


Activation of T cells relies on the availability of intracellular cholesterol for an effective response after stimulation. We investigated the contribution of cholesterol derived from extracellular uptake by the low-density lipoprotein (LDL) receptor in the immunometabolic response of T cells. By combining proteomics, gene expression profiling, and immunophenotyping, we described a unique role for cholesterol provided by the LDLR pathway in CD8+ T cell activation. mRNA and protein expression of LDLR was significantly increased in activated CD8+ compared to CD4+ WT T cells, and this resulted in a significant reduction of proliferation and cytokine production (IFNγ, Granzyme B, and Perforin) of CD8+ but not CD4+ T cells from Ldlr −/− mice after in vitro and in vivo stimulation. This effect was the consequence of altered cholesterol routing to the lysosome resulting in a lower mTORC1 activation. Similarly, CD8+ T cells from humans affected by familial hypercholesterolemia (FH) carrying a mutation on the LDLR gene showed reduced activation after an immune challenge.

Introduction
Activation of T lymphocytes requires the adaptation of cellular sterol metabolism to cope with the increased cholesterol demand required for cellular proliferation and function (Yvan-Charvet et al., 2019). This includes the activation of sterol regulatory element binding proteins (SREBPs), which control the transcription of genes involved in de novo synthesis and uptake of lipids and the repression of liver X receptors (LXRs), required for the transcription of genes promoting cellular lipid efflux. Cholesterol accumulation due to LXR deficiency leads to increased T cell proliferation (Bensinger et al., 2008), while cholesterol shortage induced by SREBP2 deficiency is associated with decreased T cell blastogenesis (Kidani et al., 2013), thus highlighting how regulation of cholesterol homeostasis is critical for T cell response.
The low-density lipoprotein receptor (LDLR) is a cell surface protein involved in receptor-mediated endocytosis of low-density lipoprotein (LDL) particles. Once bound, LDLs are taken into the cell and routed to the lysosome, where the receptor is degraded, and cholesterol is made available. Mutations in this gene cause the autosomal dominant disorder familial hypercholesterolemia (FH). Preliminary characterization of lymphocytes from FH subjects suggested that impaired extracellular cholesterol uptake from LDL can be only partially compensated by an increase of intracellular cholesterol biosynthesis (Ho et al., 1977). However, the inhibition of cholesterol synthesis following statins treatment, used in cardiovascular prevention, has a limited effect on T cell responsiveness (Cherfan et al., 2007; Fehr et al., 2004), probably due to their primary pharmacological effects in the liver, although experiments in vitro have shown that statins dampen T cell activation and proliferation (Bietz et al., 2017; Jameel et al., 2013). Collectively these observations highlight a current gap in the understanding of the relative contribution of extracellular uptake vs de novo synthesis of cholesterol to fulfill sterol requirements upon T cell activation. Understanding these aspects will contribute to clarify whether targeting selective pathways of sterol metabolism in T cells may be therapeutically exploited to improve the immune response in different pathological conditions.
By combining proteomics, gene expression profiling, and immunophenotyping, here we describe a unique role for cholesterol provided by the LDLR in CD8+ but not in CD4+ T cell activation. Specifically, we show that the LDLR plays a nonredundant role in regulating lysosomal cholesterol availability, which in turn controls mammalian target of rapamycin (mTORC1) localization at the lysosomal membrane, which is crucial for its function and subsequently CD8+ T cell activation. Defective CD8+ T cell activation in experimental models lacking the LDLR as well as in subjects carrying a loss of function mutations on the LDLR gene (FH subjects) suggests that therapeutic targeting of the LDLR–lysosome axis may be tested to improve the effector function of CD8+ T cells.
Results
LDLR is critical for CD8+ but not CD4+ T cell activation
To understand the relevance of cholesterol uptake by the LDLR on T cell responsiveness, we tested the activation of lymphocytes isolated from Ldlr −/− or WT mice following incubation with allogenic splenocytes from Balb/cJ mice (Fig. 1 A). Flow cytometry analysis of CD8+ T cells proliferation, as well as cytokines (IFNγ and TNFα) and perforin production were reduced in Ldlr −/− CD8+ T cells compared with WT cells (Fig. 1, B and C). This difference was a specific trait of LDLR deficiency in CD8+ T cells as CD4+ T cells from Ldlr −/− mice presented a similar activation (proliferation and cytokine production) compared with that of WT mice (Fig. 1, D and E). We excluded that this effect was the consequence of a different ability of dendritic cells (DCs; isolated from the spleen of Ldlr −/− and WT mice) to present antigens to T cells and to promote the proliferation of CD8+ or CD4+ T cells from Balb/cJ mice (Fig. 1, F and H). Altogether, these findings suggest that the LDLR plays a crucial role in CD8+ T cell activation.

LDLR is critical for CD8 + but not CD4 + T cell activation. (A) Workflow of in vitro proliferation of WT and Ldlr−/− T cells: T cells isolated from WT and Ldlr −/− mice were labeled with an intracellular dye and activated with allogenic splenocytes from Balb/cJ mice for 5 d; proliferation and cytokine production were analyzed by flow cytometry. (B and C) Proliferation (B) and cytokine production (C) of CD8+ T cells from WT and Ldlr−/− mice by in vitro stimulation with Balb/cJ splenocytes, n = 6/group; representative histograms from flow cytometry analysis are shown. (D and E) Proliferation (D) and cytokine production (E) of CD4+ T cells from WT and Ldlr −/− mice by in vitro stimulation with Balb/cJ splenocytes, n = 6/group; representative histograms from flow cytometry analysis are shown. (F) Workflow of in vitro proliferation of allogenic T cells: T cells isolated from allogenic Balb/cJ mice were labeled with an intracellular dye and activated with spleen-derived dendritic cells (DCs) from WT and Ldlr −/− mice for 5 d and proliferation analyzed by flow cytometry. (G and H) Proliferation of Balb/cJ CD8+ or CD4+T cells cultured for 5 d with spleen-derived dendritic cells (DCs) isolated from WT and Ldlr −/− mice, n = 6/group; representative histograms from flow cytometry analysis are shown (H). Results are presented as mean per group ± SEM; statistical analysis is performed with Mann–Whitney test; *P < 0.05 and **P < 0.01 (B–E and G–J).
LDLR is critical for CD8 + but not CD4 + T cell activation. (A) Workflow of in vitro proliferation of WT and Ldlr−/− T cells: T cells isolated from WT and Ldlr −/− mice were labeled with an intracellular dye and activated with allogenic splenocytes from Balb/cJ mice for 5 d; proliferation and cytokine production were analyzed by flow cytometry. (B and C) Proliferation (B) and cytokine production (C) of CD8+ T cells from WT and Ldlr−/− mice by in vitro stimulation with Balb/cJ splenocytes, n = 6/group; representative histograms from flow cytometry analysis are shown. (D and E) Proliferation (D) and cytokine production (E) of CD4+ T cells from WT and Ldlr −/− mice by in vitro stimulation with Balb/cJ splenocytes, n = 6/group; representative histograms from flow cytometry analysis are shown. (F) Workflow of in vitro proliferation of allogenic T cells: T cells isolated from allogenic Balb/cJ mice were labeled with an intracellular dye and activated with spleen-derived dendritic cells (DCs) from WT and Ldlr −/− mice for 5 d and proliferation analyzed by flow cytometry. (G and H) Proliferation of Balb/cJ CD8+ or CD4+T cells cultured for 5 d with spleen-derived dendritic cells (DCs) isolated from WT and Ldlr −/− mice, n = 6/group; representative histograms from flow cytometry analysis are shown (H). Results are presented as mean per group ± SEM; statistical analysis is performed with Mann–Whitney test; *P < 0.05 and **P < 0.01 (B–E and G–J).
Next, we asked whether this difference could have been the consequence of impaired CD8+ T cells priming by the hypercholesterolemic environment already observed in Ldlr −/− mice on a standard fat diet (plasma total cholesterol levels: 200.7 ± 33.2 mg/dl for Ldlr −/− and 64.2 ± 11.0 mg/dl for WT mice). To address this question, T cells were isolated from Ldlr −/− or WT mice and injected i.v. concomitantly with Balb/cJ splenocytes (i.p. injection) in C57BL/6J mice to test the in vivo allogenic response in a normocholesterolemic environment (Fig. 2 A). Five days later, a significantly reduced proliferation was still observed in CD8+ T cells from Ldlr −/− compared to those of WT mice (Fig. 2 B), while the proliferation of CD4+ T cells was not different (Fig. 2 C). In line with this, the proliferation of CD8+ T cells isolated from Ldlr −/− mice fed a high cholesterol diet was significantly reduced compared with that observed in WT mice fed the same diet (plasma total cholesterol levels: 604.8 ± 42 mg/dl for Ldlr −/− and 164.7 ± 31.3 mg/dl for WT; Fig. 2 D). Vice versa, the proliferation of CD4+ T cells was slightly increased (Fig. 2 E), in agreement with a previous observation in Ldlr −/− mice and in other models of hypercholesterolemia (Bonacina et al., 2020; Bonacina et al., 2018). These data suggest that the deficiency of LDLR in CD8+ T cells, rather than hypercholesterolemia, impairs their activation. Moreover, both thymic differentiation of T cell subsets (Fig. S1, A–D) and the distribution in naïve and memory subsets in both CD4+ and CD8+ T cell subsets (Fig. S1, E and F) were similar between Ldlr −/− and WT mice, thus excluding a major role of the LDLR in T cell generation and differentiation. In addition, a similar expression of Ki67, a marker of cell proliferation, was detected in CD8+ T cells freshly isolated from Ldlr −/− or WT mice (Fig. S2 A), suggesting that homeostatic (basal) proliferation is not affected by LDLR deficiency. This was also confirmed when T cells were isolated from Ldlr −/− or WT mice, labeled, and transferred to Rag2 −/− recipients to evaluate CD8+ T cell physiology under homeostatic conditions (Fig. S2 B); under this setting, no differences in proliferation and proinflammatory and cytotoxic cytokine production were observed in CD8+ from Ldlr −/− or WT mice (Fig. S2, C and D). Therefore, to further characterize the relevance of LDLR on CD8+ T cell activation, we tested antigen-dependent CD8+ T cell activation following immunization of Ldlr −/− mice with ovalbumin (Fig. 2 F). Under this experimental condition, both proliferation and cytokine production after ex vivo restimulation with OT-I peptide of cells isolated from lymph nodes (Fig. 2, G and H) and spleen (Fig. S2, E and F) were significantly reduced in CD8+ T cells from Ldlr −/− compared to those from WT mice, while those of CD4+ T cell, after OT-II peptide re-challenge, were not different (Fig. 2, I and J), thus confirming in vivo the critical role of the LDLR for CD8+ but not for CD4+ T cell activation. These data suggest that LDLR is involved in CD8+ T cell functional reprogramming after antigen-mediated stimulation of de novo–activated naïve and restimulated memory T cells.

Antigen-dependent activation requires LDLR expression in CD8 + T cells. (A) Workflow of in vivo alloresponse assay: T cells isolated from WT and Ldlr −/− mice were labeled with an intracellular dye and intravenously (i.v.) injected into normocholesterolemic C57BL6/J WT mice together with intraperitoneal (i.p.) injection of allogenic splenocytes from Balb/cJ mice. 5 d after, labeled cells were detected from mesenteric lymph nodes. (B and C) Proliferation of CD8+ (B) and CD4+ (C) T cells from WT and Ldlr −/− mice in response to “in vivo” alloresponse assay, n = 5/group; representative histograms from flow cytometry analysis are shown. (D and E) Proliferation of CD8+ (D) and CD4+ (E) T cells stimulated in vitro with anti-CD3/28 + IL-2 for 96 h isolated from WT and Ldlr −/− mice fed to high cholesterol rich diet (WTD) for 8 wk n = 6/group. (F) Workflow of immune response to ovalbumin vaccination and OT-I restimulation experiment: WT and Ldlr −/− mice received two immunizations with ovalbumin peptide; 1 wk after the second injection, lymph nodes were harvested and lymphocytes restimulated with OT-I peptides specific for ovalbumin-activated CD8+ T cells. (G and H) Proliferation (G) and cytokine production (H) of CD8+ T cells from lymph nodes of WT and Ldlr −/− mice immunized with ovalbumin and restimulated ex vivo with OT-I peptides for 5 d, n = 4/group; representative histograms from flow cytometry analysis are shown. (I and J) Proliferation (I) and cytokine production (J) of CD4+ T cells from lymph nodes of WT and Ldlr −/− mice immunized with ovalbumin and restimulated ex vivo with OT-II peptides for 5 d, n = 4/group; representative histograms from flow cytometry analysis are shown. Results are presented as mean per group ± SEM; statistical analysis is performed with Mann–Whitney test; *P < 0.05 and **P < 0.01 (B–E and G–J).
