


Exportin receptors are concentrated in the nucleus to transport essential cargoes out of it. A mislocalization of exportins to the cytoplasm is linked to disease. Hence, it is important to understand how their containment within the nucleus is regulated. Here, we have studied the nuclear efflux of exportin2 (cellular apoptosis susceptibility protein or CAS) that delivers karyopherinα (Kapα or importinα), the cargo adaptor for karyopherinβ1 (Kapβ1 or importinβ1), to the cytoplasm in a Ran guanosine triphosphate (RanGTP)-mediated manner. We show that the N-terminus of CAS attenuates the interaction of RanGTPase activating protein 1 (RanGAP1) with RanGTP to slow GTP hydrolysis, which suppresses CAS nuclear exit at nuclear pore complexes (NPCs). Strikingly, a single phosphomimetic mutation (T18D) at the CAS N-terminus is sufficient to abolish its nuclear retention and coincides with metastatic cellular behavior. Furthermore, downregulating Kapβ1 disrupts CAS nuclear retention, which highlights the balance between their respective functions that is essential for maintaining the Kapα transport cycle. Therefore, NPCs play a functional role in selectively partitioning exportins in the cell nucleus.
Introduction
Nuclear protein localization is a hallmark of cellular function (Hung and Link, 2011). This is underpinned by a housekeeping process in eukaryotic cells termed nucleocytoplasmic transport (NCT; Wing et al., 2022). Nuclear transport receptors known as karyopherins (or Kaps; Baade and Kehlenbach, 2019; Kalita et al., 2021) orchestrate NCT by circulating through selective conduits in the nuclear envelope (NE) known as nuclear pore complexes (NPCs) to deliver signal-specific cargoes to the nucleus or cytoplasm (Akey et al., 2022; Schuller et al., 2021; Zimmerli et al., 2021). Specifically, Kaps that usher nuclear localization signal (NLS)-cargoes from the cytoplasm into the nucleus are known as importins, whereas exportins shuttle nuclear export signal (NES)-cargoes out from the nucleus into the cytoplasm (Christie et al., 2016). Defects in NCT have known associations with neurological disorders (Kim and Taylor, 2017), cancer (Çağatay and Chook, 2018), and viral infections (Fulcher and Jans, 2011), including SARS-CoV-2 (Miorin et al., 2020).
To guard against non-specific entities, each NPC is protected by a permeability barrier that is generated by intrinsically disordered proteins called phenylalanine–glycine–repeat nucleoporins (FG Nups; Hoogenboom et al., 2021). Both importins and exportins exert multivalent interactions with the FG Nups to selectively traffic their cargoes across the NPC permeability barrier (Kapinos et al., 2014). Upon entering the nucleus, importin–NLS–cargo complexes encounter the guanosine triphosphate-bound form of GTPase Ran (RanGTP), which triggers NLS-cargo release by binding to the importin. Conversely, exportins form complexes with RanGTP and their cognate NES–cargoes (NES–cargo–exportin–RanGTP) in the nucleus for export (Fornerod et al., 1997). Following their passage through the NPC, RanGTP is hydrolyzed to RanGDP by the collective action of RanGTPase-activating protein 1 (RanGAP1), Ran-binding protein 1 (RanBP1; Plafker and Macara, 2000), and/or Ran-binding protein 2 (RanBP2, also termed Nup358; Villa Braslavsky et al., 2000), which drives NES–cargo–exportin–RanGTP disassembly that releases NES-cargoes into the cytoplasm.
Just as importins and exportins are needed to traffic cargoes across the NPC, the permeability barrier also impedes the retrograde movement of NLS- and NES-cargoes once delivery is completed, which results in their asymmetric partitioning in the nucleus and cytoplasm, respectively (Görlich et al., 1996). Thus, NCT directionality derives from the Ran-mediated release of NLS- or NES-cargoes in the nucleus and cytoplasm, respectively (Nachury and Weis, 1999). However, one peculiarity concerns how Kaps are themselves partitioned in an asymmetric manner that is directionally opposed to their cargoes (Kalita et al., 2021; Wühr et al., 2015). Evidently, standalone Kaps, Kap-cargo complexes, and Kap-RanGTP complexes all demonstrate the capacity to bind with FG Nups and traverse the NPC permeability barrier (Kapinos et al., 2017). Exportins especially are highly enriched in the nucleus under steady-state conditions despite lacking NLSs (Kalita et al., 2021; Wühr et al., 2015). Consequently, the factors that promote exportin retention in the nucleus remain to be verified. To underscore the importance of this behavior, the mislocalization of exportins to the cytoplasm is commonly associated with cancer (Çağatay and Chook, 2018; Mahipal and Malafa, 2016).
In this work, we have investigated the nuclear retention mechanism of exportin2 (XPO2), henceforth known as cellular apoptosis susceptibility protein (CAS; the human homolog of yeast chromosome segregation protein or Cse1p; Behrens et al., 2003; Brinkmann et al., 1995). CAS exclusively shuttles karyopherinα (Kapα; importinα) out of the nucleus (Kutay et al., 1997; Gilchrist et al., 2002; Matsuura and Stewart, 2004; Sun et al., 2013). Kapα itself is an essential adaptor protein that mediates the binding of diverse classical NLS-cargoes to the canonical importin karyopherinβ1 (Kapβ1; also termed importinβ1; Pumroy and Cingolani, 2015). Therefore, the nuclear import and export pathways of Kapα are linked in a closed loop by Kapβ1 and CAS, respectively. For this reason, CAS represents an important system for studying Kap partitioning and the impact of its dysregulation on NCT more generally. Overall, we find that the N-terminus of CAS suppresses RanGTP hydrolysis by RanGAP1 to prevent an uncontrolled nuclear efflux of CAS at the NPC. With this understanding, we have identified conditions that abrogate CAS nuclear retention. This includes a single phosphomimetic mutation at the CAS N-terminus that elicits cellular activity resembling metastatic behavior in melanoma and breast cancer (Chin et al., 2015; Jiang, 2016; Liao et al., 2012; Tai et al., 2010a; Wu et al., 2016). Interfering with the Ran gradient also results in the delocalization of non-phosphorylated CAS, as is observed in early apoptosis during apoptosome formation (Monian and Jiang, 2016). Our findings further shed light on how Kapα maintains the equilibrium partitioning of CAS and Kapβ1.
Results
The N-terminus of CAS is essential for its nuclear localization
Endogenous CAS (or endoCAS) accumulates in the cell nucleus (Fig. 1 A). We therefore asked if its partitioning is dependent on Kapα·CAS·RanGTP formation (henceforth termed CAS triple complex). At the structural level, CAS resembles a flexible alpha solenoid that is comprised of 20 tandem HEAT repeats (Matsuura and Stewart, 2004). In its Apo form, HEATs 1 and 2 at its N-terminus interact with HEAT 14 (Fig. 1 B). This is interrupted by RanGTP, which binds to HEAT 1, HEAT 13–14, and the HEAT 19 insertion loop. HEAT 19 is connected to HEAT 18 by a 45-aa-long flexible loop (termed C-loop). RanGTP-binding induces a concave surface in CAS that docks Kapα via interactions with the HEATs 2–7, 9, 12, and the C-loop (Cook et al., 2005; Matsuura and Stewart, 2004).

