αCXCL5. Scale bar: 50 μm. 30 representative images for each group were counted. (M) Representative fluorescence staining of neutrophils (MPO-positive cells) in KL tumors with sgTom or sgCxcl5. red: MPO; blue: DAPI. Scale bar: 20 μm. Statistical analyses are shown on the right. The statistical fields include both ADC and SCC pathologies. 30 representative images for each group were counted. (N) Representative whole lungs of KL model with sgTom or sgCxcl5. The whole lungs are reused in Fig. S3 Q. * indicates SCC. Scale bar: 1 mm. (O) Statistical analyses of the AST incidence in KL model with sgTom (n = 5) or sgCxcl5 (n = 7). Data in A–D represent one experiment of three independent repeats, and F–O represent one experiment of two independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (A, C, D, G, and J–M), Fisher’s exact test (I and O). ns: not significant. Data are represented as mean ± SD.
TET2 enhances neutrophil infiltration through CXCL5 production
Increased neutrophil infiltration is established as a typical inflammatory feature associated with AST, and depletion of neutrophils dampens squamous transition (Koyama et al., 2016; Mollaoglu et al., 2018). We then investigated how TET2 promotes neutrophil infiltration during AST. It is known that KL tumor cells can stimulate neutrophil recruitment through the production of cytokines and chemokines (Koyama et al., 2016). An appealing hypothesis is that TET2 in KL tumor cells might regulate the expression of genes involved in the neutrophil chemotaxis pathway. The CXCL subfamily (e.g., CXCL1, CXCL2, CXCL3, and CXCL5) preferentially attracts neutrophils to the inflamed site (Zlotnik and Yoshie, 2000; Palomino and Marti, 2015). We found that Cxcl3 and Cxcl5 were greatly upregulated in KL-SCC compared with KL-ADC whereas Tet2 KO only led to downregulation of Cxcl5 gene transcription in KLT tumors (Fig. S3, J and K). These findings indicate that Cxcl5 but not Cxcl3 may be one of the downstream targets of TET2, which contributes to neutrophil recruitment to tumor microenvironment.
We further confirmed the downregulation of CXCL5 protein in KLT tumors by ELISA (Fig. 2 D). Using CXCL5-neutralizing antibody treatment, we found that αCXCL5 decreased CXCL5 expression and neutrophil infiltration, as indicated by less MPO-positive staining in KL lung tumors (Fig. 2, E–G). Moreover, αCXCL5 treatment suppressed squamous transition (Fig. 2 H and Fig. S3 L). The αCXCL5 treatment showed a tendency to downregulate the AST incidence and significantly decreased the number and burden of SCC (Fig. 2, I–K). We also observed a dramatic decrease in cell proliferation in all lung tumors from the αCXCL5 treatment group (Fig. 2 L).
To further ascertain the role of CXCL5 in AST, we performed Cxcl5 KO using the CRISPR-Cas9 system in the KL model. We found that CXCL5 level and neutrophil infiltration were remarkably decreased in the sgCxcl5 group (Fig. 2 M and Fig. S3 M). Consistently, Cxcl5 KO dramatically suppressed squamous transition, as evidenced by the fact that most tumors showed ADC pathology with high TTF1 expression and no p63, SOX2, and KRT5 expression (Fig. 2 N; and Fig. S3, N and Q). As a result, Cxcl5 KO reduced the AST incidence, and the number and burden of SCC (Fig. 2 O; and Fig. S3, O and P). These data together with αCXCL5 treatment results support an important role of CXCL5 in TET2-mediated neutrophil infiltration and AST.
The TET2–STAT3–CXCL5 nexus enhances neutrophil infiltration
To elucidate how TET2 regulates CXCL5, we established two independent primary tumor cell lines derived from KL tumors (hereafter referred to as KL-#1 and KL-#2 cells). We found that Tet2 KO didn’t have a major effect on Cxcl5 expression in both cell lines (Fig. 3, A and B; and Fig. S4, A and B), in contrast to the dramatic downregulation of Cxcl5 in KLT tumors (Fig. S3 K). We noticed that the IFN pathway was enriched in KL-SCC (Fig. S3 A), indicating that the inflammatory signals may be important for the AST process. Notably, upon mouse IFNγ (mIFNγ) stimulation, Tet2 KO significantly downregulated Cxcl5 transcriptional expression in both cell lines (Fig. 3, A and B; and Fig. S4, A and B). Using hydroxymethylated DNA immunoprecipitation (hMeDIP) quantitative PCR (qPCR) assay, we found that mIFNγ treatment remarkably increased the 5hmC level at Cxcl5 proximal promoter region (−1,099 ∼ −855 bp) of the transcription start site (TSS), and such increase was completely abolished by Tet2 KO (Fig. 3, C–E). These data indicated the involvement of TET2 in Cxcl5 promoter regulation. Using chromatin immunoprecipitation (ChIP)-qPCR assay, we further found that mIFNγ treatment promoted the binding of TET2 to Cxcl5 promoter (Fig. 3 F). TET2 is known to be recruited to DNA by multiple sequence-specific transcription factors (Català-Moll et al., 2022; Wu and Zhang, 2014). Of note, the Cxcl5 proximal promoter region contained a putative binding motif for STAT3 around, −1,300 bp, near TSS (Fig. 3 G). We found that both total STAT3 and its active form (phosphorylated-STAT3, pSTAT3) were upregulated by mIFNγ treatment (Fig. S4 C). IP data further confirmed the interaction between TET2 and STAT3 (Fig. 3 H). We also found that the binding of TET2 to Cxcl5 promoter was abrogated by Stat3 KO (Fig. 3 I; and Fig. S4, D and E). Along with reduced TET2 occupancy at Cxcl5 promoter, Stat3 KO led to decreased 5hmC enrichment (Fig. 3 J) at Cxcl5 promoter and Cxcl5 expression (Fig. 3, K and L).
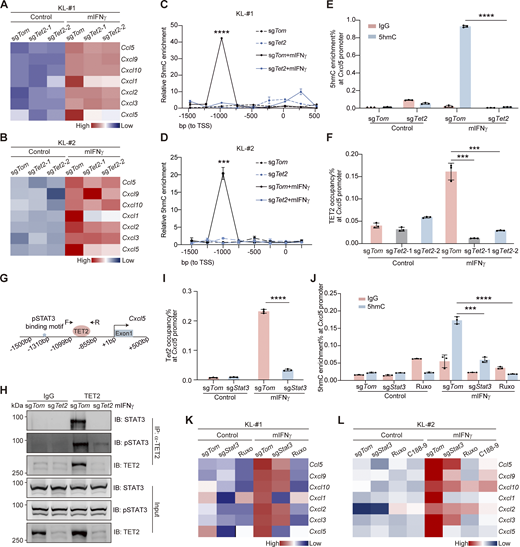
TET2 interacts with STAT3 to transactivate Cxcl5. (A and B) Heatmap of CXCL subfamily gene expression upon mIFNγ stimulation with or without Tet2 KO in KL-#1 (A) or KL-#2 cells (B). Cells were treated with 150 ng/ml mIFNγ for 24 h before RT-qPCR. (C and D) The 5hmC enrichment at the Cxcl5 promoter region was determined by hMeDIP-qPCR in KL-#1 (C) or KL-#2 cells (D). (E) The 5hmC enrichment at the Cxcl5 promoter region was determined by hMeDIP-qPCR in Tet2 KO KL cells. IgG was included as negative control. (F)Tet2 occupancy at Cxcl5 promoter region was determined by ChIP-qPCR in Tet2 KO KL cells. (G) Schematic illustration of Cxcl5 proximal promoter region, cis-regulatory elements, pSTAT3 binding motif, and binding of Tet2, as determined by ChIP assay. (H) The STAT3-TET2 interaction was confirmed by co-IP assay in KL-#1 cell line. Parallel blots using the same samples. IB, immunoblot. (I)Tet2 occupancy at Cxcl5 promoter region was determined by ChIP-qPCR in Stat3 KO KL cells. (J) 5hmC enrichment at the Cxcl5 promoter region was determined by hMeDIP-qPCR in Stat3 KO KL cells and Ruxo (2 μM)-treated KL cells. IgG was included as the negative control. (K) Heatmap of CXCL subfamily genes expression upon mIFNγ stimulation, Stat3 KO, and/or Ruxo treatment (2 μM) in KL-#1 cells. (L) Heatmap of CXCL subfamily gene expression upon mIFNγ stimulation, Stat3 KO, Ruxo treatment (2 μM), or C188-9 treatment (5 µM) in KL-#2 cells. Data in A–F and H–L represent one experiment of three independent repeats. ***P < 0.001, ****P < 0.0001 by one-way ANOVA test (C–F, I, and J). Data are represented as mean ± SD. Source data are available for this figure: SourceData F3.
