


Hypertrophic cardiomyopathy (HCM) is a genetic disease of the heart characterized by thickening of the left ventricle (LV), hypercontractility, and impaired relaxation. HCM is caused primarily by heritable mutations in sarcomeric proteins, such as β myosin heavy chain. Until recently, medications in clinical use for HCM did not directly target the underlying contractile changes in the sarcomere. Here, we investigate a novel small molecule, RLC-1, identified in a bovine cardiac myofibril high-throughput screen. RLC-1 is highly dependent on the presence of a regulatory light chain to bind to cardiac myosin and modulate its ATPase activity. In demembranated rat LV trabeculae, RLC-1 decreased maximal Ca2+-activated force and Ca2+ sensitivity of force, while it increased the submaximal rate constant for tension redevelopment. In myofibrils isolated from rat LV, both maximal and submaximal Ca2+-activated force are reduced by nearly 50%. Additionally, the fast and slow phases of relaxation were approximately twice as fast as DMSO controls, and the duration of the slow phase was shorter. Structurally, x-ray diffraction studies showed that RLC-1 moved myosin heads away from the thick filament backbone and decreased the order of myosin heads, which is different from other myosin inhibitors. In intact trabeculae and isolated cardiomyocytes, RLC-1 treatment resulted in decreased peak twitch magnitude and faster activation and relaxation kinetics. In conclusion, RLC-1 accelerated kinetics and decreased force production in the demembranated tissue, intact tissue, and intact whole cells, resulting in a smaller cardiac twitch, which could improve the underlying contractile changes associated with HCM.
Introduction
Hypertrophic cardiomyopathy (HCM) is an inherited cardiomyopathy and a leading cause of heart failure and sudden cardiac death. It is characterized by thickening of the left ventricle (LV) with reduced chamber size, hypercontractility, impaired relaxation, and increased energy consumption. It affects ∼1:500 individuals with two-thirds eventually developing obstructive HCM, where the intraventricular septum asymmetrically thickens causing obstruction of the LV outflow tract during contraction (Maron and Maron, 2013; Marian and Braunwald, 2017; Ho et al., 2018; Kawana et al., 2022). Traditional pharmacological treatments include β-blockers and calcium channel blockers. While these interventions can often manage the disease in the short term, they do not present a long-term solution as the disease progresses. More advanced options are available, such as implantation of a cardioverter-defibrillator device, myectomies for obstructive HCM, and heart transplantation, but each provides significant risk to those in heart failure.
HCM is primarily due to mutations in proteins of the sarcomere. In 1990, Geisterfer-Lowrance and colleagues identified the first mutation linked to HCM in the gene encoding β-myosin heavy chain (Geisterfer-Lowrance et al., 1990). Many more mutations in sarcomere proteins, such as myosin-binding protein C and troponin, have been linked to HCM in the intervening years (Ho et al., 2018). Hypercontractility resulting from these mutations is thought to arise from either changes in the myosin ATPase cycle, the number of myosin heads functionally available to interact with actin, the load-dependence of contractility, or a combination of these factors (Spudich, 2019). This hypercontractility of myofibrils can lead to the activation of downstream signaling pathways and result in hypertrophy, fibrosis, myofilament disarray, and maladaptive energetics (Marian and Braunwald, 2017).
Over the last 15–20 years, there has been a push to develop therapies that directly target the root cause of the disease by inhibiting contractility at the myofibril level (Lehman et al., 2022; Day et al., 2022). In 2022, the first-in-class myosin inhibitor Mavacamten (now marketed as Camzyos) was approved by the FDA for the treatment of obstructive HCM (Keam, 2022) and is now being used in clinical care. Mavacamten is a small molecule that binds directly to myosin and inhibits myosin ATPase activity, stabilizes the interacting heads motif (folded back myosin heads), and increases ADP release and crossbridge detachment (Anderson et al., 2018, Preprint; Green et al., 2016). A second myosin inhibitor, Aficamten, which is less well-studied, also binds to the myosin head and reduces the contractility of cardiac muscle (Chuang et al., 2021; Maron et al., 2023). In the phase 2 clinical trial, treatment of obstructive HCM patients with Aficamten resulted in a significant reduction in left ventricular outflow track gradients with improved patient symptoms (Maron et al., 2023).
Here, we introduce a novel myosin inhibitor (RLC-1) that is dependent on the presence of myosin regulatory light chain (RLC). RLC-1 was discovered in a high-throughput screen of bovine cardiac myofibril ATPase. RLC-1 greatly inhibits ATPase in bovine cardiac heavy meromyosin (HMM), however, not when RLC is removed. In ex vivo studies, we show that RLC-1 inhibits force production in both myofibrils and demembranated trabeculae. Interestingly, RLC-1 also moves myosin heads away from the thick filament backbone and accelerates steady-state and dynamic contractile kinetics. We hypothesize that RLC-1, while increasing the pool of available myosin heads, impairs the ability of myosin heads to bind and activate the thin filament, as well as the ability to remain bound. This results in quicker detachment and an overall reduction in the time heads are strongly bound and producing force. These changes at the level of the myofibrils contribute to a reduction of peak force and a decrease in contractile duration in both isolated cardiomyocytes and intact trabeculae.
Materials and methods
ATPase and MST protein preparation
Myosin subfragment-1 (S1) and HMM were prepared as previously described (Kawas et al., 2017). Briefly, bovine hearts were harvested, immediately placed on ice, and shipped overnight. The left ventricle and septum were then dissected and frozen in liquid nitrogen and stored at −80°C until further processing. Full-length myosin was prepared from frozen bovine cardiac left ventricle tissue followed by α-chymotryptic digestion to make myosin S1 or HMM as described previously (Margossian and Lowey, 1982). Bovine and rabbit cardiac myofibrils were prepared from left ventricular tissue as previously described (Patel et al., 2013). In brief, left ventricular tissue was treated with two homogenizations in 1 ml of a standard buffer with Triton X-100 (75 mM KCl, 10 mM imidazole, pH 7.2, 2 mM MgCl2, 2 mM EGTA, 1 mM NaN3, and 1% vol/vol Triton X-100). Following centrifugation, pellets were washed twice with 1 ml standard buffer without Triton X-100 and resuspended in the assay buffer (A-70 containing 70 mM NaCl, 10 mM MgCl2, and 40 mM MOPS, pH 7.0). Bradford assays were performed to determine the protein concentration of the sample. Actin was prepared as previously described (Spudich and Watt, 1971) from cardiac acetone powder (Pel Freez Biologicals). For depletion of RLC, HMM was treated with 1× depletion buffer (0.5% Triton X-100, 5 mM CDTA, 200 mM KCl, and 20 mM Tris, pH 7.5) for 1 h. The dissociated RLC was separated from HMM by dialysis overnight in HMM Dialysis-1 buffer (200 mM KCl, 50 mM MOPS, pH 7.2, and 1 mM DTT) using 100 K dialysis tubing. For RLC phosphorylation, recombinant RLC protein was diluted to 2 mg/ml in the following buffer: 20 mM Imidazole, pH 7.5, 4 mM MgCl2, 150 mM NaCl, 1 mM CaCl2, and 1 mM DTT. The reaction starts by adding 2 mM ATP, 0.02 mg/ml CaM (Thermo Fisher Scientific), and 10 μl of the skeletal MLCK (Thermo Fisher Scientific) to 50 μl of the protein dilution mix. This was left at room temperature for 30 min and then overnight at 4°C to ensure complete phosphorylation. Non-phosphorylated or phosphorylated RLC was then added back to RLC-depleted HMM to assess ATPase activity. Sample proteins used for ATPase and MST experiments are shown in Fig. S1.
