


Classically, striated muscle contraction is initiated by calcium (Ca2+)-dependent structural changes in regulatory proteins on actin-containing thin filaments, which allow the binding of myosin motors to generate force. Additionally, dynamic switching between resting off and active on myosin states has been shown to regulate muscle contractility, a recently validated mechanism by novel myosin-targeted therapeutics. The molecular nature of this switching, however, is not understood. Here, using a combination of small-angle x-ray fiber diffraction and biochemical assays with reconstituted systems, we show that cardiac thick filaments are directly Ca2+-regulated. We find that Ca2+ induces a structural transition of myosin heads from ordered off states close to the thick filament to disordered on states closer to the thin filaments. Biochemical assays show a Ca2+-induced transition from an inactive super-relaxed (SRX) state(s) to an active disordered-relaxed (DRX) state(s) in synthetic thick filaments. We show that these transitions are an intrinsic property of cardiac myosin only when assembled into thick filaments and provide a fresh perspective on nature’s two orthogonal mechanisms to regulate muscle contraction through the thin and the thick filaments.
Introduction
Calcium (Ca2+) signaling coordinates many different intracellular processes in plant, animal, and human physiology (Bagur and Hajnoczky, 2017). Muscle contraction is one of those biological processes regulated by Ca2+ and is propelled by the sliding of actin-containing thin filaments along myosin-containing thick filaments in the sarcomere (Hanson and Huxley, 1953). For over 50 yrs, it has generally been accepted that Ca2+ binds to the troponin complex on the thin filament to initiate and regulate this process (Tobacman, 1996). Briefly, under resting conditions, the myosin-binding sites on the thin filament are blocked by tropomyosin, thus preventing muscle contraction. Upon receiving the contraction signals, Ca2+ entering the cytosol binds to troponin-C on the thin filament, triggering a series of conformational changes that ultimately unblock the myosin-binding site on actin by physically moving the tropomyosin. Initial binding of myosin to such unblocked sites further leads to the full activation of the thin filament (Lehman et al., 1994; Risi et al., 2021).
However, this textbook Ca2+-mediated thin filament-based mechanism does not address how thick filaments are turned on and off. Under relaxed conditions, myosin heads inside the sarcomere may be found in two biochemical states: one with a relatively high ATPase (∼0.03 s−1) called the disordered-relaxed (DRX) state(s), and the other, a low ATPase (∼0.003 s−1) state known as the super-relaxed (SRX) state(s) (Stewart et al., 2010). X-ray diffraction experiments (Anderson et al., 2018; Yuan et al., 2022) have shown that myosin heads can adopt ordered configurations close to the thick filament backbone where the heads cannot interact with actin and are considered to be in an off state(s). Under other conditions, disordered heads are located away from the thick filament backbone where they are free to interact with actin, considered to be in an on state(s). The structural off-to-on transition is thought to closely correlate with the biochemical SRX to DRX transitions of myosin (Craig and Padron, 2022), but it is not yet clear whether these are strictly equivalent. Be that as it may, dynamic switching between the SRX states and the DRX states of myosin has been shown to regulate muscle contractility (Spudich, 2019; Nag and Trivedi, 2021) and has emerged as one of the underlying causes of hyper- or hypo-contractility in myopathies. Understanding the details of thick filament regulation mechanisms, therefore, has implications for understanding the etiology of many myopathies and so aid the development of rational treatment strategies (Alsulami and Marston, 2020).
Different muscle systems have developed different thick filament regulation mechanisms to fulfill their functions. Muscle myosins from invertebrates such as scallops can be activated by direct Ca2+ binding to the essential light chain (ELC; Szent-Gyorgyi, 2007), thus actively cycling cross-bridges. Tarantula skeletal muscle and vertebrate smooth muscle activate their thick filaments through Ca2+ binding to calmodulin, which activates myosin light chain kinase (MLCK), which in turn phosphorylates the myosin’s regulatory light chain (RLC; Walsh, 1994; Padron et al., 2020) thus turn the thick filament on to participate in force generation. Linari et al. (2015) proposed a strain-dependent thick filament activation model (“mechano-sensing”) for vertebrate skeletal muscle and later expanded this mechanism to rodent cardiac muscle (Reconditi et al., 2017; Caremani et al., 2019). In this mechano-sensing model, when Ca2+ is released to the cytoplasm and binds to troponin, thereby activating the thin filament, a small number of myosin heads that are constitutively on can form cross-bridges and generate sufficient force to induce strain in the thick filament. The thick filament acts as a mechanical sensor where the force exerted on the thick filament backbone induces a “structural transition” that releases myosin heads from an inactive off state(s) to an active on state(s) that increases the proportion of myosin heads competent to bind actin and generate force (Linari et al., 2015; Caremani et al., 2019). It has been shown, however, that passive stretch also can strain the thick filament backbone to a similar degree as with active force, but this strain alone cannot account for the full number of myosin heads that transition from the off-to-on states seen in actively contracting muscle (Ma et al., 2018b; Ma et al., 2019; Park-Holohan et al., 2021). This suggests that thick filament strain cannot be the only trigger for off-to-on structural transitions that result in the release of myosin heads from the thick filament backbone. One unexplored candidate for this additional trigger in contracting muscle would be Ca2+, the primary difference between active and resting muscle.