TET2 interacts with STAT3 to transactivate Cxcl5. (A and B) Heatmap of CXCL subfamily gene expression upon mIFNγ stimulation with or without Tet2 KO in KL-#1 (A) or KL-#2 cells (B). Cells were treated with 150 ng/ml mIFNγ for 24 h before RT-qPCR. (C and D) The 5hmC enrichment at the Cxcl5 promoter region was determined by hMeDIP-qPCR in KL-#1 (C) or KL-#2 cells (D). (E) The 5hmC enrichment at the Cxcl5 promoter region was determined by hMeDIP-qPCR in Tet2 KO KL cells. IgG was included as negative control. (F)Tet2 occupancy at Cxcl5 promoter region was determined by ChIP-qPCR in Tet2 KO KL cells. (G) Schematic illustration of Cxcl5 proximal promoter region, cis-regulatory elements, pSTAT3 binding motif, and binding of Tet2, as determined by ChIP assay. (H) The STAT3-TET2 interaction was confirmed by co-IP assay in KL-#1 cell line. Parallel blots using the same samples. IB, immunoblot. (I)Tet2 occupancy at Cxcl5 promoter region was determined by ChIP-qPCR in Stat3 KO KL cells. (J) 5hmC enrichment at the Cxcl5 promoter region was determined by hMeDIP-qPCR in Stat3 KO KL cells and Ruxo (2 μM)-treated KL cells. IgG was included as the negative control. (K) Heatmap of CXCL subfamily genes expression upon mIFNγ stimulation, Stat3 KO, and/or Ruxo treatment (2 μM) in KL-#1 cells. (L) Heatmap of CXCL subfamily gene expression upon mIFNγ stimulation, Stat3 KO, Ruxo treatment (2 μM), or C188-9 treatment (5 µM) in KL-#2 cells. Data in A–F and H–L represent one experiment of three independent repeats. ***P < 0.001, ****P < 0.0001 by one-way ANOVA test (C–F, I, and J). Data are represented as mean ± SD. Source data are available for this figure: SourceData F3.
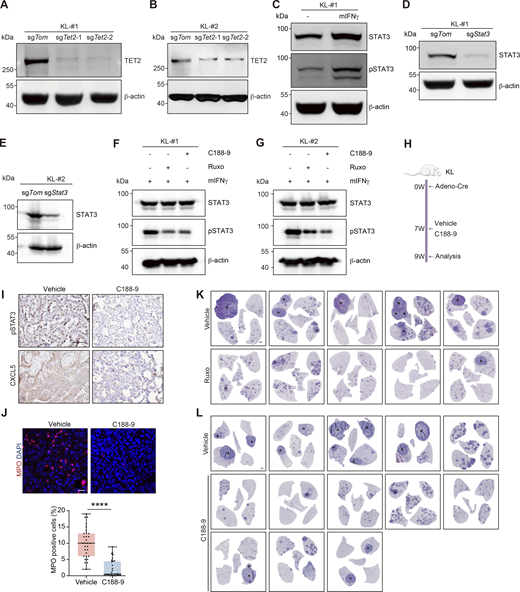
TET2 interacts with STAT3 to transactivate Cxcl5. (A and B) Western blot detection of TET2 in KL-#1 or KL-#2 cells with CRISPR-mediated Tet2 KO. β-Actin served as the internal loading control. (C) Western blot detection of STAT3 and pSTAT3 in KL cells treated with 150 ng/ml mIFNγ for 24 h β-Actin served as the internal loading control. (D and E) Western blot analysis for STAT3 in KL-#1 or KL-#2 cells with CRISPR-mediated Stat3 KO. β-Actin served as the internal loading control. (F and G) Western blot detection of STAT3 and pSTAT3 in KL cells treated with Ruxo (2 µM) and C188-9 (5 µM). β-Actin served as the internal loading control. (H) Schematic illustration of C188-9 treatment mouse experiment. W, week. (I) Representative IHC staining for pSTAT3 and CXCL5 in KL mice treated with vehicle and C188-9. Scale bar: 50 μm. (J) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL tumors. red: MPO; blue: DAPI. Scale bar: 20 μm. The statistical fields include ADC and SCC pathologies. 30 representative images for each group were counted. (K and L) Representative whole lungs of KL mice treated with Ruxo (K) and C188-9 (L). * indicates SCC. Scale bar: 1 mm. Data in A–G represent one experiment of three independent repeats and I–L represent one experiment of two independent repeats. ****P < 0.0001 by two-tailed unpaired Student’s t test (J). Data are represented as mean ± SD. Source data are available for this figure: SourceData FS4.
TET2 interacts with STAT3 to transactivate Cxcl5. (A and B) Western blot detection of TET2 in KL-#1 or KL-#2 cells with CRISPR-mediated Tet2 KO. β-Actin served as the internal loading control. (C) Western blot detection of STAT3 and pSTAT3 in KL cells treated with 150 ng/ml mIFNγ for 24 h β-Actin served as the internal loading control. (D and E) Western blot analysis for STAT3 in KL-#1 or KL-#2 cells with CRISPR-mediated Stat3 KO. β-Actin served as the internal loading control. (F and G) Western blot detection of STAT3 and pSTAT3 in KL cells treated with Ruxo (2 µM) and C188-9 (5 µM). β-Actin served as the internal loading control. (H) Schematic illustration of C188-9 treatment mouse experiment. W, week. (I) Representative IHC staining for pSTAT3 and CXCL5 in KL mice treated with vehicle and C188-9. Scale bar: 50 μm. (J) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL tumors. red: MPO; blue: DAPI. Scale bar: 20 μm. The statistical fields include ADC and SCC pathologies. 30 representative images for each group were counted. (K and L) Representative whole lungs of KL mice treated with Ruxo (K) and C188-9 (L). * indicates SCC. Scale bar: 1 mm. Data in A–G represent one experiment of three independent repeats and I–L represent one experiment of two independent repeats. ****P < 0.0001 by two-tailed unpaired Student’s t test (J). Data are represented as mean ± SD. Source data are available for this figure: SourceData FS4.
Ruxolitinib (Ruxo) is a clinically approved specific inhibitor of JAK1/2 that inhibits the JAK-STAT signaling (Lussana et al., 2018; Ajayi et al., 2018; Modi et al., 2019). We found that Ruxo treatment suppressed the effects of mIFNγ on STAT3 phosphorylation (Fig. S4, F and G), 5hmC enrichment at Cxcl5 promoter (Fig. 3 J), and Cxcl5 gene expression (Fig. 3, K and L). C188-9 (also named TTI-101) is a potent small molecule specifically targeting the Src-homology 2 domain of STAT3 currently under clinical development (Jung et al., 2017; Johnson et al., 2018; Pan et al., 2022). We found that C188-9 treatment also significantly inhibited the effects of mIFNγ on STAT3 phosphorylation (Fig. S4, F and G) and Cxcl5 gene expression (Fig. 3 L). We then treated the KL mouse model with Ruxo or C188-9 for 2 wk (Fig. 4 A and Fig. S4 H). We found that either inhibitor administration led to reduced levels of pSTAT3 and CXCL5 and suppressed neutrophil infiltration in the KL model (Fig. 4, B and C; and Fig. S4, I and J). Importantly, Ruxo or C188-9 treatment could inhibit the AST process in the KL model, as evidenced by the fact that most tumors showed ADC pathology with high TTF1 expression and no p63, SOX2, and KRT5 expression (Fig. 4, D and I; and Fig. S4, K and L). As a result, the incidence of AST exhibited a tendency to decrease, whereas the number and burden of SCC, as well as cell proliferation, were significantly reduced in the inhibitor-treated groups (Fig. 4, E–H, and J–M). These data together reaffirm the functional importance of TET2–STAT3–CXCL5 nexus in facilitating neutrophil infiltration and AST in the KL model.

