


Centrosomes play a crucial role during immune cell interactions and initiation of the immune response. In proliferating cells, centrosome numbers are tightly controlled and generally limited to one in G1 and two prior to mitosis. Defects in regulating centrosome numbers have been associated with cell transformation and tumorigenesis. Here, we report the emergence of extra centrosomes in leukocytes during immune activation. Upon antigen encounter, dendritic cells pass through incomplete mitosis and arrest in the subsequent G1 phase leading to tetraploid cells with accumulated centrosomes. In addition, cell stimulation increases expression of polo-like kinase 2, resulting in diploid cells with two centrosomes in G1-arrested cells. During cell migration, centrosomes tightly cluster and act as functional microtubule-organizing centers allowing for increased persistent locomotion along gradients of chemotactic cues. Moreover, dendritic cells with extra centrosomes display enhanced secretion of inflammatory cytokines and optimized T cell responses. Together, these results demonstrate a previously unappreciated role of extra centrosomes for regular cell and tissue homeostasis.
Introduction
Centrosomes are highly conserved cell organelles consisting of two centrioles, which are surrounded by ordered layers of pericentriolar material (PCM) that contain the functional components required for microtubule (MT) nucleation and anchoring (Bornens, 2012; Lawo et al., 2012; Mennella et al., 2012; Moritz et al., 1995). Being present as a single copy in interphase, centrosomes duplicate precisely once before a cell enters mitosis to enable bipolar spindle formation and proper distribution of the genetic material (Nigg, 2007; Tsou and Stearns, 2006a; Tsou and Stearns, 2006b). Cycling cells typically contain one centrosome in G1, consisting of a mature mother centriole that is connected to its immature daughter through a flexible linker (Paintrand et al., 1992). At the G1/S transition, a single procentriole nucleates perpendicularly to either of the two existing centrioles (Hinchcliffe et al., 1999; Lacey et al., 1999). Procentrioles remain closely connected to the parental centrioles and further elongate throughout G2 (Kohlmaier et al., 2009; Schmidt et al., 2009; Tang et al., 2009). Centriole duplication is completed by the end of G2, leading to two pairs of centrioles, each consisting of a mature parent centriole and one newly assembled immature daughter. During mitosis, the two centriole pairs form the poles of the mitotic spindle, which segregates the DNA into the two daughter cells. Consequently, each daughter cell inherits exactly one pair of centrioles.
Besides the well-established function in mitosis, centrosomes organize the MT cytoskeleton during interphase-specific processes. Efficient induction of innate and adaptive immune responses requires dynamic MT arrays, which regulate various immune cell effector functions, such as directional migration and immune synapse (IS) formation (Kopf and Kiermaier, 2021; Vicente-Manzanares and Sánchez-Madrid, 2004). Cell migration is a prerequisite for a properly operating immune system ensuring that rare cell populations meet in specialized immune compartments, such as secondary lymphoid organs (SLOs). Prior to migration, cells polarize by forming an actin-rich lamellipodium at the cell front and a contracting uropod at the back, which requires translocation of the centrosome and its associated MT cytoskeleton to the rear of the cell (Anderson et al., 1982; Eddy et al., 2002; Malech et al., 1977; Ratner et al., 1997). Centrosome polarization ensures high MT density and dynamics at the uropod resulting in stable front–back polarization (Ballestrem et al., 2000; Xu et al., 2005). After homing to SLOs, antigen-presenting cells (APCs) instruct T and B cells to recognize pathogenic microorganisms. The IS is a specialized structure at the interface of a T cell and an APC that forms downstream of antigen recognition. Formation of the IS induces drastic relocalization of the T cell’s centrosome from the uropod to the contact zone (Geiger et al., 1982; Ritter et al., 2015; Yi et al., 2013). Centrosome polarization at the IS is thought to enhance the efficiency and specificity of vesicle transport along MT tracks and subsequent secretion of vesicles into the synaptic cleft (Stinchcombe and Griffiths, 2014; Stinchcombe et al., 2006).
Dendritic cells (DCs) are leukocytes that efficiently integrate innate immune signals and initiate adaptive immune responses via antigen presentation. They reside in peripheral tissues and are characterized by a stellate morphology, high expression of major histocompatibility class (MHC) II, as well as their capacity to sense antigens via Toll-like receptors (TLRs; Banchereau et al., 2000; Medzhitov, 2001). Upon antigen recognition, DCs enter a maturation program, which triggers antigen uptake and processing and subsequent homing to SLOs (Inaba et al., 2000; Steinman et al., 1997; Turley et al., 2000). To efficiently navigate through complex 3D environments, DCs choose the path of least resistance and move without tightly adhering to the substrate, while being able to adapt their migration mode according to the environment (Lämmermann et al., 2008; Renkawitz et al., 2019; Renkawitz et al., 2009). Within lymph nodes, DCs activate antigen-specific T cells through the formation of the IS and thereby initiate adaptive immune responses (Banchereau et al., 2000).
In the following study, we investigate the role of the centrosome as a major MT organizing center (MTOC) during DC effector functions. In contrast to other highly specialized cell types such as neurons or muscle cells, which inactivate centrosomes during differentiation (Stiess et al., 2010; Zebrowski et al., 2015), we provide evidence that DCs upon antigen encounter acquire extra centrosomes, which nucleate MT filaments and confer advantageous features to the cells by promoting their ability to migrate and prime naive T cells.
Results
Primary and tissue-resident DCs possess multiple centrosomes
DCs constitute a rare population of cells in vivo, accounting for only 1–2% of total cells in most tissues (Banchereau et al., 2000). To analyze large numbers of cells, mouse bone marrow–derived DCs (BMDCs) are routinely used in cell-based assays to investigate immunomodulatory and anti-inflammatory activities of DCs (Ashley et al., 1997) and study the mechanistic basis of amoeboid cell locomotion (Maiuri et al., 2015; Renkawitz et al., 2019). Treatment of immature BMDCs with TLR agonists, such as lipopolysaccharide (LPS), results in BMDC activation and maturation, leading to a population of cells with branched and extended morphology that efficiently migrate and prime naive T cells in vitro (Lutz et al., 1999; Madaan et al., 2014).
To investigate the role of centrosomes in DC biology, we first generated BMDCs from WT mice in the presence of granulocyte–macrophage colony-stimulating factor (GM-CSF) and treated cells with LPS overnight to induce DC maturation. To assess centriole numbers, we tested various anti-centrosomal antibodies for staining discrete foci in fixed cells immobilized on ICAM/CCL21-coated cover slips. While several of the common antibodies against centriolar proteins (e.g., CEP192, CEP110, or CEP97) labeled centrioles in T cells, these antibodies did not yield a clear signal in DCs. As centrioles are ubiquitously acetylated in interphase and during cell division (Balashova et al., 2009; Bobinnec et al., 1998), an antibody against acetylated (ac)-tubulin has recently been used in cell lines to resolve single centrioles and determine centriole length (Kong et al., 2020). Immunostaining of WT BMDCs for ac-tubulin revealed discrete spots with an excellent signal-to-noise-ratio, allowing the identification of individual structures (Fig. 1 a, left panel). To confirm that ac-tubulin foci corresponded to tubulin in centriolar cylinder MTs, we generated BMDCs from either WT or Centrin-2 (CETN2)-GFP expressing mice and co-stained cells with genuine centriolar and PCM marker. Transgenic CETN2-GFP expressing cells generally display two fluorescently labeled centrioles for every organ examined (Higginbotham et al., 2004) and have been previously used for quantifying centriole numbers in a wide variety of mammalian cell types (Ching and Stearns, 2020; Durcan et al., 2008; Piel et al., 2000; Salisbury et al., 2002; Uetake et al., 2007). Ac-tubulin foci co-localized with the centriolar protein CEP135 as well as the PCM components γ-tubulin and CDK5RAP2 in WT and CETN2-GFP expressing BMDCs, indicating that they represent bona fide centrosomes (Fig. 1, a and b). Quantification of ac-tubulin– and CETN2-GFP+ foci co-localizing with either γ-tubulin or CEP135 revealed that 22% of WT and 30% of CETN2-GFP expressing BMDCs displayed four or more centrioles, corresponding to two or more centrosomes, which were all located in close proximity to the nucleus (Fig. 1 c). Centrosome numbers were slightly higher when we quantified centrioles according to CETN2-GFP+ foci. To monitor individual centrioles with nanometer precision, we further imaged mature CETN2-GFP expressing BMDCs by focused ion-beam scanning electron microscopy (FIB-SEM). High-resolution 3D reconstruction of the microtubule organizing center in CETN2-GFP expressing BMDCs confirmed the presence of cells with two, four, and more than four centrioles (Fig. 1 d and Video 1).

Visualization and quantification of centrosome numbers in BMDCs. (a) Immunostaining of centrioles and PCM in mature WT BMDCs. Merged and individual channels of ac-tubulin/DAPI (red/blue) and γ-tubulin, CEP135, and CDK5RAP2 (all displayed in green) are shown. White arrows point to cells with ≥4 ac-tubulin+ foci. Scale bars, 5 μm. (b) Immunostaining of centrioles and PCM in mature CETN2-GFP expressing BMDCs. Merged and individual channels of CETN2-GFP/DAPI (green/blue), ac-tubulin (gray), γ-tubulin, CEP135, and CDK5RAP2 (all displayed in red) are shown. White arrows point to cells with ≥4 CETN2-GFP+ foci. Scale bars, 5 μm. (c) Upper panel: Quantification of centrosome numbers in mature WT BMDCs according to ac-tubulin/γ-tubulin+ and ac-tubulin/CEP135+ foci. Graph shows mean values ± SD of 12 independent experiments (ac-tubulin/γ-tubulin) with N = 344/274/154/146/131/232/200/175/184/224/151/268 cells analyzed per experiment and 6 independent experiments (ac-tubulin/CEP135) with N = 270/254/206/207/222/225 cells analyzed per experiment. Lower panel: Quantification of centrosome numbers in mature CETN2-GFP expressing BMDCs according to CETN2-GFP/γ-tubulin+ and CETN2-GFP/CEP135+ foci. Graph shows mean values ± SD of eight independent experiments (CETN2-GFP/γ-tubulin) with N = 261/248/305/298/180/150/258/152 cells analyzed per experiment and four independent experiments (CETN2-GFP/CEP135) with N = 335/366/222/184 cells analyzed per experiment. (d) FIB-SEM of mature CETN2-GFP expressing BMDCs. Upper panel: CETN2-GFP fluorescent maximum intensity Z-stack projection images. Scale bars, 5 µm. Middle panel: Individual planes of the FIB-SEM 3D dataset containing centriole cross-section images. White arrows point to individual centrioles. Scale bars, 500 nm. Lower panel: 3D reconstruction of segmented centrioles (green). Scale bars, 500 nm. For each cell, xyz (left) and xzy (right) stacks are shown. One representative cell with one, two, and more than two centrosomes has been imaged. See also Video 1.
Visualization and quantification of centrosome numbers in BMDCs. (a) Immunostaining of centrioles and PCM in mature WT BMDCs. Merged and individual channels of ac-tubulin/DAPI (red/blue) and γ-tubulin, CEP135, and CDK5RAP2 (all displayed in green) are shown. White arrows point to cells with ≥4 ac-tubulin+ foci. Scale bars, 5 μm. (b) Immunostaining of centrioles and PCM in mature CETN2-GFP expressing BMDCs. Merged and individual channels of CETN2-GFP/DAPI (green/blue), ac-tubulin (gray), γ-tubulin, CEP135, and CDK5RAP2 (all displayed in red) are shown. White arrows point to cells with ≥4 CETN2-GFP+ foci. Scale bars, 5 μm. (c) Upper panel: Quantification of centrosome numbers in mature WT BMDCs according to ac-tubulin/γ-tubulin+ and ac-tubulin/CEP135+ foci. Graph shows mean values ± SD of 12 independent experiments (ac-tubulin/γ-tubulin) with N = 344/274/154/146/131/232/200/175/184/224/151/268 cells analyzed per experiment and 6 independent experiments (ac-tubulin/CEP135) with N = 270/254/206/207/222/225 cells analyzed per experiment. Lower panel: Quantification of centrosome numbers in mature CETN2-GFP expressing BMDCs according to CETN2-GFP/γ-tubulin+ and CETN2-GFP/CEP135+ foci. Graph shows mean values ± SD of eight independent experiments (CETN2-GFP/γ-tubulin) with N = 261/248/305/298/180/150/258/152 cells analyzed per experiment and four independent experiments (CETN2-GFP/CEP135) with N = 335/366/222/184 cells analyzed per experiment. (d) FIB-SEM of mature CETN2-GFP expressing BMDCs. Upper panel: CETN2-GFP fluorescent maximum intensity Z-stack projection images. Scale bars, 5 µm. Middle panel: Individual planes of the FIB-SEM 3D dataset containing centriole cross-section images. White arrows point to individual centrioles. Scale bars, 500 nm. Lower panel: 3D reconstruction of segmented centrioles (green). Scale bars, 500 nm. For each cell, xyz (left) and xzy (right) stacks are shown. One representative cell with one, two, and more than two centrosomes has been imaged. See also Video 1.
FIB-SEM datasets and 3D visualization of segmented centrioles (green). Arrows point to individual centrioles as present at EM data. Three cells are shown corresponding to cells in Fig. 1 d. Frame rate, 7 fps.
FIB-SEM datasets and 3D visualization of segmented centrioles (green). Arrows point to individual centrioles as present at EM data. Three cells are shown corresponding to cells in Fig. 1 d. Frame rate, 7 fps.
While two centrosomes are a regular condition in proliferating cells indicating transition through S/G2/M phases, the presence of more than two centrosomes is a rather unexpected phenomenon in untransformed somatic cells (Ganem et al., 2009) and is generally associated with tumorigenesis (Pihan et al., 1998; Carroll et al., 1999; Lingle et al., 1998; Nigg, 2002; Weber et al., 1998). To address whether extra centrosomes are restricted to primary DCs generated from bone marrow (BM), we next visualized centrioles in peripheral DCs isolated from mouse skin explants. Split ear sheets were floated on culture medium supplemented with the chemokine CCL19, which allows emigration of dermal DCs into the culture medium (Stoitzner et al., 2009). After 3 d of emigration, we collected all non-adhering cells, which expressed high cell-surface levels of typical DC markers and efficiently migrated toward chemotactic gradients in 3D collagen matrices without further antigen stimulation (Fig. S1, a and b). Quantification of centrosome numbers in WT and CETN2-GFP expressing dermal DCs revealed that 20 ± 5.5% of cells displayed four or more centrioles, similar to BMDCs (Fig. 2, a–c). To exclude that emigration of dermal DCs from split ears and subsequent culturing of cells in vitro favors the emergence of centrosomes, we used mouse skin explants and labeled centrosomes directly within tissues. An antibody against MHCII was used to stain dermal DCs and define individual cell borders. Z-sections were acquired at 200-nm steps to identify every centriole within single cells. Specific Z-section planes allowed detection of centrioles in individual cells and distinguish them from overlapping cell layers. Likewise, on emigrated DCs, we detected MHCII+ cells with two to five discrete ac-tubulin/CETN2-GFP foci, also demonstrating that tissue-resident DCs possess multiple centrosomes (Fig. 2 d and Fig. S1 c). In addition, we found similar centrosome numbers in a subset of splenic DCs (conventional DC1 [cDC1]) and Langerhans (LH) cells isolated from the epidermis of CETN2-GFP expressing mice (Fig. S1 d). Overall, our findings demonstrate that mature primary and tissue-resident DCs exhibit one, two, or more than two centrosomes.

Visualization and quantification of centrosomes in immune cells. (a) Characterization of DCs attained from skin explant culture. Skin DCs emigrated from skin explants cultured on medium supplemented with CCL19 and showed high cell surface levels of CD11c and MHCII. (b) Single-cell tracks of dermal DCs migrating in response to CCL19 gradients in 3D collagen gels. N = 63 cells pooled from two independent experiments. (c) Immunostaining of MHCII (red) and CDK5RAP2 (gray) in CETN2-GFP expressing skin explants. Left: Maximum intensity Z-stack projection of MHCII+ dermal DCs. Right: Magnification of boxed regions. Only indicated Z planes were projected. Scale bars, 5 μm. (d) Quantification of centrosome numbers in splenic DCs and LH cells prepared from epidermal sheets. Left panel: Gating strategy for splenic DCs and LH cells isolated from CETN2-GFP expressing mice. Centrosome numbers were quantified in cDC1 (MHCII+/CD11c+/CD8+) and cDC2 (MHCII+/CD11c+/CD11b+) subpopulations and LH cells (F4/80+/EpCAM+). Upper right picture: CETN2-GFP signals (green) and EdU-incorporation in cDC1. EdU+ cells (red) were excluded from the analysis as they indicate ongoing cell proliferation. White arrows point to extra centrosomes. Scale bar, 10 μm. Middle right panel: Quantification of centrosome numbers in splenic cDC subsets and LH cells sorted from epidermal sheets. Graph displays mean values ± SD of four (cDC) or two (LH cells) independent experiments. N = 242/359/337/161 cells (cDC1), 152/209/439/153 cells (cDC2) and 127/41 cells (LH cells).

Visualization and quantification of centrosomes in immune cells. (a) Characterization of DCs attained from skin explant culture. Skin DCs emigrated from skin explants cultured on medium supplemented with CCL19 and showed high cell surface levels of CD11c and MHCII. (b) Single-cell tracks of dermal DCs migrating in response to CCL19 gradients in 3D collagen gels. N = 63 cells pooled from two independent experiments. (c) Immunostaining of MHCII (red) and CDK5RAP2 (gray) in CETN2-GFP expressing skin explants. Left: Maximum intensity Z-stack projection of MHCII+ dermal DCs. Right: Magnification of boxed regions. Only indicated Z planes were projected. Scale bars, 5 μm. (d) Quantification of centrosome numbers in splenic DCs and LH cells prepared from epidermal sheets. Left panel: Gating strategy for splenic DCs and LH cells isolated from CETN2-GFP expressing mice. Centrosome numbers were quantified in cDC1 (MHCII+/CD11c+/CD8+) and cDC2 (MHCII+/CD11c+/CD11b+) subpopulations and LH cells (F4/80+/EpCAM+). Upper right picture: CETN2-GFP signals (green) and EdU-incorporation in cDC1. EdU+ cells (red) were excluded from the analysis as they indicate ongoing cell proliferation. White arrows point to extra centrosomes. Scale bar, 10 μm. Middle right panel: Quantification of centrosome numbers in splenic cDC subsets and LH cells sorted from epidermal sheets. Graph displays mean values ± SD of four (cDC) or two (LH cells) independent experiments. N = 242/359/337/161 cells (cDC1), 152/209/439/153 cells (cDC2) and 127/41 cells (LH cells).

