


Here, we report that the RTN3L–SEC24C endoplasmic reticulum autophagy (ER-phagy) receptor complex, the CUL3KLHL12 E3 ligase that ubiquitinates RTN3L, and the FIP200 autophagy initiating protein, target mutant proinsulin (Akita) condensates for lysosomal delivery at ER tubule junctions. When delivery was blocked, Akita condensates accumulated in the ER. In exploring the role of tubulation in these events, we unexpectedly found that loss of the Parkinson’s disease protein, PINK1, reduced peripheral tubule junctions and blocked ER-phagy. Overexpression of the PINK1 kinase substrate, DRP1, increased junctions, reduced Akita condensate accumulation, and restored lysosomal delivery in PINK1-depleted cells. DRP1 is a dual-functioning protein that promotes ER tubulation and severs mitochondria at ER–mitochondria contact sites. DRP1-dependent ER tubulating activity was sufficient for suppression. Supporting these findings, we observed PINK1 associating with ER tubules. Our findings show that PINK1 shapes the ER to target misfolded proinsulin for RTN3L–SEC24C–mediated macro-ER-phagy at defined ER sites called peripheral junctions. These observations may have important implications for understanding Parkinson’s disease.
Introduction
The endoplasmic reticulum (ER) is a continuous membrane structure consisting of two main domains: flat sheets that largely reside near the nucleus and a polygonal tubular network that extends to the cortex (Chen et al., 2013; Shibata et al., 2006). This unique morphology is maintained by the reticulons (RTN), atlastins (ATL), and Lunapark (LNPK), ER shaping proteins that regulate the formation of the tubular network (Chen et al., 2013; Shibata et al., 2006). The reticulons promote membrane curvature via a reticulon homology domain (RHD), while the atlastins mediate tubule fusion (Chen et al., 2013; Shibata et al., 2006). LNPK stabilizes newly formed tubule junctions that form at the site where two tubules fuse (Chen et al., 2015). In the absence of LNPK, junctions and tubules decrease, and consequently ER sheets proliferate (Chen et al., 2015; Wang et al., 2016). ER shape is important for maintaining cell health as mutations in ER shaping proteins have been linked to neurodegenerative disorders, including hereditary spastic paraplegias (HSP) (Chen et al., 2013).
The ER is the site of synthesis for secretory and membrane proteins that traffic within the cell (Chen et al., 2013). Newly synthesized proteins are packaged into COPII transport carriers at specialized ER subdomains called ER exit sites (ERES) and then transported to the Golgi (Gomez-Navarro and Miller, 2016). The formation of COPII transport carriers is initiated when the GTPase SAR1 recruits the SEC24–SEC23 complex to ER membranes to sort cargo into a prebudding complex (Gomez-Navarro and Miller, 2016). Four different SEC24 isoforms sort cargo into COPII transport carriers, SEC24A, SEC24B, SEC24C, and SEC24D (Tang et al., 1999). The SEC24–SEC23 complex also recruits a second coat complex to the prebudding complex, SEC13–SEC31, which then leads to GTP hydrolysis and the release of a COPII transport carrier (Gomez-Navarro and Miller, 2016). When defects in protein biogenesis occur, misfolded and unassembled proteins are retrotranslocated across the membrane via ER-associated protein degradation (ERAD) machinery and destroyed in the cytosol by the proteasome (Sun and Brodsky, 2019; Wu and Rapoport, 2018). Many disease-causing, aggregation-prone proteins, however, are inefficiently degraded by ERAD and a resistant population remains in the ER. ER-autophagy (ER-phagy) is an alternate degradative system that removes this ERAD-resistant population (Chino and Mizushima, 2020; Ferro-Novick et al., 2021; Mochida and Nakatogawa, 2022; Wilkinson, 2020).
ER-phagy is mediated by receptors that link ER domains in tubules or sheets to the autophagy machinery (Ferro-Novick et al., 2021). Autophagy receptors bind to lipidated Atg8 family members (called LC3 or GABARAP in mammals) via an LC3-interacting region (LIR) (Johansen and Lamark, 2020). ER-phagy can be autophagosome-mediated (macro-ER-phagy) or non-autophagosome-mediated (micro-ER-phagy and vesicular delivery) (Chino and Mizushima, 2020; Ferro-Novick et al., 2021). During macro-ER-phagy, the receptor binds to lipidated Atg8 to sequester an ER fragment into a double membrane vesicle, called an autophagosome, that is delivered to the lysosome for degradation (Chino and Mizushima, 2020; Ferro-Novick et al., 2021). In micro-ER-phagy, tubule fragments are directly engulfed by the lysosome (Liao et al., 2024; Loi et al., 2019). Recently, a trafficking pathway that delivers single membrane vesicles to the lysosome has also been described (Fregno et al., 2018; Sun et al., 2023). The FIP200-containing autophagy initiation complex, which is required for autophagosome biogenesis, is needed for macroautophagy but not microautophagy or vesicular delivery to the lysosome (Fregno et al., 2018; Hara et al., 2008; Liao et al., 2024; Loi et al., 2019; Omari et al., 2018; Sun et al., 2023).
The reticulon ER-phagy receptor RTN3L and the sheets receptor FAM134B contain an RHD (Chen et al., 2013; Shibata et al., 2006). A RHD is a hairpin membrane insertion sequence that facilitates membrane bending (Chen et al., 2013; Shibata et al., 2006). Ubiquitination within the FAM134B RHD has recently been reported to enhance its oligomerization and membrane-bending activity (González et al., 2023). FAM134B and RTN3L are also related to the yeast (Saccharomyces cerevisiae) reticulon-like ER-phagy receptor Atg40 (Khaminets et al., 2015; Mochida et al., 2015; Parashar et al., 2021). Atg40 works with the COPII coat complex, Lst1-Sec23, to package ER into autophagosomes at ER-phagy sites (ERPHS). ERPHS are distinct from the ERES where COPII vesicles bud from the ER (Cui et al., 2019).
In mammalian cells, ERPHS can be visualized in the tubular network with Akita, an ERAD-resistant mutant form of proinsulin that causes early-onset diabetes (Cunningham et al., 2019; Parashar et al., 2021). Misfolded Akita condensates colocalize with RTN3L-containing puncta that also contain SEC24C–SEC23 (mammalian Lst1-Sec23) and LC3B (Parashar et al., 2021). ERAD-resistant misfolded pro-opiomelanocortin (C28F POMC) and pro-arginine-vasopressin (G57S Pro-AVP) behave similarly to Akita (Cunningham et al., 2019; Parashar et al., 2021). While much is known about COPII-mediated cargo sorting and vesicle formation, where and how ERPHS are formed and the machinery that regulates their formation is unknown. Here, we report that ERPHS are formed at a distinct subdomain of the tubular network, LNPK-marked three-way tubule junctions. We found that FIP200, RTN3L, its binding partner SEC24C, and the E3 ligase CUL3KLHL12 are needed to retain misfolded Akita condensates in ERPHS at tubule junctions prior to their delivery to lysosomes. RTN3L is ubiquitinated by CUL3KLHL12, which is recruited to LNPK-marked junctions where RTN3L accumulates. In exploring the role of peripheral tubules and junctions in ER-phagy, we unexpectedly found that the Parkinson’s disease kinase PINK1 regulates ER-phagy by shaping the peripheral ER. PINK1, which is known to regulate mitochondrial morphology and mitophagy, localizes to the mitochondria and ER–mitochondria contact sites (Gómez-Suaga et al., 2018). Corroborating the new role for the PINK1 kinase that we describe here, PINK1 was found to associate with ER tubules. In total, these studies explain how the RTN3L–SEC24C pathway targets ERAD-resistant proteins for macro-ER-phagy at spatially defined sites at the cell periphery whose formation is regulated by PINK1.
Results
RTN3L binds to SEC24C in the absence of LC3 lipidation
To begin our analysis, we performed coprecipitation experiments to ask if the interaction of RTN3L with SEC24C requires lipidated LC3. For these studies, we used MRT68921, an autophagy inhibitor that efficiently impairs LC3 lipidation (see Fig. S10 A, right) and autophagosome maturation by disrupting ULK1 kinase activity (Petherick et al., 2015; Zachari et al., 2020). We found that the endogenous copy of SEC24C coprecipitated with RTN3L from cells stably expressing FLAG-HA-RTN3L, but not untagged cells (Fig. 1 A and Fig. S1 A, left). Furthermore, SEC24C coprecipitated equally as efficiently with RTN3L in untreated or MRT68921-treated cells (Fig. 1 B). The precipitate contained the SEC24C binding partner, SEC23A (Fig. S1 A, middle), but not SEC24A (Fig. S1 A, right), a SEC24 paralog that does not act in ER-phagy (Parashar et al., 2021). These results are consistent with previous studies showing that the induction of autophagy with the TORC1 inhibitor, Torin 2, leads to the colocalization of RTN3L puncta with SEC24C, but not other SEC24 paralogs (SEC24A) (Parashar et al., 2021). The delivery of RTN3L to lysosomes during basal cell growth was inhibited by MRT68921 and dependent on SEC24C, while SEC24C lysosomal delivery was RTN3L-dependent (Fig. S1, B–D and Fig. S2). The lysosomal delivery of the tubule marker, RTN4, was also significantly impaired in RTN3L-and SEC24C-depleted cells (Fig. S1 D and Fig. S3). As RTN3L coprecipitates with SEC24C and the lysosomal delivery of RTN3L and SEC24C are dependent on each other, these data imply that RTN3L and SEC24C are delivered together as a complex to lysosomes. Lysosomal delivery, but not RTN3L–SEC24C complex formation, appears to require LC3 lipidation.

The RTN3L–SEC24C receptor complex mediates traffic to lysosomes from ER junctions independent of COPII vesicle traffic. (A) Lysates were prepared from cells treated with Bafilomycin A1 (Baf) for 4 h and incubated with anti-HA-agarose beads. Input is 0.2% of the lysate. SEC24C inputs were equal (see Fig. S1 A). A 6.56 increase in SEC24C was present in the FLAG-HA-RTN3L precipitate compared with the untagged control. (B) Same as A only, control (Ctrl) cells were treated with Baf or Baf + MRT68921 for 4 h. There was 1.21 × more SEC24C in lane 2 when compared to lane 1 (set to 1.00). The data was normalized to RTN3L levels. (C) In vitro binding experiments performed with lysates prepared from rich or nutrient starved cells yielded the same results. Cells were grown in rich media for the data shown. Asterisk (*) marks a contaminating band. (D) siCtrl, SAR1:T39N transfected cells and siRNA-treated cells were transfected with Akita-sfGFP. The percent cells with large Akita puncta (≥0.5 µm2), and the mean size of Akita puncta are reported. Arrow marks a puncta that was quantitated. (E) Cells were transfected with LNPK–GFP and mCherry–RTN3L or mCherry–FAM134B. Arrowheads mark colocalizing puncta. (F) Cells were untreated or treated with Baf or Baf + MRT68921 (6 h). The relative pixel intensity is reported for images on the left and Fig. S5 B. Arrowheads mark LNPK-GFP puncta colocalizing with LAMP1-mCherry structures. The Ctrl for each condition was without Baf (DMSO) and set to 1.0. Error bars (D–F) represent SEM, n = 3 independent experiments. The results were quantified from 62 to 101 cells in D, 58–61 cells in E, and 63–87 cells for F and Fig. S5 B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData F1.
The RTN3L–SEC24C receptor complex mediates traffic to lysosomes from ER junctions independent of COPII vesicle traffic. (A) Lysates were prepared from cells treated with Bafilomycin A1 (Baf) for 4 h and incubated with anti-HA-agarose beads. Input is 0.2% of the lysate. SEC24C inputs were equal (see Fig. S1 A). A 6.56 increase in SEC24C was present in the FLAG-HA-RTN3L precipitate compared with the untagged control. (B) Same as A only, control (Ctrl) cells were treated with Baf or Baf + MRT68921 for 4 h. There was 1.21 × more SEC24C in lane 2 when compared to lane 1 (set to 1.00). The data was normalized to RTN3L levels. (C) In vitro binding experiments performed with lysates prepared from rich or nutrient starved cells yielded the same results. Cells were grown in rich media for the data shown. Asterisk (*) marks a contaminating band. (D) siCtrl, SAR1:T39N transfected cells and siRNA-treated cells were transfected with Akita-sfGFP. The percent cells with large Akita puncta (≥0.5 µm2), and the mean size of Akita puncta are reported. Arrow marks a puncta that was quantitated. (E) Cells were transfected with LNPK–GFP and mCherry–RTN3L or mCherry–FAM134B. Arrowheads mark colocalizing puncta. (F) Cells were untreated or treated with Baf or Baf + MRT68921 (6 h). The relative pixel intensity is reported for images on the left and Fig. S5 B. Arrowheads mark LNPK-GFP puncta colocalizing with LAMP1-mCherry structures. The Ctrl for each condition was without Baf (DMSO) and set to 1.0. Error bars (D–F) represent SEM, n = 3 independent experiments. The results were quantified from 62 to 101 cells in D, 58–61 cells in E, and 63–87 cells for F and Fig. S5 B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData F1.
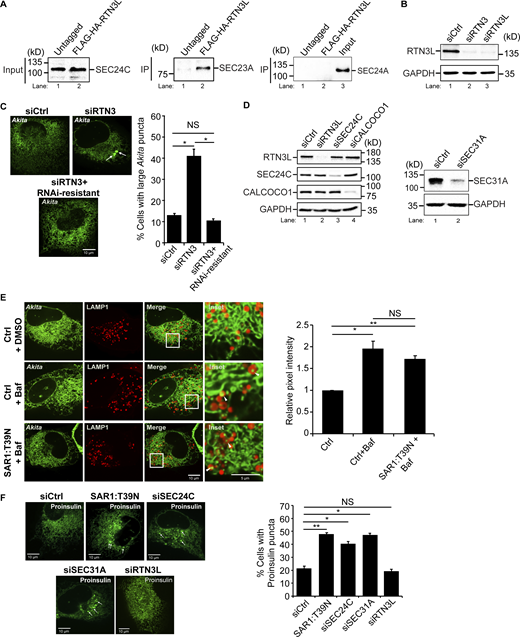
Related toFig. 1,. SAR1:T39N does not block the delivery of misfolded Akita to lysosomes. (A) SEC24C coprecipitates with RTN3L. Left, inputs showing that equal amounts of SEC24C were used for the IP in Fig. 1 A. Right, same as Fig. 1 A only the samples were blotted for SEC23A and SEC24A. (B) RTN3 depletion experiments performed in this study were done with two different siRNAs, siRTN3 (directed at a site in the reticulon domain, see details in the Materials and methods) and siRTN3L (directed at two sites in the long domain, see details in the Materials and methods). The same results were obtained with both siRNAs. Western blot analysis was performed using cell lysates treated with siRTN3 and siRTN3L. GAPDH was used as a loading control. (C) RNAi resistant mCherry-RTN3L suppresses the accumulation of large Akita-sfGFP puncta (≥0.5 µm2) in RTN3-depleted cells. Representative images (left) and quantitation of cells with large Akita puncta (right) are shown. Arrows mark large puncta. (D) Western blot analysis was performed using cells treated with siRTN3L, siSEC24C, siCALCOCO1 (left), and siSEC31A (right). GAPDH was used as a loading control. The depletion of RTN3L did not alter the expression of SEC24C or CALCOCO1. (E) Quantitation of Akita-sfGFP puncta in LAMP1-mCherry structures in DMSO treated or Baf treated (3.5 h) cells that were untransfected (Ctrl) or transfected with SAR1:T39N. Arrowheads mark Akita puncta delivered to lysosomes. The data was quantitated as described in the methods. (F) siCtrl, SAR1:T39N, and siRNA-treated cells were transfected with Proinsulin-sfGFP. The percent cells with Proinsulin-sfGFP puncta (≥0.35 µm2) are reported. Arrows mark puncta that were quantitated. Error bars in C, E, and F represent SEM, n = 3 independent experiments. The results were quantified from 29 to 53 cells in C, 65–76 cells in E, and 74–149 cells in F. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData FS1.

