


A potent γ-secretase modulator (GSM) has been developed to circumvent problems associated with γ-secretase inhibitors (GSIs) and to potentially enable use in primary prevention of early-onset familial Alzheimer’s disease (EOFAD). Unlike GSIs, GSMs do not inhibit γ-secretase activity but rather allosterically modulate γ-secretase, reducing the net production of Aβ42 and to a lesser extent Aβ40, while concomitantly augmenting production of Aβ38 and Aβ37. This GSM demonstrated robust time- and dose-dependent efficacy in acute, subchronic, and chronic studies across multiple species, including primary and secondary prevention studies in a transgenic mouse model. The GSM displayed a >40-fold safety margin in rats based on a comparison of the systemic exposure (AUC) at the no observed adverse effect level (NOAEL) to the 50% effective AUC or AUCeffective, the systemic exposure required for reducing levels of Aβ42 in rat brain by 50%.
Introduction
Alzheimer’s disease (AD) is a major public health and socioeconomic problem affecting millions of people in the United States and worldwide (Qiu et al., 2009). AD develops over decades, with accumulation of amyloid-β (Aβ) plaques that induce neuritic changes; these changes are linked to glial inflammatory responses and to accumulation of neurofibrillary tangles whose timing and regional distribution correlates with cognitive loss. Pathological and genetic evidence powerfully links Aβ to early-onset AD and to the accumulation of Aβ in neuritic plaques that serve as an early feature of late-onset AD (McDade et al., 2018). The amyloid hypothesis of AD (Hardy and Higgins, 1992) has dominated therapeutic efforts. Potential disease-modifying therapeutics for AD (Tanzi and Bertram, 2005) are currently being clinically tested, but as yet none have been proven to slow disease progression. Enzyme inhibition strategies targeting the γ-secretase enzyme (i.e., γ-secretase inhibitors [GSIs]) decrease the levels of all Aβ peptide species but in clinical trials demonstrated significant side effects, including worsening of cognitive abilities relative to placebo (Coric et al., 2015; Coric et al., 2012; Doody et al., 2013; Fleisher et al., 2008). BACE-1 inhibition (Vassar, 2014) represents a similar approach to reduce generation of all Aβ peptide species, but recent clinical trials of BACE-1 inhibitors in mild to moderate and prodromal AD were terminated prematurely because of drug-related adverse events and lack of efficacy (Egan et al., 2018; Egan et al., 2019; Henley et al., 2019).
Alternative approaches including passive immunization have mostly failed to demonstrate efficacy (Salloway et al., 2014; Sevigny et al., 2016). Biogen’s phase 1b results comparing aducanumab with a placebo showed the antibody-mediated dose-dependent reduction of Aβ plaques, as measured by amyloid positron emission tomography (PET) imaging, and attenuated cognitive decline, as measured by Mini-Mental State Examination and the Clinical Dementia Rating–Sum of Boxes in prodromal or mild AD patients (Sevigny et al., 2016). Reports of two phase 3 clinical trials of aducanumab and more recent disclosures regarding clinical trials of BAN2401 and gantenerumab lend additional support to targeting reduction/elimination of Aβ plaques for treatment of AD.
Neuritic plaques are composed predominantly of Aβ42 (Iwatsubo et al., 1994), and the most common biochemical phenotype of the >200 different early-onset familial AD (EOFAD)–linked genetic mutations is an increased ratio of Aβ42/Aβ40 (Kumar-Singh et al., 2006), which supports an approach of preferentially targeting Aβ42. Over the past several years, we synthesized and characterized small molecules that act as γ-secretase modulators (GSMs) and enhance the processivity properties of γ-secretase. Indeed, nearly 700 novel small molecules encompassing four distinct chemical scaffolds harboring a common methylimidazole moiety defined a novel and potent class of bridged heterocyclic compounds (Kounnas et al., 2010; Liu et al., 2014; Rynearson et al., 2016; Wagner et al., 2016; Wagner et al., 2014). The γ-secretase enzyme complex, which is composed of four membrane protein subunits, presenilin (PS1 or PS2), nicastrin, Pen-2, and Aph-1, proteolytically processes membrane protein substrates such as Notch and amyloid precursor protein (APP) in a sequential manner. Initially, an endoproteolytic cleavage referred to as the ε-site cleavage generates the Notch intracellular domain and the APP intracellular domain from Notch and APP, respectively. Second, the remaining membrane fragments are processed in an exopeptidase-like fashion by so-called γ-site cleavages. GSMs selectively target the γ-site cleavages of the γ-secretase enzyme complex, resulting in a shift of the secreted peptide profile (i.e., Nβ and Aβ for Notch and APP, respectively), without impairing the ε-site endoproteolytic function required for processing numerous other substrates. GSMs preferentially reduce Aβ42 over Aβ40 and concomitantly potentiate formation of shorter nonfibrillar Aβ37 and Aβ38. Thus, GSMs offer the ability to mitigate mechanism-based toxicities associated with GSIs (Kounnas et al., 2010; Liu et al., 2014; Rynearson et al., 2016; Wagner et al., 2016; Wagner et al., 2012; Wagner et al., 2014).
Primary prevention of AD in specific populations, such as those with EOFAD, Down’s syndrome, and apolipoprotein E (APOE) ε4 homozygotes, is especially attractive since it is well established that EOFAD is an autosomal dominant disorder; over 80% of Down’s syndrome patients develop AD by the age of 65; and a single APOE ε4 gene increases the risk of AD fourfold, while two genes (APOE ε4 homozygotes) increase it 14-fold compared with noncarriers. While biological agents may prove useful in certain disorders, this approach is costly, invasive, and requires repeated infusions. A more practical approach would be to use a prophylactic small molecule administered orally before the onset of AD, for example, in genetically predisposed subjects (primary prevention) or in amyloid-positive presymptomatic subjects (e.g., defined by PET imaging [secondary prevention]). Our GSM strategy is analogous to the development of statins for prevention of coronary artery disease, with initial testing in familial hypercholesterolemia patients (Mabuchi et al., 1981). Similarly, identification of high-risk populations will be used for assessing the potential clinical efficacy of GSM treatment, to specifically address the relatively elevated cerebral Aβ42 peptide levels (i.e., increased Aβ42/40 ratios) and early Aβ42 peptide deposition in EOFAD at the earliest stage of the AD pathological continuum (i.e., A+T−(N)−; Jack et al., 2018) in which a preventive approach may prove most effective. While GSM treatment for AD patients with preexisting Aβ42 pathology and pathological tau biomarkers without neurodegeneration (A+T+(N)−) may also prove effective, targeting AD patients with neurodegeneration (A+T+(N)+) for GSM therapy is less likely to offer benefit; there is abundant evidence that anti-amyloid agents are of limited effectiveness in symptomatic AD (Coric et al., 2015; Coric et al., 2012; Doody et al., 2013; Egan et al., 2018; Gilman et al., 2005; Salloway et al., 2014). The preclinical characterization of our clinical candidate GSM(s) described herein provides strong support to enable clinical development with the intention of preventing AD and, perhaps, for intervention in presymptomatic AD or secondary prevention (Jack et al., 2018; Mills et al., 2013; Reiman et al., 2011). In this study, we report the full preclinical characterization of a potent GSM capable of completely eliminating production of Aβ42 in rodents at doses as low as 5–10 mg/kg, which is >10-fold below the no observed adverse effect level (NOAEL) and >20-fold below the maximum tolerated dose (MTD). Finally, we show that our lead clinical candidate is capable of significantly attenuating cerebral amyloidosis and microgliosis in the presenilin amyloid precursor protein (PSAPP) transgenic mouse model when chronic administration is commenced either before (prophylactic administration) or after (disease-modifying administration) the time at which the appearance of substantial amyloid deposition has occurred.
Results
Integration of iterative structural activity relationship (SAR) and structural property relationship (SPR) multiparametric analyses to mitigate liabilities of a former clinical candidate GSM
Rounds of compound evolution culminated in our discovery of the pyridazine-derived class of GSMs that display exceptional activity for attenuating the production of Aβ42 as well as acceptable absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties (Rynearson et al., 2016; Rynearson et al., 2020; Wagner et al., 2016; Wagner et al., 2017; Wagner et al., 2014). Previously, we developed a potent novel pyridazine-containing GSM (BPN-15606; Wagner et al., 2017). Acute, subchronic, and chronic efficacy studies demonstrated significant lowering of Aβ42 levels in plasma, cerebrospinal fluid (CSF), and brain extracts of rats and mice at doses ∼5–10-fold lower than those required for previously developed GSMs (Kounnas et al., 2010; Wagner et al., 2017). The discovery of BPN-15606 (compound 1) demonstrated that strategic integration of heterocycles within our proprietary GSM scaffold, followed by refinement based on iterative rounds of compound optimization, enabled identification of a family of potent GSMs. However, studies on compound 1 were terminated after completion of Good Laboratory Practice (GLP)–compliant 28-d investigational new drug (IND)–enabling safety pharmacology and toxicology studies in rats and nonhuman primates (NHPs). These revealed a potentially mutagenic metabolite(s) in rat and evidence of corrected QT interval prolongation in NHPs (data not shown). In response, our medicinal chemistry and drug development efforts defined 134 novel pyridazine-derived GSM analogues and allowed for a comprehensive SAR for this new scaffold, in which we discovered compounds with improved pharmacokinetic/pharmacodynamic (PK/PD) properties that lack metabolite-based mutagenic activity (Wagner et al., 2016). Multiparametric screening of this expanded class of novel pyridazines showed that the majority of structural analogues demonstrated potent activity for attenuating Aβ42 production in vitro; 54 analogues showed IC50 (half maximal inhibitory concentration) values ≤ 10 nM for the inhibition of Aβ42 secretion in cell-based assays and without inhibiting Notch proteolysis (Rynearson et al., 2016; Wagner et al., 2016; Wagner et al., 2014). Analogues were organized into subclasses based on structural modifications for rigorous in vitro ADMET analyses, allowing for the triage of numerous compounds and the generation of SPRs. Based on all SAR and SPR data generated, two novel analogues of compound 1 emerged as the top candidates for further development, 776890 (compound 2) and 779690 (compound 3). Compounds 2 and 3 showed in vitro IC50 values of 4.1 and 5.3 nM, respectively, for inhibition of Aβ42 secretion and 80 and 87 nM IC50 values for inhibition of Aβ40 secretion, respectively, thus showing a greater activity differential between effects on Aβ42 and Aβ40 secretion (Fig. 1) compared with compound 1 (Wagner et al., 2017). The three pyridazine compounds showed in vitro 50% effective concentration (EC50) values for potentiating Aβ38 secretion of 84 nM, 18 nM, and 29 nM for compound 1 (Wagner et al., 2017), compound 2, and compound 3, respectively (Fig. 1). The three GSM pyridazine analogues all exhibited poor kinetic solubility in aqueous buffer (Table 1).

