


The primary cilium is a signaling compartment that interprets Hedgehog signals through changes of its protein, lipid, and second messenger compositions. Here, we combine proximity labeling of cilia with quantitative mass spectrometry to unbiasedly profile the time-dependent alterations of the ciliary proteome in response to Hedgehog. This approach correctly identifies the three factors known to undergo Hedgehog-regulated ciliary redistribution and reveals two such additional proteins. First, we find that a regulatory subunit of the cAMP-dependent protein kinase (PKA) rapidly exits cilia together with the G protein–coupled receptor GPR161 in response to Hedgehog, and we propose that the GPR161/PKA module senses and amplifies cAMP signals to modulate ciliary PKA activity. Second, we identify the phosphatase Paladin as a cell type–specific regulator of Hedgehog signaling that enters primary cilia upon pathway activation. The broad applicability of quantitative ciliary proteome profiling promises a rapid characterization of ciliopathies and their underlying signaling malfunctions.
Introduction
The primary cilium is a solitary, microtubule-based protrusion of the cell that organizes developmental, sensory, and homeostatic signaling pathways inside a functionally distinct compartment. Cilia defects cause multisystem pathologies, named ciliopathies, with symptoms including kidney cysts, retinal degeneration, obesity, brain malformations, and skeletal anomalies (Reiter and Leroux, 2017; Hildebrandt et al., 2011). The vast range of symptoms underscores the broad physiological importance of cilium-based signaling. Our understanding of how cilia transduce signals is based in large part on studies of the developmental morphogen Hedgehog (Hh; Gigante and Caspary, 2020; Anvarian et al., 2019; Kong et al., 2019; Tschaikner et al., 2020). In vertebrates, Hh signaling is strictly dependent on an intact primary cilium. The core Hh machinery comprises the Hh receptor Patched 1 (PTCH1), the G protein–coupled receptors (GPCRs) GPR161 and Smoothened (SMO), and the glioma-associated oncogene (GLI) transcription factors, all of which dynamically localize to primary cilia in response to Hh (Fig. 1 A). PTCH1 and GPR161, two molecules that restrain Hh pathway activation inside cilia in unstimulated cells, undergo ciliary exit upon pathway stimulation, while the central pathway activator SMO becomes enriched inside cilia when activated. It has been proposed that PTCH1 pumps a lipidic activator of SMO out of the ciliary membrane and that the PTCH1 lipid extruder activity is directly suppressed upon liganding Hh. Ciliary exit of PTCH1 further reduces the inhibitory effect exerted by PTCH1 on SMO. Downstream of SMO and GPR161 lies cAMP-dependent protein kinase (PKA), which phosphorylates GLI2 and GLI3 and commits them to processing into transcriptional repressors. Despite the recognized importance of dynamic ciliary localization in the Hh response, the extent of ciliary proteome remodeling during Hh signaling remains unknown, and key steps, such as how ciliary SMO triggers the exit of GPR161 from cilia, remain incompletely characterized.
An unbiased, systematic description of the mammalian primary cilia proteome has proven challenging because the isolation of mammalian cilia remains fraught with severe limitations (Ishikawa et al., 2012). To overcome this technical challenge, we previously fused the ascorbate peroxidase (APEX) to the ciliary targeting signal of NPHP3 (Wright et al., 2011; Nakata et al., 2012) and established Cilia-APEX, a proximity labeling–based method for cilium proteomics (Mick et al., 2015; Fig. 1 B). Cilia-targeted APEX fusions enable biotinylation and straightforward purification of ciliary proteins by streptavidin capture (Mick et al., 2015; Kohli et al., 2017). Proteomics of cilia using Cilia-APEX has contributed to our molecular understanding of Hh signaling (Mick et al., 2015), extracellular vesicles shedding from cilia (Nager et al., 2017), and regulated ciliary GPCR trafficking (Shinde et al., 2020). While proximity labeling approaches in cilia have identified several ciliary signaling proteins (Mick et al., 2015; Kohli et al., 2017), they have thus far failed to robustly detect and quantify important signaling proteins of low abundance, such as PTCH1, GPR161, and SMO. The scarcity of these factors in cilia combined with the high hydrophobicity of these multipass membrane proteins pose a considerable challenge for quantitative mass spectrometric analysis.
Here, we profile the changes of the ciliary proteome during Hh signaling in a systematic and time-resolved manner by combining improvements in the Cilia-APEX2 proximity labeling scheme with state-of-the-art quantitative mass spectrometry (MS) using tandem mass tags (TMTs; Paek et al., 2017). These technical advances enabled us to uncover the extent of the ciliary proteome remodeling in response to Hh ligand and gain novel mechanistic insights into Hh signaling.
Results
The Cilia-APEX2 expression system limits perturbations of ciliary dynamics
To investigate why Cilia-APEX had failed to identify ciliary membrane proteins of the Hh pathway, we investigated the subcellular localization of Hh signaling molecules in the inner medullary collecting duct 3 (IMCD3) cell line stably expressing Cilia-APEX by fluorescence microscopy. While endogenous GPR161 was readily detected in cilia of IMCD3 cells, ciliary GPR161 was nearly undetectable in IMCD3-[Cilia-APEX] cells (Fig. 1, C and D). Given that a similar expression system produced ∼50,000 molecules per cilium and led to a drastic lengthening of cilia (Ye et al., 2018), we suspected that the levels of Cilia-APEX required to support efficient biotinylation may have altered ciliary composition. We thus reduced expression levels of the transgene by switching to a considerably weaker promoter (CMV(Δ6); Morita et al., 2012; Ye et al., 2018) and leveraged APEX2, an APEX variant with improved labeling properties (Lam et al., 2015), to compensate for decreased Cilia-APEX expression (Fig. 1 B). Comparing the relative abundance of Cilia-APEX and Cilia-APEX2 via GFP immunoblotting revealed that Cilia-APEX2 was nearly fivefold less abundant than Cilia-APEX (Fig. 1, E and F), while the abundance of the ciliary protein IFT88 remained unchanged between the two cell lines (Figs. 1 E and S1 A). Congruently, the ciliary intensity of Cilia-APEX2 was reduced approximately fivefold compared with Cilia-APEX (Fig. 1, G and H). Despite the reduction in ciliary abundance of Cilia-APEX2 compared with Cilia-APEX, ciliary biotinylation efficiency in the presence of the APEX substrates biotin tyramide and H2O2 nearly doubled in Cilia-APEX2 compared with Cilia-APEX (Fig. 1, G and I). Importantly, expression of Cilia-APEX2 did not alter the ciliary abundance or Sonic Hh (Shh)–dependent dynamics of GPR161 (Fig. 1, C and D). Together, these results predict that Cilia-APEX2 improves sensitivity of ciliary proteomics while minimizing perturbations of ciliary protein dynamics.

Modest C ilia-APEX2 expression enables labeling of the ciliary contents without disturbing the ciliary localization of Hh signaling components. (A) Diagram of Hh signaling. Positive and negative regulators are in green and red boxes, respectively. Pharmacological agents are in oval circles and proteins in rectangles. Acronyms are defined in the text. (B) Diagrams of the Cilia-APEX and Cilia-APEX2 expression cassettes. Numbers indicate amino acid positions. The N terminus of NPHP3 is myristoylated at glycine 2 (Myr) and confers ciliary targeting. The truncated CMV promoter (pCMV(Δ6)) is considerably weaker than the EF1α promoter (pEF1α). (C) IMCD3 cells and stable clones expressing Cilia-APEX or Cilia-APEX2 were serum-starved for 24 h in the presence or absence of Shh before fixation and staining for GPR161 (white) and ARL13B (red). Cilia-APEX and Cilia-APEX2 were visualized via the intrinsic fluorescence of GFP (green). DNA in blue. (D) Box plots showing the relative GPR161 fluorescence normalized to ARL13B in the primary cilium of IMCD3, Cilia-APEX, and Cilia-APEX2 cell lines after Shh treatment as in A. n = 50 cilia per condition. In these and all subsequent box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers. RFU, relative fluorescence unit. (E) IMCD3, Cilia-APEX, and Cilia-APEX2 cell lines were immunoblotted for GFP (APEX fusions), IFT88, and actin. (F) Signals from immunoblots as in E were quantified, ratios of GFP signals in the Cilia-APEX relative to the Cilia-APEX2 cell line were calculated, and results plotted. Thick horizontal line represents the mean (n = 3). (G) IMCD3, Cilia-APEX, and Cilia-APEX2 cell lines were subjected to APEX labeling before fixation and staining for ARL13B (red) and biotin (white). The APEX fusion proteins are detected via intrinsic GFP fluorescence (green). DNA in blue. (H and I) Box plots showing background-subtracted intensities of GFP (H) and biotin (I) signals in the primary cilium from images as in G. n = 30 cilia per condition. Scale bars, 2 µm in all panels.
Modest C ilia-APEX2 expression enables labeling of the ciliary contents without disturbing the ciliary localization of Hh signaling components. (A) Diagram of Hh signaling. Positive and negative regulators are in green and red boxes, respectively. Pharmacological agents are in oval circles and proteins in rectangles. Acronyms are defined in the text. (B) Diagrams of the Cilia-APEX and Cilia-APEX2 expression cassettes. Numbers indicate amino acid positions. The N terminus of NPHP3 is myristoylated at glycine 2 (Myr) and confers ciliary targeting. The truncated CMV promoter (pCMV(Δ6)) is considerably weaker than the EF1α promoter (pEF1α). (C) IMCD3 cells and stable clones expressing Cilia-APEX or Cilia-APEX2 were serum-starved for 24 h in the presence or absence of Shh before fixation and staining for GPR161 (white) and ARL13B (red). Cilia-APEX and Cilia-APEX2 were visualized via the intrinsic fluorescence of GFP (green). DNA in blue. (D) Box plots showing the relative GPR161 fluorescence normalized to ARL13B in the primary cilium of IMCD3, Cilia-APEX, and Cilia-APEX2 cell lines after Shh treatment as in A. n = 50 cilia per condition. In these and all subsequent box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers. RFU, relative fluorescence unit. (E) IMCD3, Cilia-APEX, and Cilia-APEX2 cell lines were immunoblotted for GFP (APEX fusions), IFT88, and actin. (F) Signals from immunoblots as in E were quantified, ratios of GFP signals in the Cilia-APEX relative to the Cilia-APEX2 cell line were calculated, and results plotted. Thick horizontal line represents the mean (n = 3). (G) IMCD3, Cilia-APEX, and Cilia-APEX2 cell lines were subjected to APEX labeling before fixation and staining for ARL13B (red) and biotin (white). The APEX fusion proteins are detected via intrinsic GFP fluorescence (green). DNA in blue. (H and I) Box plots showing background-subtracted intensities of GFP (H) and biotin (I) signals in the primary cilium from images as in G. n = 30 cilia per condition. Scale bars, 2 µm in all panels.

A cilia-less cell line expressing Cilia-APEX2 serves as a specificity control for Cilia-APEX2 proteomics. (A) Dot plot showing total protein levels of IFT88 relative to actin in indicated cell lines as determined by quantitative immunoblotting (see Fig. 1 E). Mean values are indicated by horizontal lines (n = 3). (B) The Cep164 gene was sequenced from genomic DNA from Cep164−/− cells and aligned with the Cep164 gene sequence from M. musculus. A homozygous single-base-pair deletion in exon 8 leads to a frameshift mutation and protein truncation. DNA sequences were analyzed using Benchling. (C andD) Immunofluorescence micrographs of WT or CEP164-deficient (Cep164−/−) cell lines stably expressing Cilia-APEX2. Cell lines were serum-starved for 24 h before fixation. Cilia-APEX2 proteins were detected by GFP fluorescence. (C) Ninein marks centrioles and is visualized by antibody staining. (D) γ-Tubulin and CEP164 are detected by specific antibodies (Lau et al., 2012). Scale bars, 2 µm. (E) High reproducibility of Cilia-APEX2 proteomics setup. Hierarchical cluster analysis based on Ward’s minimum variance method (two-way clustering) of the relative abundances of each identified protein (rows) in the individual samples (columns). Relative scaled abundances were calculated by dividing the TMT signal to noise of an individual protein by the sum of TMT signal to noise ratios in all samples. Legend depicts color scheme for relative abundances (percentage). Brackets indicate cilia clusters (red and purple; see Fig. 2 B) and unrelated cluster (orange) without cilia proteins. (F) Zoom into the control/nonciliary cluster (orange) in the hierarchical cluster analysis shown in E.