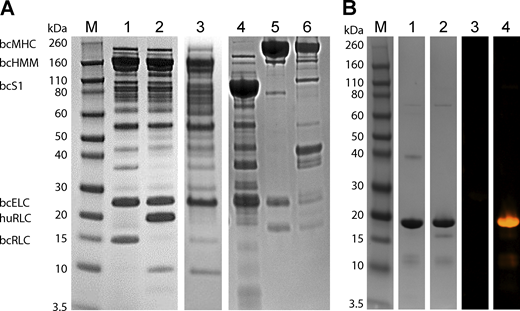
Coomassie and ProQ diamond staining of ATPase and MST protein samples. (A) Coomassie-stained gel showing lane M: Novex Sharp pre-stained protein ladder, 1: bcHMM, 2: bcHMM with human cardiac RLC, 3: bcHMM RLC-depleted, 4: bcS1, 5: bc full-length myosin used to make bcHMM and bcS1, 6: bcMF. (B) Coomassie (1, 2) and ProQ diamond stain (3, 4) showing recombinant cRLC before (1, 3) and after (2, 4) in vitro phosphorylation with MLCK. Source data are available for this figure: SourceData FS1.
Coomassie and ProQ diamond staining of ATPase and MST protein samples. (A) Coomassie-stained gel showing lane M: Novex Sharp pre-stained protein ladder, 1: bcHMM, 2: bcHMM with human cardiac RLC, 3: bcHMM RLC-depleted, 4: bcS1, 5: bc full-length myosin used to make bcHMM and bcS1, 6: bcMF. (B) Coomassie (1, 2) and ProQ diamond stain (3, 4) showing recombinant cRLC before (1, 3) and after (2, 4) in vitro phosphorylation with MLCK. Source data are available for this figure: SourceData FS1.
Compound discovery and ATPase
Steady-state ATPase measurements were conducted using a coupled enzyme system utilizing pyruvate kinase and lactate dehydrogenase. This enzyme system couples the formation of ADP to the oxidation of NADH, leading to an absorbance change at 340 nm. Unless otherwise stated, the buffering system used in all experiments was 12 mM Pipes, 2 mM MgCl2, and 2 mM BME at pH 6.8 (PM12 buffer) in the presence of BSA. All measurements were carried out in a 384-well microtiter plate at room temperature using an Envision plate reader to monitor the change in absorbance as a function of time. Dose responses of RLC-1 were conducted by monitoring the system ATPase rate in the presence of varying concentrations of RLC-1. The compound was added to the solutions from a DMSO stock and the final concentration of DMSO was kept constant at 3.3% in all experiments. In all dose-response experiments, a DMSO-only control and a 100% inhibition positive control were included to generate the normalized activity data. Calcium responses of myofibrils were conducted by measuring the steady-state ATPase rates in the presence of varying calcium concentrations. The 50% calcium activation concentration is the concentration at which the observed ATPase reached half of the maximal value. The effect of RLC-1 on the calcium sensitivity of cardiac myofibrils was evaluated using the ATPase assay as described above. Calcium response curves in the presence of DMSO or RLC-1 at multiple concentrations were plotted using GraphPad Prism.
Microscale thermophoresis (MST)
To assess the binding of RLC-1 to different myosin constructs and determine the effect of different nucleotide states, including ADP, ADP.Pi, and AMP-PNP (non-hydrolyzable form of ATP), bovine-cardiac HMM and recombinant RLC were labeled using NHS or Tris-NTA labeling kits from NanoTemper following the manufacturer’s instructions. Solutions of 50 nM labeled protein with 1 mM nucleotide were prepared in binding buffer (20 mM Tris [pH = 7.5] buffer containing 2 mM MgCl2, 25 mM KCl, 1 mM DTT, and 0.05% Tween-20). RLC-1 was 2× serial diluted in DMSO (16 points) and 1:50 further diluted in the same binding buffer. The protein/nucleotide solution and RLC-1 solutions were mixed 1:1 to perform MST measurement using Monolith (NanoTemper Technologies GmbH). The final nucleotide concentration of each measurement was 500 μM.
Animal use and ethics
All experiments followed protocols approved by the University of Washington Institutional Animal Care and Use Committee. Male Sprague–Dawley rats between 8- and 10-wk of age were used for all the following experiments. Hearts from farm pigs were collected immediately after euthanasia and rinsed in cold oxygenated Tyrode’s buffer and then the LV and septum were dissected and frozen in liquid nitrogen.
Heart extraction and tissue preparation
Rat hearts were rapidly excised via thoracotomy and rinsed in a modified Krebs buffer containing (in mM) 118.5 NaCl, 5 KCl, 1.2 MgSO4, 2 NaH2PO4, 25 NaHCO3, 1.8 CaCl2, and 10 glucose. The solution was bubbled with 95% O2/5% CO2 for at least 10 min to stabilize the pH at ∼7.2 before the addition of CaCl2. Hearts were then perfused and ventricles splayed open in oxygenated modified Krebs with 0.1 mM CaCl2 and 20 mM 2,3-butanedione 2-monoxime (BDM) to inhibit contraction and minimize damage during tissue dissection.
Demembranated tissue mechanics
Pieces of frozen porcine LV or splayed rat ventricles were transferred to a dish for permeabilization with 50:50 glycerol relaxing solution containing (in mM) 100 KCl, 10 MOPS, 5 K2EGTA, 9 MgCl2, 5 Na2ATP (adjusted to pH = 7 with KOH), 1% (by vol) Triton X-100, 1% protease inhibitor (P8340; Sigma-Aldrich), and 50% (by vol) glycerol at 4°C overnight. The solution was then changed to 50:50 glycerol relaxing solution without Triton X-100 for storage up to 1 wk at −20°C. Ventricular tissues were dissected, secured between aluminum T-clips (Aurora Scientific), and mounted between a motor (model 315C; Aurora Scientific) and a force transducer (model 400A; Aurora Scientific), as previously described (Powers et al., 2020). Muscle length was adjusted to a sarcomere length of ∼2.3 μm by lengthening the muscle 15% beyond where the muscle is just taut (∼2.0 μm). Experiments were conducted in physiological solution (pH 7.0) at 15°C (rat) or 21°C (porcine) varying calcium within a range of pCa (= −log10[Ca2+]) from 9.0 to 4.0 with and without RLC-1. Experiments with porcine tissue also included 3% dextran. Tissue was allowed to reach stead-state force (F) at each pCa before measurements were taken. F-pCa curves were collected and analyzed with custom code using LabView software and fit to the Hill equation to calculate pCa50, the pCa value at half-maximal force, and the Hill coefficient (nH), the slope at the steepest part of the curve. The rate of tension redevelopment (ktr) was calculated from the half-time of force recovery following a 15% rapid release–restretch movement. High-frequency stiffness (HFS) was measured by applying a 1,000 Hz sinusoidal length change (± 0.5% muscle length [ML]) and was calculated from the ratio of peaks of the Fourier transforms of the force and ML signals.
Myofibril mechanics
Small myofibril bundles were prepared from demembranated rat left ventricular wall tissue as previously described (Racca et al., 2013). Briefly, bundles were rinsed two times in a Rigor Buffer containing (in mM), 50 Tris, 100 KCl, 2 MgCl2, 1 EGTA, pH 7.0, and 1× protease inhibitor (Sigma-Aldrich) before being homogenized one to two times for 30 s at high speed. They were stored at 4°C and used for up to 3 days. Experiments were performed on a custom setup as previously described (Kreutziger et al., 2011). Myofibrils were mounted between two needles: one calibrated as a force transducer and the other mounted as an inflexible post attached to a piezo-electric computer-controlled motor. A dual diode system was used to measure needle displacement, and the developed force was measured based on the known stiffness of the needle. Relaxing (pCa 9.0) and activating (pCa 5.4 or pCa 4.0) solutions were delivered to the mounted myofibril using a double-barreled glass pipette. Activation and relaxation data were collected at 15°C and fit with either single-exponential curves, linear coefficients, or 50% times as previously described (Colomo et al., 1997).
