


The wound-healing process is a paradigm of the directed migration of various pools of stem cells from their niche to the site of injury where they replenish damaged cells. Two decades have elapsed since the observation that wounding activates multipotent hair follicle stem cells to infiltrate the epidermis, but the cues that coax these cells out of their niche remain unknown. Here, we report that Caspase-1, a protein classically known as an integral component of the cytosolic inflammasome, is secreted upon wounding and has a non-canonical role in the extracellular milieu. Through its caspase activation recruitment domain (CARD), Caspase-1 is sufficient to initiate the migration of hair follicle stem cells into the epidermis. Uncovering this novel function of Caspase-1 also facilitates a deeper understanding of the mechanistic basis of the epithelial hyperplasia found to accompany numerous inflammatory skin diseases.
Introduction
Upon wounding, the re-epithelialization of the skin requires the directed migration of various cutaneous stem cell pools to the site of injury and their differentiation to replenish lost or damaged cells (Gonzales and Fuchs, 2017; Rognoni and Watt, 2018). This process is enhanced by the contribution of hair follicle stem cells (HFSCs) differentiating to epidermal keratinocytes, an emergency response otherwise not observed in homeostasis (Dekoninck and Blanpain, 2019).
Wound-induced homing of HFSCs into the epidermis was first observed nearly two decades ago (Ito et al., 2005; Tumbar et al., 2004). There has been substantial effort and progress in understanding the lineage trajectory of stem cells once they reach their destination (Aragona et al., 2017; Garcin et al., 2016; Kang et al., 2020; Levy et al., 2007; Mathur et al., 2019; Page et al., 2013; Snippert et al., 2010; Vagnozzi et al., 2015); however, comparatively little is understood regarding the mechanisms guiding their chemotactic journey to the wound site. The cytoskeletal mechanisms involving GSK3β and ACF7 responsible for the migration of HFSCs after wounding were partially elucidated (Wu et al., 2011), and also Wnt ligands, Heregulin, and other EGF ligands were hypothesized as possible endogenous migratory cues (Yucel and Oro 2011). However, none of these cytokines or other known pro-migratory chemokines have since been reported to induce HFSC migration, thus suggesting a non-canonical function of a secreted protein may be accomplishing this task.
Interestingly, wound-sensing keratinocytes can secrete Caspase-1 into the extracellular milieu (Feldmeyer et al., 2007; Keller et al., 2008; Lee et al., 2009) even though this protein is classically known as a central component of the cytosolic inflammasome that regulates the unconventional secretion of many inflammatory cytokines (Keller et al., 2008). Reports have also documented that keratinocytes secrete Caspase-1 in response to stress stimuli such as UV irradiation (Feldmeyer et al., 2007; Keller et al., 2008) and in inflammatory skin diseases such as atopic dermatitis (Lee et al., 2009; Li et al., 2010). The ejection of this protein from stressed cells is speculated to serve the purpose of cytopreservation and escaping cell death (Keller et al., 2008); however, its function in the extracellular space in these various scenarios remains to be fully explored. Interestingly, the presence of extracellular Caspase-1 correlates with conditions in which HFSCs contribute to epidermal hyperplasia in inflammatory skin conditions such as sunburn due to UV exposure (Chou et al., 2013), atopic dermatitis and squamous cell carcinoma (Singh et al., 2013; White et al., 2011), and psoriasis (Braff et al., 2005).
Consequently, deciphering the mechanism of HFSC migration in a physiological process such as wound healing has the potential to provide insights toward controlling this phenomenon in pathological scenarios.
Results and discussion
Caspase-1 mediates HFSC migration during wound healing
We thus tested the hypothesis that Caspase-1 may play an unconventional role in inducing HFSC migration in a wound-healing scenario. We utilized an inducible Lgr5-tdTomato lineage reporter mouse to label HFSCs that reside in the lower bulge of the telogen hair follicle (HF) (Jaks et al., 2008; Mathur et al., 2019; Kang et al., 2020).
Full-thickness excisional wounds were administered to mice, and the hair follicles immediately adjacent to the wound and those more distal were analyzed (Fig. 1 A). The HFSC niche, known as the bulge, was identified by the intersection point between the hair follicle and the arrector pili muscle (Ji et al., 2021; Lin et al. 2022). In wild-type (WT) skin, we began to observe the emigration of Lgr5 lineage traced cells out of the bulge 48 h after wounding. This phenomenon was only observed in hair follicles immediately adjacent to the wound edge (wound proximal) (Fig. 1, B and C). By 72 h after wounding, there was a substantial number of HFSCs that migrated toward the epidermis in the wound-adjacent follicles. Interestingly, in the caspase-1−/− mouse (C1−/−), the migration of HFSCs in the wound proximal hair follicles was notably diminished compared with the WT (Fig. 1, B and C). Though the loss of caspase-1 significantly reduces the epidermal homing of HFSCs, it does not completely abrogate this phenomenon. This suggests that other factors also potentiate this behavior of HFSCs or compensate in instances where caspase-1 is absent. Given the importance of wound healing for the health of the organism, it is expected that many of the processes in wound healing have redundant mechanisms to ensure that a single malfunction does not completely cripple this important physiological process.

Caspase-1 mediates HFSC migration during wound healing. (A) Scheme of lineage tracing using the inducible Lgr5-CreER;tdTomato mice after tamoxifen injection (orange arrows) to activate the tdTomato reporter. Application of an excisional wound (red arrow) and tissue harvesting times (black arrows) are noted. (B) Representative images of Lgr5-traced cells (red) in 24, 48, and 72 h after wounded skin sections of wild-type (WT) or caspase-1 null (C1−/−) mice. Sections are stained for αSMA (cyan) to label arrector pili muscles and DAPI (blue) to label nuclei. Arrows mark the wound edge. Asterisks mark the point of contact of arrector pili muscle with the hair bulge. “IFE” refers to “inter-follicular epidermis.” White dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis. Scale bar = 100 µm. (C) Quantification of non-bulge Lgr5-traced cells in 24, 48, and 72 h post-wounded skin sections of wild-type (WT) or caspase-1 null (C1−/−) mice. The first hair follicle (HF) from the wound edge is considered “Wound proximal” and the second hair follicle from the wound edge is considered “Wound distal.” All quantifications are from at least three pooled independent replicates (Wound proximal [C1+/+, 24 h n = 11, 48 h n = 18, 72 h n = 34; C1−/−, 24 h n = 17, 48 h n = 35, 72 h n = 27], Wound distal [C1+/+, 24 h n = 11, 48 h n = 18, 72 h n = 34; C1−/−, 24 h n = 17, 48 h n = 35, 72 h n = 27]). Mean ± SEM is reported, and P values are calculated by unpaired Student’s t test.
Caspase-1 mediates HFSC migration during wound healing. (A) Scheme of lineage tracing using the inducible Lgr5-CreER;tdTomato mice after tamoxifen injection (orange arrows) to activate the tdTomato reporter. Application of an excisional wound (red arrow) and tissue harvesting times (black arrows) are noted. (B) Representative images of Lgr5-traced cells (red) in 24, 48, and 72 h after wounded skin sections of wild-type (WT) or caspase-1 null (C1−/−) mice. Sections are stained for αSMA (cyan) to label arrector pili muscles and DAPI (blue) to label nuclei. Arrows mark the wound edge. Asterisks mark the point of contact of arrector pili muscle with the hair bulge. “IFE” refers to “inter-follicular epidermis.” White dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis. Scale bar = 100 µm. (C) Quantification of non-bulge Lgr5-traced cells in 24, 48, and 72 h post-wounded skin sections of wild-type (WT) or caspase-1 null (C1−/−) mice. The first hair follicle (HF) from the wound edge is considered “Wound proximal” and the second hair follicle from the wound edge is considered “Wound distal.” All quantifications are from at least three pooled independent replicates (Wound proximal [C1+/+, 24 h n = 11, 48 h n = 18, 72 h n = 34; C1−/−, 24 h n = 17, 48 h n = 35, 72 h n = 27], Wound distal [C1+/+, 24 h n = 11, 48 h n = 18, 72 h n = 34; C1−/−, 24 h n = 17, 48 h n = 35, 72 h n = 27]). Mean ± SEM is reported, and P values are calculated by unpaired Student’s t test.
Extracellular Caspase-1 induces HFSC migration through its CARD
After establishing the crucial role of caspase-1 in the migration of HFSCs, we investigated how Caspase-1 induces HFSC migration. Interestingly, immunostaining for Caspase-1 using an unpermeabilized wounded skin section demonstrated the presence of a higher level of extracellular Caspase-1 in the wound proximal area compared with the wound distal area (Fig. S1 A). We also found extracellular Caspase-1 in the conditioned media from the epidermal explants subjected to multiple incisional wounds (Fig. S1 B). This is in agreement with our previous report on the secretion of extracellular Caspase-1 from the epidermis of a genetic model of wound healing (Lee et al., 2009). This observation suggests that the source of extracellular Caspase-1 is epidermal cells.