Antigen-dependent activation requires LDLR expression in CD8 + T cells. (A) Workflow of in vivo alloresponse assay: T cells isolated from WT and Ldlr −/− mice were labeled with an intracellular dye and intravenously (i.v.) injected into normocholesterolemic C57BL6/J WT mice together with intraperitoneal (i.p.) injection of allogenic splenocytes from Balb/cJ mice. 5 d after, labeled cells were detected from mesenteric lymph nodes. (B and C) Proliferation of CD8+ (B) and CD4+ (C) T cells from WT and Ldlr −/− mice in response to “in vivo” alloresponse assay, n = 5/group; representative histograms from flow cytometry analysis are shown. (D and E) Proliferation of CD8+ (D) and CD4+ (E) T cells stimulated in vitro with anti-CD3/28 + IL-2 for 96 h isolated from WT and Ldlr −/− mice fed to high cholesterol rich diet (WTD) for 8 wk n = 6/group. (F) Workflow of immune response to ovalbumin vaccination and OT-I restimulation experiment: WT and Ldlr −/− mice received two immunizations with ovalbumin peptide; 1 wk after the second injection, lymph nodes were harvested and lymphocytes restimulated with OT-I peptides specific for ovalbumin-activated CD8+ T cells. (G and H) Proliferation (G) and cytokine production (H) of CD8+ T cells from lymph nodes of WT and Ldlr −/− mice immunized with ovalbumin and restimulated ex vivo with OT-I peptides for 5 d, n = 4/group; representative histograms from flow cytometry analysis are shown. (I and J) Proliferation (I) and cytokine production (J) of CD4+ T cells from lymph nodes of WT and Ldlr −/− mice immunized with ovalbumin and restimulated ex vivo with OT-II peptides for 5 d, n = 4/group; representative histograms from flow cytometry analysis are shown. Results are presented as mean per group ± SEM; statistical analysis is performed with Mann–Whitney test; *P < 0.05 and **P < 0.01 (B–E and G–J).

Phenotyping of T cells in thymus ans spleen of WT and Ldlr −/− mice. (A) Gating strategy of thymus immunophenotyping: cells were gated based on dimensions (SSC-H vs. FSC-H) and single thymocytes discriminated (SSC-A vs. SSC-H). CD4+ single positive (CD4), CD8+ single positive (CD8), and CD4+ and CD8+ double positive (DP) were gated based on the fluorescence intensity for CD4+ or CD8+. (B) Percentage of CD4+ single positive (CD4), CD8+ single positive (CD8), and CD4+ and CD8+ double positive (DP) T cells in 8/10-wk-old WT and Ldlr −/− littermates, n = 3/group. (C) Gating strategy of spleen immunophenotyping: cells were gated based on dimensions (SSC-H vs. FSC-H) and single splenocytes discriminated (SSC-A vs. SSC-H). CD4+ (CD4) and CD8+ (CD8) were gated based on the intensity fluorescence for CD4+ or CD8+ respectively. T effector memory (TEM, CD44+CD62L−), T central memory (TCM, CD44+CD62L+) and T naive (CD44−CD62L+) were then identified by CD44 and CD62L fluorescence intensity. (D) Percentage of CD4+ (CD4) and CD8+ (CD8) in 8/10-wk-old WT and Ldlr −/− littermates, n = 4/group. (E and F) Percentage of CD4+ subsets (E) and CD8+ subsets (F) in the spleen of 8/10-wk-old WT and Ldlr −/− littermates, n = 4/group. Results are presented as mean per group ± SEM; statistical analysis is performed with Mann–Whitney test; (B and D) and two-way ANOVA (E and F) *P < 0.05 and **P < 0.01.
Phenotyping of T cells in thymus ans spleen of WT and Ldlr −/− mice. (A) Gating strategy of thymus immunophenotyping: cells were gated based on dimensions (SSC-H vs. FSC-H) and single thymocytes discriminated (SSC-A vs. SSC-H). CD4+ single positive (CD4), CD8+ single positive (CD8), and CD4+ and CD8+ double positive (DP) were gated based on the fluorescence intensity for CD4+ or CD8+. (B) Percentage of CD4+ single positive (CD4), CD8+ single positive (CD8), and CD4+ and CD8+ double positive (DP) T cells in 8/10-wk-old WT and Ldlr −/− littermates, n = 3/group. (C) Gating strategy of spleen immunophenotyping: cells were gated based on dimensions (SSC-H vs. FSC-H) and single splenocytes discriminated (SSC-A vs. SSC-H). CD4+ (CD4) and CD8+ (CD8) were gated based on the intensity fluorescence for CD4+ or CD8+ respectively. T effector memory (TEM, CD44+CD62L−), T central memory (TCM, CD44+CD62L+) and T naive (CD44−CD62L+) were then identified by CD44 and CD62L fluorescence intensity. (D) Percentage of CD4+ (CD4) and CD8+ (CD8) in 8/10-wk-old WT and Ldlr −/− littermates, n = 4/group. (E and F) Percentage of CD4+ subsets (E) and CD8+ subsets (F) in the spleen of 8/10-wk-old WT and Ldlr −/− littermates, n = 4/group. Results are presented as mean per group ± SEM; statistical analysis is performed with Mann–Whitney test; (B and D) and two-way ANOVA (E and F) *P < 0.05 and **P < 0.01.

Proliferation after in vivo homeostatic and in vitro antigentic stimulation of CD8+ T cells from WT and Ldlr −/− mice. (A) Percentage of Ki67+ CD8+ T cells in lymph nodes of WT and Ldlr −/− littermates under resting conditions, n = 3/group. Representative density plots from flow cytometry analysis are shown on the right. (B) Workflow of homeostatic proliferation experiment: T cells isolated from WT and Ldlr −/− mice were labelled with an intracellular dye and intravenous (i.v.) injected into Rag2 −/− mice. 5 d after, labeled cells were detected from spleens. (C and D) Homeostatic proliferation (C) and cytokine production (D) of CD8+ T cells from WT and Ldlr −/− mice i.v. injected into Rag2 −/− mice after 5 d; representative histograms from flow cytometry analysis are shown. (E and F) Proliferation (E) and cytokine production (F) of CD8+ T cells from spleens of WT and Ldlr −/− mice immunized with ovalbumin and restimulated ex vivo with OT-I peptides for 5 d, n = 4/group; representative histograms from flow cytometry analysis are shown. Results are presented as mean per group ± SEM; statistical analysis is performed with Mann-Whitney test; *P < 0.05 and **P < 0.01 (A, C, and D–F).
Proliferation after in vivo homeostatic and in vitro antigentic stimulation of CD8+ T cells from WT and Ldlr −/− mice. (A) Percentage of Ki67+ CD8+ T cells in lymph nodes of WT and Ldlr −/− littermates under resting conditions, n = 3/group. Representative density plots from flow cytometry analysis are shown on the right. (B) Workflow of homeostatic proliferation experiment: T cells isolated from WT and Ldlr −/− mice were labelled with an intracellular dye and intravenous (i.v.) injected into Rag2 −/− mice. 5 d after, labeled cells were detected from spleens. (C and D) Homeostatic proliferation (C) and cytokine production (D) of CD8+ T cells from WT and Ldlr −/− mice i.v. injected into Rag2 −/− mice after 5 d; representative histograms from flow cytometry analysis are shown. (E and F) Proliferation (E) and cytokine production (F) of CD8+ T cells from spleens of WT and Ldlr −/− mice immunized with ovalbumin and restimulated ex vivo with OT-I peptides for 5 d, n = 4/group; representative histograms from flow cytometry analysis are shown. Results are presented as mean per group ± SEM; statistical analysis is performed with Mann-Whitney test; *P < 0.05 and **P < 0.01 (A, C, and D–F).
Activated CD8+ T cells rely on cholesterol uptake by LDLR
T cell activation promotes a rapid reprogramming of cellular cholesterol metabolism that couples with the acquisition of an effector response; intriguingly, our phenotypical and functional observations point to a different role for LDLR-dependent cholesterol uptake in CD4+ and CD8+ T cells. Although T cell activation is coupled to increased sterol metabolism, we showed that LDLR expression, both at the mRNA and protein level, is largely upregulated in CD8+ as compared to CD4+ T cells (Fig. 3, A–C). Accordingly, Gene Ontology (GO) enrichment analysis of a proteomic dataset from extensive quantitative mass spectrometry analysis of activated CD4+ and CD8+ T cells (Howden et al., 2019) revealed a significant enrichment of pathways related to lipid and sterol metabolism in activated CD8+ T cells as compared to CD4+ T cells (Fig. S3 A). Key proteins involved in sterols (3-hydroxy-3-methylglutaryl-CoA reductase [HMGCR]) and fatty acids biosynthesis (fatty acid synthase [FASN]; ethylmalonyl-CoA decarboxylase 1 [ECHDC1]), as well as those involved in lipoproteins uptake, such as LDLR, were significantly increased in CD8+ T cells compared to CD4+ T cells (Fig. S3 B). These results strengthen that LDLR-dependent cholesterol uptake could play a different role in activated CD4+ compared with activated CD8+ T cells. To test this hypothesis further, we stimulated CD8+ and CD4+ T cells from Ldlr −/− or WT mice in serum-free medium in the presence or not of LDL particles. While the incubation of WT CD8+ T cells with LDL significantly increased their proliferation, the same was not true for Ldlr −/− CD8+ T cells (Fig. 3 D). Interestingly, under the same experimental conditions, WT or Ldlr −/− CD4+ T cell proliferation was not affected by LDL incubation (Fig. 3 E).
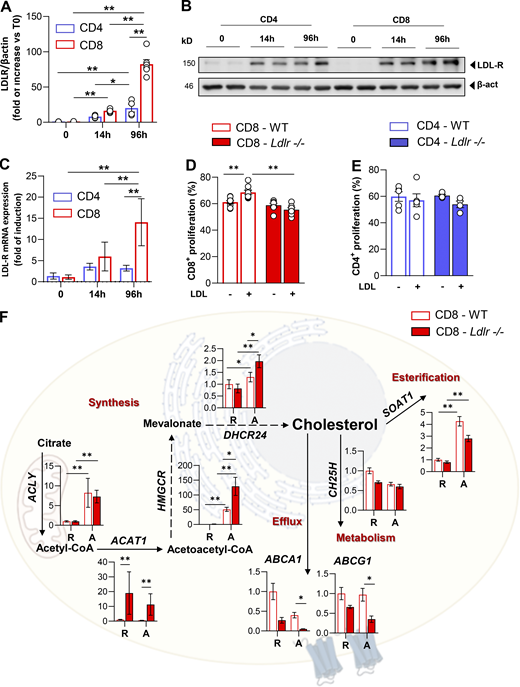
LDLR deficiency rewires sterol metabolism of activated CD8+T cells. (A and B) LDLR protein expression in WT CD4+ and CD8+ T cells under resting (T0) and activated (14- or 96 h) conditions. Representative immunoblots for LDLR and β-actin proteins are shown in B. Data are normalized on β-actin expression, n = 6/condition. (C) mRNA expression of LDLR gene in WT CD4+ and CD8+ T cells under resting (T0) and activated (14- or 96 h) conditions, data are normalized on the housekeeping gene, n = 6/condition. (D and E) Proliferation of CD8+ (D) and CD4+ (E) T cells from WT and Ldlr −/− mice stimulated in vitro for 96 h in the presence or absence of LDL particles (5 μg/ml) in serum free-medium, n = 7–8/condition. (F) mRNA expression profile of genes related to sterol metabolism in resting (R) and 14-h activated (A) CD8+ T cells isolated from WT and Ldlr −/− mice. Representative genes related to cholesterol synthesis: Acly (ATP Citrate Lyase), Acat1 (Acetyl-CoA Acetyltransferase 1); Hmgcr (3-Hydroxy-3-Methylglutaryl-CoA Reductase), Dhcr24 (24-Dehydrocholesterol Reductase); cholesterol efflux: Abca1 (ATP Binding Cassette Subfamily A Member 1), Abcg1 (ATP Binding Cassette Subfamily G Member 1); cholesterol oxidation: Ch25h (Cholesterol 25-Hydroxylase) and cholesterol esterification: Soat1 (Sterol O-Acyltransferase 1) are shown; n = 6/condition. Results are presented as mean per group ± SEM; statistical analysis is performed with 2-way Anova; *P < 0.05 and **P < 0.01 (A and C–F). Source data are available for this figure: SourceData F3.