X-ray diffraction
Muscle samples were permeabilized as described previously (Kooiker et al., 2023). Briefly, frozen porcine left ventricle wall chunks were defrosted at room temperature in skinning solution (in mM: 91 K+-propionate, 3.5 MgCl2, 0.16 CaCl2, 7 EGTA, 2.5 Na2ATP, 15 creatine phosphate, 20 Imidazole, 30 BDM, 1% Triton-X100 and 3% Dextran at pH 7) before dissection into smaller strips (∼5–10 mm long and 1–2 mm wide). The tissues were permeabilized at room temperature for 3 h or overnight at 4°C before further dissection into preparations 4 mm long with a diameter of ∼200 µm. Preparations were secured to aluminum T-clips at both ends and stored in pCa 8 solution with 3% dextran on ice for use during the day. Clipped ventricular tissues were mounted between two rigid posts, and sarcomere length was set to 2.3 μm using helium-neon laser diffraction. X-ray diffraction patterns were collected using the small-angle instrument on the BioCAT beamline 18ID at the Advanced Photon Source, Argonne National Laboratory as described previously (Kooiker et al., 2023; Ma et al., 2021). Diffraction patterns were collected in the absence and presence of 10 μM RLC-1 on EIGER2 X 9M pixel array detector (Dectris) with a 1-s exposure time at an incident flux of ∼3 × 1012 photons per second for each condition. The equatorial x-ray diffraction patterns were analyzed using the Equator module from the MuscleX software package developed at BioCAT as previously described (Kooiker et al., 2023; Ma et al., 2021). The x-ray patterns were subsequently quadrant-folded and the background subtracted to improve signal-to-noise ratio using the Quadrant Fold module of the MuscleX program suite. The meridional and layer line reflections were measured using the Projection Traces module of MuscleX program suite as previously described (Ma and Irving, 2022). Three to four patterns were collected for each condition and the x-ray reflection data extracted were averaged.
Isolated cell shortening and calcium transients
Rat cardiomyocytes were isolated by perfusion of excised hearts with a digestion buffer containing (in mM): 135 NaCl, 4 KCl, 5 Taurine, 10 HEPES, 0.3 NaH2PO4, 10 Dextrose, 0.025 Blebbistatin, and 1 MgCl2 at pH 7.4 with 0.01 CaCl2, 0.04 mg/ml Protease type XIV (Sigma-Aldrich), and 1 mg/ml Collagenase type 2 (Worthington) was added. Cells were washed in digestion buffer with 10 mg/ml BSA (Calbiochem) and 0.25 mM CaCl2. During the wash protocol, CaCl2 was slowly added to reach a final concentration of 1 mM. Cells were then rinsed and placed in HEPES-buffered Tyrode’s solution (1.8 mM CaCl2) at 25°C and visualized using a Nikon TS100 inverted microscope with a 40× brightfield objective coupled to an IonOptix video microscopy system (IonOptix). Cells were paced at 1 Hz, imaged, and sarcomere length data was collected at 1 kHz. Transients were quantitatively analyzed using the IonWizard software package (IonOptix), and a minimum of 10 traces were analyzed and averaged for each cell. Calcium transients were measured in cardiomyocytes in Tyrode’s buffer incubated with 1 μM Fura-2 AM (Thermo Fisher Scientific) and 1× Powerload (Life Technologies) for 15 min, followed by a 15-min wash in fresh Tyrode’s to allow for AM cleavage. Transients were imaged with a dual excitation LED light source for 340 nm/385 nm ratiometric fluorescence using the Ionoptix video microscopy system (IonOptix).
Intact myocardium mechanics
Unbranched, intact trabeculae or papillary muscles were dissected from rat right ventricular walls and mounted between a force transducer (model 400A; Cambridge Technology, Inc.) and a rigid post. Each end of the trabecula was sutured to custom arms, made from 22-gauge needles and attached to the post and force transducer. Mounted tissue was submerged in a custom experimental chamber continuously perfused with oxygenated modified Krebs buffer (1.8 mM CaCl2) at 33°C. Field stimulation with custom platinum plate electrodes with oscillating polarity triggered muscle twitches. Tissues were lengthened to be just taut and paced at 0.5 Hz for ∼20 min to allow BDM to wash out and tissue to equilibrate. The optimal length was set by stretching the tissue until the peak twitch height no longer increased. Continuous twitch tension traces were recorded using custom LabView software at a sampling rate of 1 kHz and were analyzed with custom code written using MATLAB software (version 2021a; The MathWorks).
Statistical analysis
GraphPad Prism 10.0.3 was used for all data presentation and statistical analysis. All values are presented as mean ± SEM. For demembranated tissue mechanics, all comparisons were paired to two-tailed t tests. For cardiac myofibrils, all comparisons were ordinary one-way ANOVA with Sidak’s multiple comparisons. For x-ray diffraction, all comparisons were paired two-tailed t tests. For single-cell IonOptix measurements, all comparisons were unpaired two-tailed t tests. For intact twitch measurements, all comparisons were repeated measures one-way ANOVA with Dunnett’s multiple comparisons.
Online supplemental material
Fig. S1 shows Coomassie and ProQ diamond staining of ATPase and MST protein samples. Fig. S2 shows the binding of RLC-1 to different myosin constructs determined by MST. Fig. S3 shows steady-state tissue mechanics in permeabilized rat cardiac trabeculae before and after the addition of 10 µM RLC-1. Fig. S4 shows x-ray diffraction of permeabilized rat cardiac muscle in resting conditions before and after incubation in 10 µM RLC-1. Fig. S5 shows calcium binding to decorated thin filaments is not affected by 10 μM RLC-1. Fig. S6 shows calcium transients in isolated rat cardiomyocytes with 10 µM RLC-1. Fig. S7 shows that Fura-2 excitation scan at various calcium concentrations was not altered over a range of RLC-1 concentrations (0–30 μM).
Results
RLC-1 inhibits cardiac HMM ATPase in an RLC–dependent manner
A small molecule screen was performed on bovine cardiac myofibrils (bcMF) to identify compounds that reduced steady-state ATPase in cardiac myosin. From this screen, RLC-1 was selected as an inhibitor of myosin ATPase in a manner dependent on the presence of the RLC. Using a coupled enzyme system utilizing both pyruvate kinase and lactate dehydrogenase, we determined the dose-response of actomyosin ATPase rates and the minimum required regions of the myosin molecule. Fig. 1 A demonstrates that myosin RLC is required for RLC-1 to inhibit myosin function. For bovine cardiac actomyosin (bcAM), both myosin S1, which does not include light chains, and HMM with RLC depleted did not respond to increasing doses of RLC-1. When RLC was present, bcAM HMM ATPase activity decreased with increasing concentrations of RLC-1 plateauing at 62% of DMSO activity. However, bcAM HMM ATPase was unchanged by the phosphorylation status of RLC, suggesting that the binding of the compound is independent of RLC phosphorylation. The most potent dose dependence was seen with bcMF, which plateaued at ∼50% of DMSO activity. Further, RLC-1 bound to bcHMM at a Kd of 1 μM and bovine skeletal HMM at a Kd of 3.34 μM. No binding was measured between RLC-1 and bovine smooth muscle HMM or recombinant RLC alone, suggesting neither RLC nor HMM alone interacts with RLC-1 (Fig. S2).

