


Notch signaling is the dominant intercellular signaling input during the earliest stages of T cell development in the thymus. Although Notch1 is known to be indispensable, we show that it does not mediate all Notch signaling in precommitment stages: Notch2 initially works in parallel to promote early murine T cell development and antagonize other fates. Notch-regulated target genes before and after T lineage commitment change dynamically, and we show that this partially reflects shifts in genome-wide DNA binding by RBPJ, the transcription factor activated by complex formation with the Notch intracellular domain. Although Notch signaling and transcription factor PU.1 can activate some common targets in precommitment T progenitors, Notch signaling and PU.1 activity have functionally antagonistic effects on multiple targets, delineating separation of pro-T cells from alternative PU.1-dependent fates. These results define a distinct mechanism of Notch signal response that distinguishes the initial stages of murine T cell development.
Introduction
Notch signaling is the main inductive signal for development of T lymphocytes in the thymus (Hozumi, 2020; Radtke et al., 2013). The Notch pathway is evolutionarily conserved and regulates differentiation and organization in diverse organs and cell types. Mammals have four Notch family members, Notch1–4, and multiple Notch ligands of delta-like (DLL) and Jagged families. Notably, Notch acts both as a cell surface receptor and as a transcriptional coactivator (Bray, 2006). Binding of cell surface Notch by Notch ligands triggers proteolytic release of the intracellular domain of Notch (ICN), which translocates to the nucleus to become a coactivator for the DNA binding protein RBPJ. In hematopoiesis, early T cell development in the thymus is the best-studied system for the roles of Notch signaling (Hozumi, 2020; Radtke et al., 2013). Conditional deletion of Notch1 or Delta-like 4 (Dll4) profoundly blocks T cell development and promotes aberrant generation of B cells in the thymus (Hozumi et al., 2008; Radtke et al., 1999). Conversely, Notch signaling driven by coculture with stromal cells expressing DLL family Notch ligands (DLL1 or DLL4) induces T cell development in vitro, and forced expression of ICN1 within fetal liver– or bone marrow (BM)–derived non-T progenitor cells does so in vivo (Hozumi et al., 2003; Pui et al., 1999; Schmitt and Zúñiga-Pflücker, 2002).
T cell developmental stages in the thymus are defined by markers such as CD4 and CD8. The immature cells are double-negative (DN; CD4− CD8−) cells, which generate double-positive (CD4+ CD8+) intermediate cells and then differentiate into mature CD4 and CD8 single-positive cells. DN thymocytes include multiple substages distinguished by expression of CD44, Kit, and CD25 (Hosokawa and Rothenberg, 2018; Yang et al., 2010; Yui and Rothenberg, 2014). The earliest intrathymic precursors are called “early T cell progenitor cells” (ETP; Kit2+CD44+CD25−) or Kit+ DN1 cells (DN1a and DN1b; Porritt et al., 2004), and these generate DN2a (Kit2+CD44+CD25+) stage pro-T cells. These stages together comprise phase 1. Through days of proliferation, phase 1 cells in ETP and DN2a stages retain potential for non–T cell fates and can switch to these alternative pathways if Notch signaling is withdrawn. Then, at the transition of DN2a to DN2b (Kitlower+CD44+CD25+) stages, pro-T cells become intrinsically committed to the T lineage, entering phase 2. They then undergo T cell receptor (TCR) gene rearrangement, with most TCRβ gene rearrangement occurring at DN3a stage (Kit−CD44−CD25+CD28−). Pre-TCR signaling next enables the cells to exit from the pro–T cell stages and progress to later TCR-expressing stages. The requirement for Notch signaling extends from the earliest stages throughout the commitment transition (Hirano et al., 2015; Wolfer et al., 2002). Phase 1 stages (ETP and DN2a) are Notch-dependent for T lineage fidelity and growth. Phase 2 (DN2b and DN3a) stages are T lineage committed but are increasingly Notch dependent for viability. Cells that pass beyond DN3a stage after pre-TCR signaling, or that differentiate to TCRγδ cells, finally become Notch independent. Of note, Notch-dependent target genes show different expression dynamics even within pro–T cell development (Rothenberg et al., 2016), suggesting that stage-dependent control mechanisms are at play.
It has been assumed that only Notch1 mediates inductive signaling for T cell specification. In single-gene knockouts (KOs), Notch1 disruption is sufficient to block T cell development, whereas Notch3-deficient or Notch2-deficient mice develop normal-seeming CD4 and CD8 populations under steady-state conditions in the thymus (Saito et al., 2003; Shi et al., 2011; Suliman et al., 2011). Several in vitro culture systems indicate that Notch2 could play a supportive role for phase 1 pro-T stages (Besseyrias et al., 2007; Hozumi et al., 2003), but this has been interpreted as an artifact of using a Notch ligand in vitro that is less biased toward Notch1 interaction than the DLL4 naturally used in the thymus. If true, then it could be assumed that Notch1-deficient thymocytes (Feyerabend et al., 2009) were completely lacking Notch signals. However, Notch2 is highly expressed from hematopoietic stem cells through phase 1, then decreases in phase 2. Furthermore, although Notch3 is virtually absent in murine hematopoietic stem cells and phase 1 cells, it is activated by Notch1 signaling in the thymus and nicely expressed in phase 2. Thus, although signaling from Notch2 and Notch3 is dispensable for T cell development in vivo, these family members could have some physiological roles, redundant or complementary with Notch1, and might contribute to select stage-specific target gene regulation in phase 1 and phase 2.
To resolve Notch family roles, we examined effects of acute and stage-specific deletion of different Notch family member genes, individually or in combination, in early T cell development. The results show that Notch1 controls its target genes in cooperation with Notch2, especially in phase 1. Notch-regulated target genes are globally distinct in the two phases, and some of these differences are linked with stage-specific shifts in RBPJ binding across the genome. Although Notch1 appears to block B lineage development from phase 1 cells, acute deletion of Notch2 along with Notch1 in phase 1 unleashes higher production of myeloid cells as well. Gene expression profiles confirm specific antagonisms between myeloid transcription factor, PU.1, and Notch signaling in phase 1 pro-T cells. Thus, Notch signaling drives T cell development not only by activating T lineage signature genes but also by repressing alternative lineage programs, including myeloid fate.
Results and discussion
Conditional deletion of Notch family genes in mice has shown an indispensable role of Notch1 for the earliest T cell development in vivo in the steady state. However, previous studies did not exclude potential accelerating or modulating roles of Notch2 and Notch3, which are also substantially expressed in phase 1 and phase 2 pro-T cells, respectively (Yoshida et al., 2019; Fig. 1 A and Fig. S1 A). Therefore, we compared single and double KOs to test whether Notch2 or Notch3 contributed at all to Notch-dependent function in early T cell development. Because available Cre mice do not delete specifically in phase 1, we exploited in vitro T cell cultures (Holmes and Zúñiga-Pflücker, 2009) to cause acute, stage-specific deletion of Notch family genes, singly or in pairs, using a CRISPR/Cas9 system (Hosokawa et al., 2018; Fig. 1 B). For deletions in phase 1, BM precursors from Cas9;Bcl2-transgenic (Cas9;Bcl2-tg) mice were cultured on OP9-DLL1 stroma to initiate T cell development (see Materials and methods). After 2 d, before most had reached DN2 stage, the cells were infected with retroviral vectors encoding single gRNA (sgRNA) against Notch1, Notch2, and/or Notch3 together with CFP (here used for mTurquoise2, a brighter version) or human nerve growth factor receptor (hNGFR) reporters, and we analyzed the sgRNA-transduced cells at 3 or 5 d postinfection (dpi; Fig. 1 B). The targeted cell surface Notch proteins were specifically depleted by 3 dpi (Fig. S1 A). As expected, deletion of Notch1, alone or in combination, sharply reduced pro–T cell recovery (Fig. S1, B and C). Deletion of Notch1 or Notch2 in phase 1 cells had minor though reproducible effects on expression of one of the most visible Notch targets, CD25, whereas Notch3 deletion had no significant phenotypic effect. Notably, however, combined Notch1 and Notch2 deletion downregulated CD25 profoundly (especially at 5 dpi; Fig. 1, Cand D; Fig. S1, D and E), a phenotype much stronger than that with deletion of Notch1 alone. Also, abundant cells expressing inappropriate lineage markers (Lin+) were generated from Notch1 and Notch2 double-knockout (DKO) cells (Fig. S1 F), more than when Notch1 alone was deleted. Thus, Notch1 alone does not account for all effects of Notch signaling on phase 1 cells.

