


The ESCRT protein CHMP2B and the RNA-binding protein TDP-43 are both associated with ALS and FTD. The pathogenicity of CHMP2B has mainly been considered a consequence of autophagy–endolysosomal dysfunction, whereas protein inclusions containing phosphorylated TDP-43 are a pathological hallmark of ALS and FTD. Intriguingly, TDP-43 pathology has not been associated with the FTD-causing CHMP2BIntron5 mutation. In this study, we identify CHMP2B as a modifier of TDP-43–mediated neurodegeneration in a Drosophila screen. Down-regulation of CHMP2B reduces TDP-43 phosphorylation and toxicity in flies and mammalian cells. Surprisingly, although CHMP2BIntron5 causes dramatic autophagy dysfunction, disturbance of autophagy does not alter TDP-43 phosphorylation levels. Instead, we find that inhibition of CK1, but not TTBK1/2 (all of which are kinases phosphorylating TDP-43), abolishes the modifying effect of CHMP2B on TDP-43 phosphorylation. Finally, we uncover that CHMP2B modulates CK1 protein levels by negatively regulating ubiquitination and the proteasome-mediated turnover of CK1. Together, our findings propose an autophagy-independent role and mechanism of CHMP2B in regulating CK1 abundance and TDP-43 phosphorylation.
Introduction
TAR DNA-binding protein 43 (TDP-43) is a DNA/RNA-binding protein participating in the assembly of various ribonucleoprotein complexes and plays an important role in regulating RNA processing and functions (Chen-Plotkin et al., 2010; Cohen et al., 2011; Lee et al., 2011). TDP-43 is also a major pathological protein linked to a spectrum of human neurodegenerative diseases, collectively known as TDP-43 proteinopathies (Arai et al., 2009; Chang et al., 2016; Hasegawa et al., 2008; Higashi et al., 2007; Neumann et al., 2009; Neumann et al., 2007; Toyoshima and Takahashi, 2014). In particular, abnormal TDP-43 protein inclusions are found in ∼97% of amyotrophic lateral sclerosis (ALS) and ∼45% frontotemporal dementia (FTD) patients (Ling et al., 2013; Tan et al., 2017). In many of these cases, the TDP-43 pathology is characterized by phosphorylation of the protein at serine 409 and 410 (pS409/410; Hasegawa et al., 2008; Neumann et al., 2009), and phosphorylated TDP-43 (pTDP-43) is associated with increased insolubility and cytoplasmic aggregation (Choksi et al., 2014; Hasegawa et al., 2008; Liachko et al., 2010; Neumann et al., 2009; Nonaka et al., 2016; Yamashita et al., 2016; Zhang et al., 2010).
Casein kinase 1 (CK1) was previously shown to phosphorylate TDP-43 in vitro and in vivo (Hasegawa et al., 2008; Kametani et al., 2009; Nonaka et al., 2016). CK1 is a family of serine/threonine-selective kinases that phosphorylate key proteins in various physiological and pathological processes such as development, metabolism, cancer, and neurodegenerative disorders (Cheong and Virshup, 2011; Cozza and Pinna, 2016; Cruciat, 2014; Jiang, 2017; Perez et al., 2012). In addition, CK1 inhibitors were shown to reduce TDP-43 phosphorylation and toxicity in a variety of models, including immortalized mammalian cell lines, fly and mouse neurons, and human cells derived from ALS and FTD patients (Salado et al., 2014; Alquezar et al., 2016; Martínez-González et al., 2020). Thus, manipulation of CK1 activity has been proposed as a potential drug target for treating ALS and FTD. However, whether and how CK1 is involved in the pathogenic pathways remain unclear.
Charged multivesicular body protein 2B (CHMP2B) is another gene associated with both ALS and FTD (Cox et al., 2010; Parkinson et al., 2006; Skibinski et al., 2005). It encodes the endosomal sorting complex required for transport (ESCRT) III subunit protein CHMP2B, which plays a vital role in endolysosomal trafficking, vesicle fusion, and autophagic degradation (Henne et al., 2011; Urwin et al., 2009). Among the disease-causing mutations in CHMP2B, the most studied is CHMP2BIntron5 in FTD linked to chromosome 3 (FTD-3). It is a single-nucleotide (G→C) mutation in exon 6, which causes aberrant splicing inclusive of the 201 bp of intron 5 that contains a stop codon and leads to a truncation of 36 amino acids from the acidic C terminus of CHMP2B. Consequently, CHMP2BIntron5 lacks the important binding site for Vps4, and its self-binding to the basic N terminus is significantly reduced, which disrupts the normal autoinhibition in CHMP2B (Isaacs et al., 2011; Krasniak and Ahmad, 2016; Urwin et al., 2009). As deficits in CHMP2B and the ESCRT complex often lead to accumulation of endosomes and autophagosomes (Clayton et al., 2015; Ghazi-Noori et al., 2012; Lee et al., 2007; Urwin et al., 2010; van der Zee et al., 2008), the pathogenicity of CHMP2B has been considered mainly a consequence of disruption of the autophagy–endolysosomal pathway. Intriguingly, CHMP2BIntron5 has not been associated with TDP-43 pathology in FTD-3 patients (Holm et al., 2007), whereas TDP-43 inclusion bodies are detected in the spinal motor neurons and glia of ALS patients carrying other CHMP2B mutations such as CHMP2BQ206H (Cox et al., 2010). In addition, manipulation of the ESCRT subunits other than CHMP2B caused TDP-43 aggregation (Filimonenko et al., 2007). Thus, it remains elusive whether CHMP2B and TDP-43 are molecularly linked in ALS and FTD.
In this study, we identify CHMP2B as a modifier of TDP-43 neurotoxicity in a fly genetic screen and reveal a previously unknown function of CHMP2B in regulating TDP-43 phosphorylation. This function is independent of the role of CHMP2B in autophagy, as disruption of the autophagy pathway does not alter pTDP-43 levels. Instead, we show that CHMP2B regulates the abundance of CK1 via the ubiquitin-proteasome system (UPS)–dependent protein turnover, which underlies the mitigating effect of knockdown (KD) of CHMP2B on TDP-43 phosphorylation and toxicity.
Results
Down-regulation of CHMP2B alleviates TDP-43 neurotoxicity and reduces TDP-43 phosphorylation levels in a Drosophila model
To identify unknown players involved in TDP-43–mediated neurodegeneration, we conducted a genetic screen for new modifiers of TDP-43 neurotoxicity using a Drosophila model that expressed human TDP-43 (hTDP-43) with the binary Gal4-UAS system. Transgenic expression of hTDP-43 in the fly eyes (by a GMR-Gal4 driver) caused age-dependent degeneration, as evidenced by rough surface, loss of pigment cells, and swelling of the compound fly eyes (Fig. 1, a–d; and Fig. S1, a and b).
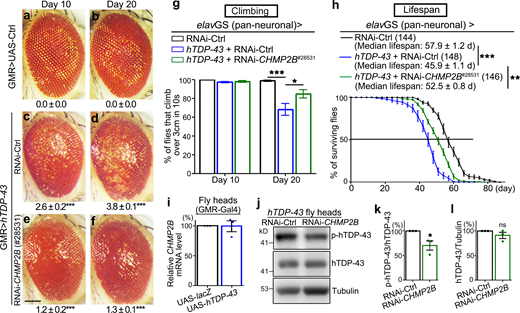
CHMP2B modifies the neurotoxicity and affects the phosphorylation levels of hTDP-43 in an in vivo Drosophila model.(a–f) KD of CHMP2B (#28531; GMR-Gal4) suppresses hTDP-43–induced eye degeneration in flies. The degeneration score (mean ± SEM) and the statistical significance compared with the UAS-lacZ (UAS-Ctrl) flies are indicated. (g and h) Adult-onset, neuronal (elavGS) down-regulation of CHMP2B suppresses hTDP-43–induced climbing deficits (g) and extends the shortened lifespan (h). RNAi-Ctrl, RNAi-luciferase flies. (i) qPCR analysis of the mRNA levels of CHMP2B in the heads of TDP-43 flies. The mRNA levels are normalized to actin and shown as the average percentage compared to that of the control flies (UAS-lacZ). (j–l) Western blot analysis (j) and quantifications of phosphorylation (p409/410; k) and total protein levels (l) of hTDP-43 in the fly heads. hTDP-43, human TDP-43; p-hTDP-43, phosphorylated hTDP-43. The protein levels are normalized to tubulin or total hTDP-43 and shown as percentages to that of the RNAi-Ctrl group. Mean ± SEM, n = ∼10 eyes/group (a–f), n = ∼10 vials/group with ∼20 flies per vial (g); the number of flies tested in each group is as indicated (h); and n = 3 (i, k, and l). Statistical significance is determined by one-way ANOVA with Tukey's HSD post-hoc test (a–f, g), two-tailed Student’s t test in (i, k, and l) and two-way ANOVA with Bonferroni’s post-hoc test in (h) at *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Scale bar, 100 µm. Source data are available for this figure: SourceData F1.
CHMP2B modifies the neurotoxicity and affects the phosphorylation levels of hTDP-43 in an in vivo Drosophila model.(a–f) KD of CHMP2B (#28531; GMR-Gal4) suppresses hTDP-43–induced eye degeneration in flies. The degeneration score (mean ± SEM) and the statistical significance compared with the UAS-lacZ (UAS-Ctrl) flies are indicated. (g and h) Adult-onset, neuronal (elavGS) down-regulation of CHMP2B suppresses hTDP-43–induced climbing deficits (g) and extends the shortened lifespan (h). RNAi-Ctrl, RNAi-luciferase flies. (i) qPCR analysis of the mRNA levels of CHMP2B in the heads of TDP-43 flies. The mRNA levels are normalized to actin and shown as the average percentage compared to that of the control flies (UAS-lacZ). (j–l) Western blot analysis (j) and quantifications of phosphorylation (p409/410; k) and total protein levels (l) of hTDP-43 in the fly heads. hTDP-43, human TDP-43; p-hTDP-43, phosphorylated hTDP-43. The protein levels are normalized to tubulin or total hTDP-43 and shown as percentages to that of the RNAi-Ctrl group. Mean ± SEM, n = ∼10 eyes/group (a–f), n = ∼10 vials/group with ∼20 flies per vial (g); the number of flies tested in each group is as indicated (h); and n = 3 (i, k, and l). Statistical significance is determined by one-way ANOVA with Tukey's HSD post-hoc test (a–f, g), two-tailed Student’s t test in (i, k, and l) and two-way ANOVA with Bonferroni’s post-hoc test in (h) at *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Scale bar, 100 µm. Source data are available for this figure: SourceData F1.

KD of CHMP2B by two independent transgenic RNAi strains (#38375 and #28531) in flies. (a–f) KD of CHMP2B by another RNAi fly line (#38375) also suppresses hTDP-43–induced eye degeneration (a–d), climbing deficits (e) and shortened lifespan (f). The degeneration score (mean ± SEM) and the statistical significance compared with the RNAi control (RNAi-mCherry) are indicated at the bottom of each group in (a–d). (g-j) KD of CHMP2B by the RNAi line #38375 (g and i) or #28531 (h and j) does not generally promote the climbing capability (g and h) or extend the lifespan (i and j) in flies. Data are shown as mean ± SEM; n = 10 eyes/group (a–d), n = 10 vials/group with ∼20 flies/vial in (e, g, and h), n of flies and the median lifespan of each group are indicated (f, i, and j). Statistical significance is determined by two-tailed Student’s t test in (a–d, g, and h), one-way ANOVA with Tukey's HSD post-hoc test (e), and two-way ANOVA with Bonferroni’s post-hoc test (f, i, and j). ***, P < 0.001. ns, not significant. Scale bar, 100 µm.
