


Intracellular trafficking is mediated by transport carriers that originate by membrane remodeling from donor organelles. Tubular carriers contribute to the flux of membrane lipids and proteins to acceptor organelles, but how lipids and proteins impose a tubular geometry on the carriers is incompletely understood. Using imaging approaches on cells and in vitro membrane systems, we show that phosphatidylinositol-4-phosphate (PI4P) and biogenesis of lysosome-related organelles complex 1 (BLOC-1) govern the formation, stability, and functions of recycling endosomal tubules. In vitro, BLOC-1 binds and tubulates negatively charged membranes, including those containing PI4P. In cells, endosomal PI4P production by type II PI4-kinases is needed to form and stabilize BLOC-1-dependent recycling endosomal tubules. Decreased PI4KIIs expression impairs the recycling of endosomal cargoes and the life cycles of intracellular pathogens such as Chlamydia bacteria and influenza virus that exploit the membrane dynamics of recycling endosomes. This study demonstrates how a phospholipid and a protein complex coordinate the remodeling of cellular membranes into functional tubules.
Introduction
Membrane-bound organelles of eukaryotic cells perform specific functions that are collectively needed for cellular physiology. The limiting membranes of endomembrane organelles are constantly remodeled to facilitate the trafficking of components required for organelle maturation and function (Jarsch et al., 2016). Such remodeling includes the biogenesis of tubular and/or vesicular carriers through bending, invagination or evagination, and scission at precise membrane subdomains (McMahon and Gallop, 2005).
Relative to vesicles, tubular transport intermediates display a high surface-to-volume ratio that is best suited for transporting lipids and membrane-associated proteins (Maxfield and McGraw, 2004). Tubular intermediates consist of elongated membrane structures that extend from largely planar membrane bilayers of many donor organelles (e.g., plasma membrane [PM], trans-Golgi network [TGN], early endosomes, lysosomes, and lysosome-related organelles [LROs]). Their biogenesis and maintenance require membrane asymmetry imposed by extrinsic and/or intrinsic factors (Stachowiak et al., 2013). Extrinsic factors include proteins that sense, impose, and/or stabilize specific membrane curvatures (e.g., BAR [Bin-amphiphysin-Rvs]-domain containing proteins) and cytoskeletal elements that exert mechanical force onto membranes. Intrinsic factors include the accumulation of membrane components with a characteristic shape (e.g., conical lipids or proteins) that can form membrane domains with a particular geometry (Jarsch et al., 2016).
Among membrane lipid species, phosphatidylinositols (PtdIns or PI) are minor components of total cellular phospholipids but play crucial roles in membrane trafficking. Up to seven different PI-Phosphate (PIXP) species can be generated through reversible phosphorylation of their myo-inositol ring at positions-3, -4, and/or -5. PIXPs contribute to the identity and function of organelles by dynamically concentrating at membrane subdomains where they aid in the recruitment and/or stabilization of specific proteins that modulate the membrane biophysical properties (Balla, 2013). For instance, PI(4,5)P2 at the PM cooperates with clathrin adaptor proteins to form carriers during endocytosis (Chang-Ileto et al., 2011); PI3P on early endosomes favors membrane binding of trafficking effectors (Gaullier et al., 2000; Xu et al., 2001) that support the formation of tubulo-vesicular endosomal carriers for cargo recycling (Wallroth and Haucke, 2018; Ketel et al., 2016; McNally and Cullen, 2018); and PI4P (a.k.a., PtdIns4P) cooperates with membrane-shaping proteins and actin-based motors to effect the formation of TGN-derived carriers (Rahajeng et al., 2019). Thus, the local concentration of PIXP defines a membrane-trafficking hotspot for membrane remodeling events during the biogenesis of transport carriers (Di Paolo and De Camilli, 2006; Saarikangas et al., 2010; Lemmon and Ferguson, 2000; Kutateladze, 2010).
Recycling endosomes (RE) are organelles that comprise a network of membrane tubules that arise from the vacuolar domain of early sorting endosomes (SE; Willingham et al., 1984; Yamashiro et al., 1984) and facilitate the sorting and trafficking of multiple cargoes to the PM and TGN of all cells and to LROs in specialized cells (Delevoye et al., 2019; Klumperman and Raposo, 2014). Cargo transport through RE regulates physiological processes at the cellular level such as nutrient uptake, cell migration, polarity, division, signaling, and autophagy, and at the tissue level such as neuronal plasticity and skin pigmentation (Grant and Donaldson, 2009; Puri et al., 2013; Delevoye et al., 2019). Accordingly, RE malfunction is associated with diseases such as neurological disorders (e.g., Huntington and Alzheimer diseases; Li et al., 2008; Zhang et al., 2006) or genetic forms of oculocutaneous albinism (e.g., Hermansky-Pudlak Syndrome [HPS]; Bowman et al., 2019). In addition, RE are hijacked during certain viral and bacterial infections (Allgood and Neunuebel, 2018; Vale-Costa and Amorim, 2016). However, it remains to be determined how specific lipids and proteins coordinate to remodel endosomal membranes for the generation and stabilization of RE tubules.
While PI4P is enriched on Golgi membranes (Graham and Burd, 2011), another PI4P pool localizes to early endosomes (Hammond et al., 2009) and endosome-derived vesicles (Ketel et al., 2016) or tubules (Jović et al., 2009). Cargo trafficking through the early endosomal system requires the spatiotemporal control of PI4P metabolism by two endosome-associated kinases (PI4KIIα and PI4KIIβ; hereafter referred to as PI4KIIs; Craige et al., 2008; Wieffer et al., 2013; Hammond et al., 2014) and at least one phosphatase (Sac2; Nakatsu et al., 2015; Hsu et al., 2015) to respectively synthesize and deplete PI4P on endosomal membranes. Loss of function or overexpression of either PI4KIIα or Sac2 results in missorting of the transferrin receptor (TfR) and the epidermal growth factor receptor (Minogue et al., 2006; Henmi et al., 2016; Nakatsu et al., 2015; Hsu et al., 2015), and PI4P contributes to stabilizing some endosomal tubules (Jović et al., 2009). Moreover, loss of function of the PI4KII orthologue in Drosophila melanogaster larvae impairs RE function in the maturation of salivary gland secretory granules (Ma et al., 2020). However, how PI4P production and consumption regulate the RE tubule dynamics is not understood.
The eight-subunit biogenesis of lysosome-related organelles complex 1 (BLOC-1) is required for the elongation and release of nascent RE tubular carriers from SE in coordination with the microtubule-based kinesin-3 motor KIF13A and actin-related machineries (Ripoll et al., 2016; Delevoye et al., 2014, 2016; Shakya et al., 2018; Thankachan and Setty, 2022). Certain BLOC-1-dependent RE carriers deliver a subset of cargoes to LROs in specialized cell types, such as melanosomes in pigment cells, and loss of BLOC-1 function underlies several subtypes of HPS (Bowman et al., 2019). The BLOC-1-dependent trafficking pathway in melanocytes requires KIF13A (Delevoye et al., 2009, 2016) and PI4KIIs (Zhu et al., 2022). In addition, PI4KIIα physically associates with BLOC-1 in other cell systems (Salazar et al., 2009; Larimore et al., 2011). Thus, BLOC-1 may be integrated with PI4P functions at endosomal membranes.
Here, we used a combination of biochemical approaches, light and electron microscopy (EM) on live and fixed cells, and in vitro cryo-EM imaging of model membranes to test whether BLOC-1 and PI4P orchestrate the biogenesis of RE tubules. We show in vitro that BLOC-1 binds to negatively charged membranes and tubulates membranes containing PI3P or PI4P. In HeLa cells, endosomal PI4P contributes to remodeling and stabilizing membranes into RE tubules. In addition to their requirement in pigment cells for cargo delivery to melanosomes (Zhu et al., 2022), the PI4KIIs are required for recycling of endosomal cargoes and for the intracellular cycles of influenza A virus and, together with BLOC-1, of the bacterium Chlamydia. Our study defines the PI4P/BLOC-1 module as a minimal machinery to generate and stabilize functional RE tubules that can be hijacked by certain infectious agents.
Results
PI4P associates with recycling endosomal tubules
To detect RE tubules by fluorescence microscopy (FM), we performed live imaging of HeLa cells transiently transfected with plasmids encoding the kinesin-3 proteins KIF13A or KIF13B fused to fluorescent proteins (i.e., KIF13A-YFP or mCherry-KIF13B [Delevoye et al., 2014; Yamada et al., 2014; Delevoye and Goud, 2015]). KIF13A is an effector of the RAB11 subfamily of small GTPases (Delevoye et al., 2014), and KIF13A-YFP expression in HeLa cells generates mCherry-RAB11A-labeled RE tubules extending toward the cell periphery (Fig. 1 A, top panels, arrowheads). Its close homologue, KIF13B, was shown to localize to RAB5-positive (+) early endosomes in mouse embryonic fibroblasts (Kanai et al., 2014) and to RAB6A+ secretory vesicles in HeLa cells (Serra-Marques et al., 2020). We found that expression of mCherry-KIF13B (pseudocolored in green) in HeLa cells also generated numerous long RE tubules positive for GFP-RAB11A (pseudocolored in magenta; Fig. 1 A, bottom panels, arrowheads), and that colocalized with KIF13A-YFP (Fig. 1 B, arrowheads). The mCherry-KIF13B+ RE tubules did not co-distribute with iRFP-RAB5+ early endosomes in these cells (pseudocolored in green), although they were, on average, within 1.94 ± 0.15 μm of each other (Fig. S1 A, arrows, and quantification). Thus, both KIF13A and KIF13B generate RE tubules.

PI4P associates with KIF13 + recycling endosomal tubules. (A) Live imaging frame of a Hela cell co-expressing KIF13A-YFP and mCh-RAB11A (green and magenta, respectively; top panels) or mCh-KIF13B and GFP-RAB11A (pseudocolored in green and in magenta, respectively; bottom panels). Magnified insets (4×) show RAB11A co-distribution with KIF13+ tubules (arrowheads). (B) Live imaging frame of a Hela cell co-expressing KIF13A-YFP (green) and mCh-KIF13B (magenta). Magnified insets (4×) show KIF13A and KIF13B co-distribution (arrowheads). (C) Live imaging frame of Hela cells expressing mCh-KIF13B (magenta) together with the GFP-coupled sensors (green) for either PI4P (SidC-GFP, top) or PI3P (GFP-FYVE, bottom). Magnified insets (4×) show the co-distribution of KIF13B+ tubules with PI4P sensor (arrowheads). (D) Line scan analyses intersecting mCh-KIF13B+ tubules captured as in C (30 tubules/condition); values are mean ± SEM (n = 3 independent experiments). Cell periphery is delimited by yellow lines. Scale bars: (main panels) 10 µm; (insets) 2.5 µm.
PI4P associates with KIF13 + recycling endosomal tubules. (A) Live imaging frame of a Hela cell co-expressing KIF13A-YFP and mCh-RAB11A (green and magenta, respectively; top panels) or mCh-KIF13B and GFP-RAB11A (pseudocolored in green and in magenta, respectively; bottom panels). Magnified insets (4×) show RAB11A co-distribution with KIF13+ tubules (arrowheads). (B) Live imaging frame of a Hela cell co-expressing KIF13A-YFP (green) and mCh-KIF13B (magenta). Magnified insets (4×) show KIF13A and KIF13B co-distribution (arrowheads). (C) Live imaging frame of Hela cells expressing mCh-KIF13B (magenta) together with the GFP-coupled sensors (green) for either PI4P (SidC-GFP, top) or PI3P (GFP-FYVE, bottom). Magnified insets (4×) show the co-distribution of KIF13B+ tubules with PI4P sensor (arrowheads). (D) Line scan analyses intersecting mCh-KIF13B+ tubules captured as in C (30 tubules/condition); values are mean ± SEM (n = 3 independent experiments). Cell periphery is delimited by yellow lines. Scale bars: (main panels) 10 µm; (insets) 2.5 µm.

PI4P and KIF13 do not associate with early sorting endosomes. (A and B) Live imaging frame of Hela cells co-expressing (A) mCh-KIF13B (magenta) and iRFP-RAB5 (pseudocolored in green), or (B) GFP-coupled PIxP sensors (green) for either PI4P (SidC-GFP, top) or PI3P (GFP-FYVE, bottom) with iRFP-RAB5 (pseudocolored in magenta). Magnified insets (4×) show RAB5+ structures overlapped with PI3P (B, bottom; arrowheads), but not with KIF13B+ tubules (A; arrows) or PI4P (B, top; arrows). Right panel shows the quantification of the shortest average distance (in µm) between mCherry-KIF13B and iRFP-RAB5+ structures. Data are the average of three independent experiments (>30 iRFP-RAB5+ structures) presented as mean ± SEM. (C) IFM of fixed Hela cells expressing the PI4P sensor (GFP-SidC, green) and labeled for markers (magenta) of TGN (TGN46; top) or plasma membrane (fluorescent-conjugated Wheat Germ Agglutinin, WGA; bottom). Arrowheads point areas of overlap. Cell periphery is delimited by yellow lines. Scale bars: (main panels) 10 µm; (insets) 2 µm.