LDLR deficiency rewires sterol metabolism of activated CD8+T cells. (A and B) LDLR protein expression in WT CD4+ and CD8+ T cells under resting (T0) and activated (14- or 96 h) conditions. Representative immunoblots for LDLR and β-actin proteins are shown in B. Data are normalized on β-actin expression, n = 6/condition. (C) mRNA expression of LDLR gene in WT CD4+ and CD8+ T cells under resting (T0) and activated (14- or 96 h) conditions, data are normalized on the housekeeping gene, n = 6/condition. (D and E) Proliferation of CD8+ (D) and CD4+ (E) T cells from WT and Ldlr −/− mice stimulated in vitro for 96 h in the presence or absence of LDL particles (5 μg/ml) in serum free-medium, n = 7–8/condition. (F) mRNA expression profile of genes related to sterol metabolism in resting (R) and 14-h activated (A) CD8+ T cells isolated from WT and Ldlr −/− mice. Representative genes related to cholesterol synthesis: Acly (ATP Citrate Lyase), Acat1 (Acetyl-CoA Acetyltransferase 1); Hmgcr (3-Hydroxy-3-Methylglutaryl-CoA Reductase), Dhcr24 (24-Dehydrocholesterol Reductase); cholesterol efflux: Abca1 (ATP Binding Cassette Subfamily A Member 1), Abcg1 (ATP Binding Cassette Subfamily G Member 1); cholesterol oxidation: Ch25h (Cholesterol 25-Hydroxylase) and cholesterol esterification: Soat1 (Sterol O-Acyltransferase 1) are shown; n = 6/condition. Results are presented as mean per group ± SEM; statistical analysis is performed with 2-way Anova; *P < 0.05 and **P < 0.01 (A and C–F). Source data are available for this figure: SourceData F3.

Adaptation of sterol metabolism in activated T cells. (A) Comparison of GO term enrichment analysis of activated CD8+ T cells compared to CD4+ T cells (data is available for interrogation using the Encyclopedia of Proteome Dynamics [EPD; Howden et al., 2019]). Data are shown as annotated pathways with a log2 of fold change (FC) > 1.35 or < −1.35 and a statistically significant t test (P < 0.05) in CD8+ T cells compared to CD4+ T cells. Data are reported as -log10 of false discovery rate (FDR). For each pathway, the number of proteins identified in the dataset compared to the total protein belonging to that pathway are reported. (B) Volcano plot showing changes in the expression of proteins related to cholesterol metabolism for activated CD8+ compared to activated CD4+ T cells (EPD; Howden et al., 2019). Proteins highlighted in red are those which are significantly modulated and with a Log2FC > 2. (C) mRNA expression profile of genes related to sterol metabolism in resting (R) and 14-h activated (A) CD4+ T cells isolated from WT and Ldlr −/− mice. Representative genes related to cholesterol synthesis: Acly (ATP Citrate Lyase), Acat1 (Acetyl-CoA Acetyltransferase 1); Hmgcr (3-Hydroxy-3-Methylglutaryl-CoA Reductase), Dhcr24 (24-Dehydrocholesterol Reductase); cholesterol effux: Abca1 (ATP Binding Cassette Subfamily A Member 1), Abcg1 (ATP Binding Cassette Subfamily G Member 1); cholesterol oxidation: Ch25h (Cholesterol 25-Hydroxylase) and cholesterol esterification: Soat1 (Sterol O-Acyltransferase 1) are shown; n = 6/condition.
Adaptation of sterol metabolism in activated T cells. (A) Comparison of GO term enrichment analysis of activated CD8+ T cells compared to CD4+ T cells (data is available for interrogation using the Encyclopedia of Proteome Dynamics [EPD; Howden et al., 2019]). Data are shown as annotated pathways with a log2 of fold change (FC) > 1.35 or < −1.35 and a statistically significant t test (P < 0.05) in CD8+ T cells compared to CD4+ T cells. Data are reported as -log10 of false discovery rate (FDR). For each pathway, the number of proteins identified in the dataset compared to the total protein belonging to that pathway are reported. (B) Volcano plot showing changes in the expression of proteins related to cholesterol metabolism for activated CD8+ compared to activated CD4+ T cells (EPD; Howden et al., 2019). Proteins highlighted in red are those which are significantly modulated and with a Log2FC > 2. (C) mRNA expression profile of genes related to sterol metabolism in resting (R) and 14-h activated (A) CD4+ T cells isolated from WT and Ldlr −/− mice. Representative genes related to cholesterol synthesis: Acly (ATP Citrate Lyase), Acat1 (Acetyl-CoA Acetyltransferase 1); Hmgcr (3-Hydroxy-3-Methylglutaryl-CoA Reductase), Dhcr24 (24-Dehydrocholesterol Reductase); cholesterol effux: Abca1 (ATP Binding Cassette Subfamily A Member 1), Abcg1 (ATP Binding Cassette Subfamily G Member 1); cholesterol oxidation: Ch25h (Cholesterol 25-Hydroxylase) and cholesterol esterification: Soat1 (Sterol O-Acyltransferase 1) are shown; n = 6/condition.
As a result of impaired cholesterol uptake, early activated Ldlr −/− CD8+ T cells upregulated the expression of genes involved in cholesterol biosynthesis (HMGCR, DHCR24, ACAT1) and reduced that of the genes involved in cholesterol efflux (ABCA1, ABCG1) and esterification (SOAT1) compared with WT CD8+ T cells, both under resting and activated conditions (Fig. 3 F). By contrast, the cholesterol biosynthetic pathway was upregulated to a lower extent in CD4+ T cells, showing no difference between WT and Ldlr −/− CD4+ T cells (Fig. S3 C).
As cholesterol is essential to build up membranes during T cell proliferation and to favor the anchoring of signal receptors in lipid rafts, we next wondered whether defective LDLR-dependent cholesterol uptake could perturb lipid raft enrichment and TCR signaling. Following T cell activation, lipid raft enrichment increased to a similar extent in CD8+ T cells from WT and Ldlr −/− mice (Fig. S4 A), as did the content of free cholesterol (Fig. S4 B). Moreover, reduced T cell proliferation (Fig. S4 E) coupled with reduced IFNγ and perforin production was observed in Ldlr −/− CD8+ T lymphocytes (Fig. S4, F and G) and also when cells were activated independently of TCR stimulation (through PMA and Ionomycin activation). Of note, no difference in lipid rafts content in CD4+ T cells from Ldlr −/− or WT mice (Fig. S4, C and D) and in cellular proliferation following stimulation with PMA/Ionomycin was observed (Fig. S4 H). To further prove that the mechanism underlying the defective response in CD8+ T cells from Ldlr −/− mice was not the consequence of a different cholesterol availability in the plasma membrane, we evaluated the activation of TCR downstream signaling pathways after T cell stimulation. Phosphorylation of proximal TCR molecules (pZAP and pLck) and NFAT1 dephosphorylation were similar in activated CD8+ T cells from WT and Ldlr −/− mice (Fig. S4, I, J, and M). Similarly, no alterations in TCR signaling were reported in CD4+ T cells from WT and Ldlr −/− mice (Fig. S4, K, L, and N). These data suggest that the impaired activation of CD8+ T cells in Ldlr −/− mice does not depend on alterations in membrane lipid rafts content and TCR signaling.
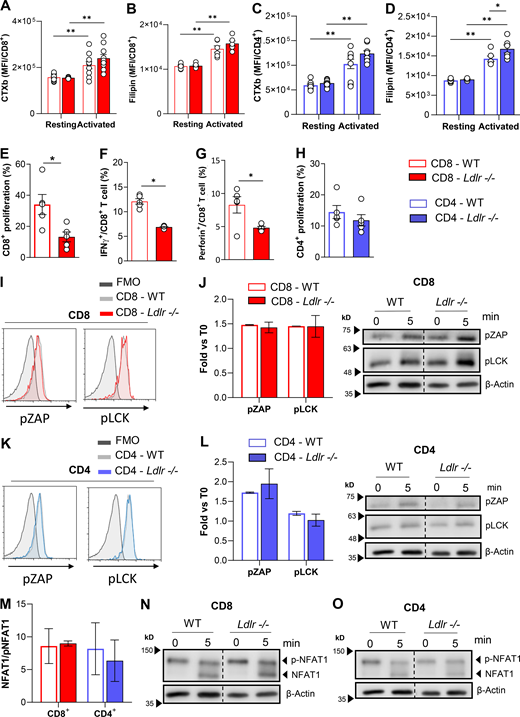
Lipid rafts quantification and TCR signalling in CD4+ and CD8+ T cells from WT and Ldlr −/− mice. (A and B) Quantification of lipid raft enrichment with ganglioside staining with cholera toxin subunit B (A) and free cholesterol staining with filipin (B) in resting and 96-h activated CD8+ T cells from WT and Ldlr −/− mice; n = 6/condition. (C and D) Quantification of lipid raft enrichment with ganglioside staining with cholera toxin subunit B (C) and free cholesterol staining with filipin (D) in resting and 96-h activated CD4+ T cells from WT and Ldlr −/− mice; n = 6/condition. (E–G) Proliferation (E), IFNγ (F) and Perforin (G) production of CD8+ T cells from WT and Ldlr −/− mice by in vitro stimulation with PMA/Ionomycin for 96-h; n = 5/condition. (H) Proliferation of CD4+ T cells from WT and Ldlr−/− mice by in vitro stimulation with PMA/Ionomycin for 96-h; n = 5/condition. (I) Representative histograms from flow cytometry analysis of pZAP and pLCK in CD8+ T cells from WT and Ldlr −/− mice in vitro activated with anti-CD3/28 for 10 min. (J) Phosphorylated ZAP and LCK Western blot analysis of in vitro activated (5-min stimulation) CD8+ T cells from WT and Ldlr −/− mice. Representative pictures from Western blot analysis are shown; n = 4/condition. (K) Representative histograms from flow cytometry analysis of pZAP and pLCK in CD4+ T cells from WT and Ldlr −/− mice in vitro activated with anti-CD3/28 for 10 min. (L) Phosphorylated ZAP and LCK Western blot analysis of in vitro activated (5-min stimulation) CD4+ T cells from WT and Ldlr −/− mice. Representative pictures from Western blot analysis are shown; n = 4/condition. (M–O) NFAT1 quantification of total (phosphorylated form, 140 kD) and de-phosphorylated (120 KD) form of in vitro activated (5-min stimulation) CD8+ and CD4+ T cells from WT and Ldlr −/− mice (M). Representative pictures from Western blot analysis are shown for CD8+ (N) and CD4+ (O) T cells; n = 4/condition. Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (A–H, J, L, and M). Source data are available for this figure: SourceData FS4.
Lipid rafts quantification and TCR signalling in CD4+ and CD8+ T cells from WT and Ldlr −/− mice. (A and B) Quantification of lipid raft enrichment with ganglioside staining with cholera toxin subunit B (A) and free cholesterol staining with filipin (B) in resting and 96-h activated CD8+ T cells from WT and Ldlr −/− mice; n = 6/condition. (C and D) Quantification of lipid raft enrichment with ganglioside staining with cholera toxin subunit B (C) and free cholesterol staining with filipin (D) in resting and 96-h activated CD4+ T cells from WT and Ldlr −/− mice; n = 6/condition. (E–G) Proliferation (E), IFNγ (F) and Perforin (G) production of CD8+ T cells from WT and Ldlr −/− mice by in vitro stimulation with PMA/Ionomycin for 96-h; n = 5/condition. (H) Proliferation of CD4+ T cells from WT and Ldlr−/− mice by in vitro stimulation with PMA/Ionomycin for 96-h; n = 5/condition. (I) Representative histograms from flow cytometry analysis of pZAP and pLCK in CD8+ T cells from WT and Ldlr −/− mice in vitro activated with anti-CD3/28 for 10 min. (J) Phosphorylated ZAP and LCK Western blot analysis of in vitro activated (5-min stimulation) CD8+ T cells from WT and Ldlr −/− mice. Representative pictures from Western blot analysis are shown; n = 4/condition. (K) Representative histograms from flow cytometry analysis of pZAP and pLCK in CD4+ T cells from WT and Ldlr −/− mice in vitro activated with anti-CD3/28 for 10 min. (L) Phosphorylated ZAP and LCK Western blot analysis of in vitro activated (5-min stimulation) CD4+ T cells from WT and Ldlr −/− mice. Representative pictures from Western blot analysis are shown; n = 4/condition. (M–O) NFAT1 quantification of total (phosphorylated form, 140 kD) and de-phosphorylated (120 KD) form of in vitro activated (5-min stimulation) CD8+ and CD4+ T cells from WT and Ldlr −/− mice (M). Representative pictures from Western blot analysis are shown for CD8+ (N) and CD4+ (O) T cells; n = 4/condition. Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (A–H, J, L, and M). Source data are available for this figure: SourceData FS4.