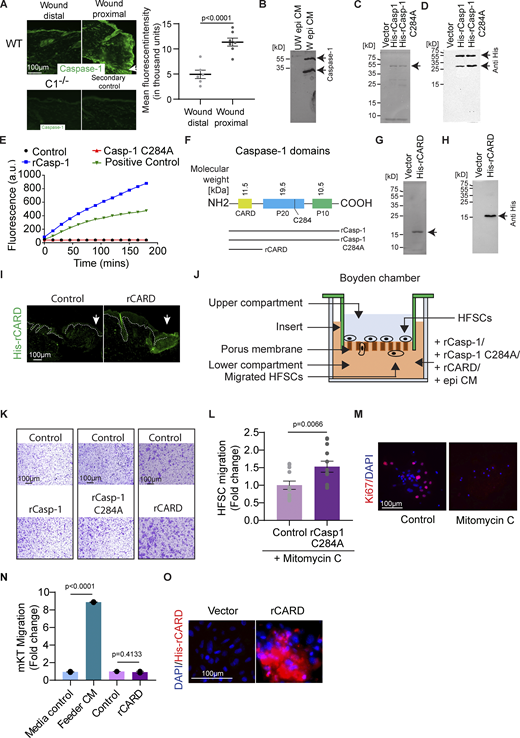
Characterization of the effect of Caspase-1 on HFSC migration. (A) Staining for extracellular Caspase-1 (green) in a wound distal and wound proximal area of WT skin sections. C1−/− skin section stained for extracellular Caspase-1 in similar conditions is presented for comparison. Secondary antibody control is presented to show the background fluorescence. Graph is the quantification of the background-subtracted mean fluorescent intensity of WT skin sections in wound distal and the wound proximal regions taken from at least three mice (Wound distal, n = 6; Wound proximal, n = 7). Mean ± SEM are reported, and P value is calculated by unpaired, one-tailed Student’s t test. Arrow marks the wound edge. (B) Full Western blot of unwounded and wounded epidermal conditioned media probed with antibody against Caspase-1. (C) Silver nitrate staining of purified histidine-tagged recombinant wild-type Caspase-1 (His-rCasp-1) and catalytically dead Caspase-1 (His-rCasp-1 C284A) on a denaturing acrylamide gel. Arrows point to the expected position of the protein of interest. (D) Full Western blot of purified recombinant histidine-tagged wild-type Caspase-1 (His-rCasp-1) and catalytically dead Caspase-1 (His-rCasp-1 C284A) probed with anti-His antibody. (E) Measurement of caspase-1 catalytic activity in a substrate cleavage assay of both the rCasp-1 and rCasp-1 C284A in comparison with HaCat cell lysate (Positive control). An increase in fluorescence upon substrate (Ac-YVAD-AMC) cleavage is plotted on the y-axis. (F) Domain structure of Caspase-1, indicating the constructs being expressed. (G) Silver nitrate staining of purified histidine-tagged rCARD (His-rCARD) on a denaturing acrylamide gel. (H) Full Western blot of purified histidine-tagged rCARD (His-rCARD) probed with anti-His antibody. Arrow points the expected position of protein of interest. (I) Immunofluorescent image of a wounded caspase-1 null mouse skin section probed with anti-His antibody to detect rCARD. Arrow marks the wound edge. (J) Schematic of Boyden chamber/transwell migration assay. (K) Representative images of migrated HFSCs in the Boyden chamber. (L) Proliferation inhibited HFSC (upon Mitomycin treatment) chemotaxis using a Boyden chamber/transwell assay towards rCasp-1 C284A in comparison with the vector control. Quantifications are from at least three pooled independent replicates (Control, n = 12; rCasp-1 C284A, n = 12). Mean ± SEM are reported, and P value is calculated by unpaired, one-tailed Student’s t test. (M) Immunofluorescence images of mitomycin C treated HFSCs probed with the proliferation marker-Ki67 (red). (N) Primary mouse keratinocyte (mKT) chemotaxis using a Boyden chamber toward rCARD in comparison with the vector control. Media control induces no migration and is the benchmark for basal migration of cells with no chemoattractant. Feeder cell-conditioned media (Feeder CM) was used as a positive control for inducing migration. Quantification is from two independent biological replicates. Mean ± SEM are reported, and P values are calculated by one-way ANOVA. (O) Binding of His-tagged rCARD onto hair follicle stem cells visualized by anti-His antibody staining (red). rCARD was applied on hair follicle stem cells grown in tissue culture plate and incubated at room temperature. Unbound CARD was washed off. Source data are available for this figure: SourceData FS1.
Characterization of the effect of Caspase-1 on HFSC migration. (A) Staining for extracellular Caspase-1 (green) in a wound distal and wound proximal area of WT skin sections. C1−/− skin section stained for extracellular Caspase-1 in similar conditions is presented for comparison. Secondary antibody control is presented to show the background fluorescence. Graph is the quantification of the background-subtracted mean fluorescent intensity of WT skin sections in wound distal and the wound proximal regions taken from at least three mice (Wound distal, n = 6; Wound proximal, n = 7). Mean ± SEM are reported, and P value is calculated by unpaired, one-tailed Student’s t test. Arrow marks the wound edge. (B) Full Western blot of unwounded and wounded epidermal conditioned media probed with antibody against Caspase-1. (C) Silver nitrate staining of purified histidine-tagged recombinant wild-type Caspase-1 (His-rCasp-1) and catalytically dead Caspase-1 (His-rCasp-1 C284A) on a denaturing acrylamide gel. Arrows point to the expected position of the protein of interest. (D) Full Western blot of purified recombinant histidine-tagged wild-type Caspase-1 (His-rCasp-1) and catalytically dead Caspase-1 (His-rCasp-1 C284A) probed with anti-His antibody. (E) Measurement of caspase-1 catalytic activity in a substrate cleavage assay of both the rCasp-1 and rCasp-1 C284A in comparison with HaCat cell lysate (Positive control). An increase in fluorescence upon substrate (Ac-YVAD-AMC) cleavage is plotted on the y-axis. (F) Domain structure of Caspase-1, indicating the constructs being expressed. (G) Silver nitrate staining of purified histidine-tagged rCARD (His-rCARD) on a denaturing acrylamide gel. (H) Full Western blot of purified histidine-tagged rCARD (His-rCARD) probed with anti-His antibody. Arrow points the expected position of protein of interest. (I) Immunofluorescent image of a wounded caspase-1 null mouse skin section probed with anti-His antibody to detect rCARD. Arrow marks the wound edge. (J) Schematic of Boyden chamber/transwell migration assay. (K) Representative images of migrated HFSCs in the Boyden chamber. (L) Proliferation inhibited HFSC (upon Mitomycin treatment) chemotaxis using a Boyden chamber/transwell assay towards rCasp-1 C284A in comparison with the vector control. Quantifications are from at least three pooled independent replicates (Control, n = 12; rCasp-1 C284A, n = 12). Mean ± SEM are reported, and P value is calculated by unpaired, one-tailed Student’s t test. (M) Immunofluorescence images of mitomycin C treated HFSCs probed with the proliferation marker-Ki67 (red). (N) Primary mouse keratinocyte (mKT) chemotaxis using a Boyden chamber toward rCARD in comparison with the vector control. Media control induces no migration and is the benchmark for basal migration of cells with no chemoattractant. Feeder cell-conditioned media (Feeder CM) was used as a positive control for inducing migration. Quantification is from two independent biological replicates. Mean ± SEM are reported, and P values are calculated by one-way ANOVA. (O) Binding of His-tagged rCARD onto hair follicle stem cells visualized by anti-His antibody staining (red). rCARD was applied on hair follicle stem cells grown in tissue culture plate and incubated at room temperature. Unbound CARD was washed off. Source data are available for this figure: SourceData FS1.
Thus, we examined whether this extracellular pool of Caspase-1 is responsible for the migration of HFSCs. To investigate this, we bacterially expressed and purified murine Caspase-1 (rCasp-1) (Fig. S1, C and D) and exogenously applied it on the wound bed of C1−/− mouse skin (Fig. 2 A), which exhibits impaired HFSC migration. Compared with control, exogenously applied rCasp-1-induced Lgr5 lineage traced HFSCs migration in the C1−/− skin (Fig. 2 B), indicating that extracellular Caspase-1 is sufficient to induce the movement of these cells out of the bulge.