PI4P and KIF13 do not associate with early sorting endosomes. (A and B) Live imaging frame of Hela cells co-expressing (A) mCh-KIF13B (magenta) and iRFP-RAB5 (pseudocolored in green), or (B) GFP-coupled PIxP sensors (green) for either PI4P (SidC-GFP, top) or PI3P (GFP-FYVE, bottom) with iRFP-RAB5 (pseudocolored in magenta). Magnified insets (4×) show RAB5+ structures overlapped with PI3P (B, bottom; arrowheads), but not with KIF13B+ tubules (A; arrows) or PI4P (B, top; arrows). Right panel shows the quantification of the shortest average distance (in µm) between mCherry-KIF13B and iRFP-RAB5+ structures. Data are the average of three independent experiments (>30 iRFP-RAB5+ structures) presented as mean ± SEM. (C) IFM of fixed Hela cells expressing the PI4P sensor (GFP-SidC, green) and labeled for markers (magenta) of TGN (TGN46; top) or plasma membrane (fluorescent-conjugated Wheat Germ Agglutinin, WGA; bottom). Arrowheads point areas of overlap. Cell periphery is delimited by yellow lines. Scale bars: (main panels) 10 µm; (insets) 2 µm.
We next investigated the distribution of some PIXP species relative to RE tubules by co-expressing endosomal PIXP-specific fluorescent sensors with KIF13 proteins. Live-cell SR-FM (spinning-disk confocal microscopy using optical photon reassignment leading to 1.4-fold increased resolution) revealed that a PI4P sensor – derived from SidC609-776 fused to GFP (SidC-GFP) – largely localized to peripheral punctate structures devoid of iRFP-RAB5 (pseudocolored in magenta, arrows; Fig. S1 B, top panels), and to perinuclear structures that corresponded to the TGN (labeled for TGN46 by immunofluorescence microscopy [IFM]; Fig. S1 C, top panels, arrowhead). The PI4P sensor also decorated to a lesser extent PM (stained using fluorescently conjugated wheat germ agglutinin [WGA]; Fig. S1 C, bottom panels, arrowheads). To analyze if a specific PIXP is enriched at RE tubules, we co-transfected HeLa cells with plasmids encoding mCherry-KIF13B and sensors of PI4P (SidC-GFP) or PI3P (GFP-FYVE). Co-expression of mCherry-KIF13B led to the appearance of numerous PI4P+ tubular structures (Fig. 1 C, top panels), many of which co-distributed with mCherry-KIF13B (arrowheads) as shown by linescan analyses of multiple tubules (Fig. 1 D, top panels). However, mCherry-KIF13B+ tubules were negative for the PI3P sensor GFP-FYVE (Fig. 1, C and D, bottom panels), which instead largely labeled iRFP-RAB5+ early endosomes (Fig. S1 B, bottom panels, arrowheads). Consistently, RE tubules in pigment cells were also positive for the PI4P sensor SidC-GFP (Zhu et al., 2022). Together, these data indicated that KIF13 expression drives the generation of RE tubules with which PI4P is specifically and abundantly associated.
BLOC-1 generates tubules from membranes in vitro
We previously showed that the biogenesis of KIF13A+ RE tubules from early endosomes relied on the BLOC-1-dependent elongation of short nascent tubules (Delevoye et al., 2016). Given the distribution of KIF13 to PI4P+ RE tubules in HeLa cells (Fig. 1, C and D), the contribution of BLOC-1 to the formation of PI4P-dependent RE tubules in pigment cells (Zhu et al., 2022), and the biochemical association of BLOC-1 with the PI4P-producing kinase PI4KIIα (Salazar et al., 2009; Ryder et al., 2013), we hypothesized that PI4P and BLOC-1 cooperate to remodel the endosomal membrane during the formation of RE tubules. We first analyzed whether BLOC-1 interacted with membranes using a minimal system composed of purified recombinant BLOC-1 and lipid tubules and/or vesicles. We expressed and purified the eight subunits of BLOC-1 tagged with GST and 6xHis using a single polycistronic plasmid in Escherichia coli (Fig. S2 A). By negative staining EM, BLOC-1 appeared as rods of ∼30 nm in length with variable curvature (Fig. 2 A), as previously described (Lee et al., 2012).
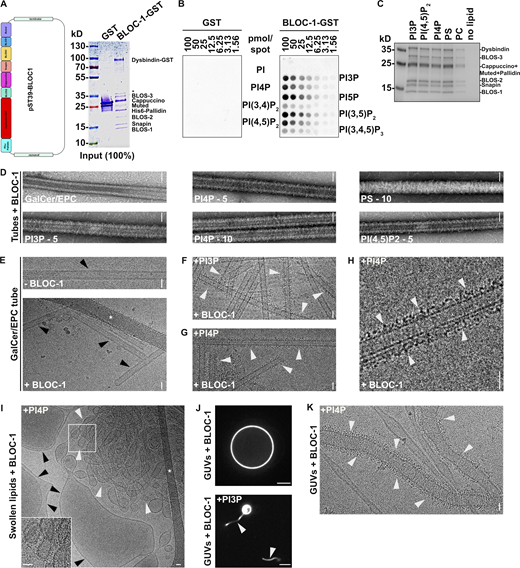
BLOC-1 binding to vesicles and formation of tubules. (A) Schematic of the polycistronic expression cassettes used for the expression and purification of recombinant BLOC-1 (left) and Coomassie Blue-stained SDS-PAGE gel of purified GST and BLOC-1-GST (right). (B) Lipid strip assay showing interaction of purified GST (left) or BLOC-1-GST (right) with PIxPs. (C) SDS-PAGE and Coommassie Blue staining analysis of BLOC-1 binding to lipid vesicles doped with negatively charged (PI3P, PI[4,5]P2, PI4P, PS) or neutral (PC) phospholipids and fractionated by flotation; only top fractions are shown. (D) Imaging by negative staining EM of BLOC-1 binding to GalCer/EPC nanotubes doped or not with PIxP or PS (5 or 10%, as indicated). BLOC-1 bound to negatively charged GalCer/EPC nanotubes (n = 100 tubes/condition, 100% of nanotubes with BLOC-1 bound per condition) compared to control tubes (n = 30 tubes, 0% bound). (E) Cryo-EM images of control GalCer/EPC nanotubes in the absence (top) or presence (bottom) of BLOC-1. No protein was bound to nanotubes lacking negatively charged lipids (arrowheads). (F and G) Cryo-EM image of BLOC-1 bound to GalCer/EPC/PI3P (F) or GalCer/EPC/PI4P (G) nanotubes (arrowheads). (H) Magnified region of G showing dark-dotted densities corresponding to BLOC-1 bound to a PI4P+ nanotube (arrowheads). (I) Cryo-EM image of a suspension of EPC/PS/PI4P vesicles and tubes incubated with BLOC-1. BLOC-1 binds to tubules (white arrowheads), but not to large vesicles (black arrowheads). Inset is the magnified boxed region. (J) Representative fluorescence images of EPC GUVs doped (bottom) or not (top) with PI3P after incubation for 30 min with BLOC-1. Membrane tubules (arrowheads) were observed in PI3P+ GUVs. (K) Cryo-EM image of tubules with non-constant diameters generated from PI4P+ GUVs upon addition of BLOC-1 (arrowheads; see also Fig. 2 G). Figures are representative of at least three independent experiments. In A, asterisk indicates a cleaved form of Dysbindin-GST in sample preparation. In E and I, asterisks indicate the carbon network of the grid. Scale bars: D–I and K, 25 nm; J, 5 µm. Source data are available for this figure: SourceData FS2.

BLOC-1 binding to vesicles and formation of tubules. (A) Schematic of the polycistronic expression cassettes used for the expression and purification of recombinant BLOC-1 (left) and Coomassie Blue-stained SDS-PAGE gel of purified GST and BLOC-1-GST (right). (B) Lipid strip assay showing interaction of purified GST (left) or BLOC-1-GST (right) with PIxPs. (C) SDS-PAGE and Coommassie Blue staining analysis of BLOC-1 binding to lipid vesicles doped with negatively charged (PI3P, PI[4,5]P2, PI4P, PS) or neutral (PC) phospholipids and fractionated by flotation; only top fractions are shown. (D) Imaging by negative staining EM of BLOC-1 binding to GalCer/EPC nanotubes doped or not with PIxP or PS (5 or 10%, as indicated). BLOC-1 bound to negatively charged GalCer/EPC nanotubes (n = 100 tubes/condition, 100% of nanotubes with BLOC-1 bound per condition) compared to control tubes (n = 30 tubes, 0% bound). (E) Cryo-EM images of control GalCer/EPC nanotubes in the absence (top) or presence (bottom) of BLOC-1. No protein was bound to nanotubes lacking negatively charged lipids (arrowheads). (F and G) Cryo-EM image of BLOC-1 bound to GalCer/EPC/PI3P (F) or GalCer/EPC/PI4P (G) nanotubes (arrowheads). (H) Magnified region of G showing dark-dotted densities corresponding to BLOC-1 bound to a PI4P+ nanotube (arrowheads). (I) Cryo-EM image of a suspension of EPC/PS/PI4P vesicles and tubes incubated with BLOC-1. BLOC-1 binds to tubules (white arrowheads), but not to large vesicles (black arrowheads). Inset is the magnified boxed region. (J) Representative fluorescence images of EPC GUVs doped (bottom) or not (top) with PI3P after incubation for 30 min with BLOC-1. Membrane tubules (arrowheads) were observed in PI3P+ GUVs. (K) Cryo-EM image of tubules with non-constant diameters generated from PI4P+ GUVs upon addition of BLOC-1 (arrowheads; see also Fig. 2 G). Figures are representative of at least three independent experiments. In A, asterisk indicates a cleaved form of Dysbindin-GST in sample preparation. In E and I, asterisks indicate the carbon network of the grid. Scale bars: D–I and K, 25 nm; J, 5 µm. Source data are available for this figure: SourceData FS2.

BLOC-1 generates tubules from PI4P-containing membranes in vitro. (A) Negatively stained image of BLOC-1 by EM. (B) Cryo-EM images of a resuspended lipid mixture of EPC/PS/PI4P before (top) or after (bottom) incubation with BLOC-1. Vesicles (black arrowheads) and tubules (white arrowheads) of different sizes and shapes are visible. (C) Cryo-EM image of PI4P+ membrane tubules with BLOC-1 bound (white arrowheads). (D) Fluorescent microscopy of EPC/DOPS/PI4P GUVs before (top) or after (bottom) addition of BLOC-1; note the formation of tubules (arrowheads) from the GUV when BLOC-1 was added (see also Video 1). (E) Cryo-EM image of the BLOC-1-containing GUVs preparation shown in D (bottom) revealing many BLOC-1-coated tubules. (F) Plot of the diameter of tubules generated with BLOC-1 from PI4P+ GUVs (n > 100 tubes analyzed with 4–5 independent measures/tube). (G) Higher magnification cryo-EM image as in E of BLOC-1-coated tubules (white arrowheads) and of vesicles devoid of BLOC-1 (black arrowheads). In B and C, asterisks indicate the carbon network of the grid. Figures are representative of at least three independent experiments. Scale bars: (A) 25 nm, (B and E) 500 nm, (C and G) 50 nm, (D) 5 μm.
BLOC-1 generates tubules from PI4P-containing membranes in vitro. (A) Negatively stained image of BLOC-1 by EM. (B) Cryo-EM images of a resuspended lipid mixture of EPC/PS/PI4P before (top) or after (bottom) incubation with BLOC-1. Vesicles (black arrowheads) and tubules (white arrowheads) of different sizes and shapes are visible. (C) Cryo-EM image of PI4P+ membrane tubules with BLOC-1 bound (white arrowheads). (D) Fluorescent microscopy of EPC/DOPS/PI4P GUVs before (top) or after (bottom) addition of BLOC-1; note the formation of tubules (arrowheads) from the GUV when BLOC-1 was added (see also Video 1). (E) Cryo-EM image of the BLOC-1-containing GUVs preparation shown in D (bottom) revealing many BLOC-1-coated tubules. (F) Plot of the diameter of tubules generated with BLOC-1 from PI4P+ GUVs (n > 100 tubes analyzed with 4–5 independent measures/tube). (G) Higher magnification cryo-EM image as in E of BLOC-1-coated tubules (white arrowheads) and of vesicles devoid of BLOC-1 (black arrowheads). In B and C, asterisks indicate the carbon network of the grid. Figures are representative of at least three independent experiments. Scale bars: (A) 25 nm, (B and E) 500 nm, (C and G) 50 nm, (D) 5 μm.
We then analyzed the interaction of purified BLOC-1 with synthetic membranes. Lipid strip assays showed that BLOC-1-GST interacted with several mono- and di-phosphorylated PIXPs (Fig. S2 B). BLOC-1 was incubated with swollen liposomes doped with negatively charged (PI3P, PI4P, PI[4,5]P2, phosphatidylserine [PS]) or neutral (phosphatidylcholine [PC]) phospholipids, and then the liposomes were recovered by a floatation assay. SDS-PAGE analysis of the buoyant fraction showed that BLOC-1 co-fractionated efficiently only with liposomes containing negatively charged phospholipids (Fig. S2 C).
We then performed a more detailed analysis of the binding of BLOC-1 to membranes of different diameter and composition. We first exploited lipid nanotubes with a narrow and constant diameter of 27 nm for lengths of several hundred nanometers. Consistent with the results of the lipid floatation assay (Fig. S2 C), BLOC-1 bound to GalactoCerebroside/Egg PhosphatidylCholine (GalCer/EPC) nanotubes doped with negatively charged phospholipids but not to GalCer/EPC nanotubes (Fig. S2 D). Because BLOC-1 functions in early endosomal tubulation (Delevoye et al., 2016), we focused on lipid nanotubes doped with the endosome-associated PI3P or PI4P for further analyses. High-resolution cryo-EM images of control GalCer/EPC lipid nanotubes incubated with or without BLOC-1 (Fig. S2 E, bottom and top panels, respectively) showed no electron density corresponding to membrane-bound BLOC-1 (black arrowheads) with the two lipid leaflets of the nanotubes readily detectable. By contrast, cryo-EM images of GalCer/EPC nanotubes doped with 5% of either PI3P (Fig. S2 F) or PI4P (Fig. S2, G and H) and incubated with BLOC-1 showed densities of black dots corresponding to BLOC-1 (white arrowheads) decorating the tubule surface, demonstrating that BLOC-1 binds to PIXPs-containing tubes.
Because PI4P decorated BLOC-1-dependent KIF13+ RE tubules (Fig. 1, C and D; and Delevoye et al., 2016), we investigated if BLOC-1 bound to PI4P+ membranes of different curvatures than the nanotubes. Lipid suspensions were prepared by swelling a dried lipid film containing EPC and 5% PI4P, to which recombinant BLOC-1 was added, and analyzed by cryo-EM. In the control condition without addition of BLOC-1 (Fig. 2 B, top panel), tubules (white arrowheads) and vesicles (black arrowheads) of varied diameters were observed (8.5 tubules/image, n = 20 images). Notably, the addition of BLOC-1 (Fig. 2 B, bottom panel) dramatically increased the number of tubules (white arrowheads; 18.6 tubules/image, n = 23 images). At higher magnification, electron densities corresponding to BLOC-1 were detected on tubules with diameters ranging from 25 to 80 nm (Fig. 2 C and Fig. S2 I, white arrowheads), but not on larger vesicles (Fig. S2 I, black arrowheads).
We next tested whether BLOC-1 tubulates membranes from giant unilamellar vesicles (GUVs), which consist of a larger reservoir of relatively flat and highly deformable membranes compared to swollen lipid vesicles. We used fluorescent GUVs that lacked PIxP or contained 5% PI4P or PI3P (Fig. 2, D–G; and Fig. S2, J and K). Control GUVs without PIxP and with BLOC-1 (Fig. S2 J, top panel) or with PI4P and without BLOC-1 (Fig. 2 D, top panel) appeared smooth and round by FM. By contrast, addition of BLOC-1 to PI4P+ or PI3P+ GUVs induced the formation of tubules from their GUV surfaces (Fig. 2 D, bottom panel, and Video 1 and Fig. S2 J, bottom panel; arrowheads). Since PI4P associated with RE tubules in cells (Fig. 1 C), we analyzed by cryo-EM the tubules derived from BLOC-1-treated PI4P+ GUVs (Fig. 2, E–G and Fig. S2 K). BLOC-1 decorated tubules with diameters centered around 40 nm (Fig. 2, F and G; and Fig. S2 K, white arrowheads). The diameter along the tubules was not always constant (Fig. 2 G and Fig. S2 K), and tubules or vesicular buds larger than 80 nm in diameter and lacking BLOC-1 were observed (Fig. 2 G, black arrowheads). Together, the results show that in vitro BLOC-1 transforms PI4P-containing vesicles into tubules by assembling and concentrating onto curved membranes.
BLOC-1 tubulates PI4P-positive GUVs. Spinning-disc confocal microscopy on PI4P+ fluorescent GUV before (left) or after (right) addition of purified BLOC-1 (0.2 µM; see Materials and methods). BLOC-1 addition led to the appearance of multiple tubular structures (arrows). Acquisition parameters: 0.2 s exposure, one z-stack (0.2 µm). Video was shown at 1 frame/s. Scale bar: 5 µm. See also Fig. 2.
BLOC-1 tubulates PI4P-positive GUVs. Spinning-disc confocal microscopy on PI4P+ fluorescent GUV before (left) or after (right) addition of purified BLOC-1 (0.2 µM; see Materials and methods). BLOC-1 addition led to the appearance of multiple tubular structures (arrows). Acquisition parameters: 0.2 s exposure, one z-stack (0.2 µm). Video was shown at 1 frame/s. Scale bar: 5 µm. See also Fig. 2.
PI4KIIs are required for the formation and stabilization of recycling tubules
Given that BLOC-1 tubulates PI4P+ membranes in vitro and BLOC-1 is required for RE tubule generation in vivo, we tested whether PI4P and its metabolism is required for RE tubule biogenesis in HeLa cells. The production of PI4P on endosomal membranes requires PI4KIIα and PI4KIIβ (PI4KIIs; Hammond et al., 2014; Minogue et al., 2006; Balla and Balla, 2006). Thus, we examined the contribution of PI4KIIs to the formation of RE tubules by siRNA-mediated knockdown. HeLa cells treated with a combination of two individual siRNAs targeting PI4KIIα and PI4KIIβ (siPI4KIIs; see Materials and methods) showed ∼90% reduction in both PI4KIIα and PI4KIIβ compared to cells treated with non-targeting control siRNA (siCTRL; Fig. 3, A and B; siPI4KIIα: 10.9 ± 0.4%; siPI4KIIβ: 6.5 ± 3.5%). By live-cell fluorescence imaging of siCTRL-treated cells transiently expressing KIF13A-YFP with or without mCherry-RAB11A, KIF13A, and RAB11A largely decorated RE tubules extending toward the cell periphery (Fig. 3 C and Fig. S3 A, left panels, arrowheads; and Delevoye et al., 2014). In contrast, RE tubules were largely absent in siPI4KIIs-treated cells, in which KIF13A-YFP and mCherry-RAB11A were instead localized to enlarged vesicular structures (Fig. 3 C and Fig. S3 A, right panels, arrows). Relative to control cells, the percentage of PI4KIIs-depleted cells that exhibited at least one KIF13A-YFP+ RE tubule was reduced by ∼30% (Fig. 3 D; see Materials and methods), and the RE tubules that were detected were ∼30% shorter (Fig. 3 E; siCTRL: 2.2 ± 0.1 μm, siPI4KIIs: 1.5 ± 0.04 μm), revealing a defect in the generation and elongation of RE tubules. In contrast, the siRNA-mediated depletion of the Golgi-associated PI4KIIIβ (Minogue, 2018) did not affect the overall formation of KIF13A+ RE tubules or the percentage of cells with at least one RE tubule (Fig. 3 F, middle panel; and Fig. 3, G and H). Furthermore, the tubulation defect observed in PI4KIIs-depleted cells reflected neither an altered microtubule network – which looked similar in siCTRL- and siPI4KIIs-treated cells (Fig. S3 B) – nor a change in the overall association of the BLOC-1 subunit pallidin to cellular membranes as shown by immunoblotting of membrane fractions prepared from cell homogenates (Fig. S3 C). Therefore, although PI4KIIs are not required for BLOC-1 association with cellular membranes, PI4KIIs are specifically required for the formation and/or elongation of KIF13+ recycling endosomal tubules.