LDLR deficiency impairs mTOR activation and metabolic reprogramming in activated CD8+ T cells
Proteomic analysis of CD8+ T cells showed that there are substantial differences in activated CD8+ T cells from Ldlr −/− compared to those of WT mice, although the profile is similar between WT and Ldlr −/− CD8+ T cells under resting conditions (Fig. 4 A and Fig. S5 A). By contrast, resting and activated Ldlr −/− and WT CD4+ T cells clustered similarly (Fig. 4 B). Among more than 3,300 proteins identified in activated CD8+ T cells, 714 were differentially expressed between activated WT and Ldlr −/− CD8+ T cells, with the majority of proteins being less upregulated in activated Ldlr −/− CD8+ T cells as compared with their WT counterpart (Fig. 4 C). Of note, only 387 proteins were differentially modulated in activated Ldlr −/− CD4+ T cells compared with WT CD4+ T cells (Fig. 4 C). KEGG enrichment analysis showed that proteins belonging to energy metabolic pathways, including oxidative phosphorylation, TCA cycle, glycolysis, and pyruvate metabolism, together with processes related to protein translation, including ribosome biogenesis and spliceosome (Fig. 4 D), were differentially modulated in activated CD8+ T cells from Ldlr −/− mice compared with WT CD8+ T cells. In contrast, the profile of resting Ldlr −/− and WT CD8+ T cells was similar (Fig. S5 B), as was the case for that of activated CD4+ T cells (Fig. 4 D), further confirming the key role of LDLR in CD8+ T cells metabolic reprogramming during activation.

mTOR pathway in differentially affected in CD8 + T cells from Ldlr −/− mice compared to WT mice. (A and B) Principal component analysis (PCA) of the proteome from CD8+ (A) and CD4+ (B) T cells under resting conditions (A: WT: dark grey ellipse and Ldlr −/−: light red ellipse; B: WT: dark grey ellipse and Ldlr −/−: light blue ellipse), and 96-h after activation (A: WT: light grey ellipse and Ldlr −/−: red ellipse; WT: light grey ellipse and Ldlr −/−: blue ellipse) conditions. (C) Total proteome quantified in activated (96-h) CD8+ and CD4+ T cells of Ldlr −/− mice compared to WT. 29 proteins upregulated and 685 downregulated in Ldlr −/− CD8+ T cells, while 186 were upregulated and 200 were downregulated in Ldlr −/− CD4+ T cells. Red indicates significantly upregulated proteins (Log2FC > 2, P value < 0.05) and blue indicated significantly downregulated proteins (Log2FC < 2, P value<0.05). N = 6/condition, samples derived from two independent experiments and were injected three times each. (D) Gene ontology (GO) enrichment analysis of top significantly modulated pathways (FDR < 0.05) in activated CD8+ (red) and CD4+ (blue) T cells from Ldlr −/− mice compared to WT mice of proteomic dataset. (E) Significant canonical pathways (z-score <−2) generated with IPA associated with mTOR activation cascade from proteomic dataset in activated CD8+ T cells from Ldlr −/− mice compared to WT mice. (F) Graphical representation of key mTORC1 downstream signaling proteins which are predicted to be either downregulated (green) or inhibited (blue) by IPA analysis in activated CD8+ T cells from Ldlr −/− mice compared to WT mice. Prediction of mTORC1 inhibition leads to reduced translation, ribosome biogenesis, energetic (glycolysis), and lipid (cholesterol and fatty acid synthesis) metabolism. Created by Biorender.com. (G) Representation of upstream analysis generated with IPA for significant up- or downregulated transcriptional regulators (Z-score >2 and −log(P Value) >1.34) in the proteomic dataset of 96-h activated CD8+ T cells from WT and Ldlr −/− mice. Prediction of mTORC1 inhibition was identified as “sirolimus treatment.”
mTOR pathway in differentially affected in CD8 + T cells from Ldlr −/− mice compared to WT mice. (A and B) Principal component analysis (PCA) of the proteome from CD8+ (A) and CD4+ (B) T cells under resting conditions (A: WT: dark grey ellipse and Ldlr −/−: light red ellipse; B: WT: dark grey ellipse and Ldlr −/−: light blue ellipse), and 96-h after activation (A: WT: light grey ellipse and Ldlr −/−: red ellipse; WT: light grey ellipse and Ldlr −/−: blue ellipse) conditions. (C) Total proteome quantified in activated (96-h) CD8+ and CD4+ T cells of Ldlr −/− mice compared to WT. 29 proteins upregulated and 685 downregulated in Ldlr −/− CD8+ T cells, while 186 were upregulated and 200 were downregulated in Ldlr −/− CD4+ T cells. Red indicates significantly upregulated proteins (Log2FC > 2, P value < 0.05) and blue indicated significantly downregulated proteins (Log2FC < 2, P value<0.05). N = 6/condition, samples derived from two independent experiments and were injected three times each. (D) Gene ontology (GO) enrichment analysis of top significantly modulated pathways (FDR < 0.05) in activated CD8+ (red) and CD4+ (blue) T cells from Ldlr −/− mice compared to WT mice of proteomic dataset. (E) Significant canonical pathways (z-score <−2) generated with IPA associated with mTOR activation cascade from proteomic dataset in activated CD8+ T cells from Ldlr −/− mice compared to WT mice. (F) Graphical representation of key mTORC1 downstream signaling proteins which are predicted to be either downregulated (green) or inhibited (blue) by IPA analysis in activated CD8+ T cells from Ldlr −/− mice compared to WT mice. Prediction of mTORC1 inhibition leads to reduced translation, ribosome biogenesis, energetic (glycolysis), and lipid (cholesterol and fatty acid synthesis) metabolism. Created by Biorender.com. (G) Representation of upstream analysis generated with IPA for significant up- or downregulated transcriptional regulators (Z-score >2 and −log(P Value) >1.34) in the proteomic dataset of 96-h activated CD8+ T cells from WT and Ldlr −/− mice. Prediction of mTORC1 inhibition was identified as “sirolimus treatment.”

Extensive proteomic analysis of CD4+ and CD8+ T cells from WT and Ldlr −/− mice. (A) Hierarchical clustering based on Pearson’s correlation and heatmap showing relative protein expression values (z-score transformed LFQ protein intensities) of significantly different proteins from GO enrichment analysis of ribosome, proteasome, and spliceosome pathway at 96-h activated CD8 T cells from WT and Ldlr −/− mice.; n = 6/condition, samples are derived from two independent experiments and were injected three times each. (B) GO enrichment analysis of significantly modulated top 10 pathways (FDR < 0.05) in resting CD8+ T cells from Ldlr −/− mice compared to WT mice. (C) mRNA expression of Rlp13a (ribosomal protein 13a) in resting and 14-h activated CD8+ T cells from WT and Ldlr −/− mice; n = 6/condition. (D) mRNA expression of Rlp13a (ribosomal protein 13a) in resting and 14-h activated CD4+ T cells from WT and Ldlr −/− mice; n = 4/condition. (E) Canonical pathways associated to energetic metabolism from proteomic dataset in activated CD8+ T cells from Ldlr −/− mice compared to WT mice obtained from Ingenuity Pathway Analysis. Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (C and D).
Extensive proteomic analysis of CD4+ and CD8+ T cells from WT and Ldlr −/− mice. (A) Hierarchical clustering based on Pearson’s correlation and heatmap showing relative protein expression values (z-score transformed LFQ protein intensities) of significantly different proteins from GO enrichment analysis of ribosome, proteasome, and spliceosome pathway at 96-h activated CD8 T cells from WT and Ldlr −/− mice.; n = 6/condition, samples are derived from two independent experiments and were injected three times each. (B) GO enrichment analysis of significantly modulated top 10 pathways (FDR < 0.05) in resting CD8+ T cells from Ldlr −/− mice compared to WT mice. (C) mRNA expression of Rlp13a (ribosomal protein 13a) in resting and 14-h activated CD8+ T cells from WT and Ldlr −/− mice; n = 6/condition. (D) mRNA expression of Rlp13a (ribosomal protein 13a) in resting and 14-h activated CD4+ T cells from WT and Ldlr −/− mice; n = 4/condition. (E) Canonical pathways associated to energetic metabolism from proteomic dataset in activated CD8+ T cells from Ldlr −/− mice compared to WT mice obtained from Ingenuity Pathway Analysis. Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (C and D).
Ingenuity pathway analysis identified mTOR activation (mTOR signaling) as the key biological pathway differentially regulated in Ldlr −/− CD8+ T cells compared with WT CD8+ T cells, with mTOR complex 1 (mTORC1) downstream signaling (HIF1α, p70S6K, EIF2, and tRNA charging) being largely affected (Fig. 4, E and F).
In parallel, also mTORC1 downstream pathways such as HIF1α, Myc, PPARγ, and SREBP1F were downregulated in Ldlr −/− CD8+ T cells compared with WT CD8+ T cells (IPA upstream regulators analysis; Fig. 4 G). Accordingly, the network of proteins associated with mTORC1 inhibition and those downstream of sirtuin 1, whose activation antagonizes that of mTORC1 in response to nutritional stressors (Wang et al., 2016), resulted increased (Fig. 4 G). These findings support the hypothesis that LDLR could play a critical role for mTORC1 activation in CD8+ T cells, which in turn controls cellular metabolic reprogramming during their activation (Chi, 2012).
Western blot analysis of phosphorylated versus the total form of mTOR (pmTOR/mTOR) and downstream signaling proteins, including ribosomal protein S6 kinase (pS6K/S6K), ribosomal protein S6 (pS6/S6), and eukaryotic translation initiation factor 4E-binding protein 1 (p4EBP1/4EBP1) showed a significantly decreased activation in stimulated Ldlr −/− CD8+ T cells compared with WT CD8+ T cells (Fig. 5, A–E) together with impaired mTORC1-dependent ribosome biogenesis as documented by reduced ribosomal protein 13a (RLP13a) expression in activated Ldlr −/− CD8+ T cells (Fig. S5 C). On the contrary, no differences in the activation of mTORC1 downstream pathway were observed in Ldlr −/− and WT CD4+ T cells (Fig. 5, F–J). Accordingly, also the expression of RLP13a showed no difference between activated CD4+ T cells from WT and Ldlr −/− mice (Fig. S5 D).
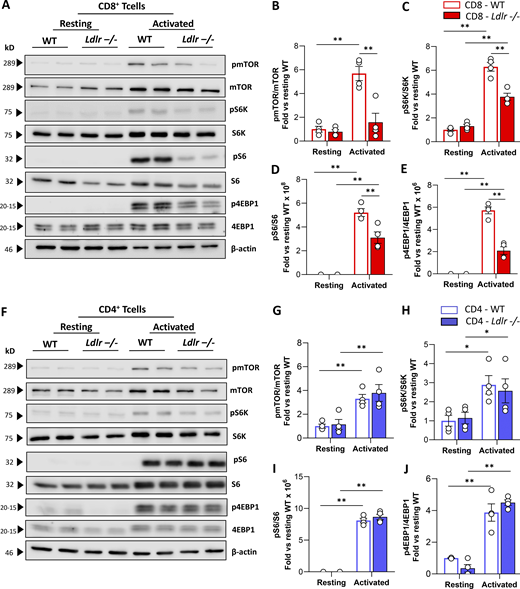
LDLR deficiency impairs mTORC1 activation in CD8+T cells. (A–E) Representative pictures (A) and quantification (B–E) of Western blot analysis of phosphorylated and total downstream proteins of mTORC1 pathway in resting and activated (14-h of stimulation) CD8+ T cells from WT and Ldlr −/− mice. Ratio between phosphorylated and total form of mTOR (B), S6K (C), S6 (D), and 4EBP-1 (E) normalized on WT cells under resting conditions are presented; (the results from four independent experiments are reported). (F–J) Representative pictures (F) and quantification (G–J) of Western blot analysis of phosphorylated and total downstream proteins of mTORC1 pathway in resting and activated (14-h of stimulation) CD8+ T cells from WT and Ldlr −/− mice. Ratio between phosphorylated and total form of mTOR (G), S6K (H), S6 (I), and 4EBP-1 (J) normalized on WT cells under resting conditions are presented; (the results from four independent experiments are reported). Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (B–E and G–J). Source data are available for this figure: SourceData F5.
