


Notch receptors control tissue morphogenic processes that involve coordinated changes in cell architecture and gene expression, but how a single receptor can produce these diverse biological outputs is unclear. Here, we employ a 3D model of a human ductal epithelium to reveal tissue morphogenic defects result from loss of Notch1, but not Notch1 transcriptional signaling. Instead, defects in duct morphogenesis are driven by dysregulated epithelial cell architecture and mitogenic signaling which result from the loss of a transcription-independent, Notch1 cortical signaling mechanism that ultimately functions to stabilize adherens junctions and cortical actin. We identify that Notch1 localization and cortical signaling are tied to apical–basal cell restructuring and discover that a Notch1–FAM83H interaction underlies control of epithelial adherens junctions and cortical actin. Together, these results offer new insights into Notch1 signaling and regulation and advance a paradigm in which transcriptional and cell adhesive programs might be coordinated by a single receptor.
Introduction
Coordinated changes in cell–cell adhesions are central to tissue morphogenesis and integrity, and thus are essential mediators in development, tissue physiology, and disease pathogenesis (Belardi et al., 2020; Borghi and Nelson, 2009). Cell–cell adhesive junctions coordinate multicellular behavior by transmitting mechanical forces between cells and by influencing the localization, duration, and cytoskeletal coupling of receptor interactions to orchestrate juxtracrine signaling (Collinet and Lecuit, 2021; Gumbiner, 1996). Yet, at a fundamental level, molecular mechanisms that orchestrate cell–cell junction assembly and stability are not well understood. Similarly, it remains unclear how junctional changes are synchronized with transcriptional programs, for example, in the context of the coupled patterning of cell lineages and movement during developmental morphogenesis (de Celis et al., 1996; Falo-Sanjuan and Bray, 2022).
The Notch family of receptors is a fundamental, conserved regulator of developmental patterning, where receptor signaling determines cell fate and patterns of gene expression amongst neighboring cells (Bray, 2016; Siebel and Lendahl, 2017). Notch receptors are integrated into the plasma membrane as noncovalent heterodimers composed of a large extracellular domain (ECD) polypeptide bound to a transmembrane fragment that consists of an extracellular sequence and a transmembrane domain (hereafter collectively referred to as the transmembrane domain [TMD]), and an intracellular domain (ICD). Notch receptors are activated via interaction with ligands presented on adjacent cells, which occurs through multiple steps that are independently gated by sequential events. Mechanical force applied to the Notch ECD causes conformational unfolding of an extracellular negative regulatory region that renders the receptor sensitive to sequential proteolytic cleavages, first at an extracellular S2 site by removing the ECD and subsequently at an intramembrane S3 site by the γ-secretase complex cleaving the ICD from the TMD (Gordon et al., 2008, 2015; Kopan and Ilagan, 2009; Kovall et al., 2017). Notch exerts transcriptional effects through cleaved ICD, which can translocate to the nucleus and form a Notch transcription activation complex with cofactors RBPJ and MAML1/2 (Borggrefe and Oswald, 2009; Wang et al., 2014). This Notch signaling mechanism is conserved across the entire animal kingdom and Notch activation is implicated during tissue morphogenesis in many developmental programs (Hellström et al., 2007; Lloyd-Lewis et al., 2019; Priya et al., 2020). However, how morphogenetic changes, which require dynamic cell architectures and cell–cell adhesions, may be regulated in the context of key developmental pathways like Notch remains largely unaddressed.
The Notch gene was discovered from a mutant allele in Drosophila causing the formation of a wing “notch” due to defects in the dorsoventral compartmentalization of the developing wing disc epithelium. Cells along the dorsoventral boundary have distinctive properties, and their specification requires Notch activity (de Celis et al., 1996). Notch-dependent, distinctive filamentous actin, non-muscle myosin II, and adherens junction phenotypes also form at the dorsoventral boundary, yet these changes are not accounted for purely by the transcriptional regulation of target genes associated with Notch activation (Major and Irvine, 2005, 2006). In mammals, Notch1 is expressed broadly, and interactions with adherens junctions and actomyosin have been linked to both Notch receptor activation and downstream function in distinct contexts (Crowner et al., 2003; Falo-Sanjuan and Bray, 2021; Hunter et al., 2019; Khait et al., 2016; Kwak et al., 2022; Lowell and Watt, 2001; Priya et al., 2020). We previously reported that hemodynamic shear stress activates the Notch1 receptor in the endothelium to enhance vascular barrier function. This process does not involve ICD-mediated transcription, but instead operates through a mechanism we refer to as Notch1 cortical signaling, where the TMD acts as a focal point of protein–protein interactions in a pathway that strengthens endothelial adherens junctions (Polacheck et al., 2017). Still, this role for Notch1 in the vasculature is a highly specialized case, and substantial structural and signaling differences exist between epithelial and endothelial cell–cell adhesions (Buckley and St. Johnston, 2022; Lampugnani et al., 2018). If, and how, Notch1 cortical signaling influences epithelial cell architecture and cell–cell adhesion remains an open question.
In the present study, we employ a tissue-engineered, microfluidic model capable of recapitulating and dissecting three-dimensional (3D) morphogenic features of a ductal epithelium. Using CRISPR-Cas9 gene editing to specifically decouple Notch1 cortical or transcriptional signaling in human epithelia, we observe distinct 3D morphogenic consequences result upon loss of Notch1 cortical signaling but not transcriptional signaling. Tissue morphogenic defects are driven by dysregulated epithelial cell architecture and mitogenic signaling, which Notch1 cortical signaling controls through the stabilization of adherens junctions and cortical actin organization. Mechanistically, we report that Notch1 receptor localization and cortical signaling function are tied to epithelial apical–basal cell columnar restructuring, and we identify a distinct Notch1–FAM83H mechanism that underlies epithelial adherens junction regulation by Notch1 cortical signaling.
Results
Notch1 influences the morphogenesis of a 3D-engineered ductal epithelium independent of Notch1 ICD transcriptional signaling
To specifically isolate Notch1 signaling functions independent of ICD transcription, we applied methods that we previously established to generate endogenous Notch1 truncation mutants in primary human endothelia (Polacheck et al., 2017). We engineered human mammary epithelial cells (MCF10A) harboring either CRISPR-Cas9-mediated ablation of Notch1 (NOTCH1KO) or truncation of the Notch1 intracellular domain (ICDKO), which preserves the ECD and TMD (Fig. 1 A). Cell lysis, heterodimer dissociation via SDS-PAGE, and Western blotting permit the visualization of Notch1 ECD or transmembrane fragment polypeptides at distinct molecular weights using respective antibodies. Western blot confirmed deletion of Notch1 in NOTCH1KO epithelia, as well as ICD truncation of the transmembrane fragment and preservation of the ECD in ICDKO epithelia. NOTCH1KO or ICDKO epithelia do not have altered E-cadherin protein levels, Notch2–4 receptor protein levels, phosphorylation levels of β-catenin, or mRNA transcripts of Notch target genes compared with a non-targeting scramble guide RNA (SCR) control (Fig. 1 A; and Fig. S1, A and B).

Notch1 influences the morphogenesis of an engineered ductal epithelium independent of ICD transcriptional signaling. (A) Left: Schematic of endogenous Notch1 CRISPR-Cas9 mutant used to truncate Notch1 ICD. Right: Western blot of lysates from scramble control (SCR), NOTCH1KO, and ICDKO MCF10A cells immunoblotted for Notch1 ECD, Notch1 ICD, E-cadherin, β-catenin (non-phosphorylated [S33/37/T41]), and GAPDH. (B) Left: Microfluidic platform consisting of an engineered 3D ductal epithelium embedded in physiologic ECM. Luminal ports (blue) used for cell seeding and perfusion of medium through the lumen, basal ports (purple) used for the delivery of medium containing growth factors, ECM injection port and ECM compartment (beige), PDMS pillars (black) used to contain hydrogel ECM (top schematic: top-down view; bottom schematic: cross-section). Right: Representative 3D oblique projection (top) and cross-section (bottom) of a 3D MCF10A duct labeled with phalloidin (white) and Hoechst (blue). Scale bars, 50 µm. (C) Left: Representative phase contrast micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML ducts. Scale bar, 150 µm. Right: Representative medial confocal slice fluorescence micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML ducts labeled with phalloidin (black). Scale bar, 100 µm. (D) Quantification of duct diameter variance measured from phase contrast micrographs as shown in C. n ≥ 7 independent ducts. (E) Quantification of lumen diameter measured from confocal micrographs of phalloidin as shown in C. Average lumen diameters from n ≥ 10 independent ducts. (F) Quantification of internuclear distances measured from confocal micrographs of Hoechst-labeled ducts. Average internuclear distance from n ≥ 7 independent ducts. Western blots are representative of three independent experiments. For all plots, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. Source data are available for this figure: SourceData F1.
Notch1 influences the morphogenesis of an engineered ductal epithelium independent of ICD transcriptional signaling. (A) Left: Schematic of endogenous Notch1 CRISPR-Cas9 mutant used to truncate Notch1 ICD. Right: Western blot of lysates from scramble control (SCR), NOTCH1KO, and ICDKO MCF10A cells immunoblotted for Notch1 ECD, Notch1 ICD, E-cadherin, β-catenin (non-phosphorylated [S33/37/T41]), and GAPDH. (B) Left: Microfluidic platform consisting of an engineered 3D ductal epithelium embedded in physiologic ECM. Luminal ports (blue) used for cell seeding and perfusion of medium through the lumen, basal ports (purple) used for the delivery of medium containing growth factors, ECM injection port and ECM compartment (beige), PDMS pillars (black) used to contain hydrogel ECM (top schematic: top-down view; bottom schematic: cross-section). Right: Representative 3D oblique projection (top) and cross-section (bottom) of a 3D MCF10A duct labeled with phalloidin (white) and Hoechst (blue). Scale bars, 50 µm. (C) Left: Representative phase contrast micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML ducts. Scale bar, 150 µm. Right: Representative medial confocal slice fluorescence micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML ducts labeled with phalloidin (black). Scale bar, 100 µm. (D) Quantification of duct diameter variance measured from phase contrast micrographs as shown in C. n ≥ 7 independent ducts. (E) Quantification of lumen diameter measured from confocal micrographs of phalloidin as shown in C. Average lumen diameters from n ≥ 10 independent ducts. (F) Quantification of internuclear distances measured from confocal micrographs of Hoechst-labeled ducts. Average internuclear distance from n ≥ 7 independent ducts. Western blots are representative of three independent experiments. For all plots, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. Source data are available for this figure: SourceData F1.
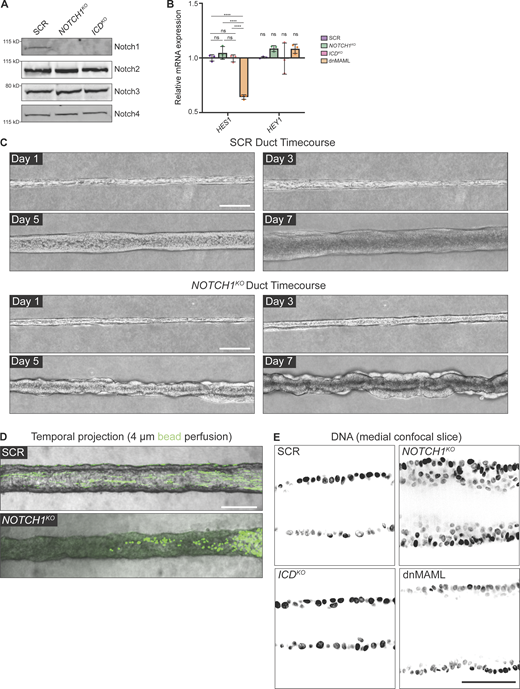
Notch1 cortical signaling influences mammary duct morphogenesis. (A) Western blot of lysates from scramble control (SCR), NOTCH1KO, and ICDKO cells immunoblotted for Notch1, Notch2, Notch3, and Notch4. (B) mRNA transcript expression of Notch1 target genes HES1 and HEY1 measured by qPCR in SCR, NOTCH1KO, ICDKO, and dnMAML-expressing cells. Average qPCR reads from n = 3 independent experiments. (C) Top: Phase contrast micrographs of SCR ducts over a 7-d timecourse shown at days 1, 3, 5, and 7 after seeding. Bottom: Phase-contrast images of NOTCH1KO ducts over a 7-d timecourse shown on days 1, 3, 5, and 7 after seeding. Scale bars, 150 µm. (D) Temporal projection micrographs of a timelapse of SCR and NOTCH1KO ducts perfused with 4 µm polystyrene beads (green). Scale bar, 200 µm. (E) Medial confocal slice micrographs from SCR, NOTCH1KO, ICDKO, and dnMAML ducts labeled with Hoechst (black). Scale bar, 50 µm. For plot B, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, ****P < 0.0001, ns denotes non-significant. Source data are available for this figure: SourceData FS1.

