


Neurotransmission is an energetically expensive process that underlies cognition. During intense electrical activity or dietary restrictions, the glucose level in the brain plummets, forcing neurons to utilize alternative fuels. However, the molecular mechanisms of neuronal metabolic plasticity remain poorly understood. Here, we demonstrate that glucose-deprived neurons activate the CREB and PGC1α transcriptional program, which induces expression of the mitochondrial deacetylase Sirtuin 3 (Sirt3) both in vitro and in vivo. We show that Sirt3 localizes to axonal mitochondria and stimulates mitochondrial oxidative capacity in hippocampal nerve terminals. Sirt3 plays an essential role in sustaining synaptic transmission in the absence of glucose by providing metabolic support for the retrieval of synaptic vesicles after release. These results demonstrate that the transcriptional induction of Sirt3 facilitates the metabolic plasticity of synaptic transmission.
Introduction
The brain requires a constant and ready source of energy to function properly. Indeed, cognitive function is particularly susceptible to metabolic perturbations, such as ischemic stroke or hypoglycemia. Glucose is considered the primary fuel for the brain, yet glucose concentration in the interstitial fluid (ISF) surrounding neurons is considerably lower (0.5–1 mM in rodents) than in the blood (Dunn-Meynell et al., 2009; Fioramonti et al., 2017; McNay and Gold, 1999). Furthermore, bouts of neuronal firing deplete the glucose content of ISF in active brain regions (McNay et al., 2000; Silver and Erecińska, 1994). Availability of glucose in the brain is further exacerbated by the relative scarcity of glucose storage as glycogen (Oz et al., 2007), forcing neurons to rely on alternative energy sources such as lactate, ketone bodies, and amino acids during hypoglycemia (Dalsgaard et al., 2003; Owen et al., 1967; van Hall et al., 2009). Lactate and its derivative pyruvate are major neuronal fuels that are supplied systemically through circulation or locally by neighboring astrocytes. Indeed, astrocytes have been shown to provide neurons with lactate produced from glutamate and other amino acids in a process known as the astrocyte-to-neuron lactate shuttle (Magistretti and Allaman, 2015; Pellerin et al., 1998). Lactate and pyruvate are oxidative fuels that bypass glycolysis and are directly broken down via mitochondrial oxidative phosphorylation (OXPHOS). Therefore, in the absence of glucose, neurons need to increase their mitochondrial capacity for OXPHOS to compensate for the lack of glycolytic ATP production. However, neuronal metabolic adaptation to low glucose and its impact on synaptic function remain poorly understood.
In most cell types, metabolic adaptation to energetic stress, such as fuel shortage, is initiated transcriptionally through coordinated control of gene expression. It is well known that neuronal electrical activity or muscle contraction activates the transcription factor family of cAMP-response element-binding proteins (CREBs; Altarejos and Montminy, 2011; Johannessen et al., 2004; Shaywitz and Greenberg, 1999). One of the target genes of the CREB transcription program is the transcriptional coactivator PGC1α (peroxisome proliferator-activated receptor gamma coactivator-1 alpha), which plays a key role in mitochondrial biogenesis and OXPHOS. In skeletal muscle, caloric restriction and exercise induce the expression of PGC1α (Akimoto et al., 2005) and its target Sirtuin 3 (Sirt3), a mitochondrial lysine deacetylase enzyme (Lombard et al., 2007). Sirt3 resides in the mitochondrial matrix and regulates mitochondrial metabolism (Jing et al., 2013) and resistance to oxidative stress (Cheng et al., 2016) in diverse cell types. Although Sirt3 is one of the most abundant sirtuins in the brain (Sidorova-Darmos et al., 2014), its function in the metabolic support of synaptic transmission remains unexplored.
In neurons, nerve terminals are loci of high energy demand due to their role in synaptic transmission. We and others have previously shown that during brief periods of glucose deprivation, mitochondrial oxidation of pyruvate can sustain the synaptic vesicle (SV) cycle (Ashrafi et al., 2020; Pathak et al., 2015), a crucial process in synaptic transmission. In the present study, we examined the metabolic adaptations of neurons to prolonged glucose deprivation and revealed a critical role for the mitochondrial deacetylase Sirt3 in ensuring the metabolic plasticity of synaptic transmission.
Results and discussion
Neuronal glucose deprivation induces transcriptional reprogramming of mitochondrial metabolism
We previously demonstrated that neurons utilize the oxidative fuels lactate and pyruvate to support synaptic transmission during acute glucose deprivation (Ashrafi et al., 2020). Other studies have also shown that neuronal activity continues to persist during prolonged glucose shortage in fasting or restrictive diets (Padamsey et al., 2022), implicating yet unknown mechanisms of metabolic plasticity in neurons. We examined the transcriptional rewiring of neuronal metabolism in primary cultures of rat cortical neurons maintained in glucose-rich or glucose-depleted (and serum-free) media for 3 h, at which point only minimal cell death was observed (Fig. S1 A). Differential gene expression under these conditions was determined by bulk RNA sequencing (Fig. 1 A; Table S1). A subset of ∼250 genes with significantly upregulated expression in the glucose-deprived conditions (fold change >1.58, adjusted P value <0.01) were selected for pathway analysis. Comparison with annotated gene sets in the Molecular Signatures Database (MSigDB) revealed significant enrichment of genes involved in the unfolded protein response mediated by PERK, ATF4, and ATF6 transcription factors (Fig. 1 B). Furthermore, plasma membrane amino acid transporters were upregulated under glucose deprivation, suggesting the reprogramming of metabolic pathways toward utilization of amino acids as an alternative energy source (Divakaruni et al., 2017). Glucose deprivation stimulated the expression of genes in the CREB transcription program (Fig. 1 C), which regulates metabolic homeostasis (Altarejos and Montminy, 2011). We confirmed activation of the CREB pathway by immunostaining primary cortical cultures with an anti-phospho-CREB antibody and found that nuclear phospho-CREB levels were elevated during glucose deprivation (Fig. 1, D and E).
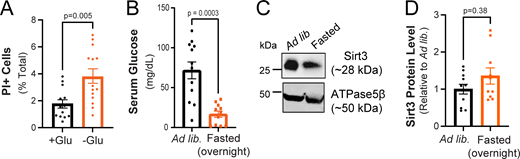
(Related toFig. 1,andFig. 2 ). Neuronal viability is sustained during glucose deprivation and hippocampal Sirt3 expression is not altered by overnight fasting. (A) Fraction of dead cells following glucose deprivation (3 h) as determined by propidium iodide staining (PI+). Mean (% total) ± SEM: +glucose (3 h), 1.76 ± 0.30; −glucose (3 h), 3.76 ± 0.53. (B) Serum glucose levels in mice fed ad lib. or fasted overnight. Serum glucose levels ± SEM (mg/dl): ad lib., 71.65 ± 10.59; fasted overnight, 16.63 ± 3.91. n = 12 mice/condition. (C) Hippocampal tissues from mice fed ad lib. or fasted overnight were immunoblotted for Sirt3 and mitochondrial ATPase5β. (D) Sirt3 band intensity normalized to ATPase5β and expressed relative to ad lib samples. Average normalized Sirt3 band intensity ± SEM: ad lib., 1.00 ± 0.13; overnight fasted, 1.354 ± 0.22. n = 11 mice/condition. Mann–Whitney U test (A, B, and D). Source data are available for this figure: SourceData FS1.
(Related toFig. 1,andFig. 2 ). Neuronal viability is sustained during glucose deprivation and hippocampal Sirt3 expression is not altered by overnight fasting. (A) Fraction of dead cells following glucose deprivation (3 h) as determined by propidium iodide staining (PI+). Mean (% total) ± SEM: +glucose (3 h), 1.76 ± 0.30; −glucose (3 h), 3.76 ± 0.53. (B) Serum glucose levels in mice fed ad lib. or fasted overnight. Serum glucose levels ± SEM (mg/dl): ad lib., 71.65 ± 10.59; fasted overnight, 16.63 ± 3.91. n = 12 mice/condition. (C) Hippocampal tissues from mice fed ad lib. or fasted overnight were immunoblotted for Sirt3 and mitochondrial ATPase5β. (D) Sirt3 band intensity normalized to ATPase5β and expressed relative to ad lib samples. Average normalized Sirt3 band intensity ± SEM: ad lib., 1.00 ± 0.13; overnight fasted, 1.354 ± 0.22. n = 11 mice/condition. Mann–Whitney U test (A, B, and D). Source data are available for this figure: SourceData FS1.

Transcriptional reprogramming of neuronal metabolism during glucose deprivation. (A) Schematic for glucose deprivation of rat cortical neurons. A subset of ∼250 genes significantly upregulated during glucose deprivation (P value <0.01) were selected for pathway analysis using annotated gene sets from the Molecular Signatures Database (MSigDB). n = 3 cortical samples from one rat litter. (B) Enrichment of genes in MSigDB Reactome pathways with adjusted P value <0.05. (C) Enrichment of the target genes of the CREB transcription factor in glucose-deprived neurons (adjusted P value <0.05). The target gene Pgc1α (PPARGC1A) is highlighted in red. (D) Immunostaining of cortical neuronal cultures (treated as in A) with an anti-phospho-CREB antibody and Hoechst nuclear stain. Arrowheads denote pCREB-positive nuclei. (E) Fraction of nuclei with positive p-CREB staining in fields of view (FOV), determined as described in Materials and methods. % total ± SEM: +glucose (3 h), 14.87 ± 2.29, −glucose (3 h), 27.41 ± 3.3. n = 17–22 FOVs. (F) Relative mRNA expression of Pgc1α in neuronal cultures treated as in A, with or without the AMPK inhibitor, dorsomorphin. Values are normalized to β-actin mRNA and expressed relative to the +glucose condition. Average normalized mRNA level ± SEM: −glucose (1 h), 2.83 ± 0.42; −glucose (3 h), 3.67 ± 0.46; −glucose + dorsomorphin (3 h), 1.32 ± 0.22. n = 3–10 cortical samples. Bar graphs are plotted as mean ± SEM. Mann–Whitney U test (E), one-way ANOVA (F). See Table 1.
