


FGF2 is a cell survival factor involved in tumor-induced angiogenesis that is secreted through an unconventional secretory pathway based upon direct protein translocation across the plasma membrane. Here, we demonstrate that both PI(4,5)P2-dependent FGF2 recruitment at the inner plasma membrane leaflet and FGF2 membrane translocation into the extracellular space are positively modulated by cholesterol in living cells. We further revealed cholesterol to enhance FGF2 binding to PI(4,5)P2-containing lipid bilayers. Based on extensive atomistic molecular dynamics (MD) simulations and membrane tension experiments, we proposed cholesterol to modulate FGF2 binding to PI(4,5)P2 by (i) increasing head group visibility of PI(4,5)P2 on the membrane surface, (ii) increasing avidity by cholesterol-induced clustering of PI(4,5)P2 molecules triggering FGF2 oligomerization, and (iii) increasing membrane tension facilitating the formation of lipidic membrane pores. Our findings have general implications for phosphoinositide-dependent protein recruitment to membranes and explain the highly selective targeting of FGF2 toward the plasma membrane, the subcellular site of FGF2 membrane translocation during unconventional secretion of FGF2.
Introduction
Beyond the ER/Golgi-dependent secretory pathway through which signal-peptide-containing secretory proteins are transported into the extracellular space (Palade, 1975; Rothman, 1994; Rothman and Wieland, 1996; Schekman and Orci, 1996), additional mechanisms of protein secretion are evolved in eukaryotic cells. These processes have collectively been termed as “unconventional protein secretion” (UPS; Rabouille, 2017; Dimou and Nickel, 2018; Pallotta and Nickel, 2020). One of the best characterized UPS cargoes is Fibroblast Growth Factor 2 (FGF2; Steringer and Nickel, 2018; Pallotta and Nickel, 2020; Sparn et al., 2022b), a potent mitogen involved in fundamental processes of pathophysiological significance such as tumor-induced angiogenesis and the generation of survival signals controlling programmed cell death (Presta et al., 2005; Beenken and Mohammadi, 2009; Akl et al., 2016). In previous work, FGF2 has been demonstrated to be secreted by a molecular mechanism that is based on direct translocation across the plasma membrane (type I UPS; Schäfer et al., 2004; Zehe et al., 2006; Rabouille, 2017; Steringer et al., 2017; Dimou and Nickel, 2018; Dimou et al., 2019; Pallotta and Nickel, 2020). Other cargo molecules making use of a type I UPS pathway include HIV-Tat, Tau and, under certain physiological conditions, Interleukin 1β (IL-1β; Rabouille, 2017; Dimou and Nickel, 2018; Sitia and Rubartelli, 2018; Pallotta and Nickel, 2020; Zhang et al., 2020), proteins with crucial roles in viral replication, neurodegenerative disorders, and inflammatory diseases.
The type I UPS pathway, by which FGF2 is secreted into the extracellular space, is initiated by FGF2 recruitment at the inner plasma membrane leaflet. At this location, FGF2 has been shown to undergo sequential physical interactions with the cytoplasmic domain of the Na,K-ATPase (Zacherl et al., 2015; Legrand et al., 2020), Tec kinase (Ebert et al., 2010; Steringer et al., 2012; La Venuta et al., 2016), and the phosphoinositide PI(4,5)P2 (Temmerman et al., 2008; Temmerman and Nickel, 2009; Steringer et al., 2012; Steringer et al., 2017; Dimou et al., 2019). While the specific functions of the Na,K-ATPase and Tec kinase in this process are only beginning to emerge (Ebert et al., 2010; Steringer et al., 2012; Zacherl et al., 2015; La Venuta et al., 2016; Dimou and Nickel, 2018; Pallotta and Nickel, 2020; Sparn et al., 2022b), the role of PI(4,5)P2 is understood in great detail. In the initial step, binding to PI(4,5)P2 triggers oligomerization of FGF2 (Steringer et al., 2012; La Venuta et al., 2015; Müller et al., 2015). This process leads to the formation of a lipidic membrane pore with a toroidal architecture accommodating membrane-spanning FGF2 oligomers (Steringer et al., 2012; Steringer et al., 2017; Dimou and Nickel, 2018). Once FGF2 oligomers become accessible from the outer leaflet of the plasma membrane, they get captured and disassembled by cell surface heparan sulfate proteoglycans resulting in the appearance of monomeric species FGF2 on cell surfaces (Zehe et al., 2006; Steringer et al., 2017; Dimou et al., 2019). This process is based on the ability of heparan sulfates to compete against PI(4,5)P2 with an about hundred fold higher affinity for FGF2 compared with PI(4,5)P2 (Steringer et al., 2017). Recently, glypican 1 (GPC1) has been identified as a heparan sulfate proteoglycan with a dedicated function in FGF2 secretion (Sparn et al., 2022a). GPC1 contains high affinity-binding sites for FGF2 within its heparan sulfate chains that are arranged in a highly membrane-proximal manner on cell surfaces, explaining its prominent role in unconventional secretion of FGF2 (Sparn et al., 2022a; Sparn et al., 2022b). The proposed mechanism has been confirmed in a fully reconstituted system using giant unilamellar vesicles (Steringer et al., 2017) and is consistent with earlier observations demonstrating that membrane translocation depends on a fully folded state of FGF2 that permits PI(4,5)P2-dependent FGF2 oligomerization and interactions with heparan sulfate chains (Backhaus et al., 2004; Torrado et al., 2009). Recently, PI(4,5)P2- and heparan-sulfate-dependent translocation of FGF2 across the plasma membrane has also been visualized in living cells using single-molecule TIRF microscopy. These studies revealed the real-time kinetics of this process with an average time interval for FGF2 membrane translocation of about 200 ms (Dimou et al., 2019; Pallotta and Nickel, 2020).
In previous studies, we observed physical interactions between FGF2 and PI(4,5)P2 to be most efficient when PI(4,5)P2 was reconstituted in a lipid environment resembling plasma membranes. In particular, reconstituting PI(4,5)P2 in a pure PC lipid background or removing cholesterol from a plasma-membrane-like lipid composition resulted in decreased binding efficiency of FGF2 to PI(4,5)P2 (Temmerman et al., 2008; Temmerman and Nickel, 2009). In the light of cholesterol being known to exert profound effects on the organization of the plasma membrane with the lateral segregation into liquid-ordered and liquid-disordered domains being one example (Yang et al., 2016; Sezgin et al., 2017; Steck and Lange, 2018; Enkavi et al., 2019; Wang et al., 2020), here we address a potential role of cholesterol in PI(4,5)P2-dependent FGF2 recruitment and translocation across the plasma membrane, the core step of its unconventional mechanism of secretion. To shed light on the high selectivity by which FGF2 is targeted to the plasma membrane, we demonstrate, using both biochemical in vitro experiments and single molecule analyses in living cells, that cholesterol enhances both FGF2 recruitment at the inner plasma membrane leaflet and translocation into the extracellular space. This phenomenon is likely to be of broad relevance as we found PI(4,5)P2-dependent membrane recruitment of the Pleckstrin Homology domain of phospholipase C δ1 (PH-PLC-δ1) to be modulated by cholesterol as well. Based on extensive atomistic molecular dynamics (MD) simulations, we find cholesterol to increase head group visibility of PI(4,5)P2 by exposing negative charges on the membrane surface in a way that promotes faster binding kinetics and a more stable interaction between FGF2 and PI(4,5)P2. Furthermore, we reveal cholesterol to induce clustering of PI(4,5)P2 molecules with the predominant appearance of trimers and tetramers. In a cellular context, at the inner plasma membrane leaflet, this phenomenon is likely to generate increased avidity enhancing PI(4,5)P2-dependent FGF2 oligomerization and membrane translocation to the cell surface. Finally, using droplet interface bilayers (DIB) inside a microfluidic setup, we measured bilayer tension as a function of the cholesterol concentration revealing a correlation between this parameter and the efficiency of PI(4,5)P2-dependent FGF2 membrane recruitment. Since an increase in bilayer tension is known to facilitate the formation of lipidic membrane pores (Tieleman et al., 2003; den Otter, 2009; Karal and Yamazaki, 2015; Cunill-Semanat and Salgado, 2019), cholesterol may also have a positive impact on PI(4,5)P2-dependent FGF2 membrane translocation in cells facilitating FGF2 oligomerization concomitant with the formation of toroidal membrane pores within the plasma membrane.
In conclusion, along with FGF2 interactions with the Na,K-ATPase (Zacherl et al., 2015; Legrand et al., 2020) and Tec kinase (Ebert et al., 2010; La Venuta et al., 2016) at the inner leaflet, our findings provide a compelling explanation for the high selectivity by which FGF2 is targeted to the plasma membrane, the subcellular site of FGF2 membrane translocation into the extracellular space. Furthermore, our findings are likely to have general implications with cholesterol being a general factor tuning membrane recruitment of proteins with a binding pocket for phosphoinositides.
Results
Cholesterol enhances PI(4,5)P2-dependent binding of FGF2 to lipid bilayers
In a previous study, we found that lipid bilayers made from a complex plasma-membrane-like (PM) lipid composition containing 2 mol% PI(4,5)P2 recruit FGF2 more efficiently than liposomes merely consisting of phosphatidylcholine (PC) and 2 mol% PI(4,5)P2 (Temmerman et al., 2008). To test a potential role for cholesterol in positively modulating PI(4,5)P2-dependent recruitment to lipid bilayers in a fully reconstituted system, we used a protein–lipid interaction assay based on analytical flow cytometry (Temmerman and Nickel, 2009). To quantify FGF2 binding to PI(4,5)P2 in the context of increasing concentrations of cholesterol, we made use of a FGF2-Halo fusion protein labeled with Alexa Fluor 488 (AF488) and normalized binding efficiency by labeling liposomes with a rhodamine-coupled derivative of phosphatidylethanolamine (PE; Temmerman et al., 2008; Temmerman and Nickel, 2009). In the first set of experiments, we analyzed the binding kinetics of FGF2-Halo-AF488 to liposomes containing 5 mol% PI(4,5)P2, 30 mol% cholesterol and 65 mol% PC (Fig. 1 A). Using FGF2 tagged with a Halo domain, binding kinetics to PI(4,5)P2 were characterized by an increase of binding efficiencies until about 6 h with a plateau being reached after about 12 h of incubation. The relatively slow binding kinetics were attributed to the highly acidic HaloTag protein domain which is repelled from membrane surfaces containing PI(4,5)P2. Nevertheless, as shown in Fig. 1 C and similar to previous studies using a FGF2-GFP fusion protein (Temmerman et al., 2008; Temmerman and Nickel, 2009), membrane recruitment of FGF2-Halo was strictly dependent on the presence of PI(4,5)P2. Furthermore, the reliability of this assay was further validated by testing a FGF2 variant form (K127Q/R128Q [FGF2-K127Q/R128Q-GFP]; Temmerman et al., 2008; Temmerman and Nickel, 2009) with a defect in PI(4,5)P2-dependent membrane recruitment (Fig. S1 A). Based on these findings, we tested the impact of increasing concentrations of cholesterol in PI(4,5)P2-dependent FGF2 recruitment to membranes at different time points of incubation ranging from 1 to 24 h (Fig. 1 B, subpanels a–d). These experiments revealed that increasing concentrations of cholesterol ranging from 10 to 50 mol% significantly enhance PI(4,5)P2-dependent FGF2 binding to membranes. This was particularly evident for incubation times of 1 and 3 h (Fig. 1 B, subpanels a and b) but was also detectable at 6 h of incubation (Fig. 1 B, subpanel c), a time span through which a steadily increasing binding behavior was observed in the experiments shown in Fig. 1 A. By contrast, following 24 h of incubation at which PI(4,5)P2-dependent FGF2 binding was found to have reached a plateau (Fig. 1 A), an impact of increasing concentrations of cholesterol could no longer be observed (Fig. 1 B, subpanel d).

