


Ceramides are essential precursors of complex sphingolipids and act as potent signaling molecules. Ceramides are synthesized in the endoplasmic reticulum (ER) and receive their head-groups in the Golgi apparatus, yielding complex sphingolipids (SPs). Transport of ceramides between the ER and the Golgi is executed by the essential ceramide transport protein (CERT) in mammalian cells. However, yeast cells lack a CERT homolog, and the mechanism of ER to Golgi ceramide transport remains largely elusive. Here, we identified a role for yeast Svf1 in ceramide transport between the ER and the Golgi. Svf1 is dynamically targeted to membranes via an N-terminal amphipathic helix (AH). Svf1 binds ceramide via a hydrophobic binding pocket that is located in between two lipocalin domains. We showed that Svf1 membrane-targeting is important to maintain flux of ceramides into complex SPs. Together, our results show that Svf1 is a ceramide binding protein that contributes to sphingolipid metabolism at Golgi compartments.
Introduction
Lipid intermediates, such as ceramide, are toxic when they accumulate in cellular membranes. Ceramides affect membrane structure and, additionally, function as signaling molecules promoting cell death. Therefore, it is not surprising that elevated ceramide levels have been linked to diseases, including neurodegenerative disorders, diabetes, and cardiovascular diseases (Alessenko and Albi, 2020; Holland et al., 2007; Pickersgill et al., 2007) This is not unique to highly specialized cells, as also in the model organism Saccharomyces cerevisiae, elevated ceramide levels have been associated with cell death (Eisenberg and Büttner, 2014). Ceramide in particular regulates various phosphatases (e.g., of the PP2A family) and kinases (including AKT, PKC, and MAP kinases) leading to decreased cellular proliferation (Bourbon et al., 2002; Teixeira et al., 2015; Dobrowsky et al., 1993).
The enzymes required for the biosynthesis of ceramide and all other SP species are known. Serine palmitoyl transferase (SPT) catalyzes the condensation of serine and palmitoyl-CoA to yield 3-keto-dihydrosphingosine. This short-lived intermediate is directly processed to dihydrosphingosine (DHS) and phytosphingosine (PHS). In a second metabolic pathway, the very long-chain fatty acids (VLCFAs) are synthesized through elongation of palmitoyl-CoA to 24 or 26 carbon chains (Dickson and Lester, 1999). A long-chain base (LCB) and a VLCFA are amide-linked to form ceramide. At the Golgi apparatus, ceramides receive various head groups to yield the complex SPs, such as sphingomyelin in mammalian cells or inositol-containing ceramides in yeast. Complex SPs are secreted from the Golgi to reach the plasma membrane by vesicular transport (Klemm et al., 2009). In addition, ceramides are also generated by the degradation of complex sphingolipids (SPs). In mammalian cells, this process is catalyzed by, for example, sphingomyelinases and glucosyl-β-glycosidases (Andrieu-Abadie and Levade, 2002; Sarmientos et al., 1986). In yeast, a single enzyme, Isc1, has been reported to hydrolyze complex SPs (Sawai et al., 2000). There is evidence that cells convert excess ceramides into other lipids to prevent toxicity. For example, the sphingomyelin synthase-related protein (SMSr) generates ceramide phosphoethanolamine from ceramide (Vacaru et al., 2009). Data from yeast and mammals suggest that ceramides can be acylated to generate acyl-ceramides that are supposedly stored in lipid droplets (Senkal et al., 2017; Voynova et al., 2012).
Besides their further processing, SP biosynthesis is highly regulated. When SP levels at the plasma membrane are low, the target of rapamycin complex 2 (TORC2) is activated and phosphorylates the yeast SGK1 homologue Ypk1. This leads to phosphorylation of the Orm proteins, negative regulators of the serine palmitoyltransferase, resulting in release of the inhibition and increased SP biosynthesis (Roelants et al., 2011). It was recently shown that upon phosphorylation, Orm2 is transported to the Golgi apparatus and degraded via the proteasome (Schmidt et al., 2019). Because of its resemblance to the ERAD pathway, this pathway has been termed Endosome/Golgi associated degradation (EGAD), and Orm2 has become the model substrate of this pathway. Moreover, recent work from our laboratory suggests that another level of regulation involves regulated uptake and direct shunting of serine into the SP metabolic pathway (Esch et al., 2020). Other mechanisms controlling SP homeostasis include the regulation of VLCFA synthesis (Olson et al., 2015) and the regulation of ceramide biosynthesis by the Ypk kinases (Muir et al., 2014). In addition, the lysosomal TOR complex has been suggested to regulate ceramide biosynthesis via control of ORM phosphorylation (Shimobayashi et al., 2013).
Although we are beginning to understand how SP metabolism is regulated, very little is known about the transfer of SPs between membranes. Other lipid classes are frequently transported between organelles via lipid transport proteins (Wong et al., 2018). Examples for this mechanism include the Oxysterol-binding protein (OSBP)-related proteins that transport both sterols and glycerophospholipids in exchange for phosphatidyl-inositol-4-phosphate (PI4P; Moser von Filseck et al., 2015; Maeda et al., 2013; Mesmin et al., 2013; Antonny et al., 2018). However, very little is known about SP transfer proteins, especially in yeast cells. In mammalian cells, ceramide is transported to the Golgi by CERT (Hanada et al., 2003). CERT binds to VAMP-associated proteins (VAPs) via its FFAT (two phenylalanines in acidic tract) motif at the ER and to PI4P at the Golgi apparatus via its pleckstrin homology (PH) domain, thereby tethering the two organelles. Ceramides are transported by a StAR-related lipid transfer (START) domain (Kumagai et al., 2019). A similar mechanism probably occurs in yeast cells, but the corresponding ceramide transfer protein has yet to be identified. One candidate protein is the Nuclear Vacuolar Junction protein 2 (Nvj2), which has been shown to be important for ceramide transfer between the ER and the Golgi apparatus (Liu et al., 2017). However, this process occurs only in cells that have high levels of ER stress. In addition, yeast tricalbins have also been implicated in ceramide transport between ER and Golgi (Ikeda et al., 2020). Ceramides in yeast cells are also transported by vesicular trafficking (Funato and Riezman, 2001). The molecular mechanism for this process remains largely elusive. The OSBP homologs Osh2, Osh3, and Osh4 together have been implicated in this process (Kajiwara et al., 2014). However, a direct role of the Osh2, Osh3, and Osh4 proteins in sorting ceramides into COP-II vesicles appears to be unlikely since their function in lipid transport has been described (Moser von Filseck et al., 2015; Mesmin et al., 2013; Maeda et al., 2013; Encinar Del Dedo et al., 2021). In summary, a complete picture of yeast ceramide transport is still lacking.
Here, we describe another putative ceramide transfer protein in yeast, survival factor 1 (Svf1). Using chemical genetic data mining, we identified overlapping functions of Svf1 with Nvj2 and the proteins Osh1 and Osh3. We used lipidomics and flux analysis to show that SVF1 mutant cells have decreased levels of complex SPs with a concomitant increase in ceramides, which is independent of the COP-II mediated transport machinery. Our analysis reveals that Svf1 is localized at the cis-Golgi apparatus and the cytoplasm. We identified an amphipathic helix (AH) at the N-terminus of Svf1 that is required for Golgi targeting. Using molecular docking studies and targeted lipidomics, we show that Svf1 harbors a hydrophobic binding pocket for ceramide that is located between its two lipocalin-like domains. Ceramide binding is abolished upon mutating two histidines in a potential cap region covering the hydrophobic pocket, which also leads to the loss of Svf1 localization at the cis-Golgi. Together, our data suggest that Svf1 is a ceramide binding protein that is involved in SP biosynthesis at the cis-Golgi most likely by transporting ceramide from the ER to the Golgi apparatus.
Results
SVF1 genetically interacts with genes involved in ceramide transport
Ceramide transport in yeast occurs by vesicular transport as well as non-vesicular transport mediated by Nvj2 (Fig. 1, a and b). Since Nvj2-mediated ceramide transport occurs only under conditions of high ER stress, we suspected that another protein might be functioning in ceramide transport. To identify this protein, we first analyzed high-throughput chemical genetics datasets (Hillenmeyer et al., 2008). Besides an effect of a gene knockout on drug resistance or sensitivity, these datasets also contain information on the similarity of genetic profiles. Highly similar profiles often indicate similar functions of genes in the cell. Our analysis revealed that the yeast gene SVF1 shows the highest correlation with NVJ2, a dubious open reading frame overlapping with NVJ2 (YPR092W) and two OSH (OSH1 and OSH3) genes, suggesting a potential role in ceramide transport within the cell (Fig. 1 c). SVF1 has been linked previously to SP homeostasis by interacting with the DHS hydroxylase SUR2. In addition, an osh2Δosh3Δosh4Δ strain has been used previously to identify the role of Nvj2 in ceramide transport (Liu et al., 2017). Tetrad analysis revealed that the additional deletion of SVF1 in an osh2Δosh3Δosh4Δ background aggravated the growth defect of the triple mutant. Similarly, the additional deletion of NVJ2 also aggravates the growth defect of the osh2Δosh3Δosh4Δ, suggesting that Svf1 might be another protein functioning in ceramide transport (Fig. 1 d; quantification in Fig. S1, a and b). Generating the quintuple mutant of OSH2, OSH3, OSH4, NVJ2, and SVF1 yields the strongest growth defect, but the cells are still viable, suggesting that some ceramides are still transported (Fig. 1 d). Two other candidate genes that could be involved in ceramide transport are the Svf1-like genes YLR225C and YDR222W. To test whether these genes have overlapping functions with SVF1, we utilized a synthetic interaction between SVF1 and SUR2, which also serves to further highlight its role in SP metabolism. Loss of SVF1 was previously shown to have a strong negative genetic interaction with deletion of the sphinganine-C4 hydroxylase SUR2 (Fig. S1 c; Brace et al., 2007). Deletion of neither YLR225C nor YDR222W alone, nor in combination with SVF1, showed an aggravated synthetic interaction with SUR2, suggesting neither gene is a functional homolog of SVF1 (Fig. S1 d).

SVF1 interacts genetically with yeast genes involved in ceramide transport. (a and b) A model for vesicular and non-vesicular ceramide transport in yeast under normal growth and ER stress conditions (b). (c) Histogram of correlation coefficients based on the genetic profiles of yeast mutants in chemical genetic screens. Data was extracted from Hillenmeyer et al. (2008). (d) Tetrad analysis of svf1Δosh3Δosh4Δ (green, yellow, and red, respectively) mutants crossed with nvj2Δosh2Δ (orange and blue, respectively). (e) Serial dilutions of WT, svf1Δ, nvj2Δ, and svf1Δnvj2Δ on YPD plates (control) and YPD plates containing 0.5 µM myriocin.
SVF1 interacts genetically with yeast genes involved in ceramide transport. (a and b) A model for vesicular and non-vesicular ceramide transport in yeast under normal growth and ER stress conditions (b). (c) Histogram of correlation coefficients based on the genetic profiles of yeast mutants in chemical genetic screens. Data was extracted from Hillenmeyer et al. (2008). (d) Tetrad analysis of svf1Δosh3Δosh4Δ (green, yellow, and red, respectively) mutants crossed with nvj2Δosh2Δ (orange and blue, respectively). (e) Serial dilutions of WT, svf1Δ, nvj2Δ, and svf1Δnvj2Δ on YPD plates (control) and YPD plates containing 0.5 µM myriocin.

Analysis of SVF1 genetic interactions with other potential ceramide transfer protein. (a) Quantification of tetrad analysis. Relative colony sizes of tetrads of diploid osh2Δosh3Δosh4Δsvf1Δ cells shown as fold change from WT tetrads. Error bars represent standard deviations. (b) Quantification of tetrad analysis. Relative colony sizes of tetrads of diploid osh2Δosh3Δosh4Δnvj2Δ shown as fold change from WT tetrads. Error bars represent standard deviations. (c) Tetrad analysis of svf1Δ (green) mutants crossed with sur2Δ (red). (d) Tetrad analysis of diploid svf1Δydr222wΔylr225cΔsur2Δ cells. svf1Δ (green); ydr222wΔ (blue); ylr225cΔ (yellow); sur2Δ (red). (e) Helical wheel representation of the first 18 amino acids of Svf1. The projection was generated by the Heliquest software (http://heliquest.ipmc.cnrs.fr). (f) Helical wheel representation of the first 18 amino acids of Ydr222w. (g) Helical wheel representation of the first 18 amino acids of Ylr225c.

Analysis of SVF1 genetic interactions with other potential ceramide transfer protein. (a) Quantification of tetrad analysis. Relative colony sizes of tetrads of diploid osh2Δosh3Δosh4Δsvf1Δ cells shown as fold change from WT tetrads. Error bars represent standard deviations. (b) Quantification of tetrad analysis. Relative colony sizes of tetrads of diploid osh2Δosh3Δosh4Δnvj2Δ shown as fold change from WT tetrads. Error bars represent standard deviations. (c) Tetrad analysis of svf1Δ (green) mutants crossed with sur2Δ (red). (d) Tetrad analysis of diploid svf1Δydr222wΔylr225cΔsur2Δ cells. svf1Δ (green); ydr222wΔ (blue); ylr225cΔ (yellow); sur2Δ (red). (e) Helical wheel representation of the first 18 amino acids of Svf1. The projection was generated by the Heliquest software (http://heliquest.ipmc.cnrs.fr). (f) Helical wheel representation of the first 18 amino acids of Ydr222w. (g) Helical wheel representation of the first 18 amino acids of Ylr225c.
To further investigate the role of Svf1 in SP metabolism and ceramide transport, we spotted WT, svf1Δ, nvj2Δ, and svf1Δnvj2Δ cells on control plates and plates containing the SP biosynthesis inhibitor myriocin. While both single mutants grow similar to a WT strain, the double deletion of SVF1 and NVJ2 is sensitive to chemical inhibition of SP biosynthesis, further supporting a role in SP homeostasis (Fig. 1 e).
Svf1 localizes to the cis-Golgi apparatus
To start dissecting the role of Svf1 in SP metabolism, we first tagged Svf1 at its C-terminus with GFP, which revealed a punctuate localization with additional cytosolic signal (Fig. 2 a). To reveal the identity of the punctuate structures, we systematically co-localized Svf1-GFP with other organelles in the cell. Svf1 co-localizes with the cis-Golgi apparatus, marked with Mnn9-mKate, but not with the late Golgi (Sec7-mKate), endosomes (Vps4-3xmCherry), or the endoplasmic reticulum marked with dsRed-HDEL (Fig. 2 a). To further confirm the co-localization of Svf1-GFP and Mnn9-mCherry, we analyzed the co-localization of both proteins over time. This analysis revealed that Svf1-GFP localizes dynamically to Mnn9-mKate positive structures (Fig. 2 b and Video 1). Together, our results demonstrate that Svf1 localizes at the cis-Golgi apparatus as well as in the cytoplasm (Fig. 2 c). This phenotype suggests an interaction of Svf1 with either a membrane protein at the cis-Golgi or directly with the membrane. In contrast to a C-terminal tagged version, an N-terminal GFP-tagged Svf1 was not sufficient to rescue the synthetic interaction with SUR2 (Fig. S2), suggesting an important role for the Svf1 N-terminus.

