


Highly sensitive and specific plasma biomarkers for Alzheimer’s disease (AD) have the potential to improve diagnostic accuracy in the clinic and facilitate research studies including enrollment in prevention and treatment trials. We recently reported CSF tau hyperphosphorylation, especially on T217, is an accurate predictor of β-amyloidosis at asymptomatic and symptomatic stages. In the current study, we determine by mass spectrometry the potential utility of plasma p-tau isoforms to detect AD pathology and investigate CSF and plasma tau isoforms’ profile relationships. Plasma tau was truncated as previously described in CSF. CSF and plasma measures of p-tau-217 and p-tau-181 were correlated. No correlation was found between CSF and plasma on total-tau levels and pS202 measures. We found p-tau-217 and p-tau-181 were highly specific for amyloid plaque pathology in the discovery cohort (n = 36, AUROC = 0.99 and 0.98 respectively). In the validation cohort (n = 92), p-tau-217 measures were still specific to amyloid status (AUROC = 0.92), and p-tau-181 measures were less specific (AUROC = 0.75).

Introduction
Tests of central nervous system (CNS) proteins found in blood plasma are being developed to enable simplified and inexpensive testing compared with the current gold standards of cerebrospinal fluid (CSF) and brain positron emission tomography (PET) imaging in Alzheimer’s disease (AD) and other neurodegenerative diseases. Brain and CSF proteins are transferred across the blood brain barrier and arachnoid granulations to the blood (Roberts et al., 2014), where the CNS proteins are diluted in a complex mixture of other biomolecules. Over the past few years, different studies have demonstrated CNS disease-associated protein alterations could be detected in blood. However, low amounts within a complex matrix, peripheral alterations, and peripheral expression of corresponding proteins may reduce the accuracy of the biomarker compared with its measurement in CSF.
Blood plasma amyloid-β (Aβ) 42/40 ratio recapitulates with accuracy change on Aβ42/40 ratio detected in CSF and associates with measures of brain amyloid plaques by amyloid PET scans (Ovod et al., 2017; Nakamura et al., 2018; Schindler et al., 2019). Neurofilament light (NfL) chain protein can be detected in blood and tracks neuronal damage in several neurological diseases (Bacioglu et al., 2016) similarly to CSF NfL (Preische et al., 2019). NfL changes are detected around the time of symptom onset, over a decade after abnormal AD amyloidosis is detectable by brain imaging or CSF Aβ 42/40 (Bateman et al., 2012; Fagan et al., 2014; Preische et al., 2019).
Tau is the second marker of AD pathology. Tau is a microtubule-binding protein that is increased and phosphorylated in AD and constitutes the main component in AD tangle and neurite pathology. Total tau (t-tau) and some phosphorylated-tau (p-tau) isoform levels are significantly increased in AD CSF. However, plasma tau and CSF tau levels poorly correlate with each other, creating a challenge in developing plasma tau as a biomarker for AD (Zetterberg et al., 2013; Mattsson et al., 2016). Recent reports using immunoassays have suggested more promising developments; for example, some reports indicated slight increases in plasma t-tau in mild cognitive impairment (MCI) and AD (Mielke et al., 2017, 2018), and several studies demonstrated plasma p-tau at threonine 181 (p-tau-181) increases in AD at MCI and moderate stages (Tatebe et al., 2017; Mielke et al., 2018). Blood p-tau-181 can differentiate AD patients from other tauopathies at symptomatic stages of AD with accuracy (Janelidze et al., 2020a; Thijssen et al., 2020).
Our laboratory developed mass spectrometry (MS) measures to accurately quantify both t-tau and multiple p-tau isoforms. Using this approach, we previously identified that certain p-tau/t-tau ratios are specifically increased in AD (Barthélemy et al., 2019, 2020a). Remarkably, we have reported that CSF tau phosphorylation measures on threonine 217 (p-tau-217) are closely associated with amyloidosis, improving identification of amyloidosis at the asymptomatic stage (Barthélemy et al., 2015, 2017,Preprint, 2018). CSF hyperphosphorylation of p-tau-T217 is more accurate than other sites, such as T181 (Barthélemy et al., 2020b; Janelidze et al., 2020b) and T205 (Barthélemy et al., 2020a), to detect the presence of amyloid plaques. Phosphorylation occupancy on T217 is also lower intracellularly than extracellularly in CSF (Barthélemy et al., 2019). Together, these findings suggest that an increase of p-tau-217 in both normal and AD CSF would be related to selective release of this isoform from the CNS to CSF and that increased p-tau-217 release is closely related to amyloid plaques. We hypothesized p-tau-217 is specific to CNS, would cross the blood brain barrier, specifically contributes to plasma level, and increases together with CSF changes. In this study, we sought to quantify blood plasma tau and p-tau species by MS, especially p-tau-217, to compare with AD pathology and assess their potential as blood-based AD biomarkers.
Results and discussion
Tau isoforms can be purified and enriched from plasma
We designed MS assays to assess plasma tau isoforms, including plasma p-tau-217. Estimated concentrations of t-tau (1–20 pg/ml; Zetterberg et al., 2013; Mattsson et al., 2016; Mielke et al., 2017) and p-tau-181 (sub pg/ml; Tatebe et al., 2017), as reported by immunoassays in plasma, are low and present a challenge for measuring plasma tau using MS techniques. Moreover, p-tau-217 in CSF is approximately five times less abundant than p-tau-181 (Barthélemy et al., 2019). The sub pg/ml range estimated for p-tau-217 measurement is far below the concentration of currently monitored plasma biomarkers (Geyer et al., 2017), including the recently assayed plasma Aβ42 peptide using MS (Ovod et al., 2017; Nakamura et al., 2018).
To overcome MS limitations in sensitivity (low attomolar range), we designed an enrichment protocol to purify and concentrate plasma tau from 20 ml of plasma to 25 µl of final extract, leading to an enrichment factor of ∼800 times. This protocol relies on an initial step of plasma protein precipitation using perchloric acid to remove a majority of plasma proteins such as albumin and immunoglobulins. Soluble tau in the supernatant was concentrated using solid phase extraction as previously reported (Barthélemy et al., 2016). The pellet obtained after drying the solid phase extract was subsequently immunopurified against N-terminus and mid-domain tau antibodies as described previously (Sato et al., 2018). This multiple-step extraction significantly decreased plasma interference on minor phosphorylated peptide signals. Both plasma and CSF were analyzed for t-tau and p-tau peptides by a highly sensitive and resolute mass spectrometer equipped with nano-flow capillary liquid chromatography interfaced with nano-electrospray ionization (Barthélemy et al., 2019).