Cholesterol enhances PI(4,5)P 2 -dependent membrane recruitment of FGF2 and PH-PLC-δ1 to lipid bilayers. FGF2-Halo-AF488 and PH-PLC-δ1-Halo-AF488 binding to PI(4,5)P2-containing liposomes were quantified using an analytical flow cytometry assay described previously (Temmerman et al., 2008; Temmerman and Nickel, 2009). (A) Kinetic analysis using liposomes containing 5 mol% PI(4,5)P2, 30 mol% cholesterol and 65 mol% PC (PC5-CHOL30). Measurements were taken after 1, 3, 6, 12, 18, and 24 h of incubation. (B) Quantitative analysis of FGF2-Halo-AF488 binding to various kinds of liposomes containing different levels of cholesterol after 1 (subpanel a), 3 (subpanel b), 6 (subpanel c), and 24 h (subpanel d) of incubation. FGF2-Halo-AF488 binding to liposomes containing 10 mol% PI(4,5)P2 and 90 mol% PC (PC10 system; positive control) and liposomes consisting of 100 mol% PC (PC0 system; negative control) were used to normalize data. (C) FGF2 binding to liposomes with a PM-like lipid composition, in the presence (30 mol%) or absence of cholesterol. The data were acquired after 1 h of incubation time. (D) Quantitative analysis of PH-PLC-δ1-Halo-AF488 membrane binding to liposomes containing either 0 or 30 mol% cholesterol after 1 h incubation. PH-PLC-δ1-Halo-AF488 binding to liposomes containing 10 mol% PI(4,5)P2 and 90 mol% PC (PC10 system; positive control) was used to normalize data. (E) PH-PLC-δ1-Halo-AF488 membrane recruitment assays using PM-like lipid compositions in the presence (30 mol%) or absence of cholesterol. The data were acquired after 1 h of incubation time. All data were corrected for background defined by binding of Halo-AF488 to the various liposomal systems indicated. Standard errors (n = 3 for A, C, and E; n = 6 for B; n = 12 for D) and P values are shown with *P ≤ 0.05; ***P ≤ 0.001. The statistical analysis was based on a one-way ANOVA test combined with Tukey’s post hoc test. Data distribution was assumed to be normal, but this was not formally tested.
Cholesterol enhances PI(4,5)P 2 -dependent membrane recruitment of FGF2 and PH-PLC-δ1 to lipid bilayers. FGF2-Halo-AF488 and PH-PLC-δ1-Halo-AF488 binding to PI(4,5)P2-containing liposomes were quantified using an analytical flow cytometry assay described previously (Temmerman et al., 2008; Temmerman and Nickel, 2009). (A) Kinetic analysis using liposomes containing 5 mol% PI(4,5)P2, 30 mol% cholesterol and 65 mol% PC (PC5-CHOL30). Measurements were taken after 1, 3, 6, 12, 18, and 24 h of incubation. (B) Quantitative analysis of FGF2-Halo-AF488 binding to various kinds of liposomes containing different levels of cholesterol after 1 (subpanel a), 3 (subpanel b), 6 (subpanel c), and 24 h (subpanel d) of incubation. FGF2-Halo-AF488 binding to liposomes containing 10 mol% PI(4,5)P2 and 90 mol% PC (PC10 system; positive control) and liposomes consisting of 100 mol% PC (PC0 system; negative control) were used to normalize data. (C) FGF2 binding to liposomes with a PM-like lipid composition, in the presence (30 mol%) or absence of cholesterol. The data were acquired after 1 h of incubation time. (D) Quantitative analysis of PH-PLC-δ1-Halo-AF488 membrane binding to liposomes containing either 0 or 30 mol% cholesterol after 1 h incubation. PH-PLC-δ1-Halo-AF488 binding to liposomes containing 10 mol% PI(4,5)P2 and 90 mol% PC (PC10 system; positive control) was used to normalize data. (E) PH-PLC-δ1-Halo-AF488 membrane recruitment assays using PM-like lipid compositions in the presence (30 mol%) or absence of cholesterol. The data were acquired after 1 h of incubation time. All data were corrected for background defined by binding of Halo-AF488 to the various liposomal systems indicated. Standard errors (n = 3 for A, C, and E; n = 6 for B; n = 12 for D) and P values are shown with *P ≤ 0.05; ***P ≤ 0.001. The statistical analysis was based on a one-way ANOVA test combined with Tukey’s post hoc test. Data distribution was assumed to be normal, but this was not formally tested.
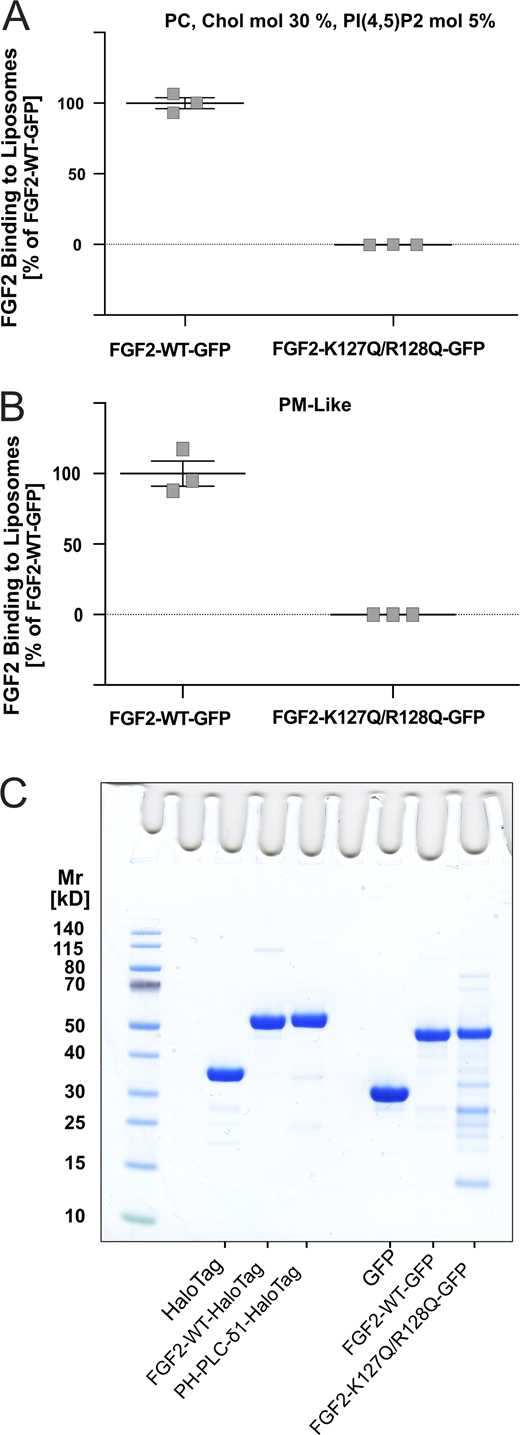
Characterization of in vitro FGF2 membrane recruitment assays using a FGF2-GFP variant form impaired in binding to PI(4,5)P2. Quantitative comparison of PI(4,5)P2-dependent binding efficiencies of FGF2-WT-GFP and a variant form known to be impaired in membrane recruitment lacking two essential amino acids of the PI(4,5)P2 binding pocket (FGF2-K127Q/R128Q-GFP [FGF2-K127Q/R128Q-GFP]; Temmerman et al., 2008, Traffic). (A) Liposomes made from a PC/Cholesterol/PI(4,5)P2 mixture. (B) Liposomes made from a complex or plasma-membrane-like lipid composition. Details on lipid compositions are given in Table S1. Data were acquired after 1 h incubation and corrected for background defined by the binding of GFP to the liposomal systems indicated. (C) Protein analysis by SDS-PAGE and Coomassie staining for all recombinant proteins used in in vitro biochemistry assays. From left to right: His-HaloTag, His-FGF2-WT-HaloTag, His- PH-PLC-δ1-HaloTag, His-GFP, His-FGF2-Y81pCMF-WT-GFP and His-FGF2-Y81pCMF-K127Q/R128Q-GFP. Data are shown as mean ± SEM (n = 3).

Characterization of in vitro FGF2 membrane recruitment assays using a FGF2-GFP variant form impaired in binding to PI(4,5)P2. Quantitative comparison of PI(4,5)P2-dependent binding efficiencies of FGF2-WT-GFP and a variant form known to be impaired in membrane recruitment lacking two essential amino acids of the PI(4,5)P2 binding pocket (FGF2-K127Q/R128Q-GFP [FGF2-K127Q/R128Q-GFP]; Temmerman et al., 2008, Traffic). (A) Liposomes made from a PC/Cholesterol/PI(4,5)P2 mixture. (B) Liposomes made from a complex or plasma-membrane-like lipid composition. Details on lipid compositions are given in Table S1. Data were acquired after 1 h incubation and corrected for background defined by the binding of GFP to the liposomal systems indicated. (C) Protein analysis by SDS-PAGE and Coomassie staining for all recombinant proteins used in in vitro biochemistry assays. From left to right: His-HaloTag, His-FGF2-WT-HaloTag, His- PH-PLC-δ1-HaloTag, His-GFP, His-FGF2-Y81pCMF-WT-GFP and His-FGF2-Y81pCMF-K127Q/R128Q-GFP. Data are shown as mean ± SEM (n = 3).
To challenge these findings, we also analyzed a potential role of cholesterol in PI(4,5)P2-dependent membrane recruitment with liposomes containing a more complex, plasma membrane-like (PM-like) lipid composition containing phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), and sphingomyelin (SM; Table S2). As shown in Fig. 1 C, omitting cholesterol from PM-like lipid compositions caused a marked and highly significant decrease in FGF2-Halo membrane-binding efficiencies. To test whether cholesterol tunes PI(4,5)P2-dependent membrane recruitment of other proteins as well, we performed the same sets of experiments with a Halo fusion protein of the Pleckstrin Homology domain of phospholipase C δ1 (PH-PLC-δ1; Ferguson et al., 1995). Similar to FGF2-Halo, we found cholesterol to increase the binding efficiency of PH-PLC-δ1-Halo to membranes containing a simple lipid composition consisting of PC, PI(4,5)P2 and cholesterol (Fig. 1 D). The other way around, removal of cholesterol from a more complex PM-like lipid composition caused PH-PLC-δ1-Halo membrane recruitment to be impaired in a highly significant manner (Fig. 1 E). These findings demonstrate under various experimental conditions, in the absence or presence of other charged membrane lipids, that cholesterol tunes membrane recruitment of proteins with binding pockets for PI(4,5)P2. Since the observed phenomenon was not restricted to FGF2 but could also be observed for PH-PLC-δ1, we concluded that cholesterol is likely to alter membrane parameters that govern the way of how the head group of PI(4,5)P2 is presented on membrane surfaces.
Cholesterol enhances head group visibility and clustering of PI(4,5)P2 on membrane surfaces
The electrostatic interaction of the headgroup of PI(4,5)P2 with the defined high affinity binding pocket in FGF2 is understood in great detail (Temmerman et al., 2008; Steringer et al., 2017). Using fully atomistic MD simulations and free energy calculations, we aimed at revealing the mechanism by which cholesterol modulates FGF2 recruitment to PI(4,5)P2-containing lipid bilayers. Based on extensive umbrella sampling simulations (Torrie and Valleau, 1974; Torrie and Valleau, 1977), we quantified the free energy barriers associated with the binding of FGF2 to POPC-based membranes containing 5 mol% PI(4,5)P2 in the presence and absence of 30 mol% of cholesterol. To avoid any bias due to different initial configurations, the systems were constructed by inserting a pre-formed FGF2-PI(4,5)P2 (1:4) complex in a bilayer with either 0 or 30 mol% of cholesterol (see Materials and methods for details). The two systems were first simulated for 1 μs at 298 K under NpT conditions using the GROMACS-2020 software (Abraham et al., 2015) and the CHARMM36m force field (Huang et al., 2017). Thirty-nine windows with 0.1 nm spacing were constructed by gently pulling the protein away from the membrane in the z-direction (along the membrane’s normal direction) at 0.1 nm/ns and a force constant of 100 kJ mol−1 nm−2. The free energy profiles were analyzed as a function of the z distance (along the membrane normal direction) to the membrane surface (Fig. 2 A), demonstrating the interaction between FGF2 and PI(4,5)P2 to represent a spontaneous process. This was true in both the presence and absence of cholesterol. However, cholesterol was found to exert a higher dissociation energy barrier shifting the energy minimum from −29 to −35 kBT. Thus, in the presence of cholesterol, FGF2 binding to PI(4,5)P2 is both faster and more stable. These findings are consistent with the biochemical reconstitution experiments as shown in Fig. 1. To reveal the mechanism underlying the positive modulation of FGF2 binding to PI(4,5)P2 by cholesterol, we investigated the effect of cholesterol on structural and electrostatic properties of PI(4,5)P2-containing lipid bilayers (Fig. 2 B and C). Seven different lipid compositions mimicking the in vitro experiments as shown in Fig. 1 were analyzed (see Table S3 for simulation details). Intriguingly, we found that cholesterol affects the charge density distribution of lipid bilayers containing PI(4,5)P2 (Fig. 2, A and B). These were calculated by summing up all charges per slice along the surface of lipid bilayers with the lipid compositions indicated (Fig. 2, A and B). In a pure POPC bilayer (Fig. 2 B, light red line), the charge density shows two peaks in the headgroup region. The positive peak at +0,6 e/nm3 originates from the positive charge of the choline headgroup of POPC. The negative peak at −0,6 e/nm3 represents the negative net charges of phosphate groups. The substitution of POPC molecules with either 5 or 10 mol% of PI(4,5)P2 (Fig. 2 B; red and dark-red lines) did not have any impact on the average charge density distribution in the phosphate region of membrane lipids. By contrast, a decrease in the positive charge density distributions in the region of the choline headgroups was observed from about +0.6 e/nm3 in the absence of PI(4,5)P2 to about +0.2 e/nm3 in the presence of 5 mol% of PI(4,5)P2 (Fig. 2 B; red line) and about −0.3 e/nm3 for 10 mol% of PI(4,5)P2 (Fig. 2 B; dark red line). The electric field calculation as shown in Fig. S2 A corroborates our hypothesis indicating the long-range effect on proteins regarding the decrease in the positive charge density distributions in the region of the choline headgroups.