A cilia-less cell line expressing Cilia-APEX2 serves as a specificity control for Cilia-APEX2 proteomics. (A) Dot plot showing total protein levels of IFT88 relative to actin in indicated cell lines as determined by quantitative immunoblotting (see Fig. 1 E). Mean values are indicated by horizontal lines (n = 3). (B) The Cep164 gene was sequenced from genomic DNA from Cep164−/− cells and aligned with the Cep164 gene sequence from M. musculus. A homozygous single-base-pair deletion in exon 8 leads to a frameshift mutation and protein truncation. DNA sequences were analyzed using Benchling. (C andD) Immunofluorescence micrographs of WT or CEP164-deficient (Cep164−/−) cell lines stably expressing Cilia-APEX2. Cell lines were serum-starved for 24 h before fixation. Cilia-APEX2 proteins were detected by GFP fluorescence. (C) Ninein marks centrioles and is visualized by antibody staining. (D) γ-Tubulin and CEP164 are detected by specific antibodies (Lau et al., 2012). Scale bars, 2 µm. (E) High reproducibility of Cilia-APEX2 proteomics setup. Hierarchical cluster analysis based on Ward’s minimum variance method (two-way clustering) of the relative abundances of each identified protein (rows) in the individual samples (columns). Relative scaled abundances were calculated by dividing the TMT signal to noise of an individual protein by the sum of TMT signal to noise ratios in all samples. Legend depicts color scheme for relative abundances (percentage). Brackets indicate cilia clusters (red and purple; see Fig. 2 B) and unrelated cluster (orange) without cilia proteins. (F) Zoom into the control/nonciliary cluster (orange) in the hierarchical cluster analysis shown in E.
TMT analysis of Cilia-APEX2 samples extend coverage of the ciliary proteome
We introduced further modifications to the Cilia-APEX workflow aimed at improving coverage and specificity of cilium proteomics. Besides proteins biotinylated by Cilia-APEX2 inside cilia, streptavidin capture of Cilia-APEX2 cell lysates recovers proteins biotinylated by Cilia-APEX2 molecules that did not reach the cilium (e.g., biogenesis intermediates), proteins biotinylated by endogenous peroxidases, and endogenously biotinylated proteins. In the past, identification of these nonciliary proteins was assessed with a nonciliary enzyme termed control-APEX2 or by withholding labeling reagents. To further control for nonciliary proteins, we generated a Cilia-APEX2 cell line devoid of cilia by deleting the centriolar distal appendage protein CEP164 (Fig. S1, B–D). Other cellular processes are unaffected by deletion of Cep164 (Daly et al., 2016; Tanos et al., 2013).
We further leveraged TMTs, a recent advance in quantitative mass spectrometry. TMTs are isobaric tags that enable precise and reproducible quantification of relative protein abundance in up to 10 different samples by synchronous precursor selection MS/MS/MS (MS3) analyses (Liu et al., 2020; Paek et al., 2017; Li et al., 2020). Triplicate APEX-labeled samples of WT Cilia-APEX2, Cep164−/− Cilia-APEX2, and control-APEX2 cells as well as a single unlabeled Cilia-APEX2 sample were subjected to streptavidin capture, tryptic digest, and TMT labeling before samples were mixed together and analyzed by MS3 (Fig. 2 A). Hierarchical clustering of each protein’s relative abundance in the 10 samples analyzed within one multiplex experiment demonstrates high reproducibility across the triplicate samples (Fig. S1 E). Proteins that are highly enriched in the Cilia-APEX2 dataset compared with the controls form two clusters of candidate ciliary proteins (Figs. S1 E and 2 B), while nonciliary proteins fall into separate clusters (Fig. S1 F).
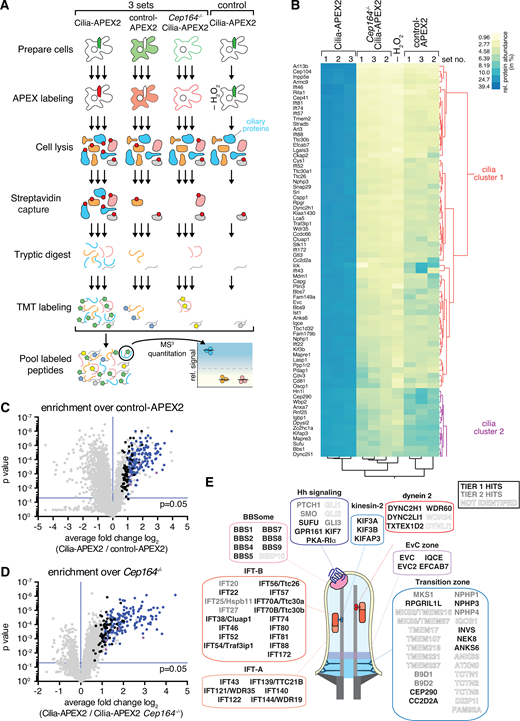
Cilia-APEX2-based deep proteomics. (A) Workflow of a Cilia-APEX2/TMT experiment. Cells were grown in serum-rich medium for 48 h and switched to low serum for 24 h before conducting APEX labeling by preincubating cells with biotin tyramide for 30 min and adding H2O2 for 2 min before quenching. Cells were lysed, biotinylated proteins (marked with red dots) were isolated on streptavidin resin, and bound material was eluted via on-bead digest with trypsin. For each individual sample, peptides were labeled with a unique TMT. All samples were then pooled and peptides were analyzed using a synchronous precursor selection MS3 method for mass spectrometric identification and quantitation. All samples were in triplicate, except for a technical control, where H2O2 was omitted. (B) Hierarchical two-way cluster analysis of a Cilia-APEX2/TMT experiment. Clustering of the relative abundances of each identified protein (rows) in the individual samples (columns) was performed based on Ward’s minimum variance method. The relative abundance of a given protein was calculated by dividing the TMT signal in one sample by the sum of TMT signals in all samples. The color scheme of relative abundances is shown on the right (in percent). Only the clusters containing cilia proteins are shown (see Fig. S1 E for full cluster analysis). (C and D) Volcano plots of statistical significance versus protein enrichment in Cilia-APEX2 compared with control-APEX2 samples (C) or in Cilia-APEX2 WT versus Cep164−/− samples (D). Calculated P values (statistical significance of enrichment calculated from unpaired Student’s t tests) for 4,836 quantified proteins were plotted against the TMT ratios of Cilia-APEX2 samples versus the respective controls. Proteins fulfilling all four significance and enrichment criteria are represented by blue dots, proteins meeting three criteria are represented by black dots, and other proteins are shown in gray (see text for details). Protein hits quantified by only one peptide are highlighted in purple. See Table S1. (E) Schematic of a primary cilium with key protein complexes, structures, or pathways. The boxes list proteins identified as tier 1 (black) or tier 2 (gray) hits of the Cilia-APEX2 proteome. Proteins not identified by Cilia-APEX2 are indicated by dashed outline lettering. Gene symbols are included when differing from conventional protein names. Note that Ttc30a1 and Ttc30a2 are grouped into IFT70A/Ttc30a.
Cilia-APEX2-based deep proteomics. (A) Workflow of a Cilia-APEX2/TMT experiment. Cells were grown in serum-rich medium for 48 h and switched to low serum for 24 h before conducting APEX labeling by preincubating cells with biotin tyramide for 30 min and adding H2O2 for 2 min before quenching. Cells were lysed, biotinylated proteins (marked with red dots) were isolated on streptavidin resin, and bound material was eluted via on-bead digest with trypsin. For each individual sample, peptides were labeled with a unique TMT. All samples were then pooled and peptides were analyzed using a synchronous precursor selection MS3 method for mass spectrometric identification and quantitation. All samples were in triplicate, except for a technical control, where H2O2 was omitted. (B) Hierarchical two-way cluster analysis of a Cilia-APEX2/TMT experiment. Clustering of the relative abundances of each identified protein (rows) in the individual samples (columns) was performed based on Ward’s minimum variance method. The relative abundance of a given protein was calculated by dividing the TMT signal in one sample by the sum of TMT signals in all samples. The color scheme of relative abundances is shown on the right (in percent). Only the clusters containing cilia proteins are shown (see Fig. S1 E for full cluster analysis). (C and D) Volcano plots of statistical significance versus protein enrichment in Cilia-APEX2 compared with control-APEX2 samples (C) or in Cilia-APEX2 WT versus Cep164−/− samples (D). Calculated P values (statistical significance of enrichment calculated from unpaired Student’s t tests) for 4,836 quantified proteins were plotted against the TMT ratios of Cilia-APEX2 samples versus the respective controls. Proteins fulfilling all four significance and enrichment criteria are represented by blue dots, proteins meeting three criteria are represented by black dots, and other proteins are shown in gray (see text for details). Protein hits quantified by only one peptide are highlighted in purple. See Table S1. (E) Schematic of a primary cilium with key protein complexes, structures, or pathways. The boxes list proteins identified as tier 1 (black) or tier 2 (gray) hits of the Cilia-APEX2 proteome. Proteins not identified by Cilia-APEX2 are indicated by dashed outline lettering. Gene symbols are included when differing from conventional protein names. Note that Ttc30a1 and Ttc30a2 are grouped into IFT70A/Ttc30a.
We assessed candidate ciliary proteins via statistical analyses of the relative enrichment between the Cilia-APEX2 samples and the controls (Fig. 2, C and D). To be scored as ciliary, proteins had to fulfill four criteria (Fig. 2, C and D, blue dots): greater than twofold enrichment in the Cilia-APEX2 samples over control-APEX2 samples and over the Cep164−/− Cilia-APEX2 samples (TMT ratio > 2.0) and statistically significant enrichment in the Cilia-APEX2 samples versus the control-APEX2 samples and the Cep164−/− Cilia-APEX2 samples (P value < 0.05). In addition, proteins that met only three out of four criteria (Fig. 2, C and D, black dots) were included if they were close to matching the fourth criterion (TMT ratio > 1.5 or P value < 0.1). This set of criteria resulted in the inclusion of 203 proteins (Table S1) and represents a compromise between inclusion of false positives and exclusion of false negatives (Fig. 2, C and D). It should be noted that these criteria select for proteins significantly enriched in cilia and that ciliary proteins found at similar levels inside and outside of cilia will be excluded.
To define a Cilia-APEX2 proteome, we leveraged two additional independent experiments conducted before the Cilia-APEX2 Cep164−/− control line was generated. We grouped the resulting candidate cilia proteins into two tiers, depending upon whether they were identified in all three experiments (tier 1) or missing from one of the three experiments (tier 2; Table S2). Application of these criteria resulted in the inclusion of 179 proteins in tier 1 and 91 proteins in tier 2. While Cilia-APEX had identified 75% of the intraflagellar transport (IFT) motor, 60% of the IFT and none of the Bardet-Biedl Syndrome complex (BBSome) subunits (Mick et al., 2015), the Cilia-APEX2 proteome comprises nearly all subunits of the IFT motors kinesin-2 and dynein 2, the IFT complexes, and the BBSome (Fig. 2 E). Most of the subunits that were not identified (e.g., BBS18 and LC8) were <10 kD in molecular weight and likely to be missed by MS because of the small number of derived tryptic peptides (Fig. 2 E). Most importantly, Cilia-APEX2 combined with TMT labeling enabled the previously elusive identification of central cilia-enriched signaling components, including most Hh signaling components known to localize to Cilia (PTCH1, SMO, GPR161, KIFf7, SUFU, and GLI3). The Hh transcription factor GLI1 was not identified by Cilia-APEX2, consistent with undetectable GLI1 expression in the absence of Hh pathway stimulation, and GLI2 could only be quantified in one experiment out of three, possibly because of its low abundance in IMCD3 cells.