Notch1, Notch2, and Notch3 cooperatively control early T cell development. (A) RPKM values for Notch1, Notch2, Notch3, and Notch4 in DN subsets (https://www.immgen.org; Yoshida et al., 2019). (B) Experimental scheme of work. (C) Representative c-Kit/CD25 profiles of sgRNA-transduced Lin−CD45+CFP+hNGFR+ phases 1 and 2 cells. (D) Percentages of CD25+ cells among sgRNA-transduced cells (C), indicated with SD. (E) Representative c-Kit/CD25 profiles in sgRNA-transduced cells cultured on OP9-DLL4. (F) The percentages of CD25+ cells in (E), indicated with SD. Results are representative of four (C) or three (E) independent experiments or compiled from four (D) or three (F) independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by one-way ANOVA followed by Dunnett’s test.
Notch1, Notch2, and Notch3 cooperatively control early T cell development. (A) RPKM values for Notch1, Notch2, Notch3, and Notch4 in DN subsets (https://www.immgen.org; Yoshida et al., 2019). (B) Experimental scheme of work. (C) Representative c-Kit/CD25 profiles of sgRNA-transduced Lin−CD45+CFP+hNGFR+ phases 1 and 2 cells. (D) Percentages of CD25+ cells among sgRNA-transduced cells (C), indicated with SD. (E) Representative c-Kit/CD25 profiles in sgRNA-transduced cells cultured on OP9-DLL4. (F) The percentages of CD25+ cells in (E), indicated with SD. Results are representative of four (C) or three (E) independent experiments or compiled from four (D) or three (F) independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by one-way ANOVA followed by Dunnett’s test.

Acute deletion of Notch accelerates generation of Lin+ cells. (A) Flow cytometric analysis of phase 1 (upper) and phase 2 (lower) pro-T cells was performed at 3 dpi. Representative Notch1, Notch2, and Notch3 profiles in Lin−CD45+CFP+ sgRNA-transduced cells are shown. Results are representative of two independent experiments. (B and C) Flow cytometric analysis of phase 1 and phase 2 DN cells was performed at 3 dpi (B) or 5 dpi (C). Relative cell numbers of Lin−CD45+CFP+hNGFR+ sgRNA-transduced cells against sgControl-transduced cells are shown with SD. Data are based on three independent experiments. (D) Flow cytometric analysis of phase 1 (upper) and phase 2 (lower) DN cells was performed at 3 dpi. Representative c-Kit/CD25 profiles in Lin−CD45+CFP+hNGFR+ sgRNA-transduced cells are shown. Results are representative of four independent experiments. (E) The percentages of CD25+ cells among Lin−CD45+CFP+hNGFR+ sgRNA-transduced phase 1 and phase 2 cells at 3 dpi (D) are indicated with SD. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA followed by Dunnett’s test. Data are based on four independent experiments. (F) Flow cytometric analysis of phase 1 pro-T cells (Fig. 1 C) was performed at 5 dpi. Representative Lin profiles in CD45+CFP+hNGFR+ sgRNA-transduced cells are shown. Numbers indicate percentages of Lin– cells. Results are representative of four independent experiments. (G) Flow cytometric analysis of phase 1 pro-T cells, cocultured on OP9-DLL4, was performed at 3 dpi. Relative cell numbers of Lin−CD45+CFP+hNGFR+ sgRNA-transduced cells against sgControl-transduced cells are shown with SD. Data are based on three independent experiments.
Acute deletion of Notch accelerates generation of Lin+ cells. (A) Flow cytometric analysis of phase 1 (upper) and phase 2 (lower) pro-T cells was performed at 3 dpi. Representative Notch1, Notch2, and Notch3 profiles in Lin−CD45+CFP+ sgRNA-transduced cells are shown. Results are representative of two independent experiments. (B and C) Flow cytometric analysis of phase 1 and phase 2 DN cells was performed at 3 dpi (B) or 5 dpi (C). Relative cell numbers of Lin−CD45+CFP+hNGFR+ sgRNA-transduced cells against sgControl-transduced cells are shown with SD. Data are based on three independent experiments. (D) Flow cytometric analysis of phase 1 (upper) and phase 2 (lower) DN cells was performed at 3 dpi. Representative c-Kit/CD25 profiles in Lin−CD45+CFP+hNGFR+ sgRNA-transduced cells are shown. Results are representative of four independent experiments. (E) The percentages of CD25+ cells among Lin−CD45+CFP+hNGFR+ sgRNA-transduced phase 1 and phase 2 cells at 3 dpi (D) are indicated with SD. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA followed by Dunnett’s test. Data are based on four independent experiments. (F) Flow cytometric analysis of phase 1 pro-T cells (Fig. 1 C) was performed at 5 dpi. Representative Lin profiles in CD45+CFP+hNGFR+ sgRNA-transduced cells are shown. Numbers indicate percentages of Lin– cells. Results are representative of four independent experiments. (G) Flow cytometric analysis of phase 1 pro-T cells, cocultured on OP9-DLL4, was performed at 3 dpi. Relative cell numbers of Lin−CD45+CFP+hNGFR+ sgRNA-transduced cells against sgControl-transduced cells are shown with SD. Data are based on three independent experiments.
For Notch deletion in phase 2, precursors were cocultured with OP9-DLL1 stroma for 10 d, when almost all of the cells had transitioned through T lineage commitment to phase 2, before they were transduced with sgRNA. Cells and RNA were again analyzed at 3 or 5 dpi (Fig. 1 B and Fig. S1 A). In phase 2, CD25 expression was partially downregulated by deletion of Notch1, but, again, stronger effects on CD25 expression were detected when Notch2 was deleted together with Notch1 (Fig. 1, C and D; and Fig. S1, D and E).
The implication that Notch1 collaborates with Notch2 in early T lineage cells was unexpected. Among Notch ligands, DLL4 is indispensable to support T cell development in vivo, and it preferentially interacts with Notch1 rather than Notch2 (Hozumi, 2020). To test whether the OP9-DLL1 system could be exaggerating the potency of Notch2, we retested the effects of Notch1 and/or Notch2 deletion using phase 1 cells cocultured on OP9-DLL4 rather than on OP9-DLL1. As expected, disruption of the Notch1 gene strongly reduced CD25 expression, whereas Notch2 deletion alone now had no significant effect. Even so, the combined loss of Notch1 and Notch2 (DKO) gave a markedly harsher effect (Fig. 1, E and F; and Fig. S1 G), supporting signaling via both Notch1 and Notch2.