KD of CHMP2B by two independent transgenic RNAi strains (#38375 and #28531) in flies. (a–f) KD of CHMP2B by another RNAi fly line (#38375) also suppresses hTDP-43–induced eye degeneration (a–d), climbing deficits (e) and shortened lifespan (f). The degeneration score (mean ± SEM) and the statistical significance compared with the RNAi control (RNAi-mCherry) are indicated at the bottom of each group in (a–d). (g-j) KD of CHMP2B by the RNAi line #38375 (g and i) or #28531 (h and j) does not generally promote the climbing capability (g and h) or extend the lifespan (i and j) in flies. Data are shown as mean ± SEM; n = 10 eyes/group (a–d), n = 10 vials/group with ∼20 flies/vial in (e, g, and h), n of flies and the median lifespan of each group are indicated (f, i, and j). Statistical significance is determined by two-tailed Student’s t test in (a–d, g, and h), one-way ANOVA with Tukey's HSD post-hoc test (e), and two-way ANOVA with Bonferroni’s post-hoc test (f, i, and j). ***, P < 0.001. ns, not significant. Scale bar, 100 µm.
In the screen, we tested ∼940 transgenic RNAi fly lines targeting genes involved in the regulation of proteostasis, including autophagy and UPS, posttranslational modifications such as ubiquitination and phosphorylation, mitochondria, and others. We isolated 32 enhancer genes and 10 suppressor genes whose loss of function (LOF) modified TDP-43–mediated neurodegeneration. Among them, two fly lines (#28531 and #38375) showed remarkable suppression of hTDP-43–induced eye degeneration (Fig. 1, e and f; and Fig. S1, c and d), which turned out to be two independent RNAi lines of the fly gene CHMP2B. Further examination revealed that down-regulation of CHMP2B with an inducible pan-neuronal elav-GeneSwitch (elavGS) driver markedly mitigated hTDP-43–induced, age-dependent climbing deficits (Figs. 1 g and S1 e). Moreover, expression of hTDP-43 in the adult fly neurons shortened the lifespan by ∼20.7%, which was increased by ∼14.4% with KD of CHMP2B (#28531) compared with transgenic expression of the control RNAi-luciferase transgene in the TDP-43 flies (Fig. 1 h). The other RNAi-CHMP2B line (#38375) also extended the lifespan of the TDP-43 flies, although to a lesser extent (Fig. S1 f). In addition, we confirmed that KD of CHMP2B in a WT background did not increase the climbing capability (Fig. S1, g and h) or extend the lifespan (Fig. S1, i and j), indicating that down-regulation of CHMP2B did not have a general beneficial effect on promoting motility or survival. Rather, it specifically suppressed TDP-43–mediated neurotoxicity in flies.
TDP-43 is known to play a vital role in regulating RNA metabolism and homeostasis (Cohen et al., 2011; Lee et al., 2011). It was reasonable to hypothesize that KD of CHMP2B rescued the TDP-43 flies, because CHMP2B was abnormally up-regulated by overexpression (OE) of hTDP-43. We hence examined the mRNA levels of CHMP2B by real-time quantitative PCR (qPCR) analysis; however, it showed no significant change of the CHMP2B mRNA levels in the TDP-43 fly heads (Fig. 1 i). Considering the known function of CHMP2B and ESCRT in the autophagy–endolysosomal pathway and proteostasis, the alternative possibility was that KD of CHMP2B might affect TDP-43 abundance. We then examined TDP-43 protein levels in the RNAi-CHMP2B flies by Western blotting. Interestingly, although the total TDP-43 abundance was unchanged, we found the disease hallmarked pS409/410 phosphorylation of TDP-43 (Hasegawa et al., 2008; Neumann et al., 2009) was reduced by RNAi-CHMP2B (Fig. 1, j–l). Transgenic OE of the FTD-3 associated mutation CHMP2BIntron5 in flies caused the fly eye to degenerate (Fig. S2 a; Ahmad et al., 2009), and we showed that OE of CHMP2BIntron5 increased the phosphorylation levels of hTDP-43 in fly neurons (Fig. S2, b–d). Neither KD of CHMP2B nor OE of CHMP2BIntron5 significantly affected the insolubility of hTDP-43 (Fig. S2, e–h), indicating that the transgenically overexpressed soluble hTDP-43 protein manifested significant toxicity in the fly model.
OE of CHMP2BIntron5 increases the phosphorylation levels but does not induce significant insolubility of hTDP-43 in flies.(a) OE of the FTD-3–associated CHMP2BIntron5 in flies (by GMR) causes the fly eye to degenerate. The degeneration score (mean ± SEM) and the statistical significance compared with the control flies (UAS-Luciferase, UAS-Luc) are indicated. (b–d) Western blot analysis (b) and quantifications of the phosphorylation (c) and total protein levels (d) of hTDP-43 in the heads of the transgenic flies overexpressing human CHMP2BIntron5. (e–h) KD of CHMP2B (e–f) or OE of CHMP2BIntron5 (g and h) in fly heads does not alter the solubility of the transgenically expressed hTDP-43 protein. S, soluble fractions (the supernatants in RIPA); I, insoluble fractions (the pellets resuspended in 9 M urea). hTDP-43, human TDP-43; p-hTDP-43, phosphorylated hTDP-43. Asterisk indicates a nonspecific band close to the Flag-CHMP2BIntron5 band in the anti-Flag blots of the fly samples. ud, undetected. All protein levels are normalized to tubulin in the soluble fraction. Mean ± SEM; n = ∼10 eyes each group (a), n = 4 (c, d, and f), and n = 3 (h). Two-tailed Student’s t test; *, P < 0.05; ***, P < 0.001. ns, not significant. Scale bar, 100 µm. Source data are available for this figure: SourceData FS2.
OE of CHMP2BIntron5 increases the phosphorylation levels but does not induce significant insolubility of hTDP-43 in flies.(a) OE of the FTD-3–associated CHMP2BIntron5 in flies (by GMR) causes the fly eye to degenerate. The degeneration score (mean ± SEM) and the statistical significance compared with the control flies (UAS-Luciferase, UAS-Luc) are indicated. (b–d) Western blot analysis (b) and quantifications of the phosphorylation (c) and total protein levels (d) of hTDP-43 in the heads of the transgenic flies overexpressing human CHMP2BIntron5. (e–h) KD of CHMP2B (e–f) or OE of CHMP2BIntron5 (g and h) in fly heads does not alter the solubility of the transgenically expressed hTDP-43 protein. S, soluble fractions (the supernatants in RIPA); I, insoluble fractions (the pellets resuspended in 9 M urea). hTDP-43, human TDP-43; p-hTDP-43, phosphorylated hTDP-43. Asterisk indicates a nonspecific band close to the Flag-CHMP2BIntron5 band in the anti-Flag blots of the fly samples. ud, undetected. All protein levels are normalized to tubulin in the soluble fraction. Mean ± SEM; n = ∼10 eyes each group (a), n = 4 (c, d, and f), and n = 3 (h). Two-tailed Student’s t test; *, P < 0.05; ***, P < 0.001. ns, not significant. Scale bar, 100 µm. Source data are available for this figure: SourceData FS2.
CHMP2B abundance regulates TDP-43 phosphorylation and toxicity in mammalian cells
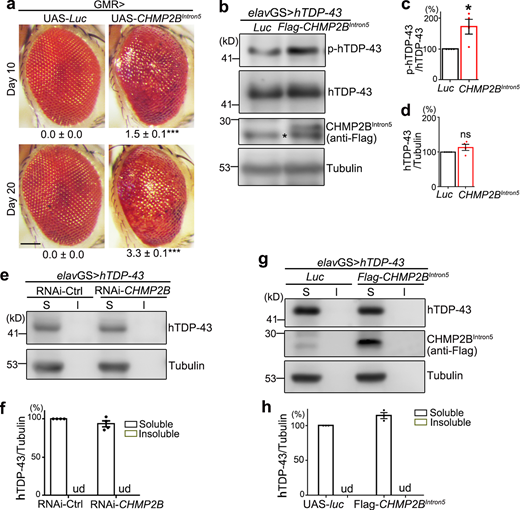
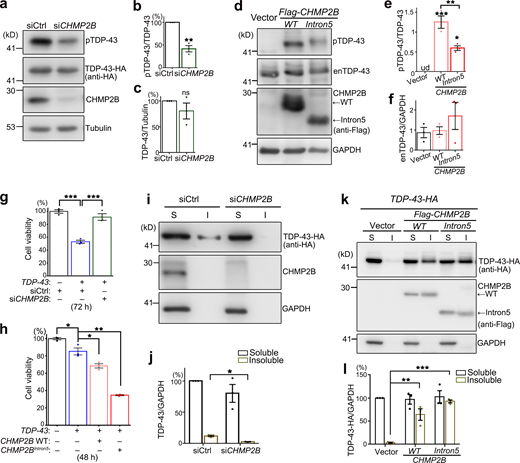
Next, we extended our study to mammalian systems to see whether the function of CHMP2B in regulating TDP-43 phosphorylation and toxicity was evolutionarily conserved. Consistent with the observations in flies, KD of CHMP2B in human 293T cells also decreased pTDP-43 levels (Fig. 2, a–c). Of note, since little endogenous TDP-43 was phosphorylated in 293T cells under normal conditions (lane 1 in Fig. 2 d), TDP-43 was overexpressed by transient transfection of TDP-43-HA in this experiment. This was to increase the basal TDP-43 phosphorylation levels in order to examine whether RNAi-CHMP2B could reduce pTDP-43 levels. Importantly, we showed that up-regulation of WT CHMP2B dramatically increased the phosphorylation levels of endogenous TDP-43 in 293T cells (Fig. 2, d and e) without affecting the TDP-43 protein abundance (Fig. 2, d and f). OE of CHMP2BIntron5 in 293T cells also increased the pTDP-43 levels compared with the vector control, but to a lesser extent compared with that by OE of WT CHMP2B (Fig. 2, d–f).
CHMP2B regulates TDP-43 phosphorylation and toxicity in 293T cells.(a–c) Representative Western blotting images (a) and quantifications (b and c) of TDP-43-HA phosphorylation (b) and protein levels (c) in 293T cells treated with the siRNA of CHMP2B (siCHMP2B). (d–f) OE of WT CHMP2B or CHMP2BIntron5 increases the phosphorylation levels of endogenous TDP-43 (enTDP-43) in 293T cells. ud, undetected. (g and h) KD of CHMP2B suppresses (g), whereas OE of WT CHMP2B or CHMP2BIntron5 enhances (h), TDP-43–induced loss of cell viability. siCtrl, scrambled siRNA. Cells are homogenized and examined using a CCK8 assay at 72 h after transfection of siRNA and the TDP-43-HA expression plasmid (g) or at 48 h after transfection of the expression plasmids only (h). (i–l) Representative Western blot images (i and k) and quantification (j and l) of TDP-43-HA proteins in the soluble (S, supernatants in RIPA) and insoluble (I, pellets resuspended in 9 M urea) fractions of the cell lysates of 293T cells treated with siCHMP2B (i and j) or transfected with WT CHMP2B or CHMP2BIntron5 (k and l). siCtrl, scrambled siRNA. All protein levels are normalized to GAPDH in the soluble fraction. Mean ± SEM; n = 3. Two-tailed Student’s t test in (b, c, and j) and one-way ANOVA with Tukey's HSD post-hoc test (e–h and l); *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Source data are available for this figure: SourceData F2.