Notch1 cortical signaling influences mammary duct morphogenesis. (A) Western blot of lysates from scramble control (SCR), NOTCH1KO, and ICDKO cells immunoblotted for Notch1, Notch2, Notch3, and Notch4. (B) mRNA transcript expression of Notch1 target genes HES1 and HEY1 measured by qPCR in SCR, NOTCH1KO, ICDKO, and dnMAML-expressing cells. Average qPCR reads from n = 3 independent experiments. (C) Top: Phase contrast micrographs of SCR ducts over a 7-d timecourse shown at days 1, 3, 5, and 7 after seeding. Bottom: Phase-contrast images of NOTCH1KO ducts over a 7-d timecourse shown on days 1, 3, 5, and 7 after seeding. Scale bars, 150 µm. (D) Temporal projection micrographs of a timelapse of SCR and NOTCH1KO ducts perfused with 4 µm polystyrene beads (green). Scale bar, 200 µm. (E) Medial confocal slice micrographs from SCR, NOTCH1KO, ICDKO, and dnMAML ducts labeled with Hoechst (black). Scale bar, 50 µm. For plot B, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, ****P < 0.0001, ns denotes non-significant. Source data are available for this figure: SourceData FS1.
We first investigated whether Notch1 influences the assembly or maintenance of an engineered 3D ductal epithelium. To maintain control over cells, extracellular matrix (ECM), and tissue architecture, we developed a microfluidic model of a 3D ductal epithelium which consists of a channel surrounded by ECM that is lined with human MCF10A mammary epithelial cells and supported by basal delivery of growth factors (Fig. 1 B). Despite the absence of a bilayered cell architecture of mammary ducts in vivo and the limited apical maturation of MCF10A cells (Qu et al., 2015), this approach resulted in an epithelial cell–lined channel, reminiscent of an anatomical ductal lumen, that allows for careful dissection of cellular behaviors contributing to overall 3D tissue architecture. Seeded SCR cells quickly populate the surface of the duct channel and form stable, non-invasive monolayers over the course of 7 d (Fig. S1 C). The resulting linear ductal tissues have a hollow central lumen that is lined by columnar epithelial cells that are growth-arrested. Seeded NOTCH1KO cells similarly populate the channel surface, but in contrast develop tortuous duct architectures with prominent tissue outgrowths that cause large variances in duct diameter (Fig. 1, C and D; and Fig. S1 C). Perfusion of duct lumens with fluorescent microbeads (4 μm) indicated that NOTCH1KO lumens are occluded compared with SCR ducts (Fig. S1 D). Furthermore, medial confocal sections of NOTCH1KO ducts revealed a failure to form an ordered monolayer and extensive lumen cell in-filling, a twofold decrease in average lumen diameter (Fig. 1, C and E), and increased cell packing as quantified by reduced internuclear distances (Fig. 1, C and F; and Fig. S1 E).
To investigate whether transcription-independent functions of Notch1 may contribute to the NOTCH1KO duct phenotype, we generated ducts from ICDKO cells or cells engineered to express a dominant-negative form of the Notch transcriptional cofactor mastermind-like protein 1 (dnMAML; Fig. S1 B; Polacheck et al., 2017; Weng et al., 2003). ICDKO or dnMAML ducts closely resemble the overall tissue architecture of SCR ducts, with slightly larger lumen diameters forming in dnMAML ducts (Fig. 1, C–E). Importantly, no evidence of lumen cell in-filling was present in either ICDKO or dnMAML ducts (Fig. 1, C–F). Taken together, NOTCH1KO cells form 3D ducts with tortuous architectures, tissue outgrowths, and occluded lumens, and these phenotypes are not present in ducts constructed from cells harboring two distinct perturbations that delete the ICD or suppress Notch1 transcriptional signaling. This identifies a specific function for transcription-independent Notch1 cortical signaling in regulating the assembly or maintenance of a 3D-engineered ductal epithelium.
Aberrant cell architecture and proliferation underlie NOTCH1KO duct defects
We next investigated which cell behaviors contribute to the NOTCH1KO tissue phenotype through a temporal analysis of duct assembly. SCR and NOTCH1KO cells similarly adhere to the channel architecture and progressively form a monolayer. Approximately 3 d after initial seeding, SCR ducts initiate expansion of duct lumens; however, lumen expansion is significantly diminished in NOTCH1KO ducts and small tissue outgrowths are detectable by phase contrast microscopy (Fig. S1 C). Using a cell-permeable fluorescent probe for filamentous actin and live confocal microscopy, we visualized cell dynamics within SCR and NOTCH1KO ducts at the onset of these morphogenic differences. Timelapse imaging revealed several areas of cell multilayering within NOTCH1KO ducts that are formed from frequent cell divisions oriented orthogonal to the basal ECM interface. Daughter cells positioned inward amassed in the lumen and typically did not reintegrate into the duct monolayer lining (Fig. 2 A and Video 1). Consistent with observations from 3D mammary epithelial acinar models (Jaffe et al., 2008), SCR and ICDKO cells instead orient spindle axes along the basal ECM interface during division (Fig. 2, A, B, and D). Pulse labeling ducts with 5-ethynyl-2′-deoxyuridine (EdU) to assess cell proliferation revealed increased EdU incorporation in NOTCH1KO ducts relative to SCR or ICDKO that is primarily localized within cell masses in the lumen (Fig. 2, C and E). These observations indicated that dysregulated epithelial architecture and proliferation may be associated with the loss of Notch1 cortical signaling in NOTCH1KO ducts.

Notch1 cortical signaling regulates epithelial cell architecture and suppresses EGFR mitogenic signaling. (A) Individual time frames from live cell movies of actin within scramble control (SCR) and NOTCH1KO ducts labeled with SPY650-FastAct (black). Inset for individual time frames outlined in magenta in the 0:00 (hour:min) frame. Parent cell is labeled with (*) and daughter cell is labeled with (**). Scale bar, 50 µm. (B) Fluorescence micrographs of dividing SCR, NOTCH1KO, and ICDKO cells in ducts labeled with Hoechst (blue) and phalloidin (white). Scale bar, 10 µm. (C) Maximum projection micrographs of 30 µm medial stacks of SCR, NOTCH1KO, and ICDKO epithelial ducts labeled with phalloidin (magenta) and EdU (green). Scale bar, 30 µm. (D) Quantification of spindle axis angle measured from independent cells in ducts during metaphase as shown in B. Spindle axis angle is measured relative to the basal ECM interface, with 0° denoting a parallel axis. n = 13 spindles from at least three independent ducts. (E) Quantification of the percentage of EdU positive nuclei in ducts. n ≥ 9 independent ducts. (F) YZ orthogonal projections from fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells labeled with phalloidin (white) and Hoechst (blue). (G) Quantification of regions of cell multilayering per field of view in fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells. n ≥ 10 fields of view from three independent experiments. (H) Quantification of cell height from SCR, NOTCH1KO, and ICDKO cells plated on hydrogels. n ≥ 10 fields of view from three independent experiments. (I) Western blot of lysates from confluent SCR, NOTCH1KO, and ICDKO cells cultured with high EGF (20 ng/ml) and immunoblotted for pEGFR (Y845), EGFR, and Notch1. (J) Quantification of Western blot intensity difference of pEGFR and total EGFR levels in cells stimulated with high (20 ng/ml) or low (2 ng/ml) EGF. n = 3 independent experiments. (K) Top: Fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells immunostained for EGFR (black). Bottom: Fluorescence EGFR (magenta) micrograph overlay with phalloidin (green). Scale bar, 10 µm. (L) Quantification of relative junctional to cytoplasmic EGFR intensity. n ≥ 20 cells from three independent experiments. (M) Representative medial confocal slice micrographs of NOTCH1KO ducts treated with DMSO or 1 µM Erlotinib labeled with phalloidin (black). Scale bar, 100 µm. (N) Quantification of duct lumen diameter. Average duct diameters from n ≥ 15 independent ducts. (O) Quantification of the percentage of EdU-positive nuclei in NOTCH1KO cells treated with DMSO or 1 µM Erlotinib. n ≥ 15 fields of view from three independent experiments. Western blots are representative of three independent experiments. For plots D, E, G, H, J, and L, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. For plots N and O, mean ± SEM; two-tailed unpaired t test, *P < 0.05, ***P < 0.001. Source data are available for this figure: SourceData F2.
Notch1 cortical signaling regulates epithelial cell architecture and suppresses EGFR mitogenic signaling. (A) Individual time frames from live cell movies of actin within scramble control (SCR) and NOTCH1KO ducts labeled with SPY650-FastAct (black). Inset for individual time frames outlined in magenta in the 0:00 (hour:min) frame. Parent cell is labeled with (*) and daughter cell is labeled with (**). Scale bar, 50 µm. (B) Fluorescence micrographs of dividing SCR, NOTCH1KO, and ICDKO cells in ducts labeled with Hoechst (blue) and phalloidin (white). Scale bar, 10 µm. (C) Maximum projection micrographs of 30 µm medial stacks of SCR, NOTCH1KO, and ICDKO epithelial ducts labeled with phalloidin (magenta) and EdU (green). Scale bar, 30 µm. (D) Quantification of spindle axis angle measured from independent cells in ducts during metaphase as shown in B. Spindle axis angle is measured relative to the basal ECM interface, with 0° denoting a parallel axis. n = 13 spindles from at least three independent ducts. (E) Quantification of the percentage of EdU positive nuclei in ducts. n ≥ 9 independent ducts. (F) YZ orthogonal projections from fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells labeled with phalloidin (white) and Hoechst (blue). (G) Quantification of regions of cell multilayering per field of view in fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells. n ≥ 10 fields of view from three independent experiments. (H) Quantification of cell height from SCR, NOTCH1KO, and ICDKO cells plated on hydrogels. n ≥ 10 fields of view from three independent experiments. (I) Western blot of lysates from confluent SCR, NOTCH1KO, and ICDKO cells cultured with high EGF (20 ng/ml) and immunoblotted for pEGFR (Y845), EGFR, and Notch1. (J) Quantification of Western blot intensity difference of pEGFR and total EGFR levels in cells stimulated with high (20 ng/ml) or low (2 ng/ml) EGF. n = 3 independent experiments. (K) Top: Fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells immunostained for EGFR (black). Bottom: Fluorescence EGFR (magenta) micrograph overlay with phalloidin (green). Scale bar, 10 µm. (L) Quantification of relative junctional to cytoplasmic EGFR intensity. n ≥ 20 cells from three independent experiments. (M) Representative medial confocal slice micrographs of NOTCH1KO ducts treated with DMSO or 1 µM Erlotinib labeled with phalloidin (black). Scale bar, 100 µm. (N) Quantification of duct lumen diameter. Average duct diameters from n ≥ 15 independent ducts. (O) Quantification of the percentage of EdU-positive nuclei in NOTCH1KO cells treated with DMSO or 1 µM Erlotinib. n ≥ 15 fields of view from three independent experiments. Western blots are representative of three independent experiments. For plots D, E, G, H, J, and L, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. For plots N and O, mean ± SEM; two-tailed unpaired t test, *P < 0.05, ***P < 0.001. Source data are available for this figure: SourceData F2.
Live-cell imaging of the medial confocal plane of a SPY650-FastAct labeled NOTCH1KOduct during assembly. Asterisks denote representative cell divisions. Scale bar, 100 μm. Time scale, hour:minute, displayed at 6,000× speed.
Live-cell imaging of the medial confocal plane of a SPY650-FastAct labeled NOTCH1KOduct during assembly. Asterisks denote representative cell divisions. Scale bar, 100 μm. Time scale, hour:minute, displayed at 6,000× speed.
To recapitulate the underlying cell morphodynamics observed in our 3D model in a setting permitting high-resolution analysis of cell architecture and adhesion, mechanical behavior, and biochemical signaling, we modeled the physiological stiffness of basement membrane and underlying ECM by plating cells on 2D-compliant composite hydrogels (Nyga et al., 2021). Consistent with increased proliferation observed in ducts, NOTCH1KO cells cultured on compliant hydrogels display elevated EdU labeling compared with SCR, ICDKO, and dnMAML cells (Fig. S2, A and B). Analysis of cell organization in the z-plane orthogonal to the substrate revealed that only NOTCH1KO cells contain regions of cell multilayering that are reminiscent of 3D duct cell lumen in-growth. In regions lacking multilayering, NOTCH1KO epithelia are twofold shorter with diminished columnar cell morphology compared with SCR and ICDKO monolayers (Fig. 2, F–H; and Fig. S2 C). Decreased cell height, impaired columnar morphology, and elevated proliferation are similarly observed in additional human intestinal, mammary, and bronchial NOTCH1KO cells relative to control (Fig. S3, A–H). Thus, loss of Notch1 cortical signaling results in elevated proliferation, cell multilayering, and impaired apical–basal cell architecture on 2D hydrogels and within 3D ducts.