To test whether different Notch family member deletions affect the same or different target genes, Lin−CD45+ sgRNA-infected cells in phase 1 or phase 2 were sorted for RNA sequencing (RNA-seq) analysis at 3 dpi (Fig. 1 B). Gene expression impacts of the single and double KOs were related to normal development by plotting expression levels of 65 indicator genes against a fixed principal component (PC) analysis framework of normal developmental stages (Zhou et al., 2019; see Materials and methods). The control phase 1 cells progressed to a state similar to early, Bcl11b-nonexpressing DN2a cells. Single Notch1 or Notch2 KOs were only slightly retarded in development (shifted rightward in PC1). In contrast, the patterns of gene expression in Notch1 and Notch2 DKO phase 1 cells were closer to reference DN1 cells (Fig. 2 A), even though they remained on the T lineage trajectory, consistent with their Kit and CD25 expression profiles (compare Fig. 1 C, upper panels). Phase 2 control cells, as expected, had a gene expression pattern near reference DN2b cells, whereas all the samples with Notch1 deletion (single or double KO) showed similarly delayed gene expression profiles, nearer to the Bcl11b+ DN2a cells (Zhou et al., 2019; Fig. 2 A). These results generally support a specific contributing role of Notch2, with Notch1, to T lineage progression in phase 1.
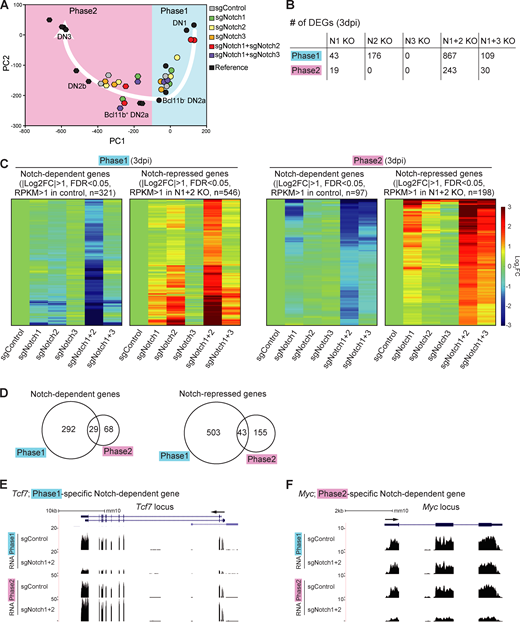
Notch signaling regulates stage-specific target genes in pro-T cells.(A) PC analysis displays RNA-seq data from sgRNA-transduced phases 1 and 2 cells (see Materials and methods). White arrow represents the T cell developmental trajectory defined from reference standards (black symbols). (B) Number of DEGs in Notch1-deficient (N1 KO), Notch2-deficient (N2 KO), Notch3-deficient (N3 KO), Notch1- and Notch2-deficient (N1+2 KO), and Notch1- and Notch3-deficient (N1+3 KO) phase 1 or phase 2 cells. (C) Heat maps show expression changes of DEGs (B; N1+2 KO) in response to acute deletion of Notch genes as indicated (Table S1 and Table S2). (D) Numbers of Notch-dependent (left) and Notch-repressed (right) DEGs in phases 1 and 2. (E and F) Representative RNA-seq tracks for control and N1+2 KO cells at the Tcf7 (E) and Myc (F) loci. Data are based on two replicate samples of RNA-seq results (A–D) or are representative of two replicate samples (E and F). FC, fold change; FDR, false discovery rate.
Notch signaling regulates stage-specific target genes in pro-T cells.(A) PC analysis displays RNA-seq data from sgRNA-transduced phases 1 and 2 cells (see Materials and methods). White arrow represents the T cell developmental trajectory defined from reference standards (black symbols). (B) Number of DEGs in Notch1-deficient (N1 KO), Notch2-deficient (N2 KO), Notch3-deficient (N3 KO), Notch1- and Notch2-deficient (N1+2 KO), and Notch1- and Notch3-deficient (N1+3 KO) phase 1 or phase 2 cells. (C) Heat maps show expression changes of DEGs (B; N1+2 KO) in response to acute deletion of Notch genes as indicated (Table S1 and Table S2). (D) Numbers of Notch-dependent (left) and Notch-repressed (right) DEGs in phases 1 and 2. (E and F) Representative RNA-seq tracks for control and N1+2 KO cells at the Tcf7 (E) and Myc (F) loci. Data are based on two replicate samples of RNA-seq results (A–D) or are representative of two replicate samples (E and F). FC, fold change; FDR, false discovery rate.
Next, we defined differentially expressed genes (DEGs) affected by Notch family member deletions, based on false discovery rate <0.05, |log2 fold change| >1, and average reads per kilobase million (RPKM) >1 across samples in phase 1 and phase 2 (Table S1 and Table S2). In this acute disruption system, despite a strong effect of Notch1 deletion on cell recovery (Fig. S1 B), the number of DEGs in the surviving Notch1 KO cells was small. In contrast, the numbers of DEGs were much higher in Notch1 and Notch2 DKO cells than in either Notch1 or Notch2 single KO cells (Fig. 2 B). The responses of the DEGs defined in Notch1 and Notch2 DKO cells (“Notch-regulated DEGs”) to each Notch family perturbation are shown, for phase 1 and phase 2, in heat maps (Fig. 2 C). The contributions of Notch1 and Notch2 to the Notch-regulated DEGs looked comparable in phase 1; the DKO caused strong supra-additive effects and on most of the same genes as in single KOs (Fig. 2 C, left). Notch3 played little role. However, in phase 2, Notch1 seemed to be the main transducer of Notch signals. There were only modestly increased effects on the target genes if Notch2 or Notch3 was disrupted together with Notch1 and little if any effects if Notch2 or Notch3 were deleted alone (Fig. 2 C, right). Importantly, we also tested the responses of Notch-regulated DEGs to Notch1 and/or Notch2 deletion in phase 1 cells cocultured with OP9-DLL4 to mimic the DLL4 in the thymic microenvironment. These results supported those from cells developing on OP9-DLL1, again showing significantly increased intensities of response of the same genes in the Notch1 and Notch2 DKO as compared with the Notch1 (or Notch2) single KO samples (Fig. S2, A and B; and Table S3). Therefore, Notch1 and Notch2 collaboratively regulate gene expression in phase 1, whereas Notch1 mainly controls the Notch-regulated DEGs in phase 2, in our experimental system.

Phase 1 cells on OP9-DLL1 and OP9-DLL4 have similar gene expression changes in response to deletion of Notch1 and Notch2. (A) Heat maps show hierarchical clustering of expression change in response to acute deletion of Notch1 and Notch2 (Notch-dependent genes, left, and Notch-repressed genes, right) in phase 1 cells cultured on OP9-DLL1 (Fig. 2 C, left), with phase 1 cells cultured on DLL4 (Fig. 1 E). (B) Number of DEGs in Notch1-deficient (N1 KO), Notch2-deficient (N2 KO), and Notch1- and Notch2-deficient (N1+2 KO) phase 1 cells cultured on OP9-DLL4 are shown. (C and D) GO annotation was performed using the DAVID analysis tool (https://david.ncifcrf.gov/). Top five GO terms for Notch-dependent (top) and Notch-repressed (bottom) target genes are shown in phase 1 (C) and phase 2 (D). (E) The top three enriched sequence motifs of RBPJ ChIP peaks in previously reported RBPJ ChIP peaks in a T-ALL cell line (Pinnell et al., 2015) are shown. (F) Venn diagrams show the numbers of reproducible RBPJ ChIP peaks in phase 2 in this study and previously reported RBPJ ChIP peaks in a T-ALL cell line. (G) ChIP-seq tracks for ICN1 and RBPJ in T-ALL cell lines (Pinnell et al., 2015; Wang et al., 2011) and RBPJ in phase 2 pro-T cells are compared around the Il2ra and Notch3 loci. (H) Representative ChIP-seq tracks for RBPJ around the Myc enhancer region are shown. The site with stronger RBPJ binding signals in phase 2 is labeled with a magenta rectangle.
