


Macroautophagy (autophagy) utilizes a serial of receptors to specifically recognize and degrade autophagy cargoes, including damaged organelles, to maintain cellular homeostasis. Upstream signals spatiotemporally regulate the biological functions of selective autophagy receptors through protein post-translational modifications (PTM) such as phosphorylation. However, it is unclear how acetylation directly controls autophagy receptors in selective autophagy. Here, we report that an ER-phagy receptor FAM134B is acetylated by CBP acetyltransferase, eliciting intense ER-phagy. Furthermore, FAM134B acetylation promoted CAMKII-mediated phosphorylation to sustain a mode of milder ER-phagy. Conversely, SIRT7 deacetylated FAM134B to temper its activities in ER-phagy to avoid excessive ER degradation. Together, this work provides further mechanistic insights into how ER-phagy receptor perceives environmental signals for fine-tuning of ER homeostasis and demonstrates how nucleus-derived factors are programmed to control ER stress by modulating ER-phagy.
Introduction
Selective autophagy is a cellular quality control pathway through which a variety of autophagy cargoes are engulfed explicitly by autophagosomes and delivered to lysosomes for degradation (Farre and Subramani, 2016; Gatica et al., 2018; Kirkin, 2019; Stolz et al., 2014; Zaffagnini and Martens, 2016). The specificity of this process is conferred by autophagy receptors that simultaneously bind to cargoes and the LC3 family members on the expanding autophagosomal membranes (Gatica et al., 2018; Khaminets et al., 2016).
Post-translational modifications (PTM) of cargo receptors relay upstream signals to initiate distinct selective autophagy pathways (Gubas and Dikic, 2021; Kirkin, 2019). For instance, p62 phosphorylation regulates its recognition of the ubiquitinated cargoes to facilitate their selective degradation (Ichimura et al., 2013; Lim et al., 2015; Matsumoto et al., 2015; Matsumoto et al., 2011). TBK1-mediated phosphorylation of OPTN or NDP52 enhances the overall xenophagy and mitophagy efficiency (Heo et al., 2015; Richter et al., 2016; Wild et al., 2011). Phosphorylation of BNIP3, FUNDC1, and NIX alter their binding affinity with LC3-family proteins to modulate mitophagy under different biological contexts (Chen et al., 2014; Rogov et al., 2017; Wu et al., 2014; Zhu et al., 2013). In yeast, the phosphorylation of cargo receptors coordinates their interaction with Atg8 and Atg11, thus increasing selective autophagy flux (Aoki et al., 2011; Farre et al., 2013; Kanki et al., 2013; Pfaffenwimmer et al., 2014; Tanaka et al., 2014). Protein lysine acetylation is an evolutionarily conserved regulatory mechanism in controlling different essential cellular pathways, including non-selective autophagy (Cheng et al., 2019; Lee and Finkel, 2009; Lin et al., 2012; McEwan and Dikic, 2011; Narita et al., 2019; Su et al., 2017; Yi et al., 2012). The critical functions of acetylation in selective autophagy just started to emerge with the finding that acetylation controls p62-mediated aggrephagy (You et al., 2019).
ER is the largest intracellular membrane system composed of tubular and sheet ER (Shibata et al., 2006). To maintain homeostasis, ER can expand itself to cope with different types of ER stresses and mitigate the toxicity derived from environmental insults (Walter and Ron, 2011). After stress, the excessive ER membrane structures have to be eliminated to avoid adverse side effects. This downsizing process is mediated by selective autophagy targeting ER, and this process was coined as ER-phagy (Bernales et al., 2006; Bernales et al., 2007). Recently, studies have shed light on how autophagy machinery recognizes redundant or damaged ER structures for lysosomal degradation by identifying distinct ER-phagy receptors (An et al., 2019; Chen et al., 2019; Chino et al., 2019; Fumagalli et al., 2016; Grumati et al., 2017; Khaminets et al., 2015; Mochida et al., 2015; Smith et al., 2017). However, it is poorly understood how ER-phagy receptors sense environmental or intracellular signals to timely trigger ER-phagy to cope with ER stress.
FAM134B is the first identified ER-phagy receptor in mammals and is conserved from yeast to humans (Khaminets et al., 2015; Mochida et al., 2015). The dysfunction of FAM134B causes hereditary sensory and autonomic neuropathy type 2 (HSAN II; Kurth et al., 2009). The Reticulon domain (RTND) of FAM134B is indispensable for ER membrane fragmentation and ER-phagy (Khaminets et al., 2015). We recently discovered that RTND-mediated FAM134B oligomerization is key to the fragmentation of the ER membrane to facilitate ER-phagy (Jiang et al., 2020). ER stress triggers CAMKII-mediated phosphorylation at the RTND of FAM134B, which is instrumental for its activity in ER membrane fragmentation and ER-phagy. In this study, we report that nucleus-derived CBP acetyltransferase and SIRT7 deacetylase constitute a regulatory circuit to control ER-phagy activity, thereby providing a more comprehensive insight into the spatiotemporal regulation of ER-phagy and ER homeostasis.
Results
FAM134B is acetylated at lysine160 (K160), which is responsive to ER stress
Mass spectrometry (MS) analysis of immunoprecipitated FAM134B identified an acetylated peptide 149SLSESWEVINSK(Ace)PDERPR166 (m/z = 533.27, MH44+). The detailed MS/MS spectrum showed alignment of a series of fragment ions including b2, y2, y4, y6, y7, y8, y9, y10, y11, y12, y13, and y14, especially fragment ions y7–y14 were detected as acetylated forms, which ambiguously verified that FAM134B was acetylated at lysine160 (K160; Fig. 1 A). Interestingly, K160 appeared to be the only acetylated lysine within the RTND of FAM134B. Next, we developed a polyclonal antibody specifically recognizing K160 acetylation (Ace-K160). Furthermore, we validated the reliability of this antibody either by mutating lysine160 into arginine (FAM134BK160R) to mimic deacetylation for a Western blot assay (Fig. 1 B) or by synthesizing the acetyl-peptide (C)WEVINSK(Ac)PDER-NH2 (amino acids 154–164; NP_001030023.1) and the unacetylated control peptide (C)WEVINSKPDER-NH2 for a dot blot assay (Fig. S1 A). As expected, this antibody was able to confirm the acetylation of both endogenous and exogenous FAM134B at K160 (Fig. 1 C and Fig. S1 B). Importantly, ER stress stimuli, including starvation by Earle’s Balanced Salt Solution (EBSS) and SERCA inhibition by Thapsigargin (Tg) treatment, enhanced the acetylation of endogenous FAM134B in a time-dependent manner (Fig. 1, D and E; and Fig. S1, C and D). Protein deacetylation is catalyzed by histone deacetylase (HDAC) and Sirtuin family members. Simultaneous administration of Trichostatin A (TSA), an HDAC inhibitor, and nicotinamide (NAM), a Sirtuin inhibitor, in the culture medium led to the accumulation of FAM134B K160 acetylation (Fig. 1 F and Fig. S1 E), indicating that a dynamic of acetylation and deacetylation of FAM134B at K160 may be involved in FAM134B regulation.
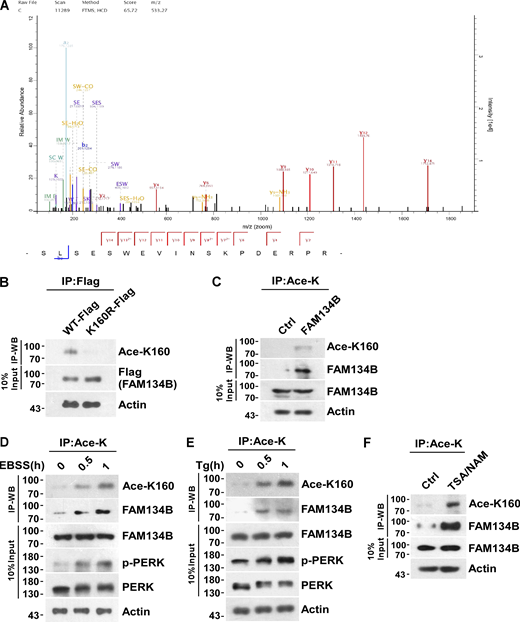
FAM134B is acetylated at lysine160 (K160), which is responsive to ER stress. (A) Representative MS/MS spectrum of acetylated peptide 149SLSESWEVINSK(Ace)PDERPR166 (m/z = 533.27, MH44+), which was acetylated at lysine160 (K160) of FAM134B. (B) Specificity verification of recognizing FAM134B K160 acetylation (~70 kD). Mutation of lysine (K) to arginine (R) mimicking deacetylation, which abolished recognition by Ace-K160) antibodies. HEK293T cells were transfected with FAM134B-Flag. IP was performed with anti-Flag beads. FAM134B K160 acetylation was analyzed by Western blot (WB) with Ace-K160 antibodies. (C) K160 acetylation of endogenous FAM134B. IP was performed with an antibody to acetylated lysine. The control was the Protein A/G agarose resin without the antibody that can recognize acetylated FAM134B. K160 acetylation and FAM134B were detected by Western blot. (D and E) K160 acetylation of endogenous FAM134B in response to ER stress. HEK293T cells were treated with EBSS starvation medium or 1 μM of Tg to trigger ER stress for 0, 0.5, 1 h. IP was performed with an antibody to acetylated lysine. K160 acetylation, FAM134B, p-PERK, and PERK were detected by Western blot. (F) K160 acetylation of endogenous FAM134B increased by treatment with deacetylase inhibitors. HEK293T cells were treated with deacetylase inhibitors TSA (1 mM) and NAM (5 mM) for 12 h. The control was treated with DMSO. IP was performed with an antibody to acetylated lysine. K160 acetylation and FAM134B were detected by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F1.
FAM134B is acetylated at lysine160 (K160), which is responsive to ER stress. (A) Representative MS/MS spectrum of acetylated peptide 149SLSESWEVINSK(Ace)PDERPR166 (m/z = 533.27, MH44+), which was acetylated at lysine160 (K160) of FAM134B. (B) Specificity verification of recognizing FAM134B K160 acetylation (~70 kD). Mutation of lysine (K) to arginine (R) mimicking deacetylation, which abolished recognition by Ace-K160) antibodies. HEK293T cells were transfected with FAM134B-Flag. IP was performed with anti-Flag beads. FAM134B K160 acetylation was analyzed by Western blot (WB) with Ace-K160 antibodies. (C) K160 acetylation of endogenous FAM134B. IP was performed with an antibody to acetylated lysine. The control was the Protein A/G agarose resin without the antibody that can recognize acetylated FAM134B. K160 acetylation and FAM134B were detected by Western blot. (D and E) K160 acetylation of endogenous FAM134B in response to ER stress. HEK293T cells were treated with EBSS starvation medium or 1 μM of Tg to trigger ER stress for 0, 0.5, 1 h. IP was performed with an antibody to acetylated lysine. K160 acetylation, FAM134B, p-PERK, and PERK were detected by Western blot. (F) K160 acetylation of endogenous FAM134B increased by treatment with deacetylase inhibitors. HEK293T cells were treated with deacetylase inhibitors TSA (1 mM) and NAM (5 mM) for 12 h. The control was treated with DMSO. IP was performed with an antibody to acetylated lysine. K160 acetylation and FAM134B were detected by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F1.
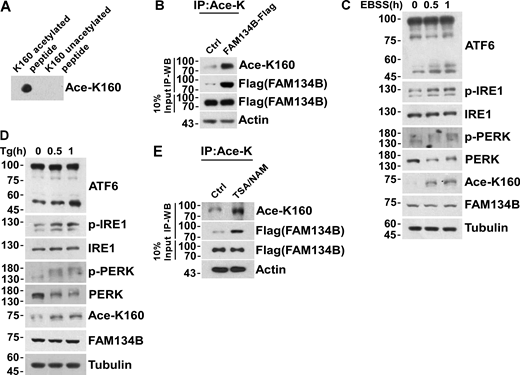
Acetylation of FAM134B K160 is responsive to ER stress. (A) The reliability of FAM134B K160 acetylation antibody was validated by performing a dot blot assay using synthesized acetyl-peptide (C)WEVINSK(Ac)PDER-NH2 (amino acids 154–164, NP_001030023.1) and the control unacetylated peptide (C)WEVINSKPDER-NH2. (B) K160 acetylation of FAM134B in FAM134B-Flag stable cell line. IP was performed with an antibody to acetylated lysine. The control was the Protein A/G agarose resin without the antibody that can recognize acetylated FAM134B. K160 acetylation and Flag were detected by Western blot (WB). (C and D) ER stress markers including ATF6, p-IRE1, and p-PERK were response to EBSS or Tg treatment. HEK293T cells were treated with EBSS or Tg (1 μM) to trigger ER stress for 0, 0.5, 1 h. The indicated proteins were detected by Western blot. (E) K160 acetylation of FAM134B in FAM134B-Flag stable cell line increased by treated with deacetylase inhibitors. Cells were treated with deacetylase inhibitors TSA (1 mM) and NAM (5 mM) for 12 h. The control was treated with DMSO. IP was performed with an antibody to acetylated lysine. K160 acetylation and Flag were detected by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS1.