Related toFig. 1,. SAR1:T39N does not block the delivery of misfolded Akita to lysosomes. (A) SEC24C coprecipitates with RTN3L. Left, inputs showing that equal amounts of SEC24C were used for the IP in Fig. 1 A. Right, same as Fig. 1 A only the samples were blotted for SEC23A and SEC24A. (B) RTN3 depletion experiments performed in this study were done with two different siRNAs, siRTN3 (directed at a site in the reticulon domain, see details in the Materials and methods) and siRTN3L (directed at two sites in the long domain, see details in the Materials and methods). The same results were obtained with both siRNAs. Western blot analysis was performed using cell lysates treated with siRTN3 and siRTN3L. GAPDH was used as a loading control. (C) RNAi resistant mCherry-RTN3L suppresses the accumulation of large Akita-sfGFP puncta (≥0.5 µm2) in RTN3-depleted cells. Representative images (left) and quantitation of cells with large Akita puncta (right) are shown. Arrows mark large puncta. (D) Western blot analysis was performed using cells treated with siRTN3L, siSEC24C, siCALCOCO1 (left), and siSEC31A (right). GAPDH was used as a loading control. The depletion of RTN3L did not alter the expression of SEC24C or CALCOCO1. (E) Quantitation of Akita-sfGFP puncta in LAMP1-mCherry structures in DMSO treated or Baf treated (3.5 h) cells that were untransfected (Ctrl) or transfected with SAR1:T39N. Arrowheads mark Akita puncta delivered to lysosomes. The data was quantitated as described in the methods. (F) siCtrl, SAR1:T39N, and siRNA-treated cells were transfected with Proinsulin-sfGFP. The percent cells with Proinsulin-sfGFP puncta (≥0.35 µm2) are reported. Arrows mark puncta that were quantitated. Error bars in C, E, and F represent SEM, n = 3 independent experiments. The results were quantified from 29 to 53 cells in C, 65–76 cells in E, and 74–149 cells in F. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData FS1.

Related to Fig. 1 . RTN3L and SEC24C are delivered to lysosomes during basal autophagy. (A) Ctrl or SEC24C-depleted cells were treated for 4 h with Baf or Baf + MRT68921. Arrowheads mark RTN3L fragments delivered to lysosomes. The relative pixel intensity for each condition is the mean intensity of YFP-RTN3L in the pixels that overlap with LAMP1-mCherry for the images shown. The control for each condition was without Baf (DMSO only) and set to 1.0. MRT68921 was used to block autophagosome formation. (B) Same as A, only cells were depleted of RTN3L and the delivery of YFP-SEC24C to lysosomes was examined. Arrowheads mark SEC24C in lysosomes. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 84 to 112 cells in A, and 88–128 in B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.

Related to Fig. 1 . RTN3L and SEC24C are delivered to lysosomes during basal autophagy. (A) Ctrl or SEC24C-depleted cells were treated for 4 h with Baf or Baf + MRT68921. Arrowheads mark RTN3L fragments delivered to lysosomes. The relative pixel intensity for each condition is the mean intensity of YFP-RTN3L in the pixels that overlap with LAMP1-mCherry for the images shown. The control for each condition was without Baf (DMSO only) and set to 1.0. MRT68921 was used to block autophagosome formation. (B) Same as A, only cells were depleted of RTN3L and the delivery of YFP-SEC24C to lysosomes was examined. Arrowheads mark SEC24C in lysosomes. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 84 to 112 cells in A, and 88–128 in B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.

Related to Fig. 1 . RTN3L and SEC24C are needed for the delivery of RTN4 fragments to lysosomes during basal autophagy. siCtrl, siRTN3L, siSEC24C, and siCALCOCO1 cells were treated for 6 h with Baf or Baf + MRT68921. The ER-phagy receptor, CALCOCO1, was used as a control for basal autophagy in this experiment. Top, representative images for the data quantitated below. Arrowheads mark RTN4 fragments delivered to lysosomes. Bottom, the relative pixel intensity for each condition is the mean intensity of RTN4-GFP in the pixels that overlap with LAMP1-mCherry. The control for each condition was without Baf (DMSO only) and set to 1.0. MRT68921 was used to block autophagosome maturation. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 36 to 66 cells. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.

Related to Fig. 1 . RTN3L and SEC24C are needed for the delivery of RTN4 fragments to lysosomes during basal autophagy. siCtrl, siRTN3L, siSEC24C, and siCALCOCO1 cells were treated for 6 h with Baf or Baf + MRT68921. The ER-phagy receptor, CALCOCO1, was used as a control for basal autophagy in this experiment. Top, representative images for the data quantitated below. Arrowheads mark RTN4 fragments delivered to lysosomes. Bottom, the relative pixel intensity for each condition is the mean intensity of RTN4-GFP in the pixels that overlap with LAMP1-mCherry. The control for each condition was without Baf (DMSO only) and set to 1.0. MRT68921 was used to block autophagosome maturation. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 36 to 66 cells. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.
To characterize the RTN3L–SEC24C interaction in more detail, in vitro binding experiments were performed with purified GST proteins and lysates that contained endogenous levels of SEC24C. Although the entire RTN3L cytoplasmic domain fused to GST was unstable, two stable fusion proteins were expressed and purified from bacteria, GST–RTN3L (amino acid [aa] 48–648) and GST-RTN3S (aa 1–47). RTN3S is a short form of RTN3 that does not act in ER-phagy (Grumati et al., 2017). GST–RTN3L (aa 48–648) bound to SEC24C, but not CALCOCO1 (Nthiga et al., 2020), another tubular ER-phagy receptor that was used as a specificity control (Fig. 1 C). SEC24C also specifically bound to GST–RTN3L but not GST-RTN3S (Fig. 1 C).
Next, we asked if the LIRs in RTN3L are needed for its interaction with SEC24C. The RTN3L cytoplasmic domain contains six binding sites for LC3 (LIRs) (Grumati et al., 2017). As GST–RTN3L (aa 48–648) lacks the sixth LIR and still binds to SEC24C, this data implies that the sixth LIR is not needed for the RTN3L–SEC24C interaction. Previous studies showed that LC3 binding to RTN3L is blocked when all six LIRs are mutated in the RTN3LΔ6LIR mutant (Grumati et al., 2017). When we treated mCherry–RTN3LΔ6LIR mutant cells with Torin, RTN3L puncta formation was dramatically impaired (Fig. S4 A). This decrease was unlikely to be the consequence of blocking the interaction of RTN3L with lipidated LC3 as no obvious decrease in RTN3L puncta formation was observed when lipidation was blocked in MRT68921-treated cells (Fig. S4 A). As RTN3L puncta formation is a key step in ER-phagy, these observations suggested that the 16-point mutations introduced into RTN3LΔ6LIR may abrogate RTN3L functions that are independent of LC3 binding. Therefore, to address if the remaining five LIRs are needed for the RTN3L–SEC24C interaction, colocalization studies were performed with two LIR mutants (RTN3LΔ3LIR and RTN3LΔ5LIR) that reduce LC3 binding (Grumati et al., 2017), but not RTN3L puncta formation (Fig. S4, B and C). We found that neither mCherry–RTN3LΔ3LIR nor mCherry–RTN3LΔ5LIR significantly affected the colocalization of YFP-SEC24C puncta with mCherry-RTN3L puncta (Fig. S4 C). In total, these studies suggest that the RTN3L LIRs are dispensable for their interaction with SEC24C. These observations are also consistent with the finding that RTN3L co-precipitates with SEC24C when LC3 lipidation is impaired.
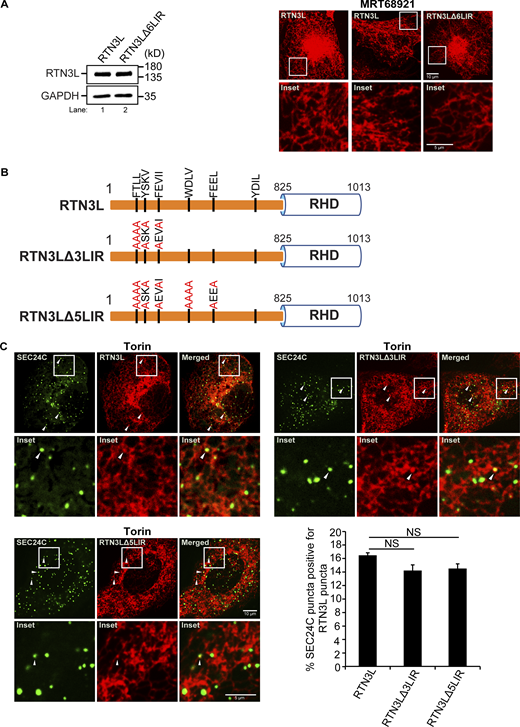
Related toFig. 1 . LIRs 1–5 are not needed for the colocalization of SEC24C puncta with RTN3L puncta. (A) Left, a western blot showing that the expression of RTN3L is the same in cells stably expressing mCherry–RTN3L or mCherry–RTN3LΔ6LIR. Right, cells stably expressing mCherry–RTN3L or mCherry–RTN3LΔ6LIR were treated with Torin 2 or MRT68921 for 3.5 h and imaged. (B) Diagram showing the LIR mutations in mCherry-RTN3L. The 6LIRs are shown as black bars and the mutated amino acids are in red. (C) Representative images for the constructs shown in B. The percent SEC24C puncta colocalizing with RTN3L puncta were quantified for the constructs shown in B. Cells were treated for 3.5 h with Torin2. Error bars in C represent SEM, n = 3 independent experiments. The results were quantified from 60 to 66 cells. NS: not significant (P ≥ 0.05), Student’s unpaired t test. Source data are available for this figure: SourceData FS4.

Related toFig. 1 . LIRs 1–5 are not needed for the colocalization of SEC24C puncta with RTN3L puncta. (A) Left, a western blot showing that the expression of RTN3L is the same in cells stably expressing mCherry–RTN3L or mCherry–RTN3LΔ6LIR. Right, cells stably expressing mCherry–RTN3L or mCherry–RTN3LΔ6LIR were treated with Torin 2 or MRT68921 for 3.5 h and imaged. (B) Diagram showing the LIR mutations in mCherry-RTN3L. The 6LIRs are shown as black bars and the mutated amino acids are in red. (C) Representative images for the constructs shown in B. The percent SEC24C puncta colocalizing with RTN3L puncta were quantified for the constructs shown in B. Cells were treated for 3.5 h with Torin2. Error bars in C represent SEM, n = 3 independent experiments. The results were quantified from 60 to 66 cells. NS: not significant (P ≥ 0.05), Student’s unpaired t test. Source data are available for this figure: SourceData FS4.
The RTN3L–SEC24C ER-phagy pathway acts independently of COPII vesicle traffic
As the lysosomal delivery of RTN3L is dependent on SEC24C, and the SEC24C–SEC23 complex also acts in COPII vesicle traffic, we asked if RTN3L–SEC24C-mediated ER-phagy is regulated by the secretory pathway. RTN3L targets a subpopulation of misfolded proinsulin Akita, which forms high molecular weight ERAD-resistant oligomers, for autophagy (Cunningham et al., 2019). Akita oligomers targeted for ER-phagy can be visualized in ER tubules with Akita-sfGFP as small (≤0.12 µm2) highly mobile puncta that behave like liquid condensates (Parashar et al., 2021). These condensates/puncta change size and shape as they move through the network (Parashar et al., 2021). Akita condensates colocalize with LC3, SEC24C, and RTN3L puncta that form on the ER when ER-phagy is induced (Parashar et al., 2021). Furthermore, the colocalization of LC3 with Akita puncta is dependent on RTN3L (Parashar et al., 2021). The RTN3L–SEC24C–LC3B-containing puncta, called ERPHS, appears to be similar in size to ERES (Parashar et al., 2021). However, RTN3L puncta do not colocalize with the COPII subunits SEC24A and SEC13 or SEC16, a marker for ERES (Parashar et al., 2021). When cells are depleted of RTN3L or the autophagy protein Beclin1, Akita forms a massive detergent insoluble complex in the ER, and large puncta (≥0.5 µm2) accumulate in a fraction (˜40%) of the cells (Chen et al., 2020; Cunningham et al., 2019; Parashar et al., 2021). These large puncta also form when cells are depleted of SEC24C (Parashar et al., 2021) but not the other SEC24 isoforms (Parashar et al., 2021). In addition to the formation of large puncta, the mean size of Akita puncta/condensates per cell also increases when ER-phagy is blocked (Fig. 1 D) (Parashar et al., 2021). Together these studies show that ERAD-resistant misfolded Akita condensates traffic from the ER to the lysosome from an ER subdomain that appears to be distinct from the site where COPII vesicles traffic to the Golgi (Cui et al., 2019; Parashar et al., 2021).
Although RTN3L–SEC24C acts at sites that are distinct from the ERES, the RTN3L-ER-phagy pathway could be regulated by COPII vesicle traffic. To directly address this possibility, we blocked secretion with the T39N SAR1 mutant and asked if large Akita puncta accumulates in the ER. SAR1-T39N is a dominant negative mutant that blocks the formation of ERES (Kuge et al., 1994). We found that the depletion of SEC24C, but not the expression of SAR-T39N, increased the percentage of cells with large Akita puncta (≥0.5 µm2), as well as the mean size of Akita puncta per cell (Fig. 1 D). Consistent with this observation, the delivery of Akita to lysosomes was not disrupted in SAR1 mutant cells (Fig. S1 E). siSEC31A-treated cells also failed to accumulate large Akita puncta (Fig. 1 D and Fig. S1 D, right). In contrast, and consistent with a block in secretion, Proinsulin-sfGFP puncta (≥0.35 µm2) accumulated in the ER (see Materials and methods for quantitation details) of SAR1-T39N transfected cells and cells lacking SEC24C or SEC31A (Fig. S1 F), but not RTN3L (Fig. S1 F). Together, these findings indicate that RTN3L–SEC24C–mediated ER-phagy acts independently of the secretory pathway and COPII vesicle traffic.
FIP200 is needed for the accumulation of misfolded Akita condensates at three-way junctions
RTN3L puncta were frequently seen colocalizing with SEC24C puncta at three-way tubule junctions in Torin-treated cells (Fig. S4 C). This observation prompted us to ask if the RTN3L–SEC24C receptor complex targets misfolded Akita condensates for ER-phagy at junctions. To begin to address this question, we first asked if RTN3L puncta accumulate at junctions. FAM134B was used as a control for these studies as it mediates ER quality control at ERES and not ERPHS (Parashar et al., 2021; Roberts et al., 2024; Sun et al., 2023). Junctions were marked by LNPK, which forms puncta at three-way tubule junctions (Chen et al., 2012, 2015). We found that LNPK puncta colocalized with RTN3L, but not FAM134B, puncta (Fig. 1 E). The colocalization of LNPK puncta with RTN3L puncta did not decrease in SEC24C-depleted cells (Fig. S5 A), indicating that RTN3L localization at junctions is not dependent on SEC24C. Consistent with these studies, RTN3L and SEC24C, but not FAM134B, were needed for the delivery of LNPK to lysosomes in a rich medium (Fig. 1 F and Fig. S5 B). LC3 lipidation also appeared to be needed for LNPK delivery as it was blocked by MRT68921 (Fig. 1 F). Interestingly, when we examined a second tubular ER-phagy receptor that acts in basal ER-phagy, CALCOCO1 (Nthiga et al., 2020), it was dispensable for the lysosomal delivery of LNPK-marked junctions (Fig. S5 B).
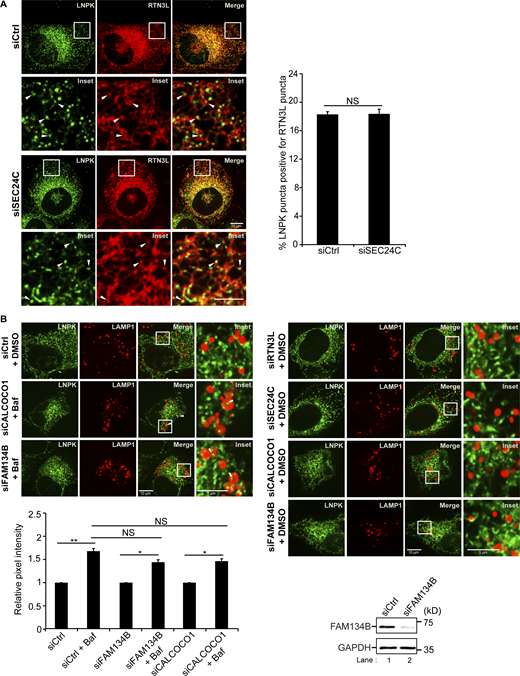
Related toFig. 1,. FAM134B and CALCOCO1 are not required for the delivery of LNPK puncta to lysosomes. (A) Left, control and siSEC24C-treated cells transfected with LNPK-GFP and mCherry-RTN3L. Arrowheads mark colocalizing puncta. Right, quantitation of percent LNPK puncta colocalizing with RTN3L puncta. (B) Bottom, same as Fig. 1 F, only siFAM134B and siCALCOCO1-treated cells were examined. The control is the same as in Fig. 1 F as the samples were all assayed at the same time. Bottom right, western blot analysis was performed with control and FAM134B-depleted lysates. GAPDH was used as a loading control. Error bars in A and B represent SEM, n = 3 independent experiments. The results were quantified from 40 to 50 cells in A, and 63–87 cells in B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData FS5.