Svf1 dynamically colocalizes with the cis-Golgi. (a) Svf1-GFP was expressed in cells expressing either Mnn9-mKate (cis-Golgi), Sec7-mKate (trans-Golgi), Vps4-3xmCherry (endosome) or HDEL-dsRed (ER). Line scans (right) of the indicated regions of Svf1-GFP (green) and the respective co-expressed organelle markers (magenta). Scale bar = 5 µM. Scale bar inlays = 1 μM. (b) Co-localization of Svf1-GFP and 3xmCherry tagged Mnn9 imaged every 0.9 s over a total time of 9.9 s to confirm the localization to the cis-Golgi. Scale bar = 5 µM. (c) A model of the two interchanging states, cytosolic and membrane bound, of Svf1 in the cell.
Svf1 dynamically colocalizes with the cis-Golgi. (a) Svf1-GFP was expressed in cells expressing either Mnn9-mKate (cis-Golgi), Sec7-mKate (trans-Golgi), Vps4-3xmCherry (endosome) or HDEL-dsRed (ER). Line scans (right) of the indicated regions of Svf1-GFP (green) and the respective co-expressed organelle markers (magenta). Scale bar = 5 µM. Scale bar inlays = 1 μM. (b) Co-localization of Svf1-GFP and 3xmCherry tagged Mnn9 imaged every 0.9 s over a total time of 9.9 s to confirm the localization to the cis-Golgi. Scale bar = 5 µM. (c) A model of the two interchanging states, cytosolic and membrane bound, of Svf1 in the cell.
Representative midsection time lapse fluorescence microscopy of yeast cells expressing Svf1-GFP and Mnn9-mCherry. Images were recorded with 0.9 s intervals.
Representative midsection time lapse fluorescence microscopy of yeast cells expressing Svf1-GFP and Mnn9-mCherry. Images were recorded with 0.9 s intervals.

Tetrad dissections of N- and C-terminal tagged SVF1. (a) Tetrad analysis of a marker less generated GFP-Svf1 strain crossed with sur2Δ. Tetrads are numbered 1–5. Spores of the tetrads are labeled A to D. (b) Tetrad analysis of a Svf1-GFP strain crossed with sur2Δ. Tetrads are numbered 1–5. Spores of the tetrads are labeled A to D.

Tetrad dissections of N- and C-terminal tagged SVF1. (a) Tetrad analysis of a marker less generated GFP-Svf1 strain crossed with sur2Δ. Tetrads are numbered 1–5. Spores of the tetrads are labeled A to D. (b) Tetrad analysis of a Svf1-GFP strain crossed with sur2Δ. Tetrads are numbered 1–5. Spores of the tetrads are labeled A to D.
An N-terminal amphipathic helix is necessary for Svf1 targeting to the cis-Golgi
To identify a potential targeting motif in the N-terminus of Svf1, we performed a HeliQuest analysis (Gautier et al., 2008) This revealed a putative AH within the first 18 amino acids (Fig. 3 a). To test whether the amphipathic nature of the N-terminal helix of Svf1 is necessary for targeting to the cis-Golgi, we mutated the hydrophobic valine residue in position 12 to a negatively charged aspartic acid, largely destroying the hydrophobic moment (Fig. 3, b and c). Expression of Svf1-GFP from an integrative plasmid under control of the endogenous promoter in a svf1Δ strain confirmed the dual localization of Svf1 to the cis-Golgi marked with Mnn9-mCherry and the cytoplasm (Fig. 3 d, upper panel). In contrast, the expression of the Svf1V12D-GFP mutant showed only cytosolic GFP signal, strongly supporting the model of the N-terminal AH in Svf1 driving membrane recruitment (Fig. 3 d, lower panels). We noted that expression of Svf1-GFP from an integrative plasmid under its endogenous promoter resulted in some overexpression of the protein as detected by microscopy as well as on Western blots (Fig. S3 a). Although we do not have an explanation for this phenotype, we continued our analyses with the appropriate controls. To confirm our results biochemically, we pelleted membranes from Svf1-GFP and Svf1V12D-GFP cells and analyzed pellet fractions and supernatant fractions by Western blot. While Svf1-GFP was present in both fractions, we were only able to detect Svf1V12D-GFP in the supernatant fraction, confirming that its inability to interact with membranes (Fig. 3 e). Next, we asked whether localization of Svf1 to the cis-Golgi is required for its function. To assess this, we dissected svf1Δsur2Δ cells expressing either Svf1-GFP or Svf1V12D-GFP from a plasmid. While the expression of Svf1-GFP was sufficient to rescue the synthetic phenotype of SVF1 and SUR2 double deletions (Fig. 3 f), the strain harboring Svf1V12D-GFP phenocopied the svf1Δsur2Δ strain (Fig. 3 g). These results show that the targeting of Svf1 via its N-terminal AH is necessary for its function (Fig. 3 h). The two proteins annotated as Svf1 homologs do not possess N-terminal AHs which might also explain why they do not function in SP homeostasis (Fig. S1 c).

Svf1 possesses an N-terminal amphipathic helix important for the targeting and function of the protein. (a and b) Helical wheel representation of the first 18 amino acids with the hydrophobic amino acids shown in yellow and the hydrophobic moment shown by the arrow and expressed in µH above the arrow for the WT sequence and (b) the V12D mutant with the exchange of the hydrophobic valine to the charged aspartate (red). (c) Graphical representation of the full-length protein (WT, left; V12D mutant, right). Shown are the AH (yellow, 1–18 aa), the rest of Svf1 (orange, 19–481 aa) and the C-terminal GFP tag (green). (d) Co-localization of GFP tagged Svf1 expressed from a plasmid under control of the endogenous promoter with mCherry tagged Mnn9 for the WT (upper panel) and the V12D mutant (lower panel). Scale bar = 5 µM. Scale bar inlays = 1 µM. (e) Samples from the membrane fractionation according to 50 µg protein concentration were analyzed by Western blot. The separation of the cell lysate (Input, I) into pellet (P) and supernatant (S) fractions shows the Svf1 localization either bound to membranes (P) or cytosolic (S) detected by an α-GFP antibody. The antibodies α-Pgk1 and α-Sec61 were used as loading controls for the cytosol and membrane fractions, respectively. (f) Tetrad analysis of the svf1Δpl.Svf1-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). (g) Tetrad analysis of the svf1Δpl.Svf1V12D-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). (h) A model of Svf1 with the folded (bound Svf1) and unfolded (cyt. Svf1) N-terminal AH. (i) Tetrad analysis of the svf1∆pl.Svf1L2E-GFP (blue and green, respectively) mutants crossed with sur2∆ (red). (j) Western blot analysis of Svf1-GFP in WT and mak3Δ mutant as described in e. (k and l) Helical wheel representation of the WT AH (k) and the G7A/G8A mutant AH (l) described as in Fig. 3 a shows no impact by the exchanges of glycines to alanines at the positions 7 and 8 on the hydrophobic moment of the AHs. (m) Co-localization of GFP tagged Svf1 expressed from a plasmid under the control of the endogenous promoter of Svf1 in a svf1Δ background (WT, upper panel; G7A/G8A mutant, lower panel), with the organelle markers Mnn9-mKate (cis-Golgi) and Sec63-Halo (ER). Scale bar = 5 µM. (n) Western blot analysis as described in e for Svf1WT-GFP and Svf1G7A/G8A-GFP. Source data are available for this figure: SourceData F3.
Svf1 possesses an N-terminal amphipathic helix important for the targeting and function of the protein. (a and b) Helical wheel representation of the first 18 amino acids with the hydrophobic amino acids shown in yellow and the hydrophobic moment shown by the arrow and expressed in µH above the arrow for the WT sequence and (b) the V12D mutant with the exchange of the hydrophobic valine to the charged aspartate (red). (c) Graphical representation of the full-length protein (WT, left; V12D mutant, right). Shown are the AH (yellow, 1–18 aa), the rest of Svf1 (orange, 19–481 aa) and the C-terminal GFP tag (green). (d) Co-localization of GFP tagged Svf1 expressed from a plasmid under control of the endogenous promoter with mCherry tagged Mnn9 for the WT (upper panel) and the V12D mutant (lower panel). Scale bar = 5 µM. Scale bar inlays = 1 µM. (e) Samples from the membrane fractionation according to 50 µg protein concentration were analyzed by Western blot. The separation of the cell lysate (Input, I) into pellet (P) and supernatant (S) fractions shows the Svf1 localization either bound to membranes (P) or cytosolic (S) detected by an α-GFP antibody. The antibodies α-Pgk1 and α-Sec61 were used as loading controls for the cytosol and membrane fractions, respectively. (f) Tetrad analysis of the svf1Δpl.Svf1-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). (g) Tetrad analysis of the svf1Δpl.Svf1V12D-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). (h) A model of Svf1 with the folded (bound Svf1) and unfolded (cyt. Svf1) N-terminal AH. (i) Tetrad analysis of the svf1∆pl.Svf1L2E-GFP (blue and green, respectively) mutants crossed with sur2∆ (red). (j) Western blot analysis of Svf1-GFP in WT and mak3Δ mutant as described in e. (k and l) Helical wheel representation of the WT AH (k) and the G7A/G8A mutant AH (l) described as in Fig. 3 a shows no impact by the exchanges of glycines to alanines at the positions 7 and 8 on the hydrophobic moment of the AHs. (m) Co-localization of GFP tagged Svf1 expressed from a plasmid under the control of the endogenous promoter of Svf1 in a svf1Δ background (WT, upper panel; G7A/G8A mutant, lower panel), with the organelle markers Mnn9-mKate (cis-Golgi) and Sec63-Halo (ER). Scale bar = 5 µM. (n) Western blot analysis as described in e for Svf1WT-GFP and Svf1G7A/G8A-GFP. Source data are available for this figure: SourceData F3.
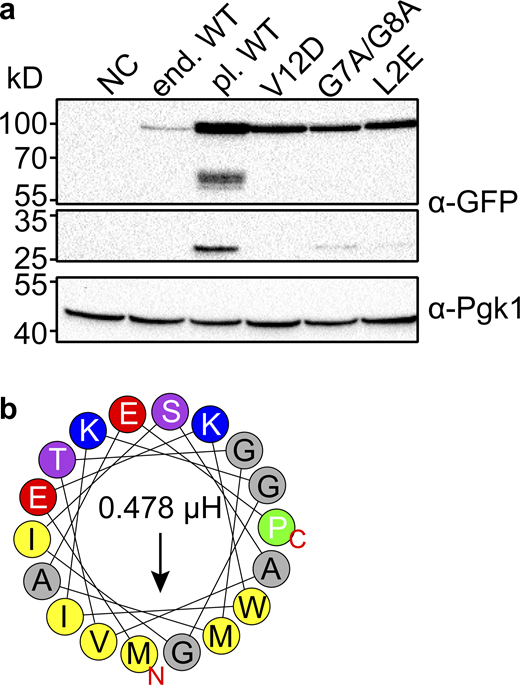
Expression levels of plasmid expressed Svf1 are slightly altered. (a) Expression levels of Svf1-GFP constructs used in this study. Equal amounts of cells were lysed and analyzed by Western blotting using antibodies against the GFP-tag or Pgk1 as a loading control. NC = negative control/ WT strain; end. WT = Svf1 GFP tagged endogenously with GFP; pl. WT = Svf1-GFP expressed under its endogenous promoter from an integrative plasmid; V12D = Svf1V12D-GFP expressed under its endogenous promoter from an integrative plasmid; G7A/G8A = Svf1G7A/G8A-GFP expressed under its endogenous promoter from an integrative plasmid; L2E = Svf1L2E-GFP expressed under its endogenous promoter from an integrative plasmid. (b) Helical wheel representation of the first 18 amino acids with the hydrophobic amino acids shown in yellow and the hydrophobic moment shown by the arrow and expressed in µH above the arrow for the L2E mutant with the exchange of the hydrophobic leucine to the charged glutamate (red). Source data are available for this figure: SourceData FS3.

Expression levels of plasmid expressed Svf1 are slightly altered. (a) Expression levels of Svf1-GFP constructs used in this study. Equal amounts of cells were lysed and analyzed by Western blotting using antibodies against the GFP-tag or Pgk1 as a loading control. NC = negative control/ WT strain; end. WT = Svf1 GFP tagged endogenously with GFP; pl. WT = Svf1-GFP expressed under its endogenous promoter from an integrative plasmid; V12D = Svf1V12D-GFP expressed under its endogenous promoter from an integrative plasmid; G7A/G8A = Svf1G7A/G8A-GFP expressed under its endogenous promoter from an integrative plasmid; L2E = Svf1L2E-GFP expressed under its endogenous promoter from an integrative plasmid. (b) Helical wheel representation of the first 18 amino acids with the hydrophobic amino acids shown in yellow and the hydrophobic moment shown by the arrow and expressed in µH above the arrow for the L2E mutant with the exchange of the hydrophobic leucine to the charged glutamate (red). Source data are available for this figure: SourceData FS3.
Another cis-Golgi localized protein, Grh1, has also been shown to target the cis-Golgi via an AH (Behnia et al., 2007). In addition, Grh1 requires N-terminal acetylation for its targeting to the cis-Golgi. We thus speculated that Svf1 also requires N-terminal acetylation for its targeting. Recently, a targeting motif for N-terminal acetylation by the yeast NatC complex has been identified (Grunwald et al., 2020). According to this prediction, Svf1 is also a target of the NatC complex. To test our hypothesis, we mutated the leucine residue following the first amino acid to glutamic acid (L2E). This barely affects the hydrophobic moment of the AH but should abolish N-terminal acetylation by NatC (Fig. S3 b). In line with this prediction, Svf1L2E-GFP does not rescue the growth of a svf1Δsur2Δ double deletion (Fig. 3 i). In addition, fractionation analysis revealed that Svf1-GFP becomes largely cytosolic in cells lacking MAK3, the catalytic subunit of NatC, compared to WT cells, suggesting that N-terminal acetylation is indeed necessary for membrane targeting of Svf1 (Fig. 3 j). Furthermore, mass spectrometric analysis confirmed the N-terminal acetylation of Svf1 (Fig. S4 a). Interestingly, we always observed both a cytosolic fraction as well as a membrane bound fraction of Svf1. The dual localization can be explained by two adjacent glycine residues (G7 and G8) in the AH of Svf1. Glycine is a common feature of AHs that is thought to prevent their folding in the absence of a membrane surface (Drin and Antonny, 2010; Cornell and Taneva, 2006). We tested whether we can increase the Svf1 membrane interaction by mutating both glycine residues to alanines (G7A/G8A), which are preferable for helix folding (Pace and Scholtz, 1998). This mutation left the hydrophobic moment of the AH largely unaffected (Fig. 3, k and l). We observed increased co-localization of Svf1G7A/G8A-GFP with the Mnn9-mKate marked cis-Golgi and recruitment of Svf1-GFP to the ER membrane marked with Sec63-Halo (Fig. 3 m). The overall membrane interaction was comparable to WT Svf1 (Fig. 3 n). Together, our results show that Svf1 can dynamically localize between the cytosol and the cis-Golgi apparatus depending on an acetylated N-terminal AH.