CAS nuclear localization involves its N-terminus. (A) CAS and Kapβ1 are asymmetrically partitioned in the nucleus and cytoplasm, respectively. Scale bar, 5 μm. (B) CAS structure adopted from Matsuura and Stewart (2004) (PDB ID: 1wa5, structure for the yeast homolog of CAS: CSE1Lp) and Cook et al. (2005) (PDB ID: 1z3h). Binding sites to RanGTP and Kapα such as HEATs 1–2 at the N-terminus (violet) and the C-loop (dark blue) are indicated. (C) Full-length CAS and truncation mutants used in this work. Binding sites for Kapα and RanGTP are indicated. (D) Immunostaining of Kapβ1 in cells expressing mCherry-CAS and associated truncation mutants. Scale bar, 5 μm. (E) N/C ratios reveal that mCherry-CAS, mCherry-CAS∆C, and mCherry-CAS∆Cloop are enriched in the nucleus but not mCherry-∆40NCAS, mCherry-∆40NCAS∆Cloop, mCherry-CASCloop, and mCherry-(1-40)NCAS. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Number of independent experiments: N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
CAS nuclear localization involves its N-terminus. (A) CAS and Kapβ1 are asymmetrically partitioned in the nucleus and cytoplasm, respectively. Scale bar, 5 μm. (B) CAS structure adopted from Matsuura and Stewart (2004) (PDB ID: 1wa5, structure for the yeast homolog of CAS: CSE1Lp) and Cook et al. (2005) (PDB ID: 1z3h). Binding sites to RanGTP and Kapα such as HEATs 1–2 at the N-terminus (violet) and the C-loop (dark blue) are indicated. (C) Full-length CAS and truncation mutants used in this work. Binding sites for Kapα and RanGTP are indicated. (D) Immunostaining of Kapβ1 in cells expressing mCherry-CAS and associated truncation mutants. Scale bar, 5 μm. (E) N/C ratios reveal that mCherry-CAS, mCherry-CAS∆C, and mCherry-CAS∆Cloop are enriched in the nucleus but not mCherry-∆40NCAS, mCherry-∆40NCAS∆Cloop, mCherry-CASCloop, and mCherry-(1-40)NCAS. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Number of independent experiments: N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
To interfere with these interactions, we first designed and characterized various CAS truncation mutants lacking specific binding sites for RanGTP or Kapα (Fig. 1 C; and Fig. S1, A–C). Thereafter, these mutants were expressed in HEK293T cells with an additional mCherry tag at their N-termini to facilitate visualization (Fig. 1 D). These were (i) mCherry-CAS as control; (ii) mCherry-CAS∆C lacking HEAT 19 and 20 at the C-terminus; (iii) mCherry-CAS∆Cloop where HEATs 19, 20, and the C-loop were deleted; (iv) mCherry-∆40NCAS that lacked the first 40 amino acids of the N-terminus; (v) mCherry-∆40NCAS∆Cloop where both the N-terminus and C-loop were deleted; (vi) mCherry-(1-40)NCAS, containing only the first 40 amino acids of CAS; and (vii) mCherry-CASCloop, which contained only the C-loop. Among these, the nuclear versus cytoplasmic (N/C) ratios of mCherry-CAS, mCherry-CAS∆C, and mCherry-CAS∆Cloop were >1, indicating enrichment in the nucleus (Fig. 1 E). Hence, C-termini deletions including the C-loop did not alter CAS nuclear localization. In comparison, nuclear localization was abolished for mCherry-∆40NCAS, mCherry-∆40NCAS∆Cloop, and mCherry-CASCloop, which showed a more even distribution between the nucleus and the cytoplasm (N/C ≈ 1). This implied that the N-terminus is required for CAS nuclear localization even though mCherry-(1-40)NCAS itself did not show any accumulation within the nucleus.
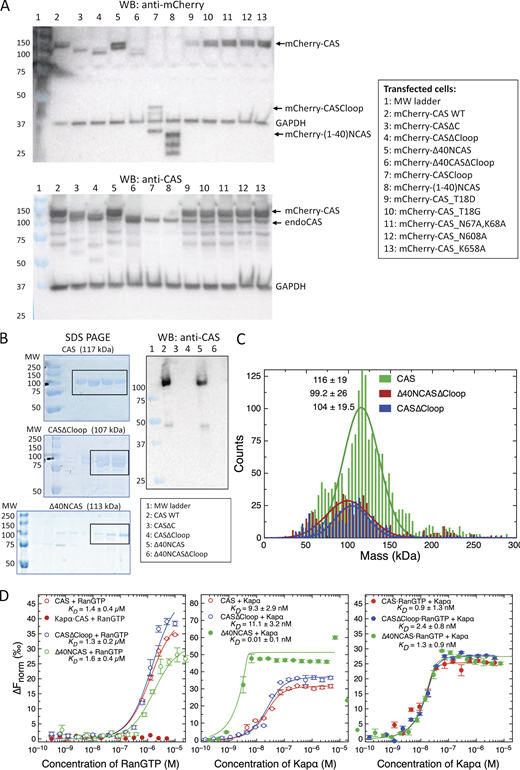
Characterization of CAS and its mutants. (A) Western blot of endoCAS and mCherry-CAS constructs expressed in HEK293T cells. All mCherry-CAS constructs were stably expressed and did not degrade. The epitope of anti-CAS is on the C-terminus of CAS, thus only CAS with non-truncated C-termini are detected. (B) Validation of CAS, CAS∆Cloop, and ∆40NCAS by SDS gel and Western blot. The fractions collected after size exclusion chromotography are indicated by a rectangle. (C) Polydispersity of CAS, CAS∆Cloop, and ∆40NCAS∆Cloop measured by mass photometry. (D) Binding of various CAS mutants with RanGTP in the presence and absence of Kapα measured by microscale thermophoresis (MST). It is worth noting that (i) preloading of Kapα to CAS inhibits subsequent RanGTP binding (left panel, solid red circles); and (ii) binding between ∆40NCAS and Kapα is enhanced. N = 3. Error bars denote standard deviation. Source data are available for this figure: SourceData FS1.

Characterization of CAS and its mutants. (A) Western blot of endoCAS and mCherry-CAS constructs expressed in HEK293T cells. All mCherry-CAS constructs were stably expressed and did not degrade. The epitope of anti-CAS is on the C-terminus of CAS, thus only CAS with non-truncated C-termini are detected. (B) Validation of CAS, CAS∆Cloop, and ∆40NCAS by SDS gel and Western blot. The fractions collected after size exclusion chromotography are indicated by a rectangle. (C) Polydispersity of CAS, CAS∆Cloop, and ∆40NCAS∆Cloop measured by mass photometry. (D) Binding of various CAS mutants with RanGTP in the presence and absence of Kapα measured by microscale thermophoresis (MST). It is worth noting that (i) preloading of Kapα to CAS inhibits subsequent RanGTP binding (left panel, solid red circles); and (ii) binding between ∆40NCAS and Kapα is enhanced. N = 3. Error bars denote standard deviation. Source data are available for this figure: SourceData FS1.
Despite their individual truncations, the in vitro binding of RanGTP to Δ40NCAS and CASΔCloop was verified by microscale thermophoresis to be comparable with CAS, whose value was consistent with previous reports (Cook et al., 2005; Matsuura and Stewart, 2004; Fig. S1 D). Likewise, the binding of Kapα to ∆40NCAS·RanGTP and CAS∆Cloop·RanGTP was equivalent to CAS·RanGTP. Hence, the observed cellular behavior was not likely caused by defects in CAS triple complex formation. When RanGTP was absent, Kapα formed a stable complex with CAS that inhibited subsequent RanGTP binding, as suggested previously (Matsuura and Stewart, 2004).
CAS triple complex binding to the FG Nups is stabilized by RanGTP and Kapα
Next, we questioned if their cellular localizations might stem from differences in their selective binding to the NPC vis-à-vis the FG Nups. As before, we used surface plasmon resonance (Kapinos et al., 2014; Schoch et al., 2012; Wagner et al., 2015) to probe the apparent binding affinity (KD) of CAS, CAS∆Cloop, and ∆40NCAS to cysteine-modified FG domains (i.e., Nup214, Nup62, Nup98, and Nup153; Materials and methods). Although CAS interacted more strongly with the FG domains than CAS∆Cloop and ∆40NCAS, their values were generally consistent with the binding range of Kapβ1 (Kapinos et al., 2014, 2017; Fig. 2). Likewise, FG domain-binding of Kapα·CAS·RanGTP, Kapα·CAS∆Cloop·RanGTP, and Kapα·∆40NCAS·RanGTP was comparable with Kapα·Kapβ1 although variations existed between different FG domains (Kapinos et al., 2017; Fig. S2, A and B). Hence, RanGTP and Kapα may stabilize CAS tertiary structure to facilitate FG Nup-binding.

Equilibrium binding analysis of CAS, CAS truncation mutants, and associated transport complexes for the selection of FG Nups. Equilibrium dissociation constants (KD) describe the apparent binding affinity of each CAS complex to Nup214, Nup62, Nup98, and Nup153 (N ≥ 4). Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Mean values are indicated below the box plots. The binding range of standalone Kapβ1 (black dashed lines) and Kapα·Kapβ1 (red dashed line) to each of the FG Nups is shown for comparison. See Kapinos et al. (2017) for details. P values were obtained using an unpaired two-tailed t test.
Equilibrium binding analysis of CAS, CAS truncation mutants, and associated transport complexes for the selection of FG Nups. Equilibrium dissociation constants (KD) describe the apparent binding affinity of each CAS complex to Nup214, Nup62, Nup98, and Nup153 (N ≥ 4). Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Mean values are indicated below the box plots. The binding range of standalone Kapβ1 (black dashed lines) and Kapα·Kapβ1 (red dashed line) to each of the FG Nups is shown for comparison. See Kapinos et al. (2017) for details. P values were obtained using an unpaired two-tailed t test.

SPR-based k inetic binding analysis of various CAS complexes with FG Nups. (A) Kinetic binding maps for CAS (blue) and Kapα·CAS·RanGTP (yellow) with Nup214, Nup62, Nup98, and Nup153. N = 4–10. (B) Kinetic maps for CAS (blue), CAS∆Cloop (yellow), and ∆40NCAS (purple) binding to FG Nups. N = 4.

SPR-based k inetic binding analysis of various CAS complexes with FG Nups. (A) Kinetic binding maps for CAS (blue) and Kapα·CAS·RanGTP (yellow) with Nup214, Nup62, Nup98, and Nup153. N = 4–10. (B) Kinetic maps for CAS (blue), CAS∆Cloop (yellow), and ∆40NCAS (purple) binding to FG Nups. N = 4.
Interfering with RanGTP and not Kapα disrupts CAS retention in the nucleus
To verify if the nuclear retention of CAS depends on its interaction with Kapα, we expressed ORF6 of SARS-CoV-2, which sequesters Kapα to the ER/Golgi membranes (Miorin et al., 2020). We generated three plasmids, namely eGFP-ORF6, ORF6-eGFP, and ORF6-eGFP-NES2 to account for the possibility that the location of eGFP on ORF6 might alter its cellular localization (Kato et al., 2021; Miorin et al., 2020). Subsequently, all three ORF6 constructs exhibited cluster-like staining in the cytoplasm, being consistent with ER membrane insertion that displaced Kapα from the nucleus to the cytoplasm (Fig. 3, A–C). In contrast, endogenous CAS (endoCAS) remained predominantly nuclear (Fig. 3 D). Taken together, this indicates that Kapα-binding to CAS·RanGTP does not play a significant role in retaining CAS in the nucleus.