Time-resolved Cilia-APEX2 proteomics reveals global alterations of the cilia proteome in response to Hh stimulation
Encouraged by the detection of the core Hh signaling machinery by Cilia-APEX2, we sought to determine the global changes of the cilia proteome in response to Hh by subjecting duplicate samples of cells exposed to Shh for 0, 1, 4, or 24 h to the Cilia-APEX2/TMT workflow (Fig. 3 A). Strikingly, comparison of the Cilia-APEX2 proteomes between untreated and Shh-treated cells revealed only a handful proteins with significantly changed abundance (TMT ratio > 2.0; Fig. 3 B). Out of the 270 cilia proteins (Table S2), the abundance of 265 proteins did not significantly change upon Shh stimulation (TMT ratio < 2.0). These proteins include the cilia trafficking components IFT88 and BBS1, inositol polyphosphate 5-phosphatase (INPP5E), and Polycystin-2 (PKD2; Fig. 4 A). Hierarchical clustering of relative protein abundance in the 10-plex experiment demonstrates high reproducibility across experimental repeats, and, for >98% of the ciliary proteome, the low variability between different time points highlights the robust and reproducible quantitation enabled by Cilia-APEX2/TMT (Fig. S2 A).

Experimental outline of time-resolved Cilia-APEX2 proteomics after Shh stimulation. (A) Workflow of a time-resolved Cilia-APEX2/TMT experiment. The workflow is identical to Fig. 2 A, except that Shh was added 24 h, 4 h, or 1 h before labeling as indicated. −Shh indicates addition of conditioned medium without Shh. Red dots mark biotinylated proteins. (B) Volcano plot of significance versus enrichment in 24 h Shh-treated compared with no-Shh samples. Hh signaling components known to change their ciliary localization are shown in blue, and proteins with newly identified changes are shown in orange. Relative quantification ratios of 5,350 proteins are plotted.
Experimental outline of time-resolved Cilia-APEX2 proteomics after Shh stimulation. (A) Workflow of a time-resolved Cilia-APEX2/TMT experiment. The workflow is identical to Fig. 2 A, except that Shh was added 24 h, 4 h, or 1 h before labeling as indicated. −Shh indicates addition of conditioned medium without Shh. Red dots mark biotinylated proteins. (B) Volcano plot of significance versus enrichment in 24 h Shh-treated compared with no-Shh samples. Hh signaling components known to change their ciliary localization are shown in blue, and proteins with newly identified changes are shown in orange. Relative quantification ratios of 5,350 proteins are plotted.

Time-resolved Cilia-APEX2 proteomics reveals the extent of ciliary proteome remodeling in response to Shh. The relative abundances of selected proteins in the Cilia-APEX2/TMT datasets are plotted against time. For each individual protein, the background signal in the control-APEX2 sample was set to 0 and the maximum average signal across all time points was set to 1. t = 0 corresponds to the −Shh sample. (A and B) 24-h time course. Data points represent averages of duplicate measurements, and error bars depict individual values. Error bars smaller than indicated datapoint symbols have been omitted. 0 h represents −Shh as in Fig. 3 A. Housekeeping ciliary proteins are shown in A. Known and newly identified Hh signaling components are shown in B. (C) 1-h time course. Normalized intensities (relative to ARL13B) were plotted over time. See also Fig. S3.
Time-resolved Cilia-APEX2 proteomics reveals the extent of ciliary proteome remodeling in response to Shh. The relative abundances of selected proteins in the Cilia-APEX2/TMT datasets are plotted against time. For each individual protein, the background signal in the control-APEX2 sample was set to 0 and the maximum average signal across all time points was set to 1. t = 0 corresponds to the −Shh sample. (A and B) 24-h time course. Data points represent averages of duplicate measurements, and error bars depict individual values. Error bars smaller than indicated datapoint symbols have been omitted. 0 h represents −Shh as in Fig. 3 A. Housekeeping ciliary proteins are shown in A. Known and newly identified Hh signaling components are shown in B. (C) 1-h time course. Normalized intensities (relative to ARL13B) were plotted over time. See also Fig. S3.

Hh pathway activation removes PKA-RIα and GPR161 from cilia, but not the AKAPs identified by Cilia-APEX2. (A) Two-way hierarchical cluster analysis (Ward’s method) of the relative protein abundances (rows) in the individual samples (columns) shows high inter-set reproducibility. Legend depicts the color scheme of relative abundances (calculated as in Fig. S1 E). SMO and GPR161 miniclusters are indicated by green and red arrows, respectively (see Fig. 5, A and B for magnified views of the miniclusters). (B and C) Ciliated NGPKA-RIα–expressing IMCD3 cells were treated with or without SAG for the indicated times. (B) Cells were fixed and stained for acetylated tubulin (ac-tub; red) and DNA (blue). NGPKA-RIα was visualized by NG fluorescence. (C) Box plot shows background-corrected NGPKA-RIα fluorescence in cilia at indicated time points after SAG addition. 50 cilia (n = 50) were analyzed per time point. (D) Ciliated IMCD3 cells expressing GPR161NG were serum-starved for 24 h in the presence of Shh for indicated times and analyzed as in B. GPR161NG was detected by NG fluorescence. For quantitative analysis, see Fig. 5 F. (E) Relative AKAP abundances assessed by mass spectrometric TMT quantitation were plotted over time. Data points represent averages of duplicate measurements, error bars depict individual values. Maximum average signal was set to 1, and background signals as assessed from control-APEX2 labeled samples were set to 0. 0 h represents −Shh. Scale bars, 2 µm. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
Hh pathway activation removes PKA-RIα and GPR161 from cilia, but not the AKAPs identified by Cilia-APEX2. (A) Two-way hierarchical cluster analysis (Ward’s method) of the relative protein abundances (rows) in the individual samples (columns) shows high inter-set reproducibility. Legend depicts the color scheme of relative abundances (calculated as in Fig. S1 E). SMO and GPR161 miniclusters are indicated by green and red arrows, respectively (see Fig. 5, A and B for magnified views of the miniclusters). (B and C) Ciliated NGPKA-RIα–expressing IMCD3 cells were treated with or without SAG for the indicated times. (B) Cells were fixed and stained for acetylated tubulin (ac-tub; red) and DNA (blue). NGPKA-RIα was visualized by NG fluorescence. (C) Box plot shows background-corrected NGPKA-RIα fluorescence in cilia at indicated time points after SAG addition. 50 cilia (n = 50) were analyzed per time point. (D) Ciliated IMCD3 cells expressing GPR161NG were serum-starved for 24 h in the presence of Shh for indicated times and analyzed as in B. GPR161NG was detected by NG fluorescence. For quantitative analysis, see Fig. 5 F. (E) Relative AKAP abundances assessed by mass spectrometric TMT quantitation were plotted over time. Data points represent averages of duplicate measurements, error bars depict individual values. Maximum average signal was set to 1, and background signals as assessed from control-APEX2 labeled samples were set to 0. 0 h represents −Shh. Scale bars, 2 µm. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
Importantly, Cilia-APEX2/TMT profiling detected changes in ciliary abundance of the three Hh signaling components known to undergo signal-dependent redistribution in or out of cilia (Fig. 4 B). Levels of the Hh receptor PTCH1 and GPR161 decreased while SMO increased within 4 h after pathway activation. The kinetics revealed by Cilia-APEX2 closely matched the kinetics previously defined by immunostaining (Rohatgi et al., 2007; Mukhopadhyay et al., 2013). Since the ciliary changes of PTCH1, SMO, and GPR161 were nearly complete after only 1 h of pathway activation, we sought to resolve the changes in proteome remodeling during the first 60 min after Shh addition (Fig. S3). The high temporal resolution and precise TMT-based quantitation of Cilia-APEX2 profiling enabled a refined characterization of the redistribution of Hh signaling components while the abundance of housekeeping ciliary proteins remained largely constant (Fig. 4 C). The levels of the Hh receptor PTCH1 in cilia started dropping 5 min after Shh addition and reached a minimum after 30 min. Meanwhile, ciliary levels of SMO steadily increased during the 60-min time course and until the 4-h time point (Fig. 4 B). The removal of GPR161 was preceded by an increase in ciliary β-arrestin2 levels (Fig. 4 C), consistent with the proposed role of β-arrestin2 in triggering signal-dependent exit of GPR161 from cilia (Pal et al., 2016).

Experimental workflow for high resolution time-resolved Cilia-APEX2 profiling of the ciliary Hh response. Related to Fig. 4 C. Cilia-APEX2 and control-APEX2 IMCD3 cells were seeded 72 h before the APEX labeling reaction. 24 h before labeling, cells were starved of serum. 5, 10, 15, 30, 45, and 60 min before APEX labeling, Shh-conditioned medium was added (as indicated). −Shh indicates addition of conditioned medium without Shh 60 min before labeling. APEX labeling and sample preparation were performed as in Fig. 2 A. In brief, biotin tyramide was added 30 min before the 2-min labeling reaction in the presence of H2O2. Samples were quenched and kept on ice for lysis, followed by streptavidin capture and on-bead tryptic digest. Peptides of each sample were labeled with individual TMTs, pooled, and fractionated offline via high-pH reverse-phase chromatography before mass spectrometric analysis. Red dots mark biotinylated proteins.
Experimental workflow for high resolution time-resolved Cilia-APEX2 profiling of the ciliary Hh response. Related to Fig. 4 C. Cilia-APEX2 and control-APEX2 IMCD3 cells were seeded 72 h before the APEX labeling reaction. 24 h before labeling, cells were starved of serum. 5, 10, 15, 30, 45, and 60 min before APEX labeling, Shh-conditioned medium was added (as indicated). −Shh indicates addition of conditioned medium without Shh 60 min before labeling. APEX labeling and sample preparation were performed as in Fig. 2 A. In brief, biotin tyramide was added 30 min before the 2-min labeling reaction in the presence of H2O2. Samples were quenched and kept on ice for lysis, followed by streptavidin capture and on-bead tryptic digest. Peptides of each sample were labeled with individual TMTs, pooled, and fractionated offline via high-pH reverse-phase chromatography before mass spectrometric analysis. Red dots mark biotinylated proteins.
To determine if any other proteins besides SMO, PTCH1, and GPR161 undergo changes in ciliary abundance in response to Shh, we searched for proteins that coclustered with SMO, PTCH1, or GPR161 during the 24-h time course in a hierarchical cluster analysis. The putative phosphatase Paladin 1 (PALD1) was the only protein that clustered closely with SMO (Fig. 5 A). It should be noted that despite tight clustering of PKD1 and GLI2, the PKD1/GLI2 cluster sits at a substantial distance from the SMO/PALD1 minicluster (Fig. 5 A). Similarly, proteins disappearing from cilia in response to Shh formed a single tight cluster composed of GPR161, PTCH1, and the PKA regulatory subunit Iα (PKA-RIα; Fig. 5 B). The ciliary levels of all three proteins reached a near minimum within 1 h of pathway induction, and manual inspection of the ciliary abundance profiles revealed nearly identical exit kinetics for GPR161 and PKA-RIα in the 60-min and 24-h time courses (Fig. 4, B and C).