Phase 1 cells on OP9-DLL1 and OP9-DLL4 have similar gene expression changes in response to deletion of Notch1 and Notch2. (A) Heat maps show hierarchical clustering of expression change in response to acute deletion of Notch1 and Notch2 (Notch-dependent genes, left, and Notch-repressed genes, right) in phase 1 cells cultured on OP9-DLL1 (Fig. 2 C, left), with phase 1 cells cultured on DLL4 (Fig. 1 E). (B) Number of DEGs in Notch1-deficient (N1 KO), Notch2-deficient (N2 KO), and Notch1- and Notch2-deficient (N1+2 KO) phase 1 cells cultured on OP9-DLL4 are shown. (C and D) GO annotation was performed using the DAVID analysis tool (https://david.ncifcrf.gov/). Top five GO terms for Notch-dependent (top) and Notch-repressed (bottom) target genes are shown in phase 1 (C) and phase 2 (D). (E) The top three enriched sequence motifs of RBPJ ChIP peaks in previously reported RBPJ ChIP peaks in a T-ALL cell line (Pinnell et al., 2015) are shown. (F) Venn diagrams show the numbers of reproducible RBPJ ChIP peaks in phase 2 in this study and previously reported RBPJ ChIP peaks in a T-ALL cell line. (G) ChIP-seq tracks for ICN1 and RBPJ in T-ALL cell lines (Pinnell et al., 2015; Wang et al., 2011) and RBPJ in phase 2 pro-T cells are compared around the Il2ra and Notch3 loci. (H) Representative ChIP-seq tracks for RBPJ around the Myc enhancer region are shown. The site with stronger RBPJ binding signals in phase 2 is labeled with a magenta rectangle.
This experimental system confirmed globally that the majority of Notch-regulated DEGs in phase 1 and phase 2 had stage-specific responses (Fig. 2 D). It was previously known that multiple genes that shared a dependence on Notch signaling, such as Hes1, Nrarp, Dtx1, Il2ra (encoding CD25), Ptrca, and Notch3, were expressed in different developmental patterns. However, our results revealed that even important regulatory genes that were strongly expressed at both stages showed stage-dependent differential Notch control. Although Tcf7 was confirmed to be Notch activated as expected (Germar et al., 2011; Weber et al., 2011), its Notch dependence was sharply confined to phase 1 (Fig. 2 E). In contrast, proto-oncogene Myc was Notch dependent in phase 2 but not in phase 1 (Fig. 2 F). Other reported Notch direct target genes, including Il2ra, Notch3, and Deptor, had significant measurable Notch dependence in both phases (“shared”). Notch signaling could repress some alternative lineage genes in the thymus, both in phase 1 and in phase 2. Innate lymphoid-related genes (Rora, Tox2, Id3, and Eomes) and myeloid-related genes (Mpo, Rarg, Ly6c1, and Ccr2) were Notch inhibited in both phases (“shared Notch repressed”; Table S1 and Table S2). However, regulatory genes Cebpa, Cebpb, Cebpd, and Cepbe, key for myeloid development, were only unleashed if Notch was deleted in phase 1 (“phase 1–specific Notch repressed”; Table S1 and Table S2). Gene Ontology (GO) analysis supported a phase-dependent shift in Notch roles. In phase 1, genes involved in “innate immune response” were enriched among “Notch-repressed” genes, and “negative regulator of pro–B cell and stem cell” categories were highly enriched among the phase 1 Notch-dependent genes (Fig. S2 C). In phase 2, instead, the Notch signaling–dependent genes were enriched for genes related to T cell homeostasis, whereas “Notch-repressed” genes were enriched for those encoding chemokine receptors and positive regulators of transcription (Fig. S2 D).
The Notch-regulated DEGs include both direct and indirect targets. We tested whether genomic regulatory sites bound by complexes of Notch–ICN with its DNA binding partner, RBPJ, were themselves changing between phase 1 and phase 2. RBPJ binding and Notch1 ICN binding may occur independently but most often coincide in mammalian systems (Castel et al., 2013), especially for positive regulation. Because of antibody limitations, T leukemias have been used for ICN and RBPJ chromatin immunoprecipitation sequencing (ChIP-seq; Geimer Le Lay et al., 2014; Herranz et al., 2014; Pinnell et al., 2015; Wang et al., 2011; Yashiro-Ohtani et al., 2014) but not primary DN pro-T cells. For RBPJ ChIP-seq analyses of primary phase 1 and phase 2 cells (two replicates each), we used a mixture of two mAbs with cross-linking conditions that preserve protein–protein as well as protein–DNA interactions. The RBPJ ChIP-seq results gave modest signal/noise ratios, limiting analysis, but they confirmed normal use of many RBPJ sites previously found in T cell acute lymphoblastic leukemia (T-ALL) cell lines (Pinnell et al., 2015; Wang et al., 2011; Fig. S2, E–G). We scored 485 reproducible RBPJ ChIP peaks in phase 1 and 4,000 in phase 2 (Fig. 3 A). The RBPJ motif was nicely enriched in these sites from both phases (Fig. 3 B). The increased RBPJ binding in phase 2 was seen at many genes scored as phase 1 DEGs as well as at phase 2 DEGs. However, stage-specific as well as shared peaks were evident at correspondingly Notch-regulated target loci, including Il2ra, Cebpa, Nrarp, and Notch3 (Fig. 3, C–F, stage-specific sites boxed). The unexpected binding to a repression target requires further study. Phase 2–specific binding was also seen at the distal enhancer (Herranz et al., 2014; Yashiro-Ohtani et al., 2014) of phase 2–specific target Myc (Fig. S2 H), whereas weak but reproducible phase 1–specific binding was also found at phase 1–expressed Deptor, Cd34, and Pdgfrb loci (not shown).

Stage-specific RBPJ binding around Notch target loci. (A) Overlap of reproducible RBPJ ChIP peaks scored in phases 1 and 2. (B) Top three enriched sequence motifs from RBPJ ChIP peaks in phase 1 (left) and phase 2 (right). (C–F) Representative ChIP-seq and RNA-seq tracks around the Il2ra (C), Cebpa (D), Nrarp (E), and Notch3 (F) loci are shown. Sites with stronger RBPJ binding signals in phase 1 or 2 are labeled with blue or magenta rectangles, respectively. Data are based on ChIP-seq peaks scored as reproducible in two replicate samples (A and B).
Stage-specific RBPJ binding around Notch target loci. (A) Overlap of reproducible RBPJ ChIP peaks scored in phases 1 and 2. (B) Top three enriched sequence motifs from RBPJ ChIP peaks in phase 1 (left) and phase 2 (right). (C–F) Representative ChIP-seq and RNA-seq tracks around the Il2ra (C), Cebpa (D), Nrarp (E), and Notch3 (F) loci are shown. Sites with stronger RBPJ binding signals in phase 1 or 2 are labeled with blue or magenta rectangles, respectively. Data are based on ChIP-seq peaks scored as reproducible in two replicate samples (A and B).