Analysis of Svf1 acetylation and analyses of Svf1 mutants. (a) Mass spectrometric analysis of the N-terminus of Svf1 showing that it is N-terminally acetylated. MS/MS spectrum extracted from MaxQuant data from a GFP pulldown of Svf1. Detected y-ions are labeled in red, b-ions are labeled in blue. The sequence showing all identified y- and b-ions is shown in the top right position. (b) Tetrad analysis of the svf1Δpl.Grh11-18aaSvf119-481-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). (c) Evaluation of the binding of the AHs from WT, V12D mutant, and G7A/G8A mutant to the ER expressed as cytosol/ER ratio in %. Intensities of the GFP signal measured in cytosolic areas were divided by the intensities of the GFP signal at the ER measured in 0.065 µm2. The signal of Sec63-Halo was used to differentiate between Cytosol and ER areas. n ≤ 48.

Analysis of Svf1 acetylation and analyses of Svf1 mutants. (a) Mass spectrometric analysis of the N-terminus of Svf1 showing that it is N-terminally acetylated. MS/MS spectrum extracted from MaxQuant data from a GFP pulldown of Svf1. Detected y-ions are labeled in red, b-ions are labeled in blue. The sequence showing all identified y- and b-ions is shown in the top right position. (b) Tetrad analysis of the svf1Δpl.Grh11-18aaSvf119-481-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). (c) Evaluation of the binding of the AHs from WT, V12D mutant, and G7A/G8A mutant to the ER expressed as cytosol/ER ratio in %. Intensities of the GFP signal measured in cytosolic areas were divided by the intensities of the GFP signal at the ER measured in 0.065 µm2. The signal of Sec63-Halo was used to differentiate between Cytosol and ER areas. n ≤ 48.
The Svf1 N-terminal AH is sufficient for membrane targeting
To further analyze the targeting mechanism of Svf1, we asked whether the AH is also sufficient for targeting. We therefore expressed fusion constructs of the first 18 amino acids of Svf1 and GFP in WT cells under the control of the Svf1 promoter (Fig. 4 a). We were able to detect membrane association of the Svf11-18-GFP construct, but this signal did not co-localize with the cis-Golgi marked by Mnn9-mKate (Fig. 4 b, upper panel). Instead, the AH of Svf1 co-localizes with the ER marked by Sec63 fused to a Halo tag. Expression of the AH containing the V12D mutation only showed a cytosolic signal, as expected from our previous finding (Fig. 4 b, lower panel). To confirm our in vivo results, we fractionated lysates from AHWT-GFP and AHV12D-GFP expressing cells into membrane and supernatant fraction and analyzed the distribution of the constructs by Western blot. This analysis revealed the partial membrane interaction of the WT AH while the V12D AH was only present in the supernatant fraction (Fig. 4 c). Expressing the GFP fused AH harboring, the glycine to alanine mutations also did not result in Golgi targeting of the construct but lead to increased ER membrane association (Fig. 4, d and e; and Fig. S4 b). Together, our results suggest that the Svf1 AH is part of a membrane targeting mechanism of Svf1, but a second mechanism is required for targeting the protein specifically to the cis-Golgi apparatus. As mentioned before, another protein, Grh1 is targeted via an AH to the cis-Golgi apparatus. To test whether the Grh1 AH is sufficient to target Svf1 to the Golgi, we exchanged the Svf1 amphipathic helix by that of Grh1 (Fig. 4 f). While the Grh1 AH has an even stronger hydrophobic moment (Fig. 4 g) than the Svf1 AH, it was not sufficient to target Svf1 to the Golgi apparatus (Fig. 4 h), and the construct was not able to rescue the synthetic phenotype between SVF1 and SUR2 (Fig. S4 c). This either suggests that the AH of Svf1 needs to be coordinated itself with other parts of the protein or that Svf1 requires additional interaction partners at the Golgi apparatus for its targeting.

The N-terminal amphipathic helix of Svf1 targets to the ER. (a) Graphical representation of the AH of Svf1 (yellow; WT, left; V12D mutant, middle; G7A/G8A mutant, right) tagged with GFP. (b) Co-localization of the GFP tagged AH expressed from a plasmid under the control of the promoter of Svf1 (WT, upper panel; V12D, lower panel) with the organelle markers Mnn9-mKate (cis-Golgi) and Sec63-Halo (ER). Scale bar = 5 µM. (c) Western blot analysis of the WT and the V12D AH (WT and AHV12D, respectively) fused to GFP. The separation of the cell lysate (Input, I) into pellet (P) and supernatant (S) fractions shows the localization of the GFP tagged AH (WT) and AHV12D either bound to membranes (P) or cytosolic (S) detected by an α-GFP antibody. The antibodies α-Pgk1 and α-Sec61 were used as loading controls for the cytosol and membrane fractions, respectively. (d) Co-localization of the GFP tagged AH expressed from a plasmid under the control of the endogenous promoter of Svf1 (WT, upper panel; G7A/G8A, lower panel) with the organelle markers Mnn9-mKate (cis-Golgi) and Sec63-Halo (ER). Scale bar = 5 µM. (e) Western blot analysis of the WT and the G7A/G8A AH (AHWT and AHG7A/G8A, respectively) fused to GFP. The separation of the cell lysate (Input, I) into pellet (P) and supernatant (S) fractions shows the localization of the GFP tagged AHWT and AHG7A/G8A either bound to membranes (P) or cytosolic (S) detected by an α-GFP antibody. The antibodies α-Pgk1 and α-Sec61 were used as loading controls for the cytosol and membrane fractions, respectively. (f and g) Helical wheel representation of the 1–18 amino acids of Grh1 as described for Svf1 in Fig. 3 a and (g) represented in the model in purple (right), which were fused to Svf1 (19–481 aa; orange) and a C-terminal GFP tag (green). (h) Co-localization of GFP tagged Svf1 expressed from a plasmid under the control of the endogenous promoter of Svf1 with mKate tagged Mnn9 (upper panel) and the fused protein with the AH of Grh1 (1–18 aa) and Svf1 (19–481 aa) expressed from a plasmid under control of the endogenous promoter of Svf1 with mKate tagged Mnn9 (lower panel). Scale bar = 5 µM. Source data are available for this figure: SourceData F4.
The N-terminal amphipathic helix of Svf1 targets to the ER. (a) Graphical representation of the AH of Svf1 (yellow; WT, left; V12D mutant, middle; G7A/G8A mutant, right) tagged with GFP. (b) Co-localization of the GFP tagged AH expressed from a plasmid under the control of the promoter of Svf1 (WT, upper panel; V12D, lower panel) with the organelle markers Mnn9-mKate (cis-Golgi) and Sec63-Halo (ER). Scale bar = 5 µM. (c) Western blot analysis of the WT and the V12D AH (WT and AHV12D, respectively) fused to GFP. The separation of the cell lysate (Input, I) into pellet (P) and supernatant (S) fractions shows the localization of the GFP tagged AH (WT) and AHV12D either bound to membranes (P) or cytosolic (S) detected by an α-GFP antibody. The antibodies α-Pgk1 and α-Sec61 were used as loading controls for the cytosol and membrane fractions, respectively. (d) Co-localization of the GFP tagged AH expressed from a plasmid under the control of the endogenous promoter of Svf1 (WT, upper panel; G7A/G8A, lower panel) with the organelle markers Mnn9-mKate (cis-Golgi) and Sec63-Halo (ER). Scale bar = 5 µM. (e) Western blot analysis of the WT and the G7A/G8A AH (AHWT and AHG7A/G8A, respectively) fused to GFP. The separation of the cell lysate (Input, I) into pellet (P) and supernatant (S) fractions shows the localization of the GFP tagged AHWT and AHG7A/G8A either bound to membranes (P) or cytosolic (S) detected by an α-GFP antibody. The antibodies α-Pgk1 and α-Sec61 were used as loading controls for the cytosol and membrane fractions, respectively. (f and g) Helical wheel representation of the 1–18 amino acids of Grh1 as described for Svf1 in Fig. 3 a and (g) represented in the model in purple (right), which were fused to Svf1 (19–481 aa; orange) and a C-terminal GFP tag (green). (h) Co-localization of GFP tagged Svf1 expressed from a plasmid under the control of the endogenous promoter of Svf1 with mKate tagged Mnn9 (upper panel) and the fused protein with the AH of Grh1 (1–18 aa) and Svf1 (19–481 aa) expressed from a plasmid under control of the endogenous promoter of Svf1 with mKate tagged Mnn9 (lower panel). Scale bar = 5 µM. Source data are available for this figure: SourceData F4.
Svf1 acts at the ER Golgi interface
Based on our hypotheses, we tried to identify interaction partners of Svf1. We immuno-purified GFP-tagged Svf1 from cells metabolically labeled with nonradioactive, stable isotope-labeled 13C615N2 lysine (stable isotope labeling with amino acids in cell culture; Ong et al., 2002). In parallel, we performed a mock purification from control cells labeled with unlabeled lysine. We mixed eluates from both purifications and analyzed them by high-resolution mass spectrometry–based proteomics (Walther and Mann, 2010). Peptides from proteins containing the isotope-labeled lysine are shifted to higher m/z values in the spectra compared with the same peptide from unlabeled protein, allowing quantitation of abundance ratios for each detected peptide and protein from the eluates. This analysis identified a set of multiple co-enriched proteins (Table S1) with many of them being membrane proteins. Amongst them are several enzymes involved in lipid biosynthesis and transport including the VLCFA elongases Elo2, Elo3, and Ifa38 (Fig. 5 a). In addition, we also identified Golgi-resident proteins such as the phosphatidylinositol-4-kinase (PI4P) Pik1 and the mannosyltransferase Mnn5. We also identified proteins of the COP machinery. Amongst them is Sec12, the guanine nucleotide exchange factor (GEF) for Sar1 (Barlowe and Schekman, 1993), the Arf1 GTPase activating protein (GAP) Glo3 (Poon et al., 1999) and Psg1, which has been identified as a component of the Golgi apparatus and COPI vesicles (Geva et al., 2017). These data further support a role for Svf1 at the interface between the ER and the Golgi apparatus, where ceramide transport is focused. It has been previously suggested that the cis-Golgi and ER exit sites contact each other in a “hug-and-kiss” behavior (Kurokawa et al., 2014). We therefore tested whether Svf1 can be co-localized with both, the cis-Golgi (marked with Mnn9-mKate) and ER-exit sites (marked with Sec31-Halo). Our analysis confirmed that ∼40% of Svf1 dots co-localized with both Mnn9 positive cis-Golgi and Sec31 containing ER-exit sites (Fig. 5, b and c). In summary, this suggests that Svf1 acts at the interface of the ER and the cis-Golgi apparatus where ceramides are likely transported from one membrane to the other. Since several proteins identified in our interaction studies are part of the COP-II machinery, we next tested whether COP-II-dependent transport is necessary for Svf1 localization to the Golgi apparatus. The dynamic localization of Svf1 to the cis-Golgi is lost when COP-II transport is inhibited. Incubation of cells containing the temperature sensitive allele of Sec12 to the restrictive temperature results in a loss of the puncta of Svf1-GFP most likely reflecting a general loss of the Golgi apparatus when COP-II transport is inhibited (Fig. 5 d). To further investigate whether COP-II is required for Svf1 targeting to the Golgi, we expressed Svf1-GFP in strains harboring temperature-sensitive alleles of either the Sar1 GTPase-activating protein Sec23 or the α-SNAP cochaperone Sec17. We used the Svf1G7A/G8A-GFP mutant since it showed a stronger affinity for the cis-Golgi apparatus and made the quantifications easier. At the permissive temperature, Svf1G7A/G8A-GFP co-localized with the Golgi marker Mnn9-mkate in sec23ts cells (Fig. 5 e, upper panels). At the restrictive temperature, Svf1G7A/G8A-GFP and Mnn9-mKate were both retained at the ER based on co-localization with Sec63-Halo (Fig. 5 e, upper panels). The increased co-localization can also be seen in the quantification of co-localization of Svf1G7A/G8A-GFP with both, Mnn9-mKate and Sec63-Halo (Fig. 5 f). In the sec17ts mutant, we already observed reduced levels of the cis-Golgi marked with Mnn9-mKate at the permissive temperature. However, Svf1G7A/G8A-GFP co-localization with some of these structures remained (Fig. 5 e, lower panels). At the restrictive temperature both, Svf1G7A/G8A-GFP and Mnn9-mKate localized to aberrant structures but the observable Mnn9 signal was very low (Fig. 5 e, lower panels). Together, our results suggest that Svf1 functions at the interface of the cis-Golgi apparatus and the ER. If Svf1 targets the Golgi apparatus via COP-II vesicles or if a general loss of the Golgi apparatus leads to loss of Svf1 localization at these structures cannot be resolved from these experiments.