CHMP2B regulates TDP-43 phosphorylation and toxicity in 293T cells.(a–c) Representative Western blotting images (a) and quantifications (b and c) of TDP-43-HA phosphorylation (b) and protein levels (c) in 293T cells treated with the siRNA of CHMP2B (siCHMP2B). (d–f) OE of WT CHMP2B or CHMP2BIntron5 increases the phosphorylation levels of endogenous TDP-43 (enTDP-43) in 293T cells. ud, undetected. (g and h) KD of CHMP2B suppresses (g), whereas OE of WT CHMP2B or CHMP2BIntron5 enhances (h), TDP-43–induced loss of cell viability. siCtrl, scrambled siRNA. Cells are homogenized and examined using a CCK8 assay at 72 h after transfection of siRNA and the TDP-43-HA expression plasmid (g) or at 48 h after transfection of the expression plasmids only (h). (i–l) Representative Western blot images (i and k) and quantification (j and l) of TDP-43-HA proteins in the soluble (S, supernatants in RIPA) and insoluble (I, pellets resuspended in 9 M urea) fractions of the cell lysates of 293T cells treated with siCHMP2B (i and j) or transfected with WT CHMP2B or CHMP2BIntron5 (k and l). siCtrl, scrambled siRNA. All protein levels are normalized to GAPDH in the soluble fraction. Mean ± SEM; n = 3. Two-tailed Student’s t test in (b, c, and j) and one-way ANOVA with Tukey's HSD post-hoc test (e–h and l); *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Source data are available for this figure: SourceData F2.
We went on to determine how manipulation of CHMP2B affected TDP-43 toxicity using a Cell Counting Kit 8 (CCK8) assay. OE of TDP-43 in 293T cells reduced the cell viability, which was suppressed by KD of CHMP2B (Fig. 2 g) and enhanced by OE of WT CHMP2B (Fig. 2 h). OE of CHMP2BIntron5 enhanced TDP-43–induced loss of cell viability even more (Fig. 2 h), although it only mildly increased TDP-43 phosphorylation compared with OE of WT CHMP2B (Fig. 2, d–f), suggesting alternative mechanisms conferring to the cytotoxicity of CHMP2BIntron5.
Since CHMP2B and the ESCRT complex play an important role in the autophagy–endolysosomal pathway and proteostasis regulation, we examined how manipulation of CHMP2B impacted on the solubility of TDP-43. Again, since endogenous TDP-43 was predominantly soluble in 293T cells, the effect on TDP-43 solubility was assessed with the transiently overexpressed TDP-43-HA in the subsequent experiments. We found that KD of CHMP2B reduced (Fig. 2, i and j), whereas OE of WT CHMP2B or CHMP2BIntron5 increased (Fig. 2, k and l), the insolubility of TDP-43-HA in 293T cells. Of note, unlike the mild effect in promoting TDP-43 phosphorylation (Fig. 2, d–f), OE of CHMP2BIntron5 increased the insolubility of TDP-43 to the similar or slightly higher level than that of OE of WT CHMP2B (Fig. 2, k and l).
The autophagy pathway does not profoundly impact on TDP-43 phosphorylation
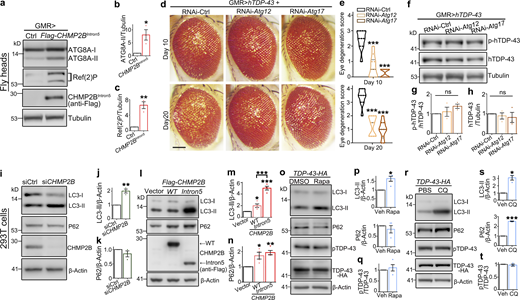
Previous studies of CHMP2BIntron5 revealed remarkable autophagy dysfunction (Krasniak and Ahmad, 2016; Lee et al., 2007; Lee et al., 2009) and inhibition of autophagy could suppress the toxicity of CHMP2BIntron5 in cultured cortical neurons (Lee and Gao, 2009). Indeed, we found that in the fly heads overexpressing CHMP2BIntron5, the protein levels of lipidated autophagy-related 8a (ATG8A-II, the Drosophila homologue of the mammalian LC3-II; Kabeya et al., 2000) and refractory to sigma P (Ref(2)P, P62 in mammals; Bjørkøy et al., 2005) were dramatically increased (Fig. 3, a–c). Inhibition of autophagy induction by KD of Atg12 or Atg17 significantly suppressed TDP-43–induced eye degeneration in flies (Fig. 3, d and e). However, the phosphorylation levels of hTDP-43 in the RNAi-Atg12 or RNAi-Atg17 flies were unaffected (Fig. 3, f–h), suggesting that the autophagy pathway did not play a major role in regulating TDP-43 phosphorylation in the fly model.
Disturbance of the autophagy–lysosomal pathway does not alter TDP-43 phosphorylation levels in flies or mammalian cells. (a–c) Western blot analysis (a) shows the levels of ATG8A-II (b) and Ref(2)P (c) are significantly increased in the fly heads transgenically expressing human CHMP2BIntron5 (GMR-Gal4). Ctrl, UAS-luciferase. (d and e) Inhibition of autophagy by RNAi-Atg12 or RNAi-Atg17 attenuates hTDP-43–induced eye degeneration in flies (d), quantified with violin plots (e). RNAi-Ctrl, RNAi-mCherry. (f–h) Western blot analysis (f) shows unaffected phosphorylation (g) and total protein (h) levels of TDP-43 in the RNAi-Atg12 and RNAi-Atg17 fly heads. hTDP-43, human TDP-43; p-hTDP-43, phosphorylated hTDP-43. (i–n) Western blot analyses (i and l) and quantifications (j, k, m, and n) of LC3-II and P62 levels in 293T cells treated with siCHMP2B (i–k) or OE of CHMP2B or CHMP2BIntron5 (l–n). siCtrl, scrambled siRNA. (o–t) Western blot analyses of LC3-II, P62, and pTDP-43 levels in 293T cells treated with Rapa (100 nM, 1 h; o–q) or CQ (10 µM, 12 h; r–t). Vehicle controls: DMSO for Rapa and PBS for CQ. Mean ± SEM, n = 3–4 in all Western blots and n = 10 flies/group (e). Two-tailed Student’s t test (b, c, j, k, p, q, s, and t) and one-way ANOVA with Tukey's HSD post-hoc test (e, g, h, m, and n); *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Scale bar, 100 µm. Source data are available for this figure: SourceData F3.
Disturbance of the autophagy–lysosomal pathway does not alter TDP-43 phosphorylation levels in flies or mammalian cells. (a–c) Western blot analysis (a) shows the levels of ATG8A-II (b) and Ref(2)P (c) are significantly increased in the fly heads transgenically expressing human CHMP2BIntron5 (GMR-Gal4). Ctrl, UAS-luciferase. (d and e) Inhibition of autophagy by RNAi-Atg12 or RNAi-Atg17 attenuates hTDP-43–induced eye degeneration in flies (d), quantified with violin plots (e). RNAi-Ctrl, RNAi-mCherry. (f–h) Western blot analysis (f) shows unaffected phosphorylation (g) and total protein (h) levels of TDP-43 in the RNAi-Atg12 and RNAi-Atg17 fly heads. hTDP-43, human TDP-43; p-hTDP-43, phosphorylated hTDP-43. (i–n) Western blot analyses (i and l) and quantifications (j, k, m, and n) of LC3-II and P62 levels in 293T cells treated with siCHMP2B (i–k) or OE of CHMP2B or CHMP2BIntron5 (l–n). siCtrl, scrambled siRNA. (o–t) Western blot analyses of LC3-II, P62, and pTDP-43 levels in 293T cells treated with Rapa (100 nM, 1 h; o–q) or CQ (10 µM, 12 h; r–t). Vehicle controls: DMSO for Rapa and PBS for CQ. Mean ± SEM, n = 3–4 in all Western blots and n = 10 flies/group (e). Two-tailed Student’s t test (b, c, j, k, p, q, s, and t) and one-way ANOVA with Tukey's HSD post-hoc test (e, g, h, m, and n); *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Scale bar, 100 µm. Source data are available for this figure: SourceData F3.
In mammalian cell cultures, we found that KD of CHMP2B increased the levels of the autophagy marker LC3-II, while OE of WT CHMP2B increased both LC3-II and P62 (Fig. 3, i–n). Both down- and up-regulation of CHMP2B impaired autophagic flux, indicating that a steady level of CHMP2B was critical for maintaining normal autophagy function. This was in contrast to the dose-dependent effect of CHMP2B on pTDP-43 levels (Fig. 2, a–f). OE of CHMP2BIntron5 caused a more severe autophagic dysfunction, as a strikingly more accumulation of LC3-II was detected even when CHMP2BIntron5 was expressed at a lower level than WT CHMP2B (Fig. 3, l–n), which suggested that the CHMP2BIntron5 mutation was likely a hypermorphic or dominant allele for the function in regulating autophagy.
Nevertheless, induction of autophagy by treating the cells with rapamycin (Rapa; Fig. 3, o and p; which mimicked the effect of siCHMP2B on autophagy in Fig. 3, i–k) or disruption of the autophagic flux with chloroquine (CQ; Fig. 3, r–s, which mimicked the effect of OE of WT CHMP2B and CHMP2BIntron5 on autophagy in Fig. 3, l–n) did not significantly alter pTDP-43 levels (Fig. 3, o, q, r, and t). Likewise, genetic inhibition of autophagy by siRNA-mediated KD of Atg12 or Atg17 also attenuated TDP-43–induced loss of cell viability in 293T cells (Fig. S3 a). KD of Atg12 or Atg17 decreased the level of the autophagy marker LC3-II and increased that of P62 (Fig. S3, b–d), indicating that autophagy was indeed disturbed. Nevertheless, pTDP-43 levels were unchanged by KD of Atg12 or Atg17 (Fig. S3, b and e). Together, both the in vivo observations in flies and the pharmacological and genetic manipulations in mammalian cell cultures indicate that autophagy does not play a major role in regulating pTDP-43 levels and that the modifying effect of CHMP2B on TDP-43 phosphorylation is independent of the autophagy pathway.
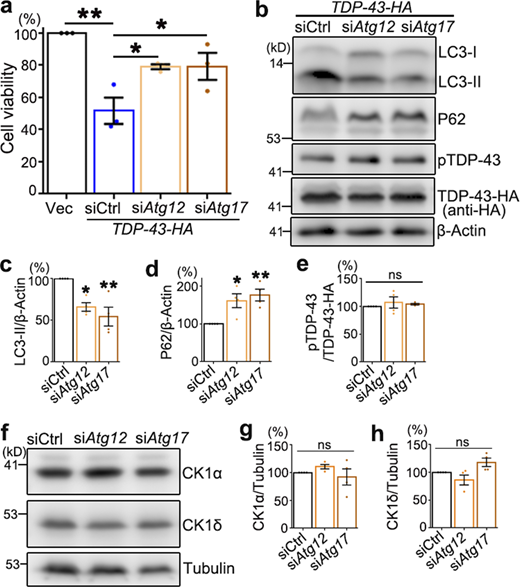
Down-regulation of Atg12 or Atg17 suppresses TDP-43–induced cytotoxicity but does not alter pTDP-43 and CK1 levels in 293T cells.(a) The viability of 293T cells transfected with the empty vector (Vec) or TDP-43-HA together with scrambled siRNA (siCtrl) or siRNA against Atg12 (siAtg12) or Atg17 (siAtg17) is assessed 72 h after transfection using the CCK8 assay. (b–e) Western blot analysis (b) and quantifications of LC3-II (c), P62 (d), and pTDP-43 (e) levels in 293T cells treated with siAtg12 or siAtg17.(f–h) Representative Western blot images (f) and quantifications of the CK1α (g) and CK1δ (h) normalized to tubulin. Mean ± SEM; n = 3–4. One-way ANOVA with Tukey's HSD post-hoc test; *, P < 0.05; **, P < 0.01. ns, not significant. Source data are available for this figure: SourceData FS3.