Structures of pyridazine-derived GSMs and the compounds’ effect on Aβ levels . (A) Compound structures are shown. The IC50 values for the inhibition of Aβ42 and Aβ40 secretion and the EC50 values for the potentiation of Aβ38 in vitro are indicated. IC50 and EC50 values represent the concentration in nanomolars of compound required for reducing either Aβ42 or Aβ40 levels or increasing Aβ38 levels by 50% and are the mean of at least four determinations. (B and C) Concentration response curves of compounds 2 and 3 using SHSY5Y-APP cell-based screening assays. Aβ42, Aβ40, and Aβ38 peptide levels were determined using Meso Scale Sector 6000 Multiplex assays. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM.
Structures of pyridazine-derived GSMs and the compounds’ effect on Aβ levels . (A) Compound structures are shown. The IC50 values for the inhibition of Aβ42 and Aβ40 secretion and the EC50 values for the potentiation of Aβ38 in vitro are indicated. IC50 and EC50 values represent the concentration in nanomolars of compound required for reducing either Aβ42 or Aβ40 levels or increasing Aβ38 levels by 50% and are the mean of at least four determinations. (B and C) Concentration response curves of compounds 2 and 3 using SHSY5Y-APP cell-based screening assays. Aβ42, Aβ40, and Aβ38 peptide levels were determined using Meso Scale Sector 6000 Multiplex assays. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM.
In vitro ADMET profile of pyridazine GSMs
| Cmpd | Aβ42 IC50a | clogPb | Kin. Aq. Sol.c | FaSSIF/ FeSSIFd | Metabolic Stabilitye | CYP450 IC50 (μM)f | % PPBg | hERGh | Pappi | Efflux Ratioj | Amesk | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | R | M | 3A4 | 1A2 | 2C9 | 2C19 | 2D6 | H | R | M | |||||||||
| 1 BPN-15606 | 7.0 | 4.40 | 6.6 | >100/ >100 | 78 | 84 | 76 | 14 (m) 8.9 (t) | 37 | 1.3 | 6.9 | 4.7 | 99.6 | 99.9 | 99.8 | 6.2 | 37.2 | 0.9 | Positive |
| 2 776890 | 4.1 | 3.61 | <1.3 | 80/79 | 71 | 76 | 91 | 42 (m) 9.4 (t) | 12 | 4.4 | 12 | 11 | 99.8 | 99.9 | 99.9 | 7.3 | 23.3 | 0.9 | Negative |
| 3 779690 | 5.3 | 3.75 | <1.3 | 79/82 | 100 | 100 | 100 | 19 (m) 17 (t) | 3.6 | 1.3 | 13 | 20 | 99.7 | 99.7 | 99.7 | 3.1 | 30.2 | 1.1 | Negative |
Cmpd, compound.
aIC50 represents the concentration in nanomolars of compound required for reducing Aβ42 levels by 50%. The IC50 values are the mean of at least four determinations.
bCalculated partition coefficient of the ratio of the compound’s concentration in octanol to the compound’s concentration in water using ChemAxon.
cKinetic solubility measured at pH 7.4 by UV/Vis (visible) absorbance in PBS buffer in micromolar.
dMeasurement of dissolved compound concentration in FaSSIF or FeSSIF via UV/Vis absorbance.
ePercent remaining after 30 min on incubation with human (H), rat (R), and mouse (M) liver microsomes (1 mg/ml) at 1-µM test compound concentration.
fFive recombinant human CYP450 isoforms were tested for inhibition using probe substrates. Percent inhibition was measured by LC-MS/MS analysis. (m), midazolam; (t), testosterone.
gPercent plasma protein binding (PPB) using human (H), rat (R), and mouse (M) plasma.
hPatch-Xpress patch-clamp assay; IC50 (in micromolar) based on five-point concentration–response curves (n = 3) in HEK-293 cells stably expressing the hERG channel.
iApparent permeability (A-B; × 10−6 cm/s) determined in MDR1-MDCK cell monolayers.
jCalculated efflux ratio determined using MDR1-MDCK cell monolayers (B-A/A-B).
kNon-GLP screening assay measuring bacterial growth due to the reversion of the histidine mutation induced by incubation with test article (5–100 µM). S. typhimurium bacterial strains, including TA98, TA100, TA1535, and TA1537, in the presence and absence of S9 fraction from Aroclor-induced rat liver.
To assess the implications for oral absorption, additional kinetic solubility assays using simulated intestinal fluids, which more accurately recapitulate the environment of an orally dosed small molecule, were employed. Significantly increased solubility was observed for compounds 1, 2, and 3 in fasted-state simulated intestinal fluid (FaSSIF) and fed-state simulated intestinal fluid (FeSSIF); the lower pH of the FaSSIF and FeSSIF (pH = 6.5 and 5.8, respectively) assay conditions resulted in a high ionization of the weakly basic compounds and led to substantial increases in observed solubility. Therefore, as with compound 1, the two novel analogues, 2 and 3, displayed more than adequate aqueous solubility for oral absorption as well as acceptable metabolic stability across three species (human, rat, and mouse), and both analogues exhibited minimal cytochrome p450 inhibition potential. Although compounds 2 and 3 showed IC50 values for inhibition of CYP450 2C9 in the low micromolar range, these values are over 200-fold above the in vitro IC50 efficacy values for Aβ42 inhibition (4.1 nM and 5.3 nM for compounds 2 and 3, respectively). Additionally, both novel analogues exceeded the 100-fold safety margin between the in vitro IC50 efficacy values for Aβ42 inhibition and the IC20 for human ether-à-go-go-related gene (hERG) inhibition (hERG IC20 = 1.3 µM for compound 2; hERG IC20 = 1.0 µM for compound 3), an in vitro assay metric commonly used to predict the in vivo potential to induce cardiac arrhythmia. The two new lead candidate compounds also exhibited favorable membrane permeability and are not P-glycoprotein substrates, thus supporting their potential for good brain penetration. Initial screening also included evaluation of compounds 2 and 3 in a mini-Ames bacterial mutagenicity assay using multiple strains of Salmonella typhimurium; neither compound 2 nor compound 3 shared the activity of the predecessor compound 1 (Table 1).
In addition to the primary pharmacology studies, potential off-target activity (secondary pharmacology) has been assessed against a broad panel of 55 targets (receptors, transporters, ion channels). The potential for compound 2 to inhibit binding of radiolabeled positive control ligands specific to each of the targets was evaluated at a concentration of 10 µM for compound 2. Greater than 50% inhibition of ligand binding was noted for eight targets: adenosine A3, dopamine D1, histamine H1, NK3, κ and μ opioid receptors, sodium channel (Site 2), and chloride channel (γ-aminobutyric acid–gated). Toxicologically significant effects of compound 2 interactions with these targets in vivowere not observed in any of the GLP safety pharmacology or toxicology studies discussed below.
In vivo PK/PD studies validate two GSMs as therapeutic candidates for advanced preclinical development
Compounds 2 and 3 were subjected to single-dose i.v./per os (po) PK studies in male CD-1 mice. Both showed highly suitable PK properties (Table S4), enabling pivotal in vivo proof of concept, dose-dependent PD, and time-course efficacy studies (Fig. 2, Fig. 3, Fig. 4, Table S1, and Table S2). Repeat-dose administration studies of compounds 2 and 3 were conducted in C57BL/6J mice to demonstrate sustained dose-dependent efficacy (modulation of Aβ peptide variant levels) following 9 consecutive d of treatment. Upon completion of the study, brain and plasma samples were obtained and evaluated for Aβ42 and Aβ40 levels. Significant dose-dependent lowering of Aβ42 and Aβ40 levels in plasma and brain was observed for both compounds (Fig. 2, A and B), and both showed dose-dependent exposures (Table S1). Importantly, robust attenuation of Aβ42 and Aβ40 levels in plasma and brain was achieved by both compounds at the lowest dose tested (10 mg/kg). In acute time course efficacy studies of compound 2, male C57BL/6J mice were administered a single oral dose of drug (5 mg/kg or 10 mg/kg) and euthanized at various times over a 48-h time period to ascertain the magnitude and duration of the therapeutic effect (Fig. 3, A and B). Following dosing, maximal reduction of Aβ42 levels in plasma occurred at the 1-h time points (Fig. 3, A and B), consistent with the observed Tmax (time to reach peak plasma concentration of compound) from the mouse PK studies (Table S4); the peak reduction in brain Aβ42 levels occurred between 6 and 12 h (Fig. 3, A and B). The duration of appreciable lowering of brain Aβ42 peptide levels was ∼24 h following a single oral dose of 10 mg/kg and ≥ 12 h but < 24 h (diurnal) following a single oral dose of 5 mg/kg (Fig. 3, A and B). Importantly, in both plasma and brain, compound 2 consistently demonstrated a preferential lowering of Aβ42 (versus Aβ40) at both doses (5 and 10 mg/kg).

Dose-dependent efficacy of GSMs compound 2 and compound 3 in mice. (A) Levels of Aβ42 and Aβ40 in plasma and brain following daily oral administration of either vehicle (V) or compound 2 to male C57BL/6J mice (n = 10/dose) were measured following a 9-d treatment course. (B) Levels of Aβ42 and Aβ40 in plasma and brain following daily oral administration of either vehicle or compound 3 to male C57BL/6J mice (n = 10/dose) were measured following a 9-d treatment course. Mouse brain Aβ42 levels were below the threshold of detection for both compounds. Aβ42 and Aβ40 peptide levels were determined using MSD multiplex assays. Data are expressed in picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. ****, P < 0.0001. ND, below detectable limit of the assay.
Dose-dependent efficacy of GSMs compound 2 and compound 3 in mice. (A) Levels of Aβ42 and Aβ40 in plasma and brain following daily oral administration of either vehicle (V) or compound 2 to male C57BL/6J mice (n = 10/dose) were measured following a 9-d treatment course. (B) Levels of Aβ42 and Aβ40 in plasma and brain following daily oral administration of either vehicle or compound 3 to male C57BL/6J mice (n = 10/dose) were measured following a 9-d treatment course. Mouse brain Aβ42 levels were below the threshold of detection for both compounds. Aβ42 and Aβ40 peptide levels were determined using MSD multiplex assays. Data are expressed in picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. ****, P < 0.0001. ND, below detectable limit of the assay.

