


T reg cells bearing a diverse antigen receptor repertoire suppress pathogenic T cells and maintain immune homeostasis during their long lifespan. How their robust function is determined genetically remains elusive. Here, we investigate the regulatory space of the cis-regulatory elements of T reg lineage–specifying factor Foxp3. Foxp3 enhancers are known as distinct readers of environmental cues controlling T reg cell induction or lineage stability. However, their single deficiencies cause mild, if any, immune dysregulation, leaving the key transcriptional mechanisms determining Foxp3 expression and thereby T reg cell suppressive capacity uncertain. We examined the collective activities of Foxp3 enhancers and found that they coordinate to maximize T reg cell induction, Foxp3 expression level, or lineage stability through distinct modes and that ablation of synergistic enhancers leads to lethal autoimmunity in young mice. Thus, the induction and maintenance of a diverse, stable T reg cell repertoire rely on combinatorial Foxp3 enhancers, suggesting broad, stage-specific, synergistic activities of cell-intrinsic factors and cell-extrinsic cues in determining T reg cell suppressive capacity.

Introduction
Regulatory T (T reg) cells with a diverse TCR repertoire are induced by tissue environmental cues (e.g., TCR agonists, IL-2, and TGF-β) in the thymus and periphery to suppress effector T cells (TE) that not only cause autoimmune diseases but also elicit antitumor immune response (Josefowicz et al., 2012a; Sakaguchi et al., 2020). Upon commitment, T reg cells rely on several pathways, including IL-2 and TCR signaling, to maintain their lineage identity, fitness, and immune suppressive function during their long lifespan. In addition, a growing list of cell-intrinsic and -extrinsic factors was discovered to modulate T reg cell development and function, thus fine-tuning immune tolerance.
T reg cell induction and lineage maintenance are governed by cues that fluctuate. Therefore, considerable stochasticity affects T reg cell clones for specific antigens or epitopes. Immune perturbations and genetic variations may further shape the T reg cell TCR repertoire, impairing immune tolerance. These adversities raise a question about the mechanisms by which a diverse TCR repertoire and stable lineage identity are programmed at the genetic and epigenetic levels to confer on T reg cells an adequate suppressive capacity that opposes the uncertainties during T reg cell development and lineage maintenance.
Investigation of the mechanisms governing the expression of T reg cell lineage–specifying factor Foxp3 has produced many clues to this question (Josefowicz et al., 2012a; Sakaguchi et al., 2020). In particular, Foxp3 enhancers were characterized as individual modules that integrate distinct environmental cues and epigenetic programs to dictate T reg cell development or lineage stability. Ablation of conserved noncoding sequence (CNS) 1 compromises immune tolerance at the mucosal surfaces because of impaired peripheral T reg cell development (Campbell et al., 2018; Josefowicz et al., 2012b; Samstein et al., 2012); CNS2 deficiency destabilizes heritable Foxp3 expression and T reg cell fate in adverse environments, causing chronic tissue inflammation in aged mice or after immune challenge (Feng et al., 2014; Li et al., 2014); and loss of CNS0 or CNS3 reduces T reg cell development in both the thymus and periphery, yet without causing noticeable immune dysregulation (Dikiy et al., 2021; Feng et al., 2015). However, in the absence of autoimmune regulator Aire, a gene required for both thymic T reg cell induction and negative selection by facilitating the expression of tissue-restricted antigens in medullary thymic epithelial cells (Klein et al., 2014), CNS0 or CNS3 deficiency abruptly disrupts T reg cell–mediated immune tolerance, leading to early-onset, multiorgan autoimmunity, in contrast to Aire single-deficient mice.
To understand the mechanisms sustaining immune tolerance in CNS0-, CNS2-, and CNS3-deficient mice, we explored the regulatory space and modes of Foxp3 enhancers. Using CRISPR screening and genetic perturbations, we observed substantial synergies among these enhancers during T reg cell induction and lineage maintenance. Our study suggests broad coordination of cell-intrinsic and -extrinsic factors acting through Foxp3 enhancers to control T reg cell suppressive capacity by modulating T reg cell induction or lineage stability, thus determining the robustness of T reg cell–dependent immune tolerance.
Results
CRISPR screening uncovers synthetic interactions of Foxp3 enhancers
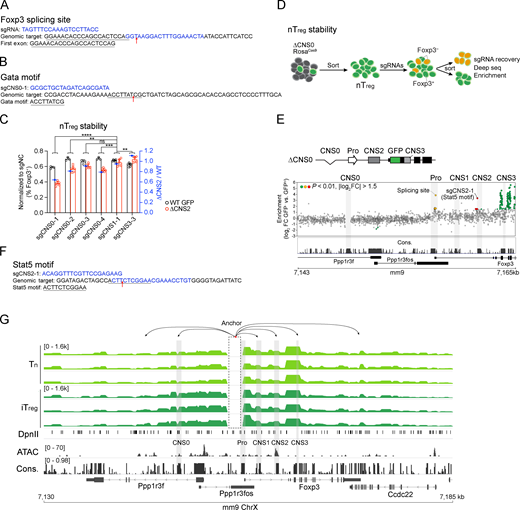
To elucidate the transcriptional mechanisms by which T reg cell repertoire diversity and lineage stability are controlled through Foxp3 expression, we examined the known regulatory circuits governing these processes. They appear to act on several distinct Foxp3 cis-regulatory elements (Josefowicz et al., 2012a), suggesting cooperative activities of the latter. In particular, enhancer CNS0 functions as an IL-2/Stat5 response element (Akamatsu et al., 2019; Dikiy et al., 2021), and CNS0 deficiency (ΔCNS0) reduces thymic T reg cells by two- to threefold without causing immune activation. To understand the CNS0-independent mechanisms of T reg cell development that confers immune tolerance in ΔCNS0 mice, we constructed a tiling library targeting all the sites bearing the Streptococcus pyogenes Cas9 protospacer-adjacent motifs (PAMs) throughout a 20-kb region surrounding the Foxp3 promoter (Table S1) to screen for the genetic elements required for in vitro T reg cell (iT reg) development from ΔCNS0 CD4 naive T cells (Tn) constitutively expressing Cas9 (Fig. 1, A and B). As expected, single guide RNAs (sgRNAs) targeting Foxp3 exons and mRNA splicing sites were highly enriched in Foxp3– cells (Fig. 1 B, Fig. S1 A, and Table S2). Although CRISPR-induced short indels might only partially disrupt enhancers’ function (Carleton et al., 2018), several sgRNAs targeting CNS3 were overrepresented in Foxp3– cells. We verified the additive effects of two of these sgRNAs with ΔCNS0 in iT reg cell induction. As a negative control, the sgRNA targeting CNS2 did not affect Foxp3 induction (Fig. 1 C). We also observed a mild reduction of Foxp3 induction when CNS1 was targeted by CRISPR in ΔCNS0 cells, suggesting a potential interaction between CNS0 and CNS1. As TCR and IL-2 signaling, two major T reg cell–inducing cues, act through CNS3 (Zheng et al., 2010) and CNS0 (Dikiy et al., 2021), respectively, and CNS0 and CNS3 are modified by enhancer marker H3K4me1 (Feng et al., 2015; Kitagawa et al., 2017; Placek et al., 2017; Zheng et al., 2010) and are bound by Satb1, whose deficiency impairs T reg cell development (Kitagawa et al., 2017), our results further suggest that CNS0 and CNS3 may coordinate to promote Foxp3 induction.