Visualization and quantification of centrosome numbers in dermal DCs. (a) Immunostaining of dermal DCs isolated from WT ear explants. Merged and individual channels of ac-tubulin/DAPI (red/blue), γ-tubulin, CEP135, and CDK5RAP2 (all displayed in green) are shown. White arrows point to cells with ≥4 ac-tubulin+ foci. Right panels: Magnification of boxed regions. Scale bars, 5 μm. (b) Immunostaining of centrioles and PCM in dermal DCs isolated from CETN2-GFP expressing ear explants. Merged and individual channels of CETN2-GFP/DAPI (green/blue), γ-tubulin, and CDK5RAP2 (both displayed in red) and ac-tubulin (gray) are shown. White arrows point to cells with ≥4 CETN2-GFP+ foci. Scale bars, 5 μm. (c) Quantification of centrosome numbers in WT and CETN2-GFP expressing dermal DCs according to ac-tubulin/γ-tubulin+, ac-tub/CEP135+, and CETN2-GFP/γ-tubulin+ foci. Graph displays mean values ± SD of three to five independent experiments. N = 96/90/109/131/108 cells (ac-tubulin/γ-tubulin), 129/285/238 (ac-tub/CEP135+). and 131/78/95/184/239 cells (CETN2-GFP/γ-tubulin) pooled from three different mice for each experiment. (d) Immunostaining of centrioles and PCM in WT skin explants. Left: Merged channels of ac-tubulin (green), CDK5RAP2 (gray) and MHCII (red) are shown. Right: Magnification of boxed regions. Only indicated Z planes were projected. Scale bars, 5 μm. All nuclei were counterstained with DAPI (blue).
Visualization and quantification of centrosome numbers in dermal DCs. (a) Immunostaining of dermal DCs isolated from WT ear explants. Merged and individual channels of ac-tubulin/DAPI (red/blue), γ-tubulin, CEP135, and CDK5RAP2 (all displayed in green) are shown. White arrows point to cells with ≥4 ac-tubulin+ foci. Right panels: Magnification of boxed regions. Scale bars, 5 μm. (b) Immunostaining of centrioles and PCM in dermal DCs isolated from CETN2-GFP expressing ear explants. Merged and individual channels of CETN2-GFP/DAPI (green/blue), γ-tubulin, and CDK5RAP2 (both displayed in red) and ac-tubulin (gray) are shown. White arrows point to cells with ≥4 CETN2-GFP+ foci. Scale bars, 5 μm. (c) Quantification of centrosome numbers in WT and CETN2-GFP expressing dermal DCs according to ac-tubulin/γ-tubulin+, ac-tub/CEP135+, and CETN2-GFP/γ-tubulin+ foci. Graph displays mean values ± SD of three to five independent experiments. N = 96/90/109/131/108 cells (ac-tubulin/γ-tubulin), 129/285/238 (ac-tub/CEP135+). and 131/78/95/184/239 cells (CETN2-GFP/γ-tubulin) pooled from three different mice for each experiment. (d) Immunostaining of centrioles and PCM in WT skin explants. Left: Merged channels of ac-tubulin (green), CDK5RAP2 (gray) and MHCII (red) are shown. Right: Magnification of boxed regions. Only indicated Z planes were projected. Scale bars, 5 μm. All nuclei were counterstained with DAPI (blue).
Mature DCs arrest during the cell division cycle
In contrast to DC progenitor cells, which still retain a certain proliferative capacity, tissue-resident cells, such as dermal DCs, represent terminally differentiated cells with a low turnover rate (Cabeza-Cabrerizo et al., 2019). Therefore, it seemed rather unlikely that the presence of two centrosomes in these cells was associated with ongoing cell proliferation. To test this hypothesis, we stained dermal DCs and mature BMDCs for 5-ethynyl-2′-deoxyuridine (EdU) and phospho-Histone3 (pH3) as markers for S-phase and G2–M transition, respectively. As a control for actively proliferating cells, we utilized immature BMDCs, which represent a population of asynchronously dividing cells. In contrast to immature BMDCs, dermal DCs and mature BMDCs were largely EdU- and pH3-negative consistent with a low proliferative capacity of mature, tissue-resident DCs (Fig. 3 a and Fig. S2 a). The absence of proliferation markers reminiscent of S (EdU) and M phases (pH3) in mature DCs suggested that cells have either exited the cell cycle or arrested during the division cycle. To distinguish between these two possibilities, we first analyzed levels of Ki67, which is expressed in G1, S, G2, and M phases but not in resting cells (G0). Nearly 100% of dermal DCs and mature BMDCs were Ki67-positive (Ki67+) demonstrating that cells are still cell-cycle committed (Fig. 3 b). To further test whether dermal DCs and mature BMDCs are retained during the cell cycle, we examined the levels of cyclins over the course of differentiation. Cyclins control cell-cycle progression from G1, S, G2, into M phase by activating cyclin-dependent kinases (Cdks) at specific time points in the cell cycle (Evans et al., 1983). Immature BMDCs harbored considerable levels of the S phase cyclin A and the mitotic cyclin B1 (Fig. 3 c), which are required for G1–S phase transition, completion of S phase, and entry into mitosis (Nurse, 2000). In proliferating cells, cyclin B1 accumulates in G2 phase ensuring high activity of Cdk1 at the G2–M transition (Brandeis et al., 1998). In contrast, these cyclins were essentially absent in dermal DCs and mature BMDCs demonstrating exit from mitosis (Fig. 3, c and d). Of note, all cells equally showed high levels of cyclin E, which in proliferating cells peaks at the G1–S transition and in complex with Cdk2 allows entry into S phase (Ohtsubo et al., 1995). To directly follow the evolution of mitotic progression upon DC activation, we stimulated immature BMDCs with LPS and determined the levels of cyclin B1 over the course of maturation. Untreated immature BMDCs showed fluctuating levels of cyclin B1 characteristic of asynchronously proliferating cells (Fig. 3 e, left panel). Prior to LPS treatment, cyclin B1 protein levels were high, which progressively declined after 1 h of stimulus application and were completely gone after 8 h of LPS treatment (Fig. 3 e, right panel). Altogether, these results demonstrate that cellular activation modifies the cell division cycle of primary DCs and limits the proliferative capacity of the cells. Consistent with the presence of arrested cells, we found high levels of the Cdk inhibitors (CKIs) p27Kip1 in dermal DCs as well as p21Waf/Cip1 in mature BMDCs, but less in proliferating immature cells (Fig. 3, f and g). In particular, p21 has been demonstrated to induce a robust G1 cell-cycle arrest, which is accompanied by destruction of cyclin A and cyclin B1, while cyclin E reaches high levels (Toettcher et al., 2009).

Mature DCs arrest during the cell cycle with cells containing two and more centrosomes. (a) EdU incorporation and pH3 staining in WT dermal DCs isolated from split ears. Left: Immunostaining of WT dermal DCs against pH3 (red) and EdU (green) incorporation. Scale bar, 10 μm. Right: Quantification of EdU+ and pH3+ dermal DCs. Graphs display mean values ± SD of six independent experiments. N = 84/119/316/181/191/183 cells, pooled from three different mice for each experiment. (b) Ki67 staining in differentiating WT BMDCs (black) and WT dermal DCs (blue). Graph displays mean values ± SD of 6 (BMDCs) and 11 (dermal DCs) independent experiments. N = 10,000 cells (BMDCs) and at least 2,100 cells (dermal DCs) analyzed per experiment. (c and d) Immunoblotting of cyclins during WT BMDC differentiation and maturation and in WT dermal DCs (d). One representative experiment out of three is shown. (e) Immunoblotting of cyclin B1 in unstimulated WT BMDCs (− LPS; left panel) and after LPS-stimulation (+ LPS; right panel). Ponceau S stained membranes represent loading controls. One representative experiment out of two is shown. (f and g) Immunoblotting of Cdk inhibitors in WT dermal DCs and immature and mature WT BMDCs (g). Samples in d and f are derived from the same experiment. Membranes have been cut after transfer and probed with the indicated antibodies. Vinculin serves as loading control for both experiments. One representative experiment out of three is shown. (g) Middle: Immunostaining and quantification (right) of nuclear p21 levels in immature and mature WT BMDCs. Graph displays mean values ± SD of N = 242 cells (immature) and 232 cells (mature). One representative experiment out of three is shown. ****, P < 0.0001 (Mann-Whitney test). im, immature; m, mature; d, day; MW, mol wt. Source data are available for this figure: SourceData F3.
Mature DCs arrest during the cell cycle with cells containing two and more centrosomes. (a) EdU incorporation and pH3 staining in WT dermal DCs isolated from split ears. Left: Immunostaining of WT dermal DCs against pH3 (red) and EdU (green) incorporation. Scale bar, 10 μm. Right: Quantification of EdU+ and pH3+ dermal DCs. Graphs display mean values ± SD of six independent experiments. N = 84/119/316/181/191/183 cells, pooled from three different mice for each experiment. (b) Ki67 staining in differentiating WT BMDCs (black) and WT dermal DCs (blue). Graph displays mean values ± SD of 6 (BMDCs) and 11 (dermal DCs) independent experiments. N = 10,000 cells (BMDCs) and at least 2,100 cells (dermal DCs) analyzed per experiment. (c and d) Immunoblotting of cyclins during WT BMDC differentiation and maturation and in WT dermal DCs (d). One representative experiment out of three is shown. (e) Immunoblotting of cyclin B1 in unstimulated WT BMDCs (− LPS; left panel) and after LPS-stimulation (+ LPS; right panel). Ponceau S stained membranes represent loading controls. One representative experiment out of two is shown. (f and g) Immunoblotting of Cdk inhibitors in WT dermal DCs and immature and mature WT BMDCs (g). Samples in d and f are derived from the same experiment. Membranes have been cut after transfer and probed with the indicated antibodies. Vinculin serves as loading control for both experiments. One representative experiment out of three is shown. (g) Middle: Immunostaining and quantification (right) of nuclear p21 levels in immature and mature WT BMDCs. Graph displays mean values ± SD of N = 242 cells (immature) and 232 cells (mature). One representative experiment out of three is shown. ****, P < 0.0001 (Mann-Whitney test). im, immature; m, mature; d, day; MW, mol wt. Source data are available for this figure: SourceData F3.
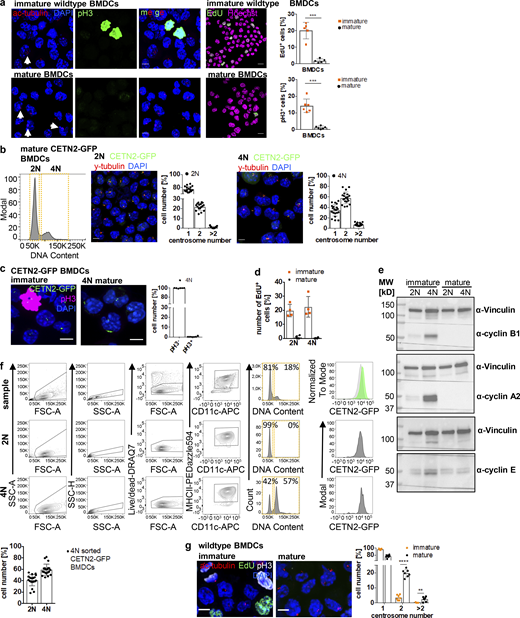
Mature DCs arrest during the cell cycle with cells containing two and more centrosomes. (a) Left: Immunostaining of ac-tubulin (red) and pH3 (green) in immature (upper panel) and mature (lower panel) WT BMDCs. White arrows point to extra centrosomes. Scale bars, 5 μm. Middle: EdU incorporation (green) in immature and mature WT BMDCs. Nuclei were counterstained with Hoechst (magenta). Scale bars, 10 μm. Right: Quantification of EdU+ and pH3+ cells in immature and mature WT BMDCs. Graph shows mean values ± SD of five (EdU) and six (pH3) independent experiments. EdU: N = 272/205/509/356/423 cells (immature) and 171/192/286/286/217 cells (mature). ***, P = 0.0009 (two-tailed, unpaired student’s t test with Welch’s correction). pH3: N = 134/98/158/125/93/124 cells (immature) and 141/203/158/204/127/128 cells (mature). ***, P = 0.0004 (two-tailed, unpaired Student’s t test with Welch’s correction). (b) Separation of 2N and 4N mature CETN2-GFP expressing BMDCs by flow cytometry. Left: Representative histogram of DNA content distribution of MHCII+/CD11c+ BMDCs. Gates for separating 2N and 4N cells are displayed in orange. Right: Immunostaining of PCM (γ-tubulin; red) and quantification of CETN2-GFP+ foci in enriched mature 2N and 4N CETN2-GFP expressing BMDCs. Scale bars, 5 μm. Graphs display mean values ± SD. N = 20 with at least 141 cells analyzed per experiment. (c) Left: Immunostaining of pH3 in immature (left panel) and sorted mature 4N (right panel) CETN2-GFP expressing BMDCs. Right: Quantification of pH3+ in sorted mature 4N cells. Graph shows mean values ± SD of six independent experiments with N = 417/485/309/411/298/497 cells analyzed per experiment. Scale bars, 5 μm. (d) Quantification of EdU incorporation in sorted immature and mature 2N and 4N BMDCs. Graph shows mean values ± SD of four independent experiments with N = 4 with at least 131 cells analyzed per experiment. (e) Immunoblotting of cyclins in immature (im) and mature (m) CD11c+ CETN2-GFP expressing BMDCs sorted on DNA content. One representative experiment out of three is shown. MW, mol wt. (f) Post-sort analysis of 2N and 4N mature CETN2-GFP expressing BMDCs separated as described in b. Graph displays mean values ± SD. N = 20 with at least 1,000 cells analyzed per experiment. (g) Left: Immunostaining of immature and mature WT BMDCs against ac-tubulin (red), EdU-incorporation (green), and pH3 (white). Scale bars, 5 μm. Right: Quantification of centrosome numbers according to ac-tubulin+ foci in EdU−/pH3− BMDCs. ****, P < 0.0001; **, P = 0.002 (unpaired Student’s t tests corrected for multiple comparisons after Holm-Sidak). Graph displays mean values ± SD of seven independent experiments. N = 134/98/158/93/124/344/274 cells (immature) and N = 141/203/158/127/128/200/175 cells (mature). Source data are available for this figure: SourceData FS2.

Mature DCs arrest during the cell cycle with cells containing two and more centrosomes. (a) Left: Immunostaining of ac-tubulin (red) and pH3 (green) in immature (upper panel) and mature (lower panel) WT BMDCs. White arrows point to extra centrosomes. Scale bars, 5 μm. Middle: EdU incorporation (green) in immature and mature WT BMDCs. Nuclei were counterstained with Hoechst (magenta). Scale bars, 10 μm. Right: Quantification of EdU+ and pH3+ cells in immature and mature WT BMDCs. Graph shows mean values ± SD of five (EdU) and six (pH3) independent experiments. EdU: N = 272/205/509/356/423 cells (immature) and 171/192/286/286/217 cells (mature). ***, P = 0.0009 (two-tailed, unpaired student’s t test with Welch’s correction). pH3: N = 134/98/158/125/93/124 cells (immature) and 141/203/158/204/127/128 cells (mature). ***, P = 0.0004 (two-tailed, unpaired Student’s t test with Welch’s correction). (b) Separation of 2N and 4N mature CETN2-GFP expressing BMDCs by flow cytometry. Left: Representative histogram of DNA content distribution of MHCII+/CD11c+ BMDCs. Gates for separating 2N and 4N cells are displayed in orange. Right: Immunostaining of PCM (γ-tubulin; red) and quantification of CETN2-GFP+ foci in enriched mature 2N and 4N CETN2-GFP expressing BMDCs. Scale bars, 5 μm. Graphs display mean values ± SD. N = 20 with at least 141 cells analyzed per experiment. (c) Left: Immunostaining of pH3 in immature (left panel) and sorted mature 4N (right panel) CETN2-GFP expressing BMDCs. Right: Quantification of pH3+ in sorted mature 4N cells. Graph shows mean values ± SD of six independent experiments with N = 417/485/309/411/298/497 cells analyzed per experiment. Scale bars, 5 μm. (d) Quantification of EdU incorporation in sorted immature and mature 2N and 4N BMDCs. Graph shows mean values ± SD of four independent experiments with N = 4 with at least 131 cells analyzed per experiment. (e) Immunoblotting of cyclins in immature (im) and mature (m) CD11c+ CETN2-GFP expressing BMDCs sorted on DNA content. One representative experiment out of three is shown. MW, mol wt. (f) Post-sort analysis of 2N and 4N mature CETN2-GFP expressing BMDCs separated as described in b. Graph displays mean values ± SD. N = 20 with at least 1,000 cells analyzed per experiment. (g) Left: Immunostaining of immature and mature WT BMDCs against ac-tubulin (red), EdU-incorporation (green), and pH3 (white). Scale bars, 5 μm. Right: Quantification of centrosome numbers according to ac-tubulin+ foci in EdU−/pH3− BMDCs. ****, P < 0.0001; **, P = 0.002 (unpaired Student’s t tests corrected for multiple comparisons after Holm-Sidak). Graph displays mean values ± SD of seven independent experiments. N = 134/98/158/93/124/344/274 cells (immature) and N = 141/203/158/127/128/200/175 cells (mature). Source data are available for this figure: SourceData FS2.
In essence, the presence of CKIs and progressive degradation of cyclin B1 demonstrates that dermal DCs and mature BMDCs arrest during the cell cycle with cells carrying one, two, and more than two centrosomes.
Persistent exposure to bacterial stimuli leads to a G1 cell-cycle arrest in primary DCs
As DNA replication and centrosome duplication are timely coordinated processes (Meraldi et al., 1999), the presence of two centrosomes in mature DCs suggested that cells replicated their DNA and subsequently got arrested in G2 phase with a tetraploid DNA content and two pairs of centrioles. To test this hypothesis, we first investigated the DNA content in mature DCs. While the majority of cells showed a diploid (2N) DNA profile, 18% of mature BMDCs and 7% of dermal DCs displayed a tetraploid (4N) DNA content (Fig. 4, a and b), confirming that these cells passed the restriction point in G1 and transited through S phase to enable DNA replication. FACS based on DNA content allowed us to separate and enrich 2N and 4N BMDCs (Fig. S2 b). Mature 4N cells stained negative for pH3 and EdU and lacked expression of cyclin A and cyclin B1, confirming again that extra centrosomes in 4N cells are not a consequence of ongoing cell proliferation (Fig. S2, c–e). Post-sort analysis of DNA content revealed that only the 2N population was of high purity (99%), while sorted 4N cells represent a mixture of 41 ± 9.8% 2N and 59 ± 9.8% 4N cells (Fig. S2 f). Accordingly, analysis of centrosome numbers in sorted 2N and 4N CETN2-GFP expressing BMDCs revealed that only 58 ± 11% of 4N cells carry two or more centrosomes (Fig. S2 b, right panel). Of note, we also found cells with two centrosomes in the sorted 2N fraction, indicating that a proportion of diploid cells carries two centrosomes as well, while the vast majority of cells with more than two centrosomes were present within the 4N fraction (Fig. S2 b).