Here, using a combination of biochemical (SRX/DRX and ATPase) assays on different myosin constructs and in synthetic thick filaments and small-angle x-ray fiber diffraction on permeabilized porcine myocardium where actin-myosin interaction is prevented using a small molecule inhibitor, we show that cardiac thick filaments are directly Ca2+-regulated. This Ca2+-mediated thick filament sensing mechanism may have broad implications for understanding the structural basis of calcium regulation of muscle.
Materials and methods
Muscle sample preparation
Frozen wild-type left ventricular wall myocardium was provided by Exemplar Genetics LLC. Humane euthanasia and tissue collection procedures were approved by the Institutional Animal Care and Use Committees at Exemplar Genetics LLC. Permeabilized tissues are prepared as described previously (Ma et al., 2021; Ma et al., 2022). Briefly, frozen wild type porcine (n = 2) left ventricle wall (about 1 cm3) is defrosted in skinning solution (91 mM K+-propionate, 3.5 mM MgCl2, 0.16 mM CaCl2, 7 mM EGTA, 2.5 mM Na2ATP, 15 mM creatine phosphate, 20 mM imidazole, 30 mM 2,3-butanedione monoxime, 1% Triton-X100, and 3% dextran at pH 7) at room temperature before dissecting into smaller strips (∼1 cm long and 2–3 mm wide). The tissues are permeabilized at room temperature for 3 h. The tissues are then washed three times, 10 min each, in pCa 8 solution (91 mM K+-propionate, 3.5 mM MgCl2, 0.16 mM CaCl2, 7 mM EGTA, 2.5 mM Na2ATP, 15 mM creatine phosphate, 20 mM imidazole, and 3% dextran at pH 7). Well-aligned tissues are further dissected into preparations of 4 mm length and a diameter of ∼200 µm before attaching aluminum T-clips to both ends.
Cardiac protein purification
Bovine cardiac actin, tropomyosin, and troponin complex are purified following modified methods previously published (Spudich and Watt, 1971). Actin is stored at −80°C as G-actin and polymerized fresh for each day of experiments by adding 50 mM KCl and 2 mM MgCl2 to the actin-containing buffer. The regulated thin filament (RTF) system is reconstituted using bovine cardiac actin:bovine cardiac tropomyosin:bovine cardiac troponin complex (1:1:1 troponin-C:troponin-T:troponin-I) in a 7:1:1 ratio. β-cardiac full-length myosin from the bovine left ventricle is isolated following established methods described elsewhere (Margossian and Lowey, 1982). Following this, proteins are dialyzed in a buffer containing 10 mM PIPES (pH 6.8), 300 mM KCl, 0.5 mM MgCl2, 0.5 mM EGTA, 1 mM NaHCO3, and 1 mM DTT and stored at −80°C. Based on densitometry analysis of SDS-PAGE, the purity of the myosin preparation varies between 90 and 95%, with negligible actin contamination. Also, the basal myosin ATPase from these preps always turns out to be 0.03 ± 0.01 s−1, suggesting negligible actin-activation.
Using bovine cardiac full-length myosin as the starting material, heavy meromyosin (HMM) and S1 subfragment are prepared according to methods described in a previous report (Margossian and Lowey, 1982).
Human β-cardiac 2-hep and 25-hep HMM are purified using methods described elsewhere (Nag et al., 2017). These HMM cDNA constructs consist of a truncated version of MYH7 (residues 1–855), corresponding to S1-subfragment and the first 2 heptads (14 amino acids) or 25 heptad repeats (175 amino acids) of S2-subfragment for the 2-hep and 25-hep HMM, respectively, followed by a GCN4 leucine zipper to ensure dimerization. This is further linked to a flexible GSG (Gly-Ser-Gly) linker, then a GFP moiety followed by another GSG linker, and finally ending with an eight-residue (RGSIDTWV) PDZ binding peptide.