Loss of Notch1 cortical signaling results in aberrant epithelial architecture and proliferation. (A) Fluorescence micrographs of scramble control (SCR), NOTCH1KO, ICDKO, and dnMAML cells labeled with EdU (green) and Hoechst (magenta). Scale bar, 20 µm. (B) Quantification of the percentage of EdU-positive nuclei in SCR, NOTCH1KO, ICDKO, and dnMAML cells. n ≥ 12 fields of view, from three independent experiments. (C) Maximum intensity projection fluorescence micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML cells labeled with Hoechst (grey). Scale bar, 10 µm. (D) Immunofluorescence micrographs of SCR and NOTCH1KO cells immunostained for YAP (white, left) and Notch1 (black, right) and labeled with DAPI (blue, right). Scale bar, 20 µm. (E) Western blot of immunoprecipitation of EGFR from SCR and NOTCH1KO cell lysates immunoblotted for E-cadherin and EGFR. Representative of three independent experiments. (F) Full field of view source fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells immunostained for EGFR (black) for representative images in Fig. 2. Scale bar, 20 µm. (G) Fluorescence micrographs of dnMAML cells immunostained for EGFR (black, left; magenta, right) and labeled with phalloidin (green, right). Scale bar, 10 µm. (H) Top: Fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells incubated with TexasRed-EGF (TR-EGF; black). Bottom: Fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells incubated with TR-EGF (magenta) and labeled with phalloidin (green). Scale bar, 20 µm. (I) Quantification of the percentage of SCR, NOTCH1KO, and ICDKO cells with internalized TR-EGF. Average internalization from n = 4 independent experiments. (J) Cell proliferation rates of SCR, NOTCH1KO, and ICDKO cells treated with and without EGF over a 3-d timecourse. n = 3 independent experiments. (K) Fluorescence micrographs of NOTCH1KO cells treated with DMSO or 1 µM Erlotinib labeled with EdU (green) and Hoechst (pink). Scale bar, 20 µm. For plots B, I, and J, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. Source data are available for this figure: SourceData FS2.

Loss of Notch1 cortical signaling results in aberrant epithelial architecture and proliferation. (A) Fluorescence micrographs of scramble control (SCR), NOTCH1KO, ICDKO, and dnMAML cells labeled with EdU (green) and Hoechst (magenta). Scale bar, 20 µm. (B) Quantification of the percentage of EdU-positive nuclei in SCR, NOTCH1KO, ICDKO, and dnMAML cells. n ≥ 12 fields of view, from three independent experiments. (C) Maximum intensity projection fluorescence micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML cells labeled with Hoechst (grey). Scale bar, 10 µm. (D) Immunofluorescence micrographs of SCR and NOTCH1KO cells immunostained for YAP (white, left) and Notch1 (black, right) and labeled with DAPI (blue, right). Scale bar, 20 µm. (E) Western blot of immunoprecipitation of EGFR from SCR and NOTCH1KO cell lysates immunoblotted for E-cadherin and EGFR. Representative of three independent experiments. (F) Full field of view source fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells immunostained for EGFR (black) for representative images in Fig. 2. Scale bar, 20 µm. (G) Fluorescence micrographs of dnMAML cells immunostained for EGFR (black, left; magenta, right) and labeled with phalloidin (green, right). Scale bar, 10 µm. (H) Top: Fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells incubated with TexasRed-EGF (TR-EGF; black). Bottom: Fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells incubated with TR-EGF (magenta) and labeled with phalloidin (green). Scale bar, 20 µm. (I) Quantification of the percentage of SCR, NOTCH1KO, and ICDKO cells with internalized TR-EGF. Average internalization from n = 4 independent experiments. (J) Cell proliferation rates of SCR, NOTCH1KO, and ICDKO cells treated with and without EGF over a 3-d timecourse. n = 3 independent experiments. (K) Fluorescence micrographs of NOTCH1KO cells treated with DMSO or 1 µM Erlotinib labeled with EdU (green) and Hoechst (pink). Scale bar, 20 µm. For plots B, I, and J, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. Source data are available for this figure: SourceData FS2.

Notch1 regulates epithelial architecture, adherens junctions, cortical actin organization, and proliferation in human epithelia. (A) Immunofluorescence micrographs of Scramble, NOTCH1KO, and ICDKO MCF7 cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Scale bar, 20 µm. (B) YZ orthogonal projections from micrographs of Scramble, NOTCH1KO, and ICDKO MCF7 cells labeled with Hoechst (white). (C) Quantification of the frequency of focal adherens junctions in scramble control (SCR), NOTCH1KO, and ICDKO MCF7 cells. n ≥ 12 fields of view from three independent experiments. (D) Relative number of SCR, NOTCH1KO, and ICDKO MCF7 cells measured at passage. n = 3 independent experiments. (E) Top: Immunofluorescence micrographs of Scramble (top row) and NOTCH1KO (bottom row) Caco-2 cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Scale bar, 20 µm. Bottom: YZ orthogonal projections from micrographs of Scramble and NOTCH1KO Caco-2 cells labeled with phalloidin (black). (F) Top: Fluorescence micrographs of Scramble (top row) and NOTCH1KO (bottom row) 16hbe14o- cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Scale bar, 20 µm. Bottom: YZ orthogonal projections from micrographs of Scramble and NOTCH1KO 16hbe14o- cells labeled with phalloidin (black). (G) Relative number of SCR and NOTCH1KO Caco-2 cells measured at passage. n = 2 independent experiments. (H) Relative number of SCR and NOTCH1KO 16hbe14o- cells measured at passage. n = 3 independent experiments. For plots C and D, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, *P < 0.05, ****P < 0.0001, ns denotes non-significant. For plot H, mean ± SEM; two-tailed unpaired t test, **P < 0.01.

Notch1 regulates epithelial architecture, adherens junctions, cortical actin organization, and proliferation in human epithelia. (A) Immunofluorescence micrographs of Scramble, NOTCH1KO, and ICDKO MCF7 cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Scale bar, 20 µm. (B) YZ orthogonal projections from micrographs of Scramble, NOTCH1KO, and ICDKO MCF7 cells labeled with Hoechst (white). (C) Quantification of the frequency of focal adherens junctions in scramble control (SCR), NOTCH1KO, and ICDKO MCF7 cells. n ≥ 12 fields of view from three independent experiments. (D) Relative number of SCR, NOTCH1KO, and ICDKO MCF7 cells measured at passage. n = 3 independent experiments. (E) Top: Immunofluorescence micrographs of Scramble (top row) and NOTCH1KO (bottom row) Caco-2 cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Scale bar, 20 µm. Bottom: YZ orthogonal projections from micrographs of Scramble and NOTCH1KO Caco-2 cells labeled with phalloidin (black). (F) Top: Fluorescence micrographs of Scramble (top row) and NOTCH1KO (bottom row) 16hbe14o- cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Scale bar, 20 µm. Bottom: YZ orthogonal projections from micrographs of Scramble and NOTCH1KO 16hbe14o- cells labeled with phalloidin (black). (G) Relative number of SCR and NOTCH1KO Caco-2 cells measured at passage. n = 2 independent experiments. (H) Relative number of SCR and NOTCH1KO 16hbe14o- cells measured at passage. n = 3 independent experiments. For plots C and D, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, *P < 0.05, ****P < 0.0001, ns denotes non-significant. For plot H, mean ± SEM; two-tailed unpaired t test, **P < 0.01.
Notch1 cortical signaling suppresses EGFR phosphorylation, internalization, and mitogenic signaling
To begin to characterize this Notch1 cortical signaling mechanism, we first focused on identifying molecular pathways leading to elevated epithelial proliferation. Notch signaling interacts with the Hippo/YAP growth control pathway (Totaro et al., 2017), but we observed no difference in nuclear YAP localization between SCR and NOTCH1KO cells cultured on compliant 2D hydrogels (Fig. S2 D). The receptor tyrosine kinase epidermal growth factor receptor (EGFR) is a critical regulator of mammary tissue expansion during development and adult life (Sternlicht, 2006), so we hypothesized that EGFR activity may be negatively regulated by Notch1 cortical signaling. NOTCH1KO cells cultured in high EGF–containing medium (20 ng/ml) have elevated levels of active, tyrosine phosphorylated EGFR (Y845) compared with SCR and ICDKO (Fig. 2 I). Given this difference in EGFR phosphorylation, we next examined EGFR phosphorylation dynamics in response to the EGF ligand. Comparing EGFR activity levels in cells cultured in low EGF–containing medium (2 ng/ml) to those stimulated with medium containing high EGF (20 ng/ml) revealed a 1.7-fold increase in pEGFR in NOTCH1KO cells (Fig. 2 J). This suggests that Notch1 cortical signaling negatively regulates EGF sensitivity and EGFR phosphorylation in mammary epithelial cells.
Localization to adherens junctions negatively regulates EGFR activity, while internalization of active EGFR can enable mitogenic signaling (Lemmon and Schlessinger, 2010; Sullivan et al., 2022). Immunoprecipitation of E-cadherin from SCR and NOTCH1KO cell lysates showed a substantial decrease in E-cadherin association with EGFR upon deletion of Notch1 (Fig. S2 E). Indeed, EGFR localizes to cell–cell interfaces in SCR, ICDKO, and dnMAML monolayers; however, NOTCH1KO cells have diminished EGFR cell–cell contact localization and increased internalized cytoplasmic localization (Fig. 2, K and L; and Fig. S2, F and G). Addition of TexasRed-labeled EGF (TR-EGF) to culture medium similarly revealed increased intracellular TR-EGF accumulation in NOTCH1KO cells (Fig. S2, H and I), consistent with ligand-activated EGFR internalization (Curto et al., 2007). Further, removal of EGF from the culture medium normalizes NOTCH1KO increases in proliferation (Fig. S2 J). To causally relate EGFR kinase activity to the 3D NOTCH1KO duct morphogenic defect, ducts were treated with the EGFR kinase inhibitor Erlotinib 3 d after cell seeding. Erlotinib treatment significantly reduces NOTCH1KO lumen cell in-filling, proliferation, and increases lumen sizes, but did not fully ameliorate disordered cell multilayering compared with vehicle control (Fig. 2, M–O; and Fig. S2 K). Altogether, these data support a model in which elevated EGFR kinase activity contributes to the aberrant proliferation observed upon the loss of Notch1 cortical signaling but suggests that EGFR activity is not responsible for defects in epithelial architecture and cell organization.
Notch1 cortical signaling stabilizes adherens junctions and cortical actin
Adherens junctions and associated actomyosin networks are critical regulators of cell–cell contact-dependent growth regulation. One mechanism by which this occurs is through suppression of EGFR mobility, internalization, and mitogenic signaling by stable adherens junctions (Chiasson-MacKenzie et al., 2015; Qian et al., 2004). Further, tension at adherens junctions is necessary for accurate orientation of epithelial cell division (Lisica et al., 2022). We therefore hypothesized that the loss of Notch1 cortical signaling causes defects in EGFR-driven proliferation and cell architecture through the alteration of adherens junctions.
Examination of SCR, NOTCH1KO, and ICDKO cells cultured on compliant ECM hydrogels revealed differences in the organization of E-cadherin-based adherens junctions, specifically within NOTCH1KO cells. SCR, ICDKO, and dnMAML adherens junctions are overall linear and continuous; however, NOTCH1KO adherens junctions are discontinuous and oriented orthogonal to the cell–cell interface (Fig. 3 A and Fig. S4, A–C). This distinct junction morphology is reminiscent of focal adherens junctions, which are immature adherens junctions that are typically associated with radially oriented actin fibers and posited to bear increased tension (Fig. 3, A and B; Oldenburg et al., 2015). Indeed, while cortical actin is tightly enriched at SCR and ICDKO cell–cell interfaces, NOTCH1KO actin fibers are less cortically compact and fail to align parallel to the cell–cell interface (Fig. 3, C and D; and Fig. S4, A and B). E-cadherin-based adherens junctions and cortical actin organization are similarly disordered in NOTCH1KO human intestinal, mammary, and bronchial epithelia (Fig. S3). Increased cell–ECM traction forces are associated with destabilized adherens junctions in epithelial monolayers (Mertz et al., 2013; Scarpa et al., 2015). Traction force microscopy identified a 1.5-fold increase in relative cell–substrate tractions in NOTCH1KO cells relative to SCR or ICDKO (Fig. 3, E and F), further indicating destabilization of adherens junctions specifically upon loss of Notch1 cortical signaling.