Targeting STAT3 suppresses CXCL5 production, neutrophil infiltration, and AST in KL model. (A) Schematic illustration of Ruxo treatment mouse experiment. W, week. (B) Representative IHC staining for pSTAT3 and CXCL5 in KL mice treated with vehicle and Ruxo. Scale bar: 50 μm. (C) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL tumors. red: MPO; blue: DAPI. Statistical analyses are shown on the right. Scale bar: 20 μm. The statistical fields include both ADC and SCC pathologies. 30 representative images for each group were counted. (D) Representative H&E staining and IHC staining in KL mice treated with vehicle or Ruxo. The whole lungs are reused in Fig. S4 K. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (E–G) Statistical analyses of the AST incidence (E), the number (F), and burden (G) of SCC in KL model treated with vehicle (n = 5) or Ruxo (n = 5). (H) Representative Ki-67 IHC staining and statistical analyses. Scale bar: 50 μm. 30 representative images for each group were counted. (I) Representative H&E staining and IHC staining in KL mice treated with vehicle or C188-9. The whole lungs are reused in Fig. S4 L. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (J–L) Statistical analyses of the AST incidence (J), the number (K), and burden (L) of SCC in KL model treated with vehicle (n = 5) or C188-9 (n = 8). (M) Representative Ki-67 IHC staining and statistical analyses. Scale bar: 50 μm. 30 representative images for each group were counted. Data in B–M represent one experiment of two independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (C, F–H, and K–M), Fisher’s exact test (E and J). ns: not significant. Data are represented as mean ± SD.
Targeting STAT3 suppresses CXCL5 production, neutrophil infiltration, and AST in KL model. (A) Schematic illustration of Ruxo treatment mouse experiment. W, week. (B) Representative IHC staining for pSTAT3 and CXCL5 in KL mice treated with vehicle and Ruxo. Scale bar: 50 μm. (C) Representative fluorescence staining of neutrophil (MPO-positive cells) infiltration in KL tumors. red: MPO; blue: DAPI. Statistical analyses are shown on the right. Scale bar: 20 μm. The statistical fields include both ADC and SCC pathologies. 30 representative images for each group were counted. (D) Representative H&E staining and IHC staining in KL mice treated with vehicle or Ruxo. The whole lungs are reused in Fig. S4 K. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (E–G) Statistical analyses of the AST incidence (E), the number (F), and burden (G) of SCC in KL model treated with vehicle (n = 5) or Ruxo (n = 5). (H) Representative Ki-67 IHC staining and statistical analyses. Scale bar: 50 μm. 30 representative images for each group were counted. (I) Representative H&E staining and IHC staining in KL mice treated with vehicle or C188-9. The whole lungs are reused in Fig. S4 L. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (J–L) Statistical analyses of the AST incidence (J), the number (K), and burden (L) of SCC in KL model treated with vehicle (n = 5) or C188-9 (n = 8). (M) Representative Ki-67 IHC staining and statistical analyses. Scale bar: 50 μm. 30 representative images for each group were counted. Data in B–M represent one experiment of two independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (C, F–H, and K–M), Fisher’s exact test (E and J). ns: not significant. Data are represented as mean ± SD.
Lipid-laden neutrophils fuel the AST process
A recent study shows that lung neutrophils are laden with lipids and able to transfer lipid to breast cancer cells to facilitate metastasis (Li et al., 2020a). Through Boron dipyrromethene (BODIPY) IF staining analyses, we found that the KL-SCC showed the highest level of lipid than KL-ADC and KLT-ADC. Moreover, KLT-ADC showed very low BODIPY staining (Fig. S5 A). These data indicated a potential link between lipid dysregulation and TET2 KO as well as AST. We next performed flow cytometry analysis to test what types of cells were laden with lipid. Indeed, we found that lipid levels were significantly increased in both neutrophils and tumor cells from KL-SCC when compared with KL-ADC or KLT-ADC (Fig. 5 A and Fig. S5 B). This was further confirmed by IF staining (Fig. 5 B and Fig. S5 C). Through applying the imaging technique of stimulated Raman scattering (SRS) microscopy, we found that KL-SCC exhibited the highest level of lipid when compared with KL-ADC and KLT-ADC (Fig. 5 C and Fig. S5, D–F). Using SRS microscopy analysis and standard samples, we were able to distinguish triacylglycerol (TG), cholesterol and phospholipids mainly containing phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and sphingomyelin (SM) (Vance, 2015) (Fig. S5 G). We found that TG, but not cholesterol or phospholipids, was the major component of increased lipid in KL-SCC (Fig. S5 H). Loss of Tet2 mainly decreased the TG level in KLT tumors, as evidenced by SRS microscopy (Fig. 5 C) and BODIPY staining analyses (Fig. S5 A).

Lipid-laden neutrophils transfer lipid to tumor cells to promote cell proliferation and AST. (A) Representative fluorescence staining of KL-ADC, KL-SCC and KLT-ADC. green: lipid (BODIPY); blue: DAPI. Scale bar: 100 μm. Statistical analyses are shown on the right. 30 representative images for each group were counted. (B) Schematic illustration for lipid detection in neutrophil (Neu) and tumor cells isolated from KL and KLT tumors using FACS. W, week. (C) Representative fluorescence staining of KL-SCC; red: MPO; green: BODIPY; gray: EpCAM; blue: DAPI. Scale bar: 50 μm. (D) Representative SRS microscope images of various intensity of lipid (relative scores: 1, 2, 3). (E and F) (E) SRS microscope analysis for lipid level in KL-ADC, KL-SCC, and KLT-ADC. Lipid signal is indicated as gray. Statistical analyses are shown (F). 300 representative images for each group were counted. (G) The standard curves of TG, cholesterol, BSA, and phospholipids including PC, PS, PE, and SM were analyzed by SRS microscope. (H) Qualitative analysis of lipid in KL-SCC (n = 6) by SRS microscope. The qualitative curves of lipid are shown on the right. Scale bar: 50 μm. (I) Cell morphology of KL-#2 cells monocultured (Ctrl) or cocultured with neutrophils (Neu) isolated from KL-SCC. EIPA was added to block lipid transport (Neu+EIPA). Scale bar: 100 μm. (J and K) (J) Representative fluorescence staining of BODIPY and Ki-67 in KL-#2 cells co-cultured with neutrophils. Scale bar: 100 μm. Statistical analyses were shown (K). 20 representative images for each group were counted. (L) KL cells were treated with TG-micelles (0.1 mM). EIPA was added to block TG transport. Lipid levels of KL cells were quantitated by FACS. (M) Diagram of the regulatory genes involved in TG hydrolysis. (N) Relative mRNA expression of Abhd5, Atgl, Hilpda, and G0s2 in neutrophils isolated from KL-ADC and KL-SCC, which derived from 8W ADC and 8W SCC of KL model, respectively. Cidec has no signal. (O) Representative whole lungs of KL mice treated with vehicle or EIPA. * indicates SCC. Scale bar: 1 mm. Data in A, C–L, and N represent one experiment of three independent repeats and O represents one experiment of two independent repeats. **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA test (A, F, and K), two-tailed unpaired Student’s t test (N). ns: not significant. Data are represented as mean ± SD.