PI4KIIs are required for the formation and stabilization of recycling endosomal tubules. (A) Western blot of HeLa cell lysates treated with control (CTRL) or PI4KIIs (PI4KIIα and PI4KIIβ) siRNAs and probed for PI4KIIα (top), PI4KIIβ (middle), and β-Actin (loading control, bottom). (B) Protein expression levels of PI4KIIα and PI4KIIβ in siCTRL- or siPI4KIIs-treated cells normalized to β-Actin levels. (C) Live imaging frame (top) and associated binary images from “skeletonize” processing (bottom) of siCTRL- or siPI4KIIs-treated HeLa cells expressing KIF13A-YFP. Arrowheads, KIF13A+ RE tubules in siCTRL cells. Arrows, KIF13A+ vesicular structures in PI4KIIs-depleted cells. (D) Quantification of the average percentage of siCTRL or siPI4KIIs cells (n > 60) with at least one KIF13A-YFP+ tubule. (E) Quantification of the average length of KIF13A-YFP+ tubules (n > 50 cells) in siCTRL or siPI4KIIs cells. (F) Live imaging frame of KIF13A-YFP expressing siCTRL (left) or siPI4KIIIβ (middle and right) HeLa cells. Right panel shows siPI4KIIIβ-treated cells treated with PAO. (G) Quantification of the PI4KIIIβ mRNA expression levels in siCTRL or siPI4KIIIβ cells by quantitative RT-PCR analysis relative to GAPDH. (H) Quantification of the average percentage of siCTRL- or siPI4KIIIβ-treated cells (n > 60) with at least one KIF13A-YFP+ tubule. Cell periphery is delimited by yellow dashed lines. Data represent the average of at least three independent experiments and are presented as mean ± SEM. (B, D, E, G, and H): two-tailed unpaired t test; ns, not significant; *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Scale bars: 10 μm. Source data are available for this figure: SourceData F3.
PI4KIIs are required for the formation and stabilization of recycling endosomal tubules. (A) Western blot of HeLa cell lysates treated with control (CTRL) or PI4KIIs (PI4KIIα and PI4KIIβ) siRNAs and probed for PI4KIIα (top), PI4KIIβ (middle), and β-Actin (loading control, bottom). (B) Protein expression levels of PI4KIIα and PI4KIIβ in siCTRL- or siPI4KIIs-treated cells normalized to β-Actin levels. (C) Live imaging frame (top) and associated binary images from “skeletonize” processing (bottom) of siCTRL- or siPI4KIIs-treated HeLa cells expressing KIF13A-YFP. Arrowheads, KIF13A+ RE tubules in siCTRL cells. Arrows, KIF13A+ vesicular structures in PI4KIIs-depleted cells. (D) Quantification of the average percentage of siCTRL or siPI4KIIs cells (n > 60) with at least one KIF13A-YFP+ tubule. (E) Quantification of the average length of KIF13A-YFP+ tubules (n > 50 cells) in siCTRL or siPI4KIIs cells. (F) Live imaging frame of KIF13A-YFP expressing siCTRL (left) or siPI4KIIIβ (middle and right) HeLa cells. Right panel shows siPI4KIIIβ-treated cells treated with PAO. (G) Quantification of the PI4KIIIβ mRNA expression levels in siCTRL or siPI4KIIIβ cells by quantitative RT-PCR analysis relative to GAPDH. (H) Quantification of the average percentage of siCTRL- or siPI4KIIIβ-treated cells (n > 60) with at least one KIF13A-YFP+ tubule. Cell periphery is delimited by yellow dashed lines. Data represent the average of at least three independent experiments and are presented as mean ± SEM. (B, D, E, G, and H): two-tailed unpaired t test; ns, not significant; *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Scale bars: 10 μm. Source data are available for this figure: SourceData F3.
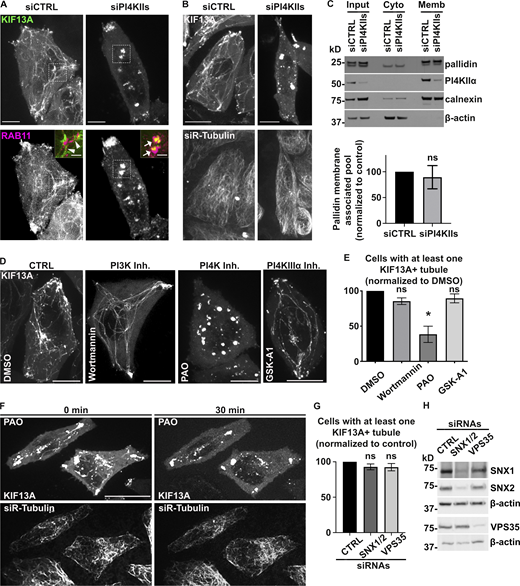
PI4KIIs enzymatic activity is required for the formation and stabilization of recycling endosomal tubules. (A) Live imaging frames of siCTRL- (left) or siPI4KIIs- (right) treated HeLa cells co-expressing KIF13A-YFP (top) and mCh-RAB11A (bottom). Note the tubular (arrowheads) or vesicular (arrows) KIF13+ structures in siCTRL- or siPI4KIIs-treated cells. Insets are magnifications of boxed areas showing KIF13A and RAB11A co-distribution. (B) Live imaging frame of siCTRL- (left) or siPI4KIIs- (right) treated HeLa cells expressing KIF13A-YFP (top) and incubated with siR-Tubulin probe (bottom) to visualize microtubules. (C) siCTRL- and siPI4KIIs-treated cells were homogenized and fractionated to yield post-nuclear membrane (Memb) and cytosolic (Cyto) fractions; input before fractionation is shown at left. Identical cell equivalents of the two fractions were analyzed by immunoblotting using antibodies to membrane-associated calnexin, cytosolic β-actin, PI4KIIα, and the pallidin subunit of BLOC-1. A representative blot is shown (top). Quantification (bottom) of the percentage of membrane-associated pallidin relative to the total cellular content and normalized to siCTRL as 100 (siPI4KIIs: 89.4 ± 22.4). (D) Live imaging frame of HeLa cells expressing KIF13A-YFP and treated for up to 30 min with DMSO vehicle (first panel), PI3K inhibitor wortmannin (10 µM, second panel), PI4K inhibitor PAO (300 nM, third panel), or PI4KIIIα inhibitor GSK-A1 (100 nM, fourth panel). (E) Quantification of the average percentage of treated cells as in D with at least one KIF13A-YFP+ tubule (n > 60 cells). (F) Time lapse images of HeLa cells expressing KIF13A-YFP (top) and treated with siR-Tubulin to visualize microtubules (bottom) before (0 min) and after (30 min) PAO (600 nM) addition. (G) Quantification of the average percentage of KIF13A-YFP+ HeLa cells treated with CTRL, SNX1 and SNX2 (SNX1/2), or VPS35 siRNAs with at least one KIF13A-YFP+ tubule (n > 30 cells). (H) Lysates of siRNA-treated HeLa cells as in G immunoblotted with antibodies to SNX1, SNX2, VPS35, or β-actin (loading control). Data represent the average of at least three independent experiments and are presented as mean ± SEM. (C, E, and G) Two-tailed unpaired t test; *, P < 0.05. ns, not significant. Scale bars: (main panels) 10 µm; (magnified inset) 2.5 µm. Source data are available for this figure: SourceData FS3.

PI4KIIs enzymatic activity is required for the formation and stabilization of recycling endosomal tubules. (A) Live imaging frames of siCTRL- (left) or siPI4KIIs- (right) treated HeLa cells co-expressing KIF13A-YFP (top) and mCh-RAB11A (bottom). Note the tubular (arrowheads) or vesicular (arrows) KIF13+ structures in siCTRL- or siPI4KIIs-treated cells. Insets are magnifications of boxed areas showing KIF13A and RAB11A co-distribution. (B) Live imaging frame of siCTRL- (left) or siPI4KIIs- (right) treated HeLa cells expressing KIF13A-YFP (top) and incubated with siR-Tubulin probe (bottom) to visualize microtubules. (C) siCTRL- and siPI4KIIs-treated cells were homogenized and fractionated to yield post-nuclear membrane (Memb) and cytosolic (Cyto) fractions; input before fractionation is shown at left. Identical cell equivalents of the two fractions were analyzed by immunoblotting using antibodies to membrane-associated calnexin, cytosolic β-actin, PI4KIIα, and the pallidin subunit of BLOC-1. A representative blot is shown (top). Quantification (bottom) of the percentage of membrane-associated pallidin relative to the total cellular content and normalized to siCTRL as 100 (siPI4KIIs: 89.4 ± 22.4). (D) Live imaging frame of HeLa cells expressing KIF13A-YFP and treated for up to 30 min with DMSO vehicle (first panel), PI3K inhibitor wortmannin (10 µM, second panel), PI4K inhibitor PAO (300 nM, third panel), or PI4KIIIα inhibitor GSK-A1 (100 nM, fourth panel). (E) Quantification of the average percentage of treated cells as in D with at least one KIF13A-YFP+ tubule (n > 60 cells). (F) Time lapse images of HeLa cells expressing KIF13A-YFP (top) and treated with siR-Tubulin to visualize microtubules (bottom) before (0 min) and after (30 min) PAO (600 nM) addition. (G) Quantification of the average percentage of KIF13A-YFP+ HeLa cells treated with CTRL, SNX1 and SNX2 (SNX1/2), or VPS35 siRNAs with at least one KIF13A-YFP+ tubule (n > 30 cells). (H) Lysates of siRNA-treated HeLa cells as in G immunoblotted with antibodies to SNX1, SNX2, VPS35, or β-actin (loading control). Data represent the average of at least three independent experiments and are presented as mean ± SEM. (C, E, and G) Two-tailed unpaired t test; *, P < 0.05. ns, not significant. Scale bars: (main panels) 10 µm; (magnified inset) 2.5 µm. Source data are available for this figure: SourceData FS3.
To assess if the enzymatic activity of PI4KIIs was required to generate RE tubules, we tested the effect of phenylarsine oxide (PAO), a PI4KIIs inhibitor that causes cellular PI4P depletion (Yue et al., 2001). Similarly to PI4KIIs depletion, treatment of HeLa cells transiently expressing KIF13A-YFP for 30 min with PAO at a low concentration (300 nM) reduced the percentage of cells having at least one KIF13+ tubule by ∼70% compared to cells treated with DMSO vehicle alone (Fig. S3, D and E). Live imaging during the 30 min PAO treatment showed that preexisting KIF13A-YFP+ RE tubules were gradually lost, concomitant with the appearance of vesicular structures (Fig. S3 F, top right panel), while the microtubule network remained intact (Fig. S3 F, bottom panels). At this concentration, PAO could inhibit in vitro the enzymatic activity of PI4KIIIα (Balla et al., 2002); however, cells similarly treated with the PI4KIIIα inhibitor GSK-A1 (100 nM) did not show defects in KIF13A-YFP+ RE tubules (Fig. S3, D and E). In contrast, cells depleted of PI4KIIIβ and similarly treated with PAO displayed a loss of the KIF13A-YFP+ RE tubules (Fig. 3 F, right panel), showing that the stability of preexisting RE tubules specifically required PI4KIIs enzymatic activity. Finally, we tested whether PI3P – which also facilitated BLOC-1 concentration on membranes and tubulation in vitro (Fig. S2, C and D; and Fig. S2, F and J) – contributed to KIF13-dependent tubulation. HeLa cells expressing KIF13A-YFP and treated with the PI3K inhibitor wortmannin (10 μM) did not show defects associated with KIF13A+ RE tubule formation (Fig. S3, D and E). Moreover, depletion of PI3P-dependent membrane binding and deformation components such as the retromer subunit VPS35 or the ESCPE-1 components SNX1 and SNX2 (Cullen and Korswagen, 2011) did not affect the number of cells with at least one KIF13A-YFP+ tubule (Fig. S3, G and H; and Delevoye et al., 2016). Taken together, these data indicate that the formation of RE tubules and their stabilization rely on cellular PI4P synthesized by PI4KIIs.
Endosomal PI4P stabilizes the recycling endosomal tubule
As PI4P is distributed among different organelles, including the Golgi apparatus and endosomes (Hammond et al., 2014), we asked which pool of PI4P contributes to generate the RE tubules. We exploited the rapamycin-inducible FRB-FKBP chemical dimerization system to trigger the recruitment of the FKBP-conjugated PI4P phosphatase enzymatic domain from Sac1 (FKBP-PJ-Sac) to early endosomal or Golgi membranes harboring FRB-conjugated RAB5 or -Giantin, respectively. Upon rapamycin addition, FKBP-PJ-Sac dimerizes rapidly with FRB-target proteins, allowing acute PI4P dephosphorylation on the cytosolic leaflet of the target organelle (Fig. 4 A; and Hammond et al., 2014). HeLa cells expressing mRFP-FKBP-PJ-Sac, KIF13A-YFP, and either iRFP-FRB-RAB5 (Fig. 4 B) or FRB-Giantin (untagged; Fig. 4 C) were analyzed by live cell imaging before and after 20 min of rapamycin treatment. As expected, rapamycin addition led to the recruitment of PJ-Sac from a primarily cytosolic localization to RAB5+ early endosomes or the Giantin+ Golgi apparatus (Fig. 4, B and C, arrows). FKBP-PJ-Sac recruitment to Golgi membranes had no impact on the appearance, number, or length of KIF13A-YFP+ RE tubules (Fig. 4 C, bottom right panel; and Fig. 4, D and E). However, FKBP-PJ-Sac recruitment to early endosomes had a dramatic effect (Fig. 4 B, bottom right panels, arrows). The KIF13A-YFP+ RE tubules were reduced in both number (Fig. 4 D) and length (Fig. 4 E), as had been observed in PI4KIIs-depleted or PAO-treated cells (Fig. 3, C–E and Fig. S3, D–F). Most of the KIF13A-YFP+ RE tubules in cells with early endosomal FKBP-PJ-Sac were suppressed and replaced by KIF13A-YFP+ vesicles, which overlapped with RAB5 and PJ-Sac signals (Fig. 4 B, arrows). These data support the conclusion that the stabilization of preexisting RE tubules requires the spatiotemporal control of PI4P levels on early endosomal membranes. Hence, together with the results presented in Fig. 3, we conclude that the pool of PI4P on early endosomal membranes functions in both the initiation and stabilization of RE tubules.