Cholesterol enhances PI(4,5)P 2 head group visibility stabilizing FGF2 membrane recruitment. (A) Potential of mean force (PMF) along with the z component of the distance between the center of masses of the protein and the P atoms of phospholipids in the closest leaflet. 39 windows with 0.1 nm spacing were simulated for 400 ns. The first 200 ns have been discarded from the PMF calculation and considered as equilibration time. (B and C) Charge density profiles along the perpendicular axis of the bilayers, averaged over the last 500 ns of the pure bilayer simulations (no FGF2) without (B) and with 30 mol% cholesterol (C). (D) Charge density profiles for all lipid composition analyzed in this study. (E) Deuterium order parameter (-SCH) of POPC sn-1 chains calculated from MD simulations for pure POPC membrane and for PI(4,5)P2-enriched (5 mol%) POPC-based membranes containing either 0, 10, 15, 30, or 50 mol% of cholesterol, respectively. SCH was evaluated as averaged over the last 500 ns of simulations by taking both leaflets into account. (F) Membrane thickness for a pure POPC membrane and PI(4,5)P2-enriched (5 mol%) POPC-based membranes containing either 0, 10, 15, 30, or 50 mol% of cholesterol, respectively. The thickness was calculated by measuring the z-component of the center of the mass distance between the two leaflets’ phosphate atoms. The data were averaged over the last 500 ns of the simulations with the error given as SD.
Cholesterol enhances PI(4,5)P 2 head group visibility stabilizing FGF2 membrane recruitment. (A) Potential of mean force (PMF) along with the z component of the distance between the center of masses of the protein and the P atoms of phospholipids in the closest leaflet. 39 windows with 0.1 nm spacing were simulated for 400 ns. The first 200 ns have been discarded from the PMF calculation and considered as equilibration time. (B and C) Charge density profiles along the perpendicular axis of the bilayers, averaged over the last 500 ns of the pure bilayer simulations (no FGF2) without (B) and with 30 mol% cholesterol (C). (D) Charge density profiles for all lipid composition analyzed in this study. (E) Deuterium order parameter (-SCH) of POPC sn-1 chains calculated from MD simulations for pure POPC membrane and for PI(4,5)P2-enriched (5 mol%) POPC-based membranes containing either 0, 10, 15, 30, or 50 mol% of cholesterol, respectively. SCH was evaluated as averaged over the last 500 ns of simulations by taking both leaflets into account. (F) Membrane thickness for a pure POPC membrane and PI(4,5)P2-enriched (5 mol%) POPC-based membranes containing either 0, 10, 15, 30, or 50 mol% of cholesterol, respectively. The thickness was calculated by measuring the z-component of the center of the mass distance between the two leaflets’ phosphate atoms. The data were averaged over the last 500 ns of the simulations with the error given as SD.

The role of electrostatics in PI(4,5)P 2 -dependent FGF2 membrane recruitment. (A) Electric field integrated from the charge density analysis of Fig. 2 C. (B) Size-aggregation frequency analysis for PM-like membrane composition systems without (gray color) and with 30 mol% of cholesterol (brown color). The analysis was averaged over the two membrane leaflets considering the last 1,000 ns of simulations. Data are shown as the average of both bilayer leaflets, and the error bar represent the standard deviation. Two monomers were considered as a dimer if any of their atoms had a distance of <1.2 nm. (C) Adaptive Poisson-Boltzmann Solver (APBS) calculations on the electrostatic surface of FGF2. (D) PI(4,5)P2 head group localization expressed as mass density for the systems containing either 0 or 30 mol% cholesterol in the presence (upper panel) or absence (lower panel) of interactions with FGF2. Data were averaged over the last 500 ns of the simulations.

The role of electrostatics in PI(4,5)P 2 -dependent FGF2 membrane recruitment. (A) Electric field integrated from the charge density analysis of Fig. 2 C. (B) Size-aggregation frequency analysis for PM-like membrane composition systems without (gray color) and with 30 mol% of cholesterol (brown color). The analysis was averaged over the two membrane leaflets considering the last 1,000 ns of simulations. Data are shown as the average of both bilayer leaflets, and the error bar represent the standard deviation. Two monomers were considered as a dimer if any of their atoms had a distance of <1.2 nm. (C) Adaptive Poisson-Boltzmann Solver (APBS) calculations on the electrostatic surface of FGF2. (D) PI(4,5)P2 head group localization expressed as mass density for the systems containing either 0 or 30 mol% cholesterol in the presence (upper panel) or absence (lower panel) of interactions with FGF2. Data were averaged over the last 500 ns of the simulations.
In further simulations, we studied the effects of substituting POPC with cholesterol using lipid membranes containing 5 mol% PI(4,5)P2 (Fig. 2 C). Under these conditions, despite the relative content of PI(4,5)P2 remained the same, the average charge density in the headgroup regions became more negative with a maximum of about −0.3 e/nm3. Furthermore, at 30 mol% cholesterol, we observed the formation of transient PI(4,5)P2 clusters with trimers and tetramers being formed at the expense of monomers and dimers (Fig. 3 A, Fig. S2 B, and Video 1). This phenomenon is documented in Fig. 3 B in which the aggregation state of PI(4,5)P2 molecules is shown as a function of time. In this way, highly negatively charged spots of PI(4,5)P2 clusters are forming on the membrane surface that we proposed to function in stabilizing FGF2 binding through increased avidity. Furthermore, when FGF2 binding to model membranes was simulated, 5 to 6 molecules of PI(4,5)P2 were observed to associate with FGF2 (Fig. S2, C and D; and Video 2), causing a strong local accumulation of negative charges that is likely to destabilize the lipid bilayer. This idea is supported by previous studies that have shown high charge-concentration gradients and electric fields across a membrane to induce pore formation (Gurtovenko and Vattulainen, 2005; Kotnik et al., 2019). The thermodynamic aspects of this phenomenon are likely to play a major role in the conversion of the lipid bilayer structure into a lipidic membrane pore during FGF2 oligomerization and membrane translocation.

Cholesterol triggers clustering of PI(4,5)P 2 molecules. (A) Size-aggregation frequency analysis (right panel) for systems without (gray color) and with 30 mol% cholesterol (brown color). The analysis was averaged over the two membrane leaflets considering the last 1,000 ns of simulations. Data are shown as the average of both bilayer leaflets, and the error is given as standard deviation. Two monomers were considered as a dimer if any of their atoms had a distance of <1.2 nm. The right panel represents a snapshot of a lateral partial density analysis with a dimer present as an example. (B) Time evolution of the cluster fractional occupation number distribution of PI(4,5)P2 head groups for systems without (subpanels a and b), and with 30 mol% cholesterol (subpanels c and d). The calculation was made for each membrane leaflet separately. Each panel represents a 1,200 ns MD simulation of a system with eight PI(4,5)P2 molecules (head groups only). The fractional occupation number was calculated every nanosecond and it is represented with a black and white scale bar.
Cholesterol triggers clustering of PI(4,5)P 2 molecules. (A) Size-aggregation frequency analysis (right panel) for systems without (gray color) and with 30 mol% cholesterol (brown color). The analysis was averaged over the two membrane leaflets considering the last 1,000 ns of simulations. Data are shown as the average of both bilayer leaflets, and the error is given as standard deviation. Two monomers were considered as a dimer if any of their atoms had a distance of <1.2 nm. The right panel represents a snapshot of a lateral partial density analysis with a dimer present as an example. (B) Time evolution of the cluster fractional occupation number distribution of PI(4,5)P2 head groups for systems without (subpanels a and b), and with 30 mol% cholesterol (subpanels c and d). The calculation was made for each membrane leaflet separately. Each panel represents a 1,200 ns MD simulation of a system with eight PI(4,5)P2 molecules (head groups only). The fractional occupation number was calculated every nanosecond and it is represented with a black and white scale bar.
Time evolution of the lateral partial mass density analysis of PI(4,5)P2head groups for membrane systems containing either 0 (left panel) or 30 mol% of cholesterol (right) in the absence of FGF2. The video is based on fully atomistic molecular dynamics simulations.
Time evolution of the lateral partial mass density analysis of PI(4,5)P2head groups for membrane systems containing either 0 (left panel) or 30 mol% of cholesterol (right) in the absence of FGF2. The video is based on fully atomistic molecular dynamics simulations.
Time evolution of the system with 30 mol% of cholesterol in presence of FGF2 (right panel) and its lateral partial mass density evolution of PI(4,5)P2head groups. The video is based on fully atomistic molecular dynamics simulations.
Time evolution of the system with 30 mol% of cholesterol in presence of FGF2 (right panel) and its lateral partial mass density evolution of PI(4,5)P2head groups. The video is based on fully atomistic molecular dynamics simulations.
Cholesterol increases lateral bilayer tension in PI(4,5)P2-containing membranes
Combining microfluidic technology with drop shape analysis, we aimed at determining the role of cholesterol on membrane mechanics and its effect on bilayer tension (; i.e., lateral membrane pressure). The presence of cholesterol is often associated with an increase in membrane tension; however, this phenomenon strongly depends on the specific membrane lipid species being analyzed (Serral Gracià et al., 2010; Takei et al., 2015; Taylor et al., 2015). In the context of this study, we analyzed potential effects of cholesterol in the context of polydisperse lipid mixtures containing PI(4,5)P2, a non-bilayer lipid known to increase lateral pressure in the acyl chain regions of lipid bilayers (van den Brink-van der Laan et al., 2004). To determine this parameter, the bilayer contact angle θ and the interfacial tension were measured for each of the lipid compositions. To obtain θ, a lipid bilayer was formed at the intersection of a microfluidic set-up (Fig. S3 A), using a two-phase system consisting of an aqueous buffer and squalene as a solvent for membrane lipids. Various lipid mixtures containing POPC, 5 mol% PI(4,5)P2, and cholesterol at either 0, 10, 15, 30, or 50 mol% were used to determine membrane tension. As shown in Fig. S3 B and Table S1, the bilayer contact angle (θ) was determined from optical micrographs as the angle between the two leaflets. To determine the interfacial tension (σ; Fig. 4, A and B), pendant buffer droplets in a squalene-lipid solution (1 mg/ml) were used as described in Materials and methods.

Analysis of bilayer contact angles using a microfluidics setup. (A) Schematic overview of the chip geometries used. Both the devices have a cross-geometry and the lipid monolayers meet at the intersection of the channels forming the bilayer. In subpanel a, fluorescence microscopy micrographs show how the chip was first filled by injection from one of the two inlets with a lipid solution in squalene stained with Rhodamine-labeled PE and then with a buffer phase using hydrostatic pressure. This setup allowed for the lipids to decorate the oil-buffer interface. In subpanel b, the chip was also filled with membrane lipids (inlet 2) as part of a squalene solution containing Rhodamine-labeled PE. Afterwards, the two buffer fingers were introduced from opposite sides (inlets 1) resulting in membrane lipid decoration of the two interfaces. After generating a close contact through hydrostatic pressure manipulation, the membrane lipids formed a bilayer as shown in the optical micrographs. The geometry of the chip in subpanel was found to form more stable bilayers due to a better control of hydrostatic pressure. The scale bar is 75 µm. (B) Optical micrographs of the bilayer contact angles for artificial membranes with different cholesterol contents increasing from top to bottom (scale bar = 50 µm).

Analysis of bilayer contact angles using a microfluidics setup. (A) Schematic overview of the chip geometries used. Both the devices have a cross-geometry and the lipid monolayers meet at the intersection of the channels forming the bilayer. In subpanel a, fluorescence microscopy micrographs show how the chip was first filled by injection from one of the two inlets with a lipid solution in squalene stained with Rhodamine-labeled PE and then with a buffer phase using hydrostatic pressure. This setup allowed for the lipids to decorate the oil-buffer interface. In subpanel b, the chip was also filled with membrane lipids (inlet 2) as part of a squalene solution containing Rhodamine-labeled PE. Afterwards, the two buffer fingers were introduced from opposite sides (inlets 1) resulting in membrane lipid decoration of the two interfaces. After generating a close contact through hydrostatic pressure manipulation, the membrane lipids formed a bilayer as shown in the optical micrographs. The geometry of the chip in subpanel was found to form more stable bilayers due to a better control of hydrostatic pressure. The scale bar is 75 µm. (B) Optical micrographs of the bilayer contact angles for artificial membranes with different cholesterol contents increasing from top to bottom (scale bar = 50 µm).