Transcriptional reprogramming of neuronal metabolism during glucose deprivation. (A) Schematic for glucose deprivation of rat cortical neurons. A subset of ∼250 genes significantly upregulated during glucose deprivation (P value <0.01) were selected for pathway analysis using annotated gene sets from the Molecular Signatures Database (MSigDB). n = 3 cortical samples from one rat litter. (B) Enrichment of genes in MSigDB Reactome pathways with adjusted P value <0.05. (C) Enrichment of the target genes of the CREB transcription factor in glucose-deprived neurons (adjusted P value <0.05). The target gene Pgc1α (PPARGC1A) is highlighted in red. (D) Immunostaining of cortical neuronal cultures (treated as in A) with an anti-phospho-CREB antibody and Hoechst nuclear stain. Arrowheads denote pCREB-positive nuclei. (E) Fraction of nuclei with positive p-CREB staining in fields of view (FOV), determined as described in Materials and methods. % total ± SEM: +glucose (3 h), 14.87 ± 2.29, −glucose (3 h), 27.41 ± 3.3. n = 17–22 FOVs. (F) Relative mRNA expression of Pgc1α in neuronal cultures treated as in A, with or without the AMPK inhibitor, dorsomorphin. Values are normalized to β-actin mRNA and expressed relative to the +glucose condition. Average normalized mRNA level ± SEM: −glucose (1 h), 2.83 ± 0.42; −glucose (3 h), 3.67 ± 0.46; −glucose + dorsomorphin (3 h), 1.32 ± 0.22. n = 3–10 cortical samples. Bar graphs are plotted as mean ± SEM. Mann–Whitney U test (E), one-way ANOVA (F). See Table 1.
Statistical data
| Fig. 1 | Sample number | Statistical test |
|---|---|---|
| Fig. 1, A–C | 1 (cultures), 3 (wells) | N/A |
| Fig. 1 E | 4 (cultures), +Glu: (6 coverslips), 17 (FOV), −Glu: (7 coverslips), 22 (FOV), | Mann–Whitney U test |
| Fig. 1 F | >4 (cultures), +Glu: 10 (wells), −Glu_1h: 7 (wells), −Glu_3h: 7 (wells), Dorsomorphin: 3 (wells) | One-way ANOVA |
| Fig. 2 | Sample number | Statistical test |
| Fig. 2 A | >4 (cultures), +Glu:11 (wells), −Glu_1h: 7 (wells), −Glu_3h: 7 (wells), Dorsomorphine: 3 (wells) | One-way ANOVA |
| Fig. 2 B | 4 (cultures), 16 (wells) per condition | Two-tailed, unpaired t test |
| Fig. 2, C and D | 4 (cultures), 7 (wells) per condition | Mann–Whitney U test |
| Fig. 2, F and G | 6 ad lib mice, 6 IF mice | Mann–Whitney U test |
| Fig. 2 I | 5 (culture), 5 (mitochondrial preps/blots) per condition | One sample t test |
| Fig. 3 | Sample number | Statistical test |
| Fig. 3 B | 3 (cultures), Ctrl: 5 (coverslips), 60 (FOV), Sirt3 KD: 4 (coverslips), 46 (FOV) | Mann–Whitney U test |
| Fig. 3 D | 3 (cultures), 29 (coverslips), 78 (FOV) | Mann–Whitney U test |
| Fig. 3 H | 2 (cultures), 14 (coverslips), 27 (FOV) | Mann–Whitney U test |
| Fig. 4 | Sample number | Statistical test |
| Fig. 4, B and C | 6 (cultures), Ctrl: 32 (coverslips/neurons), Sirt3 KD: 10 (coverslips/neurons) | Kruskal–Wallis test |
| Fig. 4 E | 3 (cultures), 9 (coverslips), Ctrl_1Hz: 2,395 (synapses), Sirt3 KD_1Hz: 1,789 (synapses), Ctrl_10Hz: 2,178 (synapses), Sirt3 KD_10Hz: 1,641 (synapses) | Two-sample t test |
| Fig. 4 G | Ctrl:7 (cultures), 10 (coverslips), 13 (neurons); Sirt3 KD_L/P:5 (cultures), 10 (coverslips), 11 (neurons); rescue L/P- 2 (cultures), 5 (coverslips), 6 (neurons), Sirt3 KD, Glu: 3 (cultures), 6 (coverslips), 10 (neurons) | Kruskal–Wallis test |
| Fig. 4 I | Ctrl: 7 (cultures), 15 (coverslips), 19 (neurons), Sirt3 KD: 5 (cultures), 14 (coverslips), 19 (neurons) | Mann–Whitney U test |
| Fig. S1 | Sample number | Statistical test |
| Fig. S1 A | 3 (cultures), +Glu: 13 (wells), 24 (FOV), −Glu: 14 (wells), 27 (FOV) | Mann–Whitney U test |
| Fig. S1 B | 12 ad lib mice, 12 overnight fasted mice | Mann–Whitney U test |
| Fig. S1 D | 11 ad lib mice, 11 overnight fasted mice | Mann–Whitney U test |
| Fig. S2 A | 3 (cultures), Ctrl: 4 (wells), Sirt3 KD: 4 (wells) | Two-tailed, unpaired t test |
| Fig. S2, B and C | 2 (cultures), Ctrl: 11 (coverslips), Sirt3 KD: 8 (coverslips), Ctrl: 31 (neurons), Sirt3 KD: 42 (neurons) | Two-tailed, unpaired t test |
| Fig. S3 A | 6 (cultures), Ctrl: 32 (coverslips/neurons), Sirt3 KD: 10 (coverslips/neurons) | N/A |
| Fig. S3 B | Ctrl: 4 (cultures), 8 (neurons), Sirt3: KD 2 (cultures), 7 (neurons) | N/A |
| Fig. S3D | 3 (cultures), 8 (neurons) | Wilcoxon test |
| Fig. S3, E and F | 5 (culture), Ctrl:19 (coverslips/neurons), Sirt3 KD:26 (coverslips/neurons) | Two-tailed, unpaired t test |
| Fig. S3, G and H | 5 (culture), Ctrl:13 (coverslips/neurons), Sirt3 KD:20 (coverslips/neurons) | Two-tailed, unpaired t test |
| Fig. S3, I and J | 5 (culture), Ctrl:13 (coverslips/neurons), Sirt3 KD:20 (coverslips/neurons) | Two-tailed, unpaired t test |
FOV: field of view.
Amino acids are catabolized by mitochondrial OXPHOS, and they can enter the tricarboxylic acid cycle as pyruvate or intermediates, such as oxaloacetate, or α-ketoglutarate. Consistent with the increased reliance on mitochondrial metabolism during glucose deprivation, we found that PGC1α (also known as PPARGC1A), the master regulator of mitochondrial energy metabolism (Spiegelman, 2007), was among the CREB target genes that were induced (Fig. 1 C, highlighted in red). The induction of Pgc1α mRNA was validated with quantitative real-time PCR (qPCR; Fig. 1 F). Energetic stress in most cell types activates the cellular energy sensor, AMP kinase, by increasing the AMP (or ADP) to ATP ratio (Fryer et al., 2002; Kurth-Kraczek et al., 1999; Mu et al., 2001). In turn, AMP kinase initiates metabolic adaptation, including the CREB transcriptional program (Thomson et al., 2008). Consistent with this upstream regulatory role, we found that inhibition of AMP kinase with dorsomorphin (Zhou et al., 2001) blocked the induction of Pgc1α expression in glucose-deprived neurons (Fig. 1 F), indicating that this transcriptional response is driven by energetic stress. We conclude that neuronal glucose deprivation activates the CREB transcriptional program resulting in the induction of PGC1α, a potent regulator of mitochondrial oxidative metabolism (Austin and St-Pierre, 2012).
Glucose deprivation stimulates the expression of Sirtuin 3 and promotes deacetylation of mitochondrial proteins
Post-translational protein modifications play a critical role in the rapid and reversible modulation of cellular homeostasis. In calorically restricted skeletal muscle, the transcription coactivator PGC1α stimulates the expression of Sirt3, a mitochondrial deacetylase enzyme, by binding to its promoter region (Kong et al., 2010). Sirt3 drives metabolic reprogramming in muscle by deacetylating mitochondrial proteins involved in OXPHOS (Jing et al., 2011, 2013). The induction of PGC1α (Fig. 1 F) in glucose-deprived neurons prompted us to investigate whether Sirt3 expression was similarly upregulated. Indeed, sirt3 mRNA level was elevated in glucose-deprived cortical neurons, and this induction was blocked by pharmacological inhibition of AMP kinase (Fig. 2 A). Furthermore, we found that overexpression of PGC1α via adenovirus (Lehman et al., 2000) was sufficient to induce sirt3 gene expression in cortical neurons even in the presence of glucose, implicating PGC1α as a transcriptional regulator of neuronal Sirt3 (Fig. 2 B). In agreement with its mRNA induction, the expression of the Sirt3 protein was similarly elevated in glucose-deprived neurons (Fig. 2, C and D).