Time course of efficacy following a single oral dose of GSM compound 2. (A and B) Male C57BL/6J mice (n = 7/time point) were administered compound 2 at 5 mg/kg (A) or 10 mg/kg (B). Following a single dose of compound 2 or vehicle (V) by oral gavage, mice were euthanized at the indicated time point (1–48 h) and the levels of Aβ42 and Aβ40 peptides were quantitated in plasma and in brain extracts using Meso Scale Sector 6000 Multiplex assays. Data are expressed in picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001. ND, below detectable limit of the assay.
Time course of efficacy following a single oral dose of GSM compound 2. (A and B) Male C57BL/6J mice (n = 7/time point) were administered compound 2 at 5 mg/kg (A) or 10 mg/kg (B). Following a single dose of compound 2 or vehicle (V) by oral gavage, mice were euthanized at the indicated time point (1–48 h) and the levels of Aβ42 and Aβ40 peptides were quantitated in plasma and in brain extracts using Meso Scale Sector 6000 Multiplex assays. Data are expressed in picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001. ND, below detectable limit of the assay.

Time course of efficacy following a single oral dose of GSM compound 3 . Male C57Bl6 mice (n = 7/time point) were dosed with 10 mg/kg of compound 3 or vehicle by oral gavage. Animals were then sacrificed at the indicated time point (1–48 h), and the levels of Aβ42 and Aβ40 peptides in plasma and brain extracts were quantified using Meso Scale Sector 6000 Multiplex assays. Data are expressed in picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; ****, P < 0.0001. ND, below detectable limit of the assay.
Time course of efficacy following a single oral dose of GSM compound 3 . Male C57Bl6 mice (n = 7/time point) were dosed with 10 mg/kg of compound 3 or vehicle by oral gavage. Animals were then sacrificed at the indicated time point (1–48 h), and the levels of Aβ42 and Aβ40 peptides in plasma and brain extracts were quantified using Meso Scale Sector 6000 Multiplex assays. Data are expressed in picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; ****, P < 0.0001. ND, below detectable limit of the assay.
An identical time course efficacy study was carried out with compound 3, with the exception that mice were only administered a single dose of 10 mg/kg (Fig. 4). At this dose (10 mg/kg), compound 3 behaved very similarly to compound 2, showing a preferential lowering of Aβ42 (versus Aβ40) and a diurnal pattern of suppressing Aβ42 and Aβ40 levels in both plasma and brain (Fig. 4). Based on the slightly superior PK and PD behavior, including the exceptionally high bioavailability (F = 99%), compound 2 was selected as the preferred preclinical candidate. However, the in vitro Aβ42-lowering activity, ADMET properties, PK, and PD profiles of compound 3 are consistent with our lead identification profile and support the potential to develop this compound as an alternative clinical candidate in the event of any unanticipated toxicity associated with the current clinical lead, compound 2.
Chronic dosing studies with the clinical candidate GSM show sustained histopathological and biochemical efficacy in PSAPP transgenic mice
Chronic efficacy studies were conducted using transgenic PSAPP mice (Borchelt et al., 1997) to evaluate the effect of compound 2 on Aβ plaque accumulation and to assess the long-term safety and tolerability of the compound. Since PSAPP transgenic mice display minimal Aβ deposition at 3 mo of age yet show significant Aβ deposition at 6 mo of age, the chronic studies were divided into two arms: one group of mice receiving treatment for 90 d starting at 3 mo of age (prophylactic group) and one group of mice receiving treatment for 90 d starting at 6 mo of age (disease-modifying group). Upon completion of the 3-mo dosing regimen, the various Aβ peptide levels were measured in plasma and brain extracts for both the prophylactic and disease-modifying groups at ages 6 mo or 9 mo, respectively. Oral treatment of 3-mo-old PSAPP transgenic mice with compound 2 at 25 mg/kg/d for 3 mo resulted in robust decreases in plasma Aβ42 and Aβ40 levels and significant potentiation of Aβ38 plasma levels (Fig. 5 A). Importantly, in both detergent-soluble (RIPA buffer) and detergent-insoluble (formic acid) brain tissue extracts, Aβ42 levels were reduced by 44% and 54%, respectively; brain Aβ40 levels were similarly reduced in both detergent-soluble and in formic acid–soluble extracts. Significant potentiation of Aβ38 levels, as observed in plasma, was also shown in the detergent-soluble brain extracts but not in the formic acid extractions where Aβ38 levels were significantly decreased, suggesting that some Aβ38 is being incorporated into plaques (Fig. 5, B and C). Treatment of 6-mo-old PSAPP transgenic mice with compound 2 (25 mg/kg/d po) for 3 mo also resulted in decreases in plasma Aβ42 and Aβ40 levels and potentiation of Aβ38 levels, although the differential effects on plasma Aβ40 and Aβ38 levels did not reach statistical significance (Fig. 5 D). Similarly, in both detergent-soluble (RIPA buffer) and -insoluble (formic acid) extracts, brain Aβ42 levels were reduced by 45% and 39%, respectively (Fig. 5, E and F). Consistent with the 3-mo-old animals treated with compound 2 (prophylactic group), Aβ40 levels were significantly reduced in both of the brain extracts, while Aβ38 levels were nonsignificantly increased in the detergent-soluble extracts and similarly decreased in the formic acid extracts of the disease-modifying group (Fig. 5, E and F).

Effects of chronic treatment with GSM compound 2 on Aβ peptides in 6-mo-old and 9-mo-old PSAPP mice. (A–F) Levels of Aβ42, Aβ40, and Aβ38 in plasma and brain following oral administration of either vehicle (Veh) or compound 2 (cmpd 2) for 3 consecutive mo to 3-mo-old or 6-mo-old APP/PS1 transgenic mice. Animals were dosed with vehicle or compound 2 (25 mg/kg/d po) and euthanized at 6 mo of age or 9 mo of age. n = 8/age vehicle; n = 8/age compound 2. The levels of Aβ42, Aβ40, and Aβ38 peptides in the 6-mo-old PSAPP mice were quantitated in plasma (A), RIPA detergent-soluble brain extracts (B), and formic acid–soluble, detergent-insoluble brain extracts (C). The levels of Aβ42, Aβ40, and Aβ38 peptides in the 9-mo-old PSAPP mice were quantitated in plasma (D), RIPA detergent-soluble brain extracts (E), and formic acid–soluble, detergent-insoluble brain extracts (F). Aβ peptides were measured using Meso Scale Sector 6000 Multiplex assays. Data are expressed as picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0005.
Effects of chronic treatment with GSM compound 2 on Aβ peptides in 6-mo-old and 9-mo-old PSAPP mice. (A–F) Levels of Aβ42, Aβ40, and Aβ38 in plasma and brain following oral administration of either vehicle (Veh) or compound 2 (cmpd 2) for 3 consecutive mo to 3-mo-old or 6-mo-old APP/PS1 transgenic mice. Animals were dosed with vehicle or compound 2 (25 mg/kg/d po) and euthanized at 6 mo of age or 9 mo of age. n = 8/age vehicle; n = 8/age compound 2. The levels of Aβ42, Aβ40, and Aβ38 peptides in the 6-mo-old PSAPP mice were quantitated in plasma (A), RIPA detergent-soluble brain extracts (B), and formic acid–soluble, detergent-insoluble brain extracts (C). The levels of Aβ42, Aβ40, and Aβ38 peptides in the 9-mo-old PSAPP mice were quantitated in plasma (D), RIPA detergent-soluble brain extracts (E), and formic acid–soluble, detergent-insoluble brain extracts (F). Aβ peptides were measured using Meso Scale Sector 6000 Multiplex assays. Data are expressed as picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0005.
Immunoblotting analyses of RIPA buffer brain extracts from vehicle-treated and compound 2–treated wild-type mice (3 mo old and 6 mo old) for 3 consecutive mo and from the aforementioned treatment groups of PSAPP transgenic mice (prophylactic and disease modifying), with an antibody recognizing full-length (FL) APP (human) or App (murine) and their corresponding carboxyl-terminal fragments (CTFs), showed that chronic compound 2 treatment of both age groups (6 mo and 9 mo) of PSAPP transgenic mice or wild-type mice (data not shown) failed to cause accumulation of APP/App FL or APP/App-CTFs (α-CTFs or β-CTFs) relative to tubulin (Fig. 6, A–D).

Lack of effect of chronic treatment with GSM compound 2 in PSAPP mice on APP (FL)/tubulin, α-CTF/tubulin, and β-CTF/tubulin ratios. (A–C) Quantitation of APP (FL)/tubulin ratios (A), α-CTF/tubulin ratios (B), and β-CTF/tubulin ratios (C) in 6-mo-old and 9-mo-old PSAPP mouse brain extracts following chronic treatment with either vehicle (V) or compound 2 (Cmpd 2) via Bio-Rad ImageLab software. Statistical analysis was performed using GraphPad Prism software, and the results are expressed as the mean of the ratio (black horizontal line) ± SEM. (D) Immunoblots of PSAPP brain extracts from 6-mo-old or 9-mo-old PSAPP mice treated with either vehicle (V) or compound 2 (2). Blots were probed for APP (FL), α-CTFs, β-CTFs, and tubulin using anti-carboxyl terminal APP and anti-tubulin antibodies.
Lack of effect of chronic treatment with GSM compound 2 in PSAPP mice on APP (FL)/tubulin, α-CTF/tubulin, and β-CTF/tubulin ratios. (A–C) Quantitation of APP (FL)/tubulin ratios (A), α-CTF/tubulin ratios (B), and β-CTF/tubulin ratios (C) in 6-mo-old and 9-mo-old PSAPP mouse brain extracts following chronic treatment with either vehicle (V) or compound 2 (Cmpd 2) via Bio-Rad ImageLab software. Statistical analysis was performed using GraphPad Prism software, and the results are expressed as the mean of the ratio (black horizontal line) ± SEM. (D) Immunoblots of PSAPP brain extracts from 6-mo-old or 9-mo-old PSAPP mice treated with either vehicle (V) or compound 2 (2). Blots were probed for APP (FL), α-CTFs, β-CTFs, and tubulin using anti-carboxyl terminal APP and anti-tubulin antibodies.