Hierarchical two-way cluster analysis reveals that PKA-RIα exits cilia together with GPR161 in response to Hh signal. (A and B) Magnified views of the hierarchical cluster analysis of the two time-resolved Cilia-APEX2 proteomics experimental replicates. (A) SMO minicluster (green) and neighboring branches. (B) GPR161 minicluster (red) and neighboring branches. Prkar1a is the gene name for PKA-RIα. Complete cluster analysis shown in Fig. S2 A. (C) Ciliated IMCD3 cells stably expressing NGPKA-RIα were treated with Shh or control medium. Cells were fixed and stained for acetylated tubulin (ac-tub; red) and DNA (blue). NGPKA-RIα was visualized via the intrinsic fluorescence of NG (green). (D) Box plot showing background-corrected NGPKA-RIα fluorescence in cilia at indicated time points after Shh addition. (E) IMCD3 cells expressing GPR161NG were treated and analyzed as in C. GPR161NG was visualized via the intrinsic fluorescence of NG. (F) Box plot showing background-corrected ciliary GPR161NG signal at indicated time points after Shh addition. (G) Box plots showing background-corrected GPR161NG fluorescence signals in the primary cilium of cells transfected with siRNA against Prkar1a or control siRNA at indicated times after Shh addition. (H) Model of the functional interaction between GPR161, PKA, and SMO. In unstimulated cells (−Hh), GPR161 keeps [cAMP]cilia high via activation of Gαs. GPR161-bound PKA-RIα releases the fully active catalytic PKA subunits (C) to phosphorylate downstream targets (GLI3). Early after pathway activation (+Hh, early), SMO begins to accumulate in cilia and lowers [cAMP]cilia via Gαi activation. This leads to the association of PKA-C with PKA-RIα to form a partially active holoenzyme that may locally phosphorylate the GPR161 C-terminal tail. GPR161 phosphorylation is a prerequisite for β-arrestin2 (Arr) recruitment, resulting in the exit of GPR161 from cilia and internalization, possibly together with PKA holoenzyme bound (+Hh, late). The removal of GPR161 from cilia eliminates the source of tonic Gαs activation, which leads to a further reduction of [cAMP]cilia. (I) HEK293T cells were transiently cotransfected with the same amount of Rlucβ-arrestin2 DNA and increasing amounts of WT or V158E, S445A/S446A (SS>AA), or S445D/S446D (SS>DD) mutants of GPR161YFP as indicated. Cells were subjected to BRET analysis 48 h after transfection (see Materials and methods for details). Titration curves are based on the means of four datasets (n = 4), and error bars indicate SEM. All scale bars represent 2 µm. n = 60 cilia analyzed per time point in all box plots. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
Hierarchical two-way cluster analysis reveals that PKA-RIα exits cilia together with GPR161 in response to Hh signal. (A and B) Magnified views of the hierarchical cluster analysis of the two time-resolved Cilia-APEX2 proteomics experimental replicates. (A) SMO minicluster (green) and neighboring branches. (B) GPR161 minicluster (red) and neighboring branches. Prkar1a is the gene name for PKA-RIα. Complete cluster analysis shown in Fig. S2 A. (C) Ciliated IMCD3 cells stably expressing NGPKA-RIα were treated with Shh or control medium. Cells were fixed and stained for acetylated tubulin (ac-tub; red) and DNA (blue). NGPKA-RIα was visualized via the intrinsic fluorescence of NG (green). (D) Box plot showing background-corrected NGPKA-RIα fluorescence in cilia at indicated time points after Shh addition. (E) IMCD3 cells expressing GPR161NG were treated and analyzed as in C. GPR161NG was visualized via the intrinsic fluorescence of NG. (F) Box plot showing background-corrected ciliary GPR161NG signal at indicated time points after Shh addition. (G) Box plots showing background-corrected GPR161NG fluorescence signals in the primary cilium of cells transfected with siRNA against Prkar1a or control siRNA at indicated times after Shh addition. (H) Model of the functional interaction between GPR161, PKA, and SMO. In unstimulated cells (−Hh), GPR161 keeps [cAMP]cilia high via activation of Gαs. GPR161-bound PKA-RIα releases the fully active catalytic PKA subunits (C) to phosphorylate downstream targets (GLI3). Early after pathway activation (+Hh, early), SMO begins to accumulate in cilia and lowers [cAMP]cilia via Gαi activation. This leads to the association of PKA-C with PKA-RIα to form a partially active holoenzyme that may locally phosphorylate the GPR161 C-terminal tail. GPR161 phosphorylation is a prerequisite for β-arrestin2 (Arr) recruitment, resulting in the exit of GPR161 from cilia and internalization, possibly together with PKA holoenzyme bound (+Hh, late). The removal of GPR161 from cilia eliminates the source of tonic Gαs activation, which leads to a further reduction of [cAMP]cilia. (I) HEK293T cells were transiently cotransfected with the same amount of Rlucβ-arrestin2 DNA and increasing amounts of WT or V158E, S445A/S446A (SS>AA), or S445D/S446D (SS>DD) mutants of GPR161YFP as indicated. Cells were subjected to BRET analysis 48 h after transfection (see Materials and methods for details). Titration curves are based on the means of four datasets (n = 4), and error bars indicate SEM. All scale bars represent 2 µm. n = 60 cilia analyzed per time point in all box plots. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
Thus, beyond detecting known changes in remodeling of the ciliary proteome in response to Hh, the time-resolved Cilia-APEX2/TMT profiling platform enabled discovery of novel dynamic factors coenriched and codepleted with known components.
Time-resolved Cilia-APEX2/TMT illuminates the mechanisms of regulated GPR161 removal from cilia
In the current models of Hh signal transduction, the Hh-dependent drop in ciliary PKA activity represents a critical step that relays SMO activation to a switch in processing of the transcription factors GLI2 and GLI3 from repressor to activator forms (Kong et al., 2019; Gigante and Caspary, 2020). We already know of three redundant mechanisms that depress ciliary PKA activity in response to Hh pathway activation. First, ciliary accumulation of the Gαi-coupled GPCR SMO brings into cilia Gαi-mediated inhibition of adenylyl cyclases (Riobo, 2014). Second, SMO may block PKA activity directly (Arveseth et al., 2020,Preprint). Third, ciliary exit of the tonically active Gαs-coupled GPCR GPR161 will lead to decreased levels of active Gαs and thus cAMP inside cilia (Mukhopadhyay et al., 2013; Pusapati et al., 2018a; Tschaikner et al., 2020). PKA-RIα exit from cilia upon Hh pathway activation may represent an additional mechanism for Hh-dependent depression of ciliary PKA activity. As PKA-RIα acts both as a stoichiometric inhibitor and a targeting partner for the catalytic PKA subunit PKA-C (Taylor et al., 2012), the exit of PKA-RIα from cilia is likely to remove PKA-C as well. To confirm that PKA-RIα undergoes Hh-dependent exit from cilia, we established a stable IMCD3 cell line that expresses PKA-RIα fused to the fluorescent protein mNeonGreen (NG; Shaner et al., 2013) and imaged NGPKA-RIα by fluorescence microscopy. While unstimulated cells exhibited robust ciliary signals of NGPKA-RIα (as previously described by Mick et al., 2015; Bachmann et al., 2016), addition of Shh triggered a decrease of NGPKA-RIα ciliary fluorescence with kinetics that mirrored those measured by Cilia-APEX2/TMT profiling (Fig. 5, C and D). To pinpoint the step in the pathway that triggers the removal of PKA-RIα from cilia, we directly activated SMO via SMO agonist (SAG; Fig. S2, B and C). The kinetics of NGPKA-RIα exit from cilia was nearly identical in cells treated with Shh or SAG, and we conclude that PKA-RIα exit from cilia lies downstream of SMO activation.
In agreement with our cluster analysis (Fig. 5 B), the kinetics of NGPKA-RIα removal from cilia upon Hh pathway activation mirrored the exit kinetics of GPR161NG (Fig. 5, E and F; and Fig. S2 D). The concomitant exit of GPR161 and PKA-RIα is consistent with the finding that the cytoplasmic tail of GPR161 harbors an atypical A kinase anchoring protein (AKAP) motif with exquisite and unprecedented specificity for PKA-RIα (Bachmann et al., 2016). A major function of the 60 different AKAPs is to direct the catalytic subunits to their substrates by recruiting the PKA regulatory subunits to discrete cellular locations (Torres-Quesada et al., 2017). We conclude that GPR161 and PKA-RIα form a stable complex that represents a functional unit, most likely together with PKA-C. Meanwhile, the abundances of other AKAPs quantified in the Cilia-APEX2 dataset (e.g., AKAP11 and AKAP9) did not change appreciably in response to Hh (Fig. S2 E).
Besides encoding an AKAP motif for PKA-RIα, the C-terminal tail of GPR161 also contains two consensus PKA phosphorylation sites (Bachmann et al., 2016). As AKAPs direct PKA to their substrates (Musheshe et al., 2018), the AKAP motif of GPR161 may direct PKA-RIα/PKA-C to phosphorylate the GPR161 C tail itself. Because phospho-mimetic mutations of the PKA site in GPR161 drastically reduce ciliary levels of GPR161 (Bachmann et al., 2016), it is conceivable that PKA-RIα/PKA-C–mediated phosphorylation of GPR161 increases in response to Hh pathway activation and that this phosphorylation event triggers exit of GPR161 from cilia. To test this hypothesis, we assessed the Hh-induced removal of GPR161 from cilia after siRNA-mediated depletion of PKA-RIα. While control siRNA did not interfere with GPR161NG exit, GPR161NG failed to exit cilia in response to Hh signal in PKA-RIα–depleted cells (Fig. 5 G).
A major conundrum then lies in how Hh pathway activation may control the PKA-RIα–dependent phosphorylation of the GPR161 C tail. In the test tube, PKA regulatory subunits inhibit the activity of the catalytic subunits until the regulatory subunits bind cAMP and release free and active PKA-C. Recent findings that intermediate concentrations of cAMP promote PKA activation without dissociation of catalytic from regulatory subunits (Smith et al., 2017) may shed light on the regulation of GPR161 C tail phosphorylation. While few measurements of [cAMP]cilia have been published, one study found that [cAMP]cilia is ∼4 µM in unstimulated cells (Moore et al., 2016), a concentration sufficient to trigger nearly complete PKA-C/PKA-R dissociation within cilia. Under these circumstances, PKA-C will freely diffuse in the cilium and phosphorylate GLI2/3 and other substrates (Fig. 5 H, left). Because SMO entry into cilia is already detectable before the onset of GPR161 exit (Fig. 4 C; Shinde et al., 2020), we consider a ciliary state where GPR161 and activated SMO coexist inside cilia. We propose that the activation of Gαi by SMO inside cilia will reduce [cAMP]cilia to a level where an active PKA holoenzyme assembles on the C tail of GPR161 and phosphorylates GPR161 (Fig. 5 H, middle). The ultimate exit of GPR161/PKA-RIα/PKA-C further amplifies the effect of ciliary Gαi activation via SMO to fully depress [cAMP]cilia to a level where GLI2/3 no longer become phosphorylated by PKA (Fig. 5 H, right).
The proposed model of regulated GPR161 exit resolves another conundrum raised previously (Pal et al., 2016). Multiple groups have found that β-arrestin2 is required for exit of GPR161 from cilia subsequent to entry of activated SMO into cilia. Consistent with the Cilia-APEX2/TMT profile of β-arrestin2 in response to Shh (Fig. 4 C), recent imaging studies found that the ciliary levels of β-arrestin2 rapidly increase upon Hh pathway activation and reach a plateau at 20 min (Shinde et al., 2020). Interestingly, β-arrestins are also recruited to cilia upon activation of the ciliary GPCR somatostatin receptor 3 (SSTR3; Green et al., 2015; Pal et al., 2016; Ye et al., 2018). Because β-arrestins are rapidly and stably recruited to activated GPCRs after they become phosphorylated, these results suggest that β-arrestin2 recognizes phosphorylated GPR161 inside cilia and instructs the ciliary export machinery to remove GPR161 from cilia. While GRK2 phosphorylation of GPR161 is already known to promote β-arrestin binding (Pal et al., 2016), we sought to test whether PKA phosphorylation of the GPR161 C tail may also affect β-arrestin recruitment. We introduced PKA phospho-mimetic and phospho-dead mutations into GPR161 fused to a YFP moiety, termed GPR161S445D/S446DYFP and GPR161S445A/S446AYFP, and assessed Rlucβ-arrestin2 recruitment by bioluminescence resonance energy transfer (BRET). While the phospho-mimetic mutant recruited β-arrestin2 as efficiently as WT GPR161, β-arrestin2 recruitment to the phospho-dead mutant was reduced (Fig. 5 I), suggesting that PKA phosphorylation of the C-terminal tail of GPR161 is a prerequisite to β-arrestin2 recruitment and subsequent ciliary exit.