Biochemical effects of RLC-1 on myosin activity and binding. (A) Dose-response effect of RLC-1 on bcAM ATPase activity with different myosin constructs and bcMF showed RLC-1 effect required the presence of myosin RLC and was not affected by RLC phosphorylation. (B) Rabbit cardiac myofibril ATPase assay showed the greatest inhibition at the lowest concentrations of calcium (pCa = −log[Ca2+]) for all doses tested. (C) Binding of RLC-1 to bovine cardiac HMM in MST did not change in the presence of different nucleotides, each at 500 μM. Data includes one replicate for two experiments and is represented as mean ± SEM.
Biochemical effects of RLC-1 on myosin activity and binding. (A) Dose-response effect of RLC-1 on bcAM ATPase activity with different myosin constructs and bcMF showed RLC-1 effect required the presence of myosin RLC and was not affected by RLC phosphorylation. (B) Rabbit cardiac myofibril ATPase assay showed the greatest inhibition at the lowest concentrations of calcium (pCa = −log[Ca2+]) for all doses tested. (C) Binding of RLC-1 to bovine cardiac HMM in MST did not change in the presence of different nucleotides, each at 500 μM. Data includes one replicate for two experiments and is represented as mean ± SEM.

Binding of RLC-1 to different myosin constructs determined by MST. Binding affinities of RLC-1 reported by the NanoTemper Kd analysis was 1 μM for bcHMM, 3.34 μM for skHMM. No binding was detected between RLC-1 and recombinant cardiac RLC or smHMM. Data includes two replicates with two repeats each.
Binding of RLC-1 to different myosin constructs determined by MST. Binding affinities of RLC-1 reported by the NanoTemper Kd analysis was 1 μM for bcHMM, 3.34 μM for skHMM. No binding was detected between RLC-1 and recombinant cardiac RLC or smHMM. Data includes two replicates with two repeats each.
Inhibition by RLC-1 was more effective at lower calcium concentrations, but not altered by different nucleotides
Calcium concentration was assessed in rabbit cardiac MF (rbcMF). Calcium affected the sensitivity of rbcMF to RLC-1 at concentrations ranging from 1 to 25 μM as measured by ATPase activity (Fig. 1 B). At each drug concentration, the inhibition of ATPase activity was most potent at lower concentrations of calcium, decreasing as calcium concentration increased. For 25 μM RLC-1, inhibition was 16% at high calcium (pCa 5.5), whereas inhibition was 98% at low calcium (pCa 8.0). MST was used to determine the binding of RLC-1 to bcHMM in the presence of various nucleotides at 500 μM, including ADP, ADP.Pi, AMP-PNP (non-hydrolysable form of ATP), and buffer alone (Fig. 1 C). None of the different nucleotides led to any changes in the binding of RLC-1 to bcHMM, suggesting there was no allosteric effect of the myosin head in response to nucleotide binding when RLC-1 was bound.
Force and calcium sensitivity were reduced by 10 μM RLC-1 in demembranated porcine tissues
We examined the effect of RLC-1 on steady-state force production and calcium sensitivity in demembranated left ventricular tissue. Each preparation underwent two force-Ca2+ (pCa) curves, one with DMSO and a second with 10 μM RLC-1 (Fig. 2 A). Maximal force (pCa 4.0) was reduced by 53% with RLC-1 versus DMSO (Fig. 2 B; 16.8 ± 2.2 mN/mm2 versus 36.1 ± 4.7 mN/mm2, P value = 0.0002). Calcium sensitivity was also significantly reduced as shown by a decrease in pCa50, the pCa value at 50% maximal force (Fig. 2 C; 5.46 ± 0.02 versus 5.73 ± 0.03, P value < 0.001). We confirmed that RLC-1 does not alter calcium sensitivity in decorated thin filaments alone (Fig. S3) and requires a more structured sarcomere and the presence of myosin. Consistent with inhibition of ATPase (Fig. 1 C), RLC-1 inhibition of force was more potent at lower levels of calcium; however, inhibition was still substantial at high levels of calcium in demembranated tissue. Passive force (pCa 9) was significantly reduced at the end of a 5-min incubation with RLC-1 (Fig. 2 D; 6.4 ± 0.9 versus 7.5 ± 1.1, P value = 0.028). The rate of tension redevelopment (ktr), which correlates with myosin crossbridge cycling, showed the same slope for the force-ktr relationship (Fig. 2 E), with a decrease in maximal ktr (Fig. 2 F; 1.36 ± 0.07 versus 0.89 ± 0.04, P value < 0.001). High-frequency sinusoidal stiffness decreased with RLC-1 (Fig. 2 G), but to a lesser extent than the decrease in force, resulting in a steeper stiffness:force relationship (Fig. 2 H), as evidenced by the steeper slope (Fig. 2 I; 0.057 ± 0.004 versus 0.067 ± 0.005, P value = 0.022), This suggests an increase in the proportion of crossbridges in a weaker binding state that produces less force per crossbridge. Similar results were observed in rat tissue (Fig. S4), except for some differences in passive force and ktr. There is no difference in passive force in the rat tissue (Fig. S4 D), and ktr showed differences relative to the level of force (Fig. S4 E) with no difference in maximal ktr (Fig. S4 F). The porcine experiments were done in the presence of 3% dextran, which compresses the lattice to physiological spacing, which could affect actomyosin interactions.

Steady-state tissue mechanics in permeabilized porcine cardiac tissue before and after addition of 10 µM RLC-1. (A) Force normalized to maximal force without the drug is substantially decreased at all levels of calcium with RLC-1. (B–D) Maximal force and (C) calcium sensitivity and (D) passive force are all decreased. (E and F)ktr is not changed relative to force (E), and (F) maximal ktr is decreased by RLC-1. (G and H) High-frequency sinusoidal stiffness is decreased at all levels of calcium with RLC-1, (H) but increased relative to force. (I) This results in a steeper slope of the stiffness–force relationship, indicating force is decreased more than stiffness. Data includes eight preps from three pigs and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND (no drug).
Steady-state tissue mechanics in permeabilized porcine cardiac tissue before and after addition of 10 µM RLC-1. (A) Force normalized to maximal force without the drug is substantially decreased at all levels of calcium with RLC-1. (B–D) Maximal force and (C) calcium sensitivity and (D) passive force are all decreased. (E and F)ktr is not changed relative to force (E), and (F) maximal ktr is decreased by RLC-1. (G and H) High-frequency sinusoidal stiffness is decreased at all levels of calcium with RLC-1, (H) but increased relative to force. (I) This results in a steeper slope of the stiffness–force relationship, indicating force is decreased more than stiffness. Data includes eight preps from three pigs and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND (no drug).

Calcium binding to decorated thin filaments is not affected by 10 μM RLC-1. Actin was purified and polymerized from bovine cardiac ether powder as previously described (Pardee and Spudich, 1982). Wild type human cardiac troponin complexes (cTnX) labeled with IANBD at C84 of cardiac troponin C (Wang et al, 2013) and cardiac tropomyosin (cTm) (Lehman et al, 2015) were made as previously described. Thin filaments were decorated by incubation of 5 μM actin: 0.67 μM cTn: 0.67 μM cTm in MOPS buffer (20 mM MOPS, 150 mM KCl, 3 mM MgCl2, 2 mM EGTA, and 1 mM DTT, pH 7.0) overnight at 4°C. Fluorescence was measured as previously described (Wang et al, 2013) as calcium was added to the reaction mixture. Data includes three runs per each condition and is represented as mean ± SEM.
Calcium binding to decorated thin filaments is not affected by 10 μM RLC-1. Actin was purified and polymerized from bovine cardiac ether powder as previously described (Pardee and Spudich, 1982). Wild type human cardiac troponin complexes (cTnX) labeled with IANBD at C84 of cardiac troponin C (Wang et al, 2013) and cardiac tropomyosin (cTm) (Lehman et al, 2015) were made as previously described. Thin filaments were decorated by incubation of 5 μM actin: 0.67 μM cTn: 0.67 μM cTm in MOPS buffer (20 mM MOPS, 150 mM KCl, 3 mM MgCl2, 2 mM EGTA, and 1 mM DTT, pH 7.0) overnight at 4°C. Fluorescence was measured as previously described (Wang et al, 2013) as calcium was added to the reaction mixture. Data includes three runs per each condition and is represented as mean ± SEM.