Lipid-laden neutrophils transfer lipid to tumor cells to promote cell proliferation and AST. (A) Representative fluorescence staining of KL-ADC, KL-SCC and KLT-ADC. green: lipid (BODIPY); blue: DAPI. Scale bar: 100 μm. Statistical analyses are shown on the right. 30 representative images for each group were counted. (B) Schematic illustration for lipid detection in neutrophil (Neu) and tumor cells isolated from KL and KLT tumors using FACS. W, week. (C) Representative fluorescence staining of KL-SCC; red: MPO; green: BODIPY; gray: EpCAM; blue: DAPI. Scale bar: 50 μm. (D) Representative SRS microscope images of various intensity of lipid (relative scores: 1, 2, 3). (E and F) (E) SRS microscope analysis for lipid level in KL-ADC, KL-SCC, and KLT-ADC. Lipid signal is indicated as gray. Statistical analyses are shown (F). 300 representative images for each group were counted. (G) The standard curves of TG, cholesterol, BSA, and phospholipids including PC, PS, PE, and SM were analyzed by SRS microscope. (H) Qualitative analysis of lipid in KL-SCC (n = 6) by SRS microscope. The qualitative curves of lipid are shown on the right. Scale bar: 50 μm. (I) Cell morphology of KL-#2 cells monocultured (Ctrl) or cocultured with neutrophils (Neu) isolated from KL-SCC. EIPA was added to block lipid transport (Neu+EIPA). Scale bar: 100 μm. (J and K) (J) Representative fluorescence staining of BODIPY and Ki-67 in KL-#2 cells co-cultured with neutrophils. Scale bar: 100 μm. Statistical analyses were shown (K). 20 representative images for each group were counted. (L) KL cells were treated with TG-micelles (0.1 mM). EIPA was added to block TG transport. Lipid levels of KL cells were quantitated by FACS. (M) Diagram of the regulatory genes involved in TG hydrolysis. (N) Relative mRNA expression of Abhd5, Atgl, Hilpda, and G0s2 in neutrophils isolated from KL-ADC and KL-SCC, which derived from 8W ADC and 8W SCC of KL model, respectively. Cidec has no signal. (O) Representative whole lungs of KL mice treated with vehicle or EIPA. * indicates SCC. Scale bar: 1 mm. Data in A, C–L, and N represent one experiment of three independent repeats and O represents one experiment of two independent repeats. **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA test (A, F, and K), two-tailed unpaired Student’s t test (N). ns: not significant. Data are represented as mean ± SD.

Lipid-laden neutrophils transfer lipid to KL tumors to fuel AST. (A) Statistical analyses of lipid levels in neutrophils and tumor cells isolated from KL-ADC (n = 7), KL-SCC (n = 7), and KLT-ADC (n = 7) tumors. (B) Representative fluorescence staining showing the colocalization of lipid (BODIPY) with the neutrophils (MPO) or cancer cells (EpCAM) isolated from KL-SCC. Scale bar: 10 μm. (C) SRS microscope analysis for lipid level in KL-ADC, KL-SCC, and KLT-ADC. The top row is formed by merging 100 smaller images, respectively. Statistical analyses are shown on the right. 900 representative images for each group were counted. Lipid and protein signals were indicated as green and blue, respectively. Scale bar: 500 μm. (D and E) (D) KL cells were co-cultured with neutrophils isolated from KL-SCC. EIPA was added to block lipid transport. Lipid levels of KL cells were quantitated by FACS and statistical analyses are shown (E). (F) Cell morphology of KL-#1 cells mono-cultured (Ctrl) or co-cultured with neutrophils (Neu) isolated from KL-SCC. EIPA was added to block lipid transport (Neu+EIPA). Scale bar: 100 μm. (G and H) (G) Representative fluorescence staining of BODIPY and Ki-67 in KL-#1 cells co-cultured with neutrophils. Scale bar: 100 μm. Statistical analyses are shown (H). 20 representative images for each group were counted. (I) KL cells were treated with TG-micelles (0.1 mM). EIPA was added to block TG transport. Lipid levels of KL cells were quantitated by FACS. (J and K) Relative growth of KL-#1 (J) or KL-#2 (K) cells after treatment with TG-micelles (0.1 mM). Cells in sextuplicate were treated with drugs over the entire experiment, with relative cell growth determined daily and normalized to day 0. Data in A–K represent one experiment of three independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA test (A, C, E, and H–K). ns: not significant. Data are represented as mean ± SD.
Lipid-laden neutrophils transfer lipid to KL tumors to fuel AST. (A) Statistical analyses of lipid levels in neutrophils and tumor cells isolated from KL-ADC (n = 7), KL-SCC (n = 7), and KLT-ADC (n = 7) tumors. (B) Representative fluorescence staining showing the colocalization of lipid (BODIPY) with the neutrophils (MPO) or cancer cells (EpCAM) isolated from KL-SCC. Scale bar: 10 μm. (C) SRS microscope analysis for lipid level in KL-ADC, KL-SCC, and KLT-ADC. The top row is formed by merging 100 smaller images, respectively. Statistical analyses are shown on the right. 900 representative images for each group were counted. Lipid and protein signals were indicated as green and blue, respectively. Scale bar: 500 μm. (D and E) (D) KL cells were co-cultured with neutrophils isolated from KL-SCC. EIPA was added to block lipid transport. Lipid levels of KL cells were quantitated by FACS and statistical analyses are shown (E). (F) Cell morphology of KL-#1 cells mono-cultured (Ctrl) or co-cultured with neutrophils (Neu) isolated from KL-SCC. EIPA was added to block lipid transport (Neu+EIPA). Scale bar: 100 μm. (G and H) (G) Representative fluorescence staining of BODIPY and Ki-67 in KL-#1 cells co-cultured with neutrophils. Scale bar: 100 μm. Statistical analyses are shown (H). 20 representative images for each group were counted. (I) KL cells were treated with TG-micelles (0.1 mM). EIPA was added to block TG transport. Lipid levels of KL cells were quantitated by FACS. (J and K) Relative growth of KL-#1 (J) or KL-#2 (K) cells after treatment with TG-micelles (0.1 mM). Cells in sextuplicate were treated with drugs over the entire experiment, with relative cell growth determined daily and normalized to day 0. Data in A–K represent one experiment of three independent repeats. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA test (A, C, E, and H–K). ns: not significant. Data are represented as mean ± SD.
Considering the highly lipid-laden phenotype of neutrophils, we hypothesized that lipid transfer from neutrophils to cancer cells might promote tumor growth. Macropinocytosis is known to assist tumor cells in ingesting neutrophil-derived TG (Li et al., 2020a). When cocultured with neutrophils isolated from KL-SCC, we observed a dramatic increase in lipid accumulation in KL cells (Fig. 5, D and E). Treatment with 5-(N-ethyl-N-isopropyl) amiloride (EIPA), which inhibits macropinocytosis (Kim et al., 2018), completely blocked the lipid acquisition of KL cells (Fig. 5, D and E). Costaining of BODIPY and Ki-67 further demonstrated that the proliferation of KL cells was increased when cocultured with KL-SCC–derived neutrophils (Fig. 5, F–H; and Fig. S5, I–K). Such an increase was profoundly blocked by EIPA treatment (Fig. 5, F–H; and Fig. S5, I–K). We further determined the effect of TG supplementation on KL cell growth. To this end, we encapsulated TG within micelles as previously described (Dai et al., 2017; Goncalves et al., 2015). Interestingly, we found that the treatment with TG-micelles dramatically promoted lipid accumulation and increased cell proliferation in KL cells, which could be diminished by EIPA treatment (Fig. 5, I–K; and Fig. S5 L). These data support the role of TG transfer in promoting KL-SCC cell proliferation.
TG is degraded via hydrolysis, a process catalyzed by adipose triglyceride lipase (ATGL), which can be activated by abhydrolase domain containing 5 (ABHD5) and suppressed by cell death inducing DFFA-like effector C (CIDEC), hypoxia inducible lipid droplet associated (HILPDA), and G1/G1 switch 2 (G0S2) (Nielsen et al., 2014) (Fig. S5 M). We then examined these genes related to TG hydrolysis in isolated neutrophils from KL-ADC and KL-SCC. We found the most significant changes were downregulation of Atgl expression and upregulation of Hilpda and G0s2 expression in KL-SCC–derived neutrophils (Fig. S5 N). Hence, these findings indicate that lipid accumulation seen in SCC is likely attributed to dysregulated TG hydrolysis in neutrophils.
We next tested if EIPA administration could inhibit AST in GEMM. We treated the KL model with EIPA for 2 wk and found that EIPA treatment dramatically blocked lipid transport (Fig. 6, A and B) and suppressed squamous transition in KL mice (Fig. 6 C and Fig. S5 O). EIPA treatment almost blocked squamous transition in KL mice and the majority of tumors exhibited ADC pathology with high TTF1 expression and no p63, SOX2, and KRT5 expression (Fig. 6 C). The AST incidence, the number, and burden of SCC were also decreased by EIPA treatment (Fig. 6, D–F). Consistently, Ki-67 staining data revealed that cell proliferation was also decreased in EIPA-treated KL tumors (Fig. 6 G). These data together support that the blockage of neutrophil–cancer cell lipid transfer can serve as a potential strategy for overcoming AST.