Conditional deletion of Notch1 or Dll4 in vivo leads to generation of B cells in the thymus (Hozumi et al., 2008; Radtke et al., 1999). As noted above, we also observed many alternative lineage marker–positive cells (“Lin+”) developing in vitro from Notch1 and Notch2 DKO cells, especially when the Notch genes were knocked out in phase 1 (Fig. S1 F). CD19+ B lineage cells emerged at similar levels in all the cultures of Notch1-disrupted cells. However, Notch2 loss substantially enhanced myeloid cell production and showed an impact even when Notch1 was intact (Fig. 4 A; Fig. S3 A). In these culture conditions, >70% of the Notch1 and Notch2 DKO phase 1 cells expressed the myeloid marker CD11b, whereas CD19+ B cells only made up ∼15% (Fig. 4 A; Mohtashami et al., 2010). Thus, Notch signaling, especially through Notch2, either blocks outgrowth of myeloid cells or blocks developmental diversion of pro-T cells to myeloid fates, and Notch2 signals can mask myeloid potential when Notch1 is disrupted alone.
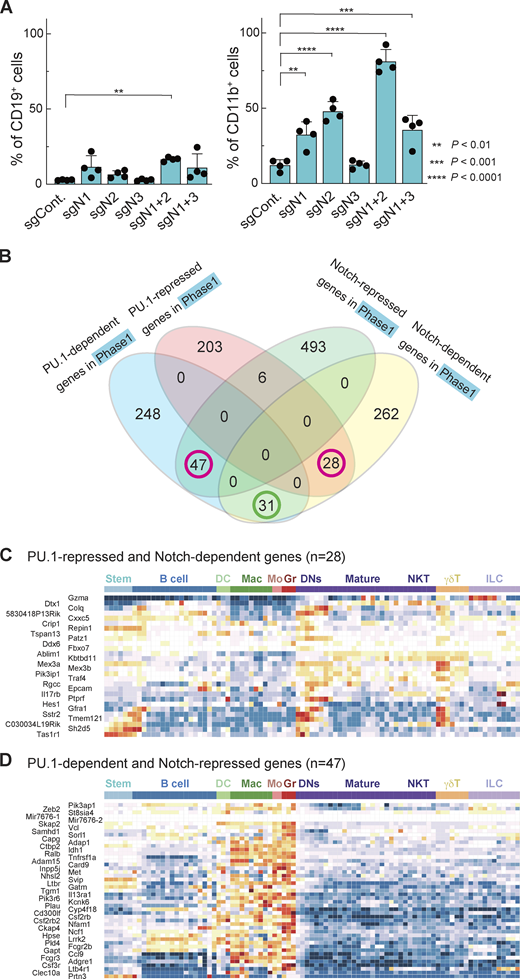
Notch signaling antagonizes PU.1 and prevents pro-T cells from adopting myeloid-like fates.(A) Percentages of CD19+ (left) and CD11b+ (right) cells among sgRNA-transduced phase 1 cells (Fig. S1 F) are indicated with SD. **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by one-way ANOVA followed by Dunnett’s test. Data are based on four independent experiments. (B) Relationships between Notch-dependent, Notch-repressed, PU.1-dependent, and PU.1-repressed genes (Hosokawa et al., 2018) defined in phase 1 cells (Table S4). Magenta circles indicate groups of genes shown in C and D. (C and D) Developmental patterns of expression of PU.1-repressed and Notch-dependent genes (n = 28; C) and PU.1-dependent and Notch-repressed genes (n = 47; D; see Materials and methods).
Notch signaling antagonizes PU.1 and prevents pro-T cells from adopting myeloid-like fates.(A) Percentages of CD19+ (left) and CD11b+ (right) cells among sgRNA-transduced phase 1 cells (Fig. S1 F) are indicated with SD. **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by one-way ANOVA followed by Dunnett’s test. Data are based on four independent experiments. (B) Relationships between Notch-dependent, Notch-repressed, PU.1-dependent, and PU.1-repressed genes (Hosokawa et al., 2018) defined in phase 1 cells (Table S4). Magenta circles indicate groups of genes shown in C and D. (C and D) Developmental patterns of expression of PU.1-repressed and Notch-dependent genes (n = 28; C) and PU.1-dependent and Notch-repressed genes (n = 47; D; see Materials and methods).

Relationship between Notch-regulated DEGs and Bcl11b-regulated DEGs. (A) Relative cell numbers of CD19+ (left) or CD11b+ (right) in CD45+CFP+hNGFR+ sgRNA-transduced cells against sgControl-transduced phase 1 cells at 5 dpi are shown with SD. Data are based on three independent experiments. (B) The heat map shows developmental patterns of expression of PU.1-dependent and Notch-dependent genes (n = 31; green circle in Fig. 4 B) among multilineage precursors and other hematopoietic cells, including B cells, dendritic cells, macrophages, monocytes, granulocytes, DN cells (thymocytes), mature αβ T cells, NKT cells, γδ T cells, and NK cells. The heat map is taken from the Immunological Genome web site (http://www.immgen.org/; Heng et al., 2008; Mingueneau et al., 2013; Yoshida et al., 2019) using the MyGeneSet tool and ImmGen ULI RNA-seq. Color scale indicates z scores, with warm colors indicating higher relative levels and cold colors indicating lower relative levels. (C) Venn diagrams show the number of Notch-dependent, Notch-repressed, Bcl11b-dependent, and Bcl11b-repressed genes (Hosokawa et al., 2018a) in phase 2 DN cells. Magenta circles indicate groups of genes characterized further in D and E. Data are based on two replicate samples of RNA-seq results. (D and E) The heat maps show developmental patterns of expression of Bcl11b-dependent and Notch-dependent genes (n = 9; D) and Bcl11b-repressed and Notch-repressed genes (n = 24; E) from gene groups in magenta circles in C. Expression is compared among multilineage precursors and hematopoietic cells as in B.
Relationship between Notch-regulated DEGs and Bcl11b-regulated DEGs. (A) Relative cell numbers of CD19+ (left) or CD11b+ (right) in CD45+CFP+hNGFR+ sgRNA-transduced cells against sgControl-transduced phase 1 cells at 5 dpi are shown with SD. Data are based on three independent experiments. (B) The heat map shows developmental patterns of expression of PU.1-dependent and Notch-dependent genes (n = 31; green circle in Fig. 4 B) among multilineage precursors and other hematopoietic cells, including B cells, dendritic cells, macrophages, monocytes, granulocytes, DN cells (thymocytes), mature αβ T cells, NKT cells, γδ T cells, and NK cells. The heat map is taken from the Immunological Genome web site (http://www.immgen.org/; Heng et al., 2008; Mingueneau et al., 2013; Yoshida et al., 2019) using the MyGeneSet tool and ImmGen ULI RNA-seq. Color scale indicates z scores, with warm colors indicating higher relative levels and cold colors indicating lower relative levels. (C) Venn diagrams show the number of Notch-dependent, Notch-repressed, Bcl11b-dependent, and Bcl11b-repressed genes (Hosokawa et al., 2018a) in phase 2 DN cells. Magenta circles indicate groups of genes characterized further in D and E. Data are based on two replicate samples of RNA-seq results. (D and E) The heat maps show developmental patterns of expression of Bcl11b-dependent and Notch-dependent genes (n = 9; D) and Bcl11b-repressed and Notch-repressed genes (n = 24; E) from gene groups in magenta circles in C. Expression is compared among multilineage precursors and hematopoietic cells as in B.