SARS-CoV-2 ORF6 sequesters Kapα to the cytoplasm but does not affect CAS nuclear retention. (A) Immunostaining of endoCAS in cells co-expressing Kapα-mCherry with eGFP-ORF6, ORF6-eGFP, or ORF6-eGFP-NES2. Scale bar, 5 μm. (B) N/C ratio showing that the localization of each ORF6 construct is largely cytoplasmic. (C) N/C ratio showing that Kapα-mCherry has a larger cytoplasmic fraction when ORF6 is present. (D) N/C ratio showing that the nuclear retention of endoCAS is largely unaffected by the sequestration of Kapα-mCherry in the cytoplasm. Dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
SARS-CoV-2 ORF6 sequesters Kapα to the cytoplasm but does not affect CAS nuclear retention. (A) Immunostaining of endoCAS in cells co-expressing Kapα-mCherry with eGFP-ORF6, ORF6-eGFP, or ORF6-eGFP-NES2. Scale bar, 5 μm. (B) N/C ratio showing that the localization of each ORF6 construct is largely cytoplasmic. (C) N/C ratio showing that Kapα-mCherry has a larger cytoplasmic fraction when ORF6 is present. (D) N/C ratio showing that the nuclear retention of endoCAS is largely unaffected by the sequestration of Kapα-mCherry in the cytoplasm. Dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
Next, we investigated how the RanGTP gradient impacts CAS retention by inhibiting ATP production using sodium azide (SA) and 2-deoxyglucose (2DG) to deplete GTP (Englmeier et al., 1999; Schwoebel et al., 2002). Whereas individual treatments of SA or 2DG were less effective, their combined usage significantly disrupted the nuclear localization of mCherry-CAS and Ran (Fig. 4, A–E). Indeed, this indicates that CAS nuclear retention is strongly linked to the RanGTP gradient. Furthermore, similarities between this observation and that of mCherry-∆40NCAS and mCherry-∆40NCAS∆Cloop (Fig. 1, D and E) implicate the N-terminus of CAS as playing a key role in maintaining its nuclear retention.

Interfering with RanGTP disrupts CAS nuclear retention. (A) Immunostaining of endoKapβ1 in cells expressing mCherry-CAS in the absence and presence of SA and DG. Scale bar, 5 μm. (B) The combined use of SA and DG leads to a marked reduction of mCherry-CAS in the nucleus. (C) Immunostaining of endoRan and endoCAS in the absence and presence of SA and DG. Scale bar, 5 μm. (D and E) (D) EndoCAS and (E) endoRan are reduced in the nucleus when SA and DG are present. (F) Immunostaining of endoCAS and endoKapβ1 in cells expressing either mCherry-Ran or its non-hydrolyzable mutant mCherry-RanQ69L. Scale bar, 5 μm. (G) Scatter plot showing a correlation between the N/C ratios of endoCAS and mCherry-Ran. The N/C ratios of endoCAS and mCherry-RanQ69L are not correlated. Each data point is obtained from an individual cell. Dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
Interfering with RanGTP disrupts CAS nuclear retention. (A) Immunostaining of endoKapβ1 in cells expressing mCherry-CAS in the absence and presence of SA and DG. Scale bar, 5 μm. (B) The combined use of SA and DG leads to a marked reduction of mCherry-CAS in the nucleus. (C) Immunostaining of endoRan and endoCAS in the absence and presence of SA and DG. Scale bar, 5 μm. (D and E) (D) EndoCAS and (E) endoRan are reduced in the nucleus when SA and DG are present. (F) Immunostaining of endoCAS and endoKapβ1 in cells expressing either mCherry-Ran or its non-hydrolyzable mutant mCherry-RanQ69L. Scale bar, 5 μm. (G) Scatter plot showing a correlation between the N/C ratios of endoCAS and mCherry-Ran. The N/C ratios of endoCAS and mCherry-RanQ69L are not correlated. Each data point is obtained from an individual cell. Dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
Subsequently, we compared endoCAS localization between cells expressing mCherry-Ran and its non-hydrolyzable mutant, mCherry-RanQ69L (Klebe et al., 1995; Stewart et al., 1998; Fig. 4, F and G). At the lowest expression levels of mCherry-Ran and mCherry-RanQ69L, endoCAS localization returned to the N/C ratio that was consistent with its value in non-transfected cells (Fig. 4, C and D). This value increased linearly with the N/C ratio of mCherry-RanGTP whereas mCherry-RanQ69L-GTP and endoCAS N/C ratios were uncorrelated. With respect to the latter, the N/C ratio of endoCAS remained at the baseline value defined by endoRan because non-hydrolysable RanQ69L does not participate in NCT. Hence, this verifies that RanGTP is required to maintain the nuclear retention of CAS.
RanGTP hydrolysis regulates CAS retention in the nucleus
At this juncture, we hypothesized that RanGTP hydrolysis might constitute the rate-limiting step that releases CAS into the cytosol. Therefore, we designed siRNA oligos against RanBP1 and RanBP2 to interfere with RanGTP hydrolysis, which in turn would impact in vivo CAS cellular localization. Indeed, silencing RanBP1 or RanBP2 led to a reduction in the N/C ratio of mCherry-CAS (Fig. 5, A–E and Fig. S3). Conversely, we did not notice a significant change in the localization of endoRan. This is likely due to the action of nuclear transport factor 2 (NTF2), the Ran importer that replenishes RanGDP in the nucleus (Ribbeck et al., 1998).

CAS nuclear retention requires RanBP1 and RanBP2. (A) Immunostaining of endoRan and RanBP1/2 in cells expressing mCherry-CAS after silencing RanBP1 or RanBP2. Scale bar, 5 μm. (B and C) Cytoplasmic-to-nuclear (C/N) ratios of (B) endoRanBP1 and (C) (endoRanBP2) after siRNA treatment. (D) Silencing RanBP1 or RanBP2 shifts the N/C ratio of mCherry-CAS toward one. (E) N/C ratio of endoRan does not show a significant change after depleting RanBP1 and RanBP2 by siRNA. Dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
CAS nuclear retention requires RanBP1 and RanBP2. (A) Immunostaining of endoRan and RanBP1/2 in cells expressing mCherry-CAS after silencing RanBP1 or RanBP2. Scale bar, 5 μm. (B and C) Cytoplasmic-to-nuclear (C/N) ratios of (B) endoRanBP1 and (C) (endoRanBP2) after siRNA treatment. (D) Silencing RanBP1 or RanBP2 shifts the N/C ratio of mCherry-CAS toward one. (E) N/C ratio of endoRan does not show a significant change after depleting RanBP1 and RanBP2 by siRNA. Dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
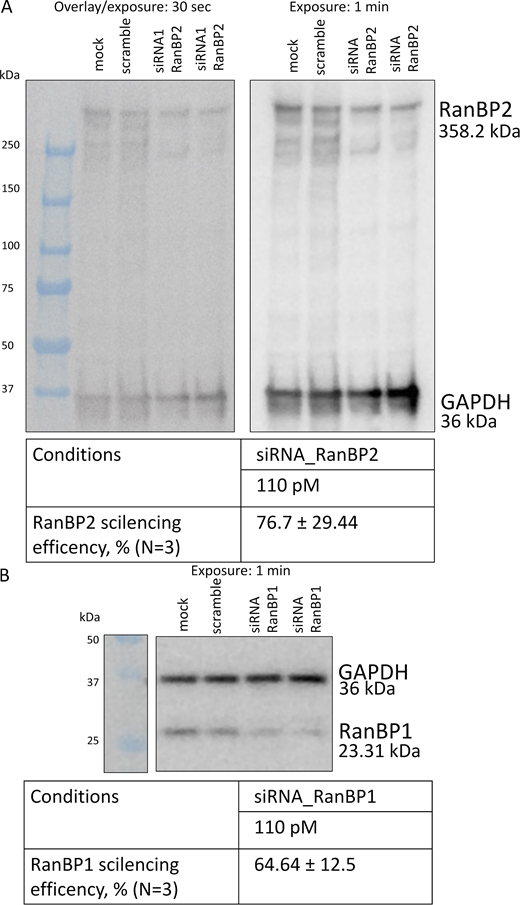
Western blot showing the depletion of RanBP1 and RanBP2 by siRNA.(A and B) Western blot analysis for RanBP2 (A) and RanBP1 (B). GAPDH was used as a loading control. Source data are available for this figure: SourceData FS3.

Western blot showing the depletion of RanBP1 and RanBP2 by siRNA.(A and B) Western blot analysis for RanBP2 (A) and RanBP1 (B). GAPDH was used as a loading control. Source data are available for this figure: SourceData FS3.
Depleting Kapβ1 disrupts CAS retention in the nucleus
Kapβ1 operates in a closed loop with CAS to regulate the nuclear import and export pathways of Kapα, respectively. We therefore postulated that depleting Kapβ1 might disturb this balance and destabilize CAS nuclear retention. As anticipated, the N/C ratio of endoCAS decreases following Kapβ1 silencing, whereas the N/C ratio of endoRan did not change significantly (Fig. 6, A–E). We hypothesize that depleting Kapβ1 reduces Kapα·Kapβ1 complexation, which facilitates the formation of Kapα·CAS complexes in the cytoplasm. Recalling that Kapα·CAS did not bind to RanGTP (Fig. S1 D), our data suggest that Kapα·Kapβ1 complexation helps to maintain CAS nuclear retention by preventing the formation of Kapα·CAS complexes in the cytoplasm.