Depletion of endosomal PI4P destabilizes recycling endosomal tubules. (A) Schematic of the rapamycin-induced FRB-FKBP system allowing the organelle-specific depletion of PI4P through the recruitment of PI4P phosphatase Sac domain (mRFP-FKBP-PJ-SAC) to membranes positive for RAB5 (iRFP-FRB-RAB5, early sorting endosomes; left) or Giantin (FRB-Giantin, Golgi apparatus; right). The recruited Sac domain catalyzes the removal of phosphate from PI4P (green ball) to generate PI (red ball). (B and C) Live imaging frames of HeLa cells co-expressing KIF13A-YFP together with (B) mRFP-FKBP-PJ-Sac and either iRFP-FRB-RAB5 or (C) FRB-Giantin before (top) or after (bottom) 20 min addition of rapamycin (1 μM) to recruit PJ-SAC to either RAB5+ endosomal (B) or Giantin+ Golgi (C) membranes (arrows). Note that acute targeting of PJ-SAC to RAB5+ membranes (B), but not to Giantin+ membranes (C), destabilizes KIF13-YFP+ RE tubules. The FRB-Giantin chimera is not fluorescently tagged, and thus not imaged. (D) Quantification of the average percentage of KIF13A-YFP+ RE tubules remaining 20 min after rapamycin addition relative to time 0 min in cells treated as in B and C (right panels). (E) Quantification of the average length (μm) of KIF13-YFP+ RE tubule before (0 min) and 20 min after rapamycin addition in cells treated as in B and C (right panels). Data presented as box-plots represent at least five independent experiments. ns, non-significant. **, P < 0.01. (D and E): two-tailed unpaired t test; endosome depletion, n = 85 tubules; Golgi depletion, n = 118 tubules. Scale bars: (main panels) 10 μm; (insets) 2.5 μm.
Depletion of endosomal PI4P destabilizes recycling endosomal tubules. (A) Schematic of the rapamycin-induced FRB-FKBP system allowing the organelle-specific depletion of PI4P through the recruitment of PI4P phosphatase Sac domain (mRFP-FKBP-PJ-SAC) to membranes positive for RAB5 (iRFP-FRB-RAB5, early sorting endosomes; left) or Giantin (FRB-Giantin, Golgi apparatus; right). The recruited Sac domain catalyzes the removal of phosphate from PI4P (green ball) to generate PI (red ball). (B and C) Live imaging frames of HeLa cells co-expressing KIF13A-YFP together with (B) mRFP-FKBP-PJ-Sac and either iRFP-FRB-RAB5 or (C) FRB-Giantin before (top) or after (bottom) 20 min addition of rapamycin (1 μM) to recruit PJ-SAC to either RAB5+ endosomal (B) or Giantin+ Golgi (C) membranes (arrows). Note that acute targeting of PJ-SAC to RAB5+ membranes (B), but not to Giantin+ membranes (C), destabilizes KIF13-YFP+ RE tubules. The FRB-Giantin chimera is not fluorescently tagged, and thus not imaged. (D) Quantification of the average percentage of KIF13A-YFP+ RE tubules remaining 20 min after rapamycin addition relative to time 0 min in cells treated as in B and C (right panels). (E) Quantification of the average length (μm) of KIF13-YFP+ RE tubule before (0 min) and 20 min after rapamycin addition in cells treated as in B and C (right panels). Data presented as box-plots represent at least five independent experiments. ns, non-significant. **, P < 0.01. (D and E): two-tailed unpaired t test; endosome depletion, n = 85 tubules; Golgi depletion, n = 118 tubules. Scale bars: (main panels) 10 μm; (insets) 2.5 μm.
PI4KIIs are required for the budding and elongation of early endosomal membranes
To form a RE tubule, the early endosomal membrane must be locally remodeled to generate a nascent tubule that is then elongated and ultimately released by membrane scission (Delevoye et al., 2014; Delevoye et al., 2016). We thus investigated if an imbalance of PI4P metabolism affects the remodeling of early endosomal membranes. We performed conventional EM in PI4KIIs-depleted cells immobilized by high-pressure freezing (HPF), which maintains the cellular membranes in a nearly native state (Hurbain et al., 2017). In siCTRL-treated cells, most early endosomes – defined as electron-lucent and round membrane-bound compartments containing a few number of intra-luminal vesicles (ILVs) – harbored one or more nascent tubule profiles at their limiting membrane (Fig. 5 A, arrowheads; quantification in Fig. 5 C; nascent tubules/endosome, siCTRL: 1.07 ± 0.17, siPI4KIIs: 0.36 ± 0.07). In PI4KIIs-depleted cells (Fig. 5 B), the limiting membranes of early endosomes harbored fewer deformations (arrowheads) as revealed by the ∼70% reduction of nascent tubules per endosomal structure (Fig. 5 C). This reduction did not reflect a defect in early endosome biogenesis, as early endosomes were frequently observed in siPI4KIIs-treated cells—although many contained few ILVs (Fig. 5 B, arrows), suggesting a potential impairment in endosomal maturation. This result shows that PI4KIIs expression is required for the initiation of nascent early endosomal tubules.

Depletion of PI4KIIs prevents the initiation and elongation of early endosomal tubules. (A and B) HeLa cells treated with control (A) or PI4KIIs siRNAs (B) were immobilized by high-pressure freezing, and ultrathin sections were analyzed by conventional EM. Arrowheads point to nascent tubular structures emerging from the limiting membranes of endosomes. Arrows (B) point to endosomes without vesicular or tubular budded structure. Insets are additional representative endosomal structures captured in cells as in A and B. (C) Quantification of the average number of nascent tubules per endosome in siCTRL- and siPIK4IIs-treated HeLa cells as in A and B. (D) Quantification of the average maximum length of endosomal nascent tubules in siCTRL- (n = 40) and siPI4KIIs- (n = 47) treated HeLa cells measured in C. Data are the average of three independent experiments presented as the mean ± SEM (siCTRL, n = 8 cells; siPI4KIIs, n = 14 cells). (C and D) Two-tailed unpaired t test; *,P < 0.05; ****, P < 0.0001. Scale bars: (main panels) 1 μm; (insets) 200 nm.
Depletion of PI4KIIs prevents the initiation and elongation of early endosomal tubules. (A and B) HeLa cells treated with control (A) or PI4KIIs siRNAs (B) were immobilized by high-pressure freezing, and ultrathin sections were analyzed by conventional EM. Arrowheads point to nascent tubular structures emerging from the limiting membranes of endosomes. Arrows (B) point to endosomes without vesicular or tubular budded structure. Insets are additional representative endosomal structures captured in cells as in A and B. (C) Quantification of the average number of nascent tubules per endosome in siCTRL- and siPIK4IIs-treated HeLa cells as in A and B. (D) Quantification of the average maximum length of endosomal nascent tubules in siCTRL- (n = 40) and siPI4KIIs- (n = 47) treated HeLa cells measured in C. Data are the average of three independent experiments presented as the mean ± SEM (siCTRL, n = 8 cells; siPI4KIIs, n = 14 cells). (C and D) Two-tailed unpaired t test; *,P < 0.05; ****, P < 0.0001. Scale bars: (main panels) 1 μm; (insets) 200 nm.
Although nascent tubules associated with endosomes were less numerous in PI4KIIs-depleted cells, three parameters were measured for those that were detected (Fig. 5, B and C, arrowheads): the maximum length, the maximum width, and the neck width (Fig. S4 A). In PI4KIIs-depleted cells, the nascent tubules were significantly shorter (Fig. 5 D; siCTRL: 98 ± 8 nm, siPI4KIIs: 76 ± 5 nm), consistent with a requirement for PI4KIIs and PI4P in tubule extension. The average maximum width of the nascent tubules was similar (Fig. S4 B; siCTRL: 60 ± 3 nm, siPI4KIIs: 61 ± 3 nm). Hence, the average length-to-width ratio of the nascent endosomal tubules in PI4KIIs-depleted cells was significantly reduced as compared to the control (Fig. S4 C; siCTRL: 1.8 ± 0.2, siPI4KIIs: 1.4 ± 0.1). Tubular membrane carriers constrict their neck prior to their release (Ripoll et al., 2018). The average neck width of the nascent tubules was marginally increased in PI4KIIs-depleted cells relative to control cells (Fig. S4 D; siCTRL: 49 ± 3 nm, siPI4KIIs: 58 ± 4 nm), although the increase did not reach the statistical significance. These data indicate that PI4KIIs mainly control the length of the nascent RE tubules.

Depletion of PI4KIIs does not alter the neck and width of endosomal nascent tubules. (A) Schematic representation of the measurements (in Fig. 5 D; and Fig. S4, B and D) of the nascent endosomal tubules captured by EM. (B–D) Quantification of the average maximum width (B), length:width ratio (C), and neck width (D) of the endosomal nascent tubules in HeLa cells treated with siCTRL (width, n = 42; length:width, n = 40; neck, n = 30) or siPI4KIIs (width, n = 50; length:width, n = 46; neck, n = 49). Data from three independent experiments are presented as the mean ± SEM (siCTRL, n = 8 cells; siPI4KIIs, n = 14 cells). (B–D) Two-tailed unpaired t test; ns, not-significant. *, P < 0.05.

Depletion of PI4KIIs does not alter the neck and width of endosomal nascent tubules. (A) Schematic representation of the measurements (in Fig. 5 D; and Fig. S4, B and D) of the nascent endosomal tubules captured by EM. (B–D) Quantification of the average maximum width (B), length:width ratio (C), and neck width (D) of the endosomal nascent tubules in HeLa cells treated with siCTRL (width, n = 42; length:width, n = 40; neck, n = 30) or siPI4KIIs (width, n = 50; length:width, n = 46; neck, n = 49). Data from three independent experiments are presented as the mean ± SEM (siCTRL, n = 8 cells; siPI4KIIs, n = 14 cells). (B–D) Two-tailed unpaired t test; ns, not-significant. *, P < 0.05.
Together, our data indicate that PI4KIIs and their PI4P product contribute to sequential steps – initiation and elongation – in the remodeling of the early endosome limiting membrane into a RE tubule.
PI4KIIs control endosomal cargo recycling
If early endosomal PI4P contributes to RE tubule initiation, elongation, and stabilization, then PI4KII depletion should impair the endosomal recycling of conventional cargoes. To test this prediction, we analyzed the dynamics of internalized transferrin (Tf) as a readout for the model recycling cargo, Tf receptor. Control- and PI4KIIs-depleted cells were pulsed for 10 min with fluorescent-Tf (Alexa Fluor 488 conjugated Tf; Tf-A488) and then chased for different time points before processing by FM (Fig. S5 A) and quantification of signal intensity (Fig. S5, B and C). First, the overall Tf-A488 intensity at time 0 of chase was ∼20% reduced in PI4KIIs-depleted cells as compared to controls (Fig. S5 A, top panels, and Fig. S5 B), showing that less Tf-A488 was internalized during the pulse. Second, the overall intracellular distribution of Tf-A488+ structures in PI4KIIs-depleted cells was distinct from the control cells (Fig. S5 A). At time 0, Tf-A488+ endosomes in control- and PI4KIIs-depleted cells were distributed throughout the cell periphery (top panels). Whereas the general peripheral distribution tended to be maintained in control cells throughout the chase, Tf-A488+ endosomes in PI4KIIs-depleted cells were more prominent in the perinuclear area after both 20 and 40 min of chase (middle and bottom right panels, arrowheads). Third, whereas control cells showed a linear decay of the average normalized Tf-A488 over the 40 min of chase, the decay of Tf-A488 intensity in PI4KIIs-depleted cells showed a significant plateau between the 20- and 40-min time points (Fig. S5 C), consistent with a recycling defect. Moreover, depletion of PI4KIIα or PI4KIIβ in pigment cells impaired the trafficking of BLOC-1-dependent cargoes from endosomes to melanosomes (Zhu et al., 2022), which occurs through RE-like tubules (Delevoye et al., 2009). Together, these results indicate that PI4KIIs are required for the recycling of endosomal cargoes.

PI4KIIs expression is required for endosomal cargo recycling. (A) FM of siCTRL- (left panels) or siPI4KIIs- (right panels) treated Hela cells pulsed with Tf-A488 (green) and chased at indicated times (0, 20, 40 min), before fixation and labeling of nuclei (DAPI, blue). Arrowheads point to perinuclear accumulation of Tf+ endosomes in siPI4KIIs-treated cells during chase. (B) Quantification of the average fluorescence intensity of Tf/cell at 0 min of chase (siCTRL, n = 50 cells; siPI4KIIs = 46 cells). (C) Quantification of the average intensity of Tf/cell at different time points of chase (siCTRL cells: 0 min, n = 50; 20 min, n = 58; 40 min, n = 49; siPI4KIIs cells: 0 min, n = 46; 20 min, n = 56; 40 min, n = 46) normalized relative to the 0 time point. Cell peripheries are delimited by yellow lines. Data from three independent experiments are presented as the mean ± SEM. (B and C)Two-tailed unpaired t test; ns, not significant. ***, P < 0.001; ****, P < 0.0001. Scale bars: 10 μm.