Cholesterol increases membrane tension in PI(4,5)P 2 -containing membranes. (A) Interfacial tensions (σ) trends measured with pendant drop experiments as a function of time for POPC + 5% PI(4,5)P2, varying the cholesterol content (0, 10, 15, 30, 50 mol%). A buffer drop (HEPES 25 mM, KCl 150 mM) is immersed in a lipid-squalene solution (1 mg/ml) and its change in shape, which corresponds to the arrangement of the lipids at the newly created buffer-oil interface, is monitored. The σ values recorded after reaching the plateau increase with higher cholesterol content. (B) Average σ values (n = 3) after reaching a plateau at t = 24 min. Errors are given as SD. (C) Values for bilayer tension ϒ of PI(4,5)P2-containing (5 mol%) membranes as a function of the cholesterol concentration. ϒ is calculated combining the bilayer angles obtained by analysis of the optical micrographs and the σ values after the plateau through the Eq. 1. Errors were calculated using error propagation.
Cholesterol increases membrane tension in PI(4,5)P 2 -containing membranes. (A) Interfacial tensions (σ) trends measured with pendant drop experiments as a function of time for POPC + 5% PI(4,5)P2, varying the cholesterol content (0, 10, 15, 30, 50 mol%). A buffer drop (HEPES 25 mM, KCl 150 mM) is immersed in a lipid-squalene solution (1 mg/ml) and its change in shape, which corresponds to the arrangement of the lipids at the newly created buffer-oil interface, is monitored. The σ values recorded after reaching the plateau increase with higher cholesterol content. (B) Average σ values (n = 3) after reaching a plateau at t = 24 min. Errors are given as SD. (C) Values for bilayer tension ϒ of PI(4,5)P2-containing (5 mol%) membranes as a function of the cholesterol concentration. ϒ is calculated combining the bilayer angles obtained by analysis of the optical micrographs and the σ values after the plateau through the Eq. 1. Errors were calculated using error propagation.
Using a microfluidic set-up (Fig. S3 A), we obtained a range of bilayer angles between 20° and 30° with a continuous decrease being observed as a function of rising concentrations of cholesterol (Fig. S3 B and Table S1). An even more pronounced effect of cholesterol was observed for the interfacial tension values (σ; Fig. 4 B). In the absence of cholesterol, the value σ was found to be (2.2 ± 0.2) mN/m measured after reaching a plateau at t 24 min (Fig. 4 A). When cholesterol was added to the lipid mixtures, σ was found to increase to (5.3 ± 0.5) mN/m, (6.8 ± 1.2) mN/m, (7.3 ± 0.3) mN/m, and (7.1 ± 0.8) mN/m for cholesterol concentrations of 10, 15, 30, and 50 mol%, respectively. As shown in Fig. 4 C, this trend is also reflected in the corresponding bilayer tension values (ϒ) combining σ and θ in Eq. 1 (see Materials and methods). The highest bilayer tension value was found for lipid bilayers made from POPC, 5 mol% PI(4,5)P2 and 30 mol% cholesterol (ϒ = 13.4 ± 0.5 mN/m). Combining σ and θ in Eq. 2 (see Materials and methods for details), the adhesion energies between the two monolayers were calculated (Table S1). When all lipid compositions were compared, a maximum value was observed for membranes containing 15 mol% cholesterol. At both lower and higher concentrations of cholesterol, the monolayer adhesion energy was found to be lower (Table S1). These observations are in line with the size of the contact areas found for different concentrations of cholesterol (Fig. S3 B and Table S1). Our findings establish a correlation between membrane tension and the cholesterol content for the lipid compositions used in the biochemical reconstitution experiments as shown in Fig. 1. Since an increase in tension is known to facilitate the formation of lipidic membrane pores (Tieleman et al., 2003; den Otter, 2009; Karal and Yamazaki, 2015; Cunill-Semanat and Salgado, 2019), we propose cholesterol to increase membrane packing and tension in PI(4,5)P2-containing membranes, fostering FGF2 recruitment and translocation.
Cholesterol, PI(4,5)P2, and FGF2 colocalize at the plasma membrane visualized by super-resolution STED microscopy
To validate the in vitro experiments and the MD simulations described above, we started a series of cell-based analyses. In a first set of experiments, we aimed at imaging cholesterol, PI(4,5)P2 and FGF2 in the plasma membrane using three-color STED microscopy (for experimental details see Materials and methods). We used the eGFP-GRAMD1b sensor that detects membrane domains characterized by high concentrations of accessible cholesterol (≥30 mol%; Ercan et al., 2021). A representative example of these experiments is shown in Fig. 5 A imaging all three components at the plasma membrane with both confocal and super-resolution STED microscopy. As emphasized in three- and two-color STED images at higher magnification, pairs of FGF2 (green) and PI(4,5)P2 (red) are in close proximity in membrane areas with elevated levels of accessible cholesterol (grey; GRAM1b-G187L-GFP cholesterol sensor; Fig. 5, B and C). These analyses demonstrated that FGF2 and PI(4,5)P2 are abundantly present in plasma membrane areas enriched in cholesterol.

FGF2 and PI(4,5)P 2 colocalize in areas enriched in cholesterol as analyzed by super-resolution STED microscopy. U2OS FGF2-Halo cells were transfected with GRAM1b-G187L-GFP to visualize membrane areas containing elevated levels of accessible cholesterol. Cells were then treated with doxycycline to induce FGF2-Halo expression 24 h prior to fixation and antibody incubation. (A) Confocal (top row) and STED (lower row) images of FGF2 (in green), PI4,5)P2 (in red) and GRAM1b-G187L-GFP (in grey). Dual or triple color merged images and cropped images showing a close-up perspective are presented. Areas with FGF2 and PI(4,5)P2 in close proximity are indicated with arrows. Scale bar = 1 µm. (B and C) Larger magnifications of the three- and two-color STED images shown in panel A with white arrows pointing at pairs of FGF2 (green) and PI(4,5)P2 (red) in membrane areas with elevated levels of accessible cholesterol (grey; GRAM1b-G187L-GFP cholesterol sensor). For experimental details, see Materials and methods. Scale bar = 1 µm.
FGF2 and PI(4,5)P 2 colocalize in areas enriched in cholesterol as analyzed by super-resolution STED microscopy. U2OS FGF2-Halo cells were transfected with GRAM1b-G187L-GFP to visualize membrane areas containing elevated levels of accessible cholesterol. Cells were then treated with doxycycline to induce FGF2-Halo expression 24 h prior to fixation and antibody incubation. (A) Confocal (top row) and STED (lower row) images of FGF2 (in green), PI4,5)P2 (in red) and GRAM1b-G187L-GFP (in grey). Dual or triple color merged images and cropped images showing a close-up perspective are presented. Areas with FGF2 and PI(4,5)P2 in close proximity are indicated with arrows. Scale bar = 1 µm. (B and C) Larger magnifications of the three- and two-color STED images shown in panel A with white arrows pointing at pairs of FGF2 (green) and PI(4,5)P2 (red) in membrane areas with elevated levels of accessible cholesterol (grey; GRAM1b-G187L-GFP cholesterol sensor). For experimental details, see Materials and methods. Scale bar = 1 µm.
Loading of cells with cholesterol enhances both FGF2 recruitment at the inner plasma membrane leaflet and FGF2 membrane translocation into the extracellular space
Using a single particle TIRF microscopy approach established previously (Dimou et al., 2019; Legrand et al., 2020), we aimed at quantifying both FGF2 recruitment at the inner plasma membrane leaflet and FGF2 translocation to the cell surface under conditions of increased cholesterol levels in living cells (Fig. 6). This was achieved using Cholesterol:Methyl-β-Cyclodextrin complexes to load cells with exogenous cholesterol (Mahammad and Parmryd, 2015; Merezhko et al., 2018). We employed both CHO K1 and U2OS cells and analyzed cellular cholesterol levels by means of filipin staining and mass spectrometry (Fig. S4). Based on filipin staining analyzed by confocal microscopy, following treatment of cells with Cholesterol:Methyl-β-Cyclodextrin complexes, a significant increase of cellular cholesterol levels by 56% in CHO K1 cells and 48% in U2OS cells (Fig. S4 A) could be observed. These findings were validated in CHO cells by mass spectrometry analyzing cholesterol levels in plasma membrane-enriched subcellular fractions. A Western blot analysis was performed to test for plasma membrane enrichment and possible contaminations by membranes derived from the endoplasmic reticulum (Fig. S4 B, subpanel a). These studies demonstrated a significant increase of plasma membrane cholesterol by 9% when cells were treated with Cholesterol:Methyl-β-Cyclodextrin complexes (Fig. S4 B, subpanel b). Thus, using two independent analytical methods, the described procedure was validated to increase cellular cholesterol levels in intact cells in a significant manner.

Increased cellular levels of cholesterol positively modulate FGF2 recruitment at the inner plasma membrane leaflet in living cells, as well as PI(4,5)P2-dependent FGF2 translocation to cell surfaces. (A) FGF2-GFP recruitment efficiency under conditions of enriched cellular cholesterol levels. CHO K1 and U2OS cell lines stably expressing FGF2-GFP in a doxycycline-dependent manner were imaged by single molecule TIRF microscopy as described previously (Dimou et al., 2019; Legrand et al., 2020). Before imaging, cells were treated with Cholesterol:Methyl-β-Cyclodextrin (1:10 M ratio) complexes for 1 h in culture conditions. Single FGF2-GFP particles were identified at the inner plasma membrane leaflet (labeled by pink circles in subpanels a and c). For each condition, a widefield image and the first frame of the corresponding TIRF video are shown (subpanels a and c; Scale bar = 6 μm). Quantification of FGF2-GFP membrane recruitment at the inner leaflet of CHO K1 and U2OS cells is shown in subpanels b and d, respectively. Time-lapse TIRF movies with a total of 80 frames (100 ms/frame) were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of GFP particles was normalized for both surface area and the relative expression levels of FGF2-GFP for each experimental condition. Mean values are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (****P ≤ 0.0001). (B) FGF2-GFP translocation efficiency under conditions of enriched cellular cholesterol levels. CHO K1 and U2OS cells were induced with doxycycline for 24 h to express FGF2-GFP. Following treatment with Cholesterol:Methyl-β-Cyclodextrin (1:10 M ratio) complexes for 1 h in culture conditions, cells were incubated on ice for 30 min with Alexa-Fluor-647-labeled anti-GFP nanobodies. After labeling of FGF2-GFP on cell surfaces, cells were fixed with 4% PFA at room temperature for 20 min and imaged by single molecule TIRF microscopy as established previously (Dimou et al., 2019). For each condition, a widefield image and the corresponding TIRF image is shown (subpanels a and c; scale bar = 6 μm). Quantification of FGF2-GFP membrane translocation in CHO K1 and U2OS for all conditions indicated is shown in subpanel b and d, respectively. Single nanobody particles were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of nanobody particles detected per cell was normalized for the surface area of the corresponding cell. The mean values for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a two-tailed unpaired t test (****P ≤ 0.0001). Data distribution was assumed to be normal, but this was not formally tested.
Increased cellular levels of cholesterol positively modulate FGF2 recruitment at the inner plasma membrane leaflet in living cells, as well as PI(4,5)P2-dependent FGF2 translocation to cell surfaces. (A) FGF2-GFP recruitment efficiency under conditions of enriched cellular cholesterol levels. CHO K1 and U2OS cell lines stably expressing FGF2-GFP in a doxycycline-dependent manner were imaged by single molecule TIRF microscopy as described previously (Dimou et al., 2019; Legrand et al., 2020). Before imaging, cells were treated with Cholesterol:Methyl-β-Cyclodextrin (1:10 M ratio) complexes for 1 h in culture conditions. Single FGF2-GFP particles were identified at the inner plasma membrane leaflet (labeled by pink circles in subpanels a and c). For each condition, a widefield image and the first frame of the corresponding TIRF video are shown (subpanels a and c; Scale bar = 6 μm). Quantification of FGF2-GFP membrane recruitment at the inner leaflet of CHO K1 and U2OS cells is shown in subpanels b and d, respectively. Time-lapse TIRF movies with a total of 80 frames (100 ms/frame) were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of GFP particles was normalized for both surface area and the relative expression levels of FGF2-GFP for each experimental condition. Mean values are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (****P ≤ 0.0001). (B) FGF2-GFP translocation efficiency under conditions of enriched cellular cholesterol levels. CHO K1 and U2OS cells were induced with doxycycline for 24 h to express FGF2-GFP. Following treatment with Cholesterol:Methyl-β-Cyclodextrin (1:10 M ratio) complexes for 1 h in culture conditions, cells were incubated on ice for 30 min with Alexa-Fluor-647-labeled anti-GFP nanobodies. After labeling of FGF2-GFP on cell surfaces, cells were fixed with 4% PFA at room temperature for 20 min and imaged by single molecule TIRF microscopy as established previously (Dimou et al., 2019). For each condition, a widefield image and the corresponding TIRF image is shown (subpanels a and c; scale bar = 6 μm). Quantification of FGF2-GFP membrane translocation in CHO K1 and U2OS for all conditions indicated is shown in subpanel b and d, respectively. Single nanobody particles were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of nanobody particles detected per cell was normalized for the surface area of the corresponding cell. The mean values for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a two-tailed unpaired t test (****P ≤ 0.0001). Data distribution was assumed to be normal, but this was not formally tested.
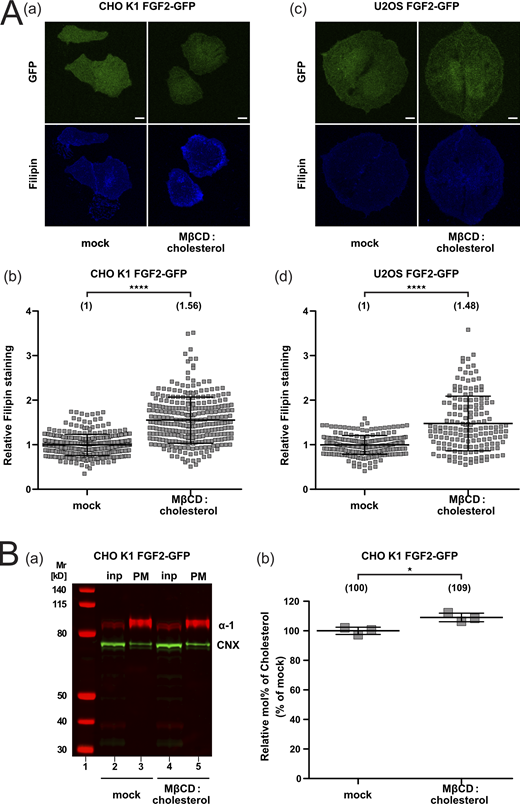
Quantification of cellular cholesterol levels using filipin imaging and mass spectrometry after cholesterol enrichment. (A) Cholesterol enrichment quantification by means of Filipin staining. Stable CHO K1 (subpanels a and b) and U2OS (subpanels c and d) cell lines were treated with Cholesterol:Methyl-β-Cyclodextrin (1:10 M ratio) complexes for 1 h in culture conditions. Cells were stained with filipin to visualize cholesterol using confocal microscopy. For each condition, both GFP and filipin fluorescence are shown (subpanels a and c; Scale bar = 6 μm). Confocal images were analyzed using ImageJ with the quantification of cholesterol levels for CHO K1 and U2OS for all conditions shown in subpanel b and d, respectively. The mean intensity values of the filipin signal detected per cell for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (****P ≤ 0.0001). (B) Cholesterol enrichment in CHO K1 plasma membrane fractions quantified via mass spectrometry. Membrane fractions were validated for membrane enrichment and ER contamination via Western blot (subpanel a). Input and membrane fractions were blotted against a plasma membrane marker (the α-1 subunit of the Na,K-ATPase) and an ER marker (calnexin). For details, see Materials and methods. For lipidic mass spectrometry, ratios of cholesterol to PC were determined for the different conditions indicated with the mock condition set to 1 (subpanel b). Data are shown as mean ± SD (n = 3). The statistical analysis was based on a two-tailed unpaired t test (*P ≤ 0.05). Data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData FS4.