To explore how Notch might block myeloid development, we focused on transcription factor PU.1, which is naturally expressed throughout phase 1 but can drive myeloid development of pro-T cells if artificially elevated. Notch signaling blocks this myeloid diversion, but the mechanism has not been clearly demonstrated (Rothenberg et al., 2019; Ungerbäck et al., 2018). PU.1 has natural proliferative roles in phase 1 pro-T cells, but elevated PU.1 can inhibit the T cell program by several mechanisms, including raising the threshold for effective Notch signaling (Del Real and Rothenberg, 2013; Hosokawa et al., 2018; Rothenberg et al., 2019). To test globally whether Notch opposition to PU.1 defines the identity of developing pro-T cells, we compared genes defined as PU.1-regulated DEGs in phase 1 pro-T cells (Hosokawa et al., 2018) with the phase 1 Notch-regulated DEGs. Responses to PU.1 and responses to Notch together were found to predict distinct associations with cell type. We identified 28 PU.1-repressed, Notch-dependent genes and 47 PU.1-dependent, Notch-repressed genes (Fig. 4 B, magenta circles; Table S4). The PU.1-repressed, Notch-dependent genes had a trend toward high expression in DN subsets and γδ T cells among hematopoietic lineages (Fig. 4 C). In contrast, the PU.1-dependent and Notch-repressed genes were selectively highly expressed in myeloid lineages (Fig. 4 D); some were markers of distinct myeloid precursors naturally found in the thymus (Zhou et al., 2019). By comparison, the 31 genes responding positively to both PU.1 and Notch signaling in phase 1 cells showed an expression pattern more biased to stem/multipotent progenitor or early pro-T cell states (Fig. S3 B). Thus, genes divergently regulated by PU.1 and Notch signaling within pro-T cells defined a split between early T and myeloid lineage fates.
The developmental lineage associations of genes divergently regulated by PU.1 and Notch were specific for this pair. The relationships between Notch targets and targets of other factors showed less of a global connection to developmental fate. For example, Bcl11b is a prominent transcription factor after commitment that occupies many open chromatin sites in phase 2, as PU.1 does in phase 1 (Hu et al., 2018). When we compared Notch-regulated DEGs and Bcl11b-regulated DEGs (Hosokawa et al., 2018a) in phase 2 (Fig. S3 C and Table S4), Notch and Bcl11b effects were more often concordant than divergent. However, genes with different response patterns showed heterogeneous expression patterns and did not clearly define any lineage separation (Fig. S3, D and E). Thus, taken together, the results suggest that Notch signaling activates T lineage genes and represses myeloid lineage genes simultaneously through antagonism of a large subset of PU.1 functions during the PU.1-expressing phase 1 stage.
In this study, we show that Notch2 can act as a potent transducer of Notch signaling that complements Notch1 signals in phase 1 pro-T cells on OP9-DLL1 and OP9-DLL4 stromal cells alike. This experimental system exposes quantitative kinetic effects with high sensitivity by minimizing homeostatic compensation. The effects of deleting Notch1 alone in this system may seem surprisingly weak. Note that to initiate T lineage differentiation in this acute deletion experimental system, the cells first receive Notch signaling for the first 2 or 3 d, before deletion of any Notch genes. Thus, we cannot rule out an obligate role for Notch1 at the earliest stage. However, it is interesting that Notch2 has previously been reported to inhibit myeloid differentiation specifically from Lin−Sca-1+Kit+ blood progenitors (Varnum-Finney et al., 2011). New evidence has increased the likelihood that prethymic Notch signaling, which might include a role for Notch2, is relevant to enabling T cell development at all (Chen et al., 2019). Studies using a mouse strain in which RBPJ could be deleted and then inducibly restored yielded strong evidence that Notch signaling via RBPJ needs to be activated in BM-resident multipotent progenitors even before they reach the thymus (Chen et al., 2019). Multipotent progenitors express less Notch1 relative to Notch2 than lymphoid-biased precursors such as Common Lymphoid Progenitors (CLP) and ETP (Kit+ DN1 pro-T cells; Yoshida et al., 2019), and our results suggest that Notch2 is important to restrain myeloid differentiation of the phase 1 cells. Hence, although T cell differentiation is most strongly promoted by Notch1–DLL4 interaction, different developmental alternatives may be excluded by complementary actions of Notch1 and Notch2.
The findings in this reductionist system indicate a broader repertoire of functional signaling receptiveness of the pro-T cells but do not contradict the evidence that Notch1–DLL4 interaction is sufficient and indispensable to support normal T cell development in vivo. An important question is why Notch2 and Notch3 cannot compensate for the effect of Notch1 deletion on T cell development in vivo. Part of the answer is likely to be quantitative because neither one is regulated in a way that sustains activity across the phase 1–phase 2 transition. However, certain gatekeeper events in the thymus in vivo could also be exquisitely dependent on high-affinity Notch1–DLL4 interactions. In fact, a specific niche of vascular endothelial cells with DLL4 and membrane-bound Kit ligand (stem cell factor) expression has been described as the initial entry domain for ETP (Kithi DN1) cells in the thymus (Buono et al., 2016).
An obstacle to complementation analysis has been the grave impact of deleting Notch genes on multiple developmental decisions in vivo. Here, using an efficient, acute deletion system with real-time longitudinal monitoring, we show that Notch family genes work synergistically to drive the earliest T cell development. Notch signaling not only activates T lineage signature genes but also represses adoption of alternative fates, particularly in phase 1 while those alternatives are still open. Therefore, Notch1 and Notch2 enable T cell development by activation and repression of gene expression, directly and indirectly.
Materials and methods
Mice
C57BL/6 (referred to as “B6”), B6.Cg-Tg(BCL2)25Wehi/J (Bcl2-tg; Strasser et al., 1991), and B6.Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (Cas9; Platt et al., 2014) mice were purchased from The Jackson Laboratory. These were crossed to generate B6-Cas9/+;+/Bcl2 heterozygotes for each experiment. All animals were bred and maintained in the California Institute of Technology Laboratory Animal Facility under specific pathogen–free conditions, and the protocol supporting animal breeding for this work was reviewed and approved by the institutional animal care and use committee of the California Institute of Technology. The Bcl2 transgene was routinely incorporated into the genetic background because it enhances survival (and thus intact RNA recovery) from pro-T cells with regulatory perturbations, without altering pro–T cell development in the control animals (Franco et al., 2006; Taghon et al., 2007; Yui et al., 2010).
Cell culture
For in vitro differentiation of pro-T cells, BM hematopoietic progenitors were used for input. BM was removed from the femurs and tibiae of 2–3-mo-old mice. Suspensions of BM cells were prepared and stained for lineage markers using biotin-conjugated lineage antibodies (CD11b, CD11c, Gr1, TER-119, NK1.1, CD19, CD3ε), then incubated with streptavidin-coated magnetic beads (Miltenyi Biotec) and passed through a magnetic column (Miltenyi Biotec). Then, hematopoietic progenitors were cultured on OP9-DLL1 or OP9-DLL4 monolayers using OP9 medium (α-MEM, 20% FBS, 50 µM β-mercaptoethanol, Pen-Strep-Glutamine) supplemented with 10 ng/ml of IL-7 (PeproTech Inc.) and 10 ng/ml of Flt3L (PeproTech Inc). On day 7, cultured cells were disaggregated, filtered through 40-µm nylon mesh, and recultured on new OP9 monolayers with medium containing 5 ng/ml of IL-7 and 5 ng/ml of Flt3L. In cultures that were continued for longer times, cells were passaged onto fresh OP9-DLL1 monolayers at day 10 and maintained up to day 15 in 1 ng/ml each of IL-7 and Flt3L.