Svf1 acts at the interface of the ER and the cis-Golgi. (a) Proteomic analysis of Svf1-GFP, expressed from a plasmid under the control of the endogenous promoter of Svf1 in a svf1Δ background, and mock treated WT cells is shown. Protein intensities are plotted against heavy/light SILAC ratios. Significant outliers are colored in red (P < 1−11), orange (P < 1−4), or steel blue (P < 0.05), other proteins are shown in light blue. (b) Co-localization of GFP tagged Svf1 with mKate tagged Mnn9 (cis-Golgi) and Halo tagged Sec31 (ER, ER exit sites). (c) Evaluation of the co-localization of Svf1 dots (n = 100, triplicates) shown in d with either the cis-Golgi (Mnn9), Sec31 (ER, COPII vesicles), or both simultaneously. Scale bar = 5 µM. (d) Co-localization of GFP tagged Svf1 with Halo tagged Sec63 (ER marker) in a sec12ts background under permissive (24°C, upper panel) and non-permissive (37°C, lower panel) temperature. Scale bar = 5 µM. (e) Co-localization of GFP tagged Svf1G7A/G8A with Halo tagged Sec31 (ER marker) and mKate tagged Mnn9 in a sec23ts background (upper panels) and in a sec17ts background (lower panels) under permissive (24°C) and non-permissive (37°C) temperature. Scale bar = 5 µM. (f) Quantification of e (n = 60, triplicates).
Svf1 acts at the interface of the ER and the cis-Golgi. (a) Proteomic analysis of Svf1-GFP, expressed from a plasmid under the control of the endogenous promoter of Svf1 in a svf1Δ background, and mock treated WT cells is shown. Protein intensities are plotted against heavy/light SILAC ratios. Significant outliers are colored in red (P < 1−11), orange (P < 1−4), or steel blue (P < 0.05), other proteins are shown in light blue. (b) Co-localization of GFP tagged Svf1 with mKate tagged Mnn9 (cis-Golgi) and Halo tagged Sec31 (ER, ER exit sites). (c) Evaluation of the co-localization of Svf1 dots (n = 100, triplicates) shown in d with either the cis-Golgi (Mnn9), Sec31 (ER, COPII vesicles), or both simultaneously. Scale bar = 5 µM. (d) Co-localization of GFP tagged Svf1 with Halo tagged Sec63 (ER marker) in a sec12ts background under permissive (24°C, upper panel) and non-permissive (37°C, lower panel) temperature. Scale bar = 5 µM. (e) Co-localization of GFP tagged Svf1G7A/G8A with Halo tagged Sec31 (ER marker) and mKate tagged Mnn9 in a sec23ts background (upper panels) and in a sec17ts background (lower panels) under permissive (24°C) and non-permissive (37°C) temperature. Scale bar = 5 µM. (f) Quantification of e (n = 60, triplicates).
Svf1 is important for the biosynthesis of complex sphingolipids
To dissect the role of Svf1 at the Golgi, we next focused on its molecular function. Based on our genetic and cell biological experiments, we hypothesized that Svf1 functions in ceramide transport at the ER Golgi interface. To test this, we measured the levels of yeast SPs using LC-MS/MS measurements. We extracted lipids from WT and svf1Δ cells as well as a knockout of the sphinganine-C4 hydroxylase SUR2 as a control. As previously observed, we detected a small but significant decrease in phytosphingosine (PHS) levels in svf1Δ cells compared to WT cells (Brace et al., 2007; Fig. 6 a). Deletion of SUR2 resulted in a complete shift from LCB 18:0;3 (equivalent of PHS) to LCB 18:0;2 (equivalent of DHS), as expected (Fig. 6 a). However, we also detected increased levels of the major ceramide species (CER 44:0;4) in svf1Δ cells compared to WT cells (Fig. 6 b). Consistent with a potential function of Svf1 in ceramide transport, we detected a decrease in the SP species IPC and MIPC in svf1Δ cells compared to WT cells. Moreover, deletion of SUR2 resulted in a shift from the 44:0;4 species to the 44:0;3 species of IPC and MIPC confirming the reliability of our measurements (Fig. 6 c). Our analysis of the localization of Svf1 in the background of mutations affecting the COP-II machinery did not allow us to determine whether Svf1 needs the COP-II machinery for its localization. Prompted by our results that Svf1 is necessary for the biosynthesis of complex SPs from ceramides, we wanted to dissect the role of Svf1 in the different pathways required for ceramide transport to the Golgi apparatus. Ceramide transport to the Golgi has been shown to depend on a functional COP-II transport machinery. We first analyzed the levels of complex SPs in a temperature-sensitive mutant of the Sec12 GEF at the restrictive temperature and compared it to WT cells, svf1Δ cells and sec12ts cells also harboring a deletion of SVF1. The levels of complex SPs were depleted in sec12ts cells and svf1Δ cells (Fig. 6, d–g). This is in line with previously measured reductions in complex SP levels in a sec12ts mutant as well as our results obtained for svf1Δ cells (Funato and Riezman, 2001). Interestingly, the additional deletion of SVF1 in a sec12ts strain resulted in even stronger depletion of complex SP, suggesting that Svf1 rather transports ceramides in a parallel pathway to the COP-II vesicular transport pathway. Next, we tested the epistatic effects of the deletion of OSH2, OSH3, OSH4, NVJ2, and SVF1 in the biosynthesis of complex SPs. Deletion of SVF1 again resulted in the reduction of all IPC and MIPC species determined (Fig. 6, h–k). Deletion of NVJ2 had a stronger effect on the IPC 44:0;4 and MIPC 44:0;4 species compared to the 44:0;3 species, harboring one less hydroxylation site. The double deletion of SVF1 and NVJ2 had an additive effect on the IPC and MIPC 44:0;3 species and was on the same level as the NVJ2 deletion alone regarding the 44:0;4 species. The similar deletion of OSH2, OSH3, and OSH4 was previously suggested to affect ceramide transport by affecting vesicular transport of ceramides (Kajiwara et al., 2014). Similar to these studies, the triple deletion resulted in reduced levels of the major IPC species (Fig. 6, h–k). However, the levels of the more complex SPs, the MIPC species were elevated in the OSH triple mutant, suggesting that the mutant does not directly affect ceramide transport but rather leads to an adapted lipid metabolism of the cell. Interestingly, the additional deletion of SVF1 in the OSH triple mutant resulted in the depletion of all analyzed complex SP species, suggesting that Svf1 is an important contributor to cellular ER to Golgi ceramide transport, acting in a parallel pathway to vesicular transport (Fig. 6, h–k). In contrast, the additional deletion of NVJ2 had no effect on the OSH2, OSH3, OSH4 deletion mutant. The quintuple mutant of OSH2, OSH3, OSH4, NVJ2, and SVF1 phenocopied the OSH SVF1 quadruple mutant, further highlighting Svf1 also functioning in a parallel pathway to NVJ2-dependent ceramide transport (Fig. 6, h–k). In line with our hypothesis, we could not detect an effect for Svf1 on the content of ceramides on in vitro budded COP-II vesicles (Fig. S5, a–c). We also did not observe any strong phenotype of the SVF1 deletion on maturation of the GPI anchored protein Gas1 under steady-state conditions (Fig. S5 d), and Svf1 has not been identified in any screens for genes important for the secretory pathway (Novick et al., 1980).

Lipidomic analysis shows an effect of Svf1 on sphingolipid metabolism. (a and b) Measurement of the different SP intermediates in WT (black), svf1Δ (gray), and sur2Δ (white) cells shows an alteration of (a) the LCB (LCB 18:0;2 and LCB 18:0;3) levels and (b) the ceramide (CER 44:0;3 and CER 44:0;4) levels represented in pmol/µg protein. (c) From the same measurement the levels of the complex SPs IPC (IPC 44:0;3 and IPC 44:0;4) and MIPC (MIPC 44:0;3 and MIPC 44:0;4) are shown as peak areas (arbitrary unit [a.u.]). (d–g) Measurements of the of the complex SPs IPC (IPC 44:0;3 [d] and IPC 44:0;4 [e]) and MIPC (MIPC 44:0;3 [f] and MIPC 44:0;4 [g]) in WT (black), svf1Δ (dark gray), sec12ts (light gray), and svf1Δsec12ts (white) cells at the restrictive temperature (37°C). Error bars represent standard deviation from mean. (h–k) Measurements of the of the complex SPs IPC (IPC 44:0;3 (h) and IPC 44:0;4 (i) and MIPC (MIPC 44:0;3 [j] and MIPC 44:0;4 [k]) in WT, svf1Δ, nvj2Δ, svf1Δnvj2Δ, osh2Δosh3Δosh4Δ, osh2Δosh3Δosh4Δ svf1Δ, osh2Δosh3Δosh4Δ nvj2Δ, and osh2Δosh3Δosh4Δ svf1Δ nvj2Δ cells. The color code is indicated below. Error bars represent standard deviation from mean. (l) Flux analysis shows the incorporation of 2H6-inositol and 13C315N-serine into IPC species (44:0;3 and 44:0;4). Unlabeled IPC, inositol only labeled IPC (+2H6), serine only labeled IPC (+13C215N) and double labeled IPC (+13C22H615N) are shown in svf1Δ, svf1Δpl.Svf1-GFP and svf1Δpl.Svf1V12D-GFP at the time point t = −15 min and t = 90 min related to the addition of the tracers at t = 0 min. The IPC levels are expressed in pmol/0.4 OD units. (m) Flux analysis shows the incorporation of 13C315N-serine into PS species (34:2 and 34:1). Unlabeled PS and serine labeled PS (+13C215N) are shown in svf1Δ, svf1Δpl.Svf1-GFP and svf1Δpl.Svf1V12D-GFP at the time point t = −15 min and t = 90 min related to the addition of the tracer at t = 0 min. The PS level is expressed in pmol/0.4 OD units. (n) Flux analysis shows the incorporation of 2H6-inositol into PI 34:1 species. Unlabeled PI and inositol labeled PI (+2H6) are shown in svf1Δ, svf1Δpl.Svf1-GFP and svf1Δpl.Svf1V12D-GFP at the time point t = −15 min and t = 90 min related to the addition of the tracers at t = 0 min. The PI levels are expressed in pmol/0.4 OD units. (o) Serial dilutions of WT, svf1Δ, nvj2Δ, svf1Δnvj2Δ, dga1Δlro1Δare1Δare2Δ (Δ4), Δ4svf1Δ, Δ4nvj2Δ, and Δ4svf1Δnvj2Δ on YPD plates (control) and YPD plates containing 20nM Auroebasidin A (Aba). Bars represent mean ± SD from three independent samples.
Lipidomic analysis shows an effect of Svf1 on sphingolipid metabolism. (a and b) Measurement of the different SP intermediates in WT (black), svf1Δ (gray), and sur2Δ (white) cells shows an alteration of (a) the LCB (LCB 18:0;2 and LCB 18:0;3) levels and (b) the ceramide (CER 44:0;3 and CER 44:0;4) levels represented in pmol/µg protein. (c) From the same measurement the levels of the complex SPs IPC (IPC 44:0;3 and IPC 44:0;4) and MIPC (MIPC 44:0;3 and MIPC 44:0;4) are shown as peak areas (arbitrary unit [a.u.]). (d–g) Measurements of the of the complex SPs IPC (IPC 44:0;3 [d] and IPC 44:0;4 [e]) and MIPC (MIPC 44:0;3 [f] and MIPC 44:0;4 [g]) in WT (black), svf1Δ (dark gray), sec12ts (light gray), and svf1Δsec12ts (white) cells at the restrictive temperature (37°C). Error bars represent standard deviation from mean. (h–k) Measurements of the of the complex SPs IPC (IPC 44:0;3 (h) and IPC 44:0;4 (i) and MIPC (MIPC 44:0;3 [j] and MIPC 44:0;4 [k]) in WT, svf1Δ, nvj2Δ, svf1Δnvj2Δ, osh2Δosh3Δosh4Δ, osh2Δosh3Δosh4Δ svf1Δ, osh2Δosh3Δosh4Δ nvj2Δ, and osh2Δosh3Δosh4Δ svf1Δ nvj2Δ cells. The color code is indicated below. Error bars represent standard deviation from mean. (l) Flux analysis shows the incorporation of 2H6-inositol and 13C315N-serine into IPC species (44:0;3 and 44:0;4). Unlabeled IPC, inositol only labeled IPC (+2H6), serine only labeled IPC (+13C215N) and double labeled IPC (+13C22H615N) are shown in svf1Δ, svf1Δpl.Svf1-GFP and svf1Δpl.Svf1V12D-GFP at the time point t = −15 min and t = 90 min related to the addition of the tracers at t = 0 min. The IPC levels are expressed in pmol/0.4 OD units. (m) Flux analysis shows the incorporation of 13C315N-serine into PS species (34:2 and 34:1). Unlabeled PS and serine labeled PS (+13C215N) are shown in svf1Δ, svf1Δpl.Svf1-GFP and svf1Δpl.Svf1V12D-GFP at the time point t = −15 min and t = 90 min related to the addition of the tracer at t = 0 min. The PS level is expressed in pmol/0.4 OD units. (n) Flux analysis shows the incorporation of 2H6-inositol into PI 34:1 species. Unlabeled PI and inositol labeled PI (+2H6) are shown in svf1Δ, svf1Δpl.Svf1-GFP and svf1Δpl.Svf1V12D-GFP at the time point t = −15 min and t = 90 min related to the addition of the tracers at t = 0 min. The PI levels are expressed in pmol/0.4 OD units. (o) Serial dilutions of WT, svf1Δ, nvj2Δ, svf1Δnvj2Δ, dga1Δlro1Δare1Δare2Δ (Δ4), Δ4svf1Δ, Δ4nvj2Δ, and Δ4svf1Δnvj2Δ on YPD plates (control) and YPD plates containing 20nM Auroebasidin A (Aba). Bars represent mean ± SD from three independent samples.
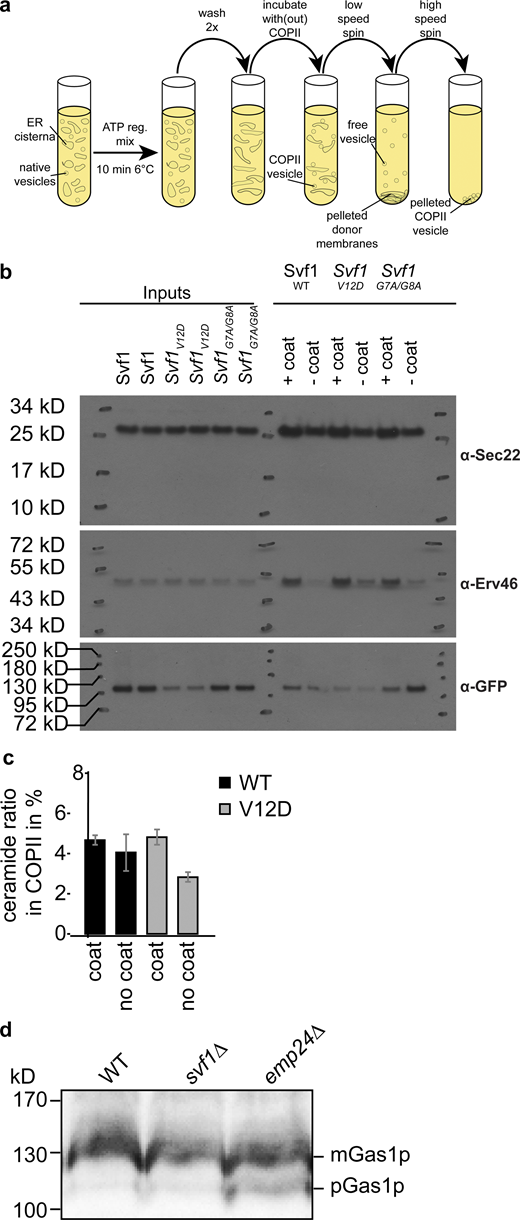
In vitro COP-II budding assays for lipidomic analysis. (a) Experimental setup of COPII budding assays. (b) Western blot analysis of COPII budding assays using antibodies against Sec22 (upper panels), Erv46 (middle panels), and GFP tagged Svf1 variants (lower panels). (c) Mass spectrometric analysis of ceramides from in vitro budded COP-II vesicles. The percentage of ceramide detected in the COPII vesicle fraction versus the sum of ceramides detected in both pelleted membranes and COPII vesicles is shown for WT Svf1 (black) and Svf1V12D (gray) for experiments with COPII coat added and without COPII coat added (n = 3). (d) Western blot analysis of Gas1 in WT, svf1Δ, and emp24Δ cells. Only in emp24Δ the pre-form of Gas1 is detected. Source data are available for this figure: SourceData FS5.

