


Marfan syndrome (MFS) is an autosomal dominant disease caused by mutations in the gene (FBN1) of fibrillin-1, a major determinant of the extracellular matrix (ECM). Functional impairment in the cardiac left ventricle (LV) of these patients is usually a consequence of aortic valve disease. However, LV passive stiffness may also be affected by chronic changes in mechanical load and ECM dysfunction. Passive stiffness is determined by the giant sarcomeric protein titin that has two main cardiac splice isoforms: the shorter and stiffer N2B and the longer and more compliant N2BA. Their ratio is thought to reflect myocardial response to pathologies. Whether this ratio and titin’s sarcomeric layout is altered in MFS is currently unknown. Here, we studied LV samples from MFS patients carrying FBN1 mutation, collected during aortic root replacement surgery. We found that the N2BA:N2B titin ratio was elevated, indicating a shift toward the more compliant isoform. However, there were no alterations in the total titin content compared with healthy humans based on literature data. Additionally, while the gross sarcomeric structure was unaltered, the M-band was more extended in the MFS sarcomere. We propose that the elevated N2BA:N2B titin ratio reflects a general adaptation mechanism to the increased volume overload resulting from the valvular disease and the direct ECM disturbances so as to reduce myocardial passive stiffness and maintain diastolic function in MFS.
Introduction
Marfan syndrome (MFS) is a systemic disease of the connective tissue linked to pathogenic variants in the fibrillin-1 (FBN1) gene, affecting 1 in 3,000–5,000 individuals worldwide (Judge and Dietz, 2005). In the majority of cases, the disorder is transmitted in an autosomal dominant manner, while up to 25% of mutations are de novo (Chiu et al., 2014). Thus, genetic variability is notably high in MFS, with over 3,000 FBN1 pathogenic variants described to date (Takeda et al., 2018). Point mutations, namely missense and nonsense, are most commonly described in MFS, accounting for approximately two-thirds of all documented mutations. Out of these, over 80% are missense mutations (Zeyer and Reinhardt, 2015). Several genotype/phenotype correlations implicating missense mutations have been identified, such as increased frequency of ectopia lentis in the presence of missense mutations altering cysteine residues within the fibrillin-1 molecule (Jondeau et al., 2011). FBN1 pathogenic variants typically interfere with the physiological function (Li et al., 2024) and morphological integrity (Godwin et al., 2023; Sulea et al., 2023) of fibrillin-1 microfibrils, which act as essential components of both the developing and the mature extracellular matrix (ECM) by orchestrating elastogenesis during tissue development and sustaining normal ECM function, respectively. Besides ensuring the structural integrity of elastic fibers, fibrillin-1 is also crucial to signal transduction within the ECM via the TGF-β pathway (Benke et al., 2013).
FBN1 mutations are classified into two groups based on their effects on fibrillin-1 molecules. While haploinsufficient (HI) variants lead to a quantitatively altered secretion of the protein in dominant negative (DN) variants, the protein is synthesized in sufficient quantity but is functionally dysregulated (Stengl et al., 2020). As fibrillin-1 is expressed in the ECM of a wide variety of connective tissues (Li et al., 2024), MFS typically presents with a systemic clinical picture. Its manifestations most commonly involve, but are not limited to, the triad of skeletal, ocular, and cardiovascular systems, as highlighted within the diagnostic criteria known as the Ghent nosology (Loeys et al., 2010). The involvement of the cardiovascular system is considered the most critical, mainly due to aortic pathology. Fibrillin-1 is highly expressed within the wall of large arteries, which explains the higher incidence of aortic dissection, the main factor that determines the life expectancy of Marfan patients (Vanem et al., 2018). Other MFS-related vascular findings include aortic and pulmonary arterial aneurysms (Loeys et al., 2010) and increased arterial tortuosity of various small- and middle-sized vessels such as the visceral, retinal, or intracranial arteries (Ágg et al., 2020; Spinardi et al., 2020; Ghoraba and Moshfeghi, 2023). Valvular pathology is also fairly common in MFS, with mitral valve prolapse being found in 65% of patients, whereas aortic and mitral valve insufficiency was identified in 40% of patients each (Sama et al., 2024). Chronic valvular dysfunction increases MFS individuals’ risk of developing chronic heart failure as a result of an incapacity to compensate for the cardiac overload (Demolder et al., 2020). However, recent findings suggest the possibility of MFS-related intrinsic myocardial dysfunction through different pathomechanisms, some directly related to the underlying genotype. A reduced quantity of fibrillin-1 in the myocardial ECM has been described to lead to dilated cardiomyopathy (DCM) via impaired mechanosignaling and contractility within the cardiac myocytes (Cook et al., 2014). Similarly, an altered fibrillin-1 microfibril network leading to the subsequent upregulation of TGF-β-related pathways was identified to cause primary contractile dysfunction in the left ventricle (LV) (Campens et al., 2015). A different approach proposed diffuse myocardial fibrosis as a cause of primary cardiac dysfunction in MFS (Karur et al., 2018).