PI4KIIs expression is required for endosomal cargo recycling. (A) FM of siCTRL- (left panels) or siPI4KIIs- (right panels) treated Hela cells pulsed with Tf-A488 (green) and chased at indicated times (0, 20, 40 min), before fixation and labeling of nuclei (DAPI, blue). Arrowheads point to perinuclear accumulation of Tf+ endosomes in siPI4KIIs-treated cells during chase. (B) Quantification of the average fluorescence intensity of Tf/cell at 0 min of chase (siCTRL, n = 50 cells; siPI4KIIs = 46 cells). (C) Quantification of the average intensity of Tf/cell at different time points of chase (siCTRL cells: 0 min, n = 50; 20 min, n = 58; 40 min, n = 49; siPI4KIIs cells: 0 min, n = 46; 20 min, n = 56; 40 min, n = 46) normalized relative to the 0 time point. Cell peripheries are delimited by yellow lines. Data from three independent experiments are presented as the mean ± SEM. (B and C)Two-tailed unpaired t test; ns, not significant. ***, P < 0.001; ****, P < 0.0001. Scale bars: 10 μm.
PI4KIIs are required for the life cycle of intracellular pathogens
Several intracellular pathogens exploit RE to establish their niche and/or to replicate. Thus, we reasoned that such pathogens might require PI4KIIs or BLOC-1 for certain steps of their developmental cycle. Among pathogen model systems, we chose to analyze (1) the influenza A virus (IAV), which exploits the RE-associated KIF13A and RAB11 to transport and propagate viral genome segments (viral ribonucleoproteins; vRNPs) during infection (Vale-Costa and Amorim, 2016; Ramos-Nascimento et al., 2017; Alenquer et al., 2019), and (2) the obligate intracellular bacterium Chlamydia trachomatis (C. trachomatis), which replicates within a membrane-bound vacuole (referred to as an inclusion) from which tubules decorated by the RE-associated components RAB11, PI4KIIα, and PI4P emerge (Rzomp et al., 2003; Moorhead et al., 2010). The IAV model was used to define whether PI4KIIs contribute to RE functions that are hijacked during viral infection, and the membrane tubules emanating from the C. trachomatis inclusion served as a proxy for PI4KIIs functions during membrane tubulation.
First, we explored the role of PI4KIIs in viral replication by IAV. In human A549 lung carcinoma cells, treatment with PI4KIIs siRNAs significantly reduced the expression of PI4KIIα and PI4KIIβ mRNAs (Fig. S6, A and B). Cells treated with siCTRL or siPI4KIIs were mock-infected or infected with IAV/Puerto Rico/8/34 (hereafter referred to as PR8 virus; see Materials and methods), and viral production was assayed. PI4KIIs depletion led to a modest but consistent ∼10% reduction of PR8 virus production at 8 h post-infection relative to siCTRL-treated cells (Fig. 6 A). Given that the synthesis of viral matrix and capsid proteins was not affected by PI4KIIs depletion (Fig. S6, C–E), this finding suggested that the formation of IAV inclusions could be impaired. Viral inclusions are viewed as sites where complete genomes assemble before packaging into nascent virions to form fully infectious particles (Amorim, 2019). The packaging of influenza segmented genomes into productive virions requires RAB11, which redistributes during infection to intracellular sites of progeny RNA accumulation and is required to form liquid viral (i.e., not membrane bound) inclusions in which RAB11+ vesicles accumulate (Alenquer et al., 2019). This is evident by IFM of PR8-infected control cells, in which labeling for RAB11 and for IAV proteins (e.g., NP) overlap in punctate structures throughout the cell (Vale-Costa et al., 2016). Whereas both siCTRL- and siPI4KIIs-treated IAV-infected cells showed that RAB11+ puncta overlapped with NP labeling, RAB11 and NP+ structures in PI4KIIs-depleted cells were substantially larger (Fig. 6, B and C). This difference in size could reflect aberrant viral inclusions with altered biophysical properties or a defect in transporting liquid viral inclusions. These data suggest that PI4KIIs contribute to the size and maintenance of liquid viral inclusions during IAV infection.
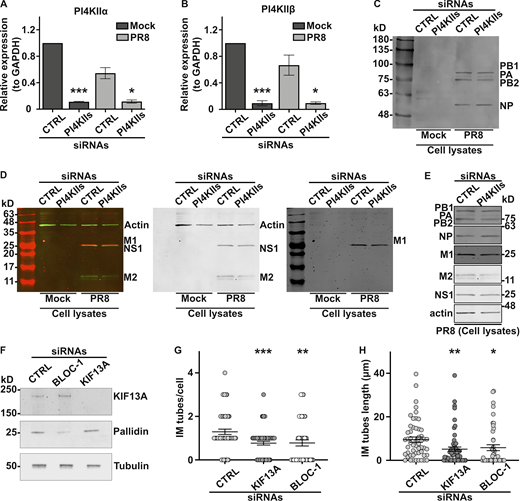
PI4KIIs, BLOC-1, and/or KIF13A expression is required for viral or bacterial infection. (A–D) A549 cells treated with control (CTRL) or PI4KIIs siRNAs for 48 h were mock-infected or infected 8 h with PR8 virus at MOI of 3. (A and B) mRNA expression levels of PI4KIIα or PI4KIIβ relative to GAPDH levels. (C and D) Immunoblot analyses of cells lysates show expression levels of viral proteins PB1 (86.5 kD), PB2 (85.7 kD), PA (84.2 kD), and NP (56.1 kD; C), or M1 (27.8 kD; red in left panel, black in right panel), NS1 (26.8 kD; green in left panel, black in middle panel), M2 (11 kD; green in left panel, black in middle panel), and actin as a loading control (42 kD; green in left panel, black in middle panel; D). (E) Immunoblots cropped from C and D showing viral protein expression in lysates of siCTRL or siPI4KIIs-treated PR8-infected cells using antibodies against indicated proteins. (F) Immunoblotting of siCTRL, siBLOC-1, or siKIF13A-treated HeLa cell lysates probed for KIF13A (top), pallidin subunit of BLOC-1 (middle) and β-tubulin as a loading control (bottom). (G and H) HeLa cells treated with siCTRL, siKIF13A, or si-BLOC-1 were infected 12 h with Ctr D, and analyzed by IFM for Cap1 to quantify the average number/cell (G) and length (H) of Cap1+ tubules emanating from bacterial inclusions. (A and B) One-way ANOVA followed by Dunn’s multiple comparisons test (*, P < 0.05; ***, P < 0.001; n = 7 independent experiments). (G and H) Data from three independent experiments are presented as the mean ± SEM; two-tailed unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData FS6.

PI4KIIs, BLOC-1, and/or KIF13A expression is required for viral or bacterial infection. (A–D) A549 cells treated with control (CTRL) or PI4KIIs siRNAs for 48 h were mock-infected or infected 8 h with PR8 virus at MOI of 3. (A and B) mRNA expression levels of PI4KIIα or PI4KIIβ relative to GAPDH levels. (C and D) Immunoblot analyses of cells lysates show expression levels of viral proteins PB1 (86.5 kD), PB2 (85.7 kD), PA (84.2 kD), and NP (56.1 kD; C), or M1 (27.8 kD; red in left panel, black in right panel), NS1 (26.8 kD; green in left panel, black in middle panel), M2 (11 kD; green in left panel, black in middle panel), and actin as a loading control (42 kD; green in left panel, black in middle panel; D). (E) Immunoblots cropped from C and D showing viral protein expression in lysates of siCTRL or siPI4KIIs-treated PR8-infected cells using antibodies against indicated proteins. (F) Immunoblotting of siCTRL, siBLOC-1, or siKIF13A-treated HeLa cell lysates probed for KIF13A (top), pallidin subunit of BLOC-1 (middle) and β-tubulin as a loading control (bottom). (G and H) HeLa cells treated with siCTRL, siKIF13A, or si-BLOC-1 were infected 12 h with Ctr D, and analyzed by IFM for Cap1 to quantify the average number/cell (G) and length (H) of Cap1+ tubules emanating from bacterial inclusions. (A and B) One-way ANOVA followed by Dunn’s multiple comparisons test (*, P < 0.05; ***, P < 0.001; n = 7 independent experiments). (G and H) Data from three independent experiments are presented as the mean ± SEM; two-tailed unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData FS6.