Related toFig. 1,. FAM134B and CALCOCO1 are not required for the delivery of LNPK puncta to lysosomes. (A) Left, control and siSEC24C-treated cells transfected with LNPK-GFP and mCherry-RTN3L. Arrowheads mark colocalizing puncta. Right, quantitation of percent LNPK puncta colocalizing with RTN3L puncta. (B) Bottom, same as Fig. 1 F, only siFAM134B and siCALCOCO1-treated cells were examined. The control is the same as in Fig. 1 F as the samples were all assayed at the same time. Bottom right, western blot analysis was performed with control and FAM134B-depleted lysates. GAPDH was used as a loading control. Error bars in A and B represent SEM, n = 3 independent experiments. The results were quantified from 40 to 50 cells in A, and 63–87 cells in B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData FS5.
We found that small Akita-sfGFP, but not wild-type Proinsulin-sfGFP, puncta colocalized with LNPK-mCherry puncta at tubule junctions (Fig. 2 A). To ask if Akita condensates accumulate in autophagosomes or preautophagosomal structures at junctions, colocalization studies were performed in FIP200-depleted cells. FIP200 is a component of the autophagy machinery that lipidates LC3 during double-membrane autophagosome biogenesis (Hara et al., 2008). Interestingly, while knocking down FIP200 did not decrease LNPK-marked junctions (Fig. S6 A), the accumulation of Akita-sfGFP puncta at LNPK junctions significantly decreased in the depleted cells (Fig. 2 B). When Akita puncta failed to accumulate at junctions in siFIP200 knock down cells, the condensates enlarged (≥0.5 µm2) and accumulated in the ER (Fig. S6 B), indicating that ER-phagy was blocked. Consistent with the proposal that RTN3L–SEC24C–mediated ER-phagy is autophagosome-mediated, the overexpression of a mCherry-SopF construct that is known to block LC3 lipidation during noncanonical autophagy (Hooper et al., 2022; Lau et al., 2019), did not lead to the accumulation of large Akita puncta/condensates in the ER (Fig. S6 C). SopF is a Salmonella effector that blocks the V-ATPase–ATG16L1–mediated LC3 lipidation of single membrane vesicles (Hooper et al., 2022; Lau et al., 2019).

FIP200 and the RTN3L–SEC24C complex are required for the retention of Akita puncta at three-way junctions. (A) Arrowheads mark colocalizing puncta in cells transfected with LNPK-mCherry and Akita-sfGFP or Proinsulin-sfGFP. (B) Same as A, only Akita puncta were examined in siCtrl and siFIP200-treated cells. (C) Cells transfected with Akita-sfGFP and LNPK-mCherry in siCtrl or siRNA-treated cells. Arrowheads mark Akita puncta that colocalize with LNPK puncta. (D) Top, percent cells with large Akita puncta (≥0.5 µm2), and mean size of Akita puncta (bottom) for the data in C. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 44 to 59 cells in A, 37–40 cells in B, 38–42 cells in C, and 115–139 cells in D. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test.
FIP200 and the RTN3L–SEC24C complex are required for the retention of Akita puncta at three-way junctions. (A) Arrowheads mark colocalizing puncta in cells transfected with LNPK-mCherry and Akita-sfGFP or Proinsulin-sfGFP. (B) Same as A, only Akita puncta were examined in siCtrl and siFIP200-treated cells. (C) Cells transfected with Akita-sfGFP and LNPK-mCherry in siCtrl or siRNA-treated cells. Arrowheads mark Akita puncta that colocalize with LNPK puncta. (D) Top, percent cells with large Akita puncta (≥0.5 µm2), and mean size of Akita puncta (bottom) for the data in C. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 44 to 59 cells in A, 37–40 cells in B, 38–42 cells in C, and 115–139 cells in D. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test.
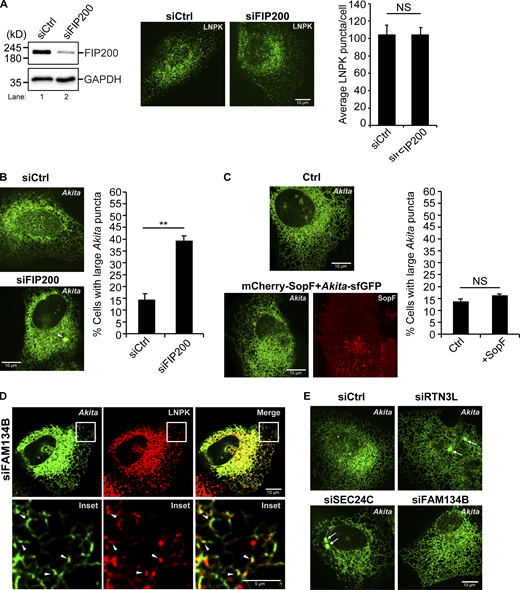
Related toFig. 2,. SopF does not inhibit RTN3L–SEC24C-mediated ER-phagy. (A) Left, a western blot showing the depletion of FIP200 in siFIP200-treated cells. GAPDH was used as a loading control. Right, quantitation of LNPK-GFP puncta in siCtrl and siFIP200-treated cells. Representative images are shown on the left. (B) Left, a representative image of a control cell and a siFIP200-treated cell showing the accumulation of a large Akita puncta (see arrowhead). Right, the percent cells with large Akita puncta in control and FIP200-depleted cells. (C) Cells were transfected with Akita-sfGFP or Akita-sfGFP and mCherry-SopF. Representative images are shown on the left and the percent cells with large Akita puncta is graphed on the right. (D) A representative image for the data quantitated in Fig. 2 C. Arrowheads mark Akita-sfGFP puncta that colocalize with LNPK-mCherry puncta. (E) Representative images for the data graphed in Fig. 2 D. Arrows mark large Akita puncta. Error bars in A–C represent SEM, n = 3 independent experiments. The results were quantified from 61 to 64 cells in A, 56–104 cells in B, and 92–95 cells in C. NS: not significant (P ≥ 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData FS6.

Related toFig. 2,. SopF does not inhibit RTN3L–SEC24C-mediated ER-phagy. (A) Left, a western blot showing the depletion of FIP200 in siFIP200-treated cells. GAPDH was used as a loading control. Right, quantitation of LNPK-GFP puncta in siCtrl and siFIP200-treated cells. Representative images are shown on the left. (B) Left, a representative image of a control cell and a siFIP200-treated cell showing the accumulation of a large Akita puncta (see arrowhead). Right, the percent cells with large Akita puncta in control and FIP200-depleted cells. (C) Cells were transfected with Akita-sfGFP or Akita-sfGFP and mCherry-SopF. Representative images are shown on the left and the percent cells with large Akita puncta is graphed on the right. (D) A representative image for the data quantitated in Fig. 2 C. Arrowheads mark Akita-sfGFP puncta that colocalize with LNPK-mCherry puncta. (E) Representative images for the data graphed in Fig. 2 D. Arrows mark large Akita puncta. Error bars in A–C represent SEM, n = 3 independent experiments. The results were quantified from 61 to 64 cells in A, 56–104 cells in B, and 92–95 cells in C. NS: not significant (P ≥ 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData FS6.
The retention of misfolded Akita condensates/puncta at three-way junctions also required RTN3L and SEC24C, but not FAM134B (Fig. 2 C and Fig. S6 D), while wild-type proinsulin localization remained unchanged in the absence of each of these three proteins (Fig. S7 A). When misfolded Akita failed to accumulate at junctions, large condensates accumulated in the ER (Fig. 2 D, top and bottom, Fig. S6 E). While SEC24C was not needed for the localization of RTN3L puncta at junctions, it was required for the association of RTN3L with Akita condensates (Fig. S7 B). This observation is consistent with the proposal that RTN3L cannot function in ER-phagy unless it is complexed to SEC24C. When RTN3L failed to associate with misfolded Akita, the condensates enlarged in the ER and accumulated (Fig. S7 C). As previous studies have shown that the association of misfolded Akita with LC3B requires RTN3L (Parashar et al., 2021), these observations imply that FIP200-dependent RTN3L–SEC24C–LC3B-containing ERPHS accumulate at ER junctions.

Related to Fig. 2 . SEC24C is needed for the association of RTN3L puncta with misfolded Akita. (A) Cells were transfected with LNPK-mCherry and Proinsulin-sfGFP and the percent proinsulin puncta that colocalize with LNPK was quantitated, bottom. Top, representative images are shown. Error bars represent SEM, n = 3 independent experiments. (B) siCtrl and siSEC24C-treated cells were transfected with Akita-sfGFP and mCherry-RTN3L and imaged. Arrowheads point to the Akita puncta that colocalize with RTN3L puncta. Right, quantification of the % Akita puncta positive for RTN3L puncta. (C) Percent cells with large Akita puncta (≥0.5 µm2) for the experiment shown in B. The large Akita puncta are marked with arrows in the images shown. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 48 to 53 cells in A, 49–56 cells in B, and 113–125 cells in C. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.

Related to Fig. 2 . SEC24C is needed for the association of RTN3L puncta with misfolded Akita. (A) Cells were transfected with LNPK-mCherry and Proinsulin-sfGFP and the percent proinsulin puncta that colocalize with LNPK was quantitated, bottom. Top, representative images are shown. Error bars represent SEM, n = 3 independent experiments. (B) siCtrl and siSEC24C-treated cells were transfected with Akita-sfGFP and mCherry-RTN3L and imaged. Arrowheads point to the Akita puncta that colocalize with RTN3L puncta. Right, quantification of the % Akita puncta positive for RTN3L puncta. (C) Percent cells with large Akita puncta (≥0.5 µm2) for the experiment shown in B. The large Akita puncta are marked with arrows in the images shown. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 48 to 53 cells in A, 49–56 cells in B, and 113–125 cells in C. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.
CUL3KLHL12 is needed for the autophagy of tubule components at junctions
We reasoned that the formation of RTN3L–SEC24C containing ERPHS may require other components that are enriched at junctions. The E3 ligase, CUL3KLHL12, is a candidate as it is recruited to membranes at ER junctions (Akopian et al., 2022; Yuniati et al., 2020). Consistent with the proposal that CUL3KLHL12 and its substrate adaptor, KLHL12, are needed for ER-phagy, we found that the lysosomal delivery of RTN3L was significantly impaired in KLHL12- or CUL3-depleted cells (Fig. 3 A and Fig. S8 A). In line with the studies described above, siFIP200-treatment also blocked the delivery of RTN3L to lysosomes (Fig. 3 A and Fig. S8 A).