Down-regulation of Atg12 or Atg17 suppresses TDP-43–induced cytotoxicity but does not alter pTDP-43 and CK1 levels in 293T cells.(a) The viability of 293T cells transfected with the empty vector (Vec) or TDP-43-HA together with scrambled siRNA (siCtrl) or siRNA against Atg12 (siAtg12) or Atg17 (siAtg17) is assessed 72 h after transfection using the CCK8 assay. (b–e) Western blot analysis (b) and quantifications of LC3-II (c), P62 (d), and pTDP-43 (e) levels in 293T cells treated with siAtg12 or siAtg17.(f–h) Representative Western blot images (f) and quantifications of the CK1α (g) and CK1δ (h) normalized to tubulin. Mean ± SEM; n = 3–4. One-way ANOVA with Tukey's HSD post-hoc test; *, P < 0.05; **, P < 0.01. ns, not significant. Source data are available for this figure: SourceData FS3.
CHMP2B promotes TDP-43 hyperphosphorylation and cytotoxicity via CK1
Seeking for an alternative mechanism to explain how CHMP2B regulated TDP-43 phosphorylation, we looked through literature and found the protein kinase CK1 was previously reported to phosphorylate TDP-43 (Hasegawa et al., 2008; Kametani et al., 2009). Consistently, we found that increase of CK1 activity by OE of CK1α or CK1δ in 293T cells promoted phosphorylation of the endogenous TDP-43 protein in 293T cells (Fig. 4, a and b). Further, KD of CK1α or CK1δ decreased the phosphorylation levels of transiently overexpressed TDP-43-HA (Fig. 4, c and d). More importantly, KD of CK1α and CK1δ significantly suppressed CHMP2B OE–induced hyperphosphorylation of endogenous TDP-43 (Fig. 4, e and f).
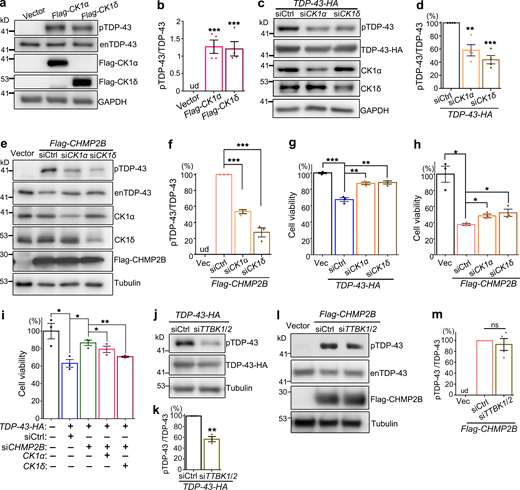
The kinase CK1 mediates the modifying effects of CHMP2B on TDP-43 phosphorylation and cytotoxicity. (a–d) OE of CK1α or CK1δ promotes hyperphosphorylation of endogenous TDP-43 (enTDP-43; a and b), whereas KD of CK1α or CK1δ reduces pTDP-43-HA levels in 293T cells (c and d). (e and f) KD of CK1α or CK1δ significantly decreases CHMP2B OE-induced hyperphosphorylation of enTDP-43. (g and h) The cell viability assay indicates that KD of CK1α or CK1δ markedly suppresses TDP-43 cytotoxicity and partially rescues CHMP2B OE–induced cell viability loss. (i) OE of CK1α or CK1δ diminishes the mitigating effect of siCHMP2B on TDP-43 cytotoxicity. (j and k) KD of TTBK1/2 reduces pTDP-43-HA levels in 293T cells. (l and m) KD of TTBK1/2 cannot suppress CHMP2B OE–induced hyperphosphorylation of enTDP-43. siCtrl, scrambled siRNA; siTTBK1/2, a mixture of siTTBK1 and siTTBK2. ud, undetected. Mean ± SEM, n = 3–4. Two-tailed Student’s t test (k) and one-way ANOVA with Tukey's HSD post-hoc test in all the rest; *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Source data are available for this figure: SourceData F4.
The kinase CK1 mediates the modifying effects of CHMP2B on TDP-43 phosphorylation and cytotoxicity. (a–d) OE of CK1α or CK1δ promotes hyperphosphorylation of endogenous TDP-43 (enTDP-43; a and b), whereas KD of CK1α or CK1δ reduces pTDP-43-HA levels in 293T cells (c and d). (e and f) KD of CK1α or CK1δ significantly decreases CHMP2B OE-induced hyperphosphorylation of enTDP-43. (g and h) The cell viability assay indicates that KD of CK1α or CK1δ markedly suppresses TDP-43 cytotoxicity and partially rescues CHMP2B OE–induced cell viability loss. (i) OE of CK1α or CK1δ diminishes the mitigating effect of siCHMP2B on TDP-43 cytotoxicity. (j and k) KD of TTBK1/2 reduces pTDP-43-HA levels in 293T cells. (l and m) KD of TTBK1/2 cannot suppress CHMP2B OE–induced hyperphosphorylation of enTDP-43. siCtrl, scrambled siRNA; siTTBK1/2, a mixture of siTTBK1 and siTTBK2. ud, undetected. Mean ± SEM, n = 3–4. Two-tailed Student’s t test (k) and one-way ANOVA with Tukey's HSD post-hoc test in all the rest; *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Source data are available for this figure: SourceData F4.
Consistent with their effects on TDP-43 phosphorylation, we found that down-regulation of CK1α or CK1δ ameliorated the cytotoxicity caused by OE of TDP-43 (Fig. 4 g) or CHMP2B (Fig. 4 h), whereas up-regulation of CK1α or CK1δ diminished the mitigating effect of siCHMP2B on TDP-43–mediated cytotoxicity (Fig. 4 i). Of note, because of the critical role of CK1 in various other cellular functions and cell survival (Cheong and Virshup, 2011; Cruciat, 2014; Jiang, 2017), the amounts of transfected siRNAs (0.8 nM of siCK1α and 0.16 nM of siCK1δ) and the expression plasmids (10 ng of CK1α and 5 ng of CK1δ) were carefully determined so that OE or KD of CK1 per se did not manifest significant toxicity in these cell viability assays (Fig. S4, a and b).
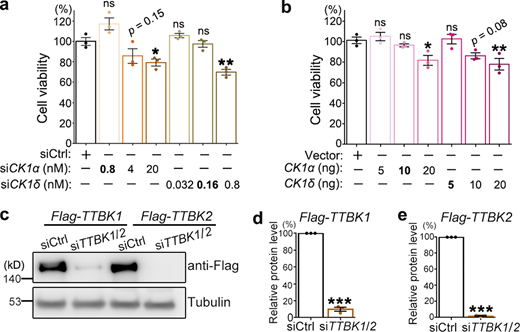
Examination of the kinases CK1 and TTBK1/2 in 293T cells.(a and b) The cell viability of 293T cells treated with siCK1α or siCK1δ (a) or transfected with Flag-CK1α or Flag-CK1δ (b) at indicated dosages is examined using the CCK8 assay. The statistical significance compared with the siCtrl (scrambled siRNA) or vector (empty vector) control is shown. KD of CK1 by transfection of 0.8 nM siCK1α or 0.16 nM siCK1δ and OE of CK1 by transfection of 10 ng CK1α or 5 ng CK1δ plasmids do not manifest significant loss of cell viability, which are used in the cell viability assays in Fig. 4, g–i. (c–e) Representative images (c) and quantifications (d and e) of the Western blot analysis confirming the down-regulation of TTBK1/2 by siTTBK1/2. Due to the lack of a good commercial anti-TTBK1/2 antibody, the transiently overexpressed Flag-TTBK1 and Flag-TTBK2 proteins are used as a readout to evaluate the KD efficiency of siTTBK1/2 (a mixture of siTTBK1 and siTTBK2 as used in Fig. 4, j–m). The siTTBK1/2 significantly reduces the protein levels of the overexpressed Flag-TTBK1 and Flag-TTBK2. Mean ± SEM; n = 3. One-way ANOVA with Tukey's HSD post-hoc test (a and b) and two-tailed Student’s t test in (d and e); *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Source data are available for this figure: SourceData FS4.
Examination of the kinases CK1 and TTBK1/2 in 293T cells.(a and b) The cell viability of 293T cells treated with siCK1α or siCK1δ (a) or transfected with Flag-CK1α or Flag-CK1δ (b) at indicated dosages is examined using the CCK8 assay. The statistical significance compared with the siCtrl (scrambled siRNA) or vector (empty vector) control is shown. KD of CK1 by transfection of 0.8 nM siCK1α or 0.16 nM siCK1δ and OE of CK1 by transfection of 10 ng CK1α or 5 ng CK1δ plasmids do not manifest significant loss of cell viability, which are used in the cell viability assays in Fig. 4, g–i. (c–e) Representative images (c) and quantifications (d and e) of the Western blot analysis confirming the down-regulation of TTBK1/2 by siTTBK1/2. Due to the lack of a good commercial anti-TTBK1/2 antibody, the transiently overexpressed Flag-TTBK1 and Flag-TTBK2 proteins are used as a readout to evaluate the KD efficiency of siTTBK1/2 (a mixture of siTTBK1 and siTTBK2 as used in Fig. 4, j–m). The siTTBK1/2 significantly reduces the protein levels of the overexpressed Flag-TTBK1 and Flag-TTBK2. Mean ± SEM; n = 3. One-way ANOVA with Tukey's HSD post-hoc test (a and b) and two-tailed Student’s t test in (d and e); *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not significant. Source data are available for this figure: SourceData FS4.
We wondered how specific the function of CK1 was in mediating the modifying effect of CHMP2B on TDP-43 phosphorylation. The tau tubulin kinases 1 and 2 (TTBK1/2) were also reported to phosphorylate TDP-43 in previous studies (Liachko et al., 2014; Taylor et al., 2018), and we showed that KD of TTBK1/2 by siRNA (Fig. S4, c–e) was indeed able to lower TDP-43 phosphorylation levels (Fig. 4, j and k). However, unlike KD of CK1, down-regulation of TTBK1/2 failed to suppress CHMP2B OE–induced hyperphosphorylation of TDP-43 (Fig. 4, l and m). The results indicated that the kinases TTBK1/2 do not underlie the function of CHMP2B in promoting TDP-43 phosphorylation and that CK1 acts as a rather specific molecular link between CHMP2B and TDP-43 phosphorylation.