PALD1 accumulates within cilia of selected cell types upon Hh stimulation
Cilia-APEX2/TMT profiling quantified a steady coaccumulation of SMO and PALD1 in cilia in response to Shh over the course of 60 min that reached a plateau after 4 h (Fig. 4, B and C; and Fig. 5 A). Staining IMCD3 cells for endogenous PALD1 revealed that the increase in ciliary PALD1 signals (Fig. 6 A) upon Shh stimulation followed very similar kinetics to those of SMO (Fig. 6 B), also observed by two-way hierarchical clustering analysis (Fig. 5 A). Given the striking Hh signal–dependent colocalization of SMO and PALD1 in cilia, we sought to determine how PALD1 responds to Hh pathway activation. Given that endogenous PALD1 and SMO accumulated efficiently in primary cilia in response to direct activation of SMO by SAG (Fig. 6, C and D), we conclude that ciliary accumulation of PALD1 lies downstream of SMO. Considering the nearly identical kinetics of PALD1 and SMO ciliary accumulation, we first hypothesized that PALD1 may piggyback on SMO entering cilia. To test this hypothesis, we leveraged cyclopamine (CYC), a SMO antagonist that promotes ciliary accumulation of SMO while blocking Hh pathway accumulation (Wilson et al., 2009; Wang et al., 2009; Rohatgi et al., 2009). While CYC promoted ciliary accumulation of SMO in IMCD3 cells, it did not increase the ciliary levels of PALD1 (Fig. 6, E and F), indicating that ciliary accumulation of SMO is not sufficient to drive PALD1 into cilia and that SMO must be activated for PALD1 to accumulate in cilia. To test whether PALD1 entry into cilia is sensitive to the reduction in [cAMP]cilia downstream of SMO, we resorted to the ciliary Gαi-coupled GPCR SSTR3. Strikingly, SSTR3 activation is sufficient to recruit PALD1 to primary cilia (Fig. 6, G and H), suggesting that, similarly to GPR161 exit from cilia, ciliary PALD1 accumulation responds to decreases in ciliary cAMP levels.

PALD1 accumulates in primary cilia in response to activation of Gαi in cilia. (A and B) Ciliated IMCD3 cells were treated with Shh for the indicated times before fixation and staining for PALD1 (A) or SMO (B). Box plots display the background-corrected signals of PALD1 and SMO in primary cilia. n = 59 cilia analyzed per condition. (C and D) Ciliated IMCD3 cells were treated with or without SAG for 24 h and immunostained for the indicated proteins. (E and F) Ciliated IMCD3 cells were treated with cyclopamine (+CYC) or SAG for 24 h and analyzed as in C and D. (E) Micrographs of representative images. (F) Box plots showing background-corrected, relative ciliary fluorescence intensities of the respective proteins normalized to acetylated tubulin signals. n = 30 analyzed per condition. Data were analyzed using two-way ANOVA with multiple comparisons (Tukey test) with a defined confidence of 95%. *, P < 0.05. (G and H) Ciliated IMCD3 cells stably expressing SSTR3NG were stimulated with 0 or 10 µM somatostatin (sst) for 24 h and analyzed as in E and F (n = 30). SSTR3NG was detected via NG fluorescence. Data were analyzed using two-way ANOVA with multiple comparisons (Sidak test) with a defined confidence of 95%. *, P < 0.05. All scale bars represent 2 µm. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
PALD1 accumulates in primary cilia in response to activation of Gαi in cilia. (A and B) Ciliated IMCD3 cells were treated with Shh for the indicated times before fixation and staining for PALD1 (A) or SMO (B). Box plots display the background-corrected signals of PALD1 and SMO in primary cilia. n = 59 cilia analyzed per condition. (C and D) Ciliated IMCD3 cells were treated with or without SAG for 24 h and immunostained for the indicated proteins. (E and F) Ciliated IMCD3 cells were treated with cyclopamine (+CYC) or SAG for 24 h and analyzed as in C and D. (E) Micrographs of representative images. (F) Box plots showing background-corrected, relative ciliary fluorescence intensities of the respective proteins normalized to acetylated tubulin signals. n = 30 analyzed per condition. Data were analyzed using two-way ANOVA with multiple comparisons (Tukey test) with a defined confidence of 95%. *, P < 0.05. (G and H) Ciliated IMCD3 cells stably expressing SSTR3NG were stimulated with 0 or 10 µM somatostatin (sst) for 24 h and analyzed as in E and F (n = 30). SSTR3NG was detected via NG fluorescence. Data were analyzed using two-way ANOVA with multiple comparisons (Sidak test) with a defined confidence of 95%. *, P < 0.05. All scale bars represent 2 µm. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
We next sought to determine whether PALD1 integration with the Hh pathway is a universal feature of the Hh response. In addition to IMCD3, PALD1 protein expression was detectable in NIH-3T3 cells, C2C12 myoblasts, MIN6 pancreatic β cells, and human embryonic kidney (HEK) cells (Fig. 7 A). Yet, unlike some Hh pathway components such as PTCH1 or GLI1, whose expression is induced by the Hh pathway, PALD1 expression did not appreciably change in response to Hh pathway stimulation (Fig. 7 A). Unexpectedly, PALD1 protein expression was not detectable in telomerase immortalized human retinal pigment epithelial cells (RPE1-hTERT). We next tested two cell types in which the Hh response has been extensively studied. NIH-3T3 cells constitute a widely used cell-based system for Hh signaling (Taipale et al., 2000). Surprisingly, activation of the Hh pathway in 3T3 cells led to the accumulation of SMO in cilia but failed to promote ciliary entry of PALD1 (Fig. 7 B). Hh signaling controls muscle differentiation (Hu et al., 2012), and the requirement for primary cilia in myoblast proliferation can be recapitulated in cultured C2C12 cells (Fu et al., 2014). While PALD1 was absent from unstimulated C2C12 cilia, PALD1 became enriched in primary cilia in response to Hh signal (Fig. 7, C and D). These results indicate that the association of PALD1 with Hh signaling can be detected in multiple cell lines but is not a universal element of the Hh response.

PALD1 accumulates in primary cilia upon Hh pathway activation in particular cell lines. (A) Lysates of different cell lines treated with or without SAG for 24 h were immunoblotted for PALD1 and GAPDH (loading control). Dot plot displaying the ratios of PALD1 to GAPDH signals with horizontal lines indicating means (n = 2 except for Pald1−/− where n = 1). (B) PALD1 does not accumulate in primary cilia of 3T3 cells after Hh pathway activation, whereas SMO does. Ciliated 3T3 cells expressing YFPSMO (Rohatgi et al., 2009) were treated with or without SAG for 24 h and stained for PALD1, acetylated tubulin, and DNA. SMO was detected via YFP fluorescence. Scale bars, 2 µm. (CandD) PALD1 is enriched in C2C12 myoblast primary cilia after Hh pathway activation. C2C12 cells were treated and analyzed as in B. Box plots show background-corrected, relative fluorescence levels normalized to acetylated tubulin signals. n = 30 cilia analyzed per condition. (E) Diagram of PALD1 showing predicted protein domains and motifs. Numbers indicate amino acid positions in M. musculus PALD1; Myr depicts the myristoylation site at Gly2. Arrowhead indicates location of missense mutation in Pald1−/− cells (see Fig. S4 A). (F) Phylogenetic analysis of PALD1 orthologues and co-conserved proteins identified by pathway clustering analysis (Li et al., 2014). Shown is a simplified taxonomic tree with crown eukaryotic groups in different colors (modified from Carvalho-Santos et al., 2010). Branch color code: purple, opisthokonts; blue, amoebozoa; green, plants; yellow, alveolates and heterokonts; orange, haptophytes; and brown, excavates. When present in the respective organism, motile cilia are shown in green and primary cilia in blue. The presence of the corresponding proteins is indicated by black circles. Conservation of IFT-B complex subunits is depicted by circles with shades of gray corresponding to percentage of subunits with orthologues (black, 100%; dark gray, <100%; light gray, <60%; white, <30%). Proteins with E-values ≤ 10−25 were scored as hits. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
PALD1 accumulates in primary cilia upon Hh pathway activation in particular cell lines. (A) Lysates of different cell lines treated with or without SAG for 24 h were immunoblotted for PALD1 and GAPDH (loading control). Dot plot displaying the ratios of PALD1 to GAPDH signals with horizontal lines indicating means (n = 2 except for Pald1−/− where n = 1). (B) PALD1 does not accumulate in primary cilia of 3T3 cells after Hh pathway activation, whereas SMO does. Ciliated 3T3 cells expressing YFPSMO (Rohatgi et al., 2009) were treated with or without SAG for 24 h and stained for PALD1, acetylated tubulin, and DNA. SMO was detected via YFP fluorescence. Scale bars, 2 µm. (CandD) PALD1 is enriched in C2C12 myoblast primary cilia after Hh pathway activation. C2C12 cells were treated and analyzed as in B. Box plots show background-corrected, relative fluorescence levels normalized to acetylated tubulin signals. n = 30 cilia analyzed per condition. (E) Diagram of PALD1 showing predicted protein domains and motifs. Numbers indicate amino acid positions in M. musculus PALD1; Myr depicts the myristoylation site at Gly2. Arrowhead indicates location of missense mutation in Pald1−/− cells (see Fig. S4 A). (F) Phylogenetic analysis of PALD1 orthologues and co-conserved proteins identified by pathway clustering analysis (Li et al., 2014). Shown is a simplified taxonomic tree with crown eukaryotic groups in different colors (modified from Carvalho-Santos et al., 2010). Branch color code: purple, opisthokonts; blue, amoebozoa; green, plants; yellow, alveolates and heterokonts; orange, haptophytes; and brown, excavates. When present in the respective organism, motile cilia are shown in green and primary cilia in blue. The presence of the corresponding proteins is indicated by black circles. Conservation of IFT-B complex subunits is depicted by circles with shades of gray corresponding to percentage of subunits with orthologues (black, 100%; dark gray, <100%; light gray, <60%; white, <30%). Proteins with E-values ≤ 10−25 were scored as hits. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
Sequence analysis revealed several key features of PALD1 (Fig. 7 E). PALD1 contains a glycine residue at amino acid position 2 that scores highly in all predictors of N-myristoylation, and expression of the N terminus of PALD1 in a cell-free system produced a protein myristoylated at Gly2 (Suzuki et al., 2010). Importantly, PALD1 was recovered in affinity purification of the myristoyl chaperone UNC119 (Wright et al., 2011) that mediates the entry of a variety of proteins (e.g., NPHP3 and Cystin) into cilia (Stephen and Ismail, 2016). PALD1’s regulated targeting to cilia is thus likely to involve the unmasking of its attached myristate. PALD1 contains two protein tyrosine phosphatase (PTP) domains with four minimal active site motifs (CX5R) and belongs to the PTP superfamily (Chen et al., 2017). While its exact substrate specificity remains to be firmly established, phosphoinositide phosphatase activity has recently been detected in PALD1 immunoprecipitates (Nitzsche et al., 2021).