Blockage of neutrophil-derived lipid transport suppresses AST in KL model. (A) Schematic illustration of EIPA treatment mouse experiment. W, week. (B) Representative BODIYP and Ki-67 fluorescence staining of KL tumors. Red: Ki-67; green: BODIPY; blue: DAPI. Scale bar: 20 μm. Statistical analyses are shown on the right. Arrows show BODIPY and Ki-67 double-positive cells. 10 representative images for each group were counted. (C) Representative H&E and IHC staining in KL tumors. The whole lungs are reused in Fig. S5 Q. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (D–F) Statistical analyses of the AST incidence (D), the number (E), and burden (F) of SCC in KL mice treated with vehicle (n = 10) or EIPA (n = 10). (G) Representative Ki-67 IHC staining and statistical analyses. Scale bar: 50 μm. 30 representative images for each group were counted. (H) Working model for TET2-mediated AST. In KL tumor cells, TET2 is recruited by STAT3 to CXCL5 promoter region and transcriptionally upregulates CXCL5, which in turn promotes infiltration of lipid-laden neutrophils to facilitate AST. Targeting the TET2–STAT3–CXCL5 nexus and/or the lipid transport could serve as potential therapeutic strategies to inhibit AST in lung cancer. Data in B–G represent one experiment of two independent repeats. **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (B and E–G), Fisher’s exact test (D). Data are represented as mean ± SD.
Blockage of neutrophil-derived lipid transport suppresses AST in KL model. (A) Schematic illustration of EIPA treatment mouse experiment. W, week. (B) Representative BODIYP and Ki-67 fluorescence staining of KL tumors. Red: Ki-67; green: BODIPY; blue: DAPI. Scale bar: 20 μm. Statistical analyses are shown on the right. Arrows show BODIPY and Ki-67 double-positive cells. 10 representative images for each group were counted. (C) Representative H&E and IHC staining in KL tumors. The whole lungs are reused in Fig. S5 Q. * indicates SCC. Scale bar for whole lung: 1 mm; scale bar for others: 50 μm. (D–F) Statistical analyses of the AST incidence (D), the number (E), and burden (F) of SCC in KL mice treated with vehicle (n = 10) or EIPA (n = 10). (G) Representative Ki-67 IHC staining and statistical analyses. Scale bar: 50 μm. 30 representative images for each group were counted. (H) Working model for TET2-mediated AST. In KL tumor cells, TET2 is recruited by STAT3 to CXCL5 promoter region and transcriptionally upregulates CXCL5, which in turn promotes infiltration of lipid-laden neutrophils to facilitate AST. Targeting the TET2–STAT3–CXCL5 nexus and/or the lipid transport could serve as potential therapeutic strategies to inhibit AST in lung cancer. Data in B–G represent one experiment of two independent repeats. **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed unpaired Student’s t test (B and E–G), Fisher’s exact test (D). Data are represented as mean ± SD.
Discussion
Previous studies have identified TET2 as an important tumor suppressor in myeloid cancers as well as solid cancer (Delhommeau et al., 2009; Langemeijer et al., 2009; Lian et al., 2012; Xu et al., 2022). Interestingly, our work here demonstrates that TET2 is required for squamous transition in the KL model and Tet2 KO significantly promotes KL mouse survival, indicative of a potential benefit to target TET2 in this specific subtype of lung cancer. Lung cancer is known to harbor strong phenotypic plasticity and display histological transformation from ADC to SCC or small cell lung cancer, which is frequently associated with malignant progression and drug resistance (Chen et al., 2021; Quintanal-Villalonga et al., 2021a, 2021b). Our previous work has revealed that Tet2 KO promotes Kras-driven lung cancer growth in GEMM (Xu et al., 2022). Consistently, we found that Tet2 KO also promotes the growth of KL-ADC. Of note, our data clearly demonstrated that KO of Tet2 impairs squamous transition in the KL model. Hence, in the LKB1-deficient context, TET2 seems important for sustaining lung cancer plasticity. We reason such functional differences of TET2 might be genetic context dependent, similar to our previous finding about the dual function of YAP in lung cancer (Zhang et al., 2015b; Gao et al., 2014). Future work is interesting to clarify the detailed mechanisms behind these observations.
Previous functional and mechanistic studies of TET2 mainly focused on immune cells (Cong et al., 2021; Zhang et al., 2015a; Banks et al., 2021). For example, in dendritic cells, TET2 interacts with STAT3 to transactivate IL-6/10 (Català-Moll et al., 2022). In macrophages, TET2 can interact with RelA to transactivate NF-κB target genes (Chen et al., 2022). In B cells, TET2 restrains IL-10 production and thereby promotes liver cancer progression (Lu et al., 2022). Except for immune cells, TET activity-dependent epigenetic pathway can increase the global 5hmC level and suppress the tumor progression of melanoma and prostate cancer (Lian et al., 2012; Zhao et al., 2020). We here find that TET2 is recruited by STAT3 to CXCL5 promoter region for transcriptional activation. Although both CXCL3 and CXCL5 are classical chemokines for recruiting neutrophils into tumor microenvironment (Mollaoglu et al., 2018), only CXCL5 is transcriptionally regulated by TET2. Although we can’t conclusively exclude the involvement of CXCL3 through protein-level analyses and there might exist other TET2-independent mechanisms involved in CXCL3 gene regulation, our data support an important role of CXCL5 in neutrophil recruitment during the AST process. Previous study shows that SOX2 or NKX2-1 directly regulates CXCL5 gene expression. Interestingly, SOX2 is known to activate STAT3 and the latter could promote CXCL5 gene expression (Mollaoglu et al., 2018; Traber et al., 2015), indicative of another potential indirect regulatory loop involved in CXCL5 regulation by SOX2. Moreover, STAT3 can also bind to TET2 to regulate downstream inflammatory signaling (Català-Moll et al., 2022). We demonstrate here that TET2 promotes CXCL5 gene expression potentially through STAT3. It seems that multiple regulatory loops might be involved in CXCL5 transcriptional regulation. Besides, our work further shows that anti-CXCL5 could effectively reduce neutrophil infiltration and inhibit AST. While the use of saline, rather than an appropriate IgG subtype, as the control is the caveat for the anti-CXCL5 treatment experiment, similar results are observed from the CRISPR/Cas9-mediated Cxcl5 KO experiment, underscoring the important role of CXCL5 in neutrophil recruitment and AST. Approaches targeting the STAT3 pathway are well-explored both preclinically and clinically, e.g., C188-9 is applied in preclinical models of inflammatory bowel disease and solid cancer (Kasembeli et al., 2018; Jung et al., 2017; Pan et al., 2022), and Ruxo has been approved for the treatment of patients with myelofibrosis, polycythemia vera, and steroid-refractory acute graft-versus-host disease treatment (Huynh et al., 2019; Wong et al., 2017; Ajayi et al., 2018; Lussana et al., 2018; Modi et al., 2019). Our data show that targeting STAT3 efficiently inhibits CXCL5 production, neutrophil infiltration, and AST in the KL model. Moreover, we find a dramatic suppression of KL cancer progression after Ruxo or C188-9 treatment, highlighting the potential benefit of JAK-STAT targeting strategy. One limitation of our study is that we didn’t perform mouse survival analyses in the experiments targeting CXCL5 or STAT3. Thus, it is currently unclear whether these intervention strategies targeting AST would benefit the mice in the long run. Nonetheless, our findings might provide a potential strategy to treat those patients with high risk for AST, which requires future detailed study.
Previous studies have highlighted the promotive role of tumor-infiltrating neutrophils in AST with unclear mechanisms (Koyama et al., 2016; Mollaoglu et al., 2018). Our data show that the neutrophils infiltrated into SCC are laden with increased lipid, with TG as the major component. Dysregulated TG hydrolysis is observed in neutrophils infiltrated into SCC, which might partially explain the increased lipid load. TG is important for energy storage and has previously been implicated in diverse pathological processes including inflammation and cancer (Wang and Dubois, 2010; Spiegelman and Flier, 2001). Interestingly, we found that lung neutrophils transfer TG to squamous cancer for promoting cell proliferation. Blockage of lipid transport by EIPA almost completely blocks AST and tumor malignant progression. These findings suggest that EIPA treatment has the capability to inhibit lipid transfer both in vitro and in vivo. While we cannot exclude the possibility that EIPA treatment may have a cytotoxic effect, our data indicate that the dose of EIPA we used did not significantly affect the KL cell proliferation. Importantly, this provides another potential strategy to inhibit AST from the metabolic communication aspect.