Plasma tau is truncated similar to CSF profiles
We used nano liquid chromatography coupled to tandem mass spectrometry (nanoLC-MS/MS) to assay plasma samples from a cohort of 34 participants enrolled during our tau stable isotope labeling kinetics (SILK) study and characterized samples for their plasma and CSF tau isoforms profile. The tau SILK protocol, previously designed for measuring the half-life of tau (Sato et al., 2018), includes the collection of large volume of blood at the time of the tracer infusion. Among all investigated samples, 15 tau peptides from residues 6–254 were detected including 0N-, 1N-, 2N-, and 3R-specific peptides. Inferred abundance of 0N/1N/2N peptides indicated similar contributions to what was previously reported in brain and CSF (∼5/5/1, respectively; Sato et al., 2018). Notably, no peptides after residues 254 (except a 3R peptide found in low abundance at residues equivalent to 306–317 residues in 2N4R tau) were detected, suggesting plasma tau does not contain detectable levels of full-length tau. Plasma tau peptide abundances dropped significantly after residue 221 (Fig. 1) as previously reported in CSF (Barthélemy et al., 2016; Sato et al., 2018; Cicognola et al., 2019), suggesting a similar tau truncation pattern in plasma as reported after release by neuronal cells (Sato et al., 2018).

Plasma tau truncation profile after chemical extraction and IP in the discovery cohort. Left: Plasma tau peptides concentration profiles obtained from the 36 individuals. Each line corresponds to the peptide profile from one participant. Right: Dots represents averaged tau peptides normalized concentration obtained from the overall cohort. Normalized concentration for each peptide is relative to the sum of the peptides concentrations measured in each participant. Bars represent SD. Label a indicates a decrease of 2N and 1N+2N peptide abundance consistent with 5/5/1 0N/1N/2N contribution in plasma tau. Label b indicates a decrease consistent with the presence of around 10% of phosphorylation on position 181. Phosphorylation on T181 induces a trypsin missed cleavage between residues 180 and –181. This contributes to a decrease of 175–180 and 181–190 peptides abundance proportional to the extent of phosphorylation on T181. Label c indicates a decrease consistent with tau truncation between residues 221 and 226. Cicognola et al. (2019) have reported CSF tau main cleavage occurring at residue 224. Label d indicates a decrease consistent with progressive C terminus degradation of plasma tau from residue 224 to microtubule binding region upstream region.
Plasma tau truncation profile after chemical extraction and IP in the discovery cohort. Left: Plasma tau peptides concentration profiles obtained from the 36 individuals. Each line corresponds to the peptide profile from one participant. Right: Dots represents averaged tau peptides normalized concentration obtained from the overall cohort. Normalized concentration for each peptide is relative to the sum of the peptides concentrations measured in each participant. Bars represent SD. Label a indicates a decrease of 2N and 1N+2N peptide abundance consistent with 5/5/1 0N/1N/2N contribution in plasma tau. Label b indicates a decrease consistent with the presence of around 10% of phosphorylation on position 181. Phosphorylation on T181 induces a trypsin missed cleavage between residues 180 and –181. This contributes to a decrease of 175–180 and 181–190 peptides abundance proportional to the extent of phosphorylation on T181. Label c indicates a decrease consistent with tau truncation between residues 221 and 226. Cicognola et al. (2019) have reported CSF tau main cleavage occurring at residue 224. Label d indicates a decrease consistent with progressive C terminus degradation of plasma tau from residue 224 to microtubule binding region upstream region.
Tau phosphorylated peptides are identified in plasma extracts by MS
Phosphorylated peptides on T181, S202, and T217 from the tau mid-domain were quantified in all plasma extracts. For samples with the lowest abundance signals for the phosphorylated peptides, liquid chromatography (LC)–MS signals obtained for p-tau-181, p-tau-202, and p-tau-217 were approximately seven, three, and two times above the lower limit of quantification (respectively at 0.2, 0.3, and 0.05 pg/ml). Phosphorylated peptide at T217 was quantified in controls at an average concentration of 0.13 pg/ml (28 amol/ml; Table 1). To our knowledge, this is the lowest concentration ever measured by MS for a protein marker in human plasma. Phosphorylated T205 peptide was inconsistently detected in 19 of the 34 samples below the lower limit of quantification (0.3 pg/ml) and was not included in further analyses. Other low abundant phosphorylated sites previously reported by MS in CSF (Barthélemy et al., 2019) at T153, T175, S199, S208, S214, and T231 were not detected in the plasma extracts.