Acetylation of FAM134B K160 is responsive to ER stress. (A) The reliability of FAM134B K160 acetylation antibody was validated by performing a dot blot assay using synthesized acetyl-peptide (C)WEVINSK(Ac)PDER-NH2 (amino acids 154–164, NP_001030023.1) and the control unacetylated peptide (C)WEVINSKPDER-NH2. (B) K160 acetylation of FAM134B in FAM134B-Flag stable cell line. IP was performed with an antibody to acetylated lysine. The control was the Protein A/G agarose resin without the antibody that can recognize acetylated FAM134B. K160 acetylation and Flag were detected by Western blot (WB). (C and D) ER stress markers including ATF6, p-IRE1, and p-PERK were response to EBSS or Tg treatment. HEK293T cells were treated with EBSS or Tg (1 μM) to trigger ER stress for 0, 0.5, 1 h. The indicated proteins were detected by Western blot. (E) K160 acetylation of FAM134B in FAM134B-Flag stable cell line increased by treated with deacetylase inhibitors. Cells were treated with deacetylase inhibitors TSA (1 mM) and NAM (5 mM) for 12 h. The control was treated with DMSO. IP was performed with an antibody to acetylated lysine. K160 acetylation and Flag were detected by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS1.
FAM134B acetylation dramatically enhances FAM134B oligomerization to induce ER fragmentation and ER-phagy
Because K160 resides in the RTND of FAM134B adjacent to Ser151 (Fig. 2 A), the phosphorylation of which promotes FAM134B homo-oligomerization (Jiang et al., 2020), we tested the hypothesis of whether K160 acetylation regulates FAM134B self-interaction by co-immunoprecipitation (co-IP) assays. Indeed, mimicking deacetylation by mutating lysine160 into arginine (FAM134BK160R) reduced FAM134B self-interaction (Fig. 2 B). In contrast, imitating permanent acetylation by substituting the lysine160 with glutamine (FAM134BK160Q) remarkably enhanced FAM134B self-association, which was comparable to that of FAM134BG216R, a putative gain-of-function mutant (Jiang et al., 2020). These observations demonstrated that FAM134B is acetylated at lysine160, which enhances FAM134B oligomerization. To measure ER membrane scission activity in vitro, we applied a previously established liposome fragmentation assay (Jiang et al., 2020; Fig. 2, C–E, and Fig. S2 A), in which the biotinylated liposomes were anchored onto streptavidin-coated glass within a chamber. Then, spinning-disk confocal microscopy was implemented to measure the dynamics of liposome fragmentation. We observed that the injection of purified recombinant protein FAM134BK160R could not induce liposome fragmentation as effectively as FAM134BWT. However, FAM134BK160Q could trigger liposome fragmentation significantly faster than FAM134BWT (Fig. 2, C–E). These observations suggested that K160 acetylation promotes membrane fragmentation in vitro. To reliably measure their ER membrane scission and ER-phagy activities in cells, we generated a serial of inducible cell lines on the basis of FAM134B knockout (KO) U2OS cells (Fig. S2 B), allowing the expression of different forms of FAM134B close to the endogenous counterpart, and we also applied Bafilomycin A1 (BafA1) treatment to accumulate EGFP-FAM134B–labeled ER membrane fragments to facilitate quantification analysis (Fig. 2, F and G). We observed that the expression of EGFP-FAM134BK160R induced fewer puncta, when compared with FAM134BWT. In contrast, EGFP-FAM134BK160Q expression yielded a significantly higher number of ER fragments. These results were confirmed under EGFP-FAM134B overexpression conditions (Fig. S2, C and D). Therefore, FAM134B acetylation at K160 is pivotal to induce ER membrane scission inside the cells. To measure ER-phagy activity, we first applied mCherry·EGFP tandem tagging strategy (Khaminets et al., 2015; Klionsky et al., 2016). The digestion of ER membrane fragments by autolysosomes lead to the biogenesis of mCherry+EGFP−puncta under confocal microscopy because of the hypersensitivity of EGFP to an acidic environment (Fig. 2, H and I). Therefore, the ratio of mCherry+EGFP can be quantified as the ER-phagy flux index. We observed that FAM134BK160Q exhibited significantly higher activities when compared with FAM134BWT. In contrast, FAM134BK160R displayed impaired activity in ER-phagy. These results were confirmed under mCherry-EGFP-FAM134B overexpression conditions (Fig. S2, E and F). Next, we assessed ER-phagy flux by GFP cleavage assay. Autophagic degradation of ER fragments produces free GFP because of the partial digestion of GFP-tagged SEC61B, an ER sheet-resident protein, FAM134BK160Q expression generated higher levels of free GFP compared with FAM134BWT (Fig. 2 J), while FAM134BK160R appeared to be less effective in inducing GFP cleavage. These results established that FAM134B K160 acetylation positively regulates ER membrane fragmentation and ER-phagy. Because overexpression of EGFP-tagged proteins may cause protein aggregations, we ruled out this possibility by performing several experiments. Using correlative light and electron microscopy (CLEM; Fig. 2 K) and immunoelectron microscopy (IEM; Fig. 2 L), we showed that the EGFP-FAM134B–labeled puncta were ER fragments, some of which were located inside autophagic vacuoles. Furthermore, the analysis of purified recombinant FAM134B proteins by size exclusion chromatography, circular dichroism, and native gel electrophoresis indicated that FAM134B proteins were not forming protein aggregates (Fig. S2, G–I), which were further confirmed by the measurement of the unfolded protein response in FAM134B-overexpressed cells using Protein kinase R (PKR)-like ER kinase (PERK) phosphorylation as an indicator (Fig. S2 J). Consistent with the CLEM and IEM results, the FAM134B and BAP31-positive ER fragments largely were colocalized with LAMP2 and LC3 (Fig. 2, M and N, and Fig. S2 K), suggesting that FAM134B faithfully labeled ER membrane structures, part of which were engulfed by autophagic vacuoles. Additionally, we also showed that FAM134B selectively regulated ER-phagy without affecting other autophagy processes (Fig. S2, L–P), and FAM134B overexpression could not alter the acetylation of ULK1 and VPS34, which are critical for autophagy initiation (Fig. S2, Q and R). Together, FAM134B acetylation facilitates FAM134B oligomerization instead of aggregation to induce ER fragmentation and ER-phagy.
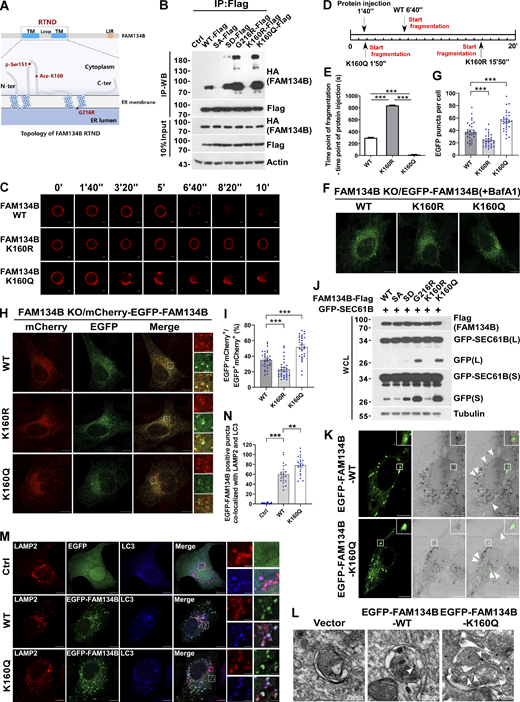
FAM134B acetylation dramatically enhances FAM134B oligomerization to induce ER fragmentation and ER-phagy. (A) Topology of FAM134B. Schematic of the full-length FAM134B; K160 resides in the RTND of FAM134B. CAMK2B-mediated FAM134B S151 phosphorylation, which further enhanced FAM134B8 oligomerization, ER fragmentation, and ER-phagy. G216R, a type II HSAN patient-derived FAM134B variant, exhibits hyperactive in FAM134B oligomerization, ER fragmentation, and ER-phagy. TM: transmembrane. (B) Comparison of self-interaction of FAM134B mutants using co-IP. Mimicking dephosphorylation by SA and deacetylation by K160R reduced FAM134B self-interaction. Mimicking permanent phosphorylation by SD enhanced FAM134B self-interaction. K160Q which mimicking permanent acetylation and G216R dramatically enhanced FAM134B self-interaction and oligomerization. HEK293T cells were transfected with mutants of HA-FAM134B and FAM134B-Flag. IP was performed with anti-Flag beads. The indicated proteins were detected by Western blot (WB). SA: mutanting Serine into Alanine. SD: mutanting Serine into Aspartic acid. (C–E) Acetylation of FAM134B enhanced liposome fragmentation in vitro. After injecting recombinant protein (25 μg) into the chamber (500 μl), the morphological changes of liposomes were monitored by live imaging for 20 min. The images at different time points as indicated were presented. The time required for liposome fragmentation was quantified in E (n = 3 experimental replicates). Scale bars, 10 μm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (F and G) FAM134B K160Q enhanced ER membrane fragmentation. FAM134B KO U2OS cells were engineered to inducibly express EGFP-FAM134B (WT), EGFP-FAM134B (K160R), or EGFP-FAM134B (K160Q) at endogenous levels. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in G (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (H and I) FAM134B K160Q enhanced ER-phagy. FAM134B KO U2OS cells were engineered to inducibly express mCherry-EGFP-FAM134B (WT), mCherry-EGFP-FAM134B (K160R), or mCherry-EGFP-FAM134B (K160Q) at endogenous levels. MCherry-positive but EGFP-negative puncta were quantified in I (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (J) Lysosomal cleavage of GFP was analyzed by Western blot for U2OS cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag mutants. WCL: whole-cell lysate. (K) Analysis of FAM134B-mediated ER membrane fragmentation by CLEM. U2OS cells grown on glass gridded coverslips were transfected with EGFP-FAM134B and imaged by Zeiss Airyscan to collect light microscopy images. Samples were then prepared for imaging by FIB-SEM. Light and electron microscope images were superimposed. Scale bars, 10 μm. (L) Analysis of FAM134B-mediated ER membrane fragmentation by IEM. U2OS cells were transfected with EGFP-FAM134B. White arrowheads indicated that the ER sheet labeled by immuno-gold was wrapped by autophagosomes. Scale bar, 200 nm. (M and N) FAM134B was colocalized with LAMP2 and LC3. U2OS cells transfected with EGFP-FAM134B were stained for endogenous LAMP2 and LC3. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in N (n = 20 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F2.
FAM134B acetylation dramatically enhances FAM134B oligomerization to induce ER fragmentation and ER-phagy. (A) Topology of FAM134B. Schematic of the full-length FAM134B; K160 resides in the RTND of FAM134B. CAMK2B-mediated FAM134B S151 phosphorylation, which further enhanced FAM134B8 oligomerization, ER fragmentation, and ER-phagy. G216R, a type II HSAN patient-derived FAM134B variant, exhibits hyperactive in FAM134B oligomerization, ER fragmentation, and ER-phagy. TM: transmembrane. (B) Comparison of self-interaction of FAM134B mutants using co-IP. Mimicking dephosphorylation by SA and deacetylation by K160R reduced FAM134B self-interaction. Mimicking permanent phosphorylation by SD enhanced FAM134B self-interaction. K160Q which mimicking permanent acetylation and G216R dramatically enhanced FAM134B self-interaction and oligomerization. HEK293T cells were transfected with mutants of HA-FAM134B and FAM134B-Flag. IP was performed with anti-Flag beads. The indicated proteins were detected by Western blot (WB). SA: mutanting Serine into Alanine. SD: mutanting Serine into Aspartic acid. (C–E) Acetylation of FAM134B enhanced liposome fragmentation in vitro. After injecting recombinant protein (25 μg) into the chamber (500 μl), the morphological changes of liposomes were monitored by live imaging for 20 min. The images at different time points as indicated were presented. The time required for liposome fragmentation was quantified in E (n = 3 experimental replicates). Scale bars, 10 μm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (F and G) FAM134B K160Q enhanced ER membrane fragmentation. FAM134B KO U2OS cells were engineered to inducibly express EGFP-FAM134B (WT), EGFP-FAM134B (K160R), or EGFP-FAM134B (K160Q) at endogenous levels. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in G (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (H and I) FAM134B K160Q enhanced ER-phagy. FAM134B KO U2OS cells were engineered to inducibly express mCherry-EGFP-FAM134B (WT), mCherry-EGFP-FAM134B (K160R), or mCherry-EGFP-FAM134B (K160Q) at endogenous levels. MCherry-positive but EGFP-negative puncta were quantified in I (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (J) Lysosomal cleavage of GFP was analyzed by Western blot for U2OS cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag mutants. WCL: whole-cell lysate. (K) Analysis of FAM134B-mediated ER membrane fragmentation by CLEM. U2OS cells grown on glass gridded coverslips were transfected with EGFP-FAM134B and imaged by Zeiss Airyscan to collect light microscopy images. Samples were then prepared for imaging by FIB-SEM. Light and electron microscope images were superimposed. Scale bars, 10 μm. (L) Analysis of FAM134B-mediated ER membrane fragmentation by IEM. U2OS cells were transfected with EGFP-FAM134B. White arrowheads indicated that the ER sheet labeled by immuno-gold was wrapped by autophagosomes. Scale bar, 200 nm. (M and N) FAM134B was colocalized with LAMP2 and LC3. U2OS cells transfected with EGFP-FAM134B were stained for endogenous LAMP2 and LC3. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in N (n = 20 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F2.