Myosin ATPase measurements from myosin synthetic myosin thick filaments reconstituted from full-length myosin
Methodologies involving the reconstitution of myosin synthetic thick filaments (STF) have been described previously (Gollapudi et al., 2021). Briefly, full-length bovine β-cardiac myosin remains fully soluble in a buffer of high ionic strength (300 mM) but spontaneously self-assembles into bipolar thick filaments at lower ionic strengths (Gollapudi et al., 2021). For each experiment, 10 µM of full-length myosin in 300 mM KCl buffer was diluted to 1 µM in KCl buffer containing 20 mM Tris-HCl (pH 7.4), 0 mM KCl, 1 mM EGTA, 3 mM MgCl2, and 1 mM DTT, to achieve a final KCL concentration of 30 mM. To allow for thick filament formation, the sample was incubated for 2 h on ice, and then used in the experiments. Basal STF ATPase activity measurements are performed at 23°C on a plate-based reader (SpectraMax 96-well) using an enzymatically coupled assay as described earlier (Kawas et al., 2017). The buffer composition used for these experiments is 12 mM PIPES (pH 6.8), 2 mM MgCl2, 10 mM KCl, and 1 mM DTT. Assuming negligible contribution from the PIPES buffer system, the ionic strength of this buffer is ∼16 mM.
Use and characterization of MYK-7660
The small molecule, MYK-7660, is a regulated thin-filament-activated myosin ATPase inhibitor. It was discovered initially from a high-throughput compound library screen at MyoKardia Inc., a wholly owned subsidiary of Bristol Myers Squibb. This article uses this small molecule as a tool compound to shut off the regulated thin filaments in the presence of high Ca2+. For all experiments, a 20 mM stock in 100% DMSO is used to attain the desired concentration in the final buffer of the experiment with a final 2% DMSO concentration. All control experiments without MYK-7660 are performed in a buffer containing 2% DMSO.
For ATPase measurements concerning the thin filament inhibitor (MYK-7660) in Fig. 1 a, actin-activated and regulated thin filament-activated ATPase measurements of the soluble S1-subfragment of the myosin molecule, as a function of increasing MYK-7660 concentrations (0–100 µM in the final buffer samples), are performed as described previously (Gollapudi et al., 2021).
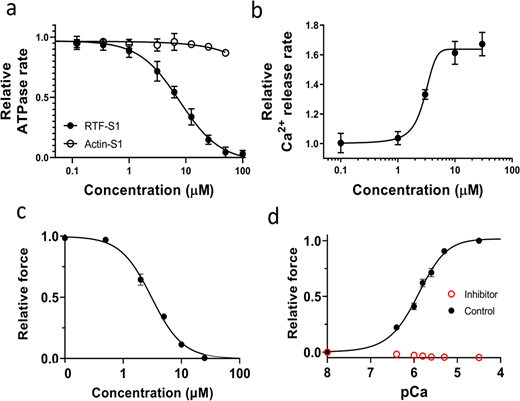
Effects of the inhibitor (MYK-7660) on the chemo-mechanical activity of different sarcomere systems. (a) The concentration-dependent steady-state ATPase activity (moles ATP used per second per mole of myosin S1 heads) of bovine cardiac myosin subfragment S1 with actin (open symbol) and with RTFs at pCa 6 (closed symbols). Inhibition (IC50 = 9 ± 3 μM) is specific to the RTF-S1 system, implicating that the compound inhibits the ATPase activity by shutting down the RTF system and not through actin and myosin. (b) Concentration-dependent transient kinetics of the Ca2+ release rate (s−1) from the RTF system. The AC50 of the increase in the Ca2+ release rate is measured as 3 ± 0.5 μM. (c) The concentration-dependent relative maximum force of permeabilized porcine myocardium. The IC50 of force inhibition is 3 ± 1 μM. IC50 and Hill slope parameters to the fits are given in Table S1. Lines in all plots are Hill fits to the data. (d) Relative force-pCa curve of permeabilized porcine myocardium in the absence or presence of 100 μM inhibitor (MYK-7660) during x-ray diffraction experiments. There is no detectable force increase above baseline generated by the myocardium in the presence of the inhibitor at all pCa values tested. Data are expressed as mean ± SEM (n = 2 for a and b; n = 6 for c; n = 12 in the control group; and n = 11 in the inhibitor group for d).