DCM is a disease of the myocardium characterized by dilated cardiac chambers and impaired ventricular function. In ∼40% of cases, it is genetic (Hershberger et al., 2013), being most frequently associated with truncating variants (TTNtv) in the gene encoding the protein titin (TTN) (Herman et al., 2012). This giant muscle filament is the largest protein in the human body (LeWinter et al., 2007) and the third most abundant myofilament after myosin and actin (Labeit et al., 1997). Within the sarcomere, it acts as a spring connecting the Z-disk and the M-line, thus maintaining the sarcomeric structure and being responsible for the passive tension of striated muscle (Granzier and Irving, 1995). Alternative mRNA splicing during development leads to the formation of several titin isoforms in different striated muscles, which differ in spring length and hence compliance (Granzier and Labeit, 2004; Linke, 2008). Mammalian hearts coexpress two distinct titin isoforms: a smaller and stiffer N2B isoform and a longer and more compliant N2BA isoform. Myocardial isoform expression ratios vary greatly between species according to cardiac passive tension requirements. Accordingly, the N2BA:N2B ratio was found to be around 0.25 in mice, in relation to the higher heart rate and lower cardiac output, contrasted with that of ∼0.66 in larger animals such as pigs (Cazorla et al., 2000). By comparison, the titin isoform expression ratio in human myocardial tissue was between 0.47 and 0.56 in normal non-ischemic transplantation donors and non-cardiac disease subjects, respectively (Neagoe et al., 2002; Nagueh et al., 2004). Furthermore, in an intraspecies context, a similar adaptive mechanism was noticed in athletes’ myocardium, where the titin N2BA isoform was upregulated in response to sustained exercise (Kellermayer et al., 2021). Further to physiological situations, in patients with left ventricular diastolic dysfunction (DCM and chronic ischemic heart disease), the expression ratio of the N2BA titin isoform was higher than in healthy hearts (Loescher et al., 2022). The findings suggest that the different ratios at which the two isoforms are expressed reflect the level of diastolic adaptation of the myocardium, making the titin isoform ratio a reliable indicator of heart failure and other cardiac conditions.
Taking into account that the N2BA:N2B titin isoform ratio is considered to reflect the myocardial response to different pathologies, here, we measured this ratio in the heart muscle samples of a cohort of MFS patients. Furthermore, the sarcomeric layout of titin was assessed with super-resolution microscopy. We found that in the MFS myocardial sarcomere the N2BA:N2B titin ratio was significantly elevated and the M-line segment of titin was in a pre-extended state. These changes likely reflect a molecular remodeling that serves as a general adaptation mechanism to the altered cardiovascular demand in MFS.
Materials and methods
Experimental setup
The study protocol was elaborated in accordance with the Declaration of Helsinki and was reviewed and approved by institutional and national ethics committees (ETT TUKEB 7891/2012/EKU [119/PI/12.], IV/10161-1/2020/EKU, and ETT TUKEB BM/17671-3/2024). The patients gave their informed consent prior to inclusion in the study. Specimens of human myocardial tissue were obtained from the Transplantation Biobank of the Heart and Vascular Center of Semmelweis University, Budapest, Hungary. LV septum samples were harvested from 12 genetically confirmed MFS patients undergoing aortic root replacement surgeries. Immediately after removal, the samples were snap-frozen in liquid nitrogen under sterile conditions and stored at −80°C until further processing. The patients’ MFS-specific clinical data and echocardiographic data were obtained from the Marfan Registry and the Transplantation Biobank database, respectively. The sample identification codes used throughout the article are the ones given to the patients in the Transplantation Biobank database (PAP and DCM for MFS and DCM patients, respectively). Septal myocardium specimens obtained from two DCM patients without titin truncation (TTNtv−) (Kellermayer et al., 2024) and one adult male Wistar rat myocardial sample (HK9) were used as control and reference samples.
Genetic testing
The genetic diagnoses of the MFS patients were retrieved from the Marfan Registry. All 12 included patients had undergone genetic testing from blood samples following informed genetic counseling and written consent (ETT TUKEB 12751-3/2017/EKU). FBN1 gene screening was performed by next-generation sequencing (NGS) with the help of a Roche GS Junior platform and Sanger sequencing using an ABI Prism 310 Genetic Analyser (Applied Biosystems). NGS-based multigene panel analysis was performed as previously described (Stengl et al., 2020). For DCM patients, the genetic data were collected from the Transplantation Biobank database. Genetic testing was performed as reported earlier (Kellermayer et al., 2024).
Protein solubilization
The solubilization protocol was conducted as previously described (Kellermayer et al., 2024). Small (10–15 mg) LV muscle pieces were homogenized in glass Kontes Dounce tissue grinders under liquid nitrogen. Following 20 min of incubation at −20°C, the samples were solubilized at 60°C for 15 min in 50% urea buffer (8 M urea, 2 M thiourea, 50 mM Tris-HCl, 75 mM DTT, 3% SDS, and 0.03% bromophenol blue, pH 6.8) and 50% glycerol containing protease inhibitors (0.04 mM E64, 0.16 mM leupeptin, and 0.2 mM PMSF). The solubilized samples were centrifuged at 13,000 rpm for 5 min, aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C.
Titin isoform analysis
For the determination of titin expression levels, 1% sodium-dodecyl-sulfate (SDS)-agarose gel electrophoresis was performed at 16 mA/gel for 3.5 h. The gels were stained overnight with SYPRO Ruby Protein Gel Stain (Thermo Fischer Scientific) before being digitized with a Typhoon-laser scanner (Amersham BioSciences). ImageJ (National Institutes of Health [NIH]) software was used to analyze the optical density of the titin bands (Schneider et al., 2012). The relative titin isoform ratio (N2BA:N2B ratio) was calculated using the integrated band densities. The relative content of full-length titin (T1), which included both N2BA and N2B, was normalized to the myosin heavy chain (MHC) (Freiburg et al., 2000; Kellermayer et al., 2024). Titin’s proteolytic degradation product (T2) and truncated proteins detected on the gels were normalized to T1. Western blot analysis was performed to identify additional bands on the electrophoresis gels using specific N- and C-terminus–specific antibodies T12 and M8M10, respectively.