Mature DCs arrest in G1 phase. (a and b) DNA staining of mature WT and CETN2-GFP expressing BMDCs (a) and WT dermal DCs (b). Left: Representative histogram of DNA content distribution of MHCII+/CD11c+ cells. Right: Quantification of 2N and 4N cells according to DNA content. Graphs display mean values ± SD of 11/17 BMDCs (WT /CETN2-GFP) and 11 (dermal DCs) independent experiments. N = 10,000 cells (BMDCs) and at least 2,100 cells (dermal DCs) analyzed per experiment. (c) Schematic representation of nuclei labeling in FUCCI-derived BMDCs. Graphic created with BioRender.com. (d) Left: Representative images of FUCCI-derived immature and mature BMDCs. Merged and individual channels of mKO2-hCdt1 (red) and mAG-hGem (green) are shown. Scale bars, 5 μm. Right: Quantification of G1, G1–S and S/G2/M phase distribution during BMDC differentiation and maturation. Graph displays mean values ± SD of three independent experiments. At least 222 cells (immature) and 206 cells (mature) have been analyzed per condition. (e) Quantification of G1, G1–S, and S/G2/M phase distribution after FUCCI-derived BMDC stimulation with LPS. Graph displays mean values ± SD of three independent experiments. At least 228 cells per condition have been analyzed. (f) Left: Gating strategy for identification of 4N cells in G1 phase. Right: Quantification of 4N cells in G1 phase. Graph displays mean values ± SD of six independent experiments. N = 10,000 cells analyzed per experiment. (g) Quantification of centrosome numbers in immature and mature FUCCI-derived BMDCs. Left: Immunostaining of immature and mature FUCCI-derived BMDCs against ac-tubulin (white). Scale bars, 5 μm. Right: Quantification of centrosome numbers according to ac-tubulin+ foci. Graph displays mean values ± SD of five independent experiments. N = 246/198/237/297/200 cells (immature) and N = 298/280/291/229/190 cells (mature). ****, P < 0.0001; **, P = 0.002 (unpaired Student’s t tests corrected for multiple comparisons after Holm-Sidak). Nuclei were counterstained with DAPI. im, immature; m, mature; d, day.
Mature DCs arrest in G1 phase. (a and b) DNA staining of mature WT and CETN2-GFP expressing BMDCs (a) and WT dermal DCs (b). Left: Representative histogram of DNA content distribution of MHCII+/CD11c+ cells. Right: Quantification of 2N and 4N cells according to DNA content. Graphs display mean values ± SD of 11/17 BMDCs (WT /CETN2-GFP) and 11 (dermal DCs) independent experiments. N = 10,000 cells (BMDCs) and at least 2,100 cells (dermal DCs) analyzed per experiment. (c) Schematic representation of nuclei labeling in FUCCI-derived BMDCs. Graphic created with BioRender.com. (d) Left: Representative images of FUCCI-derived immature and mature BMDCs. Merged and individual channels of mKO2-hCdt1 (red) and mAG-hGem (green) are shown. Scale bars, 5 μm. Right: Quantification of G1, G1–S and S/G2/M phase distribution during BMDC differentiation and maturation. Graph displays mean values ± SD of three independent experiments. At least 222 cells (immature) and 206 cells (mature) have been analyzed per condition. (e) Quantification of G1, G1–S, and S/G2/M phase distribution after FUCCI-derived BMDC stimulation with LPS. Graph displays mean values ± SD of three independent experiments. At least 228 cells per condition have been analyzed. (f) Left: Gating strategy for identification of 4N cells in G1 phase. Right: Quantification of 4N cells in G1 phase. Graph displays mean values ± SD of six independent experiments. N = 10,000 cells analyzed per experiment. (g) Quantification of centrosome numbers in immature and mature FUCCI-derived BMDCs. Left: Immunostaining of immature and mature FUCCI-derived BMDCs against ac-tubulin (white). Scale bars, 5 μm. Right: Quantification of centrosome numbers according to ac-tubulin+ foci. Graph displays mean values ± SD of five independent experiments. N = 246/198/237/297/200 cells (immature) and N = 298/280/291/229/190 cells (mature). ****, P < 0.0001; **, P = 0.002 (unpaired Student’s t tests corrected for multiple comparisons after Holm-Sidak). Nuclei were counterstained with DAPI. im, immature; m, mature; d, day.
To directly monitor the precise cell-cycle stage of mature DCs and correlate it to centrosome numbers, we generated BMDCs from fluorescent ubiquitination–based cell-cycle indicator (FUCCI) transgenic mice. FUCCI sensors identify cell-cycle phases based on the presence or absence of ectopically expressed truncated proteins of the cell-cycle regulators Cdt1 and Geminin fused to red- and green-emitting fluorescent proteins (mKO2-hCdt1 and mAG-hGem; Sakaue-Sawano et al., 2008). Cdt1 protein peaks in G1 phase just before the onset of DNA replication and declines abruptly after the initiation of S phase (Li et al., 2003). By contrast, Geminin protein levels are high during S and G2 phases, but low in G1 (McGarry and Kirschner, 1998). Accordingly, FUCCI cells residing in G1 phase display red fluorescent nuclei, while S/G2/M phase nuclei are labeled in green (Fig. 4 c). Due to the rapid degradation of Geminin in late M phase and expression of Cdt1 in early G1 phase, newborn daughter cells appear colorless. By contrast, at G1–S transition both proteins are present, leading to orange-labeled cells. We determined the proliferation pattern of BMDCs generated from FUCCI mice by analyzing red/colorless, orange-, and green-emitting populations by fluorescence confocal microscopy. During differentiation, 81–85% of immature BMDCs appeared either red or colorless, reminiscent of G1 phase, while 13–16% of cells labeled green monitoring cells that reside in G2 phase. Only a small fraction of cells (1–3%) expressed both sensors showing transition from G1 to S phase (Fig. 4 d). Surprisingly, virtually all cells imaged (98 ± 1%) turned to red fluorescence or appeared colorless after 24 h of LPS exposure, demonstrating that mature DCs arrest in G1 phase of the cell cycle. In line with these findings, LPS stimulation and subsequent analysis of cell-cycle phases over time revealed that 8 h after treatment, when cyclin B1 was gone (Fig. 3 e), the proportion of cells residing in G2 phase substantially declines, while red-emitting and colorless cells accumulate (Fig. 4 e). Analysis of DNA content in immature and mature FUCCI-derived BMDCs confirmed that 18.1 ± 1.4% of mature G1-arrested cells display a 4N DNA content while immature G1 cells were largely 2N (4.6% ± 1.4 4N cells in immature BMDCs; Fig. 4 f). Immunostaining and quantification of centrosome numbers according to ac-tubulin+ foci revealed that 22.1% of mature red-emitting and colorless cells carry either two or more centrosomes, while in immature cells only 5.6% of G1-resident cells showed more than one centrosome (Fig. 4 g). These results suggest that extra centrosomes arise during DC maturation when the cells arrest. In line with these findings, analysis of centrosome numbers in immature pH3/EdU-negative BMDCs revealed significantly fewer cells with two and more centrosomes compared to mature cells (Fig. S2 g).
Overall, these results provide evidence that DC maturation induces a robust G1 cell-cycle arrest leading to diploid and tetraploid cells containing multiple centrosomes.
DCs acquire extra centrosomes via a modified cell division cycle
We further sought to investigate the origins of the elevated centrosome phenotype in DCs. The presence of 4N cells with two centrosomes in G1 implies that these cells progressed sufficiently far in the cell cycle to allow DNA replication and centrosome duplication, but did not complete M phase. The cell division cycle typically ends with the processes of nuclear division (karyogenesis) followed by cytosolic separation (cytokinesis). Modifications of the cytokinesis process are observed in certain biological contexts and constitute a programmed step in normal development giving rise to differentiated tetraploid progeny (Davoli and de Lange, 2011).
To further explore the possibility that G1 tetraploid cells are a consequence of a modified cell division cycle, we followed mitosis of immature DCs upon antigen encounter by time-lapse microscopy. Immature BMDCs were labeled with Hoechst to visualize chromatin and condensed chromosomes and stimulated with LPS 30 min prior to imaging. Prometaphase cells were readily identified by their round shape and the typical rosette chromosome configuration (Nagele et al., 1995). Without stimulus, immature DCs predominantly progressed through mitosis normally and underwent successful divisions giving rise to diploid progenies (Fig. 5, a and b; and Fig. S3 a). After alignment of chromosomes at the metaphase plate, anaphase onset was characterized by cell elongation, followed by cleavage furrow formation and constriction of the cell membrane resulting in two daughter cells (Video 2). After LPS treatment, a significant proportion of cells exited mitosis without separating the DNA content into the two daughter cells leading to tetraploid progeny (Fig. 5 b). In 60% of cases, cells aligned their chromosomes along the metaphase plate but did not enter anaphase. After 10–20 min of metaphase arrest, chromosomes decondensed and cells started to polarize again (Fig. 5, c and d; Fig. S3 b; and Video 3). 40% of LPS-treated mitotic cells prematurely proceeded into anaphase without segregating sister chromatids to the opposing cell poles, resulting in chromatin trapped in the cleavage plane (Fig. 5, d and e; Fig. S3 c; and Video 4). In these cells, cleavage furrow formation started but then regressed resulting in cytokinesis failure.

Antigen encounter modifies the cell division cycle in DCs. (a) Time-lapse live-cell epifluorescence microscopy of unstimulated immature WT BMDCs progressing though mitosis. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. See also Video 2 and Fig. S3 a. (b) Quantification of successful mitotic divisions in WT BMDCs without (unstimulated) and after LPS (+LPS) stimulation. Graph displays mean values ± SD of three to four independent experiments. N = 15/9/6/7 mitotic cells analyzed per experiments. (c) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. See also Video 3 and Fig. S3 b. (d) Quantification of mitotic errors in WT BMDCs after LPS stimulation. N = 20 mitotic cells analyzed and pooled from three independent experiments. (e) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS showing impaired cleavage furrow ingression. Merged images of DIC (gray) and Hoechst (blue) are shown. White arrow indicates beginning of cleavage furrow ingression. White arrowheads denote regression of the cleavage furrow. Frames were collected every 15 s. See also Video 4 and Fig. S3 c. All scale bars, 5 μm. (f) Diagram illustrating the frequency of diploid and tetraploid DCs after maturation. Pictures in a, c, and e constitute key images derived from Fig. S3.
Antigen encounter modifies the cell division cycle in DCs. (a) Time-lapse live-cell epifluorescence microscopy of unstimulated immature WT BMDCs progressing though mitosis. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. See also Video 2 and Fig. S3 a. (b) Quantification of successful mitotic divisions in WT BMDCs without (unstimulated) and after LPS (+LPS) stimulation. Graph displays mean values ± SD of three to four independent experiments. N = 15/9/6/7 mitotic cells analyzed per experiments. (c) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. See also Video 3 and Fig. S3 b. (d) Quantification of mitotic errors in WT BMDCs after LPS stimulation. N = 20 mitotic cells analyzed and pooled from three independent experiments. (e) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS showing impaired cleavage furrow ingression. Merged images of DIC (gray) and Hoechst (blue) are shown. White arrow indicates beginning of cleavage furrow ingression. White arrowheads denote regression of the cleavage furrow. Frames were collected every 15 s. See also Video 4 and Fig. S3 c. All scale bars, 5 μm. (f) Diagram illustrating the frequency of diploid and tetraploid DCs after maturation. Pictures in a, c, and e constitute key images derived from Fig. S3.

Antigen encounter modifies the cell division cycle in DCs. (a) Time-lapse live-cell epifluorescence microscopy of unstimulated immature WT BMDCs progressing though mitosis. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. Only every second frame is shown in montage. See also Video 2 and Fig. 5 a. (b) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. Only every second frame is shown in montage. See also Video 3 and Fig. 5 c. (c) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS showing impaired cleavage furrow ingression. Merged images of DIC (gray) and Hoechst (blue) are shown. White arrow indicates beginning of cleavage furrow ingression. White arrowheads denote regression of the cleavage furrow. Frames were collected every 15 s. Only every fourth frame is shown in montage. See also Video 4 and Fig. 5 e. All scale bars, 10 μm. Key images from this time lapse are highlighted in Fig. 5.

Antigen encounter modifies the cell division cycle in DCs. (a) Time-lapse live-cell epifluorescence microscopy of unstimulated immature WT BMDCs progressing though mitosis. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. Only every second frame is shown in montage. See also Video 2 and Fig. 5 a. (b) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS. Merged images of DIC (gray) and Hoechst (blue) are shown. Frames were collected every 15 s. Only every second frame is shown in montage. See also Video 3 and Fig. 5 c. (c) Time-lapse live-cell epifluorescence microscopy of WT BMDCs stimulated with LPS showing impaired cleavage furrow ingression. Merged images of DIC (gray) and Hoechst (blue) are shown. White arrow indicates beginning of cleavage furrow ingression. White arrowheads denote regression of the cleavage furrow. Frames were collected every 15 s. Only every fourth frame is shown in montage. See also Video 4 and Fig. 5 e. All scale bars, 10 μm. Key images from this time lapse are highlighted in Fig. 5.
Mitotic progression of unstimulated WT BMDC. Merged channels of DIC (gray) and Hoechst (blue) are shown. Pictures were acquired every 15 s. Scale bar, 10 μm. Frame rate, 21 fps.
Mitotic progression of unstimulated WT BMDC. Merged channels of DIC (gray) and Hoechst (blue) are shown. Pictures were acquired every 15 s. Scale bar, 10 μm. Frame rate, 21 fps.
Metaphase arrest of LPS-stimulated WT BMDCs. Merged channels of DIC (gray) and Hoechst (blue) are shown. Pictures were acquired every 15 s. Scale bar, 10 μm. Frame rate, 21 fps.
Metaphase arrest of LPS-stimulated WT BMDCs. Merged channels of DIC (gray) and Hoechst (blue) are shown. Pictures were acquired every 15 s. Scale bar, 10 μm. Frame rate, 21 fps.
Impaired cleavage furrow ingression in LPS-stimulated WT BMDCs. Merged channels of DIC (gray) and Hoechst (blue) are shown. Pictures were acquired every 15 s. Scale bar, 10 μm. Frame rate, 21 fps.
Impaired cleavage furrow ingression in LPS-stimulated WT BMDCs. Merged channels of DIC (gray) and Hoechst (blue) are shown. Pictures were acquired every 15 s. Scale bar, 10 μm. Frame rate, 21 fps.
Together, these results demonstrate that upon antigen encounter, mitotic DCs suppress either karyogenesis or cytokinesis leading to 4N cells in G1 phase, which accumulate centrosomes.
PLK2-mediated centriole duplication in G1-arrested cells
About half of the cells harboring two and more centrosomes had undergone abortive mitosis (54%), leading to tetraploid cells in G1 leaving 46% of cells, which acquire extra centrosomes by other means (Fig. 5 f). Analysis of centrosome numbers in sorted mature CETN2-GFP expressing BMDCs indicated that two centrosomes are not only present in 4N but also in 2N cells (Fig. S2 b). The presence of 2N cells with two centrosomes suggested that centriole duplication proceeded during a prolonged G1 arrest giving rise to cells with four centrioles.
To distinguish between centriole duplication and accumulation of centrosomes due to abortive mitosis we carried out a CEP170 analysis in cells with extra centrosomes. CEP170 associates exclusively with subdistal appendages of mature mother centrioles (Guarguaglini et al., 2005). Cells that have completed mitosis generally contain one mature mother centriole, which stains positive for CEP170 and a corresponding immature CEP170-negative daughter centriole in the following G1 phase. In late G2–M phase, maturation of the second parental centriole takes place leading to two CEP170-positive and two CEP170-negative centrioles prior to mitosis. Analysis of CEP170 in mature CETN2-GFP expressing BMDCs with multiple centrosomes identified 46.5% of cells with two CEP170-foci and four to six centrioles (Fig. 6 a). This population corresponds to cells that had undergone incomplete mitosis without separating the DNA and centrosomal content leading to two CEP170-positive centrioles in the following G1 phase. By contrast, 53.5% of cells with extra centrosomes showed one CEP170-positive and two to three CEP170-negative centrioles (Fig. 6 a), indicative of diploid cells, which duplicated their centrioles during the G1 arrest.

LPS stimulation leads to PLK2 upregulation and untimely duplication of centrioles. (a) Left: Immunostaining of CEP170 in unsorted mature CETN2-GFP expressing BMDCs. Merged and individual images of CETN2-GFP/DAPI (green/blue) and CEP170 (red) are shown. Scale bar, 5 μm. Right: Schematic illustration of mother (red/gray) and daughter (gray) centrioles in cells that accumulate and duplicate centrosomes in G1. (b) mRNA expression levels of Plk1, Plk2, and Plk4 after LPS stimulation of immature WT BMDCs. mRNA levels were normalized to the expression of TATA-box binding protein. Graphs display mean values ± SD of three independent experiments. (c) Inhibition of PLK4 activity by Centrinone. CETN2-GFP expressing BMDCs were treated with either 250 or 500 nM Centrinone for the indicated time periods and centrosome numbers determined according to CETN2-GFP/ac-tubulin+ foci in mature BMDCs. Left: Immunostaining of ac-tubulin in Centrinone treated and control cells. Merged and individual channels of CETN2-GFP/DAPI (green/blue) and ac-tubulin (red) are shown. Right: Quantification of centriole numbers. Graphs display mean values ± SD of three and five independent experiments with at least N = 114 cells (treatment from day 6 on) and N = 146 cells (treatment during maturation) analyzed per experiment. (d) Upper panel: Immunostaininig of PCM in mature Plk2−/− and control (scramble) CETN2-GFP expressing HOXB8-derived DCs. Merged and individual channels of CETN2-GFP (green) and γ-tubulin (red) are shown. Right images: Magnification of boxed regions. Scale bars, 5 μm. Lower panel: Quantification of centrosome numbers according to CETN2-GFP/γ-tubulin+ foci in mature (left) and immature (right) Plk2−/− (cyan) and control (scramble, black) CETN2-GFP expressing HOXB8-derived DCs. Graphs display mean values ± SD of three independent experiments. N (mature) = 315/263/155 cells (scramble) and 295/268/130 cells (Plk2−/−). N (immature) = 367/342/292 cells (scramble) and 367/388/356 cells (Plk2−/−). **, P = 0.0095 (two-tailed, unpaired Student’s t test). Nuclei were counterstained with DAPI.
LPS stimulation leads to PLK2 upregulation and untimely duplication of centrioles. (a) Left: Immunostaining of CEP170 in unsorted mature CETN2-GFP expressing BMDCs. Merged and individual images of CETN2-GFP/DAPI (green/blue) and CEP170 (red) are shown. Scale bar, 5 μm. Right: Schematic illustration of mother (red/gray) and daughter (gray) centrioles in cells that accumulate and duplicate centrosomes in G1. (b) mRNA expression levels of Plk1, Plk2, and Plk4 after LPS stimulation of immature WT BMDCs. mRNA levels were normalized to the expression of TATA-box binding protein. Graphs display mean values ± SD of three independent experiments. (c) Inhibition of PLK4 activity by Centrinone. CETN2-GFP expressing BMDCs were treated with either 250 or 500 nM Centrinone for the indicated time periods and centrosome numbers determined according to CETN2-GFP/ac-tubulin+ foci in mature BMDCs. Left: Immunostaining of ac-tubulin in Centrinone treated and control cells. Merged and individual channels of CETN2-GFP/DAPI (green/blue) and ac-tubulin (red) are shown. Right: Quantification of centriole numbers. Graphs display mean values ± SD of three and five independent experiments with at least N = 114 cells (treatment from day 6 on) and N = 146 cells (treatment during maturation) analyzed per experiment. (d) Upper panel: Immunostaininig of PCM in mature Plk2−/− and control (scramble) CETN2-GFP expressing HOXB8-derived DCs. Merged and individual channels of CETN2-GFP (green) and γ-tubulin (red) are shown. Right images: Magnification of boxed regions. Scale bars, 5 μm. Lower panel: Quantification of centrosome numbers according to CETN2-GFP/γ-tubulin+ foci in mature (left) and immature (right) Plk2−/− (cyan) and control (scramble, black) CETN2-GFP expressing HOXB8-derived DCs. Graphs display mean values ± SD of three independent experiments. N (mature) = 315/263/155 cells (scramble) and 295/268/130 cells (Plk2−/−). N (immature) = 367/342/292 cells (scramble) and 367/388/356 cells (Plk2−/−). **, P = 0.0095 (two-tailed, unpaired Student’s t test). Nuclei were counterstained with DAPI.
We further investigated potential factors involved in centriole duplication in G1-arrested BMDCs. Polo-like kinase (PLK) family members are well-established kinases in the reproduction of centrosomes. PLK4 is considered the master regulator of centriole biogenesis and critically important for controlling centriole assembly in mammalian cells (Bettencourt-Dias et al., 2005). Loss of Plk4 results in failure to assemble new centrioles, while, conversely, overexpression of the kinase drives the assembly of excessive new centrioles and subsequent centrosome amplification (CA; Habedanck et al., 2005). In G2-arrested cells, activation of PLK1 induces centriole disengagement and premature centriole reduplication during interphase (Lončarek et al., 2010). Besides PLK4 and PLK1, PLK2 kinase is first activated at the G1–S phase transition, and its activity is required for centriole biogenesis (Cizmecioglu et al., 2008). Similar to PLK4, overexpression of PLK2 in cell lines results in overduplication of centrioles and CA (Cizmecioglu et al., 2008). To investigate whether PLKs are involved in centriole duplication in G1-arrested DCs, we carried out an analysis of Plk mRNA levels over the course of maturation. mRNA levels of Plk2 (and to a lesser extent of Plk4) were highly upregulated in DCs after 2 h of LPS stimulation and remained high even 16 h after stimulation (Fig. 6 b). In contrast to that, Plk1 levels progressively declined after LPS exposure. To test whether high expression levels of Plk2 and/or Plk4 account for centriole duplication in DCs, we used the selective PLK4 inhibitor Centrinone to pharmacologically interfere with PLK4 function (Wong et al., 2015). We added the drug either from day 6 on when cells were fully differentiated into the DC lineage or concomitant to LPS stimulation. Centrinone treatment did not alter DC maturation but led to a significant percentage of cells without or with only one centriole in cells treated from day 6 on, confirming successful inhibition of PLK4 (Fig. 6 c and Fig. S4 a). However, there was no difference detectable in the percentage of cells carrying two or more centrosomes with or without Centrinone in none of the conditions tested, demonstrating that PLK4 inhibition during maturation does not “rescue” centrosome numbers. Therefore, we concluded that upregulation of PLK4 after LPS stimulation does not interfere with centriole duplication in DCs. To further address the role of PLK2 in centrosome duplication in DCs, we generated CRISPR/Cas9 knockouts in precursor cell lines (Redecke et al., 2013) and differentiated them into DCs. PLK2-deficient cells differentiated into the DC lineage and upregulated MHCII molecules upon LPS stimulation (Fig. S4, b and c). Of note, Plk2−/− and control cells showed similar levels of proliferation markers and polyploidization confirming that PLK2 deficiency does not interfere with cell-cycle transition in DCs (Fig. S4 d). Analysis of centrosome numbers revealed a significant reduction of mature cells showing two and more centrosomes in Plk2−/− cells compared with control cells (Fig. 6 d). In particular, cells with more than two centrosomes were essentially absent in PLK2 knockouts, demonstrating that PLK2 plays a crucial role in centriole amplification in G1-arrested DCs. Similarly, the number of cells with two centrosomes was decreased in mature PLK2-deficient cells consistent with PLK2 function in regulating centriole duplication in late G1. Altogether our results provide evidence that G1-arrested DCs undergo untimely centriole duplication and establish a critical role for PLK2 in amplifying centrosome numbers if progression toward mitosis is blocked.
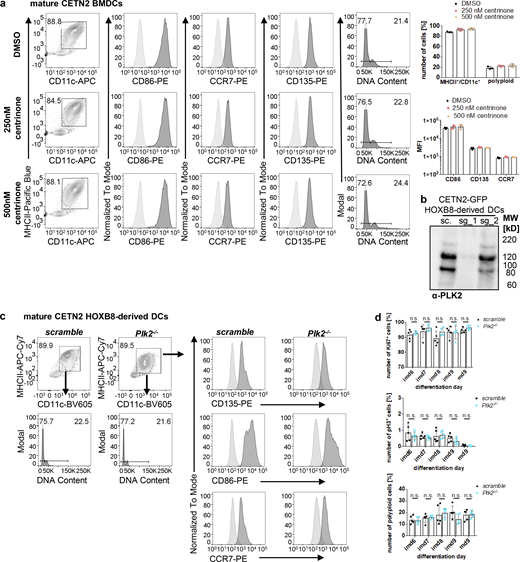
PLK2 induction after LPS stimulation leads to untimely duplication of centrioles. (a) Differentiation and maturation of BMDCs in the presence of the PLK4 inhibitor Centrinone. Left: Mature DCs were identified as MHCII+/CD11c+ cells and further analyzed for DNA content and DC-specific cell surface marker. Black bars indicate gates for 2N and 4N cells. Unstained samples served as control and were included as light gray filled line. Staining for DC marker has been conducted in parallel with PE-conjugated antibodies. Right: Quantification of CD86, CCR7, CD135 in the presence or absence of Centrinone. Graphs show mean values ± SD of three independent experiments. N = 10,000 cells per experiment. MFI, mean fluorescence intensity. (b) PLK2 depletion in CETN2-GFP expressing HOXB8-derived DCs. Immunoblotting against PLK2 in control (sc., scrambled) and Plk2−/− (sg_1 and sg_2) DCs. Note that only single guide 1 (sg_1) and not sg_2 led to efficient Plk2 knockout. MW, mol wt. (c) Differentiation and maturation of HOXB8-derived Plk2−/− and control DCs. Mature DCs were identified as MHCII+/CD11c+ cells and further analyzed for DNA content (lower panels) and DC-specific cell surface marker (CD135, CD86, CCR7; right panels). Unstained samples served as control and were included as light gray filled line. Staining for DC marker has been conducted in parallel with PE-conjugated antibodies. Representative histograms of one out of three independent experiments are shown. N = 10,000 cells per experiment. (d) Quantification of proliferation markers (Ki67, pH3, and DNA content) in Plk2−/− (blue) and control (scramble; black) HOXB8-derived DCs. Graphs display mean values of ± SD of five independent experiments. N = 10,000 cells per experiment. n.s., non-significant (multiple, two tailed, unpaired t tests). Source data are available for this figure: SourceData FS4.