LDLR deficiency impairs mTORC1 activation in CD8+T cells. (A–E) Representative pictures (A) and quantification (B–E) of Western blot analysis of phosphorylated and total downstream proteins of mTORC1 pathway in resting and activated (14-h of stimulation) CD8+ T cells from WT and Ldlr −/− mice. Ratio between phosphorylated and total form of mTOR (B), S6K (C), S6 (D), and 4EBP-1 (E) normalized on WT cells under resting conditions are presented; (the results from four independent experiments are reported). (F–J) Representative pictures (F) and quantification (G–J) of Western blot analysis of phosphorylated and total downstream proteins of mTORC1 pathway in resting and activated (14-h of stimulation) CD8+ T cells from WT and Ldlr −/− mice. Ratio between phosphorylated and total form of mTOR (G), S6K (H), S6 (I), and 4EBP-1 (J) normalized on WT cells under resting conditions are presented; (the results from four independent experiments are reported). Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (B–E and G–J). Source data are available for this figure: SourceData F5.
In line with mTORC1 impairment (Linke et al., 2017; Salmond, 2018), energetic reprogramming upon activation was less robust in activated Ldlr −/− CD8+ T cells compared to WT CD8+ T cells, with reduced activation of glycolysis and glucose degradation, oxidative phosphorylation, and fatty acid oxidation (Fig. S5 E and Fig. S6, A–F), as well as pathways associated with the synthesis of building blocks required for cellular proliferation such as (purine nucleotide de novo biosynthesis, pentose phosphate pathway, pyrimidine ribonucleotide biosynthesis, and acetylCoA biosynthesis). In agreement with the reduced energetic phenotype, the expression of PFKB3, one of the key regulators of glycolysis, was significantly decreased in activated CD8+ T cells from Ldlr −/− compared with WT mice (Fig. S6 A). By contrast, the expression of glutaminase (GLS1), involved in the conversion of glutamine to α-ketoglutarate, one of the intermediates of the TCA cycle, was significantly increased in activated Ldlr −/− CD8+ T cells compared with WT (Fig. S6 A), possibly marking the attempt to provide energy from collateral pathways (Yoo et al., 2020).

Analysis of cellular metabolism of CD8+ T cells from WT and Ldlr −/− mice from proteomic dataset. (A) Functional representation of activated CD8+ T cells protein signature from of Ldlr −/− mice compared to WT mice. Blue denotes downregulation, while red upregulation of Log2FC (fold change) from proteins (circles) and genes (rectangles). Created by Biorender.com. (B–E) Abundance distribution of differentially expressed proteins in oxidative phosphorylation (OXPHOS, B), fatty acid oxidation (FAO, C), glycolysis (D) and Kreb’s cycle (TCA, E) among activated CD8+ T cell subset from Ldlr−/− compared to WT mice. Data are from two independent experiments, each with three technical triplicates.
Analysis of cellular metabolism of CD8+ T cells from WT and Ldlr −/− mice from proteomic dataset. (A) Functional representation of activated CD8+ T cells protein signature from of Ldlr −/− mice compared to WT mice. Blue denotes downregulation, while red upregulation of Log2FC (fold change) from proteins (circles) and genes (rectangles). Created by Biorender.com. (B–E) Abundance distribution of differentially expressed proteins in oxidative phosphorylation (OXPHOS, B), fatty acid oxidation (FAO, C), glycolysis (D) and Kreb’s cycle (TCA, E) among activated CD8+ T cell subset from Ldlr−/− compared to WT mice. Data are from two independent experiments, each with three technical triplicates.
LDL-derived cholesterol regulates lysosomal response and mTORC1 activation in activated CD8+ T cells
Under physiological conditions, the LDLR contributes to cholesterol uptake via LDL binding and internalization. LDL is then directed to the lysosome where lysosomal acid lipase (LAL) hydrolyses esterified cholesterol, releasing the free form to the cell. To investigate the fate of LDL-cholesterol in T cells, we assembled a cholesterol analog, 27-alkine cholesterol, into LDL particles, and followed cholesterol distribution in the cell by tracking cholesterol fluorescence occurring after a specific chemical reaction (Click Chemistry; Hofmann et al., 2014) at the alkyne group (Fig. 6 A). Compared with activated WT cells, in Ldlr −/− CD8+ T cells we noticed reduced LDL-derived cholesterol, especially in the lysosome (Fig. 6, B and C), suggesting a defective routing of cholesterol via LDLR–lysosome axis upon stimulation. Indeed, while in WT CD8+ T cells cellular activation showed an increased lysotracker staining (Fig. 6 D), increased LAMP-1 intracellular protein content (Fig. 6 E), and increased LAMP-1 mRNA expression (Fig. S7 A), suggesting a remodeling of lysosome under stimulation, this phenomenon was significantly reduced in CD8+ T cells from Ldlr −/− mice (Fig. 6, D and E), thus pointing to a key role of the LDLR pathway in mediating the lysosomal response in CD8+ T cells. Although LDL-derived cholesterol was mainly appreciated in close proximity to lysosomes of WT CD4+ T cells compared with Ldlr −/− cells (Fig. S7, B and C), a similar increase in LAMP-1 upon cell activation between WT and Ldlr −/− CD4+ T cells (Fig. S7 D) casts for a minor impact of LDLR–lysosome axis in the immunometabolic response associated with CD4+ T cell activation. To test whether the reduced flux of cholesterol through the lysosome could affect lymphocyte cellular response, CD8+ and CD4+ T cells were treated with lalistat, an inhibitor of the lysosomal acid lipase (LAL), the only enzyme in the lysosome that hydrolyses esterified cholesterol derived from LDL (Gomaraschi et al., 2019). Under this condition, WT CD8+ T cells presented reduced IFNγ and perforin production (Fig. 6, F and G) and cell proliferation (Fig. S7 E), mirroring the profile observed in Ldlr −/− CD8+ T cells (Fig. 6, F and G and Fig. S7 E) and suggesting a critical role for the hydrolysis of LDLR-derived cholesterol during cellular activation. Of note, WT and Ldlr −/− CD4+ T cell proliferation was similar even when LAL activity was inhibited (Fig. S7 F), in agreement with the findings on CD4+ T cells described above. As it has been shown that lysosomal-derived cholesterol represents a checkpoint for mTORC1 activation, thanks to the presence of adaptor proteins that trigger the activation upon physical interaction with cholesterol in the lysosome (Castellano et al., 2017), we tested whether this mechanism could have been relevant for CD8+ T cells activation. Confocal microscopy analysis (Fig. 6 H) showed that mTOR complex localized preferentially in close proximity with cholesterol derived from lipoproteins and delivered to the cells through the LDLR pathway in WT CD8+ T cells (Fig. 6 I, yellow staining); this colocalization was appreciated to a much lower extent in Ldlr −/− CD8+ T cells (Fig. 6 J), as well as in CD4+ T cells from WT and Ldlr −/− mice (Fig. S7, G and H).
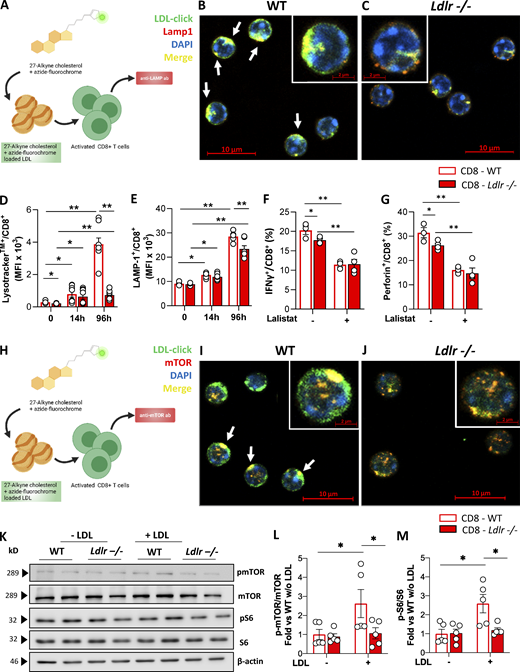
LDLR-derived cholesterol regulates lysosomal-dependent mTORC1 activation. (A) Workflow of cholesterol “click-chemistry” assay in CD8+ T cells. Isolated LDL particles were loaded with 27-alkyne cholesterol and incubated with CD8+ T cells from WT and Ldlr −/− mice that were previously stimulated with anti-CD3/28 + IL-2 in serum-free media for 24 h. Cells were then incubated with an azide conjugated to a fluorochrome to visualize cholesterol, followed by Lamp-1 staining; cells were then visualized with confocal microscopy. Created by Biorender.com. (B and C) Staining of CD8+ T cells from WT (B) and Ldlr −/− (C) mice for cholesterol (click-chol, green), Lamp1 (red), and nuclei (blue) after 24-h stimulation plus LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate colocalization. (D and E) Median fluorescence intensity for Lysotraker staining (D) and for Lamp-1 (lysosomal associated membrane protein 1) intracellular staining (E) in resting and activated (14- and 96-h) CD8+ T cells from WT and Ldlr −/− mice; n = 6/condition. (F and G) IFNγ (F) and perforin (G) production of CD8+ T cells from WT and Ldlr −/− mice by in vitro anti-CD3/28 + IL-2 stimulation for 96 h in the presence or absence of lalistat (lysosomal acid lipase inhibitor), n = 5/condition. (H) Workflow of cholesterol “click-chemistry” assay in CD8+ T cells. Isolated LDL particles were loaded with 27-alkyne cholesterol and incubated with CD8+ T cells from WT and Ldlr −/− mice that were previously stimulated with anti-CD3/28 + IL-2 in serum-free media for 24 h. Cells were then incubated with an azide conjugated to a fluorochrome to visualize cholesterol, followed by mTOR staining; cells were then visualized with confocal microscopy. Created by Biorender.com. (I and J) Staining of CD8+ T cells from WT (J) and Ldlr −/− (K) mice for click-cholesterol (click-chol, green), mTOR (orange), and nuclei (blu) after 24-h stimulation in the presence of LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar is 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate co-localization. (K–M) Representative pictures (K) and quantification (L and M) of Western blot analysis of phosphorylated and total downstream proteins of mTORC1 pathway in activated (14-h of stimulation) CD8+ T cells from WT and Ldlr −/− mice in the presence or not of LDL (5 µg/ml). Ratio between the phosphorylated and total form of mTOR (L) and S6 (M) normalized on WT cells activated in the absence of LDL are presented (the results from four independent experiments are reported). Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (D–G, L, and M). Source data are available for this figure: SourceData F6.
LDLR-derived cholesterol regulates lysosomal-dependent mTORC1 activation. (A) Workflow of cholesterol “click-chemistry” assay in CD8+ T cells. Isolated LDL particles were loaded with 27-alkyne cholesterol and incubated with CD8+ T cells from WT and Ldlr −/− mice that were previously stimulated with anti-CD3/28 + IL-2 in serum-free media for 24 h. Cells were then incubated with an azide conjugated to a fluorochrome to visualize cholesterol, followed by Lamp-1 staining; cells were then visualized with confocal microscopy. Created by Biorender.com. (B and C) Staining of CD8+ T cells from WT (B) and Ldlr −/− (C) mice for cholesterol (click-chol, green), Lamp1 (red), and nuclei (blue) after 24-h stimulation plus LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate colocalization. (D and E) Median fluorescence intensity for Lysotraker staining (D) and for Lamp-1 (lysosomal associated membrane protein 1) intracellular staining (E) in resting and activated (14- and 96-h) CD8+ T cells from WT and Ldlr −/− mice; n = 6/condition. (F and G) IFNγ (F) and perforin (G) production of CD8+ T cells from WT and Ldlr −/− mice by in vitro anti-CD3/28 + IL-2 stimulation for 96 h in the presence or absence of lalistat (lysosomal acid lipase inhibitor), n = 5/condition. (H) Workflow of cholesterol “click-chemistry” assay in CD8+ T cells. Isolated LDL particles were loaded with 27-alkyne cholesterol and incubated with CD8+ T cells from WT and Ldlr −/− mice that were previously stimulated with anti-CD3/28 + IL-2 in serum-free media for 24 h. Cells were then incubated with an azide conjugated to a fluorochrome to visualize cholesterol, followed by mTOR staining; cells were then visualized with confocal microscopy. Created by Biorender.com. (I and J) Staining of CD8+ T cells from WT (J) and Ldlr −/− (K) mice for click-cholesterol (click-chol, green), mTOR (orange), and nuclei (blu) after 24-h stimulation in the presence of LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar is 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate co-localization. (K–M) Representative pictures (K) and quantification (L and M) of Western blot analysis of phosphorylated and total downstream proteins of mTORC1 pathway in activated (14-h of stimulation) CD8+ T cells from WT and Ldlr −/− mice in the presence or not of LDL (5 µg/ml). Ratio between the phosphorylated and total form of mTOR (L) and S6 (M) normalized on WT cells activated in the absence of LDL are presented (the results from four independent experiments are reported). Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (D–G, L, and M). Source data are available for this figure: SourceData F6.