In vitro COP-II budding assays for lipidomic analysis. (a) Experimental setup of COPII budding assays. (b) Western blot analysis of COPII budding assays using antibodies against Sec22 (upper panels), Erv46 (middle panels), and GFP tagged Svf1 variants (lower panels). (c) Mass spectrometric analysis of ceramides from in vitro budded COP-II vesicles. The percentage of ceramide detected in the COPII vesicle fraction versus the sum of ceramides detected in both pelleted membranes and COPII vesicles is shown for WT Svf1 (black) and Svf1V12D (gray) for experiments with COPII coat added and without COPII coat added (n = 3). (d) Western blot analysis of Gas1 in WT, svf1Δ, and emp24Δ cells. Only in emp24Δ the pre-form of Gas1 is detected. Source data are available for this figure: SourceData FS5.
To further confirm our results, we used multi-pathway flux analysis (Martínez-Montañés et al., 2020), systematically analyzing the incorporation of serine and inositol in SPs, phosphatidylinositol (PI), and phosphatidylserine (PS; Fig. S6 a). We incubated svf1Δ cells, svf1Δ cells expressing Svf1-GFP, svf1Δ cells expressing Svf1V12D-GFP with 13C315N serine and 2H6-inositol and measured its incorporation into IPC (Esch et al., 2020; Martínez-Montañés et al., 2020). This multi-pathway flux analysis revealed a decreased synthesis rate of the inositol-containing SPs in svf1Δ and Svf1V12D expressing cells (Fig. 6 l), further supporting a role for Svf1 in ceramide transport and SP homeostasis. Importantly, we did not observe any detectable differences in the abundance of 13C315N serine-containing PS (Fig.6 m) and 2H6-inositol containing (PI (Fig. 6 n) suggesting, that indeed, ceramide transport is affected by the deletion of SVF1, and not, for example the transport of PI to the Golgi apparatus, which is required as a headgroup donor for the synthesis of SPs (Mousley et al., 2008).

Model for sphingolipid metabolism and Svf1 co-localization with the IPC synthase. (a) Overview of the multi-pathway flux analysis. 13C315N-serine and 2H6-inositol were added as tracers to exponentially growing cells. The potential incorporation into SP metabolites are shown in a simplified model of yeast SP metabolism. KDH, 3-ketodihydrosphingosine; DHS, dihydrosphingosine; PHS, phytosphingosine; PI, phosphatidylinositol; IPC, inositol phosphorylceramide; MIPC, mannosylinositol phosphorylceramide; M(IP)2C, mannosyldiinositol phosphorylceramide as well as in phosphatidylinositol and phosphatidylserine. (b) Svf1-GFP (green) was expressed in cells expressing Mnn9-mKate (cis-Golgi, red) and Aur1-Halo (mid-Golgi, blue). Scale bar = 5 µM. (c) Qunatification of Svf1 dots co-localizing with Mnn9, Aur1, Mnn9, and Aur1, only Mnn9, and only Aur1. (n = 100 Svf1 dots, triplicates).

Model for sphingolipid metabolism and Svf1 co-localization with the IPC synthase. (a) Overview of the multi-pathway flux analysis. 13C315N-serine and 2H6-inositol were added as tracers to exponentially growing cells. The potential incorporation into SP metabolites are shown in a simplified model of yeast SP metabolism. KDH, 3-ketodihydrosphingosine; DHS, dihydrosphingosine; PHS, phytosphingosine; PI, phosphatidylinositol; IPC, inositol phosphorylceramide; MIPC, mannosylinositol phosphorylceramide; M(IP)2C, mannosyldiinositol phosphorylceramide as well as in phosphatidylinositol and phosphatidylserine. (b) Svf1-GFP (green) was expressed in cells expressing Mnn9-mKate (cis-Golgi, red) and Aur1-Halo (mid-Golgi, blue). Scale bar = 5 µM. (c) Qunatification of Svf1 dots co-localizing with Mnn9, Aur1, Mnn9, and Aur1, only Mnn9, and only Aur1. (n = 100 Svf1 dots, triplicates).
According to our data, it remains possible that Svf1 affects SP metabolism at the Golgi by regulating the activity of Aur1, the yeast IPC synthase. We therefore analyzed the localization of Svf1 compared with Mnn9 at this cis-Golgi and Aur1 as a mid-Golgi marker (Fig. S6, b and c). Quantification of the data revealed that almost all Svf1 spots were positive for Mnn9, whereas only half of the Svf1 structures were also positive for Aur1. When we analyzed how many Svf1 dots were positive for only one of the two markers, Mnn9 and Aur1, we found that half of the Svf1 dots were co-localized with Mnn9, whereas only 5% were exclusively co-localized with Aur1. This reinforces our hypothesis that Svf1 is localized at the cis-Golgi. The co-localization with Aur1 most likely reflects the dynamics of Golgi maturation in yeast. Furthermore, we did not observe any physical interaction between Svf1 and Aur1 in our protein–protein interaction studies, making it unlikely that Svf1 directly regulates Aur1.
Next, we investigated the effect of increased ceramide levels resulting from SVF1 deletion on yeast physiology. Accumulation of ceramides is toxic to yeast cells (Eisenberg and Büttner, 2014). A pathway for the detoxification of ceramides is the acylation of the free hydroxyl group to yield acyl-ceramide that can be stored in lipid droplets (LDs). This process depends on the diacylglycerol acyltransferases (DGATs) Lro1 and Dga1 (Voynova et al., 2012). We spotted WT cells, svf1Δ, nvj2Δ, svf1Δnvj2Δ, dga1Δlro1Δare1Δare2Δ (Δ4), Δ4svf1Δ, Δ4nvj2Δ, and Δ4svf1Δnvj2Δ cells on control plates or plates containing the IPC synthase inhibitor Auroebasidin A (AbA) to further increase ceramide toxicity (Fig. 6 o). The deletion of both DGATs LRO1 and DGA1 together with the deletion of the acyl-CoA sterol acyltransferase (ACATs) ARE1 and ARE2 yields a strain that does not harbor any LDs (Sandager et al., 2002). While svf1Δ and Δ4 cells already showed mild growth defects in the presence of AbA, the combination of both resulted in almost no detectable cell growth (Fig. 6 o). In contrast, combining the NVJ2 deletion with the Δ4 mutant showed only mild growth defects similar to the Δ4 mutant alone (Fig. 6 o). Combining the Δ4 mutant with both svf1Δ and nvj2Δ again resulted in a strong growth defect, suggesting that ceramide toxicity is problematic for cells harboring SVF1 deletions.
Svf1 is a ceramide binding protein
Our results suggest that Svf1 is a vesicular transport independent ceramide transfer protein at the interface between the ER and the cis-Golgi apparatus. Therefore, we tested whether Svf1 is capable of binding ceramides. Svf1 harbors two lipocalin domains that are each made of anti-parallel β-sheets (Weekes et al., 2010). We first used the AlphaFold algorithm (Varadi et al., 2022; Jumper et al., 2021) to predict the structure of Svf1. These predictions yield high confidence structures for the two lipocalin domains. Two flexible loops do not yield any structure prediction and the α-helix at the N-terminus of the protein is also predicted with a low confidence score (Fig. 7 a). A different view on the predicted structure highlights the two lipocalin domains in blue and purple that harbor a hydrophobic cleft in each of the lipocalin domains, as well as in between the two domains (Fig. 7 b). To identify potential ceramide binding sites, we performed docking studies of amphiphilic ligands to assess Svf1 ligand binding sites and lipid loading configurations. Although docking methods failed to reproduce accurately experimental binding energies, their capability to compare affinities at different binding sites and to generate the most probable configurations is well known (Nguyen et al., 2020). Ceramide 18:0;3/26:0;1 (CER 44:0;4) was assayed in docking experiments with different grids comprising the whole protein, as well as the cavities at each of the β-barrels. All bonds, except for double bonds, were maintained flexible along docking assays. The most probable interaction was found to be at the inter-barrel cleft near an α-helix cap (Fig. 7 c) with binding energies of −6.4 to −5.9 kcal mol−1 for ceramide 44:0;4 for the first most favorable configuration. Both the preferred configurations and the binding energies were found to be independent of the grid size. No stable configuration was produced when forcing the binding to the barrel cavity, while a peripheral binding site to the C-terminus barrel was found near D331, S332, and F466, with a lower binding score of −4.6 and −4.16 kCal mol−1 for ceramide 44:0;4. Together, these simulations render it possible that Svf1 indeed binds ceramide 44:0;4 in the hydrophobic cleft in between the two lipocalin domains. To directly test whether Svf1 binds ceramides, we purified FLAG tagged Svf1, Svf1V12D, and Svf1G7A/G8A without detergents from yeast strains overexpressing the proteins under control of the GAL promoter. All proteins were stable and had the expected molecular weight based on SDS-PAGE and mass photometry experiments (Fig. S7). We used the purified proteins to extract lipids and determined the amount of co-purified ceramide 44:0;4 and, as a control, 34:1 phosphatidylcholine (PC 34:1) using targeted lipidomics (Fig. 7, d and e). We detected 44:0;4 ceramide co-purified with Svf1 in high amounts compared to general ceramide levels in the cell. Both Svf1 mutants yielded similar amounts of ceramide bound to the proteins, suggesting that Svf1 binds ceramide independent of its AH (Fig. 7 d). In contrast, the amounts of PC co-purified with the protein were strongly depending on the amphipathic character of the helix, suggesting that phospholipids are bound to the AH itself, consistent with its function in organelle targeting (Fig. 7 e). This suggests that Svf1 is able to extract ceramide from membranes independent of its AH.

Svf1 binds ceramide in a hydrophobic pocket between its two lipocalin domains. (a) AlphaFold prediction of the structure of Svf1. AlphaFold produces a per-residue confidence score (pLDDT) between 0 and 100 according to the color code. (b) Different visualization of the predicted structure of Svf1 with its two lipocalin domains colored in blue and in purple and the N-terminal AH color coded in orange. (c) Results of the molecular docking studies. A 44:0;4 ceramide (purple) can be accommodated in the hydrophobic cleft between the two lipocalin domains. Amino acids and the ceramide headgroup are shown as balls and sticks in the enlarged view. (d and e) Targeted lipidomic analysis of ceramide 44:0;4 and (e) PC 16:0/18:1 extracted from the purified proteins. Proteins were purified via a FLAG-tag and extracts were used for chloroform methanol extraction of co-purified lipids. Lipids co-purified with Svf1-FLAG (black), Svf1V12D-FLAG (dark gray), Svf1G7AG8A-FLAG (medium gray) and Svf1H273AH274A-FLAG (light gray) are shown. Bars represent mean ± SD from four independent samples. (f) Cartoon model of the predicted structure of Svf1 with the AH helix colored in light orange and the small α-helical cap colored in dark orange. The two side chains of the histidines H273 and H274 are shown. (g) Co-localization of GFP tagged Svf1 expressed from a plasmid under control of the endogenous promoter with mKate tagged Mnn9 and Halo tagged Sec63 for the WT (upper panel) and the H273A H274A mutant (lower panel). Scale bar = 5 µM. (h) Tetrad analysis of the svf1Δpl.Svf1H273AH274A-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). Source data are available for this figure: SourceData F7.
Svf1 binds ceramide in a hydrophobic pocket between its two lipocalin domains. (a) AlphaFold prediction of the structure of Svf1. AlphaFold produces a per-residue confidence score (pLDDT) between 0 and 100 according to the color code. (b) Different visualization of the predicted structure of Svf1 with its two lipocalin domains colored in blue and in purple and the N-terminal AH color coded in orange. (c) Results of the molecular docking studies. A 44:0;4 ceramide (purple) can be accommodated in the hydrophobic cleft between the two lipocalin domains. Amino acids and the ceramide headgroup are shown as balls and sticks in the enlarged view. (d and e) Targeted lipidomic analysis of ceramide 44:0;4 and (e) PC 16:0/18:1 extracted from the purified proteins. Proteins were purified via a FLAG-tag and extracts were used for chloroform methanol extraction of co-purified lipids. Lipids co-purified with Svf1-FLAG (black), Svf1V12D-FLAG (dark gray), Svf1G7AG8A-FLAG (medium gray) and Svf1H273AH274A-FLAG (light gray) are shown. Bars represent mean ± SD from four independent samples. (f) Cartoon model of the predicted structure of Svf1 with the AH helix colored in light orange and the small α-helical cap colored in dark orange. The two side chains of the histidines H273 and H274 are shown. (g) Co-localization of GFP tagged Svf1 expressed from a plasmid under control of the endogenous promoter with mKate tagged Mnn9 and Halo tagged Sec63 for the WT (upper panel) and the H273A H274A mutant (lower panel). Scale bar = 5 µM. (h) Tetrad analysis of the svf1Δpl.Svf1H273AH274A-GFP (blue and green, respectively) mutants crossed with sur2Δ (red). Source data are available for this figure: SourceData F7.
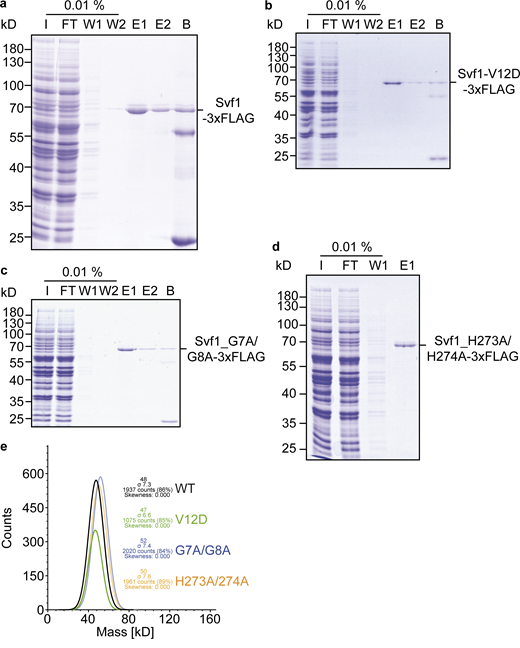
Purification of Svf1 from yeast cells. (a) Svf1-FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; W2, wash 2; E1, eluate 1; E2, eluate 2; B, beads. (b)Svf1V12D-FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; W2, wash 2; E1, eluate 1; E2, eluate 2; B, beads. (c)Svf1G7AG8A -FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; W2, wash 2; E1, eluate 1; E2, eluate 2; B, beads. (d)Svf1H273AH274A -FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; E1, eluate 1. (e) Mass photometry analysis of the purified proteins from a–d. Source data are available for this figure: SourceData FS7.

