


Posttranslational regulation of cardiac NaV1.5 channels is critical in modulating channel expression and function, yet their regulation by phosphorylation of accessory proteins has gone largely unexplored. Using phosphoproteomic analysis of NaV channel complexes from adult mouse left ventricles, we identified nine phosphorylation sites on intracellular fibroblast growth factor 13 (iFGF13). To explore the potential roles of these phosphosites in regulating cardiac NaV currents, we abolished expression of iFGF13 in neonatal and adult mouse ventricular myocytes and rescued it with wild-type (WT), phosphosilent, or phosphomimetic iFGF13-VY. While the increased rate of closed-state inactivation of NaV channels induced by Fgf13 knockout in adult cardiomyocytes was completely restored by adenoviral-mediated expression of WT iFGF13-VY, only partial rescue was observed in neonatal cardiomyocytes after knockdown. The knockdown of iFGF13 in neonatal ventricular myocytes also shifted the voltage dependence of channel activation toward hyperpolarized potentials, a shift that was not reversed by WT iFGF13-VY expression. Additionally, we found that iFGF13-VY is the predominant isoform in adult ventricular myocytes, whereas both iFGF13-VY and iFGF13-S are expressed comparably in neonatal ventricular myocytes. Similar to WT iFGF13-VY, each of the iFGF13-VY phosphomutants studied restored NaV channel inactivation properties in both models. Lastly, Fgf13 knockout also increased the late Na+ current in adult cardiomyocytes, and this effect was restored with expression of WT and phosphosilent iFGF13-VY. Together, our results demonstrate that iFGF13 is highly phosphorylated and displays differential isoform expression in neonatal and adult ventricular myocytes. While we found no roles for iFGF13 phosphorylation, our results demonstrate differential effects of iFGF13 on neonatal and adult mouse ventricular NaV channels.
Introduction
Voltage-gated Na+ (NaV) channels are key determinants of myocardial excitability, driving the fast upstroke of the action potential and the conduction of electrical impulses through the myocardium (Chen-Izu et al., 2015). While most NaV channels undergo rapid activation and inactivation to generate the transient Na+ current (INa), a fraction (∼0.5%) of the channels remains open, generating a small persistent Na+ influx, known as the late Na+ current (INaL), which contributes to determining action potential duration. In ventricular myocytes, NaV channels are composed primarily of the NaV1.5 pore-forming (α) subunit, and these channels function in macromolecular protein complexes. As such, channels are embedded within local signaling domains in which they are dynamically regulated by a rich repertoire of accessory proteins and posttranslational modifications (PTMs; Marionneau and Abriel, 2015).
Defects in NaV1.5 channel functioning and/or regulation underlie diverse forms of inherited or acquired cardiac arrhythmias (Remme and Bezzina, 2010). Impaired inactivation of NaV1.5 channels, notably, alters channel availability and/or enhances INaL, both of which can cause arrhythmia syndromes, including long QT syndrome type 3, Brugada syndrome, or conduction slowing. Leveraging the endogenous regulatory mechanisms of NaV1.5 channels is therefore essential for deciphering arrhythmogenic NaV current defects. Several recent studies demonstrated that the cardiac NaV1.5 protein is highly phosphorylated, and that phosphorylation-dependent regulation of NaV1.5 channels is critical in regulating the expression and functioning of these channels, as well as interactions between the NaV1.5 α subunit and channel accessory proteins (Marionneau et al., 2012; Lorenzini et al., 2021). This is the case, for example, of serines 1933 and 1984 in the C-terminal domain of NaV1.5, which regulate interactions with intracellular fibroblast growth factor 13 (iFGF13) and calmodulin, and modulate associated channel inactivation properties (Burel et al., 2017). While numerous phosphorylation sites have been identified on the NaV1.5 protein, phosphorylation of the other channel components and the impact of these modifications on NaV1.5 channel expression or properties remain largely unexplored.
Potential candidates for NaV channel regulation by phosphorylation are the iFGFs, also known as fibroblast growth factor homologous factors (FHFs). The iFGFs have emerged as pivotal players in controlling the inactivation properties of cardiac NaV1.5 channels, tuning both channel availability, inactivation kinetics, and INaL (Wang et al., 2011a, 2017; Park et al., 2016; Abrams et al., 2020; Gade et al., 2020; Chakouri et al., 2022; Santucci et al., 2022). The iFGF family comprises four members (iFGF11 = FHF3, iFGF12 = FHF1, iFGF13 = FHF2, and iFGF14 = FHF4), and further diversity is achieved through the generation of alternatively spliced isoforms with highly divergent N-termini (Munoz-Sanjuan et al., 2000). The iFGFs are small intracellular proteins that have been shown to interact directly with the membrane-proximal portion of the C-terminal domain of NaV channel α subunits in a 1:1 stoichiometry through their highly homologous FGF core domain (Goetz et al., 2009; Wang et al., 2011b). The structural basis by which iFGFs regulate NaV channels emerged in 2012 with the crystal structure of the ternary complex formed by a NaV C-terminal domain, an iFGF and Ca2+-free calmodulin (Wang et al., 2012), and was more recently modeled using the cryo-electron microscopy structure of the American cockroach NaV channel, NaVPas (Shen et al., 2017; Gade et al., 2020). The different iFGF isoforms demonstrate species-, age-, tissue-, and subcellular-specific expression patterns, and affect NaV channel properties distinctively (Yang et al., 2016).
In addition to their broad distribution in the nervous system, the iFGFs are prominently expressed in the mammalian heart. While iFGF13-VY is the predominant iFGF isoform in adult mouse ventricles, with in vivo Fgf13 knockout mice displaying conduction slowing (Park et al., 2016; Wang et al., 2017), iFGF12-B is the preponderant iFGF in human heart (Santucci et al., 2022) and has been linked to inherited arrhythmias including Brugada syndrome (Hennessey et al., 2013), long QT syndrome type 3 (Liu et al., 2003), idiopathic ventricular tachycardia (Li et al., 2017), and atrial and ventricular arrhythmias with sudden cardiac death (Musa et al., 2015). In addition to its ascribed function in regulating NaV channel inactivation properties, it has also been reported that iFGF13 influences the cell surface expression of NaV1.5 channels in cardiomyocytes (Wang et al., 2011a, 2017).
Evidence for a role for iFGF phosphorylation in regulating iFGF-dependent regulation of NaV channels has been provided for neuronal NaV channels, driven in large part by the identification of kinases that regulate the iFGF-NaV channel interface. Specifically, it has been reported that phosphorylation of neuronal NaV channels by glycogen synthase kinase 3 β (GSK3β; Shavkunov et al., 2013; James et al., 2015; Hsu et al., 2017), protein kinase CK2 (Hsu et al., 2016), Ca2+/calmodulin-dependent protein kinase II (CaMKII; Wildburger et al., 2015), as well as the tyrosine kinase janus kinase 2 (JAK2; Wadsworth et al., 2020), promotes binding of iFGF proteins to NaV channels and associated iFGF-mediated channel regulation.
In this study, we investigated the pattern of phosphorylation of native iFGF13 in adult mouse left ventricles and explored the effects of manipulating the identified iFGF13 phosphorylation sites in the regulation of cardiac INa. In addition, we explored iFGF13 isoform expression profiles and the consequences of iFGF13 knockdown/knockout and rescue on cardiac INa in ventricular myocytes isolated from neonatal and adult mice.
Materials and methods
Statement on the use of murine tissue
All investigations conformed to directive 2010/63/EU of the European Parliament, the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1985), and the local institutional guidelines.
Immunoprecipitation of NaV channel complexes
Immunoprecipitation (IP) of NaV channel complexes from adult mouse left ventricles was performed as described previously (Lorenzini et al., 2021). Briefly, flash-frozen left ventricles from 13-wk-old male C57BL/6J wild-type (WT) mice were homogenized individually in ice-cold lysis buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 0.5% amidosulfobetaine, 1X complete protease inhibitor cocktail tablet, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.7 μg/ml pepstatin A (Thermo Fisher Scientific), and 1X Halt phosphatase inhibitor cocktail (Thermo Fisher Scientific). All reagents were from Sigma-Aldrich unless otherwise noted. After 15 min of rotation at 4°C, 8 mg of the soluble protein fractions were precleared with 200 μl of protein G-magnetic Dynabeads (Thermo Fisher Scientific) for 1 h and subsequently used for IP with 48 μg of an anti-NaVPAN mouse monoclonal antibody (mαNaVPAN, #S8809; Sigma-Aldrich) raised against the SP19 epitope (Vassilev et al., 1988) located in the third intracellular linker loop and common to all NaV channel pore-forming subunits. Prior to the IP, antibodies were crosslinked to 200 μl of protein G-magnetic Dynabeads using 20 mM dimethyl pimelimidate (Thermo Fisher Scientific; Schneider et al., 1982). Protein samples and antibody-coupled beads were mixed for 2 h at 4°C. Magnetic beads were then collected, washed rapidly four times with ice-cold lysis buffer, and the isolated protein complexes were eluted from the beads in 1X SDS sample buffer (Bio-Rad Laboratories) at 60°C for 10 min. 99% of the immunoprecipitated mouse left ventricular NaV channel protein complexes were analyzed by MS, and the remaining 1% was used to verify NaV1.5 IP yields by Western blotting.
Peptide preparation and isobaric labeling for LC-MS
The tryptic peptides from adult mouse left ventricular NaV channel complexes were generated and labeled as described previously (Lorenzini et al., 2021). Briefly, the IP eluates were thawed on ice, reduced, and denatured by heating for 10 min at 95°C. The Cys residues were alkylated with iodoacetamide (10 mM) for 45 min at room temperature in the dark. The peptides were prepared using a modification (Erde et al., 2014) of the filter-aided sample preparation method (Wiśniewski et al., 2009). After the addition of 300 μl of 100 mM Tris buffer (pH 8.5) containing 8 M urea (UT) and vortexing, the samples were transferred to YM-30 filter units (MRCF0R030; Millipore) and spun for 14 min at 10,000 rcf (Model No. 5424; Eppendorf). The filters were washed with 200 μl of UT buffer, and the spin–wash cycle was repeated twice. The samples were then exchanged into digest buffer with the addition of 200 μl of 50 mM Tris buffer, pH 8.0, followed by centrifugation (10,000 rcf for 10 min). After transferring the upper filter units to new collection tubes, 80 μl of digest buffer was added and the samples were digested with trypsin (1 µg) for 4 h at 37°C. The digestion was continued overnight after adding another aliquot of trypsin. The filter units were then spun for 10 min (10,000 rcf) in an Eppendorf microcentrifuge. The filter was washed with 50 μl of Tris buffer (100 mM, pH 8.0) followed by centrifugation. The digests were extracted three times with 1 ml of ethyl acetate and acidified to 1% trifluoroacetic acid (TFA) using a 50% aqueous solution. The pH was <2.0 by checking with pH paper. The solid phase extraction of the peptides was performed using porous graphite carbon microtips (Chen et al., 2012). The peptides were eluted with 60% acetonitrile in 0.1% TFA and pooled for drying in a Speed-Vac (model no. Savant DNA 120 concentrator; Thermo Fisher Scientific) after adding TFA to 5%. The peptides were dissolved in 20 μl of 1% acetonitrile in water. An aliquot (10%) was removed for quantification using the Pierce Quantitative Fluorometric Peptide Assay kit (cat. no. 23290; Thermo Fisher Scientific). The remainder of the peptides from each IP sample (∼0.5–3.5 µg) and 1.16 µg of reference pool peptide were transferred into a new 0.5-ml Eppendorf tube, dried in the Speed-Vac, and dissolved in 12 μl of HEPES buffer (100 mM, pH 8.0, H3537; Sigma-Aldrich).
The samples were labeled with tandem mass tag reagents (TMT11; Thermo Fisher Scientific) according to the manufacturer’s protocol. The labeled samples were pooled, dried, and resuspended in 120 μl of 1% formic acid (FA). The TMT11 labeled sample was desalted as described above for the unlabeled peptides. The eluates were transferred to autosampler vials (cat. no. 200046; Sun-Sri), dried, and stored at −80°C for capillary liquid chromatography interfaced to a mass spectrometer (nano-LC-MS).