Demographics and biomarkers values
| 20-ml tau SILK discovery cohort (n = 34) | 4-ml Aβ SILK validation cohort (n = 92) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Young controls | Aged controls | Non-AD MCI | Preclinical AD | AD-MCI | AD-moderate | Aged controls | Non-AD MCI | Preclinical AD | AD-MCI | AD-moderate |
| n | 9 | 8 | 2 | 5 | 8 | 2 | 31 | 11 | 20 | 24 | 6 |
| Age, yr | 44 (12) | 74 (5) | 71 (3) | 75 (5) | 81 (7) | 75 (2) | 73 (5) | 75 (8) | 74 (6) | 76 (6) | 74 (8) |
| Gender (F/M) | 6/3 | 5/3 | 2/0 | 1/4 | 5/3 | 2/0 | 16/15 | 2/9 | 10/10 | 11/13 | 1/5 |
| CDR | 0 | 0 | 0.5 | 0 | 0.5 | 1 | 0 | 0.5 | 0 | 0.5 | 1 |
| CSF42/40 | 0.12 (0.01) | 0.12 (0.02) | 0.13 (0.01) | 0.07 (0.01)**** | 0.06 (0.01)**** | 0.07 (0.01)*** | 0.18 (0.02)(20) | 0.18 (0.02)(11) | 0.10 (0.02)(11)**** | 0.10 (0.02)(18)**** | 0.10 (0.03)(6)**** |
| AV45 centiloid | na | 0.95 (0.12)(7) | 0.80 (0.00)(1) | 1.98 (0.41)(4)** | 2.65 (0.38)(5)** | 2.30 (0.30)(2)ns | 1.08 (0.33)(21) | 0.85(1) | 1.86 (0.52)(13)**** | 2.20 (0.56)(8)**** | 2.41(1) |
| PiB SUVR | na | 0.05 (0.04)(4) | 0.06 (0.00)(1) | 0.44 (0.11)(4)* | 0.51 (0.12)(4)* | 0.52 (0.00)(1) | 1.08 (0.17)(25) | 0.96 (0.25)(4) | 2.17 (0.83)(16)**** | 2.92 (0.75)(11)**** | na |
| Amyloid status | Negative | Negative | Negative | Positive | Positive | Positive | Negative | Negative | Positive | Positive | Positive |
| AV1451 SUVR | na | 1.19 (0.09)(6) | 1.30 (0.06)(2) | 1.26 (0.15)(5)ns | 1.79 (0.14)(5)** | 2.36 (0.50)(2)ns | na | na | na | na | na |
| CSF | |||||||||||
| t-tau (ng/ml) | 2.11 (0.15) | 2.22 (0.16) | 2.03 (0.07)ns | 2.73 (0.61)** | 2.90 (0.62)*** | 4.27 (1.71)* | 2.21 (0.58) | 2.13 (0.09)ns | 2.81 (0.95)** | 3.28 (0.94)**** | 3.58 (1.60)ns |
| p-tau-217/T217 (%) | 2.6 (0.2) | 2.7 (0.3) | 2.7 (0.8)ns | 7.0 (1.3)**** | 8.7 (1.9)**** | 9.3 (0.8)* | 2.0 (0.5) | 2.0 (0.4)ns | 5.9 (2.6)**** | 7.8 (2.0)**** | 9.2 (2.1)**** |
| p-tau-217 level (pg/ml) | 54 (5) | 59 (5) | 56 (20)ns | 187 (42)**** | 247 (58)**** | 395 (121)* | 44 (16) | 43 (16)ns | 184 (159)**** | 248 (117)**** | 355 (206)**** |
| p-tau-181/T181 (%) | 13.5 (0.9) | 13.3 (0.8) | 14.2 (1.5)ns | 16.0 (2.0)** | 17.5 (1.9)**** | 15.9 (0.2)* | 15.6 (0.8) | 14.7 (1.0) | 19.7 (3.0)**** | 21.0 (2.1)**** | 20.7 (3.0)**** |
| p-tau-181 level (pg/ml) | 286 (33) | 295 (30) | 281 (44)ns | 438 (119)*** | 501 (93)**** | 681 (263)* | 339 (105) | 310 (96) | 574 (282)*** | 688 (241)**** | 777 (420)* |
| pS202/S202 (%) | 2.5 (1.2) | 2.0 (0.5) | 3.3 (1.1)ns | 2.1 (0.7)ns | 1.8 (0.6)ns | 1.9 (0.2)ns | na | na | na | na | na |
| Plasma | |||||||||||
| t-tau (pg/ml) | 23.3 (4.9) | 22.9 (3.3) | 18.4 (2.1)ns | 27.8 (9.6)ns | 29.8 (12.8)ns | 35.3 (10.3)ns | 20.4 (6.3) | 21.2 (5.2) | 22.0 (5.2) | 22.7 (6.9) | 23.5 (7.4) |
| p-tau-217/T217 (%) | 0.6 (0.1) | 0.6 (0.2) | 0.7 (0.1)ns | 2.0 (0.7)**** | 2.7 (1.0)**** | 4.2 (0.9)** | 0.4 (0.2) | 0.4 (0.2)ns | 1.2 (1.1)**** | 1.3 (0.7)**** | 2.2 (1.2)**** |
| p-tau-217 level (pg/ml) | 0.13 (0.02) | 0.15 (0.04) | 0.13 (0.01)ns | 0.52 (0.17)**** | 0.82 (0.52)**** | 1.57 (0.7)** | 0.07 (0.03) | 0.09 (0.02)ns | 0.26 (0.25)**** | 0.31 (0.19)**** | 0.58 (0.50)**** |
| p-tau-181/T181 (%) | 6.8 (1.5) | 8.9 (1.5) | 8.5 (1.3)ns | 12.5 (2.5)*** | 13.8 (1.8)**** | 15.1 (1.3)** | 8.5 (1.9) | 7.8 (1.0)ns | 9.7 (2.3)ns | 10.0 (2.0)** | 10.6 (1.5)* |
| p-tau-181 level (pg/ml) | 1.6 (0.6) | 2.0 (0.4) | 1.6 (0.1)ns | 3.4 (0.9)** | 4.3 (2.3)*** | 5.5 (2.0)** | 2.1 (0.7) | 2.3 (0.9)ns | 2.7 (1.2)ns | 2.9 (1.0)*** | 3.4 (1.5)* |
| pS202/S202 (%) | 4.0 (1.0) | 5.3 (1.1) | 3.9 (0.1)ns | 4.8 (1.4)ns | 4.4 (1.3)ns | 3.7 (0.4)ns | na | na | na | na | na |
Data are shown as mean (SD). Superscript parenthetical numbers, e.g., (7), indicate the number of available measures into the group; each participant has at least one measure (amyloid PET imaging or CSF42/40) used to define amyloid status. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. na, not available; ns, not significant per the Mann–Whitney test against control groups; F, female; M, male; PiB, Pittsburgh compound B.
Plasma p-tau-217 and p-tau-181 correlate with CSF tau isoforms changes
No correlation was found between CSF and plasma t-tau levels as previously reported by immunoassays (Zetterberg et al., 2013; Mattsson et al., 2016). However, a high correlation was found between CSF and plasma p-tau-217 measures (Fig. 2 A and Table 2; p-tau-217 absolute level and pT217/T217 ratio, Spearman rho 0.78 and 0.78, respectively, for all cohorts). Also, a significant correlation was found between CSF and plasma for p-tau-181 measures as previously reported (Janelidze et al., 2020a; Thijssen et al., 2020; Table 2; p-tau-181 absolute level and pT181/T181 ratio, Spearman rho 0.68 and 0.68, respectively, for all cohorts). No correlation was found between CSF and plasma measures of pS202. All significant CSF/plasma correlations were mainly driven by amyloid-positive participant values since no correlations were found in amyloid-negative groups (not shown). Consistently, plasma p-tau-217 and p-tau-181 measures recapitulated separations obtained in CSF between amyloid-positive and -negative participants regardless of their cognitive status (Fig. 1 and Table 2; area under the curve (AUC) in CSF and plasma respectively 1.00 and 0.98 for pT217/T217; 0.95 and 0.98 for p-T181/T181). Together, these data suggest that plasma p-tau-217 and p-tau-181 can act as proxies for changes in CNS-soluble tau and thus serve as useful biomarkers.