Notch1 cortical signaling stabilizes adherens junctions and cortical actin. (A) Super-resolution by optical pixel reassignment (SoRa) fluorescence micrographs of scramble control (SCR), NOTCH1KO, and ICDKO cells immunostained for E-cadherin (black). Scale bar, 10 µm. (B) Quantification of the frequency of focal adherens junctions. n ≥ 12 fields of view from three independent experiments. (C) SoRa fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells labeled with phalloidin (black). Scale bar, 10 µm. (D) The intensity of cortical actin at cell–cell junctions, quantified from phalloidin-stained micrographs. n ≥ 12 fields of view from three independent experiments. (E) Traction force microscopy traction maps averaged from 10 fields of view from SCR, NOTCH1KO, and ICDKO cells. Scale bar, 20 µm. (F) Quantification of relative integrated monolayer tractions. n ≥ 11 traction force measurements from three independent experiments. (G) Left: Domain schematics of endogenous Notch1 and two rescue constructs utilized: Notch1 ICD overexpression (ICD O/E) and a N-terminal SNAP-tagged Notch1 TMD (SNAP-TMD). Right: Fluorescence micrographs of NOTCH1KO cells expressing transduction control GFP (top row), SNAP-TMD (middle row), or ICD O/E (bottom row). Cells were immunostained with E-cadherin (black) and labeled with phalloidin (black). Scale bar, 20 µm. (H) Fluorescence micrograph of NOTCH1KO cells expressing SNAP-TMD (green) and immunostained with E-cadherin (magenta). Blue arrows indicate junctions with asymmetric expression of SNAP-TMD. Scale bar, 20 µm. (I) Quantification of the frequency of focal adherens junctions in cells with symmetric expression of SNAP-TMD and asymmetric expression of SNAP-TMD. n ≥ 12 fields of view from three independent experiments. For plots B, D, and F, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. For plot I, mean ± SEM; two-tailed unpaired t test, **P < 0.01.
Notch1 cortical signaling stabilizes adherens junctions and cortical actin. (A) Super-resolution by optical pixel reassignment (SoRa) fluorescence micrographs of scramble control (SCR), NOTCH1KO, and ICDKO cells immunostained for E-cadherin (black). Scale bar, 10 µm. (B) Quantification of the frequency of focal adherens junctions. n ≥ 12 fields of view from three independent experiments. (C) SoRa fluorescence micrographs of SCR, NOTCH1KO, and ICDKO cells labeled with phalloidin (black). Scale bar, 10 µm. (D) The intensity of cortical actin at cell–cell junctions, quantified from phalloidin-stained micrographs. n ≥ 12 fields of view from three independent experiments. (E) Traction force microscopy traction maps averaged from 10 fields of view from SCR, NOTCH1KO, and ICDKO cells. Scale bar, 20 µm. (F) Quantification of relative integrated monolayer tractions. n ≥ 11 traction force measurements from three independent experiments. (G) Left: Domain schematics of endogenous Notch1 and two rescue constructs utilized: Notch1 ICD overexpression (ICD O/E) and a N-terminal SNAP-tagged Notch1 TMD (SNAP-TMD). Right: Fluorescence micrographs of NOTCH1KO cells expressing transduction control GFP (top row), SNAP-TMD (middle row), or ICD O/E (bottom row). Cells were immunostained with E-cadherin (black) and labeled with phalloidin (black). Scale bar, 20 µm. (H) Fluorescence micrograph of NOTCH1KO cells expressing SNAP-TMD (green) and immunostained with E-cadherin (magenta). Blue arrows indicate junctions with asymmetric expression of SNAP-TMD. Scale bar, 20 µm. (I) Quantification of the frequency of focal adherens junctions in cells with symmetric expression of SNAP-TMD and asymmetric expression of SNAP-TMD. n ≥ 12 fields of view from three independent experiments. For plots B, D, and F, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns denotes non-significant. For plot I, mean ± SEM; two-tailed unpaired t test, **P < 0.01.

Loss of Notch1 cortical signaling disrupts epithelial adherens junctions and cortical actin organization. (A) Source full field of view fluorescence micrographs of scramble control (SCR), NOTCH1KO, ICDKO, and dnMAML cells immunostained for E-cadherin (black) for representative images in Fig. 3. (B) Source full field of view fluorescence micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML cells labeled with phalloidin (black) for representative images in Fig. 3. (C) SoRa fluorescence micrographs of dnMAML cells labeled with E-cadherin (black) or phalloidin (black). Scale bar, 10 µm. (D) Source full field of view fluorescence micrographs of wild type cells treated with DMSO or 10 µM DAPT for 2 h immunostained for E-cadherin (black, top row) and labeled with phalloidin (black, bottom row) for representative images in D. (E) Fluorescence micrographs of wild type cells treated with DMSO or 10 µM DAPT for 2 h and immunostained with E-cadherin (black). Scale bar, 10 µm. (F) Quantification of the frequency of focal adherens junctions in DMSO and DAPT treated cells. n ≥ 13 fields of view from three independent experiments. (G) Intensity of cortical actin at cell–cell junctions, quantified from phalloidin-stained micrographs of DMSO and DAPT treated cells. n ≥ 13 fields of view from three independent experiments. (H) Fluorescence micrographs of single frames from a timelapse movie of wild type cells labeled with SPY650-FastAct (black) and treated with 10 µM DAPT. (I) Left: Fluorescence micrographs of SCR cells or ICDKO cells treated with 10 µM DAPT for 2 h, immunostained for E-cadherin (black), and Notch1 (black) and labeled with Hoechst (blue). Right: Quantification of the frequency of focal adherens junctions in SCR + DAPT and ICDKO + DAPT treated cells. n = 6 fields of view from three independent experiments. (J) Immunofluorescence micrographs of NOTCH1KO cells treated with DMSO or 1 µM Erlotinib immunostained for E-cadherin (black). All scale bars, 10 µm. For plots F, G, and I, mean ± SEM; two-tailed unpaired t test, *P < 0.05, **P < 0.01, ****P < 0.0001.

Loss of Notch1 cortical signaling disrupts epithelial adherens junctions and cortical actin organization. (A) Source full field of view fluorescence micrographs of scramble control (SCR), NOTCH1KO, ICDKO, and dnMAML cells immunostained for E-cadherin (black) for representative images in Fig. 3. (B) Source full field of view fluorescence micrographs of SCR, NOTCH1KO, ICDKO, and dnMAML cells labeled with phalloidin (black) for representative images in Fig. 3. (C) SoRa fluorescence micrographs of dnMAML cells labeled with E-cadherin (black) or phalloidin (black). Scale bar, 10 µm. (D) Source full field of view fluorescence micrographs of wild type cells treated with DMSO or 10 µM DAPT for 2 h immunostained for E-cadherin (black, top row) and labeled with phalloidin (black, bottom row) for representative images in D. (E) Fluorescence micrographs of wild type cells treated with DMSO or 10 µM DAPT for 2 h and immunostained with E-cadherin (black). Scale bar, 10 µm. (F) Quantification of the frequency of focal adherens junctions in DMSO and DAPT treated cells. n ≥ 13 fields of view from three independent experiments. (G) Intensity of cortical actin at cell–cell junctions, quantified from phalloidin-stained micrographs of DMSO and DAPT treated cells. n ≥ 13 fields of view from three independent experiments. (H) Fluorescence micrographs of single frames from a timelapse movie of wild type cells labeled with SPY650-FastAct (black) and treated with 10 µM DAPT. (I) Left: Fluorescence micrographs of SCR cells or ICDKO cells treated with 10 µM DAPT for 2 h, immunostained for E-cadherin (black), and Notch1 (black) and labeled with Hoechst (blue). Right: Quantification of the frequency of focal adherens junctions in SCR + DAPT and ICDKO + DAPT treated cells. n = 6 fields of view from three independent experiments. (J) Immunofluorescence micrographs of NOTCH1KO cells treated with DMSO or 1 µM Erlotinib immunostained for E-cadherin (black). All scale bars, 10 µm. For plots F, G, and I, mean ± SEM; two-tailed unpaired t test, *P < 0.05, **P < 0.01, ****P < 0.0001.
Our previous work identified that TMD is the essential domain of Notch1 for regulating endothelial adherens junctions, and Notch1 cortical signaling via TMD requires removal of the ICD from TMD (Polacheck et al., 2017). To test the hypothesis that proteolytic activation of Notch1 and subsequent removal of the ICD was a necessary step for epithelial Notch1 cortical signaling, we acutely treated wild type epithelial monolayers with DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester), an inhibitor of γ-secretase that prevents cleavage of Notch1 at the S3 site to release the ICD. Acute treatment with DAPT results in focal adherens junctions and disorganized cortical actin fibers (Fig. S4, D–G). Further, visualizing live actin dynamics following treatment with DAPT revealed the dissolution of cortical actin fibers within 30 min (Fig. S4 H). Additionally, ICDKO cells maintain stable adherens junctions irrespective of whether they were treated with DAPT (Fig. S4 I). Treatment of NOTCH1KO cells with Erlotinib did not prevent focal adherens junctions, confirming that the adherens junction phenotype is independent of EGFR kinase activity (Fig. S4 J).
These findings are consistent with a model in which Notch1 cortical signaling functions through the TMD to regulate epithelial adherens junctions. To specifically test the roles of Notch1 TMD or ICD in stabilizing epithelial adherens junction and cortical actin, we expressed either ICD or a TMD in NOTCH1KO cells. To facilitate imaging and biochemical purification, we added an extracellular SNAP tag to the TMD N-terminus, hereafter called SNAP-TMD. Upon expression, ICD localizes to the nucleus and increases mRNA transcript levels of the Notch1 target HEY1. However, expression of ICD leads to significant decreases in E-cadherin expression, disorganized cortical actin, and cell multilayering (Fig. S5, A–D). In contrast, SNAP-TMD localizes robustly to cell–cell contacts, reduces focal adherens junction frequency, and enhances cortical actin organization relative to NOTCH1KO cells expressing ICD or a GFP transduction control (Fig. 3 G). Interestingly, the rescue of adherens junction and cortical actin phenotypes requires expression in both cells sharing the junction, as junctions with asymmetric SNAP-TMD expression are typically characterized by focal adherens junctions (Fig. 3, H and I, blue arrows). Altogether, these results identify that Notch1 cortical signaling functions to stabilize epithelial adherens junctions and cortical actin organization, which is meditated by Notch1 TMD.

Overexpression of the Notch1 ICD does not rescue the NOTCH1KOphenotype;loss of FAM83H leads to focal adherens junctions, depleted cortical actin, and alterations in ductal architecture. (A) Fluorescence micrographs of NOTCH1KO cells overexpressing Notch1 ICD (ICD O/E) immunostained for Notch1 ICD (white), E-cadherin (white), and labeled with Hoechst (white) and phalloidin (white). Scale bar, 20 µm. (B) YZ orthogonal projections from micrographs of NOTCH1KO cells overexpressing Notch1 ICD (ICD O/E) labeled with Hoechst (white). (C) Medial confocal slice micrographs from NOTCH1KO and NOTCH1KO cells expressing Notch1 ICD labeled with phalloidin (black). Scale bar, 50 µm. (D) mRNA expression of Notch1 target genes HES1 and HEY1 measured by qPCR in scramble control (SCR), SCR cells plated on recombinant Dll4 (rDll4), and SCR cells overexpressing Notch1 ICD. Average qPCR reads from n = 3 independent experiments. (E) Expression of Notch1-target genes HES1 and HEY1 measured by qPCR in low confluence and polarized wild type MCF10A. Average qPCR reads from n = 3 independent experiments. For plot D, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, ****P < 0.0001, ns denotes non-significant. For plot E, mean ± SEM; two-tailed unpaired t test, **P < 0.01, ***P < 0.001. (F) Representative Coomassie stained SDS-PAGE gel of Notch1 immunoprecipitation from LC and P cells. F denotes the band identified as FAM83H by mass spectrometry. (G) Medial confocal slice micrographs of SCR and FAM83HKO ducts labeled with Hoechst (black). Scale bar, 100 µm. (H) Source full field of view fluorescence micrographs of SCR and FAM83HKO cells stained with E-cadherin (black, top row) and labeled with phalloidin (black, bottom row) for representative images in Fig. 5. Scale bars, 10 µm. Source data are available for this figure: SourceData FS5.