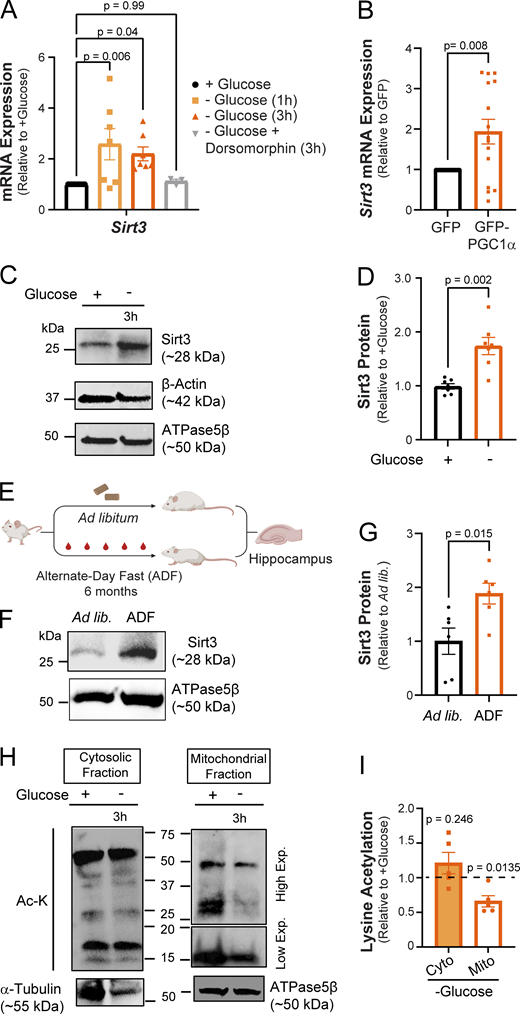
Glucose deprivation stimulates neuronal Sirt3 expression and deacetylation of mitochondrial proteins. (A) Relative Sirt3 mRNA expression in control and glucose-deprived neurons. Values are normalized to β-actin mRNA and expressed relative to control (+glucose). n = 3–10 cortical samples. Average normalized mRNA level ± SEM: −glucose (1 h), 2.58 ± 0.62; −glucose (3 h), 2.20 ± 0.27; −glucose + dorsomorphin (3 h), 1.12 ± 0.08. n = 3–11 cortical samples. (B) Relative Sirt3 mRNA expression in cultures transduced with adenoviral particles encoding GFP (control) and GFP-PGC1α, normalized to 18s rRNA and expressed relative to control. Average normalized mRNA level ± SEM: GFP-PGC1α, 1.93 ± 0.30. n = 16 cortical samples/condition. (C) Immunoblotting of Sirt3 protein expression in cortical neurons with antibodies against Sirt3 and the cytosolic and mitochondrial controls, β-actin, and ATPase5β, respectively. (D) Sirt3 band intensity normalized to the ATPase5β and expressed relative to control. Average normalized Sirt3 band intensity ± SEM: +glucose, 0.99 ± 0.05; −glucose, 1.74 ± 0.16. n = 7 cortical samples/condition. (E) A paradigm for the analysis of Sirt3 expression in hippocampi of mice fed ad libitum (ad lib) or alternate-day fasted for 6 mo (ADF). Schematic created with https://biorender.com. (F) Immunoblotting of Sirt3 protein in mouse hippocampal lysates. (G) Sirt3 band intensity normalized to the ATPase5β band and expressed relative to the ad lib mice. Average normalized Sirt3 band intensity ± SEM: ad lib, 1 ± 0.24; ADF (6 mo), 1.88 ± 0.19. n = 6 mice/condition. (H) Mitochondrial and cytosolic fractions isolated from cortical neuronal cultures maintained for 3 h with (+) or without (−) glucose. High and low exposures were 60 and 25 s, respectively. (I) Intensity of lysine acetylation bands normalized to α-tubulin or ATPase5β plotted relative to control. Average normalized Ac-K intensity: cytosolic fraction (−glucose), 1.21 ± 0.15; mitochondrial fraction (−glucose), 0.66 ± 0.08. n = 5 cortical samples. One-way ANOVA (A), two-tailed, unpaired t test (B), Mann–Whitney U test (D and G), one sample t test (I). See Table 1. Source data are available for this figure: SourceData F2.
Glucose deprivation stimulates neuronal Sirt3 expression and deacetylation of mitochondrial proteins. (A) Relative Sirt3 mRNA expression in control and glucose-deprived neurons. Values are normalized to β-actin mRNA and expressed relative to control (+glucose). n = 3–10 cortical samples. Average normalized mRNA level ± SEM: −glucose (1 h), 2.58 ± 0.62; −glucose (3 h), 2.20 ± 0.27; −glucose + dorsomorphin (3 h), 1.12 ± 0.08. n = 3–11 cortical samples. (B) Relative Sirt3 mRNA expression in cultures transduced with adenoviral particles encoding GFP (control) and GFP-PGC1α, normalized to 18s rRNA and expressed relative to control. Average normalized mRNA level ± SEM: GFP-PGC1α, 1.93 ± 0.30. n = 16 cortical samples/condition. (C) Immunoblotting of Sirt3 protein expression in cortical neurons with antibodies against Sirt3 and the cytosolic and mitochondrial controls, β-actin, and ATPase5β, respectively. (D) Sirt3 band intensity normalized to the ATPase5β and expressed relative to control. Average normalized Sirt3 band intensity ± SEM: +glucose, 0.99 ± 0.05; −glucose, 1.74 ± 0.16. n = 7 cortical samples/condition. (E) A paradigm for the analysis of Sirt3 expression in hippocampi of mice fed ad libitum (ad lib) or alternate-day fasted for 6 mo (ADF). Schematic created with https://biorender.com. (F) Immunoblotting of Sirt3 protein in mouse hippocampal lysates. (G) Sirt3 band intensity normalized to the ATPase5β band and expressed relative to the ad lib mice. Average normalized Sirt3 band intensity ± SEM: ad lib, 1 ± 0.24; ADF (6 mo), 1.88 ± 0.19. n = 6 mice/condition. (H) Mitochondrial and cytosolic fractions isolated from cortical neuronal cultures maintained for 3 h with (+) or without (−) glucose. High and low exposures were 60 and 25 s, respectively. (I) Intensity of lysine acetylation bands normalized to α-tubulin or ATPase5β plotted relative to control. Average normalized Ac-K intensity: cytosolic fraction (−glucose), 1.21 ± 0.15; mitochondrial fraction (−glucose), 0.66 ± 0.08. n = 5 cortical samples. One-way ANOVA (A), two-tailed, unpaired t test (B), Mann–Whitney U test (D and G), one sample t test (I). See Table 1. Source data are available for this figure: SourceData F2.
The induction of neuronal Sirt3 expression in vitro led us to examine whether Sirt3 levels in the brain are similarly regulated by nutrient intake. We found that hippocampal Sirt3 levels did not significantly increase in mice fasted overnight, although serum glucose content was reduced (Fig. S1, B–D). While short-term fasting limits organismal nutrient consumption, the liver can continue to supply extrahepatic tissues, such as the brain, with glucose or ketone bodies derived from glycogen and lipids. Indeed, changes in brain glucose levels are both delayed and dampened as compared with blood serum (Dunn-Meynell et al., 2009; Hwang et al., 2017). Therefore, we examined Sirt3 expression in the hippocampi of mice subjected to 6 mo of alternate-day fasting (ADF) and found significantly higher Sirt3 expression as compared with mice fed ad libitum (Fig. 2, E–G). Consistent with this finding, the induction of hippocampal Sirt3 expression has been observed within 1 mo of ADF (Liu et al., 2019). Thus, we conclude that the regulation of Sirt3 expression in the brain is an adaptive long-term response to nutrient availability.
Sirt3 abundance inversely correlates with acetylation of mitochondrial proteins as exemplified by hyperacetylation of mitochondrial proteins in Sirt3−/− mouse tissues (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Lombard et al., 2007) and deacetylation of mitochondrial proteins in exercising muscle with elevated Sirt3 expression (Jing et al., 2011). The induction of Sirt3 in glucose-deprived neurons prompted us to quantify its effects on mitochondrial protein acetylation. To this end, mitochondrial fractions prepared from neuronal lysates were immunoblotted with an antibody against acetylated lysine residues, revealing a significant reduction in mitochondrial protein acetylation in cortical neurons deprived of glucose, while acetylation of cytosolic proteins remained unchanged (Fig. 2, H and I). Overall, our findings suggest that neuronal glucose deprivation stimulates Sirt3 expression downstream of PGC1α, thereby leading to deacetylation of mitochondrial proteins.
Sirt3 localizes to neuronal mitochondria including at nerve terminals
Sirt3 has been shown to be expressed in mouse brain lysates prepared from multiple brain regions, including the cortex and hippocampus (Sidorova-Darmos et al., 2014). However, the subcellular distribution of Sirt3 in neuronal compartments remains unclear. To address this question, we performed immunocytochemistry on dissociated hippocampal neurons with antibodies against Sirt3 and markers of mitochondria, neurites, and presynaptic terminals. We first validated the specificity of Sirt3 staining by transiently knocking down Sirt3 (hereby referred to as Sirt3 KD) with a short hairpin RNA (shRNA) construct delivered by lentiviral infection. In Sirt3 KD neurons, Sirt3 immunostaining was significantly reduced as compared with control, uninfected neurons (Fig. 3, A and B). The efficiency of Sirt3 knockdown was also confirmed by qPCR of sirt3 mRNA in cortical neuronal cultures (Fig. S2 A). Coimmunostaining for Sirt3 and the mitochondrial marker TOMM20 revealed Sirt3 localization to neuronal mitochondria, as quantified by the Pearson’s correlation coefficient, a measure of the linear relationship between two signals independent of their absolute intensities (Fig. 3, C and D). Coimmunostaining neurons with Sirt3 and the neurite marker Tuj-1 (β-tubulin III) showed sparse distribution of Sirt3 along hippocampal neurites (Fig. 3 E), consistent with characteristic patterns of neuronal mitochondria (Schwarz, 2013). Furthermore, we found that Sirt3 localizes to presynaptic terminals as indicated by its colocalization with the vesicular glutamate transporter vGLUT1, which was quantified as the Pearson’s correlation coefficient between Sirt3 and vGLUT1 across fields of view (Fig. 3, F–H). We then examined the effects of Sirt3 depletion on neuronal mitochondrial morphology and found that axonal mitochondria were more fragmented in hippocampal neurons expressing Sirt3 shRNA (Fig. S2, B and C), consistent with a previous report implicating Sirt3 in mitochondrial fusion (Samant et al., 2014). Thus, we conclude that Sirt3 is expressed in neuronal mitochondria and localizes to presynaptic terminals where it may modulate presynaptic mitochondrial function.

Sirt3 is expressed in neuronal mitochondria and is present in presynaptic terminals. (A) Control hippocampal neurons and neurons transduced with lentivirus expressing Sirt3 shRNA (Sirt3 KD) were immunostained with anti-Sirt3 antibody and the Hoechst nuclear stain. (B) Quantification of Sirt3 immunofluorescence in control and Sirt3 KD neurons. Normalized mean Sirt3 intensity per coverslip ± SEM: Control, 1.02 ± 0.19; Sirt3 KD, 0.34 ± 0.04. n = 4–5 coverslips. (C) Co-immunostaining of neurons with antibodies against Sirt3 and the mitochondrial marker TOMM20. (D) Quantification of Sirt3 colocalization with TOMM20 or Hoechst in neuronal cell bodies. Mean Pearson’s correlation coefficient (r) ± SEM: Sirt3/TOMM20, r = 0.55 ± 0.01, Sirt3/Hoechst: −0.09 ± 0.02. n = 78 cell bodies. (E–G) Coimmunostaining of neurons with antibodies against the neurite marker Tuj1 (E) or presynaptic marker vGLUT1 (F and G). (G) Magnification of the boxed area in F. Arrowheads indicate colocalization of Sirt3 with presynaptic terminals. (H) Quantification of Sirt3 colocalization with vGLUT1 or Hoechst across fields of view (FOVs). Mean Pearson’s correlation coefficient ± SEM: Sirt3/vGLUT1: r = 0.60 ± 0.02, Sirt3/Hoechst: −0.01 ± 0.02. n = 27 FOVs. Scale bars, 10 μm. Mann–Whitney U test (B, D, and H). See Table 1.