The CARD of Caspase-1 is sufficient to induce HFSC migration. (A) Scheme of lineage tracing using Lgr5-CreER;tdTomato mice after tamoxifen injection (orange arrows) to activate the tdTomato reporter, excisional wounding (red arrow), topical recombinant protein application regimen (black arrows), and tissue harvesting time point (blue arrow) are noted. (B–D) Representative images of Lgr5-traced cells (red) in 72 h wounded caspase-1 null (C1−/−) mice skin treated with topical application of vector control or (B) recombinant Caspase-1 (rCasp-1), (C) catalytically dead recombinant Caspase-1 (rCasp-1 C284A), and (D) recombinant CARD of Caspase-1 (rCARD). Sections are stained for αSMA (cyan) to label arrector pili muscles and DAPI (blue) to label nuclei. Arrows indicate the wound edge and asterisks mark the point of contact of arrector pili muscle with the hair bulge. “IFE” refers to “inter-follicular epidermis” and the white dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis. Scale bar = 100 µm. (B–D) Graphs in the right panels are the quantifications of non-bulge Lgr5-traced cells in the wound-adjacent hair follicles (HF) in the respective conditions. The first HF from the wound edge are analyzed for quantification. All quantifications are from at least three pooled independent replicates ([B] Control, n = 9; rCasp-1, n = 14, [C] Control, n = 37; rCasp-1 C284A, n = 44, [D] Control, n = 27; rCARD, n = 26). Mean ± SEM are reported, and P values are calculated by unpaired Student’s t test. (E–G) In vitro HFSC chemotaxis towards (E) rCasp-1 (n = 6), (F) rCasp-1 C284A (n = 12) and (G) rCARD (n = 7) compared to their respective vector controls (n ≥ 6), using a Boyden chamber/transwell assay. All quantifications are from at least three pooled independent replicates ([E] Control, n = 6; rCasp-1, n = 6, [F] Control, n = 6; rCasp-1 C284A, n = 12, [G] Control, n = 7; rCARD, n = 7). Mean ± SEM is reported, and P values are calculated by unpaired Student’s t test.
The CARD of Caspase-1 is sufficient to induce HFSC migration. (A) Scheme of lineage tracing using Lgr5-CreER;tdTomato mice after tamoxifen injection (orange arrows) to activate the tdTomato reporter, excisional wounding (red arrow), topical recombinant protein application regimen (black arrows), and tissue harvesting time point (blue arrow) are noted. (B–D) Representative images of Lgr5-traced cells (red) in 72 h wounded caspase-1 null (C1−/−) mice skin treated with topical application of vector control or (B) recombinant Caspase-1 (rCasp-1), (C) catalytically dead recombinant Caspase-1 (rCasp-1 C284A), and (D) recombinant CARD of Caspase-1 (rCARD). Sections are stained for αSMA (cyan) to label arrector pili muscles and DAPI (blue) to label nuclei. Arrows indicate the wound edge and asterisks mark the point of contact of arrector pili muscle with the hair bulge. “IFE” refers to “inter-follicular epidermis” and the white dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis. Scale bar = 100 µm. (B–D) Graphs in the right panels are the quantifications of non-bulge Lgr5-traced cells in the wound-adjacent hair follicles (HF) in the respective conditions. The first HF from the wound edge are analyzed for quantification. All quantifications are from at least three pooled independent replicates ([B] Control, n = 9; rCasp-1, n = 14, [C] Control, n = 37; rCasp-1 C284A, n = 44, [D] Control, n = 27; rCARD, n = 26). Mean ± SEM are reported, and P values are calculated by unpaired Student’s t test. (E–G) In vitro HFSC chemotaxis towards (E) rCasp-1 (n = 6), (F) rCasp-1 C284A (n = 12) and (G) rCARD (n = 7) compared to their respective vector controls (n ≥ 6), using a Boyden chamber/transwell assay. All quantifications are from at least three pooled independent replicates ([E] Control, n = 6; rCasp-1, n = 6, [F] Control, n = 6; rCasp-1 C284A, n = 12, [G] Control, n = 7; rCARD, n = 7). Mean ± SEM is reported, and P values are calculated by unpaired Student’s t test.
We then investigated the method by which Caspase-1 can induce HFSC migration. The catalytic activity of Caspase-1 is necessary for its role in mediating unconventional protein secretion (Keller et al., 2008). This raised the question of whether the catalytic activity of extracellular Caspase-1 is required for HFSC chemotaxis. To test this, we bacterially expressed and purified mutant murine Caspase-1 (rCasp-1 C284A) (Fig. S1, C and D), which is reported to be catalytically dead (Broz et al., 2010). The catalytic activity of these different constructs was verified using a substrate cleavage assay (Fig. S1 E). Compared with control, exogenously applied rCasp-1 C284A induced Lgr5 lineage traced HFSCs in the C1−/− skin (Fig. 2 C), indicating that catalytic activity of Caspase-1 is not necessary for its migratory effect on HFSCs.
Given this unusual catalytic-independent function of Caspase-1 as an HFSC migration–inducing factor, we investigated the domain responsible for this activity. Caspase-1 is comprised of an N-terminal protein–protein interaction region (Park, 2019) called the caspase activation and recruitment domain (CARD), followed by the p20 and p10 domains (Fig. S1 F). Autoproteolysis results in the separation of these individual domains, followed by heterodimerization of the p20 and p10 domains into the catalytically active form (Broz et al., 2010). Since the catalytic activity is not required to induce HFSC migration, we focused on the CARD. The murine Caspase-1 CARD was tagged with polyhistidine, bacterially expressed and purified (Fig. S1, G and H). We examined whether the application of recombinant CARD (rCARD) on the wound beds of mice lacking caspase-1 rescued HFSC migration. Under these conditions, we can detect the CARD throughout the wound bed (Fig. S1 I). We found that compared with the control in the C1−/− skin, rCARD application induced the migration of Lgr5 lineage–traced HFSCs (Fig. 2 D). These results indicate that the extracellular Caspase-1 induces HFSC migration through its CARD.
In vivo, the wound contains a complex cocktail of factors released by wound-sensing cells. Thus, we tested if Caspase-1 alone is sufficient to induce HFSC migration in vitro using the Boyden chamber/transwell migration assay (Fig. S1 J) in response to rCasp-1. We found that recombinant rCasp-1 was sufficient to induce HFSC migration in the Boyden chamber (Fig. 2 E and Fig. S1 K). We also observed that rCasp-1 C284A likewise induced HFSC migration (Fig. 2 F and Fig. S1 K). During wound healing, HFSCs are also stimulated to proliferate (Lee et al., 2017), but the promigratory effect of caspase-1 was not dependent on HFSCs being in an actively proliferating state (Fig. S1, L and M). Similar to the sufficiency of rCasp-1 and rCasp-1 C284A in inducing HFSC migration in vitro, we observed that rCARD is also sufficient to induce HFSC migration in the Boyden chamber assay (Fig. 2 G and Fig. S1 K). Interestingly, the promigratory effect of CARD seems to be specific for HFSCs as it did not exhibit this same effect on primary epidermal keratinocytes (Fig. S1 N).
How the HFSCs sense CARD is an intriguing question. Based on the chemotactic activity of this domain of Caspase-1, we postulated that there is a cell surface cognate receptor that launches the migratory behavior following an engagement with the CARD ligand. In support of this, we observed that recombinant CARD was able to bind to the surface of HFSCs (Fig. S1 O).
Caspase-1 is a regulator of hair follicle stem cell migration in the skin during UV damage and inflammatory skin disease
There are various pathologies marked by caspase-1 secretion and HFSC appearance in the hyperplastic epidermis. For instance, cutaneous sunburns (Faustin and Reed, 2008) and other inflammatory skin diseases such as psoriasis (Bhatt et al., 2019; Johansen et al., 2007) and atopic dermatitis (Antonopoulos et al., 2001; Lee et al., 2009; Li et al., 2010) are marked by Caspase-1 in the extracellular space. Thus, we examined whether extracellular Caspase-1-mediated HFSC migration into the epidermis is a conserved phenomenon that may explain the epithelial hyperplasia associated with these pathologies. Since UV-irradiated mouse keratinocytes release Caspase-1 (Feldmeyer et al., 2007; Keller et al., 2008), we tested whether UV irradiation of mouse skin displayed epidermal homing behavior of HFSCs in a Caspase-1-dependent manner (Fig. 3 A). We observed Lgr5 lineage traced HFSCs migrating toward the epidermis in UV-irradiated WT skin. However, this phenomenon did not take place in the UV irradiated C1−/− skin (Fig. 3 B). We also observed that the conditioned media (CM) prepared from UV-irradiated epidermal keratinocytes is capable of stimulating HFSC chemotaxis (Fig. 3 C).