PALD1 is conserved among all clades of eukaryotic life, from protists to mammals (Fig. 7 F). Interestingly, a clustering analysis of pathways based on shared inferred ancestry (Li et al., 2014) grouped PALD1 with the cilia-associated proteins CFAP54 (cilia- and flagella-associated protein 54; McKenzie et al., 2015), the tubulin detyrosinases vasohibin 1 and 2 (Nieuwenhuis et al., 2017; Aillaud et al., 2017), and the IFT-B subunit IFT25. Although PALD1 is not restricted to ciliated organisms, it displays some coconservation with cilia (Fig. 7 F), and its phylogenetic conservation pattern most closely overlaps with that of IFT25. Together with the IFT27 protein, IFT25 forms a stable subcomplex of IFT-B that functions as a regulator of BBSome function and thus participates in the regulated removal of membrane proteins from cilia (Bhogaraju et al., 2011; Liew et al., 2014; Eguether et al., 2014; Dong et al., 2017). Metazoa that have lost IFT25/27 either lack a Hh response altogether (Caenorhabditis elegans) or transduce Hh signals independently of cilia (Drosophila melanogaster; Fig. 7 F). The shared phylogenetic pattern of IFT25/27 and PALD1 with Hh signaling and cilia suggests that PALD1 performs a function in cilia that supports efficient Hh signaling while not being absolutely required for either cilia assembly or Hh signaling. In agreement with the tissue-specific expression of PALD1 (Huang et al., 2009) and the integration of PALD1 with Hh signaling in a subset of cell lines, we propose that PALD1 fulfills a cell type–specific function in multicellular organisms.
PALD1 is a cell type–specific attenuator of Hh signaling
To assess the role for PALD1 in Hh signaling, we generated Pald1 knockout IMCD3 cells by CRISPR/Cas9-mediated genome editing (Fig. S4 A). The morphology of cilia, as assessed by acetylated tubulin or IFT88 staining, was indistinguishable between WT and Pald1−/− cells (Fig. 8 A). Immunoblotting confirmed the absence of PALD1 from Pald1−/− cells, while the levels of ciliary proteins PTCH1 and IFT88 were unaffected (Fig. 8 B). Consistent with a functional relationship between PALD1 and the Hh pathway, we detected a significant increase in full-length GLI3 (GLI3FL) in unstimulated Pald1−/− compared with WT cells (Figs. 8 B and S4 B). Underscoring the similarities in phylogenetic distribution between PALD1 and IFT25/27 further, the effect of Pald1 deletion on GLI3 processing is reminiscent of the Hh defect when Ift27 is deleted in mouse skin (Yang et al., 2015). The ratio between the full-length and repressor forms of GLI3 (GLI3FL/GLI3R) has been used as a proxy for Hh pathway activation, as this ratio increases sharply upon Hh pathway activation (Wang et al., 2000; Wen et al., 2010). In WT IMCD3 cells, the GLI3FL/GLI3R ratio increased twofold after addition of Hh (Fig. 8 C). Meanwhile, unstimulated Pald1−/− IMCD3 cells exhibit a GLI3FL/GLI3R ratio similar to that of Shh-treated WT cells, suggesting that PALD1 restricts Hh pathway activation in the absence of Hh ligand. The GLI3FL/GLI3R ratio increased even further after Hh stimulation in Pald1−/− cells, indicating that PALD1-deficient cells still remained responsive to Hh (Fig. 8 C). Consistent with a cell type–specific function for PALD1, Gli1 mRNA levels were increased severalfold in unstimulated and in Hh-treated Pald1−/− IMCD3 cells but remained unaffected by deletion of Pald1 in 3T3 cells (Fig. S4, D and E). The intact transcriptional Hh response in the absence of PALD1 in NIH-3T3 cells explains why PALD1 was missed in functional genomics screens for Hh signaling regulators as they were all conducted in NIH-3T3 cells (Jacob et al., 2011; Breslow et al., 2018; Pusapati et al., 2018b).

Full-length GLI3 levels are increased in Pald1−/− IMCD3 cells. (A) cDNA preparation of a CRIPSR/Cas9 genome-edited Pald1−/− IMCD3 cell clone was sequenced and aligned with the PALD1 WT gene sequence from M. musculus. Single-base-pair deletions in exon 4 lead to biallelic frameshift mutations and protein truncation. The DNA sequences have been analyzed using Benchling. (BandC) Quantitation of GLI3 full-length (FL) and repressor (R) forms. Lysates of WT and Pald1−/− IMCD3 cells cultured in the presence or absence of Shh were separated by SDS-PAGE and analyzed by immunoblotting (as in Fig. 8 B). Dot plots show relative GLI3 full-length (GLI3FL; B) and repressor (GLI3R; C) signals normalized to GAPDH, quantified from three independent experiments (n = 3). Horizontal lines indicate means. Each experiment was internally normalized to the relative signals in WT in the absence of signal (WT −Shh ratios = 1). (D) Indicated cell lines were serum-starved for 48 h in the presence or absence of Shh. Gli1 transcript levels were determined using reverse transcription quantitative real-time PCR. Shown are means of relative transcript levels normalized to Gapdh (n = 2). (E) Cell lysates of indicated WT and Pald1−/− cell lines were separated by SDS-PAGE and analyzed by Western blotting using PALD1- and GAPDH-specific antibodies.
Full-length GLI3 levels are increased in Pald1−/− IMCD3 cells. (A) cDNA preparation of a CRIPSR/Cas9 genome-edited Pald1−/− IMCD3 cell clone was sequenced and aligned with the PALD1 WT gene sequence from M. musculus. Single-base-pair deletions in exon 4 lead to biallelic frameshift mutations and protein truncation. The DNA sequences have been analyzed using Benchling. (BandC) Quantitation of GLI3 full-length (FL) and repressor (R) forms. Lysates of WT and Pald1−/− IMCD3 cells cultured in the presence or absence of Shh were separated by SDS-PAGE and analyzed by immunoblotting (as in Fig. 8 B). Dot plots show relative GLI3 full-length (GLI3FL; B) and repressor (GLI3R; C) signals normalized to GAPDH, quantified from three independent experiments (n = 3). Horizontal lines indicate means. Each experiment was internally normalized to the relative signals in WT in the absence of signal (WT −Shh ratios = 1). (D) Indicated cell lines were serum-starved for 48 h in the presence or absence of Shh. Gli1 transcript levels were determined using reverse transcription quantitative real-time PCR. Shown are means of relative transcript levels normalized to Gapdh (n = 2). (E) Cell lysates of indicated WT and Pald1−/− cell lines were separated by SDS-PAGE and analyzed by Western blotting using PALD1- and GAPDH-specific antibodies.

Hh pathway activity is derepressed in Pald1−/− IMCD3 cells. (A) Ciliated WT and Pald1−/− IMCD3 cells were stained for indicated proteins and DNA. (B) WT and Pald1−/− IMCD3 cells treated with or without Shh for 24 h were immunoblotted for the indicated proteins. GLI3 repressor (GLI3R) and full-length (GLI3FL) forms are indicated. (C) GLI3R and GLI3FL signals from three independent experiments as in B were quantified (see Fig. S4) and GLI3FL/GLI3R ratios plotted. Horizontal lines depict means (n = 3). Each experiment was internally normalized to the GLI3FL/GLI3R ratio in WT in the absence of signal (WT –Shh GLI3FL/GLI3R ratio = 1). (D) Ciliated WT and Pald1−/− IMCD3 cells were treated with or without SAG for 24 h and stained for the indicated proteins and DNA. (E and F) Box plots showing background-corrected, relative fluorescence normalized to acetylated tubulin signals. (E) Two independent experiments were performed, and 30 cilia were analyzed per condition in each experiment (n = 60). (F) 30 cilia per condition were analyzed from three independent experiments (n = 90). Data were analyzed using two-way ANOVA with multiple comparisons in a Tukey test with a defined confidence of 95%. *, P < 0.05; n.s., not significant. All scale bars represent 2 µm. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
Hh pathway activity is derepressed in Pald1−/− IMCD3 cells. (A) Ciliated WT and Pald1−/− IMCD3 cells were stained for indicated proteins and DNA. (B) WT and Pald1−/− IMCD3 cells treated with or without Shh for 24 h were immunoblotted for the indicated proteins. GLI3 repressor (GLI3R) and full-length (GLI3FL) forms are indicated. (C) GLI3R and GLI3FL signals from three independent experiments as in B were quantified (see Fig. S4) and GLI3FL/GLI3R ratios plotted. Horizontal lines depict means (n = 3). Each experiment was internally normalized to the GLI3FL/GLI3R ratio in WT in the absence of signal (WT –Shh GLI3FL/GLI3R ratio = 1). (D) Ciliated WT and Pald1−/− IMCD3 cells were treated with or without SAG for 24 h and stained for the indicated proteins and DNA. (E and F) Box plots showing background-corrected, relative fluorescence normalized to acetylated tubulin signals. (E) Two independent experiments were performed, and 30 cilia were analyzed per condition in each experiment (n = 60). (F) 30 cilia per condition were analyzed from three independent experiments (n = 90). Data were analyzed using two-way ANOVA with multiple comparisons in a Tukey test with a defined confidence of 95%. *, P < 0.05; n.s., not significant. All scale bars represent 2 µm. In box plots, crosses indicate mean values, whiskers indicate values within 1.5× interquartile range, and dots represent outliers.
To further investigate why pathway activity is increased in Pald1−/− IMCD3 cells, we assessed the cilia localization of SMO and GPR161 in response to Hh pathway stimulation. The Hh-dependent ciliary accumulation of SMO was unperturbed by deletion of Pald1 (Fig. 8, D and E). Meanwhile, GPR161 levels were reduced in cilia of unstimulated Pald1−/− IMCD3 cells compared with unstimulated WT cells (Fig. 8, D and F), consistent with a mild derepression of the Hh pathway. Nonetheless, GPR161 still exited cilia upon Hh pathway activation in the absence of PALD1. In conclusion, Pald1−/− IMCD3 cells respond to Hh stimulation; however, they display elevated basal pathway activity. Hence, unlike other negative regulators such as SUFU and PKA, PALD1 is not strictly required for Hh signaling but rather attenuates Hh signals in certain cell types to fine-tune cellular responses. Similarly, GPR161 functions as an attenuator of Hh signaling during tissue patterning (Pusapati et al., 2018a).
Discussion
Extensive coverage of the ciliary proteome by Cilia-APEX2
Improved controls and the introduction of quantitative MS have afforded a considerable increase in the coverage of the ciliary proteome between the first and second generations of Cilia-APEX. The inclusion of nearly all IFT and BBSome subunits as well as components of the Hh signaling pathway in the Cilia-APEX2 proteome lends confidence that the technique identifies a large fraction of the ciliary proteome.
Unexpectedly, half of the currently known components of the transition zone (TZ) were identified as hits in the Cilia-APEX2 proteome (Fig. 2 E). The TZ acts as a diffusion barrier that functionally separates the cilium from the rest of the cell and was initially considered to be a static structure of the ciliary base (Garcia-Gonzalo and Reiter, 2017; Gonçalves and Pelletier, 2017). The notion of a static TZ was further reinforced by the near absence of TZ components from prior ciliary APEX studies (Mick et al., 2015; Kohli et al., 2017). However, recent reports have revealed that while some TZ components are static, other TZ components dynamically cycle between the TZ and the cytoplasm or the ciliary shaft (Nachury and Mick, 2019). It is particularly striking to note that TZ components with previously documented dynamicity (e.g., NPHP4 and CEP290) were identified by Cilia-APEX2 while TZ components known to remain statically associated with the TZ over the timescale of tens of minutes (e.g., MKS2 and TMEM107) were missing from the Cilia-APEX2 proteome. Alternatively, it is conceivable that some TZ components identified by Cilia-APEX2 are positioned at the distal side of the TZ and hence in close proximity to the Cilia-APEX2 enzyme to be labeled by biotinyl radicals. This latter hypothesis is consistent with our identification of the TZ proteins NPHP3, INVS, and NEK8, which are present in the inversin compartment that extends from the TZ to the first few micrometers of the ciliary shaft (Shiba et al., 2010).