Plasma tau and p-tau changes across groups in Tau SILK discovery cohort. (A) Measures of p-tau-217/T217 ratios in plasma and CSF are highly correlated in both entire cohort and CSF p-tau-217 positive subgroup. Spearman correlations and associated P value are shown. (B–E) Separation between amyloid-negative and -positive groups are calculated using AUROC. (B and C) Consistent with CSF measurement, plasma p-tau-217/T217 ratio and p-tau-217 level distinguish amyloid-negative from amyloid-positive groups regardless of the cognitive status. Amyloid-negative groups with high CSF p-tau-217 were also separated from other amyloid-negative groups. (D) Plasma tau level is not a biomarker for amyloid status and AD dementia. (E and F) Plasma p-tau-181/T181 ratio and p-tau-181 level increase in amyloid-positive groups. YNC, young normal controls; AMC, aged-matched controls.
Plasma tau and p-tau changes across groups in Tau SILK discovery cohort. (A) Measures of p-tau-217/T217 ratios in plasma and CSF are highly correlated in both entire cohort and CSF p-tau-217 positive subgroup. Spearman correlations and associated P value are shown. (B–E) Separation between amyloid-negative and -positive groups are calculated using AUROC. (B and C) Consistent with CSF measurement, plasma p-tau-217/T217 ratio and p-tau-217 level distinguish amyloid-negative from amyloid-positive groups regardless of the cognitive status. Amyloid-negative groups with high CSF p-tau-217 were also separated from other amyloid-negative groups. (D) Plasma tau level is not a biomarker for amyloid status and AD dementia. (E and F) Plasma p-tau-181/T181 ratio and p-tau-181 level increase in amyloid-positive groups. YNC, young normal controls; AMC, aged-matched controls.
Abnormal phosphorylation status detected in AD CSF tau (Aβ+) is recapitulated in plasma tau
| n | t-tau | pT217/T217 | p-tau-217 level | pT181/T181 | p-tau-181 level | pS202/S202 | |
|---|---|---|---|---|---|---|---|
| Tau SILK discovery cohort | |||||||
| AUROC | |||||||
| CSF Aβ− vs. Aβ+ | 19 vs. 15 | 0.95**** [0.88–1.00] | 1.00**** [1.00–1.00] | 1.00**** [1.00–1.00] | 0.95**** [0.89–1.00] | 0.98**** [0.95–1.00] | 0.72* [0.55–0.90] |
| Plasma Aβ− vs. Aβ+ | 19 vs. 15 | 0.67ns | 0.98**** [0.94–1.00] | 0.99**** [0.97–1.00] | 0.98**** [0.95–1.00] | 0.95**** [0.89–1.00] | 0.58ns |
| Correlation, Spearman r | |||||||
| CSF vs. plasma: all | 34 | 0.23ns | 0.78**** [0.59–0.88] | 0.78**** [0.59–0.89] | 0.68**** [0.43–0.83] | 0.68**** [0.44–0.83] | −0.05ns |
| CSF vs. plasma: Aβ+ | 15 | 0.29ns | 0.71** [0.31–0.89] | 0.39ns [−0.16–0.76] | 0.54* [0.02–0.82] | 0.19ns | 0.04ns |
| Percent difference relative to controlsa | |||||||
| CSF | |||||||
| All Aβ+ vs. controls | 15 vs. 17 | 188 (110)ns | 215 (31)ns | 791 (110)**** | 25 (18)**** | 252 (110) | −14 (57)**** |
| Aβ+/CDR = 0 vs. controls | 5 vs. 17 | 112 (90)ns | 167 (27)ns | 450 (84)*** | 19 (19)* | 154 (97) | −6 (59)* |
| Aβ+/CDR = 0.5 vs. controls | 8 vs. 17 | 146 (83)ns | 235 (30)ns | 683 (71)**** | 30 (18)**** | 213 (79) | −19 (57)**** |
| Plasma | |||||||
| All Aβ+ vs. controls | 15 vs. 17 | 29 (58)**** | 342 (68)**** | 506 (95)**** | 74 (39)** | 128 (80) | −4 (55)**** |
| Aβ+/CDR = 0 vs. controls | 5 vs. 17 | 20 (53)ns | 228 (56)** | 285 (56)*** | 60 (43)ns | 86 (58) | −5 (55)ns |
| Aβ+/CDR = 0.5 vs. controls | 8 vs. 17 | 29 (61)ns | 282 (73)**** | 379 (93)**** | 78 (36)ns | 136 (85) | −4 (54)** |
| CSF/plasma ratio | |||||||
| All cohort | 34 | 102 (31) | 4.1 (1.2) | 418 (197) | 1.6 (0.5) | 160 (62) | 0.5 (0.2) |
| Aβ− | 19 | 99 (22) | 4.6 (1.2) | 435 (101) | 1.8 (0.5) | 178 (57) | 0.6 (0.3) |
| Aβ+ | 15 | 106 (39) | 3.6 (1.3) | 396 (272) | 1.3 (0.2) | 136 (60) | 0.5 (0.2) |
| Aβ SILK validation cohort | |||||||
| AUROC | |||||||
| CSF | |||||||
| All Aβ− vs. Aβ+ | 42 vs. 50 | 0.78**** [0.68–0.88] | 1.00**** [0.99–1.00] | 0.96**** [0.92–1.00] | 0.97**** [0.93–1.00] | 0.87**** [0.80–0.95] | |
| Aβ− vs. Aβ+ CDR0 | 31 vs. 19 | 0.74** [0.59–0.89] | 0.99**** [0.98–1.00] | 0.96**** [0.92–1.00] | 0.92**** [0.81–1.00] | 0.82*** [0.68–0.95] | |
| Aβ− vs. Aβ+ CDR0.5 | 11 vs. 22 | 0.74** [0.59–0.89] | 1.00**** [1.00–1.00] | 0.99**** [0.97–1.00] | 1.00**** [1.00–1.00] | 0.95*** [0.89–1.00] | |
| Plasma | |||||||
| Plasma Aβ− vs. Aβ+ | 42 vs. 50 | 0.59ns [0.47–0.71] | 0.92**** [0.87–0.98] | 0.93**** [0.87–0.98] | 0.75**** [0.65–0.86] | 0.72*** [0.61–0.82] | |
| Aβ− vs. Aβ+ CDR0 | 31 vs. 20 | 0.61ns [0.45–0.77] | 0.86**** [0.73–0.99] | 0.86****[0.74–0.99] | 0.66ns | 0.67ns | |
| Aβ− vs. Aβ+ CDR0.5 | 11 vs. 24 | 0.50ns [0.30–0.71] | 0.95**** [0.87–1.00] | 0.93****[0.85–1.00] | 0.86*** [0.74–0.98] | 0.68ns | |