To determine the effects of chronic compound 2 treatment on cerebral amyloidosis, immunohistochemical analyses were conducted; anti-Aβ mAb 3D6 was used to evaluate cerebral amyloid deposition, and Iba1 staining was used to assess the extent of microgliosis in the mice. Image analysis in the cortex and hippocampus of 6-mo-old PSAPP mice (prophylactic group) showed a highly significant (greater than twofold; P < 0.01) attenuation of cerebral amyloidosis by chronic treatment with compound 2 (Fig. 7, A and B). This effect on cerebral amyloidosis was also evident in the cortex and hippocampus of the compound 2–treated 9-mo-old PSAPP mice (disease-modifying group), which also showed a statistically significant decrease (P < 0.03) compared with the vehicle-treated group (Fig. 7, A and B). These immunohistochemical data using the mAb 3D6 are highly consistent with the biochemical analyses using the MSD (Meso Scale Discovery) multiplex kits showing the significant lowering of Aβ42 and Aβ40 levels in brain extracts from both the prophylactic and disease-modifying treatment groups (Fig. 5). The effects of compound 2 on microgliosis were also evident in the cortex and hippocampus of both the prophylactic and the disease-modifying treatment groups according to quantitative image analyses of Iba1 staining (Fig. 7, A and B). The effects on microgliosis were actually more pronounced in the 9-mo-old animals due to the fact that there is very little microgliosis in the 6-mo-old animals, even in the vehicle-treated group (Fig. 7 A). There was also an excellent correlation between the mAb 3D6 immunostaining and the Iba1 staining in all groups (Fig. 7 C).

Effects of chronic treatment with GSM compound 2 on amyloid deposition and microgliosis of 6-mo-old and 9-mo-old PSAPP mice. (A) Representative sagittal sections immunostained with mAb 3D6 and counterstained with Iba1 from PSAPP mice treated with either vehicle (Veh) or compound 2 (Cmpd 2) at a dose of 25 mg/kg/d for 3 mo beginning at either 3 mo of age (6-mo-old mice) or 6 mo of age (9-mo-old mice). n = 5/age group vehicle; n = 5/age group compound 2. (B) Quantitation of amyloid deposition (mAb 3D6) and microgliosis (Iba1) imaged in the cortex and hippocampus at either 6 mo of age or 9 mo of age following 3 mo of treatment with either vehicle or compound 2. Statistical analysis was performed using GraphPad Prism software, and the results are expressed as the mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Correlation analyses between mAb 3D6 immunostaining and Iba1 staining in 6-mo-old and 9-mo-old PSAPP mice following 3 mo of treatment with either vehicle or compound 2. Statistical analysis was performed with one-way ANOVA post hoc Tukey analysis.
Effects of chronic treatment with GSM compound 2 on amyloid deposition and microgliosis of 6-mo-old and 9-mo-old PSAPP mice. (A) Representative sagittal sections immunostained with mAb 3D6 and counterstained with Iba1 from PSAPP mice treated with either vehicle (Veh) or compound 2 (Cmpd 2) at a dose of 25 mg/kg/d for 3 mo beginning at either 3 mo of age (6-mo-old mice) or 6 mo of age (9-mo-old mice). n = 5/age group vehicle; n = 5/age group compound 2. (B) Quantitation of amyloid deposition (mAb 3D6) and microgliosis (Iba1) imaged in the cortex and hippocampus at either 6 mo of age or 9 mo of age following 3 mo of treatment with either vehicle or compound 2. Statistical analysis was performed using GraphPad Prism software, and the results are expressed as the mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Correlation analyses between mAb 3D6 immunostaining and Iba1 staining in 6-mo-old and 9-mo-old PSAPP mice following 3 mo of treatment with either vehicle or compound 2. Statistical analysis was performed with one-way ANOVA post hoc Tukey analysis.
Finally, full-body necropsy and histopathology analysis to identify potential target tissues showed no overt evidence of toxicity following chronic administration of compound 2, and there was no significant effect on weight gain in either of the two treatment groups (data not shown).
Repeat-dosing efficacy studies in rodents support utilization of plasma Aβ42 as a valid PD biomarker
Compound 2 was also evaluated in a single-dose i.v./po PK study (Table S5) and in a repeat-dose efficacy study in male Sprague-Dawley rats. Following a 9-d repeat administration by oral gavage regimen, plasma, CSF, and brain penetration of compound 2 demonstrated dose-proportional exposures (Table S2). The corresponding plasma, CSF, and brain samples were evaluated for Aβ42, Aβ40, and Aβ38 peptide levels. Consistent with the mouse efficacy studies with compound 2, significant dose-dependent lowering of Aβ42 and Aβ40 levels in plasma, CSF, and brain was observed. In addition, although Aβ38 was not detected in plasma or brain of either C57BL/6J mice or Sprague-Dawley rats, dose-dependent increases in Aβ38 were observed in the rat CSF, suggesting that the inability to detect this in plasma or brain, in the absence of transgenic overexpression, is possibly due to a lower sensitivity for the rodent Aβ38 ELISA. Notably, at the lowest administered dose (5 mg/kg), compound 2 significantly reduced plasma Aβ42 and Aβ40 levels by 78% and 57%, respectively; brain Aβ42 and Aβ40 levels by 54% and 29%, respectively; and CSF Aβ42 and Aβ40 levels by 41% and 29%, respectively (Table S2). Also, in agreement with the mouse studies, compound 2 had greater effects on lowering Aβ42 compared with Aβ40. Increasing the dose to 25 and 50 mg/kg further and dose dependently reduced plasma, brain, and CSF Aβ42 levels. At these higher doses, the reductions of Aβ40 in plasma, brain, and CSF were slightly less than for Aβ42 (Table S2).
The decreases in brain and plasma Aβ42 and Aβ40 levels elicited by compound 2 were significantly correlated in both rats and mice. In rats there was a statistically significant correlation coefficient (r2) between compound 2–provoked decreases in the levels in plasma, brain, and CSF of Aβ42 and Aβ40 peptides (Fig. 8, A–D). These results strongly support the posit that plasma and CSF Aβ peptide levels will serve as surrogate markers of brain Aβ peptide levels and thus allow indirect in vivo monitoring of these Aβ-modulating compounds. Availability of a readily accessible measure of biochemical efficacy of drug in brain is expected to prove very useful in clinical studies. Collectively, the in vivo results are highly promising since CSF Aβ42 is routinely used in clinical trials in order to assess target engagement (Coric et al., 2015; Doody et al., 2013; Egan et al., 2018) and has been shown to inversely correlate with Aβ plaque load in the brain demonstrated by PET imaging. Studies in EOFAD carriers suggest that CSF Aβ42 levels may begin to rise before evidence of plaque deposition and then decrease once significant plaque load develops, presumably due to incorporation of Aβ42 into diffuse Aβ deposits and neuritic Aβ plaques (Bateman et al., 2012; Sunderland et al., 2003). The vast majority of EOFAD-linked mutations are associated with an increase in the Aβ42/Aβ40 ratio, a change believed to accelerate cerebral amyloidosis, which ensues at a much younger age in EOFAD carriers—indeed, decades earlier than in sporadic AD patients. Therefore, the ability to preferentially inhibit Aβ42 (versus Aβ40) in vitro and in vivo may be a critical attribute of compound 2, especially when considering the potential for preventive therapy in EOFAD.
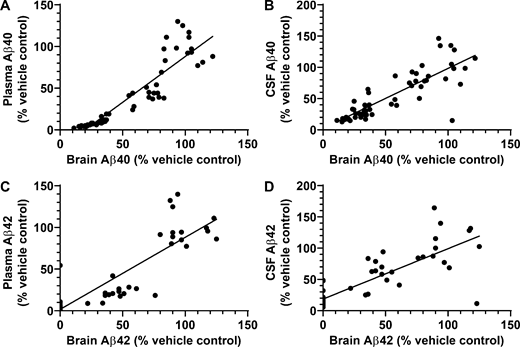
Correlation analyses of effects of GSM compound 2 treatment on Aβ peptides in brain, CSF, and plasma.(A–D) Correlation analysis of Aβ40 between brain and plasma (A) and between brain and CSF (B) in Sprague-Dawley rats treated with compound 2. Correlation analysis of Aβ42 between brain and plasma (C) and between brain and CSF (D) in Sprague-Dawley rats treated with compound 2. Data were plotted from Table S2 and analyzed by Pearson correlation analysis. (A) Scatter plot for brain Aβ40 and plasma Aβ40 from individual rats (total of 56 rats). Linear regression showed a best fit of y = 1.098*x − 21.88, r2 = 0.8326, P < 0.0001. (B) Scatter plot for brain Aβ40 and CSF Aβ40 from individual rats (total of 53 rats). Linear regression showed a best fit of y = 0.9595*x + 2.267, r2 = 0.6923, P < 0.0001. (C) Scatter plot for brain Aβ42 and plasma Aβ42 from individual rats (total of 47 rats). Linear regression showed a best fit of y = 0.8637*x + 1.723, r2 = 0.7618, P < 0.0001. (D) Scatter plot for brain Aβ42 and CSF Aβ42 from individual rats (total of 53 rats). Linear regression showed a best fit of y = 0.8056*x + 18.60, r2 = 0.6606, P < 0.0001. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect.
Correlation analyses of effects of GSM compound 2 treatment on Aβ peptides in brain, CSF, and plasma.(A–D) Correlation analysis of Aβ40 between brain and plasma (A) and between brain and CSF (B) in Sprague-Dawley rats treated with compound 2. Correlation analysis of Aβ42 between brain and plasma (C) and between brain and CSF (D) in Sprague-Dawley rats treated with compound 2. Data were plotted from Table S2 and analyzed by Pearson correlation analysis. (A) Scatter plot for brain Aβ40 and plasma Aβ40 from individual rats (total of 56 rats). Linear regression showed a best fit of y = 1.098*x − 21.88, r2 = 0.8326, P < 0.0001. (B) Scatter plot for brain Aβ40 and CSF Aβ40 from individual rats (total of 53 rats). Linear regression showed a best fit of y = 0.9595*x + 2.267, r2 = 0.6923, P < 0.0001. (C) Scatter plot for brain Aβ42 and plasma Aβ42 from individual rats (total of 47 rats). Linear regression showed a best fit of y = 0.8637*x + 1.723, r2 = 0.7618, P < 0.0001. (D) Scatter plot for brain Aβ42 and CSF Aβ42 from individual rats (total of 53 rats). Linear regression showed a best fit of y = 0.8056*x + 18.60, r2 = 0.6606, P < 0.0001. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect.