PLK2 induction after LPS stimulation leads to untimely duplication of centrioles. (a) Differentiation and maturation of BMDCs in the presence of the PLK4 inhibitor Centrinone. Left: Mature DCs were identified as MHCII+/CD11c+ cells and further analyzed for DNA content and DC-specific cell surface marker. Black bars indicate gates for 2N and 4N cells. Unstained samples served as control and were included as light gray filled line. Staining for DC marker has been conducted in parallel with PE-conjugated antibodies. Right: Quantification of CD86, CCR7, CD135 in the presence or absence of Centrinone. Graphs show mean values ± SD of three independent experiments. N = 10,000 cells per experiment. MFI, mean fluorescence intensity. (b) PLK2 depletion in CETN2-GFP expressing HOXB8-derived DCs. Immunoblotting against PLK2 in control (sc., scrambled) and Plk2−/− (sg_1 and sg_2) DCs. Note that only single guide 1 (sg_1) and not sg_2 led to efficient Plk2 knockout. MW, mol wt. (c) Differentiation and maturation of HOXB8-derived Plk2−/− and control DCs. Mature DCs were identified as MHCII+/CD11c+ cells and further analyzed for DNA content (lower panels) and DC-specific cell surface marker (CD135, CD86, CCR7; right panels). Unstained samples served as control and were included as light gray filled line. Staining for DC marker has been conducted in parallel with PE-conjugated antibodies. Representative histograms of one out of three independent experiments are shown. N = 10,000 cells per experiment. (d) Quantification of proliferation markers (Ki67, pH3, and DNA content) in Plk2−/− (blue) and control (scramble; black) HOXB8-derived DCs. Graphs display mean values of ± SD of five independent experiments. N = 10,000 cells per experiment. n.s., non-significant (multiple, two tailed, unpaired t tests). Source data are available for this figure: SourceData FS4.
Diploid cells with extra centrosomes display enhanced directional locomotion
We further sought to investigate the functional consequences of extra centrosomes in mature DCs. As cells did not enter apoptosis (Fig. S5 a), we tested whether multiple centrosomes could modulate DC effector functions. Rapid directional locomotion is a prerequisite for initiating adaptive immune responses. During DC migration, the centrosome is located on the uropod with the majority of MT filaments projecting to the back to ensure stable cell polarization (Kopf et al., 2020). To determine the behavior of individual centrosomes and their impact on cell locomotion, we followed the dynamics of mature DCs during 2D migration over time. To this aim, mature CETN2-GFP expressing BMDCs were injected under a block of agarose to mimic physiologically relevant confined cell migration and exposed to the chemokine CCL19 to attract mature DCs. Under these 2D conditions, cells display a flat morphology, which allows to readily monitor intracellular structures (Video 5). Measuring intracentrosomal (between pairs of centrioles) and intercentrosomal (between pairs of centrosomes) distances unveiled that all centrosomes were located in close proximity to each other during DC migration toward chemotactic gradients (Fig. 7, a and b; and Fig. S5 b). Similar results were also obtained in fixed samples (Fig. 7 c and Fig. S5 c). To investigate the impact of extra centrosomes on DC locomotion, we first determined the number of MT filaments emanating from individual centrosomes in migrating BMDCs by high-resolution microscopy. To distinguish between potential effects caused by differences in centrosome numbers or ploidy, we analyzed the nuclear area in cells containing one and multiple centrosomes. Frequency distribution of nuclear areas revealed two populations indicative of 2N and 4N cells (Fig. S5 d). Cell sorting according to DNA content and subsequent analysis of nuclear areas in 2N cells led to a frequency distribution that perfectly matched the distribution of nuclear areas in cells with only one centrosome. Based on this analysis, we defined a threshold of 180 µm2 for the nuclear area to distinguish between 2N and 4N cells (Fig. S5 e). Similar to cell sorting according to DNA content, 2N cells with multiple centrosomes predominantly contained two centrosomes (95%). Immunostaining against α-tubulin revealed that all centrosomes nucleated MT filaments along the axis of migration, implying that extra centrosomes act as functional MTOC (Fig. 7 d). Manual counting of MT filaments emanating from the centrosome showed that 2N cells with extra centrosomes contained a larger number of cytoplasmic MT filaments compared with 2N cells with only one centrosome (Fig. 7 e). In addition to its central role as MT nucleator, the centrosome functions as a local organizer of actin polymerization (Farina et al., 2015; Inoue et al., 2019). Phalloidin staining was significantly lower in 2N cells with extra centrosomes demonstrating reduced levels of centrosomal actin in cells containing multiple centrosomes (Fig. 7 f).

Migration in the presence of multiple centrosomes. (a) Quantification of cleaved caspase-3–positive cells (green) in mature WT BMDCs and dermal DCs after immunostaining against cleaved caspase-3. Graph displays mean values ± SD of three independent experiments with N = 222/326/346 cells (BMDCs) and N = 357/150/268 cells (dermal DCs) analyzed. (b) Left: Centriole tracks of one representative cell with multiple centrosomes during migration. Right: Quantification of intra- and intercentrosomal distances in mature CETN2-GFP expressing BMDCs with multiple centrosomes during migration. (c) Left: Illustration of intra- and intercentrosomal distances in fixed mature CETN2-GFP expressing BMDCs. Middle: Magnification of boxed region. Right: Quantification of intracentrosomal distances in cells with one (blue) and multiple (red) centrosomes during migration. Scale bar, 5 µm. For b and c, cells were tracked either manually or automatically and distances determined as shown in c. N = 130 cells (one centrosome) and 12 cells (extra centrosomes) pooled from three independent experiments. (d) Upper panel: Nuclear areas (indicated by white dotted lines) of BMDCs with one and multiple centrosomes. Scale bars, 10 µm. Lower panel: Nuclear area distribution of mature CETN2-GFP expressing BMDCs with one (blue) and multiple (red) centrosome(s) as well as sorted 2N CETN2-GFP BMDCs (gray) fixed under agarose. Black dotted line indicates threshold for distinguishing 2N and 4N cells according to nuclear areas determined in sorted mature CETN2-GFP expressing 2N cells. N = 60/67/213 cells (one/multiple/2N) analyzed from two independent experiments. (e) Representative examples of nuclear areas in 2N and 4N CETN2-GFP expressing cells migrating under agarose. White dotted lines indicate nuclear areas. Centrioles are pseudo-color coded in gray. Scale bars, 10 µm. (f) Nuclear area distribution of CETN2-GFP expressing dermal DCs with one (blue) and multiple (red) centrosome(s). Black dotted line indicates threshold for distinguishing 2N and 4N cells. N = 216/76 cells (one/multiple) analyzed from two independent experiments. (g) Quantification of migration velocity (left) and persistence (right) of 2N dermal CETN2-GFP expressing DCs migrating under agarose. Graphs display mean values ± SD. N = 28 cells (one centrosome) and 18 cells (multiple centrosomes) pooled from four independent experiments. **, P = 0.0014 (two-tailed, unpaired Student’s t test [velocity] and two-tailed, unpaired Student’s t test with Welch’s correction [persistence]). (h) msd plots of dermal DCs migrating under agarose. Blue and red circles represent experimental data sets. Curves were fitted using Fürth’s formula (see Materials and methods section) and extrapolated for longer time periods (black lines; right panel). (i) Full laser ablations in mature CETN2-GFP expressing BMDCs migrating under agarose. Left: Maximum intensity Z-stack projection of two representative cells before and after laser ablation. Centrioles are pseudo-color coded in gray. Scale bars, 10 μm. Right: Quantification of integrated CETN2-GFP signal densities in defined ROIs drawn around centrosomes. Graph displays pairs of cells before (pre-) and after (post-) ablation. Cell pairs with one centrosome are depicted in gray, cell pairs with extra centrosomes in orange. N = 5/5 cells (one/multiple centrosomes). ****, P < 0.0001 (two-tailed, paired Student’s t test). (j) Left panel: Immunostaining against α-tubulin in cells migrating under agarose after complete centrosome and control ablation. Cells were fixed immediately after the ablation process. Individual and merged channels of CETN2-GFP (green) and α-tubulin/DAPI (white/blue) are shown. Images were post-treated by deconvolution. Red circles indicate ablated areas. White lines depict cell outline. Scale bars, 10 μm. Right panel: Quantification of MT filaments emanating from the centrosome in non- and fully ablated cells with one (gray) and multiple (orange) centrosomes. Graph displays mean values. Each data point represents one cell. (k) Quantification of MT length (left) and straightness (end-to-end distance/total length of MT filament; right) in non- and fully ablated cells. Graphs show mean values ± SD. N = 413/205 filaments traced from 9 or 5 different cells (non/full ablation). ****, P < 0.0001 (Mann-Whitney test). na, nuclear area.

Migration in the presence of multiple centrosomes. (a) Quantification of cleaved caspase-3–positive cells (green) in mature WT BMDCs and dermal DCs after immunostaining against cleaved caspase-3. Graph displays mean values ± SD of three independent experiments with N = 222/326/346 cells (BMDCs) and N = 357/150/268 cells (dermal DCs) analyzed. (b) Left: Centriole tracks of one representative cell with multiple centrosomes during migration. Right: Quantification of intra- and intercentrosomal distances in mature CETN2-GFP expressing BMDCs with multiple centrosomes during migration. (c) Left: Illustration of intra- and intercentrosomal distances in fixed mature CETN2-GFP expressing BMDCs. Middle: Magnification of boxed region. Right: Quantification of intracentrosomal distances in cells with one (blue) and multiple (red) centrosomes during migration. Scale bar, 5 µm. For b and c, cells were tracked either manually or automatically and distances determined as shown in c. N = 130 cells (one centrosome) and 12 cells (extra centrosomes) pooled from three independent experiments. (d) Upper panel: Nuclear areas (indicated by white dotted lines) of BMDCs with one and multiple centrosomes. Scale bars, 10 µm. Lower panel: Nuclear area distribution of mature CETN2-GFP expressing BMDCs with one (blue) and multiple (red) centrosome(s) as well as sorted 2N CETN2-GFP BMDCs (gray) fixed under agarose. Black dotted line indicates threshold for distinguishing 2N and 4N cells according to nuclear areas determined in sorted mature CETN2-GFP expressing 2N cells. N = 60/67/213 cells (one/multiple/2N) analyzed from two independent experiments. (e) Representative examples of nuclear areas in 2N and 4N CETN2-GFP expressing cells migrating under agarose. White dotted lines indicate nuclear areas. Centrioles are pseudo-color coded in gray. Scale bars, 10 µm. (f) Nuclear area distribution of CETN2-GFP expressing dermal DCs with one (blue) and multiple (red) centrosome(s). Black dotted line indicates threshold for distinguishing 2N and 4N cells. N = 216/76 cells (one/multiple) analyzed from two independent experiments. (g) Quantification of migration velocity (left) and persistence (right) of 2N dermal CETN2-GFP expressing DCs migrating under agarose. Graphs display mean values ± SD. N = 28 cells (one centrosome) and 18 cells (multiple centrosomes) pooled from four independent experiments. **, P = 0.0014 (two-tailed, unpaired Student’s t test [velocity] and two-tailed, unpaired Student’s t test with Welch’s correction [persistence]). (h) msd plots of dermal DCs migrating under agarose. Blue and red circles represent experimental data sets. Curves were fitted using Fürth’s formula (see Materials and methods section) and extrapolated for longer time periods (black lines; right panel). (i) Full laser ablations in mature CETN2-GFP expressing BMDCs migrating under agarose. Left: Maximum intensity Z-stack projection of two representative cells before and after laser ablation. Centrioles are pseudo-color coded in gray. Scale bars, 10 μm. Right: Quantification of integrated CETN2-GFP signal densities in defined ROIs drawn around centrosomes. Graph displays pairs of cells before (pre-) and after (post-) ablation. Cell pairs with one centrosome are depicted in gray, cell pairs with extra centrosomes in orange. N = 5/5 cells (one/multiple centrosomes). ****, P < 0.0001 (two-tailed, paired Student’s t test). (j) Left panel: Immunostaining against α-tubulin in cells migrating under agarose after complete centrosome and control ablation. Cells were fixed immediately after the ablation process. Individual and merged channels of CETN2-GFP (green) and α-tubulin/DAPI (white/blue) are shown. Images were post-treated by deconvolution. Red circles indicate ablated areas. White lines depict cell outline. Scale bars, 10 μm. Right panel: Quantification of MT filaments emanating from the centrosome in non- and fully ablated cells with one (gray) and multiple (orange) centrosomes. Graph displays mean values. Each data point represents one cell. (k) Quantification of MT length (left) and straightness (end-to-end distance/total length of MT filament; right) in non- and fully ablated cells. Graphs show mean values ± SD. N = 413/205 filaments traced from 9 or 5 different cells (non/full ablation). ****, P < 0.0001 (Mann-Whitney test). na, nuclear area.
DC migration in the presence of extra centrosomes. CETN2-GFP expressing BMDCs were injected under a block of agarose and exposed to a chemokine gradient of CCL19. Left: Cell with one centrosome. Right: Cell with two centrosomes. Pictures were acquired every 2 s. Scale bar, 10 μm. Frame rate, 21 fps.
DC migration in the presence of extra centrosomes. CETN2-GFP expressing BMDCs were injected under a block of agarose and exposed to a chemokine gradient of CCL19. Left: Cell with one centrosome. Right: Cell with two centrosomes. Pictures were acquired every 2 s. Scale bar, 10 μm. Frame rate, 21 fps.

Extra centrosomes nucleate MT filaments and cluster during migration. (a) Left: Mature CETN2-GFP expressing BMDC migrating under agarose. Cell outline is indicated with white dashed line. Scale bar, 5 μm. Right: Magnification of boxed region with illustration of intra- and intercentrosomal distances in a cell with multiple centrosomes. Lines denote distances between centrioles (1; intracentrosomal distance) and centrosomes (2; intercentrosomal distance). Scale bar, 2 μm. (b) Left: Maximum projection of time frames of one representative mature CETN2-GFP expressing BMDC migrating under agarose. Scale bar, 10 μm. Right: Quantification of intra- and intercentrosomal distances during migration. Graph displays mean values ± SD Centrioles are pseudo-color coded in gray. (c) Quantification of intra- and intercentrosomal distances in fixed CETN2-GFP expressing BMDCs with multiple centrosomes. Graph displays mean values ± SD. N = 50 cells pooled from two independent experiments. (d) Immunostaining of MT filaments in mature 2N CETN2-GFP expressing BMDCs migrating under agarose. Left: Merged and individual channels of CETN2-GFP (green), γ-tubulin (cyan), and α-tubulin (red) are shown. Right: Magnification of boxed region highlighting MT-nucleating centrosomes. Images were posttreated by deconvolution. Scale bars, 10 μm. na, nuclear area. (e) Quantification of MT filaments emanating from the centrosome in mature 2N CETN2-GFP expressing BMDCs with one (blue) and multiple (red) centrosomes fixed under agarose. Graph displays mean values ± SD. N = 44 cells (2N, one centrosome) and 45 cells (2N, multiple centrosomes) pooled from seven independent experiments. **, P = 0.007 (two-tailed, unpaired Student’s t test). (f) Left: Phalloidin staining in migrating 2N CETN2-GFP expressing BMDCs fixed under agarose. Merged channels of CETN2-GFP (green), phalloidin (gray), and DAPI (blue) are shown. Scale bar, 10 μm. na, nuclear area. Insets: Magnification of boxed regions. Scale bars, 2 μm. Right: Quantification of centrosomal actin in mature 2N CETN2-GFP expressing BMDCs cells with one (blue) and multiple (red) centrosome(s). Graph displays mean values ± SD. N = 88 cells (2N, one centrosome) and 24 cells (2N, multiple centrosomes) pooled from four independent experiments. *, P = 0.039 (Mann-Whitney test), fluo. int., fluorescence intensity; cent., centrosome; cyto., cytosol.
Extra centrosomes nucleate MT filaments and cluster during migration. (a) Left: Mature CETN2-GFP expressing BMDC migrating under agarose. Cell outline is indicated with white dashed line. Scale bar, 5 μm. Right: Magnification of boxed region with illustration of intra- and intercentrosomal distances in a cell with multiple centrosomes. Lines denote distances between centrioles (1; intracentrosomal distance) and centrosomes (2; intercentrosomal distance). Scale bar, 2 μm. (b) Left: Maximum projection of time frames of one representative mature CETN2-GFP expressing BMDC migrating under agarose. Scale bar, 10 μm. Right: Quantification of intra- and intercentrosomal distances during migration. Graph displays mean values ± SD Centrioles are pseudo-color coded in gray. (c) Quantification of intra- and intercentrosomal distances in fixed CETN2-GFP expressing BMDCs with multiple centrosomes. Graph displays mean values ± SD. N = 50 cells pooled from two independent experiments. (d) Immunostaining of MT filaments in mature 2N CETN2-GFP expressing BMDCs migrating under agarose. Left: Merged and individual channels of CETN2-GFP (green), γ-tubulin (cyan), and α-tubulin (red) are shown. Right: Magnification of boxed region highlighting MT-nucleating centrosomes. Images were posttreated by deconvolution. Scale bars, 10 μm. na, nuclear area. (e) Quantification of MT filaments emanating from the centrosome in mature 2N CETN2-GFP expressing BMDCs with one (blue) and multiple (red) centrosomes fixed under agarose. Graph displays mean values ± SD. N = 44 cells (2N, one centrosome) and 45 cells (2N, multiple centrosomes) pooled from seven independent experiments. **, P = 0.007 (two-tailed, unpaired Student’s t test). (f) Left: Phalloidin staining in migrating 2N CETN2-GFP expressing BMDCs fixed under agarose. Merged channels of CETN2-GFP (green), phalloidin (gray), and DAPI (blue) are shown. Scale bar, 10 μm. na, nuclear area. Insets: Magnification of boxed regions. Scale bars, 2 μm. Right: Quantification of centrosomal actin in mature 2N CETN2-GFP expressing BMDCs cells with one (blue) and multiple (red) centrosome(s). Graph displays mean values ± SD. N = 88 cells (2N, one centrosome) and 24 cells (2N, multiple centrosomes) pooled from four independent experiments. *, P = 0.039 (Mann-Whitney test), fluo. int., fluorescence intensity; cent., centrosome; cyto., cytosol.
The efficiency of migration largely depends on two essential parameters, cellular speed and directional persistence. Cell persistence, which quantifies the ability of a cell to maintain its direction of motion, can be measured by the persistence time or the chemotactic index (defined as the ratio of displacement to trajectory length). Single-cell tracking allowed us to determine migration velocity and persistence and to directly compare the dynamic behavior of cells in dependence on the number of centrosomes. Cell persistence was significantly increased in 2N cells containing extra centrosomes, highlighting that these cells changed direction less often than 2N cells with only one centrosome (Fig. 8, a and b). By contrast, we could not detect large differences in migration velocities in 2N cells with distinct centrosome numbers. Analysis of migration velocity and directional persistence in 2N dermal DCs revealed a similar phenomenon (Video 6; and Fig. S5, f and g). To estimate how differences in directional persistence in 2N cells affect the distance that cells travel during a defined time period, we carried out a mean square displacement (msd) analysis of cells over time. The msd to time plot visualizes how far cells can move in time and can be used for quantification of cell persistence. Accordingly, higher msd values denote larger squared distances relative to the starting point. After 5 min, the difference in msd between 2N BMDCs with one and multiple centrosomes was already 86% and further increased to 106% after 10 min of migration (Fig. 8 c). These differences in squared distances correspond to 36 and 44% difference in distance, demonstrating that 2N cells with extra centrosomes make more distance within the same time period compared with cells with a single centrosome. Similarly, dermal DCs translocated larger distances in the presence of multiple centrosomes (Fig. S5 h).