Purification of Svf1 from yeast cells. (a) Svf1-FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; W2, wash 2; E1, eluate 1; E2, eluate 2; B, beads. (b)Svf1V12D-FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; W2, wash 2; E1, eluate 1; E2, eluate 2; B, beads. (c)Svf1G7AG8A -FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; W2, wash 2; E1, eluate 1; E2, eluate 2; B, beads. (d)Svf1H273AH274A -FLAG overproduced from the GAL1 promotor was purified via FLAG tag and analyzed by SDS-PAGE. I, Input; FT, flow through; W1, wash 1; E1, eluate 1. (e) Mass photometry analysis of the purified proteins from a–d. Source data are available for this figure: SourceData FS7.
Based on these data, we aimed at identifying mutations in Svf1 that interfere with ceramide binding. Most of the amino acids in the cleft between the two lipocalin domains are hydrophobic and changing them can interfere with folding of the entire protein. We were unable to identify mutations that add large hydrophobic headgroups that generate additional mass, blocking ceramide from entrance into the pocket. We therefore focused on two histidine residues located in a small α-helical stretch that lies over the potential ceramide binding pocket (Fig. 7 f). Mutation of both histidines to alanines (H273A/H274A) resulted in the purification of a stable protein (Fig. S7, d and e). However, we could barely detect any co-purified ceramide 44:0;4 showing that ceramide binding was almost completely blocked in this mutant (Fig. 7 d). Interestingly, we were also not able to detect any significant amount of co-purified PC 34:1, suggesting that the AH of Svf1 was also affected by the mutations (Fig. 7 e). In line with these results, expressing the GFP tagged Svf1H273A/H274A mutant in cells led to a complete loss of co-localization with the Mnn9 positive cis-Golgi structures compared to WT Svf1-GFP (Fig. 7 g). The Svf1H273A/H274A mutant did not rescue the negative genetic interaction between SVF1 and SUR2 (Fig. 7 h). Together, our results show that the two histidines in the cap region are required for ceramide binding and targeting of Svf1 to the Golgi apparatus. Based on the molecular docking studies, one of the histidines can potentially interact with the apolar C26 acyl chain of the ceramide. However, the other histidine is, based on the structure predictions, close to a negatively charged aspartate in the AH. It is possible that the presence of ceramide in the binding pocket of Svf1 could lead to a protein rearrangement that brings the AH in a stable conformation that is than targeting the Golgi apparatus. However, this can only be determined by solving the structure of the protein. Taken together, our data suggest that Svf1 is a ceramide-binding protein that affects the conversion of ceramides to cSPs at the cis-Golgi, most likely by the transfer of ceramides from the ER to the Golgi apparatus.
Discussion
Here we showed that the yeast protein Svf1 is a ceramide-binding protein. Svf1 harbors two lipocalin domains (Flower, 1996) with a hydrophobic cleft in between that allows the accommodation of ceramides. The deletion of SVF1 results in the accumulation of ceramides with a concomitant decrease in complex SPs suggesting that Svf1 transports ceramides between the ER, where they are synthesized, and the Golgi apparatus where they are used as a substrate for the biosynthesis of complex SPs (Körner and Fröhlich, 2022). Accordingly, we detected a Svf1 pool that is dynamically localized at the cis-Golgi apparatus as well as a cytosolic pool. In addition, immune-purification of Svf1 yields potential interaction partners from both organelles, the ER and the Golgi apparatus. For its Golgi targeting, Svf1 requires its N-terminal-AH helix as well as an N-terminal acetylation, similar to other proteins that target the Golgi via an AH (Behnia et al., 2007). In addition, we identified two histidine residues in a small predicted α-helical cap that are important for keeping ceramide in the binding pocket as well as cis-Golgi targeting of the protein.
In summary, we proposed a model where Svf1-mediated non-vesicular ceramide transport acts in parallel to the vesicular transport pathway (Fig. 8). In this model, Svf1 first picks up ceramide from the ER. The interaction of Svf1 appears to be very transient as we can only detect Svf1 at the ER when the two glycines in its AH are exchanged for alanines. The accommodation of ceramide in the binding pocket of Svf1 is directly dependent on the two histidines in the cap region of a small α-helix. Svf1 is then targeted to the Golgi apparatus via its AH. This specificity of this targeting might rely on a marker lipid at the Golgi, such as PI4P or a yet unidentified protein interaction partner of Svf1. Ceramides are released at the Golgi and used as a substrate for the biosynthesis of cSPs (Fig. 8). The AH must have some very specific properties, as the replacement of the AH of Svf1 with that of Grh1 does not allow targeting of the Golgi. The targeting of Svf1 to the cis-Golgi might also directly depend on the close proximity of ER exit sites and the cis Golgi apparatus (Ikeda et al., 2020; Kurokawa et al., 2014). In an alternative model, Svf1 is not directly transporting ceramides but rather acts as a sensor for ceramides that then regulates Aur1 activity in the Golgi apparatus. Since Svf1 mainly localizes at the cis Golgi marked by Mnn9 and not the medial Golgi marked by Aur1, we favored the first model where Svf1 acts as a ceramide transport protein.