LDLR-lysosomal-mTOR axis in T cells from WT and Ldlr −/− mice. (A) mRNA expression of Lamp-1 in resting and activated (14- or 96-h) CD8+ T cells from WT and Ldlr −/− mice; n = 5/condition. (B and C) Staining of CD4+ T cells from WT (B) and Ldlr −/− (C) mice for cholesterol (click-chol, green), Lamp1 (red) and nuclei (blue) after 24-h stimulation plus LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate co-localization. (D) Median fluorescence intensity for Lamp-1(Lysosomal Associated Membrane Protein 1) intracellular staining (E) in resting and activated (14- and 96-h) CD4+ T cells from WT and Ldlr −/− mice; n = 6/condition. (E and F) Proliferation of CD8+ (E) and CD4+ (F) T cells from WT and Ldlr −/− mice by in vitro anti-CD3/28 + IL-2 stimulation for 96 h in the presence or absence of lalistat (lysosomal acid lipase inhibitor), n = 5/condition. (G and H) Staining of CD4+ T cells from WT (G) and Ldlr −/− (H) mice for click-cholesterol (click-chol, green), mTOR (orange) and nuclei (blue) after 24-h stimulation plus LDL plus LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar is 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate co-localization. Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (A and D–F).
LDLR-lysosomal-mTOR axis in T cells from WT and Ldlr −/− mice. (A) mRNA expression of Lamp-1 in resting and activated (14- or 96-h) CD8+ T cells from WT and Ldlr −/− mice; n = 5/condition. (B and C) Staining of CD4+ T cells from WT (B) and Ldlr −/− (C) mice for cholesterol (click-chol, green), Lamp1 (red) and nuclei (blue) after 24-h stimulation plus LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate co-localization. (D) Median fluorescence intensity for Lamp-1(Lysosomal Associated Membrane Protein 1) intracellular staining (E) in resting and activated (14- and 96-h) CD4+ T cells from WT and Ldlr −/− mice; n = 6/condition. (E and F) Proliferation of CD8+ (E) and CD4+ (F) T cells from WT and Ldlr −/− mice by in vitro anti-CD3/28 + IL-2 stimulation for 96 h in the presence or absence of lalistat (lysosomal acid lipase inhibitor), n = 5/condition. (G and H) Staining of CD4+ T cells from WT (G) and Ldlr −/− (H) mice for click-cholesterol (click-chol, green), mTOR (orange) and nuclei (blue) after 24-h stimulation plus LDL plus LDL particles (200 μg/ml) loaded with 27-alkyne cholesterol, detected by confocal microscopy. Scale bar is 10 µm in the main figures and 2 µm in the magnified insets. Yellow staining and arrows indicate co-localization. Results are presented as mean per group ± SEM; statistical analysis is performed with two-way ANOVA; *P < 0.05 and **P < 0.01 (A and D–F).
To then confirm the effect of LDLR-derived cholesterol on mTORC1 activation, the incubation of cells with LDL particles added to serum-free media contributed to mTORC1 pathway activation in WT but not in Ldlr −/− CD8+ T cells as detected by the phosphorylation of mTOR and S6 (Fig. 6, K–M), further confirming the dependency of CD8+ T cells on LDLR-derived cholesterol during activation. Altogether, these data indicate that cholesterol delivered via LDLR participates in mTORC1 activation, thus highlighting LDLR as an immune checkpoint for CD8+ T cells activation.
Subjects carrying mutations on the LDLR gene show defective CD8+ T cell response
To translate these findings to humans, we investigated CD8+ T cell activation in subjects affected by familial hypercholesterolemia (FH) who present heterozygous loss of function mutations on the LDLR gene (Table S2). These subjects presented a different distribution in naïve CD8+ T cell subsets when compared to age- and sex-matched controls (Fig. 7 A) in spite of similar counts of circulating CD8+ T cells (Fig. 7 B). When activated with anti-CD3/28 antibodies, CD8+ T cells from FH subjects displayed a reduced proliferation rate compared with cells from control subjects (Fig. 7 C), thus confirming, also in humans, the impairment of CD8+ T cell response to rapid stimulation when the LDLR pathway is compromised. We also tested the ability of CD8+ T cells to mount an efficient response following rechallenging with seasonal influenza virus by treating cells from FH subjects or controls, vaccinated for 2018 seasonal influenza, with H1N1pdm09 and H3N2 specific peptides. CD8+ T cell activation (CD25+ CD8+ T cells, Fig. 7 D) and cytotoxic molecules production (Granzyme B+ CD8+ T cells, Fig. 7 E) were reduced in CD8+ T cells from FH subjects compared with vaccinated healthy controls.

Carriers of loss of function mutations on the LDLR gene present dysfunctional CD8 + T cells. (A) The proportion of CD8+ T cell subsets in carriers of loss of function mutations on the LDLR gene (FH) and in - and sex-matched controls. Tnaive (CCR7+CD45RA+), Tcentral memory (TCM, CCR7+CD45RA−), T effector memory (TEM, CCR7−CD45RA−), and Temra (CCR7−CD45RA+), n = 20/group; representative plots from flow cytometry analysis are shown. (B) Circulating CD8+ T cells (cells/μl) in FH subjects and age/sex-matched controls; n = 20/group. (C) Proliferation of CD8+ T cells in FH and matched controls following in vitro anti-CD3/28 + IL-2 stimulation for 96 h; n = 10/group. (D and E) Expression of CD25 (D) and granzyme production (E) in CD8+ T cells of FH and age/sex-matched controls vaccinated for the seasonal influenza virus (2018) and ex-vivo rechallenged with H1N1pdm09 and H3N2-specific peptides at increasing concentration (µg/ml) for 96 h in the presence of CD28 and IL-2. Results are presented as mean per group ± SEM; statistical analysis is performed with unpaired t test; *P < 0.05 and **P < 0.01 (A–E).
Carriers of loss of function mutations on the LDLR gene present dysfunctional CD8 + T cells. (A) The proportion of CD8+ T cell subsets in carriers of loss of function mutations on the LDLR gene (FH) and in - and sex-matched controls. Tnaive (CCR7+CD45RA+), Tcentral memory (TCM, CCR7+CD45RA−), T effector memory (TEM, CCR7−CD45RA−), and Temra (CCR7−CD45RA+), n = 20/group; representative plots from flow cytometry analysis are shown. (B) Circulating CD8+ T cells (cells/μl) in FH subjects and age/sex-matched controls; n = 20/group. (C) Proliferation of CD8+ T cells in FH and matched controls following in vitro anti-CD3/28 + IL-2 stimulation for 96 h; n = 10/group. (D and E) Expression of CD25 (D) and granzyme production (E) in CD8+ T cells of FH and age/sex-matched controls vaccinated for the seasonal influenza virus (2018) and ex-vivo rechallenged with H1N1pdm09 and H3N2-specific peptides at increasing concentration (µg/ml) for 96 h in the presence of CD28 and IL-2. Results are presented as mean per group ± SEM; statistical analysis is performed with unpaired t test; *P < 0.05 and **P < 0.01 (A–E).
These data confirmed in humans that impaired LDLR functionality limits CD8+ T cell activation and a proper response to immune challenges, further highlighting the role of LDLR as a crucial immuno-metabolic checkpoint in CD8+ T cell activation.
Discussion
With this work, we showed that the LDLR plays a key role in CD8+ T cells activation by providing extracellular cholesterol, which is involved in mTORC1 activation, an event that is critical for lymphocyte immunometabolic reprogramming. Accordingly, our data indicate that LDLR deficiency results in decreased CD8+ T cell proliferation and decreased production of cytotoxic and proinflammatory cytokines in experimental models and in subjects carrying mutations on the LDLR gene. Of note, these effects were restricted to CD8+ T cells, as CD4+ T cell function was not affected by LDLR deficiency, pointing out a different role for sterol metabolism in T cell subsets.
Activated T cells utilize cholesterol to build up membranes during proliferation (Kidani et al., 2013; Bensinger et al., 2008; Fessler, 2016) and to control the clustering of key effector receptors such as the TCR in lipid rafts (Yang et al., 2016; Janes et al., 2000). This results in the increase of the expression of genes involved in de novo cholesterol biosynthesis and extracellular uptake by the activation of SREBP2-mediated sterol metabolism as well as the repression of the expression of cholesterol efflux genes in both activated CD4+ and CD8+ T cells (Kidani et al., 2013; Danesh et al., 2003; Ghittoni et al., 2007). Here, we extend these findings by showing that CD8+ T cells require LDLR-dependent cholesterol uptake to achieve a proper activation. Of note, LDLR deficiency impairs the ability of CD8+ but not of CD4+ T cells to proliferate and to produce cytotoxic molecules following antigen-mediated activation, but not under homeostatic conditions.
These findings not only suggest a major role for the LDLR in the regulation of CD8+ T cell response but also raise the intriguing question of whether a differential intracellular routing of cholesterol exerts different immunometabolic functions in T cells.
Following LDL binding to LDLR, the lipoprotein is directed to the lysosome where esterified cholesterol is hydrolyzed and either exported and distributed for cell needs or involved in lysosomal dynamics (Gomaraschi et al., 2019). Lysosomes are indeed emerging as central hubs of nutrient sensing, which undergo a dynamic rearrangement in response to cell activation, leading to a continuous fusion and regeneration, as well as to movement along microtubules in a retrograde and anterograde fashion (de Araujo et al., 2017; Li et al., 2016; Sancak et al., 2010; Pu et al., 2016), which is also critical for the control of mTORC1 kinase activation (Meng et al., 2020; Thelen and Zoncu, 2017; Rogala et al., 2019). While the lysosomal hydrolysis of fatty acids (FAs) was proposed to fulfill the energetic demand of memory CD8 T cells (O’Sullivan et al., 2014), our data exclude a role for LDLR in modulating the reprogramming of FAs during CD8+ T activation, in line with a similar profile of T memory subsets detected in Ldlr −/− compared with WT mice. Instead, we showed that the uptake of LDL via the LDLR, by affecting lysosome cellular localization and, in turn, its interaction with other cellular organelles, such as mitochondria and endoplasmic reticulum, necessary for cholesterol routing within the cell (Yvan-Charvet et al., 2019), critically participates in mTORC1 activation upon cellular stimulation, which in turn coordinates several cellular pathways involved in cell effector response. Indeed, mTORC1 activation is required for the early activation of CD8+ T cells (Pollizzi et al., 2015), while CD4+ T cells are less dependent on this pathway, as also suggested by their different biosynthetic capacity and lipid metabolism compared to CD8+ T cells (Howden et al., 2019). Of note, the upregulation of cholesterol-related genes, including the LDLR, was shown to be significantly upregulated in activated CD8+ compared with CD4+ T cells. In addition, while under resting conditions, baseline mTORC1 activation in WT and Ldlr −/− CD8+ T cells is similar, following activation, the upregulation of the LDLR would rapidly support CD8+ T cells with cholesterol that is required to complement mTORC1 activation (Castellano et al., 2017). This results in reduced mTOR signaling pathway activation in activated Ldlr −/− CD8+ but not in CD4+ T cells compared with WT T cells. According to functional results, the proteomic signature was similar in activated CD4+ T cells as well as in resting CD8+ T cells between WT and Ldlr −/− mice, but was dramatically different between activated WT and Ldlr −/− CD8+ T cells. These results suggest that the cholesterol–LDLR pathway might support cell cytotoxicity, a phenotype that is a distinct trait of CD8+, while it is less pronounced in CD4+ T cells.
Mechanistically, the absence of LDLR impairs mTOR activation that depends on its clustering at the lysosomal membrane (Castellano et al., 2017), which translates into a less efficient CD8+ T cells immunometabolic reprogramming, including impaired glycolysis and oxidative phosphorylation, leading to reduced cell proliferation and cytotoxic cytokine production. CD8+ T cells from Ldlr −/− mice also showed an early upregulation of HMGCR to increase cholesterol biosynthesis, which, however, is not enough to compensate for the absence of LDLR-dependent cholesterol influx, supporting the hypothesis that a distinct cellular routing of cholesterol of either endogenous or exogenous source might impact different cellular functions in T cell subsets. In line with this, cholesterol provided by the LDLR–lysosome axis appears to be critical for boosting mTORC-1 activation in CD8+ cells, thus contributing to their effector function; other conditions changing intracellular cholesterol levels differentially impact T cells, such as ABCG1 deficiency, that affects T cell skewing toward a Treg phenotype (Cheng et al., 2016) or ACAT1 inhibition that increases TCR signaling by cholesterol enrichment in lipid raft (Schmidt et al., 2021). By showing the requirement of LDLR-cholesterol uptake upon CD8+ T cell activation, our work extends previous findings showing a role of the PCSK9/LDLR pathway in controlling the cytotoxic function of CD8+ T cells in the tumor environment via TCR signaling modulation (Yuan et al., 2021). At the same time, our findings suggest the possibility that targeting the LDLR pathway could contribute to increase the cytotoxic potential of CD8+ T cells.