Caspase-1 is a regulator of hair follicle stem cell migration during UV damage and inflammatory skin disease. (A) Scheme of lineage tracing using Lgr5-CreER;tdTomato mice after tamoxifen injection (orange arrows) to activate the tdTomato reporter, UV irradiation exposure (red arrow), and tissue harvesting time point (black arrow) are noted. (B) Representative images of Lgr5-traced cells (red) in UV treated (+UV) and untreated (−UV) skin sections of wild type (WT) and caspase-1 null mice (C1−/−) 3 days post UV exposure. Sections are stained for αSMA (cyan) to label arrector pili muscles and DAPI (blue) to label nuclei. Asterisks mark the point of contact of arrector pili muscle with the hair bulge. “IFE” refers to inter-follicular epidermis and the white dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis. Scale bar = 100 µm. Representative images of at least three biological replicates are shown. (C) In vitro HFSC chemotaxis using a Boyden chamber/transwell assay toward conditioned media from UV-treated (+UV mKT CM) (n = 9), and untreated (−UV mKT CM) (n = 9), primary mouse keratinocytes. Quantifications are from at least three pooled independent replicates ( −UV mKT CM, n = 9; +UV mKT CM, n = 9). Mean ± SEM is reported, and P value is calculated by unpaired, one-tailed Student’s t test. (D) Scheme of tamoxifen injection (orange arrows) to knockout Caspase-8 in K14-CreER; C8fl/fl mice (iC8cKO) and tissue harvesting time point (black arrow) are noted. (E) Representative immunofluorescence images of Sox9+ HFSCs (red) in the hair follicles of adult tamoxifen-injected wild type (WT), tamoxifen-induced C8cKO (iC8cKO), and caspase-8/caspase-1 double knockout (iC8cKO/C1−/−) mice. Epidermis and hair follicles are marked by K5 in cyan and brackets mark the infundibulum. “IFE” refers to interfollicular epidermis. Scale bar = 100 µm. Representative images of at least three biological replicates are shown. The number of Sox9+ HFSCs found in the infundibulum (0–50 µM from the epidermis) of hair follicles are quantified from skins of at least three mice (WT, n = 18; iC8cKO, n = 20; iC8cKO/C1−/−, n = 19). Mean ± SEM are reported, and P values are calculated by one-way ANOVA. (F) In vitro HFSC chemotaxis using a Boyden chamber/transwell assay toward epidermal conditioned media from wild-type (WT epi CM), caspase-8 conditional knockout (C8cKO epi CM) mice, and C8cKO mice in the caspase-1 null background (C8cKO/C1−/− epi CM, n = 4). At least three biological replicates have been used for the quantification (WT epi CM, n = 4; iC8cKO, n = 4; iC8cKO/C1−/− epi CM, n = 4). Mean ± SEM are reported, and P values are calculated by one-way ANOVA.
Caspase-1 is a regulator of hair follicle stem cell migration during UV damage and inflammatory skin disease. (A) Scheme of lineage tracing using Lgr5-CreER;tdTomato mice after tamoxifen injection (orange arrows) to activate the tdTomato reporter, UV irradiation exposure (red arrow), and tissue harvesting time point (black arrow) are noted. (B) Representative images of Lgr5-traced cells (red) in UV treated (+UV) and untreated (−UV) skin sections of wild type (WT) and caspase-1 null mice (C1−/−) 3 days post UV exposure. Sections are stained for αSMA (cyan) to label arrector pili muscles and DAPI (blue) to label nuclei. Asterisks mark the point of contact of arrector pili muscle with the hair bulge. “IFE” refers to inter-follicular epidermis and the white dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis. Scale bar = 100 µm. Representative images of at least three biological replicates are shown. (C) In vitro HFSC chemotaxis using a Boyden chamber/transwell assay toward conditioned media from UV-treated (+UV mKT CM) (n = 9), and untreated (−UV mKT CM) (n = 9), primary mouse keratinocytes. Quantifications are from at least three pooled independent replicates ( −UV mKT CM, n = 9; +UV mKT CM, n = 9). Mean ± SEM is reported, and P value is calculated by unpaired, one-tailed Student’s t test. (D) Scheme of tamoxifen injection (orange arrows) to knockout Caspase-8 in K14-CreER; C8fl/fl mice (iC8cKO) and tissue harvesting time point (black arrow) are noted. (E) Representative immunofluorescence images of Sox9+ HFSCs (red) in the hair follicles of adult tamoxifen-injected wild type (WT), tamoxifen-induced C8cKO (iC8cKO), and caspase-8/caspase-1 double knockout (iC8cKO/C1−/−) mice. Epidermis and hair follicles are marked by K5 in cyan and brackets mark the infundibulum. “IFE” refers to interfollicular epidermis. Scale bar = 100 µm. Representative images of at least three biological replicates are shown. The number of Sox9+ HFSCs found in the infundibulum (0–50 µM from the epidermis) of hair follicles are quantified from skins of at least three mice (WT, n = 18; iC8cKO, n = 20; iC8cKO/C1−/−, n = 19). Mean ± SEM are reported, and P values are calculated by one-way ANOVA. (F) In vitro HFSC chemotaxis using a Boyden chamber/transwell assay toward epidermal conditioned media from wild-type (WT epi CM), caspase-8 conditional knockout (C8cKO epi CM) mice, and C8cKO mice in the caspase-1 null background (C8cKO/C1−/− epi CM, n = 4). At least three biological replicates have been used for the quantification (WT epi CM, n = 4; iC8cKO, n = 4; iC8cKO/C1−/− epi CM, n = 4). Mean ± SEM are reported, and P values are calculated by one-way ANOVA.
We have previously reported that the conditional knockout of epidermal caspase-8 (C8cKO) leads to atopic dermatitis-like skin disease (Li et al., 2010) and secretion of Caspase-1 into the extracellular milieu (Lee et al., 2009). Therefore, we investigated whether the atopic dermatitis mouse model exhibits epidermal homing of HFSCs. Testing the migration of HFSCs in the C8cKO mouse model posed a technical challenge, since in this mouse model, caspase-8 is conditionally knocked out in the epidermis using a K14-driven Cre recombinase, prohibiting the use of the lineage tracing technique. To overcome this technical challenge, we developed an image analysis algorithm that can track the shift of HFSC location along the length of the hair follicle, utilizing cross-sectional time-point immunofluorescence images. Using a bonafide endogenous stem cell marker—Sox9 (Vidal et al., 2005; Wong and Reiter, 2011; Shi et al., 2013; Kadaja et al., 2014; Adam et al., 2015; Larsimont et al., 2015; Ge et al., 2017), we confirmed that similar to the Lgr5 traced-HFSCs, the Sox9-labeled HFSC exhibited the similar epidermally directed migration following wounding (Fig. S2, A–C). We observed that in the unwounded hair follicle, the Sox9+ HFSC population was predominantly concentrated at ∼200 µm from the epidermal-hair follicle junction. However, by 24 h after wounding, we found them near the epidermis (within 50 µm from the epidermis) (Fig. S2, B and C). HFSCs labeled with Sox9 were also consistent with the Lgr5 traced HFSCs in their dependence on caspase-1 for wound-induced epidermal homing. In the WT skin, Sox9+ cells migrate in the wound adjacent hair follicles, whereas C1−/− skin shows significantly lower migration of Sox9+ cells (Fig. S2, D and E). Moreover, topical application of rCARD on the wounds of caspase-1 null mice also increased the mobilization of Sox9+ HFSCs toward the epidermis in the wound proximal hair follicles (Fig. S2, F and G). We observed that even though Lgr5 and Sox9 label different subpopulations of HFSCs in the bulge (Jaks et al., 2008; Kadaja et al., 2014), both respond to a wound stimulus to migrate toward the epidermis (Fig. S2 H).