Inhibition of CK1 alleviates CHMP2B-mediated TDP-43 hyperphosphorylation and cell death in neuroblastoma cells
CK1 inhibitors have been proposed as a drug candidate for treating TDP-43–associated ALS/FTD (Salado et al., 2014; Alquezar et al., 2016; Martínez-González et al., 2020). Here, we explored the therapeutic potential of CK1 inhibition in CHMP2B-related neurodegeneration. First, we showed that OE of WT CHMP2B in murine neuroblastoma N2a cells caused significant cell death in the propidium iodide (PI) staining assay and OE of CHMP2BIntron5 caused even more (Fig. 5, a and b). Consistent with the results in 293T cells (Fig. 2, d–f), they both promoted hyperphosphorylation of endogenous TDP-43, although OE of CHMP2BIntron5 was markedly less effective than OE of WT CHMP2B (Fig. 5, c–e). Next, we treated N2a cells with a commonly available CK1 inhibitor D4776 (Rena et al., 2004), which significantly decreased TDP-43 phosphorylation levels and did not alter its protein levels (Fig. 5, f–h). We showed that inhibition of CK1 by D4776 in N2a cells almost completely suppressed the cell death induced by OE of WT CHMP2B and partially suppressed the toxicity of CHMP2BIntron5 (Fig. 5, i and j).
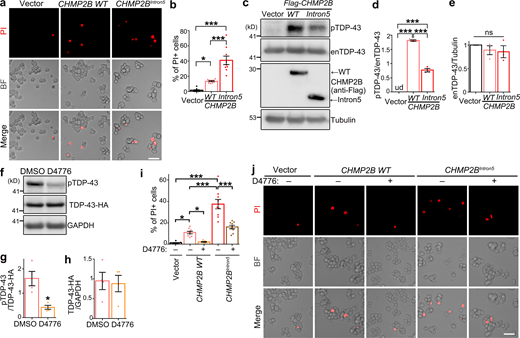
CK1 inhibition diminishes CHMP2B-mediated cell death in neuroblastoma N2a cells. (a and b) Representative PI staining images (a) and quantification (b) of the cell death of N2a cells transfected with WT CHMP2B or CHMP2BIntron5. (c–e) OE of WT CHMP2B or CHMP2BIntron5 promotes hyperphosphorylation of endogenous TDP-43 (enTDP-43) in N2a cells, examined by Western blot (c) and quantified in (d and e). (f–h) The CK1 inhibitor D4776 (5 µM, 12 h) potently suppresses phosphorylation of TDP-43-HA in N2a cells. (i and j) Quantification (i) and representative PI staining images (j) show remarkable suppression of CHMP2B-mediated cell death by the CK1 inhibitor D4776 in N2a cells. Vehicle control, DMSO. BF, brightfield. Mean ± SEM, n = ∼500 cells each group of pooled results from three independent repeats (b and i) and n = 3–4 (d, e, g, and h). One-way ANOVA with Tukey's HSD post-hoc test (b, d, e, and i) and two-tailed Student’s t test (g and h); *, P < 0.05; ***, P < 0.001. ns, not significant. Scale bars, 50 µm. Source data are available for this figure: SourceData F5.
CK1 inhibition diminishes CHMP2B-mediated cell death in neuroblastoma N2a cells. (a and b) Representative PI staining images (a) and quantification (b) of the cell death of N2a cells transfected with WT CHMP2B or CHMP2BIntron5. (c–e) OE of WT CHMP2B or CHMP2BIntron5 promotes hyperphosphorylation of endogenous TDP-43 (enTDP-43) in N2a cells, examined by Western blot (c) and quantified in (d and e). (f–h) The CK1 inhibitor D4776 (5 µM, 12 h) potently suppresses phosphorylation of TDP-43-HA in N2a cells. (i and j) Quantification (i) and representative PI staining images (j) show remarkable suppression of CHMP2B-mediated cell death by the CK1 inhibitor D4776 in N2a cells. Vehicle control, DMSO. BF, brightfield. Mean ± SEM, n = ∼500 cells each group of pooled results from three independent repeats (b and i) and n = 3–4 (d, e, g, and h). One-way ANOVA with Tukey's HSD post-hoc test (b, d, e, and i) and two-tailed Student’s t test (g and h); *, P < 0.05; ***, P < 0.001. ns, not significant. Scale bars, 50 µm. Source data are available for this figure: SourceData F5.
We noted that OE of CHMP2BIntron5 induced significantly more cell death in N2a cells (Fig. 5, a and b) and more severe impediment of the autophagic flux in 293T cells (Fig. 3, l and m) than WT CHMP2B, whereas OE of WT CHMP2B caused a much greater increase of pTDP-43 levels than that of CHMP2BIntron5 (Fig. 2, d and e; and Fig. 5, c and d). As CHMP2B appeared to regulate autophagy and TDP-43 phosphorylation through different mechanisms, we speculated that the CHMP2BIntron5 mutation might have dominant or hypermorphic activity in the autophagy–endolysosomal pathway but was a hypomorphic mutation with regard to the function in regulating TDP-43 phosphorylation. CHMP2BQ206H is a single-nucleotide (A→C) replacement causing a Glu-to-His mutation, which is associated with TDP-43 pathology in the spinal motor neurons and glia of ALS patients (Cox et al., 2010). We found that OE of CHMP2BQ206H increased TDP-43 phosphorylation (Fig. S5, a and b) and CK1 levels (Fig. S5, a, c, and d) to a similar extent as OE of WT CHMP2B. Thus, not all disease-linked CHMP2B mutations are associated with impaired function in promoting TDP-43 phosphorylation.
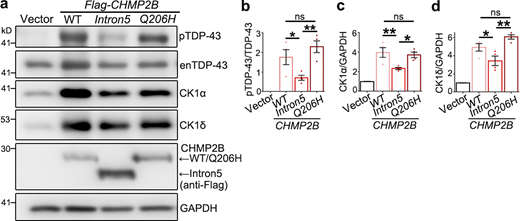
The effects of different ALS/FTD-associated CHMP2B mutations on TDP-43 phosphorylation and CK1 levels.(a–d) Representative Western blot images (a) and quantification of TDP-43 phosphorylation levels (b) and protein abundance of CK1α (c) and CK1δ (d) in 293T cells transfected with WT CHMP2B, CHMP2BIntron5, or CHMP2BQ206H. Cells transfected with the empty vector are used as a control. enTDP-43, endogenous TDP-43. Means ± SEM; n = 4. One-way ANOVA with Tukey's HSD post-hoc test; *, P < 0.05; **, P < 0.01. ns, not significant. Source data are available for this figure: SourceData FS5.
The effects of different ALS/FTD-associated CHMP2B mutations on TDP-43 phosphorylation and CK1 levels.(a–d) Representative Western blot images (a) and quantification of TDP-43 phosphorylation levels (b) and protein abundance of CK1α (c) and CK1δ (d) in 293T cells transfected with WT CHMP2B, CHMP2BIntron5, or CHMP2BQ206H. Cells transfected with the empty vector are used as a control. enTDP-43, endogenous TDP-43. Means ± SEM; n = 4. One-way ANOVA with Tukey's HSD post-hoc test; *, P < 0.05; **, P < 0.01. ns, not significant. Source data are available for this figure: SourceData FS5.
CHMP2B regulates CK1 abundance via the UPS-mediated protein turnover
Earlier in this study, we noticed that even a slight OE or KD of CK1α or CK1δ was sufficient to alter pTDP-43 levels (Fig. 4, a–d; and Fig. S4, a and b), suggesting that the protein abundance of CK1 closely controlled TDP-43 phosphorylation levels in cells. It prompted us to test whether CHMP2B affected CK1 protein levels. Indeed, KD of CHMP2B reduced the protein levels of CK1α and CK1δ by ∼50% (Fig. 6, a and b), whereas OE of CHMP2B increased their levels by approximately two- to threefold in 293T cells (Fig. 6, c and d). In contrast, KD of Atg12 or Atg17 did not alter the protein levels of CK1α or CK1δ (Fig. S3, f–h). Furthermore, we confirmed that lentiviral expression of the shRNA of CHMP2B significantly decreased the protein abundance of CK1α and CK1δ in primary mouse cortical neurons (Fig. 6, e and f). We then conducted a pulse-chase assay with cycloheximide to inhibit protein translation to examine the protein turnover rate. We found that KD of CHMP2B accelerated (Fig. 6, g and h), whereas OE of CHMP2B hindered (Fig. 6, i and j) the turnover of CK1α and CK1δ.
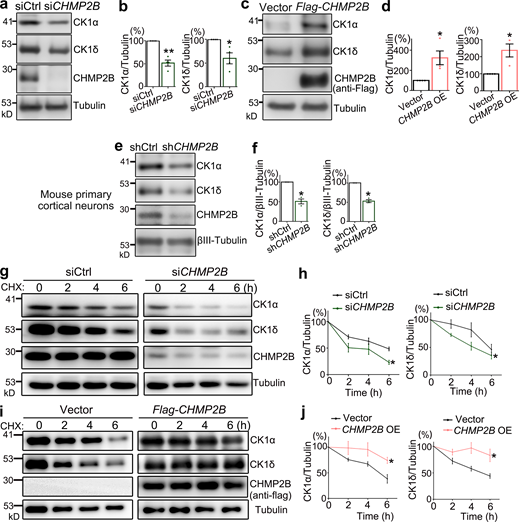
CHMP2B controls the abundance of CK1α and CK1δ by regulating their protein turnover. (a–d) Western blot analyses of the protein levels of CK1α and CK1δ with KD (a and b) or OE (c and d) of CHMP2B in 293T cells. (e and f) Lentiviral shRNA KD of CHMP2B decreases CK1α and CK1δ levels in mouse primary cortical neurons. (g–j) The pulse-chase assay to examine the protein turnover rates of CK1α and CK1δ with KD (g and h) or OE (i and j) of CHMP2B. All proteins are normalized to tubulin, and the relative level at 0 h is set to 100%. Mean ± SEM, n = 3–4. Two-sided Student’s t test (b, d, and f) and two-way ANOVA with Bonferroni’s post-hoc test (h and j); *, P < 0.05; **, P < 0.01. Source data are available for this figure: SourceData F6.
CHMP2B controls the abundance of CK1α and CK1δ by regulating their protein turnover. (a–d) Western blot analyses of the protein levels of CK1α and CK1δ with KD (a and b) or OE (c and d) of CHMP2B in 293T cells. (e and f) Lentiviral shRNA KD of CHMP2B decreases CK1α and CK1δ levels in mouse primary cortical neurons. (g–j) The pulse-chase assay to examine the protein turnover rates of CK1α and CK1δ with KD (g and h) or OE (i and j) of CHMP2B. All proteins are normalized to tubulin, and the relative level at 0 h is set to 100%. Mean ± SEM, n = 3–4. Two-sided Student’s t test (b, d, and f) and two-way ANOVA with Bonferroni’s post-hoc test (h and j); *, P < 0.05; **, P < 0.01. Source data are available for this figure: SourceData F6.
Next, we determined whether CK1 was degraded mainly through the UPS or the autophagy pathway. Blocking the proteasome-mediated degradation with MG132, but not the autophagy–lysosomal pathway with CQ, significantly slowed down the protein turnover of CK1α and CK1δ (Fig. 7, a–c). This was confirmed with another proteasome inhibitor bortezomib (Fig. 7, d–g). Together, these data indicated that the protein turnover of CK1 was mediated through the UPS, but not the autophagy pathway. This raised the possibility that CHMP2B might generally regulate the function of the proteasomal degradation machinery, thereby affecting the turnover of CK1 nonspecifically. To test this possibility, we conducted an in vitro proteasome activity assay and found that neither KD nor OE of CHMP2B significantly altered the proteasome function in cells (Fig. 7, h and i). These results indicate that manipulation of CHMP2B does not alter the overall proteasomal activity but rather modulates the turnover of CK1 specifically.