Preliminary toxicity and safety pharmacology testing in rats and NHPs
Initial toxicity testing of compound 2 in male and female Sprague-Dawley rats demonstrated an ∼10–20-fold safety margin over the efficacious dose (5 mg/kg) based on the 7-d dose range-finding (DRF) toxicity and toxicokinetic study that showed an MTD ≥ 100 mg/kg and an NOAEL ≥ 50 mg/kg. Similarly, compound 2 has also undergone a single-dose i.v./po PK study (Table S5) and a single escalating dose study followed by a 7-d repeat-dosing phase at the MTD in a DRF toxicity and toxicokinetic study in male and female cynomolgus macaques (NHPs). In the single escalating dose phase, NHP plasma samples collected at 4 h after dose showed that single doses of 10 mg/kg, 30 mg/kg, 100 mg/kg, or 200 mg/kg of compound 2 demonstrated dose-dependent exposures from 10 mg/kg up to 100 mg/kg; however, all doses were equally efficacious at reducing Aβ42 and Aβ40 levels in plasma, showing a maximal (∼60–70%) reduction (Fig. 9 A). These data are consistent with the rodent data that also showed maximal lowering of plasma Aβ levels at 10 mg/kg (Fig. 2 A). The single-dose time course study of the ability of compound 2 to reduce NHP plasma Aβ42 and Aβ40 levels, conducted on day 1 (200 mg/kg) of the repeat-dosing phase of the DRF, showed a sustained lowering (∼70% reduction for over 24 h) in the levels of plasma Aβ42 and Aβ40 peptides, as well as time-dependent and sustained plasma drug exposures from 1–24 h (Fig. 9 B). Interestingly, compound 2 did not affect levels of Aβ38 in the plasma of NHPs (Fig. 9, A and B). This profile of selectively lowering levels of Aβ42 and Aβ40, as opposed to shorter Aβ peptides (Aβ37 and Aβ38), is consistent with a GSM mechanism as opposed to that of a GSI, which equally lower levels of all Aβ peptide variants. Collectively, these DRF toxicity and toxicokinetic studies determined the NOAEL to be ≥ 50 mg/kg in rats and the MTD to be ≥ 100 mg/kg in both rats and NHPs. This is consistent with similarities in a number of PK parameters between rats and NHPs (Table S5).

Efficacy of GSM compound 2 on various Aβ peptide levels in plasma of NHPs. (A) Plasma levels of Aβ38, Aβ40, and Aβ42 in male and female NHPs (n = 4/dose) at 4 h following a single nasogastric dose of 0 mg/kg (vehicle), 10 mg/kg (mean plasma [drug] = 753 ng/ml), 30 mg/kg (mean plasma [drug] = 1,235 ng/ml), 100 mg/kg (mean plasma [drug] = 2,895 ng/ml), or 200 mg/kg (mean plasma [drug] = 2,475 ng/ml). (B) Plasma levels of Aβ38, Aβ40, and Aβ42 in male and female NHPs (n = 4/time point) following a single nasogastric dose (200 mg/kg) on day 1 of the repeat-dosing phase in the DRF toxicokinetic study measured at 0 h (mean plasma [drug] = LLOQ), 1 h (mean plasma [drug] = 109 ng/ml), 2 h (mean plasma [drug] = 460 ng/ml), 4 h (mean plasma [drug] = 2,300 ng/ml), 8 h (mean plasma [drug] = 2,560 ng/ml), or 24 h (mean plasma [drug] = 1,705 ng/ml) after drug or vehicle administration. Aβ38, Aβ40, and Aβ42 peptides were quantitated using Meso Scale Sector 6000 Multiplex assays. Data are expressed as picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Efficacy of GSM compound 2 on various Aβ peptide levels in plasma of NHPs. (A) Plasma levels of Aβ38, Aβ40, and Aβ42 in male and female NHPs (n = 4/dose) at 4 h following a single nasogastric dose of 0 mg/kg (vehicle), 10 mg/kg (mean plasma [drug] = 753 ng/ml), 30 mg/kg (mean plasma [drug] = 1,235 ng/ml), 100 mg/kg (mean plasma [drug] = 2,895 ng/ml), or 200 mg/kg (mean plasma [drug] = 2,475 ng/ml). (B) Plasma levels of Aβ38, Aβ40, and Aβ42 in male and female NHPs (n = 4/time point) following a single nasogastric dose (200 mg/kg) on day 1 of the repeat-dosing phase in the DRF toxicokinetic study measured at 0 h (mean plasma [drug] = LLOQ), 1 h (mean plasma [drug] = 109 ng/ml), 2 h (mean plasma [drug] = 460 ng/ml), 4 h (mean plasma [drug] = 2,300 ng/ml), 8 h (mean plasma [drug] = 2,560 ng/ml), or 24 h (mean plasma [drug] = 1,705 ng/ml) after drug or vehicle administration. Aβ38, Aβ40, and Aβ42 peptides were quantitated using Meso Scale Sector 6000 Multiplex assays. Data are expressed as picograms per milliliter. Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. ANOVA was used to detect a significant effect. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Importantly, a recently completed GLP-compliant IND-enabling cardiopulmonary safety pharmacology study in NHPs (compliant with International Conference on Harmonisation [ICH] S7A Safety Pharmacology Guidance) showed that compound 2 had no significant effect on electrocardiogram parameters, including corrected QT interval, at any of the doses tested (10, 30, and 60 mg/kg). In addition to showing excellent in vivo potency, compound 2 has undergone extensive preclinical evaluation of in vitro properties that may predict its potential for drug–drug interactions, including metabolite profiling in five species (mouse, rat, dog, NHP, and human), which showed no major metabolites and confirmed the absence of any unique human metabolites (Table S3). However, a recent GLP-compliant Ames bacterial mutagenicity assay with compound 2 unexpectedly demonstrated a positive result in two S. typhimurium strains (T98 and T100), but only in the presence of metabolic activation using Aroclor-induced rat liver S9 extracts, suggesting the presence of one or more mutagenic metabolite(s) in this Ames assay. This finding was not consistent with results from previous non-GLP screening Ames assays performed with distinct lots of compound 2.
In an attempt to mitigate this recent Ames finding, we performed additional GLP genetic toxicity studies, as performed with compound 1 (BPN-15606), which also showed highly variable Ames assay results in the same two S. typhimurium TA98 and TA100 strains and only in the presence of Aroclor-induced rat liver S9 extracts (data not shown). Results of a GLP-compliant in vitro micronucleus evaluation and a GLP-compliant 28-d repeat-dose toxicity and in vivo micronucleus study in rats (compliant with ICH S7A Safety Pharmacology Guidance) were both negative (data not shown). To further explore the unanticipated Ames result, a GLP-compliant 28-d in vivo Pig-a gene mutation study was conducted in rats (compliant with ICH S7A Safety Pharmacology Guidance) to determine whether compound 2 demonstrates mutagenicity in vivo. Compound 2 was tested at doses of 0, 6, 20, and 60 mg/kg/d (males) and 0, 3, 10, and 30 mg/kg/d (females) and was shown not to induce Pig-a mutant mature RBCs or Pig-a mutant immature reticulocytes (RETs) in the peripheral blood of male and female rats following 28 d of administration at any dose tested, thus confirming that compound 2 is negative as an in vivo mutagen in rats. The doses selected for the experiment were designed to correct for the twofold greater exposures of compound 2 observed in female versus male rats identified during the DRF study. Importantly, these results were consistent with a GLP-compliant 28-d rat toxicity study that determined the NOAEL to be 60 mg/kg, thus establishing safe dosing levels in rats.
Human equivalent 50% effective dose projection is 100 mg qd (quaque die; once daily) based on predicted human PK parameters obtained from in vitro and in vivo animal studies
In mice, a single oral dose of compound 2 (10 mg/kg) reduced Aβ42 levels in brain by > 70% and in plasma by > 90% at peak reduction (Fig. 3 B); repeated dosing (10 mg/kg/d) for 9 d resulted in complete (100%) elimination of detectable Aβ42 in brain and > 90% reduction in plasma levels (Fig. 2 A). In rats, repeated dosing of 5 mg/kg/d for 9 d reduced Aβ42 levels in CSF and brain by 41% and 54%, respectively, and in plasma by 78% (Table S2). Allometric scaling in both species (mouse and rat) equates to a human equivalent 50% effective dose of 1.5 mg/kg or 100 mg for a 70-kg human. The clinically effective dose can also be estimated using predicted human PK parameters derived from in vitro and in vivo rodent studies, such as clearance (CL) and bioavailability (F), as well as an effective area under the curve (AUCeffective) as indicated by Eq. 1: Effective dose = CL × AUCeffective/F. Applying this calculation predicts the same 1.5 mg/kg dose as above.
Prediction of human oral bioavailability (F): Compound 2 is a low-CL compound in mice, rats, and NHPs. The CL of compound 2 was 483, 357, and 331 ml/h/kg (8, 6, and 5.5 ml/min/kg) in mice, rats, and NHPs, respectively (Table S5). The values of CL were < 10% of the reported hepatic blood flow of these species (95, 75, and 50 ml/min/kg, respectively). Thus, hepatic first-pass metabolism of compound 2 is expected to be insignificant (much < 10%) for these species. On the other hand, compound 2 exhibited good oral bioavailability (F) in mice and rats (> 95% for both species). Based on kinetic analysis, the fraction of dose absorbed from intestinal lumen (fabs) is estimated to be ∼90% in mice and rats. Since it is well accepted that the fabs value of a test compound is expected to be species independent, the fabs value of compound 2 in humans is also expected to be ∼90%. With an fabs of 90%, the oral bioavailability (F) of compound 2 in humans is expected to be ∼80%.
Prediction of human CL: Using an allometric scaling approach, the data from the mouse, rat, and NHP PK experiments predict the human CL of compound 2 to be ∼300 ml/h/kg. Since it is recognized that NHPs are a reliable animal model for predicting human CL, the predicted value is quantitatively similar to NHP CL (331 ml/h/kg; Table S5). In a retrospective study, the CL of more than 100 drugs in rats, dogs, NHPs, and humans was identified and compared; the data pointed to NHPs as providing the most qualitatively and quantitatively accurate predictions for human CL (Ward et al., 2004).