Kapβ1 depletion leads to CAS mis-localization. (A) Steady-state distribution of endoCAS and endoKapβ1 in control MDCK cells and transfected with either scramble siRNA-Cy5 or siRNA specific for Kapβ1 (siRNA1 and siRNA2). Scale bar, 5 μm. (B) Western blot showing the depletion of Kapβ1 by siRNA1 and siRNA2. (C) N/C ratios of endoKapβ1 and endoCAS under siRNA conditions. Only cells labeled with siRNA-Cy5 were analyzed. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. (D) Immunostaining of endoRan and endoCAS in control MDCK cells and transfected with either scramble siRNA-Cy5 or siRNA specific for the Kapβ1 (siRNA1 and siRNA2). Scale bar, 5 μm. (E) N/C ratios of endoRan obtained at each of these conditions. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3 or 4 in all experiments. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using unpaired two-tailed t test. Source data are available for this figure: SourceData F6.
Kapβ1 depletion leads to CAS mis-localization. (A) Steady-state distribution of endoCAS and endoKapβ1 in control MDCK cells and transfected with either scramble siRNA-Cy5 or siRNA specific for Kapβ1 (siRNA1 and siRNA2). Scale bar, 5 μm. (B) Western blot showing the depletion of Kapβ1 by siRNA1 and siRNA2. (C) N/C ratios of endoKapβ1 and endoCAS under siRNA conditions. Only cells labeled with siRNA-Cy5 were analyzed. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. (D) Immunostaining of endoRan and endoCAS in control MDCK cells and transfected with either scramble siRNA-Cy5 or siRNA specific for the Kapβ1 (siRNA1 and siRNA2). Scale bar, 5 μm. (E) N/C ratios of endoRan obtained at each of these conditions. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3 or 4 in all experiments. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using unpaired two-tailed t test. Source data are available for this figure: SourceData F6.
The N-terminus of CAS is required for its nuclear retention
Overall, our results indicate that the nuclear localization of CAS is most dramatically affected by its N-terminus and RanGTP hydrolysis under WT conditions. But how sensitive is it? Previously, it had been shown that activating the Ras/MEK1/ERK pathway delocalizes CAS to the cytoplasm in vivo, while inhibiting MEK1 rescues it (Behrens et al., 2003; Jiang, 2016; Scherf et al., 1998). This is associated with CAS phosphorylation at its N-terminus at threonine 18 (T18) and tyrosine 20 (Y20) in malignant melanoma but not in benign tissue (Chin et al., 2015). Likewise, our own results suggested that oxidative stress resulted in a cytoplasmic mislocalization of mCherry-CAS, whereas inhibiting MEK1 returned it to the nucleus (Fig. S4, A and B).

Phosphorylation of the CAS N-terminus reduces the nuclear retention of CAS, which colocalizes with F-actin. (A) Immunostaining of Kapβ1 in cells expressing mCherry-CAS in the presence and absence of oxidative stress (H2O2) and MEK1 inhibitor. (B) Oxidative stress reduces the N/C ratio of mCherry-CAS. This is rescued by adding MEK1 inhibitor, which has a null effect in the absence of H2O2. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test. (C) Representative images of HEK293T cells expressing mCherry-CAS, mCherry-CAS_T18G, or mCherry-CAS_T18D. Filamentous actin (F-actin) is stained with mEos2-Lifeact-7 peptide (Addgene 54809; green). mCherry-CAS_T18D colocalizes with F-actin at invadopodia but does not show enrichment in the nucleus. Scale bar, 5 µm.

Phosphorylation of the CAS N-terminus reduces the nuclear retention of CAS, which colocalizes with F-actin. (A) Immunostaining of Kapβ1 in cells expressing mCherry-CAS in the presence and absence of oxidative stress (H2O2) and MEK1 inhibitor. (B) Oxidative stress reduces the N/C ratio of mCherry-CAS. This is rescued by adding MEK1 inhibitor, which has a null effect in the absence of H2O2. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test. (C) Representative images of HEK293T cells expressing mCherry-CAS, mCherry-CAS_T18G, or mCherry-CAS_T18D. Filamentous actin (F-actin) is stained with mEos2-Lifeact-7 peptide (Addgene 54809; green). mCherry-CAS_T18D colocalizes with F-actin at invadopodia but does not show enrichment in the nucleus. Scale bar, 5 µm.
Thereafter, we overexpressed an mCherry–CAS_T18D mutant (Fig. 7 A) that uses aspartic acid (D) as a phosphomimic to test for its impact on CAS localization (Pérez-Mejías et al., 2020). As anticipated, the N/C ratio of mCherry-CAS_T18D was close to one and was insensitive to the MEK1 inhibitor (Fig. 7 B), being comparable with mCherry-∆40NCAS (Fig. 1, D and E). In contrast, a neutral mutation at this position (T18G) did not affect the nuclear localization of mCherry-CAS. Hence, the phosphorylation of a single amino acid at its N-terminus is sufficient to impair CAS nuclear retention. Curiously, mCherry-CAS_T18D cells exhibited increased agitation accompanied by (i) the presence of mCherry-CAS_T18D-enriched microvesicles, (ii) invadopodia-like membrane extensions (i.e., F-actin), and (iii) a two times higher mobility than non-transfected cells in wound-healing assays (Fig. 7, C and D; Fig. S4; and S5; and Videos 1, 2, 3, and 4). This behavior appears consistent with cancer cells that exhibit phosphoCAS-enriched microvesicles and invadopodia (Jiang, 2016; Jiang et al., 2013; Liao et al., 2012; Tai et al., 2010a, 2010b; Wu et al., 2016), and may be attributed to the emergence of a more metastatic phenotype.

A single phosphomimetic mutation (T18D) at the CAS N-terminus is sufficient to abolish its nuclear retention. (A) Immunostaining of Kapβ1 in cells expressing mCherry-CAS_T18D in the presence and absence of MEK1 inhibitor. Cells expressing mCherry-CAS_T18G, which has a neutral mutation at the same position is shown for comparison. Scale bar, 5 μm. (B) N/C ratios for mCherry-CAS_T18G and mCherry-CAS_T18D in the presence and absence of MEK1 inhibitor. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3 or 4. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using unpaired two-tailed t test. (C) Filamentous actin (F-actin) is stained using mEos2-Lifeact-7 peptide in cells expressing mCherry-CAS, mCherry-CAS_T18G, and mCherry-CAS_T18D. Only mCherry-CAS_T18D lacks retention in the nucleus and enriches with cytoplasmic micro-vesicles and invadopodia (indicated by arrows). Magnification of the bounded region (red) in Row 3 is shown in Row 4. Colocalization of F-actin and mCherryCAS_T18D gives a Pearson’s coefficient of 0.767. Scale bar, 5 µm. (D) The wound healing rate of mCherry-CAS_T18D cells (green) is the fastest overall. This is followed by mCherry-CAS (black) and mCherry-CAS_T18G (blue) cells. Non-transfected cells are shown to be slowest (red). Note: The slope of mCherry-CAS is omitted due to its similarity to mCherry-CAS_T18G. N = 3.
A single phosphomimetic mutation (T18D) at the CAS N-terminus is sufficient to abolish its nuclear retention. (A) Immunostaining of Kapβ1 in cells expressing mCherry-CAS_T18D in the presence and absence of MEK1 inhibitor. Cells expressing mCherry-CAS_T18G, which has a neutral mutation at the same position is shown for comparison. Scale bar, 5 μm. (B) N/C ratios for mCherry-CAS_T18G and mCherry-CAS_T18D in the presence and absence of MEK1 inhibitor. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. Median values are indicated next to the box plots. N = 3 or 4. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using unpaired two-tailed t test. (C) Filamentous actin (F-actin) is stained using mEos2-Lifeact-7 peptide in cells expressing mCherry-CAS, mCherry-CAS_T18G, and mCherry-CAS_T18D. Only mCherry-CAS_T18D lacks retention in the nucleus and enriches with cytoplasmic micro-vesicles and invadopodia (indicated by arrows). Magnification of the bounded region (red) in Row 3 is shown in Row 4. Colocalization of F-actin and mCherryCAS_T18D gives a Pearson’s coefficient of 0.767. Scale bar, 5 µm. (D) The wound healing rate of mCherry-CAS_T18D cells (green) is the fastest overall. This is followed by mCherry-CAS (black) and mCherry-CAS_T18G (blue) cells. Non-transfected cells are shown to be slowest (red). Note: The slope of mCherry-CAS is omitted due to its similarity to mCherry-CAS_T18G. N = 3.

Wound healing assays. (A) Schematic description of a wound healing assay and its analysis. (B) Representative images are shown for HEK293T cells expressing mCherry-CAS, mCherry-CAS_T18G, or mCherry-CAS_T18D. Scale bar, 100 µm.

Wound healing assays. (A) Schematic description of a wound healing assay and its analysis. (B) Representative images are shown for HEK293T cells expressing mCherry-CAS, mCherry-CAS_T18G, or mCherry-CAS_T18D. Scale bar, 100 µm.
Fluorescence recovery after photobleaching (FRAP) of mCherry-CAS in the nucleus of a HEK293T cell. The measurement was conducted 24 h after transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. These data were used in the nuclear FRAP analysis shown in Fig. 8 C.
Fluorescence recovery after photobleaching (FRAP) of mCherry-CAS in the nucleus of a HEK293T cell. The measurement was conducted 24 h after transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. These data were used in the nuclear FRAP analysis shown in Fig. 8 C.
FRAP of mCherry-∆40NCAS in the nucleus of a HEK293T cell. The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. These data were used in the nuclear FRAP analysis shown in Fig. 8 C.
FRAP of mCherry-∆40NCAS in the nucleus of a HEK293T cell. The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. These data were used in the nuclear FRAP analysis shown in Fig. 8 C.
FRAP of mCherry-CAS_T18G in the nucleus of a HEK293T cell. The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. The data were used in the nuclear FRAP analysis shown in Fig. 8 C.
FRAP of mCherry-CAS_T18G in the nucleus of a HEK293T cell. The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. The data were used in the nuclear FRAP analysis shown in Fig. 8 C.
FRAP of mCherry-CAS_T18D in the nucleus of a HEK293T cell. The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. Selected frames of this movie are shown in Fig. 8 A. These data were used in the nuclear FRAP analysis shown in Fig. 8 C.
FRAP of mCherry-CAS_T18D in the nucleus of a HEK293T cell. The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s. Selected frames of this movie are shown in Fig. 8 A. These data were used in the nuclear FRAP analysis shown in Fig. 8 C.
CAS nuclear efflux is significantly faster than its influx
Further, we questioned if the nuclear retention of triple complexes might be linked to their kinetic behavior. Using fluorescence recovery after photobleaching (FRAP), we found that >90% mCherry-CAS in the nucleus was mobile and exhibited a half-time of recovery (τ) of 143 ± 93 s (Fig. 8, A–C; and Table S1). This is consistent with values obtained for GFP-CRM1 and NLS-cargoes of comparable mass (Bizzarri et al., 2012; Cardarelli et al., 2007, 2009; Daelemans et al., 2005; Wei et al., 2003), but significantly faster than non-specific cargoes of similar mass (>500 s; Timney et al., 2016). To our surprise, however, mCherry-CAS required half the amount of time to recover in the cytoplasm (τ = 51.5 ± 43.0 s; faster). This likely results from (i) RanGAP1 activity that hydrolyses RanGTP at a rate of 2.1 s−1 (Klebe et al., 1995); (ii) the smaller mass of CAS in comparison to the triple complex; (iii) an increase in diffusive flux down the concentration gradient (Fig. 8 B; i.e., Fick’s Law) that underlies the transition of triple complexes in the nucleus to free CAS in the cytoplasm; or (iv) combinations thereof.