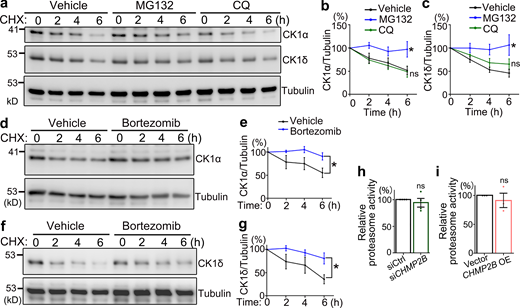
CK1α and CK1δ are degraded through the proteasome-mediated pathway. (a–c) The pulse-chase assay shows that inhibition of the proteasome activity by MG132 (10 µM), but not the autophagy pathway by CQ (10 µM), significantly delays the protein turnover of CK1α and CK1δ. (d–g) Inhibition of the proteasome function by bortezomib (10 µM) also suppresses the turnover of CK1α and CK1δ. CHX, cycloheximide. Vehicle control, DMSO. All proteins are normalized to tubulin and the relative levels at 0 h of each group are set to 100%. (h and i) The effects of KD (h) or OE (i) of WT CHMP2B on the proteasome activity in 293T cells are assessed using an in vitro fluorogenic peptide cleavage assay. The relative proteolytic activities are shown as average percentages to the total fluorescence intensity of the control group at the end of the assay (set to 100%). Means ± SEM; n = 3–4. Two-way ANOVA with Bonferroni’s post-hoc test (b, c, e, and g) and two-tailed Student’s t test (h and i); *, P < 0.05. ns, not significant. Source data are available for this figure: SourceData F7.
CK1α and CK1δ are degraded through the proteasome-mediated pathway. (a–c) The pulse-chase assay shows that inhibition of the proteasome activity by MG132 (10 µM), but not the autophagy pathway by CQ (10 µM), significantly delays the protein turnover of CK1α and CK1δ. (d–g) Inhibition of the proteasome function by bortezomib (10 µM) also suppresses the turnover of CK1α and CK1δ. CHX, cycloheximide. Vehicle control, DMSO. All proteins are normalized to tubulin and the relative levels at 0 h of each group are set to 100%. (h and i) The effects of KD (h) or OE (i) of WT CHMP2B on the proteasome activity in 293T cells are assessed using an in vitro fluorogenic peptide cleavage assay. The relative proteolytic activities are shown as average percentages to the total fluorescence intensity of the control group at the end of the assay (set to 100%). Means ± SEM; n = 3–4. Two-way ANOVA with Bonferroni’s post-hoc test (b, c, e, and g) and two-tailed Student’s t test (h and i); *, P < 0.05. ns, not significant. Source data are available for this figure: SourceData F7.
Protein ubiquitination plays an important role in regulating protein degradation, which can serve as a signal for protein disposal via the UPS pathway (Khaminets et al., 2016; Kirkin et al., 2009). Since CHMP2B did not appear to regulate the overall proteasomal activity in cells, we tested the possibility that CHMP2B might regulate the turnover of CK1 by modulating its ubiquitination levels. Our data showed that KD of CHMP2B increased (Fig. 8, a and b), whereas OE of CHMP2B reduced (Fig. 8, c and d), the ubiquitination levels of CK1α and CK1δ in 293T cells. The same effects were observed when testing KD or OE of CHMP2B in neural N2a cells (Fig. 8, e–h). Together, these data indicate that CHMP2B regulates the ubiquitination and turnover of CK1 via the UPS-dependent degradation pathway.
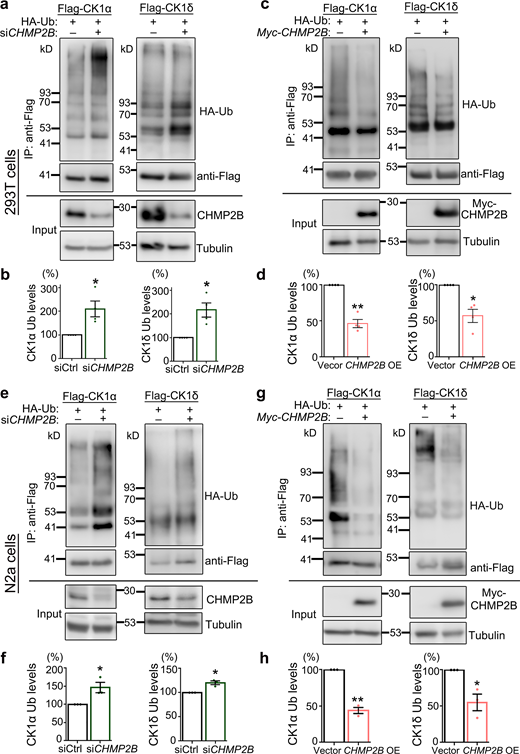
CHMP2B modulates the ubiquitination levels of CK1. (a–h) Representative Western blot images (a and c) and quantifications (b and d) of the impacts of KD (a and b) or OE (c and d) of CHMP2B on the ubiquitination levels of CK1α and CK1δ in 293T cells. The Flag-tagged CK1 proteins are immunoprecipitated using an anti-Flag antibody, and the ubiquitination levels (HA-Ub) are examined by Western blotting. IP, immunoprecipitation. (e–h) The effects of KD (e and f) or OE (g and h) of CHMP2B on the ubiquitination levels of CK1α and CK1δ in N2a cells. Mean ± SEM, n = 3–4. Two-tailed Student’s t test; *, P < 0.05; **, P < 0.01. Source data are available for this figure: SourceData F8.
CHMP2B modulates the ubiquitination levels of CK1. (a–h) Representative Western blot images (a and c) and quantifications (b and d) of the impacts of KD (a and b) or OE (c and d) of CHMP2B on the ubiquitination levels of CK1α and CK1δ in 293T cells. The Flag-tagged CK1 proteins are immunoprecipitated using an anti-Flag antibody, and the ubiquitination levels (HA-Ub) are examined by Western blotting. IP, immunoprecipitation. (e–h) The effects of KD (e and f) or OE (g and h) of CHMP2B on the ubiquitination levels of CK1α and CK1δ in N2a cells. Mean ± SEM, n = 3–4. Two-tailed Student’s t test; *, P < 0.05; **, P < 0.01. Source data are available for this figure: SourceData F8.
Discussion
Rare mutations in CHMP2B are associated with ALS and FTD (Skibinski et al., 2005; Parkinson et al., 2006; Cox et al., 2010). Previous studies of CHMP2B mutations in cell and animal models point to a major deficit in the autophagy–endolysosomal pathway (Lee et al., 2007; van der Zee et al., 2008; Ghazi-Noori et al., 2012; Clayton et al., 2015; Vernay et al., 2016). In this study, we uncover a previously unknown function of CHMP2B in regulating TDP-43 phosphorylation. Interestingly, we show that genetic and pharmacological inhibition of autophagy does not alter TDP-43 phosphorylation levels in flies or mammalian cells, suggesting that the function of CHMP2B in regulating TDP-43 phosphorylation is independent of its well-known function in the autophagy–endolysosomal pathway.
Another line of evidence supporting the independent, dual functions of CHMP2B in regulating autophagy and TDP-43 phosphorylation comes from the study of the disease-causing CHMP2B mutations. It is noted that OE of WT CHMP2B causes a much greater increase of TDP-43 phosphorylation than that of CHMP2BIntron5, while the latter causes a much stronger cytotoxicity and impediment of autophagy than WT CHMP2B. It appears that CHMP2BIntron5 is hypermorphic or dominant mutation with regard to the function in autophagy but is a hypomorph in regulating TDP-43 phosphorylation (Fig. 9), which may underlie the lack of TDP-43 pathology in CHMP2BIntron5-linked FTD. In contrast, CHMP2BQ206H, an ALS-causing CHMP2B mutation that is associated with TDP-43 pathology and regarded as a LOF in autophagy (Cox et al., 2010), exhibits normal capability of increasing CK1 and TDP-43 phosphorylation levels. Thus, the functions of CHMP2B in regulating autophagy and TDP-43 phosphorylation can be disjointedly affected; a mutation in CHMP2B can make the allele a hypermorph or LOF for one function (autophagy) but a hypomorph or WT-like for the other (TDP-43 phosphorylation).

A schematic model of the dual functions of CHMP2B in regulating autophagy and TDP-43 phosphorylation. (a) A normal level and function of CHMP2B are required to maintain the autophagy function and suppress ubiquitination and protein turnover of the kinase CK1 that phosphorylates TDP-43. (b) KD of CHMP2B releases the suppression of CK1 ubiquitination, which promotes CK1 turnover, leading to decreased CK1 abundance and reduced TDP-43 phosphorylation levels. Meanwhile, down-regulation of CHMP2B causes autophagy impediment, leading to the cytotoxicity. (c) OE of CHMP2B also impairs the autophagy pathway. At the same time, it enhances the suppression of CK1 ubiquitination, which reduces CK1 degradation, increases CK1 levels, and promotes TDP-43 hyperphosphorylation. In this case, both impaired autophagy and increased TDP-43 phosphorylation contribute to the cytotoxicity. (d) The FTD-3–associated CHMP2BIntron5 is a hypermorphic or dominant mutation that manifests strikingly more severe autophagy dysfunction and cytotoxicity than WT CHMP2B but is a hypomorph with regard to the function of CHMP2B in regulating TDP-43 phosphorylation. Together, CHMP2B plays the dual functions in regulating autophagy and TDP-43 phosphorylation, and the two functions may be executed independently through different mechanisms.
A schematic model of the dual functions of CHMP2B in regulating autophagy and TDP-43 phosphorylation. (a) A normal level and function of CHMP2B are required to maintain the autophagy function and suppress ubiquitination and protein turnover of the kinase CK1 that phosphorylates TDP-43. (b) KD of CHMP2B releases the suppression of CK1 ubiquitination, which promotes CK1 turnover, leading to decreased CK1 abundance and reduced TDP-43 phosphorylation levels. Meanwhile, down-regulation of CHMP2B causes autophagy impediment, leading to the cytotoxicity. (c) OE of CHMP2B also impairs the autophagy pathway. At the same time, it enhances the suppression of CK1 ubiquitination, which reduces CK1 degradation, increases CK1 levels, and promotes TDP-43 hyperphosphorylation. In this case, both impaired autophagy and increased TDP-43 phosphorylation contribute to the cytotoxicity. (d) The FTD-3–associated CHMP2BIntron5 is a hypermorphic or dominant mutation that manifests strikingly more severe autophagy dysfunction and cytotoxicity than WT CHMP2B but is a hypomorph with regard to the function of CHMP2B in regulating TDP-43 phosphorylation. Together, CHMP2B plays the dual functions in regulating autophagy and TDP-43 phosphorylation, and the two functions may be executed independently through different mechanisms.
Although protein inclusions containing hyperphosphorylated TDP-43 are associated with ALS and FTD, there has been controversy whether phosphorylation of TDP-43 promotes toxicity or is only a consequence of TDP-43 deposition or even protective (Liachko et al., 2010; Li et al., 2011). In this study, we show that TDP-43 exhibits remarkable neurotoxicity and phosphorylation in fly neurons but is unassociated with significant insolubility, indicating that the phosphorylation levels and aggregation of TDP-43 are not always correlated. Furthermore, we demonstrate in mammalian 293T and N2a cells that decrease of TDP-43 phosphorylation levels by inhibition of CK1 with siRNA or small-molecule D4776 markedly suppresses TDP-43– and CHMP2B-induced cytotoxicity. Together, our data lend support to the view that TDP-43 phosphorylation is toxic and pathogenic and that inhibition of TDP-43 phosphorylation may be a druggable target for treating ALS and FTD.