Finally, the observation that the proliferation of CD8+ T cells is not influenced by an increase in plasma cholesterol levels, in contrast to what occurs for CD4+ T cells, further supports the concept of a different regulation of the response within T cell subsets, which depends on a distinct activation of pathways involved in the regulation of cellular cholesterol levels. Intriguingly, this reflects a different involvement of T cell subsets in diseases associated with the loss of cholesterol homeostasis as atherosclerosis. Indeed, while activated CD4+ T cells contribute to the extent and severity of atherosclerosis in animal models and in humans (Kita et al., 2014; Emeson et al., 1996; Saigusa et al., 2020), the role of CD8+ T cells is still less clear. CD8+ T cell depletion results in either increased or decreased atherosclerosis (Schafer and Zernecke, 2020; van Duijn et al., 2019; Cochain and Zernecke, 2016), and the transplantation of bone marrow from WT mice into Ldlr −/− mice does not affect atherosclerosis development as compared with full Ldlr −/− mice (Herijgers et al., 1997; Fazio et al., 1997; Linton et al., 1999; Ouweneel et al., 2021). Also, in patients with heterozygous familial hypercholesterolemia (HeFH), while CD4+ T cells are more activated compared with not affected matched individuals (Bonacina et al., 2020) and contribute to disease progression, data on the activation status for CD8+ T cells are missing. This casts for testing whether the blunting of antigen-specific activation in CD8+ T cells under LDLR loss of function conditions would affect atherosclerotic disease progression in FH subjects.
In summary, by showing that LDLR delivers cholesterol involved in mTORC1 complex clustering on the lysosomal membrane, we shed light on a key mechanism guiding CD8+ T cell immunometabolic reprogramming and function. These findings have important translational relevance as CD8+ T cells from subjects carrying loss of function mutations on the Ldlr gene (FH subjects) presented reduced activation and proliferation following an immune rechallenge and support the intriguing hypothesis that strategies aimed at improving LDLR expression and/or activity in CD8+ T cells could improve their cytotoxic efficacy.
Materials and methods
Mice
WT and Ldlr −/− male and female mice on C57BL/6J background, Balb/cJ, and Rag2 −/− mice were purchased from The Jackson Laboratory. Five mice were housed per cage and kept in a temperature-controlled environment (20 ± 2°C, 50 ± 5% relative humidity) with a 12-h light/dark cycle and free access to food and water. In some experimental conditions, 6- to 8-wk old mice were fed a high cholesterol diet for 8 wk. For blood, lymph node, and spleen collection, mice were euthanized by an overdose of CO2. All animal procedures performed conformed to the guidelines from 2010/63/EU directive of the European Parliament on the protection of animals used for scientific purposes and were approved by the Ethical Committee of the University of Milan and Italian Ministry of Health (Progetto di Ricerca 2012/02, 426/2019, 271/2020) and of Queen Mary University (London, UK).
Human samples
Patients with a genetic diagnosis of familial hypercholesterolemia were recruited to the Centre for the Study of Atherosclerosis at Bassini Hospital, Cinisello Balsamo, Italy, within the Lipigen study, an integrated network aimed at improving the identification of patients with genetic dyslipidaemias, including FH, in Italy (Casula et al., 2018). The human study was approved by the Ethical Committee of IRCCS Multimedica “LIPIGEN—LIpid TransPort Disorders Italian GEnetic Network—Italian Registry of Familial Dyslipidemias (N.13/2011/Cardiovascolare). All participants signed an informed consent in accordance with the Declaration of Helsinki. Clinical characteristics are presented in Table S2.
In vitro proliferation assay and cytokine production
PBMC from humans or lymphocyte suspension from murine lymphoid organs were stained with 5 µM of CFSE (Merck) incubated for 10 min at RT in the dark, diluted 10 times in PBS/FBS 2%/2 mM EDTA, washed three times, and resuspended in warm media. A total of 0.2 × 106 cells were plated with 200 μl of complete RPMI medium or either of complete TexMACS medium (with or not isolated LDL according to experimental conditions) in the presence of IL-2 in a 96-well plate (U-bottom wells), previously coated with 0.5 µg/ml anti-CD3 and 2.5 µg/ml CD28 overnight (14 h) or for 4/5 d (depending on experimental conditions, see figure legends for details) at 37°C with 5% of CO2. Additionally, T cells were activated with 1 ng/ml PMA and 20 ng/ml ionomycin for 4 d at 37°C with 5% of CO2. Some experiments were performed with the addition of 100 µM Lalistat. Proliferation was assessed as CFSE dilution by flow cytometry.
For cytokine production, 96 h after the culture, cells were pulsed with 0.1 µg/ml PMA and 1 µg/ml ionomycin for 4 h at 37°C with 5% of CO2 in the presence of Brefeldin A (1:1,000). Cytokine analysis was performed by flow cytometry following the instructions from the fixation/permeabilization kit. Antibodies are listed in Table S1.
Mixed lymphocyte reaction
For in vitro assay, 106 Cell Trace Violet–labeled T cells from WT and Ldlr −/− mice were plated in a 24-well plate (U-bottom wells) in the presence of Balb/c splenocytes for 5 d at 37°C with 5% of CO2. For in vivo assay, Cell Trace Violet–labeled T cells from WT and Ldlr −/− mice were injected in the caudal vein of a C57BL6/J recipient together with an intraperitoneal injection of 25 × 106 Balb/c splenocytes inactivated with mitomycin C. Mesenteric lymph nodes were collected after 5 d for proliferation, restimulated in vitro for cytokine assay, and analyzed by flow cytometry.
Homeostatic proliferation
Cell Trace Violet–labeled T cells from WT and Ldlr −/− mice were injected in the caudal vein of an immunodeficient Rag2 −/− mice. Spleen was collected after 5 d for proliferation and cytokine production and analyzed by flow cytometry.
In vivo ovalbumin vaccination
WT and Ldlr −/− mice received the first injection of ovalbumin (0.5 µg) together with the adjuvant GLA (Glucopyranosyl lipid adjuvant) in the footpad and, a second, after 4 wk. After 1 wk later, mice were sacrificed and footpad draining lymph nodes (popliteal LNs) and spleen were collected. The cell suspension was stimulated with 1 µg/ml of OT-I (Ova 257-264) and OT-II peptides (Ova 323-339) for analysis of antigen-specific proliferation and cytokine production by flow cytometry.
In vitro stimulation of human PBMC with virus peptides
A total of 0.25 × 106 PBMC were cultured in the presence of increasing concentration from 2.5 to 50 µg/ml of virus peptides (VAXIGRIP TETRA, SANOFI PASTEUR EUROPE, cod aic 044898) in the presence of soluble 2.5 µg/ml anti-CD28. After 4 d of stimulation, cells were stained for CD4, CD8, CD25, and Granzyme B antibodies.
Flow cytometry
Lymphocytes were analyzed for the expression of extracellular markers, intracellular cytokines, proliferation, cellular viability, and lysosomes’ abundance by flow cytometry.
Immunophenotyping was performed on blood (100 μl for human, 50 μl for mice) or cell suspensions (from thymus, spleen, lymph nodes, and cultured cells). Blood was incubated at RT for 30 min with the specific antibody mixture, and thereafter samples were lysed and fixed according to manufacturer instructions. Single-cell suspensions were incubated with antibody mixtures at 4°C for 30 min and then washed with PBS/FBS 2%/EDTA 2 mM. For intracellular staining, first cells were fixed and permeabilized according to manufacturer instructions and then incubated with specific antibodies at 4°C for 30 min, washed, and analyzed.
For live and dead cell discrimination, the cell suspension was stained prior to antibody mix with 100 μl of a 1:1,000 dilution of the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit in PBS at 4°C for 30 min according to manufacturer instruction.
For detection of lipid rafts, cells were incubated with 2 μg/ml of Cholera Toxin-FITC conjugated for 30 min at 4°C or with 25 μg/ml 100 filipin for 30 min at 30°C, washed, and analyzed.
For detection of lysosomes, cells were incubated with 0.1 μM LysoTracker Green (Thermo Fisher Scientific) for 30 min at 30°C, washed, and analyzed.
Samples were acquired with Novocyte 3,000 (ACEA Biosciences) and BD LSRII Fortessa, and analyzed with Novoexpress software (ACEA Bioscience). Antibodies used are listed in Table S1.
Flow cytometry for phosphorylated proteins
A total of 1 × 106 T cells from lymph nodes of WT and Ldlr −/− mice were stimulated with plate-bound anti-CD3 (0.5 µg/ml) and CD28 (2.5 µg/ml) for 15 min. Activation was stopped by adding 4% PFA (final volume) and left at RT for 20 min. Cells were then washed in PBS 1× and permeabilized with 90% MeOH (final volume) on ice for 15 min. Cells were then washed two times with PBS 1×, stained with antibodies against phosphoresidues following dilution suggested on datasheet in BSA 5% for 1 h at RT. Cells were then washed in PBS 1× and stained with secondary anti-rabbit AlexaFluor 488 antibody plus anti-CD4 and anti-CD8 antibodies, for 30 min at RT. Cells were washed in PBS 1× and immediately acquired at BD LSRII flow cytometer. Antibodies are listed in Table S1.
Quantitative real time PCR (qRT-PCR)
For gene expression analysis, total RNA was extracted using the Monarch Total RNA Miniprep Kit (New England BioLabs). RNA was assessed for quality and quantity using absorption measurements (NanoDrop 1,000 Spectrophotometer, Thermo Fisher Scientific) and transcripted in cDNA with iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad). Gene expression analysis was performed using SYBR Green Supermix (Thermo Fisher Scientific) in CFX connect light cycler (Cat#1708841; BioRad). Expression was calculated using the ΔCt method and normalized to a housekeeping gene. Primers for qPCR were designed with the help of online tools (https://wop.metabion.com/wop/). The thermal cycling profile was a two-step amplification process (95°C for 5 min, followed by 45 cycles of 95°C for 10 s and 55°C for 30 s). The sequences of the qPCR primers are listed in Table S1.
Western blotting
Frozen cell pellets from resting or stimulated CD8+ T cells were resuspended into 60 μl of RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% sodium dodecyl sulfate, SDS, 1.0% NP-40, 0.5% sodium deoxycholate) with 0.6 μl of proteinase/phosphatase inhibitor (100X), left to homogenize 15 min in ice, and then vortexed again followed by a second incubation of 15 min in ice. Samples were then centrifuged, and the supernatant was collected into new vials. Protein content was assessed by Lory Protein Assay. The same amount of protein (between 15 and 40 µg/ml) per sample (previously denatured by the addition of Laemmli buffer and β-mercaptoethanol and incubation for 5 min at 100°C) was loaded on polyacrylamide gel and thereafter transferred to a nitrocellulose sheet. Unspecific binding sites were saturated after 1-h incubation in agitation with 5% nonfat milk or 5% Bovine Serum Albumin (BSA) in TBS-T 1X depending on the specificities of the primary antibody. Incubation with primary antibodies was performed overnight at 4°C in agitation. Specific antibodies against LDLR, p-mTOR, pS6, and β-actin were used. Membranes were then incubated with secondary antibodies conjugated to the horseradish peroxidase (HRP) for 1 h at RT and acquired with Odissey Imaging system. Densiometric analysis of the blots was performed using Image Studio Lite Version 3.1 (LI-COR) program, and the intensities of the target proteins were normalized on the respective value of β-actin, the housekeeping gene.
For the detection of phosphorylated proteins, 2/3 × 106 purified CD4+ and CD8+ T cells were stimulated with plate-bound anti-CD3 (0.5 µg/ml) and CD28 (2.5 µg/ml) for 5 min, or not for resting control. After incubation, the plate was immediately centrifuged and the cells were washed in cold PBS before being collected in RIPA buffer + proteases and phosphatases inhibitor. Protein content was assessed by Lory Protein Assay. A total of 20 µg/ml of protein per sample (previously denatured by the addition of Laemmli buffer and β-mercaptoethanol and incubated for 5 min at 100°C) were loaded on polyacrylamide gel and thereafter transferred to a nitrocellulose sheet. Unspecific binding sites were saturated after 1-h incubation in agitation with 5% BSA in TBS-T 1X. Incubation with primary antibodies was performed overnight at 4°C in agitation. Specific antibodies for TCR signaling were used. Membranes were then incubated with secondary antibodies conjugated to the horseradish peroxidase (HRP) for 1 h at RT and acquired with Odissey Imaging system.