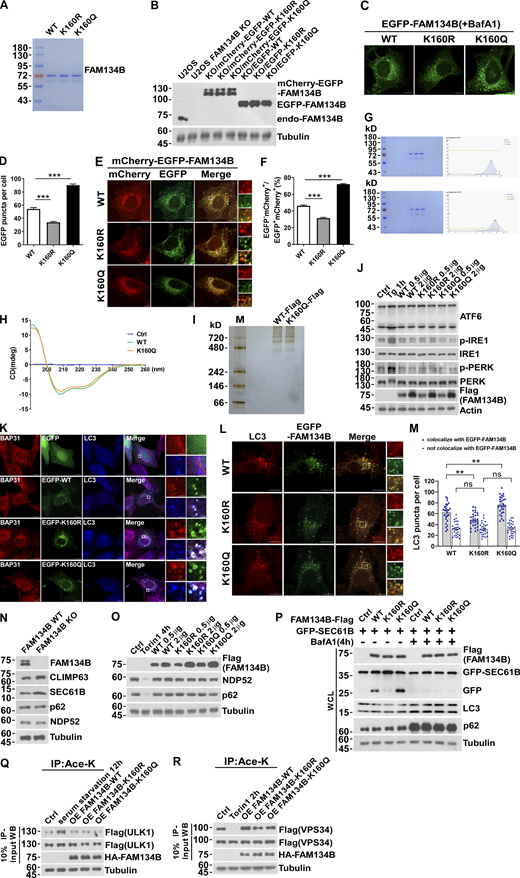
FAM134B acetylation dramatically enhances FAM134B oligomerization to induce ER fragmentation and ER-phagy. (A) Purified recombinant protein FAM134B for in vitro liposome fragmentation assay (Fig. 2 C). (B) Induction of expression of mCherry-EGFP-FAM134B or EGFP-FAM134B to endogenous levels. Construction of mCherry-EGFP-FAM134B or EGFP-FAM134B inducible cell line in FAM134B KO U2OS cells. Cells were induced with 0.5 µg Doxycycline for 24 h. Western blot was performed using anti-FAM134B antibody. (C and D) FAM134B K160Q enhanced ER membrane fragmentation. U2OS cells transiently expressed 0.2 µg EGFP-FAM134B (WT), EGFP-FAM134B (K160R), or EGFP-FAM134B (K160Q). Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in D (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (E and F) FAM134B K160Q enhanced ER-phagy. U2OS cells transiently expressed 0.2 µg mCherry-EGFP-FAM134B (WT), mCherry-EGFP-FAM134B (K160R), or mCherry-EGFP-FAM134B (K160Q). MCherry-positive but EGFP-negative puncta were quantified for in F (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (G) Analysis of the oligomer size of recombinant FAM134B protein (WT and K160Q) using Superose6 SEC. (H) Analysis of the secondary structure of FAM134B protein by circular dichroism assay. FAM134B-WT and FAM134B-K160Q protein at peaks collected by SEC were analyzed by circular dichroism (JASCO, J-1500-150ST). (I) Analysis of the oligomer size of FAM134B (WT and K160Q) using native PAGE. FAM134B-Flag purified from HEK293T was eluted with 3× Flag peptide, then loaded into a linear 3–12% gradient native PAGE gel. (J) ER stress markers including ATF6, p-IRE1, and p-PERK were responsive to Tg treatment but not overexpression of FAM134B. HEK293T cells were treated with 1 μM of Tg for 1 h or transfected with FAM134B-Flag. The indicated proteins were detected by Western blot. (K) FAM134B was colocalized with LC3 and BAP31. U2OS cells transfected with EGFP-FAM134B were stained for endogenous LC3 and BAP31, which was as a marker for ER-derived membrane structure. Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. (L and M) More ER membrane fragments were induced by K160Q, which were colocalized with LC3. The number of LC3 puncta not involved in ER-phagy (not colocalized with FAM134B) were the same in each group. LC3-positive puncta were quantified in M (n = 27 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ns means no significance. (N) ER-phagy substrates instead of macroautophagy substrates were accumulated in FAM134B KO U2OS cells. The indicated proteins were detected by Western blot. (O) Overexpression of FAM134B failed to activate macroautophagy. HEK293T cells treated with Torin1 (250 nM) for 4 h or transfected with FAM134B-Flag were analyzed by Western blot for NDP52 and p62 degradation. (P) Lysosomal cleavage of GFP was analyzed by Western blot in U2OS cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag. Cells were treated with 10 nM BafA1 for 4 h or DMSO. WCL: whole-cell lysate. (Q) ULK1 acetylation increased in response to serum starvation but not overexpression of FAM134B. HEK293T cells were serum starved for 12 h or transfected with HA-FAM134B, which simultaneously expressed ULK1-Flag. IP was performed with an antibody to acetylated lysine. ULK1-Flag was detected by Western blot (WB). The experiments were performed twice. (R) VPS34 acetylation decreased in response to Torin1 treatment but not overexpression of FAM134B. HEK293T cells were treated with Torin1 (250 nM) for 2 h or transfected with HA-FAM134B, which simultaneously expressed VPS34-Flag. IP was performed with an antibody to acetylated lysine. VPS34-Flag was detected by Western blot. The experiments were performed twice. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS2.
FAM134B acetylation dramatically enhances FAM134B oligomerization to induce ER fragmentation and ER-phagy. (A) Purified recombinant protein FAM134B for in vitro liposome fragmentation assay (Fig. 2 C). (B) Induction of expression of mCherry-EGFP-FAM134B or EGFP-FAM134B to endogenous levels. Construction of mCherry-EGFP-FAM134B or EGFP-FAM134B inducible cell line in FAM134B KO U2OS cells. Cells were induced with 0.5 µg Doxycycline for 24 h. Western blot was performed using anti-FAM134B antibody. (C and D) FAM134B K160Q enhanced ER membrane fragmentation. U2OS cells transiently expressed 0.2 µg EGFP-FAM134B (WT), EGFP-FAM134B (K160R), or EGFP-FAM134B (K160Q). Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in D (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (E and F) FAM134B K160Q enhanced ER-phagy. U2OS cells transiently expressed 0.2 µg mCherry-EGFP-FAM134B (WT), mCherry-EGFP-FAM134B (K160R), or mCherry-EGFP-FAM134B (K160Q). MCherry-positive but EGFP-negative puncta were quantified for in F (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (G) Analysis of the oligomer size of recombinant FAM134B protein (WT and K160Q) using Superose6 SEC. (H) Analysis of the secondary structure of FAM134B protein by circular dichroism assay. FAM134B-WT and FAM134B-K160Q protein at peaks collected by SEC were analyzed by circular dichroism (JASCO, J-1500-150ST). (I) Analysis of the oligomer size of FAM134B (WT and K160Q) using native PAGE. FAM134B-Flag purified from HEK293T was eluted with 3× Flag peptide, then loaded into a linear 3–12% gradient native PAGE gel. (J) ER stress markers including ATF6, p-IRE1, and p-PERK were responsive to Tg treatment but not overexpression of FAM134B. HEK293T cells were treated with 1 μM of Tg for 1 h or transfected with FAM134B-Flag. The indicated proteins were detected by Western blot. (K) FAM134B was colocalized with LC3 and BAP31. U2OS cells transfected with EGFP-FAM134B were stained for endogenous LC3 and BAP31, which was as a marker for ER-derived membrane structure. Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. (L and M) More ER membrane fragments were induced by K160Q, which were colocalized with LC3. The number of LC3 puncta not involved in ER-phagy (not colocalized with FAM134B) were the same in each group. LC3-positive puncta were quantified in M (n = 27 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ns means no significance. (N) ER-phagy substrates instead of macroautophagy substrates were accumulated in FAM134B KO U2OS cells. The indicated proteins were detected by Western blot. (O) Overexpression of FAM134B failed to activate macroautophagy. HEK293T cells treated with Torin1 (250 nM) for 4 h or transfected with FAM134B-Flag were analyzed by Western blot for NDP52 and p62 degradation. (P) Lysosomal cleavage of GFP was analyzed by Western blot in U2OS cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag. Cells were treated with 10 nM BafA1 for 4 h or DMSO. WCL: whole-cell lysate. (Q) ULK1 acetylation increased in response to serum starvation but not overexpression of FAM134B. HEK293T cells were serum starved for 12 h or transfected with HA-FAM134B, which simultaneously expressed ULK1-Flag. IP was performed with an antibody to acetylated lysine. ULK1-Flag was detected by Western blot (WB). The experiments were performed twice. (R) VPS34 acetylation decreased in response to Torin1 treatment but not overexpression of FAM134B. HEK293T cells were treated with Torin1 (250 nM) for 2 h or transfected with HA-FAM134B, which simultaneously expressed VPS34-Flag. IP was performed with an antibody to acetylated lysine. VPS34-Flag was detected by Western blot. The experiments were performed twice. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS2.
CBP acetylates FAM134B at K160
We next searched for the enzymes mediating FAM134B acetylation at K160. Protein acetylation is catalyzed by lysine acetyltransferases, which were classified into three families, including GNAT, MYST, and p300/CBP (Allis et al., 2007; Berndsen and Denu, 2008). To identify the acetyltransferase that mediates FAM134B acetylation at K160, we collected some members from these families and conducted a co-IP screen, which resulted in the identification of CBP as the candidate lysine acetyltransferase for FAM134B (Fig. 3 A). Accumulative evidence has demonstrated that the nuclear acetyltransferases can translocate to cytosol to exert autophagy-modulating functions (Cheng et al., 2019; Lee and Finkel, 2009; Lin et al., 2012; Wan et al., 2017; Yi et al., 2012). CBP was initially identified as a crucial transcription regulator in the nucleus (Chrivia et al., 1993). However, previous studies suggested that CBP also plays an important role in the cytoplasm (Ma et al., 2010; Tang et al., 2007). Indeed, we observed that CBP translocated to cytoplasm from nucleus in response to Tg-triggered ER stress (Fig. 3, B–E).
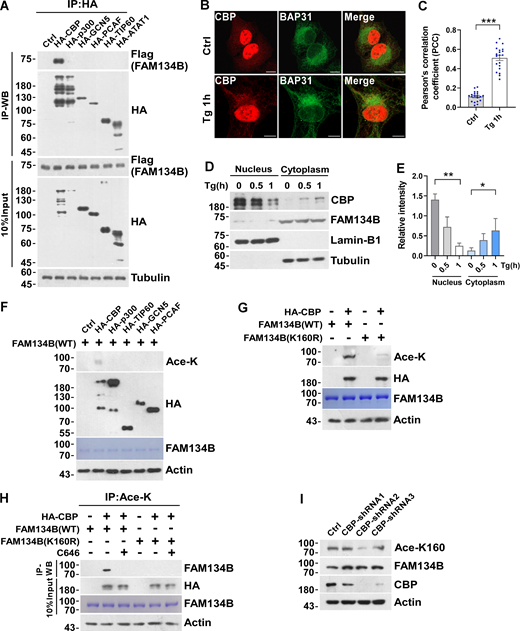
FAM134B is acetylated by CBP at K160. (A) The interaction of FAM134B with lysine acetyltransferase CBP. HA-tagged lysine acetyltransferases CBP, p300, GCN5, PCAF, TIP60, and ATAT1 were expressed individually in HEK293T cells, which simultaneously expressed FAM134B-Flag. IP was performed with anti-HA beads, which was followed by Western blot for FAM134B-Flag. The experiments were performed twice. (B and C) Endogenous CBP was translocated from nucleus to ER labeled by BAP31 upon Tg treatment (1 μM, 1 h). Scale bars, 10 µm. The colocalization was analyzed by Pearson’s correlation coefficient in C (n = 20 cells per group). Data are shown as means ± SEM and analyzed with Student’s t test (two-tailed, unpaired). ***P < 0.001. (D and E) CBP was translocated from nucleus to cytoplasm under Tg treatment. HEK293T cells treated with Tg (1 μM) for 0, 0.5, 1 h were performed with nuclear and cytoplasmic protein extraction assay. Nucleus was labeled with Lamin-B1. Cytoplasm was labeled with Tubulin. The indicated proteins were detected by Western blot. Quantification of CBP is shown in E (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. *P < 0.05, **P < 0.01. (F) FAM134B K160 was acetylated by CBP in vitro. Recombinant protein FAM134B WT purified from E. coli was incubated with lysine acetyltransferase HA-tagged CBP, p300, TIP60, GCN5, PCAF purified from HEK293T cells in HAT buffer at 30°C for 3 h. FAM134B acetylation was analyzed by Western blot. (G) FAM134B K160 was acetylated by CBP in vitro. Recombinant protein FAM134B WT or FAM134B K160R purified from E. coil was incubated with HA-CBP purified from HEK293T cells in HAT buffer at 30°C for 3 h. FAM134B acetylation was analyzed by Western blot. (H) FAM134B K160 acetylation by CBP was abolished by CBP/p300 inhibitor C646 in vitro. Recombinant protein FAM134B WT or FAM134B K160R purified from E. coli was incubated with HA-CBP purified from HEK293T cells in HAT buffer in the presence or absence of C646 at 30°C for 3 h. FAM134B acetylation was analyzed by Western blot. (I) CBP KD downregulated K160 acetylation. HEK293T cells were transfected with control, shRNA1, shRNA2, shRNA3 targeting CBP. K160 acetylation, FAM134B, and CBP were detected by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F3.