Overexpression of the Notch1 ICD does not rescue the NOTCH1KOphenotype;loss of FAM83H leads to focal adherens junctions, depleted cortical actin, and alterations in ductal architecture. (A) Fluorescence micrographs of NOTCH1KO cells overexpressing Notch1 ICD (ICD O/E) immunostained for Notch1 ICD (white), E-cadherin (white), and labeled with Hoechst (white) and phalloidin (white). Scale bar, 20 µm. (B) YZ orthogonal projections from micrographs of NOTCH1KO cells overexpressing Notch1 ICD (ICD O/E) labeled with Hoechst (white). (C) Medial confocal slice micrographs from NOTCH1KO and NOTCH1KO cells expressing Notch1 ICD labeled with phalloidin (black). Scale bar, 50 µm. (D) mRNA expression of Notch1 target genes HES1 and HEY1 measured by qPCR in scramble control (SCR), SCR cells plated on recombinant Dll4 (rDll4), and SCR cells overexpressing Notch1 ICD. Average qPCR reads from n = 3 independent experiments. (E) Expression of Notch1-target genes HES1 and HEY1 measured by qPCR in low confluence and polarized wild type MCF10A. Average qPCR reads from n = 3 independent experiments. For plot D, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, ****P < 0.0001, ns denotes non-significant. For plot E, mean ± SEM; two-tailed unpaired t test, **P < 0.01, ***P < 0.001. (F) Representative Coomassie stained SDS-PAGE gel of Notch1 immunoprecipitation from LC and P cells. F denotes the band identified as FAM83H by mass spectrometry. (G) Medial confocal slice micrographs of SCR and FAM83HKO ducts labeled with Hoechst (black). Scale bar, 100 µm. (H) Source full field of view fluorescence micrographs of SCR and FAM83HKO cells stained with E-cadherin (black, top row) and labeled with phalloidin (black, bottom row) for representative images in Fig. 5. Scale bars, 10 µm. Source data are available for this figure: SourceData FS5.
Localization and cleavage of Notch1 at lateral cell–cell contacts
Phenotypes in NOTCH1KO ducts present during lumen expansion (Fig. 1) and NOTCH1KO cells have impaired columnar morphology (Fig. 2), indicating that Notch1 cortical signaling may play a role in stabilizing adherens junctions as cells reach confluence and undergo apical–basal restructuring. To further understand the mechanistic contribution of Notch1 cortical signaling to this morphodynamic change, we assessed the localization of endogenous Notch1 within MCF10A cultured on compliant ECM hydrogels at distinct morphogenic timepoints ranging from low confluence (LC), where cells are surrounded by other cells yet remained elongated, to high confluence (HC), where cells are surrounded and cuboidal but still flat, to polarized (P), where cells had adopted a columnar morphology. During this transition, immunofluorescence staining revealed Notch1 progressively accumulates at cell–cell interfaces (Fig. 4, A and B). High magnification confocal micrographs further showed that E-cadherin most strongly localizes to apical domains in the polarized state, while Notch1 and cortical actin intensity is highest at lateral cell membranes (Fig. 4 C). This lateral localization is consistent with a recent study reporting that Notch1 activity is limited by the formation of lateral membrane contacts and adherens junctions during cellularization of the embryonic Drosophila syncytium (Falo-Sanjuan and Bray, 2021).

Localization and proteolytic activation of Notch1 at lateral cell–cell contacts. (A) Immunofluorescence micrographs of Notch1 (black) in MCF10A in low confluence (LC), high confluence (HC), and polarized (P) states. Scale bar, 20 µm. (B) Quantification of relative Notch1 intensity across the width of the cell. n = 8 cells from three independent experiments. (C) Representative SoRa immunofluorescence micrographs of wild type cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Top row: Apical domain. Bottom row: Lateral domain. Scale bar, 20 µm. (D) Top: Western blot of wild type lysates from the indicated monolayer states, immunoblotted for cleaved Notch1 V1754 (N1 V1754), total Notch1, and GAPDH. Bottom: Quantification of fold change in N1 V1754 and total Notch1 band intensities. n = 3 independent experiments. (E) Western blot of cytosolic and nuclear fractions from wild type monolayer lysates in LC and P states, immunoblotted for total Notch1, cleaved Notch1 V1754 (N1 V1754), Lamin B1, and GAPDH. (F) Left: Schematic of Notch1 transcriptional destabilized GFP reporter (d1GFP). Right: Western blot of lysates from wild type cells treated with DMSO, DMSO + rDLL4, DAPT + rDLL4, or overexpressing a constitutively active form of Notch1 (TMD-ICD-mApple), immunoblotted for GFP, N1 V1754, and GAPDH. Western blots are representative of three independent experiments. For plot in D, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, **P < 0.01, ***P < 0.001, ns denotes non-significant. Source data are available for this figure: SourceData F4.
Localization and proteolytic activation of Notch1 at lateral cell–cell contacts. (A) Immunofluorescence micrographs of Notch1 (black) in MCF10A in low confluence (LC), high confluence (HC), and polarized (P) states. Scale bar, 20 µm. (B) Quantification of relative Notch1 intensity across the width of the cell. n = 8 cells from three independent experiments. (C) Representative SoRa immunofluorescence micrographs of wild type cells immunostained for E-cadherin (black) and Notch1 (black) and labeled with phalloidin (black). Top row: Apical domain. Bottom row: Lateral domain. Scale bar, 20 µm. (D) Top: Western blot of wild type lysates from the indicated monolayer states, immunoblotted for cleaved Notch1 V1754 (N1 V1754), total Notch1, and GAPDH. Bottom: Quantification of fold change in N1 V1754 and total Notch1 band intensities. n = 3 independent experiments. (E) Western blot of cytosolic and nuclear fractions from wild type monolayer lysates in LC and P states, immunoblotted for total Notch1, cleaved Notch1 V1754 (N1 V1754), Lamin B1, and GAPDH. (F) Left: Schematic of Notch1 transcriptional destabilized GFP reporter (d1GFP). Right: Western blot of lysates from wild type cells treated with DMSO, DMSO + rDLL4, DAPT + rDLL4, or overexpressing a constitutively active form of Notch1 (TMD-ICD-mApple), immunoblotted for GFP, N1 V1754, and GAPDH. Western blots are representative of three independent experiments. For plot in D, mean ± SEM; one-way ANOVA with Tukey’s post-hoc test, **P < 0.01, ***P < 0.001, ns denotes non-significant. Source data are available for this figure: SourceData F4.
Our data suggest that Notch1 activation and cleavage are necessary for cortical signaling function, so we next examined whether Notch1 junctional accumulation correlated with functional changes in ICD S3 cleavage or Notch1 transcriptional activity. Western blot analysis of monolayers transitioning between low confluence and polarized states revealed that Notch1 junctional accumulation is coincident with a sixfold increase in γ-secretase-mediated cleavage of ICD (cleavage-specific Notch1 V1754 antibody) with no significant increase in total Notch1 protein levels (Fig. 4 D). Despite substantial increases in cleaved ICD, the amount of ICD within isolated nuclear fractions does not significantly change between low confluence and polarized states (Fig. 4 E), and mRNA transcript levels of Notch1 transcriptional targets HES1 and HEY1 decrease (Fig. S5 E). Interestingly, ICD within nuclear fractions presents as two lower molecular weight bands relative to ICD in cytosolic fractions, which is consistent with intracellular posttranslational regulatory mechanisms directing ICD function after cleavage (Antfolk et al., 2019).
To further investigate if increased ICD cleavage in the polarized state leads to ICD-dependent transcription, we generated cells stably expressing a fluorescent protein-based Notch transcription reporter consisting of twelve CSL-binding motifs coupled to a destabilized GFP (d1GFP) that has an approximate halflife of 1–2 h (Hansson et al., 2006). Despite increased levels of cleaved ICD, polarized monolayers have no detectable GFP expression. Coating hydrogels with recombinant Notch ligand Delta Like Canonical Notch Ligand 4 (rDll4) further increases ICD cleavage but does not increase HES1 or HEY1 transcript levels (Fig. S5 D) and similarly lacks reporter GFP expression. However, expressing a constitutively active form of Notch1 lacking the ECD (TMD-ICD-mApple; Chiang et al., 2006; Polacheck et al., 2017) at levels approximately twofold endogenous Notch1 is sufficient to stimulate reporter GFP expression (Fig. 4 F). Altogether, these data indicate that as cells reach confluence and initiate apical–basal restructuring, Notch1 localizes to lateral cell–cell contacts and the ICD is proteolytically removed from the TMD. This increase in cleaved ICD does not lead to higher levels of ICD in nuclear fractions or the expression of a Notch transcriptional reporter, which can be engaged by overexpression of a constitutively active form of Notch1. Along with the truncation of the ICD and expression of dnMAML, this observed elevated proteolytic activation absent from a robust transcriptional response further affirms an important role for Notch1 cortical signaling in stabilizing adherens junctions in epithelial monolayers.
Notch1 cortical signaling functions through FAM83H to stabilize adherens junctions and regulate tissue architecture
To identify molecular pathways associated with the localization and/or adherens junction stabilizing function of Notch1 cortical signaling at lateral cell contacts, we unbiasedly profiled differential Notch1 interacting proteins from low confluence and polarized monolayer lysates using Notch1 immunoprecipitation, SDS-PAGE and Coomassie staining, and mass spectrometry (Fig. 5 A and Fig. 5 F). Notably, while this approach identified several distinct interactions, Notch transcriptional effectors MAML1/2 and RBPJ were not identified, supporting the observed lack of increase in nuclear ICD within polarized monolayers (Fig. 4 E). One prominent band (∼150 kD) isolated from polarized monolayers was identified as FAM83H from the FAM83 family of oncogenes (Snijders et al., 2017). Coimmunoprecipitation and Western blot confirmed a FAM83H–Notch1 interaction that increases threefold as monolayers progress from low confluence to polarized states (Fig. 5 B).