CAS transport kinetics reveals a distinct mechanism controlling its nuclear retention. (A) Successive image frames capture the photobleaching and fluorescence recovery of mCherry-CAS, mCherry-Δ40NCAS, and mCherry-CAS_T18D within the nucleus. Note: Only mCherry-CAS shows nuclear retention. Scale bar, 5 μm. (B) The gray value profile highlights the concentration gradient (i.e., slope) of mCherry-CAS that exists between the nucleus and cytoplasm but is absent in mCherry-Δ40NCAS and mCherry-CAS_T18D. (C) The recovery half-times for mCherry-CAS, mCherry-Δ40NCAS, mCherry-CAS_T18D, and mCherry-CAS_T18G are summarized in the boxplots for nuclear and cytoplasmic FRAP, respectively. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
CAS transport kinetics reveals a distinct mechanism controlling its nuclear retention. (A) Successive image frames capture the photobleaching and fluorescence recovery of mCherry-CAS, mCherry-Δ40NCAS, and mCherry-CAS_T18D within the nucleus. Note: Only mCherry-CAS shows nuclear retention. Scale bar, 5 μm. (B) The gray value profile highlights the concentration gradient (i.e., slope) of mCherry-CAS that exists between the nucleus and cytoplasm but is absent in mCherry-Δ40NCAS and mCherry-CAS_T18D. (C) The recovery half-times for mCherry-CAS, mCherry-Δ40NCAS, mCherry-CAS_T18D, and mCherry-CAS_T18G are summarized in the boxplots for nuclear and cytoplasmic FRAP, respectively. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test.
To investigate this further, we carried out FRAP experiments with cells expressing mCherry-∆40NCAS and mCherry-CAS_T18D, respectively, recalling that neither entity accumulates in the nucleus (Fig. 8, A and B). In both cases, we obtained τ values that were similar in the nucleus and cytoplasm, although, between the two mutants, mCherry-∆40NCAS exhibited overall smaller τ values due to its smaller mass. Their values were also comparable with the recovery of mCherry-CAS in the cytoplasm. This indicates that the regulation of RanGTP hydrolysis by RanGAP1 and RanBP1/2 at NPCs brings about a steep concentration gradient of CAS in the nucleus. Indeed, this is further validated by mCherry-CAS_T18G whose τ values were comparable with mCherry-CAS (Fig. 8 C).
CAS nuclear retention is regulated by its N-terminus interaction with RanGTP via SWITCHI
Finally, we sought to confirm if interfering with the interaction between CAS and RanGTP impaired the nuclear localization of CAS. It was shown that RanGTP utilizes two conformational switches termed SWITCHI and SWITCHII to stabilize its interactions between Cse1p and Kap60 (yeast homologs of CAS and Kapα, respectively; Matsuura and Stewart, 2004). We then used site-directed mutagenesis to interfere with the key binding sites on CAS that engage with SWITCHI and SWITCHII (Fig. 9 A). Evidently, pairing K67A and N68A mutations on the N-terminus of CAS that bind to SWITCHI resulted in a significant reduction in its nuclear accumulation (Fig. 9, B and C). In contrast, N608A and E658A mutations close to the C-terminus of CAS that binds to SWITCHII did not affect CAS nuclear retention. However, expressing a CAS mutant with K67A/N68A/N608A/E658A mutations present incurred an equal reduction in its nuclear accumulation as for the K67A/N68A mutant. Thus, this indicates that the interaction between the CAS N-terminus and RanGTP via SWITCHI is a primary determinant in regulating the nuclear retention of CAS.