FAM134B is acetylated by CBP at K160. (A) The interaction of FAM134B with lysine acetyltransferase CBP. HA-tagged lysine acetyltransferases CBP, p300, GCN5, PCAF, TIP60, and ATAT1 were expressed individually in HEK293T cells, which simultaneously expressed FAM134B-Flag. IP was performed with anti-HA beads, which was followed by Western blot for FAM134B-Flag. The experiments were performed twice. (B and C) Endogenous CBP was translocated from nucleus to ER labeled by BAP31 upon Tg treatment (1 μM, 1 h). Scale bars, 10 µm. The colocalization was analyzed by Pearson’s correlation coefficient in C (n = 20 cells per group). Data are shown as means ± SEM and analyzed with Student’s t test (two-tailed, unpaired). ***P < 0.001. (D and E) CBP was translocated from nucleus to cytoplasm under Tg treatment. HEK293T cells treated with Tg (1 μM) for 0, 0.5, 1 h were performed with nuclear and cytoplasmic protein extraction assay. Nucleus was labeled with Lamin-B1. Cytoplasm was labeled with Tubulin. The indicated proteins were detected by Western blot. Quantification of CBP is shown in E (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. *P < 0.05, **P < 0.01. (F) FAM134B K160 was acetylated by CBP in vitro. Recombinant protein FAM134B WT purified from E. coli was incubated with lysine acetyltransferase HA-tagged CBP, p300, TIP60, GCN5, PCAF purified from HEK293T cells in HAT buffer at 30°C for 3 h. FAM134B acetylation was analyzed by Western blot. (G) FAM134B K160 was acetylated by CBP in vitro. Recombinant protein FAM134B WT or FAM134B K160R purified from E. coil was incubated with HA-CBP purified from HEK293T cells in HAT buffer at 30°C for 3 h. FAM134B acetylation was analyzed by Western blot. (H) FAM134B K160 acetylation by CBP was abolished by CBP/p300 inhibitor C646 in vitro. Recombinant protein FAM134B WT or FAM134B K160R purified from E. coli was incubated with HA-CBP purified from HEK293T cells in HAT buffer in the presence or absence of C646 at 30°C for 3 h. FAM134B acetylation was analyzed by Western blot. (I) CBP KD downregulated K160 acetylation. HEK293T cells were transfected with control, shRNA1, shRNA2, shRNA3 targeting CBP. K160 acetylation, FAM134B, and CBP were detected by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F3.
Next, we immunoprecipitated these recombinant acetyltransferases from HEK293T cells and performed in vitro acetylation assay using recombinant FAM134B as the substrate, we showed that only CBP exhibited acetyltransferase activity toward FAM134B (Fig. 3 F). Furthermore, recombinant protein of FAM134BWT but not that of FAM134BK160R was selectively acetylated by IP-purified CBP (Fig. 3 G). Therefore, CBP is responsible for FAM134B acetylation at K160. This conclusion was further solidified by using a small molecule inhibitor of CBP/p300, C646 (Bowers et al., 2010), which was able to abolish CBP-mediated FAM134B K160 acetylation (Fig. 3 H). Furthermore, shRNA-mediated CBP depletion reduced FAM134B acetylation (Fig. 3 I). Altogether, these results demonstrated that CBP acetylates FAM134B at K160.
CBP is required for optimal ER-phagy under ER-stress conditions
To assess whether CBP regulates ER-phagy through FAM134B, we produced a recombinant protein of FAM134B WT with K160 acetylation by performing a CBP-mediated in vitro acetylation reaction, followed by a liposome fragmentation assay. CBP-mediated FAM134B acetylation enhances membrane fragmentation activity in vitro (Fig. 4, A–C). In U2OS cells either overexpressing EGFP-FAM134B or expressing endogenous level of EGFP-FAM134B on a FAM134B KO background (Fig. S2 B), CBP knockdown (KD) significantly impaired ER membrane fragmentation in both unstimulated and Tg-treated conditions (Fig. 4, D and E, and Fig. S3, A and B), and the defects were rescued by re-expression of CBP. In CBP-depleted cells, Tg-induced FAM134B acetylation at K160 was abolished, which was accompanied by impaired degradation of SEC61B, indicating that CBP is required for Tg-stimulated ER-phagy (Fig. 4, F and G). This conclusion is further supported by the results that CBP KD diminished ER-phagy under FAM134B overexpression conditions (Fig. 4, H–K). Consistently, the overexpression of CBP WT but not CBP mutant was able to significantly stimulate autophagic degradation of FAM134B-labeled ER membrane (Fig. S3, C and D). To further demonstrate the physiological role of CBP-mediated FAM134B acetylation, we set up different assays to show that CBP facilitates the clearance of toxic protein aggregates, which are the substrates of FAM134B-mediated ER-phagy, to maintain ER fitness (Forrester et al., 2019; Fregno et al., 2018; Schultz et al., 2018). As indicated below, a CBP/p300 inhibitor, C646, blocked FAM134B acetylation and significantly diminished FAM134B-mediated degradation of ATZ, collagen I, and an NPC1 mutant (Fig. 4, L–N). These data demonstrate that the CBP–FAM134B axis has important functions in both physiological and pathological situations, and that targeting this pathway may have therapeutic values in alleviating the diseases caused by these toxic protein aggregations. Together, these data suggested that CBP acetylates FAM134B at K160 to promote ER-phagy.
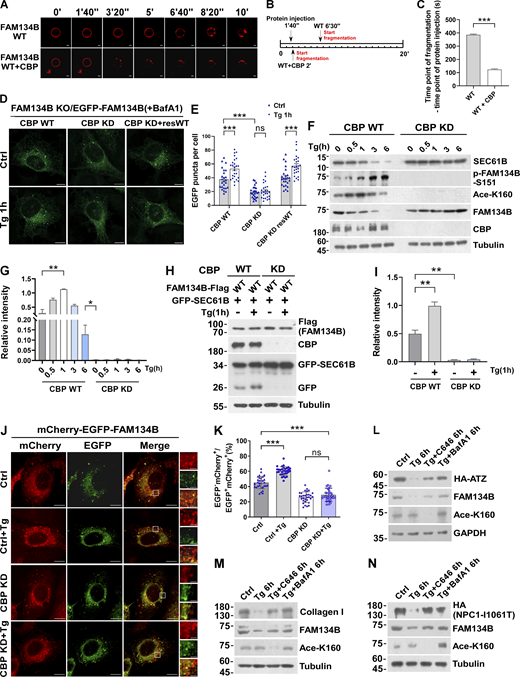
CBP is required for optimal ER-phagy under ER-stress conditions. (A–C) CBP-mediated acetylation for FAM134B is important for liposome fragmentation. Purified recombinant FAM134B protein was acetylated by CBP in vitro, followed by liposome fragmentation assay. After injecting recombinant proteins (25 μg) into the chamber (500 μl), the morphological changes of liposomes were monitored by live imaging for 20 min. The images at different time points as indicated were presented. The time required for liposome fragmentation was quantified in C (n = 3 experimental replicates). Scale bars, 10 μm. Data are shown as means ± SEM and analyzed with Student’s t test (two-tailed, unpaired). ***P < 0.001. (D and E) Overexpression of CBP enhanced FAM134B-mediated ER membrane fragmentation. FAM134B KO U2OS cells were engineered to inducibly express EGFP-FAM134B (WT) at endogenous levels. Cells were treated with Tg (1 μM, 1 h) or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in E (n = 25 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001, ns means no significance. (F and G) ER membrane protein degradation analysis in CBP WT or KD cells. Cells were treated with Tg (1 μM) for 0, 0.5, 1, 3, 6 h. The indicated proteins were detected by Western blot. Quantification of K160 acetylation was shown in G (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. *P < 0.05, **P < 0.01. (H and I) Lysosomal cleavage of GFP was analyzed by Western blot in CBP WT or KD cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag. Cells were treated with Tg (1 μM) for 1 h or DMSO. Quantification of cleavaged GFP is shown in I (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01. (J and K) CBP KD impaired FAM134B-mediated ER-phagy. CBP WT or KD U2OS cells transiently expressing 0.2 µg mCherry-EGFP-FAM134B (WT). MCherry-positive but EGFP-negative puncta were quantified for in K (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001, ns means no significance. (L) Acetylation of FAM134B-K160 by Tg treatment promotes the degradation of ER resident protein ATZ. HEK293T cells transfected with HA-ATZ were treated with Tg (1 µM), C646 (10 µM), or BafA1 (10 nM) for 6 h. The indicated proteins were detected by Western blot. The experiments were performed twice. (M) Acetylation of FAM134B-K160 by Tg treatment promotes the degradation of collagen I. Mouse embryonic fibroblast cells were treated with Tg (1 µM), C646 (10 µM) or BafA1 (10 nM) for 6 h. The indicated proteins were detected by Western blot. The experiments were performed twice. (N) Acetylation of FAM134B-K160 by Tg treatment promotes the degradation of ER resident protein NPC1 mutant (I1061T). HEK293T cells transfected with HA-NPC1(I1061T) were treated with Tg (1 µM), C646 (10 µM), or BafA1 (10 nM) for 6 h. The indicated proteins were detected by Western blot. The experiments were performed twice. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F4.
CBP is required for optimal ER-phagy under ER-stress conditions. (A–C) CBP-mediated acetylation for FAM134B is important for liposome fragmentation. Purified recombinant FAM134B protein was acetylated by CBP in vitro, followed by liposome fragmentation assay. After injecting recombinant proteins (25 μg) into the chamber (500 μl), the morphological changes of liposomes were monitored by live imaging for 20 min. The images at different time points as indicated were presented. The time required for liposome fragmentation was quantified in C (n = 3 experimental replicates). Scale bars, 10 μm. Data are shown as means ± SEM and analyzed with Student’s t test (two-tailed, unpaired). ***P < 0.001. (D and E) Overexpression of CBP enhanced FAM134B-mediated ER membrane fragmentation. FAM134B KO U2OS cells were engineered to inducibly express EGFP-FAM134B (WT) at endogenous levels. Cells were treated with Tg (1 μM, 1 h) or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in E (n = 25 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001, ns means no significance. (F and G) ER membrane protein degradation analysis in CBP WT or KD cells. Cells were treated with Tg (1 μM) for 0, 0.5, 1, 3, 6 h. The indicated proteins were detected by Western blot. Quantification of K160 acetylation was shown in G (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. *P < 0.05, **P < 0.01. (H and I) Lysosomal cleavage of GFP was analyzed by Western blot in CBP WT or KD cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag. Cells were treated with Tg (1 μM) for 1 h or DMSO. Quantification of cleavaged GFP is shown in I (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01. (J and K) CBP KD impaired FAM134B-mediated ER-phagy. CBP WT or KD U2OS cells transiently expressing 0.2 µg mCherry-EGFP-FAM134B (WT). MCherry-positive but EGFP-negative puncta were quantified for in K (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001, ns means no significance. (L) Acetylation of FAM134B-K160 by Tg treatment promotes the degradation of ER resident protein ATZ. HEK293T cells transfected with HA-ATZ were treated with Tg (1 µM), C646 (10 µM), or BafA1 (10 nM) for 6 h. The indicated proteins were detected by Western blot. The experiments were performed twice. (M) Acetylation of FAM134B-K160 by Tg treatment promotes the degradation of collagen I. Mouse embryonic fibroblast cells were treated with Tg (1 µM), C646 (10 µM) or BafA1 (10 nM) for 6 h. The indicated proteins were detected by Western blot. The experiments were performed twice. (N) Acetylation of FAM134B-K160 by Tg treatment promotes the degradation of ER resident protein NPC1 mutant (I1061T). HEK293T cells transfected with HA-NPC1(I1061T) were treated with Tg (1 µM), C646 (10 µM), or BafA1 (10 nM) for 6 h. The indicated proteins were detected by Western blot. The experiments were performed twice. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F4.