CRISPR/Cas9-mediated acute deletion of Notch in T cell development cultures
To generate input cells, Cas9 mice were first bred to Bcl2-tg mice to generate heterozygotes for both transgenes. BM cells from these Cas9;Bcl2-tg animals were then used to seed in vitro differentiation cultures as above. At day 2 (phase 1) or 10 (phase 2), the cells were transduced with retroviral vectors encoding reporters (CFP and hNGFR) and the indicated sgRNAs as detailed below and then returned to OP9-DLL1 culture. The methods used to generate the virus supernatant and for infection were described previously (Hosokawa et al., 2018a). Cells were analyzed after another 3 or 5 d of culture. For RNA-seq, retrovirus-infected Lin−CD45+c-KithiCFP+hNGFR+ (phase 1) or Lin−CD45+c-KitloCFP+hNGFR+ (phase 2) cells were sorted on a BD FACSAria FACS.
Cloning
The sgRNA expression vector (E42-dTet) we used was described previously (Hosokawa et al., 2018). 19-mer sgRNAs were designed using the CHOPCHOP web tool (https://chopchop.cbu.uib.no/) and inserted into the empty sgRNA expression vector by PCR-based insertion. Three sgRNA expression vectors were generated for one gene, and pooled retroviral plasmids were used to make retroviral supernatant. Sequences of sgRNAs used in this study are as follows: control (luciferase): ggcatttcgcagcctaccg; Notch1 no. 1: ctggcgagcaggcatgcca; Notch1 no. 2: tggggccatggaagccagg; Notch1 no. 3: tacctcttgcggcgagcgc; Notch2 no. 1: ccaacgcttgtcagaatgg; Notch2 no. 2: ctttccccaacatgccctg; Notch2 no. 3: agtaccaccattctgacag; Notch3 no. 1: ggccatcaggcgacgacgg; Notch3 no. 2: tgggcttccatccagacaa; and Notch3 no. 3: caacgacaggagaatcggg.
Flow cytometric analysis
For cell-surface staining of sgRNA-introduced BM cells, antibodies against Notch1, conjugated to phycoerythrin (Notch1 PE; catalog no. 130607; BioLegend), Notch2 PE (catalog no. 130707), Notch3 PE (catalog no. 130507), CD45 PE-cyanine 7 (catalog no. 25-0451-82; eBioscience), c-Kit allophycocyanin (catalog no. 17-1171-82), CD25 allophycocyanin-e780 (catalog no. 47-0251-82), human-NGFR PE (catalog no. 12-9400-42), and a biotin-conjugated lineage cocktail [CD8α (catalog no. 13-0081-86), CD11b (catalog no. 13-0112-86), CD11c (catalog no. 13-0114-85), Gr-1 (catalog no. 13-5931-86), TER-119 (catalog no. 13-5921-85), NK1.1 (catalog no. 13-5941-85), CD19 (catalog no. 13-0193-85), TCRβ (catalog no. 13-5961-85), and TCRγδ (catalog no. 13-5711-85)] were used for staining. Prior to cell surface staining, cells were treated with 2.4G2 cell supernatant. All of the cells were analyzed using a flow cytometer, MACSQuant 10 (Miltenyi Biotec), FACSAria Fusion (BD Biosciences), or LSRFortessa (BD Biosciences), with FlowJo software (FlowJo LLC).
ChIP and ChIP-seq
5–10 × 106 cells were fixed with 1 mg/ml DSG (Thermo Fisher Scientific) in PBS for 30 min at RT followed by an additional 10 min with addition of formaldehyde up to 1%. The reaction was quenched by addition of a 1:10 volume of 0.125 M glycine, and the cells were washed with Gibco HBSS (Thermo Fisher Scientific). Pelleted nuclei were dissolved in lysis buffer (0.5% SDS, 10 mM EDTA, 0.5 mM EGTA, 50 mM Tris-HCl [pH 8] and Protease Inhibitor Cocktail [Roche #11873580001]) and sonicated on a Bioruptor (Diagenode) for 18 cycles of 30-s sonication followed by 30-s rest, with maximum power. Five micrograms of anti-RBPJ mAbs (a mixture of 2.5 µg of 5313 [Cell Signaling Technology] and 2.5 µg of ab180588 [Abcam]) were prebound to Dynabeads antirabbit Ig (Invitrogen) and then added to the diluted chromatin complexes in parallel aliquots. The samples were incubated overnight at 4°C, then washed and eluted for 6 h at 65°C in ChIP elution buffer (20 mM Tris-HCl, pH 7.5, 5 mM EDTA, 50 mM NaCl, 1% SDS, and 50 µg/ml proteinase K). Precipitated chromatin fragments were cleaned up using ChIP DNA Clean & Concentrator (Zymo Research). ChIP-seq libraries were constructed using the NEBNext ChIP-Seq Library Preparation Kit (catalog no. E6240; New England Biolabs) and sequenced on an Illumina HiSeq 2500 system in single-read mode with a read length of 50 nt. Analysis pipelines used are described below under the “ChIP-seq analysis” and “RNA-seq analysis” subheadings. All analyses are based on results from at least two biologically separate replicates.
mRNA preparation and RNA-seq
Total RNA was isolated from samples of 1–2 × 105 cultured cells using the RNeasy Micro Kit (Qiagen). Libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (catalog no. E7530; New England Biolabs) from ∼1 µg of total RNA following the manufacturer’s instructions. Libraries were sequenced on the Illumina HiSeq 2500 system in single-read mode with a read length of 50 nt. Base calls were performed with RTA 1.13.48.0 followed by conversion to FASTQ with bcl2fastq 1.8.4 and produced ∼30 million reads per sample.
ChIP-seq analysis
Base calls were performed with RTA 1.13.48.0 followed by conversion to FASTQ with bcl2fastq 1.8.4 and produced ∼30 million reads per sample. ChIP-seq data were mapped to the mouse genome build NCBI37/mm10 using Bowtie (version 1.1.1; http://bowtie-bio.sourceforge.net/index.shtml) with “-v 3 -k 11 -m 10 -t--best --strata” settings, and HOMER tag directories were created with makeTagDirectory and visualized in the UCSC Genome Browser (http://genome.ucsc.edu; Speir et al., 2016). ChIP peaks were identified with findPeaks.pl against a matched control sample using the settings “-P .1 -LP .1 -poisson .1 -style factor.” The identified peaks were annotated to genes with the annotatePeaks.pl command against the mm10 genomic build in the HOMER package. Peak calls were always based on data from at least two independent biological replicates. Peak reproducibility was determined by a HOMER adaptation of the IDR (Irreproducibility Discovery Rate) package according to ENCODE guidelines (https://sites.google.com/site/anshulkundaje/projects/idr). Only reproducible high-quality peaks, with a normalized peak score ≥15, were considered for further analysis. Motif enrichment analysis was performed with the findMotifsGenome.pl command in the HOMER package using a 200-bp window. Notch IC and RBPJ ChIP-seq data in a T-ALL cell line, 8946, used in this study were previously published (GEO accession no. GSE66147; Pinnell et al., 2015).