Intracellular pathogens exploit PI4KIIs and BLOC-1 to achieve their life cycle. (A) Viral production assessed by plaque assay was plotted as plaque forming units per milliliter (PFU/ml). A549 cells treated with siCTRL or siPI4KIIs were infected with PR8 at MOI of 3 for 8 h. A single experiment composed of six samples is shown and is representative of three independent experiments. (B) Cells treated with siCTRL or siPI4KIIs were infected or mock-infected with PR8 at MOI of 3 for 8 h and processed by IFM. Cytosolic viral inclusions were identified using antibodies against RAB11 (green) and viral NP (magenta) proteins. Insets show RAB11 and NP co-localization. Nuclei (blue) and cell periphery were delimited by yellow-dashed lines. (C) Frequency distribution of NP viral inclusions within the three area categories (in µm2) was plotted for each condition. (D) HeLa cells treated with siCTRL or siPI4KIIs were infected 12 h with C. trachomatis serovar D (Ctr D) expressing mCherry (top panels), then fixed and analyzed by IFM using antibodies against the bacterial inclusion protein Cap1 (bottom panels). Arrowheads point to Cap1+ tubules elongating from the inclusion membrane. Cell periphery is delimited by yellow lines. (E and F)Ctr D-infected cells were analyzed for the average number/cell (E) and the mean length (F) of Cap1+ tubules observed in D. (G) Working model of the role of PI4P, PI4KIIs, and BLOC-1 during recycling endosomal tubule formation from the early sorting endosomal membrane. (1) At PI3P+ early endosomal membrane subdomains, PI4P locally produced by PI4KIIs is quickly depleted by SAC2 phosphatase activity. (2) In PI3P-deficient domains, endosomal PI4P locally synthesized by PI4KIIs accumulates. (3) BLOC-1 concentration on PI4P+ domains facilitates the formation of nascent tubules with diameter and curvature compatible with BLOC-1 association/stabilization to membrane. (4) Nascent tubules accumulate PI4P, produced by local PI4KIIs, that sustains elongation through continuous BLOC-1 concentration to the PI4P+ tubular membrane and concomitant pulling by KIF13 motors along microtubules (Delevoye et al., 2016). (5) Eventually, nascent-extended RE tubules are severed at their neck through BLOC-1 cooperation with actin-polymerizing machinery (Delevoye et al., 2016) to release PI4P+ tubules required for endosomal cargo (e.g., Tf) recycling. The machineries required for the biogenesis of RE tubules can be exploited by pathogens to either use RE membranes (e.g., IAV) or tubulate parasitophorous vacuoles (e.g., Chlamydia). (B) Non-parametric two-way ANOVA test, followed by Sidak’s multiple comparisons test (***, P < 0.001); ∼45 cells/condition from three independent experiments. (A, E, and F) Data are the average of three independent experiments presented as the mean ± SEM; two-tailed unpaired t test (**, P < 0.01; ****, P < 0.0001). Scale bars: (main panels) 10 μm; (insets) 2.5 μm.
Intracellular pathogens exploit PI4KIIs and BLOC-1 to achieve their life cycle. (A) Viral production assessed by plaque assay was plotted as plaque forming units per milliliter (PFU/ml). A549 cells treated with siCTRL or siPI4KIIs were infected with PR8 at MOI of 3 for 8 h. A single experiment composed of six samples is shown and is representative of three independent experiments. (B) Cells treated with siCTRL or siPI4KIIs were infected or mock-infected with PR8 at MOI of 3 for 8 h and processed by IFM. Cytosolic viral inclusions were identified using antibodies against RAB11 (green) and viral NP (magenta) proteins. Insets show RAB11 and NP co-localization. Nuclei (blue) and cell periphery were delimited by yellow-dashed lines. (C) Frequency distribution of NP viral inclusions within the three area categories (in µm2) was plotted for each condition. (D) HeLa cells treated with siCTRL or siPI4KIIs were infected 12 h with C. trachomatis serovar D (Ctr D) expressing mCherry (top panels), then fixed and analyzed by IFM using antibodies against the bacterial inclusion protein Cap1 (bottom panels). Arrowheads point to Cap1+ tubules elongating from the inclusion membrane. Cell periphery is delimited by yellow lines. (E and F)Ctr D-infected cells were analyzed for the average number/cell (E) and the mean length (F) of Cap1+ tubules observed in D. (G) Working model of the role of PI4P, PI4KIIs, and BLOC-1 during recycling endosomal tubule formation from the early sorting endosomal membrane. (1) At PI3P+ early endosomal membrane subdomains, PI4P locally produced by PI4KIIs is quickly depleted by SAC2 phosphatase activity. (2) In PI3P-deficient domains, endosomal PI4P locally synthesized by PI4KIIs accumulates. (3) BLOC-1 concentration on PI4P+ domains facilitates the formation of nascent tubules with diameter and curvature compatible with BLOC-1 association/stabilization to membrane. (4) Nascent tubules accumulate PI4P, produced by local PI4KIIs, that sustains elongation through continuous BLOC-1 concentration to the PI4P+ tubular membrane and concomitant pulling by KIF13 motors along microtubules (Delevoye et al., 2016). (5) Eventually, nascent-extended RE tubules are severed at their neck through BLOC-1 cooperation with actin-polymerizing machinery (Delevoye et al., 2016) to release PI4P+ tubules required for endosomal cargo (e.g., Tf) recycling. The machineries required for the biogenesis of RE tubules can be exploited by pathogens to either use RE membranes (e.g., IAV) or tubulate parasitophorous vacuoles (e.g., Chlamydia). (B) Non-parametric two-way ANOVA test, followed by Sidak’s multiple comparisons test (***, P < 0.001); ∼45 cells/condition from three independent experiments. (A, E, and F) Data are the average of three independent experiments presented as the mean ± SEM; two-tailed unpaired t test (**, P < 0.01; ****, P < 0.0001). Scale bars: (main panels) 10 μm; (insets) 2.5 μm.
Second, we investigated the impact of PI4KIIs on the formation of membrane tubules from the surface of bacterial inclusions during C. trachomatis serovar D (Ctr D) infection of HeLa cells. CTRL- or PI4KIIs-depleted cells were infected with Ctr D expressing mCherry for 12 h before analysis by IFM. In both control cells and cells depleted of PI4KIIs, Ctr D (Fig. 6 D, top panels) were internalized and developed within an inclusion (detected using an antibody to the Cap1 inclusion membrane protein) of comparable size and shape (Fig. 6 D, bottom panels). However, relative to control cells, PI4KIIs-depleted cells harbored ∼60% fewer elongated inclusion membrane (IM) tubes decorated by Cap1 (Fig. 6 D, arrowheads; and Fig. 6 E; IM tube/cell, siCTRL: 1.3 ± 0.1; siPI4KIIs: 0.6 ± 0.1). Moreover, the remaining IM tubes in PI4KIIs-depleted cells were ∼70% shorter than in controls (Fig. 6 F; siCTRL: 9.5 ± 1.2 μm, siPI4KIIs: 2.3 ± 0.5 μm). Similar results were observed in Ctr D infected cells that had been depleted of KIF13A or of BLOC-1 (Fig. S6 F). Compared to control cells, the IM tubes per cell in KIF13A- or BLOC-1–depleted cells were fewer in number (Fig. S6 G; siKIF13A: 0.8 ± 0.1, siBLOC-1: 0.8 ± 0.1) and shorter in length (Fig. S6 H; siKIF13A: 5.1 ± 1.0 μm, siBLOC-1: 5.8 ± 1.3 μm). These data reveal that components required to form RE tubules are also used to form tubules from the Chlamydia inclusion membrane.
Discussion
Recycling endosomal tubules are critical in the trafficking of essential physiological cargoes from early endosomes to other intracellular compartments. Here, by exploring the way lipids and proteins shape the early endosomal membrane to form RE tubules, we identified the phospholipid PI4P and the protein complex BLOC-1 as a unit that tubulates endosomal membranes. In vitro analysis with a minimal set of lipids shows that BLOC-1 binding to negatively charged membranes, such as those containing PI3P or PI4P, generates tubules. As a working model (Fig. 6 G), we propose that (1) the PI phosphatase Sac2 activity maintains a low level of PI4P at the PI3P-rich early sorting endosomal membrane by counteracting the activity of PI4KIIs. While BLOC-1 could associate with early endosomes through binding to PI3P+ membranes, (2) the local elevation of PI4P by PI4KIIs would favor (3–4) BLOC-1 concentration on endosomal membrane subdomains that would elongate through the combination of curvature induction and microtubule-based motor engagement (e.g., KIF13A [Delevoye et al., 2014]). While the RE tubule would be (5) stabilized at least in part by membrane-associated KIF13 bound to microtubules, BLOC-1 together with branched actin filaments would contribute to the constriction of the RE tubule neck prior to its release by membrane fission (Delevoye et al., 2016). Together, we conclude that BLOC-1 and PI4P play a central role in the initiation, elongation, and stabilization of RE tubules.
PI4P metabolism relies on specific kinases (PI4KIIs and -IIIs, each comprising two members, α and β) and phosphatases (Sac1 and Sac2) that control PI4P abundance on the cytosolic leaflet of membranes of various organelles (e.g., Golgi apparatus, endosomes, and lysosomes) and transport carriers derived therefrom. PI4KIIα can associate with both vacuolar sorting endosomes (Zhu et al., 2022) and/or endosome-derived tubules in Drosophila and mammalian cells (Burgess et al., 2012; Henmi et al., 2016; Minogue et al., 2006). This indicates that PI4P production and consumption at early endosomal membranes are differently balanced between the vacuolar and tubular domains. Indeed, PI4P extensively decorates RE tubules in different cell types of different species such as human HeLa cells (this study) or mouse pigment cells (Zhu et al., 2022). The relative PI4P abundance on RE tubules over RAB5+ early endosomes might thus reflect a preferential sorting of PI4KIIs to RE tubules (Zhu et al., 2022), and a prominent Sac2 activity at the sorting endosomal membrane, where it is restricted to vacuolar domains (Nakatsu et al., 2015; Hsu et al., 2015), that counteracts the PI4KIIs activity. Hence, a PI3P-to-PI4P gradient would be implemented along the sorting-to-recycling pathway (Ketel et al., 2016). Interestingly, PI4P hydrolysis at RAB5+ early endosomes (via targeting of PJ-Sac) destabilizes the RE tubules, suggesting together that PI3P+ sorting and PI4P+ recycling endosomal membranes may form a continuum, as previously observed in pigment cells (Dennis et al., 2015; Delevoye et al., 2009).
Along such a continuum, local production of PI4P at the sorting endosomal membrane might be a signal to initiate the formation of RE tubules. Depleting PI4KIIs expression abrogates the biogenesis of RE tubules and leads to the accumulation of early vacuolar endosomes with fewer nascent tubules. This suggests that PI4P must first be produced at the sorting endosomal membrane to concentrate locally a curvature-inducing component, like BLOC-1, that contributes to initiate and/or to stabilize membrane curvature prior to elongation to form a RE tubular carrier. The PI4P contribution to the biogenesis of tubular transport carriers might not be restricted to endosomal membranes, as PI4P together with curvature-inducing proteins could aid their formation from various membrane sources, including the Golgi apparatus (Dippold et al., 2009; Rahajeng et al., 2019), autolysosomes (McGrath et al., 2021), or phagolysosomes (Levin-Konigsberg et al., 2019; López-Haber et al., 2020).
We show that acute inhibition of PI4KIIs activity or rapid hydrolysis of endosomal PI4P destabilizes preexisting RE tubules. Moreover, the few tubules that still emerged from vacuolar sorting endosomes in cells depleted of PI4KIIs were unusually short, suggesting that PI4P contributes not only to the initiation of RE tubules but also to their elongation and/or stabilization. The elongating/stabilizing function might not be limited to KIF13+ RE tubules, as PI4P decorates and maintains other extended endosomal tubules with cargo recycling or trafficking functions (Jović et al., 2009; Ma et al., 2020). Given that PI4P, together with BLOC-1 (Delevoye et al., 2016), contributes first to initiate the RE tubule and second to stabilize it, this implies that one or more PI4Ks must be required to supply PI4P along the tubules. This is consistent with data in melanocytes that both PI4KIIα and PI4KIIβ are present on melanosome-bound RE tubules and that both are required for their stability (Zhu et al., 2022). Because we show that the depletion of PI4KIIIβ, which interacts with RAB11 (Burke et al., 2014), or the inhibition of PI4KIIIα enzymatic activity, does not affect the formation or stability of KIF13+ RE tubules, we propose that only PI4KIIs (α and/or β) produce PI4P (1) at the early sorting endosomal membrane to facilitate the biogenesis of RE tubules, and (2) along the RE tubules to support their elongation and/or stabilization.
We also show that BLOC-1 tubulates relatively curved (i.e., small liposomes) or flat (i.e., GUVs) membranes provided they are negatively charged, such as those containing PI4P or PI3P. As shown biochemically, the lack of PI4KIIs expression does not impact the ability of BLOC-1 to bind to cellular membranes (Fig. S3 C; and Zhu et al., 2022). Thus, in cells, BLOC-1 could associate with PI3P-rich vacuolar domains of early endosomes, where local elevation of PI4P would allow its concentration and/or recruitment of PI4P-binding components contributing to RE tubulogenesis, such as AP-1 (Wang et al., 2003), a KIF13A partner (Nakagawa et al., 2000; Delevoye et al., 2009; Campagne et al., 2018) that could thus stabilize the kinesin on PI4P-rich domains. Therefore, BLOC-1 concentration on the PI4P-rich endosomal domain would be sufficient to form and/or stabilize tubules. This is consistent with observations in cells, in which depleting BLOC-1 expression led to the accumulation of early sorting endosomes harboring numerous buds that did not elongate (Delevoye et al., 2016). We, therefore, propose that BLOC-1 is a membrane-shaping complex (Johannes et al., 2014) that facilitates tubulation of PI4P+ cellular membranes.
Several membrane remodeling components, such as dynamin (Kong et al., 2018), the N-BAR protein endophilin (Mim et al., 2012), ESCRT-III complex proteins (Nguyen et al., 2020), retromer (Kovtun et al., 2018), or COP-II complexes (Hutchings et al., 2021), form supramolecular assemblies on membranes. Interestingly, our data suggest that BLOC-1 can be densely packed on PI4P+ or PI3P+ nanotubes without forming an obvious periodic arrangement. Given its 30-nm length (Lee et al., 2012), several BLOC-1 molecules are likely needed to surround RE tubules. By organizing into various architectures, BLOC-1 assemblies could then stabilize tubules of different curvatures (and hence different diameters). Indeed, as shown by EM of purified BLOC-1, the linear chain made by the eight subunits can bend as much as 45° (Lee et al., 2012), together suggesting that BLOC-1 may tune its 3D architecture and/or assembly of molecules on curved membranes.
As PI4P and BLOC-1 function as a module in endosomal membrane remodeling, loss of their functions should have similar consequences. As expected from previous studies (Henmi et al., 2016; Jović et al., 2009; Hsu et al., 2015) and as also observed in cells depleted of BLOC-1 or KIF13A (Delevoye et al., 2014; Setty et al., 2007), cells depleted of PI4KIIs display slower Tf recycling associated with reduced Tf uptake, likely reflecting decreased cell surface expression of TfR. BLOC-1-deficient cells also missort additional cargoes (Monis et al., 2017; Setty et al., 2007; Di Pietro et al., 2006; Delevoye et al., 2016). Similarly, Zhu et al. (2022) document missorting of melanosome cargoes in mouse melanocytes depleted of PI4KIIα or β. Thus, by initiating RE tubulogenesis, BLOC-1 and PI4KIIs together maintain the trafficking and sorting of cargoes along the endocytic pathway.
The requirement for PI4KIIs in RE biogenesis was validated in our analyses of PI4KIIs function during the developmental cycle of two intracellular pathogens. We show that the bacterium C. trachomatis uses PI4KIIs (as well as BLOC-1 and KIF13A) to build the parasitophorous inclusion-derived tubules, while the virus influenza A used PI4KIIs to form adequately sized IAV liquid viral inclusions and to control viral progeny production. The role of PI4KIIs in Chlamydia inclusion maturation is consistent with reports showing the recruitment of RAB11 (Rzomp et al., 2003) and of PI4KIIα-, PI4P-, or PI4P-binding proteins (Moorhead et al., 2010) to the inclusion. Such redistribution of components allows the internalized bacteria to manipulate intracellular pathways such as slow endosomal recycling (Ouellette and Carabeo, 2010) and to complete the Chlamydia infectious cycle (Mölleken and Hegemann, 2017; Leiva et al., 2013). The role of PI4KIIs in IAV assembly is also consistent with reports of the exploitation of RE function by IAV. Cells infected by IAV redistribute RAB11 to vRNPs+ liquid structures, which relocalize to the cell periphery upon KIF13A overexpression (Vale-Costa and Amorim, 2016; Ramos-Nascimento et al., 2017; Amorim et al., 2011; Vale-Costa and Amorim, 2017; Alenquer et al., 2019). Given the requirement of PI4P and BLOC-1 during the building of functional RE tubules, it is likely that many other intracellular pathogens exploit these components to hijack the host RE system to their own benefit (Yong et al., 2021).