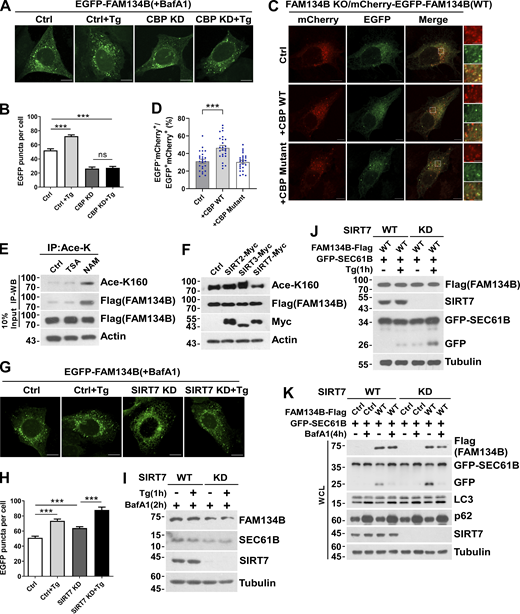
CBP and SIRT7 regulate FAM134B-mediated ER fragmentation and ER-phagy. (A and B) CBP KD attenuated FAM134B-mediated ER fragmentation. CBP WT or KD U2OS cells transiently expressing 0.2 µg EGFP-FAM134B (WT) were treated with Tg (1 μM) for 1 h or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in B (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001, ns means no significance. (C and D) CBP WT but not the catalytic activity-dead mutant enhanced FAM134B-mediated ER-phagy. FAM134B KO U2OS cells were engineered to inducibly express mCherry-EGFP-FAM134B (WT) at endogenous levels. CBP was transfected into cells. MCherry-positive but EGFP-negative puncta were quantified in D (n = 25 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (E) K160 acetylation of FAM134B in FAM134B-Flag stable cell line was increased by being treated with deacetylase inhibitor NAM (5 mM) for 12 h. IP was performed with an antibody to acetylated lysine. K160 acetylation and Flag were detected by Western blot (WB). (F) K160 acetylation accelerated by SIRT7. Myc-tagged Ctrl, SIRT2, SIRT3, or SIRT7 was expressed individually in FAM134B-Flag stable cell line. The indicated proteins were detected by Western blot. (G and H) SIRT7 KD enhanced FAM134B-mediated ER fragmentation. SIRT7 WT or KD U2OS cells transiently expressing 0.2 µg EGFP-FAM134B (WT) were treated with Tg (1 μM) for 1 h or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in H (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (I) Protein levels in G. SIRT7 WT and KD U2OS cells were treated with 1 μM of Tg for 1 h or DMSO. The indicated proteins were detected by Western blot. (J) Lysosomal cleavage of GFP was analyzed by Western blot in SIRT7 WT or KD cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag. Cells were treated with Tg (1 μM) for 1 h or DMSO. (K) Lysosomal cleavage of GFP was analyzed by Western blot in SIRT7 WT or KD cells transfected with 1.5 µg GFP-SEC61B and 0.5 µg FAM134B-Flag. Cells were treated with 10 nM BafA1 for 4 h or DMSO. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS3.
CBP and SIRT7 regulate FAM134B-mediated ER fragmentation and ER-phagy. (A and B) CBP KD attenuated FAM134B-mediated ER fragmentation. CBP WT or KD U2OS cells transiently expressing 0.2 µg EGFP-FAM134B (WT) were treated with Tg (1 μM) for 1 h or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in B (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001, ns means no significance. (C and D) CBP WT but not the catalytic activity-dead mutant enhanced FAM134B-mediated ER-phagy. FAM134B KO U2OS cells were engineered to inducibly express mCherry-EGFP-FAM134B (WT) at endogenous levels. CBP was transfected into cells. MCherry-positive but EGFP-negative puncta were quantified in D (n = 25 cells per group). Scale bars, 10 µm. The scale bars in the magnification boxes are 2 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (E) K160 acetylation of FAM134B in FAM134B-Flag stable cell line was increased by being treated with deacetylase inhibitor NAM (5 mM) for 12 h. IP was performed with an antibody to acetylated lysine. K160 acetylation and Flag were detected by Western blot (WB). (F) K160 acetylation accelerated by SIRT7. Myc-tagged Ctrl, SIRT2, SIRT3, or SIRT7 was expressed individually in FAM134B-Flag stable cell line. The indicated proteins were detected by Western blot. (G and H) SIRT7 KD enhanced FAM134B-mediated ER fragmentation. SIRT7 WT or KD U2OS cells transiently expressing 0.2 µg EGFP-FAM134B (WT) were treated with Tg (1 μM) for 1 h or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in H (n = 30 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (I) Protein levels in G. SIRT7 WT and KD U2OS cells were treated with 1 μM of Tg for 1 h or DMSO. The indicated proteins were detected by Western blot. (J) Lysosomal cleavage of GFP was analyzed by Western blot in SIRT7 WT or KD cells transfected with 1.5 μg GFP-SEC61B and 0.5 μg FAM134B-Flag. Cells were treated with Tg (1 μM) for 1 h or DMSO. (K) Lysosomal cleavage of GFP was analyzed by Western blot in SIRT7 WT or KD cells transfected with 1.5 µg GFP-SEC61B and 0.5 µg FAM134B-Flag. Cells were treated with 10 nM BafA1 for 4 h or DMSO. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS3.
SIRT7 deacetylates FAM134B at K160 to reduce ER-phagy
Acetyltransferases and deacetylases control the homeostasis of protein acetylation, and the latter consists of HDAC and the Sirtuin (SIRT) family deacetylases (Drazic et al., 2016; Gallinari et al., 2007). Therefore, acetylation in general is followed by deacetylation, which is also implied by the alteration of FAM134B acetylation levels (Fig. 1 F and Fig. S1 E). To identify the putative deacetylases for FAM134B, we treated the HEK293T cells with TSA, an inhibitor for HDAC family deacetylases, and NAM, an inhibitor of SIRT family deacetylases, respectively. We observed that NAM but not TSA treatment significantly increased acetylation of endogenous and exogenous FAM134B, which suggested that FAM134B deacetylation was likely mediated by SIRT family members (Fig. 5 A and Fig. S3 E). Next, we conducted a co-IP screen, and we found that FAM134B interacted with SIRT2, SIRT3, and SIRT7 (Fig. 5 B). In the second screen, we observed that the overexpression of SIRT7 but not that of SIRT2 or SIRT3 resulted in a significant decrease of FAM134B acetylation (Fig. 5, C and D; and Fig. S3 F). To further confirm that SIRT7 deacetylates FAM134B, we conducted an in vitro deacetylation assay (Tong et al., 2016). The recombinant protein of FAM134B-Flag that purified from Tg-treated HEK293T cells exhibited increased K160 acetylation, while the addition of SIRT7 in vitro effectively diminished this acetylation. Importantly, the in vitro activity of recombinant SIRT7 was blocked by its inhibitor 97491, which led to the maintenance of K160 acetylation levels (Fig. 5 E). These results established that SIRT7 deacetylates acetyl-K160 of FAM134B. Previous studies showed that SIRT7 has important functions in the nucleus (Barber et al., 2012; Ryu et al., 2014), it is not clear whether this enzyme has a role in the cytoplasm. By performing fractionation assay, we observed that SIRT7 predominantly located in the nucleus; however, under ER-stress conditions, SIRT7 started to accumulate in ER membrane fraction (Fig. 5 F), indicating that SIRT7, similar to CBP, may translocate to ER compartment from nucleus during ER stress. This conclusion was further solidified by the confocal microscopy analysis, which showed increased colocalization of SIRT7 and CLIMP63 under Tg treatment (Fig. 5, G and H). Next, we asked whether SIRT7 regulates ER-phagy. SIRT7 KD increased fragmentation under different EGFP-FAM134B expression conditions (Fig. 5, I and J, and Fig. S3, G–I). Consistently, silencing SIRT7 expression significantly enhanced ER-phagy activity, which was demonstrated by an increased ratio of mCherry+EGFP-ER membrane fragments (Fig. 5, K and L). As expected, SIRT7 depletion enhanced ER-phagy, which was shown by enhanced degradation of exogenous and endogenous ER sheet protein, SEC61B (Fig. 5, M and N, and Fig. S3 J). Additionally, we also showed SIRT7 KD selectively upregulated ER-phagy without altering bulk autophagy process (Fig. S3 K). These results demonstrated that SIRT7 is an inhibitor of ER-phagy by deacetylating FAM134B.
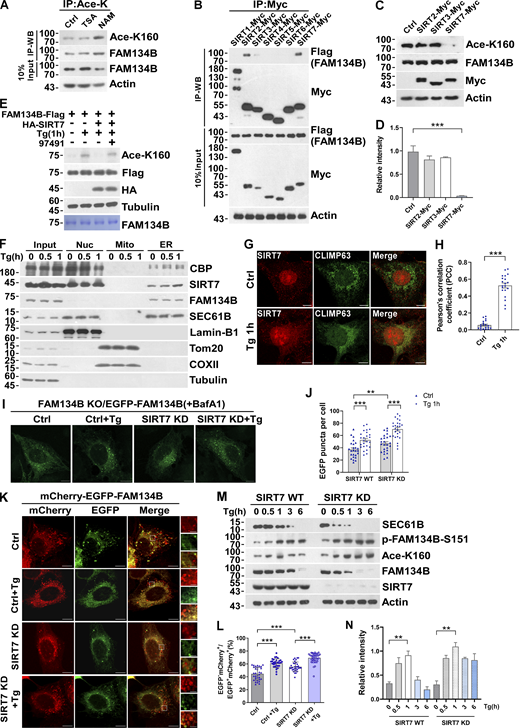
SIRT7 deacetylates FAM134B at K160 to reduce ER-phagy. (A) K160 acetylation of endogenous FAM134B increased by being treated with deacetylase inhibitor NAM (5 mM) for 12 h. IP was performed with an antibody to acetylated lysine. K160 acetylation and FAM134B were detected by Western blot (WB). (B) The interaction of FAM134B with deacetylase SIRT. Myc-tagged SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, or SIRT7 were expressed individually in HEK293T cells, which simultaneously expressed FAM134B-Flag. IP was performed with anti-Myc magnetic beads, which was followed by Western blot for FAM134B-Flag. (C and D) K160 acetylation accelerated by SIRT7. Myc-tagged Ctrl, SIRT2, SIRT3, or SIRT7 was expressed individually in HEK293T. The indicated proteins were detected by Western blot. Quantification of K160 acetylation was shown in D (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (E) FAM134B K160 deacetylation by SIRT7 in vitro. FAM134B-Flag purified from HEK293T with anti-Flag beads were incubated with HA-SIRT7 in deacetylation buffer at 37°C for 2 h. FAM134B acetylation was analyzed by Western blot. The experiments were performed twice. (F) CBP and SIRT7 were translocated from nucleus to ER under Tg treatment. HEK293T cells treated with Tg (1 μM) for 0, 0.5, 1 h were performed with ER isolation assay. ER components were labeled with SEC61B. Nucleus was labeled with Lamin-B1. Mitochondria were labeled with COXII. The indicated proteins were detected by Western blot. The experiments were performed twice. (G and H) Endogenous SIRT7 was translocated from nucleus to ER labeled by CLIMP63 upon Tg treatment (1 μM, 1 h). Scale bars, 10 µm. The colocalization was analyzed by Pearson’s correlation coefficient in H (n = 20 cells per group). Data are shown as means ± SEM and analyzed with Student’s t test (two-tailed, unpaired). ***P < 0.001. (I and J) SIRT7 KD enhanced FAM134B-mediated ER membrane fragmentation. FAM134B KO U2OS cells were engineered to inducibly express EGFP-FAM134B (WT) at endogenous levels. Cells were treated with Tg (1 μM, 1 h) or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in J (n = 25 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001. (K and L) SIRT7 KD accelerated FAM134B-mediated ER-phagy. SIRT7 WT or KD U2OS cells transiently expressing 0.2 µg mCherry-EGFP-FAM134B (WT). MCherry-positive but EGFP-negative puncta were quantified for in L (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (M and N) ER membrane protein degradation analysis in SIRT7 WT or KD cells. Cells were treated with Tg (1 μM) for 0, 0.5, 1, 3, 6 h. The indicated proteins were detected by Western blot. Quantification of K160 acetylation was shown in N (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F5.