Estimation of effective AUC: Compound 2 is a potent GSM with an in vitro IC50 of 4.1 nM (1.8 ng/ml). Assuming in vitro potency (IC50) can be extrapolated to in vivo, the EC50 in vivo can be calculated from the plasma unbound fraction (fu) as shown in Eq. 2: C50 = IC50/fu. Moreover, the AUCeffective over a period of 24 h can be calculated from Eq. 3: AUCeffective = EC50•24 h. Compound 2 is highly lipophilic and binds extensively to plasma proteins. The bound fraction of compound 2 in plasma was estimated to be 99.8%, 99.9%, and 99.9%, in humans, rats, and mice, respectively (Table 1). This equates to a plasma unbound fraction of 0.2%, 0.1%, and 0.1% in humans, rats, and mice, respectively. Due to its lipophilic nature, it is highly likely that a significant fraction of unbound compound 2 binds to (adsorbs onto) the devices (i.e., tubing) used for plasma protein-binding measurements, resulting in underestimation of the unbound fraction. In fact, the rat plasma unbound fraction of 0.1% predicts a smaller effect than is registered via compound 2–induced Aβ42 reductions in rodents. For example, compound 2 significantly reduced plasma and brain Aβ42 levels by ∼70% and 50%, respectively, in mice after a single oral dose 5 mg/kg 6–12 h after administration (Fig. 3 A). Using the AUCeffective (13,847 h•ng/ml) and rat plasma unbound fraction (0.1%), the unbound average Ceffective (50% effective concentration) of compound 2 is calculated to be ∼0.58 ng/ml. Obviously, the calculated unbound average Ceffective of compound 2 (0.58 ng/ml) is significantly lower than the reported IC50 (4.1 nM; 1.8 ng/ml) by a factor of ∼3. In other words, the calculated unbound average Ceffective cannot account for the extent of efficacy observed if the plasma unbound fraction of 0.1% is used. In contrast, if a plasma unbound fraction of 1.0% is used, the unbound average Ceffective is 5.8 ng/ml, and the efficacy of compound 2 can be reasonably explained. For this reason, a human plasma unbound fraction of 1.0%, instead of 0.1%, was used to estimate the 50% AUCeffective. With the human plasma unbound fraction of 1.0%, the AUCeffective of compound 2 in humans is calculated to be 4,320 h•ng/ml (with the knowledge of CL [300 ml/h/kg], F [80%], and 50% AUCeffective [4,320 h•ng/ml]), the human 50% effective dose can be estimated to be ∼1.5 mg/kg by using Eq. 1: 50% effective dose = AUC × CL/F. Assuming a body weight of 70 kg, the human 50% effective dose is predicted to be ∼100 mg).
Thus, compound 2 has a predicted wide margin of safety. The systemic exposure (AUC) of compound 2 at the NOAEL (60 mg/kg) in male rats was 586,000 h•ng/ml. At the predicted 50% effective equivalent human dose (100 mg), the AUC of compound 2 is predicted to be 4,320 h•ng/ml. The analysis points to a more than 130-fold safety margin for compound 2 based on comparison of systemic exposures; however, the ability of this promising candidate to be evaluated in the clinic will depend on the outcome of the Food and Drug Administration’s review of the recently completed GLP-compliant 28-d IND-enabling safety and toxicity studies.
Discussion
In this report, we detail the preclinical pharmacology of a potent and efficacious pyridazine-based GSM referred to as compound 2. The robust in vivo potencies exhibited by compound 2 across species, combined with the preliminarily acceptable safety profile based on both the efficacy and the DRF toxicology studies, strongly suggest the possibility of this compound being capable of providing a wide therapeutic index in humans. Commencement of clinical studies with compound 2 will obviously require successful IND filing and Food and Drug Administration clearance. This drug discovery effort, which entailed a highly focused medicinal chemistry campaign involving the iterative optimization of our previous clinical candidate BPN-15606 (compound 1; Wagner et al., 2017) was inspired by liabilities observed during the 28-d IND-enabling studies of compound 1 in rats and NHPs and a critical need for disease-modifying therapies for neurodegenerative diseases such as AD. Fortunately, the combination of SAR and SPR multiparametric studies of well over 100 novel pyridazine-based analogues of compound 1 identified two promising candidates, referred to as compound 2 and compound 3 (Fig. 1 A). Both molecules demonstrated suitable PK properties, enabling evaluation in acute and repeat-dosing PD studies as well as additional in vivo efficacy studies in rodents. Mouse efficacy studies comparing compounds 2 and 3 following either an acute or repeated subchronic dosing regimen demonstrated unprecedented in vivo potencies and dose-dependent efficacies with both candidate compounds.
Based on slightly superior overall pharmacological properties, compound 2 was prioritized for a number of additional preclinical experiments. These included chronic (3-mo) daily repeat administration studies in PSAPP transgenic mice, commencing at either the preplaque (prophylactic group) or postplaque (disease-modifying group) age, which showed significant biochemical and pathological efficacy in the absence of any notable toxicity at necropsy. Chronic treatment of either wild-type or PSAPP transgenic mice failed to show accumulation of App-CTFs or APP-CTFs, respectively, thus indicating preservation of γ-secretase function. In repeated subchronic dosing studies in rats, compound 2 demonstrated dose-dependent efficacies for lowering Aβ42 and Aβ40 levels in plasma, brain, and CSF along with dose-proportional exposures in plasma, CSF, and brain. In rat CSF, there was a dose-dependent increase in Aβ38 levels that was not observed in rat plasma or brain; however, there was significant potentiation of Aβ38 levels in plasma and in the detergent-soluble brain extracts of the chronically treated PSAPP transgenic mice, which may suggest that a lack of sensitivity of the rodent Aβ38 ELISA is the reason why this peptide is not observed in wild-type rodent plasma or brain extracts. The decreases in brain and plasma Aβ42 and Aβ40 levels elicited by compound 2 were significantly correlated in both rats and mice. In rats, there was also a significant correlation between decreases in the levels in brain and CSF of Aβ42 and Aβ40. These results strongly support the posit that plasma and CSF Aβ peptide levels will serve as surrogate markers for changes in brain Aβ peptide levels and thus allow indirect in vivo monitoring of our Aβ-targeting compounds. Availability of a readily accessible measure of biochemical efficacy of the drug in brain is expected to prove very useful in clinical studies of PK/PD and for dose selection.
A number of previous efforts to develop a safe and effective small molecule able to decrease the production of Aβ peptides have failed in clinical trials. Several trials have relied on enzyme inhibition strategies such as GSIs (Coric et al., 2015; Coric et al., 2012; Doody et al., 2013) or BACE inhibitors (Egan et al., 2018; Salloway et al., 2014; Sevigny et al., 2016) and exposed intolerable side effect profiles and, in most cases, appeared to cause cognitive worsening relative to placebo (Coric et al., 2015; Doody et al., 2013; Knopman, 2019). The dose-limiting adverse effects appear to be related to the mode of action of these enzyme inhibitors by affecting critical proteolytic functions of either γ-secretase or BACE-1, both of which have important normal functions involving the proteolytic cleavage of multiple endogenous substrates. In the case of GSIs, gastrointestinal adverse events and nonmelanoma skin cancer are possibly the result of inhibition of γ-secretase–mediated ε-site proteolysis of the Notch 1 receptor, required for release of the Notch intracellular domain, an activator of a transcription factor required for proper cellular differentiation (De Strooper et al., 1999). The methylimidazole-containing class of GSMs, which includes compound 2, has been shown not to affect proteolysis of the Notch 1 receptor and does not cause accumulation of CTFs (Kounnas et al., 2010; Liu et al., 2014; Rynearson et al., 2016; Wagner et al., 2016; Wagner et al., 2017; Wagner et al., 2012; Wagner et al., 2014). In fact, chronic treatment studies of compound 2 not only prevented an increase in APP/App CTFs in the older age group of PSAPP mice; there was a trend toward lowering of these CTFs (Fig. 7, B and C), suggesting that additional chronic studies may point to the possibility that lowering Aβ may secondarily impact processing of CTFs—APP products that others have argued contribute to AD pathogenesis (Kwart et al., 2019).
Other GSM programs have been developed and tested in the clinic. R-flurbiprofen has weak in vitro GSM activity and, although safe, did not show efficacy in phase 3 human trials (Green et al., 2009). More recent discovery efforts have yielded several newer GSMs that were tested in early-phase clinical trials, primarily single ascending dose (SAD) and multiple ascending dose (MAD) phase 1 studies, and yet none of these has moved beyond these primary safety evaluations (Ahn et al., 2020; Kounnas et al., 2019; Soares et al., 2016; Toyn et al., 2016; Yu et al., 2014). Lack of clinical development of prior GSM drug candidates could be due to the lack of establishing a suitable therapeutic index for dosing regimens able to substantially lower CSF Aβ42 levels without eliciting significant adverse effects. Whether or not this is the case for E2212 is uncertain, but this compound was shown to maximally lower plasma Aβ42 levels by 44.1% (AUC0–24 h) at a dose of 250 mg in a SAD study (without CSF measurements reported; Yu et al., 2014). BMS-932481 was shown to be capable of maximally lowering CSF Aβ42 and Aβ40 levels by 77% and 74%, respectively, at a dose of 200 mg in a 2-wk MAD study (Soares et al., 2016). Unfortunately, stage 3 elevations in alanine amino transferase occurred in two of six subjects receiving this dose, and elevations in other liver enzymes were observed in both the 100-mg and 200-mg dosed groups compared with placebo, which halted further development of this GSM (Soares et al., 2016). Another MAD study involved the GSM NGP555, which showed a trend toward increasing ratios of Aβ37/Aβ42 and Aβ38/Aβ42 in CSF at doses of 200 mg and 400 mg, but without demonstrating statistical significance (Kounnas et al., 2019). The most recently published clinical studies involved both SAD and MAD regimens with the GSM PF-06648671 (Ahn et al., 2020). Study designs differed between SAD and MAD stages. In the SAD study serial PD samples did not provide a clear exposure–response relationship in modulating CSF Aβ42 levels due to placebo baseline drift and large variability; however, in the MAD study, robust dose–response relationships were reported with estimates of CSF Aβ42 levels being lowered by 44%, 59%, and 65% at doses of 100 mg, 200 mg, and 360 mg, respectively. Importantly, PF-06648671 exhibited a good safety profile at doses up to 360 mg qd for 14 d in both young and elderly healthy normal subjects; however, further development plans for this GSM, if any, are currently unknown. Based on a limited number of comparative analyses of preclinical in vivo studies, compound 2 appears to be significantly more potent than any of these previously tested GSMs. The superior aggregate in vivo PK and PD data for compound 2 used to project the human 50% effective dose as being ∼100 mg is based on the predicted human PK calculations outlined above.