CUL3KLHL12is needed for ER-phagy and the ubiquitination of RTN3L. (A) YFP-RTN3L in LAMP1-mCherry structures in siCtrl and siRNA-treated cells incubated with Baf (4 h). Representative images are shown on the left and in Fig. S8 A. The control for each condition was set to 1.0. (B) RTN3L coprecipitates with KLHL12, but not CALCOCO1. Immunoprecipitations were performed as in Fig. 1 A only the precipitates were eluted using HA peptide as described in the methods. Left, input is 2% of the lysate. Right, equal KLHL12 inputs were used for the immunoprecipitates. (C) Same as B, except the immunoprecipitates were eluted in sample buffer. 4.8 x more PEF1 was precipitated in the tagged sample compared to the untagged control. Input is 2% of the lysate. Equal amounts of PEF1 and SEC24C were used for the immunoprecipitates (see Fig. S8 B). (D) Left, high (asterisks) and low (brackets) molecular weight forms of ubiquitinated RTN3L were detected with ubiquitin antibody. Right, cells were untreated (DMSO) or treated with Baf or Torin 2 + Baf (4 h). (E) The ubiquitination of RTN3L is dependent on KLHL12 and CUL3. Error bars represent SEM, n = 3–6 independent experiments. The results in A were quantified from 59 to 154 cells. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test. Source data are available for this figure: SourceData F3.
CUL3KLHL12is needed for ER-phagy and the ubiquitination of RTN3L. (A) YFP-RTN3L in LAMP1-mCherry structures in siCtrl and siRNA-treated cells incubated with Baf (4 h). Representative images are shown on the left and in Fig. S8 A. The control for each condition was set to 1.0. (B) RTN3L coprecipitates with KLHL12, but not CALCOCO1. Immunoprecipitations were performed as in Fig. 1 A only the precipitates were eluted using HA peptide as described in the methods. Left, input is 2% of the lysate. Right, equal KLHL12 inputs were used for the immunoprecipitates. (C) Same as B, except the immunoprecipitates were eluted in sample buffer. 4.8 x more PEF1 was precipitated in the tagged sample compared to the untagged control. Input is 2% of the lysate. Equal amounts of PEF1 and SEC24C were used for the immunoprecipitates (see Fig. S8 B). (D) Left, high (asterisks) and low (brackets) molecular weight forms of ubiquitinated RTN3L were detected with ubiquitin antibody. Right, cells were untreated (DMSO) or treated with Baf or Torin 2 + Baf (4 h). (E) The ubiquitination of RTN3L is dependent on KLHL12 and CUL3. Error bars represent SEM, n = 3–6 independent experiments. The results in A were quantified from 59 to 154 cells. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test. Source data are available for this figure: SourceData F3.
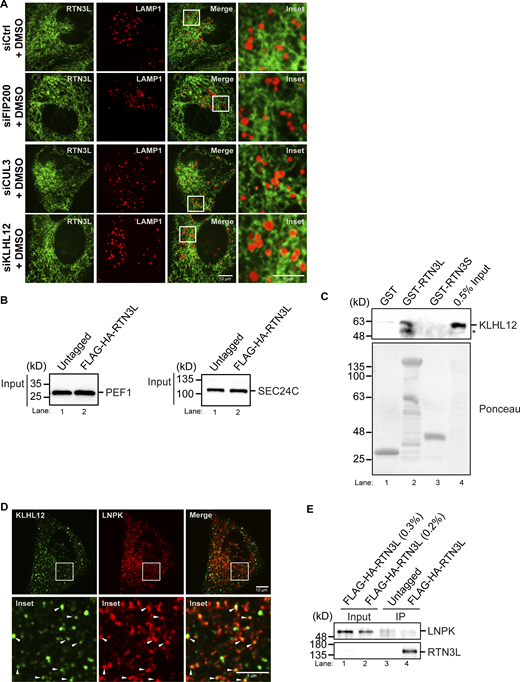
Related toFig. 3,. KLHL12 colocalizes with LNPK at junctions. (A) Representative DMSO control images of YFP-RTN3L puncta with LAMP1-mCherry structures in siCtrl, siFIP200, siCUL3, and siKLHL12-treated cells for the data shown in Fig. 3 A. (B) Inputs for PEF1 and SEC24C for the precipitates that are shown in Fig. 3 C. (C) The in vitro binding experiment was done as in Fig. 1 C only the samples were blotted for the presence of KLHL12. The asterisk marks a breakdown fragment of KLHL12. (D) An image of U2OS cells that were transfected with pHAGE2-LNPK-mCherry and GFP-KLHL12 is shown. Arrowheads mark KLHL12 puncta that colocalize with LNPK puncta. (E) Same as Fig. 3 B, only blotted for the presence of LNPK. Source data are available for this figure: SourceData FS8.

Related toFig. 3,. KLHL12 colocalizes with LNPK at junctions. (A) Representative DMSO control images of YFP-RTN3L puncta with LAMP1-mCherry structures in siCtrl, siFIP200, siCUL3, and siKLHL12-treated cells for the data shown in Fig. 3 A. (B) Inputs for PEF1 and SEC24C for the precipitates that are shown in Fig. 3 C. (C) The in vitro binding experiment was done as in Fig. 1 C only the samples were blotted for the presence of KLHL12. The asterisk marks a breakdown fragment of KLHL12. (D) An image of U2OS cells that were transfected with pHAGE2-LNPK-mCherry and GFP-KLHL12 is shown. Arrowheads mark KLHL12 puncta that colocalize with LNPK puncta. (E) Same as Fig. 3 B, only blotted for the presence of LNPK. Source data are available for this figure: SourceData FS8.
Interestingly, mass spectrometry studies identified KLHL12 as an RTN3L interactor and nine ubiquitination sites were identified in RTN3L (Grumati et al., 2017; Pedersen et al., 2021; Sarraf et al., 2013; Stes et al., 2014; Stukalov et al., 2021). Corroborating these findings, the endogenous copy of KLHL12, but not CALCOCO1, was found to coprecipitate with FLAG-HA-RTN3L (Fig. 3 B). The CUL3KLHL12 co-adaptor, PEF1, was also present in the precipitate, as was SEC24C (Fig. 3 C and Fig. S8 B). Since SEC24C does not appear to be a KLHL12 binding partner (McGourty et al., 2016), the SEC24C present in the precipitate is likely bound to RTN3L. In addition, in vitro–binding studies confirmed that the endogenous copy of KLHL12 bound to GST–RTN3L (aa 48–648), but not RTN3S (Fig. S8 C). While we could confirm that KLHL12 puncta extensively colocalize with LNPK-marked junctions (Fig. S8 D), RTN3L did not coprecipitate with LNPK (Fig. S8 E), a known binding partner for KLHL12 at junctions (Akopian et al., 2022; Yuniati et al., 2020). These findings imply that RTN3L binds directly to CUL3KLHL12.
Consistent with mass spectrometry studies, ubiquitinated RTN3L was detected in FLAG-HA-RTN3L, but not untagged, precipitates (Fig. 3 D, left). Strikingly, RTN3L ubiquitination was only observed in the presence of Baf or Baf + Torin 2 (Fig. 3 D, right), implying that the ubiquitinated form of the receptor is preferentially degraded in lysosomes. RTN3L ubiquitination was dependent on CUL3KLHL12, as it decreased in KLHL12 and CUL3-depleted cells (Fig. 3 E). Two forms of ubiquitinated RTN3L were observed, one form that migrates between the 135 and 180 kD markers (Fig. 3, D and E), and a high molecular weight species that migrates near the 245 kD marker (Fig. 3, D and E). The ubiquitination of both forms partially decreased in CUL3 KLHL12-depleted cells (Fig. 3 E). The ubiquitinated RTN3L high molecular weight species is similar in size to an SDS-resistant oligomer described below (Fig. S9 D). MLN4924, which blocks CUL3 ligase activity and CUL3 neddylation (Fig. S9 A) (Soucy et al., 2009), also partially decreased the ubiquitination of RTN3L (Fig. S9 A), while the potent ubiquitination inhibitor, TAK243, completely blocked ubiquitination (Fig. S9 B). These observations suggest that RTN3L may be ubiquitinated by additional E3 ligases. RTN3L ubiquitination did not lead to proteasomal degradation, as no significant increase in RTN3L was seen in U2OS cells treated with the proteasome inhibitor, MG132 (Fig. S9 C).

Related to Fig. 3,. The ubiquitination of RTN3L increases in the presence of Baf. (A) Left, CUL3 blot showing that the neddylation of CUL3 is blocked 4 h after treatment with 1 µM MLN4924. Right, same as Fig. 3 E only the sample in lane 4 was treated for 4 h with Baf with 1 µM MLN4924. (B) Same as Fig. 3 D only the sample was treated with 10 µM TAK243 for 4 h. (C) U2OS cells were treated for 4 h with 10 μM MG132 before lysates were prepared for western blot analysis. GAPDH was used as a loading control. (D) Membranes were prepared from control, siCUL3 and siSEC24C-treated cells and crosslinked with the indicated concentration of EGS as described in the methods. (E) Top, the position of the known RTN3L ubiquitination sites are shown. Five of the nine sites are in the C-terminus of the RHD. Bottom, the five sites shown are predicted to be at the end of the second hairpin in the RHD. Right, images of mCherry-RTN3L and mCherry-RTN3L-5KR. The mCherry-RTN3L-5KR mutant contains five mutations (K977R, K979R, K995R, K999R and K1003R) that were found to disrupt the tubular ER network. (F) Representative control images of LNPK-GFP puncta in LAMP1-mCherry structures in siCUL3 and siKLHL12-treated cells for the data shown in Fig. 4 A. (G) Representative control images of Akita-sfGFP puncta in LAMP1-mCherry structures in siCtrl and siRNA-treated cells for the data shown in Fig. 4 B. The siCtrl was also treated with Baf + MRT68921 for 3.5 h to block autophagosome maturation.

Related to Fig. 3,. The ubiquitination of RTN3L increases in the presence of Baf. (A) Left, CUL3 blot showing that the neddylation of CUL3 is blocked 4 h after treatment with 1 µM MLN4924. Right, same as Fig. 3 E only the sample in lane 4 was treated for 4 h with Baf with 1 µM MLN4924. (B) Same as Fig. 3 D only the sample was treated with 10 µM TAK243 for 4 h. (C) U2OS cells were treated for 4 h with 10 μM MG132 before lysates were prepared for western blot analysis. GAPDH was used as a loading control. (D) Membranes were prepared from control, siCUL3 and siSEC24C-treated cells and crosslinked with the indicated concentration of EGS as described in the methods. (E) Top, the position of the known RTN3L ubiquitination sites are shown. Five of the nine sites are in the C-terminus of the RHD. Bottom, the five sites shown are predicted to be at the end of the second hairpin in the RHD. Right, images of mCherry-RTN3L and mCherry-RTN3L-5KR. The mCherry-RTN3L-5KR mutant contains five mutations (K977R, K979R, K995R, K999R and K1003R) that were found to disrupt the tubular ER network. (F) Representative control images of LNPK-GFP puncta in LAMP1-mCherry structures in siCUL3 and siKLHL12-treated cells for the data shown in Fig. 4 A. (G) Representative control images of Akita-sfGFP puncta in LAMP1-mCherry structures in siCtrl and siRNA-treated cells for the data shown in Fig. 4 B. The siCtrl was also treated with Baf + MRT68921 for 3.5 h to block autophagosome maturation.
As recent studies suggested that the ubiquitination of the RHD in FAM134B increases its oligomerization and enhances ER-phagy (González et al., 2023), we asked if CUL3 KLHL12 enhances the oligomerization of RTN3L. RTN3L oligomers can be visualized by irreversibly crosslinking intact membranes with the crosslinker EGS (Shibata et al., 2008). When control membranes were crosslinked, two high-molecular weight RTN3L species were observed (Fig. S9 D). Both species formed as efficiently in CUL3 or SEC24C-depleted cells as in control cells (Fig. S9 D), indicating that CUL3KLHL12 does not facilitate RTN3L oligomerization and may have other roles in ER-phagy (see Discussion). To ask if ubiquitination of the RHD plays a role in RTN3L function, we mutated the five known ubiquitin sites (amino acids K977, K979, K995, K999 and K1003, RTN3L-5KR) that are clustered in the second hairpin of the RHD (Fig. S9 E, top and bottom) (Jumper et al., 2021; Varadi et al., 2022) by substituting lysine for arginine. However, the combination of these five mutations severely disrupted the tubular ER network and decreased cell viability (Fig. S9 E, right). This observation suggested that the RTN3L-5 KR mutations may be impairing RHD functions that are independent of ubiquitination, and for this reason, we decided not to characterize these mutations any further.
As the CUL3KLHL12 substrate, RTN3L, is needed for the FIP200-dependent accumulation of Akita at junctions, we asked if CUL3KLHL12 targets tubule components for ER-phagy at junctions. When we knocked down KLHL12 or CUL3, LNPK was no longer delivered to lysosomes (Fig. 4 A and Fig. S9 F), indicating that CUL3KLHL12 is needed for the delivery of junction components to lysosomes. Additionally, misfolded Akita failed to be delivered to lysosomes in CUL3KLHL12 -depleted cells (Fig. 4 B and Fig. S9 G) and as a consequence, large Akita puncta accumulated in the ER (Fig. 4 C). Interestingly, Akita-sfGFP also failed to colocalize with LNPK-mCherry in ligase-deficient cells (Fig. 4 D). Therefore, like RTN3L, CUL3KLHL12 is needed to retain Akita condensates/puncta at junctions. The failure of Akita to localize to junctions in the depleted cells was not the consequence of decreased levels of RTN3L, SEC24C (Fig. S10 A, left and middle) or impaired autophagosome formation, as lipidated LC3 levels were unaltered (Fig. S10 A, right). Instead, and consistent with a defect in lysosomal delivery, RTN3L levels were reproducibly higher in siCUL3 cells (Fig. S10 A, left). SEC24C levels did not increase, likely because only a small fraction of the total SEC24C is devoted to ER-phagy. While the loss of CUL3KLHL12 blocked the delivery of junction components to lysosomes, the colocalization of RTN3L puncta with LNPK puncta (Fig. S10 B, left) and SEC24C puncta (Fig. S10 C) were unaltered in the absence of ligase. LNPK still appeared to localize to junctions in CUL3-depleted cells, as the number of LNPK puncta was the same as the control (Fig. S10 B, right). This finding is consistent with the observation that ER structure is not disrupted in CUL3-depleted cells (Yuniati et al., 2020).