Steady-state tissue mechanics in permeabilized rat cardiac trabeculae before and after addition of 10 µM RLC-1. (A) Force normalized to maximal force without drug is substantially decreased at all levels of calcium with RLC-1. (B and C) Maximal force (B) and calcium sensitivity (C) are both decreased. (D) Passive force is unchanged. (E and F) I ktr is elevated relative to force (E), while maximal ktr (F) is unaffected by RLC-1. (G and H) High-frequency sinusoidal stiffness is decreased at all levels of calcium with RLC-1 (G), but increased faster at equivalent force (H). (I) The ratio of force over stiffness is decreased at maximal calcium, indicating force is decreased more than stiffness. Data include seven trabeculae from three rats and are represented as mean ± SEM. All P values were calculated using a paired t test versus ND (no drug).
Steady-state tissue mechanics in permeabilized rat cardiac trabeculae before and after addition of 10 µM RLC-1. (A) Force normalized to maximal force without drug is substantially decreased at all levels of calcium with RLC-1. (B and C) Maximal force (B) and calcium sensitivity (C) are both decreased. (D) Passive force is unchanged. (E and F) I ktr is elevated relative to force (E), while maximal ktr (F) is unaffected by RLC-1. (G and H) High-frequency sinusoidal stiffness is decreased at all levels of calcium with RLC-1 (G), but increased faster at equivalent force (H). (I) The ratio of force over stiffness is decreased at maximal calcium, indicating force is decreased more than stiffness. Data include seven trabeculae from three rats and are represented as mean ± SEM. All P values were calculated using a paired t test versus ND (no drug).
Maximal and submaximal rates of relaxation were significantly increased, while activation was slowed by RLC-1 in rat cardiac myofibrils
To look at the effect of RLC-1 on dynamic contraction and relaxation kinetics, we activated rat cardiac myofibrils in maximal (pCa 4.0) and submaximal (pCa 5.4) calcium in the presence of either DMSO or 10 μM RLC-1. Force at both pCa 4.0 and 5.4 was significantly reduced with the addition of the drug by 47% and 58%, respectively (Fig. 3 B). The rate of activation (kACT) was slower only at maximal calcium (Fig. 3 C). Relaxation, however, was substantially faster. The rate constant of the fast phase of relaxation, which represents relaxation when the thin filament is deactivating, was 121% faster at pCa 4.0 and 75% faster at pCa 5.4 (Fig. 3 D). The rate constant of the slow phase of relaxation, which represents myosin head detachment from actin, was 108% faster at pCa 4.0 and 61% faster at pCa 5.4 (Fig. 3 E). The duration of the slow phase, which represents the time it takes for the thin filament to turn off (Kreutziger et al., 2011), was shorter at both maximal and submaximal calcium (Fig. 3 F).

Effects of 10 µM RLC-1 force and kinetics of isolated cardiac myofibrils. (A) Example maximal calcium (pCa 4.0) trace of DMSO (black) versus 10 μM RLC-1 (blue) focusing on activation (top) and relaxation (bottom) with inset zoomed in on the slow phase of relaxation. (B) Absolute force was inhibited both at maximal (pCa 4.0) and submaximal (pCa 5.4) levels of calcium. (C–E) The rate of activation was inhibited by the drug (C), while the rate of fast (D) and slow (E) relaxation were significantly faster. (F) The duration of slow relaxation was significantly shorter after the addition of RLC-1. Data includes a total of 23–27 myofibrils per group from three different rats and is represented as mean ± SEM. All P values were calculated using one-way ANOVA with Sidak’s multiple comparisons versus ND.
Effects of 10 µM RLC-1 force and kinetics of isolated cardiac myofibrils. (A) Example maximal calcium (pCa 4.0) trace of DMSO (black) versus 10 μM RLC-1 (blue) focusing on activation (top) and relaxation (bottom) with inset zoomed in on the slow phase of relaxation. (B) Absolute force was inhibited both at maximal (pCa 4.0) and submaximal (pCa 5.4) levels of calcium. (C–E) The rate of activation was inhibited by the drug (C), while the rate of fast (D) and slow (E) relaxation were significantly faster. (F) The duration of slow relaxation was significantly shorter after the addition of RLC-1. Data includes a total of 23–27 myofibrils per group from three different rats and is represented as mean ± SEM. All P values were calculated using one-way ANOVA with Sidak’s multiple comparisons versus ND.
Myosin heads moved away from the thick filament and were less ordered in relaxed tissue with RLC-1
We performed x-ray diffraction studies on skinned porcine cardiac trabeculae in relaxing conditions before and after the addition of 10 µM RLC-1. Analysis of the equatorial diffraction pattern (Fig. 4 A) showed decreased distance between the 1,0 reflections (d1,0) in the presence of 10 μM RLC-1, indicating compression of the lattice spacing of the myofilaments (Fig. 4 B; 36.0 ± 0.2 versus 34.7 ± 0.3, P value = 0.008). The relative intensities of the 1,1 and 1,0 reflections, or the intensity ratio (I1,1/I1,0) were increased after the addition of RLC-1 (Fig. 4 C; 0.34 ± 0.02 versus 0.58 ± 0.05, P value = 0.008). A higher ratio suggests myosin heads are moving away from thick filament backbones and closer to thin filaments. The meridional axis provides information about the spacing and ordering of myosin heads (Fig. 4 D). The third-order meridional myosin reflection (M3) represents the axial spacing of neighboring myosin heads as they wind around the backbone while the myosin layer line 1 (MLL1) results from the quasi-helical ordering of resting myosin heads around the thick filament backbone. The average intensity of the M3 reflection (IM3) decreased in the presence of RLC-1 (Fig. 4 E; 2.1 ± 0.4 versus 0.7 ± 0.1 a.u.; P value = 0.008). The average intensity of MLL1 (IMLL1) also decreased after RLC-1 (Fig. 4 F; 3.3 ± 0.5 versus 1.2 ± 0.3 a.u.; P value = 0.008). Both decreases in IM3 and IMLL1 suggest myosin heads are less ordered, which is consistent with the increased intensity ratio. Similar results were observed in rat tissue (Fig. S5). Porcine myocardium was used for these experiments to take advantage of the improved quality of the diffraction patterns relative to skinned rodent myocardium (Ma et al., 2021).

X-ray diffraction of permeabilized porcine cardiac muscle in resting conditions before and after 10 µM RLC-1 incubation. (A) Example x-ray diffraction patterns before and after incubation in 10 μM RLC-1. (B) Schematic showing how the positions of actin and myosin relate to equatorial measurements. (C and D) The distance between 1,0 reflections related to the interfilament lattice spacing (d1,0) decreased, whereas (D) the ratio of the 1,0 and 1,1 reflection intensities (I1,1/I1,0) showed myosin to be closer to the thin filament in the presence of RLC-1. (E) Schematic showing how the positions of neighboring myosin heads relate to meridional measurements. (F and G) The intensity of the (F) M3 (IM3) and (G) MLL1 (IMLL1) meridional reflections showed myosin head became less ordered along the myosin backbone. The schematics (B and E) were modified from Kooiker et al. (2023). Data includes eight preps from one pig and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND (no drug).