The dysregulation of PI4KIIs expression or genetic mutations of BLOC-1 subunits are associated with risks of development of cancers or neurological disorders (Minogue, 2018; Hartwig et al., 2018), and inactivating BLOC-1 subunit mutations are found in subtypes of HPS (Bowman et al., 2019), an inherited syndromic disorder characterized by albinism, excessive bleeding, and other defects. Given that an altered biogenesis and/or function of RE has been characterized in these different disorders (Delevoye et al., 2019; O’Sullivan and Lindsay, 2020), and in order to better understand the mechanisms of membrane dynamics in physiology and their malfunction in pathophysiology, it needs to be defined whether PI4P and BLOC-1 consist of a universal and minimal machinery reshaping membranes into tubules.
Materials and methods
Cell culture, transfections, and infections
Cell culture
Hela cells were grown in DMEM with GlutaMAX (Invitrogen), supplemented with 10% FBS at 37°C under 5% CO2. For viral infection, the epithelial cells A549 human alveolar basal carcinoma were kind gifts of Pr. Paul Digard (Roslin Institute, Edinburgh, UK), cultured as previously described (Amorim et al., 2011), and regularly tested for mycoplasma contamination with the LookOut mycoplasma PCR detection kit (MP0035; Sigma-Aldrich), using JumpStart Taq DNA Polymerase (D9307; Sigma-Aldrich).
Transfection
For plasmid transfections in 6-well plate, cells (2.5 × 105/well or fluorodish [World Precision Instrument]) were seeded at day 1, transfected on day 2 with respective plasmids (300–1,000 ng) using jetPRIME reagent (Polyplus-transfection), and processed on day 3 for IFM, live-cell imaging or immunoblotting (IB). For rapamycin-inducible-sac system, live-cell imaging was carried out by co-transfecting plasmids coding for mRFP-PJ-Sac-FKBP (600 ng) and KIF13A-YFP (1 μg) together with FRB-ECFP(W66A)-Giantin (400 ng) or iRFP-FRB-RAB5 (400 ng), and imaging was carried out before and after addition of rapamycin (1 mM, 20 min; see Hammond et al. [2014] for details). For siRNA transfections in HeLa cells, cells were seeded in 6-well plates on day 1 (1.5 × 105/well), transfected with respective siRNAs (200 pmol/well) on day 2 and day 4 using Oligofectamine (10 μl/well; Invitrogen), and processed on day 6 for IFM or biochemical analyses. For live-cell imaging, siRNA-treated cells were transfected in fluorodish on day 5 with respective plasmids and imaged at day 6. For siRNA transfections in A549 cells, cells were grown in 6-well plates to ∼50% confluency the day before transfection, and transfected (100 pmol/well) using DharmaFECT (Dharmacon) for 48 h, and infected or mock-infected (i.e., non-infected; similar protocols as for infection, but without virus addition) with PR8 at a multiplicity of infection (MOI) of 3 for 8 h.
Infection with pathogens
For bacterial infection, 1.5 × 105 HeLa cells were seeded in 12-well plates before transfection with siRNA by Lipofectamine RNAimax (Invitrogen) for 24 h following the manufacturer’s instructions. Transfected cells were infected with C. trachomatis serovar D stably transformed with the p2TK2-SW2 plasmid expressing mCherry (Agaisse and Derré, 2013) in triplicates at a MOI of 0.2, with a 30-min centrifugation step at 270 g at 37°C. For viral infection, reverse-genetics derived A/Puerto Rico/8/34 (PR8 WT; H1N1) was used as a model virus and titrated according to reference (Vale-Costa et al., 2016). Virus infections were performed at a MOI of 3 for 8 h. After 45 min, cells were overlaid with DMEM containing 10% fetal bovine serum (10500-064; Gibco Life Technologies) and 1% penicillin/streptomycin mix (L0022-100; Biowest).
Plasmids
The KIF13A-YFP and mCherry-RAB11A were obtained by recombination using the GATEWAY cloning system (Invitrogen; Delevoye et al., 2014); GFP-RAB11A was a kind gift from Dr. Bruno Goud (Institut Curie, Paris, France); mCherry-KIF13B plasmid was a kind gift from Pr. Kaori Horiguchi Yamada (University of Illinois, Chicago, IL); plasmid coding for the PI4P sensor (pEGFP-SidC609-776) was previously described (Domingues et al., 2016); plasmid coding for the PI3P sensor (pEGFP-2x-FYVE domain) was a kind gift from Pr. Harald Stenmark (Department of Molecular Cell Biology, Institute of Cancer Research, The Norwegian Radium Hospital, Oslo, Norway). The plasmids coding for the following protein products were obtained from Addgene: non-fluorescent FRB-ECFP(W66A)-Giantin (#67903, pEGFP-C1; Pr. Dorus Gadella, Swammerdam Institute for Life Sciences, Amsterdam, The Netherlands; [van Unen et al., 2015]); mRFP-PJ-Sac-FKBP (Pseudojanin [PJ]-Sac is a mutated chimeric phosphatase which dephosphorylates PI4P to produce PI) (#38000, pmRFP-C1; Dr. Robin Irvine, University of Cambridge, Cambridge, UK [Hammond et al., 2012]); iRFP-FRB-RAB5 (#51612, pEGFP-C1; Pr. Tamas Balla, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD [Hammond et al., 2014]). The BLOC-1 expression plasmid (pST39-BLOC-1) is described in Lee et al. (2012). The GST expression plasmid (pGST-parallel-1) is described in Sheffield et al. (1999).
siRNAs
The sense strand of the following siRNAs was synthesized by Qiagen: (1) Control: 5′-AATTCTCCGAACGTGTCACGT-3′; (2) PI4KIIα: SMARTpool (Flexitube siRNA, GS55361); (3) PI4KIIβ: 5′-CAGAGTACTGGCCTTGTTCAA-3′ (PI4KIIβ#7) and 5′-TCGGATTGTCCACCTGAGCAA-3′ (PI4KIIβ#9); (4) PI4KIIIβ: SMARTpool (Flexitube siRNA, GS5298); (5) BLOC-1 (individual siRNAs against the following BLOC-1 subunits): 5′-AATGCTGGATTCGGGAATTTA-3′ (snapin), 5′-ATGGTCCATGTTAAATGTAAA-3′ (muted), 5′-AAAAGTGCATGTACGGGAAAT-3′ (pallidin#1), 5′-AACTGCAGCAGAAGAGGCAAA-3′ (pallidin#2); (6) KIF13A: 5′-CTGGCGGGTAGCGAAAGAGTA-3′ (KIF13A#2), 5′-CCGCAACAACTTGGTAGGAAA-3′ (KIF13A#3); (7) VPS35: 5′-CACCATACTCCTTTCCATGTA-3′ (VPS35#5), 5′-CACCATACTCCTTTCCATGTA-3′ (VPS35#6); (8) SNX1: 5′-AAGAACAAGACCAAGAGCCAC-3′; (9) SNX2: 5′-AAGTCCATCATCTCCAGAACC-3′. For all PI4KIIs knockdown experiments, expressions of PI4KIIα and PI4KIIβ were extinguished using a mix of individual siRNAs targeting PI4KIIα or PI4KIIβ.
Antibodies and other reagents
Primary antibodies
Sheep polyclonal anti-TGN46 (1:200; IFM; BioRad); mouse monoclonal anti-β-actin (A5316; 1:1,000; Sigma-Aldrich; IB); rabbit polyclonal anti-PI4KIIβ (1:1,000; IB) and -PI4KIIα (1:1,000; IB) were generous gifts from Pr. Pietro De Camilli (Yale School of Medicine, New Haven, CT); rabbit polyclonal anti-β-tubulin (ab6046; 1:1,000; Abcam; IB); rabbit polyclonal anti-pallidin (10891-1-AP; 1:1,000; Proteintech; IB); goat polyclonal anti-VPS35 (ab1099; 1:1,000; Abcam; IB); mouse monoclonal anti-SNX1 (#611482; 1:1,000; BD Bioscience; IB); mouse monoclonal anti-SNX2 (#611308; 1:1,000; BD Bioscience; IB); rabbit polyclonal anti-α-calnexin (ADI-SPA-860D; 1:1,000; Enzo Life Sciences; IB); rabbit polyclonal anit-RAB11A (#15903-1-AP; 1:200; IFM; Proteintech); mouse monoclonal anti-NP (ab20343; 1:1,000; Abcam, IF); rabbit polyclonal anti-NP (1:1,000), -PB1 (v19/6; 1:500), -PB2 (2N580; 1:500), -PA (1:500), and -NS1 (v29; 1:500) for IB were kindly provided by Pr. Paul Digard (Roslin Institute, Edinburgh, UK); mouse monoclonal anti-M2 (ab5416, clone 14C2; 1:500; Abcam; IB); mouse monoclonal anti-actin (#A5441; 1:1,000; Sigma-Aldrich; IB); goat anti-M1 (ab20910; 1:500; Abcam; IB); goat polyclonal anti-GST (#27-4577-01; 1:2,000; GE healthcare, IB); and rabbit polyclonal anti-Cap1 (1 μg/ml, prepared as described in Hamaoui et al. [2020]).
Secondary antibodies
HRP-conjugated goat anti-mouse, anti-rabbit, and anti-donkey (ab6721 and ab6789; 1:10,000; Abcam, and A15999; 1:2,000; Thermo Fisher Scientific; IB), or donkey anti-goat (A15999; 1:2,000; Thermo Fisher Scientific; IB in PIP binding assay); fluorescent antibodies from IRDye range (1:10,000; LI-COR Biosciences; IB); Alexa Fluor (AF)-488, −555 or −647-conjugated anti-rabbit, anti-sheep, or anti-mouse (1:200; Invitrogen; IFM).
Other reagents
AF-488 (1:500) labelled-Tf and AF-647 labelled WGA (1:200) were from Invitrogen (Thermo Fisher Scientific); siR-Tubulin (1:1,000) was from Spirochrome. PAO (PhenylArsineOxide), wortmannin, GSK-A1, and rapamycin were from Sigma Aldrich, and stock solutions were dissolved in dimethyl sulfoxide (DMSO; Sigma Aldrich). The following reagents for in vitro studies were from Avanti Polar Lipids: Egg phosphatidylcholine, brain phosphatidylserine, and 1,2-dioleoyl-sn-glycero-3-phospho-(1'-myo-inositol-3'-phosphate) 18:1 (EPC, bPS, and PI3P, respectively; in chloroform); brain phosphatidylinositol-4-phosphate (bPI4P; in chloroform/methanol/water [20:9:1]); brain phosphatidylinositol-4,5-bisphosphate, C24:1 Galactosyl(β) Ceramide, and 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (bPI[4,5]P2, GalCer, and DOPS; in chloroform/methanol [90/10% v/v]). Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (Texas Red DHPE) and BODIPY Texas Red ceramide (BODIPY TR ceramide) was from Thermo Fisher Scientific.
Drug treatments
Cells were incubated for up to 30 min with PAO (300–600 nM), or with wortmannin (10 mM), or with GSK-A1 (100 nM), or 20 min with rapamycin (1 mM) prior to analyses by live-cell imaging. Control cells were treated with the same volume of DMSO alone.
Preparation of purified BLOC-1
Preparation of BLOC-1 for in vitro experiments using membrane systems
BLOC-1 was prepared according to Lee et al. (2012) with some modifications. The pST39-BLOC-1 plasmid, encoding human BLOC-1 full-length subunits except that a tobacco etch virus (TEV) cleavage site was introduced between dysbindin and GST to generate dysbindin 1-259, was transformed into BL21gold(DE3)plysS cells (230134; Agilent). Several colonies from the plate were inoculated into a starter culture of 10 ml LB supplemented with 34 μg/ml chloramphenicol and 100 μg/ml ampicillin that was grown overnight at 37°C with moderate shaking. Next day, the starter culture was transferred to 1 liter Terrific Broth medium supplemented with antibiotics and grown at 37°C, 200 rpm for ∼7 h. Cultures were induced with 0.8 mM IPTG and grown overnight at 16°C, 200 rpm. Cells were centrifuged at 4°C, 6,000 g for 15 min, and bacterial pellets were resuspended in buffer containing 50 mM Tris (pH 7.4), 300 mM NaCl, 5 mM β-mercaptoethanol supplemented with lysozyme, DNase I, and complete-EDTA-free protease inhibitor tablets (1183617001; Roche) and incubated at 4°C with rotation for 30 min. The sample was further sonicated, and the soluble fraction was separated by centrifugation at 4°C, 35,267 g for 45 min. The supernatant was loaded on a gravity chromatography column preloaded with glutathione-Sepharose 4B (17-0756-05; GE Healthcare) and incubated for 2 h at 4°C with gentle rotation in batch mode. Following incubation, the column was extensively washed with buffer (50 mM Tris [pH 7.4], 300 mM NaCl, and 5 mM β-mercaptoethanol) and on-column cleavage of the GST and 6xHis tags using TEV (His6-TEV) S219V protease (∼0.5 mg/liter) was done overnight at 4°C. The flow-through fraction containing the cleaved BLOC-1 was collected and loaded onto a Ni-NTA column. Flow-through material was collected, concentrated, and further purified by size exclusion chromatography on a Superose 6 10/300 column in buffer containing 50 mM Tris (pH 8.0), 0.4 M NaCl, 2 mM DTT, 5% glycerol and 1 mM EDTA. Fractions containing BLOC-1 were pooled together, aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C. The integrity of the complex was analyzed by SDS-PAGE.
Preparation of GST and BLOC-1-GST
Plasmids encoding GST and BLOC-1-GST were expressed and purified as described above except that after the binding of the protein to glutathione Sepharose 4B and following the washing step, the bound GST proteins were eluted from the beads with elution buffer containing 50 mM Tris (pH 8.0) and 10 mM glutathione. 21 nM of GST or BLOC-1-GST were used for the PIP array assays.
PIP array assay
PIP array (P-6100; Echelon Biosciences)-binding assays were carried out according to the manufacturer’s protocol. Briefly, PIP membranes were blocked in PBS-T (PBS supplemented with 0.1% v/v Tween-20) supplemented with 3% fatty acid free BSA for 1 h at room temperature (RT) with gentle shaking. Following blocking, membranes were incubated with the proteins that were diluted in blocking buffer and incubated for 1 h with gentle agitation. Membranes were washed 3 times in 5 ml PBS-T for 5 min and then incubated with anti-GST antibody for 1 h at RT with gentle agitation. Following incubation, membranes were washed 3 times in 5 ml PBS-T for 5 min and incubated with HRP antibodies. Membranes were washed again 3 times in 5 ml PBS-T for 5 min and an additional final wash of PBS. Membranes were blotted with Pierce ECL Western Blotting Substrate (cat#185698) and signal was detected with Bio-Rad ChemiDoc MP imaging system.
Liposome floatation assay
Dried films of EPC/bPI4P (95/5 mol/mol), EPC/18:1 PI3P (95/5 mol/mol), EPC/bPI(4,5)P2 (95/5 mol/mol), or EPC/bPS (90/10 mol/mol), doped with 1% Texas Red DHPE, were made by evaporation of organic solution under vacuum for 1 h. The lipid film was resuspended in HK buffer (50 mM Hepes [pH 7.4], 120 mM potassium acetate) at 1 mM while vortexing. Liposomes (0.2 mM) and BLOC-1 (0.5 μM) were incubated for 15 min in HK buffer at RT in a Beckman polycarbonate centrifuge tube. A 150 μl mixture was mixed with HK buffer supplemented with 60% sucrose (100 μl), overlaid with an intermediate sucrose solution (HK buffer with 24% sucrose; 150 μl), and a layer of HK buffer (100 μl). After centrifugation (55,000 rpm, TLS 55 Beckman swing rotor) for 90 min at 4°C, three fractions (bottom, medium, top) of 250, 150, and 100 μl were collected. Top fractions that contain fluorescent vesicles were analyzed by SDS-PAGE and Coomassie blue staining.
In vitro experiments using membrane systems
Preparation of lipid nanotubes and binding assay with BLOC-1
GalCer/EPC nanotubes doped or not with PIxP or PS were prepared according to Wilson-Kubalek et al. (1998). Briefly, a mixture of GalCer, EPC, and bPI4P, 18:1 PI3P, bPI(4,5)P2 or bPS (in chloroform/methanol 90/10% v/v) was dried under vacuum. GalCer/EPC (80/20 mol/mol), GalCer/EPC/bPI4P (80/15/5 or 10 mol/mol), GalCer/EPC/18:1 PI3P (80/15/5 mol/mol), GalCer/EPC/bPI(4,5)P2 (80/15/5 mol/mol), or GalCer/EPC/bPS (80/10/10 mol/mol) tubes were formed at RT after resuspension of the dried film at 5 mg/ml total lipid concentration in HK buffer followed by 5 cycles of 10 min vortex, 2 min at 40°C. For experiments, tubes were aliquoted and stored at −20°C. Tubes were diluted in HK buffer at 50 μM and BLOC-1 was added at a lipid/BLOC-1 ratio of 300 mol/mol. After 5 min incubation, sample was used for EM analysis.
Preparation of lipid suspension and binding assay with BLOC-1
Dried films made of EPC/bPS/PI4P (85/10/5 mol/mol) were made by evaporation of organic solution under vacuum for 1 h. The lipid film was resuspended in HK buffer at 1 mM while vortexing. This led to the formation of various populations of vesicles and tubules. For cryo-EM, the lipid suspension was diluted to 50 μM and BLOC-1 was added at a lipid/BLOC-1 ratio of 300 mol/mol and incubated 15 min at RT before grid preparation for cryo-EM.
Giant unilamellar vesicle (GUV) preparation
GUVs were generated using a lipid mixture composed of EPC/DOPS/bPi4P (85/10/5 mol/mol); EPC/DOPS/bPI3P (85/10/5 mol/mol); or EPC only at 1 mg/ml dissolved in chloroform. BODIPY TR ceramide (0.5 mol/mol) was added to visualize GUVs by confocal fluorescence microscopy. The buffer (named inner buffer) used to generate GUVs is 50 mM NaCl, 20 mM sucrose, and 20 mM Tris (pH 7.5). GUVs were prepared using the polyvinyl alcohol (PVA) gel-assisted vesicle formation method as previously described in Weinberger et al. (2013). Briefly, a PVA gel solution (5%, w/w, dissolved in 280 mM sucrose and 20 mM Tris, pH 7.5) warmed up to 50°C was spread on clean coverslips (20 × 20 mm). Before use, the coverslips were washed once with ethanol and twice with ddH2O. For experiments, the PVA-coated coverslips were incubated at 50°C for 30 min, and then ∼5 μl of the mixture of lipids dissolved in organic solution was spread on the PVA-coated coverslips. The coverslips were then placed under vacuum for 30 min at RT, and then in a petri dish. Approximately 500 μl of the inner buffer was pipetted on top of the coverslips. The coverslips were kept at RT for at least 45 min, allowing GUVs to grow. Once done, we gently “ticked” the bottom of the petri dish to detach GUVs from the PVA gel. GUVs were collected using a 1-ml pipette tip with its tip cut to prevent breaking GUVs.