Sirt3 is expressed in neuronal mitochondria and is present in presynaptic terminals. (A) Control hippocampal neurons and neurons transduced with lentivirus expressing Sirt3 shRNA (Sirt3 KD) were immunostained with anti-Sirt3 antibody and the Hoechst nuclear stain. (B) Quantification of Sirt3 immunofluorescence in control and Sirt3 KD neurons. Normalized mean Sirt3 intensity per coverslip ± SEM: Control, 1.02 ± 0.19; Sirt3 KD, 0.34 ± 0.04. n = 4–5 coverslips. (C) Co-immunostaining of neurons with antibodies against Sirt3 and the mitochondrial marker TOMM20. (D) Quantification of Sirt3 colocalization with TOMM20 or Hoechst in neuronal cell bodies. Mean Pearson’s correlation coefficient (r) ± SEM: Sirt3/TOMM20, r = 0.55 ± 0.01, Sirt3/Hoechst: −0.09 ± 0.02. n = 78 cell bodies. (E–G) Coimmunostaining of neurons with antibodies against the neurite marker Tuj1 (E) or presynaptic marker vGLUT1 (F and G). (G) Magnification of the boxed area in F. Arrowheads indicate colocalization of Sirt3 with presynaptic terminals. (H) Quantification of Sirt3 colocalization with vGLUT1 or Hoechst across fields of view (FOVs). Mean Pearson’s correlation coefficient ± SEM: Sirt3/vGLUT1: r = 0.60 ± 0.02, Sirt3/Hoechst: −0.01 ± 0.02. n = 27 FOVs. Scale bars, 10 μm. Mann–Whitney U test (B, D, and H). See Table 1.

(Related to Fig. 3 ). Sirt3 regulates mitochondrial morphology in hippocampal axons. (A) Relative expression of sirt3 mRNA in control and Sir3 KD cortical neurons. Values are normalized to β-actin mRNA and expressed relative to the control. Average normalized mRNA level ± SEM: Sirt3 KD, 0.19 ± 0.07. n = 4 cortical samples. (B) Hippocampal axons from control and Sirt3 KD neurons expressing the mitochondrial marker mito4x-RFP. (C) Mitochondria length in control and Sirt3 KD axons. Mean length per neuronal axon (μm) ± SEM: control, 2.0 ± 0.03, Sirt3 KD, 1.6 ± 0.03. n = 31–42 neurons. Two-tailed, unpaired t test (A and C).
(Related to Fig. 3 ). Sirt3 regulates mitochondrial morphology in hippocampal axons. (A) Relative expression of sirt3 mRNA in control and Sir3 KD cortical neurons. Values are normalized to β-actin mRNA and expressed relative to the control. Average normalized mRNA level ± SEM: Sirt3 KD, 0.19 ± 0.07. n = 4 cortical samples. (B) Hippocampal axons from control and Sirt3 KD neurons expressing the mitochondrial marker mito4x-RFP. (C) Mitochondria length in control and Sirt3 KD axons. Mean length per neuronal axon (μm) ± SEM: control, 2.0 ± 0.03, Sirt3 KD, 1.6 ± 0.03. n = 31–42 neurons. Two-tailed, unpaired t test (A and C).
Sirt3 stimulates mitochondrial ATP synthesis and sustains synaptic transmission
Presynaptic terminals are loci of high energy demand, and we have previously shown that synaptic function can be sustained by mitochondrial oxidation of pyruvate in the absence of glucose. Since Sirt3 modulates mitochondrial metabolism in non-neuronal cells and is expressed in presynaptic mitochondria (Fig. 3, F and G), we examined the metabolic function of Sirt3 in terminals that were deprived of glucose. To this end, we monitored presynaptic ATP levels in dissociated hippocampal neurons before, during, and after electrical stimulation using Syn-ATP, a genetically encoded ATP indicator targeted to nerve terminals (Dehkharghanian et al., 2022; Rangaraju et al., 2014; Fig. 4 A). This indicator has been previously validated for use in hippocampal neurons without causing major impairment of synaptic function (Rangaraju et al., 2014). Neurons were supplied with a 1:1 mixture of lactate and pyruvate, and no glucose, before and during image acquisition totaling ∼30 min of glucose deprivation. The presynaptic ATP level prior to electrical stimulation (prestimulation) was significantly lower in nerve terminals expressing Sirt3 shRNA (Sirt3 KD) as compared with control terminals (Fig. 4, B and C; and Fig. S3, A and B). Robust action potential (AP) firing (600 AP at 10 Hz) did not cause further ATP depletion, although, after stimulation, ATP remained lower in Sirt3 KD terminals than in control (Fig. 4, B and C). These findings indicate that Sirt3 stimulates mitochondrial capacity for oxidative ATP production in nerve terminals.

Sirt3 stimulates oxidative ATP production and sustains synaptic transmission in hippocampal nerve terminals. (A) Luminescence and fluorescence (physin-mCherry) images of hippocampal nerve terminals expressing Syn-ATP. (B) Normalized and pH-corrected presynaptic ATP traces in control and Sirt3 KD neurons were supplied with lactate and pyruvate and stimulated with 600 AP at 10 Hz. (C) Normalized ATP level before and after electrical stimulation. Average ATP level: control (prestimulation), 1.00 ± 0.06, Sirt3 KD (prestimulation), 0.61 ± 0.19, control (poststimulation), 1.10 ± 0.35, Sirt3 KD (poststimulation), 0.65 ± 0.18. n = 10–32 neurons. (D) Schematic for determination of SV release probability (Pr) using vGLUT1-pH in neurons stimulated with 100 AP at 1 Hz, or 10 Hz. Representative images from successful and failed release events are shown in green and grey, respectively. (E) Pr in control and Sirt3 KD terminals at different stimulation frequencies. Average Pr ± SEM: control (1 Hz), 0.079 ± 0.005, Sirt3 KD (1 Hz), 0.081 ± 0.003, control (10 Hz), 0.112 ± 0.007, Sirt3 KD (10 Hz), 0.116 ± 0.011. n = 9 coverslips. (F) Sample vGLUT1-pH traces of control, Sirt3 KD, and neurons expressing shRNA-resistant Sirt3 (rescue) stimulated with 100 AP at 10 Hz in the presence of glucose or lactate/pyruvate. Inset: Semi-log plot of vGLUT1-pH traces following stimulation. (G) Fractional retrieval block of vGLUT1-pH was calculated as described in Materials and methods. n = 6–13 neurons. Average retrieval block ± SEM: control (lact+pyr), 0.14 ± 0.02, Sirt3 KD (lact+pyr), 0.40 ± 0.05, rescue (lact+pyr), 0.19 ± 0.01, Sirt3 KD (glucose), 0.18 ± 0.04. n = 6–13 neurons. (H) Sample vGLUT1-pH traces of control and Sirt3 KD neurons stimulated with 100 AP at 10 Hz in the presence of β-hydroxybutyrate (BHB). (I) Fractional retrieval block of vGLUT1-pH. n = 19 neurons. Average retrieval block ± SEM: control, 0.13 ± 0.03, Sirt3 KD, 0.36 ± 0.05. Gray bars and arrows denote electrical stimulation. The box-whisker plots denote median (line), 25th–75th percentile (box), and min-max (whiskers). Kruskal–Wallis test (C and G), two-sample t test (E), Mann–Whitney U test (I). See Table 1.
Sirt3 stimulates oxidative ATP production and sustains synaptic transmission in hippocampal nerve terminals. (A) Luminescence and fluorescence (physin-mCherry) images of hippocampal nerve terminals expressing Syn-ATP. (B) Normalized and pH-corrected presynaptic ATP traces in control and Sirt3 KD neurons were supplied with lactate and pyruvate and stimulated with 600 AP at 10 Hz. (C) Normalized ATP level before and after electrical stimulation. Average ATP level: control (prestimulation), 1.00 ± 0.06, Sirt3 KD (prestimulation), 0.61 ± 0.19, control (poststimulation), 1.10 ± 0.35, Sirt3 KD (poststimulation), 0.65 ± 0.18. n = 10–32 neurons. (D) Schematic for determination of SV release probability (Pr) using vGLUT1-pH in neurons stimulated with 100 AP at 1 Hz, or 10 Hz. Representative images from successful and failed release events are shown in green and grey, respectively. (E) Pr in control and Sirt3 KD terminals at different stimulation frequencies. Average Pr ± SEM: control (1 Hz), 0.079 ± 0.005, Sirt3 KD (1 Hz), 0.081 ± 0.003, control (10 Hz), 0.112 ± 0.007, Sirt3 KD (10 Hz), 0.116 ± 0.011. n = 9 coverslips. (F) Sample vGLUT1-pH traces of control, Sirt3 KD, and neurons expressing shRNA-resistant Sirt3 (rescue) stimulated with 100 AP at 10 Hz in the presence of glucose or lactate/pyruvate. Inset: Semi-log plot of vGLUT1-pH traces following stimulation. (G) Fractional retrieval block of vGLUT1-pH was calculated as described in Materials and methods. n = 6–13 neurons. Average retrieval block ± SEM: control (lact+pyr), 0.14 ± 0.02, Sirt3 KD (lact+pyr), 0.40 ± 0.05, rescue (lact+pyr), 0.19 ± 0.01, Sirt3 KD (glucose), 0.18 ± 0.04. n = 6–13 neurons. (H) Sample vGLUT1-pH traces of control and Sirt3 KD neurons stimulated with 100 AP at 10 Hz in the presence of β-hydroxybutyrate (BHB). (I) Fractional retrieval block of vGLUT1-pH. n = 19 neurons. Average retrieval block ± SEM: control, 0.13 ± 0.03, Sirt3 KD, 0.36 ± 0.05. Gray bars and arrows denote electrical stimulation. The box-whisker plots denote median (line), 25th–75th percentile (box), and min-max (whiskers). Kruskal–Wallis test (C and G), two-sample t test (E), Mann–Whitney U test (I). See Table 1.