Nano-LC-MS
The mass spectrometric analysis of adult mouse left ventricular NaV channel complexes was performed as described previously (Lorenzini et al., 2021). Briefly, the samples in formic acid (1%) were loaded (2.5 μl) onto a 75 µm i.d. × 50 cm Acclaim PepMap 100 C18 RSLC column (Thermo Fisher Scientific) on an EASY nano-LC (Thermo Fisher Scientific). The column was equilibrated using constant pressure (700 bar) with 20 μl of solvent A (0.1% FA). The peptides were eluted using the following gradient program with a flow rate of 300 nl/min and using solvents A and B (acetonitrile with 0.1% FA): solvent A containing 5% B for 1 min increased to 25% B over 87 min, to 35% B over 40 min, to 70% B in 6 min, and constant 70% B for 6 min, to 95% B over 2 min and constant 95% B for 18 min. The data were acquired in data-dependent acquisition (DDA) mode. The MS1 scans were acquired with the Orbitrap mass analyzer over m/z = 375–1,500, and resolution was set to 70,000. 12 data-dependent high-energy collisional dissociation spectra (MS2) were acquired from each MS1 scan with a mass resolving power set to 35,000, a range of m/z = 100–1,500, an isolation width of 2 Th, and a normalized collision energy setting of 32%. The maximum injection time was 60 ms for parent-ion analysis and 120 ms for product-ion analysis. The ions that were selected for MS2 were dynamically excluded for 20 s. The automatic gain control (AGC) was set at a target value of 3e6 ions for MS1 scans and 1e5 ions for MS2. Peptide ions with charge states of 1 or ≥7 were excluded for higher-energy collision-induced dissociation (HCD) acquisition.
MS data analysis
Peptide identification from raw MS data was performed using PEAKS Studio 8.5 (Bioinformatics Solutions, Inc.; Zhang et al., 2012). The Uni-mouse-Reference-20131008 protein database was used for spectral matching. The precursor and product ion mass tolerances were set to 20 ppm and 0.05 D, respectively, and the enzyme cleavage specificity was set to trypsin, with a maximum of three missed cleavages allowed. Carbamidomethylation (Cys) and TMT tags (Lys and/or peptide N-terminus) were treated as fixed modifications, while oxidation (Met), pyro-glutamination (Gln), deamidation (Asn and/or Gln), methylation (Lys and/or Arg), dimethylation (Lys and/or Arg), acetylation (Lys), and phosphorylation (Ser, Thr, and/or Tyr) were considered variable modifications. The definitive annotation of each iFGF13 phosphopeptide-spectrum match was obtained by manual verification and interpretation. The phosphorylation site assignments were based on the presence or absence of the unphosphorylated and phosphorylated b- and y-ions flanking the site(s) of phosphorylation, ions referred to as site-discriminating ions throughout this study. Peptide sequences, m/z, charge states, mass errors of parent ions (in ppm), PEAKS -10lgP and A scores, and charge state confirmations of site-discriminating b- and y-ions are presented in Tables S1 and 1.
Label-free quantitative analysis of the areas of extracted MS1 chromatograms of phosphorylated and non-phosphorylated peptide ions covering the phosphorylation site(s) of interest was used to evaluate the proportion of phosphorylated to non-phosphorylated peptides at each position, as well as the relative abundance of phosphopeptides.
Plasmids and adenoviruses
The iFGF13 and control shRNA sequences were subcloned behind a U6 promoter into the pDUAL-U6 plasmid (Vector Biolabs). The sequence for iFGF13 shRNA was 5′-CAGCACTTACACTCTGTTTAACTCGAGTTAAACAGAGTGTAAGTGCTG-3′, which targets nucleotides corresponding to mouse iFGF13-VY amino acids 106–113. The sequence for control shRNA was 5′-GCGCGATAGCGCTAATAATTTCTCGAGAAATTATTAGCGCTATCGCGC-3′, which does not correspond to any known sequence in the mouse genome. The iFGF13-VY phosphomutant rescue constructs were generated by mutating the serine(s)/threonine(s) to alanine(s) (A) or glutamate(s) (E) by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis kit (Agilent) of a pDUAL2-CCM(−) plasmid (Vector Biolabs) containing the CMV promoter in front of the human iFGF13-VY cDNA (NCBI Reference Sequence NM_001139500, full-length cDNA clone purchased from Origene) silently mutated in the sequence targeted by the iFGF13 shRNA. The mutated iFGF13-VY constructs were then digested with restriction endonucleases to excise the mutated fragments, which were then subcloned into the original pDUAL2-CCM(−) plasmid. The pDUAL plasmids containing the shRNA or iFGF13-VY constructs were then provided to Vector Biolabs for the generation, purification, and titration of recombinant (human type 5, dE1/E3) adenoviruses, which also contain the red (RFP) or green fluorescent proteins (GFP), respectively, as markers of infection, under the control of a CMV promoter. All plasmid and adenoviral constructs were sequenced to ensure that no unintentional mutations were introduced.
Isolation, culture, and adenoviral infection of neonatal mouse ventricular myocytes
Single cardiomyocytes were isolated from the ventricles of C57BL/6J WT mouse neonates aged from postnatal day 0 to 3 by enzymatic and mechanical dissociation in a semiautomated procedure by using the Neonatal Heart Dissociation kit and the GentleMACS dissociator (Miltenyi Biotec). Briefly, hearts were harvested and the ventricles were separated from the atria and digested in the GentleMACS dissociator. After termination of the program, the digestion was stopped by adding a medium containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% horse serum, 5% fetal bovine serum, and 100 U/ml penicillin and 100 μg/ml streptomycin. The cell suspension was filtered to remove undissociated tissue fragments and centrifugated. The cell pellet was resuspended in a culture medium and the cells were plated in 60 mm-diameter Petri dishes at 37°C for 1.5 h. The non-plated cardiomyocytes were then resuspended, plated on laminin-coated dishes at a density of 5,000 or 500,000 cells per 35 mm-diameter plate for patch-clamp and molecular biology/biochemical analyses, respectively, and incubated in 37°C, 5% CO2: 95% air incubator. After 24 h-plating, the medium was replaced by DMEM supplemented with 1% fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin, in the presence or absence of the shRNA- and iFGF13-VY-expressing adenoviruses at a multiplicity of infection (MOI) of 50 and 1, respectively. The culture medium was then changed 24 and 48 h after adenoviral infection with DMEM supplemented with 1% fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin without adenoviruses.
Fgf13fl/y, αMHC-Cre, and cFgf13KO mice
Cardiac-specific Fgf13 knockout (cFgf13KO) and control Fgf13 floxed (Fgf13fl/y) adult (8–16-wk-old) male C57BL/6J mice (Angsutararux et al., 2023) were used in the studies here. The Fgf13fl/y C57BL/6J mouse line, in which the Fgf13 locus is floxed, was obtained from Dr. Jeanne Nerbonne, and the αMHC-Cre C57BL/6J mouse line, expressing the Cre-recombinase driven by the cardiac-specific α myosin heavy chain (αMHC) promoter, was purchased from The Jackson Laboratory (Tg(Myh6-cre)2182Mds/J mouse line). To obtain cardiac-specific Fgf13 knockout (cFgf13KO) mice, Fgf13fl/y female mice were crossed with αMHC-Cre male mice. The Fgf13fl/y male littermates were used as controls.
Isolation, culture, and adenoviral infection of adult mouse ventricular myocytes
Single cardiomyocytes were isolated from the ventricles of cFgf13KO and Fgf13fl/y adult (8–16-wk-old) male C57BL/6J mice (Angsutararux et al., 2023) by enzymatic dissociation and mechanical dispersion according to a modified procedure of established methods. All reagents were from Sigma-Aldrich unless otherwise noted. Briefly, mice were injected with heparin (5,000 U/kg body weight) 30 min before sacrifice by cervical dislocation. Hearts were quickly excised and perfused retrogradely through the aorta with a solution at 37°C containing (in mM) NaCl, 113; KCl, 4.7; MgSO4, 1.2; KH2PO4, 0.6; NaH2PO4, 0.6; HEPES, 10; NaHCO3, 1.6; taurine, 30; and glucose, 20 (pH 7.4 with NaOH). Hearts were subsequently digested for 11 min with the same solution supplemented with 0.08 mg/ml Liberase Research Grade. Following digestion, the perfusion was stopped, the atria were removed, and the ventricles were dispersed by gentle trituration. The resulting cell suspension was filtered to remove large undissociated tissue fragments and resuspended in solutions containing 10 mg/ml bovine serum albumin and Ca2+ concentrations successively increasing from nominally 0 to 0.2, 0.5, and 1 mM. Isolated cardiomyocytes were then resuspended in medium-199 supplemented with 5% fetal bovine serum, 10 mM 2,3-Butanedione monoxime, 100 U/ml penicillin, and 100 μg/ml streptomycin, plated on laminin-coated dishes, and incubated in 37°C, 5% CO2, 95% air incubator. After 1-h plating, the culture medium was replaced by medium-199 supplemented with 0.1% bovine serum albumin, 10 mM 2,3-Butanedione monoxime, 1X insulin/transferrin/sodium selenite, 1X chemically defined lipid concentrate (Thermo Fisher Scientific), 0.5 µM cytochalasine D, 100 U/ml penicillin, and 100 μg/ml streptomycin in the presence or absence of the different iFGF13-VY-expressing adenoviruses at a MOI of 1.
RNA preparation and SYBR Green quantitative RT-PCR
Total RNA was isolated from cultured ventricular myocytes and analyzed using standard methods previously described in detail (Marionneau et al., 2008). Briefly, cells were washed twice in ice-cold PBS (pH 7.4) and lysed in a buffer provided in the Nucleospin RNA kit (Machery-Nagel). Total RNA was isolated and DNase-treated following the kit instructions. The quality of total RNA in each sample was examined by gel electrophoresis. Genomic DNA contamination was assessed by PCR amplification of each total RNA sample without prior cDNA synthesis; no genomic DNA was detected.
First-strand cDNA was synthesized from 200 ng of total RNA from each sample using the High-Capacity cDNA Archive kit (Thermo Fisher Scientific). The relative expression levels of transcripts encoding the different Fgf isoforms, including Fgf11, Fgf12, Fgf13-VY, Fgf13-V, Fgf13-Y, Fgf13-S, Fgf13-U, and Fgf14, as well as the hypoxanthine guanine phosphoribosyl transferase I (Hprt) used as an endogenous control, were determined by quantitative RT-PCR using 1X SYBR Green PCR Master Mix (Thermo Fisher Scientific). PCR reactions were performed on 10 ng of cDNA in the ABI PRISM 7900HT Sequence Detection System (Thermo Fisher Scientific) using isoform-specific primer pairs giving 90–100% efficacy and a single amplicon at the appropriate melting temperature and size (Table S2). The cycling conditions included a hot start at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Results for each sample were normalized to Hprt and expressed according to the 2−ΔCt method as a relative transcript expression compared with Hprt.
Preparation of cardiomyocyte lysates and Western blot analyses
Cultured ventricular myocytes were lysed and Western blot analyses of myocyte lysates were completed as described previously (Lorenzini et al., 2021). Briefly, cells were washed twice in ice-cold PBS (pH 7.4) and lysed in ice-cold lysis buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 0.5% amidosulfobetaine, 1X complete protease inhibitor cocktail tablet, 1 mM PMSF, 0.7 μg/ml pepstatin A (Thermo Fisher Scientific), and 1X Halt phosphatase inhibitor cocktail (Thermo Fisher Scientific). All reagents were from Sigma-Aldrich unless otherwise noted. After a 15-min rotation at 4°C, protein concentrations in detergent-soluble cell lysates were determined using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific), and proteins were subsequently analyzed by Western blot. The mouse iFGF13 isoforms (all included), the human iFGF13-VY isoform, and the mouse iFGF13-S isoform were specifically detected using an anti-iFGF13 rabbit polyclonal antibody (1:1,000), given by Dr. Cecilia Lindskog (Human Protein Atlas, Uppsala University, Uppsala, Sweden), the anti-iFGF13 mouse monoclonal antibody (NeuroMab clone N91/27, 1:300; Antibodies Incorporated), and the anti-Pan-iFGF-A mouse monoclonal antibody (NeuroMab clone N235/22, 1:300; Antibodies Incorporated), respectively. The anti-transferrin receptor mouse monoclonal antibody (TransR, clone H68.4, 1:1,000; Thermo Fisher Scientific) and the anti-α 1 Na+/K+-ATPase mouse monoclonal antibody (Na+/K+-ATPase α1, #ab7671, 1:1,000; Abcam) were used to verify equal protein loading. Bound primary antibodies were detected using horseradish peroxidase-conjugated goat anti-mouse or -rabbit secondary antibodies (Cell Signaling Technology, Inc.), and protein signals were visualized using the SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific). Bands corresponding to iFGF13 were normalized to bands corresponding to TransR from the same sample, and relative iFGF13 protein expression is expressed relative to TransR protein expression.