Model for the proposed function of Svf1 in ceramide transport at the ER-Golgi interface. A model for vesicular and non-vesicular ceramide transport in yeast under normal growth conditions. Ceramides are transported by vesicular transport while Nvj2 resides in the NVJ. Under these conditions Svf1 transports ceramides in a non-vesicular pathway. Svf1 first picks up ceramide in the ER in a transient interaction with the organelle. Svf1 targets the cis-Golgi apparatus and releases ceramides that is than metabolized into cSPs.
Model for the proposed function of Svf1 in ceramide transport at the ER-Golgi interface. A model for vesicular and non-vesicular ceramide transport in yeast under normal growth conditions. Ceramides are transported by vesicular transport while Nvj2 resides in the NVJ. Under these conditions Svf1 transports ceramides in a non-vesicular pathway. Svf1 first picks up ceramide in the ER in a transient interaction with the organelle. Svf1 targets the cis-Golgi apparatus and releases ceramides that is than metabolized into cSPs.
Another important question is how ceramide is released from the hydrophobic pocket of Svf1. At the moment, we can only speculate about this without having structural data on Svf1 available. It is important to highlight that our results depend on structure predictions based on the AlphaFold algorithm (Varadi et al., 2022; Jumper et al., 2021). However, one possible model is that ceramide is exchanged via another lipid at the Golgi apparatus. An attractive candidate would be diacylglycerol (DAG) as a product of complex SP biosynthesis. It remains after the phosphoinositol headgroup of PI is transferred to ceramide. DAG, similar to ceramide only harbors a hydroxyl group as a headgroup, allowing both of the molecules to flip between the two leaflets of the Golgi membrane (Holthuis and Levine, 2005). Determining the mechanism of ceramide release at the Golgi will be an important point to address in the future.
As mentioned before, several different pathways contribute to ceramide transport in yeast. Non-vesicular transport depends on the Nvj2 protein. This pathway is mainly active under conditions of artificial ER stress and transports ceramides preferably to the medial-Golgi (Liu et al., 2017). In addition, yeast tricalbins have also been implicated in non-vesicular ceramide transport (Ikeda et al., 2020). In mammalian cells, the majority of ceramides are transported by CERT (Hanada et al., 2003). Some ceramides that are substrates for the biogenesis of glycosyl-ceramides appear to be also transported by vesicular transport (Halter et al., 2007). Vesicular transport in yeast depends on the COP-II machinery and accounts for about 60–80% of ceramides transported (Funato and Riezman, 2001). In yeast, the Osh proteins have also been implicated in vesicular ceramide transport, but the molecular mechanism remains unknown (Kajiwara et al., 2014). Osh4 is clearly described as lipid transfer protein that exchanges sterols for PI4Ps between the ER and the Golgi apparatus (de Saint-Jean et al., 2011). Osh2 is the homolog of Osh1, which localizes at the nuclear vacuolar junction (NVJ; Olkkonen, 2015; Kvam and Goldfarb, 2004), but its exact role here has not been determined. Osh3 is mainly found at ER plasma membrane contact sites where also the tricalbins play an important role (Olkkonen, 2015; Stefan et al., 2011). In summary, the effects of the triple knockout of OSH2, OSH3, and OSH4 on ceramide transport are more likely the result of an overall metabolic adaptation of yeast cells to changes in lipid transport and or metabolism. Based on our lipidomics experiments, Svf1 acts in a parallel pathway to COP-II-dependent vesicular transport and Nvj2 mediated non-vesicular transport (Fig. 8). Especially the latter could be viewed as a detoxification mechanism that becomes active once ceramides accumulate in the ER and cause ER stress. Alternatively, Nvj2 could also transport ceramides at the NVJ. Another protein localizing to the NVJ, yeast Cvm1, has recently been suggested to function in SP and ceramide homeostasis (Bisinski et al., 2022). However, yeast cells have multiple mechanisms to transport ceramides between the ER and the Golgi and potentially between other organelles. One explanation could be that ceramides are toxic to cells when they accumulate (Eisenberg and Büttner, 2014). This is also supported by the detoxification mechanism resulting in the acylation of ceramides allowing the cell to store them in lipid droplets (Voynova et al., 2012).
Finally, is the function of Svf1 conserved? Svf1 belongs to the family of lipocalins (Flower, 1996; Lu et al., 2020). While the lipocalin fold is a highly conserved structure, its sequence conservation is very low (Flower, 1996). This makes it difficult to identify homologs of Svf1 in higher eukaryotes. Based on our data, proteins harboring two lipocalin domains are likely candidates for ceramide transporting Svf1 homologs. However, lipocalins have been implicated in the transport and solubilization of small hydrophobic molecules, e.g., fatty acids and other lipids. Prominent examples are the fatty acid binding proteins (Smathers and Petersen, 2011) that are described as transport proteins for a variety of fatty acids (McArthur et al., 1999). Since we also detect interactions between Svf1 and the VLCFA elongation complex, a handover of activated VLCFAs to the ceramide synthases would be an attractive model for the molecular function of Svf1. While the lack of Svf1 would then also result in lower levels of complex SPs, this model is contrary to the increase of ceramides we are observing in svf1Δ cells. A direct interaction of ceramides and a lipocalin protein have also been observed for the tear lipocalin protein (Glasgow and Abduragimov, 2018). It thus remains possible that a lipocalin protein has a similar function to Svf1 in higher eukaryotes. Along this line, it is especially interesting that ceramides transported by CERT are usually containing C16 or C18 acyl chains (Loizides-Mangold et al., 2012). Sorting of VLCFA-containing ceramides by lipocalin domain containing proteins could thus be an evolutionary conserved mechanism.
Material and methods
Yeast strains and plasmids
Growth conditions and media
Tetrad dissections were performed on standard YPD plates and incubated at 30°C for 72 h. Yeast strains were grown according to normal growth conditions. For spotting assays, myriocin or Aureobasidin A were added at concentrations as indicated and the plates were grown at 30°C for 48 h. For SILAC experiments SDC medium without lysine with 2% glucose, 6.7 g/liter yeast nitrogen base without amino acids and yeast synthetic dropout without lysine (Sigma-Aldrich) was used. Normal or heavy lysine (L-lysine 13C615N2; Cambridge Isotope Laboratories) at the concentration of 30 g/liter were added. Cells were diluted from an overnight pre-culture to an OD600 = 0.2 and grown at 30°C. Cells were harvested at an OD600 = 1.0–1.4.
Colony size quantification
For the quantification of colony size, ImageJ was used. The obtained image was converted to a binary. A circle was placed around each tetrad, and the mean intensity values were measured.
FastCloning
For an insertion of a DNA fragment into a vector, the FastCloning method established by Li et al. (2011) was used. In short, fragments were amplified with 15–17 bp homologous overlapping overhangs at both ends to the opposite fragment. After digestion of the initial methylated DNA by DpnI at 37°C for 1 h, the fragments were transformed in a 1:1 M ratio followed by homologous recombination in E. coli DH5α competent cells.
Q5 mutagenesis
The Q5 mutagenesis method was used to generate deletions or point mutations in a desired DNA locus. Primers were designed using NEBaseChanger (New England BioLabs, Inc.; version 1.3.2; Table 1). After amplification of the desired DNA fragment, the initial DNA was digested by DpnI at 37°C for 1 h. After a PCR clean up (NucleoSpin Gel and PCR Clean-up; Macherey-Nagel) the PCR fragment was phosphorylated by the T4 PNK Polynucleotide Kinase (New England BioLabs Inc.) for 30 min at 37°C and afterwards ligated by the T4 DNA ligase over night at 4°C.
Primer list
| Name | Sequence |
|---|---|
| Svf1 S1 | 5′-TGCATCGAATCAAGAAAAAAGAAACGAATAAGCTTTTGTATAATATAATATAATGCGTACGCTGCAGGTGCAC-3′ |
| Svf1 S2 | 5′-GTAGTATTAAATCACAGATAGGAAAAAGCATACGAGACCTTCCCACTTTTGCTCAATCGATGAATTCGAGCTCG-3′ |
| Svf1 S3 | 5′-CATTCATTTCTGAATCAGATGTGATTAGTGAGGAGTCTTACAATGAAGCACGTACGCTGCAGGTCGAC-3′ |
| Svf1_pom_n_GFP_fo | 5′-AATCAAGAAAAAAGAAACGAATAAGCTTTTGTATAATATAATATAATGTGCAGGTCGACAACCCTTAAT-3′ |
| Svf1_pom_n_GFP_re | 5′-AGCCATACCAGTGACTGCTGAGATCCCACCTTTTATCCACTTCAAGCGGCCGCATAGGCCACT-3′ |
| SUR2_S1 | 5′-TAACATTCTAGTCCGAAGAGGGTGTATACGAAAAGAAAATATACGATGCGTACGCTGCAGGTCGAC-3′ |
| SUR2_S2 | 5′-GACATTGCCTTTACCCAGCAATTGAACGGGAGGTATGCAAAAGGGTTAATCGATGAATTCGAGCTCG-3′ |
| Nvj2_S1 | 5′-CATCGAAGAGCAGAACAGCAAGAGAAAAGTAGCATTAAAAGACCATAATGCGTACGCTGCAGGTCGAC-3′ |
| Nvj2_S2 | 5′-GCTTCAAGTGATATTTATTTATTTTTAATATAGTACCGTGGACTCAATCGATGAATTCGAGCTCG-3′ |
| Mak3_S1 | 5′-GATTACAAGATAAAAAAGCCACTACTACAGAAAAGGCGTTGGGTCAGGACATGCGTACGCTGCAGGTCGAC-3′ |
| Mak3_S2 | 5′-GTATTATTAATATATATTTTATCATCATCGAGTGTTTTTCCTTTTAATCGATGAATTCGAGCTCG-3′ |
| Svf1_V12D_for | 5′-ATCTCAGCAGACACTGGTATG-3′ |
| Svf1_V12D_rev | 5′-CCCACCTTTTATCCACTTC-3′ |
| Ydr222w_S1 | 5′-GTGAGGGCGTATAAGATACATCGTACATACATAGAGACTCATTTAGTGATGCGTACGCTGCAGGTCGAC-3′ |
| Ydr222w_S2 | 5′-GAAAAAAAAGGACAAAAAACGAATTTCATGTGGAAGTGTTCACGCTTTTGTTCAATCGATGAATTCGAGCTCG-3′ |
| Ylr225c_S1 | 5′-GGGCAACGAACAGTTTAATTAACATAAAAATCATAGAGCAAAGAAAAAATGCGTACGCTGCAGGTCGAC-3′ |
| Ylr225c_S2 | 5′-GATATAATATTCTTGCAATGATGAAAATATGTATAGTCAAATTTCATTTTTTTCAATCGATGAATTCGAGCTCG-3′ |
| Svf1 G7AG8A Q5 for | 5′-TGGATAAAAGCTGCGATCTCAGCAGTC-3′ |
| Svf1 G7AG8A Q5 rev | 5′-CTTCAACATTATATTATATTATACAAAAGC-3′ |
| Osh2_S1 | 5′-ATATTTCATCGATAATCATAAGCTTAAGCTCGCCAACTGAAAACTTCACAATATGCGTACGCTGCAGGTCGAC-3′ |
| Osh2_S2 | 5′-CATTCAAAATACATACAAGTACCAGGAAAAAAGCTCGCATAAAAAAGGCGTGTTAATCGATGAATTCGAGCTCG-3′ |
| Osh3_S1 | 5′-ATTTGCTGGAGAGTAGTAAAACGAGCTTGAATAGCAGCCTGCGAATCGAATTATGCGTACGCTGCAGGTCGAC-3′ |
| Osh3_S2 | 5′-ATAAATAGAGTACAAAGGGGGGAAAAAAGTTCACTTGATGTCATCAAGGCATTCAATCGATGAATTCGAGCTCG-3′ |
| Osh4_S1 | 5′-CCTAGAAACAAATCGAAAAATTTATAAGATTTAGTCTCAAGAATTTCAAGTCATGCGTACGCTGCAGGTCGAC-3′ |
| Osh4_S2 | 5′-GATAGATAATATATTAGTGCAACGGTAACAAGTTGTTACTTTATCGTTCTCCTTAATCGATGAATTCGAGCTCG-3′ |
| Sec31 S2 | 5′-CAAGGCCAATACGCCACTTTTTGTACTGAAAGTTTTGAGACTGAATTAATCGATGAATTCGAGCTCG-3′ |
| Sec31 S3 | 5′-CTGGCTGACAGGAGTGAAGAGGTTGATTGGCATAGCTGAAGCGACTTTGAATCGTACGCTGCAGGTCGAC-3′ |
| Nvj2_pRS_KO_for | 5′-GGTTTTGAACACATCGAAGAGCAGAACAGCAAGAGAAAAGTAGCATTAAAAGACCAGATTGTACTGAGAGTGCAC-3′ |
| Nvj2_pRS_KO_rev | 5′-GCATATAGCTTCAAGTGATATTTATTTATTTTTAATATAGTACCGTGGACCTGTGCGGTATTTCACACCG-3′ |
| Q5_L2E_for | 5′-TAATATAATGGAGAAGTGGATAAAAGGTG-3′ |
| Q5_L2E_rev | 5′-TATTATACAAAAGCTTATTCGTTTC-3′ |
| HAHA_for | 5′-CATGAAGCCTGCTGCCGCAGCTAAGG-3′ |
| HAHA_rev | 5′-CCTTGGAAGGCCATTATAAATG-3′ |
| AUR1_S2 | 5′-ATATGTATCTACATAAGACCAACCGTATCCGTAATTGCAGATAAAATACTCATTAATCGATGAATTCGAGCTCG-3′ |
| AUR1_S3 | 5′-TTCTGTTTCTCGTTCGTCCGCCACGTCTATAACGTCACTAGGTGTAAAGAGGGCTCGTACGCTGCAGGTCGAC-3′ |
| Svf1_19aa_for_Grh1_AH | 5′-GGACTTTTGAGCAGAGCGTGCAAGATGAATATGGGAAAGATTACATTCATAGTG-3′ |
| Svf1_pr_rev_oh_Grh1AH | 5′-GTACGAGGTTTTTAGCTATTCTAAACATTATATTATATTATACAAAAGCTTATTCGTTTC-3′ |
| GFP_for_oh_Svf1 | 5′-AGTCTTACAATGAAGCACGTACGCTGCAGGTCGACGG-3′ |
| GFP_rev_oh_Svf1_t | 5′-GACCTTCCCACTTTTGCTTATTTGTACAATTCATCCATACC-3′ |
| pRS405Svf1_term_f | 5′-ATGAATTGTACAAATAAGCAAAAGTGGGAAGGTCTCG-3′ |
| pRS405Svf1_rev_oh | 5′-TCGACCTGCAGCGTACGTGCTTCATTGTAAGACTCCTCAC-3′ |
| pRS405Svf1_AH_GFP_Q5mut_for | 5′-CGTACGCTGCAGGTCGAC-3′ |
| pRS405Svf1_AH_GFP_Q5mut_rev | 5′-AGGCTCAGCCATACCAGTG-3′ |
| Galpr_for_oh_pRS | 5′-CCGGGCCCCCCCTCGAGTGAAGTACGGATTAGAAGCC-3′ |
| Galpr_rev_oh_Svf1 | 5′-TTTATCCACTTCAACATGTTTTTTCTCCTTGACGTTAAAG-3′ |
| Svf1_for_oh_Galpr | 5′-CGTCAAGGAGAAAAAACATGTTGAAGTGGATAAAAGGTG-3′ |
| pRS_rev_oh_Galpr | 5′-TTCTAATCCGTACTTCACTCGAGGGGGGGCCCGG-3′ |
| Svf1_term_oh_3xFlag | 5′-TACAAAGATGACGACGATAAGGATTATAAGGACGACGACGACAAATAATCGTATGCTTTT-3′ |
| Svf1_rev_3xFLAG_oh | 5′-TCGTCGTCATCTTTGTAGTCCTTGTCATCATCATCCTTGTAATCTGCTTCATTGTAAGAC-3′ |
| Mnn9_S2 | 5′-TCTTTCAATAACGCTATAGCTTCTGTATGCTTTTTGCTCAGTTGCTCAATCGATGAATTCGAGCTCG-3′ |
| Mnn9_S3 | 5′-TTTGGCTTACCAAACTATTTGGTTTATCACATAGAGGAAGAGAACCATCGTACCGCTGCAGGTCGAC-3′ |
| Sec63_S2 | 5′-ATACGTCTAAGAGCTAAAATGAAAAACTATACTAATCACTTATATCTAATCGATGAATTCGAGCTCG-3′ |
| Sec63_S3 | 5′-ACTGATATCGATACGGATACAGAAGCTGAAGATGATGAATCACCAGAACGTACCGCTGCAGGTCGAC-3′ |
| Sec7_S2 | 5′-TTCTACAACTAAGCATATTTTAATCTGCTGGACCATTCAACAAAGCCTTAATCGATGAATTCGAGCTCG-3′ |
| Sec7_S3 | 5′-CTATAAAACAATTTCTAAGCAGAGTTGGTGAATTATACCTTTCTACTGATCGTACCGCTGCAGGTCGAC-3′ |
| Vps4_S2 | 5′-TATTTTCATGTACACAAGAAATCTACATTAGCACGTTAATCAATTGACTAATCGATGAATTCGAGCTCG-3′ |
| Vps4_S3 | 5′-GACTTGCTGAAGCAAGAACAGTTCACTAGAGATTTTGGTCAAGAAGGTAACCGTACCGCTGCAGGTCGAC-3′ |
| Lys2_S1 | 5′-GAAAAACTGCTAATTATAGAGAGATATCACAGAGTTACTCACTAATGCGTACCGCTGCAGGTCGAC-3′ |
| Lys2_S2 | 5′-CATATTTAATTATTGTACATGGACATATCATACGTAATGCTCAACCTTAATCGATGAATTCGAGCTCG-3′ |
Fluorescence microscopy
Cells were grown to logarithmic growth phase in synthetic medium supplemented with essential amino acids (SDC). For the visualization of the Halo tag, one OD unit cells were centrifuged and resuspended in 50 μl SDC medium. Then, 0.625 μl Janelia Fluor646 HaloTag Ligand (stock concentration: 200 µM; Promega) were added and the cells were incubated for 30 min at 30°C shaking. Afterwards, the cells were washed 10 times with 1 ml SDC medium, transferred to a new tube and were used for imaging. Cells were imaged live in SDC medium on an Olympus IX-71 inverted microscope equipped with 100× NA 1.49 and 60× NA 1.40 objectives, a sCMOS camera (PCO), an InsightSSI illumination system 4′,6-diamidino-2-phenylindole, GFP, mCherry, and CY5 filters, and SoftWoRx software (Applied Precision). We used constrained-iterative deconvolution (SoftWoRx). ImageJ (National Institutes of Health; RRID:SCR_003070) was used for the processing and quantification of the images.
Statistical analysis
Statistical analysis was based on two-sided t tests when two conditions were compared: *, P < 0.05; **, P < 0.01; ***, P < 0.001. For microscopy experiments, the statistical comparisons were performed among the means of individual experiments, not based on data points for individual cells. For SILAC plots (ratio versus intensity), the P value was defined as significance A as described before (Cox and Mann, 2008). Significant outliers are colored in red (P < 1e−14), orange (P < 0.0001), or steelblue (P < 0.05); other proteins are shown in light blue.
Western blot
Samples prepared from cell lysates or in the membrane fractionation assay were used for Western blot analysis. GFP-tagged proteins were detected with a mouse anti-GFP (RRID:AB_390913; Roche) antibody diluted 1:1,000. Pgk1 was detected with a 1:10,000 diluted mouse anti-Pgk1 (RRID:AB_2532235; Thermo Fisher Scientific) antibody using a horseradish peroxidase-coupled mouse IgG kappa binding protein (RRID:AB_2686626; Santa Cruz Biotechnology). Sec61 was detected with a 1:10,000 diluted rabbit anti-Sec61 (Ungermann lab; University of Osnabrück, Osnabrück, Germany; Gao et al., 2018) and Gas1 with a polyclonal rabbit anti-Gas1 (Miller lab; University of Cambridge, Cambridge, UK). The lumenal domain of Gas1 was expressed as a His-tagged fusion in E. coli and purified by Ni-affinity chromatography. Purified protein was injected into rabbits, pre-screened for low cross-reactivity to yeast lysate. Polyclonal sera were collected and tested by immunoblotting and radioactive immunoprecipitation from yeast lysates, both yielding specific recognition of the appropriate protein. Antibodies from the Schekman lab prepared similarly were used as a benchmark (Doering and Schekman, 1997). Antibody using a horseradish peroxidase coupled mouse anti-rabbit IgG binding protein (RRID:AB_628497; Santa Cruz Biotechnology). Western blots were acquired on a BioRad (www.biorad.com) Chemidoc XRS + imager and quantified using the ImageJ software package ImageJ (RRID:SCR_003070; National Institutes of Health, Bethesda, MD) or via exposure on film.
Proteomics