(Related to Fig. 4 ). Quantification of presynaptic ATP, cytosolic pH, mitochondrial and cytosolic calcium dynamics in control and Sirt3 KD nerve terminals. (A) Average traces of raw luminescence/fluorescence values of control and Sirt3 KD hippocampal nerve terminals expressing Syn-ATP, electrically stimulated with 600 AP at 10 Hz. n = 10–32 neurons. (B) Average traces of cytosolic pH in control and Sirt3 KD terminals expressing cyto-pHluorin, electrically stimulated with 600 AP at 10 Hz. n = 7–8 neurons. (C) Representative vGLUT1-pH traces of a neuron supplied with 5 mM glucose, with or without incubation with 2 µM oligomycin. (D) Fractional retrieval block of vGLUT1-pH. Average retrieval block ± SEM: Glucose, 0.14 ± 0.02, Glucose + Oligomycin 0.40 ± 0.05. n = 8 neurons. (E) Average traces of mito4xi-GCaMP6f showing mitochondrial Ca2+ uptake in control and Sirt3 KD axons stimulated with 20 AP at 20 Hz. (F) Peak responses of Mito4xi - GCaMP6f (ΔF/F) following stimulation. Mean ΔF/F ± SEM: control, 0.34 ± 0.04, Sirt3 KD, 0.26 ± 0.02. n = 19–26 neurons. (G) Average traces of physin-GCaMP6f showing cytosolic Ca2+ uptake in control and Sirt3 KD axons stimulated with 1AP. (H) Peak responses of physin-GCaMP6f (ΔF/F) to stimulation. Mean ΔF/F ± SEM: control, 0.27 ± 0.06, Sirt3 KD, 0.21 ± 0.04. n = 13–20 neurons. (I) Average traces of physin-GCaMP6f showing cytosolic Ca2+ uptake in control and Sirt3 KD axons stimulated with 10 AP at 10 Hz. (J) Peak responses of physin-GCaMP6f (ΔF/F). Mean ΔF/F ± SEM: control, 2.5 ± 0.7, Sirt3 KD, 2.7 ± 0.4. n = 13–20 neurons. Grey bars or arrows denote electrical stimulation. Error bars are SEM. Wilcoxon test (D), Two-tailed, unpaired t test (F, H, and J).
(Related to Fig. 4 ). Quantification of presynaptic ATP, cytosolic pH, mitochondrial and cytosolic calcium dynamics in control and Sirt3 KD nerve terminals. (A) Average traces of raw luminescence/fluorescence values of control and Sirt3 KD hippocampal nerve terminals expressing Syn-ATP, electrically stimulated with 600 AP at 10 Hz. n = 10–32 neurons. (B) Average traces of cytosolic pH in control and Sirt3 KD terminals expressing cyto-pHluorin, electrically stimulated with 600 AP at 10 Hz. n = 7–8 neurons. (C) Representative vGLUT1-pH traces of a neuron supplied with 5 mM glucose, with or without incubation with 2 µM oligomycin. (D) Fractional retrieval block of vGLUT1-pH. Average retrieval block ± SEM: Glucose, 0.14 ± 0.02, Glucose + Oligomycin 0.40 ± 0.05. n = 8 neurons. (E) Average traces of mito4xi-GCaMP6f showing mitochondrial Ca2+ uptake in control and Sirt3 KD axons stimulated with 20 AP at 20 Hz. (F) Peak responses of Mito4xi - GCaMP6f (ΔF/F) following stimulation. Mean ΔF/F ± SEM: control, 0.34 ± 0.04, Sirt3 KD, 0.26 ± 0.02. n = 19–26 neurons. (G) Average traces of physin-GCaMP6f showing cytosolic Ca2+ uptake in control and Sirt3 KD axons stimulated with 1AP. (H) Peak responses of physin-GCaMP6f (ΔF/F) to stimulation. Mean ΔF/F ± SEM: control, 0.27 ± 0.06, Sirt3 KD, 0.21 ± 0.04. n = 13–20 neurons. (I) Average traces of physin-GCaMP6f showing cytosolic Ca2+ uptake in control and Sirt3 KD axons stimulated with 10 AP at 10 Hz. (J) Peak responses of physin-GCaMP6f (ΔF/F). Mean ΔF/F ± SEM: control, 2.5 ± 0.7, Sirt3 KD, 2.7 ± 0.4. n = 13–20 neurons. Grey bars or arrows denote electrical stimulation. Error bars are SEM. Wilcoxon test (D), Two-tailed, unpaired t test (F, H, and J).
We previously demonstrated that mitochondrial Ca2+ uptake is essential for maintaining presynaptic ATP levels during electrical activity. To determine whether Sirt3 regulates mitochondrial Ca2+ dynamics, we monitored mitochondrial Ca2+ uptake in nerve terminals using mitochondrially targeted GCaMP6f (mito4xi-GCaMP6f). We failed to detect changes in mitochondrial Ca2+ uptake in response to brief bursts of AP firing in Sirt3 KD terminals (Fig. S3, E and F), consistent with the ATP level remaining stable after stimulation (Fig. 4, B and C). Altogether, Ca2+ and ATP measurements in nerve terminals suggest that Sirt3 stimulates neuronal mitochondrial capacity for oxidative ATP production without altering mitochondrial Ca2+ dynamics during electrical stimulation.
Several studies have shown that mitochondrial oxidation of lactate or pyruvate can replace glucose to power energy-sensitive steps in the SV cycle (Ashrafi et al., 2020; Pathak et al., 2015). Given that Sirt3 modulates mitochondrial ATP synthesis in nerve terminals, we investigated the effects of Sirt3 depletion on the SV cycle in the absence of glucose (Fig. 4, D–G). We monitored AP-driven release and retrieval of SVs using vGLUT1-pHluorin (vGLUT1-pH) in nerve terminals supplied with lactate and pyruvate but no glucose. We first examined SV release in terminals that were electrically stimulated with trains of 100 AP at low (1 Hz) or high (10 Hz) activity levels (Fig. 4 D). We observed no significant differences between control and Sirt3 KD terminals in SV release probability in response to 1 or 10 Hz stimulation (Fig. 4 E). In contrast, following stimulation with 100 AP at 10 Hz, SV retrieval (which hereby refers to SV endocytosis and reacidification) was significantly less efficient in Sirt3 KD terminals supplied with lactate and pyruvate, but this defect could be rescued by re-expression of shRNA-resistant Sirt3 (Fig. 4, F and G). This is consistent with our previous study where we found that in neurons supplied with lactate and pyruvate, SV retrieval was highly sensitive to mitochondrial inhibition with oligomycin (Ashrafi et al., 2020), but the mitochondrial susceptibility of SV retrieval can be rescued by the addition of glucose (Fig. S3, C and D). We similarly found that incubating neurons with glucose instead of lactate and pyruvate was sufficient to restore SV retrieval in Sirt3 KD terminals (Fig. 4, F and G), thus validating the functional integrity of synapses in Sirt3-deficient neurons.
During starvation, ketone bodies released from the liver are a major energy source for extrahepatic tissues including the brain. Since Sirt3 has been shown to deacetylate multiple enzymes involved in ketone oxidation (Dittenhafer-Reed et al., 2015), we examined the effects of Sirt3 depletion on the SV cycle in the presence of the ketone body β-hydroxybutyrate (BHB). Indeed, we found that SV retrieval was significantly slower in Sirt3 KD terminals compared with control terminals supplied with BHB (Fig. 4, H and I). Therefore, we conclude that Sirt3 sustains SV retrieval in glucose-deprived nerve terminals by stimulating mitochondrial ATP generation from multiple fuel types, namely, lactate, pyruvate, and ketone bodies.
Sirt3 has been implicated in buffering cytosolic Ca2+ during excitotoxicity (Cheng et al., 2016). Since Ca2+ modulates SV endocytosis (Balaji et al., 2008), we examined whether or not the SV retrieval defect in Sirt3 KD neurons was secondary to impaired Ca2+ dynamics. To this end, we monitored cytosolic Ca2+ dynamics during electrical activity using (synapto-) physin-GCaMP6f in which the Ca2+ sensor GCaMP6f is anchored to SVs at nerve terminals. Presynaptic Ca2+ spikes evoked by a single AP or a short train of 10 APs were not found to be significantly different in control and Sirt3 KD terminals (Fig. S3, G–J). Therefore, we conclude that the effects of Sirt3 depletion on the SV cycle are not driven by changes in cytosolic Ca2+.
In nerve terminals, the SV cycle is one of the most energy-consuming processes of synaptic transmission (Jang et al., 2016; Rangaraju et al., 2014), which includes multiple events, namely, the release of SV through fusion with the plasma membrane, SV retrieval via endocytosis, reacidification, and refilling of SVs with neurotransmitter. While SV retrieval has been known to be most susceptible to energetic imbalances caused by mitochondrial or glycolytic inhibition (Ashrafi et al., 2017, 2020; Rangaraju et al., 2014), the molecular underpinnings of such susceptibility remain poorly understood. The metabolic sensitivity of SV retrieval may be due to energy consumption by dynamin during endocytosis (Bashkirov et al., 2008; Boissan et al., 2014; Yamashita et al., 2005) or the vacuolar-type ATPase, which re-establishes the proton gradient across SVs (Pulido and Ryan, 2021). We found that the release of SVs via exocytosis, however, is not strongly impacted by ATP depletion in Sirt3-deficient nerve terminals (Fig. 4 E), suggesting that the docking and release of SVs have a lower energetic barrier than SV retrieval. Our findings reinforce the idea that different cellular processes have distinct ATP demands and are differentially susceptible to metabolic perturbations in disease states.
While our study elucidates the function of Sirt3 in hippocampal (mainly excitatory) synapses, Sirt3 may regulate synaptic transmission differently in other types of neurons, such as inhibitory interneurons, which have distinct activity patterns and metabolic demands. Furthermore, given the critical function of astrocytes in regulating synaptic transmission and plasticity (Sancho et al., 2021), future studies are needed to uncover the glial functions of Sirt3.