| Correlation, Spearman r | |||||||
| CSF vs. plasma: all | 92 | 0.03ns | 0.79**** [0.70–0.86] | 0.70**** [0.57–0.79] | 0.45**** [0.27–0.60] | 0.21ns [−0.01 to 0.41] | |
| CSF vs. plasma: Aβ+ | 50 | 0.03ns | 0.59**** [0.36–0.75] | 0.25ns | 0.22ns | 0.08ns | |
| Aβ PET vs. plasma: all | 66 | −0.02ns [−0.26 to 0.23] | 0.70**** [0.54–0.81] | 0.67**** [0.51–0.79] | 0.45*** [0.22–0.63] | 0.26* [0.00–0.48] | |
| Aβ PET vs. plasma: Aβ+ | 34 | −0.20ns [−0.51 to 0.16] | 0.38* [0.04–0.64] | 0.30ns [−0.05–0.59] | 0.25ns [−0.12–0.55] | −0.04ns [−0.39 to 0.32] | |
Data are shown as mean (SD) for % of increase compared to control value and ratio and as value (95%, CI) for AUROC and Spearman r. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. ns, not significant per the Mann–Whitney test against aged and normal control groups (amyloid-negative participants without cognitive symptoms). Numbers in brackets indicate confidence intervals for AUROC.
Statistical significance (t test using the linear step-up procedure of Benjamini, Krieger, and Yekutieli) of the change compared to p-tau-181 level change as reference.
Changes in plasma p-tau-217 have a larger dynamic range than p-tau-181 in AD
We calculated the magnitude of change in plasma tau isoforms between controls and different amyloid clinical groups (Table 2). In CSF, the highest difference in amplitude between amyloid-positive individuals and controls was found for the level of p-tau-217 (+800%) group followed by the p-tau-181 (+250%) group. However, these changes were partially due to the concomitant contributions of CSF tau isoform increases as measured for t-tau level (+190%). When normalized from tau variation using the p-tau/t-tau ratio, pT217/T217 demonstrated a greater change than pT181/T181 (+220% vs. +25%). In plasma, the high magnitude of increase for p-tau-217 in the amyloid-positive group remained higher than p-tau-181 measurements in all of the clinical groups (from +230% to +340% for pT217/T217 and from +60% to +80% for pT181/T181). Importantly, CSF and plasma p-tau-217 measures (Table 1 and Fig. 2, B and C) distinguished amyloid-positive, tau PET–negative participants from controls. This suggests that p-tau biomarkers are changed before detectable tau aggregation and reflect abnormal soluble tau metabolism occurring concomitantly with brain Aβ pathology.
Plasma p-tau modifications are detected in a validation cohort using lower plasma volume
To validate the discovery results in a larger cohort, we modified the plasma extraction method to downscale the volume of plasma needed to 4 ml. This assay provided sufficient sensitivity and reproducibility for measuring endogenous plasma p-tau levels (Fig. S1 and Table S1). We measured CSF and plasma for t-tau, p-tau-181, and p-tau-217 in 92 participants enrolled in Aβ SILK studies and selected according to their amyloid and cognitive status (Patterson et al., 2015; Ovod et al., 2017). Measures of p-tau-217 remained specific to amyloid status (Fig. 3, B and C; and Table 2; area under receiver operating curve [AUROC] 0.92 and 0.93 for ratio and level, respectively) and correlated with CSF (Fig. 3 A and Table 2; r = 0.79 and 0.70) and PET amyloid (Table 2; r = 0.70 and 0.67). PT217/T217 provided reasonable separation of amyloid-positive and -negative groups cognitively unimpaired (AUC 0.86). The p-tau-181 measures were less specific than in the discovery cohort in differentiating amyloid groups (Fig. 3, E and F; AUROC 0.75 and 0.72 for ratio and level, respectively). Again, no significant increase of plasma t-tau was observed (Fig. 3 D). For this larger cohort, the better performance for amyloid detection of p-tau-217 measures over p-tau-181 observed in CSF (Fig. 3 G) was recapitulated in plasma (Fig. 3 H). Plasma pT217/T217 was inversely correlated with CSF Aβ 42/40 as expected with normal Aβ levels associated with low pT217 phosphorylation (Fig. 3 I).

Spike and recovery experiment assessing assay performance using 4 ml of plasma volume. Plasma pool or 5% solution of recombinant HSA were spiked with increased volumes of AD or non-AD CSF pools. Plasma was extracted using the 4-ml protocol described in the Materials and methods. HSA samples were only immuno-purified (IP) before digestion and analysis.
Spike and recovery experiment assessing assay performance using 4 ml of plasma volume. Plasma pool or 5% solution of recombinant HSA were spiked with increased volumes of AD or non-AD CSF pools. Plasma was extracted using the 4-ml protocol described in the Materials and methods. HSA samples were only immuno-purified (IP) before digestion and analysis.