Time-resolved Cilia-APEX2 proteomic profiling reveals novel insights into Hh signaling
Starting with the discovery that Hh signaling requires cilia for signal transduction in vertebrates (Huangfu et al., 2003), the past 15 yr have revealed an elaborate choreography of signaling factors entering and exiting cilia: one protein (SMO) that accumulates in cilia upon Hh pathway activation, two proteins (PTCH1 and GPR161) that undergo Hh-dependent exit from cilia and four proteins (GLI2, GLI3, SUFU, and KIF7) that accumulate at the tip of cilia in Hh-stimulated cells. Because these findings relied on imaging the core Hh signaling components defined by Drosophila genetics, and since Drosophila transduce Hh signals independently of cilia, the question remained how many proteins redistributing in response to Hh were left to discover. Cilia-APEX2/TMT proteomics addressed this question by enabling a systematic quantitation of individual signaling factors in cilia in a time-resolved manner. Our analysis of the cilia proteome remodeling during Hh signaling correctly identified the Hh-dependent redistribution of SMO, PTCH1, and GPR161 and revealed two additional factors that undergo Hh-induced ciliary redistribution.
We note that the transcription factors GLI2 and GLI3, the GLI chaperone SUFU, and the GLI regulator KIF7, which all become enriched at the tip of cilia in response to Hh (Wen et al., 2010; Tukachinsky et al., 2010; Endoh-Yamagami et al., 2009; Liem et al., 2009), increased by <20% during the time course of Shh treatment in the Cilia-APEX2/TMT quantitative profiles (Fig. 4 B; although GLI2 failed the stringent inclusion criteria for the Cilia-APEX2 proteome, Cilia-APEX2/TMT did allow for robust quantitation in the 24 h time course). These findings suggest that GLI2, GLI3, KIF7, and SUFU become mobilized to the tip of cilia from a preexisting ciliary pool in response to Hh signal. Similarly, while the BBSome becomes enriched at the tip of cilia upon Hh pathway activation (Ye et al., 2018), Cilia-APEX2 signals of BBSome subunits do not change appreciably during the 24-h time course. The discovery of novel “tipping” proteins will require the development of cilia tip–APEX2 or similar methodologies.
Role of the PALD1 phosphatase inside cilia
Our analysis revealed that PALD1 functions as a cell type–specific attenuator of Hh signaling. The biochemical task performed by PALD1 inside cilia remains to be determined. At the primary sequence level, PALD1 is most closely related to bacterial phytases, a class of phosphatases that hydrolyze inositol hexakisphosphate (Alonso and Pulido, 2016). Although the activity of PALD1 against inositol hexakisphosphate remains to be tested, a recent publication found that PALD1 immunoprecipitates from mammalian cells can release phosphate from the phosphoinositides phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) and phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3). Given that hydrolysis of PI(4,5)P2 inside cilia is functionally important to maintain the balance of ciliary trafficking and hence Hh signaling (Garcia-Gonzalo et al., 2015; Chávez et al., 2015), it is conceivable that PALD1 may participate in the homeostasis of ciliary phosphoinositides.
In addition to uncovering a number of cilia-related proteins, the phylogenetic pathway clustering analysis of PALD1 identified two phosphatases co-conserved with PALD1: Phospho1, an ancient phosphoethanolamine and phosphocholine phosphatase with roles in bone mineralization in vertebrates (Dillon et al., 2019), and Phospho2, a pyridoxal-5-phosphate phosphatase related to Phospho1 (Roberts et al., 2005). Phospho1/2 belong to the haloacid dehalogenase phosphatase superfamily and bear no resemblance to the PTP family or PALD1. The coretention of Phospho1, Phospho2, and PALD1 in a variety of organism is remarkable, as the only common thread between these proteins is the release of Pi. A role for the local production of Pi in ciliary processes is an intriguing possibility.
GPR161 removal from cilia amplifies drops in ciliary PKA activity
Similar to the dual inhibition of ciliary PTCH1 by Hh ligands that block its transporter activity and promote its exit from cilia, our model of PKA regulation (Fig. 5 H) proposes that PKA activity in cilia is reduced via a four-pronged mechanism. First, activation of Gαi lowers ciliary cAMP levels; second, depression of Gαs also lowers ciliary cAMP levels; third, SMO directly inhibits PKA inside cilia; and fourth, GPR161 removes the PKA holoenzyme from cilia. Our hypothesis of ciliary Gαi activation promoting exit of GPR161 is supported by our previous observation that activation of the ciliary SSTR3 leads to the exit of GPR161 from cilia (Ye et al., 2018). Definitive support for these mechanisms awaits the development of well-validated ciliary cAMP biosensors and the direct detection of ciliary PKA-C.
Our model further proposes two levels of β-arrestin binding to GPR161. First, a labile association of β-arrestin to tonically active GPR161 in the absence of Hh pathway stimulation is required but insufficient to productively remove GPR161 from cilia. Second, our BRET data suggest increased recruitment of β-arrestin2 to GPR161 when the entry of SMO into cilia promotes PKA-mediated phosphorylation of the GPR161 C tail. The finding that mutations in GPR161 that mimic PKA phosphorylation drastically reduce the ciliary levels of GPR161 in dorsal midline cells of zebrafish (Bachmann et al., 2016) further supports that PKA phosphorylation may trigger the removal of GPR161 from cilia. We note that removing the PKA phosphorylation sites from GPR161 results in a modest but significant decrease in the ciliary levels of GPR161 (Tschaikner et al., 2021), thus pointing to a greater complexity of GPR161 ciliary trafficking regulation by PKA.
While our model accounts for Hh pathway regulation by ciliary PKA-RIα, it is important to consider the extraciliary functions of PKA-RIα when interpreting the drastic overproduction of GLI3R in unstimulated Prkar1a−/− mouse embryonic fibroblasts (Jacob et al., 2011). PKA-RIα is the only one of four PKA regulatory subunits essential for embryonic development, and partial loss of PKA-RIα leads to a wide range of disease states, including cystic kidneys (Veugelers et al., 2004; Amieux et al., 2002; Ye et al., 2017). Accordingly, Taylor and colleagues describe PKA-RIα as the “master regulator of PKA signaling” and conclude that PKA-RIα contributes the bulk of PKA activity restriction in cells (Lu et al., 2019). Cytoplasmic PKA-C activity therefore becomes greatly elevated in the absence of PKA-RIα, and GLI2/3 phosphorylation by PKA may take place outside of cilia when PKA-RIα is missing. In line with our interpretation, overexpression of a PKA-C variant that shows reduced binding to the regulatory subunits potently inhibits Hh signaling, presumably by phosphorylating GLI2/3 in the cytoplasm (Hammerschmidt et al., 1996).
Our observation that the constitutively active GPR161 fails to exit cilia in the absence of PKA-RIα suggests that GPR161 may represent the first instance of a GPCR that is not controlled by extracellular ligands but instead monitors changes in second messenger concentrations in primary cilia. The GPR161/PKA-RIα module may thus act as a simple amplifier of the state of ciliary SMO activity.
Materials and methods
Cell line generation, cultivation, and manipulation
C2C12 myoblasts and NIH-3T3 and HEK293T cells were cultured in DMEM and RPE1-hTERT (described as RPE in the text), and all IMCD3 cell lines were grown in DMEM/F12 (all supplemented with 10% FBS). The pancreatic β cell line MIN6 was cultured in DMEM supplemented with 15% FBS. Ciliation was induced by reducing the growth media to 0.2% FBS for 24 h. Transfections were performed using XtremeGene9 (Roche) or FuGene6 (Promega) according to manufacturers guidelines. Cep164 and Pald1 genes were disrupted in IMCD3 FlpIn cells using CRISPR/Cas9-mediated genome editing with gRNAs targeting exons 8 and 4, respectively (Ran et al., 2013). Clones of each cell line were obtained by limited dilution (Cep164) or single-cell sorting (Pald1). Clones with disrupted genes were screened by immunofluorescence (IF) microscopy and Western blotting using protein-specific antibodies. Selected positive clones were further characterized by sequencing, confirming missense mutation leading to early termination of translation.
IMCD3 cell lines stably expressing Cilia-APEX2, control-APEX2, and NGPKA-RIα were generated using the FlpIn system as described previously (Breslow and Nachury, 2015). Plasmids encoding Cilia-APEX2 and control-APEX2 were created by site-directed mutagenesis of Cilia-APEX and control-APEX plasmids and confirmed by sequencing. A plasmid encoding NGPKA-RIα was generated using the Gateway cloning system (Life Technologies) by LR clonase reaction of pEF5/FRT/NG-DEST with pENTR-PRKAR1A (Addgene; #23741). IMCD3 cells stably expressing GPR161NG have previously been described (Ye et al., 2018). All cell lines used in this study are listed in Table S3.
To induce Hh signaling, growth media were supplemented with either 200 nM SAG or Shh-N conditioned medium (10–16% [vol/vol] depending on batch) produced with EcR-ShhN cells (gift from Phil Beachy; Stanford University, Stanford, CA). To block Hh signaling, CYC was added to the growth medium to a final concentration of 10 µM.
APEX labeling experiments
Cells were incubated in the presence of 0.5 mM biotin tyramide for 30 min before the addition of H2O2 to a final concentration of 1 mM. For nonlabeling samples, water was added instead of H2O2. After 2 min of incubation at room temperature, the medium was aspirated quickly, and cells were washed three times with quenching buffer (1× PBS supplemented with 10 mM sodium ascorbate, 10 mM sodium azide, and 5 mM Trolox). For fluorescence microscopy, cells were immediately fixed. For proteomic and Western blot analyses, cells were lysed by scraping them off the growth surface in ice-cold lysis buffer (0.5% [vol/vol] Triton X-100, 0.1% [wt/vol] SDS, 10% [wt/vol] glycerol, 300 mM NaCl, 100 mM Tris/HCl, pH 7.5, and protease inhibitors) supplemented with 10 mM sodium ascorbate, 10 mM sodium azide, and 5 mM Trolox. After collecting the lysate in a reaction tube, the lysate was vortexed, incubated on ice for 15 min, and cleared by centrifugation (16,000 ×g for 20 min at 4°C).
Streptavidin capture
After determining protein concentrations of lysates from APEX labeling experiments, they were adjusted to equal concentrations and volumes as starting material, from which samples were taken as loading control for SDS-PAGE and Western blot analysis. Samples were added onto washed and equilibrated Streptavidin-Sepharose beads (GE Healthcare, #17-5113-01), and biotinylated proteins were allowed to bind for 1 h at room temperature. Unbound material was removed, and samples taken for Western blot (WB) analysis. Beads with bound proteins were washed extensively with lysis buffer, then with urea wash buffer (4 M urea and 10 mM Tris/HCl, pH 7.5) and finally with urea wash buffer supplemented with 50 µM biotin. For mass spectrometric analyses, bound proteins were alkylated and digested with endopeptidase Lys-C (Wako) for 3 h and trypsin (Promega) on beads overnight at 37°C.
MS
Tryptic digests were directly labeled in 200 mM Hepes, pH 8.5, with TMT 10-plex reagents (Thermo Fisher Scientific; #90406). After efficient labeling was checked by MS, peptides were subjected to alkaline reversed phase fractionation as described previously (Paek et al., 2017). Pooled fractions were analyzed on a Fusion Lumos Orbitrap mass spectrometer coupled to a Proxeon EASY-nLC 1000 liquid chromatography system (Thermo Fisher Scientific) using a synchronous precursor selection MS3 method (McAllister and Gygi, 2013). Capillary columns had an inner diameter of 100 µm and were packed with 2.6-µm Accucore beads (Thermo Fisher Scientific). Peptides were analyzed on acidic acetonitrile gradients for 5 h with MS1 (Orbitrap; resolution 120,000) scans, MS2 scans after collision-induced dissociation (CE-35) in the ion trap and MS3 precursor fragmentation by high-energy collision–induced dissociation. Reporter ions were analyzed by MS3 in the orbitrap at a resolution of 50,000. Further details on liquid chromatography and MS parameters can be found in (Paek et al., 2017).