Electron microscopy
Negatively stained images of BLOC-1
BLOC-1 (25 μg/ml) was deposited on a carbon-coated EM grid previously glow-discharged and negatively stained with 2% uranyl acetate in water. Negatively stained images of BLOC-1 were acquired with a QUEMESA CCD camera (Olympus) on a Tecnai Spirit (Thermo Fisher Scientific) operating at 80 kV.
Cryo-EM experiments, imaging, and analysis
Lacey carbon 300 mesh grids (Ted Pella) were used in all cryo-EM experiments. Blotting was carried out on the opposite side from the liquid drop and samples were plunge frozen in liquid ethane (EMGP, Leica). Cryo-EM images were acquired with a Tecnai G2 (Thermo Fisher Scientific) Lab6 microscope operated at 200 kV and equipped with a 4 × 4 k CMOS camera (F416; TVIPS). Image acquisition was performed under low dose conditions of 10 e−/Å2 at a magnification of 50,000 or 6,500 with a pixel size of 0.21 or 1.6 nm, respectively. The diameter of the tubes depicted in Fig. 2 F was measured from >100 tubes in 17 images from two experiments. Diameters were measured every 60 nm along a tube leading to a total of 408 values.
High pressure freezing, sample preparation, and conventional EM imaging
HeLa cells were seeded on carbonated sapphire discs and grown for 2 d prior to transfection with either control or PI4KIIs siRNAs. After two consecutive siRNA transfections, cells were immobilized by HPF using either a HPM 100 (Leica Microsystems) or a HPM Live µ (CryoCapCell), and freeze substituted in anhydrous acetone containing 1% OsO4/2% H2O for 64 h using the Automatic Freeze Substitution unit (AFS; Leica Microsystems). The samples were included in Epon. Ultrathin sections were contrasted with uranylacetate/H2O and Reynold’s lead citrate solution. Electron micrographs were acquired using a Transmission Electron Microscope (Tecnai Spirit; Thermo Fisher Scientific) operated at 80 kV and equipped with a 4 k CCD camera (Quemesa; EMSIS).
Fluorescence microscopy
Live cell
Hela cells were grown on fluorodish and imaged at 37°C in their respective media supplemented with 20 mM Hepes (GIBCO; Thermo Fisher Scientific).
Sample preparation of GUVs and fluorescent imaging
Experimental chambers were assembled by sandwiching two coverslips using parafilm. Before use, the chambers were passivated with a β-casein solution at a concentration of 5 mg/ml in PBS for at least 5 min at RT. GUVs were incubated with 0.2 μM BLOC-1 in the experimental chambers for at least 15 min at RT before observation. To mix GUVs with BLOC-1, we first added 8 μl of the GUVs in 15 μl of an “outer buffer” (60 mM NaCl, 20 mM Tris, pH 7.5), followed by adding 2 μl of BLOC-1 (2.5 μM in stock). Samples were observed using a spinning disk confocal Nikon eclipse Ti-E microscope equipped with Yokogawa CSU-X1 confocal head, 100× CFI Plan Apo VC objective (Nikon) and a CMOS camera, Prime 95B (Photometrics). After formation of tubules, sample was pipetted off and flash-frozen for cryo-EM experiments.
Immunofluorescence (IF)
Hela cells grown on coverslips were first fixed with 4% PFA in PBS for 15 min at RT and then subsequently incubated for 5 min each with PBS/2% glycine and PBS/0.2% BSA (blocking buffer) and PBS/0.2% BSA/0.1% Saponin (permeabilization buffer). The cells were washed three times with permeabilization buffer and incubated 45 min with primary antibodies, washed again and incubated 45 min with secondary antibodies diluted in permeabilization buffer before mounting with Prolong Gold Antifade (Invitrogen). For viral infection studies, A549 cells were similarly fixed with 4% PFA, permeabilized with 0.2% Triton X-100, and stained in PBS/1% PBS and processed as described above. For bacterial inclusion studies, 20 h after infection, HeLa cells were fixed and permeabilized for immunofluorescence using 0.05% saponin, 5 mg/ml BSA in PBS.
Fluorescence imaging
HeLa cells were imaged on an inverted Eclipse Ti-E (Nikon) + Spinning disk CSU-X1 (Yokogawa) integrated in Metamorph software (Molecular Devices, version 7.8.13) with an EMCCD camera (iXon 897; Andor Technology), a 100× 1.4 NA Plan-Apo objective lens and Z-images were taken at 0.2 mm with the piezoelectric motor (Nano z100, Mad City Lab). For imaging of virus-infected cells, single optical sections were imaged with a SP5 live confocal microscope (Leica). Images were post-processed using ImageJ/FIJI (National Institutes of Health).
Super-resolution (SR) microscopy
Images were acquired on a spinning-disk system equipped with an SR module (Live-SR; Gataca Systems) leading to SR spinning-disk confocal microscopy using optical photon reassignment (as described in Azuma and Kei [2015]). This method allows the doubling of resolution together with the physical optical sectioning of confocal microscopy. The maximum resolution is 128 nm with a pixel size in SR mode of 64 nm.
Transferrin recycling assay
Cells starved for 30 min in serum-free medium supplemented with Hepes (20 mM, pH 7.4; Invitrogen) and 0.5% (w/v) BSA medium were pulsed for 10 min with Tf-A488. Cells were then quickly incubated with cold acid buffer (0.5 M Glycine, pH 2.2) to dissociate surface bound Tf, extensively washed with PBS, and chased at 37°C with complete media (containing 20 mM Hepes) for various time points prior to chemical fixation with 4% PFA (Thermo Fisher Scientific) and processing by FM.
Cytosol vs. membrane cell fractionation
Two 10-cm dishes of siRNA-treated Hela cells per condition (∼12 × 106 cells) were washed, scraped, and pelleted in cold PBS. Pellets were incubated in cold hypotonic buffer (20 mM Tris [pH 7.5], 5 mM MgCl2, 1 mM EDTA, 250 mM sucrose supplemented with protease inhibitor cocktail [Roche]) for 15 min and homogenized by passage through a 27 G syringe (30×/sample). Cell homogenates were centrifuged (10,000 rpm, 10 min), and a fraction of post-nuclear supernatants (inputs) were collected and fractionated by ultracentrifugation (80,000 rpm, 60 min). The corresponding supernatants (cytosolic fractions) were collected. The pellets were washed with hypotonic buffer and repelleted by ultracentrifugation (80,000 rpm, 30 min). The resulting pellets were homogenized in hypotonic buffer and collected (membrane fractions). The same volume of each fraction was then subjected to immunoblotting.
Immunoblotting
Hela cells were trypsinized, pelleted, washed with cold PBS and incubated in cold lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% Triton X-100, 10 mM EDTA, pH 7.2, supplemented with protease inhibitor cocktail [Roche]). After the cell lysis, post-nuclear supernatants were obtained by centrifugation. Cell lysates (∼5 μg) or cell fractions as described above were incubated with sample buffer, boiled for 5–10 min before the loading on 4–12% Bis-Tris gels (Nu-PAGE, Invitrogen) and then transferred onto nitrocellulose membranes (GE Healthcare). The membranes were blocked in PBS (with 0.5% Tween-20, 5% non-fat-dried milk) for 30 min and incubated with respective primary antibodies for 8–12 h and 30 min with secondary antibodies diluted in PBS/0.1% Tween 20. Membranes were washed twice with PBS/0.1% Tween-20 for 30 min and developed using ECL SuperSignal West (Pico or Dura, Thermo Fisher Scientific) and visualized using x-ray films (Kodak developer). For virus-infected cell lysates, IB was performed as above and imaged using a LI-COR Biosciences Odyssey near-infrared platform.
Quantitative real-time reverse-transcription PCR (RT-qPCR)
RNA was extracted from samples in NZYol (MB18501; NZYtech) using Direct-zol RNA minipreps (R2052; Zymo Research). Reverse transcription (RT) was performed using the NZY MuLV first strand cDNA synthesis kit (MB17302; NZYTech). Real-time RT-PCR to detect GAPDH, PI4KIIα, PI4KIIβ, and PI4KIIIβ was prepared in 384-well, white, thin-walled PCR Plates (12164142; ABgene) using SYBR Green Supermix (172–5124; BioRad), 10% (v/v) of cDNA and 0.4 µM of each primer. The reaction was performed on an ABI QuantStudio-384 (Applied Biosciences) under the following PCR conditions: Cycle 1 (1 repeat): 95 °C for 2 min; Cycle 2 (40 repeats): 95 °C for 5 s and 60 °C for 30 s; Cycle 3: 95 °C for 5 s and melt curve 65–95 °C (increment 0.05 °C each 5 s). Data were analyzed using QuantStudioTM Real Time PCR software (v1.1., Applied Biosciences). Primer sequences used for real-time RT-qPCR were the following: (1) GAPDH — Fw: 5′-CTCTGCTCCTCCTGTTCGAC-3′, Rv: 5′-ACCAAATCCGTTGACTCCGAC-3′; (2) PI4KIIα — Fw: 5′-TCTTTCCCGAGCGCATCTAC-3′, Rv: 5′-GCAGCCACTTGGTCCACTTA-3′; (3) PI4KIIβ — Fw: 5′-AAGCGGGTGCCTATCTTGTG-3′, Rv: 5′-TTTTGCACGGTCAATCGCAT-3′; (4) PI4KIIIβ — Fw: 5′-TCAGCAGCAACCTGAAACGA-3′, Rv: 5′-CAGTCGAACAGGGGAACTGA-3′.
Image analyses and quantification
KIF13 tubule quantification
Quantification of the average number of cells with at least one KIF13A+ tubule was done manually (Fig. 3, D and H; and Fig. S3, E and G). Quantification of the average length of KIF13A+ tubules was carried out using ImageJ/Fiji as described previously (Delevoye et al., 2016; Dennis et al., 2015). In short, live cell fluorescent images were converted into binary images and skeletonized (Fig. 3 C). The resulting skeletonized images were then analyzed using the integrated plugin “AnalyzeSkeleton.” The skeletonized images were manually compared with the raw images to exclude false-positive tubules from the analysis. The length and number of tubules were determined after applying a threshold of 8 px (1.28 μm). Only tubules longer than 8 px were included in the analysis (Figs. 3 E and 4 E).
Maximum length, width, and neck width of endosomal nascent tubules
EM images were analyzed using the iTEM software that was used to manually draw lines along the long axes of the length, width, or neck of the endosomal nascent tubules. The obtained values were collected in nm and statistical analyses were performed using GraphPad Prism software (version 9; Fig. 5 D; and Fig. S4, B and D).
Measurement of fluorescence intensity
Fluorescence intensity of Tf labeling was measured using the “Measure” function in ImageJ/Fiji on average z-projections from the raw images (Fig. S5, B and C). For each condition, the intensities were normalized to their respective mean at 0 min of chase (Fig. S5 C).
Line scan analysis
Hela cells co-expressing mCherry-KIF13B and SidC-EGFP or EGFP-FYVE were analyzed by live SR-FM. Using the image of the mCherry channel, a line of 30 pixels was drawn crossing the mCh-KIF13B tubule and the fluorescence intensities of the mCherry and GFP were measured per pixel using the “Plot Profile” function in ImageJ/Fiji, then set in nm, and normalized to the maximum value (Fig. 1 D).
Distance analysis
Hela cells co-expressing mCherry-KIF13B and iRFP-RAB5 were analyzed by live SR-FM. Using the merged image of the two channels, the shortest line was drawn between the mCh-KIF13B+ tubule and iRFP-RAB5+ vesicle and the distance in μm was measured by the “Line” function in ImageJ/Fiji (Fig. S1 A).
Image processing
Immunoblot quantification
Protein expression levels were quantified using ImageJ/Fiji software and intensities were normalized to the respective bands of the loading control or using the Image Lab software (version 5.2.1, BioRad). Quantifications were performed on at least three independent experiments.
Bacterial inclusion analyses
5–10 pictures were taken randomly for each coverslip using ImageJ/Fiji. The scale was set from pixels to micrometers; the length in µm of tubules was measured using the ImageJ/Fiji tracing tool and the number of tubules was counted using the multi-point tool. We calculated the mean number of tubules and the average tubular length per cell for each condition. Each condition was analyzed in blinded from three individual coverslips with a minimum of 50 inclusions per coverslips.
Statistical analysis
Statistical data are represented as mean ± SEM. GraphPad (Prism 7 or 9) was used for all statistical analyses. Either unpaired two-tailed Student’s t test or ordinary ANOVA test was used, as indicated in the figure legends. The significant differences noted are between the control and test condition, and only P < 0.05 was considered significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Online supplemental material
Fig. S1 shows by fluorescence imaging that PI4P and KIF13B do not associate with RAB5+ early sorting endosomes. Fig. S2 shows in vitro that BLOC-1 binds to membranes doped with negatively charged phospholipids and tubulates GUVs containing PI3P or PI4P. Fig. S3 shows in HeLa cells that the formation and stabilization of KIF13A-dependent recycling endosomal tubules specifically require the expression or enzymatic activity of PI4KIIs. Fig. S4 shows at the ultrastructural level that the neck and width of nascent recycling endosomal tubules are unaffected in HeLa cells depleted of PI4KIIs. Fig. S5 shows by fluorescence imaging of PI4KIIs-depleted HeLa cells that transferrin internalization is reduced and its recycling is delayed. Fig. S6 shows by biochemistry that the decrease of PI4KIIs expression does not alter the production of PR8 viral proteins, and, by quantifications of fluorescence imaging, that the expression of KIF13A or BLOC-1 is required for the production of long tubules emanating from the bacterial inclusion of C. trachomatis. Video 1 shows by fluorescence imaging that the addition of BLOC-1 to PI4P+ GUVs generates tubules.
Acknowledgments
We thank the Cell and Tissue Imaging core facility (PICT IBiSA, Institut Curie), CryoCapCell (France) for providing equipment (HPM Live µ) and expertise in high pressure freezing, as well as Alexis Canette from the IBPS (Institut de Biologie Paris-Seine) electron microscopy core facility with the support of Sorbonne-Université and CNRS. We thank J. Manzi (BMBC, UMR CNRS 168) for technical support on biochemical analysis of BLOC-1, and Drs. Bruno Antonny and Bruno Mesmin (Université Nice Sophia Antipolis, France) for insightful discussions.
This work was supported by National Institutes of Health grants R01 EY015625 (to M.S. Marks and G. Raposo), GENESPOIR (to C. Delevoye), Institut National de la Santé et de la Recherche Médicale (INSERM), Institut Curie, the Centre National de la Recherche Scientifique (CNRS), Institut Pasteur, the Intramural Program of NICHD, NIH (ZIA-HD001607, to J.S. Bonifacino), the LabEx CellTisPhyBio (ANR-10-LABX-0038/Labex Celltisphybio part of the Idex PSL/International) and the LabEx Cell(n)Scale (ANR-11-LABX-0038, ANR-10-IDEX-0001-02), and the French National Research Infrastructure France-BioImaging (ANR10-INBS-04). M.J. Amorim is funded by The European Research Council consolidator grant under the European Union’s Horizon 2020 research and innovation program (No 101001521 - LOFlu) and by the Portuguese Fundação para a Ciência e a Tecnologia (CEECIND/02373/2020). S. Vale-Costa is funded by a Junior Researcher working contract from Fundação para a Ciência e a Tecnologia (FCT) and Instituto Gulbenkian de Ciência (IGC, Portugal). M. Di Marco is funded by a doctoral contract from the Ligue contre le Cancer.
The authors declare no competing financial interests.
Author contributions: R.A. Jani and C. Delevoye conceived the study; M.J. Amorim, J.S. Bonifacino, A. Subtil, M.S. Marks, D. Lévy, G. Raposo, and C. Delevoye contributed to fund the project; R.A. Jani, A. Di Cicco, T. Keren-Kaplan, S. Vale-Costa, D. Hamaoui, F.-C. Tsai, A. Subtil, D. Lévy, G. Raposo, and C. Delevoye designed the research; R.A. Jani, A. Di Cicco, T. Keren-Kaplan, S. Vale-Costa, D. Hamaoui, I. Hurbain, F.-C. Tsai, M. Di Marco, and D. Lévy performed the experiments; R.A. Jani, A. Di Cicco, T. Keren-Kaplan, S. Vale-Costa, D. Hamaoui, F.-C. Tsai, M. Di Marco, and D. Lévy collected the data; R.A. Jani, A. Di Cicco, T. Keren-Kaplan, S. Vale-Costa, D. Hamaoui, M. Di Marco, A.-S. Macé, A. Subtil, D. Lévy, and C. Delevoye analyzed the data; R.A. Jani, A. Di Cicco, T. Keren-Kaplan, S. Vale-Costa, D. Hamaoui, I. Hurbain, F.-C. Tsai, M. Di Marco, Y. Zhu, M.J. Amorim, P. Bassereau, J.S. Bonifacino, A. Subtil, M.S. Marks, D. Lévy, G. Raposo, and C. Delevoye interpreted the results; R.A. Jani and C. Delevoye wrote the manuscript with contribution from A. Di Cicco, T. Keren-Kaplan, S. Vale-Costa, D. Hamaoui, F.-C. Tsai, M.J. Amorim, P. Bassereau, J.S. Bonifacino, A. Subtil, M.S. Marks, D. Lévy, and G. Raposo. All authors contributed intellectual capital into the study and edited versions of the manuscript.