In summary, we describe the pharmacological characterization of a potent GSM that, based on the preclinical attributes detailed herein, appears to equal or exceed the potency of any previously tested GSMs that have reached early-phase clinical trials. The chronic efficacy studies conducted using the PSAPP transgenic mouse model demonstrated that compound 2 administered daily for 3 consecutive mo was effective when the treatment was initiated either before or after the appearance of significant cerebral amyloidosis. This suggests the possibility of testing compound 2 in secondary prevention studies, in high-risk subjects determined by amyloid PET to harbor detectable levels of cerebral amyloid. Future studies will decide the fate of this promising GSM and whether it can and should be tested as a potential treatment or preventive therapy for AD.
Materials and methods
Compounds
The novel GSMs (S)-N-(1-(4-fluorophenyl)ethyl)-6-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-4-methylpyridazin-3-amine (BPN-15606; compound 1), (S)-N-(1-(3,5-difluorophenyl)ethyl)-6-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-4-methylpyridazin-3-amine (776890; compound 2), and (S)-6-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-4-methyl-N-(1-(2,4,6-trifluorophenyl)ethyl)pyridazin-3-amine (779690; compound 3) were synthesized at Albany Molecular Research Institute (Albany, NY), using the methods reported in the Patent Cooperation Treaty WO2016070107 (Wagner et al., 2016).
SHSY5Y-APP neuroblastoma cell-based Aβ42, Aβ40, Aβ38, and Aβtotal peptide ELISAs
Briefly, human SHSY5Y neuroblastoma cells stably overexpressing wild-type human APP751 (SHSY5Y-APP) have been described previously (Wagner et al., 2014). Aβ peptide variants in conditioned medium from SHSY5Y-APP cells following treatment for 24 h with either vehicle or GSM compounds were quantitated using sandwich ELISAs for human Aβ42, human Aβ40, and total human Aβ as described previously (Wagner et al., 2014). Aβ42, Aβ40, and Aβ38 peptides from stably transfected SHSY5Y-APP cells treated for 24 h were quantitated in conditioned medium by an ELISA using Meso Scale Sector 6000 multiplex technology. Total Aβ peptide levels were quantitated in conditioned medium by a sandwich ELISA as described previously (Wagner et al., 2014).
In vitro ADMET assays
All in vitro ADMET assays, including kinetic aqueous solubility, liver microsomal stability, CYP inhibition, functional hERG, and multi-drug resistance gene 1–Madin Darby canine kidney (MDR1-MDCK) permeability, were performed as described previously (Wagner et al., 2017). CEREP profiling (Eurofins-Cerep) for potential off-target activity (secondary pharmacology) was assessed against a broad panel of 55 targets (receptors, transporters, ion channels). The potential for compound 2 at a concentration of 10 µM to inhibit binding of radiolabeled positive control ligands specific to each of the targets was evaluated. Assays were performed as described previously (Wagner et al., 2017).
Plasma protein binding–equilibrium dialysis
Equilibration of test compound dissolved in plasma or tissue homogenate against buffer across semipermeable membrane was measured by liquid chromatography (LC)/mass spectrometry (MS) analysis of compound concentrations in both compartments. The plasma protein binding of each compound was evaluated using human, rat, and mouse plasma as described previously (Wagner et al., 2017).
Metabolite identification
Compound 2 (1 µM) was incubated at 37°C for 4 h with cryopreserved mouse, rat, dog, monkey, or human hepatocytes. Identification of metabolites’ molecular weights and relative abundance of the specific metabolites was based on HPLC (high-performace LC)/MS/MS peak areas, and structural characterization of metabolites was accomplished by LC/MS/MS/Q-Trap as described previously (Wagner et al., 2017).
Bioanalytical assays
Mouse, rat, and NHP plasma and brain extract calibration standards and quality control samples for compound 2 and compound 3 (mouse only) were prepared in untreated plasma or brain extracts, as described previously for BPN-15606 (compound 1); N-(2-ethyl-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (BPN-3783) was used as the internal standard (Wagner et al., 2017). Briefly, a primary stock solution of 2 mg/ml analyte (compound 2, compound 3, or BPN-3783 [internal standard]) in DMSO was prepared. Dilutions were performed to make spiking solutions of analyte in DMSO at various concentrations. One volume of spiking solution was added to 99 volumes of untreated plasma or brain extract to attain nominal concentrations of standards with a final non = plasma matrix concentration of 1.0%. Plasma extractions were performed as follows: to a 50-µl sample in a 2-ml microfuge tube was added 0.9 ml acetonitrile containing 1,600 ng/ml BPN-3783 (internal standard). Tubes were vortexed at maximum speed on a plate vortexer for 5 min and centrifuged 10 min at ∼18,000 g, and 20 µl supernatant was removed and placed into a glass HPLC vial containing 880 µl 85/15/0.1:water/acetonitrile/formic acid (vol/vol/vol). Brain extractions were performed with rat or mouse brain samples as follows: Brains were stored frozen at −70°C and thawed to room temperature before homogenization in PBS using a probe sonicator (15 s at 50% maximal amplitude), using 4 ml PBS per 1 g brain tissue. To a 50-µl brain homogenate aliquot was added 0.3 ml acetonitrile containing 100 ng/ml BPN-3783 (internal standard). Tubes were vortexed and centrifuged 10 min at ∼18,000 g, and 75 µl supernatant was removed to a glass HPLC vial containing 800 µl 85/15/0.1:water/acetonitrile/formic acid (vol/vol/vol). The vials were briefly vortexed before LC/MS/MS analysis via the ultraperformance method using Shimadzu LC-20AD Pumps, Leap Technologies CTC HTS PAL Autosampler, a Phenomenex Luna C18(2), 50 × 2-mm, 5-mm column operated at 20°C, and an AB Sciex QTrap 5500 mass spectrometer with multiple reaction monitoring as described previously for BPN-15606 (Wagner et al., 2017).
Animal studies
All animal experiments were conducted following National Institutes of Health guidelines and were in compliance with the policies of University of California, San Diego Institutional Animal Care and Use Committee and the SRI International (Palo Alto, CA) Institutional Animal Care and Use Committee.
PK and analysis in mice, rats, and NHPs
Single-dose PK studies and PK analyses for compounds 2 and 3 were conducted in CD-1 mice, Sprague-Dawley rats, and cynomolgus monkeys (NHPs) as described previously (Wagner et al., 2017) for BPN-15606 (compound 1). Briefly, CD-1 mice and Sprague-Dawley rats were administered a single dose of either compound 2 or compound 3 (mice only) by i.v. or oral gavage (po) dose routes to assess oral bioavailability. Mice received either compound 2 or compound 3 as a single i.v. dose at 1 mg/kg (n = 12) or a single po dose at 5 mg/kg (n = 12). Rats received either compound 2 as a single i.v. dose at 1 mg/kg (n = 13) or a single po dose at 5 mg/kg (n = 3).
Blood was collected from mice at 5 (i.v. only), 15, and 30 min, and 1, 2, 4, 8, and 12 h after dose for processing to plasma, and brains were collected from both dose groups (i.v. and po) at 1, 4, 8, and 12 h after dose. For rats, blood was collected at 5 (i.v. only), 15, and 30 min, and 1, 2, 4, 8, 12, 24, and 48 h (po only) after dose for processing to plasma. Rat brains and CSF samples were collected from those same animals at the 1-, 2-, and 24-h time points for the i.v. group and at the 48-h (brain only) time point for the po group. All samples were analyzed by LC/MS/MS; for BPN-15606 (compound 1) levels, using a bioanalytical method that had a lower limit of quantitation (LLOQ) of 1 ng/ml in plasma and CSF and 5 ng/g in brain tissue; for compound 2 levels, using a bioanalytical method that had an LLOQ of 10 ng/ml in plasma and 50 ng/g in brain tissue; and for compound 3 levels, using a bioanalytical method that had an LLOQ of 5 ng/ml in plasma and 26 ng/g in brain tissue. Non-naive cynomolgus monkeys (NHPs) were administered compound 2 as a single nasogastric (po; 2 mg/kg) or i.v. (1 mg/kg) dose to assess oral bioavailability. Whole-blood samples were collected at specific time points and processed to plasma. Study in-life was conducted at Charles River Laboratories (Reno, NV). Plasma samples were shipped to SRI International for determination of drug concentrations. Samples were analyzed at SRI International using the bioanalytical method described above and validated for NHP plasma. Clinical observations were performed immediately after dose. All animals appeared normal throughout the study.
Repeat-dose subchronic efficacy studies in mice and rats
Male C57BL/6J mice (n = 10/dose level) and male Sprague-Dawley rats (n = 14/dose level) were administered either vehicle (80% polyethylene glycol 400 vol/vol; 20% sterile water vol/vol, and 0.1% Tween 20 vol/vol), compound 2 (10, 25, and 50 mg/kg), or compound 3 (10, 25, and 50 mg/kg) once daily po for 9 consecutive d (mice) or administered either vehicle or compound 2 (5, 25, and 50 mg/kg) once daily po for 9 consecutive d (rats). Clinical observations were performed immediately following dosing on each day, and all animals appeared normal throughout the study. All animals were euthanized 4 h following dosing on day 9 of the 9-d oral treatment course. Plasma and brain extracts were prepared (mice and rats); CSF was aspirated from the cisterna magnum with minimal to no blood contamination (rats only); and tissues were frozen at −70°C. Aβ peptides were quantitated in brain extracts and plasma (mice) or CSF, brain extracts, and plasma (rats), as described previously, using MSD multiplex kits and the Meso Scale Sector Imager 6000 (Kounnas et al., 2010). Statistical analysis was performed using GraphPad Prism software, and results are expressed as mean ± SEM. Analysis of variance was used to detect a significant effect. Drug levels of compounds were measured in plasma and brain extracts using the bioanalytical methods detailed above.