CRISPR screening reveals synthetic interactions of Foxp3 enhancers during T reg cell induction or lineage maintenance in vitro. (A) CRISPR-tiling screening for genetic elements regulating Foxp3 induction. After activation, ΔCNS0 CD4 Tn cells were transduced with the sgRNA library. GFP-Foxp3+ and GFP-Foxp3– cells were sorted 5 d later to assess sgRNA representation. (B) Enrichment of sgRNAs in GFP-Foxp3– versus GFP-Foxp3+ cells. The sgRNA targets and Stat5 chromatin immunoprecipitation sequencing (ChIP-seq) in nT reg cells (Dikiy et al., 2021) are aligned with DNA sequence conservation (Cons.) in placental mammals. The yellow, red, and blue dots highlight the sgRNAs targeting Foxp3 promoter, CNS3, and exons, respectively. n = 7 replicates. (C) Validation of the effects of sgRNAs targeting CNS1 and CNS3 on iT reg cell induction. The percentages of GFP-Foxp3+ among live transduced cells were normalized to those of sgNC (nontargeting control)–transduced cells of the same genotypes (left y axis). The ratios of normalized GFP-Foxp3+ percentages of ΔCNS0 cells to those of WT cells were calculated (right y axis). Technical replicates are shown. sgCNS1 and sgCNS3 represent three experiments, and sgCNS2-1 represents one experiment. The differences between WT and ΔCNS0 were compared between sgCNS2-1 and other sgRNAs by two-way ANOVA. *, P < 0.05; **, P < 0.01. (D) CRISPR-tiling screening for genetic elements maintaining Foxp3 expression. ΔCNS2 CD4 Tn cells were cultured in T reg cell–inducing conditions with ASC for 3 d and then transduced with the sgRNA library. After an additional 3-d culture, the top 10% of GFP-Foxp3+ cells and GFP-Foxp3– cells were sorted to assess sgRNA representation. (E) Enrichment of sgRNAs in ΔCNS2 GFP-Foxp3– versus GFP-Foxp3hi cells. sgRNAs targeting Gata, Egr2, Yy1, and E2F1 motifs at CNS0 (sgCNS0-1, -2, -3, and -4, respectively) were enriched. Red dots highlight the sgRNAs targeting the CNS0 region. n = 4 replicates. (F) Validation of the effects of sgRNAs targeting CNS0 on Foxp3 expression in ASC-treated iT reg cells. Results were analyzed as described in C. Data show technical replicates and represent three experiments. ***, P < 0.001; ****, P < 0.0001. (G) Chromatin looping between Foxp3 promoter and enhancers assessed by Capture-C. One end of the reads mapped to the bait region were filtered for clarity (dashed box). Arrows indicate potentially looped regions. Data represent two replicates. Foxp3 ChIP-seq (nT reg cells; Kitagawa et al., 2017), ATAC-seq (nT reg cells), and DNA conservation are aligned. ChrX, X chromosome; FC, fold change; Pro, promoter.
CRISPR screening reveals synthetic interactions of Foxp3 enhancers during T reg cell induction or lineage maintenance in vitro. (A) CRISPR-tiling screening for genetic elements regulating Foxp3 induction. After activation, ΔCNS0 CD4 Tn cells were transduced with the sgRNA library. GFP-Foxp3+ and GFP-Foxp3– cells were sorted 5 d later to assess sgRNA representation. (B) Enrichment of sgRNAs in GFP-Foxp3– versus GFP-Foxp3+ cells. The sgRNA targets and Stat5 chromatin immunoprecipitation sequencing (ChIP-seq) in nT reg cells (Dikiy et al., 2021) are aligned with DNA sequence conservation (Cons.) in placental mammals. The yellow, red, and blue dots highlight the sgRNAs targeting Foxp3 promoter, CNS3, and exons, respectively. n = 7 replicates. (C) Validation of the effects of sgRNAs targeting CNS1 and CNS3 on iT reg cell induction. The percentages of GFP-Foxp3+ among live transduced cells were normalized to those of sgNC (nontargeting control)–transduced cells of the same genotypes (left y axis). The ratios of normalized GFP-Foxp3+ percentages of ΔCNS0 cells to those of WT cells were calculated (right y axis). Technical replicates are shown. sgCNS1 and sgCNS3 represent three experiments, and sgCNS2-1 represents one experiment. The differences between WT and ΔCNS0 were compared between sgCNS2-1 and other sgRNAs by two-way ANOVA. *, P < 0.05; **, P < 0.01. (D) CRISPR-tiling screening for genetic elements maintaining Foxp3 expression. ΔCNS2 CD4 Tn cells were cultured in T reg cell–inducing conditions with ASC for 3 d and then transduced with the sgRNA library. After an additional 3-d culture, the top 10% of GFP-Foxp3+ cells and GFP-Foxp3– cells were sorted to assess sgRNA representation. (E) Enrichment of sgRNAs in ΔCNS2 GFP-Foxp3– versus GFP-Foxp3hi cells. sgRNAs targeting Gata, Egr2, Yy1, and E2F1 motifs at CNS0 (sgCNS0-1, -2, -3, and -4, respectively) were enriched. Red dots highlight the sgRNAs targeting the CNS0 region. n = 4 replicates. (F) Validation of the effects of sgRNAs targeting CNS0 on Foxp3 expression in ASC-treated iT reg cells. Results were analyzed as described in C. Data show technical replicates and represent three experiments. ***, P < 0.001; ****, P < 0.0001. (G) Chromatin looping between Foxp3 promoter and enhancers assessed by Capture-C. One end of the reads mapped to the bait region were filtered for clarity (dashed box). Arrows indicate potentially looped regions. Data represent two replicates. Foxp3 ChIP-seq (nT reg cells; Kitagawa et al., 2017), ATAC-seq (nT reg cells), and DNA conservation are aligned. ChrX, X chromosome; FC, fold change; Pro, promoter.
Extended CRISPR-tiling screening and Capture-C experiments. (A) sgRNA targeting a Foxp3 mRNA splicing site identified in Fig. 1 B. (B) sgRNA targeting a Gata motif identified in Fig. 1 E. (C) Verification of the effect of sgRNAs targeting CNS0 on the stability of WT GFP and ΔCNS2 nT reg cells. nT reg cells were cocultured with mouse T-Activator CD3/CD28 beads in the presence of 500 U/ml recombinant IL-2 and 10 ng/ml IL-4 after sgRNA transduction for 4 d before analysis. GFP-Foxp3+ cells were analyzed as described in Fig. 1 C. Data represent one experiment. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by two-way ANOVA. See Table S3 for sgRNA sequences. (D) CD4+GFP+ nT reg cells were double sorted from ΔCNS0 RosaCas9 mice and cultured with mouse T-Activator CD3/CD28 beads in the presence of 500 U/ml recombinant IL-2 for 3 d. Retroviral sgRNA library was then transduced. Cells were grown for an additional 3 d with mouse T-Activator CD3/CD28 beads, 500 U/ml recombinant IL-2, and 10 ng/ml recombinant IL-4. Within the live-cell gating, the top 50% of GFP-Foxp3hi cells and GFP-Foxp3– cells were sorted to assess the representation of sgRNAs. (E) Enrichment scores of sgRNAs in Foxp3– versus Foxp3+ cells. Dots represent individual sgRNA targets. Colored dots represent the sgRNAs significantly enriched or depleted in GFP-Foxp3– cells (P < 0.01, |log2FC| > 1.5), among which the yellow and red dots highlight the sgRNAs targeting Foxp3 promoter and CNS2 region, respectively. n = 3 replicates. (F) sgRNA targeting a Stat5 motif identified in E. (G) Capture-C of CD4 Tn and ASC-treated iT reg cells, aligned with ATAC-seq of ASC-treated iT reg cells and DNA sequence conservation. Several looping regions are shown. Data represent three replicates. ChrX, X chromosome; Con., conservation; FC, fold change; Pro, promoter.
Extended CRISPR-tiling screening and Capture-C experiments. (A) sgRNA targeting a Foxp3 mRNA splicing site identified in Fig. 1 B. (B) sgRNA targeting a Gata motif identified in Fig. 1 E. (C) Verification of the effect of sgRNAs targeting CNS0 on the stability of WT GFP and ΔCNS2 nT reg cells. nT reg cells were cocultured with mouse T-Activator CD3/CD28 beads in the presence of 500 U/ml recombinant IL-2 and 10 ng/ml IL-4 after sgRNA transduction for 4 d before analysis. GFP-Foxp3+ cells were analyzed as described in Fig. 1 C. Data represent one experiment. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by two-way ANOVA. See Table S3 for sgRNA sequences. (D) CD4+GFP+ nT reg cells were double sorted from ΔCNS0 RosaCas9 mice and cultured with mouse T-Activator CD3/CD28 beads in the presence of 500 U/ml recombinant IL-2 for 3 d. Retroviral sgRNA library was then transduced. Cells were grown for an additional 3 d with mouse T-Activator CD3/CD28 beads, 500 U/ml recombinant IL-2, and 10 ng/ml recombinant IL-4. Within the live-cell gating, the top 50% of GFP-Foxp3hi cells and GFP-Foxp3– cells were sorted to assess the representation of sgRNAs. (E) Enrichment scores of sgRNAs in Foxp3– versus Foxp3+ cells. Dots represent individual sgRNA targets. Colored dots represent the sgRNAs significantly enriched or depleted in GFP-Foxp3– cells (P < 0.01, |log2FC| > 1.5), among which the yellow and red dots highlight the sgRNAs targeting Foxp3 promoter and CNS2 region, respectively. n = 3 replicates. (F) sgRNA targeting a Stat5 motif identified in E. (G) Capture-C of CD4 Tn and ASC-treated iT reg cells, aligned with ATAC-seq of ASC-treated iT reg cells and DNA sequence conservation. Several looping regions are shown. Data represent three replicates. ChrX, X chromosome; Con., conservation; FC, fold change; Pro, promoter.
Next, we tested whether T reg cell lineage stability is controlled by the coordination of Foxp3 enhancers. Because ΔCNS2 partially impairs the stability of Foxp3 expression, we performed CRISPR-tiling screening to search for the genetic elements maintaining Foxp3 expression in ΔCNS2 T reg cells (Fig. 1, D and E). We transduced the same sgRNA library into ascorbic acid (ASC)–pretreated iT reg cells that recapitulate T reg cell lineage commitment in vivo via Tet-mediated DNA demethylation (Yue et al., 2016). Several sgRNAs targeting CNS0 (e.g., sgCNS0-1 against a Gata motif) were overrepresented in Foxp3– cells (Fig. 1 E, Fig. S1 B, and Table S2). We verified this result with individual sgRNAs targeting CNS0 in ASC-pretreated iT reg cells and natural T reg cells (nT reg), with sgRNAs targeting CNS1 and CNS3 as negative controls (Fig. 1 F and Fig. S1 C). We also performed CRISPR-tiling screening with ex vivo–isolated ΔCNS0 nT reg cells and found that sgRNAs targeting CNS2, particularly a Stat5 motif, were highly enriched in Foxp3– cells (Fig. S1, D–F; and Table S2). Thus, CNS0 may coordinate with CNS2 to maintain Foxp3 expression, consistent with their Stat5 binding in nT reg cells (Fig. 1 B, Stat5 chromatin immunoprecipitation sequencing track) and the important role of IL-2/Stat5 signaling in maintaining Foxp3 expression (Feng et al., 2014).
Notably, sgRNAs targeting several nonconserved regions were also enriched or depleted in our CRISPR screening (Fig. 1, B and E; and Fig. S1 E), which requires further investigation. In addition, there are a few limitations of CRISPR-tiling screening: (1) DNA motifs might not be fully covered by available PAMs, (2) short indels may only partially disrupt enhancer’s function, (3) CRISPR-induced DNA breaks might influence Foxp3 transcription by reported mechanisms (Vitelli et al., 2017), and (4) in vitro assays only partially recapitulate T reg cell development and lineage maintenance in vivo. Nonetheless, our results, together with the epigenetic markers and regulatory circuits described above, suggest the coordination of Foxp3 enhancers during T reg cell induction and lineage maintenance.
To understand the mechanistic basis of this coordination, we assessed the three-dimensional chromatin architecture around the Foxp3 locus with Capture-C (Davies et al., 2016; Kim and Dekker, 2018) in precursor cells (i.e., CD4 Tn cells), ASC-treated iT reg cells, and nT reg cells. We observed reproducible, abundant loops between the Foxp3 promoter and enhancers as well as other novel chromatin regions (Fig. 1 G and Fig. S1 G). Interestingly, these interactions appear to be independent of T reg cell development. The looping of DNA segments depicts the chromatin architecture that supports Foxp3 enhancers’ function in regulating transcription.
Deficiencies of enhancers CNS0/CNS3 and CNS0/CNS2 cause fatal autoimmunity
To reveal the biological significance of the coordination of Foxp3 enhancers, we generated CNS0/CNS3 and CNS0/CNS2 double-deficient (ΔCNS0,3 and ΔCNS0,2, respectively) mice expressing the same GFP-Foxp3 fusion protein as WT Foxp3GFP and single-deficient mice to facilitate T reg cell isolation (Fig. 2, A–C; Fontenot et al., 2005). Heterozygous female ΔCNS0,2 and ΔCNS0,3 mice were indistinguishable from WT mice, and hemizygous male ΔCNS0,2 and ΔCNS0,3 mice were born at Mendelian ratios equal to those of WT littermates. ΔCNS0,3 mice had a lifespan as short as Foxp3null mice, suggesting abolished T reg cell–mediated immune tolerance (Fig. 2 D). ΔCNS0,2 mice died at age 2–4 mo, whereas no fatality was observed in ΔCNS0 and ΔCNS2 mice up to 7 mo, consistent with previous reports (Feng et al., 2014; Li et al., 2014). The longer lifespan of ΔCNS0,2 mice than that of Foxp3null mice implies that ΔCNS0,2 leads to residual T reg cell function. Overall, the synthetic lethal autoimmunity of ΔCNS0,2 and ΔCNS0,3 suggests that these enhancers have significant overlapping or compensatory roles such that their single deficiencies are largely tolerated.

Deficiencies of enhancers CNS0/CNS3 and CNS0/CNS2 cause fatal autoimmunity. (A) Mouse strains used in this study. ΔCNS0,2 and ΔCNS0,3 mice were generated from Foxp3ΔCNS0-GFP or ΔCNS0 mice (Dikiy et al., 2021) by the CRISPR/Cas9 approach. (B and C) Verification of genetic deletions. The regions deleted in previous ΔCNS2 mice (Zheng et al., 2010) and ΔCNS0,2 founders (B) or those deleted in previous ΔCNS3 mice (Zheng et al., 2010) and ΔCNS0,3 founders (C) were compared. Horizontal bars represent deleted regions. (D) Survival curves of male WT GFP, Foxp3null, ΔCNS0, ΔCNS2, ΔCNS3, ΔCNS0,2, and ΔCNS0,3 mice. P values by log-rank (Mantel-Cox) test. (E) Representative body sizes of 5-wk-old male mice. Arrowheads indicate dermatitis. Scale bars, 1 cm. (FandH) Comparison of littermate WT GFP and ΔCNS0,2 mice 3–4 mo old. Scale bars, 1 cm. (G) Average scores of tissue inflammation assessed by histopathology in 3–5-wk-old male mice. n = 6 WT, ΔCNS2, and ΔCNS3; n = 4 ΔCNS0, ΔCNS0,2, ΔCNS0,3, and Foxp3null. (I) Representative images of tissue sections after H&E staining in G. Ear skin: bounded lines, thickened dermis due to inflammatory cell infiltrates; arrows, epidermal ulcers. Lung: arrows, perivascular inflammatory cell aggregates. Liver: arrows, perivascular inflammatory cell aggregates. Pancreas: arrows, inflammatory cell aggregates; asterisks, residual or remaining acinar cell foci. Stomach: bounded lines, the region of lamina propria; arrows, representative inflammatory cell aggregates. Brown adipose: arrows, perivascular inflammatory aggregates; the color difference is due to brown fat hypertrophy. Scale bars, 200 µm (ear skin), 100 µm (lung, liver, pancreas, and brown adipose), and 20 µm (stomach). chrX, X chromosome; Cons, conservation; ES, embryonic stem; Pro, promoter.
Deficiencies of enhancers CNS0/CNS3 and CNS0/CNS2 cause fatal autoimmunity. (A) Mouse strains used in this study. ΔCNS0,2 and ΔCNS0,3 mice were generated from Foxp3ΔCNS0-GFP or ΔCNS0 mice (Dikiy et al., 2021) by the CRISPR/Cas9 approach. (B and C) Verification of genetic deletions. The regions deleted in previous ΔCNS2 mice (Zheng et al., 2010) and ΔCNS0,2 founders (B) or those deleted in previous ΔCNS3 mice (Zheng et al., 2010) and ΔCNS0,3 founders (C) were compared. Horizontal bars represent deleted regions. (D) Survival curves of male WT GFP, Foxp3null, ΔCNS0, ΔCNS2, ΔCNS3, ΔCNS0,2, and ΔCNS0,3 mice. P values by log-rank (Mantel-Cox) test. (E) Representative body sizes of 5-wk-old male mice. Arrowheads indicate dermatitis. Scale bars, 1 cm. (FandH) Comparison of littermate WT GFP and ΔCNS0,2 mice 3–4 mo old. Scale bars, 1 cm. (G) Average scores of tissue inflammation assessed by histopathology in 3–5-wk-old male mice. n = 6 WT, ΔCNS2, and ΔCNS3; n = 4 ΔCNS0, ΔCNS0,2, ΔCNS0,3, and Foxp3null. (I) Representative images of tissue sections after H&E staining in G. Ear skin: bounded lines, thickened dermis due to inflammatory cell infiltrates; arrows, epidermal ulcers. Lung: arrows, perivascular inflammatory cell aggregates. Liver: arrows, perivascular inflammatory cell aggregates. Pancreas: arrows, inflammatory cell aggregates; asterisks, residual or remaining acinar cell foci. Stomach: bounded lines, the region of lamina propria; arrows, representative inflammatory cell aggregates. Brown adipose: arrows, perivascular inflammatory aggregates; the color difference is due to brown fat hypertrophy. Scale bars, 200 µm (ear skin), 100 µm (lung, liver, pancreas, and brown adipose), and 20 µm (stomach). chrX, X chromosome; Cons, conservation; ES, embryonic stem; Pro, promoter.
Consistent with survival, ΔCNS0,3 mice exhibited an aggressive autoimmune syndrome comparable to that of Foxp3null mice at an early age (5 wk old; Fig. 2 E). In contrast, relatively milder inflammation was observed in 3–4-mo-old ΔCNS0,2 mice (Fig. 2 F). Despite different onsets, both ΔCNS0,2 and ΔCNS0,3 mice developed severe lymphoproliferative disease (Fig. 2, G–I). Conventional T (T con) cells were markedly activated in these mice characterized by mixed T helper 1 (Th1; IFNγ+), Th2 (IL-13+), and Th17 (IL-17+) responses (Fig. 3). To test whether T reg cells in ΔCNS0,2 mice suppress any specific autoimmunity, we performed single-cell RNA sequencing (scRNA-seq) of the lymphocytes isolated from the spleen and lung of WT and ΔCNS0,2 mice, which revealed widespread immune activation as observed in T reg cell–deficient mice (Fig. 4 and Fig. S2; Sakaguchi et al., 2020). Therefore, ΔCNS0,2 may nonselectively impair T reg cell function.

Activation of pathogenic T cells in ΔCNS0/CNS2 and ΔCNS0/CNS3 mice. (A) Frequencies of CD44hiCD62Llo in CD4+Foxp3– cells from 3–4-wk-old male mice. n ≥ 5 per group. Data were pooled from two experiments. (B–K) Activation status of T con cells in 3–4-wk-old WT GFP, ΔCNS0, ΔCNS3, and ΔCNS0,3 mice (B–F) or WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 mice (G–K). Frequencies of CD8 effector memory T cells (CD44hiCD62Llo) (B and G) and IFN-γ–, IL-13–, or IL-17a–producing T con cells (C–F and H–K) were quantified. n = 7–10 per group. Data represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by Mann-Whitney test.
Activation of pathogenic T cells in ΔCNS0/CNS2 and ΔCNS0/CNS3 mice. (A) Frequencies of CD44hiCD62Llo in CD4+Foxp3– cells from 3–4-wk-old male mice. n ≥ 5 per group. Data were pooled from two experiments. (B–K) Activation status of T con cells in 3–4-wk-old WT GFP, ΔCNS0, ΔCNS3, and ΔCNS0,3 mice (B–F) or WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 mice (G–K). Frequencies of CD8 effector memory T cells (CD44hiCD62Llo) (B and G) and IFN-γ–, IL-13–, or IL-17a–producing T con cells (C–F and H–K) were quantified. n = 7–10 per group. Data represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by Mann-Whitney test.