Extra centrosomes promote persistent locomotion. (a) Left: Maximum projection of time frames of mature CETN2-GFP expressing BMDCs migrating under agarose. Right: Single-cell tracks of mature CETN2-GFP expressing BMDCs migrating under agarose. (b) Quantification of migration velocity (left) and persistence (right). Graphs display mean values ± SD. N = 31 cells (2N, one centrosome) and 22 cells (2N, multiple centrosomes) pooled from six independent experiments. *, P = 0.030 (two-tailed, unpaired Student’s t test). (c) msd plots of mature CETN2-GFP expressing BMDCs migrating under agarose. msd values were calculated from single-cell tracks used for velocity and persistence analysis in b (upper graph). Lower graph: Extrapolated msd plots of BMDCs migrating under agarose. Blue and red circles represent experimental data sets. Curves were fitted using Fürth’s formula (see Materials and methods section) and extrapolated for longer time periods (black lines). (d) Illustration of partial laser ablation of centrosomes. Left: Pre- and post-ablation images of a CETN2-GFP expressing cell with two centrosomes migrating under agarose. Insets show magnification of boxed regions. Dotted line indicates the ablated area. Right: Maximum intensity projection over time of one representative cell before and after partial laser ablation. Inset: Magnification of centrosome tracks. Red arrow indicates time point of extra centrosome ablation. Scale bars, 10 μm. (e) Quantification of migration velocity (left) and persistence (right) before and after laser ablation. Graphs display changes as mean values ± SD. N = 14/19/9 cells (non/complete/partial ablation) pooled from six independent experiments. *, P = 0.015 and **, P = 0.009 (Kruskal-Wallis with Dunn’s test). Centrioles in a and d are pseudo-color coded in gray.
Extra centrosomes promote persistent locomotion. (a) Left: Maximum projection of time frames of mature CETN2-GFP expressing BMDCs migrating under agarose. Right: Single-cell tracks of mature CETN2-GFP expressing BMDCs migrating under agarose. (b) Quantification of migration velocity (left) and persistence (right). Graphs display mean values ± SD. N = 31 cells (2N, one centrosome) and 22 cells (2N, multiple centrosomes) pooled from six independent experiments. *, P = 0.030 (two-tailed, unpaired Student’s t test). (c) msd plots of mature CETN2-GFP expressing BMDCs migrating under agarose. msd values were calculated from single-cell tracks used for velocity and persistence analysis in b (upper graph). Lower graph: Extrapolated msd plots of BMDCs migrating under agarose. Blue and red circles represent experimental data sets. Curves were fitted using Fürth’s formula (see Materials and methods section) and extrapolated for longer time periods (black lines). (d) Illustration of partial laser ablation of centrosomes. Left: Pre- and post-ablation images of a CETN2-GFP expressing cell with two centrosomes migrating under agarose. Insets show magnification of boxed regions. Dotted line indicates the ablated area. Right: Maximum intensity projection over time of one representative cell before and after partial laser ablation. Inset: Magnification of centrosome tracks. Red arrow indicates time point of extra centrosome ablation. Scale bars, 10 μm. (e) Quantification of migration velocity (left) and persistence (right) before and after laser ablation. Graphs display changes as mean values ± SD. N = 14/19/9 cells (non/complete/partial ablation) pooled from six independent experiments. *, P = 0.015 and **, P = 0.009 (Kruskal-Wallis with Dunn’s test). Centrioles in a and d are pseudo-color coded in gray.
Migration of dermal DCs isolated from split ears. CETN2-GFP expressing dermal DCs were injected under agarose and DC migration followed toward soluble gradients of CCL19. Left: Cell with one centrosome. Right: Cell with multiple centrosomes. Pictures were acquired every 2 s. Scale bars, 10 μm. Frame rate, 21 fps.
Migration of dermal DCs isolated from split ears. CETN2-GFP expressing dermal DCs were injected under agarose and DC migration followed toward soluble gradients of CCL19. Left: Cell with one centrosome. Right: Cell with multiple centrosomes. Pictures were acquired every 2 s. Scale bars, 10 μm. Frame rate, 21 fps.
Overall, these findings demonstrate that cells with extra centrosomes are stably polarized and move straight toward a chemokine source without frequently changing their direction of migration. Increased directional persistence allows the cells to bypass longer distances within a given time period compared with cells with only one centrosome.
Extra centrosomes are sufficient to promote persistent locomotion
To further address whether enhanced directional locomotion is causally linked to the presence of extra centrosomes, we removed extra centrosomes during BMDC migration by laser ablation and measured migration velocity and directional persistence before and after the ablation process. To this aim, we first determined the settings required for efficient centriole ablation. To distinguish between mere bleaching of the CENT2-GFP signal or centrosome destruction, we used gridded glass coverslips that allow the relocation of cells after ablation and determined MT integrity in cells after full centrosomal ablation. We randomly ablated cells, and immediately after the ablation process fixed the cells under agarose and stained them against α-tubulin to quantify MT numbers and architecture. Maximum intensity Z-stack projections of CETN2-GFP signals before and after full centrosomal ablation revealed a loss of fluorescence signal at the irradiated region after laser exposure (Fig. S5 i). Immunostaining against α-tubulin showed fewer MT filaments nucleating from ablated centrioles compared with non-treated cells (Fig. S5 j). Overall, MT filaments in ablated cells were shorter and displayed a disorganized structure with a more bent configuration compared with control cells (Fig. S5 k). From this, we concluded that our laser setup allows for efficient destruction of centrosomes and their MT nucleation capacity.
To assess the impact of non-centrosome targeted laser exposure on cell behavior, we exposed random areas in close proximity to centrosomes with identical laser settings. As we aim to compare migration parameters within the same cell on a timescale of minutes before and after laser ablation, we did not distinguish between 2N and 4N cells. Cells experiencing control, non-centrosome targeted ablations retained their polarized shape and continued to migrate throughout the imaging period. Migration velocity was slightly decreased, whereas directional locomotion was unaffected (Video 7; and Fig. 8, d and e). Complete ablation of centrosomes also slightly decreased migration velocity, but strongly impaired persistent movement compared with control ablated cells (Video 8; and Fig. 8 e). These results demonstrate a critical role of the centrosome for directional DC locomotion. Similar to complete ablations, partial destruction of extra centrosomes, where only one centrosome was ablated while the other centrosome stays intact, significantly reduced locomotion persistence of cells (Video 9; and Fig. 8, d and e). These results confirm that enhanced directional locomotion is causally linked to the presence of extra centrosomes.
Non-centrosome targeted laser ablation in CETN2-GFP expressing BMDCs. Cells were injected under agarose and migration followed toward CCL19 gradients. Non-centrosome targeted control ablation was carried out next to centrosomes. Region of ablation is indicated with a white circle. Pictures were acquired every 5 s. Scale bar, 10 μm. Frame rate, 21 fps.
Non-centrosome targeted laser ablation in CETN2-GFP expressing BMDCs. Cells were injected under agarose and migration followed toward CCL19 gradients. Non-centrosome targeted control ablation was carried out next to centrosomes. Region of ablation is indicated with a white circle. Pictures were acquired every 5 s. Scale bar, 10 μm. Frame rate, 21 fps.
Full centrosome ablation in CETN2-GFP expressing BMDCs. Cells were injected under agarose and migration followed toward CCL19 gradients. Region of ablation is indicated with a white circle. Pictures were acquired every 5 s. Scale bar, 10 μm. Frame rate, 21 fps.
Full centrosome ablation in CETN2-GFP expressing BMDCs. Cells were injected under agarose and migration followed toward CCL19 gradients. Region of ablation is indicated with a white circle. Pictures were acquired every 5 s. Scale bar, 10 μm. Frame rate, 21 fps.
Partial laser ablation of extra centrosomes in CETN2-GFP expressing BMDCs. Cells were injected under agarose and migration followed toward CCL19 gradients. Note that the non-targeted centrosome stays intact after partial ablation. Region of ablation is indicated with a white circle. Pictures were acquired every 5 s. Scale bar, 10 μm. Frame rate, 21 fps.
Partial laser ablation of extra centrosomes in CETN2-GFP expressing BMDCs. Cells were injected under agarose and migration followed toward CCL19 gradients. Note that the non-targeted centrosome stays intact after partial ablation. Region of ablation is indicated with a white circle. Pictures were acquired every 5 s. Scale bar, 10 μm. Frame rate, 21 fps.
Altogether, our data unequivocally demonstrate that extra centrosomes promote efficient locomotion along chemotactic cues.
Directional persistence determines the efficiency of cells entering the lymphatic vasculature
Directional migration enables cells to efficiently explore their surroundings and home to SLOs upon antigen encounter. Within the dermis of the skin, the lymphatic vasculature forms a dense network of highly permeable initial lymphatic capillaries and larger collecting lymphatic vessels (Oliver et al., 2020). To estimate the mean distance that cells have to travel to reach the lymphatic vessels in the skin, we labeled the lymphatic vasculature in split ear sheets (Fig. 9 a). 2D projection of the 3D vessel system illustrates that distances between adjacent borders vary and reach up to 300 µm, which is roughly 10–15 times the diameter of a single DC (Fig. 9 b). To quantify the size of the 2D-projected 3D vessel system in more detail, we detected the topological space of locally connected areas. Connected areas were further analyzed by measuring the diameters in four directions (Fig. 9 c). We found a long-tailed distribution of diameters (<d> = 167 μm), highlighting that small areas appear more frequently compared with big areas (Fig. 9 d). By contrast, big areas will contain more DCs compared with small areas, and importantly, lead to DC positions with long distances (>300 µm) to the nearest lymph vessel. To better understand how directional persistence in a lymphatic system of various topological area sizes affect arrival times of cells at the capillary system, we developed Monte Carlo Simulations of interstitial cell migration (Fig. 9 e). To exactly mimic the experimentally found persistence and velocities of 2N cells, we fitted a persistent random walk model to the data (Fig. 9 f). Persistent random walks are characterized by directional movements during short time scales and random walk behavior for long time scales. The time to cross from the persistent to the random regime constitutes the persistence time Pt. Tracks of simulated cells with low and high Pt values confirmed that cells that move straight for longer time periods (red tracks) reach defined boundaries faster compared with cells with a low intrinsic persistency (blue tracks; Fig. 9 e). To test whether our model recapitulates the experimental data, we compared the msd–time plot for 2N cells with multiple (Pt = 11 min) and one centrosome (Pt = 6 min) with simulated data of the same process (Fig. 9, f and g). We were able to precisely recapitulate msd–time values when applying a persistent random walk model to our experimental data.

Cell migration persistence affects first passage times to reach the lymphatic system. (a) Maximum Z-stack projection of two exemplary areas of ear dermis stained for lvye1 (green). Scale bar, 100 µm. (b) Binary images from ear dermis shown in a. (c) Quantification of ear dermis area size (light gray) in between lymph vessels (dark gray). Contiguous regions in binary images were separated and individually analyzed for their diameters. Red lines show four diameters (horizontal, vertical, +45°, −45°) starting at the nearest lymph vessel entry. (d) Quantification of ear dermis free area diameters according to c. N = 9 areas pooled from ear sheets of three different mice. (e) Square design of three simulation areas (200, 300, and 400 µm) for persistent random walks using the speed and persistence values from experimental data. For comparison the start value for both cases (Pt = 11 min [red] and Pt = 6 min [blue]) were set to (0,0). (f and g) Experimental (f) and simulated (g) msd to time plots showing that the simulated persistent random walks (light red and light blue) closely resemble the experimental data (red and blue). (h and i) Efficiency time as a function of persistence time from persistent random walks with v = 5 µm/min (h) and v = 3 µm/min (i). Efficiency time represents the time that >99% of the persistent random walkers reached a boarder of the square. The start points are set randomly inside the square.
Cell migration persistence affects first passage times to reach the lymphatic system. (a) Maximum Z-stack projection of two exemplary areas of ear dermis stained for lvye1 (green). Scale bar, 100 µm. (b) Binary images from ear dermis shown in a. (c) Quantification of ear dermis area size (light gray) in between lymph vessels (dark gray). Contiguous regions in binary images were separated and individually analyzed for their diameters. Red lines show four diameters (horizontal, vertical, +45°, −45°) starting at the nearest lymph vessel entry. (d) Quantification of ear dermis free area diameters according to c. N = 9 areas pooled from ear sheets of three different mice. (e) Square design of three simulation areas (200, 300, and 400 µm) for persistent random walks using the speed and persistence values from experimental data. For comparison the start value for both cases (Pt = 11 min [red] and Pt = 6 min [blue]) were set to (0,0). (f and g) Experimental (f) and simulated (g) msd to time plots showing that the simulated persistent random walks (light red and light blue) closely resemble the experimental data (red and blue). (h and i) Efficiency time as a function of persistence time from persistent random walks with v = 5 µm/min (h) and v = 3 µm/min (i). Efficiency time represents the time that >99% of the persistent random walkers reached a boarder of the square. The start points are set randomly inside the square.
Persistent random walk motility of DCs in a complex lymph vessel system predicts a strong dependence on persistence to reach the capillary. To validate this hypothesis, we simulated DC trajectories starting at randomly set positions inside a square of edge length of 200, 300, and 400 µm with various persistence times. We measured the time until >99% of all cells have reached the border, defined as efficiency time, for two distinct velocities (5 and 3 µm/min; Fig. 9, h and i). For distances of 200 µm and a velocity of 5 µm/min, cells with a Pt of 6 min need 180 min, while cells with a Pt of 11 min require only 130 min and consequently reach the boundaries 27% faster (Fig. 9 h). At larger distances, the difference in efficiency becomes even more prominent: for distances up to 300 µm, 99% of all cells that move with a Pt of 11 min reach the boundaries within 251 min, while cells with a Pt of 6 min travel 406 min (38% slower). These data highlight the significant decrease in efficiency time whenever the persistence of a cell is enhanced. In essence, quantification of lymph vessel geometry and size in combination with DC persistence and speed allowed us to explore efficiency times important for fast and effective arrival of cells at the lymphatic vasculature.
Cells with extra centrosomes are more potent in eliciting T cell responses
Due to the beneficial effects of extra centrosomes during DC migration, we next addressed their impact on adaptive immune responses. To induce T cell immunity, DCs present antigenic peptides by MHC complexes on their cell surface. Immature DCs sequester internalized antigens in lysosomes, process them into small peptides, and load them on MHCII molecules for presentation to CD4+ T cells (Turley et al., 2000; Wubbolts, 1996). To test whether cells with extra centrosomes are more potent in eliciting T cell responses, we first sought to enrich BMDC subpopulations of different centrosomal content to directly compare cells with one and multiple centrosomes. To this aim, we separated mature CETN2-GFP expressing BMDCs according to DNA content as described above and further sorted 2N cells based on CETN2-GFP signal intensities in either CETN2-GFP–high or –low expressing cells (Fig. S6 a). Post-sort analysis of BMDC subpopulations confirmed that CETN2-GFP signals were shifted to higher values in the CETN2-GFPhigh population, while both populations showed a 2N DNA profile (Fig. S6 b). Analysis of centrosome numbers in sorted 2N BMDC subpopulations by confocal microscopy revealed that CETN2-GFP expression levels strongly correlated with the number of centrosomes (Fig. 10 a, left). After cell sorting, 99% of 2N cells contained either one or two centrosomes (see also Fig. S2 b). Within the CETN2-GFPlow population, the percentage of cells carrying two centrosomes ranged from 8–42 and 21–70% within the CETN2-GFPhigh population, leading to an enrichment of cells with two centrosomes by a factor of at least 1.5 (Fig. 10 a, middle and right). For simplicity, we refer to the CETN2-GFPlow population as 2N1C and the CETN2-GFPhigh population as 2N2C cells. Both 2N subpopulations expressed classical DC markers, while monocyte, macrophage, and granulocyte markers were absent, confirming that both subpopulations consisted of DCs (Fig. S6 c). Similarly, expression levels of cyclins were indistinguishable in 2N1C and 2N2C cells (Fig. S6 d). These data provide evidence that CETN2-GFP expression levels correlate with the number of centrosomes present in BMDCs, thus allowing to separate and enrich cells with different centrosome numbers.
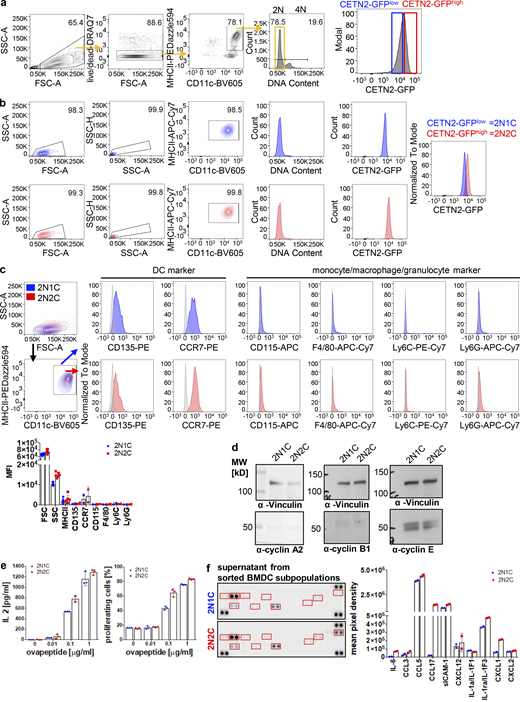
Cells with extra centrosomes show enhanced T cell activation and cytokine secretion. (a) Separation of CETN2-GFPlow (blue) and CETN2-GFPhigh (red) expressing mature BMDCs. MHCII+/CD11c+ cells were gated on 2N and further separated according CETN2-GFP signal intensities. Black bars indicate gates for 2N and 4N cells. (b) Post-sort analysis of CETN2-GFPlow (2N1C) and CETN2-GFPhigh (2N2C) expressing cells for DNA content and CETN2-GFP signal intensities. (c) Cell-surface expression of DC, monocyte, macrophage, and granulocyte markers on enriched MHCII+/CD11c+ 2N1C and 2N2C subpopulations. Unstained controls were included in the histograms as gray filled lines. Representative histograms of one out of at least two independent experiments are shown. Graph below shows quantification of cell-surface markers indicated as mean fluorescence intensity (MFI). Mean values ± SD are depicted. (d) Immunoblotting of cyclins in sorted 2N1C and 2N2C BMDC subpopulations. One representative experiment out of three is shown. MW, mol wt. (e) Left: Quantification of IL-2 levels after co-culture of OVA-peptide-pulsed sorted 2N1C and 2N2C BMDC subpopulations with OT-II CD4+ T cells. Supernatants were analyzed for IL-2 by ELISA. Right: Quantification of OT-II CD4+ T cells that divided after co-culture with OVA-peptide-pulsed sorted 2N1C and 2N2C BMDC subpopulations. Graphs display mean values ± SD of one representative out of five (IL-2) and four (T cell proliferation) independent experiments. Data points represent technical replicates. (f) Cytokine array analysis of conditioned medium harvested from sorted 2N1C and 2N2C DC subpopulations. Sorted 2N1C and 2N2C cells were cultured and supernatants harvested after 16 h. Left: Cytokine array of supernatants harvested from 2N1C and 2N2C cells. Quantified cytokine signals are highlighted in red. Right: Quantification of cytokine signal intensities. Graph shows mean pixel intensities ± SD of one out of two independent experiments. Data points represent technical replicates. Source data are available for this figure: SourceData FS6.