Sox9 tracing of HFSC migration. (A) Three points at the junction between the epidermis and hair follicle were marked (reference points) and the positions of individual Sox9+ HFSCs were marked manually. The distance of individual Sox9+ HFSCs from the three reference points (D; represented by white dashed lines) and the frequency of localization along the length of the hair follicle was calculated (see Materials and methods). (B) Cross-sectional time-point immunofluorescence images of localization of Sox9+ HFSCs (red) in hair follicles of the unwounded and wounded skins at 0 (unwounded) 8, 16, and 24 h post wounding. K5 marks the epidermis and hair follicles in cyan. The arrow indicates the wound edge, and the bracket denotes the bulge. Insets are magnified views of the infundibulum marked by the dashed box. Representative images of at least three biological replicates have been presented. (C) Probability distribution of Sox9+ HFSCs along the hair follicle in unwounded (UW) and wounded skin at 8, 16, and 24 h after wounding, represented as a heat-map with distance from the epidermis represented on the y-axis. Quantification is performed using at least three biological replicates (UW, n = 18; 8 h, n = 14; 16 h, n = 30; 24 h, n = 14). The cyan-colored box drawn on the heat map represents the portions that are statically different between unwounded (UW) condition and the respective time points. (D) Immunofluorescence images of localization of Sox9+ HFSCs in the hair follicles of unwounded and wound proximal region of wild-type (WT) and caspase-1 null (C1−/−) mice at 24 h post wounding. Lower panel is the magnified view of the infundibulum marked by the dashed box. K5 marks the epidermis and hair follicles in cyan, and arrows indicate the wound edge. Representative images of at least three biological replicates have been presented. (E) Probability distribution of Sox9+ HFSCs along the hair follicle of unwounded (UW) and wounded wild-type (WT) and caspase-1 null (C1−/−) skins at 8, 16, and 24 h post wounding, represented as a heat-map with distance from the epidermis represented on the y-axis. Quantification is performed using at least three biological replicates ([WT (UW, n = 18; 16 h, n = 30; 24 h, n = 14], C1−/− [UW, n = 27; 16 h, n = 13; 24 h, n = 15]) The cyan-colored box drawn on the heat map represents the portions that are statically different between unwounded (UW) and the respective time points. Note, UW, 16 and 24 h of (C and E) are same. (F) Immunofluorescence images of the localization of Sox9+ HFSCs in the 24 h wound proximal hair follicles of caspase-1 null mice skin upon topical application of recombinant CARD (rCARD) compared with the vector control. Lower panel is the magnified view of the infundibulum marked by the dashed box. K5 marks epidermis and hair follicles in cyan. Arrow indicates the wound edge. Representative images of at least three biological replicates have been presented. (G) Probability distribution of Sox9+ HFSCs along the hair follicle of control and rCARD applied C1−/− skins 24 h post wounding, represented as a heat-map with distance from the epidermis represented on the y-axis. Quantification is performed using at least three biological replicates (Control, n = 16; rCARD, n = 14). The cyan-colored box drawn on the heat map represents the portions that are statically different between control and rCARD-treated conditions. (C, E, and G) p/pmax represents the normalized frequencies to their maxima. (H) Immunofluorescence images of skin sections 72 h post wound with Sox9+ (cyan) Lgr5-traced (red). The arrow indicates the wound edge. White dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis.
Sox9 tracing of HFSC migration. (A) Three points at the junction between the epidermis and hair follicle were marked (reference points) and the positions of individual Sox9+ HFSCs were marked manually. The distance of individual Sox9+ HFSCs from the three reference points (D; represented by white dashed lines) and the frequency of localization along the length of the hair follicle was calculated (see Materials and methods). (B) Cross-sectional time-point immunofluorescence images of localization of Sox9+ HFSCs (red) in hair follicles of the unwounded and wounded skins at 0 (unwounded) 8, 16, and 24 h post wounding. K5 marks the epidermis and hair follicles in cyan. The arrow indicates the wound edge, and the bracket denotes the bulge. Insets are magnified views of the infundibulum marked by the dashed box. Representative images of at least three biological replicates have been presented. (C) Probability distribution of Sox9+ HFSCs along the hair follicle in unwounded (UW) and wounded skin at 8, 16, and 24 h after wounding, represented as a heat-map with distance from the epidermis represented on the y-axis. Quantification is performed using at least three biological replicates (UW, n = 18; 8 h, n = 14; 16 h, n = 30; 24 h, n = 14). The cyan-colored box drawn on the heat map represents the portions that are statically different between unwounded (UW) condition and the respective time points. (D) Immunofluorescence images of localization of Sox9+ HFSCs in the hair follicles of unwounded and wound proximal region of wild-type (WT) and caspase-1 null (C1−/−) mice at 24 h post wounding. Lower panel is the magnified view of the infundibulum marked by the dashed box. K5 marks the epidermis and hair follicles in cyan, and arrows indicate the wound edge. Representative images of at least three biological replicates have been presented. (E) Probability distribution of Sox9+ HFSCs along the hair follicle of unwounded (UW) and wounded wild-type (WT) and caspase-1 null (C1−/−) skins at 8, 16, and 24 h post wounding, represented as a heat-map with distance from the epidermis represented on the y-axis. Quantification is performed using at least three biological replicates ([WT (UW, n = 18; 16 h, n = 30; 24 h, n = 14], C1−/− [UW, n = 27; 16 h, n = 13; 24 h, n = 15]) The cyan-colored box drawn on the heat map represents the portions that are statically different between unwounded (UW) and the respective time points. Note, UW, 16 and 24 h of (C and E) are same. (F) Immunofluorescence images of the localization of Sox9+ HFSCs in the 24 h wound proximal hair follicles of caspase-1 null mice skin upon topical application of recombinant CARD (rCARD) compared with the vector control. Lower panel is the magnified view of the infundibulum marked by the dashed box. K5 marks epidermis and hair follicles in cyan. Arrow indicates the wound edge. Representative images of at least three biological replicates have been presented. (G) Probability distribution of Sox9+ HFSCs along the hair follicle of control and rCARD applied C1−/− skins 24 h post wounding, represented as a heat-map with distance from the epidermis represented on the y-axis. Quantification is performed using at least three biological replicates (Control, n = 16; rCARD, n = 14). The cyan-colored box drawn on the heat map represents the portions that are statically different between control and rCARD-treated conditions. (C, E, and G) p/pmax represents the normalized frequencies to their maxima. (H) Immunofluorescence images of skin sections 72 h post wound with Sox9+ (cyan) Lgr5-traced (red). The arrow indicates the wound edge. White dotted line denotes the basement membrane separating the epidermis and hair follicle from the dermis.
Since the Sox9 labeling of HFSC reproduced similar results as the lineage reporter mouse, we utilized this approach to determine the contribution of caspase-1 in HFSC migration in the mouse model of atopic dermatitis (tamoxifen-induced C8cKO [iC8cKO]) (Fig. 3 D). We observed a noticeable accrual of Sox9+ HFSC in the infundibulum of the hair follicles of iC8cKO adult mice compared with that of the tamoxifen-injected WT mice (Fig. 3 E and Fig. S3 A). Further, the migration of Sox9+ HFSCs was significantly reduced in the iC8cKO/caspase-1 double-knockout skin (Fig. 3 E and Fig. S3 A). Supporting this, we also found that in the constitutive epidermal caspase-8 knockout neonatal mice, there was a gradual increase in the number of Sox9+ HFSCs in the epidermis (Fig. S3 B) as the atopic dermatitis phenotype progressively increases with age (Li et al., 2010).
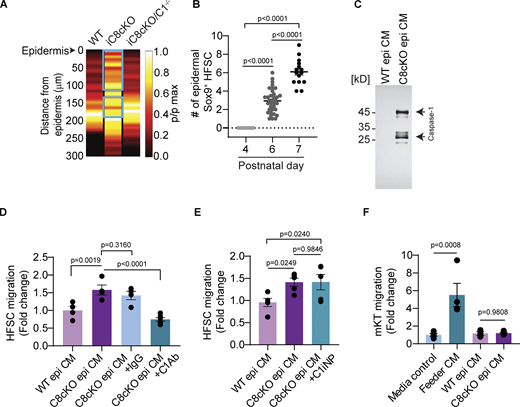
Role of Caspase-1 in inflammatory skin conditions. (A) Probability distribution of Sox9+ HFSCs in the hair follicles of adult wild-type (WT), tamoxifen-induced C8cKO (iC8cKO), and caspase-8/caspase-1 double knockout (iC8cKO/C1−/−) mice. The distance from the epidermis is represented on the y-axis. Quantification is performed using at least three biological replicates (UW, n = 18; 8 h, n = 14; 16 h, n = 30; 24 h, n = 14). Quantification is performed using at least three biological replicates (WT, n = 18; iC8cKO, n = 20; iC8cKO/C1−/−, n = 19). The cyan-colored box drawn on the heat map represents the portions that are statically different between represented conditions compared with WT. p/pmax represents the normalized frequencies to their maxima. (B) Number of Sox9+ HFSCs present in the interfollicular epidermis (within a 50 × 50 µm area) quantified from at least three C8cKO mice at postnatal day 4, 6, and 7 (p4, n = 23; p6, n = 88; p7, n = 32). Mean ± SEM are reported, and P value is calculated by one-way ANOVA. (C) Full Western blot of epidermal conditioned media prepared from WT and C8cKO mice probed with antibody against Caspase-1. (D) In vitro HFSC chemotaxis using a Boyden chamber assay towards epidermal conditioned media from wild-type (WT epi CM), C8cKO epidermis (C8cKO epi CM). Conditioned media from the C8cKO epidermis was immunodepleted with IgG control antibody (C8cKO epi CM+IgG) or Caspase-1 antibody (C8cKO epi CM+C1Ab). (E) HFSC migration in a Boyden chamber assay in response to inhibition of catalytic activity of extracellular Caspase-1 using the chemical inhibitor (Ac-YVAD-CHO) (C1iNP) in the C8cKO epi CM (C8cKO epi CM+C1iNP) compared with the vehicle-treated C8cKO (C8cKO epi CM) and WT epidermal conditioned media (WT epi CM). (F) Primary mouse keratinocyte (mKT) chemotaxis using a Boyden chamber assay toward WT epidermal conditioned media (WT epi CM) or C8cKO epidermal conditioned media (C8cKO epi CM). Media control induces no migration and is used as the benchmark for the basal migration of cells with no chemoattractant. Feeder cell-conditioned media (Feeder CM) was used as a positive control for inducing migration. (D–F) Quantification is done from at least three distinct biological replicates ([D] WT epi CM, n = 4; C8cKO epi CM, n = 4; C8cKO epi CM+IgG, n = 4; C8cKO epi CM+C1Ab, n = 6, [E] WT epi CM, n = 5; C8cKO epi CM, n = 5; C8cKO epi CM+C1iNP, n = 5, [F] Media control, n = 4; Feeder CM, n = 4; WT epi CM, n = 3; C8cKO epi CM, n = 4). Mean ± SEM are reported and P values are calculated by one-way ANOVA. Source data are available for this figure: SourceData FS3.