CUL3 KLHL12 is needed for the accumulation of Akita condensates at junctions. (A) Quantitation of LNPK-GFP puncta in LAMP1-mCherry structures in siCtrl and siRNA-treated cells incubated with Baf (6 h). Representative images on the left and in Fig. S9 F. The control for each condition was set to 1.0. (B) Quantitation of Akita-sfGFP puncta in LAMP1-mCherry structures in siCtrl and siRNA-treated cells with Baf (3.5 h). Representative images on the left and in Fig. S9 G(C) Images and the percent cells with large Akita puncta (≥0.5 µm2) (left), and the mean size of Akita puncta (right) in Ctrl and siRNAi-treated cells. Arrows mark large Akita puncta. (D) Left, images of cells transfected with Akita-sfGFP and LNPK-mCherry in siCtrl or siCUL3-treated cells. Arrowheads mark Akita puncta that colocalize with LNPK puncta. Quantitation is on the right. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 62 to 78 cells in A, 59–77 cells in B, 112–154 cells in C, and 39–44 cells in D. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.
CUL3 KLHL12 is needed for the accumulation of Akita condensates at junctions. (A) Quantitation of LNPK-GFP puncta in LAMP1-mCherry structures in siCtrl and siRNA-treated cells incubated with Baf (6 h). Representative images on the left and in Fig. S9 F. The control for each condition was set to 1.0. (B) Quantitation of Akita-sfGFP puncta in LAMP1-mCherry structures in siCtrl and siRNA-treated cells with Baf (3.5 h). Representative images on the left and in Fig. S9 G(C) Images and the percent cells with large Akita puncta (≥0.5 µm2) (left), and the mean size of Akita puncta (right) in Ctrl and siRNAi-treated cells. Arrows mark large Akita puncta. (D) Left, images of cells transfected with Akita-sfGFP and LNPK-mCherry in siCtrl or siCUL3-treated cells. Arrowheads mark Akita puncta that colocalize with LNPK puncta. Quantitation is on the right. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 62 to 78 cells in A, 59–77 cells in B, 112–154 cells in C, and 39–44 cells in D. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), Student’s unpaired t test.
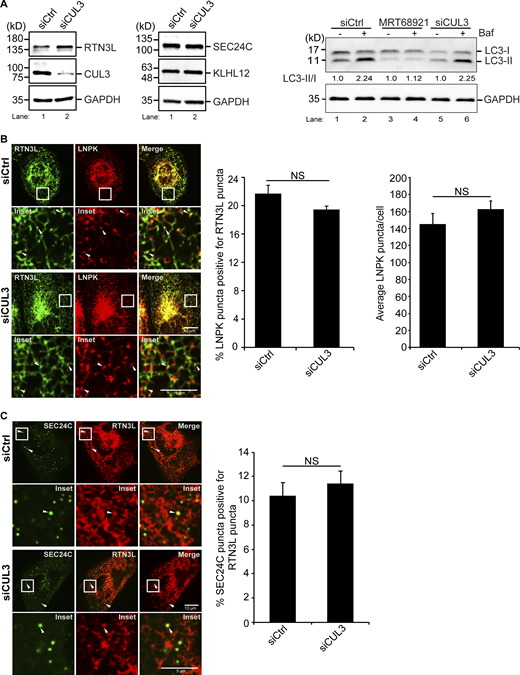
Related toFig. 4 . The localization of RTN3L puncta to three-way junctions is not dependent on CUL3KLHL12. (A) Left, the level of RTN3L, but not SEC24C or KLHL12, increases in CUL3-depleted U2OS cells. Western blot analysis was performed with lysates from Ctrl and CUL3-depleted cells. Quantitation of the blot revealed there was approximately a 1.5 times increase of RTN3L in siCUL3-treated cells compared with siCtrl cells, when the RTN3L levels were normalized to GAPDH. Right, western blot analysis was performed with lysates prepared from cells that were treated with DMSO or Baf for 3.5 h. The LC3-II/I ratio is reported in the figure. The data was normalized to GAPDH. (B) Left, cells were transfected with LNPK-GFP and mCherry-RTN3L. Arrowheads mark colocalizing puncta. Middle, quantification of percent LNPK puncta colocalizing with RTN3L puncta, and average LNPK puncta/cell (right) in siCtrl and siCUL3-treated cells. (C) Representative images of the colocalization of mCherry-RTN3L puncta with YFP-SEC24C puncta in Ctrl and CUL3-depleted cells that were treated with Torin 2 for 3.5 h. Arrowheads mark colocalizing puncta. Error bars in B and C represent SEM, n = 3 independent experiments. The results were quantified from 42 to 43 cells in B, and 38–41 cells in C. NS: not significant (P ≥ 0.05), Student’s unpaired t test. Source data are available for this figure: SourceData FS10.

Related toFig. 4 . The localization of RTN3L puncta to three-way junctions is not dependent on CUL3KLHL12. (A) Left, the level of RTN3L, but not SEC24C or KLHL12, increases in CUL3-depleted U2OS cells. Western blot analysis was performed with lysates from Ctrl and CUL3-depleted cells. Quantitation of the blot revealed there was approximately a 1.5 times increase of RTN3L in siCUL3-treated cells compared with siCtrl cells, when the RTN3L levels were normalized to GAPDH. Right, western blot analysis was performed with lysates prepared from cells that were treated with DMSO or Baf for 3.5 h. The LC3-II/I ratio is reported in the figure. The data was normalized to GAPDH. (B) Left, cells were transfected with LNPK-GFP and mCherry-RTN3L. Arrowheads mark colocalizing puncta. Middle, quantification of percent LNPK puncta colocalizing with RTN3L puncta, and average LNPK puncta/cell (right) in siCtrl and siCUL3-treated cells. (C) Representative images of the colocalization of mCherry-RTN3L puncta with YFP-SEC24C puncta in Ctrl and CUL3-depleted cells that were treated with Torin 2 for 3.5 h. Arrowheads mark colocalizing puncta. Error bars in B and C represent SEM, n = 3 independent experiments. The results were quantified from 42 to 43 cells in B, and 38–41 cells in C. NS: not significant (P ≥ 0.05), Student’s unpaired t test. Source data are available for this figure: SourceData FS10.
The loss of PINK1 decreases peripheral ER junctions and blocks ER-phagy
The PINK1 kinase was recently shown to be required for ER-phagy during drosophila intestinal development (Wang et al., 2023). PINK1 is a mitochondrial kinase that regulates mitochondrial morphology and stimulates mitophagy by phosphorylating Parkin and ubiquitin (Pickrell and Youle, 2015). It was proposed that PINK1 determines whether ER or mitochondria are degraded by balancing two E3 ligases that associate with mitochondria, KEAP1 and Parkin (Wang et al., 2023). KEAP1 promotes ER-phagy (Wang et al., 2023), while Parkin regulates mitophagy and directs PINK1-dependent phosphorylation of ubiquitin to influence which organelle is degraded (Wang et al., 2023). To begin to address if mammalian PINK1 is needed for RTN3L–SEC24C-mediated ER-phagy, we examined the delivery of ER to lysosomes during basal autophagy and found a block in the lysosomal delivery of RTN3L and LNPK in siPINK1-treated cells (Fig. 5, A and B; and Fig. S11, A–C). Supporting these experiments, we found that the constitutively expressed ER-phagy reporter, ssRFP-GFP-KDEL, accumulated fewer red dots in siPINK1-depleted cells (Fig. 5 C). GFP is quenched in the lysosome, and as a consequence, ssRFP-GFP-KDEL only forms red dots in this compartment (Chino et al., 2019). Consistent with these findings, we observed PINK1-containing puncta in close proximity to ER junctions (Fig. S11 D). Additionally, some PINK1 puncta were associated with ER tubules as they could be seen moving with the ER network (see Fig. 5 D; Fig. S11 E; and Video 1). We also found that PINK1 is needed for ER proteostasis as large Akita puncta/condensates accumulated in the ER of PINK1-depleted cells (Fig. 5 E). The accumulation of Akita condensates was rescued with RNAi-resistant mCherry-PINK1, but not RNAi-resistant kinase dead PINK1 (Fig. 5 F and Fig. S11 E), indicating that the kinase activity of PINK1 was needed for rescue. When large Akita puncta accumulated in PINK1-deficient cells, Akita failed to be delivered to lysosomes (see below, Fig. 7 B). The block in ER-phagy in PINK1-deficient cells was not mediated by Parkin or KEAP1, as Akita condensates did not accumulate in the ER when cells were depleted of either ligase (Fig. 5, E and G; and Fig. S11 F).

PINK1 is needed to target tubule components for ER-phagy at three-way junctions. (A and B) YFP-RTN3L (A) and LNPK-GFP (B) were quantitated in LAMP1-mCherry marked structures. YFP-RTN3L samples were treated with Baf for 4 h, while LNPK-GFP samples were treated for 6 h. (C) PINK1 and FIP200 are needed for the lysosomal delivery of the ER-phagy reporter ssRFP-GFP-KDEL. The number of red dots per cell are reported for the indicated samples. (D) Stills (3.6, 8.1 and 9.6 s) from Video 1 of cells transfected with mCherry-KDEL and YFP-PINK1. (E–G) Quantitation and representative images used to calculate the percent cells with large Akita puncta (≥0.5 µm2) in Ctrl and siRNA-treated cells. In E, the siPINK1-treated cells were transfected with mCherry-RNAi-resistant PINK1 or mCherry-RNAi-resistant PINK1 kinase dead (KD). Arrows mark large Akita puncta. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 58 to 69 cells in A, 47–59 cells in B, 69–85 cells in C, 99–111 cells in E, 79–105 cells in F, and 79–101 cells in G. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test.
PINK1 is needed to target tubule components for ER-phagy at three-way junctions. (A and B) YFP-RTN3L (A) and LNPK-GFP (B) were quantitated in LAMP1-mCherry marked structures. YFP-RTN3L samples were treated with Baf for 4 h, while LNPK-GFP samples were treated for 6 h. (C) PINK1 and FIP200 are needed for the lysosomal delivery of the ER-phagy reporter ssRFP-GFP-KDEL. The number of red dots per cell are reported for the indicated samples. (D) Stills (3.6, 8.1 and 9.6 s) from Video 1 of cells transfected with mCherry-KDEL and YFP-PINK1. (E–G) Quantitation and representative images used to calculate the percent cells with large Akita puncta (≥0.5 µm2) in Ctrl and siRNA-treated cells. In E, the siPINK1-treated cells were transfected with mCherry-RNAi-resistant PINK1 or mCherry-RNAi-resistant PINK1 kinase dead (KD). Arrows mark large Akita puncta. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 58 to 69 cells in A, 47–59 cells in B, 69–85 cells in C, 99–111 cells in E, 79–105 cells in F, and 79–101 cells in G. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test.
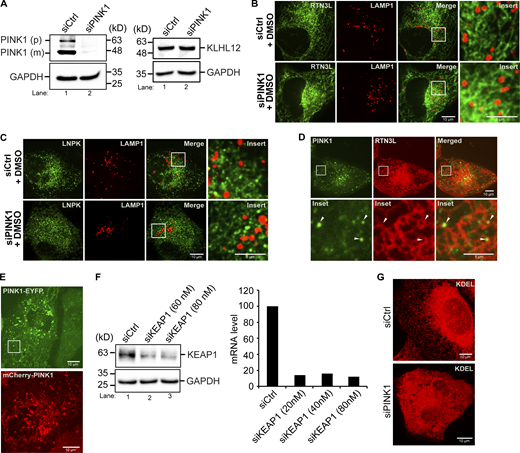
Related toFigs. 5 and 6,. PINK1-containing puncta are in close proximity to ER junctions. (A) Left, western blot analysis was performed using lysates from control and PINK1-depleted cells. The precursor (p) and mature (m) forms of PINK1 are marked in siCtrl samples. Right, KLHL12 was blotted in Ctrl and siPINK1-treated cells. GAPDH was used as a loading control. (B) Representative control images of YFP-RTN3L puncta in LAMP1-mCherry structures in siCtrl and siPINK1-treated cells for the graph shown in Fig. 5 A. (C) Representative control images of LNPK-GFP puncta in LAMP1-mCherry structures in siPINK1-treated cells for the graph shown in Fig. 5 B. (D) A representative image of a cell transfected with PINK1-YFP and mCherry-RTN3L. Arrowheads mark PINK1-YFP puncta near junctions. (E) Representative PINK1-YFP (used in Fig. 5 D) and mCherry-PINK1 (used in Fig. 5 F) images. The inset in the PINK1-YFP image was used to make Video 1 and the stills in Fig. 5 D. This area was chosen as it is largely devoid of mitochondria. (F) Left, western blot showing KEAP1 levels in control and siRNA-treated cells. Right, qPCR was performed with cells treated with siKEAP1. (G) Compressed Z stacks of Ctrl and siPINK1-treated cells that were transfected with mCherry-KDEL. A total of 14 slices were compressed for Ctrl cells, and 22 slices for siPINK1-treated cells. Source data are available for this figure: SourceData FS11.

Related toFigs. 5 and 6,. PINK1-containing puncta are in close proximity to ER junctions. (A) Left, western blot analysis was performed using lysates from control and PINK1-depleted cells. The precursor (p) and mature (m) forms of PINK1 are marked in siCtrl samples. Right, KLHL12 was blotted in Ctrl and siPINK1-treated cells. GAPDH was used as a loading control. (B) Representative control images of YFP-RTN3L puncta in LAMP1-mCherry structures in siCtrl and siPINK1-treated cells for the graph shown in Fig. 5 A. (C) Representative control images of LNPK-GFP puncta in LAMP1-mCherry structures in siPINK1-treated cells for the graph shown in Fig. 5 B. (D) A representative image of a cell transfected with PINK1-YFP and mCherry-RTN3L. Arrowheads mark PINK1-YFP puncta near junctions. (E) Representative PINK1-YFP (used in Fig. 5 D) and mCherry-PINK1 (used in Fig. 5 F) images. The inset in the PINK1-YFP image was used to make Video 1 and the stills in Fig. 5 D. This area was chosen as it is largely devoid of mitochondria. (F) Left, western blot showing KEAP1 levels in control and siRNA-treated cells. Right, qPCR was performed with cells treated with siKEAP1. (G) Compressed Z stacks of Ctrl and siPINK1-treated cells that were transfected with mCherry-KDEL. A total of 14 slices were compressed for Ctrl cells, and 22 slices for siPINK1-treated cells. Source data are available for this figure: SourceData FS11.
Related toFig. 5 . PINK1 associates with ER tubules. U2OS cells were transfected with PINK1-YFP and mCherry-KDEL. Puncta containing PINK1-YFP can be seen associating with ER tubules marked with mCherry-KDEL. The time period of recording was 31.5 s. Images were analyzed using Nikon Elements software. Playback speed is 30 frames per second.
Related toFig. 5 . PINK1 associates with ER tubules. U2OS cells were transfected with PINK1-YFP and mCherry-KDEL. Puncta containing PINK1-YFP can be seen associating with ER tubules marked with mCherry-KDEL. The time period of recording was 31.5 s. Images were analyzed using Nikon Elements software. Playback speed is 30 frames per second.
Unexpectedly, we found that the loss of PINK1 altered ER shape. Approximately, 72% of the PINK1-deficient cells had abnormal ER: ∼32% of the cells mostly contained expanded ER sheets, while 40% of the cells appeared to have fewer tubules and junctions at the cell periphery (Fig. 6 A). A 3D reconstruction of the ER network in siPINK1-treated cells illustrates this network defect (see Fig. 6 B; and Videos 2 and 3). The ER morphology defect in PINK1-depleted cells was also confirmed with two ER markers that were commonly used to analyze ER structure, GFP-SEC61 (Fig. 6 C) and mCherry-KDEL (Fig. S11 G). Although the morphology of mitochondria was impaired in siParkin-treated cells (Fig. S12 A), ER organization was similar to control cells (Fig. 5 E), indicating that PINK1 does not indirectly affect ER structure via Parkin. The observation that PINK1-containing puncta associate with the ER, in combination with the structural defects found in PINK1-deficient cells, suggests that mammalian PINK1 directly regulates ER-phagy from the ER and not the mitochondria via Parkin and KEAP1.