CAS nuclear retention depends on its N-terminus interaction with RanGTP vis-à-vis SWITCHI. (A) Structure of the yeast homolog of CAS (CSE1; purple) in complex with Kap60 (yeast Kapα; green) and Gsp1pGTP (yeast RanGTP; yellow) (PDB ID: 1wa5). The T18D mutation (red arrow) at the CAS N-terminus is also indicated (see Fig. 7 and main text for details). Its conformation was modeled using the ELASPICwebserver (Witvliet et al., 2016). K67A and N68A mutations (red) on the CAS N terminus and N609A and E656A mutations (black) are shown. (B) Immunostaining of Kapβ1 in cells expressing mCherry-CAS and mCherry-CAS mutants shown. Scale bar, 5 μm. (C) N/C ratios reveal that CAS nuclear accumulation is significantly reduced for mCherry-CAS mutants carrying the K67A and N68A mutations. N608A and E658A mutations in the C-terminus of CAS have no impact on CAS nuclear localization. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3 or 4. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test. (D) Schematic model illustrating the closed loop of Kapβ1 and CAS that regulates Kapα nuclear import and export, respectively. CAS nuclear retention follows from a “hold” and “release” mechanism at RanGAP1. RanGTP hydrolysis is suppressed by the CAS N-terminus in the “hold” state and achieves “release” by the action of RanBP1 (or RanBP2). See text for details. SWITCH I (I) and SWITCHII (II) on RanGTP are indicated in the cartoon.
CAS nuclear retention depends on its N-terminus interaction with RanGTP vis-à-vis SWITCHI. (A) Structure of the yeast homolog of CAS (CSE1; purple) in complex with Kap60 (yeast Kapα; green) and Gsp1pGTP (yeast RanGTP; yellow) (PDB ID: 1wa5). The T18D mutation (red arrow) at the CAS N-terminus is also indicated (see Fig. 7 and main text for details). Its conformation was modeled using the ELASPICwebserver (Witvliet et al., 2016). K67A and N68A mutations (red) on the CAS N terminus and N609A and E656A mutations (black) are shown. (B) Immunostaining of Kapβ1 in cells expressing mCherry-CAS and mCherry-CAS mutants shown. Scale bar, 5 μm. (C) N/C ratios reveal that CAS nuclear accumulation is significantly reduced for mCherry-CAS mutants carrying the K67A and N68A mutations. N608A and E658A mutations in the C-terminus of CAS have no impact on CAS nuclear localization. The dashed line indicates N/C = 1. Box plots denote the median, first, and third quartiles. The whiskers represent the minimum and maximum values. N = 3 or 4. The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test. (D) Schematic model illustrating the closed loop of Kapβ1 and CAS that regulates Kapα nuclear import and export, respectively. CAS nuclear retention follows from a “hold” and “release” mechanism at RanGAP1. RanGTP hydrolysis is suppressed by the CAS N-terminus in the “hold” state and achieves “release” by the action of RanBP1 (or RanBP2). See text for details. SWITCH I (I) and SWITCHII (II) on RanGTP are indicated in the cartoon.
Discussion
We have elucidated a mechanism at NPCs that prevents the uncontrolled efflux of CAS from the nucleus (Fig. 9 D). Under WT conditions, Kapα·CAS·RanGTP complexes dominate the nucleus, and in this form, the CAS N-terminus obstructs the Ran catalytic center that prevents RanGAP1 binding and GTP hydrolysis. In other words, the nuclear retention of triple complexes derives from a suppression of RanGTP hydrolysis. Thus, the Ran-binding domain (RanBD) of RanBP1 or RanBP2 is required to bind to the C-terminus of Ran to increase the accessibility of the Ran catalytic center to RanGAP1 (Güttler and Görlich, 2011; Seewald et al., 2003). Moreover, CAS nuclear exit is very likely rate limited by interactions between RanBD and RanGTP (kon = 5 × 105 M−1 s−1 and koff = 4.3 × 10−4 s−1), which is markedly slower than RanGTP binding to RanGAP1 in the absence of CAS (kon = 4.5 × 108 M−1 s−1 and koff = 900 s−1; Seewald et al., 2003). Evidently, these interactions are also essential for triple complex disassembly (Matsuura and Stewart, 2004; Seewald et al., 2002; Vetter et al., 1999), and CAS nuclear exit (Kutay et al., 1997; Sun et al., 2013).
Accordingly, CAS nuclear retention is abolished by interfering with the reactions that regulate RanGTP hydrolysis. We show that it is vulnerable to (i) deletions in the CAS N-terminus (Fig. 1 D); (ii) depletion of GTP (Fig. 4, A–E); (iii) impaired RanGTP hydrolysis (Fig. 4, F and G); (iv) depletion of RanBP1 and RanBP2 (Fig. 5); and (v) impaired binding of the CAS N-terminus to RanGTP (Figs. 7 and 9). As NCT operates in a nexus of different transport receptors, we have also shown that perturbations to Kapβ1 can also lead to systematic shifts in CAS localization (Fig. 6). To rationalize this, consider that Kapα·CAS·RanGTP represents the most stable dominant form of CAS in the nucleus under normal conditions (KD = 0.9 ± 0.3 nM; Fig. S1 C). Following triple complex export and disassembly, free Kapα binds to Kapβ1 in the cytoplasm to form (NLS-cargo)·Kapα·Kapβ1 (KD = 210 ± 77 nM; Kapinos et al., 2017). However, if the concentration of Kapβ1 was depleted, the equilibrium would be shifted toward the formation of Kapα·CAS (KD = 9.3 ± 2.9 nM) in the cytoplasm, which does not bind to RanGTP (Fig. S1 D). This might explain the decrease in the N/C ratio of CAS upon silencing Kapβ1 (Fig. 6 C). In this state, the data suggests that the highly stable Kapα·CAS complexes may no longer bind RanGTP to form functional triple complexes in the nucleus. Subsequently, the number of RanGTP·Kapβ1 and Kapα·CAS·RanGTP complexes in the nucleus would diminish. As a consequence, less Kapα would be available in the cytoplasm for Kapβ1-dependent transport of NLS-cargoes. Hence, we hypothesize that constraining CAS to the nucleus prevents Kapα·CAS formation in the cytoplasm to maximize its competition with Kapβ1. On this note, the same equilibrium shift is not observed when Kapα was sequestered by SARS-CoV-2 ORF6 in the cytoplasm (Fig. 3). In this case, the retention of CAS in the nucleus might be explained by the predominance of nuclear CAS·RanGTP (KD = 1.4 ± 0.4 µM; Fig. S1 C), which is still able to undergo RanGTP hydrolysis (Bischoff and Görlich, 1997).
But how general is such a mechanism? In the absence of RanBP1 and RanBP2, the RanGTP active site that interacts with RanGAP1 can also be obstructed by the N-termini of CRM1, Kapβ1, and transportin, respectively (Bischoff and Görlich, 1997; Koyama and Matsuura, 2010; Lounsbury and Macara, 1997; Matsuura and Stewart, 2004; Sarić et al., 2007). In line with this observation, we show here that both RanBP1 and RanBP2 play essential roles in controlling the nuclear retention and release of CAS (Fig. 5, A–E). This regulatory mechanism may also extend to other exportins, including CRM1 and exportin-t, as mentioned (Kuersten et al., 2002), among others. Given that RanGAP1 and RanBP2 are located at the cytoplasmic filaments, it also implies that the NPC central channel remains unobstructed and accessible for nuclear import. Still, this brings into question why Kapβ1 is more enriched in the cytoplasm rather than the nucleus. This peculiarity may stem from the contrasting behaviors of Kapα, which could slow the nuclear exit of CAS triple complexes but expedite the nuclear exit of Kapβ1·RanGTP (by increasing the GTPase activity of Ran; Bischoff and Görlich, 1997). Recalling that the concentration of CAS is approximately twofold lower than Kapβ1 (Geiger et al., 2012) also implies that a sufficiently large pool of Kapα is free to engage Kapβ1 in the cytoplasm (to form NLS-cargo·Kapα·Kapβ1 complexes). Nevertheless, it is unclear how other importins that do not bind Kapα maintain their cytoplasmic localization.
CAS is usually dephosphorylated in the nucleus (Chin et al., 2015). However, mitogen-induced stress or changes in cell metabolism can activate the MEK1/ERK pathway to phosphorylate CAS at T18 and Y20 at its N-terminus (Chin et al., 2015; Lee et al., 2015), followed by delocalization to the cytoplasm (Scherf et al., 1998; Wu et al., 2016; Fig. S4). Strikingly, we also find that a single phosphomimetic point mutation (T18D), but not a neutral one (T18G), is sufficient to disrupt CAS nuclear retention (Fig. 7, C and D). Furthermore, this suggests that the loss of CAS partitioning in the nucleus as observed in specific cancers may also be attributed to phosphorylation at its N-terminus (Behrens et al., 2003; Jiang, 2016; Lorenzato et al., 2013; Scherf et al., 1998), which compromises the interaction between CAS and RanGTP. Coincidentally, T18D cells also demonstrated invadopodia formation and enhanced cell motility (Fig. 7, C and D), which are phenotypic of tumor invasion and metastatic activity in melanoma and breast cancer (Jiang, 2016; Liao et al., 2008, 2012; Tai et al., 2010a, 2010b). Importantly, future experimentation will be required to ascertain the role of phosphorylated CAS in the cytoplasm.
Our attempts at interfering with the Ran gradient have also resulted in the delocalization of non-phosphorylated CAS into the cytoplasm (Fig. 4, A–E). This is reminiscent of early apoptosis where a decrease in the expression levels of RanGAP1 leads to a reduction in the N/C ratio of Ran (Lin et al., 2015; Wong et al., 2009). Hence, the nuclear retention of CAS can be abrogated by different means to arrive at different cellular outcomes e.g., cancer versus apoptosis. Taken together, these findings highlight important intersection points between CAS mislocalization, defective NCT, and disease that warrants further investigation.
Conclusion
We have clarified the nuclear retention mechanism of CAS. This is functionally independent of the NPC permeability barrier and highlights a separate control system that NPCs might use to regulate exportin localization. In this regard, CAS nuclear retention acts as a counterbalance against Kapβ1 to sustain the bidirectional exchange of Kapα. Still, the details governing such a “balancing act” remain to be explored, such as how fluctuating Kap levels might impact the feedback loop(s) governing NCT altogether. Clearly, such insights will shed light on the asymmetric partitioning of Kaps or lack thereof. Still, because of its implications in disease, resolving the CAS nuclear retention mechanism could be beneficial for developing specific inhibitors of nuclear export (SINE) as anticancer drugs, such as in the case of CRM1-mediated NES-cargo export (Azmi et al., 2021; Sun et al., 2016).
Materials and methods
Cloning, expression, and purification of recombinant proteins
CAS