Cells with extra centrosomes show enhanced T cell activation and cytokine secretion. (a) Separation of CETN2-GFPlow (blue) and CETN2-GFPhigh (red) expressing mature BMDCs. MHCII+/CD11c+ cells were gated on 2N and further separated according CETN2-GFP signal intensities. Black bars indicate gates for 2N and 4N cells. (b) Post-sort analysis of CETN2-GFPlow (2N1C) and CETN2-GFPhigh (2N2C) expressing cells for DNA content and CETN2-GFP signal intensities. (c) Cell-surface expression of DC, monocyte, macrophage, and granulocyte markers on enriched MHCII+/CD11c+ 2N1C and 2N2C subpopulations. Unstained controls were included in the histograms as gray filled lines. Representative histograms of one out of at least two independent experiments are shown. Graph below shows quantification of cell-surface markers indicated as mean fluorescence intensity (MFI). Mean values ± SD are depicted. (d) Immunoblotting of cyclins in sorted 2N1C and 2N2C BMDC subpopulations. One representative experiment out of three is shown. MW, mol wt. (e) Left: Quantification of IL-2 levels after co-culture of OVA-peptide-pulsed sorted 2N1C and 2N2C BMDC subpopulations with OT-II CD4+ T cells. Supernatants were analyzed for IL-2 by ELISA. Right: Quantification of OT-II CD4+ T cells that divided after co-culture with OVA-peptide-pulsed sorted 2N1C and 2N2C BMDC subpopulations. Graphs display mean values ± SD of one representative out of five (IL-2) and four (T cell proliferation) independent experiments. Data points represent technical replicates. (f) Cytokine array analysis of conditioned medium harvested from sorted 2N1C and 2N2C DC subpopulations. Sorted 2N1C and 2N2C cells were cultured and supernatants harvested after 16 h. Left: Cytokine array of supernatants harvested from 2N1C and 2N2C cells. Quantified cytokine signals are highlighted in red. Right: Quantification of cytokine signal intensities. Graph shows mean pixel intensities ± SD of one out of two independent experiments. Data points represent technical replicates. Source data are available for this figure: SourceData FS6.