Role of Caspase-1 in inflammatory skin conditions. (A) Probability distribution of Sox9+ HFSCs in the hair follicles of adult wild-type (WT), tamoxifen-induced C8cKO (iC8cKO), and caspase-8/caspase-1 double knockout (iC8cKO/C1−/−) mice. The distance from the epidermis is represented on the y-axis. Quantification is performed using at least three biological replicates (UW, n = 18; 8 h, n = 14; 16 h, n = 30; 24 h, n = 14). Quantification is performed using at least three biological replicates (WT, n = 18; iC8cKO, n = 20; iC8cKO/C1−/−, n = 19). The cyan-colored box drawn on the heat map represents the portions that are statically different between represented conditions compared with WT. p/pmax represents the normalized frequencies to their maxima. (B) Number of Sox9+ HFSCs present in the interfollicular epidermis (within a 50 × 50 µm area) quantified from at least three C8cKO mice at postnatal day 4, 6, and 7 (p4, n = 23; p6, n = 88; p7, n = 32). Mean ± SEM are reported, and P value is calculated by one-way ANOVA. (C) Full Western blot of epidermal conditioned media prepared from WT and C8cKO mice probed with antibody against Caspase-1. (D) In vitro HFSC chemotaxis using a Boyden chamber assay towards epidermal conditioned media from wild-type (WT epi CM), C8cKO epidermis (C8cKO epi CM). Conditioned media from the C8cKO epidermis was immunodepleted with IgG control antibody (C8cKO epi CM+IgG) or Caspase-1 antibody (C8cKO epi CM+C1Ab). (E) HFSC migration in a Boyden chamber assay in response to inhibition of catalytic activity of extracellular Caspase-1 using the chemical inhibitor (Ac-YVAD-CHO) (C1iNP) in the C8cKO epi CM (C8cKO epi CM+C1iNP) compared with the vehicle-treated C8cKO (C8cKO epi CM) and WT epidermal conditioned media (WT epi CM). (F) Primary mouse keratinocyte (mKT) chemotaxis using a Boyden chamber assay toward WT epidermal conditioned media (WT epi CM) or C8cKO epidermal conditioned media (C8cKO epi CM). Media control induces no migration and is used as the benchmark for the basal migration of cells with no chemoattractant. Feeder cell-conditioned media (Feeder CM) was used as a positive control for inducing migration. (D–F) Quantification is done from at least three distinct biological replicates ([D] WT epi CM, n = 4; C8cKO epi CM, n = 4; C8cKO epi CM+IgG, n = 4; C8cKO epi CM+C1Ab, n = 6, [E] WT epi CM, n = 5; C8cKO epi CM, n = 5; C8cKO epi CM+C1iNP, n = 5, [F] Media control, n = 4; Feeder CM, n = 4; WT epi CM, n = 3; C8cKO epi CM, n = 4). Mean ± SEM are reported and P values are calculated by one-way ANOVA. Source data are available for this figure: SourceData FS3.
We have previously established that the conditional knockout of Caspase-8 from the epidermis is sufficient to mimic the wound-healing response throughout the skin of the animal in the absence of any injury (Lee et al., 2009). Thus, in this mouse model, unlike the excisional wound, a large number of keratinocytes exhibit the wound response. Consistent with our previous report (Lee et al., 2009), we confirmed that the conditioned media from C8cKO epidermis contains Caspase-1 (Fig. S3 C) and is capable of inducing HFSC migration in the Boyden chamber/transwell migration assay (Fig. 3 F). Removing caspase-1 from the C8cKO background impaired the ability of the CM to induce HFSC migration (Fig. 3 F). Supporting this finding, C8cKO-conditioned media immunodepleted of Caspase-1 failed to induce HFSC migration in the Boyden chamber (Fig. S3 D). Also, upon inhibiting Caspase-1 activity in the conditioned media using a cell impermeable pharmacological inhibitor, we found no reduction in migration (Fig. S3 E) in accordance with the previous data showing that the Caspase-1 induces HFSC migration independent of its catalytic activity (Fig. 2, C and F). Consistent with the specificity of rCARD toward HFSC migration (Fig. S1 N), the CM from iC8cKO epidermis had no effect on epidermal keratinocyte migration in the Boyden chamber assay (Fig. S3 F).
Our findings provide new insights into the underlying mechanisms driving wound-healing and inflammatory skin diseases (Fig. 4). This may open up new avenues of research into the development of more effective treatments to accelerate the repair of cutaneous wounds and therapies for pathologies marked by epidermal hyperplasia such as UV-induced sunburn, atopic dermatitis, and psoriasis. It would also be interesting to determine whether the epidermal hyperplasia in squamous cell carcinoma and basal cell carcinoma are fueled by extracellular Caspase-1. This is stimulated by reports demonstrating that this disease is partly due to a mixture of epidermal stem cells and HFSCs in the epidermis (Latil et al., 2017; Peterson et al., 2015; White et al., 2016).

Model of Caspase-1 mediated HFSC homing to the epidermis upon wounding. Stressed epidermal keratinocytes secret Caspase-1 to the extracellular milieu upon wounding, UV exposure or genetic ablation of Caspase-8. Via caspase activation and recruitment domain (CARD) of caspase-1, hair follicle stem cells are coaxed to migrate to the epidermis.
Model of Caspase-1 mediated HFSC homing to the epidermis upon wounding. Stressed epidermal keratinocytes secret Caspase-1 to the extracellular milieu upon wounding, UV exposure or genetic ablation of Caspase-8. Via caspase activation and recruitment domain (CARD) of caspase-1, hair follicle stem cells are coaxed to migrate to the epidermis.
Materials and methods
Ethics statement
All animal work was approved by the Institutional Animal Ethics Committee in the Colin Jamora lab (INS-IAE-2019/06[R1]). Experiments on mice followed the norms specified by the Committee for the Purpose of Control and Supervision of Experiments on Animals (Government of India). All experimental work was approved by the Institutional Biosafety Committee of inStem (inStem/G-141(3)/2012 and inStem/G-141(3)-06/2016).
Mouse models
Lgr5-tdTom mice were created by crossing B6.129P2-Lgr5tm1(cre/ERT2)Cle/J (#008875; JAX) with B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (#007914; JAX). Lgr5-tdTom/C1−/− null mice were created by mating Lgr5-tdTom mice with caspase-1 null mice (#016621; JAX). The caspase-1 null mice have been described in previous reports (Li et al., 1995; Lee et al., 2015). Cre-driven recombination was induced in these animals by intraperitoneal injection of 1.5 mg tamoxifen (#T5648; Sigma-Aldrich) dissolved in corn oil (S5007; Sigma-Aldrich) to 7-wk-old animals for five consecutive days. Animals were used for wounding experiments after a waiting period of 5 days.
Atopic dermatitis mouse model (epidermal specific caspase-8 knockout) C8cKO is described in previous reports (Lee et al., 2009; Li et al., 2010). Inducible K14-CreER C8fl/fl were created by crossing K14-CreER (Vasioukhin et al., 1999) with C8fl/fl (Beisner et al., 2005) mice. K14-CreER C8fl/fl/C1−/− were created by crossing K14-CreER C8fl/fl with Caspase-1 null mice (#016621; JAX). Cre-driven recombination was induced in these animals by intraperitoneal injection of 5 mg tamoxifen (#T5648; Sigma-Aldrich) dissolved in corn oil (S5007; Sigma-Aldrich) to 7-wk-old animals for three alternative days. Following a waiting period of 2 wk, the mice were sacrificed for histological analysis of the skin.