Electrophysiological recordings
Whole-cell NaV currents were recorded at room temperature from neonatal and adult mouse ventricular myocytes using an Axopatch 200B amplifier (Axon Instruments, Molecular Devices) 48 h following adenoviral infection. Voltage-clamp protocols were applied using the pClamp 10.4 software package (Axon Instruments) interfaced to the electrophysiological equipment using a Digidata 1440A digitizer (Axon Instruments). Current signals were filtered at 10 kHz prior to digitization at 50 kHz and storage. Patch-clamp pipettes were fabricated from borosilicate glass (OD: 1.5 mm, ID: 0.86 mm, Sutter Instrument) using a P-97 micropipette puller (Sutter Instrument) to obtain a resistance between 0.8 and 1.5 MΩ when filled with an internal solution. For both neonatal and adult cardiomyocytes, the internal solution contained (in mM) NaCl 5, CsF 115, CsCl 20, HEPES 10, and EGTA 10 (pH 7.35 with CsOH, ∼300 mosM). The external solution used to patch neonatal cardiomyocytes contained (in mM) NaCl 20, CsCl 103, TEA-Cl (tetraethylammonium chloride) 25, HEPES 10, glucose 5, CaCl2 1, MgCl2 2, and CoCl2 2.5 (pH 7.4 with CsOH, ∼300 mosM) and the external solution used to patch adult cardiomyocytes contained (in mM) NaCl 10, CsCl 5, N-methyl-D-glucamine (NMDG) 104, TEA-Cl 25, HEPES 10, glucose 5, CaCl2 1, MgCl2 2, and CoCl2 2.5 (pH 7.4 with CsOH, ∼300 mosM). All chemicals were purchased from Sigma-Aldrich. After establishing the whole-cell configuration, 5 min were allowed to ensure stabilization of voltage dependence of activation and inactivation properties, at which time 25 ms voltage steps to ±10 mV from a holding potential (HP) of −70 mV were applied to allow measurement of whole-cell membrane capacitances, input and series resistances. Only cells with access resistance <7 MΩ were used and input resistances were typically >1 GΩ. After compensation of series resistance (80%), the membrane was held at an HP of −120 mV and the voltage-clamp protocols were carried out as indicated below. Leak currents were always <300 pA at HP (−120 mV) and were corrected offline. Cells exhibiting peak current amplitudes <500 or >5,000 pA were excluded from analyses of biophysical properties because of errors associated with leak or voltage-clamp (Montnach et al., 2021), respectively, but were conserved in analyses of peak current density to avoid bias in the evaluation of current densities.
Data were compiled and analyzed using ClampFit 11.2 (Axon Instruments), Microsoft Excel, and Prism (GraphPad Software). Whole-cell membrane capacitances (Cm) were determined by analyzing the decays of capacitive transients elicited by brief (25 ms) voltage steps to ±10 mV from the HP (−70 mV). Input resistances were calculated from the steady-state currents elicited by the same ±10 mV steps (from the HP). Series resistances were calculated by dividing the decay time constants of the capacitive transients (fitted with single exponentials) by the Cm. To determine peak Na+ current-voltage relationships, currents were elicited by 50-ms depolarizing pulses to potentials ranging from −80 to +40 mV (presented at 5-s intervals in 5-mV increments) from an HP of −120 mV. Peak current amplitudes were defined as the maximal currents evoked at each voltage. Current amplitudes were leak-corrected, normalized to the Cm, and current densities are presented.
To analyze voltage dependence of current activation properties, conductances (G) were calculated and conductance-voltage relationships were fitted with the Boltzmann equation , in which V1/2 is the membrane potential of half-activation and k is the slope factor. The time courses of inactivation of macroscopic currents were determined by fitting the current decay with the biexponential function , in which Afast and Aslow are the amplitudes of the fast and slow inactivating current components, respectively, and τfast and τslow are the decay time constants of Afast and Aslow, respectively. To visually inspect changes in current decay kinetics, overlays of INa recordings were obtained after normalization by the peak current amplitude and representative current traces are presented. A standard two-pulse protocol was used to examine the voltage dependences of steady-state inactivation. From an HP of −120 mV, 1-s conditioning pulses to potentials ranging from −120 to −35 mV (in 5-mV increments) were followed by 20-ms test depolarizations to 0 mV (for neonatal myocytes) or −20 mV (for adult myocytes; interpulse intervals were 5-s). Current amplitudes evoked from each conditioning voltage were measured and normalized to the maximal current (Imax) evoked from −120 mV, and normalized currents were plotted as a function of the conditioning voltage. The resulting steady-state inactivation curves were fitted with the Boltzmann equation , in which V1/2 is the membrane potential of half-inactivation and k is the slope factor. To examine the rates of recovery from inactivation, a three-pulse protocol was used. Cells were first depolarized to −20 mV (from an HP of −120 mV) to inactivate the channels and subsequently repolarized to −120 mV for varying times (ranging from 1 to 200 ms) followed by test depolarizations to −20 mV to assess the extent of recovery (interpulse intervals were 5-s). The current amplitudes at −20 mV, measured following each recovery period, were normalized to the maximal current amplitude and plotted as a function of the recovery time. The resulting plot was fitted with a double exponential function to determine the time constants for fast (τfast) and slow (τslow) recovery from inactivation. For each of these biophysical properties, data from individual cells were first fitted and then averaged.
In experiments aimed at recording the tetrodotoxin (TTX)-sensitive late Na+ current (INaL), adult mouse ventricular myocytes were bathed in external solution containing (in mM) NaCl 120, TEA-Cl 25, HEPES 10, glucose 5, CaCl2 1, MgCl2 2, CoCl2 2.5 (pH 7.4 with CsOH, ∼300 mosM). Repetitive 350-ms test pulses to −20 mV from an HP of −120 mV (at 5-s intervals) were applied to cells to record Na+ currents in the absence of TTX. Cells were then superfused locally with the external solution supplemented with 60 μM TTX (Bio-Techne SAS). Cells exhibiting differences in leak current amplitudes before and after TTX application >5 pA at −20 mV (calculated from leak currents at −120 mV) were excluded from analyses. TTX-sensitive currents from individual cells were determined by offline digital subtraction of average leak-subtracted currents obtained from five recordings in the absence and the presence of TTX after achieving steady state. The amplitude of TTX-sensitive INaL was defined as the mean steady-state current amplitude of macroscopic TTX-sensitive current measured from 150 to 350 ms. For each cell, the TTX-sensitive INaL amplitude was normalized to the Cm and INaL current densities are presented.
Statistical analyses
Results are expressed as means ± SEM. Data were first tested for normality using the D’Agostino and Pearson normality test. Depending on the results of normality tests, statistical analyses were then performed using the Mann-Whitney nonparametric test or the ordinary one-way ANOVA followed by Tukey’s multiple comparisons post-hoc test, as indicated in the figures and tables. All these analyses, as well as plots and graphs, were performed using Prism (GraphPad Software).
Online supplemental material
Results
Identification and stoichiometry of nine iFGF13 phosphorylation sites in adult mouse left ventricular NaV1.5 channel complexes
The identification of the iFGF13 protein from adult mouse left ventricles was obtained from the mass spectrometric analysis of mouse left ventricular NaV channel complexes purified by IP using an anti-NaVPAN mouse monoclonal (mαNaVPAN) antibody using previously described methods (Lorenzini et al., 2021). Among the 52 unique (169 total) iFGF peptides detected in the mαNaVPAN-IPs, 38 (117 total) peptides were specific for iFGF13 and conserved across the 5 iFGF13 isoforms (Fig. 1 A and Table S1). While 8 additional unique (25 total) peptides located in the alternatively spliced N-terminus of the iFGF13-VY isoform and 4 unique (24 total) peptides common to the iFGF13-VY and iFGF13-Y N-termini could be discriminated, no peptides specific for the 3 other iFGF13 isoforms (iFGF13-V, iFGF13-S, and iFGF13-U) were detected. Hence, as highlighted in yellow in Fig. 1 A, 84% of the iFGF13-VY amino acid sequence was covered by mass spectrometry, representing most of the protein sequence. In addition to iFGF13 peptides, only one peptide specific for the iFGF12 and/or iFGF14 sequences was identified. Altogether, these observations confirm that iFGF13-VY is the predominant iFGF isoform in adult mouse left ventricular NaV1.5 channel complexes, and suggest minor representations by the iFGF13-Y, iFGF12, and/or iFGF14 isoforms.
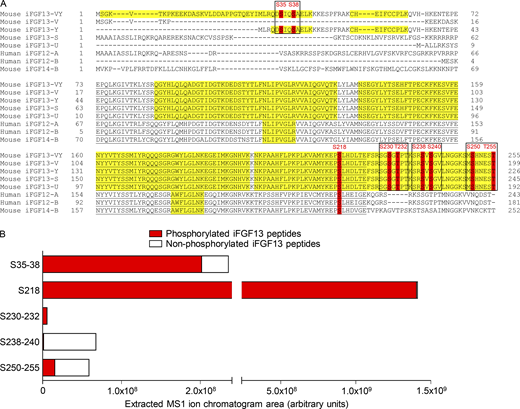
Mass spectrometric identification and stoichiometry of nine iFGF13 phosphorylation sites from adult mouse left ventricular NaV1.5 channel complexes. (A) The mouse iFGF13-VY, iFGF13-V, iFGF13-Y, iFGF13-S, iFGF13-U, and iFGF14-B, and human iFGF12-A and iFGF12-B sequences are aligned, and the phosphorylation sites identified by MS on mouse iFGF13-VY and conserved in the other iFGF isoforms are highlighted in red. The MS-covered sequence is highlighted in yellow; and the FGF core domain is underlined in black. Amino acid sequences, masses, and MS quality indicators of detected iFGF peptides are provided in Tables 1 and S1. The four phosphorylation clusters analyzed electrophysiologically are boxed in black. (B) The areas of extracted MS1 ion chromatograms, corresponding to MS2 spectra assigning phosphorylated (in red) and non-phosphorylated (in white) iFGF13 peptides at indicated site(s), in mαNaVPAN-IPs from adult mouse left ventricles are indicated. Phosphosite stoichiometry is analyzed individually (S218) or by pairs (S35–38, S230–232, S238–240, and S250–255) as corresponding phosphosites are identified from the same phosphopeptides.
Mass spectrometric identification and stoichiometry of nine iFGF13 phosphorylation sites from adult mouse left ventricular NaV1.5 channel complexes. (A) The mouse iFGF13-VY, iFGF13-V, iFGF13-Y, iFGF13-S, iFGF13-U, and iFGF14-B, and human iFGF12-A and iFGF12-B sequences are aligned, and the phosphorylation sites identified by MS on mouse iFGF13-VY and conserved in the other iFGF isoforms are highlighted in red. The MS-covered sequence is highlighted in yellow; and the FGF core domain is underlined in black. Amino acid sequences, masses, and MS quality indicators of detected iFGF peptides are provided in Tables 1 and S1. The four phosphorylation clusters analyzed electrophysiologically are boxed in black. (B) The areas of extracted MS1 ion chromatograms, corresponding to MS2 spectra assigning phosphorylated (in red) and non-phosphorylated (in white) iFGF13 peptides at indicated site(s), in mαNaVPAN-IPs from adult mouse left ventricles are indicated. Phosphosite stoichiometry is analyzed individually (S218) or by pairs (S35–38, S230–232, S238–240, and S250–255) as corresponding phosphosites are identified from the same phosphopeptides.
Among these 166 total iFGF13 peptides, 62 peptides were phosphorylated at one or two positions, which represents more than a third of the detected iFGF13 peptides (Tables 1 and S1). The annotation of MS/MS spectra obtained for each phosphopeptide allowed the unambiguous identification of nine phosphorylation sites on the iFGF13 protein at positions S35, S38, S218, S230, T232, S238, S240, S250, and T255 (Fig. 1 A). Table 1 lists the phosphopeptides enabling the best phosphorylation site assignment(s) for each phosphorylation site. The identification of two N-terminal phosphorylation sites at positions S35 and S38 arises from 2 (6 total) phosphopeptides specific for iFGF13-VY and 3 (16 total) phosphopeptides common to iFGF13-VY and iFGF13-Y, suggesting that these N-terminal phosphosites are localized on the most represented ventricular iFGF13-VY isoform. Interestingly, the phosphorylation site at position S218 is conserved across all mouse iFGF (iFGF11–14) isoforms, as well as the human iFGF12-A and iFGF12-B isoforms, while the six C-terminal iFGF13 phosphorylation sites are specific for the mouse iFGF13 isoforms. It is also interesting to note that, excluding S218, the identified phosphosites are clustered in pairs, which may indicate concomitant phosphorylation and coregulation.