STED measurements
STED microscopic measurements were carried out on MFS LV muscle as reported recently (Kellermayer et al., 2024). Briefly, pieces of flash-frozen samples were dissected in a relaxing solution and then skinned overnight at 4°C after adding 1% Triton X-100 and protease inhibitors. Small muscle strips containing cardiomyocyte bundles were prepared and stretched from slack length to various degrees (∼40–70%) and then fixed, embedded, frozen, and cryosectioned. The sections were permeabilized in 0.2% Triton X-100/PBS, blocked with 2% BSA and 1% normal donkey serum, then labeled with anti-titin antibodies (MIR and A170; Myomedix) and phalloidin, washed with PBS, and then incubated with secondary antibody (STAR580 goat anti-rabbit IgG; Abberior GmbH). STED microscopy was performed with an Abberior Expert Line microscope (Abberior GmbH). Images were acquired using a Nikon CFI PL APO 100× (NA = 1.45) oil immersion objective coupled to avalanche photodiode detectors with spectral detection capabilities. The images were deconvoluted with Huygens Professional software (v.23.04; SVI) by using the maximum likelihood estimation (CMLE) image restoration method. The likelihood of an estimate of the object in the measured image was computed by a quality criterion with the assumption that the photon noise is governed by Poisson statistics. Intensity plot profiles were collected on the true, deconvolved images without any post-processing; hence, the profile background was zero. The intensity plot profiles were generated with Fiji (based on ImageJ v.1.52, NIH) (Schneider et al., 2012) and fitted with Gaussians to determine the epitope peak position, height, and full width at half maximum (FWHM) using Fityk 1.3.0 software.
Statistics
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, Inc.). The normality of data was checked with the Kolmogorov–Smirnov test. STED data were compared with regression analysis. Correlation studies were performed using the Pearson or Spearman tests. Results were considered to be statistically significant at P < 0.05. Data are reported as mean ± standard deviation (SD) or median (95% confidence interval).
Results
The Marfan syndrome patient cohort
The investigated MFS cohort comprised 12 individuals (six females and six males) with a mean age of 33 ± 10.92 years and a mean body mass index of 21.55 ± 3.70 kg/m2 at the time of surgery and sample collection. In all cases, the MFS diagnosis was confirmed by genetic analyses of the FBN1 gene. Table 1 summarizes the MFS-specific characteristics of the cohort and presents the FBN1 pathogenic variants identified. Six patients (50%) had missense variants with DN effect, while in the other six patients, different HI mutations were identified: two nonsense, two splice sites, one copy number variation (CNV), and one frameshift variant. According to the modified Ghent nosology (Loeys et al., 2010), systemic involvement with a score of ≥7 was established in 10 patients (83.33%), with a median systemic score of 8 (7–9). Aortic involvement in the form of ascending aortic aneurysms was present in all cases (n = 12, 100%), with one patient having a previous history of chronic Stanford type B aortic dissection. Familial history was present in five cases (41.66%). Ectopia lentis (EL) was diagnosed in four cases (30%), all associated with missense variants affecting cysteine residues (Table 1).
MFS-specific diagnostic criteria and results of FBN1 genetic testing
| Patient | Systemic score | Family history | Ectopia lentis | FBN1 gene pathogenic variant | |||||
|---|---|---|---|---|---|---|---|---|---|
| Exon | Nucleotide change | Amino acid change | Mutation effect | Type | Pathogenicity | ||||
| PAP33 | 9 | 55 | c.6697C>T | p.Pro2233Ser | Missense | DN | Likely pathogenic | ||
| PAP37 | 8 | Present | 32 | c.3959G>C | p.Cys1320Ser | Missense | DN | Likely pathogenic | |
| PAP39 | 8 | 56 | c.6856G>T | p.Gly2286Ter | Nonsense | HI | Pathogenic | ||
| PAP46 | 7 | Present | Intron 38 | c.4748-3T>G | Splice site | HI | Pathogenic | ||
| PAP57 | 13 | Present | 8 | c.762delC | p.Leu256Serfs*74 | Frameshift | HI | Pathogenic | |
| PAP70 | 9 | Present | 58 | c.7168T>C | p.Cys2390Arg | Missense | DN | Pathogenic | |
| PAP87 | 5 | Intron 35 | c.4337-2A>G | Splice site | HI | Pathogenic | |||
| PAP90 | 9 | Present | 2–4 | CNV | HI | Pathogenic | |||
| PAP93 | 7 | Present | 14 | c.1693C>T | p.Arg565Ter | Nonsense | HI | Pathogenic | |
| PAP100 | 5 | c.1282_1283delCCinsTG | p.Pro428Cys | Missense | DN | Likely pathogenic | |||
| PAP101 | 9 | Present | Present | 19 | c.2287T>G | p.Cys763Gly | Missense | DN | Likely pathogenic |
| PAP104 | 7 | Present | 24 | c.2809T>C | p.Cys937Arg | Missense | DN | Likely pathogenic | |
All patients underwent open heart surgery for aortic root replacement via valve replacement (Bentall procedure, n = 4), valve-sparing (Yacoub procedure, n = 5), or valve reimplantation (Tirone-David procedure, n = 3). Concomitant mitral valve replacement and aortic arch replacement were performed in one and three cases, respectively. The indication for surgery was annuloaortic ectasia in all cases, with associated ascending aortic aneurysm, aortic valve insufficiency, and mitral valve insufficiency in nine, five, and one cases, respectively. Seven of the surgeries were performed in an emergency setting, while the other five were elective. The median grade of regurgitation for both the aortic and mitral valves was one (0–2). In all cases, the aortic valves were tricuspid. All patients had normal LV ejection fraction (EF), with an overall mean of 60.83 ± 3.97%. No wall motion abnormalities were observed within the cohort. Mitral valve prolapse was identified in seven (58.33%) of the cases. Evidence of coronary artery disease was found in preoperative coronary angiography in two of the patients. Hypertension was present in three patients (25%). Two patients (PAP33 and PAP57) had symptomatic cardiac failure (New York Heart Association class II). Table 2 summarizes the preoperative echocardiographic characteristics of the studied cohort. Altogether, the patient cohort well covered the characteristic symptomatology of MFS.