Notch1 cortical signaling functions through FAM83H to stabilize adherens junctions and regulate duct architecture. (A) Schematic of mass spectrometry workflow to identify monolayer state-dependent, differential Notch1 protein–protein interactions. (B) Western blot of immunoprecipitated Notch1 from low confluence (LC) or polarized (P) cells immunoblotted for FAM83H and Notch1. (C) Western blot of lysates from SCR or FAM83HKO cells immunoblotted for FAM83H, cleaved Notch1 V1754 (N1 V1754), and E-cadherin. (D) Representative medial confocal slice fluorescence micrographs of SCR and FAM83HKO ducts labeled with phalloidin (black). Scale bar, 100 µm. (E) Quantification of duct lumen diameter. Average lumen diameters from n ≥ 9 independent ducts. (F) Quantification of internuclear distances measured from Hoechst labeled ducts. Average internuclear distances from n ≥ 9 independent ducts. (G) Immunofluorescence micrographs of SCR and FAM83HKO cells immunostained with E-cadherin (black) and the corresponding quantification of the frequency of focal adherens junctions. Scale bar, 10 µm. n ≥ 9 fields of view from three independent experiments. (H) Fluorescence micrographs of SCR and FAM83HKO cells labeled with phalloidin (black) and the corresponding quantification of cell–cell junction cortical actin intensity. Scale bar, 10 µm. n ≥ 9 fields of view from three independent experiments. (I) Western blot of immunoprecipitation of E-cadherin from SCR, NOTCH1KO, and ICDKO cells immunoblotted for FAM83H and E-cadherin. (J) Left: Immunofluorescence micrographs of methanol fixed SCR, NOTCH1KO, and ICDKO cells immunostained with FAM83H (white, top row), and E-cadherin (magenta, bottom row). Scale bar, 10 µm. Right: Quantification of relative FAM83H intensity along a seven-micron line centered on and drawn orthogonal to cell–cell junctions of SCR, NOTCH1KO, and ICDKO cells (representative yellow dashed line). n = 6 junction profiles from three independent experiments. (K) Western blot of SNAP-Capture affinity purification from NOTCH1KO and NOTCH1KO + SNAP-TMD cells immunoblotted for E-cadherin, FAM83H, and SNAP. (L) Western blot of immunoprecipitation of E-cadherin from NOTCH1KO and NOTCH1KO + SNAP-TMD cells immunoblotted for E-cadherin, FAM83H, and SNAP. Western blots are representative of three independent experiments. For all plots, mean ± SEM; two-tailed unpaired t test, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F5.
Notch1 cortical signaling functions through FAM83H to stabilize adherens junctions and regulate duct architecture. (A) Schematic of mass spectrometry workflow to identify monolayer state-dependent, differential Notch1 protein–protein interactions. (B) Western blot of immunoprecipitated Notch1 from low confluence (LC) or polarized (P) cells immunoblotted for FAM83H and Notch1. (C) Western blot of lysates from SCR or FAM83HKO cells immunoblotted for FAM83H, cleaved Notch1 V1754 (N1 V1754), and E-cadherin. (D) Representative medial confocal slice fluorescence micrographs of SCR and FAM83HKO ducts labeled with phalloidin (black). Scale bar, 100 µm. (E) Quantification of duct lumen diameter. Average lumen diameters from n ≥ 9 independent ducts. (F) Quantification of internuclear distances measured from Hoechst labeled ducts. Average internuclear distances from n ≥ 9 independent ducts. (G) Immunofluorescence micrographs of SCR and FAM83HKO cells immunostained with E-cadherin (black) and the corresponding quantification of the frequency of focal adherens junctions. Scale bar, 10 µm. n ≥ 9 fields of view from three independent experiments. (H) Fluorescence micrographs of SCR and FAM83HKO cells labeled with phalloidin (black) and the corresponding quantification of cell–cell junction cortical actin intensity. Scale bar, 10 µm. n ≥ 9 fields of view from three independent experiments. (I) Western blot of immunoprecipitation of E-cadherin from SCR, NOTCH1KO, and ICDKO cells immunoblotted for FAM83H and E-cadherin. (J) Left: Immunofluorescence micrographs of methanol fixed SCR, NOTCH1KO, and ICDKO cells immunostained with FAM83H (white, top row), and E-cadherin (magenta, bottom row). Scale bar, 10 µm. Right: Quantification of relative FAM83H intensity along a seven-micron line centered on and drawn orthogonal to cell–cell junctions of SCR, NOTCH1KO, and ICDKO cells (representative yellow dashed line). n = 6 junction profiles from three independent experiments. (K) Western blot of SNAP-Capture affinity purification from NOTCH1KO and NOTCH1KO + SNAP-TMD cells immunoblotted for E-cadherin, FAM83H, and SNAP. (L) Western blot of immunoprecipitation of E-cadherin from NOTCH1KO and NOTCH1KO + SNAP-TMD cells immunoblotted for E-cadherin, FAM83H, and SNAP. Western blots are representative of three independent experiments. For all plots, mean ± SEM; two-tailed unpaired t test, ***P < 0.001, ****P < 0.0001. Source data are available for this figure: SourceData F5.
The cellular function of FAM83H is not well understood, despite reported roles in developmental morphogenesis, cancer progression, and intermediate filament dynamics (Kim et al., 2008, 2019; Kuga et al., 2016). We first investigated whether FAM83H contributes to the morphogenic phenotypes associated with loss of Notch1 cortical signaling by engineering 3D ducts from MCF10A depleted of FAM83H by CRISPR-Cas9 (FAM83HKO; Fig. 5 C). FAM83HKO phenocopies key morphologic signatures associated with NOTCH1KO ducts, namely a tortuous duct architecture that is driven by lumen cell in-filling, multilayering, and increased cell packing (Fig. 5, D–F; and Fig. S5 G). FAM83HKO does not abolish Notch1 ICD cleavage, Notch1 localization to cell–cell contacts, or E-cadherin expression levels (Fig. 5 C), suggesting FAM83H functions as a downstream arm of the Notch1 cortical pathway. Indeed, FAM83HKO results in focal adherens junctions and disordered cortical actin, resembling phenotypes observed upon loss of Notch1 cortical signaling in NOTCH1KO (Fig. 5, G and H; and Fig. S5 H).
FAM83H coimmunoprecipitates with E-cadherin, consistent with identification in the E-cadherin adhesome (Guo et al., 2014) and a function downstream of Notch1 cortical signaling, and this interaction is significantly reduced in NOTCH1KO cells relative to SCR or ICDKO cells (Fig. 5 I). Interestingly, methanol, but not paraformaldehyde, fixation of polarized epithelia revealed an accumulation of endogenous FAM83H at and proximal to lateral cell–cell contacts in SCR and ICDKO cells that are lost in NOTCH1KO cells (Fig. 5 J). Affinity precipitation of SNAP–TMD expressed in NOTCH1KO cells revealed that SNAP–TMD complexes with FAM83H and E-cadherin (Fig. 5 K). Importantly, the expression of SNAP–TMD in NOTCH1KO cells increases coimmunoprecipitation between E-cadherin and FAM83H (Fig. 5 L). Altogether, these data provide evidence for a novel interaction between Notch1 and FAM83H and identify FAM83H as a new Notch1 cortical signaling effector that functions in the stabilization of epithelial adherens junctions and cortical actin organization.
Discussion
Notch signaling broadly controls developmental and homeostatic morphogenic processes that involve coordinated changes in cell architecture and gene expression; however, how a singular canonical transcriptional pathway is able to produce such diverse biological output is unclear (Bray, 2016). The possibility of direct links from Notch to cell–cell adhesion and the actin cytoskeleton has been suggested in past studies on axon guidance, keratinocyte motility, Drosophila embryogenesis, sprouting angiogenesis (Crowner et al., 2003; Lowell and Watt, 2001; Major and Irvine, 2005; Zakirov et al., 2021), and our previous work describing a shear stress-dependent Notch1 cortical signaling pathway that regulates vascular barrier function in endothelial cells (Polacheck et al., 2017). Our study here reveals a new context by which Notch1 can influence the tissue architecture of an engineered 3D human ductal epithelium through a mechanism distinct from Notch1 ICD transcriptional signaling.
We report that Notch1 controls epithelial cell architecture and proliferation by regulating adherens junctions independent of ICD-mediated transcription. This is supported by genetic perturbation that specifically truncates the endogenous Notch1 ICD, a dominant negative approach (dnMAML) to globally suppress Notch transcription, and phenotypic rescue by Notch1 TMD but not ICD in NOTCH1KO cells. Unlike sender–receiver models of Notch lateral induction or inhibition that pattern the fates of proximal cells via transcription, here, within a homogenous epithelial monolayer, Notch1 cortical signaling is uniformly engaged and coordinates changes in cell–cell adhesions via the TMD. This unappreciated function of Notch1 may provide key insights into how Notch1 can exert not only its known transcriptional effects but also the structural changes associated with the many morphogenetic contexts where Notch1 is critical, such as cooperative Notch1-dependent changes in cell fate and differential adhesion necessary for mammalian sensory hair cell patterning within the cochlea (Cohen et al., 2023). Indeed, we demonstrate adherens junction and actin defects result from the loss of cortical Notch1 signaling in several human epithelial cell types in addition to MCF10A, which also show associated tissue morphogenic consequences in a 3D tissue-engineered model of a simple ductal epithelium. Still, despite these important morphogenic insights, the marked absence of a requirement for Notch1 transcriptional signaling in our model does not allow for careful dissection of cooperativity between Notch1 cortical and transcriptional signaling. For instance, in vivo, the mammary ductal epithelium is a bilayered architecture consisting of outer basal and inner luminal cells. Notch1 is expressed in luminal cells and Notch1 activity is a key determinant of luminal cell fate (Bouras et al., 2008). Interestingly, FAM83H is similarly expressed in luminal cells (Bach et al., 2017), and it is therefore plausible that Notch1 cortical and transcriptional signals coordinate luminal fate, positioning, and barrier function in the mammary gland. Future work will focus on defining the coordination and relative contributions of these two pathways to tissue development and homeostasis.
While offering important insight into an unappreciated arm of Notch1 signaling, this study also illuminates a new mechanism by which cell–cell adhesion may be dynamically regulated. There is considerable evidence in vivo and in vitro that modulation of Notch1 activation converges on adherens junction dynamics (Bentley et al., 2014; Falo-Sanjuan and Bray, 2021; Grammont, 2007; Polacheck et al., 2017), and we are just beginning to define the contexts and molecular underpinnings. Here, Notch1 cortical signaling via TMD mediates changes in adherens junctions independent of E-cadherin expression levels, which could provide an important new mechanism for morphogenetic patterning by differential adhesion that might apply to other developmental contexts. This work also advances our understanding of transcription-independent Notch1 function by identifying a previously unappreciated interaction with FAM83H, whereby FAM83H recruitment to E-cadherin is dependent on Notch1 TMD. We find that FAM83H localizes to cell–cell interfaces in polarized epithelial monolayers. This is in agreement with studies that show FAM83H localizes to cell–cell interfaces in vivo (Kuga et al., 2016) and posit FAM83H functions as a peripheral membrane protein (Ding et al., 2009). Interestingly, while TMD is sufficient to complex with FAM83H and increase FAM83H association with E-cadherin, coimmunoprecipitation of FAM83H and E-cadherin is reduced in ICDKO cells relative to SCR, suggesting ICD may cooperate with TMD for complex formation. Still, the nature of FAM83H interaction and recruitment within the context of Notch1 cortical signaling remains to be determined.
Notch signaling depends on the size and geometry of the contact sites in sender–receiver cell models (Shaya et al., 2017). We demonstrate that Notch1 cortical signaling is uniformly engaged as Notch1 accumulates at lateral cell–cell interfaces and is proteolytically cleaved during epithelial apical–basal columnar restructuring. Notch1 accumulation at lateral interfaces independent of protein level change suggests active recruitment through mechanisms linked to this change in cellular architecture, and is consistent with positive regulation of Notch activation by the polarity protein Par3 in vivo (Wu et al., 2022, Preprint) and in vitro (Williams et al., 2017). Reciprocally, our model predicts that Notch1 activation and cortical signaling in turn maintains apical–basal cell architecture and cell–cell adhesion. Indeed, histological sections of Notch1−/− murine intestinal epithelia show altered epithelial polarity and cell–cell adhesion, coincident with compromised intestinal barrier function (Dahan et al., 2011; Dunkin et al., 2018). Moreover, we observe a substantial increase in γ-secretase-cleaved ICD during this transition, yet this increase in cleaved ICD does not lead to more nuclear-localized ICD or transcription, suggesting intricate, still-undetermined mechanisms gate transcriptional activity of cleaved ICD. While the distinct molecular weights observed in nuclear fractions indicate posttranslational ICD regulation, another potential mechanism is an additional requirement for nuclear mechanotransduction. In this model, forces on the nucleus gate ICD transcription, either through regulation of nuclear pore transport or altered chromatin accessibility, as was proposed for Notch during mesoderm invagination in Drosophila gastrulation (Falo-Sanjuan and Bray, 2022).