Phosphorylation sites, phosphopeptides, and site-discriminating ions identified in iFGF13 proteins from NaV channel complexes purified from adult mouse left ventricles using MS
| Phosphorylation site(s) | Phosphopeptide sequence | m/z (charge) | Ascore | b ion | Phospho b ion | y ion | Phospho y ion |
|---|---|---|---|---|---|---|---|
| S35 | 33-QD(pS)IQSAELK | 828.936 (+2) | 10 | b2 (+1) | b5 | y7 (+1) | y8 (+1) |
| S38 | 33-QDSIQ(pS)AELK | 828.933 (+2) | 38 | b5 (+1) | (−) | y4 (+1) | (−) |
| S35 + S38 | 33-QD(pS)IQ(pS)AELK | 868.917 (+2) | 1,000; 1,000 | b2 (+1) | b5 | y4 (+1) | y5; y8 |
| S218 | 216-EP(pS)LHDLTEFSR | 870.414 (+2) | 54 | b1 (+1) | b7 | y9 (+1) | (−) |
| S230 + T232 | 228-SG(pS)G(pT)PTKSR | 532.593 (+3) | 11; 14 | b2 | (−) | y5 (+2) | (−) |
| T232 | 228-SGSG(pT)PTKSR | 505.936 (+3) | 8 | b4 | (−) | y5 (+2) | (−) |
| S238 | 236-SR(pS)VSGVLNGGK | 566.982 (+3) | 9 | b2 | b3 (+2) | y9 | (−) |
| S240 | 238-SV(pS)GVLNGGK | 728.401 (+2) | 1000 | b2 | (−) | y7 | (−) |
| S250 | 248-SM(pS)HNEST | 609.238 (+2) | 2 | b2 | b5 (+2) | (−) | (−) |
| T255 | 248-SMSHNES(pT) | 601.240 (+2) | 12 | b7 (+2) | (−) | (−) | (−) |
| S250 + S255 | 248-SM(pS)HNES(pT) | 641.226 (+2) | 0; 0 | b2 | (−) | (−) | (−) |
The site-discriminating ions observed in MS/MS spectra of each annotated iFGF13 phosphopeptide support the assignment of the indicated phosphorylation site(s). The PEAKS Ascore is a quality indicator of site localization. The manually verified charge state of unphosphorylated and phosphorylated site-discriminating b and y ions is reported in parentheses. The (−) symbol indicates that the ion was not detected.
Concordant with a large number of detected phosphorylated, compared with non-phosphorylated, iFGF13 peptides, further label-free quantitative analysis of the areas of extracted MS1 peptide ion chromatograms demonstrated a greater relative abundance of phosphorylated iFGF13 peptides (summed area = 1.6E+09 AU) compared with non-phosphorylated iFGF13 peptides (summed area = 1.5E+08 AU, Fig. 1 B). This analysis also revealed large differences in the relative abundances of the individual iFGF13 phosphopeptides. While phosphorylation at position S218 is the most abundant (area = 1.4E+09 AU), followed by phosphorylation at S35–38 (area = 2.0E+08 AU), phosphorylation at the six C-terminal sites at positions S250–255 (area = 1.6E+07 AU), S230–232 (area = 5.9E+06 AU) and S238–240 (area = 1.6E+06 AU) is less represented. Of note, the phosphorylated peptides assigning S35–38, S218, and S230–232 are more abundant than their non-phosphorylated counterparts, suggesting that these sites are mostly phosphorylated in mouse left ventricular NaV1.5 channel complexes. Taken together, these quantitative phosphoproteomic analyses identified nine phosphorylation sites on iFGF13-VY from adult mouse left ventricular NaV1.5 channel complexes, among which one site at position S218 is conserved across iFGF isoforms and species, and three sites at positions S35, S38, and S218 are heavily phosphorylated.
iFGF13 knockdown/knockout and rescue in neonatal and adult mouse ventricular myocytes
To explore the possible roles of the newly identified iFGF13 phosphorylation sites in regulating the expression and/or the gating properties of the cardiac NaV1.5 channels, two models were developed: freshly isolated neonatal and adult mouse ventricular myocytes. Neonatal ventricular myocytes were isolated from WT mouse pups, and adult ventricular myocytes were isolated from cardiac-specific Fgf13 knockout (cFgf13KO) or control Fgf13 floxed (Fgf13fl/y) adult male mice, generated as described in Angsutararux et al. (2023). The knockdown of iFGF13 expression in isolated WT neonatal cardiomyocytes in culture was achieved using iFGF13 shRNA-expressing adenoviruses and was compared directly to cardiomyocytes exposed to adenoviruses expressing control shRNA. The expression of iFGF13 in both neonatal and adult cardiomyocytes was then “rescued” using adenoviruses expressing WT iFGF13-VY or one of the phosphosilent or phosphomimetic iFGF13-VY variants. Note that the human iFGF13-VY cDNA sequence was used in these rescue experiments as it only differs from one amino acid (leucine 146) compared with the mouse sequence (histidine 146). With the exception of the S218 phosphosite which was mutated individually, all the other iFGF13 phosphosites were mutated and analyzed in pairs of two residues (35 and 38, 230 and 232, 238 and 240, and 250 and 255) as indicated by the black boxes in Fig. 1 A. In the phosphosilent constructs, mutations were introduced to replace serine(s)/threonine(s) (S/T) with alanines (A), whereas, in the phosphomimetic constructs, mutations were introduced to substitute glutamate(s) (E) for serine(s)/threonine(s) to mimic phosphorylation. An additional adenovirus expressing iFGF13-VY phosphosilent at all nine identified sites (iFGF13-VY-9A) was also generated and used as a rescue.
Quantitative RT-PCR analyses of the four Fgf genes and the five Fgf13 isoforms (Fgf13-VY, -V, -Y, -S, and -U) as well as iFGF13 Western blot analyses were performed to verify the specific knockdown and rescue of iFGF13 in the two cardiomyocyte models. As illustrated in Fig. 2 A, the application of iFGF13 shRNA-expressing adenoviruses on neonatal mouse ventricular myocytes provided ∼90% knockdown of the transcripts encoding the Fgf13-VY, Fgf13-V, and Fgf13-S isoforms (P < 0.0001), whereas no significant changes in the expression of the transcripts encoding Fgf12, Fgf13-Y, or Fgf14 were obtained. Consistent with previous reports (Wang et al., 2011a), the transcript expression levels of the Fgf13-U and Fgf11 isoforms were not detected in neonatal or adult mouse ventricular myocytes (data not shown). Accordingly, Western blot analyses showed 99% knockdown in iFGF13 protein expression (P < 0.01) in iFGF13, compared with control, shRNA-treated neonatal cardiomyocytes (Fig. 2, C and D). In addition, although readily detected in lysates prepared from Fgf13 floxed (Fgf13fl/y) myocytes, iFGF13 protein expression was undetectable in lysates of ventricular myocytes isolated from adult cFgf13KO mice (Fig. 2 F).
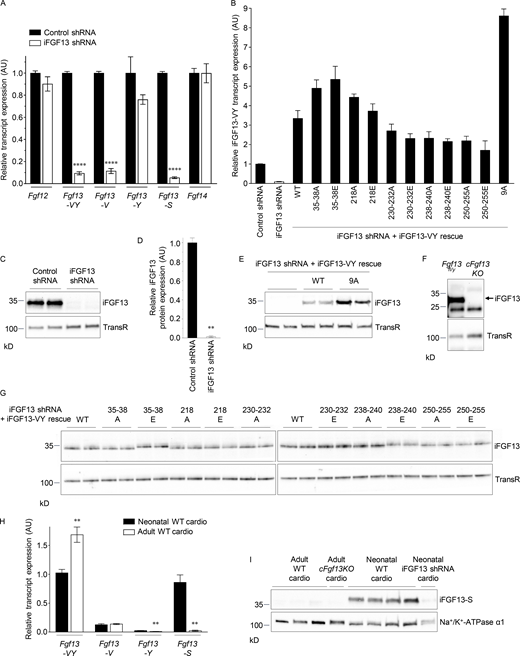
iFGF13 expression in WT, knockdown/knockout, and rescued neonatal and adult mouse ventricular myocytes. Neonatal ventricular myocytes were freshly isolated from WT mouse pups. Adult ventricular myocytes were freshly isolated from Fgf13 floxed (Fgf13fl/y) or cardiac-specific Fgf13 knockout (cFgf13KO) mice. The knockdown of iFGF13 in neonatal cardiomyocytes was obtained using iFGF13 shRNA-expressing adenoviruses, and the expression of iFGF13 in both neonatal and adult cardiomyocytes was rescued using adenoviruses expressing WT (iFGF13-VY-WT), phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY at indicated sites. (A) Mean ± SEM relative transcript expression of Fgf12 (n = 12 in each group), Fgf13-VY (n = 28 in control and 24 in iFGF13 shRNA samples), Fgf13-V (n = 12 in each group), Fgf13-Y (n = 4 in each group), Fgf13-S (n = 12 in each group), and Fgf14 (n = 12 in each group) isoforms in neonatal mouse ventricular myocytes infected with control or iFGF13 shRNA-expressing adenoviruses. (B) Mean ± SEM relative transcript expression of Fgf13-VY in neonatal mouse ventricular myocytes infected with adenoviruses expressing control shRNA (n = 28), iFGF13 shRNA alone (n = 24), or with iFGF13-VY-WT (n = 16), iFGF13-VY-35–38A (n = 6), iFGF13-VY-35–38E (n = 4), iFGF13-VY-218A (n = 4), iFGF13-VY-218E (n = 4), iFGF13-VY-230–232A (n = 4), iFGF13-VY-230–232E (n = 4), iFGF13-VY-238–240A (n = 4), iFGF13-VY-238–240E (n = 4), iFGF13-VY-250–255A (n = 4), iFGF13-VY-250–255E (n = 4), or iFGF13-VY-9A (n = 2). (C and D) Representative Western blot (C) and mean ± SEM relative protein expression (D) of iFGF13 (all isoforms) in neonatal mouse ventricular myocytes infected with adenoviruses expressing control (n = 6) or iFGF13 (n = 6) shRNA. (E) Representative Western blot of the rescued human iFGF13-VY isoform in neonatal mouse ventricular myocytes infected with adenoviruses expressing iFGF13 shRNA alone (n = 4) or with iFGF13-VY-WT (n = 4) or iFGF13-VY-9A (n = 4). (F) Representative Western blot of iFGF13 (all isoforms) in ventricular myocytes isolated from Fgf13fl/y (n = 3) and cFgf13KO (n = 3) adult mice. (G) Representative Western blots of the rescued human iFGF13-VY isoform in neonatal mouse ventricular myocytes infected with adenoviruses expressing iFGF13 shRNA and iFGF13-VY-WT (n = 14), iFGF13-VY-35-38A (n = 10), iFGF13-VY-35-38E (n = 8), iFGF13-VY-218A (n = 6), iFGF13-VY-218E (n = 4), iFGF13-VY-230-232A (n = 2), iFGF13-VY-230-232E (n = 4), iFGF13-VY-238-240A (n = 4), iFGF13-VY-238-240E (n = 4), iFGF13-VY-250-255A (n = 4), or iFGF13-VY-250-255E (n = 4). (H) Mean ± SEM relative transcript expression of Fgf13-VY, Fgf13-V, Fgf13-Y, and Fgf13-S isoforms in neonatal and adult ventricular myocytes isolated from WT mice (n = 6 in each group). (I) Representative Western blot of iFGF13-S in ventricular myocytes isolated from WT (n = 6) and cFgf13KO (n = 2) adult mice, and WT neonatal mouse ventricular myocytes infected (n = 2) or not (n = 8) with iFGF13 shRNA-expressing adenoviruses. Note that the iFGF13-S band is absent in neonatal cardiomyocytes knockdown for iFGF13, validating the specificity of the detection. All Western blots were probed in parallel with the anti-transferrin receptor (TransR) or the anti-Na+/K+-ATPase α1 antibodies to verify equal protein loading. **P < 0.01, ****P < 0.0001 versus control shRNA (A and D) or neonatal WT mouse ventricular myocytes (H), Mann–Whitney test. Source data are available for this figure: SourceData F2.