Fewer ER junctions are present at the cell periphery in PINK1-depleted cells. (A) siCtrl and siPINK1-treated cells transfected with Akita-sfGFP were quantitated as described in the methods. (B) Frames from representative 3D reconstructions of Z stacks of siCtrl (22 s in Video 2) and siPINK1-treated (23 s in Video 3) cells. (C) Compressed Z stacks of siCtrl and siPINK1-treated cells transfected with GFP-SEC61. A total of 27 slices were compressed for the siCtrl, and 19 slices for siPINK1-treated cells. (D) Left, representative images of LNPK-GFP in siCtrl and siPINK1-treated cells. Middle, the average number of LNPK puncta/cell. The data was normalized as described in the methods. Right, blots showing LNPK levels. GAPDH was used as a loading control. (E) Left, representative images used in the quantitation shown on the right. (F) Left, representative images of cells containing Akita-sfGFP with insets used in the quantitation (right) of junctions. (G) Same as E only, cells were transfected with mCherry-Sec61. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 93 to 116 cells in A, 78–82 cells in D, 84–85 cells in E, 27–32 cells in F, and 51–59 cells in G. * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData F6.
Fewer ER junctions are present at the cell periphery in PINK1-depleted cells. (A) siCtrl and siPINK1-treated cells transfected with Akita-sfGFP were quantitated as described in the methods. (B) Frames from representative 3D reconstructions of Z stacks of siCtrl (22 s in Video 2) and siPINK1-treated (23 s in Video 3) cells. (C) Compressed Z stacks of siCtrl and siPINK1-treated cells transfected with GFP-SEC61. A total of 27 slices were compressed for the siCtrl, and 19 slices for siPINK1-treated cells. (D) Left, representative images of LNPK-GFP in siCtrl and siPINK1-treated cells. Middle, the average number of LNPK puncta/cell. The data was normalized as described in the methods. Right, blots showing LNPK levels. GAPDH was used as a loading control. (E) Left, representative images used in the quantitation shown on the right. (F) Left, representative images of cells containing Akita-sfGFP with insets used in the quantitation (right) of junctions. (G) Same as E only, cells were transfected with mCherry-Sec61. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 93 to 116 cells in A, 78–82 cells in D, 84–85 cells in E, 27–32 cells in F, and 51–59 cells in G. * (P < 0.05), **(P < 0.01), Student’s unpaired t test. Source data are available for this figure: SourceData F6.
Related toFig. 6 . The tubular ER in control cells. A 3D reconstruction of the ER in control U2OS cells transfected with Akita-sfGFP. The reconstruction contains 28 slices taken with 0.20 µm steps. The image was prepared using Richardson-Lucy algorithm with automatic parameter setting in NIS Elements. Playback speed is 30 frames per second.
Related toFig. 6 . The tubular ER in control cells. A 3D reconstruction of the ER in control U2OS cells transfected with Akita-sfGFP. The reconstruction contains 28 slices taken with 0.20 µm steps. The image was prepared using Richardson-Lucy algorithm with automatic parameter setting in NIS Elements. Playback speed is 30 frames per second.
Related toFig. 6 . ER sheets proliferate at the cell periphery in siPINK1-treated cells. A 3D reconstruction of the ER in siPINK1-treated U2OS cells transfected with Akita-sfGFP. The reconstruction contains 32 slices taken with 0.20 µm steps. ER sheet expansion is visible at the cell periphery. The image was prepared using Richardson-Lucy algorithm with automatic parameter setting in NIS Elements. Playback speed is 30 frames per second.
Related toFig. 6 . ER sheets proliferate at the cell periphery in siPINK1-treated cells. A 3D reconstruction of the ER in siPINK1-treated U2OS cells transfected with Akita-sfGFP. The reconstruction contains 32 slices taken with 0.20 µm steps. ER sheet expansion is visible at the cell periphery. The image was prepared using Richardson-Lucy algorithm with automatic parameter setting in NIS Elements. Playback speed is 30 frames per second.
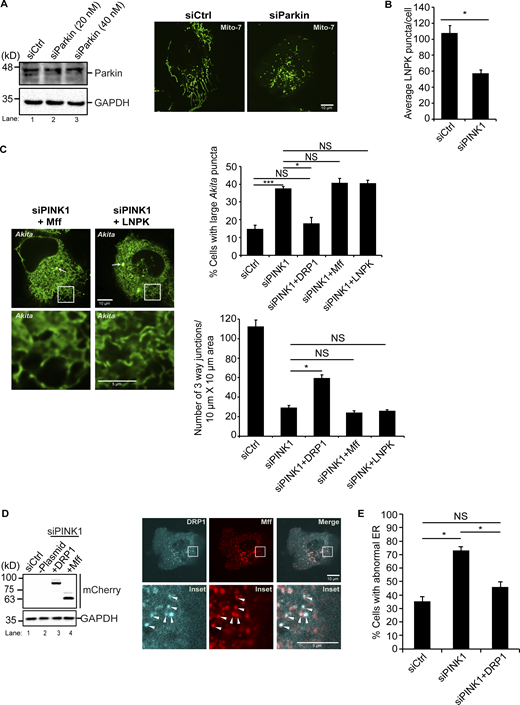
Related toFigs. 5, 6, and 7,. mCherry-DRP1 suppresses the ER morphology defect in siPINK1-treated cells. (A) Left, western blot analysis was performed using cell lysates depleted of Parkin. Samples were blotted for Parkin and GAPDH. Right, representative images of GFP-Mito7 in Ctrl and siParkin-treated cells. (B) Same as Fig. 6 D middle, only the data was not normalized. (C) siCtrl and siPINK1-treated cells containing Akita-sfGFP were transfected with mCherry-Mff, or LNPK-mCherry. Left, representative images for the data graphed on the right. The remaining images are shown in Fig. 7 A. (D) Left, siCtrl and siPINK1-treated cells were transfected with mCherry-DRP1 (lane 3) and mCherry-Mff (lane 4) and blotted for the expression of the mCherry fusion proteins. Lane 2 does not contain plasmid. Right, Cells were transfected with mTurquoise2-DRP1 and mCherry-Mff. Arrowheads mark colocalizing puncta on mitochondrial membranes. (E) The percentage of cells with abnormal ER was quantitated as described in the methods. Error bars in B, C, and E represent SEM, n = 3 independent experiments. The results were quantified from 78 to 82 cells in B, 100–195 cells in C, top, 33–38 cells in C, bottom, and 78–154 cells in E. NS: not significant (P ≥ 0.05), * (P < 0.05), *** (P < 0.001), Student’s unpaired t test. Source data are available for this figure: SourceData FS12.

Related toFigs. 5, 6, and 7,. mCherry-DRP1 suppresses the ER morphology defect in siPINK1-treated cells. (A) Left, western blot analysis was performed using cell lysates depleted of Parkin. Samples were blotted for Parkin and GAPDH. Right, representative images of GFP-Mito7 in Ctrl and siParkin-treated cells. (B) Same as Fig. 6 D middle, only the data was not normalized. (C) siCtrl and siPINK1-treated cells containing Akita-sfGFP were transfected with mCherry-Mff, or LNPK-mCherry. Left, representative images for the data graphed on the right. The remaining images are shown in Fig. 7 A. (D) Left, siCtrl and siPINK1-treated cells were transfected with mCherry-DRP1 (lane 3) and mCherry-Mff (lane 4) and blotted for the expression of the mCherry fusion proteins. Lane 2 does not contain plasmid. Right, Cells were transfected with mTurquoise2-DRP1 and mCherry-Mff. Arrowheads mark colocalizing puncta on mitochondrial membranes. (E) The percentage of cells with abnormal ER was quantitated as described in the methods. Error bars in B, C, and E represent SEM, n = 3 independent experiments. The results were quantified from 78 to 82 cells in B, 100–195 cells in C, top, 33–38 cells in C, bottom, and 78–154 cells in E. NS: not significant (P ≥ 0.05), * (P < 0.05), *** (P < 0.001), Student’s unpaired t test. Source data are available for this figure: SourceData FS12.
A role for PINK1 in regulating ER structure has not been previously reported. To begin to assess how PINK1 shapes the ER, we analyzed LNPK-marked ER junctions. LNPK marks most but not all ER junctions (Chen et al., 2015). When junctions are diminished, fewer LNPK puncta are present throughout the ER network. As junctions decrease, the tubular network is reduced and ER sheets are expanded (Chen et al., 2015; Wang et al., 2016). To ask if the loss of PINK1 leads to a decrease in LNPK-marked junctions, control and siPINK1-treated cells were transfected with LNPK-GFP and puncta numbers were quantified (Fig. 6 D). Interestingly, a significant decrease in LNPK puncta was observed in the absence of PINK1 (Fig. 6 D). Similar results were obtained whether or not the data was normalized to account for differences in transfection efficiency (see Materials and methods and compare Fig. 6 D, middle to Fig. S12 B), indicating that transfection was comparable in both samples. The decrease in puncta was not due to a reduction in the level of LNPK as the endogenous expression of LNPK was unchanged in the depleted cells (Fig. 6 D, right). As LNPK sequesters KLHL12 at junctions (Akopian et al., 2022; Yuniati et al., 2020), we also analyzed GFP-KLHL12 puncta in siPINK1-treated cells. GFP-KLHL12 puncta significantly decreased in PINK1-depleted cells, while the cytosolic pool appeared to increase, and the endogenous KLHL12 level remained unchanged (Fig. 6 E and Fig. S11 A). Together these findings imply that fewer LNPK-marked junctions are present in the ER network of PINK1-depleted cells.
As the reduction in ER tubules and junctions in siPINK1-treated cells was most obvious at the cell periphery, we quantitatively assessed ER junctions by scoring the number of three-way junctions in a 10 × 10 µm boxed area at the cell periphery (see Materials and methods for details). First, we measured the number of junctions in PINK1-depleted cells expressing Akita-sfGFP and compared the data with control cells. Consistent with the observed defect in peripheral ER tubules, we found a dramatic decrease in peripheral ER junctions in the absence of PINK1 (Fig. 6 F). Similar results were obtained when junctions were measured in control and depleted cells that were transfected with mCherry-SEC61 (Fig. 6 G). These findings suggest that the defect in RTN3L–SEC24C–mediated ER-phagy in PINK1-depleted cells is due to a dramatic loss of peripheral tubular ER junctions.
DRP1 induces ER tubulation in PINK1-deficient cells and suppresses the ER-phagy defect
If the defect in ER-phagy in PINK1-deficient cells is the consequence of disrupting peripheral ER junctions, ER-phagy should be restored as junctions are replenished. The PINK1 kinase substrate, DRP1, is a dynamin-related GTPase that is recruited from the cytosol to the ER where it promotes peripheral ER tubulation to facilitate ER–mitochondria interactions and mitochondrial fission (Adachi et al., 2020). Interestingly, the tubulating activity of DRP1 is independent of GTP hydrolysis and is contained within an 18-amino acid peptide (called D-octadecapeptide) (Adachi et al., 2020). As the overexpression of DRP1 has been shown to suppress the mitochondrial morphology defect in PINK1 mutant cells (Poole et al., 2008), we asked if DRP1 overexpression suppresses the ER structural defect in PINK1 knockdown cells. Membrane-bound DRP1 forms two distinct pools of puncta on the ER (Adachi et al., 2020). One pool associates with Mff to mediate DRP1-dependent mitochondrial fission, while the second pool acts independently of Mff to promote ER tubulation (Adachi et al., 2020; Ji et al., 2017). In addition, DRP1 associates with Mff on mitochondria (Ji et al., 2017). Previous studies have shown that the role of DRP1 in ER tubulation is independent of its role in mitochondrial fission (Adachi et al., 2020).
To ask if DRP1 overexpression suppresses the ER-phagy and ER structural defect in PINK1-depleted cells, we quantified the accumulation of Akita condensates/puncta and peripheral ER junctions in siPINK1-treated cells transfected with mCherry-DRP1 and mCherry-DRP1 D-octadecapeptide (554–571 aa). As controls, we also overexpressed two other fusion proteins that are known to be active, mCherry-Mff and LNPK-mCherry (Bertolin et al., 2018; Wang et al., 2016). Mff promotes mitochondrial fission but not ER tubulation (Adachi et al., 2020), while LNPK regulates the abundance of ER junctions (Chen et al., 2015; Wang et al., 2016). LNPK-mCherry did not suppress the ER-phagy defect in PINK1-depleted cells, even though it was used at a concentration that is sufficient to suppress the LNPK KO mutant defect (Fig. S12 C). While DRP1 and its binding partner Mff were expressed at comparable levels and mCherry-Mff recruited DRP1 to membranes (Fig. S12 D), only DRP1 and the DRP1 D-octadecapeptide suppressed the accumulation of Akita condensates in siPINK1-treated cells (Fig. 7 A). DRP1 suppressed the accumulation of large Akita puncta (Fig. 7 A, left), as well as the mean size of Akita puncta per cell in PINK-depleted cells (Fig. 7 A, middle). In addition to the suppression of the Akita accumulation phenotype, the ER morphology defect was suppressed (Fig. 7 A and Fig. S12 E), the number of ER junctions significantly increased (Fig. 7 A, right), and Akita puncta/condensates were efficiently delivered to lysosomes (Fig. 7 B). Interestingly, a partial restoration of junctions appeared to be sufficient to largely restore ER-phagy in siPINK1-treated cells (Fig. 7 B and Fig. S12 C). This observation suggests that cells normally contain an excess of junctions for ER-phagy; however, a minimal number appears to be needed for the RTN3L–SEC24C pathway to effectively clear ERAD-resistant proteins from the ER.

The overexpression of DRP1 increases peripheral junctions in PINK1-depleted cells and suppresses the ER-phagy defect. (A) siCtrl and siPINK1-treated cells containing Akita-sfGFP were transfected with mCherry-DRP1 or mCherry-DRP1 D-octadecapeptide (554–571 aa). Top, representative images for the data graphed below. (B) Quantification of Akita-sfGFP puncta in LAMP1-mCherry structures in siCtrl and siPINK1-treated cells that were untreated or incubated with Baf (3.5 h) in the absence and presence of mCherry-DRP1. The control for each condition was set to 1.0. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 100 to 195 cells in A, left, 67–149 cells in A, middle, 33–47 cells in A, right, 60–97 cells in B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test.
The overexpression of DRP1 increases peripheral junctions in PINK1-depleted cells and suppresses the ER-phagy defect. (A) siCtrl and siPINK1-treated cells containing Akita-sfGFP were transfected with mCherry-DRP1 or mCherry-DRP1 D-octadecapeptide (554–571 aa). Top, representative images for the data graphed below. (B) Quantification of Akita-sfGFP puncta in LAMP1-mCherry structures in siCtrl and siPINK1-treated cells that were untreated or incubated with Baf (3.5 h) in the absence and presence of mCherry-DRP1. The control for each condition was set to 1.0. Error bars represent SEM, n = 3 independent experiments. The results were quantified from 100 to 195 cells in A, left, 67–149 cells in A, middle, 33–47 cells in A, right, 60–97 cells in B. NS: not significant (P ≥ 0.05), * (P < 0.05), **(P < 0.01), *** (P < 0.001), Student’s unpaired t test.
While knocking down DRP1 decreased peripheral junctions, the number of junctions in the knockdown cells was comparable with siPINK1-treated cells overexpressing mCherry-DRP1 (Fig. S13 A). In either case, large Akita puncta/condensates did not accumulate in the ER (Fig. 7 A and Fig. S13 B), implying that sufficient junctions were present for ER-phagy. As DRP1 is dispensable for ER-phagy, but contributes to tubulation, other tubulating factors appear to be needed for ER autophagy.
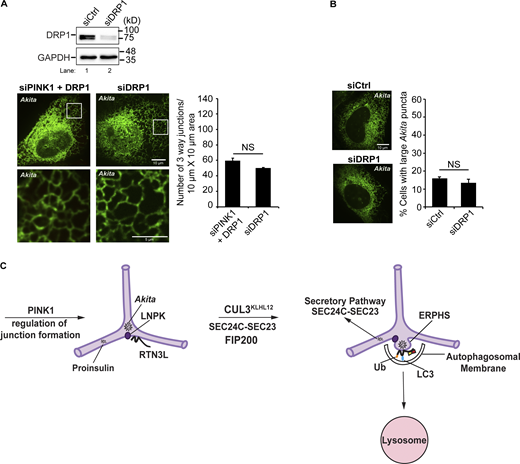
Related toFig. 7 . RTN3L–SEC24C, CUL3KLHL12 and PINK1 target ERAD resistant misfolded proteins for ER-phagy at LNPK-marked junctions. (A) Top, siCtrl and siDRP1-treated cells were blotted for the presence of DRP1. GAPDH was used as a loading control. Bottom, siCtrl and siDRP1-treated cells were transfected with Akita-sfGFP. Left, representative images with 10 × 10 µm insets used in the quantitation (right) of ER junctions at the cell periphery (see Materials and methods). (B) Percentage of cell with large Akita puncta/condensates (≥0.5 µm2) were quantified. Error bars in A and B represent SEM, n = 3 independent experiments. The results were quantified from 34 to 38 cells in A, and 73–103 cells in B. NS: not significant (P ≥ 0.05), Student’s unpaired t test. (C) An Akita puncta associates with RTN3L (left) and enlarges in the sheet-like domain of an LNPK-marked junction where the E3 ligase that ubiquitinates RTN3L, CUL3KLHL12, is recruited (right). SEC24C–SEC23 and FIP200 are needed to retain Akita condensates in LC3-containing autophagic structures called ERPHS (right) that are delivered to lysosomes (right) during ER-phagy. The PINK1 kinase, DRP1, and other ER tubulating proteins regulate the formation of peripheral junctions where ERPHS are formed. Key: Ub, ubiquitinated RTN3L. Source data are available for this figure: SourceData FS13.