Glucose is considered the preferred fuel for the brain (Peters et al., 2004), yet its availability varies in time and space within various brain regions due to intense circuit activity or dietary restrictions (McNay et al., 2000). Thus, compensatory metabolic pathways are essential for sustaining neuronal function when glucose availability becomes limited. Here, we show that neuronal glucose deprivation activates the CREB transcriptional program and induces the expression of the mitochondrial master regulator PGC1α and the mitochondrial deacetylase Sirt3. The activation of CREB signaling during glucose deprivation is noteworthy given its well-established role in learning and memory (Deisseroth et al., 1996; Won and Silva, 2008), which are also energetically intensive processes (Harris et al., 2012; Karbowski, 2019). Therefore, it is tempting to speculate that the activation of CREB signaling during memory formation results from local hypoglycemia caused by sustained neuronal activity.
At present, it is unclear whether Sirt3 activation occurs in a cell-wide or compartment-specific manner. Given that Sirt3 is an NAD+-dependent deacetylase, localized modulation of its activity could be mediated by changes in the mitochondrial NAD+ content as neuronal NAD+/NADH ratios are known to fluctuate during electrical activity (Díaz-García et al., 2021). Our discovery that Sirt3 stimulates mitochondrial ATP production to facilitate neurotransmission under oxidative conditions further supports the emerging evidence for the metabolic flexibility of neurons in utilizing alternative energy sources (Camandola and Mattson, 2017), such as ketones and lactate, which are utilized by several organs (Hui et al., 2017). The role of Sirt3 in mammalian synapses is relevant to many disease states, such as obesity and aging, where Sirt3 expression has been reported to decline (He et al., 2012; Kendrick et al., 2011). Indeed, our study suggests that neuronal Sirt3 deficiency may contribute to synaptic dysfunction or cognitive impairments in these conditions, highlighting the importance of determining whether restoring Sirt3 activity in the brain is therapeutic in disease states.
Materials and methods
Animals
Animal-related experiments were performed with wild-type rats of the Sprague-Dawley strain (RRID: RGD_734476) using protocols that were ethically approved by the Washington University Institutional Animal Care and Use Committee IACUC board. In fasting experiments, hippocampi were dissected from female C57BL/6J (RRID:IMSR_JAX:000664) mice fed ad libitum or alternate-day fasted on a 24-h cycle for 6 mo, starting at 6 wk of age. All animals were placed on aspen bedding for the duration of the study and cages were changed for both mouse groups on fasting days mice fed ad libitum. Mice were sacrificed during the fasting phase. In overnight fasting experiments, hippocampi were dissected from 11-wk-old C57BL/6NJ (RRID:IMSR_JAX:005304) male mice that were fed ad libitum or fasted overnight for ∼16 h.
Hippocampal neuronal cultures
Hippocampi were dissected from 1- to 3-d-old rat pups of mixed sex from the Sprague–Dawley strain. Tissues were dissociated and mixed cultures of neurons and glia were plated on coverslips coated with polyornithine and transfected 6–8 d after plating with calcium phosphate as previously described (Ryan, 1999). Hippocampal neurons were maintained in culture media composed of MEM (51200038; Thermo Fisher Scientific), 0.6% glucose, 0.1 g/l bovine transferrin (616420; Millipore), 0.25 g/l insulin, 0.3 g/l GlutaMAX supplement (35050-061; Thermo Fisher Scientific), 5% fetal bovine serum (S11510; R&D Systems), 2% N-21 (AR008; R&D Systems), and 4 μm cytosine β-d-arabinofuranoside (Ara-C), added after 2–3 d in vitro (DIV) to limit glial proliferation. Cortical neurons were maintained in Neurobasal-A media (NB-A; # A24775-01; Gibco) supplemented with 2% N-21, 5% fetal bovine serum, and 4 μm Ara-C (added after 1 DIV). Cultures were incubated at 37°C in a 95% air and 5% CO2 humidified incubator for 10–14 DIV (cortical neurons) or 14–21 DIV (hippocampal neurons) prior to use.
Plasmid constructs
Key reagents and plasmids used in this paper are listed in Table 2. The following previously published DNA constructs were used: vGLUT1-pHluorin (Voglmaier et al., 2006), Syn-ATP (Rangaraju et al., 2014), (synapto-)physin-GCaMP6f (de Juan-Sanz et al., 2017), mito4x-RFP (gift of T.A. Ryan, Weill Cornell Medicine, New York, NY, USA), and mito4xi-GCaMP6f construct (#192075; Addgene). Human Sirt3-RFP driven by the CaMKII promoter was constructed by PCR amplification of the human Sirt3 sequence from Sirt3-FLAG (plasmid 13814; Addgene; North et al., 2003) and insertion into the BamHI and AgeI sites of CaMKII-MICU3-RFP plasmid (Ashrafi et al., 2020) resulting in the linker sequence RPVVA joining Sirt3 and RFP sequences.
Key resources
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-vGLUT1 | Millipore Sigma | CAT#: AB5905 |
| Anti-Tuj1 (β-tubulin III) antibody | R&D Systems | MAB1195 |
| Anti-ATPase5β antibody | Sigma-Aldrich | CAT#: HPA001520 |
| Anti-Sirt3 antibody | Cell Signaling Technology | CAT#: 5490S |
| Anti-β actin antibody | Bio-Rad | CAT#: HCA147P |
| Anti-acetylated lysine antibody | Cell Signaling Technology | CAT#: 9441S |
| anti-α tubulin antibody | Sigma-Aldrich | CAT#: T6074 |
| Anti-TOMM20 antibody | Cell Signaling Technology | CAT#: 42406S |
| Anti-pCREB antibody | Cell Signaling Technology | CAT#: 9198S |
| Biological samples | ||
| rSirt3 shRNA lentivirus | Hope center viral vectors core at Washington University in St. Louis | N/A |
| PGC1α AAV | Gift of B. Finck | N/A |
| GFP AAV | Gift of B. Finck | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dorsomorphin | Sigma-Aldrich | CAT#: 866405-64-3 |
| Deacetylation inhibition cocktail | Santa Cruz | CAT#: sc-362323 |
| 6-Cyano-7nitroquinoxalibe-2, 3-dione (CNQX) | Sigma-Aldrich | CAT#: C239-25 MG |
| D,L-2-amino-5phosphonovaleric acid (APV) | Sigma-Aldrich | CAT#: A5282 |
| Propidium iodide | Invitrogen | CAT#: P1304MP |
| Critical commercial assays | ||
| All-in-one 5X RT master mix | Applied Biosytems | CAT#: ABM-G592 |
| PowerUp SYBR green master mix | Thermo Fisher Scientific | CAT#: A25742 |
| TaqMan fast advanced master mix | Applied Biosytems, | CAT#: 4444557 |
| Deposited Data | ||
| RNA-seq data | Dryad repository | Tiwari et al., 2023 Preprint |
| Syn-ATP analysis code | GitHub | https://github.com/ashrafilab/SynATP-Analysis |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/Strains | ||
| Sprague-Dawley rat | Charles River | Strain code: CD IGS SD # 001 RRID: RGD_734476 |
| C57BL/6NJ mice (overnight starvation) | Jackson Labs | Strain code: 005304 RRID:IMSR_JAX:005304 |
| C57BL/6J mice (alternate day fasting) | Jackson Labs | Strain code: 000664 RRID:IMSR_JAX:000664 |
| Recombinant DNA | ||
| vGLUT1-pHluorin | Voglmaier et al. (2006) | N/A |
| Syn-ATP | Rangaraju et al. (2014) | N/A |
| Synaptophysin-GCaMP6f | de Juan-Sanz et al. (2017) | N/A |
| mito4x-RFP | Gift of T.A. Ryan | N/A |
| mito4xi-GCaMP6f | Ashrafi et al. (2020) | #192075; Addgene |
| pLKO.1 vector | Moffat et al. (2006) | #10878; Addgene |
| hSirt3-RFP | This paper | |
| Sirt3-FLAG | North et al. (2003) | #13814; Addgene |
| Sequence-based reagents | ||
| rSirt3 shRNA (pLKO.1 vector) | This paper | Target sequence: 5′-CTTACTACATGTGGCTGAT-3′ |
| qPCR primers | Source | Sequence (5′-3′) |
| Mouse Pgc1α forward primer | Thermo Fisher Scientific | TGAAAAAGCTTGACTGGCGT |
| Mouse Pgc1α reverse primer | Thermo Fisher Scientific | CAACCAGAGCAGCACACTCTA |
| Rat Pgc1α forward primer | Thermo Fisher Scientific | TGAAAAAGCTTGACTGGCGT |
| Rat Pgc1α reverse primer | Thermo Fisher Scientific | CAGGGCAGCACACTCTATGT |
| Rat Sirt3 forward primer | Thermo Fisher Scientific | TGTGGGGTCCGGGAGTATTA |
| Rat Sirt3 reverse primer | Thermo Fisher Scientific | ACAGCTTCTTCTCACTGTGTCC |
| Rat Gapdh forward primer | Thermo Fisher Scientific | TGTGCAGTGCCAGCCTC |
| Rat Gapdh reverse primer | Thermo Fisher Scientific | AGAGAAGGCAGCCCTGGTAA |
| Rat 18 s Taqman probe | Thermo Fisher Scientific, #4331182 | N/A |
| Rat Sirt3 Taqman probe | Thermo Fisher Scientific, #4331182 | N/A |
| Rat β-actin forward primer | Thermo Fisher Scientific | GTCGAGTCCGCGTCCAC |
| Rat β-actin reverse primer | Thermo Fisher Scientific | TATCGTCATCCATGGCGAACTGG |
| Software and algorithms | ||
| ImageJ | National Institute of Health | https://imagej.nih.gov/ij/ |
| GraphPad prism 9.2.0 | GraphPad Software | http://www.graphpad.com/scientific-software/prism/ |
| Jupyter notebook | Project Jupyter | https://jupyter.org/ |
| Adobe illustrator | Adobe, Inc. | https://www.adobe.com |
shRNA against rat Sirt3 (target sequence: 5′-CTTACTACATGTGGCTGAT-3′) was cloned into the pLKO.1 vector (plasmid 10878; Addgene; Moffat et al., 2006).