Quantification of cellular cholesterol levels using filipin imaging and mass spectrometry after cholesterol enrichment. (A) Cholesterol enrichment quantification by means of Filipin staining. Stable CHO K1 (subpanels a and b) and U2OS (subpanels c and d) cell lines were treated with Cholesterol:Methyl-β-Cyclodextrin (1:10 M ratio) complexes for 1 h in culture conditions. Cells were stained with filipin to visualize cholesterol using confocal microscopy. For each condition, both GFP and filipin fluorescence are shown (subpanels a and c; Scale bar = 6 μm). Confocal images were analyzed using ImageJ with the quantification of cholesterol levels for CHO K1 and U2OS for all conditions shown in subpanel b and d, respectively. The mean intensity values of the filipin signal detected per cell for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (****P ≤ 0.0001). (B) Cholesterol enrichment in CHO K1 plasma membrane fractions quantified via mass spectrometry. Membrane fractions were validated for membrane enrichment and ER contamination via Western blot (subpanel a). Input and membrane fractions were blotted against a plasma membrane marker (the α-1 subunit of the Na,K-ATPase) and an ER marker (calnexin). For details, see Materials and methods. For lipidic mass spectrometry, ratios of cholesterol to PC were determined for the different conditions indicated with the mock condition set to 1 (subpanel b). Data are shown as mean ± SD (n = 3). The statistical analysis was based on a two-tailed unpaired t test (*P ≤ 0.05). Data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData FS4.
Based on the described procedures and experimental conditions, we quantified FGF2-GFP recruitment at the inner plasma membrane leaflet in intact CHO K1 and U2OS cells (Fig. 6 A). For all conditions, both widefield and TIRF images were taken (Fig. 6 A, subpanels a and c). While the widefield images allowed for the analysis of total expression levels of FGF2-GFP, the TIRF images were processed for the quantification of individual FGF2-GFP particles per surface area in the direct vicinity of the plasma membrane. These experiments demonstrated that treating cells with Cholesterol:Methyl-β-Cyclodextrin complexes to increase cellular cholesterol levels (Fig. S4) results in enhanced FGF2-GFP recruitment at the inner plasma membrane leaflet. This phenomenon could be observed in both cell lines being investigated with a significant increase of 39% in CHO K1 cells and 33% in U2OS cells, respectively (Fig. 6 A, subpanels b and d). Similarly, under conditions of increased cellular cholesterol levels, we found FGF2-GFP translocation to the cell surface to be enhanced in a highly significant manner by 83% (CHO K1) and 74% (U2OS), respectively (Fig. 6 B, subpanels b and d). These findings in intact cells are consistent with the biochemical reconstitution experiments as shown in Fig. 1, demonstrating that cholesterol positively modulates FGF2 membrane recruitment by PI(4,5)P2, a process that triggers FGF2 translocation across the plasma membrane during unconventional secretion.
Depletion of cellular cholesterol impairs both FGF2 recruitment at the inner plasma membrane leaflet and FGF2 membrane translocation to cell surfaces
As a complement to the experiments analyzing FGF2 membrane recruitment and translocation at increased cholesterol levels (Fig. 6 and Fig. S4), we also studied these processes in CHO and U2OS cells under conditions characterized by reduced cellular levels of cholesterol (Fig. 7 and Fig. S5). To experimentally achieve this, we treated cells by a combination of mevastatin and mevalonate, compounds that inhibit cholesterol biosynthesis (Cheng et al., 2006). To monitor cholesterol levels in plasma membranes of CHO and U2OS cells under various experimental conditions, we again used a combination of confocal microscopy and cholesterol imaging based on filipin. For both cell types, a significant reduction in cellular cholesterol levels by 47% (CHO) and 43% (U2OS) was observed (Fig. S5 A, subpanels b and d). In addition, we prepared subcellular fractions from CHO cells enriched in plasma membrane vesicles and analyzed their cholesterol contents using mass spectrometry. Plasma membrane-enriched fractions were subjected to a Western analysis to validate the actual enrichment of plasma membranes and possible contaminations (Fig. S5 B, subpanel a). This analysis revealed a significant reduction of cholesterol in the plasma membrane of CHO cells by 14% (Fig. S5 B, subpanel b). Thus, two independent methods revealed a highly significant reduction of cholesterol levels in the plasma membranes of cells treated with mevastatin and mevalonate. Under these conditions, we measured both FGF2-GFP recruitment to the inner plasma membrane leaflet and FGF2 membrane translocation to cell surfaces using TIRF microscopy (Dimou et al., 2019; Legrand et al., 2020). For both CHO and U2OS cells, a moderate reduction in FGF2 recruitment to the inner plasma membrane leaflet could be observed under conditions of reduced cholesterol levels (Fig. 7 A). This phenomenon translated into a strong decrease of FGF2 membrane translocation to cell surfaces with 70% inhibition in CHO and 49% inhibition in U2OS cells when cellular cholesterol levels were downregulated by treating cells with mevastatin and mevalonate (Fig. 7 B). Of note, neither increased nor decreased levels of cellular cholesterol were found to affect protein transport along the classical, ER/Golgi-dependent secretory pathway (Fig. 8). Quantifying trafficking of CD4, an integral membrane protein that is inserted into the ER and transported through the Golgi toward the plasma membrane, no differences in the transport efficiencies could be observed between mock conditions and either increased cholesterol levels (Fig. 8 A) or reduced cholesterol levels (Fig. 8 B). These findings demonstrate that the experimental conditions used in this study to manipulate cellular cholesterol levels do not cause pleiotropic effects affecting a broad range of cellular processes.

Cholesterol depletion from cells reduces FGF2 recruitment at the inner plasma membrane leaflet in living cells, as well as PI(4,5)P2-dependent FGF2 translocation to cell surfaces. (A) FGF2-GFP recruitment efficiency under conditions of decreased cellular cholesterol levels. CHO K1 and U2OS cell lines stably expressing FGF2-GFP in a doxycycline-dependent manner were imaged by single molecule TIRF microscopy as described previously (Dimou et al., 2019; Legrand et al., 2020). Cells were treated for 24 h with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum (Cheng et al., 2006). Single FGF2-GFP particles were identified at the inner plasma membrane leaflet (A; labeled by pink circles in subpanels a and c). For each condition, a widefield image and the first frame of the corresponding TIRF video are shown (subpanels a and c; scale bar = 6 μm). Quantification of FGF2-GFP membrane recruitment at the inner leaflet of CHO K1 and U2OS cells is shown in subpanels b and d, respectively. Time-lapse TIRF movies with a total of 80 frames (100 ms/frame) were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of GFP particles was normalized for both surface area and the relative expression levels of FGF2-GFP for each experimental condition. Mean values are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (*P ≤ 0.05). (B) FGF2-GFP translocation efficiency under conditions of decreased cellular cholesterol levels. CHO K1 and U2OS cells were induced with doxycycline for 24 h to express FGF2-GFP. Following treatment for 24 h with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum, cells were incubated on ice for 30 min with Alexa-Fluor-647-labeled anti-GFP nanobodies. After labeling of FGF2-GFP on cell surfaces, cells were fixed with 4% PFA at room temperature for 20 min and imaged by single molecule TIRF microscopy as established previously (Dimou et al., 2019). For each condition, a widefield image and the corresponding TIRF image is shown (subpanels a and c; scale bar = 6 μm). Quantification of FGF2-GFP membrane translocation in CHO K1 and U2OS for all conditions indicated is shown in subpanel b and d, respectively. Single nanobody particles were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of nanobody particles detected per cell was normalized for the surface area of the corresponding cell. The mean values for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a two-tailed unpaired t test (****P ≤ 0.0001). Data distribution was assumed to be normal, but this was not formally tested.
Cholesterol depletion from cells reduces FGF2 recruitment at the inner plasma membrane leaflet in living cells, as well as PI(4,5)P2-dependent FGF2 translocation to cell surfaces. (A) FGF2-GFP recruitment efficiency under conditions of decreased cellular cholesterol levels. CHO K1 and U2OS cell lines stably expressing FGF2-GFP in a doxycycline-dependent manner were imaged by single molecule TIRF microscopy as described previously (Dimou et al., 2019; Legrand et al., 2020). Cells were treated for 24 h with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum (Cheng et al., 2006). Single FGF2-GFP particles were identified at the inner plasma membrane leaflet (A; labeled by pink circles in subpanels a and c). For each condition, a widefield image and the first frame of the corresponding TIRF video are shown (subpanels a and c; scale bar = 6 μm). Quantification of FGF2-GFP membrane recruitment at the inner leaflet of CHO K1 and U2OS cells is shown in subpanels b and d, respectively. Time-lapse TIRF movies with a total of 80 frames (100 ms/frame) were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of GFP particles was normalized for both surface area and the relative expression levels of FGF2-GFP for each experimental condition. Mean values are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (*P ≤ 0.05). (B) FGF2-GFP translocation efficiency under conditions of decreased cellular cholesterol levels. CHO K1 and U2OS cells were induced with doxycycline for 24 h to express FGF2-GFP. Following treatment for 24 h with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum, cells were incubated on ice for 30 min with Alexa-Fluor-647-labeled anti-GFP nanobodies. After labeling of FGF2-GFP on cell surfaces, cells were fixed with 4% PFA at room temperature for 20 min and imaged by single molecule TIRF microscopy as established previously (Dimou et al., 2019). For each condition, a widefield image and the corresponding TIRF image is shown (subpanels a and c; scale bar = 6 μm). Quantification of FGF2-GFP membrane translocation in CHO K1 and U2OS for all conditions indicated is shown in subpanel b and d, respectively. Single nanobody particles were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of nanobody particles detected per cell was normalized for the surface area of the corresponding cell. The mean values for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a two-tailed unpaired t test (****P ≤ 0.0001). Data distribution was assumed to be normal, but this was not formally tested.
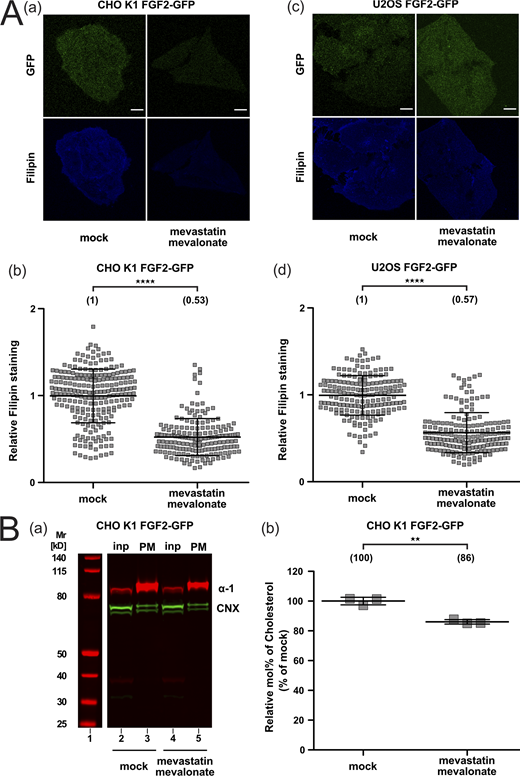
Quantification of cellular cholesterol levels using filipin imaging and mass spectrometry after cholesterol depletion. (A) Cholesterol depletion quantification by means of Filipin staining. Stable CHO K1 (subpanels a and b) and U2OS (subpanels c and d) cell lines were treated for 24 h with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum. Cells were stained with filipin to visualize cholesterol using confocal microscopy. For each condition, both GFP and filipin fluorescence are shown (subpanels a and c; scale bar = 6 μm). Confocal images were analyzed using ImageJ with the quantification of cholesterol levels for CHO K1 and U2OS for all conditions shown in subpanel b and d, respectively. The mean intensity values of the filipin signal detected per cell for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (****P ≤ 0.0001). (B) Cholesterol depletion in CHO K1 plasma membrane fractions quantified via mass spectrometry. Membrane fractions were validated for membrane enrichment and ER contamination via Western blot (subpanel a). Input and membrane fractions were blotted against a plasma membrane marker (the α-1 subunit of the Na,K-ATPase) and an ER marker (calnexin). For details, see Materials and methods. For lipidic mass spectrometry, ratios of cholesterol to PC were determined for the different conditions indicated with the mock condition set to 1. Data are shown as mean ± SD (n = 3). The statistical analysis was based on a two-tailed unpaired t test (**P ≤ 0.01). Data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData FS5.