Finally, we show that CHMP2B negatively regulates ubiquitination and the turnover of CK1 through the UPS-mediated pathway. It is not completely surprising that the ESCRT complex also functions in regulating protein ubiquitination. For example, CHMP5, another component of the ESCRT-III complex, was shown to interact with the deubiquitinase USP8 and regulate protein ubiquitination in immune cells (Adoro et al., 2017; Son et al., 2019). It will be interesting to investigate how CHMP2B regulates CK1 ubiquitination in the future. Nevertheless, our data indicate that CHMP2B suppresses ubiquitination and degradation of CK1, which regulates the protein abundance of CK1 and subsequently affects the phosphorylation levels of TDP-43. Collectively, our findings propose a model of the dual functions of CHMP2B in regulating autophagy and TDP-43 phosphorylation, which may act through different mechanisms and be distinctly affected by the same disease-causing mutation (Fig. 9).
Materials and methods
Drosophila strains
The following strains were obtained from the Bloomington Drosophila Stock Center: UAS-lacZ (#8529, a control for UAS transgene expression), RNAi-mCherry (#35785, a control for short hairpin RNAi KD), RNAi-luciferase (#31603, a control for long hairpin RNAi KD), elavGS (#43642), RNAi-CHMP2B (#28531), UAS-RNAi-Atg12 (#34675), and UAS-RNAi-Atg17 (#36918). The RNAi-CHMP2B (#38375) strain was obtained from the Tsinghua Fly Center. For the long hairpin RNAi line of CHMP2B (#28531), a copy of UAS-Dcr2 was coexpressed to boost the KD efficiency (Ni et al., 2008).
The transgenic fly carrying the human CHMP2BIntron5 mutation (UAS-Flag-CHMP2BIntron5) was generated by ΦC31 integrase–mediated, site-specific integration, and the attP landing site stock used was UAS-phi2b2a;VK5 (75B1). The UAS-hTDP-43 flies (Sun et al., 2018) and the UAS-luciferase fly (Cao et al., 2017) were similarly generated using the same landing site, and the latter was used as a control.
Flies tested in this study were raised on standard cornmeal media and maintained at 25°C and 60% relative humidity. For adult onset, neuronal expression of the UAS, or RNAi transgenes using the elavGS driver (Osterwalder et al., 2001), flies were raised on regular fly food supplemented with 80 µg/ml RU486 (Tokyo Chemical Industry; M8046).
Fly eye degeneration, lifespan, and climbing assays
The severity of the eye degeneration was assessed by rough surface, swelling, and loss of pigment cells of the compound eyes. Each fly eye was single-blindly scored in a scale of 0 to 5, with 0 indicating no degeneration and 5 indicating full degeneration.
For the lifespan experiment, 20 flies per vial and 6–10 vials per group were tested. Flies were transferred to fresh fly food every 3 d, and the number of dead flies of each vial was recorded. Flies lost before natural death through escape or accidental death were excluded from the final analysis. The median lifespan was calculated as the mean of the medians of each vial belonging to the same group, whereas the “50% survival” shown on the survival curves was derived from compilation of all vials of the group.
For the climbing assay, 20 flies were transferred into an empty polystyrene vial and gently tapped down to the bottom of the vial. The number of flies that climbed over a distance of 3 cm within 10 s was recorded. The test was repeated three times for each vial, and five to eight vials of each genotype were assessed.
RNA extraction and real-time qPCR
For qPCR, total RNA was isolated from fly heads or cell culture using TRIzol (Invitrogen; 15596018) according to the manufacturer’s instruction. After DNase (Promega; M6101) treatment to remove genomic DNA, the reverse transcription reactions were performed using All-in-One cDNA Synthesis SuperMix kit (Bimake; B24403). The cDNA was then used for real-time qPCR using 2x SYBR Select Master Mix (Thermo Fisher; 4472908) with the QuantStudio 6 Flex Real-Time PCR system (Life Technologies). The mRNA levels of actin were used as an internal control to normalize the mRNA levels of genes of interest. The following qPCR primers were used in this study: dActin-forward (F), 5′-GAGCGCGGTTACTCTTTCAC-3′; dActin-reverse (R), 5′-GCCATCTCCTGCTCAAAGTC-3′; dCHMP2B-F: 5′-GAAAGAAACCCACCGTGAAG-3′; dCHMP2B-R: 5′-TCCTCCTCCTCCATTTTCCT-3′.
Plasmids and siRNAs
The pCAG-hTDP-43-HA plasmid was described previously (Sun et al., 2018). In brief, DNA sequence encoding TDP-43 was amplified from a pcDNA3.1-TDP-43-myc plasmid (Jiang et al., 2013) by PCR using the primers containing the HA tag sequence. The PCR product was then subcloned into a pCAG vector (Chen et al., 2014) using the XhoI–EcoRI sites. The pcDNA3.1-HA-Ub plasmid was a kind gift from Z. Zhang (Chinese Academy of Sciences, Shanghai, China; Shi et al., 2019). To generate pCAG-Flag-CHMP2B, pCAG-Flag-CK1α, pCAG-Flag-CK1δ, pCAG-Flag-TTBK1, and pCAG-Flag-TTBK2 plasmids, DNA fragments encoding human CHMP2B, CK1α, CK1δ, TTBK1, and TTBK2 were amplified from pCMV3-Flag-CHMP2B (Sino Biological; #HG14596-NF) or cDNA from 293T or SH-SY5Y cells by PCR using the primers containing the Flag tag sequence. The PCR products were then subcloned into a pCAG vector (Chen et al., 2014) using the XhoI–EcoRI sites. The following PCR primers were used: Flag-CHMP2B-F, 5′-CATCATTTTGGCAAAGAATTCGCCACCATGGATTACAAGGAT-3′; Flag-CHMP2B-R, 5′-GCTCCCCGGGGGTACCTCGAGTTAATCTACTCCTAA-3′; Flag-CK1α-F, 5′-CATCATTTTGGCAAAGAATTCGCCACCATGGATTACAAGGATGACGACGATAAGATGGCGAGTAGCAGC-3′; Flag-CK1α-R, 5′-GCTCCCCGGGGGTACCTCGAGTTAGAAACCTTTCATGTTAC-3′; Flag-CK1δ-F, 5′-CATCATTTTGGCAAAGAATTCGCCACCATGGATTACAAGGATGACGACGATAAGATGGAGCTGAGAGTC-3′; Flag-CK1δ-R, 5′-GCTCCCCGGGGGTACCTCGAGTCATCGGTGCACGAC-3′; Flag-TTBK1-F, 5′-CATCATTTTGGCAAAGAATTCGCCACCATGGATTACAAGGATGACGACGATAAGATGCAGTGCCTAGCGGCCGC-3′; Flag-TTBK1-R, 5′-TGAGCTCCCCGGGGGTACCTCGAGTTATCTGGCCCCAGCCCGGC-3′; Flag-TTBK2-F, 5′-ATTTTGGCAAAGAATTCGCCACCATGGATTACAAGGATGACGACGATAAGATGAGTGGGGGAGGAGAGC-3′; Flag-TTBK2-R, 5′-GAGCTCCCCGGGGGTACCTCGAGCTATCTGCTGAGTTTACTGG-3′.
The pCAG-Flag-CHMP2BIntron5, pCAG-Flag-CHMP2BQ206H, and pBID-UASC-Flag-CHMP2BIntron5 plasmids were generated by homologous recombination. Briefly, the DNA fragment of Flag-CHMP2BIntron5 or Flag-CHMP2BQ206H was amplified by PCR and inserted into the cloning vector using ClonExpress MultiS One Step Cloning Kit (Vazyme; C113). The construct was then subcloned into the pCAG or pBID-UASC vector as above. The following PCR primers were used in this study: Flag-CHMP2BIntron5-F, 5′-CATCATTTTGGCAAAGAATTCGCCACCATGGATTACAAGGAT-3′; Flag-CHMP2BIntron5-R, 5′-GCTCCCCGGGGGTACCTCGAGTTACACCTTTCCAGA-3′; Flag-CHMP2BQ206H-F, 5′-GAGATTGAACGGCACCTCAAGGCTTTAGGAGTAGATTAACTCG-3′; Flag-CHMP2BQ206H-R, 5′-CCTAAAGCCTTGAGGTGCCGTTCAATCTCTTCATCTGAG-3′.
To generate the pLKO-shRNA-CHMP2B plasmid, the following sense and antisense oligos (GenePharma) of the mouse CHMP2B were annealed and then ligated with the digested pLKO.1 vector using the T4 DNA ligase (New England Biolabs; M0202S): sh-CHMP2B-F, 5′-CCGGGCCTTAAATAGCACGAACATACTCGAGTATGTTCGTGCTATTTAAGGCTTTTTG-3′; sh-CHMP2B-R, 5′-AATTCAAAAAGCCTTAAATAGCACGAACATACTCGAGTATGTTCGTGCTATTTAAGGC-3′.
The negative control vector containing scrambled shRNA was obtained from Addgene (1864), and the sequence of the hairpin was 5′-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′.
The siRNA oligonucleotides to CHMP2B, CK1α, CK1δ, TTBK1 and TTBK2 were purchased from GenePharma. A mixture of three independent siRNAs (1:1:1) was used for each targeted gene, and the sequences are as follows: siCtrl (scrambled siRNA), 5′-ACGUGACACGUUCGGAGAA-3′; si-hCHMP2B#1, 5′-UUUAUUACAUCAUCCACGG-3′; si-hCHMP2B#2, 5′-AGAUGCACAAGUUGUUUGG-3′; si-hCHMP2B#3, 5′-CAGAUGGUAAGCUUCGAGC-3′; si-CK1α#1, 5′-UUCUACUGAUCAUCUGGUC-3′; si-CK1α#2, 5′-UAACUGGUUUAAUCCUGAG-3′; si-CK1α#3, 5′-UUUCUGCUUUAACAUUGUC-3′; si-CK1δ#1, 5′-CAUUUGGUCAGCAAGCAGC-3′; si-CK1δ#2, 5′-UUGUUCAAUUCCAAGGUGC-3′; si-CK1δ#3, 5′-AUUUCUGUCUCUUGGUGGC-3′; si-TTBK1#1, 5′-AAUGAACCUGCACACAUGG-3′; si-TTBK1#2, 5′-UUGCCCAGCCGCAAUGUGG-3′; si-TTBK1#3, 5′-AUGAUCAACUGGUAGUCGG-3′; si-TTBK2#1, 5′-AAUUGGUAAAUUGUCGAGC-3′; si-TTBK2#2, 5′-AUCAUUUCCAGUCUUCUCC-3′; si-TTBK2#3, 5′-AAGAGCUCAGGUUAACAGC-3′; si-Atg12#1, 5′-GUUGCAGCUUCCUACUUCA-3′; si-Atg12#2, 5′-CUGGCUGAAUACCUCAAAU-3′; si-Atg12#3, 5′-GAGACUAAGACUGUAUAAA-3′; si-Atg17#1, 5′-GGAGGACUGUUCAAAUUCA-3′; si-Atg17#2, 5′-CGACCAUUUAUAGCAGAAU-3′; si-Atg17#3, 5′-CAGCUUGCAUUGGAAAUGU-3′; si-mCHMP2B#1, 5′-GGACGAUGUCAUAAAGGAA-3′; si-mCHMP2B#2, 5′-CCACAGAAGACACUACAAA-3′; si-mCHMP2B#3, 5′-GGGAUUGAAAUCUCUGGAA-3′.