Preoperative echocardiographic findings in the studied MFS cohort
| Measured values | Normal range | |
|---|---|---|
| Ejection fraction (%) | 60.83 ± 3.97 | 52–72 |
| LV end-systolic diameter (mm) | 35.60 ± 8.75 | 25.0–39.8 |
| LV end-diastolic diameter (mm) | 50.08 ± 7.75 | 42.0–58.4 |
| Aortic annulus diameter (mm) | 23.75 ± 1.91 | 26 ± 3 |
| Diameter at the sinuses of Valsalva (mm) | 50.42 ± 7.54 | 34 ± 3 |
| Ascending aorta diameter (mm) | 40.20 ± 9.85 | 30 ± 4 |
| Aortic valve regurgitation >1 (n) | 4 (30%) | Absent |
| Mitral valve regurgitation >1 (n) | 4 (30%) | Absent |
| Mitral valve E/A ratio | 1.32 ± 0.36 | 0.73–2.33 |
| Deceleration time (ms) | 205.11 ± 51.91 | 138–219 |
Data are shown as mean ± standard deviation. Reference values are stated according to the Recommendations of the European Society of Cardiology and The American Society of Echocardiography (Nagueh et al., 2004; Lang et al., 2015).
Proteomic analysis
To investigate the expression of titin in the MFS myocardium, we carried out proteomics on the LV muscle samples obtained from the patients. As a control, we used samples from DCM patients’ TTNtv− samples (Kellermayer et al., 2024). As a reference, we used rat LV myocardium (Kellermayer et al., 2021). The SDS–agarose electrophoresis gel showed distinguishable N2BA and N2B bands for both the MFS and DCM samples, whereas in the case of the rat reference sample, the N2B band dominated as expected (Fig. 1) (Kellermayer et al., 2021). Titin degradation products in the form of T2 bands were diffusely present. In many of the MFS samples, most notably in samples PAP100 and PAP104, the gel revealed additional, prominent bands (marked with red squares in Fig. 1) above the ones corresponding to the MHC.
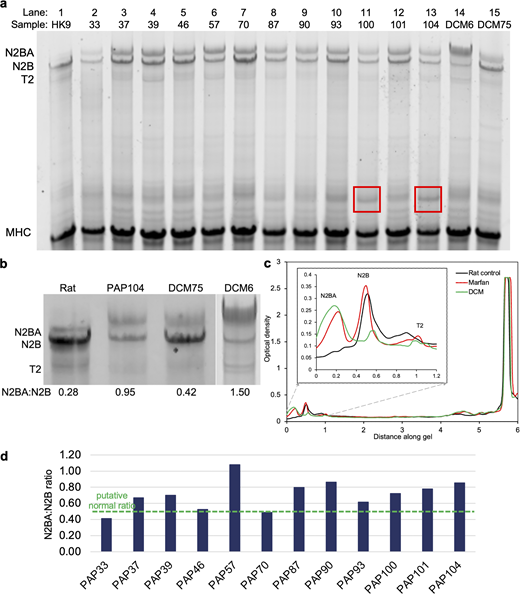
Proteomic analysis of titin. (a) 1% SDS-agarose gel analysis of the samples including left ventricular muscle (PAP) from MFS patients and samples used as control. The lanes from 1 to 15 are as follows: HK9 rat control, PAP33, PAP37, PAP39, PAP46, PAP57, PAP70, PAP87, PAP90, PAP93, PAP100, PAP101, PAP104, and DCM6 and DCM75 as controls (Kellermayer et al., 2024). (b) Magnified regions of gels showing the titin isoforms N2BA and N2B, and titin’s proteolytic cleavage product T2. (c) Typical gel profile plot, which was used to calculate the titin isoform ratios from the area under the curves. The MFS profile is that of the PAP39 sample, the DCM profile demonstrates the DCM6 sample. The two minor bands sometimes observed near the N2BA isoform were integrated to determine the total N2BA content (Freiburg et al., 2000; Kellermayer et al., 2024). (d) N2BA:N2B titin isoform ratios across the MFS patient cohort. The green segmented line indicates the average conceived normal ratio (0.5) (Nagueh et al., 2004). The mean value of the MFS N2BA:N2B ratio is 0.71 (±0.18 SD, see also Table 3). Source data are available for this figure: SourceData F1.
Proteomic analysis of titin. (a) 1% SDS-agarose gel analysis of the samples including left ventricular muscle (PAP) from MFS patients and samples used as control. The lanes from 1 to 15 are as follows: HK9 rat control, PAP33, PAP37, PAP39, PAP46, PAP57, PAP70, PAP87, PAP90, PAP93, PAP100, PAP101, PAP104, and DCM6 and DCM75 as controls (Kellermayer et al., 2024). (b) Magnified regions of gels showing the titin isoforms N2BA and N2B, and titin’s proteolytic cleavage product T2. (c) Typical gel profile plot, which was used to calculate the titin isoform ratios from the area under the curves. The MFS profile is that of the PAP39 sample, the DCM profile demonstrates the DCM6 sample. The two minor bands sometimes observed near the N2BA isoform were integrated to determine the total N2BA content (Freiburg et al., 2000; Kellermayer et al., 2024). (d) N2BA:N2B titin isoform ratios across the MFS patient cohort. The green segmented line indicates the average conceived normal ratio (0.5) (Nagueh et al., 2004). The mean value of the MFS N2BA:N2B ratio is 0.71 (±0.18 SD, see also Table 3). Source data are available for this figure: SourceData F1.