Acute single-dose time course studies in mice
Male C57BL/6J mice (n = 7 per group) were administered a single dose of either vehicle (80% polyethylene glycol 400 vol/vol, 20% sterile water vol/vol, and 0.1% Tween 20 vol/vol), compound 2 (5 or 10 mg/kg), or compound 3 (10 mg/kg) by oral gavage (po) and euthanized at 0.5, 1, 2, 4, 8, 12, 24, and 48 h following the dosing. Plasma and brain extracts were prepared, and Aβ peptides were quantitated as described previously (Prikhodko et al., 2020). Statistical analysis was performed as described above for the repeat-dosing studies.
Chronic repeat-dose efficacy studies in PSAPP transgenic mice
35 female double-transgenic mice, model PSAPP, purchased from The Jackson Laboratory (B6.C3-Tg [APPswe, PSEN1dE9] 85Dbo/Mmjax, stock no. 0034829) and then bred in-house were used in the study at either 90 or 180 d of age. The mice included PSAPP transgenic mice and wild-type (C57BL/6J) mice that were housed in a temperature-controlled room at a constant 22°C in a 12:12 h light/dark cycle (lights off at 18:00), with food and water available ad libitum. Age-matched mice were housed by treatment group in groups of two to four per cage. All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at University of California, San Diego. Subjects were randomly assigned to either the drug group (compound 2) or vehicle (80% polyethylene glycol 400 vol/vol; 20% sterile water vol/vol, and 0.1% Tween 20 vol/vol), with individual groups ranging from 9 to 10 mice. Mice were treated for 3 mo with either vehicle or compound 2 at a dose of 25 mg/kg/d by oral gavage (po). All animals were weighed three times weekly to assess any adverse effects on normal weight gain during the 3-mo treatment period. Food consumption was determined by weighing the metal cage, including the chow, to the nearest 0.1 g. Plasma and brain extracts were prepared, and Aβ peptides were quantitated as described previously (Prikhodko et al., 2020). Immunoblotting of holo-APP/holo-App and APP/App-CTFs was performed as described previously using an antibody directed against the APP carboxyl-terminus (Kounnas et al., 2010).
Immunohistochemical and histochemical analyses of chronically treated PSAPP transgenic mice
To determine Aβ plaque load, 40-µm-thick sagittal sections were immunostained using anti-Aβ mAb 3D6 (1:2,000) and anti-Iba1 (1:500) and fluorescence-labeled secondary antibodies. The sections were imaged on an automated Nikon Ti2 microscope fitted with the Yokogawa spinning-disk field scanning confocal system. Large area scans of the entire sections (17 × 13 fields, each 0.427 mm2) were acquired using a 20× objective as z-stacks (seven optical sections with 0.9-µm step size). The image panels were processed using NIS elements (Nikon Inc.) to generate stitched image stacks and converted into a single 2D image using maximum-intensity projection. The amyloid burden was quantified using Fiji as the cortex and hippocampus area covered by mAb 3D6 staining. Iba1 staining was quantified similarly, and the contribution of homeostatic microglia (calculated from the amyloid-free midbrain) was subtracted to yield the percentage of area associated with amyloid-induced gliosis. Two sections from each animal, administered with vehicle (n = 5) or compound 2 (n = 5), were analyzed.
Necropsy analysis of chronically treated PSAPP transgenic mice
Formalin-fixed tissues representing major organ systems were submitted to the Pathology Core of the Animal Care Program Diagnostic Laboratory at University of California, San Diego for light microscopic examination of hematoxylin and eosin–stained sections and for assessment for histopathologic abnormalities that might be related to the experimental treatment as described previously (Prikhodko et al., 2020).
GLP Salmonella–Escherichia coli/microsome plate incorporation assay
The ability of compound 2 to induce genetic damage as detected by the Salmonella–E. coli/microsome assay was evaluated. The assay was performed using the plate incorporation procedure with S. typhimurium strains TA1535, TA1537, TA98, and TA100 and E. coli strain WP2 (uvrA) in both the presence and absence of a metabolic activation mixture containing Aroclor 1254–induced rat liver microsomes (S9) according to previously published protocols (Mortelmans and Riccio, 2000; Mortelmans and Zeiger, 2000).
Repeat-dose toxicity and micronucleus evaluation of compound 2 in rats
DRF 7-d and GLP-compliant 28-d repeat-dose toxicity and micronucleus evaluation studies were performed with compound 2 as described previously (Wagner et al., 2017) for BPN-15606 (compound 1).
Repeat-dose Pig-a gene mutation study of compound 2 in rats
A GLP-compliant 28-d repeat-dose Pig-a gene mutation study was performed with compound 2 as described previously (Raschke et al., 2016). Briefly, the objective of this study was to evaluate compound 2 for its ability to induce Pig-a mutant mature RBCs and Pig-a mutant immature RETs in peripheral blood of male and female Sprague-Dawley rats after daily oral dose administration for 28 d. Compound 2 formulated in 15% Labrasol:85% sterile water was administered via oral gavage (po) for 28 consecutive d at 0, 6, 20, or 60 mg/kg/d to males (n = 5/dose) and at 0, 3, 10, and 30 mg/kg/d to females (n = 5/dose). The doses evaluated corresponded to doses tested in the pivotal 28-d GLP toxicology and toxicokinetics study of compound 2. The study was designed to administer twofold higher dose levels (and volumes) to males than females because prior studies showed this twofold difference resulted in comparable tolerance and exposure levels of compound 2 in male and female rats. A positive control group composed of three males and three females was administered 20 mg/kg N-Nitroso-N-ethylurea po once daily on days 1–3. Following 28 d of dosing, peripheral blood was collected from each animal and analyzed using flow cytometric analysis to determine for each animal the number of RETs (expressed as percent of total RBCs), the frequency of mutant phenotype RBCs, and the frequency of mutant phenotype RETs.
Dose escalation/7-d oral gavage toxicity and toxicokinetics study of compound 2 in NHPs
Studies aimed at determining the MTD of compound 2, characterizing potential toxicity, and calculating toxicokinetic parameters in adult male and female cynomolgus macaques following daily po dose administration at a near-MTD dose for 7 consecutive d were performed as described previously (Wagner et al., 2017). Briefly, the study was conducted in two phases. Phase A was the escalation phase wherein all NHPs (two males and two females; 4–5 yr of age) were administered a single, nasogastric oral dose of compound 2 at the same dose level (10 mg/kg). After a 2- or 4-d washout period, the same NHPs were administered the next higher dose (30 mg/kg). This cycle was continued at the next dose (100 mg/kg) until an MTD was achieved (200 mg/kg). After the observation period for the last dose, the NHPs went through a 7-d rest/washout period before they were assigned to phase B. In phase B, all NHPs were treated with compound 2 (200 mg/kg) for 2 d and then reduced to 100 mg/kg (due to emesis at the MTD) for the remaining 5 consecutive d of the study. At the conclusion of the study, the MTD after single-dose administration was detemined, the potential toxicity was characterized, toxicokinetic parameters were calculated, and target tissues of compound 2 were identified.
Acute single-dose escalation and time course efficacy studies in NHPs
During the dose escalation (phase A), blood was collected at 4 h after administration at each dose (0, 10, 30, 100, and 200 mg/kg) from femoral, cephalic, or saphenous veins into tubes containing K3EDTA, processed to plasma, and then stored frozen at or below −60°C. During the repeat dosing (phase B), blood samples were collected on days 1 and 7 at predose (0 h), 1, 2, 4, 8, and 24 h after dose. Levels of Aβ38, Aβ40, and Aβ42 peptides were quantitated in plasma samples using Meso Scale Sector 6000 Multiplex assays as described previously (Kounnas et al., 2010).
Statistical analysis
All experiments were performed blind coded. Values in the figures are expressed as means ± SEM. To determine the statistical significance, values were compared using ANOVA or Student’s t test. The differences were considered to be significant if P values were <0.05. Data were analyzed using GraphPad Prism (GraphPad Software).
Online supplemental material
Table S1 depicts the dose-dependent drug level exposures in plasma and brain of C57BL/6J mice following a 9-d treatment course (po) of either compound 2 or compound 3 and the corresponding brain/plasma ratios. Table S2 details the repeat-dose PD (effects on Aβ38, Aβ40, and Aβ42 peptides and the drug levels) for compound 2 in plasma, brain, and CSF of Sprague-Dawley rats following a 9-d treatment course (po) at doses of 5, 25, and 50 mg/kg. Table S3 depicts the metabolite profile of compound 2 in hepatocyte cultures of mouse, rat, dog, and monkey following 4-h incubation at a concentration of 1 µM. Table S4 details the single-dose i.v./po PK parameters for compounds 1, 2, and 3 in male CD-1 mice. Table S5 depicts the PK parameters of compound 2 in mice, rats, and NHPs.
Acknowledgments
We acknowledge SRI International for support in conducting the pharmacokinetic and repeat-dose toxicological studies. We also thank Albany Molecular Research International for support in medicinal chemistry and analytical chemistry and in conducting the in vitro absorption, distribution, metabolism, excretion, and toxicity screening. We acknowledge the vital contribution of Dr. Charles Cywin of the National Institute of Neurological Disorders and Stroke for guidance and insights throughout the course of this research program.
This work was supported by the Cure Alzheimer’s Fund, the National Institute of Neurological Disorders and Stroke, and the National Institute on Aging; National Institutes of Health/National Institute of Neurological Disorders and Stroke Blue Print and National Institute on Aging, U01 grants NS 074501 and U01AG048986, R01AG055523, R01AG054223, and R01AG056061. We also acknowledge support from the Chen Foundation (R-86U55A).
Author contributions: K.D. Rynearson designed medicinal chemistry, designed experiments, analyzed data, and wrote the manuscript. O. Prikhodko, Y. Xie, C. Zhang, P. Nguyen, B. Hug, M. Sawa, A. Becker, J. Florio, M. Mante, and B. Salehi performed the animal experiments analyzing drug effects. M. Ponnusamy, B. Spencer, and C. Arias carried out the histological analyses and performed the microscopic quantitation. D. Galasko helped to analyze the acute and subchronic efficacy biomarker experiments and wrote aspects of the manuscript. G. Johnson helped with the design of the medicinal chemistry and wrote portions of the manuscript. J.H. Lin designed the pharmacokinetic studies, analyzed the data, and wrote portions of the manuscript. S.K. Duddy designed the toxicological experiments, analyzed the data, and wrote portions of the manuscript. B.P. Head, G. Thinakaran, R.A. Rissman, W.C. Mobley, and R.E. Tanzi designed the chronic pathological biomarker studies and wrote aspects of the manuscript. S.L. Wagner supervised the entire project and wrote the manuscript.