X-ray diffraction of permeabilized porcine cardiac muscle in resting conditions before and after 10 µM RLC-1 incubation. (A) Example x-ray diffraction patterns before and after incubation in 10 μM RLC-1. (B) Schematic showing how the positions of actin and myosin relate to equatorial measurements. (C and D) The distance between 1,0 reflections related to the interfilament lattice spacing (d1,0) decreased, whereas (D) the ratio of the 1,0 and 1,1 reflection intensities (I1,1/I1,0) showed myosin to be closer to the thin filament in the presence of RLC-1. (E) Schematic showing how the positions of neighboring myosin heads relate to meridional measurements. (F and G) The intensity of the (F) M3 (IM3) and (G) MLL1 (IMLL1) meridional reflections showed myosin head became less ordered along the myosin backbone. The schematics (B and E) were modified from Kooiker et al. (2023). Data includes eight preps from one pig and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND (no drug).

X-ray diffraction of permeabilized rat cardiac muscle in resting conditions before and after incubation in 10 µM RLC-1. (A and B) The distance between 1,0 reflections related to the interfilament lattice spacing (d1,0) did not change (A), whereas the ratio of the 1,0 and 1,1 reflection intensities (I1,1/I1,0) increased (B), indicating myosin heads to be closer to the thin filament in the presence of RLC-1. (C and D) The intensity of the M3 (IM3) meridional reflection decreased, indicating less axial order of myosin heads, while (D) the intensity of the MLL1 (IMLL1) reflection did not significantly decrease. Muscle samples were permeabilized as described previously (Powers et al, 2019). Briefly, rat ventricles were demembranated as described above overnight at 4°C. Trabeculae were dissected and clipped with aluminum T-clips and subsequently stored in a cold relaxing solution at 4°C for the day’s experiments. Clipped trabeculae were mounted between two rigid posts and sarcomere length was set to ∼2.3 μm by stretching the tissue 15% beyond taut. X-ray diffraction patterns were collected using the small-angle instrument on the BioCAT beamline 18ID at the Advanced Photon Source, Argonne National Laboratory as described in the Materials and methods. Data include seven preparations from three rats and is represented as mean ± SEM. All P values were calculated using a paired t test versus ND (no drug).
X-ray diffraction of permeabilized rat cardiac muscle in resting conditions before and after incubation in 10 µM RLC-1. (A and B) The distance between 1,0 reflections related to the interfilament lattice spacing (d1,0) did not change (A), whereas the ratio of the 1,0 and 1,1 reflection intensities (I1,1/I1,0) increased (B), indicating myosin heads to be closer to the thin filament in the presence of RLC-1. (C and D) The intensity of the M3 (IM3) meridional reflection decreased, indicating less axial order of myosin heads, while (D) the intensity of the MLL1 (IMLL1) reflection did not significantly decrease. Muscle samples were permeabilized as described previously (Powers et al, 2019). Briefly, rat ventricles were demembranated as described above overnight at 4°C. Trabeculae were dissected and clipped with aluminum T-clips and subsequently stored in a cold relaxing solution at 4°C for the day’s experiments. Clipped trabeculae were mounted between two rigid posts and sarcomere length was set to ∼2.3 μm by stretching the tissue 15% beyond taut. X-ray diffraction patterns were collected using the small-angle instrument on the BioCAT beamline 18ID at the Advanced Photon Source, Argonne National Laboratory as described in the Materials and methods. Data include seven preparations from three rats and is represented as mean ± SEM. All P values were calculated using a paired t test versus ND (no drug).
RLC-1 decreased cardiomyocyte shortening and lengthened resting sarcomere length
To assess the effect of RLC-1 on individual cardiomyocytes, we plated isolated rat cardiomyocytes in Tyrode’s buffer with or without RLC-1 and paced at 1 Hz. In the presence of 10 µM RLC-1, fractional shortening was reduced to 3.6 ± 0.9% from 11.2 ± 1.2% (control), a 68% reduction in shortening (Fig. 5, A and B). Resting sarcomere length was significantly longer with RLC-1 (Fig. 5 C; 1.92 ± 0.02 versus 2.00 ± 0.01; P value = 0.010), suggesting RLC-1 promotes a more relaxed myofilament in unloaded conditions at low calcium. The time to peak shortening (Fig. 5 D; 0.14 ± 0.02 versus 0.08 ± 0.01; P value = 0.005) and time to 50% relaxation (Fig. 5 E; 0.07 ± 0.01 versus 0.05 ± 0.003; P value = 0.049) were both faster, while the 90% relaxation time was not (Fig. 5 F). Together, these lead to a shorter overall twitch duration to 90% relaxation (Fig. 5 G; 0.26 ± 0.04 versus 0.19 ± 0.02; P value = 0.015). We also saw a small decrease in the baseline (Fig. S6 D) and maximal (Fig. S6 C) ratiometric Fura-2 calcium fluorescence in isolated rat cardiomyocytes, without significant changes in calcium transient kinetics (Fig. S6, E–G). We confirmed that the decreases at baseline and maximal calcium were not due to interaction between the compound and RLC-1 (Fig. S7). This suggests that RLC-1 might have subtle effects on calcium handling within the cell.

Fractional shortening in isolated rat cardiomyocytes with 10 µM RLC-1. (A) Average twitches from rat cardiomyocytes paced at 1 Hz either in Tyrode’s solution with DMSO or with RLC-1 show substantial inhibition of sarcomere shortening and twitch duration. (B and C) Percent fractional shortening was reduced in cardiomyocytes exposed to RLC-1 and (C) resting sarcomere length was significantly longer. (D–F) Time to peak tension (D) and time to 50% relaxation (E) were both shorter, with no significant difference in time to 90% relaxation (F). (G) Twitch duration to 90% relaxation was also significantly shorter in the presence of RLC-1. Data include 12–15 cells per condition for each of the five rats and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND.
Fractional shortening in isolated rat cardiomyocytes with 10 µM RLC-1. (A) Average twitches from rat cardiomyocytes paced at 1 Hz either in Tyrode’s solution with DMSO or with RLC-1 show substantial inhibition of sarcomere shortening and twitch duration. (B and C) Percent fractional shortening was reduced in cardiomyocytes exposed to RLC-1 and (C) resting sarcomere length was significantly longer. (D–F) Time to peak tension (D) and time to 50% relaxation (E) were both shorter, with no significant difference in time to 90% relaxation (F). (G) Twitch duration to 90% relaxation was also significantly shorter in the presence of RLC-1. Data include 12–15 cells per condition for each of the five rats and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND.

Calcium transients in isolated rat cardiomyocytes with 10 µM RLC-1. (A) Average transients from rat cardiomyocytes normalized to baseline Fura-2 ratio (R0) paced at 1 Hz either in Tyrode’s solution with DMSO or with RLC-1. (B–D) Ca2+ transient amplitude (ΔR/R0) showed a small but insignificant change (B), while the (C) maximum ratio (Rmax) and (D) baseline ratio (R0) were both significantly decreased. (E–G) Time to peak calcium (E), time to 50% relaxation (F), and time to 90% relaxation (G) were not different in the presence of RLC-1. Data include 16–26 cells per condition for each of the three rats and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND (no drug).
Calcium transients in isolated rat cardiomyocytes with 10 µM RLC-1. (A) Average transients from rat cardiomyocytes normalized to baseline Fura-2 ratio (R0) paced at 1 Hz either in Tyrode’s solution with DMSO or with RLC-1. (B–D) Ca2+ transient amplitude (ΔR/R0) showed a small but insignificant change (B), while the (C) maximum ratio (Rmax) and (D) baseline ratio (R0) were both significantly decreased. (E–G) Time to peak calcium (E), time to 50% relaxation (F), and time to 90% relaxation (G) were not different in the presence of RLC-1. Data include 16–26 cells per condition for each of the three rats and is represented as mean ± SEM. All P values were calculated using paired t tests versus ND (no drug).