Proteomics analysis was performed as described previously (Fröhlich et al., 2013). Briefly, cells were grown in SDC medium containing either light or heavy lysine to a concentration of 30 mg/ml. Main cultures were inoculated in 500 ml SDC medium to an OD600 0.1 from a pre-culture containing light or heavy lysine (30 mg/ml) and grown over day to an OD600 0.7–1.2. The same amount of OD units from both cultures were harvested at 4,000 rpm, 4°C for 5 min, washed with ice cold water, and snap-frozen as pellets. Cells were lysed with 500 μl acid-washed glass beads in 500 μl GFP-Trap pull-down buffer (20 mM HEPES, pH 7.4, 150 mM potassium acetate, 5% glycerol, 1% n-octyl-β-D-glucoside and 1 pill/10 ml buffer cOmplete Protease Inhibitor Cocktail EDTA free (Roche) using the FastPrep (MP biomedicals). After a centrifugation step at 14,000 rpm and 4°C for 10 min, the supernatants of light and heavy lysine-cultured strains were incubated for 10 min at RT on a turning wheel with 12.5 μl in GFP-Trap pull-down buffer equilibrated GFP Trap beads (Chromotek). The beads were washed in total six times first with the pull-down buffer and then with the washing buffer (20 mM HEPES pH 7.4, 150 mM potassium acetate, 5% glycerol). Afterwards, the beads were combined and the proteins on the beads were reduced, alkylated and digested with LysC at 37°C overnight. Resulting peptides were transferred to a glass vial and 10 μl were used to perform reversed-phase chromatography on a Thermo Ultimate 3000 RSLCnano system connected to a QExactivePLUS mass spectrometer (Thermo Fisher Scientific) through a nano-electrospray ion source. Peptides were separated on a 50-cm PepMap C18 easy spray column (Thermo Fisher Scientific) with an inner diameter of 75 µm. The column temperature was kept at 40°C. The peptides were eluted from the column via a linear gradient of acetonitrile from 12 to 35% in 0.1% formic acid for 80 min at a constant flow rate of 250 nl/min followed by a 20 min increase to 60% and finally 10 min to reach 90% buffer B. Eluted peptides from the column were directly electro sprayed into the mass spectrometer. Mass spectra were acquired on the Q ExactivePlus in a data-dependent mode to automatically switch between full scan MS and up to ten data-dependent MS/MS scans. The maximum injection time for full scans was 50 ms, with a target value of 3,000,000 at a resolution of 70,000 at m/z 200. The 10 most intense multiply charged ions (z ≥ 2) from the survey scan were selected with an isolation width of 1.6 Th and fragment with higher energy collision dissociation (Olsen et al., 2007) with normalized collision energies of 27. Target values for MS/MS were set at 100,000 with a maximum injection time of 80 ms at a resolution of 17,500 at m/z 200. To avoid repetitive sequencing, the dynamic exclusion of sequenced peptides was set at 20 s. The resulting MS and MS/MS spectra were analyzed using MaxQuant (version 2.0.1.0, www.maxquant.org/; Cox and Mann, 2008; Cox et al., 2011). All calculations and plots were performed using the R software package (www.r-project.org/; RRID:SCR_001905). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2022) partner repository with the dataset identifier PXD039395 and http://www.ebi.ac.uk/pride/archive/projects/PXD039395.
Lipidomics
For the LC-MS/MS analysis of LCB and ceramides, cells were grown in YPD to exponential growth phase. As an internal standard for normalization and quantification ceramide (CER d18:1/17:0; Avanti) was spiked prior to the lipid extraction. Lipids were extracted from lysed yeast cells corresponding to 200 µg of protein by chloroform/methanol extraction (Ejsing et al., 2009). External standard lipids (PE 17:0/14:1; PS 17:0/14:1; PI 17:0/14:1; PC 17:0/14:1; LCB d17:1; CER d18:1 17:0; all Avanti) were used for the calculation of a standard curve and calculation of lipid amounts. Dried lipid films were dissolved in a 65:35 (v/v) mixture of the mobile phases A (60:40 water/acetonitrile, 10 mM ammonium formate and 0.1% formic acid) and B (88:10:2 2-propanol/acetonitrile/water, 2 mM ammonium formate and 0.02% formic acid). A C30 reverse-phase column (Thermo Acclaim, C30, 2.1 × 250 mm, 3 µm, operated at 50°C; Thermo Fisher Scientific) connected to Shimadzu Nexera HPLC system and a ExactivePlus Orbitrap mass spectrometer equipped with a heated electrospray ionization (HESI) probe was used, as previously described (Eising et al., 2019). Peaks were analyzed using the Lipid Search algorithm (MKI). Peaks were defined through raw files, accurate m/z values of product ions and precursor ions. Candidate molecular species were identified by database (>1,000,000 entries) search of positive (+H+; +NH4+) or negative (−H−; +HCOO−) adducts. Mass tolerance was set to ±5 ppm for the precursor and fragment m/z values. Samples were aligned within a retention time window of 0.5 min and results combined in a single report. From the intensities of the lipid standard, the absolute values for each lipid in pmol/µg protein were calculated. For complex SPs, no standards are commercially available. Therefore, IPC and MIPC data are represented as area under the peak quantified with Thermo freestyle or Skyline (Pino et al., 2020; Peng et al., 2020; MacLean et al., 2010) software. A PI standard also measured in negative ion mode was used to normalize the values between the different samples.
COPII budding assay
Yeast microsomal membranes were prepared as previously described (Wuestehube and Schekman, 1992). Briefly, exponential growth phase yeast cells grown in YPD (1% w/v yeast extract, 2% w/v peptone, and 2% w/v glucose) were harvested and resuspended in 100 mM Tris, pH 9.4, 10 mM DTT to 40 OD600/ml and incubated at RT for 10 min. Cells were pelleted and resuspended to 40 OD600/ml in lyticase buffer (0.7 M sorbitol, 0.75× YPD, 10 mM Tris, pH 7.4, and 1 mM DTT + 2 μl/OD600 lyticase) and then incubated for 30 min at 30°C with gentle agitation. Cells were pelleted, washed once with 2× JR buffer (0.4 M sorbitol, 100 mM potassium acetate, 4 mM EDTA, 40 mM HEPES, pH 7.4, 2 mM DTT, and 2 mM PMSF) at 100 OD600/ml and then resuspended in 2× JR buffer at 400 OD600/ml before freezing at −80°C. Spheroplasts were thawed on ice and an equal volume of ice cold dH2O (with 2 mM DTT and 2 mM PMSF) was added before disruption with a motor-driven Potter Elvehjem homogenizer at 4°C. The homogenate was cleared by low-speed centrifugation (750 g, 5 min, 4°C) and crude membranes collected by centrifugation of the low-speed supernatant at 27,000 g. The membrane pellet was resuspended in 1 ml/l of cells used of buffer B88 (20 mM HEPES, pH 6.8, 250 mM sorbitol, 150 mM potassium acetate and 5 mM magnesium acetate) and loaded onto a step sucrose gradient composed of 1 ml 1.5 M sucrose in B88 and 1 ml 1.2 M sucrose in B88. Gradients were subjected to ultracentrifugation at 190,000 g for 1 h at 4°C. Microsomal membranes were collected from the 1.2 M/1.5 M sucrose interface, diluted 10-fold in B88, and collected by centrifugation at 27,000 g. The microsomal pellet was resuspended in a small volume of B88, aliquoted in 1 mg total protein aliquots and snap-frozen in liquid nitrogen.
Budding reactions were adapted from Melero et al. (2018). Briefly, 0.66 mg of microsomal membranes per reaction were incubated in B88/ATP regeneration mix (final concentration of ATP regeneration mix is: 1 mM ATP, 50 µM GDP-mannose, 40 mM creatine phosphate, and 200 µg/ml creatine phosphokinase) for 15 min at 15°C. The samples were then mixed with 1 ml cold B88, pelleted (19,300 g, 3 min, 4°C) and washed twice with 1 ml cold B88. Budding reactions were set up in B88 to a final volume of 250 μl at the following concentrations: 40 µg/ml Sar1, 10 µg/ml Sec23–Sec24, 30 µg/ml Sec13–Sec31, and 0.2 mM GTP and ATP regeneration mix. Low salt B88 (100 mM potassium acetate) was used for the coat-containing reactions and coat proteins were omitted in the negative reactions. Reactions were incubated for 25 min at 25°C and then on ice for 5 min. A 10-μl aliquot was collected as the total fraction and the vesicle-containing supernatant was collected after pelleting the donor membrane (19,300 g 3 min, 4°C). Vesicle fractions were then collected by centrifugation in a Beckman TLA-55 rotor (55,000 rpm, 25 min, 4°C). The supernatant was removed, and the pelleted vesicles and the separated supernatant were snap-frozen in liquid nitrogen.
Targeted lipidomics analysis of COP-II vesicles and purified proteins from S. cerevisiae
Ceramide and phosphatidylcholine (PC) were measured specifically in COPII vesicles (only ceramide) and in Svf1 proteins purified from yeast. Prior to measurement lipids were extracted from different stages of the COPII budding assay or according to 20 µg of purified protein by chloroform/methanol extraction as described previously (Ejsing et al., 2009). 150 mM ammonium formate was added ad 200 μl and in addition the COP-II vesicle containing samples were shaken for 20 min. Ceramide (CER d17:1/24:0; Avanti) was added prior to lipid extraction as an internal standard. Dried lipid films were dissolved in a 50:50 (v/v) mixture of the mobile phases A (50:50 water/acetonitrile, 10 mM ammonium formate and 0.1% formic acid) and B (88:10:2 2-propanol/acetonitrile/water, 2 mM ammonium formate and 0.02% formic acid). External standard curves were prepared using phytoceramide (CER t18:0/24:0; Cayman Chemical) and PC (PC 16:0/18:1; Avanti). Samples were run using an Accucore C30 LC column (150 × 2.1 mm 2.6 µm Solid Core; Thermo Fisher Scientific Scientific) connected to a Shimadzu Nexera HPLC system and a QTRAP 5500 LC-MS/MS (SCIEX) mass spectrometer. Different lipid species (CER d17:1/24:0 as a control (Avanti Polar lipids), CER 42:0;3, CER 42:0;4, CER 42:0;5, CER 44:0;3, CER 44:0;4, CER 44:0;5 and PC 34:1) were detected in a positive MRM mode with optimized transition settings within a 6 min HPLC run. For peak integration, the SciexOS software was used. The concentrations of all ceramide species were calculated using the external standard curve. The ceramide concentrations were expressed in pmol/μl and represented in a percentage ratio of ceramide in COPII vesicles to input. The lipid concentrations extracted from purified proteins were expressed in pmol/µg protein.
Lipidomic flux analysis
Yeast strains were grown in YPD. At an OD600 of 0.8 (t = 0), cell suspensions were spiked with a tracer cocktail resulting in a final concentration of 4.59 mM [13C315N]- serine 0.22 mM [2H6]- inositol, as previously described (Martínez-Montañés et al., 2020). At time points t = −15 min and t = 90 min, cell suspensions were incubated at 4°C for 10 min with 1 M perchloric acid. The cells were centrifuged and washed with 4°C 155 mM ammonium formate, frozen in liquid nitrogen and stored at −80°C. In-depth lipidome analysis was performed as described previously (Martínez-Montañés et al., 2020). Shortly, yeast cell pellets (appr. 4 OD units) were resuspended in 1 ml 4°C 155 mM ammonium formate and lysed at 4°C with 400 μl of acid-washed glass beads using a Mini-Beadbeater (Biospec). Lysates corresponding with 0.4 OD units were spiked with internal lipid standards and subjected to two-step lipid extraction (Ejsing et al., 2009). Lipid extracts were vacuum evaporated and re-dissolved in 100 μl chloroform/methanol (1:2; v/v) and analyzed by MSALL (Martínez-Montañés et al., 2020) using an Orbitrap Fusion Tribid (Thermo Fisher Scientific) equipped with a TriVersa NanoMate robotic nanoflow ion source (Advion Biosciences). Lipid identification and quantification was performed using ALEX123 software (Pauling et al., 2017; Ellis et al., 2018; Husen et al., 2013). The results were expressed in mol % per all detected lipids.
Membrane fractionation
For the membrane fractionation, 100 OD600 units from yeast strains in exponential growth phase were harvested at 4°C, 4,000 rpm for 5 min. The pellets were washed with ice cold water, snap frozen and stored at −80°C. Cell lysis was performed in 250 μl acid-washed glass beads and 500 μl lysis buffer (50 mM HEPES pH 6.8, 150 mM potassium acetate, 2 mM magnesium acetate, 1 mM calcium chloride, 200 mM sorbitol, and 1 pill/10 ml buffer cOmplete Protease Inhibitor Cocktail EDTA free (Roche) using the FastPrep (MP biomedicals). Two consecutive centrifugation steps at 1,000 g for 20 min were performed and after each step the supernatant was transferred into a new tube. After the second centrifugation the protein concentration was determined using Bradford reagent 1:5 diluted. Cell lysate according to 500 µg protein was taken as input and the same amount of lysate was then spun down at 45,935 g, 4°C for 30 min. The supernatant and pellet were separated, and the pellet was resuspended in the same volume as the supernatant in lysis buffer. Loading dye was added and the samples were boiled for 10 min at 95°C.
Molecular docking studies
For the molecular docking study, Svf1 structure was obtained from the Uniprot database (https://www.uniprot.org) [uniprot] where the three-dimensional structure prediction was obtained through the neural network-based model AlphaFold [afold]. The predicted structure has very high confidence score (per-residue confidence score pLDDT > 90) along most of the core structure, with only a few lower confidence score regions (pLDDT > 70) at the N and C terminal α-helixes, and at the interbarrel loops (residues 202–245 and 362–393). These regions may have an unstructured preferred conformation in water, as confirmed by molecular dynamics simulations of an atomistic CHARMM36 model of the protein solvated in TIP4P water. Ceramide 18:0;3/26:0;1 (44:0;4) and DAG 16:0/18:1 (34:1) structures were generated and optimized by Pymol [pymol] and Avogadro (https://avogadro.cc) packages respectively. The UCSF Chimera program (http://www.cgl.ucsf.edu/chimera) and AutoDockTools 4 (http://mgltools.scripps.edu) were used to prepare the protein and ligand structures for input to the docking procedure by adding Gasteiger charges. The structures were then saved in PDBQT file format, for input into AutoDock Vina version 1.1.2 (http://vina.scripps.edu; Trott and Olson, 2009; Eberhardt et al., 2021) Both ceramide and DAG torsions were made active (flexible) for the docking search, prioritizing the terminal parts of each chain in order not to surpass the 32 active bonds allowed by Vina. The search space was defined as a cube 75 Å on each side, centered on the protein geometric center (X, Y, Z coordinates: −4.11, 4.84, −6.17). This search space encompasses the whole protein space, as required for first blind docking. Once the blind docking was validated, the search was restricted in order to probe each barrel cavity. Cubes of 27 Å of side were placed around each barrel center (X, Y, Z coordinates: 1.5, 17.0, 10.0; and −17.0, −3, −9 respectively).
Protein purification of 3xFLAG tagged Svf1 protein from S. cerevisiae
Yeast cells were collected after grown for 24 h in yeast peptone (YP) medium containing 2% galactose (v/v), washed in lysis buffer (150 mM NaCl, 50 mM HEPES-NaOH [pH 7.4], 1.5 mM MgCl2, 5% [v/v] glycerol), and resuspended in a 1:1 ratio (w/v) in lysis buffer supplemented with 1 mM phenylmethylsulfonylfluoride (PMSF) and 1× FY protease inhibitor mix (Serva). Resuspended cells were frozen in a drop-by-drop fashion in liquid nitrogen, pulverized in 15 × 2 min cycles at 12 CPS in a 6875D Freezer/Mill Dual-Chamber Cryogenic Grinder (SPEX SamplePrep) and thawed in lysis buffer with 1 mM PMSF, 1× FY, and 1 mM dithiothreitol (DTT). After two centrifugation steps at 5,000 and 15,000 g at 4°C for 10 and 20 min, respectively, the supernatant was added to 1.5 ml α-Flag resin (Sigma-Aldrich) and nutated for 45 min at 4°C. Beads were washed twice with 20 ml lysis buffer and bound proteins were eluted on a turning wheel for 45 min at 4°C with 3xFLAG peptide. The eluate was collected by centrifugation at 1,800 rpm, 4°C for 30 s.
Mass photometry analysis
Molecular mass photometry measurements were performed using a Refeyn TwoMP mass photometer (Refeyn Ltd.) and carried out in silicone gaskets placed on clean coverslips. Immediately prior to mass photometry measurements, protein samples were diluted directly in lysis buffer to 100 nM working concentration. For each acquisition, 2 μl of diluted protein were added to 18 μl lysis buffer on the gasket resulting in a final concentration of 10 nM. After autofocus stabilization, images were acquired for 60 s. Data acquisition was performed using AcquireMP and then analyzed using DiscoverMP (both Refeyn Ltd).
Online supplemental material
Fig. S1, a–d are related to Fig. 1. Fig S1, e–g are related to Fig. 3. Fig. S2 shows tetrad dissections to test functionality of N- and C-terminal tagging of Svf1. Fig. S3 shows expression checks of all Svf1 constructs used in this study. Fig. S4 is related to Fig. 4. Fig. S5 shows the results of in vitro COPII budding assays and lipidomic analysis. Fig. S6 a is related to Fig. 6, l–n. Fig S6, b and c show co-localization of Svf1 with the mid-Golgi marker Aur1. Fig. S7 is related to Fig. 7. Table S1 shows the results from the GFP pulldown MS experiment in Fig 5 a. Table S2 shows S. cerevisiae strains used in this study. Table S3 shows plasmids used in this study. Table S3 shows antibodies used in this study. Video 1 shows representative midsection time lapse fluorescence microscopy of yeast cells expressing Svf1-GFP and Mnn9-mCherry.
Acknowledgments
We thank members of the Fröhlich and Ungermann labs for critical comments on the manuscript, and Alejandro Melero Carrillo for advice on COPII budding for lipidomics.
This research was supported by the VILLUM Foundation (VKR023439 to C.S. Ejsing), the VILLUM Center for Bioanalytical Sciences (VKR023179 to C.S. Ejsing), the Lundbeckfonden (R54-A5858, C.S. Ejsing) and the UK Medical Research Council (MRC_UP_12-1/10 to E.A. Miller). Sergej Limar is a member of the UOS funded graduate school “EvoCell.” Florian Fröhlich is supported by the DFG grants FR 3647/3-1, FR 3647/4-1, and SFB944.
Author contributions: Investigation: S. Limar, C. Körner, F. Martínez-Montañés, V.G. Stancheva, V.N. Wolf, S. Walter, E.A. Miller, C.S. Ejsing, V.V. Galassi, F. Fröhlich. Formal analysis: S. Limar, C. Körner, F. Martínez-Montañés, V.G. Stancheva, V.N. Wolf. Visualization: S. Limar, C. Körner, F. Martínez-Montañés, V.G. Stancheva, V.N. Wolf, F. Fröhlich. Conceptualization: S. Limar, F. Fröhlich. Funding acquisition: E.A. Miller, C.S. Ejsing, F. Fröhlich. Writing—original draft:: S. Limar, F. Fröhlich. Writing—review and editing: All authors.