Wounding and tissue processing
The adult mice were shaved, and their back skin was wounded under isoflurane-induced anesthesia using 5-mm punch biopsy to make full-thickness excisional wounds. Animals were sacrificed at specific timepoints to collect skin samples for histological analysis. Skin samples were embedded in tissue freezing solution and 10–20 µm thick sections were obtained.
Immunostaining, image acquisition, and analysis
For histology, skin sections were fixed using 4% PFA at room temperature for 10 min. Sections were permeabilized using 0.2% Triton X-100 in 1× PBS for 10 min to probe for intracellular proteins like αSMA, Sox9, and K5. Sections or cells were not permeabilized while probing for extracellular Caspase-1 or to detect surface-bound recombinant CARD (rCARD). Sections or cells were probed with primary antibody overnight at 4°C (anti-Sox 9 [ab185230, rabbit, 1:100; Abcam], anti-Ki67 [ab16667, rabbit, 1:200; Abcam], anti-αSMA [ab5694, rabbit, 1:100; Abcam], anti-Keratin 5 [raised in lab, chicken, 1:100], and anti His [EPR20547, rabbit, 1:200; Abcam] and Anti Caspase-1 [ab1871, rabbit, 1:200; Abcam]). Primary signals were amplified with secondary antibodies conjugated with fluorophores (Alexa-488 anti-chicken [1:400], Alexa-568 anti-rabbit [1:400], and Alexa-647 anti-rabbit [1:400] from Life Technologies). To detect the cell surface–bound rCARD, HFSCs were incubated with rCARD for 4 h at 37°C and fixed with 4% PFA at room temperature and probed for anti-His antibody (Anti-His [EPR20547, rabbit, 1:200; Abcam]). Fluorescent images of tissue samples were obtained on the Olympus FV3000 confocal microscope (UPLFLN oil immersion 40×/1.3). Images were tiled using FV31S-SW FLUOVIEW, Zeiss Axio observer 7 (Plan-APOCHROMAT 20×/0.8, camera model—Hamamatsu Photonics ORCA Flash 4.0). Images were tiled using ZEN 3.8 pro and Olympus IX73 (C Plan N 10×/0.25 PhC or LUCPlanFL N 20×/0.45 Ph1). The images were acquired using the Cellsens software, camera model Olympus DP80. All images were acquired at room temperature. For image processing, ImageJ software (Fiji) was used. Immunofluorescence images were z-projected as a maximum intensity projection wherever applicable. The number of non-bulge Lgr5-tdT cells was manually counted. Graphs were plotted using Graphpad Prism software.
Cross-sectional time point study of Sox9+ HFSC migration using image analysis algorithm
To analyze the distribution of Sox9+ HFSCs along the hair follicles, the positions of Sox9+ nuclei and the hair follicle epidermis junction were marked manually in Fiji (Fig. S2 A). The spatial coordinates of all the marked nuclei were exported to MATLAB for further analysis. The MATLAB routine we developed calculates the distance vector, D of individual Sox9+ nuclei in a hair follicle from the epidermal junction. It then performs the binning of distance from the epidermis (bin size [b]: 8 um was chosen based on the average size of nuclei in the hair follicles). It then calculates the number (N) of Sox9+ nuclei in each bin. The algorithm then calculates the empirical probability for finding Sox9+ nuclei in a given bin, pi = Ni/ΣNi. Then based on data similarly collected from several hair follicles across multiple biological samples, the probability of occurrence of these nuclei along the length of hair follicles was estimated. The spatial frequency was calculated from 8-μm bins along the length of hair follicles. For the heatmap, the frequencies were normalized to their maxima (p/pmax) and plotted such that the white color represents 1 and the darker shades until black represent lower values until 0. Statistics were performed on pi values. Differences in localization frequency were considered significant for P < 0.05 with at least n = 3 mice for each condition and represented by a green box (Fig. S1 C). In the wounded samples, the first hair follicle immediately adjacent to the wound, particularly within 300 µm, was considered. The MATLAB code used for data analysis of the frequency distribution of the HFSC migration in the epidermis can be found here: https://github.com/skinlab-sunnyk/HFSCs.
Cloning, expression, and purification of recombinant proteins
The murine Caspase-1 wild-type and catalytic dead constructs were gifted to us in a retroviral vector, pMScV (Broz et al., 2010). The genes for procaspase-1 were cloned into a pET-28a vector by engineering flanking Xho-1 and Nco-1 sites. For purification from bacterial lysates, the protein sequence was followed by C-terminal 6x-His tag expression from the vector backbone. The constructs were expressed in BL21 pLysS under IPTG (#0487; Amresco) induction at 18°C for 16 h. The cells were lysed by sonication in lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 10% glycerol, 5 mM DTT, 100 mM sodium malonate dibasic monohydrate [#M4795; Sigma-Aldrich], pH 8.0 with EDTA free Protease Inhibitor Cocktail [11836145001; Roche], DNase I and lysozyme). Following centrifugation at 16,000 g, the supernatant was incubated with Ni-NTA beads for 3 h at 4°C in the presence of 10 mM imidazole. After washing with 20 and 40 mM imidazole, the recombinant proteins were eluted at 120 mM imidazole at 4°C in the lysis buffer without additives. Following dialysis to imidazole-free buffer, the proteins were snap-frozen for future use. Substrate cleavage assay was performed by incubating purified recombinant proteins with Caspase-1 15 µM substrate Ac-YVAD-AMC (ALX-260-024; Enzo Lifesciences) at room temperature and measuring the emitted fluorescence over time. The CARD of the caspase-1 gene synthesized (Geneart; Thermo Fisher Scientific) after codon optimization with N-terminal Strep-II tag with Enterokinase cleavage site and C-terminal 10x-His tag was then cloned into pET-28a vector using the same restriction sites. The cell pellet was lysed by probe sonication in a denaturing buffer composed of 50 mM Tris-HCl, pH 8.0 with 6.5 M GdnHCl, 300 mM NaCl, and 50 mM imidazole; following centrifugation at 16,000 g, the supernatant was incubated with Ni-NTA beads (#88221; Thermo Fisher Scientific). Following on-column refolding using a reducing urea gradient in Tris-buffer with imidazole, the final elution was performed at 500 mM imidazole at 4°C. Following dialysis into an imidazole-free buffer, the proteins were snap-frozen for future use. “Control” or “Vector” in Fig. 2, Fig. S1, and Fig. S2 refers to BL21 pLysS transfected with empty vector (pET-28a) but which underwent a similar treatment along with the cells transfected with the vector containing the gene of interest. The purity of the proteins was tested by silver staining of the eluates, and the identity was confirmed by Western blotting by probing with anti-His antibody (EPR20547, rabbit, 1:5,000; Abcam).
Topical application of recombinant proteins
10 µl recombinant proteins (300 µg/ml of rCasp-1, rCasp-1 C284A, and 100 µg/ml of rCARD) were topically applied to the wound beds every 12 h from the time of wounding until the time of skin harvesting.
Preparation of cell culture and conditioned media
HFSCs were isolated from 8-wk-old wild-type mice skin by published methods and cultured on feeders for eight passages in HFSC culture media (E-media with 0.3 mM calcium) (Nowak and Fuchs, 2009), or the 3C culture method was used as reported by Chacón-Martínez et al. (2017).
Briefly, 8-wk-old mice were sacrificed following approved protocols, and their hairs were removed using an electric shaver. Skins were washed with 70% ethanol and blot dried. The skins were harvested using forceps and scissors. The fat and blood vessels were removed by scraping the skin with a scalpel and the skin containing only the dermis and epidermis was floated on 10 ml PBS containing 0.24% trypsin overnight at 4°C. 16 h later, the epidermis was separated from the dermis and minced. The minced epidermis was triturated using a 10-ml pipette until the single-cell suspension was formed. The suspension was passed through 70-µm and subsequently 45-µm cell strainers. The cells were washed with 0 mM calcium E media (Dulbecco’s modified Eagle’s medium/Ham’s F-12 Nutrient Medium (3:1 Mix) 8% with 8% fetal calf serum (calcium free) (Gibco), 5 µg/ml insulin (#I5500; Sigma-Aldrich), 5 µg/ml transferrin (#T8158; Sigma-Aldrich), and 20 pM triiodothyronine (#T6397; Sigma-Aldrich)). The cells were pelleted at 250 g for 10 min and stained with FITC (CD34) (11-0341-82; eBioscience) and PE (CD49f) (561894; BD), and the HFSCs were FACS-isolated. Isolated HFSCs were plated on mytomycin c-treated 3T3-J2 cells. The HFSCs were supplied with 0.3 mM calcium E media. The culture was maintained at 37°C, 5% CO2. E media leads to the death of 3T3-J2 cells; thus, to propagate the HFSC culture, continuously fresh 3T3-J2 cells have to be supplied.