Effects of the inhibitor (MYK-7660) on the chemo-mechanical activity of different sarcomere systems. (a) The concentration-dependent steady-state ATPase activity (moles ATP used per second per mole of myosin S1 heads) of bovine cardiac myosin subfragment S1 with actin (open symbol) and with RTFs at pCa 6 (closed symbols). Inhibition (IC50 = 9 ± 3 μM) is specific to the RTF-S1 system, implicating that the compound inhibits the ATPase activity by shutting down the RTF system and not through actin and myosin. (b) Concentration-dependent transient kinetics of the Ca2+ release rate (s−1) from the RTF system. The AC50 of the increase in the Ca2+ release rate is measured as 3 ± 0.5 μM. (c) The concentration-dependent relative maximum force of permeabilized porcine myocardium. The IC50 of force inhibition is 3 ± 1 μM. IC50 and Hill slope parameters to the fits are given in Table S1. Lines in all plots are Hill fits to the data. (d) Relative force-pCa curve of permeabilized porcine myocardium in the absence or presence of 100 μM inhibitor (MYK-7660) during x-ray diffraction experiments. There is no detectable force increase above baseline generated by the myocardium in the presence of the inhibitor at all pCa values tested. Data are expressed as mean ± SEM (n = 2 for a and b; n = 6 for c; n = 12 in the control group; and n = 11 in the inhibitor group for d).
Ca2+ dissociation rate from the RTF system in the presence of increasing MYK-7660 concentrations (0–100 µM in the final buffer samples) in Fig. 1 b is measured in a stopped-flow instrument (KinTek Model AutoSF-120) using transient kinetic measurements. Briefly, the RTFs (with a final concentration of 7 μM actin, 2 μM tropomyosin, and 2 μM troponin) are preincubated with ∼1 μM Ca2+ (pCa 6) and are rapidly mixed with a fluorescent Ca2+-chelator, Quin-2, of a final concentration of 50 μM in a buffer containing 20 mM Tris-HCL (pH 7.4), 10 mM KCl, 3 mM MgCl2, and 1 mM DTT. Experiments are performed at 25°C by exciting Quin-2 at 310 nm and monitoring the emission at 450 nm. Under these conditions, no significant contribution to the fluorescence is measured from MYK-7660 alone.
The dose-dependent maximally activated force data are collected in the presence of increasing MYK-7660 concentrations (0, 0.5, 2, 5, 10, and 25 μM in both pCa 8 and pCa 4.5 solutions) on skinned porcine myocardium (Fig. 1 c). The tissues are relaxed in pCa 8 solution with the same concentration of MYK-7660 as the following pCa 4.5 solution. Forces are normalized against the force generated with no inhibitor.
Biochemical SRX measurements
Single ATP turnover kinetic experiments using a fluorescent 2′/3′-O-(N-Methylanthraniloyl) (mant)-ATP are conducted in a 96-well plate fluorescence plate reader at 25°C. A protocol involving this method has been described in previous studies (Gollapudi et al., 2021). This assay measures fluorescent nucleotide (excitation is at 385 nm, and emission is acquired using a long-pass filter with a cutoff at 450 nm) release rates following incubation of myosin preparations with mant-ATP and chased with excess unlabeled ATP. Briefly, in the first step, 100 μl of 0.8 μM myosin is combined with 50 μl of 3.2 μM mant-ATP in a UV-transparent fluorescence plate, and the reaction is aged for 60 s to allow binding and hydrolysis of mant-ATP to inorganic phosphate and mant-ADP. In the second step, mant-nucleotides are chased with non-fluorescent ATP by adding 50 μl of 16 mM non-fluorescent ATP to the above mixture, and the resulting fluorescence decay due to mant-nucleotide dissociation from myosin is monitored over time. The final buffer composition is as follows: 20 mM Tris-HCl (pH 7.4), 30 mM KCl, 1 mM EGTA, 3 mM MgCl2, and 1 mM DTT, unless otherwise mentioned. The ionic strength of this buffer is ∼39 mM, assuming negligible contribution from the buffer system. The concentrations of myosin, mant-ATP, and non-fluorescent ATP in the final mixture are 0.4 μM, 0.8 μM, and 4 mM, respectively. The fluorescence decay profile obtained during the chase phase characteristically depicts two phases, a fast phase followed by a slow phase. Therefore, a bi-exponential function is fitted to each trace to estimate four parameters corresponding to amplitudes and rates of the fast and slow phases. The fast and slow phases correspond to the myosin activity in the DRX and SRX states, respectively (Gollapudi et al., 2021).