To quantitate the expression of the titin isoforms and their ratio, we carried out gel densitometry analysis (Fig. 1, b and c). The N2BA:N2B ratios in the specific patient samples are shown in Fig. 1 d. The gel densitometry analysis revealed that the mean N2BA:N2B titin isoform ratio in the MFS myocardium was 0.71 ± 0.19, which was significantly higher than in the control rat heart (0.28), but somewhat smaller than in patients with DCM (0.80 ± 0.02) (Kellermayer et al., 2024). All in all, the obtained values for the MFS samples were significantly higher than the ones reported in the literature for normal heart tissue (Nagueh et al., 2004). The expression levels of the N2BA and N2B isoforms in the MFS myocardium were rather similar to DCM samples, which showed a marked dominance of the N2BA isoform, and the control rat heart sample, where the stiffer N2B isoform was dominant (Fig. 1 c). The total titin (T1) to MHC ratio was significantly lower in MFS than in DCM patients (0.15 ± 0.01 and 0.22 ± 0.01, respectively, P < 0.05). Titin degradation, reflected by the T2:MHC ratio, did not vary significantly between the two groups (0.05 ± 0.002 versus 0.08 ± 0.02). Both parameters were comparable with values previously reported in the literature (Nagueh et al., 2004). Nevertheless, the total titin content was reduced in the studied MFS samples compared with DCM patients. The MFS samples also showed an increased mean expression level of the more compliant N2BA isoform compared with normal heart tissue, as previously reported in DCM samples (Kellermayer et al., 2024) (Table 3). In sum, the most important observation is that the N2BA:N2B ratio was markedly increased, while the amount of full-length titin was reduced in MFS.
Proteomic analysis of titin isoforms in the MFS samples
| N2BA/N2B | N2BA/MHC | N2B/MHC | T1/MHC | TT/MHC | T2/MHC | |
|---|---|---|---|---|---|---|
| PAP33 | 0.41 | 0.02 | 0.06 | 0.08 | 0.13 | 0.05 |
| PAP37 | 0.67 | 0.07 | 0.11 | 0.18 | 0.23 | 0.05 |
| PAP39 | 0.70 | 0.07 | 0.10 | 0.17 | 0.22 | 0.05 |
| PAP46 | 0.53 | 0.06 | 0.11 | 0.16 | 0.21 | 0.05 |
| PAP57 | 1.08 | 0.08 | 0.07 | 0.15 | 0.19 | 0.04 |
| PAP70 | 0.49 | 0.06 | 0.13 | 0.19 | 0.24 | 0.04 |
| PAP87 | 0.80 | 0.07 | 0.08 | 0.15 | 0.19 | 0.04 |
| PAP90 | 0.87 | 0.06 | 0.07 | 0.12 | 0.16 | 0.03 |
| PAP93 | 0.62 | 0.06 | 0.09 | 0.15 | 0.20 | 0.05 |
| PAP100 | 0.72 | 0.05 | 0.07 | 0.11 | 0.15 | 0.04 |
| PAP101 | 0.78 | 0.07 | 0.09 | 0.16 | 0.21 | 0.05 |
| PAP104 | 0.86 | 0.07 | 0.08 | 0.14 | 0.20 | 0.06 |
| Average | 0.71 | 0.06 | 0.09 | 0.15 | 0.19 | 0.05 |
| SEM | 0.05 | 0.004 | 0.01 | 0.01 | 0.01 | 0.002 |
| SD | 0.19 | 0.01 | 0.02 | 0.03 | 0.03 | 0.01 |
Relationship between titin isoform expression and LV function
To elucidate whether the titin isoform expression ratio in MFS LV myocardium correlates with cardiac performance, we searched for possible correlations between the N2BA:N2B titin isoform expression ratio and various clinical and echocardiographic structural and functional parameters (Fig. 2). The parameters included the MFS systemic score (Loeys et al., 2010) (Fig. 2 a), LV EF (Fig. 2 b), LV end-systolic (Fig. 2 c) and -diastolic (Fig. 2 d) diameters, aortic root (Fig. 2 e), sinus Valsalva (Fig. 2 f), and ascending aorta (Fig. 2 g) diameters, aortic (Fig. 2 h) and mitral valve (Fig. 2 i) regurgitation, mitral valve E/A ratio (Fig. 2 j), and deceleration time (Fig. 2 k). Overall, three patients (PAP33, PAP90, and PAP100) had dilated LV based on measured end-systolic and -diastolic diameters (ESD and EDD). However, presumably due to the limited sample size and the otherwise generally normal cardiovascular status of the patients, no conclusive associations could be found between the clinical (MFS Systemic Score), structural (aortic root, sinus Valsalva, ascending aorta diameters), and functional (EF, ESD, EDD, aortic and mitral valve regurgitation, mitral valve E/A ratio, deceleration time) parameters and the titin N2BA:N2B ratio.

Comparison of the N2BA:N2B isoform ratio with clinical parameters. (a) MFS systemic score as a function of the N2BA:N2B ratio. The systemic score takes into account the presence of typical MFS-related body deformities and its maximal value is 20 (Loeys et al., 2010). (b–k) Echocardiographic parameters as a function of the N2BA:N2B ratio. Pearson’s correlation coefficient r2 varied between 0.001 (for ejection fraction) and 0.362 (for mitral valve E/A ratio).
Comparison of the N2BA:N2B isoform ratio with clinical parameters. (a) MFS systemic score as a function of the N2BA:N2B ratio. The systemic score takes into account the presence of typical MFS-related body deformities and its maximal value is 20 (Loeys et al., 2010). (b–k) Echocardiographic parameters as a function of the N2BA:N2B ratio. Pearson’s correlation coefficient r2 varied between 0.001 (for ejection fraction) and 0.362 (for mitral valve E/A ratio).