SIRT7 deacetylates FAM134B at K160 to reduce ER-phagy. (A) K160 acetylation of endogenous FAM134B increased by being treated with deacetylase inhibitor NAM (5 mM) for 12 h. IP was performed with an antibody to acetylated lysine. K160 acetylation and FAM134B were detected by Western blot (WB). (B) The interaction of FAM134B with deacetylase SIRT. Myc-tagged SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, or SIRT7 were expressed individually in HEK293T cells, which simultaneously expressed FAM134B-Flag. IP was performed with anti-Myc magnetic beads, which was followed by Western blot for FAM134B-Flag. (C and D) K160 acetylation accelerated by SIRT7. Myc-tagged Ctrl, SIRT2, SIRT3, or SIRT7 was expressed individually in HEK293T. The indicated proteins were detected by Western blot. Quantification of K160 acetylation was shown in D (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (E) FAM134B K160 deacetylation by SIRT7 in vitro. FAM134B-Flag purified from HEK293T with anti-Flag beads were incubated with HA-SIRT7 in deacetylation buffer at 37°C for 2 h. FAM134B acetylation was analyzed by Western blot. The experiments were performed twice. (F) CBP and SIRT7 were translocated from nucleus to ER under Tg treatment. HEK293T cells treated with Tg (1 μM) for 0, 0.5, 1 h were performed with ER isolation assay. ER components were labeled with SEC61B. Nucleus was labeled with Lamin-B1. Mitochondria were labeled with COXII. The indicated proteins were detected by Western blot. The experiments were performed twice. (G and H) Endogenous SIRT7 was translocated from nucleus to ER labeled by CLIMP63 upon Tg treatment (1 μM, 1 h). Scale bars, 10 µm. The colocalization was analyzed by Pearson’s correlation coefficient in H (n = 20 cells per group). Data are shown as means ± SEM and analyzed with Student’s t test (two-tailed, unpaired). ***P < 0.001. (I and J) SIRT7 KD enhanced FAM134B-mediated ER membrane fragmentation. FAM134B KO U2OS cells were engineered to inducibly express EGFP-FAM134B (WT) at endogenous levels. Cells were treated with Tg (1 μM, 1 h) or DMSO. Lysosomal degradation of EGFP-FAM134B was blocked by 10 nM BafA1 for 2 h. EGFP-positive puncta were quantified in J (n = 25 cells per group). Scale bars, 10 µm. Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001. (K and L) SIRT7 KD accelerated FAM134B-mediated ER-phagy. SIRT7 WT or KD U2OS cells transiently expressing 0.2 µg mCherry-EGFP-FAM134B (WT). MCherry-positive but EGFP-negative puncta were quantified for in L (n = 30 cells per group). Scale bars, 10 µm. The scale bars in the magnification. Data are shown as means ± SEM and analyzed with one-way ANOVA. ***P < 0.001. (M and N) ER membrane protein degradation analysis in SIRT7 WT or KD cells. Cells were treated with Tg (1 μM) for 0, 0.5, 1, 3, 6 h. The indicated proteins were detected by Western blot. Quantification of K160 acetylation was shown in N (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F5.
CBP-mediated acetylation facilitates FAM134B phosphorylation by CAMKII to further boost ER-phagy
We recently revealed that CAMKII, in response to ER stress, phosphorylates FAM134B at serine 151 to facilitate oligomerization, ER membrane fragmentation, and ER-phagy (Jiang et al., 2020). Therefore, we asked whether there is a functional interplay between CBP-mediated acetylation and CAMKII-conferred phosphorylation. We observed the mutant FAM134BK160R mimicking deacetylation exhibited reduced Ser151 phosphorylation (Fig. 6 A). In contrast, the mutant FAM134BK160Q imitating permanent acetylation showed enhanced Ser151 phosphorylation. Consistently, lowering CBP expression levels by shRNA KD (Fig. 6, B and C) or reducing CBP enzymatic activity using its inhibitor C646 (Fig. 6, D and E) were able to abolish both K160 acetylation and Ser151 phosphorylation. Importantly, CBP WT but not the enzymatic-dead mutant was able to rescue the defects of acetylation and phosphorylation caused by CBP depletion (Fig. 6 F). These results suggested that K160 acetylation may facilitate the interaction between FAM134B and CAMKII, which enabled Ser151 phosphorylation. Indeed, the co-IP experiment showed that the mutant mimicking K160 acetylation (FAM134B K160Q) increased the interaction of FAM134B with CAMKII, while the mutant FAM134BK160R mimicking deacetylation exhibited reduced CAMKII binding (Fig. 6, G and H). Therefore, FAM134B K160 acetylation enhanced the association of CAMKII with FAM134B to facilitate FAM134B S151 phosphorylation. However, co-IP experiments indicated that FAM134B S151 phosphorylation failed to affect FAM134B interaction with CBP or SIRT7 (Fig. S4, A and B). Accordingly, the FAM134B phosphorylation status had no significant effects on FAM134B acetylation, as the acetylation levels were not unaltered among the FAM134B WT and phosphor-mimetic mutants (Fig. S4, C and D). Furthermore, CAMKII KD reduced FAM134B phosphorylation without altering its acetylation (Fig. S4 E). Therefore, CAMKII-mediated phosphorylation occurs after CBP-mediated acetylation. We further investigated the dynamics of CBP and SIRT7 during FAM134B activation, and we observed that under Tg-treated conditions, the acetylation levels of FAM134B K160 peaked around 1 h after treatment (Fig. 6 I), which expectedly lagged behind the timing of the maximal interaction between CBP and FAM134B (Fig. 6 J). The binding between SIRT7 and FAM134B that culminated around 1 h after treatment (Fig. 6 K) was about 30 min after the climax of CBP and FAM134B interaction. Together, the dynamic acetylation and phosphorylation of FAM134B within its RTND are designed for optimal activation of ER-phagy to cope with ER stress (Fig. 6 L).
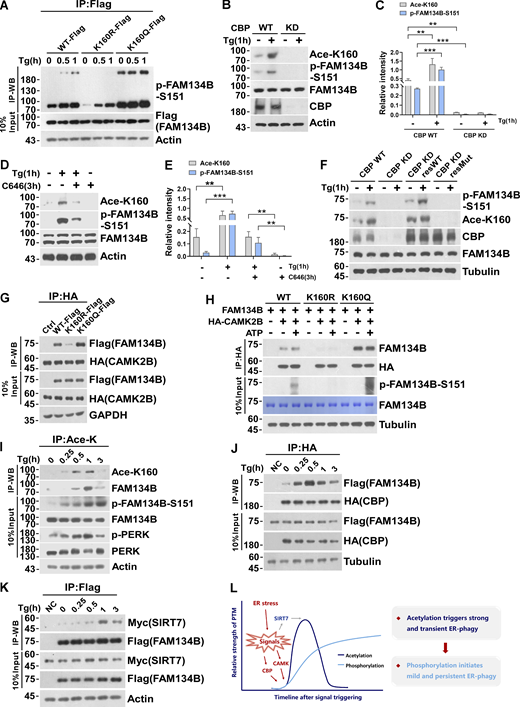
CBP-mediated acetylation facilitates FAM134B phosphorylation by CAMKII to further boost ER-phagy. (A) Mimicking permanent acetylation by K160Q enhanced S151 phosphorylation. HEK293T cells transfected with FAM134B (WT)-Flag, FAM134B (K160R)-Flag and FAM134B (K160Q)-Flag were treated with Tg (1 μM) for 0, 0.5, 1 h. IP was performed with anti-Flag beads, which was followed by Western blot (WB) for S151 phosphorylation. (B–E) CBP inhibition by small compounds or shRNA KD significantly reduced FAM134B phosphorylation at S151. Cells were treated with Tg (1 μM) for 1 h or C646 (10 μM) for 3 h. Cells were collected and analyzed for proteins as indicated by Western blot. Quantification of K160 acetylation and S151 phosphorylation is shown in C and E (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001. (F) CBP WT instead of catalytic activity-dead mutant rescued FAM134B K160 acetylation in CBP KD cells. The indicated proteins were detected by Western blot. (G) Mimicking permanent acetylation by K160Q facilitated the interaction with CAMK2B. FAM134B (WT)-Flag, FAM134B (K160R)-Flag, and FAM134B (K160Q)-Flag were expressed individually in HEK293T cells, which simultaneously expressed HA-CAMK2B. IP was performed with anti-HA beads, which was followed by Western blot for FAM134B-Flag. The experiments were performed twice. (H) FAM134B K160 acetylation promotes S151 phosphorylation. Purified recombinant FAM134B proteins were pulled down by HA-CAMK2B which purified from HEK293T cells. In vitro kinase assay was performed. The mixture was incubated with 400 µM ATP and 1× CAMK2B kinase buffer at 30°C for 10 min. The indicated proteins were detected by Western blot. (I) Dynamic K160 acetylation and S151 phosphorylation of FAM134B. HEK293T cells were treated with Tg (1 μM) for different time points as indicated and BafA1 (10 nM) to block degradation. IP was performed with an antibody to acetylated lysine. The indicated proteins were detected by Western blot. (J) Measurement of interaction between HA-CBP and FAM134B-Flag by Western blot in HEK293T cells treated with 1 μM of Tg for different time points or DMSO as indicated in the presence of BafA1 (10 nM). The experiments were performed twice. (K) Measurement of interaction between SIRT7-Myc and FAM134B-Flag by Western blot in HEK293T cells treated with 1 μM of Tg for different time points or DMSO as indicated in the presence of BafA1 (10 nM). The experiments were performed twice. (L) Schematic representation of dynamic acetylation and phosphorylation of FAM134B to cope with ER stress. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F6.
CBP-mediated acetylation facilitates FAM134B phosphorylation by CAMKII to further boost ER-phagy. (A) Mimicking permanent acetylation by K160Q enhanced S151 phosphorylation. HEK293T cells transfected with FAM134B (WT)-Flag, FAM134B (K160R)-Flag and FAM134B (K160Q)-Flag were treated with Tg (1 μM) for 0, 0.5, 1 h. IP was performed with anti-Flag beads, which was followed by Western blot (WB) for S151 phosphorylation. (B–E) CBP inhibition by small compounds or shRNA KD significantly reduced FAM134B phosphorylation at S151. Cells were treated with Tg (1 μM) for 1 h or C646 (10 μM) for 3 h. Cells were collected and analyzed for proteins as indicated by Western blot. Quantification of K160 acetylation and S151 phosphorylation is shown in C and E (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001. (F) CBP WT instead of catalytic activity-dead mutant rescued FAM134B K160 acetylation in CBP KD cells. The indicated proteins were detected by Western blot. (G) Mimicking permanent acetylation by K160Q facilitated the interaction with CAMK2B. FAM134B (WT)-Flag, FAM134B (K160R)-Flag, and FAM134B (K160Q)-Flag were expressed individually in HEK293T cells, which simultaneously expressed HA-CAMK2B. IP was performed with anti-HA beads, which was followed by Western blot for FAM134B-Flag. The experiments were performed twice. (H) FAM134B K160 acetylation promotes S151 phosphorylation. Purified recombinant FAM134B proteins were pulled down by HA-CAMK2B which purified from HEK293T cells. In vitro kinase assay was performed. The mixture was incubated with 400 µM ATP and 1× CAMK2B kinase buffer at 30°C for 10 min. The indicated proteins were detected by Western blot. (I) Dynamic K160 acetylation and S151 phosphorylation of FAM134B. HEK293T cells were treated with Tg (1 μM) for different time points as indicated and BafA1 (10 nM) to block degradation. IP was performed with an antibody to acetylated lysine. The indicated proteins were detected by Western blot. (J) Measurement of interaction between HA-CBP and FAM134B-Flag by Western blot in HEK293T cells treated with 1 μM of Tg for different time points or DMSO as indicated in the presence of BafA1 (10 nM). The experiments were performed twice. (K) Measurement of interaction between SIRT7-Myc and FAM134B-Flag by Western blot in HEK293T cells treated with 1 μM of Tg for different time points or DMSO as indicated in the presence of BafA1 (10 nM). The experiments were performed twice. (L) Schematic representation of dynamic acetylation and phosphorylation of FAM134B to cope with ER stress. Molecular weight measurements are in kD. Source data are available for this figure: SourceData F6.