It’s worth noting that multiple AST or SCC mouse models have been established previously (Perera et al., 2009; Xiao et al., 2013; Mukhopadhyay et al., 2014; Xu et al., 2014; Liu et al., 2015, 2019; Camolotto et al., 2018; Mollaoglu et al., 2018; Ruiz et al., 2019; Ferone et al., 2016; Tata et al., 2018). It’ll be interesting to check if our mechanistic findings also apply to various models based on the strong link between neutrophils and squamous pathology. Similarly, it’ll be interesting to test our targeting strategies proposed here in various AST or SCC models. Somerville et al. have previously suggested a link between neutrophils and squamous transition in pancreatic cancer (Somerville et al., 2020). Future studies would be interesting to test if the contribution of lipid transfer to squamous transition we identified here can be applied to other cancer types. Besides neutrophils, we also found a significant increase of TAM upon Tet2 KO in the KL model, which might require future efforts to clarify its potential contribution to AST.
In summary, our work here identifies TET2 as an important regulator of AST and unveils a novel mechanism by which TET2–STAT3–CXCL5 nexus promotes lipid-laden neutrophil recruitment to facilitate AST (Fig. 6 H). We provide proof-of-principle for developing therapies targeting the TET2–STAT3–CXCL5 nexus and/or the lipid transport from tumor-associated neutrophils. Further investigations are warranted to confirm these findings, especially in clinical samples, and to prospectively evaluate the effect of the above-mentioned potential therapeutic strategies on overcoming squamous transition.
Materials and methods
Mouse study
KrasLSL-G12D/+, Lkb1L/L, and Tet2L/L C57BL/6 mice were generously provided by Dr. Tyler Jacks (Jackson et al., 2001), Ronald Depinho (Bardeesy et al., 2002), and Guoliang Xu (Dai et al., 2016), respectively. All mice were kept in a specific pathogen–free environment of the Center for Excellence in Molecular Cell Science and treated in strict accordance with protocols (SIBCB-2101008) approved by the Institutional Animal Care and Use Committee of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Mice at 6–8 wk of age were treated with Adeno-Cre (2 × 106 p.f.u.) via nasal inhalation and analyses were performed after 10 wk. Mice at 6–8 wk of age were treated with lentivirus (2 × 105 p.f.u.) via nasal inhalation and pathological analyses were performed 24 wk afterward.
Ruxo (HY-50856; MedChemExpress), C188-9 (HY-112288; MedChemExpress), or EIPA (HY-101840; MedChemExpress) were formulated in 10% DMSO, 40% PEG300, and 50% saline. CXCL5-neutralizing antibody (αCXCL5) (mab433; R&D) was formulated in 50% saline. Treatment of Ruxo (45 mg/kg) and vehicle (10% DMSO: 40% PEG300: 50% saline) was conducted daily for 2 wk through intraperitoneal injection in KL mice, as previously reported (Siersbæk et al., 2020). Treatment of C188-9 (75 mg/kg) was conducted daily for 2 wk through intraperitoneal injection in KL mice as previously described (Zhao et al., 2021). Treatment of αCXCL5 (5 mg/kg) and saline was conducted weekly for 2 wk through intraperitoneal injection in KL mice, as previously reported (Li et al., 2020b). Treatment of EIPA (10 mg/kg) was conducted daily for 2 wk through intraperitoneal injection in KL mice, as previously reported (Li et al., 2020a). Mice were then sacrificed for histopathological examination. Tumor incidence and number and burden were analyzed using ImageJ software as previously described (Chen et al., 2021). All treatment experiments were conducted using littermate mice as controls.
CRISPR-Cas9 plasmids construction and lentivirus production
Single guide RNAs (sgRNAs) against Tet1, 2, or 3 were previously described (Zuo et al., 2017). We employed the CRISPR-related plasmid pSECC for lentivirus packaging and delivered it via nasal inhalation to KL mice, achieving targeted gene KO in vivo as previously reported (DuPage et al., 2009). For in vivo viral infection, lentivirus supernatant was collected 48 h after transfection and concentrated by ultracentrifugation at 20,000 rpm for 2 h and resuspended overnight in an appropriate volume of OptiMEM (Gibco). All sgRNAs were as follows: sgTomato, 5′-GGCCACGAGTTCGAGATCGA-3′; sgCxcl5, 5′-ATGGCGAGATGGAACCGCTG-3′; sgTet1-1, 5′-GGCATGCTGGACTTCATTCT-3′; sgTet1-2, 5′-GATGTCCATGCCGGTTACAC-3′; sgTet1-3, 5′-GAAGCCAGAGGCCACCTCAC-3′; sgTet2-1, 5′-CAGGGAGCAAGAGATTCCGA-3′; sgTet2-2, 5′-TCAGTCCTCCACTCTCAGAC-3′; sgTet2-3, 5′-GTGAACCAAGGACCGTCTCC-3′; sgTet3-1, 5′-CCCTACTTCCACAGAGCCTC-3′; sgTet3-2, 5′-GCCTGTTAGGCAGATTGTTC-3′; sgTet3-3, 5′-ACAAGCTGGAGGAGCTCATC-3′; and sgStat3, 5′-GATCGTGGCCCGATGCCTGT-3′. Primers used for detecting sgRNA efficiency were as follows: sgTet1 test-F, 5′-AGCCTCCAAACACAGTTGCT-3′; sgTet1 test-R, 5′-TGGCTATGTCTGAGGAAGCG-3′; sgTet2 test-F, 5′-ATGATTGTGGCTCACCGAGC-3′; sgTet2 test-R, 5′-GTCTCTGTGGCCTGTTGACT-3′; sgTet3 test-F, 5′-GGCAGAACTGGAGCAGCTAT-3′; sgTet3 test-R, 5′-GGTGTGAGTGGGTTCTCAGC-3′.
Genotyping primers used for detecting Tet2 KO were as follows: Tet2-C, 5′-ACACAGAGAAAAGGGTACGTGAA-3′; Tet2-F, 5′-ACTCATTAGTGAAATATGTGAGTG-3′; Tet2-R, 5′-CTGCTTAGTTCAATGCCAACC-3′.
Histologic, IHC, and IF analyses
Lung tissues were freshly dissected and fixed with 10% formalin buffer and then embedded in paraffin and cut into 5-μm sections. Hematoxylin and Eosin (H&E) staining, and IHC and IF staining were performed as described previously (Wang et al., 2021). For IHC staining, the stained sections were evaluated using H-score as previously described (Detre et al., 1995; Wang et al., 2019). In brief, staining intensity was scored in the following categories: negative staining (scored as 0), weak positive staining (scored as 1), intermediate positive staining (scored as 2), and strong positive staining (scored as 3). The IHC score was calculated with the formula: cell percentage × staining intensity score ×100. The final IHC score was on a scale of 0–300. To calculate the percentages of cells positive for Ki-67 or MPO and the relative scores for 5mC or 5hmC, at least 30 high-power fields were randomly selected from each group for statistical analysis as previously described (Li et al., 2015). IF images were captured using the laser confocal microscope with a Leica TCS SP8 system with an HC PL APO CS2 63x/1.40 OIL objective as previously described (Guo et al., 2022). The following antibodies used for IHC and IF: TTF1 (ab133638; Abcam), p63 (ab124762; Abcam), SOX2 (ab92494; Abcam), Cytokeratin 5 (KRT5) (BS1208; Bioworld), 5mC (BI-MECY-0100; Eurogentec), 5hmC (39792; Active Motif), Ki-67 (NB500; Novus), pSTAT3 (9145; CST), MPO (AF3667; R&D system), BODIPY (GC42959; GLPBIO), EpCAM (2929; CST), and DAPI (4083; CST).