Assessment of immune cell activation in WT GFP and ΔCNS0,2 mice with scRNA-seq. scRNA-seq of CD45+ cells sorted from pooled spleens and lungs of WT GFP and ΔCNS0,2 mice. Transcriptional clusters of CD4 and CD8 T cells were identified for further analysis. (A–C) Expression (log2 transformed) of resting state markers Ccr7, Lef1, and Ly6c1 (A); activation markers Ctla4, Ikzf2, and Tigit (B); and inflammatory cytokines Ifng, Il4, and Il13 (C) in CD4 T cells. (D–F) Expression (log2-transformed) of resting state markers Ccr7 and Lef1 (D); activation markers Nr4a1, Nkg7, and Mki67 (E); and effector molecules Gzma, Ccl5, and Ifng (F) in CD8 T cells.
Assessment of immune cell activation in WT GFP and ΔCNS0,2 mice with scRNA-seq. scRNA-seq of CD45+ cells sorted from pooled spleens and lungs of WT GFP and ΔCNS0,2 mice. Transcriptional clusters of CD4 and CD8 T cells were identified for further analysis. (A–C) Expression (log2 transformed) of resting state markers Ccr7, Lef1, and Ly6c1 (A); activation markers Ctla4, Ikzf2, and Tigit (B); and inflammatory cytokines Ifng, Il4, and Il13 (C) in CD4 T cells. (D–F) Expression (log2-transformed) of resting state markers Ccr7 and Lef1 (D); activation markers Nr4a1, Nkg7, and Mki67 (E); and effector molecules Gzma, Ccl5, and Ifng (F) in CD8 T cells.

Immune cells in WT GFP and ΔCNS0,2 mice assessed by scRNA-seq. (A and B) Transcriptional clusters and the frequencies of immune cell types. CD45+ cells were sorted from pooled spleens (A) or lungs (B) of WT GFP and ΔCNS0,2 mice. B cells are not shown. Numbers of cells sequenced: GFP WT spleen, 6,543; lung, 5,863; ΔCNS0,2 spleen, 7,246; and lung, 6,425. (C) Normalized cell counts of individual clusters. UMAP, uniform manifold approximation and projection.
Immune cells in WT GFP and ΔCNS0,2 mice assessed by scRNA-seq. (A and B) Transcriptional clusters and the frequencies of immune cell types. CD45+ cells were sorted from pooled spleens (A) or lungs (B) of WT GFP and ΔCNS0,2 mice. B cells are not shown. Numbers of cells sequenced: GFP WT spleen, 6,543; lung, 5,863; ΔCNS0,2 spleen, 7,246; and lung, 6,425. (C) Normalized cell counts of individual clusters. UMAP, uniform manifold approximation and projection.
Systemic, fatal autoimmune inflammation in ΔCNS0,2 and ΔCNS0,3 mice demonstrates the significance of the coordination of Foxp3 enhancers, revealing that almost the entire T reg cell suppressive capacity relies on synergistic Foxp3 enhancers.
An additive effect of CNS0 and CNS3 on thymic T reg cell development and T reg cell TCR diversity
Our CRISPR screening result suggests that CNS0 and CNS3 coordinate to promote Foxp3 induction. To determine its role in T reg cell development in vivo, we examined T reg cells in 3–4-wk-old male mice. ΔCNS0 or ΔCNS3 resulted in a two- to threefold reduction of thymic T reg cell frequencies compared with WT mice (Fig. 5, A and B). In contrast, ΔCNS0,3 decreased thymic T reg cells by ∼10-fold, indicating a substantial cooperation between CNS0 and CNS3.

An additive effect of CNS0 and CNS3 on thymic T reg cell development and T reg cell TCR diversity. (A and B) T reg frequencies in 3–4-wk-old mice. Data were pooled from three experiments. n > 5 per genotype. ****, P < 0.0001 by Mann-Whitney test. (C) Experimental procedures for generating mixed bone marrow chimeric mice. (D–J) T reg cell frequencies and the ratios (underlined numbers) of median T reg cell frequencies of the comparison groups (D), frequencies of Ki67+ in T reg cells (E), ratios of T reg cell percentages in the periphery to those in the thymuses of individual mice (F), GFP-Foxp3 median fluorescence intensity (MFI) of T reg cells (G), CD25 MFI of T reg cells (H), frequencies of T reg precursor cells (CD4+CD25+Foxp3–) among CD4+CD8– thymocytes (I), and frequencies of CD44hiCD62Llo among CD4+Foxp3– cells (J) in the CD45.1– compartment of mixed bone marrow chimeras. n = 5–6 per genotype. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 by two-way ANOVA. (K and L) Inverse Simpson indexes of TCR repertoire and total unique TCR clones of T reg cells in 16-d-old mice. T reg cells were sorted from pooled spleen, skin draining LN, and mLN of individual mice. Ratios of mean inverse Simpson indexes or total unique TCR clones of WT GFP to other genotypes are shown. n = 5: WT GFP; n = 3: ΔCNS0, ΔCNS3, and ΔCNS0,3. Data are mean ± SD. Significance by unpaired t test.
An additive effect of CNS0 and CNS3 on thymic T reg cell development and T reg cell TCR diversity. (A and B) T reg frequencies in 3–4-wk-old mice. Data were pooled from three experiments. n > 5 per genotype. ****, P < 0.0001 by Mann-Whitney test. (C) Experimental procedures for generating mixed bone marrow chimeric mice. (D–J) T reg cell frequencies and the ratios (underlined numbers) of median T reg cell frequencies of the comparison groups (D), frequencies of Ki67+ in T reg cells (E), ratios of T reg cell percentages in the periphery to those in the thymuses of individual mice (F), GFP-Foxp3 median fluorescence intensity (MFI) of T reg cells (G), CD25 MFI of T reg cells (H), frequencies of T reg precursor cells (CD4+CD25+Foxp3–) among CD4+CD8– thymocytes (I), and frequencies of CD44hiCD62Llo among CD4+Foxp3– cells (J) in the CD45.1– compartment of mixed bone marrow chimeras. n = 5–6 per genotype. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 by two-way ANOVA. (K and L) Inverse Simpson indexes of TCR repertoire and total unique TCR clones of T reg cells in 16-d-old mice. T reg cells were sorted from pooled spleen, skin draining LN, and mLN of individual mice. Ratios of mean inverse Simpson indexes or total unique TCR clones of WT GFP to other genotypes are shown. n = 5: WT GFP; n = 3: ΔCNS0, ΔCNS3, and ΔCNS0,3. Data are mean ± SD. Significance by unpaired t test.
Reduced T reg cell induction in ΔCNS0 or ΔCNS3 mice was followed by elevated proliferation of T reg cells in the periphery through homeostatic expansion (Dikiy et al., 2021; Feng et al., 2015). In addition, inflammation also drives T reg cell proliferation (Kim et al., 2007). However, in the presence of both homeostatic and inflammatory drivers, ΔCNS0,3 T reg cells in the periphery remained significantly less than those in WT, ΔCNS0, or ΔCNS3 mice. If ΔCNS0,3 predominantly affected Foxp3 induction, T reg cell expansion was likely constrained by available antigens, which is comparable to the limited T reg cells derived from monoclonal transgenic TCRs against self-antigens (Bautista et al., 2009). Therefore, T reg cell clones for many self-antigens might be absent in ΔCNS0,3 mice.
To assess the cell-intrinsic effects of ΔCNS0,3, we generated a competitive setting with mixed bone marrow chimeric mice (Fig. 5 C) in which congenically marked WT T reg cells suppressed aberrant immune activation. ΔCNS0 or ΔCNS3 reduced thymic T reg cell frequencies by three- to fourfold (Fig. 5 D). Remarkably, ΔCNS0,3 caused a >50-fold reduction of thymic T reg cells, indicating a strong synergistic effect of ΔCNS0 and ΔCNS3. ΔCNS0,3 T reg cells expressed higher Ki67, and their abundance in the periphery mildly increased relative to that in the thymus (Fig. 5, D–F), suggesting that ΔCNS0,3 T reg cells were more self-reactive as also observed in ΔCNS0 and ΔCNS3 T reg cells (Dikiy et al., 2021; Feng et al., 2015). Thus, Foxp3 induction in precursor cells bearing lower-affinity TCRs for self-antigens might be preferentially affected by ΔCNS0,3. Notably, ΔCNS0,3 reduced Foxp3 expression levels per cell by ∼50% (Fig. 5 G), suggesting that CNS0 and CNS3 play redundant roles in maintaining high levels of Foxp3 expression. ΔCNS0,3 also decreased CD25 expression (Fig. 5 H), probably because of attenuated Foxp3-mediated positive regulation (Gavin et al., 2007) and/or activation-dependent downregulation of CD25 (van der Veeken et al., 2016).
Thymic T reg cell development appears to undergo two steps: TCR agonists triggering CD25 expression followed by IL-2 signaling–dependent Foxp3 induction (Lio and Hsieh, 2008). CD4+CD8–CD25+Foxp3– thymocytes were significantly accumulated in ΔCNS0,3 mice (Fig. 5 I), suggesting that ΔCNS0,3 blocks IL-2–induced precursor-to–T reg cell transition. This defect might convert many T reg precursor cells to autoreactive TE cells. However, CD4 TE cells in WT GFP, ΔCNS0, ΔCNS3, and ΔCNS0,3 compartments were equally suppressed by WT T reg cells (Fig. 5 J). Therefore, the lethal autoimmunity in ΔCNS0,3 mice was primarily caused by the dysregulation of Foxp3 expression.
To understand to what extent the coordination between CNS0 and CNS3 determines the diversity of T reg cell TCR repertoire, we profiled the TCRα and TCRβ chains of T reg cells isolated from the lymphoid organs of age-matched male WT, ΔCNS0, ΔCNS3, and ΔCNS0,3 mice. We observed a 0.7- to twofold reduction of the diversity intensities (inverse Simpson indexes) of ΔCNS0 and ΔCNS3 T reg cells (Fig. 5, K and L). Similarly, ΔCNS0 and ΔCNS3 reduced the total unique TCRs of T reg cells in each animal. Markedly, ΔCNS0,3 decreased the diversity intensities and total unique TCRs of T reg cells by >40-fold. Given the varying immunological consequences of ΔCNS0, ΔCNS3, and ΔCNS0,3, this TCR sequencing result defines the dynamic range of the T reg cell TCR diversity related to nearly the entire spectrum of T reg cell suppressive capacity determined by CNS0 and CNS3, individually and collectively.
Additive effects of CNS0 and CNS3 on T reg cell induction in vitro and T reg cell lineage stability
We then compared the effects of ΔCNS0, ΔCNS3, and ΔCNS0,3 on iT reg cell induction. To avoid the influence of autoimmune inflammation, we isolated CD4 Tn cells from mixed bone marrow chimeric mice described above. We observed a mild additive effect of ΔCNS0 and ΔCNS3 on iT reg cell induction (Fig. 6 A), probably because of prolonged exposure to T reg cell–inducing cues (Materials and methods). Unlike nT reg cells, Foxp3 expression levels in iT reg cells were not influenced by the additive effect of ΔCNS0 and ΔCNS3 (Fig. 6 B, compare ΔCNS0 and ΔCNS3 with ΔCNS0,3).