Western blot analysis
To investigate the nature of the additional protein bands observed on the electrophoresis gel (see Fig. 1 a), we performed western blot analyses using the T12 and M8M10 titin-specific antibodies that bind to the N- and C-termini of titin, respectively (Fig. 3). Only the T12 antibody labeled, albeit minimally, the additional protein bands (Fig. 3 a); with the M8M10 antibody, we did not find any labeling (Fig. 3 b). The results suggest that the additional bands that were observed most vividly in the cases of the PAP100 and PAP104 MFS samples were probably the N-terminal part of titin complementary to T2.
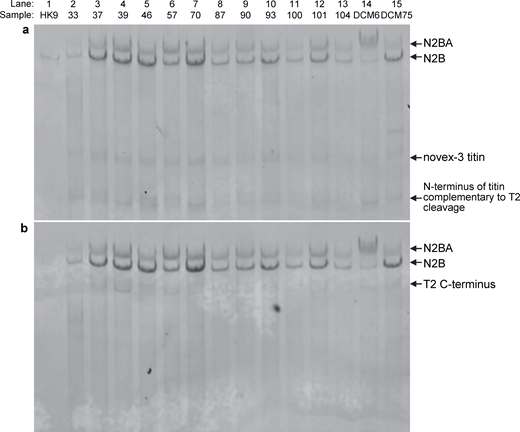
Western blot analysis. The lanes are identical to the ones in Fig. 1. (a) Western blot using the T12 anti-titin antibody, which labels titin towards its N-terminus. (b) Western blot using the M8M10 anti-titin antibody, which labels titin near its C-terminus. The smaller or partial titins are also indicated. Source data are available for this figure: SourceData F3.
Western blot analysis. The lanes are identical to the ones in Fig. 1. (a) Western blot using the T12 anti-titin antibody, which labels titin towards its N-terminus. (b) Western blot using the M8M10 anti-titin antibody, which labels titin near its C-terminus. The smaller or partial titins are also indicated. Source data are available for this figure: SourceData F3.
Super-resolution microscopy
The sarcomeric layout of titin in the MFS myocardium was investigated with super-resolution (STED) microscopy on samples labeled with sequence-specific anti-titin antibodies (MIR and A170). As a control, we used DCM samples in which titin truncation could not be detected (Kellermayer et al., 2024). To explore the sarcomeric arrangement of titin, the samples were exposed to stretch so as to reveal the position of the titin epitopes as a function of sarcomere length (SL). The results are shown in Fig. 4. The overall structural arrangement of the MFS (“Marfan+”) sarcomere was regular, and the epitopes and their sarcomeric positions could be well identified and quantified (Fig. 4 b). The distance between the A-band-spanning MIR epitopes, which reflects the length of the A-band titin section, increased monotonically as a function of SL (Fig. 4, c and d). While the slope of the increment was identical in the control and MFS, the intercept was greater in the MFS data, indicating that the A-band titin section is ∼43 nm longer throughout the SL range investigated (Fig. 4 d). We observed an even greater difference between the control and MFS in the behavior of the M-line to titin kinase distance (Fig. 4, e and f). While in the control this distance increased monotonically, in MFS it remained invariant as a function of SL (Fig. 4 f). Although A-band width is somewhat lower than expected (∼1.6 µm) (Tonino et al., 2017), it is similar to that observed recently in DCM sarcomeres (Kellermayer et al., 2024) and might be related to the increased cardiomyocyte stiffness found in MFS samples (Aalders et al., 2020). Altogether, we found significant differences in the sarcomeric layout and stretch response of titin in the MFS samples.

STED microscopic analysis of LV muscle samples from MFS patients. The samples were labeled with the MIR and A170 anti-titin antibodies. (a) Representative STED image of a control sarcomere from a non-MFS (TTNtv− DCM) sample used as control (top). Intensity plot profile of the image (bottom). (b) Representative STED image from a MFS sample (top). Intensity plot profile of the image (bottom). (c) MIR–MIR distance, corresponding to the width of the A-band in the control sample as a function of sarcomere length. (d) Magnified view of the data in the sarcomere-length range of 1.8–2.6 µm, with a linear fit to the data points (nCTRL = 884, nMFS = 568). The slope and intercept of the lines are 0.24 and 0.22 µm/µm, and 0.97 and 1.04 µm, for the control and MFS, respectively. The slopes are statistically indifferent (P = 0.18). The intercepts are significantly different (P < 0.0001). Thus, at a slack sarcomere length of 1.6 µm, the MFS A-band is longer by 43 nm than the control. (e) M-line to titin kinase distance as a function of sarcomere length. (f) Magnified view of the data in the sarcomere-length range of 1.8–2.6 µm, with a linear fit to the data points (nCTRL = 879, nMFS = 571). The slope and intercept of the lines are 21.4 and 3.961 nm/µm, and 20.61 and 86.29 nm, for the control and MFS, respectively. The slopes are significantly different (P < 0.0001), and the slope of the MFS data is not significantly different from zero (P = 0.0927). Thus, at a slack sarcomere length of 1.6 µm, the MFS titin kinase region is longer by 38 nm than the control. (g) Schematic model of the structural and mechanical changes in the MFS cardiac sarcomere. Gaussian functions above the sarcomere schemes indicate the intensity profiles of the anti-titin epitopes used in this work. The red arrowheads indicate the epitope positions in the respective titin molecules. The blue double arrow indicates the increase in the MIR–MIR distance upon stretch. The red flat-head double arrow indicates the essentially nonvariant M-line-to-A170 distance during the sarcomere stretch. The jagged M-line in the pre-stretch MFS sarcomere is shown merely to illustrate, in an exaggerated way, the structural–functional problem in this region.