X-ray diffraction
X-ray diffraction experiments are performed at the BioCAT beamline 18ID at the Advanced Photon Source, Argonne National Laboratory (Fischetti et al., 2004). The x-ray beam energy is set to 12 keV (0.1033 nm wavelength) at an incident flux of ∼5 × 1012 photons per second. The specimen to detector distance is ∼3 m. The preparation is then attached to a hook on a force transducer (model 402B, Aurora Scientific, Inc.) and a static hook. The muscle is incubated in a customized chamber whose bottom is attached to a heat exchanger, so the solution is kept between 28 and 30°C. For remote solution changes, the chamber is connected to a multiway valve syringe pump (Hamilton model 500). The muscles are stretched to a sarcomere length of 2.3 µm using micromanipulators attached to the hooks while monitoring light diffraction patterns from a helium-neon laser (633 nm) on a screen. The x-ray patterns are collected sequentially at seven increasing Ca2+ concentrations (pCa 8, pCa 6.4, pCa 6, pCa 5.8, pCa 5.6, pCa 5.3, and pCa 4.5) in the absence or presence of 100 μM of MYK-7660, well below its solubility in the PBS buffer (180 μM), on a MarCCD 165 detector (Rayonix, Inc.) with a 1 s exposure time. To minimize radiation damage, the muscle samples are oscillated along their horizontal axes at a velocity of 1–2 mm/s. The irradiated areas are moved vertically after each exposure to avoid overlapping x-ray exposures. The force-pCa data are collected during the x-ray experiment (Fig. 1 d). Force measurements are normalized against the force generated at pCa 4.5 in the control experiment. There is no detectable increase in force above baseline in the inhibitor group in the presence of 100 μM MYK-7660.
X-ray data analysis
The data are analyzed using data reduction programs from the open-source MuscleX software package developed at BioCAT (Jiratrakanvong et al., 2018). One to two patterns are collected under each condition, and reflection spacings and intensities extracted from these patterns are averaged. As described previously, the equatorial reflections are measured by the “Equator” routine in MuscleX (Ma et al., 2018a). For subsequent analyses, the four quadrants, divided by meridian and equator, of x-ray patterns are averaged together to improve the signal-to-noise ratio, and the diffuse scatterings are subtracted from the x-ray diffraction patterns with the “Quadrant Folding” routine in MuscleX. The intensities and spacings of meridional and layer line reflections are measured by the “Projection Traces” routine in MuscleX, as described previously (Ma et al., 2020). The spacings of targeted reflections are estimated by measuring the distance from the beam center to the peak position as the centroid of the intensity in profile, considering only the top half of the diffraction peak (Huxley et al., 1994; Kiss et al., 2018). The center of mass of the cross-bridges (Rm) is estimated based on modeling the layer line as a J3 Bessel function with the argument 4.2 = 2 × π × r × Rm, where r is a reciprocal space coordinate as described (Ma et al., 2021; Ma and Irving, 2022). To compare the intensities under different conditions, the measured intensities of x-ray reflections are normalized to the sixth-order actin-based layer line intensities. This reflection is chosen because its intensity did not change significantly from its value at pCa 8 with changes of contractile state in cardiac muscle (see Fig. S1)

Thin filament-based x-ray reflections in the presence and absence of inhibitor (MYK-7660). (a and b) The intensity of the sixth-order actin-based layer line (a) and the third-order troponin meridional reflection (b) at pCa 4.5 normalized to its value at pCa 8 (C: control; I: inhibitor).
Thin filament-based x-ray reflections in the presence and absence of inhibitor (MYK-7660). (a and b) The intensity of the sixth-order actin-based layer line (a) and the third-order troponin meridional reflection (b) at pCa 4.5 normalized to its value at pCa 8 (C: control; I: inhibitor).
Statistics
Statistical analyses are performed using GraphPad Prism 9 (GraphPad Software). The results are given as mean ± SEM. One-way repeated measures ANOVA with the Geisser-Greenhouse correction and Tukey’s multiple comparisons test with individual variances computed for each comparison is performed on bar graphs in Fig. 2 c. A nested t-test was performed for the bar graphs in Fig. 3 and Fig. S1. The relative changes versus pCa curves are fit to a four-parameter modified Hill equation (minimum response + [maximum response − minimum response] / [1 + 10h (pCa50 − pCa)]; Walker et al., 2010), where pCa50 is the Ca2+ concentration yielding a response halfway between the minimum and maximum values reported in the article. Symbols on figures: *: P < 0.05, **: P < 0.01, ***: P < 0.001. For Fig. 4, each experiment is repeated at least twice with a minimum of two replicates per experiment, and a two-tailed Student’s t-test is used to differentiate the changes in parameters among groups (P < 0.01).