How FAM83H contributes to adherens junction stability is an outstanding question. One of the few prescribed functions of FAM83H is the regulation of keratin intermediate filament dynamics through casein kinase I (Kim et al., 2008, 2019; Tokuchi et al., 2021). A recent report described a role for keratin-desmosome networks in organizing the cortical actin cytoskeleton to control epithelial cohesion and limit tensile stress on adherens junctions (Prechova et al., 2022), which aligns with adherens junction phenotypes upon loss of Notch1 cortical signaling. Interestingly, loss of function mutations in Fam83h and Notch1 both cause amelogenesis imperfecta and a failure to form desmosomes between ameloblast and stratum intermedium layers in the mouse incisor (Jheon et al., 2016; Kim et al., 2008). Fam83h−/− mice die by postnatal day 14–21 and have visible skin defects (Wang et al., 2015). Similarly, Notch1−/− keratinocytes impair epidermal barrier integrity (Blanpain et al., 2006; Demehri et al., 2008, 2009), which is a tissue property that is regulated by desmosome–keratin networks (Johnson et al., 2014). Keratin filaments interact with Notch1 to regulate colonic epithelial proliferation and differentiation through an unknown mechanism (Lähdeniemi et al., 2017). It is therefore plausible that reciprocal interactions between Notch1 and desmosome–keratin networks, which involve FAM83H, may contribute to the maintenance of epithelial cell–cell adhesion and differentiation.
Together, our work offers new insights into Notch1 signaling and regulation, how cell–cell adhesions are dynamically regulated, and a model in which transcriptional and adhesive programs might be coordinated. The convergence of transcription-independent Notch1 cortical signaling on cell–cell adhesion regulation may explain skin barrier defects associated with the tumor suppressive function of Notch1, as well as tissue jamming/fluidization events occurring during developmental morphogenesis. Identifying ways to isolate the Notch1 cortical pathway from the transcriptional pathway may therefore provide new opportunities to instruct development and treat associated complications.
Materials and methods
Cell culture
Human mammary epithelial cells (MCF10A; ATCC) were maintained in a growth medium consisting of DMEM/F12 (Gibco), 5% horse serum (Invitrogen), 20 ng/ml EGF (PeproTech), 0.5 mg/ml hydrocortisone (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 10 μg/ml insulin (Sigma-Aldrich). Human bronchial epithelial cells (16HBE14o-; Sigma-Aldrich) were maintained in a medium consisting of α-MEM (Sigma-Aldrich), 10% fetal bovine serum (Peak), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 2 mM L-glutamine (Sigma-Aldrich). Human colorectal adenocarcinoma cells (Caco-2; ATCC) were maintained in a medium consisting of DMEM (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 20% fetal bovine serum (Peak). Human mammary epithelial cells (MCF7; ATCC) were maintained in a medium consisting of DMEM (Sigma-Aldrich), 10% fetal bovine serum (Peak), 10 μg/ml insulin (Sigma-Aldrich), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Human HEK-293T cells (Clonetech) were maintained in DMEM (Sigma-Aldrich), 10% fetal bovine serum (Peak), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 1 mM sodium pyruvate (Gibco). MCF10A, 16HBE14o-, and Caco-2 cells were used at passages 2–12 and all cell types were maintained at 37°C in 5% CO2 in a humidified incubator. Cell numbers were counted at passage using the Countess 3 automated cell counter (Invitrogen). Cell-line authentication (performance, differentiation, and STR profiling) was provided by ATCC, Sigma-Aldrich, and Clonetech. All cells were routinely tested for Mycoplasma via PCR test (Applied Biological Materials).
Antibodies and reagents
Antibodies against Notch1 ICD (D1E11, 1:100 IF, 1:1,000 WB), Notch1 V1744 (D3B8, 1:500 WB), EGFR (D38B1, 1:100 IF, 1:1,000 WB), pEGFR (D7A5, 1:1,000 WB), GFP (D5.1, 1:1,000 WB), non-phospho (active) β-catenin (D13A1, 1:1,000 WB), GAPDH (14C10, 1:10,000 WB), and YAP (D8H1X, 1:200 IF) were from Cell Signaling Technologies. β-catenin antibody (14, 1:1,000 WB) was from BD Biosciences. E-cadherin antibody (HECD-1, 1:1,000 IF, 1:1,000 WB) was from Takara Bio. Notch1 ECD (ABS90, 1:1,000) was from Millipore. FAM83H antibody (1:1,000 WB) was from Bethyl Laboratories. Lamin B1 antibody (12987-1-AP, 1:1,000) was from Proteintech. TexasRed-EGF, rhodamine phalloidin, and Alexa Fluor 488, 568, and 647 goat anti-mouse and anti-rabbit IgG secondary antibodies (1:400) were from Invitrogen. Alexa Fluor 647 azide and EdU cell proliferation kit were from Invitrogen. Hoescht and DAPT were from Sigma. Anti-SNAP (P9310S, 1:1,000), SNAP-Capture, and SNAP-Surface 488 were from New England Biolabs.
Lentiviral-mediated CRISPR-Cas9 editing
Stable CRISPR-modified primary MCF10A, 16HBE14o-, and Caco-2 cell lines were generated using the lentiCRISPRv2 system using our previously established protocols (Kutys et al., 2020; Polacheck et al., 2017). Specific guide RNAs were cloned into the BsmBI site of plentiCRISPRv2: SCR, 5′-GTATTACTGATATTGGTGGG-3′; NOTCH1KO, 5′-CGTCAGCGTGAGCAGGTCGC-3′; ICDKO, 5′-TGCTGTCCCGCAAGCGCCGG-3′; and FAM83HKO, 5′-GGACAACCCACTGGCACCC-3′. sgRNA-containing plentiCRISPRv2 plasmids were cotransfected with psPAX2 (plasmid #12260; Addgene) and pMD2.G (plasmid #12259; Addgene) packaging plasmids into HEK-293T cells using calcium phosphate transfection. After 48 h, viral supernatants were collected from the culture dish, concentrated using 4× lentivirus concentrator (PEG-IT), and resuspended in PBS. MCF10A, 16HBE14o-, MCF7, and Caco-2 cells were transduced in their corresponding growth medium overnight and given fresh medium the following morning. At 48 h after transduction, cells were passaged and plated in 6-well plates at 1.25 × 105 cells per well and selected with 2 μg/ml puromycin for 4 d. All CRISPR modifications were verified by Western blot.
Microfluidic device design and fabrication
The microfluidic device contains four main ports (two basal ports and two luminal ports) along with a central hydrogel-containing compartment. The basal ports functioned as media reservoirs containing all supplementary components of the MCF10A culture medium and luminal ports functioned as inlet and outlet reservoirs to maintain lumen pressure. Between the hydrogel compartment and the basal ports is a row of PDMS pillars that spans the height of the hydrogel compartment and function to contain the hydrogel within that central region. The silicon master was fabricated using photolithography methods previously described (Polacheck et al., 2019); however, UV exposure steps were performed on an Alvéole PRIMO micropatterning system. Individual microfluidic devices were generated using soft lithography. Polydimethylsiloxane (PDMS; Sylgard 184; Dow-Corning) was mixed at a ratio of 10:1 (base:curing agent) and cured at 60°C on a silicon master. The PDMS was cut from the silicon master, trimmed, and surface-activated by plasma treatment for 30 s at 300 mTorr. Devices were then bonded to glass coverslips and surface-treated with 0.01% poly-L-lysine for 2 h and 1% glutaraldehyde for 15 min. Devices were then washed three times with water and sterilized in 70% ethanol for 30 min. Steel acupuncture needles (160 μm diameter, Tai-Chi) were inserted into each device and devices were placed in a vacuum desiccator for 60 min. An unpolymerized ECM mixture containing 70% neutralized collagen type I (Dow-Corning) and 30% growth factor reduced Matrigel (Dow-Corning) was injected into the hydrogel compartment via the ECM-loading port. Collagen type I solution was buffered with 10× DMEM, 10× reconstitution buffer containing 0.2 M HEPES, and 0.26 M sodium bicarbonate, titrated to a pH of 7.6 with NaOH, and brought to a final concentration of 2.5 mg/ml collagen I in PBS. Once the unpolymerized hydrogel was injected into the central compartment, the devices were incubated at 37°C for 25 min. Following polymerization of the hydrogel, all ports were filled with sterile PBS and the devices were left overnight at 37°C. Steel acupuncture needles were removed from the devices to create 160-μm diameter channels in the hydrogel and the outer edges of the PDMS were sealed with vacuum grease (Dow-Corning). Devices were kept hydrated with sterile PBS and maintained at 37°C in 5% CO2 humidified air.
Engineered 3D duct tissue model fabrication and culture
MCF10A were harvested with 0.05% trypsin-EDTA (Gibco) and centrifuged at 200 g for 3 min. Cells were resuspended at 2 × 106 cells/ml in assay medium containing DMEM/F12 (1:1; Gibco) supplemented with 5% horse serum (Invitrogen), 5 ng/ml EGF (PeproTech), 0.5 µg/ml hydrocortisone (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 10 μg/ml insulin (Sigma-Aldrich). Following resuspension, 70 μl of cell suspension was added to the two luminal ports to allow for perfusion and adherence of cells to the channel surface. Cells were perfused with occasional device flipping to coat the top of the channel for ∼15 min or until 70% cell coverage prior to exchanging with the growth medium. Luminal and basal ports were then filled with fresh assay medium, and the devices were incubated at 37°C in 5% CO2 humidified air. The assay medium was replenished in all ports for days 1 and 2 after seeding. After a confluent tissue formed (typically day 3), the assay medium was introduced to basal ports and the lumen ports were changed to a base medium daily containing only DMEM/F12 (Invitrogen) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Dead cells accumulating in the lumen were removed by perfusion and medium changes were performed every 24 h.
Immunofluorescence
MCF10A duct tissues were fixed in 4% paraformaldehyde in PBS supplemented with calcium and magnesium (PBS++) for 15 min on a rocker at 37°C, rinsed three times with PBS, and permeabilized in 0.1% Triton X-100 for 1 h on a rocker. MCF10A ducts were rinsed three times with PBS and blocked with 2% BSA in PBS overnight on a rocker at 4°C. Primary and secondary antibodies were added to all device ports in 2% BSA in PBS and devices were placed on a rocker overnight at 4°C. MCF10A ducts were rinsed three times over 3 h with PBS between primary and secondary antibody treatments.
For immunofluorescence of 2D monolayers on hydrogels, cells (MCF10A, 16HBE14o-, and Caco-2) were plated on 18-mm glass coverslips coated with an 80:20 collagen type I:Matrigel composite hydrogel. Coverslips were surface activated by plasma treatment for 30 s, coated with 0.01% poly-L-lysine for 45 min, and washed three times with PBS. Collagen type I solution and Matrigel mixture were prepared as described in device fabrication methods. Once prepared, 40 μl of hydrogel was added and spread around the surface of each coverslip and incubated at 37°C for 20 min. Coverslips with polymerized hydrogel were added to 12-well plates and the medium was added to each well. All epithelial cell types were resuspended at 2 × 106 cells/ml in their corresponding culture medium and 1.25 × 105 cells were added to each well. Once cells reached a confluent monolayer, coverslips were fixed in 4% paraformaldehyde in PBS++ for 15 min at 37°C, rinsed three times with PBS, and permeabilized in 0.1% Triton X-100 for 30 min. For methanol fixation and permeabilization, coverslips were submerged in ice-cold methanol for 5 min and then stored in PBS. Coverslips were washed three times with PBS and blocked in 2% BSA in PBS for 1 h. Primary and secondary antibodies were applied in 2% BSA in PBS for 1–2 h at room temperature or overnight at 4°C and rinsed three times over 30 min with PBS between each treatment. For immunofluorescence imaging of MCF10A epithelial ducts, images were acquired on a Yokogawa CSU-X1 spinning disk confocal on a Nikon Ti-E microscope with a 40 × 1.25 water immersion lens (Nikon) using an Evolve EMCCD camera (Photometrics). For immunofluorescence imaging of epithelial monolayers, images were acquired on the Yokogawa CSU-W1/SoRa spinning disk confocal system in SoRa mode with a 60 × 1.49 NA oil immersion lens (Nikon) and BT Fusion camera (Hamamatsu). Fluorescence images were adjusted for contrast and brightness using ImageJ.
Live-cell imaging
Cells were labeled for 2 h with SPY650-FastAct (Cytoskeleton, Inc.) by adding 1× of the probe to the growth medium. Immediately prior to imaging, DAPT (10 µM) was added to the culture medium. For the MCF10A epithelial ducts, 1× SPY650-FastAct (Cytoskeleton, Inc.) was applied for 4 h prior to imaging. All images were acquired on a Yokogawa CSU-X1 spinning disk confocal on a Nikon Ti-E microscope equipped with an imaging chamber equilibrated to 37°C in 5% CO2 humidified air.
Image processing and analysis