Quantification of cellular cholesterol levels using filipin imaging and mass spectrometry after cholesterol depletion. (A) Cholesterol depletion quantification by means of Filipin staining. Stable CHO K1 (subpanels a and b) and U2OS (subpanels c and d) cell lines were treated for 24 h with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum. Cells were stained with filipin to visualize cholesterol using confocal microscopy. For each condition, both GFP and filipin fluorescence are shown (subpanels a and c; scale bar = 6 μm). Confocal images were analyzed using ImageJ with the quantification of cholesterol levels for CHO K1 and U2OS for all conditions shown in subpanel b and d, respectively. The mean intensity values of the filipin signal detected per cell for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a t test (****P ≤ 0.0001). (B) Cholesterol depletion in CHO K1 plasma membrane fractions quantified via mass spectrometry. Membrane fractions were validated for membrane enrichment and ER contamination via Western blot (subpanel a). Input and membrane fractions were blotted against a plasma membrane marker (the α-1 subunit of the Na,K-ATPase) and an ER marker (calnexin). For details, see Materials and methods. For lipidic mass spectrometry, ratios of cholesterol to PC were determined for the different conditions indicated with the mock condition set to 1. Data are shown as mean ± SD (n = 3). The statistical analysis was based on a two-tailed unpaired t test (**P ≤ 0.01). Data distribution was assumed to be normal, but this was not formally tested. Source data are available for this figure: SourceData FS5.