Additive effects of CNS0 and CNS3 on T reg cell induction in vitro and T reg cell lineage stability. (A and B) T reg cell induction in vitro. CD4 Tn cells sorted from mixed bone marrow chimeric mice were used for iT reg cell induction. Frequencies of GFP-Foxp3+ cells (A) and GFP-Foxp3 expression levels (MFI) of iT reg cells (B) are shown. Data are mean ± SEM of triplicates and represent two experiments. *, P < 0.05; ***, P < 0.001 by unpaired t test. (C–E) Experimental procedures for assessing the stability of Foxp3 expression (C). CD4 Tn cells from mixed bone marrow chimeras were cultured in T reg cell–inducing conditions with or without ASC. iT reg cells were sorted and cultured in fresh media for 4 d before analysis (D and E). Numbers indicate the ratios of the average percentages of Foxp3+ cells between WT and Foxp3-enhancer deficiencies. Data are mean ± SEM of triplicates and represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by two-way ANOVA. (F and G) Experimental procedures for assaying the stability of nT reg cells in Rag1−/− mice (F). Frequencies of Foxp3+ in CD45.2+CD4+TCRβ+ cells were analyzed (G). n = 5–6 per genotype. Data represent two experiments. **, P < 0.01 by Mann-Whitney test. (H and I) Assessment of the immunosuppressive function of nT reg cells in vitro. nT reg cells sorted from male mice (H) or mixed bone marrow chimeric mice (I) were cocultured with antigen-presenting cells and cell trace–labeled WT CD4 Tn cells for 4 d. Dilution of cell trace was quantified to assess the proliferation of T con cells. Data represent two experiments. **, P < 0.01; ****, P < 0.0001 by two-way ANOVA. (J) Percentages of CD44hiCD62Llo in nT reg cells from mixed bone marrow chimeric mice. n = 5–6 per genotype. Data represent two experiments. *, P < 0.05; **, P < 0.01 by two-way ANOVA. (K–M) Schematic illustration of the coverage of T reg cell TCR repertoire for antigens bearing multiple epitopes (K). Numbers of T reg cells against individual epitopes are shown by box plots. In ΔCNS0 or ΔCNS3 mice, impaired T reg cell induction leads to underrepresentation of TCR clones for particular epitopes without significantly affecting overall immune tolerance. Further reduction of T reg cell development in ΔCNS0,3 mice depletes all the clones for many antigens, breaking down immune tolerance. Numbers of plus signs indicate the levels of immune tolerance (K). CNS3 and CNS0 indirectly or directly integrate TCR and IL-2/Stat5 signaling, respectively, to promote Foxp3 induction (L). A hypothetical model of CNS0 and CNS3 in determining T reg cell induction efficiency and T reg cell suppressive capacity (M).
Additive effects of CNS0 and CNS3 on T reg cell induction in vitro and T reg cell lineage stability. (A and B) T reg cell induction in vitro. CD4 Tn cells sorted from mixed bone marrow chimeric mice were used for iT reg cell induction. Frequencies of GFP-Foxp3+ cells (A) and GFP-Foxp3 expression levels (MFI) of iT reg cells (B) are shown. Data are mean ± SEM of triplicates and represent two experiments. *, P < 0.05; ***, P < 0.001 by unpaired t test. (C–E) Experimental procedures for assessing the stability of Foxp3 expression (C). CD4 Tn cells from mixed bone marrow chimeras were cultured in T reg cell–inducing conditions with or without ASC. iT reg cells were sorted and cultured in fresh media for 4 d before analysis (D and E). Numbers indicate the ratios of the average percentages of Foxp3+ cells between WT and Foxp3-enhancer deficiencies. Data are mean ± SEM of triplicates and represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by two-way ANOVA. (F and G) Experimental procedures for assaying the stability of nT reg cells in Rag1−/− mice (F). Frequencies of Foxp3+ in CD45.2+CD4+TCRβ+ cells were analyzed (G). n = 5–6 per genotype. Data represent two experiments. **, P < 0.01 by Mann-Whitney test. (H and I) Assessment of the immunosuppressive function of nT reg cells in vitro. nT reg cells sorted from male mice (H) or mixed bone marrow chimeric mice (I) were cocultured with antigen-presenting cells and cell trace–labeled WT CD4 Tn cells for 4 d. Dilution of cell trace was quantified to assess the proliferation of T con cells. Data represent two experiments. **, P < 0.01; ****, P < 0.0001 by two-way ANOVA. (J) Percentages of CD44hiCD62Llo in nT reg cells from mixed bone marrow chimeric mice. n = 5–6 per genotype. Data represent two experiments. *, P < 0.05; **, P < 0.01 by two-way ANOVA. (K–M) Schematic illustration of the coverage of T reg cell TCR repertoire for antigens bearing multiple epitopes (K). Numbers of T reg cells against individual epitopes are shown by box plots. In ΔCNS0 or ΔCNS3 mice, impaired T reg cell induction leads to underrepresentation of TCR clones for particular epitopes without significantly affecting overall immune tolerance. Further reduction of T reg cell development in ΔCNS0,3 mice depletes all the clones for many antigens, breaking down immune tolerance. Numbers of plus signs indicate the levels of immune tolerance (K). CNS3 and CNS0 indirectly or directly integrate TCR and IL-2/Stat5 signaling, respectively, to promote Foxp3 induction (L). A hypothetical model of CNS0 and CNS3 in determining T reg cell induction efficiency and T reg cell suppressive capacity (M).
To test whether ΔCNS0,3 could also destabilize Foxp3 expression, we assessed the stability of Foxp3 expression in control and ASC-treated iT reg cells, which stabilize Foxp3 expression through Tet-dependent DNA demethylation (Yue et al., 2016). We used CD4 Tn cells from mixed bone marrow chimeras to induce iT reg cells with or without supplemented ASC in culture media and then sorted iT reg cells and cultured them in fresh media without TGF-β for 4 d before analysis (Fig. 6 C). Without ASC treatment, high levels of IL-2 maintained Foxp3 expression in 40%–50% WT and ΔCNS3 but not in ΔCNS0 or ΔCNS0,3 iT reg cells (Fig. 6, D and E), consistent with the role of CNS0 in maintaining Foxp3 expression before DNA demethylation (Dikiy et al., 2021). When IL-2 was blocked by neutralization antibodies, Foxp3 expression was silenced in >80% of all iT reg cells. Upon ASC treatment, iT reg cells of all the genotypes stabilized Foxp3 expression, despite minor differences, indicating that ΔCNS0,3 did not affect Tet-dependent stabilization of Foxp3 expression.
Next, we assessed the stability of Foxp3 expression in nT reg cells isolated from aged-matched male mice by cotransferring them with CD45.1+ WT CD4 T con and T reg cells into Rag1−/− mice (Fig. 6 F). We recovered the cells to analyze Foxp3 expression 3–4 wk later. In this adverse environment, 5%–20% of WT, ΔCNS0, and ΔCNS3 nT reg cells and 40%–60% of ΔCNS0,3 nT reg cells lost Foxp3 expression (Fig. 6 G), indicating that ΔCNS0,3 nT reg cells had a defect in maintaining Foxp3 expression. Because nT reg cells in ΔCNS0,3 mice had undergone extensive expansion and had been exposed to high levels of proinflammatory cytokines, both destabilizing Foxp3 expression (Feng et al., 2014; Komatsu et al., 2014; Zhou et al., 2009), the effect of ΔCNS0,3 was likely overestimated in this assay.
To assess the effect of ΔCNS0,3 on nT reg cell function on a per-cell basis, we performed T reg cell in vitro suppression assays with nT reg cells sorted from aged-matched male WT GFP and ΔCNS0,3 mice. In the presence of invariant TCR agonists (i.e., anti-CD3 antibodies), ΔCNS0,3 nT reg cells exhibited significantly stronger suppression of CD4 T con cell proliferation than WT nT reg cells did (Fig. 6 H). To test if this result was caused by the preexposure of nT reg cells to inflammatory conditions in ΔCNS0,3 mice, we performed similar assays with nT reg cells sorted from WT GFP and ΔCNS0,3 mixed bone marrow chimeric mice. ΔCNS0,3 nT reg cells displayed a milder, but significantly higher suppressive activity (Fig. 6 I). This result was consistent with elevated activation of ΔCNS0,3 nT reg cells in mixed bone marrow chimeras (Fig. 6 J). Our data suggest that ΔCNS0,3 does not dramatically impair T reg cell function per cell, despite reduced Foxp3 expression level and stability.
CNS0 and CNS3 coordinate to promote Foxp3 induction efficiency, expression level, and maintenance, among which Foxp3 induction is affected most by ΔCNS0,3 in terms of the fold changes. We propose that reduced T reg cell development decreases the representation of TCR clones for certain epitopes in ΔCNS0 or ΔCNS3 mice (Fig. 6 K). Although this defect is insufficient to cause immune dysregulation, it sensitizes ΔCNS0 and ΔCNS3 mice to other impaired tolerance mechanisms, e.g., Aire deficiency (Dikiy et al., 2021; Feng et al., 2015). Further reduction of T reg cell development by ΔCNS0,3 may deplete T reg cell clones for all the epitopes of many self-antigens, resulting in severe autoimmune inflammation. Mechanistically, CNS3 and CNS0 integrate TCR (Feng et al., 2015; Zheng et al., 2010) and IL-2/Stat5 signaling (Dikiy et al., 2021), respectively, to enhance Foxp3 expression (Fig. 6 L). Involvement of additional factors (Feng et al., 2015; Kitagawa et al., 2017; Placek et al., 2017) suggests that the coordination of CNS0 and CNS3 relies on multiple molecular mechanisms. Overall, these two distinct regulatory modules cooperate to maximize T reg cell suppressive capacity (Fig. 6 M).
CNS0 and CNS2 coordinate to confer stable T reg cell lineage identity
CNS2 maintains heritable Foxp3 expression, and CNS0 facilitates Foxp3 induction, raising a possibility of their combined defects in causing fatal autoimmunity in ΔCNS0,2 mice. However, our CRISPR screening results suggest that CNS0 and CNS2 may coordinate to maintain Foxp3 expression (Fig. 1, E and F). To distinguish between these two models, we first examined T reg cells in 3–4-wk-old mice. We observed comparable reduction of thymic T reg cells in ΔCNS0 and ΔCNS0,2 mice (Fig. 7 A). In the periphery, homeostatic expansion largely compensated for the T reg cell developmental defect in ΔCNS0 mice but not in ΔCNS0,2 mice. This result, together with decreased Foxp3 protein per cell (Fig. 7 B) and constrained activation (shown by CD44, CTLA-4, and Ki67 expression) of ΔCNS0,2 T reg cells (Fig. S3, A–C) in the presence of inflammation, suggests that CNS0 and CNS2 coordinate to maintain Foxp3 expression during activation-dependent proliferation.

CNS0 and CNS2 coordinate to confer stable T reg cell lineage identity. (A and B) T reg cell frequencies (A) and T reg cell Foxp3 MFI (B) in 3–4-wk-old mice. n = 7–10 per genotype. Data represent two experiments. (C–G) T reg cell frequencies (C); ratios of T reg cells in the periphery versus those in the thymus of the same mice (D); and Foxp3 MFI (E), CTLA-4 MFI (F), and CD44 MFI (G) of T reg cells in mixed bone marrow chimeric mice. n = 6 per genotype. Data represent two experiments. (H) Assessment of the lineage stability of nT reg cells in Rag1−/− mice. CD45.1+ Tn and T reg cells were cotransferred with CD45.1–CD45.2+ T reg cells sorted from WT GFP, ΔCNS0, ΔCNS2, or ΔCNS0,2 mice; 3–4 wk later, cells were recovered to analyze Foxp3 expression. n = 5–6 per group. Data represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by Mann-Whitney test. LPL, lamina propria lymphocyte; PP, Peyer’s patch.
CNS0 and CNS2 coordinate to confer stable T reg cell lineage identity. (A and B) T reg cell frequencies (A) and T reg cell Foxp3 MFI (B) in 3–4-wk-old mice. n = 7–10 per genotype. Data represent two experiments. (C–G) T reg cell frequencies (C); ratios of T reg cells in the periphery versus those in the thymus of the same mice (D); and Foxp3 MFI (E), CTLA-4 MFI (F), and CD44 MFI (G) of T reg cells in mixed bone marrow chimeric mice. n = 6 per genotype. Data represent two experiments. (H) Assessment of the lineage stability of nT reg cells in Rag1−/− mice. CD45.1+ Tn and T reg cells were cotransferred with CD45.1–CD45.2+ T reg cells sorted from WT GFP, ΔCNS0, ΔCNS2, or ΔCNS0,2 mice; 3–4 wk later, cells were recovered to analyze Foxp3 expression. n = 5–6 per group. Data represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by Mann-Whitney test. LPL, lamina propria lymphocyte; PP, Peyer’s patch.

Assessment of the activation status of ΔCNS0,2 T reg cells. (A–C) CD44 MFI (A), CTLA-4 MFI (B), and frequencies of Ki67+ (C) in WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 T reg cells isolated from 3–4-wk-old mice. n = 7–10 per genotype. Data represent two experiments. (D) Frequencies of Ki67+ in WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 T reg cells isolated from mixed bone marrow chimeric mice. n = 6 per genotype. Data represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by Mann-Whitney test. LPL, lamina propria lymphocyte; PP, Peyer’s patch.
Assessment of the activation status of ΔCNS0,2 T reg cells. (A–C) CD44 MFI (A), CTLA-4 MFI (B), and frequencies of Ki67+ (C) in WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 T reg cells isolated from 3–4-wk-old mice. n = 7–10 per genotype. Data represent two experiments. (D) Frequencies of Ki67+ in WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 T reg cells isolated from mixed bone marrow chimeric mice. n = 6 per genotype. Data represent two experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by Mann-Whitney test. LPL, lamina propria lymphocyte; PP, Peyer’s patch.
To reveal the cell-intrinsic effects of ΔCNS0,2, we generated chimeric mice with CD45.2 WT GFP, ΔCNS0, ΔCNS2, or ΔCNS0,2, and CD45.1 WT bone marrow. ΔCNS0 and ΔCNS0,2 led to a similar reduction in thymic T reg cells (Fig. 7 C). Unlike ΔCNS0 T reg cells, ΔCNS0,2 T reg cells remained at lower frequencies in the periphery. When the extent of expansion was estimated by the ratios of peripheral-to-thymic T reg cell numbers in individual mice, ΔCNS0 T reg cells expanded significantly more than WT T reg cells did (Fig. 7 D). In contrast, ΔCNS2 T reg cells expanded considerably less, which was further reduced in ΔCNS0,2 mice. As ΔCNS0,2 T reg cells expressed lower levels of Foxp3, CTLA-4, CD44, and Ki67 (Fig. 7, E–G; and Fig. S3 D), their reduced peripheral expansion suggests a crucial role of CNS0,2’s coordination in maintaining T reg cell identity.
Next, we directly assessed T reg cell lineage stability as described in Fig. 6 F. To alleviate the potential influence of different activation statuses, we transferred equal numbers of activated (CD44hiCD62Llo) and resting (CD44loCD62Lhi) T reg cells for each genotype into Rag1−/− mice. 4 wk later, 10%–30% of WT and ΔCNS0 nT reg cells and 40%–60% of ΔCNS2 T reg cells lost Foxp3 expression (Fig. 7 H). In contrast, >90% ΔCNS0,2 T reg cells silenced Foxp3 expression, demonstrating an essential role of CNS0,2’s coordination in maintaining T reg cell identity. Impaired T reg cell function upon Foxp3 loss, together with the effector-like exT reg cells (or "former" T reg cells; Feng et al., 2014; Komatsu et al., 2014; Zhou et al., 2009), may contribute to the progressive, lethal autoimmune lesions in ΔCNS0,2 mice.
scRNA-seq reveals the functional states of ΔCNS0,2 T reg cells
To assess T reg cell lineage stability and functional states in a competitive, lymphocompetent environment, we performed scRNA-seq with T reg cells sorted from mixed bone marrow chimeric mice, which revealed 17 Seurat clusters associated with differentially expressed genes (Fig. 8, A–C). Among them, substantially fewer cells were identified in activation-related clusters in ΔCNS2 and ΔCNS0,2 T reg cells than in WT and ΔCNS0 counterparts (Fig. 8, D and E; and Fig. S4 A). Pseudotime is widely used to reconstruct the dynamic processes experienced by cells in a heterogeneous population (Trapnell et al., 2014). Applying this algorithm to T reg cells revealed that ΔCNS0,2 mainly impaired T reg cells at the terminally activated state (Fig. 8, F and G; and Fig. S4 B, state S4), whereas T reg cells at the resting (S1 and S7) and intermediate (S2, S3, S5, and S6) states were less affected, if any. Together with impaired T reg cell stability observed in the in vivo assay (Fig. 7 H), these results suggest that ΔCNS0,2 mainly disrupted T reg cell lineage stability during activation-dependent clonal expansion.

scRNA-seq reveals the functional states of ΔCNS0,2 T reg cells. (A) scRNA-seq identified 17 Seurat clusters among all T reg cells. Numbers of cells in each genotype: 9,607 (GFP), 9,003 (ΔCNS2), 9,649 (ΔCNS0), and 8,368 (ΔCNS0,2). (BandC) Top 20 differentially expressed genes in each cluster. Genes in red and green are activation and resting state markers, respectively. Clusters C15 and C16 are not shown because of their small number of cells. Columns represent individual cells. (D) Normalized cell counts of Seurat clusters. (E) Comparison of activation-associated clusters C2, C6, C7, and C11 across genotypes. (F) Pseudotime states of all T reg cells. (G) Normalized cell counts of individual pseudotime states. (H) Frequencies of duplicate TCR clones. (I) Superimposition of duplicate TCRs (two or more copies) and Seurat clusters. tSNE, t-distributed stochastic neighbor embedding.
scRNA-seq reveals the functional states of ΔCNS0,2 T reg cells. (A) scRNA-seq identified 17 Seurat clusters among all T reg cells. Numbers of cells in each genotype: 9,607 (GFP), 9,003 (ΔCNS2), 9,649 (ΔCNS0), and 8,368 (ΔCNS0,2). (BandC) Top 20 differentially expressed genes in each cluster. Genes in red and green are activation and resting state markers, respectively. Clusters C15 and C16 are not shown because of their small number of cells. Columns represent individual cells. (D) Normalized cell counts of Seurat clusters. (E) Comparison of activation-associated clusters C2, C6, C7, and C11 across genotypes. (F) Pseudotime states of all T reg cells. (G) Normalized cell counts of individual pseudotime states. (H) Frequencies of duplicate TCR clones. (I) Superimposition of duplicate TCRs (two or more copies) and Seurat clusters. tSNE, t-distributed stochastic neighbor embedding.