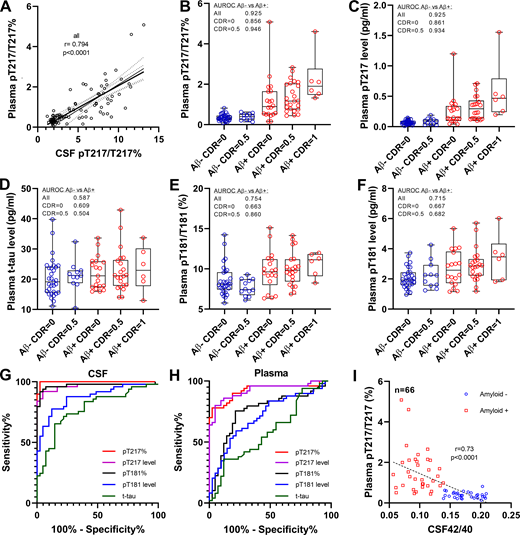
Plasma tau and p-tau changes across groups in Aβ SILK validation cohort. (A) Phosphorylation occupancies on T217 in plasma and CSF correlate. Spearman correlations and associated P value are shown. (B and C) As found in the discovery cohort, plasma p-tau-217/T217 ratio and p-tau-217 level distinguish amyloid-negative from amyloid-positive groups regardless of the cognitive status. Amyloid-negative with high CSF p-tau-217 were also separated from other amyloid-negative groups. Separations between groups are calculated using AUROC. (D) Plasma tau level is not a biomarker for amyloid status and AD dementia. (E and F) Plasma p-tau-181/T181 ratio and p-tau-181 level increase in amyloid-positive groups but are less accurate than p-tau-217 measures to detect abnormal tau phosphorylation. (G and H) Receiver operating characteristic curves discriminating amyloid-positive from amyloid participants using CSF and plasma tau measures. Corresponding AUCs are summarized in Table 2. AUC comparison between p-tau-217 and p-tau-181 measures is described in Table S3. (I) Plasma p-tau-217/T217 ratio associates with Aβ42/40 ratio measured in CSF.
Plasma tau and p-tau changes across groups in Aβ SILK validation cohort. (A) Phosphorylation occupancies on T217 in plasma and CSF correlate. Spearman correlations and associated P value are shown. (B and C) As found in the discovery cohort, plasma p-tau-217/T217 ratio and p-tau-217 level distinguish amyloid-negative from amyloid-positive groups regardless of the cognitive status. Amyloid-negative with high CSF p-tau-217 were also separated from other amyloid-negative groups. Separations between groups are calculated using AUROC. (D) Plasma tau level is not a biomarker for amyloid status and AD dementia. (E and F) Plasma p-tau-181/T181 ratio and p-tau-181 level increase in amyloid-positive groups but are less accurate than p-tau-217 measures to detect abnormal tau phosphorylation. (G and H) Receiver operating characteristic curves discriminating amyloid-positive from amyloid participants using CSF and plasma tau measures. Corresponding AUCs are summarized in Table 2. AUC comparison between p-tau-217 and p-tau-181 measures is described in Table S3. (I) Plasma p-tau-217/T217 ratio associates with Aβ42/40 ratio measured in CSF.
Peripheral tau phosphorylation status is different from both control and AD CNS tau
The absence of correlation between CSF and plasma t-tau levels (Table 2) suggests that the main origin of plasma t-tau is from peripheral sources, not the CNS. From previous reports, plasma t-tau levels reflect CNS tau changes only when significantly increased compared with baseline, for example, in patients with acute stroke, brain injury (Bulut et al., 2006; Neselius et al., 2013; Bogoslovsky et al. 2017; Rubenstein et al., 2017), brain metastases, (Darlix et al., 2019) and likely AD patients with high CNS t-tau release (Kasai et al., 2017; Mielke et al., 2018). Thus, tau released in the CNS would contribute to significant plasma t-tau increases only when its contribution becomes much higher than the peripheral tau contribution.
The higher contribution of peripheral tau over CNS tau in plasma is also supported by the difference observed in p-tau/tau ratios between CSF and plasma (Table 2). Both pT217/T217 and pT181/T181 ratios are significantly decreased in plasma compared with CSF. This observation supports the dilution of CNS tau in peripheral tau, with much lower p-tau-217 and slightly lower p-tau-181 abundance (Table 2, a decrease of 4.6 and 1.8 times, respectively) in peripheral tau compared with CSF from amyloid-negative controls. For p-tau-202, the significant increase of pS202/S202 ratio in plasma (1.7 times) suggests that peripheral tau is more phosphorylated at this site than in CSF. We consistently found that the less the site is phosphorylated in peripheral tau compared with normal CSF, the greater the CNS tau change would impact plasma levels for the corresponding tau isoform. This may explain why CSF and plasma p-tau-217 levels better correlate than p-tau-181 levels and the absence of correlation found for pS202.
Finally, by hypothesizing that peripheral tau does not contain significant amounts of p-tau-217, we estimated that CNS tau contributes to ∼20% of the overall plasma t-tau level in controls. Assuming CNS tau release could increase by three times in AD (Fagan et al., 2007; Zetterberg, 2017), a higher CNS AD tau contribution is unlikely to increase AD plasma t-tau levels by >40%. Moreover, peripheral tau levels could be affected by different biological factors, potentially interfering with CNS tau contributions to changes in plasma. This important contribution of peripheral tau on plasma tau isoforms is a potential source of interference for assessing effects of tau-directed therapies using plasma tau biomarkers.
Together, these findings support the use of more specific CNS tau isoforms such as p-tau-217 and p-tau-181 over t-tau or p-tau-202 to overcome the contribution of peripheral tau and to monitor CNS tau in plasma. Plasma measures of p-tau-181 have been reported to significantly increase in AD particularly at symptomatic stages, detecting AD with good accuracy (Mielke et al., 2018; Janelidze et al., 2020a; Thijssen et al., 2020). Our results suggest that, as reported in CSF (Barthélemy et al., 2020a; Barthélemy et al., 2020b; Janelidze et al., 2020b), p-tau-217 in plasma would be more accurate than p-tau-181 for detecting abnormal CNS tau metabolism.