For 3C culture, 8 × 104 cells were suspended in 100 μl ice-cold 1:1 mixture of KGM (MEM [Spinners modification, #M8167; Sigma-Aldrich] supplied with 5 μg/ml insulin [#I5500; Sigma-Aldrich], 10 ng/ml EGF [#E9644; Sigma-Aldrich], 10 μg/ml transferrin [#T8158; Sigma-Aldrich], 10 μM phosphoethanolamine [#P0503; Sigma-Aldrich], 10 μM ethanolamine [#E0135; Sigma-Aldrich], 0.36 μg/ml hydrocortisone, [#386698; Calbiochem], 5 μM Y27632 [130-103-922; Miltenyi Biotec], 20 ng/ml mouse recombinant VEGF [130-094-087; Miltenyi Biotec], 20 ng/ml human recombinant FGF-2 [130-093-837; Miltenyi Biotec], 2 mM glutamine [Gibco], 100 U/ml penicillin and 100 μg/ml streptomycin [Gibco], 8% calcium free fetal calf serum [Gibco], and 50 µM CaCl2) and Matrigel (CLS354277; Corning) was dispensed as a droplet in 24-well cell culture dishes. The suspension was allowed to solidify for 15 min at 37°C. 500 μl of KGM media was added to each well. The culture was maintained at 37°C, 5% CO2. The media was changed the next day after initial seeding, and subsequently, every second day the media was changed. Cells were passaged every 10–14 days. For cell passaging, cells were extracted from Matrigel by pipetting and incubation in 0.5% Trypsin (25300054; Gibco) and 0.5 mM EDTA for 5 min at 37°C.
Wild-type primary mouse keratinocyte cultures were established by isolating cells from the epidermis of newborn pups and cultured in low-calcium (0.05 mM) E-media as described previously (Lee et al., 2009). To isolate keratinocytes and prepare conditioned media, the epidermis was chemically dissociated from the dermis by dispase treatment (1 h, at 37°C) from the skin of newborn pups aged p0–p3. For primary keratinocyte isolation, the epidermis was briefly treated with 0.25% Trypsin-EDTA, followed by culturing the isolated cells on mitomycin C-treated J2-3T3 feeder cells in low-calcium (0.05 mM Calcium) E-media (Nowak and Fuchs, 2009). The low calcium media leads to the death of any other cell type in the culture, leading to the propagation of a highly pure population of keratinocytes. For preparing epidermal conditioned media (epi CM), the isolated epidermis was floated in serum-free E-media for 16 h. To prepare conditioned media from the wounded epidermis, isolated epidermal explants were subjected to multiple incisional wounds and were subsequently floated on serum-free E-media for 16 h.
Western blotting
Conditioned media or purified proteins were loaded on SDS-polyacrylamide gels and transferred to the nitrocellulose membrane. Extracellular Caspase-1 found in the conditioned media was detected by anti-Caspase-1 antibody (PAB592 Mu01 Cloud Clone, rabbit, 1:4,000), and recombinant Caspase-1 proteins and recombinant CARD were detected using anti-His antibody (EPR20547, rabbit, 1:5,000; Abcam).
Chemotaxis assays and quantification
HFSCs or mouse primary keratinocytes were trypsinized and plated (30,000 cells per well) onto the top chamber of either 96-well transwell plates with a membrane having 8-μm pore size. The conditioned media or proteins being tested (3 µg/ml of rCasp-1 and rCasp-1 C284A and 1 µg/ml of rCARD) were dissolved in HFSC culture media or low Calcium E-media and added to the bottom chamber. Cell migration was assayed after 12 h of incubation, in which the cells were fixed by PFA and stained with Crystal Violet. The top of the transwell was cleaned before imaging on an Olympus IX73 (C Plan N 10×/0.25 PhC). The images were acquired using the Cellsens software, camera model—Olympus DP80. Cells were counted using the Cell Counter plugin on ImageJ-Fiji.
UV irradiation experiments
Shaved back skin of WT and C1−/− mice were exposed to UV using Narrowband 311 nm NBUVB phototherapy lamp under isoflurane-induced anesthesia for 15 min. The skin was harvested after 3 days of UV exposure.
To prepare the UV-exposed mKT conditioned media (UV mKT CM), differentiated keratinocyte culture was exposed to 50 mJ of UV radiation in phosphate buffer saline followed by the addition of serum-free keratinocyte culture media. Conditioned media was collected after 4 h.
Statistical analysis
For all statistical analyses, at least three independent replicates were used. Data were pooled from the independent replicates. ±SEM were reported, and P values were calculated by one-tailed unpaired Student’s t test or ordinary one-way ANOVA. Data distribution was assumed to be normal but was not formally tested.
In “Cross-sectional time point study of Sox9+ HFSC migration using image analysis algorithm,” the statistical significance of difference was calculated using one one-tailed Student’s t test. Graphical representation shows individual data points. Statistical analysis and graphical representations were done using Graphpad Prism version 8.4. Data distribution was assumed to be normal but was not formally tested.
Online supplemental material
Supplemental material includes additional data investigating the presence of extracellular Caspase-1 near the wound edge and in the conditioned media collected from the wounded epidermis (Fig. S1, A and B), characterization of purified recombinant WT and mutant Caspase-1 and the CARD domain (Fig. S1, C–H), and characterization of the penetration of topically applied rCARD in the wound bed (Fig. S1 I). Fig. S1 J is a diagram of the Boyden chamber/transwell and Fig. S1 K are representative images of migrated HFSCs in the Boyden chambers. Fig. S1 L is the quantification of migrating, non-proliferating HFSCs in response to catalytically dead rCasp-1 and Fig. S1 M is the characterization of the effect of Mytomycin C on the proliferative state of HFSCs. Fig. S1 N is the quantification of the migratory response of mouse primary keratinocytes to rCARD. Fig. S1 O is the characterization of the rCARD bound to the surface of unpermeabilized HFSCs. Fig. S2, A–G demonstrates the standardization of the image analysis algorithm on cross-sectional time point study of Sox9+ HFSC migration in WT, C1−/−, and on topical application of rCARD on C1−/− wound bed. Fig. S2 H demonstrates Lgr5+ and Sox9+ as distinct HFSC subpopulations. Fig. S3, A and B are the quantification of Sox9+ HFSC distribution in atopic dermatitis mouse models. Fig. S3 C is a Western blot image for extracellular Caspase-1 in the conditioned media prepared from the epidermis of the atopic dermatitis mouse model. Fig. S3, D and E are the quantification of HFSC migration in the Boyden chamber/transwell migration assay in response to conditioned media immunodepleted of Caspase-1 and conditioned media with catalytic activity inhibited extracellular Caspase-1. Fig. S3 F is the quantification of mouse primary keratinocyte migration in the Boyden chamber/transwell migration assay in response to epidermal conditioned media from the atopic dermatitis mouse model.
Data availability
The data underlying the figures in the manuscript are available in the published article and its online supplemental material.
Acknowledgments
We thank Professor Petr Broz (Department of Biochemistry, University of Lausanne, Lausanne, Switzerland) for his gift of the caspase-1 constructs.
This work is supported by the Wellcome-DBT India Alliance Early Career Fellowship awarded to S. Ghosh and grants to C. Jamora from the Department of Biotechnology of the Government of India (BT/PR8738/AGR/36/770/2013 and DBT/PR32539/BRB/10/1814/2019); and inStem Core funds. Animal work in the National Centre for Biological Sciences/inStem Animal Care and Resource Center was partially supported by the National Mouse Research Resource BT/PR5981/MED/31/181/2012;2013–2016 and 102/IFD/SAN/5003/2017–2018 from DBT. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank the Central Imaging and Flow Cytometry Facility of the Bangalore Life Sciences Cluster for flow cytometry and image acquisition support.
Author contributions: A. Hegde and S. Ghosh conceptualized, investigated, conducted formal analysis, and administered the project. A. Hegde and S. Ghosh wrote the original draft. A. Hegde reviewed and edited the manuscript. A.SHP. Ananthan, A. Dutta, S. Kataria, S. Prabhu, and S.U. Khedkar performed investigation and formal analysis. S. Kataria developed the image analysis algorithm and performed formal analysis. S. Ghosh and C. Jamora acquired the funding. C. Jamora conceptualized and administered the project, and reviewed and edited the manuscript.