Cell culture and transfection
293T (American Type Culture Collection; CRL-11268) and N2a (American Type Culture Collection; CCL-131) cells were cultured in DMEM (Sigma-Aldrich; D0819) supplemented with 10% (vol/vol) FBS (Biowest; S1710) and GlutaMAX (Invitrogen; 35050061) at 37°C in 5% CO2. Transient transfection of siRNA oligonucleotides or plasmids was performed using Lipofectamine RNAiMAX (Invitrogen; 13778075) in Opti-MEM (Invitrogen; 11058021) or the In Vitro DNA Transfection Reagent PolyJet (SignaGen Laboratories; SL100688) in DMEM (Sigma-Aldrich; D0819). Cells were transfected with expression plasmids for 48 h or with siRNAs for 72 h before harvest.
Lentivirus production, primary neuron culture, and infection
To generate lentivirus for infecting primary neurons, 293T cells were cotransfected with pLKO-shRNA-CHMP2B, psPAX2, and pMD2.G with a ratio of 4:3:1 in DMEM (Sigma-Aldrich; D0819) using PolyJet (SignaGen Laboratories; SL100688). The supernatant of the cell culture was collected at 48 h after transfection and passed through a 0.45-µm filter (Merck Millipore; SLHVR33RB). Fresh supernatant containing viral particles was used or stored at 4°C for less than 1 wk before infection of the primary neurons.
Primary cortical neurons were isolated from C57BL/6 mouse cortex at embryonic day 16 and cultured in serum-free Neurobasal medium (Invitrogen; 21103049) supplemented with 2% B27 (Invitrogen; 17504044), GlutaMAX (Invitrogen; 35050061), and penicillin–streptomycin (Invitrogen; 10378016). At 5 d in vitro, neurons were infected with pLKO-shRNA-CHMP2B for 7 d before harvest.
Pharmacological experiments
For the pulse-chase assay, cycloheximide (Sigma-Aldrich; 01810) was added into the culture medium at a final concentration of 25 ng/ml. For inhibition of the proteasome or the autophagy–lysosome pathway, MG132 (Beyotime; S1748) or bortezomib (Selleck; S1013) was added at a final concentration of 10 µM, Rapa (Selleck; S1039) was added at a final concentration of 100 nM, and CQ (Sigma-Aldrich; C6628) was added at a final concentration of 10 µM. For CK1 inhibition, D4776 (Selleck; S7642) was added at a final concentration of 5 µM. The cells were treated with the above drugs for indicated durations and then harvested for the subsequent Western blotting.
Cell viability analysis and PI staining assay
293T cells were plated in 24-well plates at a density of 200,000 cells/ml and transfected with siRNAs or plasmids 12–24 h later. 6–8 h after transfection, the cells were seeded at 10,000 cells/well into 96-well plates at a volume of 100 µl/well. Cell viability was assessed by measuring the reduction of WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2, 4-disulfophenyl)-2H-tetrazolium) into formazan using the CCK8 assay (Dojindo; CK04) by adding 10 µl of CCK8 solution to the cells at specific time points. Thereafter, the cells were incubated for 2 h at 37°C before measurement of the OD values at 450 nm. Cell viability was quantified according to the manufacturer’s instruction. PI staining to detect cell death was performed using the PI Staining Kit (Sangon Biotech; E607306) at 1 µl/well of a 24-well plate at 37°C for 20 min according to the manufacturer’s instruction.
Microscopic image acquisition
For PI staining, cells were imaged within the 24-well plate using a Leica DMi8 inverted live-cell microscopy system with a 20×/0.7 NA dry objective at room temperature. Images were captured using a 1.4 MP monochrome digital camera (Leica; DFC310FX) and processed using the Leica Application Suite X software. The images of the fly eyes were captured using an Olympus SZX16 stereomicroscope equipped with a 5 MP CCD color digital camera (Olympus; DP26-CU) at room temperature. The focus stacking was acquired and processed using Helicon Focus 6 and the cellSens Dimension software (Olympus). All images were assembled into figures using Adobe Photoshop CS6.
Antibodies
The following antibodies were used in Western blotting, immunoprecipitation, and immunofluorescence assays: rabbit anti–phospho-TDP-43 (Ser409/Ser410; Sigma-Aldrich; SAB4200225), mouse anti–pTDP-43 (pSer409/Ser410; COSMO BIO CO, TIP-PTD-M01), mouse anti-FLAG (Sigma-Aldrich; F3165), rabbit anti-Flag (Proteintech; 20543–1-AP), mouse anti-HA (Proteintech; 66006–1), rabbit anti-HA (Cell Signaling Technology; 3724), rabbit anti–TDP-43 (Proteintech; 10782–2-AP), mouse anti–TDP-43 (Abcam; ab57105), mouse anti-P62/SQSTM1 (Proteintech; 66184–1-Ig), rabbit anti-LC3B (Abcam; ab48394), rabbit anti-ATG8A/GABARAP (Abcam; ab109364), rabbit anti-Ref(2)P (Abcam; ab178440), rabbit anti-CK1α (Proteintech; 55192–1-AP), rabbit anti-CK1δ (Proteintech; 14388–1-AP), mouse anti-GAPDH (Proteintech; 60004–1), rabbit anti-tubulin (MBL; PM054), mouse anti–β-actin (Cell Signaling Technology; 3700), and rabbit anti–βIII-tubulin (Sigma-Aldrich; T2200). HRP-conjugated secondary antibodies included anti-mouse (Sigma-Aldrich; A4416) and anti-rabbit IgG (Sigma-Aldrich; A9169).
Protein extraction and Western blotting
Fly heads or cultured cells were lysed in 2% SDS lysis buffer (100 mM Tris-HCl, pH 6.8, 2% SDS, 40% glycerol, 10% β-mercaptoethanol, and 0.04% bromophenol blue) or tissue extraction reagent I (50 mM Tris, pH 7.4, 250 mM NaCl, 5 mM EDTA, 2 mM Na3VO4, 1 mM NaF, 20 mM Na4P2O7, and 0.02% NaN3) containing protease (Roche; 5892791001) and phosphatase inhibitor cocktails (Roche; 04693132001).
All protein samples were boiled at 95°C for 5 min. Equal amounts of lysates were resolved by electrophoresis using a 10% Bis-Tris SDS-PAGE (Invitrogen; NP0303BOX). The 0.45-µm Immobilon-P polyvinylidene fluoride transfer membrane for Western blotting (Merck Millipore; IPVH00010) were used, which were then probed with the primary and secondary antibodies listed above. Detection was performed using the High-sig ECL Western Blotting Substrate (Tanon; e168230). Images were captured using an Amersham Imager 600 (GE Healthcare) and the densitometry was measured using ImageQuant TL Software (GE Healthcare) and ImageJ. The contrast and brightness were optimized equally in Adobe Photoshop CS6. Tubulin, GAPDH, or β-actin was used as a loading control for normalization as indicated in the figures. The loading control is selected mainly according to the molecular weight of the proteins to be detected, the origin of each antibody, and the source of the protein samples.
Immunoprecipitation
293T cells were lysed in an IP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, and 5% glycerol) containing protease inhibitor cocktails (Roche; 5892791001) and N-ethylmaleimide (Sigma-Aldrich; E3876). Following centrifugation at 15,000 × g for 15 min at 4°C, the supernatants were collected in new vials and incubated with mouse anti-Flag beads on a rotary shaker at 4°C overnight. The beads were then collected and eluted in the 2X SDS buffer for the subsequent Western blotting assay.
In vitro proteasome activity assay
Proteasomal activity was measured using a proteasome activity assay kit (Abcam; ab107921). 293T cells were transfected with indicated plasmids for 48 h and siRNA for 72 h, followed by trypsin digestion and quantification. The cells were subsequently lysed in 90 µl lysis buffer (0.5% NP-40 in PBS), and the lysates were centrifuged at 16,000 × g for 15 min at 4°C; then, the supernatants were collected in new vials. The proteasomal activity of the supernatants was determined by assaying the cleavage of a fluorogenic peptide substrate Suc-LLVY-AMC according to the manufacturer’s instruction. The substrate peptides were incubated with the cell lysates at 37°C for 1 h, and the fluorescence intensity was measured at the end of the assay using a BioTek microplate reader (excitation/emission = 350/440 nm).
Statistical analysis
Statistical significance in this study is determined by one-way ANOVA with Tukey's honestly significant difference (HSD) post-hoc test, two-way ANOVA with Bonferroni’s post-hoc test, or unpaired, two-tailed Student’s t test with equal variance (*, P < 0.05; **, P < 0.01; ***, P < 0.001) as indicated. Error bars represent the SEM. Data distribution was assumed to be normal, but this was not formally tested.
Online supplemental material
Fig. S1 shows the effects of CHMP2B KD by two independent transgenic RNAi strains (#38375 and #28531) in flies. Fig. S2 demonstrates that OE of CHMP2BIntron5 increases the phosphorylation levels but does not induce significant insolubility of hTDP-43 in flies. Fig. S3 indicates that down-regulation of Atg12 or Atg17 suppresses TDP-43–induced cytotoxicity but does not alter pTDP-43 and CK1 levels in 293T cells. Fig. S4 displays the dose-dependent effect of KD or OE of CK1 on the cell viability and confirms the KD efficiency of si-TTBK1/2 in 293T cells. Fig. S5 shows the effects of different ALS/FTD-associated CHMP2B mutations on TDP-43 phosphorylation and CK1 levels.
Acknowledgments
We thank the Bloomington Drosophila Stock Center and Tsinghua Fly Center for providing the fly strains, J. Yuan for careful proofreading, Z. Zhang (Chinese Academy of Sciences) for the pcDNA3.1-HA-Ub plasmid, S. Zhang for technical support, and members of the Fang laboratory for helpful discussion.
This work is supported by the National Natural Science Foundation of China (grant 31970697), the Science and Technology Commission of Shanghai Municipality (grants 2019SHZDZX02, 20490712600, and 201409003300; to Y. Fang), the National Natural Science Foundation of China (grant 82071372), Natural Science Foundation of Guangdong Province of China (grant 2021A1515011231), the Outstanding Scholar Program of Bioland Laboratory (Guangzhou Regenerative Medicine and Health Guangdong Laboratory; grant 2018GZR110102002), the Science and Technology Program of Guangzhou (grant 202007030012), and the Guangdong Key Laboratory of Non-human Primate Research (grant 2020B121201006; to A. Li).
The authors declare no competing financial interests.
Author contributions: X. Sun, A. Li, and Y. Fang conceived the research; X. Deng, X. Sun, A. Li, and Y. Fang designed the experiments; X. Deng, X. Sun, W. Yue, Y. Duan, R. Hu, J. Ni, J. Cui, and Q. Wang performed the experiments; X. Deng, X. Sun, W. Yue, Y. Duan, R. Hu, and Y. Chen contributed important new reagents; X. Deng, X. Sun, W. Yue, K. Zhang, and Y. Fang analyzed the data and interpreted the results; X. Deng, X. Sun, and Y. Fang prepared the figures; and X. Deng, X. Sun, A. Li, and Y. Fang wrote the paper. All authors read and approved the final manuscript.