iFGF13 expression in WT, knockdown/knockout, and rescued neonatal and adult mouse ventricular myocytes. Neonatal ventricular myocytes were freshly isolated from WT mouse pups. Adult ventricular myocytes were freshly isolated from Fgf13 floxed (Fgf13fl/y) or cardiac-specific Fgf13 knockout (cFgf13KO) mice. The knockdown of iFGF13 in neonatal cardiomyocytes was obtained using iFGF13 shRNA-expressing adenoviruses, and the expression of iFGF13 in both neonatal and adult cardiomyocytes was rescued using adenoviruses expressing WT (iFGF13-VY-WT), phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY at indicated sites. (A) Mean ± SEM relative transcript expression of Fgf12 (n = 12 in each group), Fgf13-VY (n = 28 in control and 24 in iFGF13 shRNA samples), Fgf13-V (n = 12 in each group), Fgf13-Y (n = 4 in each group), Fgf13-S (n = 12 in each group), and Fgf14 (n = 12 in each group) isoforms in neonatal mouse ventricular myocytes infected with control or iFGF13 shRNA-expressing adenoviruses. (B) Mean ± SEM relative transcript expression of Fgf13-VY in neonatal mouse ventricular myocytes infected with adenoviruses expressing control shRNA (n = 28), iFGF13 shRNA alone (n = 24), or with iFGF13-VY-WT (n = 16), iFGF13-VY-35–38A (n = 6), iFGF13-VY-35–38E (n = 4), iFGF13-VY-218A (n = 4), iFGF13-VY-218E (n = 4), iFGF13-VY-230–232A (n = 4), iFGF13-VY-230–232E (n = 4), iFGF13-VY-238–240A (n = 4), iFGF13-VY-238–240E (n = 4), iFGF13-VY-250–255A (n = 4), iFGF13-VY-250–255E (n = 4), or iFGF13-VY-9A (n = 2). (C and D) Representative Western blot (C) and mean ± SEM relative protein expression (D) of iFGF13 (all isoforms) in neonatal mouse ventricular myocytes infected with adenoviruses expressing control (n = 6) or iFGF13 (n = 6) shRNA. (E) Representative Western blot of the rescued human iFGF13-VY isoform in neonatal mouse ventricular myocytes infected with adenoviruses expressing iFGF13 shRNA alone (n = 4) or with iFGF13-VY-WT (n = 4) or iFGF13-VY-9A (n = 4). (F) Representative Western blot of iFGF13 (all isoforms) in ventricular myocytes isolated from Fgf13fl/y (n = 3) and cFgf13KO (n = 3) adult mice. (G) Representative Western blots of the rescued human iFGF13-VY isoform in neonatal mouse ventricular myocytes infected with adenoviruses expressing iFGF13 shRNA and iFGF13-VY-WT (n = 14), iFGF13-VY-35-38A (n = 10), iFGF13-VY-35-38E (n = 8), iFGF13-VY-218A (n = 6), iFGF13-VY-218E (n = 4), iFGF13-VY-230-232A (n = 2), iFGF13-VY-230-232E (n = 4), iFGF13-VY-238-240A (n = 4), iFGF13-VY-238-240E (n = 4), iFGF13-VY-250-255A (n = 4), or iFGF13-VY-250-255E (n = 4). (H) Mean ± SEM relative transcript expression of Fgf13-VY, Fgf13-V, Fgf13-Y, and Fgf13-S isoforms in neonatal and adult ventricular myocytes isolated from WT mice (n = 6 in each group). (I) Representative Western blot of iFGF13-S in ventricular myocytes isolated from WT (n = 6) and cFgf13KO (n = 2) adult mice, and WT neonatal mouse ventricular myocytes infected (n = 2) or not (n = 8) with iFGF13 shRNA-expressing adenoviruses. Note that the iFGF13-S band is absent in neonatal cardiomyocytes knockdown for iFGF13, validating the specificity of the detection. All Western blots were probed in parallel with the anti-transferrin receptor (TransR) or the anti-Na+/K+-ATPase α1 antibodies to verify equal protein loading. **P < 0.01, ****P < 0.0001 versus control shRNA (A and D) or neonatal WT mouse ventricular myocytes (H), Mann–Whitney test. Source data are available for this figure: SourceData F2.
The expression of iFGF13 was then rescued using adenoviruses expressing the human iFGF13-VY isoform in its WT, phosphosilent, or phosphomimetic forms. The results from quantitative RT-PCR analyses showed that the adenoviral-mediated expression of the human Fgf13-VY constructs were, on average, two- to threefold greater than the expression of the endogenous mouse Fgf13 transcript (Fig. 2 B). No direct comparison of the endogenous mouse and rescued human iFGF13 protein expression levels could be performed, however, because the anti-iFGF13 antibodies used to detect mouse or human iFGF13 only allowed the specific and exclusive detection of the mouse or the human iFGF13 proteins, respectively (data not shown). Additionally, although the averaged rescued Fgf13-VY transcript expression varied between the different adenovirus constructs, from 1.7- (for Fgf13-VY-250-255E) to 5.4- (for Fgf13-VY-35-38E) fold the level of the endogenous mouse Fgf13-VY expression (Fig. 2 B), no significant differences in iFGF13-VY protein expression were observed following rescue with the WT and the various phosphosilent or phosphomimetic iFGF13-VY variants (Fig. 2 G). Interestingly, expression of the Fgf13-VY-9A transcript was substantially (8.6-fold) higher than the endogenous mouse Fgf13 transcript as well as any of the other (WT or mutant) Fgf13-VY rescue transcripts (Fig. 2 B). Expression of the iFGF13-VY-9A protein was also threefold higher than the expression of the other WT or phosphomutant iFGF13 proteins, an observation that was also apparent when these constructs were transfected (using plasmids) in heterologous cells (data not shown). Altogether, therefore, these molecular analyses validated our ability to manipulate the expression of endogenous and rescued iFGF13 proteins, and thus, the possibility to examine the effects of iFGF13 phosphorylation using phosphosilent or phosphomimetic iFGF13-VY constructs in both neonatal and adult mouse ventricular myocytes.
Regulation of INa by iFGF13 knockdown and rescue in neonatal mouse ventricular myocytes
The densities, voltage dependences, and kinetic properties of voltage-gated Na+ (NaV) currents (INa) following the knockdown and rescue of iFGF13 were evaluated in neonatal mouse ventricular myocytes 48 h after adenoviral infections using whole-cell voltage-clamp analyses. As illustrated in Fig. 3 B, and consistent with previous studies (Wang et al., 2011a, 2017; Hennessey et al., 2013; Park et al., 2016; Santucci et al., 2022), these analyses showed that knockdown of iFGF13 significantly (P < 0.0001) shifts the voltage dependence of steady-state INa inactivation toward more hyperpolarized potentials compared with cardiomyocytes exposed to control shRNA-expressing adenoviruses (see distributions at −10 mV, detailed properties and statistics in Fig. 4 B and Table 2). Consistent with this effect, an acceleration of the inactivation kinetics was also observed upon iFGF13 knockdown (Fig. 3, A and E), with a significant (P < 0.001) decrease in the time constant of fast inactivation (τfast; Fig. 3 F and Fig. 4 D) and an increase (P < 0.01) in the proportion of fast to slow inactivating components (Afast/Aslow; Fig. 3 H and Fig. 4 F). No significant differences in the time constant of the slow component of inactivation (τslow; Fig. 3 G and Fig. 4 E), peak INa density (Fig. 3, A and D; and Fig. 4 C), time to peak INa, or time for recovery from inactivation, however, were observed with iFGF13 knockdown (Table 2). Interestingly, these analyses also revealed for the first time that the knockdown of iFGF13 in neonatal mouse ventricular myocytes results in a significant (P < 0.001) shift in the voltage dependence of INa activation toward more hyperpolarized potentials (Fig. 3 C, Fig. 4 A, and Table 2). Noteworthy, while the rescue of iFGF13 expression with the WT iFGF13-VY isoform partially, but significantly restored INa inactivation properties (Figs. 3 and 4, and Table 2), no rescue of the shift in INa activation was observed (Fig. 3 C, Fig. 4 A, and Table 2).

The increased closed-state inactivation rate of Na V channels induced by iFGF13 knockdown is partially rescued by iFGF13-VY while no rescue of the shift in the voltage dependence of activation (toward hyperpolarized potentials) is obtained in neonatal mouse ventricular myocytes. (A) Representative whole-cell voltage-gated Na+ currents were recorded 48 h following infection of neonatal WT mouse ventricular myocytes with adenoviruses expressing control shRNA, iFGF13 shRNA alone, or with WT iFGF13-VY (iFGF13-VY-WT) using the protocols illustrated in each panel. Scale bars are 1 nA and 3 ms. (B and C) Voltage dependences of steady-state current inactivation (B) and activation (C). (D) Mean ± SEM peak Na+ current (INa) densities plotted as a function of test potential. (E) Superimposed representative current traces were recorded at 0 mV (HP = −120 mV) from cardiomyocytes infected with adenoviruses expressing control shRNA, iFGF13 shRNA alone, or with iFGF13-VY-WT. (F–H) Mean ± SEM time constants of fast (τfast, F) and slow (τslow, G) inactivation, and proportions of fast to slow inactivating components (Afast/Aslow, H) are plotted as a function of test potential. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Fig. 4 and Table 2.
The increased closed-state inactivation rate of Na V channels induced by iFGF13 knockdown is partially rescued by iFGF13-VY while no rescue of the shift in the voltage dependence of activation (toward hyperpolarized potentials) is obtained in neonatal mouse ventricular myocytes. (A) Representative whole-cell voltage-gated Na+ currents were recorded 48 h following infection of neonatal WT mouse ventricular myocytes with adenoviruses expressing control shRNA, iFGF13 shRNA alone, or with WT iFGF13-VY (iFGF13-VY-WT) using the protocols illustrated in each panel. Scale bars are 1 nA and 3 ms. (B and C) Voltage dependences of steady-state current inactivation (B) and activation (C). (D) Mean ± SEM peak Na+ current (INa) densities plotted as a function of test potential. (E) Superimposed representative current traces were recorded at 0 mV (HP = −120 mV) from cardiomyocytes infected with adenoviruses expressing control shRNA, iFGF13 shRNA alone, or with iFGF13-VY-WT. (F–H) Mean ± SEM time constants of fast (τfast, F) and slow (τslow, G) inactivation, and proportions of fast to slow inactivating components (Afast/Aslow, H) are plotted as a function of test potential. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Fig. 4 and Table 2.

Functional effects of WT iFGF13-VY and iFGF13-VY phosphomutants on Na + currents in neonatal mouse ventricular myocytes. (A–F) Distributions and means ± SEM membrane potentials for half-activation (A) and half-inactivation (B), peak Na+ current (INa) densities (C), time constants of fast (τfast, D) and slow (τslow, E) inactivation, and proportions of fast to slow inactivating components (Afast/Aslow, F) from neonatal WT mouse ventricular myocytes infected with adenoviruses expressing control shRNA, iFGF13 shRNA alone or with WT (iFGF13-VY-WT), phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY at indicated sites. Currents were recorded as described in the legend to Fig. 3. The INa, τfast, τslow, and Afast/Aslow values presented were determined from analyses of records obtained on depolarizations to −10 mV (HP = −120 mV). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus control shRNA; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 versus iFGF13 shRNA; one-way ANOVA. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Table 2.
Functional effects of WT iFGF13-VY and iFGF13-VY phosphomutants on Na + currents in neonatal mouse ventricular myocytes. (A–F) Distributions and means ± SEM membrane potentials for half-activation (A) and half-inactivation (B), peak Na+ current (INa) densities (C), time constants of fast (τfast, D) and slow (τslow, E) inactivation, and proportions of fast to slow inactivating components (Afast/Aslow, F) from neonatal WT mouse ventricular myocytes infected with adenoviruses expressing control shRNA, iFGF13 shRNA alone or with WT (iFGF13-VY-WT), phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY at indicated sites. Currents were recorded as described in the legend to Fig. 3. The INa, τfast, τslow, and Afast/Aslow values presented were determined from analyses of records obtained on depolarizations to −10 mV (HP = −120 mV). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus control shRNA; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 versus iFGF13 shRNA; one-way ANOVA. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Table 2.