MS data analysis
Mass spectra were processed and peptide-spectrum matches were obtained using SEQUEST (V.28, rev. 12)–based software. Searches used a size-sorted forward and reverse database of the Mus musculus proteome (UniProt accession no. UP000000589, from July 2014) with a mass tolerance of 20 ppm for precursors and a fragment ion tolerance of 0.9 D. Oxidized methionine residues were dynamically searched (+15.9949 D). A false discovery rate of 1% was set for peptide-spectrum matches following linear discriminant analysis, and the false discovery rate for final collapsed proteins was 1% as well.
Relative protein quantification used summed MS3 TMT signal/noise per protein filtered for summed signal/noise > 180 over all channels per peptide and an isolation specificity >70% for each peptide. Details of the TMT intensity quantification method can be found in Paulo et al. (2016).
Sequence analysis
The PALD1 sequence was analyzed with InterPro 83.0 and the NMT predictors (Bologna et al., 2004; Maurer-Stroh et al., 2002; Xie et al., 2016). The presence of orthologues was determined via pathway clustering by inferred models of evolution (Li et al., 2014), except for Batrachochytrium dendrobatidis, Chlorella sp., and Emiliania huxleyi, which were analyzed by BLASTp (https://blast.ncbi.nlm.nih.gov).
IF microscopy
For microscopic analyses, all cells were grown on round 12-mm #1.5 coverslips and fixed in 4% PFA for 10–15 min at room temperature. After fixation, cells were permeabilized in −20°C cold methanol for 5 min and rehydrated in 1× PBS at room temperature. After extensive washing in 1× PBS, fixed cells were blocked in blocking buffer (3% BSA and 5% serum in 1× PBS) for 30 min. After blocking, cells were incubated with primary antibody dilutions in blocking buffer for 1 h at room temperature or 4°C overnight, washed three times with 1× PBS over 15 min, and incubated with Alexa Fluor 488–, Cy3-, or Cy5-coupled secondary antibodies (Jackson ImmunoResearch; catalog numbers 115–175-206, 115–545-206, and 115–165-207; Invitrogen; catalog numbers A21131 and A31573) or Alexa Fluor 647–conjugated streptavidin (Invitrogen; S21374) in blocking buffer for 30 min. Finally, cells were washed five times in PBS and mounted on glass slides using Roti-Mount FluorCare DAPI (Carl Roth; Fig. 1; Fig. 6, C and D; Fig. 7; and Fig. 8) or DNA stained with Hoechst 33258 and mounted on glass slides using Fluoromount G (Electron Microscopy Sciences; Fig. 5; Fig. 6, E and G; and Fig. S1). APEX enzymes and YFPSMO were detected by GFP and YFP fluorescence and NGPKA-RIα and GPR161NG by NG fluorescence.
Prepared specimens were imaged on an AxioImager.M1 microscope (Carl Zeiss; Slidebook 6.0 software; Fig. 5, C, E, and G) or a Leica DMi8 (LAS X software, version 3.7.0.20979) with PlanApochromat oil objectives (63×, 1.4 NA) using appropriate filters. Images were captured using a CoolSNAP HQ (Photometrics) or Leica DFC3000 G camera system, respectively. Images were processed using ImageJ (v2.1.0/1.53c).
SDS-PAGE and Western blotting
Standard techniques were used for SDS-PAGE and Western blotting. Cell lysates were generated after washing cells with 1× PBS and scraping cells of the growth surface in solubilization buffer (25 mM Tris/HCl, 300 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100 [vol/vol], 0.1% SDS [wt/vol], 1 mM PMSF, and proteinase inhibitors [Roche; Complete EDTA-free]). Lysates were cleared by centrifugation (20,000 ×g at 4°C for 45 min), and 25 µg protein was separated on 10% Bis-Tris polyacrylamide gels and transferred onto nitrocellulose membranes. After blocking in 5% milk or Intercept (TBS) Protein-Free Blocking Buffer (LI-COR) and specific antibody decoration, membranes were washed and primary antibodies visualized using IRDye800-conjugated and IRDye680-conjugated secondary antibodies on a LI-COR Odyssey CLx laser scanner. Quantitation of bands was performed using the Image Studio Lite software (version 5.2.5).
BRET assay
For intermolecular BRET assays, we adapted published procedures (Pal et al., 2016). In brief, 800,000 HEK293T cells were grown in 6-cm culture dishes for 24 h to reach 60–70% confluence. Cells were then cotransfected with 0.1 µg Rlucβ-arrestin2 plasmid and varying amounts (0, 0.05, 0.1, and 0.2 µg) of WT or mutant GPR161YFP constructs using Jetprime DNA and siRNA transfection reagent (Polyplus Transfection) according to the manufacturer’s protocol. Total DNA amount for each transfection was adjusted to 0.6 µg by adding empty vector and complexed with 1.2 µl Jetprime reagent. 4 h after adding the transfection mix to the cells, the medium was replaced for growth medium, and cells were grown for an additional 24 h. After removal of the growth medium, cells were resuspended in 3 ml fresh growth medium and transferred into a 96-well flat black (chimney) plate (Greiner Bio-One) at 10,000 cells in 100 µl medium per well. For each condition, cells were seeded in triplicate and grown for an additional 24 h. Medium was then removed, cells carefully washed twice with 1× PBS, after which 100 µl of a 5 µM coelenterazine-h/PBS solution was added to start the BRET reaction. After 10-min incubation in the dark, bioluminescence emissions (Em) at 475 nm (Rluc) and 535 nm (YFP) were detected in a plate reader (TECAN; Spark 20M). BRET ratios (Em535/Em475) were determined and means calculated from two consecutive readings of three wells per condition. By subtracting the BRET ratio of cells transfected with Rlucβ-arrestin2 alone, net BRET ratios were calculated and plotted. Titration curves were generated by nonlinear curve fitting using the one-site specific binding option in GraphPad Prism (version 8.4.2).
Reverse transcription quantitative real-time PCR
For reverse transcription quantitative real-time PCR analysis 150,000 cells were grown in 12-well plates in regular growth medium for 24 h, followed by serum starvation to induce ciliation and addition of 12% (vol/vol) Shh-N conditioned (or control) medium to induce Hh signaling. After 48 h, cells were washed with PBS and RNA extracted using the microRNeasy kit (Qiagen) according to the manufacturer’s protocol. Isolated RNA was further cleaned up by ethanol precipitation, and 200 ng was used for RT-PCR using the QuantiTect reverse transcription kit (Qiagen). Quantitative PCR was performed on 4 ng cDNA per reaction using the StepOnePlus Real-Time PCR System (Applied Biosystems) with custom primers for Gli1 and GAPDH as described previously (Everson et al., 2018).
Statistical analyses
Statistical analyses were performed with GraphPad Prism v8.3.1. For Western blot analyses, mean values from independent experiments (exact n stated in figure legends) were calculated and are shown with either SD or SEM as described in the figure legends. For GLI3 analysis, each experiment was internally normalized to the GLI3FL/GLI3R ratio in WT cells in the absence of Shh-N. For IF experiments, at least 30 cilia were analyzed (exact n stated in figure legends). To compare biological replicates, relative fluorescence values were normalized to the average relative fluorescence signal in WT cells in the absence of inducing reagents. In all statistically analyzed experiments, significance was assessed by two-way ANOVA assuming normal distribution, and multiplicity adjusted P values were obtained by Holm–Sidak post-hoc or Tukey testing (P < 0.05 was considered statistically significant). For volcano plot graphs, Student’s t test were used and statistical analyses were performed using Prism 8 (GraphPad Software). Hierarchical cluster analyses were performed according to Ward’s minimum variance method using JMP software (Statistical Analysis System; v15.1.0).
Antibodies and reagents
Antibodies against the following proteins were used at indicated dilutions: anti-acTub (Sigma-Aldrich; T7451, mouse 1:2,000), anti-ARL13B (Proteintech; 17711–1-AP, rabbit 1:2,000), anti-IFT88 (Proteintech; 13967–1-AP, rabbit 1:200 in IF, 1:1,000 in WB), anti-GFP (raised against 6His-tagged eGFP, rabbit 1:1,000), anti-GPR161 (gift from S. Mukhopadhyay; University of Texas Southwestern, Dallas, TX; rabbit 1:500), anti-GLI3 (R&D Systems; AF3690, goat 1:1,000) anti-actin (self-made; rabbit 1:5,000), anti-GAPDH (Proteintech; 60004–1-Ig, mouse 1:2,000), anti-PALD1 (Sigma-Aldrich; HPA017343, rabbit, IF: 1:250, WB: 1:1,000), anti-SMO (Santa Cruz Biotechnology; sc-166685, mouse IgG2a in IF 1:200; Abcam; ab236465; for Fig. 5, B, D, and E), anti-PTCH1 (Abcam; ab53715, rabbit, WB 1:1,000), anti-ninein (gift from M. Bornens; Institute Curie, Paris, France; rabbit 1:10,000), anti-CEP164 (gift from T. Stearns; Stanford University; rabbit, IF 1:2,000), and γ-tubulin (Proteintech; 66320–1-AP, rabbit 1:1,000). Streptavidin-pHRP (Thermo Fisher Scientific; #21140, 1:1,000), Streptavidin-AF647 (Invitrogen; S21374, 1:1,000), SAG (Abcam; ab142160), Shh (N terminus of Shh, produced in cell line EcR-ShhN cells, a gift from Phil Beachy; Stanford University), CYC (Merck Millipore; #239806), biotin tyramide (Iris Biotech), and somatostatin (Alfa Aesar; J66168) were also used.
Online supplemental material
Fig. S1 shows characterization of the Cep164−/− cell line and the full two-way hierarchical cluster analysis of Cilia-APEX2 proteomic data presented in Fig. 2. Fig. S2 displays the full two-way hierarchical cluster analysis of time-resolved Cilia-APEX2 proteomics and representative micrographs related to Fig. 5. Fig. S3 illustrates experimental workflow for the Cilia-APEX2 profiling of the ciliary Hh response at high temporal resolution. Fig. S4 presents additional validation and characterization of the Pald1−/− cell lines. Table S1 shows Cilia-APEX2/TMT deep proteomics data. Table S2 presents the Cilia-APEX2 cilia proteome. Table S3 lists all cell lines used in this study.
Acknowledgments
We thank J. Goldstein and A. von der Malsburg for assistance in generating knockout IMCD3 cell lines; E. Krause for fluorescence-activated cell sorting; K.V. Anderson (Sloan Kettering Institute, New York, NY), J.K. Chen (Stanford University), E. Ampofo (Saarland University, Homburg, Germany), J.M. Gerdes (Helmholtz Center Munich, Munich, Germany), T. Stearns (Stanford University), S. Mukhopadhyay (University of Texas Southwestern), P.A. Beachy (Stanford University), and M.P. Scott (Stanford University) for reagents; N. Ludwig, E. Maldener, and L.K. Sanwald for experimental assistance; D.K. Breslow, S. Schuch, and S.S. Taylor for helpful discussions; and M. von Zastrow and M. Delling for comments on the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (grant SFB894/TPA-22 to D.U. Mick) and the National Institutes of Health (grant GM67945 to S.P. Gygi and grants R01GM089933 and R21HD087126 to M.V. Nachury). This work was made possible, in part, by the National Eye Institute (EY002162 Core Grant for Vision Research to M.V. Nachury) and Research to Prevent Blindness (unrestricted grant to M.V. Nachury).
The authors declare no competing financial interests.
Author contributions: E.A. May performed and analyzed most experiments. D.U. Mick and M.V. Nachury conceptualized and wrote the manuscript with support from all authors. I.G. D’Auriac conducted and analyzed the PKA-related experiments. P.S. Schuster performed and analyzed the BRET assays. M. Kalocsay and D.U. Mick established and performed the proximity labeling, TMT labeling, and MS experiments and analyzed respective data together with S.P. Gygi.