Assessment of the functional states of T reg cells by scRNA-seq. (A) Heatmaps of the top 20 differentially expressed genes across Seurat clusters C0–C14 of WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 T reg cells. Columns represent individual cells. Arrowheads indicate underrepresented clusters in ΔCNS2 or ΔCNS0,2 T reg cells. (B) Cross comparison of T reg cells distributed among 17 Seurat clusters and seven pseudotime states.
Assessment of the functional states of T reg cells by scRNA-seq. (A) Heatmaps of the top 20 differentially expressed genes across Seurat clusters C0–C14 of WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 T reg cells. Columns represent individual cells. Arrowheads indicate underrepresented clusters in ΔCNS2 or ΔCNS0,2 T reg cells. (B) Cross comparison of T reg cells distributed among 17 Seurat clusters and seven pseudotime states.
To test this possibility, we profiled TCR clonotypes using scRNA-seq. Although only 8,000–10,000 T reg cells were sampled, this experiment uncovered profound effects of Foxp3 enhancer deficiencies on the clonotypes of T reg cell TCRs without influencing their usages of V, D, and J gene segments (data not shown); while ΔCNS0 T reg cells exhibited elevated clonal expansion, ΔCNS2 and ΔCNS0,2 T reg cells had markedly fewer duplicate TCRs (Fig. 8 H). This result was strikingly consistent with the expansion potential of T reg cells estimated in mixed bone marrow chimeras (Fig. 7 D). T reg cells with multiple copies were located in the clusters expressing higher levels of activation markers, e.g., Tigit, Ctla-4, IL-10, and Cxcr3 (Fig. 8, C, E, and I). These clusters were depleted in ΔCNS2 and ΔCNS0,2 T reg cells. Our scRNA-seq results support the crucial role of CNS0,2’s coordination in maintaining T reg cell identity during activation-dependent expansion.
Epistasis between CNS0/CNS2 and the epigenetic programs controlling Foxp3 expression
Whereas CNS2’s function is activated by Tet-mediated DNA demethylation (Yue et al., 2016), CNS0 does not show differential DNA methylation during T reg cell development. Stat5 binds to CNS0 upon IL-2 stimulation in both differentiating and mature T reg cells (Dikiy et al., 2021). The dramatic effects of ΔCNS0,3 and ΔCNS0,2 indicate that CNS0 contributes to both T reg cell development and T reg cell lineage stability, with the latter being shielded by CNS2’s function in WT T reg cells.
Tet-dependent DNA demethylation is a key mechanism securing Foxp3 expression (Sakaguchi et al., 2020). As IL-2/Stat5 signaling was proposed to recruit Tet enzymes (Yang et al., 2015), CNS0 and CNS2 may act via a partially redundant mechanism to promote this process. To test this notion, we performed whole-genome bisulfite sequencing (WGBS) with T reg cells isolated from mixed bone marrow chimeric mice that were derived from male donors because the Foxp3 gene is located on the X chromosome. We confirmed T reg cell–specific DNA demethylation around the Foxp3 locus. Surprisingly, except for some upstream, less conserved regions, ΔCNS0,2 did not cause significant DNA hypermethylation in the remaining T reg cell–specific demethylated regions downstream of Foxp3 promoter (Fig. 9 A), suggesting that Tet recruitment or enzymatic activity at these regions is independent of the two major IL-2/Stat5 response elements.

Epistasis between CNS0/CNS2 and the epigenetic programs controlling Foxp3 expression. (A) CpG methylation (mCpG) levels around the Foxp3 locus. CD4 Tn, TE, T reg, iT reg, ASC-treated iT reg cells were derived from male WT GFP mice, and nT reg cells were sorted from mixed bone marrow chimeras (MBC). Cells were subjected to WGBS with 40× coverage. Vertical bars represent individual CpG sites, and their heights indicate mCpG (5-methylcytosine and 5-hydroxymethylcytosine) levels ranging from 0% to 100%. CpG sites covered by fewer than five reads were marked as –0.2. (B–D) CNS2 is activated by Tet-induced DNA demethylation, and CNS0 is constitutively hypomethylated (B). Coordination between CNS0 and CNS2 maximizes T reg cell lineage stability during activation and proliferation (C), thus maintaining T reg cell suppressive capacity against genetic variations and immune perturbations (D). ChrX, X chromosome; Cons., conservation; Pro, promoter.
Epistasis between CNS0/CNS2 and the epigenetic programs controlling Foxp3 expression. (A) CpG methylation (mCpG) levels around the Foxp3 locus. CD4 Tn, TE, T reg, iT reg, ASC-treated iT reg cells were derived from male WT GFP mice, and nT reg cells were sorted from mixed bone marrow chimeras (MBC). Cells were subjected to WGBS with 40× coverage. Vertical bars represent individual CpG sites, and their heights indicate mCpG (5-methylcytosine and 5-hydroxymethylcytosine) levels ranging from 0% to 100%. CpG sites covered by fewer than five reads were marked as –0.2. (B–D) CNS2 is activated by Tet-induced DNA demethylation, and CNS0 is constitutively hypomethylated (B). Coordination between CNS0 and CNS2 maximizes T reg cell lineage stability during activation and proliferation (C), thus maintaining T reg cell suppressive capacity against genetic variations and immune perturbations (D). ChrX, X chromosome; Cons., conservation; Pro, promoter.
To understand how CNS0 and CNS2 coordinate to stabilize Foxp3 transcription, we examined active promoter and enhancer markers with CUT&RUN sequencing (Skene and Henikoff, 2017). We did not observe differences of H3K4me3 and H3K27ac at the Foxp3 locus in WT, ΔCNS0, ΔCNS2, and ΔCNS0,2 nT reg cells (Fig. S5 A), although RNA polymerase II (Pol II) association was drastically reduced in ΔCNS0,2 nT reg cells (Fig. S5 B). As ΔCNS0, ΔCNS2, and ΔCNS0,2 did not change the local chromatin accessibility as assessed by assay for transposase-accessible chromatin using sequencing (ATAC-seq; Fig. S5 C; Buenrostro et al., 2013), CNS0 and CNS2 probably stabilize Foxp3 transcription through their associated factors.