Voltage-gated Na+ current densities and properties in neonatal mouse ventricular cardiomyocytes infected with control shRNA, iFGF13 shRNA alone, or with WT or phosphomutant iFGF13-VY-expressing adenoviruses
| INa (pA/pF) | Time to peak (ms) | Time course of inactivation | Voltage dependence of activation | Voltage dependence of inactivation | Recovery from inactivation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| tfast (ms) | tslow (ms) | Afast/Aslow | V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | tfast (ms) | tslow (ms) | |||
| Control shRNA | −51.4 ± 5.1 (23) | 1.03 ± 0.05 (19) | 0.99 ± 0.06 (18) | 4.3 ± 0.3 (18) | 15.5 ± 2.6 (18) | −32.6 ± 0.5 (19) | 7.0 ± 0.2 (19) | −70.7 ± 0.8 (18) | 5.1 ± 0.2 (18) | 3.1 ± 0.3 (16) | 51.1 ± 8.7 (16) |
| iFGF13 shRNA | −54.9 ± 5.7 (23) | 0.92 ± 0.04 (19) | 0.73 ± 0.05 (19) *** | 5.5 ± 0.5 (19) | 27.8 ± 1.9 (19) ** | −36.7 ± 0.8 (19) *** | 7.5 ± 0.3 (19) | −82.8 ± 0.5 (19) **** | 4.9 ± 0.1 (19) | 4.0 ± 0.3 (19) | 45.4 ± 7.5 (19) |
| iFGF13 shRNA + iFGF13-VY-WT | −53.5 ± 4.6 (24) | 0.97 ± 0.03 (23) | 0.84 ± 0.04 (22) | 3.7 ± 0.2 (22) | 16.8 ± 2.6 (22) # | −36.2 ± 0.5 (22) ** | 6.5 ± 0.2 (22) | −74.6 ± 0.8 (22) #### | 4.5 ± 0.1 (22) | 2.7 ± 0.1 (19) | 56.8 ± 8.5 (19) |
| iFGF13 shRNA + iFGF13-VY-35-38A | −56.0 ± 4.8 (21) | 0.90 ± 0.02 (17) | 0.82 ± 0.04 (17) | 3.9 ± 0.2 (17) | 8.4 ± 1.1 (17) #### | −36.1 ± 0.6 (17) * | 6.8 ± 0.2 (17) | −74.6 ± 0.7 (17) #### | 4.9 ± 0.2 (17) | 2.9 ± 0.2 (16) | 44.8 ± 10.0 (16) |
| iFGF13 shRNA + iFGF13-VY-35-38E | −70.1 ± 7.1 (20) | 0.75 ± 0.02 (14) | 0.66 ± 0.03 (12) **** | 3.6 ± 0.2 (12) | 15.2 ± 1.4 (12) # | −36.8 ± 0.7 (14) ** | 7.1 ± 0.2 (14) | −76.9 ± 1.3 (14) ### | 4.8 ± 0.2 (14) | 2.8 ± 0.2 (14) | 61.1 ± 7.1 (14) |
| iFGF13 shRNA + iFGF13-VY-218A | −53.7 ± 5.5 (25) | 0.91 ± 0.03 (20) | 0.86 ± 0.03 (19) | 4.3 ± 0.2 (19) | 18.2 ± 2.3 (19) | −35.0 ± 0.5 (20) | 7.1 ± 0.1 (20) | −75.5 ± 0.8 (20) #### | 4.5 ± 0.1 (20) | 3.4 ± 0.2 (14) | 46.6 ± 3.9 (14) |
| iFGF13 shRNA + iFGF13-VY-218E | −65.9 ± 5.2 (22) | 1.00 ± 0.04 (17) | 0.89 ± 0.05 (17) | 4.5 ± 0.2 (17) | 19.2 ± 2.1 (17) | −34.2 ± 0.5 (17) | 6.6 ± 0.2 (17) | −73.3 ± 0.8 (17) #### | 4.6 ± 0.1 (17) | 3.2 ± 0.2 (15) | 52.7 ± 8.6 (15) |
| iFGF13 shRNA + iFGF13-VY-230-232A | −44.1 ± 4.8 (19) | 0.78 ± 0.04 (19) | 0.83 ± 0.03 (16) | 4.6 ± 0.3 (16) | 22.2 ± 2.9 (16) | −35.4 ± 0.8 (19) | 7.7 ± 0.2 (19) | −76.8 ± 1.0 (16) #### | 4.8 ± 0.2 (16) | 3.1 ± 0.2 (14) | 59.3 ± 10.5 (14) |
| iFGF13 shRNA + iFGF13-VY-230-232E | −55.9 ± 6.1 (20) | 0.74 ± 0.03 (20) | 0.75 ± 0.02 (19) *** | 3.8 ± 0.2 (19) | 21.4 ± 2.9 (19) # | −36.4 ± 0.6 (20) ** | 6.7 ± 0.2 (20) | −74.8 ± 0.6 (18) #### | 4.4 ± 0.1 (18) | 2.4 ± 0.2 (19) | 55.1 ± 4.8 (19) |
| iFGF13 shRNA + iFGF13-VY-238-240A | −58.1 ± 4.9 (26) | 0.73 ± 0.02 (24) | 0.77 ± 0.03 (23) ** | 4.2 ± 0.3 (23) | 18.8 ± 1.9 (23) | −36.9 ± 0.7 (24) *** | 7.2 ± 0.2 (24) | −77.8 ± 0.9 (20) ### | 4.5 ± 0.1 (20) | 2.8 ± 0.1 (18) | 64.6 ± 5.0 (18) |
| iFGF13 shRNA + iFGF13-VY-238-240E | −58.3 ± 6.6 (22) | 0.77 ± 0.02 (20) | 0.82 ± 0.03 (16) | 4.6 ± 0.4 (16) | 17.2 ± 1.8 (16) # | −33.7 ± 0.5 (20) | 7.3 ± 0.1 (20) | −76.1 ± 1.0 (17) #### | 4.4 ± 0.1 (17) | 2.5 ± 0.2 (12) | 64.1 ± 13.2 (12) |
| iFGF13 shRNA + iFGF13-VY-250-255A | −71.8 ± 7.0 (16) | 0.75 ± 0.03 (16) | 0.73 ± 0.02 (16) *** | 3.9 ± 0.3 (16) | 15.8 ± 1.4 (16) # | −36.4 ± 0.7 (16) ** | 6.9 ± 0.2 (16) | −75.0 ± 1.0 (14) #### | 4.9 ± 0.2 (14) | 2.4 ± 0.1 (14) | 65.3 ± 7.1 (14) |
| iFGF13 shRNA + iFGF13-VY-250-255E | −52.4 ± 7.0 (19) | 0.79 ± 0.02 (17) | 0.82 ± 0.04 (17) | 5.0 ± 0.3 (17) | 19.6 ± 1.8 (17) | −34.0 ± 0.9 (17) | 7.4 ± 0.2 (17) | −75.0 ± 1.0 (15) #### | 4.6 ± 0.2 (15) | 2.6 ± 0.1 (15) | 49.4 ± 5.5 (15) |
| iFGF13 shRNA + iFGF13-VY-9A | −64.1 ± 7.7 (15) | 0.75 ± 0.02 (15) | 0.75 ± 0.03 (13) ** | 3.3 ± 0.1 (13) | 12.7 ± 1.8 (13) ### | −36.2 ± 0.8 (15) * | 7.2 ± 0.3 (15) | −77.4 ± 0.6 (10) ## | 4.2 ± 0.1 (10) | 4.4 ± 0.4 (8) | 78.5 ± 7.6 (8) |
Whole-cell voltage-gated Na+ currents were recorded 48 h following infection of neonatal WT mouse ventricular cardiomyocytes with adenoviruses expressing control shRNA, iFGF13 shRNA alone, or with WT (iFGF13-VY-WT), phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY cDNA constructs using the protocols described in the Materials and methods section. The peak Na+ current (INa) density, time to peak INa, and time course of inactivation properties presented were determined from analyses of records obtained on depolarizations to −10 mV (HP = −120 mV). All values are means ± SEM. The number of cells analyzed is provided in parentheses. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus control shRNA; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 versus iFGF13 shRNA; one-way ANOVA.
To explore the possible functional roles of the newly identified iFGF13 phosphorylation sites in regulating cardiac NaV1.5 channels, iFGF13 expression was rescued with the different iFGF13-VY phosphomutant adenoviruses, and INa properties and densities were compared with those obtained with the WT iFGF13-VY rescue. Importantly, the phosphosilent and phosphomimetic constructs for each phosphosite subgroup were compared directly to the WT iFGF13-VY rescue obtained on the same days of patch-clamp analyses, and for the sake of clarity, a single representative subset of this later condition was chosen and presented in Fig. 4 and Table 2. To our surprise, no significant differences in the voltage dependences, kinetic properties, or peak INa densities were obtained for any of the 10 phosphosilent or phosphomimetic iFGF13-VY constructs compared with the WT iFGF13-VY rescue. An iFGF13-VY construct phosphosilent at the nine identified phosphorylation sites was thus generated, but the expression of this construct also did not result in any significant differences in INa properties nor densities compared with the WT iFGF13-VY rescue.
Regulation of INa by Fgf13 knockout and rescue in adult mouse ventricular myocytes
One possible caveat with interpreting the results of the experiments described above is that these experiments were conducted on neonatal mouse ventricular myocytes, and the iFGF13 phosphorylation sites were identified in adult mouse left ventricles. To determine directly if the identified iFGF13 phosphosites affect the properties of INa in adult mouse ventricular myocytes, electrophysiological experiments similar to those described above were conducted on ventricular myocytes isolated from adult cFgf13KO (and control Fgf13fl/y) mice (Angsutararux et al., 2023) 48 h following culture and infection with the different phosphomutant (or WT) iFGF13-VY adenoviruses. Similar to findings obtained in neonatal cardiomyocytes, as well as in adult cardiomyocytes isolated from the same mouse lines in the Nerbonne laboratory (Angsutararux et al., 2023), the knockout of Fgf13 in adult mouse ventricular myocytes significantly increases the rate of NaV channel inactivation (Fig. 5, A, B, and E). The membrane potential of half inactivation (V1/2; Fig. 5 B and Fig. 6 B), the time constant of fast inactivation (τfast; Fig. 5 F and Fig. 6 D), and the proportion of fast to slow inactivating components (Afast/Aslow; Fig. 5 H and Fig. 6 F) were significantly (P < 0.0001) different in INa recordings from cFgf13KO and Fgf13fl/y cardiomyocytes (see distributions at −20 mV, detailed properties and statistics in Fig. 6 and Table 3). Contrary to findings obtained in neonatal cardiomyocytes, however, these changes were completely restored on expression of WT iFGF13-VY, demonstrating the exclusive role of this iFGF13 isoform in regulating adult mouse ventricular NaV currents. Also, in contrast with the results in neonatal cells (Fig. 3 C), the voltage dependence of INa activation was not affected by the knockout (or subsequent rescue) of iFGF13 expression (Fig. 5 C and Fig. 6 A). Interestingly, however, the rescue of iFGF13 expression with WT iFGF13-VY markedly (P < 0.0001) increased the density of the peak INa, whereas there were no differences in peak INa densities in cFgf13KO compared with Fgf13fl/y, ventricular myocytes (Fig. 5, A and D; Fig. 6 C; and Table 3). The roles of iFGF13 phosphorylation sites were then explored in adult mouse ventricular myocytes using the different phosphosilent or phosphomimetic iFGF13-VY adenoviruses. Similar to results obtained in neonatal cardiomyocytes, however, we did not detect any significant differences in INa densities or properties between the different phosphomutant and the WT adenoviral rescues, including with the iFGF13-VY-9A rescue (Fig. 6 and Table 3).

The increased closed-state inactivation rate of Na V channels induced by Fgf13 knockout is rescued by iFGF13-VY in adult mouse ventricular myocytes. (A) Representative whole-cell voltage-gated Na+ currents recorded 48 h following isolation of Fgf13 floxed (Fgf13fl/y) or cardiac-specific Fgf13 knockout (cFgf13KO) adult mouse ventricular myocytes and/or infection with WT iFGF13-VY (iFGF13-VY-WT)-expressing adenoviruses using the protocols illustrated in each panel. Scale bars are 1 nA and 3 ms. (B and C) Voltage dependences of steady-state current inactivation (B) and activation (C). (D) Mean ± SEM peak Na+ current (INa) densities plotted as a function of test potential. (E) Superimposed representative current traces recorded at 0 mV (HP = −120 mV) from Fgf13fl/y or cFgf13KO adult mouse ventricular myocytes infected or not with iFGF13-VY-WT-expressing adenoviruses. (F–H) Mean ± SEM time constants of fast (τfast, F) and slow (τslow, G) inactivation and proportions of fast to slow inactivating components (Afast/Aslow, H) plotted as a function of test potential. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Fig. 6 and Table 3.
The increased closed-state inactivation rate of Na V channels induced by Fgf13 knockout is rescued by iFGF13-VY in adult mouse ventricular myocytes. (A) Representative whole-cell voltage-gated Na+ currents recorded 48 h following isolation of Fgf13 floxed (Fgf13fl/y) or cardiac-specific Fgf13 knockout (cFgf13KO) adult mouse ventricular myocytes and/or infection with WT iFGF13-VY (iFGF13-VY-WT)-expressing adenoviruses using the protocols illustrated in each panel. Scale bars are 1 nA and 3 ms. (B and C) Voltage dependences of steady-state current inactivation (B) and activation (C). (D) Mean ± SEM peak Na+ current (INa) densities plotted as a function of test potential. (E) Superimposed representative current traces recorded at 0 mV (HP = −120 mV) from Fgf13fl/y or cFgf13KO adult mouse ventricular myocytes infected or not with iFGF13-VY-WT-expressing adenoviruses. (F–H) Mean ± SEM time constants of fast (τfast, F) and slow (τslow, G) inactivation and proportions of fast to slow inactivating components (Afast/Aslow, H) plotted as a function of test potential. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Fig. 6 and Table 3.

Functional effects of WT iFGF13-VY and iFGF13-VY phosphomutants on Na + currents in adult mouse ventricular myocytes. (A–F) Distributions and means ± SEM membrane potentials for half-activation (A) and half-inactivation (B), peak Na+ current (INa) densities (C), time constants of fast (τfast, D) and slow (τslow, E) inactivation, and proportions of fast to slow inactivating components (Afast/Aslow, F) from Fgf13 floxed (Fgf13fl/y) or cardiac-specific Fgf13 knockout (cFgf13KO) adult mouse ventricular myocytes infected or not with WT (iFGF13-VY-WT), phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY-expressing adenoviruses at indicated sites. Currents were recorded as described in the legend to Fig. 5. The INa, τfast, τslow, and Afast/Aslow values presented were determined from analyses of records obtained on depolarizations to −20 mV (HP = −120 mV). **P < 0.01, ****P < 0.0001 versus Fgf13fl/y; ###P < 0.001, ####P < 0.0001 versus cFgf13KO; one-way ANOVA. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Table 3.