Antibodies are listed in Table S1.
Proteomics
Proteomics analysis was performed as previously described (Bonacina et al., 2020) on 2 × 106 purified CD8+ T lymphocytes from four WT and Ldlr −/− mice resting or after 96-h of stimulation.
Sample preparation
Samples (n = 6) were pooled into two groups per condition, washed with ice-cold sterile PBS 1X, and centrifuged at low speed (1,000 g) twice before being resuspended into 100 μl of urea 8 M, Tris-HCl 0.1 M pH 8.5 with 0.1 of protease inhibitor. Samples were homogenized for 30 min at 4°C in agitation and centrifuged at 14,000 g for 30 min at 4°C. Next, the supernatant containing the proteins extracted was collected and quantified with a Lowry protein assay as previously described.
In-solution trypsin digestion
A total of 10 μg of samples were dried completely using a vacuum concentrator at 45°C for 45 min and later resuspended in 10 μl of water with the addition of 10 μl of Ambic 50 mM. The pH was checked to be 8.5.
Proteins were reduced by adding 4 μl of 100 mM dithiothreitol (DTT, to obtain a final concentration of 5 mM) and incubating for 30 min at 55°C.
After cooling the sample to room temperature, the alkylation reaction was started by the addition of iodoacetamide 150 mM (4 μl, 15 mM final concentration), followed by 30 min of incubation in the dark.
Trypsin was added with an enzyme-to-protein ratio of 1:20, and digestion occurred overnight at 37°C, stopped the following day by acidifying the samples with 2 μl of 50% trifluoroacetic acid (TFA) to have 1% TFA as the final concentration.
The final volume was 30 μl with a protein concentration of 0.33 μg/μl.
LC-MS/MS analysis
Samples were analyzed at UNITECH OMICs (University of Milan, Milan, Italy) using a Dionex Ultimate 3,000 nano-LC system connected to an orbitrap Fusion Tribrid Mass Spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray ion source operating in positive ion mode. The peptide mixtures were preconcentrated onto an Acclaim PepMap C18 5 µm, 100 Å, 100 µm ID × 2 cm (Thermo Fisher Scientific) and separated at 35°C on an EASY-Spray PepMap RSLC C18 column: 3 µm, 100 Å, 75 µm ID × 15 cm (Thermo Fisher Scientific). Each group was injected three times.
Elutions were run in gradient mode from 96% buffer A (0.1% aqueous formic acid) to 40% buffer B (0.1% aqueous formic acid/acetonitrile [2:8]). Run total length: 110 min, flow rate: 300 nl min−1. MS spectra were collected over an m/z range of 375—1,500 Da at a resolution of 120.000 in the data-dependent mode, cycle time 3 s between master scans. Fragmentation was induced by higher-energy collisional dissociation (HCD) with collision energy set at 35 eV.
Data processing and analysis
Before data analysis, each LC-MS raw file was converted from the Thermo.raw format to mzML format in centroid mode using the MSconvert tool of the software ProteoWizard (version 3.0.1957).
The mzML files were analyzed using a pipeline built using OpenMS nodes operating within the open-source software platform KNIME (Konstanz Information Miner, version 3.7.2).
Peptide identification was done by combining the search engines X!Tandem (Craig and Beavis, 2004), MS-GF+, (Kim et al., 2008), Novor (Ma, 2015), and SpectraST (Svecla et al., 2021) against the mouse Uniprot FASTA database (uniprot-mus + musculus.fasta, downloaded at www.uniprot.org in September 2019), and a common contaminant proteins database (Lam et al., 2007). The spectral library required by the SpectraST search engine was downloaded from the website www.peptideatlas.org (file NIST_mouse_IT_2012-04-21_7AA.splib).
Fragment mass tolerance was set at 0.02 Da and precursor mass tolerance at 5.0 ppm. Peptide sequences were indexed through the OpenMS PeptideIndexer node, setting leucine/isoleucine equivalence (or nonequivalence). Protein inference was carried out using the protein inference analysis (PIA) algorithm using the default parameters set by the developers.
Protein abundance estimates were calculated with the prior generation of spectral feature by the node FeatureFinderMultiplex followed by PIA-assisted FDR-scores estimation and filtering (<0.01), their ID mapping and combination with peptide IDs, their subsequent alignment, grouping, and normalization (e.g., MapAlignerIdentification, FeatureUnlabeledQT, and ConsensusmapNormalizer nodes). Proteins and peptides label-free quantification (LFQ) was then computed with the OpenMS ProteinQuantifier node based on intensities of the n = 2 most abundant identified peptides.
The relative output files, read as tables of the CSVreader node output, were finally exported in Microsoft Office Excel 2016 for further formatting and statistical elaboration. To compare proteome in between groups, the mean intensity of six technical replicates per experimental were grouped and the LFQ values were log2 -transformed. For further analysis, proteins with less than three peptides were imputed.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD030365.
Low-density lipoproteins (LDL) isolation
LDL particles were isolated from human plasma through sequential ultracentrifuge as described. The volume of the remaining solution was then measured, and its density brought to 1,063 d with the correction solution and centrifuged again for 24 h at 40,000 rpm and 4°C. At the end of this passage, LDLs floated enabling their collection. LDL solution was dialysate leaving it to percolate through a PD-10 Desalting Column (GE Healthcare) thus allowing to remove excess salts and exchange buffer with PBS 1X. Finally, LDL solution in PBS 1X was sterilized by using a 0.22 μm strainer and preserved at 4°C. LDLs were quantified by Lowry protein assay based on the amount of apolipoprotein B (the main protein of LDL) abundance.
Click chemistry
The cholesterol analog 27-alkyne cholesterol was complexed with LDL particles and incubated with stimulated CD8+ or CD4+ T cells, and then cells were seeded to polylisinated coverslips and the click-chemistry reaction was performed.
A total of 20 μl of sterile 27-alkyne cholesterol and 0.5 μg/μl in EtOH (Avanti Polar Lipids) were added to 200 μl of sterile LDL in PBS 1X. The proportion of 1:10 was chosen to avoid protein precipitation due to EtOH. The solution was left overnight at 4°C and the following day was percolated through a PD-10 desalting column to separate LDL complexed to 27-alkyne cholesterol from free 27-alkyne cholesterol. Several fractions of 200 μl were collected, and the protein content was quantified by Lowry protein assay. Fractions with at least 0.2 μg/μl of proteins were pooled together and filtered with a 0.22-μm strainer of 13 mm of diameter to reduce the minimum volume loss, and the resulting concentration was calculated. Sterile LDL-27-alkyne cholesterol solution was stored at 4°C until use.
Purified CD8+ or CD4+ T cells (0.2 × 106) were stimulated with coated anti-CD3/28 + IL-2 as previously described in TexMacs medium with 200 μg/ml of LDL-27-alkyne cholesterol solution for 24 h. Then cells were seeded on glass coverslips treated with poly-L-lysine (Merck) and left to adhere for 30 min at RT. Cells were washed twice with sterile PBS 1X and then fixed in 3.7% formaldehyde in A buffer (0.1 M Hepes pH 7.4) overnight at 4°C. Samples were washed once with 155 mM ammonium acetate and twice with A buffer in agitation for 5 min each. A solution of 5 μM Azide-fluor 488 (Merck) plus a solution of CuBF4 in 4% acetonitrile was added to coverslips and left in incubation at 43°C under agitation for 30 min. Here, the presence of an omega-terminal alkyne on the 27-alkyne cholesterol reacts with the azide group, catalyzed by the presence of copper ion, in a reaction called click chemistry. Next, samples were washed three times in A buffer for 5 min, at RT, in agitation before proceeding with antibody staining.
Immunofluorescence
Primary antibody staining was performed overnight at 4°C in agitation using anti-mouse mTOR or anti-mouse Lamp1, both of them diluted 1:200 in blocking solution. The following day, coverslips were washed with cold PBS 1X and stained with 2 mg/ml of goat anti-rabbit IgG (H+L) crossadsorbed secondary antibody, Alexa Fluor 555 diluted 1:1,000 in blocking solution for 1 h, at RT, in agitation. Then, samples were washed three times with PBS 1X for 5 min under agitation, and the nuclei were stained through incubation with Hoechst diluted 1:20,000 for 8 min at RT. Samples were washed for 5 min, at RT, under agitation, and the coverslips sealed up over microscope slides with 2 μl of Fluoroshield histology mounting medium. Samples were visualized and acquired at the Zeiss confocal microscopy by LSM software (Zeiss ZEN), and images were taken at 63× magnification.
Statistics
For proteomic interpretation, pathways analysis for the data sets were analyzed using the use of QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City). Further analyses for enrichment of functional and biological functions were performed using STRING and KEGG databases. Heatmaps were constructed using the web interface of Morpheus. The columns were clustered by the average linkage method, and the web interface of ClustVis (Metsalu and Vilo, 2015) was used to perform principal component analysis (PCA). QPCR data were analyzed using the delta CT method by taking the CT values of the genes of interest from the housekeeping gene followed by normalization to the WT control sample.
Results are presented as mean per group ± SEM, and statistical analysis was performed using nonparametric test (Mann–Whitney test) with a 95% confidence interval or two-way ANOVA by GraphPad Prism software (details in figure legends).
Online supplemental material
Fig. S1 presents a characterization of T cell subsets in the thymus and spleen of WT and Ldlr −/− mice; Fig. S2 shows different assays to test proliferative potential of CD8+ T cells from WT and Ldlr −/− mice; Fig. S3 provides an extensive analysis of a proteomic dataset from activated CD4+ and CD8+ T cells (data are available for interrogation using the Encyclopedia of Proteome Dynamics [EPD; Howden et al., 2019]) and gene expression analysis of cholesterol metabolism in CD4+ T cells from WT and Ldlr −/− mice; Fig. S4 shows a characterization of lipid raft enrichment and TCR signaling activation of CD4+ and CD8+ from WT and Ldlr −/− mice; Fig. S5 lists a characterization of metabolic energetic pathways from proteomic and gene expression analyses of CD4+ and CD8+ T cells from WT and Ldlr −/− mice; Fig. S6 provides an extensive characterization of proteomic dataset of activated CD8+ T cells from WT and Ldlr −/− mice; Fig. S7 shows additional data on lysosome reorganization in CD8+ T cells from Ldlr −/− compared to WT mice and cholesterol routing in activated CD4+ from WT and Ldlr −/− mice. Table S1 contains all the models (transgenic mice), antibodies, reagents, chemicals, information on proteomic dataset repository, instruments, and software used in the manuscript. Table S2 reports the clinical and biochemical characteristics of patients with heterozygous familial hypercholesterolemia compared to healthy controls.
Data availability
Data are available via ProteomeXchange with identifier PXD030365. The raw data underlying this article will be shared upon reasonable request to the corresponding author.
Acknowledgments
The help of Dr. Monica Gomaraschi and technicians Dr. Fiorenza Farè, Dr. Giulia Garrone from the proteomic facility of the University of Milan (Unitech OMICS) for sample LC-MS/MS processing is kindly acknowledged.
The work is supported by: Telethon Foundation (GGP19146 to G.D. Norata), Fondazione Cariplo 2019-1560 (F. Bonacina), Progetti di Rilevante Interesse Nazionale (PRIN 2017 K55HLC to G.D. Norata and PRIN 2017 H5F943 to A.L. Catapano), Ricerca Finalizzata, Ministry of Health (RF-2019-12370896, G.D. Norata e A.L. Catapano). F.M. Marelli-Berg is a recipient of the British Heart Foundation Chair of Cardiovascular Immunology (CH/15/2/32064).
The authors declare no competing financial interests.
Author contributions: Conceived and designed the experiments: F. Bonacina, A.L. Catapano, F.M. Marelli-Berg, G.D. Norata—Performed the experiments: F. Bonacina, A. Moregola, D. Coe, S. Fraire, S. Beretta, and P. Uboldi—Analyzed the data: F. Bonacina, A. Moregola, D. Coe, S. Fraire, S. Beretta, F.M. Marelli-Berg, G.D. Norata—Performed proteomic analysis: M. Svecla, S. Beretta, G. Beretta—Recruited healthy and FH subjects: F. Pellegatta—Wrote the paper: F. Bonacina, G.D. Norata—Review and edit the paper: F. Bonacina, F.M. Marelli-Berg, A.L. Catapano, and G.D. Norata.