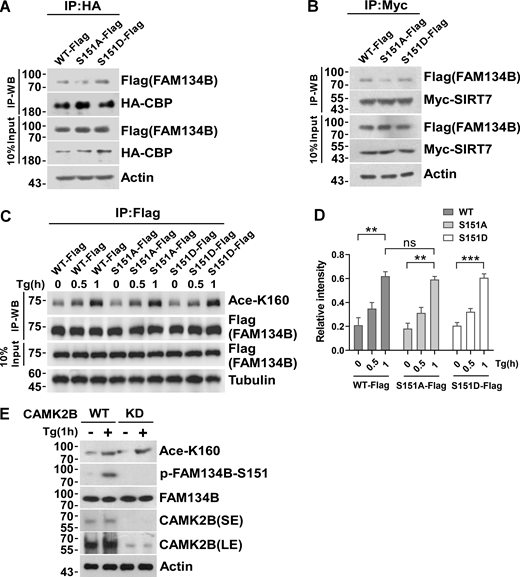
Inhibition of S151 phosphorylation does not affect K160 acetylation. (A) Measurement of interaction between HA-CBP and FAM134B (WT)-Flag, FAM134B (S151A)-Flag, and FAM134B (S151D)-Flag. IP was performed with anti-HA beads, which was followed by Western blot (WB) for FAM134B-Flag. (B) Measurement of interaction between Myc-SIRT7 and FAM134B (WT)-Flag, FAM134B (S151A)-Flag, and FAM134B (S151D)-Flag. IP was performed with anti-Myc magnetic beads, which was followed by Western blot for FAM134B-Flag. (C and D) K160 acetylation was not affected by expression of S151D mimicking permanent phosphorylation. HEK293T cells transfected with FAM134B (WT)-Flag, FAM134B (S151A)-Flag, and FAM134B (S151D)-Flag were treated with Tg (1 μM) for 0, 0.5, 1 h. IP was performed with anti-Flag beads, which was followed by Western blot for K160 acetylation. Quantification of K160 acetylation was shown in D (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001, ns means no significance. (E) K160 acetylation was not affected by CAMK2B KD. CAMK2B WT or KD cells were treated with Tg (1 μM) for 1 h or DMSO. Cells were collected and analyzed for proteins as indicated by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS4.
Inhibition of S151 phosphorylation does not affect K160 acetylation. (A) Measurement of interaction between HA-CBP and FAM134B (WT)-Flag, FAM134B (S151A)-Flag, and FAM134B (S151D)-Flag. IP was performed with anti-HA beads, which was followed by Western blot (WB) for FAM134B-Flag. (B) Measurement of interaction between Myc-SIRT7 and FAM134B (WT)-Flag, FAM134B (S151A)-Flag, and FAM134B (S151D)-Flag. IP was performed with anti-Myc magnetic beads, which was followed by Western blot for FAM134B-Flag. (C and D) K160 acetylation was not affected by expression of S151D mimicking permanent phosphorylation. HEK293T cells transfected with FAM134B (WT)-Flag, FAM134B (S151A)-Flag, and FAM134B (S151D)-Flag were treated with Tg (1 μM) for 0, 0.5, 1 h. IP was performed with anti-Flag beads, which was followed by Western blot for K160 acetylation. Quantification of K160 acetylation was shown in D (n = 3 experimental replicates). Data are shown as means ± SEM and analyzed with one-way ANOVA. **P < 0.01, ***P < 0.001, ns means no significance. (E) K160 acetylation was not affected by CAMK2B KD. CAMK2B WT or KD cells were treated with Tg (1 μM) for 1 h or DMSO. Cells were collected and analyzed for proteins as indicated by Western blot. Molecular weight measurements are in kD. Source data are available for this figure: SourceData FS4.
Discussion
Non-histone protein acetylation emerges as an essential regulatory mechanism for many cellular processes (Narita et al., 2019). Our study provides one piece of evidence that acetylation of cargo receptors regulates selective autophagy. It would be interesting to investigate whether this mechanism is also involved in other selective autophagy pathways. Notably, previously characterized PTM of cargo receptors predominantly affects their interaction affinity with Atg8/LC3 family members. While we show that these modifications regulate FAM134B oligomerization, thereby fragmenting ER subdomains into suitable size to facilitate autophagosomal engulfment. Therefore, our work demonstrates an alternative mechanism involved in cargo receptors’ biochemical and functional modulation during selective autophagy. Because the structure of FAM134B was not solved, it remains unclear how the acetylation and phosphorylation regulate its self-interaction. One possible explanation is that the negative charged by phosphorylation or the neutralization of positively charged lysine residues through acetylation that might facilitate RTND-mediated interactions.
In this study, we showed that sequential PTM of FAM134B is implicated in regulating the receptor’s activity. The reason behind these observations could be that the cells need to initiate more robust ER-phagy activity driven by acetylation in the early stage of ER stress. Indeed, the mutant FAM134B K160Q exhibited the same level of ER-phagy activity as FAM134B G216R (Fig. 2 B), which is a gain-of-function variant from an HSAN-II patient (Jiang et al., 2020). Therefore, acetylation-mediated ER-phagy might be too harsh to the cell. This inevitably necessitates a switch from an acute and intense mode of ER-phagy to a mild one controlled by phosphorylation, which induces less intensified self-interaction (Fig. 2 B) and modest increase of ER-phagy activity (Fig. 2 J). Intertwined with the sequential acetylation and phosphorylation is SIRT7-mediated K160 deacetylation, which can slow down or inhibit ER-phagy in unstimulated and stimulated conditions. We show that both CBP and SIRT7 translocate from the nucleus to the cytosol upon ER stress. However, due to technical limitations, it remains unclear how their respective catalytic activities are coordinated on the FAM134B-anchored ER subdomains. However, the value of our findings is that we identify previously uncharacterized functional connections among CBP, SIRT7, CAMKII, and FAM134B in ER-stress response. It is unclear how the dynamic recruitment of SIRT7 and CBP is regulated during ER stress. It remains elusive how SIRT7 and CBP cooperate to regulate the acetylation and deacetylation circuit of FAM134B. We observed the dynamic interaction between FAM134B and CBP or SIRT7, but it is not clear how the dynamic interaction is regulated. Furthermore, how CBP and SIRT7 coordinate their function in different subcellular compartments remains unclear. It is noteworthy that the CBP, SIRT7, and FAM134B regulatory circuit may be implicated in colorectal cancer. FAM134B was reported to be a candidate tumor suppressor in colorectal cancer (Islam et al., 2016; Kasem et al., 2014a; Kasem et al., 2014b) and high CBP expression levels (staining index > 16.6) indicated long-term survival (Ishihama et al., 2007). On the contrary, SIRT7 crucially contributes to the development and progression of human colorectal cancer and functions as a valuable marker of colorectal cancer prognosis (Yu et al., 2014). In the future, it would be necessary to investigate whether this pathway can be targeted for colorectal cancer diagnosis and therapy. A previous study reported that acetylation of RTN-1C in the nucleus regulates its pro-apoptosis activity in neuroectodermal tumors (Fazi et al., 2009). It would be interesting to investigate whether the regulatory circuit comprising CBP and SIRT7 regulates FAM134B function in the nucleus in an ER-phagy–independent fashion as well.
Materials and methods
Cell culture
U2OS, HEK293T cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, and 1% penicillin-streptomycin in a humidified incubator at 37°C with 5% CO2. FAM134B-Flag stable cell line, CBP KD cell line, and SIRT7 KD cell line were constructed in our lab.
Antibodies
Anti-β-Actin (M1210-2; Huaan Hangzhou); Anti-Flag-Tag-HRP-direct (M185-7; MBL); Anti-PERK (3192; Cell Signaling); Anti-p-PERK (3179; Cell Signaling); Anti-HA-Tag-HRP-direct (M180-7; MBL); Anti-GFP (11814460001; Roche); Anti-GFP (ab6556; Abcam); Anti-β-Tubulin (M1305-2; Huaan Hangzhou); Anti-Ac-lysine (32268; Santa Cruz); Anti-CBP (7389S; Cell Signaling); Anti-Acetylated lysine (9441S; Cell Signaling); Anti-Lamin-B1 (12987-1-AP; Proteintech); Anti-SEC61B (15087-1-AP; Proteintech); Anti-Myc (R1208-1; Huaan Hangzhou); Anti-GAPDH (60004-1; Proteintech); Anti-SIRT7 (365344; Santa Cruz); Anti-MTCO2(COXII; 55070-1-AP; Proteintech); Anti-Tom20 (42406S; CST); Anti-Collagen I (14695-1-AP; Proteintech); Anti-CAMKII (4436S; Cell Signaling); Anti-NDP52 (ab68588; Abcam); Anti-p62 (66184-1; Proteintech); Anti-CKAP4(CLIMP63; 16686-1-AP; Proteintech); Anti-BAP31 (393810; Santa Cruz); Anti-LC3 (PM036; MBL); Anti-LAMP2 (sc-18822; Santa Cruz); Anti-Flag (F1840; Sigma-Aldrich); Alexa Fluor 488 (A11001; Thermo Fisher Scientific); Alexa Fluor 546 (A11003; Thermo Fisher Scientific); Alexa Fluor 594 (A11005; Thermo Fisher Scientific); Alexa Fluor 405 (A31556; Thermo Fisher Scientific); ATF6, p-IRE1, and IRE1 were gifts from Xiaojian Wang (Zhejiang University, Hangzhou, China). All antibodies used in this study are listed in Table S1.
Antibody preparation and purification
FAM134B rabbit polyclonal antibody was raised by DJ Biotech, and p-FAM134B (Ser151) rabbit polyclonal antibody was raised by Wenyuange Biotech (Jiang et al., 2020). Acetylated-FAM134B (K160) rabbit polyclonal antibody was generated by immunizing four rabbits using the synthesized acetyl-peptide (C)WEVINSK(Ac)PDER-NH2 (amino acids 154–164; NP_001030023.1) and the control peptide (C)WEVINSKPDER-NH2 (Youke Biotech). The target peptide was coupled with KLH as an immune antigen (1 mg/ml). The target peptide/control polypeptide was, respectively, coupled with BSA as the detection antigen. On the first day, 2 ml of each immune antigen was added into 2 ml Freund’s complete adjuvant and emulsified and four New Zealand white rabbits were immunized for each antigen. The immunization was repeatedly conducted on the 15th/29th/43rd days, respectively. On the 53rd day, carotid artery blood was taken and centrifuged for 30 min (4°C, 10,000 rpm), the supernatant was collected. The target peptide or the control peptide was conjugated to the activated Sulfolink resin, respectively, to prepare the affinity columns. The liquid was first passed through the control peptide column to remove the antibodies against unacetylated antigen, and the flowthrough solution was collected and was passed through the target peptide column, and the elution was collected, which was the antibody specific for K160 acetylation. After dialysis, protein concentration was detected by UV absorbance, and the antibody titer was determined by ELISA. The specificity of the antibody was further verified via dot blot and Western blot.
Chemicals
TSA was purchased from Selleck. NAM was purchased from ALADDIN. C646 was purchased from Selleck. Tg was purchased from J&K Scientific. Torin1 was purchased from Selleck. BafA1 was purchased from Selleck. Acetyl-coenzyme A was purchased from Sigma-Aldrich. SIRT7 inhibitor 97491 was purchased from MCE. All chemicals used in this study are listed in Table S1.
Stable cell lines construction
FAM134B-Flag/293FRT stable cell line was constructed by using Flp-In System from Invitrogen. POG44 and pcDNA5/FRT-FAM134B vectors were co-transfected into Flp-In 293FRT cells and selected with 75 μg/ml of Hygromycin B (V900372; Sigma-Aldrich).
Inducible stable cell lines construction
A tet-on system was used in FAM134B−/− U2OS cell line (Jiang et al., 2020). Cells were co-transfected with HP216 vector and HP138-FAM134B–related vectors. Cell lines were obtained through 1 µg/ml puromycin selection.
shRNA KD
Annealed oligonucleotides were cloned into pLKO-shRNA-Puro using AgeI and EcoRI. Target sequences are 5′-CCCGATAACTTTGTGATGTTT-3′ (CBP shRNA1), 5′-TAACTCTGGCCATAGCTTAAT-3′ (CBP shRNA2), 5′-CCGTTTACCATGAGATCCTTA-3′ (CBP shRNA3), 5′-GCCTGAAGGTTCTAAAGAAGT-3′ (SIRT7 shRNA). The KD stable cell lines were obtained by lentivirus infection and selected with 1 µg/ml puromycin. Recombinant lentiviruses were produced following the lentiviral packaging protocol.
IP and Western blot
Cells were lysed in TAP buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 10 nM MG132, Protease cocktail, Phosphatase cocktail) and incubated on ice for 30 min. The cell lysates were centrifuged at 14,000 rpm for 10 min. The supernatant was incubated with antibody-conjugated beads and rotated for 4 h at 4°C. After incubation, the beads were washed 3 times with TAP buffer and eluted with 1× SDS loading buffer. Samples were separated with SDS-PAGE, transferred to Polyvinylidene Fluoride Membrane and probed with the corresponding antibody. For each representative figure which was shown, at least two different experiments were performed. Protein A/G agarose resin was purchased from Yeasen (36403ES03). Anti-Flag Affinity Gel (B23102), Anti-HA Affinity Gel (B23302), and Anti-Myc magnetic beads (B26302) were purchased from Bimake.