Related toFig. 7 . RTN3L–SEC24C, CUL3KLHL12 and PINK1 target ERAD resistant misfolded proteins for ER-phagy at LNPK-marked junctions. (A) Top, siCtrl and siDRP1-treated cells were blotted for the presence of DRP1. GAPDH was used as a loading control. Bottom, siCtrl and siDRP1-treated cells were transfected with Akita-sfGFP. Left, representative images with 10 × 10 µm insets used in the quantitation (right) of ER junctions at the cell periphery (see Materials and methods). (B) Percentage of cell with large Akita puncta/condensates (≥0.5 µm2) were quantified. Error bars in A and B represent SEM, n = 3 independent experiments. The results were quantified from 34 to 38 cells in A, and 73–103 cells in B. NS: not significant (P ≥ 0.05), Student’s unpaired t test. (C) An Akita puncta associates with RTN3L (left) and enlarges in the sheet-like domain of an LNPK-marked junction where the E3 ligase that ubiquitinates RTN3L, CUL3KLHL12, is recruited (right). SEC24C–SEC23 and FIP200 are needed to retain Akita condensates in LC3-containing autophagic structures called ERPHS (right) that are delivered to lysosomes (right) during ER-phagy. The PINK1 kinase, DRP1, and other ER tubulating proteins regulate the formation of peripheral junctions where ERPHS are formed. Key: Ub, ubiquitinated RTN3L. Source data are available for this figure: SourceData FS13.
Discussion
Here, we show that the RTN3L–SEC24C pathway regulates ER proteostasis by locally promoting ER autophagy at specific ER subdomains, ER junctions. The RTN3L–SEC24C receptor complex works with the E3 ligase, CUL3KLHL12, and the PINK1 kinase to target ERAD-resistant misfolded Akita condensates for ER-phagy at ERPHS, autophagic sites at junctions that are distinct from ERES. Previous studies have shown that the COPII coat subunit, SEC24C, is needed for the lysosomal delivery of the ER-phagy receptors RTN3L and FAM134B (Cui et al., 2019). However, our findings and recent studies reported by others, indicate that these two receptors work with SEC24C in very different ways (Roberts et al., 2024; Sun et al., 2023). Specifically, while RTN3L binds to SEC24C, FAM134B and its splice variant FAM134B-II do not (Roberts et al., 2024; Sun et al., 2023). Instead, FAM134B and FAM134B-II associate with SEC24C-containing COPII vesicles at ERES to deliver misfolded α1-antitrypsin to lysosomes (Roberts et al., 2024; Sun et al., 2023). In the case of FAM134B-II, the association with COPII vesicles was shown to be mediated by LC3C (Sun et al., 2023). Although the RTN3L–SEC24C pathway uses a COPII coat subunit (SEC24C), it does not use ERES or transport vesicles. These observations separate this ER-phagy pathway from FAM134B-mediated ER-phagy and also distinguishes it from a recently described micro-ER-phagy pathway that delivers ERES to lysosomes during nutrient stress (Liao et al., 2024). Furthermore, while RTN3L–SEC24C-mediated ER-phagy is autophagosome-mediated, FAM134B-II-mediated ER-phagy is not (Sun et al., 2023).
We found that Akita condensates accumulate at LNPK-marked junctions where RTN3L accumulates (Fig. S13 C, left). The E3 ligase, CUL3KLHL12, is recruited to these junctions and ubiquitinates RTN3L (Fig. S13 C, left and middle). RTN3L, SEC24C–SEC23, and FIP200 are required to retain misfolded Akita in ERPHS, autophagic sites that form at junctions (Fig. S13 C, middle and right). These autophagic sites were previously shown to contain LC3 (Parashar et al., 2021). Additional studies will be needed to determine if ERPHS also contain other components of the autophagy machinery. After ERPHS are formed, they are delivered to lysosomes where their contents are degraded (Fig. S13 C, right). The ubiquitination of other junction components may also contribute to autophagic clearance. Consistent with this proposal, LNPK was recently reported to be ubiquitinated at junctions by CUL3KLHL12 (Yuniati et al., 2020).
ER junctions have a unique architecture that may make these specialized ER subdomains ideally suited for ER-phagy. The central area of the junction consists of a small flat sheet, and lysosomes have been reported to make contact with junctions (Obara et al., 2023; Shemesh et al., 2014; Yuniati et al., 2020). Junctions also contain regions of negative curvature that balance the positive curvature of tubules to relieve membrane tension. This may make the membrane surface of junctions more amenable to fission (Obara et al., 2023). Additionally, recent studies have shown that condensates, which are targeted for autophagy, must expand to a certain size before they can undergo autophagy (Feng et al., 2022). The small triangular sheets that form at junctions would allow for this expansion and for the accumulation of misfolded proteins. In total, our findings demonstrate the importance of junctions in the formation of ERPHS. Furthermore, they explain why the loss of LNPK, which dramatically decreases the number of three-way tubule junctions (Chen et al., 2015; Wang et al., 2016), blocks the RTN3L–SEC24C pathway in mammals (Parashar et al., 2021) and the Atg40-Lst1 ER-phagy pathway in yeast (Chen et al., 2018). In total, our studies have revealed that ER-phagy does not take place randomly in tubules. It occurs at a specific subdomain where the CUL3KLHL12 ligase is recruited. A possible role for CUL3KLHL12 in ER-phagy may be to locally enhance autophagic clearance by ubiquitinating components, such as RTN3L and LNPK, at the target site. This suggestion is supported by the observation that RTN3L ubiquitination is only observed in the presence of bafilomycin. There is also precedence for this proposal, as the ubiquitination of mitochondria has been reported to enhance its degradation during mitophagy (McLelland et al., 2018; Tanaka et al., 2010; Wang et al., 2011).
Unexpectedly, our studies have revealed a novel role for the Parkinson’s disease kinase, PINK1, in controlling ER shape. When cells are depleted of PINK1, ER junctions decrease at the cell cortex (Fig. S13 C, left), and as a consequence, ER-phagy is blocked. The overexpression of DRP1, a PINK1 target that tubulates the ER to promote ER–mitochondria interactions and mitochondria fission (Adachi et al., 2020), increases peripheral ER junctions and restores ER-phagy. As the loss of DRP1 does not reduce peripheral junctions sufficiently to block ER-phagy, additional PINK1 targets that promote ER tubulation appear to be needed for autophagy. Interestingly, the region in DRP1 that is needed for ER tubulation (D-octadecapeptide) is not conserved in yeast, flies, and worms (Adachi et al., 2020). This could explain why the role we describe here for mammalian PINK1 in ER-phagy is different from its role during drosophila development (Wang et al., 2023). Our findings imply that PINK1 regulates RTN3L–SEC24C–mediated ER-phagy by promoting the formation of tubule junctions at the cell periphery. Future work will be needed to determine the mechanism by which PINK1 and DRP1 tubulate the ER.
It is well documented that loss of PINK1 leads to mitochondrial dysfunction (Pickrell and Youle, 2015). The link between abnormalities in mitochondrial function and the pathogenesis of Parkinson’s disease has prompted a flurry of studies on the role of PINK1 in regulating mitochondrial homeostasis. The studies we report here indicate that PINK1 also regulates ER shape. Consistent with a direct role for PINK1 in regulating ER structure, we observed PINK1-containing puncta associating with ER tubules. PINK1 has also been observed at ER–mitochondria contact sites and on mitochondria (Gómez-Suaga et al., 2018). Interestingly, several recent studies have shown that the loss of peripheral ER tubules disrupts mitochondrial fission and alters mitochondria function (Carter et al., 2022; Jang et al., 2022). These studies suggest that mitochondrial health is connected to ER structure and function. In total, our findings raise the intriguing possibility that ER dysfunction may play an important role in Parkinson’s disease.
Materials and methods
Plasmid constructions
To construct the RTN3L RNAi-resistant clone, pC1–mCherry–RTN3L (SFNB 2558), human RTN3L was PCR amplified from SFNB 2455 (pHAGE-mCherry-RTN3L) using primer pair #1 5′-TTTTCGGTAATTTCGGTAGTAAGTTATCTCATCCTGGCTCTTCTCTC-3′, 5′-GTGGTATGGCTGATTATGATCAGTTATTCTGCCTTTTTTTTGGCGATTCCAG-3′ and primer pair #2 5′-GGTGGTAGTGGTGGATCCGGAGGTTCTGCGGAGCCGTCGG-3′ 5′-ATAACTTACTACCGAAATTACCGAAAAAGCTGCCAGGGAAAGCAGC-3′. The silent mutations incorporated into the PCR fragments were cloned into a pC1 mammalian expression vector using the Gibson assembly method (Gibson et al., 2009).
To construct pC1–mCherry–RTN3LΔ3LIR or pC1–mCherry–RTN3LΔ5LIR, human RTN3LΔ3LIR or RTN3LΔ5LIR PCR fragments were amplified from pHAGE–EGFP–RTN3LΔ6LIR (SFNB 2529) and pC1–mCherry–RTN3L (SFNB 2481). The PCR fragments were cloned into a pC1 mammalian expression vector using the Gibson assembly method (Gibson et al., 2009). To construct pC1–GFP–KLHL12, the KLHL12 coding sequence was PCR-amplified from SFNB2597 and, using the Gibson assembly method, cloned into a pC1 mammalian expression vector together with a PCR fragment that contained GFP. To construct pC1–mCherry–RTN3L-5KR, the lysine to arginine mutations (mCherry–RTN3L K977R K979R, K995R K999R K1003R) were constructed by PCR. The point mutations were incorporated into the PCR fragments and then cloned into a pC1 mammalian expression vector using the Gibson assembly method.
GST–RTN3L (aa 48–624) (SFNB2546) and GST–RTN3S (aa 1–47) (SFNB2547) were generated by PCR amplification using pHAGE–mCherry-RTN3L (SFNB2455) as a template and then the PCR fragments were cloned into a pGEX-4T-2 vector at the BamHI/SalI restriction site.
pC1–mCherry–PINK-RNAi–resistant (SFNB2643) was constructed using two primer pairs #1 5′-GCTGTGTATGAGGCAACAATGCCGACTCTACCCCAGAACCTGG-3′, 5′-CTGGGGTAGAGTCGGCATTGTTGCCTCATACACAGCAGCACTGC-3′ and primer pair #2 5′-GGCCATGGCCGAACCCTCTTTCTGGTGATGAAGAACTATCCCTGTAC-3′ 5′-GATAGTTCTTCATCACCAGAAAGAGGGTTCGGCCATGGCCCAGGCC-3′. SFNB 2624 (pEYFP-N1-PINK1) was used as a template. The silent mutations that were incorporated into the PCR fragments were cloned into a pC1 mammalian expression vector using the Gibson assembly method. Three sequential rounds of PCR were needed to make pC1-mCherry-PINK1 K219D/D362A/D384A (PINK1 kinase dead (KD) (SFNB2645). The lysine (K) to aspartic acid (D), and aspartic acid (D) to alanine (A) point mutations at K219D/D362A/D384A (bold below) in human PINK1 were PCR amplified using the following pairs of primers. For K219D 5′-CTTGGCCATCGACATGATGTGGAACATC-3′′and 5′-CCACATCATGTCGATGGCCAAGGGGAAGGC-3′, for D362A 5′-CGCGCACAGAGCTCTGAAATCCGACAAC-3′′and 5′-GGATTTCAGAGCTCTGTGCGCGATGCCCTG-3′, for D384A 5′-GGTGATCGCAGCTTTTGGCTGCTGCCTGG-3′ and 5′-CAGCCAAAAGCTGCGATCACCAGCCAGG-3′, pC1-mCherry-PINK-RNAi-resistant (SFNB2643) was used as a template. The point mutations were incorporated into the PCR fragments and then cloned into a pC1 mammalian expression vector using the Gibson assembly method. All plasmids used in this study are listed in the Table S1.
Transfection, siRNA knockdowns, and complementation assay
U2OS cells were transiently transfected with plasmid DNA using TransIT-LT1 transfection reagent following the manufacturer’s instructions, and imaged 46–48 h after transfection. RNAi oligonucleotides were transfected into cells using HiPerFect transfection reagent as described before (Parashar et al., 2021). The concentrations and sequence of siRNA used were as follows; siCtrl (10 nM; D-001206-13-05; Dharmacon), siRTN3 (20 nM; 5′-UCAGUGUCAUCAGUGUGGUUUCUUADTDT-3′), siRTN3L (40 nM; 5′-GACAGGUUGAGAAGUCAACDTDT, 5′-GUCCAGAUGCGUGAAUGAUAUDTDT-3′), siCALCOCO1 (40 nM; 5′-GAAGCUGAGUGCAGAGAUADTDT-3′, 5′-GUGAGAAAGUGCUGACGAADTDT-3′), siSEC24C (20 nM; M-008467-01-0007; Dharmacon), siSEC31A (100 nM; 5′-GCUUCUCUUCCACUCAAUADTDT-3′), siKLHL12 (40 nM; 5′-GGAAGGUGCCGGACUCGUADTDT-3′, 5′-GCAGGGAUCUGGUUGAUGADTDT-3′), siCUL3 (40 nM; 5′-AACAACUUUCUUCAAACGCUADTDT-3′), siPINK1 (60–80 nM; 5′-GGACGCUGUUCCUCGUUAUDTDT-3′, 5′-AAGCCACCAUGCCUACAUUDTDT-3′), siParkin (60–80 nM; 5′-GCCACGUGAUUUGCUUAGADTDT-3′, 5′-CUUGGCUACUCCCUGCCUUDTDT-3′), siFAM134B (80 nM; 5′-CAAAGATGACAGTGAATTADTDT-3′), siDRP1 (80 nm; 5′-CAGGAGCCAGCUAGAUAUUAADTDT-3′), siKEAP1 (40–80 nm; 5′-GGGCGUGGCUGUCCUCAAUDTDT-3′, 5′-CAGCAGAACUGUACCUGUUDTDT-3′), and siFIP200 (80 nm; 5′-GGAAAUGUAUGAAGUUGCCAAGAAADTDT-3′).
To perform complementation assays, Akita-sfGFP (SFNB2468) and the RNAi-resistant clone were co-transfected in siRNA knockdown cells and imaged after 46–48 h. The cells with large Akita puncta (≥0.5 μm2) were quantified as described below.
Fluorescence microscopy and image analysis
For live cell imaging experiments, cells were cultured on glass-bottom dishes (D35-20-1.5-N; Cellvis) and imaged on a Nikon Eclipse Ti2-E microscope. The Nikon microscope possesses a Yokogawa X1 spinning disk confocal system (CSU-X1-50 µm pinholes) with a SR HA APO TIRF100x NA 1.49 WD 120 µm objective, MLC400B 4-line (405, 488, 561, and 647 nm) dual-filter laser combiner (Agilent), Prime 95B back-thinned sCOMS camera (Teledyne photometrics), and piezo Z-stage (200 µm; Mad City Lab Inc.) with Perfect Focus System (PFS) and running NIS-Elements software (version 5.21.03 High content analysis package). Band-pass emission filters were employed in each channel (432/36, 515/30, 595/30, and 755/189 nm) to separate distinct flour emissions. Imaging was performed in an automated stage-top environmental chamber (Tokai Hit) equipped with an enclosure that is regulated for temperature (30–40°C), CO2, and humidity.
To induce ER-phagy with Torin, the cells were treated for 3.5 h with 250 nM Torin 2 that was solubilized in DMSO. DMSO treated cells were used as a control. To inhibit autophagy, cells were treated with 2 µM of MRT68921 that was solubilized in water. The incubation time for each experiment is indicated in the legends.