RNA-seq analysis
RNA-seq reads were mapped onto the mouse genome build NCBI37/mm10 with STAR (version 2.4.0; Dobin et al., 2013) and post-processed with RSEM (version 1.2.25; http://deweylab.github.io/RSEM/; Li and Dewey, 2011) according to the settings in the ENCODE long-rna-seq-pipeline (https://github.com/ENCODE-DCC/long-rna-seq-pipeline/blob/master/DAC/STAR_RSEM.sh) with the minor modifications that settings “--output-genome-bam --sampling-for-bam” was added to rsem-calculate-expression. STAR and RSEM reference libraries were created from genome build NCBI37/mm10 together with the Ensembl gene model file Mus_musculus.NCBIM37.66.gtf. The resulting bam-files were used to create HOMER (Heinz et al., 2010) tag directories (makeTagDirectory with -keepAll setting). For analysis of statistical significance among DEGs, the raw gene counts were derived from each tag directory with analyzeRepeats.pl with the -noadj -condenseGenes options followed by the getDiffExpression.pl command using EdgeR (version 3.6.8; http://bioconductor.org/packages/release/bioc/html/edgeR.html; Robinson et al., 2010). For data visualization, RPKM normalized reads were derived using the analyzeRepeats.pl command with the options -count exons -condenseGenes -rpkm followed by log transformation. The normalized datasets were hierarchically clustered with “average” linkage and visualized in MATLAB (clustergram). GO analysis was performed using the DAVID analysis tool (https://david.ncifcrf.gov).
UCSC Genome Browser Bigwig visualization
BigWigs were generated from the aligned SAM or BED file formats using Samtools (Hosoya et al., 2009), Bedtools (Quinlan and Hall, 2010), and the UCSC genomeCoverageBed and bedGraphToBigWig and normalized to 1 million reads. For visualization of RNA-seq tracks, bamToBed and genomeCoverageBed were used with the “-split” setting enabled. BigWig files were uploaded to the UCSC Genome Browser (http://genome.ucsc.edu; Speir et al., 2016) for visualization.
PC analysis
PC loading for 65 curated T cell developmental trajectory genes was obtained from a previous single-cell RNA-seq study (Zhou et al., 2019). After the PC analysis of single-cell RNA-seq, the PC loadings of PCs 1 and 2 were projected to bulk RNA-seq data. Reference bulk RNA-seq results for in vivo thymocytes (DN1, DN2a, DN2b, and DN3) were reported previously (GEO accession no. GSE130812 and GEO accession no. GSE115744; Hosokawa et al., 2018a; Zhou et al., 2019). PC analysis displays RNA-seq data from sgRNA-transduced Lin−CD45+CFP+hNGFR+ phase 1 and phase 2 cells at 3 dpi. In the plot, colored circles represent phase 1 samples, and colored hexagons represent phase 2 samples. Black symbols indicate the reference unperturbed DN subsets from previous studies (Hosokawa et al., 2018a; Zhou et al., 2019). A curved arrow represents the T cell developmental trajectory defined in controls.
Analysis of developmental expression patterns of selected genes
Expression is characterized among multilineage precursors and hematopoietic cells, including B cells, dendritic cells, macrophages, monocytes, granulocytes, DN cells (thymocytes), mature αβ T cells, natural killer (NK) T cells, γδ T cells, and NK cells. The heat map display is taken from the Immunological Genome web site (http://www.immgen.org/; Heng et al., 2008; Mingueneau et al., 2013; Yoshida et al., 2019) using the MyGeneSet tool and ImmGen ULI RNA-seq. Color scale indicates z scores, with warm colors indicating higher relative levels and cold colors indicating lower relative levels. Venn diagrams were generated using the Bioinformatics & Evolutionary Genomics webtool (http://bioinformatics.psb.ugent.be/webtools/Venn/). PU.1-regulated and Bcl11b-regulated DEGs were reported previously and were defined in primary pro-T cells by concordant (opposite) responses in gain and loss of PU.1 function (Table S6 B in Ungerbäck et al., 2018) or by concordant responses to Cas9 and Cre-mediated deletion for Bcl11b (Hosokawa et al., 2018a; Table S3). The same PU.1 and Bcl11b target gene lists and their statistical cutoffs were described previously (Zhou et al., 2019).
Statistical analysis
DEGs were defined using EdgeR, typically with false discovery rate <0.05, |log2 fold change| >1, and RPKM >1, except when otherwise indicated, on the basis of measurements from at least two biologically independent replicates for each sample type. The statistical significance of differences between datasets was determined by one-way ANOVA, followed by Dunnett’s multiple comparisons test using Prism 8 software (GraphPad Software). Statistical details of experiments can be found in the figure legends. In all figures, error bars indicate SD.
Data availability
The accession number for all the new deep-sequencing data reported in this paper is GEO accession no. GSE148441.
Online supplemental material
Fig. S1 shows conformation of the KO of Notch family molecules by flow cytometry, cell recovery of Notch KO cells, and CD25 and non–T lineage marker expression profiles. Fig. S2 shows heat maps for gene expression changes in response to Notch family KO in pro-T cells cultured on OP9-DLL4, results of GO analysis for Notch-regulated DEGs, and comparison of RBPJ ChIP-seq results with previously published RBPJ ChIP-seq data in T-ALL cell lines. Fig. S3 shows cell numbers of CD19+ and CD11b+ cells in Notch KO phase 1 cells and the relationship between Bcl11b-regulated DEGs and Notch-regulated DEGs in phase 2. Table S1 lists the Notch-regulated DEGs in phase 1. Table S2 shows the list of Notch-regulated DEGs in phase 2. Table S3 lists the Notch-regulated DEGs in phase 1 cells cocultured on OP9-DLL4. Table S4 shows the genes regulated in different patterns by Notch signaling, PU.1, and Bcl11b for Fig. 4 B and Fig. S3 C.
Acknowledgments
We thank D. Perez, J. Tijerina, P. Cannon, and R. Diamond for cell sorting and advice; I. Soto for mouse colony care; V. Kumar for library preparation and sequencing; H. Amrhein and D. Trout for computational assistance; and I. Antoshechkin for sequencing management (all at the California Institute of Technology); members of the Support Center for Medical Research and Education at Tokai University for technical help; and members of the Rothenberg group (California Institute of Technology) for valuable discussion and reagents.
This work was supported by grants to E.V. Rothenberg from the U.S. Public Health Service (R01AI135200, R01AI083514, and R01HD076915) and by grants to H. Hosokawa from the Japan Society for the Promotion of Science (KAKENHI grant JP19H03692), the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Naito Foundation, and the Takeda Science Foundation. Support was also received by M. Romero-Wolf from the California Institute for Regenerative Medicine Bridges to Stem Cell Research Program (Pasadena City College and California Institute of Technology) and by E.V. Rothenberg from the Louis A. Garfinkle Memorial Laboratory Fund, the Al Sherman Foundation, and the Albert Billings Ruddock Professorship.
The authors declare no competing financial interests.
Author contributions: M. Romero-Wolf designed the study, performed experiments, analyzed data, and wrote the manuscript. B. Shin performed experiments, analyzed data, and edited the manuscript. W. Zhou analyzed data and edited the manuscript. M. Koizumi performed experiments and analyzed data. E.V. Rothenberg designed and supervised the study, analyzed data, and wrote the manuscript. H. Hosokawa designed and supervised the study, performed experiments, analyzed data, and wrote the manuscript.