Western blot
Tissues or cell lysates were prepared for western blot analysis with the following antibodies: TTF1 (ab133638; Abcam), p63 (12143-I-AP; Proteintech), Cytokeratin 5 (KRT5) (BS1208; Bioworld), TET2 (18950S; CST), STAT3 (9132; CST), pSTAT3 (9145; CST), and β-actin (AC026; ABclonal).
Real-time qPCR (RT-qPCR)
Total RNA was isolated by TRIzol reagent and retrotranscribed into first-strand cDNA using the first-strand synthesis kit (Invitrogen). cDNA was subjected to RT-qPCR with SYBR-Green Master PCR mix (Roche). Primers used for RT-qPCR were as follows: Tet2, 5′-GCTCCAATATACAAGAAGCTTGCAC-3′ and 5′-TATTGAGGGTGACCACCACTGTACT-3′; p63 (DNp63), 5′-ATGTTGTACCTGGAAAACAATG-3′ and 5′-GATGGAGAGAGGGCATCAAA-3′; Sox2, 5′-CACAACTCGGAGATCAGCAA-3′ and 5′-TCCGGGAAGCGTGTACTT-3′; Krt5, 5′-CAGAGCTGAGGAACATGCAG-3′ and 5′-CATTCTCAGCCGTGGTACG-3′; Krt14, 5′-ATCGAGGACCTGAAGAGCAA-3′ and 5′-TCGATCTGCAGGAGGACATT-3′; Abhd5, 5′-TGGTGTCCCACATCTACATCA-3′ and 5′-CAGCGTCCATATTCTGTTTCCA-3′; Atgl, 5′-GGATGGCGGCATTTCAGACA-3′ and 5′-CAAAGGGTTGGGTTGGTTCAG-3′; Cidec, 5′-ATGGACTACGCCATGAAGTCT-3′ and 5′-CGGTGCTAACACGACAGGG-3′; Hilpda, 5′-TGCTGGGCATCATGTTGACC-3′ and 5′-TGACCCCTCGTGATCCAGG-3′; G0s2, 5′-GTGAAGCTATACGTGCTGGG-3′ and 5′-CCGTCTCAACTAGGCCGAG-3′; Actin, 5′-CAGCCTTCCTTCTTGGGTAT-3′ and 5′-GGTCTTTACGGATGTCAACG-3′.
RNA-sequencing (RNA-seq) data analyses
RNA-seq data of mouse KL lung tumors were obtained from our previous study (GSE78220) (Fang et al., 2023). RNA-seq data were aligned to mouse genome reference using STAR (v2.6.0) (Dobin et al., 2013). To comparatively analyze the gene expression, RNA-seq data of TCGA-LUAD (tumor: n = 526, normal: n = 59) and TCGA-LUSC (tumor: n = 550, normal: n = 49) were downloaded from UCSC Xena (https://xenabrowser.net), from which samples with tumor purity >0.5 were used (Aran et al., 2015) (LUAD: n = 430, LUSC: n = 433). Combat function in the SVA package was used to correct the batch effects (Leek et al., 2012). “GSVA” package was used to evaluate the expression scores of signal pathways (Hänzelmann et al., 2013).
Establishment of primary cell lines
To obtain the primary cell line of KrasG12D; Lkb1−/−, lung tumor tissues from KrasLSL-G12D/+; Lkb1L/L mice infected with Adeno-Cre were isolated in a sterile manner. Tissues were washed extensively with 1× PBS with 200 μg/ml penicillin (Sigma-Aldrich) and 200 μg/ml streptomycin (Sigma-Aldrich). Next, tissues were mechanically dissociated using a scalpel with removal of vascular material and plated in RPMI (Gibco) with 10% FBS, penicillin/streptomycin for further experiments.
Establishment of squamous organoids
To obtain the squamous organoids, the tissues of KL-SCC were mechanically dissociated using a scalpel and digested with collagenase Type 1 (LS004196; Worthington-Biochem). Then single cells were mixed with matrigel and plated in mouse medium as previously described (Hu et al., 2018). The full-length wild-type or inactive mutant (H1295R1296D1297 mutated to Y1295R1296A1297) (Xu et al., 2022) Tet2 was overexpressed in SCC organoid cells as previously described (Hai et al., 2020).
hMeDIP-qPCR analysis
The hMeDIP-qPCR assay was performed as previously described (Ito et al., 2010). Briefly, genomic DNA was collected (2 μg) and denatured, sonicated for 15 min (70% power, 30 s on, 30 s off; Bioruptor), and immunoprecipitated with anti-5hmC (39769; Active Motif) or IgG antibody (DA1E; CST) for 3.5 h. The antibody–DNA complexes were pulled down using protein G beads (17061801; GE) for 1.5 h. Beads were washed three times and treated with proteinase K (TaKaRa) at 65°C for 4 h. DNA was isolated by PCR Purification Kit (QIAGEN) and analyzed by RT-qPCR using the primers as follows: Cxcl5-1, 5′-AAGTTCGAAAGTCTACCACC-3′ and 5′-GTGGTGCCCTGGCTTTCTTT-3′; Cxcl5-2, 5′-GTTATAACCGACTGCCTTGG-3′ and 5′-CAGCTGAGGGACTCATCCGT-3′; Cxcl5-3, 5′-CACCAACCTCCTTTAGCTTC-3′ and 5′-TAAAAGCTGCTTACTGGAAG-3′; Cxcl5-4, 5′-GAAGGCCATTCATGCAGCTG-3′ and 5′-CGAAGACTGTTTTTATTCAG-3′; Cxcl5-5, 5′-CACACATTTCAAATGTATTT-3′ and 5′-ACCTGGGAGATGGCTCAACC-3′; Cxcl5-6, 5′-GTCAGCAATAGTGTTTGTAG-3′ and 5′-GAGTGGCTCTTTGCCAAATC-3′; Cxcl5-7, 5′-TCCTAGCCATCCTCCCGCCC-3′ and 5′-CTCATTGTGGACAAGATCCC-3′; Cxcl5-8, 5′-CCTCCAGCTCCGCAGCTCCG-3′ and 5′-AAGGGGCAGCTGTGGAAGAG-3′; and Cxcl5-9, 5′-CCTCAGTCATAGCCGCAACG-3′ and 5′-GAGGATGCACAGGTGAGCCC-3′.
ChIP-qPCR assays
ChIP-qPCR assay was performed as previously described (Lan et al., 2007). Briefly, cells were crosslinked with 1% paraformaldehyde for 10 min and stopped by glycine. The chromatin was sonicated for 30 min (30% power, 30 s on, 30 s off; Bioruptor) and immunoprecipitated with anti-TET2 antibody (Shanghai Youke) or IgG for 3.5 h as previously described (Chen et al., 2018). The antibody–chromatin complexes were pulled down using protein A beads for 1.5 h. Beads were washed three times with high salt buffer, two times with low salt buffer, and once with TE buffer. After crosslink reversal and proteinase K (TaKaRa) treatment, DNA was isolated by PCR Purification Kit (QIAGEN) and analyzed by RT-qPCR.
Co-IP
KL cells were washed with cold PBS and lysed in NP-40 buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 0.5% NP-40, and protease inhibitor cocktail (Biotool) with rotation at 4°C for 45 min. Then the cell lysate was centrifuged at 13,000 rpm for 15 min and the supernatant was incubated with IgG antibody (DA1E; CST) or TET2 antibodies (18950S; CST) for 2.5 h, followed by incubation with Protein A for another 1.5 h at 4°C. Beads were washed three times with cold NP-40 buffer and the proteins were denatured by SDS loading buffer containing dithiothreitol.
ELISA
Tumors derived from KL and KLT mice at 7 wk after Adeno-Cre infection were randomly collected in different volumes of cell lysis buffer according to their weight. Then, the whole tumor extract was centrifuged at 14,000 rpm for 15 min and the supernatant was collected. The level of CXCL5 was detected in eight tumors in each group using a mouse CXCL5 ELISA kit according to the manufacturer’s instructions.