Glucose deprivation experiments
Cortical neurons at 10–14 DIV were treated with Neurobasal-A (NB-A) media without glucose (#A24775-01; Gibco) or with media supplemented with 5-mM glucose (control condition). After 1–3 h of incubation at 37°C, the cells were lysed for either RNA or protein extraction or immunostaining.
RNA isolation and quantitative RT-PCR
Total RNA was isolated from primary dissociated cortical neuron cultures using RNeasy Mini Kits (74104; QIAGEN). cDNA was synthesized by reverse transcription using an All-In-One 5X RT Master Mix (ABM-G592) following the manufacturer’s protocol. qPCR reaction was set up in three technical replicates with cDNA as a template using PowerUp SYBR Green Master Mix (A25742; Thermo Fisher Scientific) or TaqMan Fast Advanced Master Mix (4444557; Applied Biosystems). Relative mRNA expression was normalized to housekeeping genes (Gapdh, 18s rRNA, or β-actin) using the ΔΔCt method.
Production and application of lentiviral particles
Lentivirus was produced by the Hope Center Viral Vectors Core at Washington University School of Medicine. Viral aliquots were functionally titrated, and the exact volume of lentivirus required to achieve maximal transduction of hippocampal or cortical cultures was determined using BFP expression, driven by a CMV promoter on the pLKO.1 backbone. For imaging experiments, lentiviral particles were added to neurons at 5 DIV, and after 2 d of infection, media exchange was performed. All experiments were performed at least 7 d after viral transduction to maximize knockdown efficiency.
Western blotting
Cultured cortical cells and brain tissues were solubilized in the neuronal extraction buffer (87792; Thermo Fisher Scientific) supplemented with 1X protease inhibitor cocktail and 0.1 mM PMSF. Protein concentrations were quantified using a BCA assay kit (BioVision) and BioTek Synergy plate reader and Gen5 software (BioTek Instruments). Immunoblotting was conducted using 4–10% SDS gradient polyacrylamide gel followed by transfer on PVDF membrane. The following primary antibodies were used: Sirt3 (1:1,000, 5490S; Cell Signaling Technology, rabbit, reactivity for human, mouse, and rat), anti-acetyl-lysine (1:1,000, 9441S; Cell Signaling Technology rabbit, all species expected), ATPase5β (1:1,000, HPA001520; Sigma-Aldrich, rabbit, reactivity for rat, mouse, and humans), β-actin (1:5,000; HCA147P; Bio-Rad, Human Combinatorial Antibody Library/HuCAL, reactivity for rat, mouse, and humans), α-tubulin (1:2,000, T6074; Sigma-Aldrich, mouse, reactivity for mouse, chicken, Chlamydomonas, African green monkey, human, rat, bovine, sea urchin, and kangaroo rat). Blots were developed by chemiluminescence using ECL (34579; Thermo Fisher Scientific) and images were acquired on ChemiDoc XRS+ molecular imager (Bio-Rad). Band intensity was quantified using ImageJ.
Mitochondrial fractionation
Cultured cortical cells were homogenized in mitochondrial isolation buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, pH 8.0, 250 mM Sucrose, 10 mM KCl, 1× Protease inhibitor, 0.1 mM PMSF and deacetylase inhibitor [1;200, sc-362323; Santa Cruz]) and centrifuged at 4°C for 10 min. The supernatant was collected and centrifuged for 20 min at 4°C. The mitochondrial pellet was washed thrice with mitochondrial isolation buffer and the supernatant containing the cytosolic fraction was concentrated using Amicon Ultra 0.5 centrifugal filter devices. The mitochondrial and cytosolic samples were prepared using 1× Lammelli buffer (1610747; BioRad).
Immunofluorescence and confocal microscopy
Hippocampal neurons were fixed with 4% PFA, permeabilized with 0.5% Triton, blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature (RT), and incubated at room temperature for 2 h or overnight at 4°C with the following primary antibodies: Sirt3 (1:200, 5490S; Cell Signaling Technology, rabbit, reactivity for human, mouse, and rat), TOMM20 (1:500, ab56783; abcam, mouse, reactivity for human, mouse, and rat), vGLUT1 (1:500, AB5905; Sigma-Aldrich, guinea pig, reactivity for rat), Tuj1 (1:1,000, MAB1195; R&D Systems, mouse, reactivity for mammals and chicken), phospho-CREB (1:500, 9198S; Cell Signaling Technology, rabbit, reactivity for human, mouse, and rat). Coverslips were then incubated with the following secondary antibodies: goat anti-rabbit Alexa-fluor555 (1:500, A21428; Thermo Fisher Scientific), goat anti-mouse Alexa-fluor488 (1:500, A11001; Thermo Fisher Scientific), and goat anti-guinea Alexa-fluor488 (1:500, A11073; Thermo Fisher Scientific) for 1 h at RT. For staining of nuclei, Hoechst (1:2,000, H3570; Invitrogen) was used at RT for 10 min and coverslips were then mounted with anti-fade mounting media (P36965; Thermo Fisher Scientific) and kept at 4°C until imaged. Confocal images were collected on a Zeiss LSM 880 confocal microscope using a 40X oil immersion objective (NA 1.3) at the Washington University Center for Cellular Imaging, which was purchased with support from the Office of Research Infrastructure Programs (ORIP) as part of the NIH Office of the Director under grant OD021629.
Live imaging of neurons
Imaging experiments were performed on a custom-built laser-illuminated epifluorescence microscope with a U Plan Fluorite 40X Oil Objective (NA 1.30). Images were acquired using an Andor iXon Ultra 897 camera cooled to −80°C to −95°C. Coverslips were mounted in a laminar flow perfusion chamber and perfused with Tyrodes buffer (pH 7.4) containing (in mM) 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 50 HEPES, 5 glucose, or 2.5 β-hydroxybutyrate, or 1.25 lactate and 1.25 pyruvate, supplemented with 10 μM 6-cyano-7nitroquinoxalibe-2, 3-dione (CNQX) and 50 μM D,L-2-amino-5phosphonovaleric acid (APV; both from Sigma-Aldrich) to inhibit post-synaptic responses. Neurons were maintained at 37°C using an Okolab stage top incubator. Action potentials were evoked in neurons with 1-ms pulses creating field potentials of ∼10 V/cm via platinum–iridium electrodes. Live imaging in Tyrodes buffer lacking glucose exposed neurons to a maximum of 30–60 min of glucose deprivation. This experimental condition was selected to ensure both PGC1α/Sirt3 transcriptional induction and reliable neuronal activity.
Analysis of live imaging data
Images were analyzed using the ImageJ plugin Time Series Analyzer where ∼20–50 regions of interest (ROIs) of ∼2 μm corresponding to responding synaptic boutons were selected for measurement of fluorescence intensity.
Quantification of presynaptic ATP level
Luminescence and fluorescence imaging of the presynaptic ATP reporter, Syn-ATP, and data analysis were performed using a semi-automated platform, as previously described (Dehkharghanian et al., 2022). Luminescence/fluorescence (L/F) values were corrected for pH changes using the cytosolic pH sensor cyto-pHluorin as shown in Fig. S3 B and as previously described (Rangaraju et al., 2014). It should be noted that due to the nonlinearity of the Syn-ATP indicator, L/F values do not reflect absolute ATP concentrations.
Quantitative analysis of release probability
vGLUT1-pH responses in single hippocampal terminals were recorded by stimulating cultures at 1 Hz for 100 s, followed by 100 AP at 10 Hz, separated by a 10-s rest period. Single fusion events were determined during the 1-Hz period using a MATLAB custom code (Maschi and Klyachko, 2017) based on the change in fluorescence. Release at high frequency was calculated as the relation between total fluorescence change during the 100-Hz stimulation period and the quantum size of a single SV fusion event. The quantal size was determined as the average fluorescence change of the events detected at the same terminal at 1 Hz. A minimum of five single events were taken for quantification.
Quantification of SV retrieval block
SV endocytosis in nerve terminals of hippocampal neurons was examined using vGLUT1-pHluorin as previously described by Ashrafi et al. (2020). Neurons were stimulated with trains of 100 AP at 10 Hz, and images were acquired at a frame rate of 2 Hz. For alkalization of pHluorin-containing vesicles, NH4Cl solution with a similar composition as Tyrodes buffer except containing 50 mM NH4Cl and 66.5 mM NaCl was used. Endocytic time constants (τ) were calculated by fitting fluorescent change after the stimulus to a single exponential decay as described earlier (Armbruster and Ryan, 2011). The fractional retrieval block in endocytosis of vGLUT1-pH was calculated as the fraction of ΔF remaining after two times the average endocytic time constant of the control (2τ), divided by the maximum ΔF at the end of stimulation (ΔF2τ/ΔFmax).
Cytosolic and mitochondrial Ca2+ measurements
Cytosolic Ca2+ experiments were performed using physin-GCaMP6f in which GCaMP6f is fused to the C-terminus of synaptophysin as described earlier (de Juan-Sanz et al., 2017). Neurons were stimulated with trains of 10 AP at 10 Hz, and the images were acquired at a frame rate of 10 Hz. Single AP responses were determined by averaging responses to 10 AP fired at 1 Hz. Mitochondrial Ca2+ signaling was measured using mito4x-GCaMP6f as previously described (Ashrafi et al., 2020). Neurons were stimulated with trains of 20 AP at 20 Hz, images were acquired at 5 Hz, and responses were averaged from three trials. In both physin-GCaMP6f and Mito4xi-GCaMP6f traces, 30 image frames were recorded before the stimulus train was triggered and peak ΔF/F0 values at the end of the stimulus were calculated and plotted.
Quantification of nuclear phospho-CREB intensity
Analysis of phospho-CREB immunostaining of neuronal cultures was performed with ImageJ. Briefly, ROIs corresponding to Hoechst-stained nuclei (7–500 μm in diameter) were selected with the Analyze Particles function. ROIs were transferred to the FITC channel corresponding to phospho-CREB staining to calculate average fluorescence intensity, normalized to the mean of control coverslips. Nuclei were considered phospho-CREB-positive nuclei if their normalized intensity value exceeded 1.24 (which represents the 75th percentile of normalized p-CREB intensity in glucose-deprived neurons).