Functional effects of WT iFGF13-VY and iFGF13-VY phosphomutants on Na + currents in adult mouse ventricular myocytes. (A–F) Distributions and means ± SEM membrane potentials for half-activation (A) and half-inactivation (B), peak Na+ current (INa) densities (C), time constants of fast (τfast, D) and slow (τslow, E) inactivation, and proportions of fast to slow inactivating components (Afast/Aslow, F) from Fgf13 floxed (Fgf13fl/y) or cardiac-specific Fgf13 knockout (cFgf13KO) adult mouse ventricular myocytes infected or not with WT (iFGF13-VY-WT), phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY-expressing adenoviruses at indicated sites. Currents were recorded as described in the legend to Fig. 5. The INa, τfast, τslow, and Afast/Aslow values presented were determined from analyses of records obtained on depolarizations to −20 mV (HP = −120 mV). **P < 0.01, ****P < 0.0001 versus Fgf13fl/y; ###P < 0.001, ####P < 0.0001 versus cFgf13KO; one-way ANOVA. Current densities, time- and voltage-dependent properties, as well as statistical comparisons across groups, are provided in Table 3.
Voltage-gated Na+ current densities and properties in Fgf13fl/y and cFgf13KO adult mouse ventricular cardiomyocytes infected or not with WT or phosphomutant iFGF13-VY-expressing adenoviruses
| INa (pA/pF) | Time to peak (ms) | Time course of inactivation | Voltage dependence of activation | Voltage dependence of inactivation | Recovery from inactivation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| τfast (ms) | τslow (ms) | Afast/Aslow | V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | τfast (ms) | τslow (ms) | |||
| Fgf13fl/y | −31.8 ± 1.4 (58) | 1.09 ± 0.03 (33) | 1.42 ± 0.05 (33) | 5.3 ± 0.3 (33) | 8.6 ± 1.0 (33) | −28.0 ± 0.5 (33) | 7.5 ± 0.1 (33) | −77.3 ± 0.7 (30) | 6.3 ± 0.2 (30) | 2.9 ± 0.1 (27) | 37.3 ± 1.2 (27) |
| cFgf13KO | −33.9 ± 2.0 (47) | 0.97 ± 0.03 (20) | 0.90 ± 0.02 (20) **** | 5.9 ± 0.2 (20) | 21.9 ± 1.1 (20) **** | −28.1 ± 0.4 (20) | 7.8 ± 0.1 (20) | −85.9 ± 0.6 (19) **** | 6.1 ± 0.2 (19) | 3.0 ± 0.1 (18) | 41.9 ± 2.6 (18) |
| cFgf13KO + iFGF13-VY-WT | −58.5 ± 5.3 (39) **** | 1.02 ± 0.03 (28) | 1.48 ± 0.06 (28) #### | 4.4 ± 0.2 (28) | 5.2 ± 0.4 (28) #### | −31.2 ± 0.5 (28) | 7.1 ± 0.2 (28) | −76.7 ± 0.6 (28) #### | 5.2 ± 0.1 (28) | 3.2 ± 0.2 (27) | 45.1 ± 2.5 (27) |
| cFgf13KO + iFGF13-VY-35-38A | −50.2 ± 3.3 (37) ** | 1.01 ± 0.02 (27) | 1.41 ± 0.05 (27) #### | 4.6 ± 0.1 (27) | 5.8 ± 0.6 (27) #### | −30.4 ± 0.7 (27) | 7.2 ± 0.2 (27) | −77.2 ± 0.7 (21) #### | 6.0 ± 0.2 (21) | 3.4 ± 0.2 (15) | 40.2 ± 1.8 (15) |
| cFgf13KO + iFGF13-VY-35-38E | −46.5 ± 2.5 (38) | 1.09 ± 0.04 (22) | 1.48 ± 0.05 (22) #### | 4.7 ± 0.2 (22) | 6.4 ± 0.7 (22) #### | −29.5 ± 0.4 (22) | 6.8 ± 0.1 (22) | −74.3 ± 0.7 (20) #### | 5.6 ± 0.2 (20) | 2.7 ± 0.1 (18) | 39.9 ± 2.8 (18) |
| cFgf13KO + iFGF13-VY-218A | −49.5 ± 2.4 (49) ** | 1.04 ± 0.02 (27) | 1.42 ± 0.04 (27) #### | 4.5 ± 0.1 (27) | 5.2 ± 0.4 (27) #### | −31.5 ± 0.4 (27) | 7.1 ± 0.1 (27) | −75.6 ± 0.5 (20) #### | 5.7 ± 0.1 (20) | 3.0 ± 0.2 (20) | 38.1 ± 2.1 (20) |
| cFgf13KO + iFGF13-VY-218E | −49.9 ± 2.3 (39) ** | 0.93 ± 0.04 (18) | 1.14 ± 0.07 (18) | 3.9 ± 0.2 (18) | 6.2 ± 1.3 (18) #### | −32.5 ± 0.5 (18) | 7.0 ± 0.16 (18) | −78.9 ± 1.6 (14) #### | 6.6 ± 0.4 (14) | 3.0 ± 0.2 (11) | 39.4 ± 2.5 (11) |
| cFgf13KO + iFGF13-VY-230-232A | −39.8 ± 2.7 (24) | 1.25 ± 0.05 (11) | 1.52 ± 0.11 (11) #### | 4.8 ± 0.2 (11) | 8.6 ± 1.6 (11) #### | −30.7 ± 0.5 (11) | 6.8 ± 0.2 (11) | −74.9 ± 0.6 (11) #### | 5.0 ± 0.1 (11) | 2.8 ± 0.1 (11) | 38.2 ± 3.3 (11) |
| cFgf13KO + iFGF13-VY-230-232E | −44.2 ± 2.6 (35) | 1.17 ± 0.03 (18) | 1.57 ± 0.06 (18) #### | 4.7 ± 0.2 (18) | 5.4 ± 0.5 (18) #### | −30.9 ± 0.5 (18) | 7.0 ± 0.2 (18) | −75.9 ± 0.5 (18) #### | 5.1 ± 0.1 (18) | 3.1 ± 0.1 (18) | 40.9 ± 2.3 (18) |
| cFgf13KO + iFGF13-VY-238-240A | −56.5 ± 2.4 (31) **** | 1.01 ± 0.03 (16) | 1.42 ± 0.04 (16) #### | 4.5 ± 0.2 (16) | 5.7 ± 0.7 (16) #### | −31.2 ± 0.4 (16) | 7.4 ± 0.2 (16) | −77.3 ± 0.9 (16) #### | 5.5 ± 0.2 (16) | 3.3 ± 0.3 (16) | 39.6 ± 2.6 (16) |
| cFgf13KO + iFGF13-VY-238-240E | −55.8 ± 3.5 (35) **** | 0.94 ± 0.03 (26) | 1.30 ± 0.06 (26) ### | 4.1 ± 0.2 (26) | 5.6 ± 0.6 (26) #### | −33.8 ± 0.9 (26) | 7.2 ± 0.2 (26) | −75.5 ± 0.9 (26) #### | 5.4 ± 0.2 (26) | 2.6 ± 0.2 (26) | 35.3 ± 1.8 (26) |
| cFgf13KO + iFGF13-VY-250-255A | −47.7 ± 3.0 (25) | 1.08 ± 0.04 (18) | 1.63 ± 0.07 (18) #### | 5.0 ± 0.3 (18) | 6.7 ± 0.9 (18) #### | −30.9 ± 0.7 (18) | 7.1 ± 0.2 (18) | −76.7 ± 0.7 (18) #### | 5.3 ± 0.2 (18) | 3.3 ± 0.3 (16) | 51.1 ± 4.0 (16) |
| cFgf13KO + iFGF13-VY-250-255E | −50.9 ± 3.0 (27) ** | 1.01 ± 0.02 (22) | 1.42 ± 0.08 (22) #### | 4.4 ± 0.4 (22) | 6.2 ± 1.7 (22) #### | −31.1 ± 0.5 (22) | 6.9 ± 0.2 (22) | −75.9 ± 0.5 (22) #### | 5.0 ± 0.1 (22) | 2.8 ± 0.1 (20) | 38.7 ± 2.0 (20) |
| cFgf13KO + iFGF13-VY-9A | −63.6 ± 2.9 (26) **** | 0.98 ± 0.03 (20) | 1.38 ± 0.08 (20) #### | 4.3 ± 0.2 (20) | 4.5 ± 0.4 (20) #### | −34.1 ± 0.5 (20) | 6.6 ± 0.2 (20) | −77.2 ± 0.5 (20) #### | 5.1 ± 0.1 (20) | 3.1 ± 0.2 (20) | 37.2 ± 1.5 (20) |
Whole-cell voltage-gated Na+ currents were recorded 48 h following isolation of control Fgf13 floxed (Fgf13fl/y) or cardiac specific Fgf13 knockout (cFgf13KO) adult mouse ventricular cardiomyocytes infected or not with WT, phosphosilent (mutation to alanine), or phosphomimetic (mutation to glutamate) iFGF13-VY-expressing adenoviruses using the protocols described in the Materials and methods section. The peak Na+ current (INa) density, time to peak INa, and time course of inactivation properties presented were determined from analyses of records obtained on depolarizations to −20 mV (HP = −120 mV). All values are means ± SEM. The number of cells analyzed is provided in parentheses. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus Fgf13fl/y; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 versus cFgf13KO; one-way ANOVA.
Because it was previously shown that iFGF13 also plays a crucial role in regulating the late Na+ current (INaL; Abrams et al., 2020; Gade et al., 2020; Chakouri et al., 2022), additional voltage-clamp experiments were designed to investigate whether the simultaneous mutation of the nine identified iFGF13 phosphorylation sites to alanine, using the iFGF13-VY-9A phosphomutant rescue, affects the density of TTX-sensitive INaL in adult mouse ventricular myocytes. In accordance with previous studies, these analyses demonstrated that the averaged INaL density is significantly (P < 0.05) increased in cFgf13KO compared with Fgf13fl/y, cardiomyocytes (Fig. 7). Similar to the other NaV current inactivation properties, however, comparable rescues in INaL density were obtained with the iFGF13-VY-WT and iFGF13-VY-9A adenoviruses.

The increased I NaL density induced by Fgf13 knockout is rescued by the WT or the phosphosilent iFGF13-VY isoform in adult mouse ventricular myocytes. (A) Representative TTX-sensitive late Na+ current (INaL) recordings evoked by prolonged depolarizations (350 ms at −20 mV) from a HP of −120 mV obtained from Fgf13 floxed (Fgf13fl/y) or Fgf13 knockout (cFgf13KO) adult mouse ventricular myocytes infected or not with iFGF13-VY-WT or -9A-expressing adenoviruses. Scale bars are 1 pA/pF and 100 ms. (B) Distributions and means ± SEM TTX-sensitive INaL densities. *P < 0.05 versus Fgf13fl/y; ####P < 0.0001 versus cFgf13KO; one-way ANOVA.
The increased I NaL density induced by Fgf13 knockout is rescued by the WT or the phosphosilent iFGF13-VY isoform in adult mouse ventricular myocytes. (A) Representative TTX-sensitive late Na+ current (INaL) recordings evoked by prolonged depolarizations (350 ms at −20 mV) from a HP of −120 mV obtained from Fgf13 floxed (Fgf13fl/y) or Fgf13 knockout (cFgf13KO) adult mouse ventricular myocytes infected or not with iFGF13-VY-WT or -9A-expressing adenoviruses. Scale bars are 1 pA/pF and 100 ms. (B) Distributions and means ± SEM TTX-sensitive INaL densities. *P < 0.05 versus Fgf13fl/y; ####P < 0.0001 versus cFgf13KO; one-way ANOVA.