Human WT CAS and its mutants were cloned into pPEP-TEV vector using BamH1 and Xho1 restriction sites. All proteins were expressed in Escherichia coli BL21 cells overnight at 20–22°C after induction with 0.5 M IPTG (3 L culture). Cells were collected by centrifugation at 4°C at 5,422 × g and lysed for 1 h at 4°C in the following buffer: 20 mM Tris pH 8.0, 300 mM NaCl, 2 mM β-mercaptoethanol, 5 mM imidazole, DNase, lysozyme, pefabloc, and a complete protease inhibitor cocktail (Roche) followed by an additional sonication step for 5 min on ice. The supernatant was then collected after lysate centrifugation at 25,000 × g for 1 h at 4°C and transferred into a Ni-NTA column (Roche). Purified WT CAS and its mutants were eluted using the following buffer: 20 mM Tris pH 8.0, 300 mM NaCl, 2 mM β-mercaptoethanol, and 500 mM imidazole. This was followed by additional purification using a Superdex 200 size exclusion column. The collected fraction was validated using a 12% SDS gel (see Fig. S1 B) and flash-frozen at −80°C. For a list of all resources, see Table S2.
FG Nups
FG Nups (specifically the FG domains of Nup62, Nup153, Nup98, and Nup214), Ran, and Kapα (importinα1; KPNA2) were purified as described previously (Kapinos et al., 2014, 2017; Kapinos and Lim, 2022). Briefly, the FG domains of Nup62, Nup153, and Nup98 with additional cysteines at the C- or N-terminus were cloned into a pPEP-TEV expression vector, while the FG domain of Nup214 was cloned into a pETM-11 plasmid. All constructs were expressed in E. coli BL21 (DE3) cells (Novagen). The FG domains were purified under denaturing conditions (8 M urea, 100 mM Na2HPO4, and 10 mM Tris-HCl, pH 8.5) using a Ni-NTA column (Roche) and eluted by lowering the pH. The His-tag was then removed by TEV protease. His-tag-free protein fragments were purified again using a Ni-NTA column (Roche) and their purity was analyzed by 12% PAGE at 0.1% SDS. All FG Nups were dialyzed against PBS buffer (pH 7.2; Invitrogen) for usage in binding assays.
Ran
WT Ran provided in pQ70 vector was purified using a Ni-NTA column (Roche) over an imidazole gradient (10–500 mM) and dialyzed into a 10 mM HEPES buffer pH 7.2 with 100 mM NaCl. Purified Ran was incubated for 30 min at 4°C with 10 mM EDTA. Subsequently, 25 mM MgCl2 was added together with 1 mM GTP or GDP, which ensured its binding to nucleotide-free Ran. GTP- or GDP-loaded Ran was dialyzed into PBS buffer, pH 7.2 (Invitrogen) supplemented with 1 mM MgCl2, and additionally purified using a size-exclusion column (Superdex75 HiLoad 16/60; Cytiva). The protein fractions were validated using 15% PAGE at 1% SDS and subsequently snap-frozen at −80°C. Prior to its use in binding assays, Ran was dialyzed against PBS buffer (pH 7.2; Invitrogen) and its concentration was measured.
Kapα
A full-length human Kapα construct was cloned using EcoRI and BamII restriction enzymes into the pQE70 vector with a His-tag at its C-terminus and expressed in E. coli BL21 (DE3) cells (Novagen) at 24°C. Cells were collected by centrifugation at 4°C at 5,422 × g and lysed for 1 h at 4°C in the following buffer: 50 mM phosphate buffer pH 7.5, 200 mM NaCl, 2 mM β-mercaptoethanol, 10 mM imidazole, 10% glycerol, DNase, lysozyme, pefabloc, and a complete protease inhibitor cocktail (Roche) followed by sonication step for 5 min on ice. The supernatant was collected by centrifugation at 25,000 × g for 1 h at 4°C and applied to a Ni-NTA column (Roche). Kapα was eluted using a stepped imidazole gradient in a buffer containing 20 mM Tris HCl (pH 7.5), 200 mM NaCl, 2 mM DTT, and 10% glycerol. This was followed by an additional purification step on a size-exclusion column (Superdex75 HiLoad 16/60; Cytiva). The protein fractions were verified using 12% PAGE at 1% SDS and subsequently snap-frozen at −80°C. Kapα was dialyzed against PBS buffer (pH 7.2; Invitrogen) prior to its use in binding assays.
Design, cloning, and expression of mCherry-CAS and its mutants in mammalian cells
The sequences encoding mCherry-CAS and its mutants (mCherry-CAS∆C, mCherry-CAS∆Cloop, mCherry-∆40NCAS, mCherry-∆40NCAS∆Cloop, mCherry-(1-40)NCAS, mCherry-CAS_T18D, mCherry-CAS_T18G, mCherry-CAS_K67A,N68A mCherry-CAS_N608A, mCherry-CAS_E658A, and mCherry-CAS_K67A,N68A,N608A,E658A) were cloned into a mammalian expression vector, pcDNA3.1(-; puromycin) plasmid. First, mCherry was inserted using EcoR1/BamH1 cloning sites and then the sequence encoding CAS or its mutants was implemented into this plasmid using the BamH1 restriction site. Point mutations (T18D and T18G) were introduced into the mCherry-CAS encoding plasmid using site-directed mutagenesis with the following primers:
T18D
Forward primer: 5′-CAGAATATTTAAAGAAAGACCTTGATCCTGATCCTGCCCAS-3′.
Reverse primer: 5′-GGCAGGATCAGGATCAAGGTCTTTCTTTAAATATTCTG-3′.
T18G
Forward primer: 5′-CAGAATATTTAAAGAAAGGACTTGATCCTGATCCTGCC-3′.
Reverse primer: 5′-GGCAGGATCAGGATCAAGTCCTTTCTTTAAATATTCTG-3′.
Other mutations (K67A, N68A, N609A, and E656A) were introduced using the following primers:
K67A, N68A
Forward primer: 5′-GTATGTGCTTCAGTAACATTCGCAGCCTATATTAAAAGGAACTGGAG-3′.
Reverse primer: 5′-CTCCAGTTCCTTTTAATATAGGCTGCGAATGTTACTGAAGCACATAC-3′.
N608A
Forward primer: 5′-GCTATTAGCTGTTAGTAAGGCCCCAAGCAAACCTCACTTTAATC-3′.
Reverse primer: 5′-GATTAAAGTGAGGTTTGCTTGGGGCCTTACTAACAGCTAATAGC-3′.
E658A
Forward primer: 5′-CTTACAAAATGATGTGCAAGCATTTATTCCATACGTCTTTC-3′.
Reverse primer: 5′-GAAAGACGTATGGAATAAATGCTTGCACATCATTTTGTAAG-3′.
Each plasmid was then used for transient transfection in HEK293T cells (CRL-3216; ATCC) using jetOptimus (Polyplus) following standard protocols. Successful transfection was validated by detecting the expression of mCherry-targeted proteins using a Cell Imager PAULA system (Leica Microsystems). Additionally, the stability of mCherry-CAS constructs was validated by Western blotting. Transfected HEK293T cells were cultured for 24 h in a six-well plate and then lysed using radioimmunoprecipitation assay buffer (89900; Thermo Fisher Scientific) supplemented with complete protease inhibitor cocktail (ll873580001; Roche), benzonase (Novagen), and phosphatase inhibitors (P5726 and P0044, respectively; Sigma-Aldrich). Lysates were spun down for 15 min at 15,000 × g at 4°C, and total protein concentration was determined using a Pierce bicinchoninic acid assay (23227; Thermo Fisher Scientific). For each sample, 20 µg total protein amount was resolved by SDS-PAGE (12% PAGE at 1% SDS) and transferred onto nitrocellulose membranes using a Trans-Blot Turbo transfer system (Trans-Blot Turbo Midi 0.2 µm, 1704158; Bio-Rad Laboratories). Then membranes were blocked with 0.1% (wt/vol) blocking reagent (11096176001; Roche) prepared in TBS with Tween 20 (TBST) solution for 1 h at RT and targeted with anti-mCherry mouse monoclonal Antibody (4B3; Invitrogen) or anti-CAS rabbit polyclonal antibody (ab96755; 1:1,000 dilution; Abcam; Fig. S1 A). Simultaneously, anti-GAPDH antibodies (rabbit, PAl-987; Thermo Fisher Scientific or mouse, 60004-1-Ig; ProteinTech) were used to allow for signal normalization and quantification. Finally, secondary antibodies (goat anti-mouse ab97020 or goat anti-rabbit, ab6722; Abcam) were used for immunodetection. All membranes were developed in a Fusion FX (Vilber Lourmat) system, and the resulting chemiluminescent signals were quantified using Fiji (ImageJ).
Design of eGFP-ORF6, ORF6-eGFP, and ORF6- eGFP-NES2
The ORF6 sequence from SARS-CoV-2 was cloned into a pcDNA3.1(-) (puromycin) plasmid using Nhe1/Sac1 cloning sites. An eGFP sequence was added at its N-terminus using a Not1/EcoRV restriction site. A pcDNA3.1(-) (puromycin) plasmid encoding ORF6-eGFP at its C-terminus was constructed by cloning the ORF6 sequence using Nhe1/Sac1 cloning sites in front of eGFP. Afterward 2×NES was introduced into this plasmid following the Sac1 cloning site to obtain ORF6-eGFP-NES2. The ORF6 sequence in these constructs is as follows: MFHLVDFQVTIAEILLIIMRTFKVSIWNLDYIINLIIKNLSKSLTENKYSQLDEEQPMEID, where the NLS-like sequence that is recognized by Kapα is underlined (Miorin et al., 2020). The amino acids that make up the ORF6 intermembrane domain are indicated in bold.
Kapβ1 silencing
siRNAs against dog Kapβ1 were designed and used in MDCK cells as previously described (Kalita et al., 2022). siRNA nucleotides were modified with Cy5 at the end of their antisense strand to visualize the cells affected by siRNA treatment.
Immunofluorescence
Primary anti-CAS rabbit polyclonal antibody (ab96755; 1:200 diluted in PBS buffer with 1% BSA; Abcam) and monoclonal mouse anti-Kapβ1 antibody (3E9; 1:200 dilution in PBS buffer with 1% BSA; Abcam) were used (1 h in the dark at room temperature). Cells were incubated with donkey anti-rabbit labeled with AlexaFluor-568 (A10042; 1:400 dilution; Invitrogen), donkey anti-mouse labeled with AlexaFluor-488 (A21207; 1:400 dilution; Invitrogen), and DAPI (D9542; 1:1,000 dilution; Sigma-Aldrich) after washing with blocking solution (3 × 5 min each). An additional control verified for unspecific binding of the secondary antibody. After washing, the coverslips were mounted on microscopy slides inverted into a drop of Vectashield (∼50 μl) and sealed with nail polish. These slides were stored in darkness at 4°C.
Immunofluorescence imaging and analysis
The fluorescent images were recorded using an LSM880 inverted confocal microscope (Zeiss) with an oil-immersed 63×/1.4 NA PLAN APO objective equipped with a widefield camera and an Airyscan detector (Zeiss) at RT. Mean nuclear and cytoplasmic fluorescence signals were quantified using CellProfiler software (Kamentsky et al., 2011). DAPI channel was used for initial image segmentation by the Otsu method. The nuclear envelope, nucleus, and cytoplasm regions of interest (ROIs) were created by shrinking or expanding the original DAPI-defined regions. The resulting nucleocytoplasmic ratios were then calculated in ProFit 7.3 and plotted as box-plots, where the number of the analyzed cells (technical replicates, n) is indicated together with the number of independent experiments (N).
Silencing of RanBP1 and RanBP2
siRNAs were designed and synthesized as follows: 5′-GGACCATGATACTTCCACTG-3′ for RanBP1 and 5′-CACAGACAAAGCCGUUGAA-3′ for RanBP2 (Wälde et al., 2012). Silencing efficiency was verified by Western blot using a standard protocol (Kalita et al., 2022). Chemiluminescence was measured using Fusion FX (Witec AG). Prior to imaging, the HEK293T cells were first transfected with an mCherry-CAS expressing plasmid. After 24 h, the cells were washed and cotransfected with 110 pmol siRNA for either RanBP1 or RanBP2 silencing. Control cells were transfected with scramble siRNA (Microsynth AG) using a standard jetOptimus protocol. After this, cells were incubated for another 24 h followed by immunostaining of RanBP1 and RanBP2 as described above. Primary anti-RanBP1 polyclonal rabbit antibody (ab97659; 1:200 dilution in 1% BSA/PBS buffer; Abcam) and anti-RanBP2 polyclonal rabbit antibody (ab64276; 1:2,000 dilution in 1% BSA/PBS buffer; Abcam) were used, respectively. The respective secondary antibodies were donkey anti-rabbit antibody labeled with AlexaFlour-647 (A31573; dilution 1:400; Invitrogen) and donkey anti-mouse antibody labeled with AlexaFlour-488 (A21207; dilution 1:400; Invitrogen). Cells were costained with Ran using a monoclonal mouse antibody (cat.610341; BD Transduction Laboratories). Only cells expressing mCherry-CAS were analyzed.