The transient and stable expression levels of fluorescently tagged SEC24C, RTN3L, and RTN4 was previously compared with the endogenous levels by immunofluorescence (Parashar et al., 2021). This analysis revealed that for fluorescently tagged transfected RTN3L, SEC24C, and RTN4, there was no more than twofold overexpression. Highly overexpressing cells were excluded in all colocalization studies and therefore also excluded from the estimates reported in this study.
To determine the size of large Akita puncta that accumulate when ER-phagy is blocked, a binary black-and-white image was generated and a mask of the image (size criteria of ≥0.5 µm2 and circularity criteria of 0.5–1) was created. The size of proinsulin puncta (see Fig. S1 F) was determined the same way, but a different size criteria (≥0.35 µm2 and circularity criteria of 0.5–1) was created. Cells highly overexpressing Akita or proinsulin were not used in the calculation.
To calculate the mean size of Akita puncta per cell, puncta of all sizes were identified using the Yen thresholding algorithm. Then, the Analyze Particle command in Image J was used to calculate the number and area of Akita puncta. An average puncta size was calculated for each cell using the following equation: total area of all puncta/total number of puncta per cell.
A customized MATLAB script (see below), that was described before (Wu and Voeltz, 2021), was used to calculate the number of ER junctions in a 10 × 10 µm area at the cell periphery. A large tubular peripheral area distal from the nucleus was measured for each cell. For PINK1-depleted cells, the number of ER junctions was calculated manually. The calculation for Akita-sfGFP cells was done by two independent observers and the numbers obtained were divided by the total number of cells analyzed.
MATLAB script
I = imread([PathName FileName]);
T = I;
level = graythresh(T); Threshold = imbinarize(T,level);
BW_skel = bwmorph(Threshold,“skel,”Inf);
BW_3WJ = bwmorph(BW_skel,“branchpoints”);
[L,NUM] = bwlabel(BW_3WJ);
BW_Merge = imfuse(BW_skel,BW_3WJ,“falsecolor,”“ColorChannels,”[1 2 0]);
imshow(BW_Merge).
Cells were blindly scored as abnormal if they mostly contained sheets or reduced peripheral tubules throughout most of the cell periphery. Examples of each category are shown in Fig. 6 A. The percentage of cells with abnormal ER is the sum of the percentage of cells that are mostly sheets and the percentage of cells with reduced peripheral tubules.
Measurement of ER delivery to lysosomes
To assess the delivery of RTN4, RTN3L, SEC24C, Akita, and LNPK puncta to lysosomes marked by LAMP1-mCherry, transfected cells were untreated (DMSO) or treated with an inhibitor of lysosomal degradation, 100 nM Bafilomycin A1 (Baf) that was solubilized in DMSO. The time of treatment is indicated in each figure legend. To inhibit autophagy, the cells were treated with either 2 µM MRT68921, or siFIP200 and incubated with Baf as indicated in the legends. Image J was used to calculate the relative pixel intensity. Briefly, the LAMP1 image (red channel) was subjected to autothresholding to produce a binary image. The analyze particles command was then used to obtain a mask of the object that was superimposed on the ER marker (green channel), and the median intensity of the green channel was measured. The DMSO sample was set to 1.0. The value obtained for each condition was divided by the mean pixel intensity from the control condition to produce the relative pixel intensity.
ER-phagy flux assay
To examine ER autophagic flux, imaging was performed using a U2OS stable cell line constitutively expressing the tandem reporter ssRFP-GFP-KDEL (Chino et al., 2019). Control and siRNAi-treated cells were grown in rich medium (DMEM supplemented with 10% FBS), and the number of red dots per cell was calculated using ImageJ v2.0 (Fiji). Briefly, the two-channel image (GFP and RFP) was split into two images, as described before (Parashar et al., 2021). Each image was then threshold adjusted by choosing the Yen algorithm to produce black and white images with black objects that display the labeling pattern for each channel. The image calculator was then used to generate an intersection image of the red channel with the green channel using the SUBTRACT operator tool. The binary image that was generated showed the non-overlapping red dots. The analyze particle command was used to acquire the red dots.
Cell lysate preparation and immunoprecipitations
TRex U2OS untagged cells or cells stably expressing FLAG-HA-RTN3L were grown to 50–60% confluency in a 10-cm diameter petri dish and then treated for 24 h with doxycycline (1 µg/ml) to induce the expression of RTN3L. For the ubiquitination studies, the cells were treated with doxycycline for 4 or 24 h. The results from both time points were the same. All cells were treated with 100 nM Baf for the last 4 h of the incubation, harvested, pelleted at 1,000 rpm, washed three times with ice cold 1x phosphate-buffered saline, and the pellet was resuspended in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP40, 1 mM DTT, and protease inhibitor). The resuspended cells were incubated at 4°C for 20 min with gentle shaking and the lysate was cleared by centrifugation at 13,200 rpm for 20 min. The protein concentration was measured by the BCA method following the manufacturer’s protocol, and 1.5 mg of lysate was incubated overnight at 4°C with monoclonal anti-HA-agarose antibody. The anti-HA–agarose beads were pelleted at 3,000 rpm for 1 min, washed three times with ice cold lysis buffer, and then washed three times with ice cold wash buffer (50 mM Tris, pH 7.5, 150 mM NaCl). The resin was heated to 70°C in sample buffer for 10 min. The eluted protein was subjected to 10% SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked for 1 h at room temperature in 5% milk (for SEC24C, SEC24A, SEC23A, RTN3L, LNPK, PEF1 and KLHL12) or 5% BSA (for CALCOCO1) made with PBST (0.1% Tween-20 in PBS) and incubated either overnight at 4°C or 1 h at room temperature with one of the following antibodies: anti-SEC24C, anti-SEC24A, anti-SEC23A, anti-RTN3, anti-LNPK, anti-PEF1, anti-KLHL12 and anti-CALCOCO1. Because KLHL12 and LNPK are close in molecular weight to the heavy chain of IgG, to probe for these proteins, the resin was eluted with HA-peptide (250 µg/ml) during a 30-min incubation at 37°C. The supernatant was collected and heated to 70°C in sample buffer for 10 min and immunoblotted. All antibodies used in this study are listed in Table S2.
The detection of ubiquitinated RTN3L
To probe for ubiquitinated RTN3L, TRex U2OS untagged cells or cells stably expressing FLAG-HA-RTN3L were immunoprecipitated as described above with the following modifications. The cells were treated with 10 μM MG132 plus 100 nM Baf for 4 h and then lysed in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP40, 1 mM DTT, 10 mM NEM, 2 mM EDTA, 2 mM EGTA, and protease inhibitor). The eluted protein was subjected to SDS-PAGE using a 4–15% gradient gel and then transferred to a nitrocellulose membrane. The membrane was autoclaved (30 min liquid cycle followed by a 15 min dry cycle), blocked for 1 h at room temperature in 20% FBS prepared with PBST (0.1% Tween-20 in PBS), and then incubated for 1 h at room temperature with ubiquitin antibody (1:500) that was prepared in 20% FBS (Swerdlow et al., 1986).
Crosslinking studies
Crosslinking experiments were performed with U2OS cells or cells stably expressing FLAG-HA-RTN3L. The protocol of Shibata et al. (2008) was followed (Shibata et al., 2008). Cells were treated with Baf for 4 h before lysis. Briefly, cells were cultured to 90% confluency in a 10-cm diameter petri dish, harvested, and washed three times with ice cold PBS (pH 7.4). The cells were resuspended and lysed in hypotonic buffer (10 mM HEPES, pH 7.8, 10 mM potassium acetate, 1.5 mM magnesium acetate, 2 mM PMSF and 5x protease inhibitor) by incubating on ice for 20 min. Subsequently, the lysed cells were passed 10 times through a 25-gauge needle and centrifuged at 3,000 × g for 5 min. The supernatant was centrifuged at 100,000 × g for 10 min to obtain a membrane fraction. The membranes were washed with HKM buffer (25 mM HEPES, pH 7.8, 150 mM potassium acetate, 2.5 mM magnesium acetate, 2 mM PMSF and 5x protease inhibitor) and resuspended in HKM buffer. The protein concentration of the membrane fraction was estimated using the BCA method and 40 µg of protein was cross-linked with EGS during a 30 min incubation at room temperature. Reactions were quenched using SDS-PAGE loading dye and heated at 70°C for 10 min before they were electrophoresed on a 4–15% gradient gel and western blot analysis was performed.
In vitro binding assays and western blot analysis
Lysates were prepared from U2OS cells that were grown in DMEM medium (10% FBS) in a 10-cm diameter petri dish to 90–100% confluency, treated with 100 nM Baf for 4 h, and lysed in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP40, 1 mM DTT, and 5x protease inhibitor cocktail). Subsequently, the lysate was diluted with wash buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM DTT, and 5X protease inhibitor cocktail) to a final concentration of 0.3% NP-40. Equimolar amounts (1 µM) of purified GST fusion protein, immobilized on glutathione–Sepharose beads, were incubated with 0.75 mg of lysate overnight at 4°C with rotation. The beads were pelleted at 3,000 rpm for 1 min, washed five times with ice cold wash buffer, and the resin was heated to 70°C in sample buffer for 10 min. The eluted protein was subjected to 10% SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked for 1 h at room temperature in 5% milk (for SEC24C), or 5% BSA (for CALCOCO1) made with PBST (0.1% Tween-20 in PBS) and incubated overnight at 4°C with anti-SEC24C, and anti-CALCOCO1 antibodies.
LC3 processing assay
Cells were grown in 6-well plates in DMEM media and left untreated (DMSO) or were treated with 100 nM Baf for 4 h. To block LC3 processing, cells were treated with 2 µM MRT68921 with and without Baf for 4 h. The cells were harvested, washed three times with ice cold PBS, resuspended in 1 X lysis buffer (50 nM Tris, pH 7.5, 150 mM NaCl, 0.5% NP40, 1 mM DTT, and protease inhibitor), and incubated for 30 min on ice. During the 30-min incubation, the cells were resuspended every 10 min with gentle pipetting. The lysate was centrifuged at 14,000 rpm for 12 min and protein was measured by the BCA method. Samples were heated at 65°C for 10 min, electrophoresed on a 13.5% SDS polyacrylamide gel, and then processed for western blot analysis using the ECL method.
qPCR analysis
siRNA transfected cells were harvested, washed with PBS, and lysed in 1 ml of TRIzol reagent (15596026; Thermo Fisher Scientific). Subsequently, 200 μl of chloroform was added to the lysed cells and samples were centrifuged at 12,000 rpm for 10 min at room temperature. The aqueous phase was added to 500 μl of 2-propanol and centrifuged at 12,000 rpm for 10 min at 4°C. The resulting pellet was washed in 500 μl of 70% ethanol and solubilized in water. The RNA concentration was estimated and 1 µg of RNA was used to synthesize cDNA with a Thermo Script RT-PCR kit. For real-time PCR (qPCR), 1 μl of cDNA was used with gene-specific primers. The gene amplifications were quantified by the Ct method (2-ΔΔCt) and GAPDH was used as an internal control (Livak and Schmittgen, 2001).
Statistical analysis
To calculate statistical significance, at least three independent biological replicates were employed. Error bars in graphs are reported as SEM. Probability values (P values) were calculated using the unpaired two-tailed Student’s t test and plotted and analyzed in Excel. P values <0.05 were reported to be significant. For imaging studies, a minimum of 20 cells were quantitated for each replicate, and representative images are shown. For ER tubule three-way junction quantification, at least 10 cells were calculated in each replicate. Densitometric analysis of western blots was carried out using Image Lab software (BIO-RAD). Data distribution was assumed to be normal, but this was not formally tested.
Online supplemental material
Supplemental data Figs. S1, S2, S3, S4, and S5 are related to Fig. 1. Fig. S6 and Fig. S7 are related to Fig. 2. Figs. S8, S9, and S10 are related to Fig. 3 and Fig. 4. Figs. S11, S12, and S13 are related to Figs. 5, 6, and 7. Video 1 is related to Fig. 5 D. Video 2 and Video 3 are related to Fig. 6 B. Table S1 lists the reagents used in this study and Table S2 lists the antibodies used in this work.
Data availability
All reagents and data generated in this study will be available upon request from the corresponding author. Requests for reagents and/or data should be directed to Susan Ferro-Novick ([email protected]).
Acknowledgments
We thank Drs. Randy Schekman and Jesse Hay for antibodies, Drs. Paolo Grumati, Wade Harper, Michael Rape, Jesse Hay, and Peter Arvan for plasmids and cells, and Drs. Michael Rape, Eric Bennett, Jesse Hay, and Geoffrey Chang for stimulating discussions. Microscopy and image analysis was performed at the Nikon Imaging Center at UC San Diego (UCSD). We thank Peng Guo for his help with the MATLAB analysis. We also acknowledge Peng Guo and the Nikon Imaging Center at UCSD for their support, Plaboni Sen for assistance in data analysis, and Sophia Wang and Nancy Xing for help in preparing the manuscript.
This research was supported by National Institutes of Health grant R01NS117440 awarded to S. Ferro-Novick.
Author contributions: R. Chidambaram: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, K. Kumar: Conceptualization, Formal analysis, Investigation, Methodology, Resources, Validation, Visualization, S. Parashar: Formal analysis, Investigation, Methodology, Visualization, G. Ramachandran: Data curation, Formal analysis, Investigation, Methodology, Validation, S. Chen: Formal analysis, S. Ferro-Novick: Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing.