Limitations and perspectives
One of the limitations of this study is the moderate size of the study cohort (n = 126 in discovery and validation cohorts). Although the validation cohort reduced the sample volume needed to 4 ml, this volume remains a restriction. Although clinical plasma samples of 4 ml are routinely obtained in single collection tubes, research biorepositories typically share 0.5 ml with requesters, limiting our current access. In addition, the volume of plasma currently needed for the plasma p-tau MS assay may prevent its application for retrospective analyses of plasma cohorts from existing biobanks with limited plasma resources. Although the protocol used for the validation study using 4 ml of plasma (∼8 ml whole blood) would be challenging with current plasma biobanks, changes in plasma collection volume would be compatible for future studies. In its current form, the assay would still be significantly less costly and invasive than a PET scan or lumbar puncture. Our data suggest that plasma t-tau may not be an accurate AD biomarker with current methods due to the lower correlation with CSF t-tau. However, significantly higher plasma t-tau levels in AD were detected in another study (Mielke et al., 2017). Nevertheless, the magnitude of p-tau-217 changes detected and its high correlation with CSF results demonstrate promising clinical utility for a plasma test specific for p-tau-217 epitope. We observed plasma p-tau-181 accuracy was lower in the validation cohort than the discovery cohort and recent studies using immunoassays (Janelidze et al., 2020a; Thijssen et al., 2020). We hypothesize the CSF collection (6 ml/h collected by catheter for Aβ SILK monitoring) occurring in parallel of the plasma collection would have contributed to a reduction of CNS tau release to the periphery for this particular cohort. No concomitant CSF collection occurred at the time of the plasma collection used for the discovery cohort. Thus, the increase of CNS p-tau isoforms would have been attenuated, affecting mainly p-tau-181, which is more subject to peripheral tau interference than p-tau-217.
This plasma tau MS assay may be applicable beyond AD to a wider array of neurodegenerative diseases, especially if combined with a plasma Aβ MS assay. The use of plasma p-tau biomarkers in combination with direct measurement of plasma amyloid 42/40 ratio would improve the specificity of the test and enable differential diagnosis of pure tauopathies versus diseases with mixed pathologies. This clinical application has recently been reported for plasma p-tau-181 measured by immunoassay (Janelidze et al., 2020a; Thijssen et al., 2020). Alternatively, this plasma p-tau assay could be used as a highly sensitive screening tool to identify a high risk of amyloidosis in normal subjects, replacing costly PET imaging. Further work is needed to determine the relationship and timing of plasma p-tau changes compared with plasma Aβ (Ovod et al., 2017; Nakamura et al., 2018; Schindler et al., 2019) and other biomarkers (Preische et al., 2019) for more accurate staging of the disease. For diagnostic validation, confirmation for amyloid status or diagnosis for AD could then be obtained by CSF biomarkers, amyloid and tau PET measurements, or brain neuropathology.
Conclusion
We demonstrate that measuring attomolar concentrations of tau isoforms in plasma is feasible using an enrichment protocol and MS. Our results indicate that changes in plasma p-tau, especially p-tau-217, mirror highly specific modifications in CSF to detect phosphorylation changes in soluble tau and amyloidosis. We provide strong evidence of a peripheral contribution to blood tau in plasma, which has a different phosphorylation profile compared with CSF. These findings support blood p-tau isoforms being potentially useful for detecting AD pathology, staging disease, and diagnosis.
Material and methods
Participants
The tau SILK studies had enrolled 58 participants for tau kinetic measurements at the time of our study (Sato et al., 2018). These studies were approved by the Washington University in St. Louis Institutional Review Board. Written informed consent was obtained from all participants before inclusion in the study. Participants were identified by number, not by name. Control participants were referred from Volunteer for Health at Washington University. AD and age-matched control participants with cognitive measures and amyloid and tau PET scans were referred from the Knight Alzheimer’s Disease Research Center and Memory Diagnostic Center at Washington University in St. Louis. During the tau SILK study, participants received 16 h of labeled 13C-leucine infusion followed by five lumbar punctures to collect CSF over the next 4 mo (Sato et al., 2018). To monitor leucine enrichment in the periphery, ∼10 ml of plasma was collected at each of the 12 time points performed over 19.5 h during the infusion procedure (Potter et al., 2013).
Discovery cohort
CSF from the 58 SILK participants was analyzed by MS for CSF Aβ42/40 and tau isoforms as previously reported (Patterson et al., 2015; Barthélemy et al., 2019; Sato et al., 2018). Plasma samples from 34 participants were selected based on their amyloid and cognitive status, as described in Table 1. 11 of these participants were identified as amyloid-positive using amyloid AV45 PET (cutoff 1.3). Four additional amyloid-positive participants with no amyloid PET data were identified by CSF42/40 ratio (cutoff 0.086 established using results from participants with AV45 PET data). The 15 amyloid-positive participants had clinical dementia rating (CDR) scores of 0 (5 participants), 0.5 (8 participants), and 1 (2 participants). All preclinical AD participants (amyloid-positive, CDR = 0) had tau PET AV-1451 standardized uptake value (SUVR) measures not significantly different from amyloid-negative participants (Table 1). The 41 remaining participants were amyloid-negative by AV45 PET when available or by CSF Aβ42/40. 17 of these participants were selected as controls (9 young controls and 8 randomly selected aged controls). Two participants identified as MCI were also included. The number of participants for this plasma cohort was 34 total. Of note, all amyloid-positive participants were abnormal for CSF pT217/T217 ratio (cutoff 4.6%), though this variable was not considered for sample selection.
Validation cohort
CSF and plasma from 92 participants collected from Aβ SILK studies (Patterson et al., 2015; Ovod et al., 2017) were analyzed for tau isoforms as the validation cohort. The Aβ SILK protocol involved blood and CSF collection at baseline, followed by a leucine bolus and infusion over 9 h. Blood (12 ml) and CSF (6 ml) samples were obtained hourly over 36 h, aliquoted at 1 ml in polypropylene tubes, and stored at −80°C CSF until use. Aliquots collected at hour 32 were used for the MS measurement of tau isoforms. For plasma, 1-ml aliquots collected after hour 16 were combined for constituting 4-ml plasma samples. Amyloid status was defined by CSF Aβ42/40 (cutoff 0.139), when available (n = 66), and amyloid PET centiloid from Pittsburgh compound B tracer or AV45 SUVR. Under this definition, all amyloid-positive participants (n = 50) had elevated CSF pT217/T217 ratio (cutoff 2.75%) and 41 of the 42 amyloid-negative participants had CSF pT-217/T217 below this cutoff. Amyloid groups were divided into clinical groups according to their CDR status.