Poison assay
HEK293T cells were plated on 0.17-mm coverslips in 12-multiwell plates in DMEM with 10% FBS for 24 h at 37°C and 5% CO2. Following a media exchange, the cells were transiently transfected with the plasmid encoding mCherry-CAS using jetOptimus. After 24 h and a media exchange, the cells were grouped as follows: (i) untreated control cells; (ii) 6 µM 2D-glucose; (iii) 10 µM sodium azide; and (iv) 6 µM 2D-glucose and 10 µM sodium azide. Treatments were added for 30 min each. The cells were then washed, fixed, and stained for endogenous Kapβ1. The primary antibodies were monoclonal mouse anti-Kapβ1 antibody (3E9; 1:200 dilution; Abcam). The secondary antibody was donkey anti-mouse labeled with AlexaFlour-488 (A21207; 1:400; Invitrogen). The nucleus was stained with DAPI.
Mass photometry
The molecular mass of CAS and its mutants was measured using Refeyn OneMP mass photometry instrument following standard protocols (Young et al., 2018). Measurements were analyzed using Refeyn software. Native PAGE markers (NativeMark) were used for the instrument calibration.
Microscale thermophoresis
A Nanotemper Monolith NT.115 (Nanotemper Technology) system was used. WT CAS, CAS∆C, CAS∆Cloop, ∆40NCAS, ∆40NCAS∆Cloop, CAS_T18D, and CAS_T18G were labeled at one of the available cysteines with Alexa Fluor 488 C5 Maleimide (A10254; Molecular Probes). Excess dye was removed using spin columns (Princeton Separations). The labeling efficiency was measured using a UV–Vis spectrophotometer. Then 16 Kapα 1:1 dilution was prepared starting from 4.9 µM as the highest concentration and 16 RanGTP 1:1 dilution starting from 36 µM. To measure the equilibrium dissociation constant (KD) of CAS (or mutants) and human Kapα, 20 μl of Kapα (each of 16 dilutions) was mixed with 20 μl of 25 nM CAS-488 (or mutants). To measure the CAS (or mutants) interaction with RanGTP, 20 μl of RanGTP (16 dilutions) was mixed with 20 μl of 20 nM CAS-488 (or mutants). And finally, to measure the interaction of CAS·RanGTP complexes (or CAS mutant complexes) with human Kapα, 20 μl of Kapα (16 dilutions) was mixed with 20 μl of 25 nM CAS-488 (or its labeled mutants) complex with RanGTP, where Ran was always in excess (i.e., 50 nM). The resulting solution was then filled into the capillaries (Monolith NT.115 Premium Capillaries MO-K025), and the obtained binding curves were analyzed using Nanotemper MST analysis software (see Fig. S1 C).
Surface plasmon resonance
Surface plasmon resonance (SPR) was used to measure the interaction of CAS (its mutants and their complexes) with the FG domains of Nup214, Nup153, Nup98, and Nup98, as described previously (Kapinos et al., 2014, 2017). Briefly, SPR measurements were performed at 25°C on gold sensor chips (SIA Kit Au; Cytiva) using Biocore T200 (Cytiva). FG Nups were immobilized in chambers 3 and 4, whereas chambers 1 and 2 (used as a reference) were passivated with hydroxyl-terminated triethylene glycol undecanethiol or HS-[CH2]11-[OCH2CH2]3-OH (abbreviated as PUT; 673110; Sigma-Aldrich). Thereafter, binding sensograms were recorded at a flow rate of 10 μl/min. The maximal CAS concentration was 8 µM. All solutions were prepared in PBS (Invitrogen), filtered, and degassed.
FRAP measurements and analysis
Measurements
HEK293T cells were plated in an eight-well µ-Slide (ibidi) and transfected with CAS-mCherry mutants using jetOptimus the next day. About 24 h later, FRAP measurements were conducted using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss) built on a Zeiss Axio Observer stand (Intelligent Imaging Innovations GmbH). The system is equipped with a 1.4NA 63× plane apochromat objective (Plan-Apochromal 63×/1.4 Oil DIC M27), EMCCD camera (Evolve(R) 512; Photometrics), and a humidified climate control system at 37°C supplemented with 5% CO2 (Okolab). For nuclear FRAP, a round 8 μm2 region within each nucleus was chosen and bleached by application of three 10-ms pulses per pixel using a 561-nm laser (3.4 mW). Sequential images (including five images before and 595 images after the bleaching event) were collected every second by illuminating the sample with a 555 nm laser at 30% of its power (10 mW). Movies from three independent experiments were collected and analyzed (see Videos 1, 2, 3, and 4). For cytoplasmic FRAP, a round 8 μm2 region was chosen within the cytoplasm.
Analysis
For each movie frame, a time-stamp and fluorescent intensities of the nucleus, whole cell, and background were exported and collected in an .xls file. These data were used to plot and analyze recovery curves using easyFRAP software (Rapsomaniki et al., 2012). All the data points were normalized following a double-scale normalization and the resulting curves were fit using a single exponent equation. The data were tested for normality using a built-in function of GraphPad Prism software. An unpaired two-tailed t test (GraphPad Prism software) was used to perform the statistical analysis on the resulting half-time recovery values (τ).
Invadopodia staining
HEK293T cells (∼200,000) were plated on 0.17-mm coverslips in 12-multiwell plates in DMEM with 10% FBS for 24 h at 37°C and 5% CO2. Following a medium exchange, the cells were cotransfected with mCherry-CAS, mCherry-CAS_T18D, or mCherry-CAS_T18G together with plasmids expressing mEos2-Lifeact-7 peptide (Addgene 54809), which stains specifically filamentous actin (F-actin). After 24 h, the cells were fixed and stained following standard protocols. Representative images are shown in Fig. S4 C and Fig. 7 C. To obtain movies the cells were plated into four wells of “µ-Slide 4 Well” (Ibidi) and imaged after 24 h using a “LSM880 Inverted” microscope with Airyscan (Zeiss). 150 frames obtained at a frame rate of 5 s per frame were recorded (Videos 5, 6, 7, and 8).
HEK293T cells expressing mCherry-CAS (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
HEK293T cells expressing mCherry-CAS (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
HEK293T cells expressing mCherry-∆40NCAS (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
HEK293T cells expressing mCherry-∆40NCAS (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
HEK293T cells expressing mCherryCAS_T18G (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
HEK293T cells expressing mCherryCAS_T18G (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
HEK293T cells expressing mCherryCAS_T18D (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
HEK293T cells expressing mCherryCAS_T18D (red) and mEos2-Lifeact-7 peptide (Addgene 54809; green). The measurement was conducted 24 h post transfection using a point scanning confocal “LSM880 Inverted” microscope with Airyscan (Zeiss). Frame rate, 0.2 fps (one frame per 5 s). In total, 120 frames were recorded within 600 s.
Wound healing assay
HEK293T cells (∼100,000 cells/ml) were plated into cell culture inserts (Ibidi) and placed in each well of the µ-Slide (high Glass Bottom, 8 Well). After 24 h, cells were transfected with plasmids expression mCherryCAS, mCherryCAS_T18D, or mCherryCAS_T18G using jetOptimus. Cell culture inserts were removed once the cells reached confluency. Subsequent cell growth was recorded using a Paula system (Leica Microsystems) for the next 40 h. The area between the opposing fronts was measured in several positions along the separation line to calculate a mean value (see Fig. S5). This value was normalized by the mean area measured at 0 h and plotted as a function of time (Fig. 8 E).
Quantification and statistical analysis
The data were tested for normality using a built-in function of GraphPad Prism software. P values were obtained using an unpaired two-tailed t test (GraphPad Prism software).
Online supplemental material
Fig. S1 shows the characterization of CAS and its mutants, and their interaction with RanGTP and Kapα1 in vitro. Fig. S2 shows kinetic binding maps of CAS, its mutants, and their complexes with Nup214, Nup62, Nup98, and Nup153. Fig. S3 shows RanBP1 or RanPB2 abundance in HEK293T cells after siRNA treatment. Fig. S4 shows the effect of the CAS N-terminus phosphorylation on CAS nuclear retention and its colocalization with F-actin in the cytoplasm. Fig. S5 schematically describes a wound healing assay and its analysis. Table S1 lists FRAP settings used in the experiments together with the mean values obtained for half-time recovery. Table S2 lists all reagents and resources that were used in this study. Video 1 shows a representative time-lapse clip of fluorescence recovery after photobleaching (FRAP) of mCherry-CAS in the nucleus of HEK293T cells. Video 2 shows a representative time-lapse clip of mCherry-∆40NCAS FRAP in the nucleus of HEK293T cells. Video 3 shows a representative time-lapse clip of mCherry-CAS_T18G FRAP in the nucleus of HEK293T cells. Video 4 shows a representative time-lapse clip of mCherry-CAS_T18D FRAP in the nucleus of HEK293T cells. Video 5 shows a representative time-lapse clip of HEK293T cells expressing mCherry-CAS and mEos2-Lifeact-7 peptide. Video 6 shows a representative time-lapse clip of HEK293T cells expressing mCherry-∆40NCAS and mEos2-Lifeact-7 peptide. Video 7 shows a representative time-lapse clip of HEK293T cells expressing mCherryCAS_T18G and mEos2-Lifeact-7 peptide. Video 8 shows a representative time-lapse clip of HEK293T cells expressing mCherryCAS_T18D and mEos2-Lifeact-7 peptide.
Data availability
Raw image data files are available in the Zenodo public repository under the identifier 10.5281/zenodo.10463692. All other supporting data are available in the main text or supplemental material.
Acknowledgments
This work is supported by the Biozentrum and the Swiss Nanoscience Institute. R.Y.H. Lim acknowledges funding support from the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation; grant no. 310030_201062).
We also thank the Imaging Core Facility and the Biophysics Facility at the Biozentrum for their support and assistance.
Author contributions: L.E. Kapinos and R.Y.H. Lim conceptualized the project, scrutinized data, and wrote the manuscript. L.E. Kapinos, J. Kalita, and E. Kassianidou designed and performed experiments as well as analyzed data. C. Rencurel expressed and purified proteins. All authors commented on the manuscript.