Immunofluorescence
U2OS grown on coverslips were transfected with EGFP-FAM134B, mcherry-EGFP-FAM134B, or FAM134B-Flag plasmids, then fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and permeabilized with 0.2% Triton X-100 (T0694; Sangon Biotech) in PBS for 20 min. Cells were blocked in a blocking buffer containing 5% BSA and 0.1% Triton X-100 for 1 h at room temperature. Cells were stained with primary antibodies diluted in blocking buffer overnight at 4°C and washed with PBS three times. Cells were then stained with Alexa Fluor–conjugated secondary antibody diluted in blocking buffer for 1 h at room temperature. Images were acquired using a laser scanning confocal microscope (Zeiss LSM 800) equipped with plan-apochromat 63×/1.4-NA oil-immersion objective, and photomultiplier tube detector. ImageJ was used to quantify the number of puncta, fluorescence intensity, and calculate the Pearson correlation coefficient. Unless otherwise stated, images were processed using thresholding for quantification. The number of cells included for analysis for each condition and the number of independent experiments are indicated in the figure legends accordingly.
Recombinant protein purification
GST-TEV-FAM134B was expressed in Escherichia coli BL21 (DE3) cells by induction with 0.25 mM IPTG at 16°C for 24 h. Cells were lysed by sonication in lysis buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1 mM EDTA, 0.5% Triton-X100) and centrifuged at 14,000 rpm for 20 min. Proteins were purified using GST-Sepharose resin (GE Healthcare) and then eluted by incubating with 10 µg/ml TEV protease in TEV protease cleavage buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 0.1% NP-40; 1 mM EDTA) at 4°C overnight to release from the resin. Purified untagged recombinant proteins were further fractionated by using a Mono-Q column. Proteins were quantified by the Bradford method and analyzed by SDS-PAGE and Coomassie-blue staining.
In vitro acetylation assay
CBP was purified from HEK293T with anti-HA antibody-conjugated beads. 50 µl reaction mix contains 38 µl 1× HAT buffer (20 mM Tris HCl, pH 8.0; 250 mM KCl; 5 mM DTT; 0.5 mM EDTA; 25% Glycerol), 1 mg/ml acetyl-CoA, and 10 µg purified recombinant FAM134B protein. After incubation at 30°C for 3 h, the reaction was terminated by the addition of 5× SDS loading buffer and subjected to SDS-PAGE analysis.
In vitro deacetylation assay
In vitro deacetylation assay was performed as followings (Tong et al., 2016). FAM134B-Flag was purified from HEK293T with anti-Flag beads, and eluted from anti-Flag beads with 3× Flag peptide (F4799; Sigma-Aldrich). HA-SIRT7 was purified from HEK293T with anti-HA antibody-conjugated beads. Eluted FAM134B-Flag was incubated with HA-SIRT7 in 50 µl deacetylation buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 2 mM NAD+, 1 mM DTT). After incubated at 37°C for 2 h, the reaction was terminated by addition of 5× SDS loading buffer. The samples were then subjected to SDS-PAGE.
Liposome assay
One glass slide was coated with 200 µl 1% agarose overnight, and the other glass slide was coated with 7 µg streptavidin protein. Next day, prepare lipid solution 100 µl, including 40 µl chloroform, 30 µl cholesterol, 15 µl phosphatidylcholine, 12.5 µl phosphatidylethanolamine (PE), 1.5 µl 16:0–18:1 phosphatidyiserine, 0.5 µl rhodamine-PE. Use a glass syringe (HAMILTON), add the lipid solution to the agarose-coated glass slide, and allow the sample to dry under Argon gas for 3 h. 500 µl 1× PBS was gently added to agarose glass to suspend liposomes. The liposomes were added to the streptavidin-coated glass slide and anchored to glass slides through biotin-streptavidin interaction for incubation 20 min. The recombinant protein was injected into the chamber, and the morphological change of liposomes was observed using a laser scanning confocal microscope (Olympus IX2). For each representative image that was shown, three different experiments were performed to ensure reproducibility.
Nuclear and cytoplasmic protein extraction assay
Nuclear and cytoplasmic protein extraction assay was performed as the protocol from Beyotime (Cat#P0027). Cells were collected and washed with PBS. Add 200 µl of PMSF-added cytoplasmic protein extraction reagent A per 20 µl of cell pellet. Vortex for 5 s until the cell pellet was wholly suspended and dispersed, then incubated on ice for 10–15 min. Add cytoplasmic protein extraction reagent B 10 µl per 20 µl of cell pellet, vortex for 5 s. After being incubated on ice for 1 min, the Eppendorf tube was vortexed for 5 s again and centrifuged at 12,000–16,000 g 4°C for 5 min. The supernatant contained cytoplasmic proteins. For the precipitation, add 50 µl of PMSF-added nuclear protein extraction reagent. Vortex 15–30 s until the cell pellet was suspended entirely and dispersed. Then put it back on the ice, vortex for 15–30 s every 1–2 min for 30 min, and centrifuge at 12,000–16,000 g 4°C for 10 min. The supernatant contained nuclear proteins.
ER isolation assay
The ER isolation assay was performed as the protocol from Endoplasmic Reticulum Isolation Kit (ER0100; Sigma-Aldrich). Detached cells were collected at 600 g for 5 min; wash the cells with 10 volumes of PBS and centrifuge to obtain the packed cell volume (PCV). PCV was measured and suspended in a volume of 1× Hypotonic Extraction Buffer equivalent to 3× the PCV for 20 min at 4°C to allow the cells to swell. The cells were collected by centrifugation. The “new” PCV was measured and resuspended in 1× Isotonic Extraction Buffer equivalent to 2× the new PCV. The cells were then treated with 10 strokes of the Dounce homogenizer and then proceeded to the differential centrifugation steps. Centrifuge the homogenate at 1,000 g for 10 min at 4°C. Carefully remove the thin floating lipid layer by aspiration, being careful not to aspirate the post-nuclear supernatant. Transfer the supernatant to another centrifuge tube using a pipette and discard the pellet. Centrifuge at 12,000 g for 15 min at 4°C. Carefully remove the thin floating lipid layer by aspiration, aspirating the post mitochondrial supernatant. Transfer the supernatant to another tube using a pipette and discard the pellet. This supernatant fraction, the post mitochondrial fraction, is the source for ER.
EGFP-cleavage assay
1.5 µg EGFP-SEC61B and 0.5 µg FAM134B-Flag plasmids were transiently co-transfected into U2OS cells. The cell lysates were immunoblotted with antibodies against Flag, GFP, and β-tubulin.
Size-exclusion chromatography (SEC)
SEC with Superose 6 10/300 GL was performed at 4°C using an AKTA PURE according to the Handbooks from GE Healthcare Life Sciences (https://www.gehealthcare.com/products/life-sciences). Equilibrating the column with two column volumes of buffer (20 mM Hepes, pH 7.5, 100 mM NaCl, filtration, and ultrasonic defoaming) at a flow rate of 0.25 ml/min 500 µg/500 µl FAM134B protein was injected into the column. Samples were collected and subjected to SDS-PAGE analysis and Coomassie blue staining. The FAM134B oligomer size was deduced from the chromatograms of six standard proteins on Superose 6 10/300 GL.
Native PAGE
Cells transfected with FAM134B-Flag were lysed in TAP buffer (mentioned above) and incubated with anti-Flag beads, then eluted with 3× Flag peptide (F4799; Sigma-Aldrich). A mixture containing 20 µl protein + 2 µl 50% glycerol + 3 µl 0.1% Coomassie blue G-250 was incubated for 15 min at room temperature and loaded into a linear 3–12% gradient native PAGE gel (Life Technologies) running in blue cathode/anode buffer for 2 h at 150 V in an ice bath. The gel was stained using the Pillsin silver staining kit.
IEM
U2OS cells overexpressed with EGFP-FAM134B were fixed with IEM fixing buffer (4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer [PB]) overnight at 4°C. The fixed cells were washed three times with 0.1 M PB, and terminated with 50 mM glycine, then followed by permeabilization for 15 min with 0.1% Triton X-100 and 5% fetal bovine serum in 1× PBS. Cells were incubated with anti-GFP antibody (ab6556, 1:400; Abcam) overnight at 4°C, followed by nanogold-labeled Fab’goat anti-mouse IgG(H + L) antibody (34C918, 1:50; Nanoprobes) overnight at 4°C. Cells were silver enhanced for 2 min and then fixed with 1% aqueous osmium tetraoxide for 40 min. Cells were dyed with 2% uranyl acetate for 40 min and dehydrated through graded ethanol (50–100%) and 100% acetone twice each for 15–20 min. Samples were embedded in EPON 812 resin. Ultrathin (90 nm) sections were obtained by ultrathin slicer machine. EM images of the samples were taken using Tecnai G2 Spirit transmission electron microscope (FEI Company).
CLEM
U2OS cells grown on glass gridded coverslips (Cellvis, D35-14-1.5GI) were transfected with EGFP-FAM134B plasmids. Cells were fixed with 4% paraformaldehyde for 20 min after transfected for 24 h and imaged by Zeiss Airyscan to collect light microscopy images. And cells were fixed with 2.5% glutaraldehyde at 4°C overnight. Then cells were postfixed in 2% osmium tetroxide–3% potassium ferrocyanide in cacodylate buffer for 1 h followed by 1% thiocarbohydrazide dissolved in water for 20 min and incubated in 2% osmium in cacodylate buffer for 30 min. Samples were then dehydrated with a graded ethanol series (20, 50, 70, 90, and 100%) each for 15 min and processed for Epon embedding. The samples were cut (30 KV and 2.5 nA) and imaged (2 KV and 0.2 nA) by focused ion beam–SEM (Helios UC G3).
Circular dichroism assay
FAM134B-WT and FAM134B-K160Q protein at peaks collected by SEC were concentrated using 30 kD ultrafiltration centrifugal tubes in phosphate buffer (0.1 M Na2HPO4 and NaH2PO4, pH 7.6). The secondary structure of FAM134B-WT and FAM134B-K160Q protein (200 µl 0.2 mg/ml) was analyzed by circular dichroism (J-1500-150ST; JASCO).
In vitro kinase assay
In vitro kinase assay was performed as followings (Rose et al., 2006). HA-CAMK2B was purified from HEK293T with anti-HA antibody-conjugated beads. Purified recombinant FAM134B proteins that interact with CAMK2B can be pulled down by HA-CAMK2B. Then the HA-beads mixture was incubated with 400 µM ATP and 1× CAMK2B kinase buffer (10 mM Hepes, pH 7.2, 1 mM EGTA, 5 mM MgCl2, 2 mM CaCl2). After incubated at 30°C for 10 min, the reaction was terminated by addition of 5× SDS loading buffer. The samples were then subjected to SDS-PAGE.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software). Student’s t test (two-tailed, unpaired) was performed for comparisons between two groups. For multiple comparisons, one-way ANOVA followed by Tukey’s post hoc test was performed. Data are presented as mean ± SEM of at least three independent experiments, unless otherwise noted. Data distribution was assumed to be normal but this was not formally tested. The levels of statistical significance are indicated by asterisks. NS, no significance.
Online supplemental material
Fig. S1 shows FAM134B is acetylated at lysine160 (K160), which is responsive to ER stress, related to Fig. 1. Fig. S2 shows FAM134B acetylation dramatically enhances FAM134B oligomerization to induce ER fragmentation and ER-phagy, related to Fig. 2. Fig. S3 shows CBP and SIRT7 regulates FAM134B-mediated ER fragmentation and ER-phagy, related to Figs. 3, 4, and 5. Fig. S4 shows relationship between phosphorylation and acetylation of FAM134B, related to Fig. 6. Table S1 lists reagents and resources used in this study.
Acknowledgments
We thank the Center of Cryo-Electron Microscopy, Zhejiang University for assistance with transmission electron microscopy. We thank the Imaging Center of Zhejiang University School of Medicine for assistance with confocal microscopy.
This study was supported by the National Natural Science Foundation under grants 32025012, 92254307, 31970695, 31771525 to Q. Sun; 31770938 and 91854113 to D. Neculai, 32000521 and 3227050033 to X. Jiang; and 32200600 to X. Wang; Ministry of Science and Technology of the People’s Republic of China under grants 2017YFA0503402 and 2021YFC2700901 (to Q. Sun); and the National Science Foundation for Post-doctoral Scientists of China under grants 2020M671698 and BX20200294 to X. Jiang.
Author contributions: Q. Sun and D. Neculai designed the experiments. X. Wang, X. Jiang, and B. Li performed major experiments and analyzed data. J. Zheng, M. Du, and X. Weng assisted in experiments. T. Liu, W. Liu, and L. Wang contributed reagents and discussion. L. Li, S. Chen, J. Zhang, and L. Fang performed the MS. L. Gao and J. Guo assisted in CLEM. Q. Sun and X. Wang wrote the manuscript. All authors discussed the results and commented on the manuscript.