Fura-2 excitation scan at various calcium concentrations was not altered over a range of RLC-1 concentrations (0–30 μM). Fura-2 pentapotassium salt (Invitrogen F1200) was dissolved in MOPS buffer (20 mM MOPS, 150 mM KCl, 3 mM MgCl2, 2 mM EGTA, and 1 mM DTT, pH 7.0) to a final concentration of 1 μM at 30°C. (A) Excitation scans on a PerkinElmer FL 6500 fluorescence spectrophotometer between 300 and 400 nm showed the standard shift from longer to shorter excitation wavelength at calcium concentration of 0, 0.09, and 0.79 μM CaCl2. At each calcium concentration, RLC-1 was titrated to 0, 1, 3, 10, and 30 μM, and a new scan was taken. (B) Summary of Fura-2 ratios (335/363 nm) at each calcium and RLC-1 concentration. Data include three runs per condition and is represented as mean ± SEM.
Fura-2 excitation scan at various calcium concentrations was not altered over a range of RLC-1 concentrations (0–30 μM). Fura-2 pentapotassium salt (Invitrogen F1200) was dissolved in MOPS buffer (20 mM MOPS, 150 mM KCl, 3 mM MgCl2, 2 mM EGTA, and 1 mM DTT, pH 7.0) to a final concentration of 1 μM at 30°C. (A) Excitation scans on a PerkinElmer FL 6500 fluorescence spectrophotometer between 300 and 400 nm showed the standard shift from longer to shorter excitation wavelength at calcium concentration of 0, 0.09, and 0.79 μM CaCl2. At each calcium concentration, RLC-1 was titrated to 0, 1, 3, 10, and 30 μM, and a new scan was taken. (B) Summary of Fura-2 ratios (335/363 nm) at each calcium and RLC-1 concentration. Data include three runs per condition and is represented as mean ± SEM.
RLC-1 inhibited twitch force and slowed kinetics in intact rat trabeculae
To understand the combined result of the effects presented thus far in multicellular tissue with fully intact cellular membranes, we paced intact trabeculae at 1 Hz in a modified Krebs buffer with DMSO, 1, 3, or 10 µM RLC-1. Fig. 6 A shows that the average peak twitch tension was inhibited by 30%, 57%, and 80%, respectively. Both activation and relaxation kinetics were also inhibited but to a lesser degree than twitch force. At 10 µM, time to peak tension, time to 50%, and time to 90% relaxation were all inhibited between 25% and 30% (Fig. 6, D–F). The overall effect of RLC-1 on the twitch was assessed using the twitch-tension index (TI), which describes the difference in the area under the twitch curve under different conditions, normalized to control (Davis et al., 2016; Powers et al., 2020). Here, we compare Krebs buffer with DMSO (control) versus RLC-1 to assess the effects of RLC-1. The TI for DMSO was, by definition, zero, and the TI became progressively more negative as the concentration of RLC-1 increased, up to −1.44 × 104 at 10 µM (Fig. 6 C). This indicates that RLC-1 progressively inhibited the overall twitch force and kinetics in a concentration-dependent manner (Fig. 6 A).

Dose-response of RLC-1 on wild-type rat intact trabeculae. (A and B) Averaged normalized twitches (A) and peak twitch tension (B) show increasing inhibition with increasing drug concentration. (C) The tension index, indicative of overall twitch shape and size, shows progressively negative values that reflect twitch inhibition. (D–F) Time to peak (D), 50% (E), and 90% (F) relaxation time are all shorter in duration indicating faster twitch kinetics. Data include one to three trabeculae from each of the seven rats and are represented as mean ± SEM. All P values were calculated using one-way ANOVA with Dunnett’s multiple comparisons versus ND (no drug).
Dose-response of RLC-1 on wild-type rat intact trabeculae. (A and B) Averaged normalized twitches (A) and peak twitch tension (B) show increasing inhibition with increasing drug concentration. (C) The tension index, indicative of overall twitch shape and size, shows progressively negative values that reflect twitch inhibition. (D–F) Time to peak (D), 50% (E), and 90% (F) relaxation time are all shorter in duration indicating faster twitch kinetics. Data include one to three trabeculae from each of the seven rats and are represented as mean ± SEM. All P values were calculated using one-way ANOVA with Dunnett’s multiple comparisons versus ND (no drug).
Discussion
Here, we present a new class of myosin inhibitor that is dependent on the presence of myosin RLC for effective inhibition of myosin function. Other myosin inhibitors, such as Mavacamten and Aficamten, interact directly with the myosin head to alter function (Green et al., 2016; Chuang et al., 2021). Three key findings arose from this work. First, we found that RLC-1 inhibited function in demembranated tissues, including myofibril ATPase rate, myofibril force, and dynamic kinetics of activation and relaxation, as well as steady-state force and calcium sensitivity in trabeculae. Key to this is that in ATPase assays utilizing HMM, the presence of RLC was necessary for inhibition; however, RLC phosphorylation status did not have an effect. Second, under relaxing conditions, myosin heads moved away from the thick filament backbone and became less ordered in the presence of RLC-1. This is contrary to the myosin inhibitor, Mavacamten, which potently moves myosin heads toward the thick filament to reduce the number of available heads for force production (Anderson et al., 2018, Preprint). Third, in both loaded and unloaded intact preparations, RLC-1 substantially reduced the magnitude of contraction and shortened the overall duration of contraction. Together, these data suggest RLC-1 could be effective in reducing contractility in a hypercontractile heart and potentially restoring normal function.
Targeting of myosin RLC has been proposed as a therapeutic target for the treatment of HCM
Mutations in RLC are implicated in HCM, as well as dilated and restrictive cardiomyopathies. Multiple mutations have been shown to modulate both myosin motor activity and myosin structure (Yadav et al., 2019; Yuan et al., 2015, 2018). Additionally, RLC phosphorylation increased resting and maximal calcium-activated force and calcium sensitivity as well as crossbridge cycling (through ktr) (Colson et al., 2010). Structurally, RLC phosphorylation decreased lattice spacing with an increased intensity ratio, which likely contributed to the increased function (Colson et al., 2010). Work done in the Szczesna-Cordary lab (Yuan et al., 2015; Liang et al., 2024) has demonstrated how crossing RLC D166V HCM mice with mice either heterozygous or homozygous for phosphomimetic RLC (S15D) was able to rescue the hypercontractile structure and function. It follows from this work that a compound, such as RLC-1, which targets myosin function in a way that is dependent on the presence of RLC, could substantially alter both the structure and function of cardiac contraction. The work presented here strongly supports this.
RLC-1 inhibited force production, calcium sensitivity, and crossbridge cycling kinetics
In both demembranated trabeculae (Fig. 2, A and B) and myofibrils (Fig. 3 A), force at maximal calcium and submaximal calcium was inhibited. In demembranated trabeculae, the passive force was also inhibited (Fig. 2 D). The rate constant of force redevelopment (ktr) was not different at equivalent levels of force (Fig. 2 E); however, it was lower at comparable levels of calcium with a decrease in maximal ktr (Fig. 2 F). We showed that high-frequency sinusoidal stiffness, which is indicative of the number and force of bound crossbridges, is decreased at all levels of calcium (Fig. 2 G). However, stiffness increases with calcium faster than force, resulting in a steeper stiffness:force relationship (Fig. 2 H), suggesting an increase in the number or the proportion of crossbridges in a weakly bound state.