Epigenetic markers and chromatin accessibility in T reg and CD4 Tn cells. (A and B) CUT&RUN sequencing of H3K4me3 (A), H3K27ac (A), and RNA Pol II (B) around the Foxp3 locus in T reg cells isolated from mixed bone marrow chimeric mice. Data represent one replicate. (C) Chromatin accessibility assessed by ATAC-seq in CD4 Tn and T reg cells isolated from mixed bone marrow chimeric mice. Samples processed in parallel were grouped. Data represent two replicates. ChrX, X chromosome; Cons., conservation; Pro, promoter.
Epigenetic markers and chromatin accessibility in T reg and CD4 Tn cells. (A and B) CUT&RUN sequencing of H3K4me3 (A), H3K27ac (A), and RNA Pol II (B) around the Foxp3 locus in T reg cells isolated from mixed bone marrow chimeric mice. Data represent one replicate. (C) Chromatin accessibility assessed by ATAC-seq in CD4 Tn and T reg cells isolated from mixed bone marrow chimeric mice. Samples processed in parallel were grouped. Data represent two replicates. ChrX, X chromosome; Cons., conservation; Pro, promoter.
Despite nonoverlapping roles, distinct epigenetic regulation, and temporal activities, CNS0 and CNS2 coordinate to maintain T reg cell lineage identity, especially during activation and expansion, conferring persistent immune tolerance (Fig. 9, B–D). This process appears to be independent or downstream of the mechanisms governing the deposition of permissive histone markers and DNA demethylation.
Discussion
We found that three Foxp3 enhancers, besides their nonoverlapping roles, substantially coordinate to promote T reg cell development and lineage stability. The coordination between CNS0 and CNS3 maximizes T reg cell suppressive capacity by promoting T reg cell induction. A three- to fourfold reduction of T reg cell development in ΔCNS0 or ΔCNS3 mice does not cause immune dysregulation but exacerbates autoimmune pathology in Aire-deficient background (Dikiy et al., 2021; Feng et al., 2015). Further reduction of T reg cell induction by 50- to 100-fold in ΔCNS0,3 mice leads to a minimal T reg cell TCR repertoire and lethal autoimmunity. These results define the role of T reg cell induction efficiency in controlling T reg cell repertoire diversity and the robustness of immune tolerance. Notably, these Foxp3 enhancer deficiencies led to a relative enrichment of T reg cells with stronger reactivity to self-antigens. Future experiments are required to determine the causal role of T reg cell repertoire diversity in determining T reg cell suppressive capacity.
The coordination between CNS0 and CNS2 produces new insights into the mechanisms controlling T reg cell lineage identity. Although ΔCNS2 impairs T reg cell lineage stability in a cell-intrinsic manner, reduced T reg cells were mainly observed in the periphery of aged ΔCNS2 mice (Feng et al., 2014; Li et al., 2014), suggesting that other factors compensate for unstable T reg cell lineage, e.g., continuous T reg cell development and peripheral expansion. However, these factors fail to sufficiently counteract the effect of ΔCNS0,2 in young mice. Future investigations are required to determine the contributions of these opposing factors at different postnatal stages.
Tet-mediated DNA demethylation was proposed as a key mechanism stabilizing Foxp3 expression through CNS2. However, ΔCNS2 only partially destabilizes Foxp3 expression compared with the drastic effect of Tet2/Tet3 deletions (Feng et al., 2014; Li et al., 2014; Nakatsukasa et al., 2019; Yue et al., 2019; Yue et al., 2016; Zheng et al., 2010). Thus, Tet enzymes may act on CNS2 and other genetic elements to stabilize Foxp3 transcription. Because many of the Foxp3 cis-regulatory elements remain hypomethylated in ΔCNS0,2 T reg cells, Tet enzymes likely are regulated by a mechanism upstream or independent of CNS0/CNS2. Permissive histone modifications H3K27ac and H3K4me3 are deposited at the Foxp3 cis-regulatory elements independently of CNS0 and CNS2. These results suggest that T reg cell lineage stability relies on multiple cross-regulatory mechanisms acting through Foxp3 enhancers, involving DNA demethylation, histone modifications, sensing of environmental cues, and associated nuclear factors. Revelation of their context-dependent roles will generate insights into T reg cell–dependent immune tolerance.
The coordination of CNS0/CNS3 and CNS0/CNS2 suggests that other collective activities of Foxp3 enhancers might exist. Exploration of these interactions would uncover novel mechanisms controlling T reg cell repertoire diversity, lineage stability, or immune suppressive function. Coordination of Foxp3 enhancers also reflects the interactions among cell-intrinsic and -extrinsic factors. How these factors act in combinatorial ways to affect T reg cell–mediated immune tolerance remains to be fully explored.
In summary, our study reveals the cooperating Foxp3 enhancers that maximize the induction efficiency and lineage stability of T reg cells to suppress autoimmunity and oppose genetic variations and immune perturbations. Thus, regulation of Foxp3 expression by these synergistic enhancers in a stage-specific manner exemplifies the genetic control of the functional robustness of differentiated cells.
Materials and methods
Mice
All mice were maintained in the Animal Resources Center at St. Jude Children’s Research Hospital under specific pathogen–free conditions, and the experiments were approved by the institutional animal care and use committee (approval no. 612). Foxp3GFP, Foxp3ΔCNS2-GFP, Foxp3ΔCNS3-GFP, Foxp3ΔCNS0-GFP, and Foxp3YFP-Cre on a C57BL/6 background have been previously described (Dikiy et al., 2021; Rubtsov et al., 2008; Zheng et al., 2010). Our recently generated Foxp3null mice (unpublished data) develop lethal autoimmunity as the scurfy mice do (Brunkow et al., 2001; Fontenot et al., 2003). Rag1−/− C57BL/6 and CD45.1 C57BL/6 mice were purchased from The Jackson Laboratory. Homozygous RosaCas9 mice constitutively expressing Cas9 (Platt et al., 2014) were bred with Foxp3ΔCNS2-GFP or Foxp3ΔCNS0-GFP mice to generate RosaCas9/+ Foxp3ΔCNS2-GFP or RosaCas9/+ Foxp3ΔCNS0-GFP mice for isolating T cells for CRISPR experiments. Because the Foxp3 gene is located on the X chromosome and male Foxp3null, ΔCNS0,2 and ΔCNS0,3 mice developed lethal, early-onset autoimmunity that causes infertility, only male WT GFP, Foxp3null, ΔCNS0, ΔCNS2, ΔCNS3, ΔCNS0,2, and ΔCNS0,3 mice were used in this study. All these mice carry the same GFP–Foxp3 fusion protein engineered at the endogenous Foxp3 locus.
Generation of ΔCNS0,2 and ΔCNS0,3 mice
ΔCNS0,2 and ΔCNS0,3 mice were generated on a C57BL/6 background by using the CRISPR/Cas9 platform (Qin et al., 2016). Briefly, the pronuclei of Foxp3ΔCNS0-GFP (ΔCNS0) zygotes were injected with several picoliters per reagent mix of 60 ng/µl Cas9 protein (St. Jude Protein Production Core) and 15 ng/µl each of 5′ and 3′ chemically modified sgRNA (Synthego). CNS2 and CNS3 deletion reagents were injected independently. Approximately 25 injected zygotes were transferred into an infundibulum of a 0.5-d postconception pseudopregnant CD-1 foster mother and carried to term. Founder mice were genotyped via targeted high-throughput sequencing on a MiSeq sequencer (Illumina) and analyzed by using CRIS.py (Connelly and Pruett-Miller, 2019). Editing construct sequences and relevant primers are listed in Table S3. After the deletion was confirmed, mice were backcrossed to C57BL/6 mice at least three times.
Tissue lymphocyte preparation and flow cytometry analysis
Lymphocytes from lymphoid and nonlymphoid organs were prepared as previously described, with a few modifications (Feng et al., 2014; Feng et al., 2015). To isolate lymphocytes from the thymus, spleen, and LNs, lymphoid organs were dissected after euthanasia, mechanically dissociated, and passed through a 100-µm strainer. To isolate lymphocytes from the liver and lung, mice were first perfused with 1× PBS after euthanasia. Tissues were then cut into small fragments of <1 mm in diameter and digested with 1 mg/ml collagenase IV (Worthington) and 0.25 mg/ml DNase I (Sigma) in DMEM, 10% newborn calf serum (NCS), and 15 mM Hepes at 37°C for 30 min with shaking. Digested samples were passed through a 100-µm strainer, washed once with complete RPMI1640, and then fractionated by centrifugation in 40% Percoll (GE Healthcare) in 1× PBS supplemented with 10% NCS to remove debris. The resulting cell pellets were resuspended in complete RPMI1640. To isolate lymphocytes from small intestine, Peyer’s patches were removed, and the remaining tissue was cut into 5-mm fragments and washed with 5% NCS in 1× PBS. Intraepithelial lymphocytes were released by incubating the tissue with 1× PBS, 5% NCS, and 5 mM EDTA at 37°C for 20 min with shaking. After EDTA was removed, lamina propria lymphocytes were released by digesting the tissue with 125 U/ml or 1 mg/ml collagenase IV and 0.25 mg/ml DNase I in DMEM, 10% NCS, and 15 mM Hepes at 37°C for 45 min with shaking. Digested tissue was then filtered through a 100-µm strainer, pelleted by centrifugation, and fractionated by two layers of Percoll (42% and 70%) in 1× PBS and 5% NCS with centrifugation. The resulting cells between the two layers of Percoll were pelleted and resuspended in complete RPMI1640 with 10% FBS for downstream analyses. All samples were washed by centrifugation in 5–10 ml wash buffer.
For the flow cytometric analysis of cytokine production, lymphocytes were stimulated in vitro with 50 ng/ml phorbol 12-myristate 13-acetate (Sigma) and 0.5 μg/ml ionomycin (Sigma) in the presence of 2 µM monensin (Sigma) at 37°C for 4 h. Cells were stained with antibodies against indicated cell surface markers; then, cytokines were stained by using an intracellular staining kit (BD Biosciences).
Cell staining and flow cytometric analyses were performed as we previously described (Feng et al., 2014; Feng et al., 2015). Cells were stained with a fixable viability dye, incubated with indicated antibodies against cell surface markers, and fixed/permeabilized with a Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and then stained for intracellular Foxp3, if needed. Cells were fixed in 1% paraformaldehyde for 10 min after staining. The following cell-surface proteins were detected with the indicated fluorophore-conjugated antibodies: CD4 (RM4-5; eBioscience), CD8 (5H10; eBioscience), CD25 (PC61.5; eBioscience), CD44 (IM7; eBioscience), CD62L (MEL-14; eBioscience), CTLA-4 (UC10-4B9; eBioscience), KLRG1 (2F1; eBioscience), TCRβ (BioLegend), CD45.1 (A20; eBioscience), and CD45.2 (104; eBioscience). The following intracellular proteins were detected with indicated antibodies: Foxp3 (FJK-16s; eBioscience), Ki67 (B56; BD Biosciences), IL-13 (eBio13A; eBioscience), IL-17 (eBio17B7; eBioscience), IFN-γ (XMG1.2; eBioscience), and IL-2 (JES6-5H4; eBioscience). To track cell division, cells were labeled with CellTrace CFSE (Thermo Fisher Scientific) according to the manufacturer’s instructions.
FACS analyses were performed on LSR II or LSRFortessa (BD Biosciences) flow cytometers. Data were analyzed via FlowJo software (BD Biosciences).
T cell sorting and culture
CD4 T cells were enriched by using EasySep Mouse CD4 T Cell Isolation Kits (STEMCELL) to process cells from LNs and spleens. CD4 Tn cells (CD4+CD25–CD44loCD62Lhi) and nT reg cells (CD4+GFP+) were further FACS sorted from Foxp3GFP, ΔCNS0, ΔCNS2, ΔCNS3, ΔCNS0,2, or ΔCNS0,3 mice. CD4 Tn and nT reg cells were isolated from ΔCNS2 RosaCas9/+ or ΔCNS0 RosaCas9/+ mice for CRISPR experiments. nT reg cells were double sorted to ensure >99% purity. T cells were cultured at 37°C, 5% CO2 in RPMI1640 supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1% nonessential amino acid, 10 mM Hepes, 20 µM 2-mercaptoethanol, 100 U/ml penicillin, 100 mg/ml streptomycin (complete RPMI1640), and indicated cytokines and compounds.
T reg cell induction and stability assay
Induction of T reg cell differentiation in vitro was conducted according to published protocols (Feng et al., 2015; Yue et al., 2016). Briefly, cell culture plates or dishes were precoated with 1 µg/ml anti-CD3 (clone 145-2C11; Bio X Cell) and anti-CD28 (clone 37.51; Bio X Cell) antibodies in PBS at 37°C for 2 h and washed with PBS once before cell culture. CD4 Tn cells were grown in complete RPMI1640 supplemented with 100 U/ml recombinant human IL-2 (PeproTech) and 1 ng/ml recombinant human TGF-β (R&D Systems) with or without 0.25 mM ASC-2-phosphate (Sigma) for 4 d.
To assess the stability of Foxp3 expression in vitro, iT reg cells sorted based on GFP-Foxp3 expression after 4 d of induction were further cultured on plates coated with both anti-CD3 (1 µg/ml) and anti-CD28 (1 µg/ml) antibodies and either recombinant human IL-2 or 25 µg/ml IL-2 neutralization antibodies (JES6-54H and S4B6-1; Bio X Cell) for 4 d more. To culture nT reg cells, FACS-sorted nT reg cells were cultured in complete RPMI1640 supplemented with 10% FBS in the presence of Dynabeads Mouse T-Activator CD3/CD28 Beads (Thermo Fisher Scientific) with 500 U/ml recombinant human IL-2.
Assessment of T reg cell lineage stability in lymphopenic mice
Activated T reg cells (CD4+GFP+CD44hiCD62Llo) and resting T reg cells (CD4+GFP+CD44loCD62Lhi) were sorted from the spleen and LNs of CD45.1–CD45.2+Foxp3GFP, ΔCNS0, ΔCNS2, or ΔCNS0,2 mice and mixed at a 1:1 ratio of aTreg and rTreg cells. To assess the stability of ∆CNS0,3 nT reg cells, CD4+GFP+ nT reg cells were sorted from the spleens and LNs of CD45.1–CD45.2+ Foxp3GFP, ∆CNS0, ∆CNS3, ∆CNS0,3 mice. Then, 100,000 CD45.1–CD45.2+ T reg cells were cotransferred into each Rag1−/− recipient mouse via intravenous injection with 1 million CD4 Tn cells and 1 million CD4 T cells isolated from WT CD45.1+CD45.2– or CD45.1+CD45.2+ male mice. The lymphocyte subsets were analyzed by flow cytometry 3–4 wk later.
CRISPR-tiling library construction
A retroviral sgRNA library was designed targeting all the sites bearing PAMs throughout a 20-kb region surrounding the Foxp3 locus (from 12.5 kb upstream to 7.5 kb downstream of the transcription start site). Specifically, we first identified all PAM sequences (NGG) within the X chromosome: 7143000–7165000 (mm9). To ensure the target specificity, we filtered the targets with low complexity sequences, such as repetitive ACG and TTTT. We then selected the targets ending with PAM and bearing unique sequence specificity in the mouse genome. In addition, 100 nontargeting negative controls were added to the library (Table S1). A library containing 75-nt single-stranded DNA oligonucleotides was synthesized by using a high-throughput method (GenScript) and amplified by PCR with primers binding to the flanking arms according to standard protocols. PCR product was then assembled with BbsI-linearized pSIR-BbsI-Thy1.1 vector backbone (modified from pSIR-hCD2, Cat51143; Addgene) by using Gibson Assembly Master Mix (New England Biolabs). The resulting product was electroporated into Escherichia coli (New England Biolabs) and selected by ampicillin on 16 25-cm2 dishes. Transformation efficiency was quantified to ensure sufficient coverage. Plasmid was extracted by using the CompactPrep Plasmid Maxi Kit (QIAGEN), and the sgRNA coverage was validated by performing high-throughput sequencing.
Retroviral packaging and transfection
Packaging of retrovirus and transduction of mouse primary T cells were conducted according to our published protocols, with a few modifications (Feng et al., 2015). Specifically, platinum-E cells were used for packaging the retrovirus per the manufacturer’s instructions (Cell Biolabs). Cells were grown in DMEM supplemented with 10% FBS, 10 mM Hepes, 1% nonessential amino acid, 100 U/ml penicillin, and 100 mg/ml streptomycin. To maintain the packaging vectors, cells were cultured in 1 µg/ml puromycin and 10 µg/ml blasticidin. 1 d before transfection, cells were seeded on new dishes or plates in medium without puromycin or blasticidin. Then, sgRNA-expressing constructs and pCL-Eco plasmid (Addgene) were cotransfected into these cells with TransIT-293 Transfection Reagent (Mirus). Approximately 18 h after transfection, cells were given fresh complete DMEM. Virus-containing medium was collected 48 h and 72 h after transfection, aliquoted, and stored at –80°C.