Quantification of colocalization in confocal images
Colocalization of Sirt3 immunostaining with vGLUT1 and TOMM20 was calculated in ImageJ using Costes’s automatic threshold and reported as the Pearson’s correlation coefficient which measures how the variability in one image can be explained by the variability in another image, independent of signal intensity. Pearson’s coefficient varies from −1 to 1, where −1 denotes total negative correlation, 0 stands for random correlation, and 1 denotes total positive correlation. For Sirt3 colocalization with TOMM20, neuronal cell bodies were cropped for correlation analysis to exclude signals from surrounding glial cells that express Sirt3 at low levels. To quantify Sirt3 colocalization with vGLUT1, a field of view including cell bodies and neurites was analyzed. For comparison, Sirt3 colocalization with Hoechst was calculated in the same images used for colocalization analysis with TOMM20 and vGLUT1.
Quantification of Sirt3 knockdown in confocal images
Control hippocampal neurons and neurons transduced with lentiviral particles encoding Sirt3 shRNA were immunostained with anti-Sirt3 antibody and the Hoechst nuclear stain. Confocal images of Sirt3 in control and transduced neurons were thresholded using the same values and integrated densities limited to the threshold were calculated for each field of view/image, divided by the total number of Hoechst-stained nuclei. Values were normalized to the mean of control samples from each immunostaining experiment and plotted per coverslip.
Quantification of cell death by Hoechst and propidium iodide staining
Cortical neurons from control (5 mM glucose) and 3-h glucose-deprived conditions were stained with propidium iodide (5 μg/ml, P1304MP; Invitrogen) and Hoechst (1 μg/ml, 33342; Invitrogen) for 5 min at 37°C, as previously described (Jiajia et al., 2017), and imaged using 40X objective under Olympus CKX53 epifluorescence microscope (camera ORCA-spark, model C1144-36U) at room temperature. The total number of cells in each field of view was quantified by counting the number of Hoechst-stained nuclei (7–500 μm in diameter) using the Analyze Particles function of ImageJ. Similarly, the total number of dead cells was quantified by counting the number of PI-stained nuclei. The average percentage of dead cells in each well was plotted.
Quantification of mitochondrial length in neuronal axons
Hippocampal neurons expressing mito4x-RFP were infected with lentiviral particles expressing Sirt3 shRNA, fixed, and imaged with a confocal microscope. Uninfected cultures were used as the control. ImageJ was used to threshold images and measure mitochondrial size in distal axons. At least 20 individual mitochondria per axon were randomly selected for analysis.
Quantification of blood glucose in overnight-fasted mice
Blood was collected by terminal submandibular bleeding with a 21G × 1½ needle, left to clot for 2 h at room temperature, and centrifuged for 10 min at 4°C at 2,000 g. The supernatant was then collected for further analysis. Serum glucose levels were measured using a commercial colorimetric assay (#10009582; Cayman chemical) according to the manufacturer’s instructions.
RNA sequencing and analysis
Cortical neuronal cultures 13 DIV were treated with NB-A media containing 5 mM glucose (control) or NB-A without glucose (glucose deprivation) for 3 h. Total RNA was isolated using RNeasy Mini Kits (74104; QIAGEN) and triplicate samples were submitted to the GTAC core at McDonnell Genome Institute, Washington University. A subset of ∼250 genes whose expression was significantly upregulated in glucose-deprived samples (fold change >1.58, adjusted P value <0.01) were selected for pathway analysis by the FUMA GWAS (Functional Mapping and Annotation of Genome-Wide Association Studies) platform (Watanabe et al., 2017) using the Molecular Signatures Database C2 and C3 gene sets and thresholded at P value <0.05. Differentially expressed genes overlapping with these datasets were plotted in Fig. 1, B and C. A detailed description of the RNA sequencing protocol is included below:
Samples were prepared according to the library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovaSeq 6000. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq software and a custom python demultiplexing program with a maximum of one mismatch in the indexing read. RNA-seq reads were then aligned to the Ensembl release 76 primary assembly with STAR version 2.5.1a (Bakken et al., 2020,Preprint). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.6-p5 (Liao et al., 2014). Isoform expression of known Ensembl transcripts was estimated with Salmon version 0.8.2 (Patro et al., 2017). Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, and features detected. The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC version 2.6.2 (Wang et al., 2012).
All gene counts were then imported into the R/Bioconductor package EdgeR (Robinson et al., 2010) and TMM normalization size factors were calculated to adjust for samples for differences in library size. Ribosomal genes and genes not expressed in the smallest group size minus one sample greater than one count-per-million were excluded from further analysis. The TMM size factors and the matrix of counts were then imported into the R/Bioconductor package Limma (Ritchie et al., 2015). Weighted likelihoods based on the observed mean-variance relationship of every gene and sample were then calculated for all samples and the count matrix was transformed to moderated log2 counts-per-million with Limma’s voom WithQualityWeights (Liu et al., 2015). The performance of all genes was assessed with plots of the residual standard deviation of every gene to their average log-count with a robustly fitted trend line of the residuals. Differential expression analysis was then performed to analyze for differences between conditions and the results were filtered for only those genes with Benjamini-Hochberg false-discovery rate adjusted P values ≤0.05.
For each contrast extracted with Limma, global perturbations in known Gene Ontology (GO) terms, MSigDb, and KEGG pathways were detected using the R/Bioconductor package GAGE (Luo et al., 2009) to test for changes in expression of the reported log2 fold-changes reported by Limma in each term versus the background log2 fold-changes of all genes found outside the respective term. The R/Bioconductor package heatmap3 (Zhao et al., 2014) was used to display heatmaps across groups of samples for each GO or MSigDb term with a Benjamini-Hochberg false-discovery rate adjusted P value ≤0.05. Perturbed KEGG pathways where the observed log2 fold-changes of genes within the term were significantly perturbed in a single-direction versus background or in any direction compared with other genes within a given term with P values ≤0.05 were rendered as annotated KEGG graphs with the R/Bioconductor package Pathview (Luo and Brouwer, 2013).
To find the most critical genes, the Limma voom Wit QualityWeights transformed log2 counts-per-million expression data was then analyzed via weighted gene correlation network analysis with the R/Bioconductor package WGCNA (Langfelder and Horvath, 2008). Briefly, all genes were correlated across each other by Pearson correlations and clustered by expression similarity into unsigned modules using a power threshold empirically determined from the data. An eigengene was then created for each de novo cluster, and its expression profile was then correlated across all coefficients of the model matrix. Because these clusters of genes were created by expression profile rather than known functional similarity, the clustered modules were given the names of random colors where grey is the only module that has any pre-existing definition of containing genes that do not cluster well with others. These de novo clustered genes were then tested for functional enrichment of known GO terms with hypergeometric tests available in the R/Bioconductor package clusterProfiler (Yu et al., 2012). Significant terms with Benjamini-Hochberg adjusted P values <0.05 were then collapsed by similarity into clusterProfiler category network plots to display the most significant terms for each module of hub genes to interpolate the function of each significant module. The information for all clustered genes for each module was then combined with their respective statistical significance results from Limma to determine whether or not those features were also found to be significantly differentially expressed.
Statistical analysis
All statistical analysis was performed with GraphPad Prism v9.0 (Table 1). P values were calculated with two-tailed, unpaired t test or nonparametric Mann–Whitney U test when comparing two sets of data or with two-way ANOVA when comparing >2 sets of data. P values <0.05 were considered statistically significant. Sample size were selected based on power analysis using estimates of variability in the data based on previous results and preliminary studies. Outliers were selected and excluded from further analysis using the Grubb’s test in GraphPad prism. Where possible, experimenters were blinded to genotypes and/or experimental conditions. Error bars represent standard error in the graphs.
Online supplemental material
Fig. S1 shows (related to Fig. 1 and Fig. 2) that hippocampal Sirt3 expression is not altered by overnight fasting. Fig. S2 shows (related to Fig. 3) that Sirt3 regulates mitochondrial morphology in hippocampal axons. Fig. S3 shows (related to Fig. 4) the quantification of presynaptic ATP, cytosolic pH, mitochondrial and cytosolic calcium dynamics in control, and Sirt3 KD nerve terminals. Table S1 shows (related to Fig. 1) the list of differentially expressed genes in glucose-deprived versus control neuronal cultures.
Data availability
All the data underlying this study are available in the published article and its online supplemental material.
Material availability
Plasmids generated in this study have been deposited to Addgene. RNA-seq data are deposited on DRYAD. Syn-ATP analysis code is available on GitHub. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ghazaleh Ashrafi at [email protected].
Acknowledgments
We thank T. Roostaei and A. Nazeri for their help with RNA sequencing analysis, B. Finck for kindly providing PGC1α adenovirus, and J. Myeong for help with figure preparation.
We also thank Washington University Center for Cellular Imaging and the Genome Technology Access Center at the McDonnell Genome Institute, which is partially supported by National Cancer Institute Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by Institute of Clinical and Translational Sciences (ICTS) Grant #UL1TR002345 from the National Center for Research Resources, and National Institutes of Health (NIH) Roadmap for Medical Research. This work was funded by the Washington University Institute of Clinical and Translational Sciences (G. Ashrafi), the McDonnell Institute for Cellular Neurobiology Small Grant Program (G. Ashrafi), the Klingenstein-Simons Fellowship in Neuroscience (G .Ashrafi), the Whitehall Foundation (G. Ashrafi), National Institute of General Medical Sciences R35GM147222 (G. Ashrafi), R01HL147884 (C. Bergom), S10OD020136-01 (C. Bergom), and National Institute of Neurological Disorders and Stroke R35 NS111596 (V. Klyachko), National Heart, Lung, and Blood Institute K08HL138262 and 1R01HL155344 (A. Javaheri), the Children’s Discovery Institute and St. Louis Children’s Hospital MC-FR-2020-919 (A. Javaheri), the Diabetes Research Center P30DK020579 (A. Javaheri), NIH grant P30DK056341 (A. Javaheri), and the Longer Life Foundation (A. Javaheri).
Author contributions: G. Ashrafi: Conceptualization, funding acquisition, supervision, and writing (original draft, review, and editing), A. Tiwari: Investigation, visualization, formal analysis, and writing (original draft), A. Hashemiaghdam: Investigation and formal analysis, M.A. Laramie: Investigation, D. Maschi: Investigation and formal analysis, T. Haddad: Investigation, M.I. Stunault: Investigation, V. Klyachko: Conceptualization, funding acquisition, and supervision, C. Begom and A. Javaheri: funding acquisition, supervision.