To quantify average lumen size, confocal micrographs of a midline slice of MCF10A duct tissues stained with phalloidin were processed in ImageJ and the width of the lumen was measured at 10 distinct locations along the MCF10A duct. MCF10A epithelial duct internuclear distances were determined by measuring the distance from the midpoint of one nucleus to the midpoint of its neighboring nuclei in ImageJ. Variance in diameter was quantified by binarizing phase images of MCF10A ducts through global intensity thresholding in MATLAB. Outer boundaries of each MCF10A duct were determined by marking the first and last white pixel in each column of the image matrix, and the diameter along the MCF10A duct was compared with the mean diameter. To quantify the number of EdU positive nuclei, the fractional percentage (% positive nuclei) was calculated by dividing the number of EdU positive nuclei by the total number of nuclei. Regions of multilayering were defined by the number of regions within a field of view that had three or more multilayered nuclei above the monolayer plane. To quantify the number of focal adherens junctions, confocal micrographs of epithelial monolayers immunostained for E-cadherin were processed in ImageJ and the total number of cell–cell junctions was measured. Focal adherens junctions were defined as cell–cell junctions that had a discontinuous, jagged, and nonlinear phenotype, and the fractional percentage was calculated by dividing the number of focal adherens junctions by the total number of junctions. To quantify relative cortical actin intensity, line profiles were drawn through the short axis of the cell, passing through the nucleus. The intensity of phalloidin-labeled cells was plotted along the lines. Cortical actin was defined as the area under the peak at cell–cell junctions normalized to the total area under the curve. Cell height was determined by measuring the distance in ImageJ from the basal surface to the apical surface in orthogonal projection fluorescence micrographs of monolayers stained with phalloidin. To quantify Notch1 localization, line profiles were drawn through the short axis of the cell, passing through the nucleus. Notch1 signal intensity was plotted along this line and intensity values and length were normalized to their respective maxima. To quantify EGFR localization, fluorescence micrographs of monolayers immunostained for EGFR were segmented into individual cells and thresholded by Otsu’s method in MATLAB. Cytoplasmic to junctional EGFR ratio was defined as the total number of pixels within the cell interior divided by those at cell–cell interfaces.
Cloning and qPCR
Cells were lysed with cold TRI reagent (Sigma-Aldrich) following the prescribed protocol. RNA extraction was performed according to the manufacturer’s protocol (Zymogen Direct-zol RNA MiniPrep Kit). pMSCV-dnMAML-GFP was a gift from Martin Schwartz (Yale University, New Haven, CT). GFP control, SNAP tag TMD (SNAP-TMD), IRES-ICD, and TMD-ICD-mApple constructs were cloned into pRRL lentiviral vectors and introduced via stable transduction. FAM83H cDNA sequence was verified by whole-plasmid sequencing. Realtime PCR was performed in 20 μl reactions using the SYBR Green Master Mix protocol (Thermo Fisher Scientific) and BioRad thermocycler. The following qPCR primers were obtained from IDT: hPSMB2_F 5′-ACTATGTTCTTGTCGCCTCCG-3′, hPSMB2_R 5′-CTGTACAGTGTCTCCAGCCTC-3′, hHES1_F 5′-CCAAGTGTGCTGGGGAAGTA-3′, hHES1_R 5′-CACCTCGGTATTAACGCCCT-3′, hHEY1_F 5′-CTGAGCAAAGCGTTGACA-3′, and hHEY1_R 5′-TCCACCAACACTCCAAA-3′. Relative gene expression levels were calculated by −∆∆CT in which ∆CT is the difference between the CT value of the gene of interest (HES1 or HEY1) and the CT value of the housekeeping gene (PSMB2), and ∆∆CT is the difference between the experimental condition and the control.
Immunoblotting
For Western blot of monolayers transitioning between low confluence and polarized states, cells were plated into individual wells of a 6-well plate in the assay medium at 1.25 × 105 cells/well. The assay medium was changed daily. Cells were lysed in the buffer described below at 24 h for low confluence, 48 h for high confluence, and 72 h for polarized monolayer states. Monolayers cultured in assay medium (unless otherwise noted) were rinsed with PBS and lysed with 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, and 1.5× protease and cOmplete Protease Inhibitor (Thermo Fisher Scientific). Lysates were passed through a 21G syringe 10 times and incubated on ice for 10 min prior to centrifugation at 4°C for 10 min at 13,000 × g. Lysate protein content was normalized using a BCA protein assay kit (Prometheus) and samples were denatured with 1× NuPAGE LDS Sample Buffer (Life Technologies) containing 5% β-mercaptoethanol. Denatured lysates were analyzed by SDS-PAGE and gels were transferred to PVDF membranes using a Mini Trans-Blot Cell (Bio-Rad). Membranes were blocked in 5% non-fat dry milk in TBS containing 0.1% Tween-20 (TBST) for 1 h at room temperature. Primary antibodies were applied to membranes in a blocking buffer overnight at 4°C. Membranes were washed three times over 30 min with TBST. IRDye donkey anti-rabbit and anti-mouse IgG secondary antibodies (1:10,000; LI-COR) were incubated in a blocking buffer for 2 h at room temperature. Membranes were washed three times over 30 min with TBST. All immunoblots were imaged using an Odyssey CLx LI-COR Imaging System and quantified using ImageJ. Immunoblots were adjusted for brightness and contrast using ImageJ, and (unless otherwise noted) intensity values were normalized to the GAPDH loading control. All uncropped Western blots are provided in source data figures.
Traction force microscopy
Polyacrylamide gels of desired stiffness were made by adjusting acrylamide and bisacrylamide stock solution (Bio-Rad Laboratories) concentrations (Chopra et al., 2018). A solution of 40% acrylamide, 2% bisacrylamide, and 1× PBS was polymerized by adding tetramethylethylene diamine (Fisher BioReagents) and 1% ammonium persulfate. A droplet of the gel solution supplemented with 0.2 μm fluorescent beads solution (Molecular Probe, Thermo Fisher Scientific) was deposited on a quartz slide (Thermo Fisher Scientific) and covered with a 25-mm glass (Thermo Fisher Scientific) coverslip pretreated with 3-aminopropyltrimethoxysilane (Sigma-Aldrich) and glutaraldehyde (Sigma-Aldrich). After polymerization, the gel surface attached to the quartz slide was functionalized with fibronectin via EDC-NHS chemistry. Briefly, the gel surface was activated in a UV-Ozone cleaner (Jelight) for 2 min, detached from the quartz slide, soaked in a solution with EDC and NHS for 15 min, and incubated with 50 μg/ml fibronectin solution at 37°C for 2 h. The gel was sterilized and stored in 1× PBS before cell seeding. The traction forces exerted by epithelial monolayers on the polyacrylamide gel substrates were computed by measuring the displacement of fluorescent beads embedded within the gel. Briefly, images of bead motion near the substrate surface, distributed in and around the contact region of a single cell (before and after cell detachment with 10% sodium dodecyl sulfate) were acquired with Yokogawa CSU-21/Zeiss Axiovert 200M inverted spinning disk microscope with a Zeiss LD C-Apochromat 40×, 1.1 N.A. water-immersion objective, and an Evolve EMCCD camera (Photometrics). The traction stress vector fields were generated using an open-source package of FIJI plugins (Tseng et al., 2012).
Nuclear/cytosolic fractionation
Monolayers cultured in 10-cm plates in the growth medium were rinsed once with PBS++ and then scraped into cold PBS++. Cells were pelleted by centrifugation at 500 × g, and the supernatant was aspirated. Cells were resuspended and swollen using ice-cold hypotonic lysis buffer (20 mM Tris-HCl, pH 7.4, 10 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.5 mM DTT, 1.5× cOmplete Protease Inhibitor). Cells were then lysed by adding Triton X-100 to 0.1% and incubated on ice for 3 min. Lysate was centrifuged at 1,000 g to pellet nuclei and the supernatant (cytoplasmic fraction) was collected. The supernatant was clarified by centrifugation at 15,000 g for 3 min. Nuclear pellet was resuspended in cold isotonic buffer (20 mM Tris-HCl, pH 7.4, 150 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.3% Triton X-100, 0.5 mM DTT, and cOmplete Protease Inhibitor) and incubated for 7 min. Nuclei were centrifuged at 1,000 g for 3 min and the supernatant was aspirated. Nuclei were resuspended in cold RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, cOmplete Protease inhibitor) and were incubated on ice for 30 min. Nuclei were then centrifuged at 2,000 g for 3 min and supernatant (nucleosol fraction) was collected. Lysate protein content was normalized for each fraction using a BCA protein assay kit (Prometheus) and samples were reduced with 4× NuPAGE LDS Sample Buffer (Life Technologies) containing 10% β-mercaptoethanol. Samples were analyzed via SDS-PAGE and immunoblotting as stated above.
Immunoprecipitation and mass spectrometry
Cells cultured in growth medium were rinsed with PBS++ and lysed with cold 25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 5 mM MgCl2, 2× protease, and cOmplete Protease Inhibitor (Thermo Fisher Scientific). Lysates were needle-passed through a 21G syringe 10 times and incubated on ice for 20 min prior to being centrifuged at 4°C for 10 min at 13,000 × g. Lysate volume and protein content were equalized using a BCA protein assay kit (Prometheus). Lysates were incubated for 2 h with 2 µg of anti-Notch1, 2 µg of anti-EGFR, or 2 µg of anti-E-cadherin antibodies at 4°C with rotation. Pierce Protein A/G beads (Thermo Fisher Scientific) were equilibrated in cold lysis buffer prior to incubation with antibody complexes for 2 h at 4°C with rotation. Bead pellets were rinsed three times with cold lysis buffer and reduced with 1× NuPAGE LDS sample buffer (Life Technologies) containing 5% β-mercaptoethanol. Samples were analyzed via SDS-PAGE and Western blot.
Single, excised Coomassie-stained bands for protein identification were analyzed by MS Bioworks as follows. In-gel digestion was performed using a ProGest robot (DigiLab). Gel bands were washed with 25 mM ammonium bicarbonate followed by acetonitrile, reduced with 10 mM dithiothreitol at 60°C followed by alkylation with 50 mM iodoacetamide at room temperature, digested with trypsin (Promega) at 37°C for 4 h, and quenched with formic acid, and the supernatant was analyzed directly without further processing. Half of each digested sample was analyzed by nano LC-MS/MS with a Waters M-Class HPLC system interfaced with a ThermoFisher Fusion Lumos mass spectrometer. Peptides were loaded on a trapping column and eluted over a 75-μm analytical column at 350 nl/min; both columns were packed with Luna C18 resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 60,000 FWHM and 15,000 FWHM for MS and MS/MS, respectively.
Statistical analysis
Sample sizes and P values are reported in each of the corresponding figure legends. Statistical analyses were performed in GraphPad Prism 8. Data distribution was assumed to be normal but this was not formally tested. Unless otherwise noted, graphs show mean ± SEM. When experiments involved only a single pair of conditions, statistical differences between the two sets of data were analyzed with a two-tailed, unpaired Student t test assuming unequal variances. For data sets containing more than two samples, one-way ANOVA with a classical Bonferroni multiple-comparison post-test was used to determine adjusted P values. Images are representative of at least three independent experiments. Experiments were not randomized, and the investigators were not blinded during data analysis. Source data for all graphs are provided in the Data S1.
Online supplemental material
Fig. S1 shows that Notch1 cortical signaling influences mammary duct morphogenesis. Fig. S2 shows that the loss of Notch1 cortical signaling results in aberrant epithelial architecture and proliferation. Fig. S3 shows that Notch1 regulates epithelial architecture, adherens junctions, cortical actin organization, and proliferation in human epithelia. Fig. S4 shows that the loss of Notch1 cortical signaling disrupts epithelial adherens junctions and cortical actin organization. Fig. S5 shows that the overexpression of the Notch1 ICD does not rescue the NOTCH1KO phenotype. Loss of FAM83H leads to focal adherens junctions, depleted cortical actin, and alterations in ductal architecture. Video 1 shows live cell imaging of the medial confocal plane of a SPY650-FastAct labeled NOTCH1KO duct during assembly. Data S1 contains source data for all of the graphs.
Data availability
The data underlying all representative images and graphs are available in the source data table, source data figures, and online supplemental material. Raw mass spectrometry data and repeats of representative images are available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank Diane Barber, Torsten Wittmann, and Yi Liu for critical assistance and feedback on the manuscript.
This work was supported by the National Institutes of Health (NIH) grants R00CA226366 (to M.L. Kutys), R35GM150987 (to M.L. Kutys), R21AG072232 (to M.L. Kutys, Y.-w. Jun), R35GM134948 (to Y.-w. Jun), and the UCSF Program for Breakthrough Biomedical Research. The Nikon CSU-W1 SoRa microscope was obtained through the NIH shared equipment grant S10OD028611-01.
Author contributions: Conceptualization: M.L. Kutys; Methodology: M.L. Kutys and M.J. White. Formal analysis: M.L. Kutys and M.J. White. Investigation: M.L. Kutys, M.J. White, K.A. Jacobs, T. Singh, A. Lin, L.N. Mayo. C.S. Chen, and Y.-W. Jun. Writing—original draft: M.L. Kutys and M.J. White. Writing—review and editing: all authors. Funding acquisition: M.L. Kutys.