Retrovirus transduction
The titers of retroviral preparations were quantified before experiments to achieve expected transduction efficiency. To this end, 4 × 104 CD4 Tn cells were seeded in one well of 96-well plates coated with 1 µg/ml anti-CD3 antibodies and 1 µg/ml anti-CD28 antibodies in T reg cell–inducing conditions (i.e., complete RPMI1640 supplemented with recombinant human IL-2 and recombinant human TGF-β). After 72 h, 10 µg/ml Polybrene (Sigma), 10 mM Hepes, and titrated amounts of viral preparations were mixed and added to the cells. Transduction was performed by centrifuging the cells at 1,200 g at 35°C for 90 min. After transduction, the culture medium was replaced with fresh complete RPMI1640 supplemented with recombinant IL-2 and TGF-β. 18 h after transduction, cells were resuspended, and the culture was split by reseeding onto uncoated plates. Transduction efficiency and viral titers were determined 3 d later by quantifying Thy1.1- or dsRed-expressing cells via flow cytometry.
CRISPR-tiling screening for genetic elements controlling T reg cell induction
CD4 Tn cells sorted from ΔCNS0 RosaCas9/+ mice were seeded on culture plates precoated with 1 µg/ml anti-CD3 and 1 µg/ml anti-CD28 antibodies in complete RPMI1640 supplemented with 10% FBS and 100 U/ml recombinant human IL-2. 2 d later, cells were transduced with the retroviral library with 10%–20% anticipated efficiency. Transduction was performed as described above in the presence of 6 µg/ml Polybrene and 10 mM Hepes by centrifuging at 1,200 g at 35°C for 90 min. After transduction, cells were cultured in T reg cell–inducing medium (i.e., complete RPMI1640 supplemented with 10% FBS, 100 U/ml recombinant human IL-2, 1 ng/ml recombinant human TGF-β, and 0.25 mM ASC-2-phosphate; Mitsumoto et al., 1994). On day 3, cell cultures were split onto uncoated plates with fresh T reg cell–inducing medium. Cells were harvested on day 6 and stained with Fixable Viability Dye (eBioscience). GFP-Foxp3+ and GFP-Foxp3– cells within the live-cell gating were sorted to assess sgRNA representation.
CRISPR-tiling screening for genetic elements controlling T reg cell lineage stability
CD4 Tn cells sorted from ΔCNS2 RosaCas9/+ mice were seeded on cell culture plates precoated with 1 µg/ml anti-CD3 antibodies and 1 µg/ml anti-CD28 antibodies in T reg cell–inducing medium (with 0.25 mM ASC-2-phosphate). 3 d later, cells were transduced with the retroviral library with 10%–20% anticipated efficiency. After transduction, the culture medium was replaced with a fresh T reg cell–inducing medium (with 0.25 mM ASC-2-phosphate). 18 h later, cell cultures were split onto uncoated plates with fresh T reg cell–inducing medium (with 0.25 mM ASC-2-phosphate). Cells were harvested on day 6 and stained with Fixable Viability Dye. The top 10% of GFP-Foxp3hi and GFP-Foxp3– cells within the live-cell gating were sorted to compare sgRNA representation.
To screen for genetic elements controlling T reg cell stability in ΔCNS0 nT reg cells, nT reg cells double sorted from ΔCNS0 RosaCas9/+ mice were seeded on 24-well plates at 106–2 × 106 cells per well with Mouse T-Activator CD3/CD28 Beads and 500 U/ml recombinant human IL-2. Cells were transduced with the retroviral library 3 d later, with 10%–20% anticipated efficiency. After transduction, cells were cultured in fresh complete RPMI1640 supplemented with 10% FBS and 500 U/ml recombinant human IL-2. Cells were split onto 12-well plates on day 4 and cultured with fresh Mouse T-Activator CD3/CD28 Beads in complete RPMI1640 supplemented with 500 U/ml recombinant human IL-2 and 10 ng/ml recombinant murine IL-4. Cells were harvested on day 7 and stained with Fixable Viability Dye. The top 10% of GFP-Foxp3hi and GFP-Foxp3– cells within the live-cell gating were sorted for downstream analysis.
sgRNA recovery
To retrieve sgRNA information, genomic DNA was extracted from sorted T cells by proteinase K digestion, phenol:chloroform:isoamyl extraction, and 2-propanol precipitation. The sgRNA-coding sequences were amplified from genomic DNA by using a two-step PCR protocol. The first PCR (PCR1) amplified the region covering the sgRNA cassette by using primers targeting the retroviral vector backbone and containing the adaptor sequences for the second indexing PCR. The second PCR (PCR2) added barcodes with Illumina i5 and i7 indexing primers. PCR and DNA purification were conducted according to previously published protocols (Yau and Rana, 2018). Specifically, 5 µg genomic DNA was loaded into each 100-µl PCR1 reaction with Q5 Hot Start High-Fidelity DNA Polymerase (New England Biolabs), with the following parameters: 98°C for 2 min, 18 cycles of 98°C for 20 s, 55°C for 30 s, and 60°C for 45 s followed by 65°C for 5 min. Products of PCR1 were purified by using a NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel) and eluted in 100 µl of 10 mM Tris, 1 mM EDTA, pH 8.0, per 100 µl PCR1 reaction. Every 10 µl of eluted PCR1 product was used as a template for a 50-µl PCR2 reaction with the following parameters: 98°C for 2 min, 18 cycles of 98°C for 20 s, 55°C for 30 s, and 65°C for 45 s followed by 65°C for 5 min. Products of PCR2 were separated by electrophoresis through a 1.5% agarose gel, and DNA bands of 261 bp were excised and retrieved by using a NucleoSpin Gel and PCR Clean-up Kit. Purified PCR2 products were then sequenced by using MiSeq kits (Illumina) with single-end sequencing for 100 cycles. More than 106 reads were obtained for each sample.
Validation of CRISPR-tiling screening results
To validate the effects of sgCNS1-1, sgCNS3-1, sgCNS3-2 (Table S3) on iT reg cell induction, we used sgNC as a nontargeting negative control and sgCNS2-1 as a neutral reference based on the T reg cell induction screening result. CD4 Tn cells sorted from WT GFP RosaCas9/+ and ΔCNS0 RosaCas9/+ mice were transduced with individual sgRNAs and treated in the same way as in the iT reg cell induction CRISPR-tiling screening.
To validate the effects of sgCNS0-1, sgCNS0-2, sgCNS0-3, and sgCNS0-4 (Table S3) on the stability of ASC-pretreated iT reg cells, sgNC was used as a nontargeting negative control and sgCNS1-1 and sgCNS3-3 as neutral references based on the T reg cell stability screening result. CD4 Tn cells sorted from WT GFP RosaCas9/+ and ΔCNS2 RosaCas9/+ mice were transduced with individual sgRNAs and treated in the same way as in iT reg cell stability CRISPR-tiling screening. To validate their effects on the stability of nT reg cells, nT reg cells double sorted from WT GFP RosaCas9/+ and ΔCNS2 RosaCas9/+ mice were transduced with individual sgRNAs and treated in the same way as in nT reg cell stability CRISPR-tiling screening.
The percentages of GFP-Foxp3+ among the live, transduced cells (dsRed+) were quantified and normalized to those of sgNC-transduced cells of the same genotypes. The ratios of normalized percentages of GFP-Foxp3+ of ΔCNS0 or ΔCNS2 cells to those of WT GFP cells were further calculated.
Generation of mixed bone marrow chimeras
Mixed bone marrow chimeras were generated as previously described (Feng et al., 2014). Briefly, recipient mice (CD45.1+CD45.2+) were irradiated (950 Rad) 24 h before intravenous injection of 10 × 106 bone marrow cells from CD45.1+CD45.2+ WT codonor mice and CD45.1–CD45.2+ WT GFP, ΔCNS0, ΔCNS2, ΔCNS3, ΔCNS0,2, or ΔCNS0,3 mice mixed at a 1:1 ratio. After bone marrow transfer, the recipient mice were administered with 2 mg/ml neomycin in drinking water for 3 wk. Mice were analyzed 8–10 wk later.
T reg cell in vitro suppression assay
nT reg cells were FACS sorted from pooled spleens and LNs of 3–4-wk-old WT GFP and ΔCNS0,3 male littermates or mixed bone marrow chimeric mice of WT GFP and ΔCNS0,3 described in Fig. 5 C. CD4 Tn cells were sorted from the spleens of CD45.1 mice and stained with CellTrace CFSE according to the manufacturer’s manuals (Thermo Fisher Scientific). Antigen-presenting cells were prepared from the splenocytes of male CD45.1 mice by depleting CD90.2+ T cells and TER-119+ red blood cells followed by lethal irradiation (20 Gy). T reg cell suppression assay was conducted in 96-well U-bottom plates, each well containing 200 µl complete RPMI1640 supplemented with 1 µg/ml anti-CD3 antibody, 105 antigen-presenting cells, and 4 × 104 CD4 Tn cells. T reg cells were added at different ratios to CD4 Tn cells. Cells were harvested 3 d later for flow cytometric analysis after being stained with viability dye (Tonbo Biosciences) and antibodies against CD4 and CD45.1.
scRNA-seq and TCR V(D)J clonotype profiling
To profile differential gene expression of innate and adaptive immune cells, spleens or lungs from two Foxp3GFP and four ΔCNS0,2 mice were pooled. Spleens were ground mechanically, and lungs were digested with 1 mg/ml collagenase IV and 0.25 mg/ml DNase I as described above. The resulting cells were stained with viability dye followed by surface markers. Approximately 90% of B220+ cells were depleted. The remaining live single cells were FACS sorted for 10× scRNA-seq. To profile gene expression and TCR V(D)J clonotypes of T reg cells, CD4+GFP+ cells were sorted from pooled spleens and LNs of mixed bone marrow chimeric mice of WT GFP (n = 5), ΔCNS0 (n = 5), ΔCNS2 (n = 5), and ΔCNS0,2 (n = 22, because of the lower frequencies of T reg cells). Cell density and viability of sorted cells were further determined by trypan blue staining and hemocytometer. All the processed samples had cell viability >85%. Sample processing for single-cell gene expression and TCR V(D)J clonotypes was conducted by using the Chromium Single Cell 5′ Library and Gel Bead Kit (10x Genomics) according to the manufacturer’s manuals, with an anticipated recovery of 10,000 cells per sample. Altogether, 13 cycles of PCR were used to amplify the cDNA library to produce sufficient material to prepare RNA and TCR V(D)J libraries. TCR V(D)J enrichment was conducted according to the manufacturer’s protocols for mouse T cell Chromium Single Cell V(D)J Enrichment Kit (10x Genomics). RNA- and V(D)J-seq libraries were prepared by following the manufacturer’s manual (10x Genomics) and profiled by TapeStation (Agilent Technologies). Sequencing was performed on a NovaSeq 6000 sequencer (Illumina) with an expected 500 million clusters for each scRNA-seq library and 50 million clusters for each scTCR V(D)J library, according to the manufacturer’s specifications (10x Genomics).
Capture-C
Capture-C experiments were performed as previously described (Davies et al., 2016), with minor modifications (Zhang et al., 2019). In brief, 107 mouse primary T cells were fixed in 1% formaldehyde for 10 min at room temperature with gentle rotation. The reaction was quenched by 0.125 M glycine. Cells were then resuspended in 5 ml lysis buffer and incubated on ice for 20 min. Cells were pelleted and resuspended in 1 ml of 1× DpnII digestion buffer and transferred to a 2-ml tube for homogenization followed by centrifugation at 14,000 rpm for 5 min at 4°C. Complete homogenization was confirmed under the microscope, and the supernatant was removed. The remaining pellets were subjected to DpnII (New England Biolabs) digestion, T4 DNA ligation (Thermo Fisher Scientific), and de-crosslinking. The resulting 3C library DNA was precipitated, and a small portion was separated by electrophoresis with an agarose gel to assess the digestion and ligation efficiency. Then, 5 µg of the 3C DNA library was sheared to ∼200 bp followed by end repair, adaptor ligation, and PCR enrichment of adaptor-ligated DNA. Two rounds of captures were conducted at 47°C by using 2 µg of DNA, 5 µg mouse Cot-1 DNA (Thermo Fisher Scientific), biotinylated DNA oligonucleotide probes targeting the Foxp3 promoter region (Table S3), 1 µl xGen Universal Blocking Oligo i7, and 1 µl xGen Universal Blocking Oligo i5 (Integrated DNA Technologies) in a 1.5-ml tube. The constructed library was sequenced with paired ends of 150 cycles each.
Paired-end reads from Capture-C were mapped and processed by HiC-Pro (v2.11.4; Servant et al., 2015) to mm9 (MGSCv37 from Sanger) with fragments for DpnII sites. Validated contacts were used to generate BIGWIG files with make_viewpoints.py script (HiC-Pro). Reads were normalized to 100 thousand contacts within 5 million bp of the bait region. For suitable data visualization, the BIGWIG files were uploaded to http://epigenomegateway.wustl.edu/browser/ and exported as SVG format.
CUT&RUN
nT reg cells (CD4+GFP+) were sorted from the spleens and LNs of mixed bone marrow chimeric mice. H3K4me3, H3K27ac, and pan-RNA Pol II CUT&RUN was performed according to published protocols, with a few modifications (Skene and Henikoff, 2017). Briefly, 0.2 × 106–0.5 × 106 T reg cells were attached to concanavalin A–coated magnetic beads (Bangs Laboratories) for each experiment. Cells were permeabilized with digitonin (Sigma) in a wash buffer (20 mM Hepes, pH 7.5, 150 mM NaCl, 0.5 mM spermidine, 0.01% digitonin, and protease inhibitors) and then incubated with primary antibodies against H3K4me3 (Active Motif), H3K27ac (Abcam), or pan-Pol II (Abcam) at 4°C for 2 h on a rotator. The beads–cells mixture was washed three times with digitonin wash buffer, resuspended in 200 µl protein A-MNase (Addgene), and incubated at 4°C for 1 h on a rotator. After three rounds of washing, beads were resuspended in 150 µl digitonin wash buffer and chilled to 0°C for 5 min. Then, 3 µl of 100 mM CaCl2 was added into the tubes with gentle vortexing, and the tubes were placed in an ice-water bath. After 30 min of incubation, 150 µl of 2× STOP buffer (170 mM NaCl, 20 mM EDTA, 20 mM EGTA, 0.05% digitonin, 20 mg/ml GlycoBlue, and 25 mg/ml RNase A) was added. Beads were incubated at 37°C for 30 min and then placed on a magnet stand for 2 min. The clarified liquid was transferred to a new tube for phenol:chloroform extraction followed by DNA precipitation by ethanol. The sequencing library was prepared by using KAPA HyperPrep Kit (Kapa Biosystems) according to the manufacturer’s instructions.
WGBS
nT reg cells (CD4+GFP+) were sorted from the spleens and LNs of mixed bone marrow chimeric WT GFP, ΔCNS0, ΔCNS2, or ΔCNS0,2 mice. CD4 Tn, TE, and T reg cells were sorted from male WT GFP mice (Tn cells: CD4+GFP–CD25–CD44loCD62Lhi; TE cells: CD4+GFP–CD44hiCD62Llo; T reg cells: CD4+GFP+). iT reg and ASC-treated iT reg cells were induced from CD4 Tn cells of male WT GFP mice in vitro with or without ASC (as described above), and GFP+ T reg cells were sorted on day 4 of in vitro culture. Genomic DNA was prepared from sorted cells via proteinase K digestion followed by phenol:chloroform:isoamyl alcohol extraction and 2-propanol precipitation. More than 100 ng of genomic DNA in each sample was converted by using EpiTect Bisulfite Kits (QIAGEN). Libraries were prepared from converted DNA by using the Accel-NGS Methyl-Seq DNA Library Kit (Swift Biosciences). Libraries were analyzed for insert-size distribution on a 2100 BioAnalyzer after processing using a High Sensitivity DBA Kit (Agilent Technologies), on an Agilent 4200 TapeStation system after processing using a D1000 ScreenTape assay, or on a Caliper LabChip GX microfluidic instrument after processing using a DNA High Sensitivity Reagent Kit (PerkinElmer) and then quantified by using the Quant-iT PicoGreen dsDNA assay (Life Technologies) or low-pass sequencing with a MiSeq Nano Kit (Illumina). Paired-end, 150-cycle sequencing was performed on a NovaSeq 6000 sequencer (Illumina) to achieve an average 40× coverage.
ATAC-seq
ATAC-seq was performed as previously reported (Buenrostro et al., 2013), with a few modifications. Briefly, 5 × 104 cells were FACS sorted, washed once with ice-cold PBS, and lysed in 300 µl lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, and 0.1% NP-40) by gently pipetting up and down. After centrifugation, the supernatant was removed. Then, 50 µl of reaction mix containing 25 µl TD Buffer, 2.5 µl TDE1 (Illumina Nextera DNA Library Prep Kit), and 22.5 µl nuclease-free water was immediately added to set up a transposition reaction at 42°C for 40 min. DNA was immediately purified afterward by using the NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel). The transposed DNA was amplified by PCR for 10–12 cycles with the Nextera DNA Library Prep Kit and Nextera XT Indexing Kit (Illumina). The library DNA within the 150–500-bp range was enriched by one round of negative selection with 0.6 volume AMPure XP beads (Beckman Coulter) and two rounds of positive selection with 1 volume AMPure XP beads. The libraries were quantified by NEBNext Library Quant Kit for Illumina (New England Biolabs) and sequenced, with paired-end 100-cycle sequencing performed on a HiSeq 4000 or HiSeq 2500 sequencer (Illumina).