Cellular cholesterol manipulation does not interfere with CD4 conventional secretion from cells. (A and B) CD4-GFP translocation efficiency under conditions of either increased (A) or decreased (B) cellular cholesterol levels. CHO cells stably expressing CD4-GFP in a doxycycline-dependent manner were imaged by single molecule TIRF microscopy as described previously (Dimou et al., 2019). Following treatment with either Cholesterol:Methyl-β-Cyclodextrin to increase cellular cholesterol levels (A), or 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum to decrease cholesterol levels (Cheng et al., 2006; B), cells were incubated on ice for 30 min with Alexa-Fluor-647-labeled anti-GFP nanobodies (for details, see Materials and methods). For each condition, a widefield image and the corresponding TIRF image is shown (A and B, subpanel a; Scale bar = 6 μm). Quantification of CD4-GFP transport to the plasma membrane in CHO cells for all conditions indicated is shown in subpanel b of both A and B. Single nanobody particles were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of nanobody particles detected per cell was normalized for the surface area of the corresponding cell. The mean values for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a two-tailed unpaired t test. Data distribution was assumed to be normal, but this was not formally tested.
Cellular cholesterol manipulation does not interfere with CD4 conventional secretion from cells. (A and B) CD4-GFP translocation efficiency under conditions of either increased (A) or decreased (B) cellular cholesterol levels. CHO cells stably expressing CD4-GFP in a doxycycline-dependent manner were imaged by single molecule TIRF microscopy as described previously (Dimou et al., 2019). Following treatment with either Cholesterol:Methyl-β-Cyclodextrin to increase cellular cholesterol levels (A), or 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum to decrease cholesterol levels (Cheng et al., 2006; B), cells were incubated on ice for 30 min with Alexa-Fluor-647-labeled anti-GFP nanobodies (for details, see Materials and methods). For each condition, a widefield image and the corresponding TIRF image is shown (A and B, subpanel a; Scale bar = 6 μm). Quantification of CD4-GFP transport to the plasma membrane in CHO cells for all conditions indicated is shown in subpanel b of both A and B. Single nanobody particles were analyzed using the Fiji plugin TrackMate (Tinevez et al., 2017). The number of nanobody particles detected per cell was normalized for the surface area of the corresponding cell. The mean values for each condition are shown in brackets, with the mock condition set to 1. Data are shown as mean ± SD (n = 4). The statistical analysis was based on a two-tailed unpaired t test. Data distribution was assumed to be normal, but this was not formally tested.
Discussion
The current study originated from earlier observations suggesting the lipid environment to be an important modulator of PI(4,5)P2-dependent FGF2 recruitment to membrane surfaces (Temmerman et al., 2008; Temmerman and Nickel, 2009). Based on (i) biochemical in vitro experiments, (ii) membrane tension analyses, (iii) extensive atomistic MD simulations, and (iv) cell-based FGF2 recruitment and secretion experiments using single-molecule TIRF microscopy, we propose these effects to be primarily linked to cholesterol, an abundant membrane lipid of plasma membranes.
We found PI(4,5)P2-dependent membrane recruitment of FGF2 to be affected by cholesterol in at least two ways. First, the presence of cholesterol has an impact on how negative charges of PI(4,5)P2 headgroups are presented on the membrane surface, promoting increased head group visibility for the PI(4,5)P2 binding site in FGF2. This effect is due to (i) an enrichment of cholesterol at the expense of PC molecules carrying a positively charged choline headgroup, mimicking the cholesterol gradient along the secretory pathway toward the plasma membrane (van Meer et al., 2008; Brugger, 2014; Harayama and Riezman, 2018) and (ii) the condensing effect of cholesterol on membrane surfaces that leads to an increased packing of lipids and a more dense arrangement of negative charges derived from PI(4,5)P2 (Leonard and Dufourc, 1991; Ikonen, 2008; Rog et al., 2009; Magarkar et al., 2014; Doktorova et al., 2017; Harayama and Riezman, 2018). In both MD simulations and in vitro experiments presented in this study, cholesterol was added to membrane systems at the expense of POPC, a zwitterionic molecule which carries a positive charge in the headgroup surface region. Under these conditions, cholesterol affects membrane packing and tension, resulting in a phase transition from liquid-disordered to liquid-ordered arrangements as observed in MD simulations contained in this study. The lateral segregation of membrane lipids with saturated and unsaturated fatty acids, respectively, and cholesterol into regions with high lipid packing (liquid-ordered domains) and less lipid packing (liquid-disordered domains) could be one principle that leads to a local enrichment of PI(4,5)P2 molecules (van Meer et al., 2008; Harayama and Riezman, 2018). Therefore, we propose that, in the presence of cholesterol, the negative charges of the headgroups of PI(4,5)P2 molecules might be packed more densely, facilitating electrostatic interactions with the PI(4,5)P2 binding pocket of FGF2. As demonstrated by free energy calculations, the described effects are linked to faster binding kinetics and a substantial stabilization of the interaction of FGF2 with PI(4,5)P2 with the free energy of binding increasing from −20 to −35 kBT in the presence of cholesterol. Interestingly, we made similar observations for another PI(4,5)P2 binding protein, PH-PLC-δ1. These observations support the above-described hypothesis and suggest cholesterol to be of general importance in tuning the interaction of proteins with phosphoinositides. Thus, we propose cholesterol to have a general impact on the physico-chemical properties of the lipid bilayer resulting in profound changes on how PI(4,5)P2 is presented to phosphoinositide-binding proteins.
Second, we found cholesterol to induce clustering of PI(4,5)P2 molecules, a process that is likely to strengthen interactions between FGF2 and PI(4,5)P2 through increased avidity. In particular, in the presence of cholesterol, PI(4,5)P2 tends to form trimers and tetramers that we propose to enhance PI(4,5)P2-dependent FGF2 membrane recruitment. This process appears to be fostered by the ability of a single FGF2 molecule to bind five to six PI(4,5)P2 molecules through both the main high affinity binding site (defined by K127, R128 and K133; Temmerman et al., 2008; Müller et al., 2015; Steringer et al., 2017) and additional low affinity PI(4,5)P2 binding sites on the positively charged surface of FGF2. Under these conditions, PI(4,5)P2-dependent oligomerization of FGF2 will result in a massive local accumulation of PI(4,5)P2 molecules at sites of FGF2 recruitment, a process that, due to the nature of PI(4,5)P2 as a cone-shaped membrane lipid, is likely to exert membrane stress (van den Brink-van der Laan et al., 2004). This condition may facilitate FGF2 membrane translocation since, through the formation of a lipidic membrane pore with a toroidal architecture that allows for the accommodation of cone-shaped PI(4,5)P2 molecules (Steringer and Nickel, 2018; Pallotta and Nickel, 2020), this process is likely to relax bilayer stress at sites of PI(4,5)P2-dependent FGF2 recruitment and membrane translocation. This phenomenon might further be linked to variations of bilayer tension induced by cholesterol, a stress condition that is known to be relieved by the formation of a lipidic membrane pore (Tieleman et al., 2003; den Otter, 2009; Karal and Yamazaki, 2015; Cunill-Semanat and Salgado, 2019). This is because the activation energy of tension-induced pore formation decreases with an increase in bilayer tension (Karal et al., 2016). Furthermore, the localized accumulation of negative charges derived from PI(4,5)P2 will increase the transmembrane electric field and, therefore, facilitate the emergence of lipidic membrane pores triggered by PI(4,5)P2-dependent FGF2 oligomerization (Gurtovenko and Vattulainen, 2005; Kotnik et al., 2019). In a cellular context, once membrane-spanning FGF2 oligomers have been inserted into the plasma membrane in a PI(4,5)P2-dependent manner, they will be captured and disassembled by cell surface heparan sulfates at the outer plasma membrane leaflet, thereby completing this unconventional secretory pathway with FGF2 being exposed on cell surfaces (Zehe et al., 2006; Steringer et al., 2017; Dimou and Nickel, 2018; Pallotta and Nickel, 2020; Sparn et al., 2022a; Sparn et al., 2022b).
In the specific context of FGF2 membrane recruitment and translocation, the results presented in this study offer a sound explanation for the high specificity by which the process of FGF2 membrane translocation into the extracellular space is physically linked to the plasma membrane, a subcellular structure that is enriched in both PI(4,5)P2 and cholesterol. Based on similar observations with PH-PLC-δ1 reported in this study, our findings are likely to be relevant in general for protein-phosphoinositide interactions. Therefore, cholesterol is likely to also tune other types of UPS type I pathways of UPS regarding cargo proteins that either directly or indirectly depend on PI(4,5)P2 (Rabouille, 2017; Dimou and Nickel, 2018; Merezhko et al., 2020; Pallotta and Nickel, 2020). This includes HIV-Tat and Tau proteins that make direct physical contacts to PI(4,5)P2 as a prerequisite to enter their pathways of unconventional secretion (Rayne et al., 2010; Katsinelos et al., 2018). In addition, under certain physiological conditions, unconventional secretion of IL-1β has been reported to follow a type I mechanism of UPS (Evavold et al., 2017). Here, although IL-1β does not bind to PI(4,5)P2 directly (Martin-Sanchez et al., 2016), membrane ruffles enriched in PI(4,5)P2 appear to play an important role in Gasdermin-dependent secretion of IL-1β (Monteleone et al., 2018), suggesting that so far unidentified interaction partners of PI(4,5)P2 play a role in unconventional secretion of IL-1β. Since the secretory mechanisms of HIV-Tat and Tau as well as Gasdermin-dependent secretion of IL-1β are all linked to the plasma membrane as the subcellular site of membrane translocation, the described role of cholesterol in positively modulating PI(4,5)P2-dependent FGF2 membrane recruitment is likely to extend to a larger group of UPS type I cargoes secreted by unconventional means. Furthermore, our findings are likely to have broad implications for the highly dynamic processes of phosphoinositide-dependent protein recruitment to membranes in general. This family of membrane lipids with differentially phosphorylated inositol head groups has been demonstrated to be distributed in a highly specific manner across different subcellular organelles (Di Paolo and De Camilli, 2006). In this way, they have been implicated to exert functions in many cellular processes as diverse as endo- and exocytosis (Martin, 2015; Posor et al., 2015), cell migration (Tsujita and Itoh, 2015), and organellar contact sites (Raiborg et al., 2016), among others. Therefore, the highly dynamic interconversion of different types of phosphoinositides combined with different levels of cholesterol in various subcellular organelles might allow cells to fine-tune protein recruitment to membranes in a highly sophisticated manner to enable phosphoinositides to serve a broad variety of biological processes.
Materials and methods
Recombinant proteins
His-tagged variants of FGF2-Halo, PH-PLC-δ1-Halo, and FGF2-Y81pCMF-GFP fusion proteins were expressed in Escherichia coli BL21 Star cells, and they were purified to homogeneity in three steps using Ni-NTA affinity chromatography, heparin chromatography (except K127Q/R128Q), and size exclusion chromatography using a Superdex 75 column (Steringer et al., 2017). Desalting, buffer exchange, and protein concentration were achieved by 30 K ultrafiltration (Amicon). In the case of FGF2-Halo and PH-PLC-δ1-Halo, desalting was performed using Nap-5 columns (GE Healthcare). Protein purity was determined by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). For each protein, 3 µg were loaded using a reducing sample buffer. Protein patterns were analyzed by Coomassie staining (Fig. S1 C).
Preparation of large unilamellar vesicles (LUVs)
All membrane lipids were purchased from Avanti Polar Lipids that were either purified from natural extracts (bovine liver phosphatidylcholine [PC], bovine liver phosphatidylethanolamine [PE], porcine brain phosphatidylserine [PS], bovine liver phosphatidylinositol [PI], porcine brain PI(4,5)P2, ovine wool cholesterol, and chicken egg sphingomyelin [SM]) or were made as a synthetic product (Rhodamine-labeled PE). Liposomes were prepared as described previously (Temmerman et al., 2008; Temmerman and Nickel, 2009; Steringer et al., 2012; Müller et al., 2015). In brief, chloroform-dissolved lipid mixtures containing (750 µl containing 3 µmol total lipids) were dried in round-bottomed flasks at 25°C under vacuum to yield a homogeneous lipid film. To obtain a final lipid concentration of 4 mM, dried lipid films were slowly resuspended in 750 µl HK buffer (150 mM KCl and 25 mM HEPES, pH = 7.4) containing 10% (w/v) sucrose. The obtained lipid mixtures were subjected to 10 freeze/thaw cycles (50°C/liquid nitrogen) to produce unilamellar liposomes. A size-distribution of 250–300 nm in diameter was obtained by 27 size-extrusion steps using a mini-extruder (Avanti Polar Lipids) equipped with a 400-nm filter. The final size distribution of liposomes was found to have an average diameter of 259 ± 32 nm as analyzed by dynamic light scattering (Wyatt). Different lipid compositions were used to prepare liposomes, all of which contained 1 mol% Rhodamine-PE (Table S2).
Quantification of protein–lipid interactions using analytical flow cytometry
Using a FGF2-Halo fusion protein fluorescently labeled with AF488, protein binding to PI(4,5)P2-containing liposomes was analyzed by analytical flow cytometry (Becton-Dickinson FACS Calibur along with data analysis using CellQuest Pro software) as described previously (Temmerman et al., 2008; Temmerman and Nickel, 2009). Briefly, liposomes (final lipid concentration = 1 mM) were blocked with 4% (w/v) fatty-acid-free BSA (Roche) in HK buffer for 1 h at 25°C. FGF2-Halo-AF488 (5 µM) was incubated at 25°C for the time indicated. Following extensive washing with HK buffer (150 mM KCl and 25 mM HEPES, pH = 7.4), FGF2-Halo-AF488 binding to PI(4,5)P2-containing lipid bilayers was recorded for 30,000 individual liposomes detected by rhodamine-PE fluorescence and light scattering. Primary data were corrected for background fluorescence defined by the unspecific binding of AF488-labeled Halo (lacking FGF2) to liposomes. FGF2-Halo-AF488 binding signals derived from liposomes containing 10 mol% of PI(4,5)P2 and 90 mol% PC (PC10) were set to 100% and used as a reference value, when normalization was required (Fig. 1, B, E, and F). Signals derived from PC-only containing liposomes (PC0) were used as the negative control.
Cell culture
CHO and CHO K1 cells were cultured in α-MEM medium and U2OS cells in DMEM, both supplemented with 10% FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 95% humidity in the presence of 5% CO2. To increase cellular cholesterol levels, cells were treated with Cholesterol:Methyl-β-Cyclodextrin complexes (final cholesterol concentration = 0.2 mM) for 1 h at 37°C (Merezhko et al., 2018). To decrease cholesterol levels, cells were incubated for 24 h in the presence of de-lipidized serum, 5 μM mevastatin, and 50 μM mevalonate (Cheng et al., 2006).
Imaging of cholesterol, PI(4,5)P2 and FGF2-Halo by three-color STED microscopy
U2OS MT cells overexpressing FGF2-Halo in a doxycycline-dependent manner were cultivated in µ-Slide 8-well glass bottom dishes for co-localization experiments. 24 h before fixation cells were transfected with EGFP-GRAM1b-G187L (Ercan et al., 2021) using FuGENE HD (Promega) according to manufacturer’s recommendation and FGF2-Halo expression was induced through addition of 1 µg/ml doxycycline. Cells were washed for 10 min with 0.5 mg/ml heparin (H3149; Sigma-Aldrich) in Live-Cell Imaging Solution (Thermo Fisher Scientific) prior to 20 min fixation with 4% PFA and 10 min permeabilization with 0.05% saponin. Following 30 min blocking in 1% BSA-PBS, cells were incubated for 1 h at RT with primary antibodies directed against FGF2 (Engling et al., 2002; Zehe et al., 2006) and PI(4,5)P2 (2C11, ab11039; Abcam). After washing, cells were incubated for 1 h with secondary antibodies (goat anti-rabbit IgG coupled to abberior STAR 635P and goat anti-mouse IgG coupled to abberior STAR 580, both from Abberior) and GFP-Booster Alexa Fluor 488 (Chromotek). Cells were imaged on a Leica TCS SP8 STED 3× microscope using the HC PL APO 100×/1.40 NA STED White Oil objective at room temperature in PBS. Images were acquired using Leica Application Suite X (LASX) and −2 Hybrid GaAsp detectors (HyD) from Leica. Images were processed using Huygens Professional Deconvolution Software using standard deconvolution in express deconvolution option (Scientific Volume Imaging, http://svi.nl) and Fiji (Schindelin et al., 2012).
Preparation of Cholesterol:Methyl-β-Cyclodextrin complexes
Chloroform-dissolved cholesterol was dried under a stream of nitrogen and, subsequentially, under vacuum at room temperature in glass tubes. A Methyl-β-Cyclodextrin solution (200 mM) in Live-Cell Imaging Solution (Thermo Fisher Scientific) was added to the cholesterol lipid film at a molar ratio of 10 to 1. The mixture was sonicated and vortexed for 1 h at room temperature, resulting in a complete dissolution of the cholesterol lipid film. Aliquots were stored at −20°C. Cells were treated with Cholesterol:Methyl-β-Cyclodextrin complexes (final cholesterol concentration = 0.2 mM) for 1 h at 37°C.
Single-molecule TIRF microscopy
To quantify both FGF2-GFP recruitment at the inner plasma membrane leaflet and translocation to cell surfaces, a previously established single particle TIRF assay was employed (Dimou et al., 2019; Legrand et al., 2020). Widefield fluorescence and TIRF images were acquired using an Olympus IX81 xCellence TIRF microscope equipped with an Olympus PLAPO 100×1.45 NA Oil DIC objective lens and a Hamamatsu ImagEM Enhanced (C9100-13) camera. Data were recorded with the Olympus xCellence software and exported in Tagged Image File Format (TIFF) and analyzed through Fiji (Schindelin et al., 2012). GFP fluorescence was excited with an Olympus 488 nm and 100 mW diode laser. For the quantification of FGF2-GFP recruitment at the inner leaflet of the plasma membrane, CHO K1 and U2OS cells were seeded in μ-Slide 8-well glass bottom plates (ibidi). Before imaging was started, cells were incubated with either Cholesterol:Methyl-β-Cyclodextrin complexes for 1 h, or in the presence of de-lipidized serum, 5 μM mevastatin, and 50 μM mevalonate for 24 h in culture conditions. Imaging was performed in Live-Cell Imaging Solution (Thermo Fisher Scientific) at room temperature. The quantification of FGF2-GFP particles recruitment to the plasma membrane was achieved through the analysis of time-lapse TIRF movies. The frame of each cell was selected by widefield imaging. The number of FGF2-GFP particles was normalized to the cell surface area (μm2) and to the expression level of FGF2-GFP. The latter was quantified for each individual cell at the first frame of each time-lapse TIRF movie using ImageJ. The total number of FGF2-GFP particles per cell was quantified employing the Fiji plugin TrackMate (Tinevez et al., 2017).