Cells with extra centrosomes show enhanced T cell activation and cytokine secretion. (a) Left: Representative images of 2N BMDCs sorted for CETN2-GFPlow and CETN2-GFPhigh expressing cells. Sorted BMDC subpopulations were immunostained against γ-tubulin (white) and CEP135 (red) to assess centrosome numbers. Left graph: Quantification of percentage of cells with ≥2 centrosomes in CETN2-GFPlow (2N1C, blue) and CETN2-GFPhigh (2N2C, red) expressing cells. Centrosome numbers of sorted DC subpopulations were determined by confocal microscopy according to CETN2-GFP/γ-tubulin+ foci. Each data point represents one experiment. ****, P < 0.0001 (two-tailed, paired Student’s t test). Right graph: Ratio of cells with multiple centrosomes between 2N2C and 2N1C cells. (b) Quantification of IL-2 levels after co-culture of OVA-pulsed 2N BMDC subpopulations with OT-II CD4+ T cells. Graph displays mean values ± SD of one representative out of six independent experiments. Data points represent technical replicates. 2N1C, cells enriched for one centrosome (blue); 2N2C, cells enriched for multiple centrosomes (red). (c) Left: Representative histograms of T cell proliferation of co-cultured OT-II CD4+ T cells with enriched BMDC subpopulations pulsed with OVA. Unprimed T cells (−OVA) served as negative control and are displayed as gray filled line. Right: Graph shows quantification of OT-II CD4+ T cells that divided after co-culture with OVA-pulsed enriched BMDC subpopulations and displays mean values ± SD of one representative out of four independent experiments. Data points represent technical replicates. Black bars indicate gates for proliferating (left) and non-proliferating (right) cells. (d) Quantification of cell-surface expression levels of co-stimulatory molecules on sorted 2N1C and 2N2C BMDC subpopulations. Left: Representative histograms of co-stimulatory molecules. Unstained controls were included in the histograms as gray filled lines. Right: Graph displays mean values of mean fluorescence intensities (MFI) ± SD. Each data point represents one independent experiment. *, P = 0.027 (all two-tailed, paired Student’s t test). (e) Levels of indicated cytokines in conditioned medium harvested from sorted DC subpopulations were quantified by ELISA. Graphs show mean values ± SD of at least nine independent experiments. **, P = 0.002 (CCL17; Wilcoxon test); ***, P = 0.0005 (IL-6); ****, P < 0.0001 (CCL5) and **, P = 0.0057 (CXCL1; all two-tailed, paired Student’s t test). (f) mRNA levels of indicated cytokines in sorted 2N1C and 2N2C DC subpopulations. Graph displays mean values ± SD of at least three independent experiments. *, P = 0.012 (two-tailed, paired Student’s t test). (g) Transmigration of CD4+ T cells toward supernatant harvested from 2N1C and 2N2C cells and toward control media. Graph displays mean values ± SD from 12 independent experiments. *, P = 0.033 (one-way ANOVA followed by two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli).
Cells with extra centrosomes show enhanced T cell activation and cytokine secretion. (a) Left: Representative images of 2N BMDCs sorted for CETN2-GFPlow and CETN2-GFPhigh expressing cells. Sorted BMDC subpopulations were immunostained against γ-tubulin (white) and CEP135 (red) to assess centrosome numbers. Left graph: Quantification of percentage of cells with ≥2 centrosomes in CETN2-GFPlow (2N1C, blue) and CETN2-GFPhigh (2N2C, red) expressing cells. Centrosome numbers of sorted DC subpopulations were determined by confocal microscopy according to CETN2-GFP/γ-tubulin+ foci. Each data point represents one experiment. ****, P < 0.0001 (two-tailed, paired Student’s t test). Right graph: Ratio of cells with multiple centrosomes between 2N2C and 2N1C cells. (b) Quantification of IL-2 levels after co-culture of OVA-pulsed 2N BMDC subpopulations with OT-II CD4+ T cells. Graph displays mean values ± SD of one representative out of six independent experiments. Data points represent technical replicates. 2N1C, cells enriched for one centrosome (blue); 2N2C, cells enriched for multiple centrosomes (red). (c) Left: Representative histograms of T cell proliferation of co-cultured OT-II CD4+ T cells with enriched BMDC subpopulations pulsed with OVA. Unprimed T cells (−OVA) served as negative control and are displayed as gray filled line. Right: Graph shows quantification of OT-II CD4+ T cells that divided after co-culture with OVA-pulsed enriched BMDC subpopulations and displays mean values ± SD of one representative out of four independent experiments. Data points represent technical replicates. Black bars indicate gates for proliferating (left) and non-proliferating (right) cells. (d) Quantification of cell-surface expression levels of co-stimulatory molecules on sorted 2N1C and 2N2C BMDC subpopulations. Left: Representative histograms of co-stimulatory molecules. Unstained controls were included in the histograms as gray filled lines. Right: Graph displays mean values of mean fluorescence intensities (MFI) ± SD. Each data point represents one independent experiment. *, P = 0.027 (all two-tailed, paired Student’s t test). (e) Levels of indicated cytokines in conditioned medium harvested from sorted DC subpopulations were quantified by ELISA. Graphs show mean values ± SD of at least nine independent experiments. **, P = 0.002 (CCL17; Wilcoxon test); ***, P = 0.0005 (IL-6); ****, P < 0.0001 (CCL5) and **, P = 0.0057 (CXCL1; all two-tailed, paired Student’s t test). (f) mRNA levels of indicated cytokines in sorted 2N1C and 2N2C DC subpopulations. Graph displays mean values ± SD of at least three independent experiments. *, P = 0.012 (two-tailed, paired Student’s t test). (g) Transmigration of CD4+ T cells toward supernatant harvested from 2N1C and 2N2C cells and toward control media. Graph displays mean values ± SD from 12 independent experiments. *, P = 0.033 (one-way ANOVA followed by two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli).
We next used our sorted 2N BMDC subpopulations, pulsed them with the model antigen ovalbumin (OVA), and incubated the cells with OVA-specific CD4+ OT-II T cells. The formation of MHCII–OVA323–337 peptide complexes was determined by assessing T cell stimulation via IL-2 cytokine secretion by ELISA. Cells enriched for two centrosomes displayed a marked increase in CD4+ T cell activation compared with cells with only one centrosome at all OVA concentrations tested (Fig. 10 b). To directly address T cell activation, we measured T cell expansion by proliferation-mediated dilution of the fluorescent dye carboxyfluorescein succinimidyl ester (CFSE; Quah et al., 2007). According to elevated IL-2 levels, T cell proliferation was increased after co-culture of CFSE-labeled OT-II T cells with BMDCs that exhibit extra centrosomes compared with cells with only one centrosome (Fig. 10 c). To address whether increased T cell activation was a consequence of enhanced intracellular processing of antigens, we directly loaded sorted BMDC subpopulations with OVA329-337 peptide to bypass the processing step. Similar to OVA protein, we detected differences in IL-2 secretion and T cell proliferation (Fig. S6 e), suggesting that enhanced T cell activation was not solely a consequence of improved intracellular antigen processing. Similarly, we excluded T cell co-stimulation via CD40, CD70, and CD86 as reason for optimized T cell activation since cell-surface levels were largely indistinguishable on both DC subpopulations (Fig. 10 d).
Taken together, these results demonstrate that DCs with extra centrosomes are more potent in activating antigen-specific CD4+ T cells compared with cells with only a single centrosome.
Cells with extra centrosomes show increased cytokine secretion and T cell attraction
Recent data in macrophages suggest an important role of centrosomes for cytokine production in response to inflammatory stimuli (Vertii et al., 2016). Moreover, the extra centrosome-associated secretory pathway has been identified as a distinct secretory phenotype in cells with extra centrosomes (Arnandis et al., 2018). In DCs, cytokines are stored in endosomes and/or lysosomes and released via the secretory pathway (Blott and Griffiths, 2002; Semino et al., 2005; Verboogen et al., 2019). MTs emanating from centrosomes act as major tracks, which orchestrate long-range intracellular vesicle trafficking of cargos to their destination compartment (Fourriere et al., 2020). As MT numbers were increased in 2N cells with extra centrosomes (Fig. 7 e), we investigated whether cytokine release is altered in cells with distinct centrosome numbers.
To quantify cytokine levels, we collected supernatants of sorted 2N BMDC subpopulations and monitored cytokines by antibody arrays and ELISA, respectively. Both approaches revealed that cytokine levels were increased in supernatants harvested from cells with two centrosomes. In particular, chemokines that attract and activate naive T cells and neutrophils, such as CCL17, CCL5, IL-6, and CXCL1 (Dienz and Rincon, 2009; Fülle et al., 2017; Griffith et al., 2014; Ritzman et al., 2010), were elevated in supernatants collected from 2N2C cells (Fig. 10 e and Fig. S6 f). Except for CCL17, which induces T cell chemotaxis (Imai et al., 1996), mRNA levels of CCL5, IL-6, and CXCL1 were indistinguishable in both 2N DC subpopulations (Fig. 10 f), pointing out that extra centrosomes do not alter the synthesis of these cytokines. To link increased cytokine secretion to enhanced T cell attraction and stimulation, we followed the transmigration of T cells toward conditioned medium harvested from cells with different centrosome numbers. As expected, CD4+ T cells efficiently transmigrated in the presence of CCL19, while under control conditions only a few cells passed the membrane (Fig. 10 g). Of note, we found significantly more CD4+ T cells transmigrating toward the supernatant collected from cells containing two centrosomes, confirming that 2N2C cells are more potent in attracting T cells. In summary, these results demonstrate that DCs with extra centrosomes have a higher capacity for secretion of inflammatory cytokines and T cell attraction, which directly correlates with their ability to activate antigen-specific CD4+ T cells.
Discussion
CA, a state in which more than two centrosomes are present within a cell, is found in virtually all types of human cancers (Chan, 2011) and has been demonstrated to cause chromosomal instability and cell transformation (Basto et al., 2008; Ganem et al., 2009; Levine et al., 2017; Sabino et al., 2015). Moreover, amplified centrosomes have been described to confer advantageous features, such as enhanced invasion, to some tumor cells indicating that multiple centrosomes promote certain aspects of cancer cell motility (Godinho et al., 2014).
Beyond this adverse relationship between amplified centrosomes and cell transformation, multiple centrosomes have long been recognized in mammalian epithelial tissues such as those lining the inner surface of the trachea, the oviduct, and the brain ventricles (Afzelius, 1976; reviewed in Brooks and Wallingford, 2014). In these non-dividing cells, the centrioles migrate to the cell surface where the mother centriole forms a basal body that organizes the generation of motile cilia, which are critically important for mucus clearance, ovum transport, or cerebrospinal fluid circulation. More recently, multiple centrosomes have been identified in cycling progenitors of olfactory sensory neurons suggesting that cells with amplified centrosomes can also be part of physiological development and differentiation programs (Ching and Stearns, 2020).
Here, we provide evidence that multiple centrosomes naturally appear in certain tissues of the immune compartment supporting the concept of a potential physiological role of extra centrosomes during regular cell and tissue homeostasis. We demonstrate that activation of primary DCs with bacterial compounds leads to a robust G1 cell-cycle arrest during which cells acquire two and more centrosomes. Upon antigen encounter, DCs undergo incomplete mitosis and suppress either karyogenesis or cytokinesis leading to accumulation of centrosomes and duplication of DNA content in G1 phase. In contrast to other differentiated cell types such as hepatocytes and megakaryocytes, which can undergo several rounds of such alternative cell division cycles as part of a programmed step during regular development (Fox and Duronio, 2013), DCs arrest in the subsequent G1 phase.
Conventional DCs originate from cDC-committed hematopoietic progenitors in the BM, referred to as the common DC progenitor (Guilliams et al., 2014; Merad et al., 2013). Common DC progenitors give rise to pre-cDCs, which exit the BM via the blood and subsequently populate lymphoid and non-lymphoid tissues (Liu et al., 2009). Under homeostatic conditions, pre-cDCs proliferate, while differentiated cDCs have only a residual proliferative capacity (Cabeza-Cabrerizo et al., 2019). In our studies, we observed that about 20–30% of activated BMDCs as well as dermal DCs carry either two or more centrosomes, while pH3/EdU-negative immature cells mostly contain one centrosome. As immature DCs are a population of asynchronously proliferating cells, we speculate that depending on the initial cell-cycle phase before cells encountered a pathogen, only those cells which passed the restriction point in G1 undergo a modified cell division cycle after activation, resulting in a restricted number of cells which accumulate centrosomes due to suppression of mitosis. In addition to bacterial stimuli, lentiviruses that infect primates have been demonstrated to perturb the cell division cycle of human cells (He et al., 1995). The human immunodeficiency virus type 1 vpr gene induces a robust cell-cycle block leading to a proportion of cells arrested at the G2/M boundary as a consequence of inhibition of the CDK1/cyclin B kinase activity (Planelles et al., 1996). How bacterial infections affect the cell cycle in DCs and whether viral stimuli similarly increase centrosome numbers in DCs still has to be determined.
Besides the accumulation of centrosomes due to incomplete mitosis, our study revealed the presence of two centrosomes in diploid cells. When exposed to microbial stimuli, DCs react with a transcriptional program, which supports antigen processing and presentation, migration, and T cell co-stimulation (Mellman and Steinman, 2001). PLK2 expression levels markedly increased after LPS stimulation, leading to replication of centrioles and overduplication of centrosomes in G1-arrested cells. PLK2 has a well-established role in ensuring the timely coordination between centrosome duplication and cell-cycle progression (Cizmecioglu et al., 2012). Besides its function in centriole replication in proliferating cells, PLKs were recently implicated in TLR signaling thereby regulating inflammatory and anti-viral programs in primary DCs (Chevrier et al., 2011). Pharmacological inhibition of PLKs substantially affects the TLR phosphoproteome, including several known regulators of antiviral pathways, indicating that components that are required for regular centriole duplication promote the host defense against pathogens. The precise mechanism(s) as to how PLKs contribute to downstream TLR signaling are not yet known.
So far, extra centrosomes have been mostly ascribed to pathological processes such as tumorigenesis and metastasis (Chan, 2011; Godinho et al., 2014; Levine et al., 2017). This study extends our knowledge by demonstrating a beneficial effect of multiple centrosomes during directional locomotion along gradients of chemotactic cues. Extra centrosomes in DCs nucleate MT filaments and tightly cluster during migration. Efficient locomotion requires an asymmetric morphology with defined leading and trailing edges. It is well recognized that centrosomes act as a crucial regulator of cell polarity. While actin-rich lamellipodia are formed at the cell front and constantly explore the cells’ surrounding, the MTOC reorients to the rear and generally follows the turning leading edge (Ueda et al., 1997; Xu et al., 2005). During DC chemotaxis, increased MT dynamics at the uropod generate local contraction forces that pull the cell body forward thereby maintaining the orientation of migration (Kopf et al., 2020). In line with these findings, we found that cells with extra centrosomes are stably polarized and show increased persistent locomotion toward chemotactic cues. Enhanced persistent movement results in a larger net displacement of cells allowing them to travel longer distances in shorter time periods. Simulating DC migration in the presence of extra centrosomes highlights the importance of persistence for the overall efficiency of eukaryotic cells to enter the lymphatic vasculature. Similar to DCs, amplified centrosomes in cancer cells have been proposed to facilitate directional movement by enhancing focused trafficking of Golgi vesicles containing migration-promoting factors to the leading edge of the cell (Ogden et al., 2013).
In endothelial cells (ECs) of tumor blood vessels, extra centrosomes were found to be scattered throughout the cytoplasm when cultured in 2D environments and showed fluctuating distances relative to each other (Kushner et al., 2014). Laser ablation of extra centrosomes in tumor ECs partially rescued centrosome organization and led to improved EC migration, suggesting that clustering of extra centrosomes is a prerequisite for directional 2D locomotion. In line with these findings, we found that in DCs all centrosomes tightly clustered during 2D locomotion along gradients of soluble chemokine. A similar phenomenon of centrosomal clustering has been described in mitotic cancer cells to allow formation of a bipolar spindle and separation of DNA content into two daughter cells (Quintyne et al., 2005). The molecules that promote centrosomal clustering have been of general interest as they may offer novel routes for anti-cancer therapies (Castiel et al., 2011; Kwon et al., 2008; Pannu et al., 2014). Centrosome declustering in mitotic cells leads to chromosome segregation errors as a consequence of multipolar spindle formation and subsequent cell death, thereby suppressing tumor cell growth (Raab et al., 2012). Yet, effects of declustering agents on interphase-specific processes in cells that naturally possess extra centrosomes have not been examined. They require further investigations to estimate whether centrosome clustering is a prerequisite for immune cell migration and to determine potential side effects of declustering agents on immune cell trafficking.
In addition to the positive effects on cell locomotion, we found enhanced secretion of T cell–stimulating cytokines of cells containing extra centrosomes. The secretion of cytokines and chemokines from innate immune cells serves as a link to communicate with the adaptive immune system and orchestrate the expansion of B and T cells. Recent studies suggest a crucial role of the centrosome for cytokine release in immune and non-immune cells. Centrosome maturation in interphase was reported in macrophages after treatment of cells with proinflammatory stimuli (Vertii et al., 2016). LPS stimulation induced PCM recruitment at the centrosome, which is accompanied by increased centrosomal MT nucleation. Pharmacological disruption of centrosomes did not alter IL-6 synthesis but impaired IL-6 secretion, suggesting that centrosome integrity per se is required for cytokine secretion. In support of these data, our study demonstrates that enrichment of cells with extra centrosomes strongly correlates with their ability to secrete inflammatory cytokines and the capacity for T cell activation. In most tissues, DCs reside in an immature state, unable to activate T cells. However, they are well equipped to capture antigens, which triggers full maturation and antigen presentation via MHC complexes. Mature DCs upregulate MHCII molecules, which bind to peptides that are derived from proteins processed in the endocytic pathway (Neefjes et al., 2011). Several studies highlight the importance of the MT cytoskeleton as scaffold for delivering T cell stimulatory molecules toward the IS (Pulecio et al., 2010; Wubbolts, 1996; Wubbolts et al., 1999). Upon specific DC–T cell contact, tubular endosomes extend in DCs and polarize toward the area of contact. 3D reconstruction of DC–T cell conjugates revealed the presence of MHCII-positive tubules associated with the MT network, which was abolished after MT depolymerization with nocodazole (Boes et al., 2002; Vyas et al., 2007). Although we observe a correlation between increased MT numbers in cells with extra centrosomes and enhanced cytokine secretion, direct evidence of such interdependence remains to be determined. Interestingly, artificial induction of centrosome amplification in B lymphoma cells has been shown to increase the antigen presentation capacity of cells, indicating that multiple MTOCs can also modify stimulatory properties of B cells (Yuseff et al., 2011).
Overall, our study demonstrates that extra centrosomes in terminally differentiated immune cells can serve as a strategy to enhance centrosome and MT-associated immune processes. They challenge the current paradigm of cells having precisely one centrosome in interphase and support a model in which multiple centrosomes contribute to regular cell and tissue homeostasis.
Materials and methods
Mice
All mice used in this study were bred on a C57BL/6J background and maintained at the institutional animal facility in accordance with the German law for animal experimentation. Permission for all experimental procedures involving animals was granted and approved by the local authorities (Landesamt für Natur, Umwelt und Verbraucherschutz North Rhine-Westphalia [under AZ81-02.05.40.19.022]). CETN2-GFP mice were purchased from Jackson. OVA-specific OT-II mice were a gift from Sven Burgdorf, Life and Medical Sciences Institute, Cellular Immunology, University of Bonn, Bonn, Germany.
DC culture
Cultures were started from freshly isolated bone marrow of 8–12-wk-old mice with C57BL/6J background (WT, CETN2-GFP). DC differentiation was induced by plating 2 × 106 cells in 10 ml complete medium (RPMI 1640 supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM ß-mercaptoethanol; all purchased from Thermo Fisher Scientific) containing 10% GM-CSF (supernatant from hybridoma culture). Cells were fed on days 3 and 6 with complete medium supplemented with 20% GM-CSF. To induce maturation, cells were stimulated overnight with 200 ng/ml LPS from E.coli 0127:B8 (Sigma-Aldrich) and used for experiments on days 6 and 7 (immature DCs) and days 8 and 9 (mature DCs). For Centrinone experiments, cells were treated with 250 or 500 nM Centrinone (Tocris), which was added directly to the culture medium on day 6 or together with LPS to induce maturation overnight. BM from FUCCI mice was kindly provided by Andreas Villunger, Institute for Developmental Immunology, Biocenter, Medical University of Innsbruck, Innsbruck, Austria.
Flow cytometry
Before staining, 1–2 × 106 cells were incubated for 15 min at 4°C with blocking buffer (1xPBS, 1% BSA, 2 mM EDTA) containing 5 mg/ml anti-CD16/CD32 antibody (2.4G2; BD Biosciences). For cell surface staining, cells were incubated for 30 min at 4°C with conjugated monoclonal antibodies diluted 1:500 (if not stated otherwise) in blocking buffer. For intranuclear staining, eBioscience Foxp3/Transcription Factor Staining Buffer Set was used according to the manufacturer’s protocol. The following antibodies were used: mouse anti-mouse CCR7-PE (4B12), rat anti-mouse I-A/I-E-eFluor450 (M5/114.15.2, 1:1,000), hamster anti-mouse CD11c-APC (N418), rat anti-mouse CD45-Brilliant Violet (30-F11), rat anti-mouse CD3 APC-Cy7 (17A2), rat anti-mouse CD19 APC-Cy7 (6D5), rat anti-mouse TER-119/Erythroid cells APC-Cy7 (TER-119), rat anti-mouse CD49b APC-Cy7 (DX5), rat anti-mouse Ly-6G APC-Cy7 (1A8), anti-mouse CD64 PE-Cy7 (X54-5/7.1), anti-mouse Ly-6C PE-Cy7 (HK1.4), rat anti-mouse F4/80 BV605 (BM8), anti-mouse/human CD45R/B220 Alexa Flour 700 (RA3-6B2), rat anti-mouse I-A/I-E PE/Dazzle 594 (M5/114.15.2), Armenian hamster anti-mouse CD11c BV711 (N418), rat anti-mouse CD8α PE (53-6.7), anti-mouse/human CD11b APC (M1/70), rat anti-mouse CD8α APC (53-6.7), Armenian hamster anti-mouse CD11c BV421 (N418), rat anti-mouse CD19 BV421 (6D5), anti-mouse Ly6G eFlour450 (RB6-8C5), rat anti-mouse/human CD11b BV421 (M1/70), rat anti-mouse I-A/I-E APC-Cy7 (M5/114.15.2), rat anti-mouse CD19 APC-Cy7 (HIB19), rat anti-mouse CD4 Pacific Blue (RM4-5), rat anti-mouse CD3 Pe-Cy7 (17A2), anti-mouse CD86 PE (GL1), rat anti-mouse CD40 PE/Dazzle594 (3/2.3), Armenian hamster anti-mouse CD80 PE-Cy7 (16-10A1), rat anti-mouse CD70 APC (FR70), Armenian hamster anti-mouse-CD11c BV605 (N418), anti-mouse CD135 PE (A2F10), rat anti-mouse CD115 APC (AFS98), rat anti-mouse F4/80 APC-Cy7 (BM8), rat anti-mouse/human Ki-67 BV421 (11F6), mouse anti-Histone H3 Phospho(Ser10) Alexa Flour 647 (11D8), rat anti-human CD4 Pe-Cy7, rat anti-mouse CD8a FITC (53-6.7), rat anti-mouse CD25 PE (PC61), Armenian hamster anti-mouse CD69 APC (H1-2F3), rat anti-mouse/humanCD44 PE-Cy7 (IM7), rat-anti mouse CD45 PerCP/Cy5.5 (I3/2.3, 1:200), rat anti-mouse CD326 (Ep-CAM, 1:200) BV711 (G8.8), and rat anti-mouse F4/80 PE/Dazzle 594 (BM8, 1:200).
For quantification of DNA content, cells were incubated for 30 min at 37°C with Vybrant DyeCycle Violet Stain (1:1,000; Invitrogen), according to the manufacturer’s instructions, or propidium iodide (1:100; Invitrogen) after intranuclear fixation. For live dead staining, the cells were incubated for 15 min at RT with the Live/Dead Fixable Dead Cell Stain Kit (1:1,000; Invitrogen) or DRAQ7 (1:1,000; Biolegend) in 1xPBS. Flow cytometry was performed on an LSR flow cytometer (BD Biosciences) and ARIAIII Sorter (BD Bioscience). Data analysis was carried out using FlowJo X 10.0.7r2.
Isolation of dermal DCs from skin explants
Ears of 8–12-wk-old mice were cut off with a scissor and separated into dorsal and ventral ear sheets using forceps. Skin explant culture represents an inflammatory setting, and mechanical rupture of the ear is sufficient to induce maturation of skin DC. Ventral ear sheets were placed in 48-well plates and floated upside down on complete medium supplemented with 1 μl CCL19 (R&D; 25 μg/ml) for 3 d to allow emigration of dermal DCs into the culture medium. Cells were fed with 500 μl complete medium on days 1 and 2. On day 3, only cells in suspension, but not adherent cells, were harvested and used for further experiments.
Isolation of splenic DCs and LH cells
To assess centrosome numbers in splenic DCs, spleens from 8–10-wk-old CETN2-GFP expressing mice were isolated, cut into pieces, and treated for 30 min at 37°C with digestion buffer (HBSS containing Ca2+, Mg2+; 0.1 mg/ml deoxyribonuclease I; and 0.05 mg/ml collagenase from Clostridium histolyticum; all purchased from Sigma-Aldrich). Samples were further mechanically crushed, and erythrocyte lysis was performed using ACK Lysing Buffer (Invitrogen). Cells were stained for DC cell-surface markers and sorted using an ARIAIII Sorter (BD Biosciences). Sorted DC subpopulations (MHCII+/CD11c+/CD8+ [cDC1] and MHCII+/CD11c+/CD11b+ [cDC2]) were incubated in 10 μM EdU in complete medium for 60 min. Afterward, cells were immobilized and fixed. EdU was detected using the Click-iT EdU Imaging Kit (Invitrogen) according to the manufacturer’s protocol. For analyzing centrosome numbers only EdU− cells were considered.
LH cells were prepared from epidermal ear sheets and sorted as previously described (Liu et al., 2020). Briefly, ears were split into two skin halves and treated with Dispase II (Gibco) at 4°C overnight. Epidermis separated from dermis was digested with collagenase IV and DNase I (both Sigma-Aldrich) at 37°C for 1 h. Cell suspension prepared from digested epidermis was labeled with a fixable viability dye and stained against cell surface markers. LH cells were sorted as singlet live CD45+Epcam+F4/80+ cells.
Immunofluorescence
For fixation experiments, round-shaped coverslips were placed in 24-well plates and coated with 1:1 (vol:vol) mixed ICAM-1 (Thermo Fisher Scientific; 100 μg/ml) and CCL21 (R&D; 25 μg/ml) for 10 min at RT. Cells were incubated on coated coverslips for 5 min at 37°C and subsequently fixed with 3% PFA diluted in 1xPBS. Migrating cells were fixed by adding 4% PFA directly on top of the agarose. After fixation, the agarose pad was carefully removed using a coverslip-tweezer. Cells were permeabilized with 0.5% Triton X-100 in 1xPBS for 20 min and washed 3 × 10 min with 1xPBS. Samples were blocked to prevent unspecific binding by incubating for 60 min in blocking solution (5% BSA in 1xPBS). Primary antibodies were diluted in blocking solution and incubated either for 30 min at RT (CEP135) or overnight at 4°C. The following antibodies were used: rat anti-α-tubulin (YL1/2; 1:500; AbD Serotec), rabbit anti-phospho-H3 (pH3; D2C8; 1:600; Cell Signaling), mouse anti-γ-tubulin (GTU-88; 1:500; Sigma-Aldrich), mouse anti-ac-tubulin (6-11B-1; 1:500; Sigma-Aldrich) and rabbit anti-CEP170 (Novus Biology; 1:400), rabbit anti-CEP135 (Abcam; 1:600), rabbit anti-CDK5RAP2 (Millipore; 1:600), rabbit anti-cleaved caspase-3 (1:400; all Cell Signaling), and rabbit anti-p21 (EPR18021; 1:100; Abcam).