Bulk TCR repertoire profiling
Bulk TCR sequencing was performed as previously described (Dash et al., 2015; Egorov et al., 2015; Feng et al., 2015). Briefly, CD4+GFP-Foxp3+ nT reg cells were sorted from the spleen, skin draining LN, and mesenteric LN (mLN) of individual WT GFP, ΔCNS0, ΔCNS3, and ΔCNS0,3 mice at 16 d of age. Total RNA was extracted by using TRIzol Reagent (Life Technologies). All the RNA was used for cDNA synthesis and TCR sequencing library preparation. cDNA was synthesized by SMARTScribe reverse transcription (Takara), with primers targeting the constant regions of mouse TCRα (5′-AGTCAAAGTCGGTGAAC-3′) or TCRβ (5′-ATCTCTGCTTTTGATG-3′) chains and template switch adapter integrated with unique molecular identifiers (5′-AAGCAGUGGTAUCAACGCAGAGUNNNNUNNNNUNNNNUCTTrGrGrGrG-3′). The TCRα and TCRβ chains were amplified with a two-step protocol: First, a mixture of primers (5′-CACTCTATCCGACAAGCAGTGGTATCAACGCAG-3′ and 5′-CACTCTATCCGACAAGCAGT-3′) bound at the template switch adapter and primers targeting the constant regions of TCRα or TCRβ chains (TCRα: 5′-GCTGTCCTGAGACCGAGGAT-3′; TCRβ: 5′-ATGGCTCAAACAAGGAGACC-3′) were used. Second, indexing primer 5′-(N)2–4(XXXXX)CAGTGGTATCAACGCAGAG-3′ annealing on the template switch adapter was used together with TCRα 3′ indexing primer 5′-(N)2–4(XXXXX)CAGGTTCTGGGTTCTGGATGT-3′ or TCRβ 3′ indexing primer 5′-(N)2–4(XXXXX)AGTCACATTTCTCAGATCCT-3′. Sequencing adapters were then added using the KAPA HyperPrep Kit. Samples were pooled and sequenced on a NovaSeq 6000 sequencer (Illumina) with paired-end reads of 150 cycles each. Reads with at least 10× coverage relative to the input cell numbers were acquired. Note: (N)2–4 indicates two to four random nucleotides for efficient separation of clusters during sequencing; U, uracil for degradation of template switch adaptor by uracil-DNA glycosylase (New England Biolabs) after reverse transcription; rG, riboguanosine for template switch during reverse transcription; and XXXXX, 5-nt barcodes for multiplexing samples.
Histological analysis
Tissue samples were fixed in 10% neutral buffered formalin and processed for H&E staining. Stained slides were scored to indicate the grades of tissue inflammation.
Data analysis
CRISPR-tiling screening
To quantify the representation of sgRNAs, the raw FASTQ data were first de-barcoded and mapped to the original reference sgRNA library. The differentially enriched sgRNAs were defined by comparing normalized counts between sorted GFP+ and GFP– cells. Normalized counts for each sgRNA were extracted and used to identify differentially enriched sgRNA by DESeq2 (Love et al., 2014).
Processing, dimensionality reduction, and clustering of 10× data
More than 730 and 260 million reads were acquired for each scRNA-seq library and scTCR V(D)J library, respectively. Single-cell gene expression was obtained by using 5′ kits from 10x Genomics (v1.1). Libraries were sequenced on the Illumina NovaSeq platform at 100 × 100 bp to obtain at least 50,000 reads per cell on average. For a subset of samples, TCR libraries were also generated from the barcoded single-cell cDNA by using the mouse V(D)J enrichment kit (10x Genomics) and sequenced at 150 × 150 bp to obtain at least 5,000 reads per cell on average. Read processing, read alignment, cell–barcode demultiplexing, and gene-specific unique molecular identifier (UMI) counting was performed by using CellRanger (v3.1.0) and the corresponding mouse references (mm10 v3.0.0 for gene expression, vdj-GRCm38-alts-ensembl-3.1.0 for TCR). Gene expression data obtained from distinct samples within the same experiment were subsequently aggregated by CellRanger and normalized by read depth across libraries by subsampling to obtain a similar average number of mapped reads per cell across cells. The average number of reads per cell after normalization was 92,531 for Fig. 4 and Fig. S2 and 66,111 for Fig. 8 and Fig. S4.
Downstream analyses of filtered feature barcode matrices were conducted separately for each experiment using Seurat (Stuart et al., 2019). Genes observed in <0.1%–0.2% of cells were excluded from downstream analysis. Cells that exhibited extremes in the percentage of mitochondrial expression, number of genes expressed, or number of transcripts expressed were excluded. Data were normalized by using default parameters, and the top 2,000 variable genes were detected by using the ‘vst’ method after excluding clonotype-specific V(D)J gene segments. The cell-cycle phase was inferred by using markers from Tirosh et al. (2016). Variable genes were then scaled and centered by their variation across the dataset while regressing out the effects of the number of UMIs, percentage of mitochondrial gene expression, and the cell-cycle phase scores per cell. We then conducted principal component (PC) analysis on the scaled genes, selecting the PCs to use for downstream dimensionality reduction by either considering the variation accounted for in the PC analysis plot (Fig. 4 and Fig. S2, PCs 1–22) or the significance of the PCs after random permutation (Fig. 8 and Fig. S4, PCs 1–63). These PCs were used to construct shared nearest neighbor graphs, define transcriptional clusters, and generate two-dimensional t-distributed stochastic neighbor embedding plots by using default parameters or uniform manifold approximation and projection plots.
Cell-type identification in 10× data
We applied Seurat (Stuart et al., 2019) for scRNA-seq analysis and data visualization. High mitochondrial RNA (>8%) and low-UMI cells (<400) were removed from our single-cell dataset. Cellular identities were determined by the marker genes found by the Seurat R package. We excluded erythroid cells (Hba-a1+ and Hba-a2+), endothelial cells (Pecam1+ and Epcam+), and fibroblasts (Col3a1+ and Col1a1+) because these cells were CD45– cells irrelevant to our studies. Further cell identification was based on gene expression of known cell markers for each cluster: Cd3e, Cd4, CD8a, Foxp3, Sell, Ccr7, Gzma, Gzmb, Cd44, Cd163l1, Gata3, Ncr1, Klrb1c, Igta2, Klra7, Zbtb16, Ly6g, Ly6c1, Itgax, Itgam, Cd63, Cd200r3, Lyz2, Cd14, Adgre1, and Fcgr4. We obtained 4,199 cells from spleens of WT GFP mice, 3,696 cells from the spleens of ΔCNS0,2 mice, 4,639 cells from lungs of WT GFP mice, and 5,546 from lungs of ΔCNS0,2 mice. Violin plots were constructed with the Seurat package’s implementation function VlnPlot() to show individual gene expression in different cell subsets.
Analysis of differential gene expression of 10× data
The FindMarkers function from Seurat (Stuart et al., 2019) was used to identify the differentially expressed genes for each cluster compared with other clusters. The top 20 significant genes (min.pct = 0.001, logfc.threshold = 0.1) ordered by false discovery rate–corrected P values (<0.01) were extracted to plot the heatmaps (DoHeatmap function). We excluded low-confidence clusters C15 and C16 because not all genotypes had >50 cells.
Pseudotime modeling of T reg cell heterogeneity
Processed results from Seurat were first imported into monocle (v2.9.0; Qiu et al., 2017). Based on the elbow of variance–explained plot (plot_pc_variance_explained function), 10 PCs were chosen to construct the spanning tree (max_components = 2; num_dim = 10) with DDRTree (v0.1.5). Cell states assigned by monocle were extracted for comparison among genotypes. Cell trajectories were plotted by monocle and colored by pseudotime (plot cell trajectory function).
TCR clonotype analysis of 10× data
TCR or V(D)J sequences were extracted by CellRanger (v3.1.0). TCR clones were counted across genotypes. To visualize T reg cell activation states and associated TCR copy numbers, TCR clonotypes were overlaid with Seurat clusters by using Loupe Browser (10x Genomics), and TCRs with different copy numbers were highlighted.
Analysis of bisulfite sequencing data
WGBS data were aligned to mouse genome mm9 assembly by using BSMAP2.74 (Xi and Li, 2009). The methylation ratio for each CpG site was extracted by methratio.py from BSMAP2.74. The methylation ratio was then converted to a BW file for visualization. Regions covered by fewer than five reads were marked as –0.2.
ATAC-seq
Paired-end reads of 100 bp were trimmed by Cutadapt (v1.9, paired-end mode, default parameter with “-m 25 -O 6”; Martin, 2011) and aligned to mouse genome mm9 (MGSCv37 from Sanger) by BWA (v0.7.12-r1039, default parameter; Li and Durbin, 2009). Duplicate reads were marked with biobambam2 (v2.0.87; Tischler and Leonard, 2014); only nonduplicate, properly paired reads were kept by the samtools (parameter “-q 1 -F 1804” v1.2; Li et al., 2009). After adjusting the Tn5 shift (reads were offset by +4 bp for the sense strand and –5 bp for the antisense strand), we used fragment size to separate reads into nucleosome-free, mononucleosome, dinucleosome, and trinucleosome as previously described (Buenrostro et al., 2013) and generated BIGWIG files by using the center 80 bp of fragments and scale to 20 million nucleosome-free reads. We observed acceptable nucleosome-free peaks and patterns of mono-, di-, and tri-nucleosomes on IGV (v2.4.13; Robinson et al., 2011). We next merged both replicates to enhance the peak calling on nucleosome-free reads by MACS2 (v2.1.1.20160309 default parameters with “--extsize 200 --nomodel”; Zhang et al., 2008). We considered all important nucleosome-free regions called if a sample had >15 million nucleosome-free reads after merging. To ensure reproducibility, we merged peaks from different cell types to create a set of reference chromatin-accessible regions. We then used bedtools (v2.24.0; Quinlan and Hall, 2010) to count nucleosome-free reads from each sample to overlay with the reference regions. As a confirmation of reproducibility, the Spearman correlation coefficient between replicates was larger than that between samples from different groups. To identify the differentially accessible regions, we first normalized raw nucleosome-free reads counts by using trimmed mean of M-values and then applied the empirical Bayes statistics test after linear fitting with the voom package (R 3.23, edgeR 3.12.1, limma 3.26.9; Law et al., 2014).
CUT&RUN sequencing
Paired-end reads of 50 bp were mapped to mouse genome mm9 (MGSCv37 from Sanger) by BWA (v0.7.12-r1039, default settings; Li and Durbin, 2009) after adapters were trimmed by cutadapt (v1.9, paired-end mode, parameter “-m 25 -O 6”). Duplicated reads were marked with biobambam2 (v2.0.87; Tischler and Leonard, 2014), and unique reads were kept by samtools (v1.2, parameter “-q 1 -F 1804”; Li et al., 2009). All samples had >5 million fragments as suggested by the CUT&RUN protocol (Skene and Henikoff, 2017) after replicates were merged. We generated BIGWIG files using the center 80 bp of fragments <2,000 bp and normalized to 10 million fragments. We compiled reproducible reference peaks for each cell type if the peaks called with stringent cutoff overlapped with the peaks called with relaxed cutoff in the other replicate. We merged reproducible peaks between cell types to create the reference peak set and counted fragments (fragment size <2,000 bp) overlapping with these merged reference peaks using bedtools (v2.24.0; Quinlan and Hall, 2010) for each sample. After trimmed mean of M-values normalization (R 3.4.0, edgeR 3.18.1), voom from R package limma 3.34.9 (Law et al., 2014) was used to identify differential binding sites.
Bulk TCR sequencing
To analyze TCR diversity, bulk TCR sequencing data were de-multiplexed by using MIGEC v1.2.9 software (Shugay et al., 2014). V(D)J gene assignments were calculated and filtered with MIGEC software. VDJtools v1.2.1 software (Shugay et al., 2015) was used to filter nonfunctional TCR and decontaminate samples. Immunarch package (https://immunarch.com) was used to explore the T cell repertoires and compute TCR diversity.
Statistical analysis
Statistical tests were performed using GraphPad Prism (GraphPad Software) or the R statistical environment. As indicated in the figure legends, for data with a small sample size, a robust nonparametric Mann-Whitney test was used; otherwise, an unpaired t test or two-way ANOVA with Tukey’s multiple comparisons test was applied. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
Online supplemental material
Fig. S1 shows three representative sgRNA target sequences identified by CRISPR-tiling screening, verification of the effects of sgRNAs targeting CNS0 on the stability of nT reg cells, CRISPR-tiling screening for genetic elements maintaining Foxp3 expression in ΔCNS0 nT reg cells, and the regions looping to the Foxp3 promoter in Tn and ASC-treated iT reg cells assessed by Capture-C. Fig. S2 shows the UMAP plots and the frequencies and normalized numbers of different types of immune cells revealed by scRNA-seq of CD45+ cells sorted from pooled spleens or lungs of WT GFP and ΔCNS0,2 mice. Fig. S3 compares the CD44 and CTLA-4 expression per cell and percentages of Ki67+ in T reg cells among WT GFP, ΔCNS0, ΔCNS2, ΔCNS0,2, or mixed bone marrow chimeric mice. Fig. S4 shows the heatmaps of the top 20 differentially expressed genes across Seurat clusters C0–C14 of WT GFP, ΔCNS0, ΔCNS2, and ΔCNS0,2 T reg cells. The Seurat clusters are also cross compared with pseudotime states. Fig. S5 shows the epigenetic markers (i.e., H3K4me3 and H3K27ac), RNA Pol II, and chromatin accessibility around the Foxp3 locus in Foxp3 enhancer-deficient T reg or CD4 Tn cells. Table S1 lists the sgRNA sequences of the CRISPR-tiling library. Table S2 lists the read numbers of sgRNAs recovered from CRISPR screening experiments. Table S3 lists the DNA sequences used for Capture-C, mouse gene targeting, or validation of CRISPR screening results.
Data availability
scRNA-seq data have been deposited in the Sequence Read Archive under BioProject PRJNA661257. The Capture-C, bulk TCR sequencing, WGBS, CUT&RUN sequencing, and ATAC-seq data have been deposited in the Gene Expression Omnibus under accession no. GSE158223.
Acknowledgments
We thank Alexander Rudensky (Memorial Sloan Kettering Cancer Center, New York, NY) for providing mouse strains, Jack Sublett (St. Jude Children's Research Hospital, Memphis, TN) for generating ΔCNS0,2 and ΔCNS0,3 mice, the Hartwell Center for sequencing, the Flow Cytometry Core Facility for cell sorting, and Cherise Guess for editing the manuscript.
X. Zong and X. Hao were supported by Special Postdoctoral Fellowships of the Academic Programs Office of St. Jude Children’s Research Hospital. This study was supported by the American Lebanese Syrian Associated Charities and National Institutes of Health grants R01 AI153138 and R21 AI146614 (Y. Feng) and cancer center support grant P30 CA021765 (St. Jude Children's Research Hospital). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions: X. Zong and Y. Feng conceived the project, designed the experiments, and interpreted the results. Y. Zhang and C. Li designed the sgRNA library and analyzed the screening results. S. Wright, Y. Zhang, and C. Li performed Capture-C. X. Zong and X. Hao performed CRISPR screening. J. Li conducted T reg cell in vitro suppression assays, iT reg cell induction, and ATAC-seq. X. Zong, L. Bai, X. Hao, and M. Jiang performed the flow cytometric analysis. M. He assisted with epigenetic experiments. J.C. Crawford, B. Xu, and X. Hao analyzed scRNA-seq data. Y. Fan analyzed WGBS data. J.C. Crawford edited the manuscript. J.P. Connelly, H. Berns, and S.M. Pruett-Miller engineered mouse strains. L. Janke performed histopathological analysis. X. Zong and Y. Feng wrote the manuscript.