Measurements in demembranated trabeculae look at crossbridge function when the thin filament has reached a steady-state level of activation. To look at the dynamics of activation and relaxation, we use isolated myofibrils. These data point to slowed contractile activation (Fig. 3 B), which includes both thin filament activation and crossbridge binding and substantially increased fast and slow relaxation phases (Fig. 3, C–E), which include thin filament deactivation and crossbridge detachment. The decreased maximal ktr measurements in demembranated trabeculae (Fig. 2 F) are consistent with the decreased rate constant of contractile activation in myofibrils (Fig. 3 C), suggesting that slower cycling crossbridges develop force more slowly. This could also involve weakly bound crossbridges that are not strong enough to push open the troponin–tropomyosin complex on thin filament, which would also slow activation. This would be consistent with the faster relaxation kinetics (Fig. 3, D–F), where the thin filament can more easily deactivate and crossbridges easily detach. Together with the ktr and stiffness data in permeabilized trabeculae, myofibril data indicate that RLC-1 hampered the ability of crossbridges to bind and stay bound to the thin filament leading to lower force and slower thin filament activation with faster crossbridge cycling and relaxation kinetics.
RLC-1 caused myosin heads to move away from the thick filament backbone in resting conditions, which is contrary to expectations for a myosin inhibitor
One of the main mechanisms for the inhibitory effect of Mavacamten is its ability to stabilize myosin heads in a more “closed” state, removing heads from the pool of actively cycling heads resulting in a reduction in force production (Gollapudi et al., 2021). RLC-1 increased the equatorial intensity ratio (Fig. 4 D) and reduced the intensity of the meridional M3 reflection (Fig. 4 F), which are both reflective of less ordered myosin heads that are further away from the thick filament backbone. It is possible that RLC-1 not only destabilizes or weakens cycling crossbridges but also the ability of myosin heads to form the interacting heads motif. This is plausible considering that RLC-1 requires the presence of RLC to function and RLC is known to be involved in the stability of myosin head structure and its position relative to the thick filament backbone (Padrón et al., 2020; Yuan et al., 2015; Colson et al., 2010). It is also possible that even though there are more myosin heads available to form crossbridges, the weaker thin filament binding negates the increase in the number of heads.
RLC-1 decreased the size and duration of twitches in unloaded cardiomyocytes and loaded trabeculae
In intact cells and trabeculae, cellular membranes are fully functional, and contraction is initiated by calcium influx into the cell after electrical stimulation. We paced both cells and tissues at 1 Hz and saw consistent effects of RLC-1 in both. With RLC-1 present, the cells shortened less (Fig. 5, A and B), and in trabeculae, the peak force decreased (Fig. 6, A and B). Interestingly, the resting sarcomere length measured in cells increased from 1.9 to 2.0 μm with RLC-1 (Fig. 6, A and C), which is consistent with a similar magnitude change in resting sarcomere length after treatment with Mavacamten (Sparrow et al., 2020). We are unable to measure diastolic force in our intact trabeculae but we did see a decrease in the passive force in demembranated trabeculae (Fig. 2 D). While the amplitude of the Fura-2 transients showed a subtle but insignificant decrease (Fig. S6, A and B), both the baseline (Fig. S6 D) and maximal (Fig. S6 C) ratiometric Fura-2 calcium fluorescence decreased, suggesting the transient shifted downward and that overall intracellular calcium concentrations might be lower in the presence of RLC-1, which could contribute to the decreased twitches in both isolated cells and intact trabeculae. This is also consistent with cytosolic calcium measurements with Mavacamten (Sparrow et al., 2020).
The duration of contraction and relaxation was shorter in both cells and trabeculae (Fig. 5, D–G; and Fig. 6, D–F). These results are similar to the effects of Mavacamten on engineered heart tissues, including decreased peak tension, faster time-to-peak contraction, and time to 50% relaxation (Sewanan et al., 2021).To look at the overall effect of RLC-1, we calculated the tension index, which identifies a value of zero as a normal twitch, a positive value as hypercontractile, and a negative value as hypocontractile. Tension index is a strong predictor of cardiac remodeling and function in rodent and hiPSC-CM models of cardiomyopathy (Davis et al., 2016) and has been used to assess the rescue of dilated cardiomyopathy mouse models with both genetic and therapeutic treatments (Kooiker et al., 2023; Powers et al., 2020). As the concentration of RLC-1 increases, the tension index becomes increasingly negative, suggesting it is moving toward a hypocontractile state. If this effect was extrapolated to hypercontractile tissue, such as what is expected for tissue from hearts with mutations causing HCM, this model would predict RLC-1 to reduce the hypercontractility. Typically, HCM tissue both increases the peak force and prolongs relaxation, contributing to the characteristic diastolic dysfunction. Based on the results presented here, RLC-1 has the capability to correct both the increased force and prolonged relaxation common to HCM tissue and normalize the twitch to look like one from a normal heart.
A potentially interesting future study would be to investigate how RLC-1 affects hypercontractility from different causes. Ideally, the ability of RLC-1 to substantially reduce twitch force and duration would be beneficial to many different etiologies. However, it is possible that RLC-1 could be less effective against specific mechanisms of HCM. For example, the P710R myosin mutation was shown to cause hypercontractility through destabilization of the myosin OFF state and is even inhibitory in ATPase and filament sliding velocity (Vander Roest et al., 2021). Considering RLC-1 also destabilizes the OFF state as shown with the increase in x-ray intensity ratio, it is possible that this would negatively affect this mutation. Whereas for a mutation where the primary mechanism is to alter thin filament activation or calcium sensitivity, it is possible that RLC-1 would be particularly beneficial.
In conclusion, we use a variety of techniques to understand the effect of the RLC-dependent myosin inhibitor, RLC-1. We propose that it significantly weakens myosin function, slows contractile activation, and then quickens crossbridge detachment to reduce the amount of time in a strongly bound force-producing state. Together, these effects at the myofilament level result in a reduced tension index in a cardiac twitch. It is important to consider that myosin is a very allosteric molecule, so even though RLC-1 is likely interacting with myosin at or near RLC, its effects propagate through the catalytic head to affect function. Future studies should look at how RLC-1 alters the interaction between actin and myosin and if that can extend to how the thin filament is activated.
Data availability
Data used to make all figures can be found in FigShare at https://doi.org/10.6084/m9.figshare.24354289. The RLC-1 compound will be made available upon request.
Acknowledgments
Henk L. Granzier served as editor.
Support for this study was provided by National Institutes of Health (NIH) grants R01 HL128368 (to M. Regnier), K08 HL128826 (to F. Moussavi-Harami), and R01 HL157169 (to F. Moussavi-Harami). Additional support was provided by the University of Washington Center for Translational Muscle Research (UW CTMR) to M. Regnier under Award Number P30AR074990 by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health. Partial support was provided by a sponsored research agreement (SRA) between Myocardia, Inc., now Bristol Myers Squibb (MyoKon 11456), and the UW CTMR. This research used resources from the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357 and supported by grant P30 GM138395 to T.C. Irving from the National Institute of General Medical Sciences of the National Institutes of Health. The content is solely the authors’ responsibility and does not necessarily reflect the official views of the National Institute of General Medical Sciences, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, or the National Institutes of Health.
Author contributions: K. Kooiker: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Validation, Writing—original draft, Writing—review & editing, Q.-F. Gan: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing—review & editing, M. Yu: Formal analysis, Investigation, Validation, N. Sa: Formal analysis, Investigation, Methodology, Resources, Writing—review & editing, S. Mohran: Formal analysis, Investigation, Visualization, Writing—review & editing, Y. Cheng: Conceptualization, Formal analysis, Methodology, G. Flint: Formal analysis, Investigation, Writing—review & editing, S. Neys: Formal analysis, Investigation, C. Gao: Methodology, D. Nissen: Investigation, T. McMillen: Investigation, A. Asencio: Investigation, Writing—review & editing, W. Ma: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing—review & editing, T.C. Irving: Funding acquisition, Supervision, Writing—review & editing, F. Moussavi-Harami: Funding acquisition, Resources, Supervision, Writing—review & editing, M. Regnier: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing—review & editing.