STED microscopic analysis of LV muscle samples from MFS patients. The samples were labeled with the MIR and A170 anti-titin antibodies. (a) Representative STED image of a control sarcomere from a non-MFS (TTNtv− DCM) sample used as control (top). Intensity plot profile of the image (bottom). (b) Representative STED image from a MFS sample (top). Intensity plot profile of the image (bottom). (c) MIR–MIR distance, corresponding to the width of the A-band in the control sample as a function of sarcomere length. (d) Magnified view of the data in the sarcomere-length range of 1.8–2.6 µm, with a linear fit to the data points (nCTRL = 884, nMFS = 568). The slope and intercept of the lines are 0.24 and 0.22 µm/µm, and 0.97 and 1.04 µm, for the control and MFS, respectively. The slopes are statistically indifferent (P = 0.18). The intercepts are significantly different (P < 0.0001). Thus, at a slack sarcomere length of 1.6 µm, the MFS A-band is longer by 43 nm than the control. (e) M-line to titin kinase distance as a function of sarcomere length. (f) Magnified view of the data in the sarcomere-length range of 1.8–2.6 µm, with a linear fit to the data points (nCTRL = 879, nMFS = 571). The slope and intercept of the lines are 21.4 and 3.961 nm/µm, and 20.61 and 86.29 nm, for the control and MFS, respectively. The slopes are significantly different (P < 0.0001), and the slope of the MFS data is not significantly different from zero (P = 0.0927). Thus, at a slack sarcomere length of 1.6 µm, the MFS titin kinase region is longer by 38 nm than the control. (g) Schematic model of the structural and mechanical changes in the MFS cardiac sarcomere. Gaussian functions above the sarcomere schemes indicate the intensity profiles of the anti-titin epitopes used in this work. The red arrowheads indicate the epitope positions in the respective titin molecules. The blue double arrow indicates the increase in the MIR–MIR distance upon stretch. The red flat-head double arrow indicates the essentially nonvariant M-line-to-A170 distance during the sarcomere stretch. The jagged M-line in the pre-stretch MFS sarcomere is shown merely to illustrate, in an exaggerated way, the structural–functional problem in this region.
Discussion
In the present work, we investigated the isoforms and sarcomeric layout of titin in the MFS myocardium. MFS is a heritable systemic connective tissue disorder with important consequences on the cardiovascular system, among which aortic root dilation and mitral valve prolapse are the most common primary manifestations, with estimated prevalences of up to 80% and 40%, respectively (Milewicz et al., 2005; Rybczynski et al., 2010). Tricuspid valve degeneration has also been described in MFS, occurring in 12% of patients undergoing aortic or valvular surgery (Gu et al., 2015). Significant multivalvular regurgitation may lead to LV dilation and dysfunction through chronic volume overload. In 5% of MFS patients with mitral valve regurgitation, cardiac failure can develop (Rybczynski et al., 2011). However, ventricular dysfunction and underlying myocardial disease seem to occur in MFS patients even in the absence of significant valvular involvement and during childhood (Alpendurada et al., 2010; Muiño-Mosquera et al., 2020; Weigand et al., 2024).
In the myocardium, the giant elastic protein titin is responsible for the passive stiffness of the cardiac sarcomere, functioning as a molecular spring between the Z-disk and the M-band (Granzier and Irving, 1995; Granzier and Labeit, 2004; Linke, 2008). As a regulatory mechanism, alternative splicing in a specific region of titin generates the two cardiac-specific isoforms, namely the shorter and stiffer N2B and the longer and more compliant N2BA, which determine the titin-dependent passive tension in the heart muscle (Trombitás et al., 2000). Therefore, the expression levels of the two isoforms vary according to tensile requirements not only in physiological contexts, during pre- and postnatal development, and between species, but were also related to cardiac disease (Cazorla et al., 2000; Warren et al., 2004). In a tachycardia-induced canine DCM model, the N2BA:N2B titin ratio was found to shift toward the N2B isoform, accompanied by an increase in passive stiffness (Wu et al., 2002). Similar effects were observed in a hypertensive rat model, in response to the increased afterload (Warren et al., 2003). Alternatively, an increased N2BA:N2B isoform ratio emerges in human DCM hearts, reflecting a loss in passive stiffness (Makarenko et al., 2004; Nagueh et al., 2004). As recent advances support the existence of intrinsic myocardial dysfunction in MFS, it is thus reasonable to investigate the role of sarcomeric titin isoforms in the development of MFS cardiomyopathy.
Here, we found that the total amount of titin (i.e., the sum of N2BA, N2A, and T2) normalized to MHC was 0.19 (±0.03) in the MFS cohort. This value is comparable with those found earlier in the normal human myocardium (0.20 ± 0.02 and 0.23 ± 0.03) (Morano et al., 1994; Nagueh et al., 2004) and to that observed in the TTNtv− DCM myocardium (0.19 ± 0.05), which we used for comparison in the present work. The relative amount of full-length titin (T1/MHC) (0.15 ± 0.03), however, was smaller than that found recently in the DCM myocardium (0.22 ± 0.08) (Kellermayer et al., 2024), suggesting that titin in the MFS sarcomere might be exposed to proteolytic degradation. While a relationship between MFS and the activation of protein degradation pathways needs to be investigated, the extra bands found in the electrophoretograms (Fig. 1 a) may reflect this possibility. Importantly, we found that the mean N2BA:N2B titin isoform expression ratio was 0.71 ± 0.19, a value significantly higher than those reported for human control myocardial tissue (0.56 ± 0.06) and normal donor hearts (∼0.4) (Neagoe et al., 2002; Nagueh et al., 2004), albeit comparable with that observed in TTNtv− DCM (0.8 ± 0.24) (Kellermayer et al., 2024). We observed no association of the elevated N2BA:N2A isoform ratio with any of the relevant clinical, structural, and functional diagnostic parameters (Fig. 2), but this is likely due to the relatively small size of the cohort which puts a limit on statistical power. Conceivably, however, the shift in titin expression toward the longer isoform reflects a general adaptive and compensatory molecular remodeling response mandated by the changes in the mechanical status of the MFS myocardium.