Coverslips were washed 3 × 10 min with 1xPBS and incubated with secondary antibodies for 60 min at RT in the dark. The following secondaries were used: Donkey Anti-Mouse Alexa Fluor 488 AffiniPure F(ab')2 Fragment IgG (H+L), Donkey Anti-Mouse Alexa Fluor 647 AffiniPure F(ab')2 Fragment IgG (H+L), Donkey Anti-Mouse Cy3 AffiniPure F(ab')2 Fragment IgG (H+L), Donkey Anti-Rat Cy3 AffiniPure IgG (H+L), Goat Anti-Rabbit Cy3 AffiniPure IgG (H+L; all Jackson ImmunoResearch; all diluted 1:400 in blocking solution), and Goat Anti-Rabbit IgG (H+L) Alexa Flour 488 (Invitrogen). After incubation, cells were washed 3 × 10 min with 1xPBS. Samples were conserved in non-hardening mounting medium with DAPI (Invitrogen) and stored at 4°C in the dark.
Centrosome and lymphatic vessel labeling in skin explants
Ears of 4–6-wk-old mice were cut off with a scissor and separated into dorsal and ventral ear sheets using forceps. Ears were fixed with 4% PFA overnight and permeabilized with 0.5% Triton X-100 in 1xPBS for 30 min. After washing 3 × 10 min with 1xPBS, samples were blocked by incubating for 60 min in blocking solution (1% BSA in 1xPBS). Centrosome staining was carried out as described in Immunofluorescence. Lymphatic vessels were stained against Lyve-1 (rat anti-mouse Lyve-1 [ALY7; 1:200] and dermal DCs against MHCII [rat anti-mouse I-A/I-E-bio (M5/114.15.2; 1:300]). Ears were conserved in non-hardening mounting medium with or without DAPI (Invitrogen) and stored at 4°C in the dark.
Correlative light and electron microscopy
Cells were grown on gridded sapphire discs coated with poly-L-lysine for 2 h. Samples were immediately frozen on a Leica EM HPM100 high-pressure freezing machine. Freeze substitution was done as recently described (Müller et al., 2021) with some modifications. Briefly, samples were substituted in acetone containing 2% osmium tetroxide, 1% uranyl acetate, 0.5% glutaraldehyde, 5% water, and 1% methanol at −90°C for 46 h. Afterward, the temperature was raised to 0°C over 15 h. At this temperature, the substitution medium was changed to pure acetone and the temperature was increased to 22°C in four 15-min steps. Between steps, the acetone was exchanged for the new one at the same temperature. Afterward, samples were incubated in 0.2% thiocarbohydrazide in 80% methanol at RT for 1 h and washed four times for 15 min in acetone, followed by 2% osmium tetroxide for 1 h, brief washing in acetone, 1% uranyl acetate in 10% methanol for another hour, and washing for 4 × 15 min in acetone. Then the samples were embedded into resin with increasing concentrations (Araldite 502/Embed 812; 25, 50, 75, and 100%), 1 h each step, and left overnight in the freshly made 100% resin. After 48 h of polymerization at 60°C, the sapphire discs were removed and samples were coated with 25 nm of the platinum and imaged by FIB-SEM (FEI Helios NanoLab 660 G3 UC).
Segmentation of centrioles from FIB-SEM data
Centrioles were segmented by image thresholding and converted to masks in Fiji. Segmentation masks were exported to Microscopy Image Browser and thresholded using black-white thresholding. Parameters were set to match the voxel size of the FIB-SEM data, and the models were exported as .am files for 3D rendering in Amira for images in Fig. 1 and Fig. S1 and as .mod files for 3Dmod, where the model was rendered and Video 1 created.
EdU incorporation assay
Cells were incubated in 10 μM EdU in complete medium for 60 min. EdU was detected using the Click-iT EdU Imaging Kit (Invitrogen) according to the manufacturer’s protocol.
Immunoblotting
For the analysis of cell-cycle parameters and PLK2-knockout control, equal amounts of cells were lysed in radioimmunoprecipitation assay buffer (Cell Signaling) mixed with protease and phosphatase inhibitor cocktail (Roche) according to the manufacturer’s instruction. After adding Laemmli sample buffer containing 10% β-mercaptoethanol, lysates were boiled for 5 min at 95°C. Equal volumes were loaded on precast gradient gels (4–20% Mini-PROTEAN TGX Stain-Free Protein Gels; Bio-Rad), and proteins were separated by SDS-PAGE. Using Trans-Blot Turbo Transfer System (Bio-Rad), proteins were transferred to 0.2-µm nitrocellulose membranes (Bio-Rad) in a semi-dry manner. Following staining with Ponceau S and subsequent destaining, membranes were blocked with 5% non-fat dry milk (Bio-Rad) or 5% BSA (Sigma-Aldrich) in 0.1% TBS-Tween for 1 h at RT. The following primary antibodies were used: monoclonal mouse anti-GAPDH (GA1R, 1:4.000; Abcam), monoclonal mouse anti-vinculin (hVIN-1, 1:40,000; Sigma-Aldrich), monoclonal rabbit anti-cyclin A2 (EPR17351, 1:2,000; Abcam), monoclonal rabbit anti-cyclin B1 (EPR17060, 1:1,000; Abcam), monoclonal rabbit anti-cyclin E1 (D7T3U, 1:1,000; Cell Signaling), monoclonal rabbit anti-p21 (EPR18021, 1:1,000; Abcam), polyclonal rabbit anti-p27 (1:1,000, Cell Signaling), and polyclonal rabbit anti-PLK2 (1:500; Abcam). For the detection of primary antibodies, HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG antibodies were used (1:10.000, Bio-Rad). Proteins were visualized by enhanced chemiluminescence (Pierce ECL Plus Substrate; Thermo Fisher Scientific) detection using ChemiDoc MP Imaging System (Bio-Rad) or/and x-ray films (Amersham Hyperfilm ECL; GE Healthcare).
Analysis of cell-cycle distribution
BMDCs were generated from FUCCI mice and immobilized on CCL21/ICAM-coated coverslips on days 6–9 (immature) and after LPS stimulation overnight on day 9 (mature d9). Cells were fixed with 3% PFA and conserved in non-hardening mounting medium with DAPI (Invitrogen). For analysis of cell-cycle distribution after LPS activation, cells were treated with 200 ng/ml LPS, and samples were collected 0, 1, 2, 4, 6, 8,10, and 25 h after LPS treatment.
Cells were imaged with a confocal microscope (LSM880; Zeiss), equipped with an Airyscan module, a Plan-Apochromat 63×/1.4 oil differential interference contrast (DIC) objective, 488, 561, and 633 laser lines and a photomultiplier tube (all Zeiss). For FACS analysis cells were fixed with 70% EtOH and stained against CD11c, MHCII, and Hoechst (1:1,000; Invitrogen).
Generation of immortalized hematopoietic progenitor reporter cell lines
Hematopoietic progenitor cell lines were generated by retroviral delivery of an estrogen-regulated form of Hoxb8 as described recently (Leithner et al., 2018; Redecke et al., 2013). Briefly, BM of 8–12-wk-old CETN2-GFP expressing mice was isolated and retrovirally transduced with an estrogen-regulated form of the HOXB8 transcription factor. Cells were cultured in estradiol-containing medium. After about 10 d, cells were frozen in liquid nitrogen or differentiated into DCs by washing out estradiol and growing in estradiol-free medium.
CRISPR knockout generation
To generate specific knockouts of immortalized hematopoietic progenitor reporter cell lines, a lentiviral CRISPR/Cas9 approach was applied. Single guide RNAs (PLK2sg1a_fw: 5′CACCGGATTATAGTCGACCCCACGA-3′; PLK2sg1b_rv: 5′AAACTCGTGGGGTCGACTATAATCC-3′; scramble_fw: 5′ CACCGGCCGTGGCGCATGGGTAGCA-3′; and scramble_rv: 5′ AAACTGCTACCCATGCGCCACGGC-3′) were cloned into the lentiCRISPR v2 plasmid (52961; Addgene) as described recently (Sanjana et al., 2014; Shalem et al., 2014). 5.4 μg of the cloned plasmid was co-transfected together with 2.7 μg envelope plasmid pCMV-VSV-G (8454; Addgene) and 4 μg packaging plasmid pCMV-dR8.2 dvpr (8455; Addgene) into 6 × 106 Lenti-X 293T HEK cells in DMEM using Lipofectamine2000 and Opti-MEM medium (all materials purchased by Gibco; Invitrogen). Transfection mix was aspirated after 6 h, and 10 ml of DMEM was added. 48 h after transfection, cells were fed with 10 ml DMEM medium. 96 h after transfection, supernatants containing respective lentiviruses were harvested. 3 × 105 immortalized hematopoietic precursor cells were spin-infected with lentivirus and 10 mg/ml polybrene beads for 60 min at 1,500 g. 72 h after infection, cells were washed to remove the remaining viruses, and a selection medium containing 3 μg/ml puromycin-dihydrochloride was added.
In vitro 3D collagen migration assay
For 3D in vitro migration, 2 × 105 dermal DCs were suspended in a medium-collagen I mixture (PureCol bovine collagen [INAMED]) in 1× MEM (Invitrogen) and 0.4% sodium bicarbonate (Sigma-Aldrich) at a volume ratio of 1:2 yielding a final collagen concentration of 1.73 mg/ml. Collagen gel mixtures were cast into custom-made migration chambers as previously described (Kopf et al., 2020) and incubated for 45 min at 37°C to allow polymerization of the gel. CCL19 was suspended in full medium to a final concentration of 0.33 μM and placed on top of the gel. To prevent drying-out of the gels, migration chambers were sealed with Paraplast X-tra (Sigma-Aldrich). Gels that failed to polymerize were excluded from the analysis.
Image acquisition was performed with a Nikon Eclipse widefield microscope and a C-Apochromat 20×/0.5 PH1 air objective. Images were acquired in 120-s intervals for 5 h at 37°C, 5% CO2. Cells were tracked manually, using the “Manual tracking Plug-in” for ImageJ. The ImageJ Chemotaxis tool was used to determine average (frame-to-frame) speed and persistence (distance in gradient direction/total distance).
In vitro under-agarose migration assay
For 2D migration assays, 4% Ultra-Pure Agarose (Invitrogen) was diluted in nuclease-free water (Gibco) and mixed with a combination of phenol red-free RPMI1640 Medium (Gibco) supplemented with 20% FBS and 1% penicillin 100 U/ml/streptomycin 100 μg/ml (Invitrogen) and 1× HBSS, pH 7.2, in a ratio of 1:4. Ascorbic acid was added to a final concentration of 50 μM to catch free radicals during fluorescent imaging. 500 µl of agarose-mix was cast into custom-made migration chambers (glass-bottom dishes [MatTek] with a 1-cm plastic ring glued with Paraplast X-tra [Sigma-Aldrich] into the middle of the dish). After polymerization, a 2-mm hole was punched into the agarose pad and 2.5 μg/ml CCL19 (R&D Systems) was placed into the hole to generate a soluble chemokine gradient. Outer parts of the dish were filled with water and incubated for 45–60 min at 37°C, 5% CO2 to equilibrate the agarose. The cell suspension was injected under agarose opposite of the chemokine hole. Prior to acquisition, dishes were incubated at least 2 h at 37°C, 5% CO2 to allow recovery and persistent migration of cells. Dishes with failed confinement, assessed by cell morphology, were excluded from analysis. For fixation experiments, 4% PFA solution was placed on top of the agarose pad and incubated overnight at 4°C. Agarose was removed carefully using a forceps and cells were washed three times with 1xPBS before immunostaining.
Microscopy
Confocal microscopy of fixed samples was performed on a motorized stage at RT with an inverted microscope equipped with an Airyscan module; a Plan-Apochromat 63×/1.4 oil DIC objective; 488, 561, and 633 laser lines; and a photomultiplier tube (all Zeiss). For quantification of centrosome numbers, cells were immobilized on ICAM/CCL21-coated coverslips or injected under agarose. 200-nm sections were acquired and maximum intensity Z-stack projections were obtained from all stacks. Due to the flat nature of the cells under these experimental conditions, images were taken as Z-stacks in the range of 4–8 µm. Centrioles were identified using either ac-tubulin/CETN2-GFP and γ-tubulin staining or ac-tubulin/CETN2-GFP and CEP135 staining. Only structures where two different markers co-localized were considered as centriole. Centriole numbers were counted manually on 2D projections. 3D reconstructions of Z-stacks were generated exemplary to validate centriole numbers in 3D and led to the same results as in 2D due to the flat nature of DCs under agarose as well as on ICAM/CCL21-coated coverslips.
To analyze MT filament numbers, images were acquired using the Airy module and posttreated by deconvolution. Maximum intensity Z-stack projections were generated and MT filaments emanating from centrosomes were counted manually. 2N and 4N cells were distinguished by measuring nuclear areas after DAPI staining. The same setup was used to determine migration parameters of DCs with different numbers of centrosomes. During live-cell acquisition of CETN2-GFP expressing dermal DCs and BMDCs, dishes were held at 37°C in a humid chamber. Migrating cells under agarose were imaged in 2-s intervals for 10 min. 2N and 4N cells were distinguished by measuring nuclear areas in CETN2-GFP expressing cells migrating under agarose. Nuclear areas were identified by the absence of CETN2-GFP signal in live migrating cells. For all experiments, imaging software ZEN Black 2.3 SP1 was deployed. Image processing and data analysis were performed using ImageJ. For tracking migrating cells, each cell was tracked manually using ImageJ’s Manual Tracking Plugin. Cell velocity and directional persistence were quantified using the “Chemotaxis and Migration Tool.” Cells with contact with other cells were excluded from the analysis.
For actin visualization, mature CETN2-GFP expressing BMDCs were used and co-stained against γ-tubulin and DAPI. After permeabilization and blocking (for details see section Immunofluorescence), cells were treated with Alexa Fluor 546 Phalloidin (Thermo Fisher Scientific/Invitrogen diluted in blocking solution) overnight at 4°C. Samples were conserved in non-hardening mounting medium with DAPI (Invitrogen). Images were acquired using the Airy module and posttreated by deconvolution. For quantifying integrated actin fluorescence intensity, images were analyzed in ImageJ using a defined area that marked centrosomal actin of every cell and normalized to a second area of non-centrosomal actin. The quotient of centrosomal actin signal/non-centrosomal actin signal was built for every cell.
For measuring inter- and intracentrosomal distances, migrating cells were imaged with an inverted wide-field Nikon Eclipse Ti-2E microscope in a humidified and heated chamber at 37°C and 5% CO2 (Ibidi Gas Mixer), equipped with a Plan-Apochromat 40×/0.95 air objective, a DS-Qi2 camera, and a Lumencor Spectra X light source (390, 475, 542/575 nm; Lumencor). For analysis of intracentrosomal distances in cells with one centrosome, centrioles were segmented based on the pixel classification workflow of ilastik v1.3.3 (https://www.ilastik.org/about.html). For each time point, the centriole locations were found by segmenting the centriole-probability output, performing particle size filtering, and calculating the center of mass. The centrioles were then tracked over time by solving the linear assignment problem using the Hungarian algorithm (Munkres), which can deal with gaps in the detection. Completed tracks were filtered by minimum track length and track duration. Pairs of centrioles were automatically identified by imposing the condition that they move mostly in parallel (due to cell migration) and are in close proximity during the entire length of the track. Cells with extra centrosomes were then filtered out by calculating the distance to the closest third centriole. The output of this automated analysis was validated by comparison with manual tracking. For analysis of intra- and intercentrosomal distances of extra centrosomes, cells were identified and their centrioles were tracked manually in ImageJ using the Manual Tracking plugin. Tracks were exported and the intercentrosomal distance, defined as the distance between the centers of pairs of centrioles, was calculated.
Mitotic imaging
To follow mitotic progression of BMDCs, immature BMDCs (day 6 or 7) were labeled with NucBlue (Invitrogen; 1 drop per 20 ml cell suspension) 30 min prior to imaging and injected under agarose to avoid cells that float away during the imaging period. Mitotic cells were imaged with an inverted wide-field Nikon Eclipse Ti-2E microscope in a humidified and heated chamber at 37°C and 5% CO2 (Ibidi Gas Mixer), equipped with a Plan-Apochromat 40×/0.95 air objective, a DS-Qi2 camera, and a Lumencor Spectra X light source (390, 475, 542/575 nm; Lumencor). For DC activation, cells were treated with LPS 15–30 min prior to imaging. Frames were collected every 15 s for 2–3 h.
Laser ablation of centrosomes
Laser ablation experiments were performed on a spinning-disc confocal system (Yokogawa CSU-X1, iXon897, Andor) installed on an inverted Axio observer microscope (Zeiss) using a C-Apochromat 63×/1.2 W Korr UV-VIS-IR objective (Zeiss) and a 50-mW 488-nm laser (LP emission filter). Centrosome ablation was carried out during 2D under agarose migration using a 355-nm pulsed laser (pulse length: 350 ps, intensity: 1 [≙10.8 µW during shot]; pulses/shot: 5; pulse rate: 1 kHz; shots/µm2: 1) after defining a region of interest (ROI) drawn around the centrosome. The same ROI was used for all ablations. Control cells received non-centrosomal laser shots into the cytoplasm in close proximity to the centrosome. Migrating cells were imaged at 5-s intervals for at least 4 min prior to centrosome or control ablation and 4 min after the ablation process. Migration velocity and persistence represent mean values over the time of imaging before and after ablation. To test for efficient centriole depletion, Z-stacks were recorded before and after the ablation process. To identify ablated cells after immunostaining for MT filaments, gridded glass coverslips (grid repeat distance: 50 μm, Ibidi) have been used to relocate ablated cells. During the experiment, cells were kept at 37°C in a chamber with 80% humidity and 5% CO2 generated by a gas incubation system (Ibidi). Images were obtained with Andor software. Cells were tracked manually using ImageJ and the Manual Tracking plugin. Cell velocity and persistence were quantified using the Chemotaxis and Migration Tool. MTs were traced manually using the “NeuroJ” Plugin, and straightness was defined as end-to-end distance divided by the total length of the MT.
msd analysis
Distance analysis of lymph vessels in split ear sheets
Maximum intensity Z-stack projections of confocal images from ear dermis stained with Lyve-1 were used to create binary images using Fiji. Binary images were analyzed using custom-made scripts in Matlab (Mathworks 2019). In brief, connected pixel areas in the binary image were detected, and the corresponding midpoint was calculated. The midpoint was used to calculate maximum distances (diameters) to the next lymph vessel in four directions (horizontal, vertical, +45°, −45°).
Simulation of persistent random walks
Monte Carlo Simulations of persistent random walks were done using custom-made scripts in Matlab (Mathworks 2019). To create persistence random walks, the current position (xi/yi) and last track position (xi−1/yi−1) were used to calculate actual directions Ki. From actual directions, we allowed for normal distributed directional changes. Depending on the SD of the normal distributed directional change and the set step size—in accordance with cell speed (µm/min) and (min)—a persistent random trajectory was created. The msd to time plots of persistent random walks were analyzed according to Fürths formula (see above) to read out the persistence time .
Simulation of persistent random walks in square lattices to calculate efficiency times
Monte Carlo Simulations of persistent random walks with various were used to calculate efficiency times. We defined efficiency time as the time when more than 99% of the random walkers have already reached the border of the square. We set >1e4 persistent random walkers at random start positions inside squares with sizes of 200, 300, and 400 µm.
Sorting of DC subpopulations
Mature CETN2-GFP expressing BMDCs were harvested, counted, and stained for live cells, DNA content, and cell-surface markers as described above. Samples were sorted using the ARIAIII Sorter (BD Bioscience). Mature CETN2-GFP expressing BMDCs (MHCII+/CD11c+) were gated on DNA content to get rid of 4N cells. CETN2-GFP signal distribution of 2N cells was separated into CETN2-GFPlow and CETN2-GFPhigh expressing cells. For determining the efficiency of separation, sorted DC subpopulations were immobilized, fixed, and centrosome numbers were assessed by confocal microscopy. The absolute numbers of cells with ≥2 centrosomes were determined in sorted CETN2-GFPlow and CETN2-GFPhigh expressing cells, and the ratio between these numbers was built to evaluate the efficiency of enrichment. CETN2-GFPlow expressing cells were referred to as 2N1C, and CETN2-GFPhigh expressing cells as 2N2C. For all experiments, the ratio was at least 1.5 and ranged from 8 to 42% for the 2N1C population and 21–70% for the 2N2C population.
Mixed lymphocyte reactions and IL-2 ELISA
104 cells from sorted BMDC subpopulations (2N1C, 2N2C) were seeded in 100-μl full medium into 96-well U-bottom plates and kept at 37°C, 5% CO2. 30 min after seeding, OVA (1,000; 500; 100 and 10 μg/ml) or ova-peptide (OVA323-339 specific for CD4 T cell responses: 1; 0.1 and 0.01 μg/ml) was added in 100 μl full medium and incubated for 2 h at 37°C, 5% CO2. In the meantime, splenocytes were isolated and pooled from two to three OT-II+/+ mice. Cells were processed through a 70- and 40-μm cell strainer, and erythrocyte lysis was performed using ACK Lysing Buffer (Invitrogen). After antigen loading, supernatants were discarded and 5 × 104 splenocytes were added to 200 μl full medium. 24 h after co-culture, supernatants were harvested and IL-2 levels were determined using the Quantakine ELISA Kit (R&D Systems) according to the manufacturer’s instructions. Colorimetric measurements were carried out on Infinite M200 spectrophotometer (Tecan).
T cell proliferation
104 cells from sorted DC subpopulations (2N1C, 2N2C) were seeded in 100-μl full medium in 96-well U-bottom plates and kept at 37°C, 5% CO2. 30 min after seeding, OVA (1,000; 500; 100 and 10 μg/ml) or ova-peptide (OVA323-339 specific for CD4 T cell responses: 1; 0.1 and 0.01 μg/ml) was added in 100 μl full medium. After 2 h, supernatants were discarded and 5 × 104 CFSE-labeled splenocytes were added in 200 μl full medium. CFSE labeling was performed using 2 × 106 cells/ml with a concentration of 0.5 μM Celltrace CFSE (Invitrogen) in PBS for 7 min at 37°C. 62 h after co-culture, samples were stained (splenocytes from OT-II+/+ mice: CD11c−, CD19−, live cells, CD3+, CD4+), and flow cytometric analysis was performed on an LSRII flow cytometer (BD Biosciences). Data analysis was carried out using FlowJo X 10.0.7r2.
Cytokine array and ELISA
For secretome analysis, 1 × 106 sorted DC subpopulations (2N1C, 2N2C) were seeded into a 6-well plate in 3 ml full medium. After 16 h, 500 μl supernatant was harvested and incubated with the mouse cytokine antibody array (Panel A, R&D Systems) according to the manufacturer’s instructions. Chemiluminescence was acquired using a ChemiDoc Imaging System (BioRad). Data analysis was carried out with Image Lab 6.1 Software (BioRad).
For quantification of cytokine levels via ELISA, 0.8 × 106 sorted DC subpopulations were seeded into a 6-well plate in 3 ml full medium. After 16 h, supernatants were harvested and incubated with the respective mouse ELISA Kit (CCL17, CCL5, IL-6, CXCL1; all Invitrogen) according to the manufacturer’s instructions.
mRNA expression levels
For mRNA quantification of 2N1C and 2N2C, 1 × 106 sorted cells were harvested in 350 μl Lysis Buffer (RNeasy Lysis Buffer + 1% ß-Mercaptoethanol), and RNA isolation was carried out using the RNeasy Mini Kit (all products purchased from Qiagen). Gene expression was assessed using the TaqMan RNA-to-CT 1-Step Kit (Thermo Fisher Scientific) with a reaction volume of 20 μl containing 250 ng RNA template and 1 μl of Taq Man Gene Expression Assay (Thermo Fisher Scientific; triplicates performed). Samples were run on a CFX96 Real-Time System (BioRad) according to the manufacturer’s instructions. Data were normalized according to the expression of a housekeeping gene in DCs (TATA-binding protein). Analysis of relative gene expression was carried out using the CFX Manager Software Version 3.1 (BioRad).
Transmigration assay of CD4+ T cells
For the isolation of naive CD4 T cells, spleens and lymph nodes from C57BL/6J mice were homogenized and washed. Enrichment was achieved by negative selection with magnetic beads using the naive CD4+ T Cell Isolation Kit (stem cell) according to the manufacturer’s instruction. Naive CD4+ T cells were activated for 3 d in full medium (RPMI 1640 supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM ß-mercaptoethanol, all purchased from Thermo Fisher Scientific) with Dynabeads Mouse T-Activator CD3/CD28, according to the manufacturer’s instructions. T cell activation was assessed by flow cytometry and staining against CD3ε, CD4 CD8, CD25, CD69, and CD44. After cell activation, cells were used for transmigration assays. 2 × 105 cells in 250 µl full medium were loaded to the upper chamber of a transwell apparatus (Costar, 24 well plate, 3 μm pore size). The lower chamber was loaded with either 600 µl medium, medium supplemented with 200 nM CCL19, or supernatant of sorted 2N1C and 2N2C DC subpopulations (see Cytokine array and ELISA). Cells were harvested from the lower chamber after 2 h of transmigration at 37°C and analyzed by flow cytometry on a LSRII flow cytometer (BD Biosciences) using absolute counting beads (Invitrogen). Data analysis was carried out using FlowJo X 10.0.7r2.
Statistics
Data analysis was carried out with GraphPad Prism 7 (GraphPad Software). Samples were tested for Gaussian distribution using D’Agostino-Pearson omnibus normality test to fulfill the criteria for performing Student’s t tests. Welch’s correction was applied when two samples had unequal variances. When data distribution was not normal, Mann–Whitney test was carried out. For small data sets, Gaussian distribution was assumed but could not be formally tested. For analysis of cytokine secretion, 2N1C and 2N2C samples from individual experiments were paired. When data distribution was not normal (CCL17), Wilcoxon test was applied. For multiple comparisons where data distribution was normal, one-way ANOVA was used followed by two-stage linear step-up method by Benjamini, Krieger, and Yekutieli as post-hoc test. When data distribution was not normal, Kruskal–Wallis test with Dunn’s multiple comparisons was used. All graphs display mean values ± SD (95% confidence interval). No statistical method was used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment. Individual experiments were validated separately and only pooled if showing the same trend. The level of significance was denoted as *, P < 0.05; **, P < 0.01; ***, P < 0.001 and ****, P < 0.0001 as indicated in the figure legends.
Online supplemental material
Online supplemental material includes additional data covering visualization of centrosomes in vivo (Fig. S1), characterization of cell-cycle arrest in mature DCs (Fig. S2), complete time-lapse images of mitotic live-cell imaging (Fig. S3), DC differentiation and proliferation in the absence of PLK2 (Fig. S4), as well as additional data on centrosome configuration during migration (Fig. S5), separation of centrosomes according to CETN2-GFP signal intensities, and the capacity of cells with multiple centrosomes to enhance T cell activation (Fig. S6). Video 1 shows 3D visualization of segmented centrioles of FIB-SEM datasets. Videos 2, 3, and 4 illustrate impaired mitotic division of DCs after antigen encounter compared to untreated cells. Videos 5, 6, 7, 8, and 9 highlight centrioles during DC migration and provide supporting information about their role for persistent locomotion.
Data availability
Data that support the findings of this study are available within the article and its supplementary information or on request from the corresponding author.
Acknowledgments
We thank Markéta Dalecká and Irena Krejzová for their support with FIB-SEM imaging, the Imaging Methods Core Facility at BIOCEV supported by the Ministry of Education, Youth and Sports Czech Republic (Large RI Project LM2018129 Czech-BioImaging), and European Regional Development Fund (project No. CZ.02.1.01/0.0/0.0/18_046/0016045) for their support with obtaining imaging data presented in this paper. The authors further thank Andreas Villunger, Florian Gärtner, Frank Bradke, and Sarah Förster for helpful discussions; Andy Zielinski for help with statistics; and Björn Weiershausen for assisting with figure illustration.
This work was funded by a fellowship of the Ministry of Innovation, Science and Research of North-Rhine-Westphalia (AZ: 421-8.03.03.02-137069) to E. Kiermaier and the Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany’s Excellence Strategy – EXC 2151 – 390873048. R. Hauschild was funded by grant number 2020-225401 from the Chan Zuckerberg Initiative Donor-Advised Fund, an advised fund of Silicon Valley Community Foundation. M. Hons is supported by Czech Science Foundation GACR 20-24603Y and Charles University PRIMUS/20/MED/013.
The authors declare no competing financial interests.
Author contributions: A.-K. Weier, M. Homrich, S. Ebbinghaus, L. Zhang, and E. Kiermaier performed experiments. P. Juda, E. Miková, and M. Hons performed FIB-SEM. E. Mass, A. Schlitzer, W. Kolanus, S. Burgdorf, and O.J. Gruß gave technical support and conceptual advice. R. Hauschild developed tools for image analysis, and T. Quast helped with image analysis. S. Wieser carried out msd analysis and simulations of cell migration. E. Kiermaier designed and supervised the research and wrote the manuscript. All authors discussed the results and implications and commented on the manuscript at all stages.