For the quantification of FGF2-GFP translocation to cell surfaces, CHO K1 and U2OS cells were seeded in μ-Slide 8-well glass bottom plates (ibidi) followed by incubation for 24 h in the presence of 1 μg/ml doxycycline to induce FGF2-GFP expression. To study the effect of increased cholesterol levels, prior the addition of Cholesterol:Methyl-β-Cyclodextrin complexes, cells were washed with heparin (500 μg/ml; Sigma-Aldrich) to remove FGF2-GFP particles from cell surfaces. Cells were then treated with Cholesterol:Methyl-β-Cyclodextrin complexes (final cholesterol concentration = 0.2 mM) for 1 h at 37°C. To study the effect of decreased cholesterol levels, cells were incubated with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum for 24 h in culture conditions. Afterwards, the medium was removed, and cells were rinsed three times with Live-Cell Imaging Solution (Thermo Fisher Scientific). Cells were further incubated on ice with membrane impermeable Alexa Fluor 647–labeled anti-GFP nanobodies (Chromotek) for 30 min. Afterwards, they were rinsed three times with Live-Cell Imaging Solution and fixed with 4% PFA (Electron Microscopy Sciences) for 20 min at room temperature. Imaging was performed in PBS at room temperature. Nanobody fluorescence was excited with an Olympus 640 nm, 140 mW diode laser. The quantification of FGF2-GFP on cell surfaces was achieved through a quantitative analysis of TIRF images. The frame of each cell was selected by widefield imaging. The number of nanobody particles was normalized to the cell surface area (μm2). The total number of nanobody particles per cell was quantified employing the Fiji plugin TrackMate (Tinevez et al., 2017). Background fluorescence was subtracted for all representative images shown.
Quantification of cellular cholesterol levels based on filipin imaging
Cellular cholesterol levels were quantified by filipin (Sigma-Aldrich) staining and confocal imaging. CHO K1 and U2OS cells were seeded in μ-Slide 8-well glass bottom plates (ibidi) 1 d before imaging. Cells were incubated for either 1 h with Cholesterol:Methyl-β-Cyclodextrin complexes or for 24 h with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum, in culture conditions. Cells were then rinsed three times with PBS and fixed with 3% PFA for 1 h at room temperature. Afterwards, to quench PFA, cells were rinsed three times with PBS and incubated with 1.5 mg/ml glycine in PBS for 10 min at room temperature. A stock solution of filipin (25 mg/ml in DMSO) was prepared and diluted to prepare a working solution of 50 μg/ml in PBS, supplemented with 10% FCS. Cells were stained with filipin for 2 h at room temperature and rinsed three times with PBS. Imaging was performed in PBS at room temperature using a confocal laser scanning microscope (LSM800; Zeiss) with a 63× 1.4 NA Oil DIC objective lens (Zeiss). Images were acquired using the software ZEN 2.6 (blue edition). In addition to Filipin fluorescence, GFP fluorescence (coming from FGF2-GFP) was acquired as a reference of the cells. Confocal images were then analyzed through Fiji (Schindelin et al., 2012).
Quantification of cellular cholesterol levels using mass spectrometry
Plasma membrane fractions of CHO K1 cells were collected using the Minute Plasma Membrane Protein Isolation and Cell Fractionation Kit (Invent Biotechnologies). Cells were seeded in 15-cm dishes (Thermo Fisher Scientific), 24 h before treating them with either Cholesterol:Methyl-β-Cyclodextrin complexes for 1 h, or with 5 μM mevastatin and 50 μM mevalonate in the presence of de-lipidized serum for 24 h. Cells were then detached with 1 mM EDTA and pelleted at 500 g for 5 min. Pellets were rinsed with 1 ml cold PBS, and supernatant was removed completely. Each pellet was then resuspended in 500 µl buffer A and incubated for 5–10 min on ice and vortexed for 10–30 s. After that, cell suspensions were transferred to the filter cartridge and centrifuged at 16,000 g for 30 s. Pellets were resuspended and passed through the same filter at 16,000 g for 30 s. Pellets were resuspended by vortexing for 10 s and suspensions were centrifuged at 700 g for 1 min and supernatants were transferred to fresh 1.5 ml tubes and centrifuged at 16,000 g for 15 min at 4°C. Pellets were resuspended in 200 µl buffer B by vortexing and centrifuged at 7,800 g for 20 min. Supernatants were transferred into fresh 2 ml tubes and 1.6 ml cold PBS was added. Suspensions were centrifuged at 16,000 g for 20 min and supernatant was removed. Pellets contain plasma membrane fractions. As a supplementary validation for successful plasma membrane enrichment, 10% of the input and 50% of plasma membrane fractions were subjected to Western blot analysis, after resuspension in SDS sample buffer and warmed up at 60°C for 7 min. They were stained for the α-1 subunit of the Na,K-ATPase, as plasma membrane marker, and calnexin, as endoplasmic reticulum marker.
Cells were subjected to lipid extractions using an acidic (Bligh and Dyer, 1959). Lipid standards were added prior to extractions, using a master mix containing 50 pmol phosphatidylcholine (PC)-standard mix (PC 13:0/13:0, 14:0/14:0, 2.0:0/20:0; 21:0/21:0; Avanti Polar Lipids) and 150 pmol D7-cholesterol (Avanti Polar Lipids). The final CHCl3 phases were evaporated under a gentle stream of nitrogen. Lipid extracts were resuspended in 60 µl 10 mM ammonium acetate in methanol and diluted 1:4 in 96-well plates (Eppendorf twin.tec 96; Sigma-Aldrich) prior to measurement of PC species, applying precursor ion scanning (prec) in positive ion mode (+prec184, mass range: m/z 644-880). For cholesterol determination, the remaining lipid extract was evaporated and subjected to acetylation as described (Liebisch et al., 2006). Following acetylation, samples were evaporated and resuspended in 60 µl 10 mM ammonium acetate in methanol, diluted 1:10 in 96-well plates (Eppendorf twin.tec 96; Sigma-Aldrich). Cholesterol measurements were performed in positive ion mode, applying neutral loss (NL) scanning (+NL 77, mass range: m/z 442-480, for MS parameters see Table 1) on a QTRAP 5500 (Sciex; Canada) mass spectrometer using chip-based (HD-D ESI Chip; Advion Biosciences; USA) electrospray infusion and ionization via a Triversa Nanomate (Advion Biosciences; Ozbalci et al., 2013).
MS parameters.
| Lipid class | DP (eV) | EP (eV) | CE (eV) | CXP (eV) | CUR (psi) | IHT (°C) | CAD (psi) | MCA | Q1 | Q3 |
|---|---|---|---|---|---|---|---|---|---|---|
| PC | 100 | 12 | 32 | 13 | 20 | 60 | 5 | 300 | High | unit |
| Chol | 100 | 10 | 12 | 14 | 20 | 60 | 5 | 600 | unit | unit |
CAD, collisionally-activated dissociation (N2); CE, collision energy; Chol, cholesterol; CUR, curtain gas (N2); CXP, collision cell exit potential; DP, declustering potential; EP, entrance potential; IHT, interface heater temperature; MCA, multi-channel acquisition; PC, phosphatidylcholine; Q, quadrupole.
The scan rate was set to 200 Da/s with a step size of 0.1 Da. Data evaluation was done using LipidView 1.3 (Sciex) and a software developed in-house (ShinyLipids). One sample (Kon A) contained only the standards and was used for background subtraction. Amounts of endogenous lipid species were calculated on the basis of the intensities of internal standards. PC is a bulk lipid of cellular membranes and was used as reference for cholesterol determinations.
Molecular dynamics simulations
Atomistic MD simulations were performed using the CHARMM36m force field for lipids and proteins, the CHARMM TIP3P force field for water, and the standard CHARMM36 force field for ions (Huang et al., 2017). The GROMACS 2020 simulation package (Abraham et al., 2015) was used in all simulations. For FGF2, we used structural information based on PDB accession no. 1BFF that covers the crystal structure of residues 26–154 of the monomeric form of FGF2 (Kastrup et al.,1997). The N- and C-termini were modeled as charged residues. To match the experimental membrane systems used in Fig. 1, the CHARMM-GUI webserver (Lee et al., 2016) was used to model seven different lipid compositions (Table S2) with 300 lipids each. All membrane systems were first simulated for 1.2 μs. The final snapshot for each membrane lipid composition was used to build the protein-membrane system. FGF2 was placed 2 nm far from the membrane surface at 10 different random orientations (see Table S3 for simulations details). The protein-membrane systems were first energy-minimized in vacuum and later hydrated and neutralized by an appropriate number of counter-ions, followed by the addition of 150 mM potassium chloride to mimic the experimental conditions. All systems were first energy-minimized and an equilibration step was used to keep the temperature, pressure, and number of particles constant (NpT ensemble). During this step, proteins were restrained in all dimensions, whereas the first heavy-atom of each lipid was restrained in the xy-plane of the membrane with a force constant of 1,000 kJ/mol.
The Nose-Hoover thermostat (Evans and Holian, 1985) was used to maintain the temperature at 298 K with a time constant of 1.0 ps. The pressure of 1 atm was kept constant using the Parrinello–Rahman barostat (Parrinello and Rahman, 1981) with a time constant set to 5.0 ps and isothermal compressibility to a value of 4.5 × 10−5 bar−1. The semi-isotropic pressure-coupling scheme was used. For neighbor searching, we used the Verlet scheme (Verlet, 1967) with an update frequency of once every 20 steps. Electrostatic interactions were calculated using the Particle Mesh Ewald method (Darden et al., 1993). Periodic boundary conditions were applied in all directions. The simulations were carried out using an integration time step of 2 fs until 1,200 ns were reached. The first 500 ns of all analyses were considered as equilibration time and excluded from the analysis. The total time scale of the simulations (including free energy calculations) was about 115 μs.
Free energy calculations
The calculation of the potential of mean force (PMF) for the interaction between FGF2 and PI(4,5)P2 in POPC-based membranes was calculated using the umbrella sampling technique (Torrie and Valleau, 1974; Torrie and Valleau, 1977). The systems were built by inserting, using Javanainen’s method (Javanainen, 2014), a pre-formed FGF2-PI(4,5)P2 (1:4) complex in two membranes with 0 and 30 mol% of cholesterol, respectively. FGF2-PI(4,5)P2 (1:4) complexes were built from an unbiased MD simulation of FGF2 bound to PI(4,5)P2 in its high-affinity orientation (Steringer et al., 2017), using a POPC-based bilayer containing 5 mol% of PI(4,5)P2. Before starting the pulling simulations, the systems were minimized and simulated for 500 ns to stabilize the FGF2-PI(4,5)P2 (1:4) complex bound to the lipid bilayer. Thirty-nine windows with 0.1 nm spacing were constructed by pulling FGF2 on the z-axis (along the membrane normal direction) with a pulling rate of 0.1 nm/ns and a force constant of 100 kJ mol−1 nm−2. To minimize membrane deformation during the pulling simulations, the head groups and tails of all membrane lipids were restrained on the z-axis using harmonic position restraints with a force constant of 10,000 kJ mol−1 nm−2. However, for the actual umbrella sampling simulations, the restraints on lipids were released. Each window was simulated for 400 ns with the first 200 ns being considered an equilibration phase that was discarded from the free energy calculations. The PMF was reconstructed using the weighted histogram analysis method implemented in GROMACS (Meng and Roux, 2015). The reconstructed free energy profiles’ statistical errors were estimated with the Bayesian bootstrap method (Rubin, 1981) using 200 bootstrap iterations.
Charge density and electric field calculations
Since the position of the center of mass of the membrane, which is the origin of the charge density distribution, fluctuates in time, the center of mass of the membrane was first centered in all frames of the trajectory before performing the charge density calculation (Gurtovenko and Vattulainen, 2009). Furthermore, the new versions of GROMACS (>5.0) perform dynamic histogram binning, calculating the center of the membrane of each frame and bin in (−bZ/2,bZ/2) before the charge density calculation. The obtained charge density profiles were integrated to calculate the electric field profiles.
Microfluidics
To form a freestanding lipid membrane, two microfluidic cross-channel geometries previously tested as a platform to produce stable bilayers were employed (Thutupalli et al., 2011; Vargas et al., 2014; Hahl et al., 2017; Hahl et al., 2018; Khangholi et al., 2020). In both geometries, a continuous phase consisting of lipids dissolved in squalene solution (5 mg/ml) separates two fingers of HEPES buffer (25 mM, KCl 150 mM). Once buffer fingers are injected in the device with the lipid solution previously injected, the lipids will decorate the interface between the two phases forming two monolayers that are not in contact (Fig. S3 A). A slow flow is created by hydrostatic pressure such that the lipid decorated buffer fingers are brought in close vicinity until they make a direct contact. When the two monolayers face each other, a zipping phenomenon driven by attractive intermolecular forces between the hydrophobic chains will occur and the bilayer will form (Hahl et al., 2018). In this setup, the control of pressure and lipid concentrations is necessary to avoid rupture of the lipid bilayer (Schlicht and Zagnoni, 2015). The lipid bilayer contact angle θ was then obtained by analyzing optical micrographs. To guarantee the stability of the fingers, a good wettability of the phases is required. Therefore, a hydrophobic chip made of PDMS was used and prepared as described previously (Hahl et al., 2017; Guo et al., 2018). Briefly, silicon elastomer base was mixed with curing agent (Sylgard 184; Dow Corning) were mixed in a ratio of 10:1 and were casted on a silicon wafer, reproducing negative structures fabricated by photolithography with a SU-8 photoresist, and cured for 6 h at 65°C. The prepared chip and a PDMS coated glass slide were then exposed to a nitrogen plasma treatment (Diener electronic GmbH), sealed and heated at 125°C for 1–2 h to restore the hydrophobic properties. Optical images of bilayer formation occurring in the chip were acquired with SciDAVis or Zeiss software at 10× magnification (Zeiss observer Z1 and Z7 with an Imager pro X camera from LaVision).
Interfacial tension measurements
Interfacial tension σ of a single lipid monolayer was measured through the standard pendant drop method using a OCA20 goniometer (OCA 20, DataPhysics Instruments GmbH, Filderstadt, Germany). Briefly, a 1 mg/ml solution of lipid in squalene was stirred at 45°C for 3 h and transferred into an optical glass chamber. A drop of HEPES buffer (25 mM, KCl 150 mM) was formed at the end of a hanging needle that was immersed in the lipid solution. The shape change of the pendant drop was recorded as function of time. The interfacial tension σ was obtained from a quantitative analysis of the drop contour. The decrease of σ due to the adsorption of lipids to the newly created oil-water interface of the drop was recorded over 20–30 min until a plateau was reached.
Bilayer tension and adhesion energy
Statistical analyses
The statistical analyses were based on either a one-way ANOVA test combined with Tukey’s post hoc test or on a two-tailed unpaired t test. Data distributions were assumed to be normal, but this was not formally tested. The number of independent data points (n) is given in the figure legends, as well as the P values. The mean ± SD is displayed, unless otherwise stated. For the measurements of FGF2 binding to liposomes as shown in Fig. 1 and in Fig. S1, error bars were based on SEM.
Online supplemental material
Fig. S1 shows of in vitro FGF2 membrane recruitment assays using a FGF2-GFP variant form impaired in binding to PI(4,5)P2. In particular, Fig. S1 A shows the quantification of variant FGF2-GFP forms (wild-type and K127Q/R128Q) binding to liposomes containing 30 mol% cholesterol and 5 mol% PI(4,5)P2. Fig. S1 B shows the binding of the same variant forms of FGF2-GFP on liposomes with a plasma membrane-like lipid composition. Fig. S1 C shows the analysis through SDS-PAGE of the proteins used in this study. Fig. S2 shows the role of electrostatics in PI(4,5)P2-dependent FGF2 membrane recruitment. In particular, Fig. S2 A shows the electric field calculation derived from unbiased MDS. Fig. S2 B shows the PI(4,5)P2 clustering in a complex lipid mixture. Fig. S2 C shows the solvent-accessible surface calculation of FGF2 monomer in water. Fig. S2 D shows the PI(4,5)P2 lateral density profile before and after interacting with FGF2. Fig. S3 shows an analysis of bilayer contact angles using a microfluidics setup. In particular, Fig. S3 A illustrates the geometry of the used microfluidic setups. Fig. S3 B shows optical micrographs of the bilayer contact angles for artificial membranes with different cholesterol contents. Fig. S4 includes validation for the cholesterol enrichment method in both cell lines included in this study (CHO K1 and U2OS). Fig. S4 A shows quantification of cholesterol levels in both cell lines using Filipin staining, whereas Fig. S4 B shows quantification of cholesterol levels in CHO K1 through mass spectrometry analysis. Fig. S5 shows validation for the cholesterol depletion method in both cell lines included in this study (CHO K1 and U2OS). Fig. S5 A shows quantification of cholesterol levels in both cell lines using Filipin staining. Fig. S5 B shows quantification of cholesterol levels in CHO K1 through mass spectrometry analysis. The Video 1 shows the time evolution of the lateral partial mass density analysis of PI(4,5)P2 head groups for membrane systems containing either 0 (left panel) or 30 mol% of cholesterol (right) in the absence of FGF2. The video is based on fully atomistic MD simulations. The Video 2 shows the time evolution of the system with 30 mol% of cholesterol in presence of FGF2 (right panel) and its lateral partial mass density evolution of PI(4,5)P2 head groups. The video is based on fully atomistic MD simulations. Table S1 corresponds to Fig. 4 and provides the values of the bilayer contact angle (θ), interfacial tension (σ), bilayer tension (ϒ), and adhesion energy (E) for 5 lipid mixtures with different concentrations of cholesterol. Table S2 provides the liposome compositions used for preparing unilamellar liposomes. Table S3 provides details on the atomistic simulations contained in this study. Lipid names correspond to the CHARMM-GUI lipid model. PI(4,5)P2 corresponds to SAPI25, Cholesterol to CHL1, and water to TIP3P.
Acknowledgments
We thank André Nadler (MPI-CBG Dresden, Germany) for providing an expression construct encoding a fusion protein of a PH domain and the Halo tag. We thank Jaime Fernandez Sobaberas (Heidelberg University Biochemistry Center, Germany) for providing the U2OS FGF2-Halo cell line. F. Lolicato and I. Vattulainen wish to acknowledge the CSC - IT Center for Science (Espoo, Finland) for computational resources. We would like to further acknowledge help from Holger Lorenz (ZMBH imaging facility) and Monika Langlotz (ZMBH FACS facility).
This work was supported by grants from the Deutsche Forschungsgemeinschaft (W. Nickel; SFB/TRR 83, SFB/TRR 186 and DFG Ni 423/10-1, B. Brügger; SFB/TRR 186 and A. Griffo, J-B. Fleury, H. Hähl, R. Seemann, K. Jacobs; SFB1027, projects B1 and B4). A. Griffo and K. Jacobs thanks the Max Planck School “Matter of Life” supported by by the German Federal Ministry of Education and Research (BMBF) in collaboration with the Max Planck Society. I. Vattulainen thanks the Sigrid Juselius Foundation, the Academy of Finland (project no. 331349, 346135), Human Frontier Science Program (project no. RGP0059/2019), and the Helsinki Institute of Life Science (HiLIFE) Fellow program for financial support. F. Scollo wishes to acknowledge HPC-EUROPA3 (INFRAIA-2016-1-730897), with the support of the EC Research Innovation Action under the H2020 Programme and the computer resources and technical support provided by Universität Stuttgart - Höchstleistungsrechenzentrum Stuttgart (HLRS). F. Scollo. and M. Hof acknowledge the support from the Czech Science Foundation GA CR EXPRO (grant 19-26854X).
The authors declare no competing financial interests.
Author contributions: F. Lolicato: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Writing—original draft, Writing—review, and editing; R. Saleppico: Conceptualization, Methodology, Validation, Investigation, Data curation, Writing—original draft, Writing—review, and editing; A. Griffo: Methodology, Investigation; A. Meyer, Methodology, Investigation; F. Scollo, Investigation; B. Pokrandt, Investigation, Data Curation; H.-M. Müller, Methodology; H. Ewers, Methodology, Data curation; H. Hähl, formal analysis; J.-B. Fleury: methodology; R. Seemann, Methodology, Funding acquisition; M. Hof, Methodology, Data curation; B. Brügger: Methodology and Funding acquisition; K. Jacobs: Funding acquisition; I. Vattulainen: Funding acquisition; W. Nickel: Conceptualization, Methodology, Formal analysis, Supervision, Funding acquisition, Project administration, Writing—original draft, and Writing—review and editing.