The STED microscopic data indicated that beyond the titin isoform shift, there is also an interesting alteration in the way titin is arranged in the MFS sarcomere. It appears as if the A-band section of the MFS sarcomeric titin is in a pre-extended (by about 43 nm) state at the expense of an over-stretched region near the M-line and the kinase domain (Fig. 4), even though the extensibility of the A-band titin is similar in the control and MFS sarcomere. Whether the structural arrangement of titin might also be altered in the I-band needs to be investigated in the future by using antibodies specific to this region of the molecule. While the structural origin of titin’s pre-extension in the M-line–titin kinase region and its relation to the isoform shift are yet to be uncovered, the structural changes associated with the M-line region of the sarcomere in MFS may reflect an altered mechanosensory status of titin (Fig. 4 g). The relationship with titin’s mechanosensory function and the altered ECM is an intriguing possibility. First, diffuse fibrotic structural changes have been recently shown in the myocardium of a few MFS patients by using cardiac magnetic resonance imaging (Demolder et al., 2024). Screening for fibrosis in biopsies is certainly valuable and needs to be investigated in future experiments. Second, we speculate that in MFS the dysfunctional ECM indirectly alters titin’s function through mechanical stress redistribution and excessive TGF-β signaling. A structurally impaired fibrillin/elastin network along with fibrotic changes may increase ECM stiffness, thus exacerbating titin’s mechanical load and leading to the compensatory shift in titin isoforms. At the same time, the defects in fibrillin-1 lead to reduced TGF-β sequestering capability of the fibrillin-1 microfibrils and, consequently, to the overactivation of the TGF-β pathway (Benke et al., 2013), which can ultimately influence titin phosphorylation, therefore indirectly modulating its elasticity. Evidence linking changes in ECM with M-line stiffness exists but is limited (Biquand et al., 2021) and needs to be explored further.
We need to emphasize that the relatively small size and large clinical heterogeneity of the cohort place limitations on the present study, which hinders the analysis of potential correlations between molecular and clinical parameters. Despite this, the overall consistency of the proteomic data and the surprising shift in titin’s sarcomeric layout warrant further investigation into the relationship between MFS and the role titin may play in the pathogenesis of this severe disease.
In conclusion, here we demonstrated that an isoform shift occurs toward the longer and more compliant N2BA titin isoform in the MFS myocardium. The N2BA:N2B isoform ratio was 0.71 in the studied MFS population, much higher than that in the normal human myocardium. Furthermore, as revealed by super-resolution microscopy, there appears to be a structural shift in the M-line region of titin. We speculate that the changes in titin isoform expression and its sarcomeric layout reflect a general adaptive response (molecular remodeling) to the MFS-specific altered mechanical status.
Data availability
All data are available from the corresponding author upon reasonable request.
Acknowledgments
Henk L. Granzier served as editor.
The authors express gratitude toward the Laboratory of Molecular Genetics, Central Hospital of Southern Pest, National Institute of Hematology and Infectious Diseases, Budapest, Hungary, the Institute of Genomic Medicine and Rare Disorders, Semmelweis University, Budapest, Hungary, and the Department of Pathology and Experimental Cancer Research, Semmelweis University, Budapest, Hungary, for help with sequence analysis.
This research was supported by grants from the Hungarian National Research, Development and Innovation Office (K135076 to B. Merkely, K135360 to M. Kellermayer, FK135462 to B. Kiss, and FK145928 to K. Benke), the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00314/24/5 to D. Kellermayer and BO/00242/24 to K. Benke), and the New National Excellence Program of the Ministry for Culture and Innovation from the source of the National Research, Development, and Innovation Fund (ÚNKP-19-3-I to D. Kellermayer, ÚNKP-23-3-II-SE-22 to C.M. Șulea). Projects no. RRF-2.3.1-21-2022-00003 (National Cardiovascular Laboratory) and no. RRF-2.3.1-21-2022-00004 (Artificial Intelligence National Laboratory) were implemented with the support provided by the European Union. TKP2021-EGA-23 and TKP2021-NVA-15 have been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA and TKP2021-NVA funding schemes, respectively. This project has received funding from the HUN-REN Hungarian Research Network.
Author contributions: D. Kellermayer: Conceptualization, Formal analysis, Investigation, Visualization, Writing - original draft, Writing - review & editing, C.M. Șulea: Formal analysis, Visualization, Writing - original draft, Writing - review & editing, H. Tordai: Investigation, Writing - review & editing, K. Benke: Investigation, Resources, Supervision, Writing - original draft, M. Pólos: Writing - review & editing, B. Ágg: Data curation, Project administration, Software, Writing - review & editing, R. Stengl: Data curation, Investigation, Methodology, Writing - review & editing, M. Csonka: Data curation, Visualization, Writing - original draft, Writing - review & editing, T. Radovits: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing - review & editing, B. Merkely: Data curation, Formal analysis, Investigation, Supervision, Writing - review & editing, Z. Szabolcs: Conceptualization, Funding acquisition, Supervision, Writing - review & editing, M. Kellermayer: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing, B. Kiss: Data curation, Formal analysis, Investigation, Resources, Software, Visualization, Writing - original draft, Writing - review & editing.
References
This work is part of a special issue on Myofilament Structure and Function.