Plasma and CSF tau immunopurification (IP)-LC-MS analysis
Frozen plasma aliquots of 1 ml were thawed at 4°C overnight. For the discovery study, ∼20 ml of plasma was pooled by combining 20 tubes of 1-ml plasma aliquots from the same participant from the 16-h infusion. After 16,000 g centrifugation for 30 min at 4°C, 20 ml was transferred into a new tube, and samples were spiked with 1 ng of 15N-labeled recombinant 2N4R tau internal standard (gift from Guy Lippens, Centre national de la recherche scientifique, Université de Lille, Villeneuve-d’Ascq, France). Plasma proteins were precipitated with perchloric acid (3.5% vol/vol final), vortexed to homogenize, and incubated for 30 min on ice. Samples were then centrifuged for 30 min at 4°C at 16,000 g. Supernatants containing soluble tau (Barthélemy et al., 2016) were transferred to new tubes and spiked with trifluoroacetic acid (TFA) to a final concentration of 1%. Samples were loaded on an Oasis HLB VAC RC 30-mg extraction cartridge (Waters) initially conditioned with 1 ml MeOH and 1 ml 0.1% TFA. After loading, samples were desalted with 1 ml 0.1% TFA and then eluted with 700 µl 27.5% acetonitrile-0.1% TFA solution. Eluates were lyophilized by speed-vac and then reconstituted in 1 ml 1× PBS, 1× protease inhibitor cocktail (Roche), 1% NP-40, and 5 mM guanidine. After this step, both plasma and thawed CSF (spiked with 15N-tau and IP reagents) were IP using similar protocol as previously reported for CSF (Sato et al., 2018; Barthélemy et al., 2019). Briefly, CSF and plasma tau were immunoprecipitated with Tau1 and HJ8.5 antibodies, then digested with trypsin. Digests were spiked with absolute quantification peptides (Life Technologies) to 50 and 5 fmol for each unphosphorylated and phosphorylated peptide, respectively. Tryptic digest was purified by solid phase extraction on C18 TopTip. The eluate was lyophilized and resuspended in 25 µl before nano-LC-MS/high resolution MS analysis on nanoAcquity ultra performance liquid chromatography system (Waters) coupled to an Orbitrap Tribrid Eclipse mass spectrometer (Thermo Fisher Scientific) operating as previously reported (Barthélemy et al., 2020a). Tandem MS (MS/MS) transitions from ionized peptides (Table S2) were recorded using parallel reaction monitoring and extracted at 5 ppm using Skyline software (MacCoss Lab, University of Washington, Seattle, WA).
The internal calibration of tau and p-tau levels was performed in two steps. First, the addition of 15N-tau internal standard to plasma before the first extraction step allows the measurement of absolute level of each unmodified tau peptide in plasma using LC-MS area ratio between each endogenous tau peptide and 15N-tau peptide. Second, the phosphorylation occupancy for each modified residues was obtained by comparison of the endogenous p-tau/tau ratio with p-tau/tau ratio measured on corresponding unphosphorylated and phosphorylated absolute quantification synthetic peptide standard labeled at lysine or arginine on the C-terminal position of the tryptic peptide. The determination of p-tau level was obtained by combination of t-tau level and p-tau/tau phosphorylation occupancy.
Performance of the 4 ml starting volume was assessed using a spike and recovery experiment on plasma pools from healthy volunteers. Coefficients of variation measured on plasma biological replicates without CSF spikes for t-tau, p-tau-217 and p-tau-181 were all below 7% (Fig. S1 and Table S1). LC-MS/MS responses for the different plasma tau measures were proportional to the amount of control and AD CSF successively added. We measured 15N-tau protein internal standard recovery obtained after plasma extraction (chemical extraction + IP) and compared with the recovery obtained after IP of the same amount of standard spiked in recombinant human serum albumin (HSA) 5% solution. We found the plasma protocol recovered from 30 to 50% of the amount recovered by IP only. This protein recovery is similar to previous assessment on tau chemical extraction protocol in CSF (Barthélemy et al., 2016).
Statistics
All statistical analyses were performed using GraphPad Prism software (v8.3.0.) The Mann–Whitney test was used for comparing p-tau/tau ratios and levels across subgroups of AD and control. Spearman correlations were used to analyze correlations between plasma and CSF tau. Paired t was used to test the differences in the percentage change relative to control group between p-tau-181 (reference biomarker) and other biomarkers. P values were adjusted using the two-stage step-up method of Benjamini, Krieger, and Yekutieli to control the false discovery rate. Data are represented as mean ± SD unless otherwise specified. Significance of the AUROC was assessed using the DeLong test.
Online supplemental material
Fig. S1 shows spike and recovery experiment validating the analytical performance for the plasma assay using 4 ml of sample. Table S1 shows replication in non-spiked plasma pool from healthy volunteers and method performance measured in the spike and recovery experiment. Table S2 shows peptides sequences and corresponding MS/MS transitions used for the quantitation of plasma tau peptides. Table S3 shows AUC comparison by DeLong test on p-tau217 and p-tau181 performance in separating amyloid positive from amyloid negative.
Acknowledgments
We thank the participants and their families for their contributions to this study. We thank Melody Li and Kathleen Schoch for their helpful input on the revision of the manuscript and Yan Li for discussion on statistical analysis. We thank Dr. David Holtzman and Ms. Hong Jiang (Washington University in St. Louis School of Medicine, St. Louis, MO) for HJ8.5 antibody; Dr. Nicholas Kanaan (Michigan State University, Grand Rapids, MI) for Tau1 antibody; Drs. Guy Lippens and Isabelle Huvent for 15N-recombinant tau (Université des Sciences et Technologies de Lille 1, Lille); and Chloe Yingxin He and Andrew Espeland for their help with sample retrieval and processing.
Funding for this work includes the Alzheimer’s Association Research Fellowship AARF-16-443265 (to N.R. Barthélemy), the Rainwater Charitable Foundation (to R.J. Bateman and N.R. Barthélemy), the National Institutes of Health, National Institute of Neurological Disorders and Stroke grant R01NS095773 (R.J. Bateman), the Tau SILK Consortium (AbbVie, Biogen, and Eli Lilly and Company; principal investigator, R.J. Bateman), and the Coins for Alzheimer’s Research Trust grant (to C. Sato).
Author contributions: N.R. Barthélemy and R.J. Bateman conceived the project. N.R. Barthélemy and C. Sato conducted CSF tau profile analyses. N.R. Barthélemy and K. Horie conducted plasma tau measurements by MS, interpreted data, and prepared figures. N.R. Barthélemy, K. Horie, C. Sato, and R.J. Bateman wrote the manuscript.