Co-culture KL cells with neutrophils
Freshly isolated neutrophils from KL-SCC by FACS and KL cells were mixed and then seeded in 12-well plates with 10% FBS/DMEM medium in a direct cell–cell contact manner (the ratio of neutrophils to KL cells was 20:1). EIPA (50 μM) was added to block the lipid transport. For lipid analysis in KL cells by FACS, all cells were cultured for 24 h and then stained with BODIPY 493/503 (GC42959; GLPBio) and EpCAM (563216; BD). For cell proliferation analysis in KL cells, all cells were cultured for 48 h and then stained with Ki-67 (NB500; Novus) and DAPI (4083; CST).
SRS microscope analysis
In the setup of SRS microscope, the pulsed Stokes (1,040 nm) and pump (804 nm) beams were directed from a commercial optical parametric oscillator (80 MHz; Insight DS+), chirped by SF57 glass rods, coupled into an inverted laser-scanning microscope (FV1200; Olympus), and focused onto the tissue sections with an objective lens (UPLSAPO 60 × W, NA = 1.2; Olympus). The Stokes beam was modulated at 20 MHz by an electronic optic modulator. The forward-going pump and Stokes beams were collected by a high NA oil condenser (NA = 1.4) after passing through samples. Transmitted through a lowpass filter, the stimulated Raman loss signal was detected by a reverse-biased photodiode followed by demodulation with a lock-in amplifier (HF2LI; Zurich Instruments) to feed the analog input of the microscope to generate images. For the unmixing of the two chemical components, we first acquired SRS images at 2,845 cm−1 and 2,930 cm−1 (the vibration frequency of CH2 and CH3 bonds, respectively). The relative concentrations of protein (aP) and lipid (bL) could be decomposed by linear algebra with the preknowledge of their SRS spectra (He et al., 2017).
For lipid detection, the tissue cryosections (20 μm) of KL and KLT mice were analyzed with SRS microscope.
For qualitative analysis of lipid, the standard curves of TG (G107438; Aladdin), cholesterol (A90286; Innochem), PC (T19514; Topscience), PE (P856654; Macklin), PS (T21437; Topscience), and SM (S877036; Macklin) were analyzed and then six fields of KL-SCC tissue cryosections (20 μm) were detected with SRS microscopy. 300 high-power fields were randomly selected for score analyses using the formula similar to H-score: cell percentage × imaging intensity score ×100. The final score was on a scale of 0–300.
TG-micelle preparation and treatment
TG-micelles were prepared as previously described (Dai et al., 2017) to obtain the final following concentration: 3 mg/ml (4.695 mM). In detail, we first dissolved 39 mg mPEG5000-b-PCL5000 and 39 mg TG in 3 ml tetrahydrofuran solution. The mixed solution was slowly added dropwise to 8 ml of ultrapure water at a suitable stirring speed. The emulsion was dialyzed against phosphate buffer (PBS) for 24 h by using a 3,500 g/mol molecular weight cutoff dialysis membrane to remove residual organic solvent. The final TG-micelles were made up to 3 ml and filtered through a 0.45-μm filter before use.
To evaluate TG-micelle effects in vitro, 5 × 105 cells per well were seeded into 12-well plates and starved for 3 h in basal medium (without FBS) after attachment. The TG-micelles (final concentration at 0.1 mM) were added into the basal medium. EIPA (50 µM) was added to block lipid transportation. After incubation for 24 h, KL cells were stained with BODIPY 493/503 (GC42959; GLPBio) and EpCAM (563216; BD). FACS (BD LSRII flow cytometer) was used to detect the lipid signal.
For cell proliferation detection, 3,000 cells were seeded in sextuplicate in 96-well plates and starved for 3 h in basal medium (without FBS) after attachment. Next, TG-micelles (0.1 mM) were added into the complete medium with FBS. EIPA (50 µM) was added to block lipid transport. The cells were stained with 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide and assessed with Epoch multivolume spectrophotometer system (570 nm/630 nm) at indicated time points.
FACS analysis
To prepare the single-cell suspension from mouse whole lung tissues or ex vivo cultured cells, the samples were incubated with fluorescently labeled antibodies. All the antibodies are listed as follows: EpCAM (563216; BD), CD45 (563709; BD), CD3 (553062; BD), CD4 (553051; BD), CD8 (551162; BD), Ly6G (551460; BD), CD11B (550993; BD), F4/80 (565410; BD), CD49b (563063; BD), and Foxp3 (12-4771-82; eBioscience). The population of indicated immune cells was analyzed by using the BD LSRII flow cytometer and FlowJo software as described previously (Jin et al., 2022).
Statistical analyses
Differences were compared using two-tailed unpaired Student’s t test or one-way ANOVA. P value < 0.05 was considered statistically significant. Error bars were represented with SD. Differences in AST incidence were calculated by Fisher’s exact test. Survival analysis was performed using the Kaplan Meier method as previously described (Wu et al., 2018).
Online supplemental material
Fig. S1 shows heatmap of indicated gene expression from TCGA, additional IHC data of the KL model, and data from CRISPR/Cas9-mediated Tet (Tet1/2/3) gene KO model (related to Fig. 1). Fig. S2 shows validation of Tet2 KO, the whole lung of the KL or KLT model, and the gene expression of squamous lineage markers (related to Fig. 1). Fig. S3 demonstrates that TET2 promotes neutrophil infiltration potentially through the upregulation of CXCL5 expression (related to Fig. 2). Fig. S4 shows validation of TET2/STAT3 inhibition in KL tumor cell lines, and additional data of Ruxo/C188-9 treatment in KL model (related to Figs. 3 and 4). Fig. S5 demonstrates that neutrophils transfer lipids to tumor cells to promote cell proliferation and AST (related to Figs. 5 and 6).
Data availability
RNA-seq data of mouse tumors of the KL model used in this study are openly available in the National Omics Data Encyclopedia under accession no. OEP002019 (Fang et al., 2023).
H. Ji is the lead contact.
Acknowledgments
We thank Dr. T. Jacks (Massachusetts Institute of Technology, Cambridge, MA, USA) for providing KrasLSL-G12D/+ mouse, Dr. Ronald DePinho (The University of Texas MD Anderson Cancer Center, Houston, TX, USA) for Lkb1L/L mouse, and Dr. Guoliang Xu (Center for Excellence in Molecular Cell Science, Shanghai, China) for Tet2L/L mouse and plasmids.
This work was supported by the National Key Research and Development Program of China (grants 2022YFA1103900 to H. Ji, 2020YFA0803300 to H. Ji); the National Natural Science Foundation of China (grants 82341002 to H. Ji, 82030083 to H. Ji, 32293192 to H. Ji, 31821002 to D. Ye, 31871431 to D. Ye, 82173340 to L. Hu, 32100593 to X. Tong, 82372763 to X. Wang, 82303039 to Z. Qin, and 82303575 to S. Tang); the Basic Frontier Scientific Research Program of Chinese Academy of Science (ZDBS-LY-SM006 to H. Ji); the Innovative Research Team of High-Level Local Universities in Shanghai (SSMU-ZLCX20180500 to H. Ji); the Science and Technology Commission of Shanghai Municipality (21ZR1470300 to L. Hu); and the Shanghai Sailing Program (23YF1452900 to Z. Qin).
Author contributions: Y. Xue: Conceptualization, Investigation, Validation, Writing—original draft, Writing—review and editing, Yuting Chen: Investigation, Validation, S. Sun: Investigation, Writing—review and editing, X. Tong: Resources, Yujia Chen: Formal analysis, Investigation, Validation, S. Tang: Formal analysis, X. Wang: Funding acquisition, Investigation, S. Bi: Validation, Y. Qiu: Resources, Q. Zhao: Validation, Z. Qin: Resources, Validation, Q. Xu: Investigation, Y. Ai: Validation, Leilei Chen: Resources, B. Zhang: Resources, Software, Writing—review and editing, Z. Liu: Software, M. Ji: Methodology, Supervision, M. Lang: Conceptualization, Investigation, Supervision, Luonan Chen: Data curation, Funding acquisition, Investigation, Methodology, Supervision, G. Xu: Conceptualization, Supervision, Writing—review and editing, L. Hu: Funding acquisition, Supervision, Writing—original draft, Writing—review and editing, D. Ye: Conceptualization, Project administration, Resources, Supervision, Writing—review and editing, H. Ji: Conceptualization, Funding acquisition, Project administration, Supervision, Writing—original draft, Writing—review and editing.