Differential transcript and protein expression of iFGF13 isoforms in neonatal and adult mouse ventricular myocytes
To begin to explore the molecular determinants of the differential regulation of neonatal and adult ventricular NaV channels by iFGF13, we examined the expression levels of the various iFGF13 isoforms in freshly isolated neonatal and adult mouse ventricular myocytes. Quantitative RT-PCR analyses using isoform-specific primers demonstrated a greater (P < 0.01) expression of the Fgf13-VY isoform in adult, compared with neonatal, cardiomyocytes (Fig. 2 H). Conversely, significantly (P < 0.01) higher expression of the Fgf13-Y and Fgf13-S isoforms was measured in neonatal, compared to adult, cardiomyocytes. Although direct comparison of relative expression of different transcripts cannot reliably be achieved using the employed relative quantitative RT-PCR method, it is interesting to underscore that the Fgf13-VY isoform is ∼10-fold more abundant than the three other Fgf13 isoforms combined in adult cardiomyocytes, a result consistent with the exclusive mass spectrometric identification of peptides specific for the iFGF13-VY (or iFGF13-Y) N-terminus (Fig. 1 A and Table S1) and the complete rescue of inactivation properties with the iFGF13-VY isoform in adult cardiomyocytes (Figs. 5, 6, and 7, and Table 3). In neonatal cardiomyocytes, however, the expression levels of the Fgf13-VY and Fgf13-S isoforms are ∼6- to ∼30-fold greater than Fgf13-V and Fgf13-Y, respectively (Fig. 2 H). Nonetheless, it is important to note here that the iFGF13-Y isoform is not of interest in the context of the studies on neonatal cardiomyocytes reported here as the expression level of this isoform is not affected by the iFGF13 shRNA used in these studies (Fig. 2 A). Consistent with transcript expression data, the use of an antibody specific for the iFGF13-S isoform in Western blot analyses demonstrated that iFGF13-S is expressed in neonatal mouse ventricular myocytes, whereas no expression could be detected in adult mouse ventricular myocytes (Fig. 2 I). Together, therefore, these molecular analyses suggest that the iFGF13-dependent regulation of NaV1.5 channels in adult mouse ventricular myocytes is exclusively mediated by iFGF13-VY, while the iFGF13-VY and iFGF13-S together regulate neonatal NaV1.5 channels.
Discussion
The results presented here provide the first phosphorylation map of the iFGF13 protein isolated from native adult mouse left ventricular NaV1.5 channel complexes. No roles for the nine newly identified iFGF13 phosphorylation sites in the regulation of the basal expression or gating properties of NaV1.5 channels, however, were identified in voltage-clamp recordings of INa in adult or neonatal mouse ventricular myocytes. A comparison of the two models, however, revealed the differential regulation of INa in neonatal and adult mouse ventricular myocytes. While iFGF13-VY appears to be the sole iFGF13 isoform involved in regulating the inactivation properties of NaV channels in adult mouse ventricular myocytes, both iFGF13-VY and iFGF13-S are expressed in neonatal mouse ventricular myocytes, and iFGF13-mediated regulation of NaV1.5 channels in these cells also affects the voltage dependence of INa activation.
Differential expression of iFGF13 isoforms in neonatal and adult mouse ventricular myocytes
Consistent with previous studies (Wang et al., 2011a), our mass spectrometric and transcript expression analyses confirmed that, of the four Fgf genes and the five distinct Fgf13 isoforms generated by N-amino terminus alternative splicing (Fgf13-VY, -V, -Y, -S, and -U) previously reported (Munoz-Sanjuan et al., 2000), the expression of the Fgf13-VY isoform is preponderant in adult mouse ventricular myocytes, while the Fgf13-U and Fgf11 isoforms are not detected in either neonatal or adult mouse ventricular myocytes. Additionally, the present transcript expression findings are consistent with the differential expression of the Fgf13 isoforms in the developing and adult mouse hearts, with adult mouse ventricular myocytes expressing mainly the Fgf13-VY and neonatal cardiomyocytes expressing both Fgf13-VY and Fgf13-S. Importantly, the differential transcript expression profile of the Fgf13-S isoforms observed in neonatal and adult mouse ventricular myocytes was confirmed at the protein level by Western blot analysis, demonstrating the specific expression of iFGF13-S in neonatal ventricular lysates.
Proteomic and functional mapping of mouse left ventricular iFGF13-VY phosphorylation sites
The present phosphoproteomic analysis identified a total of nine phosphorylation sites in the native iFGF13 protein purified from adult mouse left ventricular NaV1.5 channel complexes. Three of these sites, at positions S35, S38, and S218, are heavily phosphorylated in adult mouse left ventricles. Interestingly, the S218 phosphosite is conserved across iFGF isoforms and species, while the two N-terminal phosphosites at positions S35 and S38 are specific for iFGF13-VY and iFGF13-Y. Having established that the transcript expression level of the Fgf13-VY isoform is predominant in adult mouse ventricular myocytes, especially compared with the much lower expression of Fgf13-Y, these two N-terminal phosphosites have most likely been detected from the iFGF13-VY isoform. In contrast, the six other C-terminal iFGF13 sites, at positions S230, T232, S238, S240, S250, and T255 that are common to the five iFGF13 isoforms, are less abundantly phosphorylated and show a much lower stoichiometry compared with the N-terminal and S218 phosphosites. The simplest interpretation of these differences in phosphosite abundance and stoichiometry is that the first group may contribute to the basal iFGF13-dependent regulatory mechanisms of cardiac NaV1.5 channels, while the latter may participate in more local or transient roles. Interestingly, three of the nine iFGF13 phosphorylation sites identified at positions S35, S38, and S240 have previously been reported in a large-scale phosphoproteomic analysis of guinea pig left ventricles (Dey et al., 2018). While the Laezza group identified three phosphoserines on iFGF14 at positions S226, S228, and S230 (Hsu et al., 2016, 2017), no alignment of these three phosphoserines with the newly identified iFGF13 phosphoserines here could be obtained as the surrounding amino acid sequences are not conserved (Fig. 1 A). Additionally, while phosphorylation at Y158 was previously identified in iFGF14 using in silico and in vitro analyses (Wadsworth et al., 2020), no phosphorylation was detected at the corresponding conserved iFGF13 tyrosine in the present mass spectrometric analysis, likely reflecting the distinctly low level of tyrosine phosphorylation compared with phosphoserines and phosphothreonines, tissue specificity, and/or differences in the approaches used (in situ versus in silico/in vitro approaches).
With the exception of the S218 phosphosite, which is more isolated in the iFGF13 amino acid primary sequence, the distribution of phosphosites by pairs led us to explore the possible functional roles of these sites by four clusters of two phosphosilent or phosphomimetic mutations each (35–38, 230–232, 238–240, and 250–255). To our surprise, however, no differential effects on the density or biophysical properties of INa were revealed in cells expressing the various phosphomutant, compared with WT, iFGF13-VY proteins in either neonatal or adult mouse ventricular myocytes. In addition, the complete phosphosilent iFGF13-VY construct in which the nine phosphosites were mutated to alanines (iFGF13-VY-9A) did not reveal any differential effects on INa compared with WT iFGF13-VY. These combined results suggest that iFGF13 phosphorylation does not affect the association of the iFGF13 protein with NaV1.5 and/or the modulatory effects of this association on assembled INa channels under basal conditions. Notably, the nine iFGF13 phosphorylation sites identified here are not located in the conserved NaV binding surface in the FGF core domain (Goetz et al., 2009), suggesting that they may not be expected to play a direct regulatory role in mediating the iFGF13-NaV1.5 interaction and/or the functional impact of this interaction. It is certainly also possible, however, that the high levels of the iFGF13 proteins (on average two to threefold, and ninefold greater for iFGF13-VY-9A compared with endogenous iFGF13), expressed in the rescue experiments, obscured the physiological effects of phosphorylation or that we have simply not selected/mutated the right combinations of physiologically relevant phosphosites. We also cannot eliminate the possibility that there are additional phosphosites on iFGF13 not identified in our analysis for technical reasons. Finally, we cannot eliminate the possibility that, in spite of the fact that we used two native cardiomyocyte models, there are sine qua none molecular details involved in iFGF13-dependent regulation of INa that we are missing in these preparations. To begin to explore this possibility, we probed the kinases that may be involved in phosphorylating the iFGF13 sites herein identified by native phosphoproteomics using three different commonly used phosphorylation site prediction algorithms (Scansite 4.0, NetPhos-3.1b, and PhosphoMotif Finder; Table S3). Future studies focused on exploring these predictions directly will certainly be of interest.
iFGF13 affects cardiac NaV current properties in an age-specific manner
This study took advantage of the use of two distinct ventricular myocyte models, from neonatal and adult mice, to explore the differential regulation of NaV1.5 channels in developing and adult mouse hearts. In agreement with previous studies (Wang et al., 2011a, 2017; Park et al., 2016; Santucci et al., 2022), the effects of iFGF13 knockout and rescue on NaV current properties demonstrate that the iFGF13-VY isoform is predominant, if not the sole, iFGF13 isoform involved in destabilizing closed state inactivation of NaV1.5 channels in adult mouse ventricular myocytes. The present study also confirms the role of iFGF13-VY in decreasing the late Na+ current in adult mouse ventricular myocytes. A differential regulation schema with effects on current activation as well as inactivation was observed in neonatal mouse ventricular myocytes. As the molecular analyses revealed the expression of iFGF13-S, in addition to iFGF13-VY, in neonatal mouse ventricular myocytes, it is tempting to speculate whether the iFGF13-S isoform underlies the differences in endogenous and exogenous iFGF13-mediated effects observed here on INa in neonatal mouse ventricular myocytes. However, it is possible that iFGF13-VY does not exert the same effects on the neonatal and adult NaV1.5 channel isoforms (Onkal et al., 2008). Alternatively, the iFGF13 knockdown in neonatal cardiomyocytes may change some other channel regulatory components and, therefore, affect channel functioning differently compared with adult cells. Underscoring the role of the cellular and channel complex environment on channel functioning and regulation, another recent study from the Silva and Nerbonne laboratories (Angsutararux et al., 2023) revealed differential effects of two iFGF proteins, iFGF12-B and iFGF13-VY, on NaV current properties in native adult mouse ventricular myocytes and heterologously expressed NaV1.5 channels in Xenopus oocytes. In addition, although the diversity of the roles of the iFGF13 isoforms, including iFGF13-S, in regulating NaV channels has previously been queried in several heterologous expression systems, as well as in neurons, no specific roles for iFGF13-S in the regulation of NaV channel activation have been reported (Rush et al., 2006; Yang et al., 2016; Effraim et al., 2019). Indeed, the most consistent finding from these studies was that iFGF13-S hastens the rate of NaV channel entry into the slow inactivation state and induces slowing of recovery from inactivation, resulting in a large current decrease upon repetitive stimulations at both low and high frequencies. Together with these previous studies, therefore, the results presented here demonstrate that the iFGF13 proteins are robust cellular factors controlling the inactivation properties of INa in both neonatal and adult mouse ventricular myocytes, while the newly identified role for iFGF13 in the regulation of the voltage dependence of INa activation is specific for neonatal mouse ventricular NaV channels.
Data availability
The data are available from the corresponding author upon reasonable request.
Acknowledgments
Olaf S. Andersen served as editor.
The expert technical assistance of Agnès Tessier, Bérangère Evrard, Petra Erdmann-Gilmore, Dr. Yiling Mi, Alan Davis, and Rose Connors is gratefully acknowledged.
This work was supported by the Agence Nationale de la Recherche (grants ANR-15-CE14-0006-01 and ANR-16-CE92-0013-01 to C. Marionneau) and the National Institutes of Health (grants R01-HL148803 to J.R. Silva and C. Marionneau; R01-HL034161 and R01-HL142520 to J.M. Nerbonne; and R01-HL150637 to J.M. Nerbonne and J.R. Silva). The proteomic experiments were performed at the Washington University Proteomics Shared Resource (WU-PSR), R.R. Townsend MD. PhD., Director, and Robert W. Sprung and Qiang Zhang, PhD., Co-Directors. The WU-PSR is supported in part by the WU Institute of Clinical and Translational Sciences (NCATS UL1 TR000448), the Mass Spectrometry Research Resource (NIGMS P41 GM103422), and the Siteman Comprehensive Cancer Center Support Grant (NCI P30 CA091842). M Lorenzini was supported by a Groupe de Réflexion sur la Recherche Cardiovasculaire-Société Française de Cardiologie predoctoral fellowship (SFC/GRRC2018). S. Burel was supported by a Lefoulon-Delalande postdoctoral fellowship. The content of the research reported is solely the responsibility of the authors and does not necessarily represent the official view of the funding agencies.
Author contributions: C. Marionneau designed the study and wrote the paper. D. Maloney, R.R. Townsend, J.M. Nerbonne, and C. Marionneau designed, performed, and/or analyzed the mass spectrometry experiments. A. Lesage, M. Lorenzini, S. Burel, M. Sarlandie, F. Bibault, J.R. Silva, J.M. Nerbonne, and C. Marionneau designed, performed, and/or analyzed the functional analyses. J.M. Nerbonne provided the Fgf13fl/y C57BL/6J mouse line. C. Lindskog (Human Protein Atlas) provided the anti-iFGF13 rabbit polyclonal antibody. All authors reviewed the results and approved the final version of the manuscript.
References
This work is part of a special issue on Structure and Function of Ion Channels in Native Cells and Macromolecular Complexes.
Data availability
The data are available from the corresponding author upon reasonable request.