Online supplemental material
Fig. S1 shows thin filament-based x-ray reflections in the presence and absence of the inhibitor (MYK-7660). Table S1 includes parameters obtained from MYK-7660 characterization. Table S2 includes parameters obtained from fitting x-ray datasets to the modified Hill equation. Supplemental text at the end of the PDF provides additional information about Ca2+-induced thin filament reflections in the presence and absence of MYK-7660.
Results
Characterization of MYK-7660
Here, we use a small-molecule inhibitor of the thin filament system (MYK-7660) to decouple Ca2+-mediated thin filament–based regulation from thick filament–based regulation. Actin-activated (Actin-S1) and regulated thin filament (RTF-S1)–activated ATPase activity at pCa 6 of bovine cardiac myosin subfragment S1 was measured in response to increasing concentrations of the inhibitor (MYK-7660; Fig. 1 a and Table S1). The ATPase rates were normalized against the basal ones in the absence of the drug for these two systems, 0.02 ± 0.002 s−1 (n = 3). The compound inhibited the RTF-S1 system (IC50 = 9 ± 3 μM) but not the Actin-S1 in a dose-dependent manner, suggesting that the mechanism of inhibition is through shutting down the regulated thin filament system and not through actin and myosin. The Ca2+ release rate is enhanced as the concentration of MYK-7660 increases (AC50 = 3 ± 0.5 μM; Fig. 1 b and Table S1). This can explain the ATPase inhibition of MYK-7660 without hampering Ca2+ binding to troponin.
The inhibitor inhibited the force production of permeabilized porcine myocardium in a dose-dependent manner with an IC50 of 3 ± 1 (Fig. 1 c and Table S1; n = 6). The force dropped to zero beyond 25 μM inhibitor concentration. The force produced by permeabilized porcine myocardium at different pCa values during the x-ray experiment is presented in Fig. 1 d. Permeabilized porcine myocardium produces a classic sigmoidal force-pCa curve at a sarcomere length of 2.3 μm in the absence of the inhibitor with a pCa50 of 5.91 ± 0.1 (Fig. 1 d, black symbols; Table S2; n = 12). However, in the presence of saturating levels of the inhibitor (100 μM), no active contraction is detected at all pCa values (Fig. 1 d, red symbols; n = 11). 100 μM of MYK-7660 was chosen for all x-ray diffraction experiments to ensure complete inhibition of active force.
Radial movement of myosin heads
Small-angle x-ray diffraction is used to examine the structural transitions of permeabilized porcine cardiac myocardium at different Ca2+ concentrations in the presence of the inhibitor. At pCa 8 the x-ray diffraction patterns of permeabilized porcine cardiac myocardium show characteristic relaxed patterns similar to those reported previously (Anderson et al., 2018; Ma et al., 2021). With increasing Ca2+ concentration, the intensities of all the myosin-based reflections (M3, M6, MLLs in Fig. 2 a) become weaker. The intensities of the sixth-order actin-based layer line (ALL6, Fig. 2 a) are relatively stable throughout the experiment indicating that the changes in the thick filament myosin-based reflections are due to the direct effects of Ca2+-binding and not a result of radiation damage or by strong binding of cross-bridges to the thin filament.

Myosin heads move radially away from the thick filament backbone. (a) Representative x-ray diffraction patterns from permeabilized porcine myocardium in different Ca2+ in the absence of force. (b) Change of equatorial intensity ratio (Δ I1,1/I1,0) at different Ca2+concentrations in the presence (red) and absence (black) of inhibitor (MYK-7660). (c) The radius of the average mass of myosin heads (Rm) at different Ca2+ concentrations in the presence (red) and absence (black) of inhibitor. Ca2+ shifts the distribution of myosin heads away from the thick filament backbone towards the thin filaments. Data are expressed as mean ± SEM (n = 12 in the control group and n = 11 in the inhibitor group).
Myosin heads move radially away from the thick filament backbone. (a) Representative x-ray diffraction patterns from permeabilized porcine myocardium in different Ca2+ in the absence of force. (b) Change of equatorial intensity ratio (Δ I1,1/I1,0) at different Ca2+concentrations in the presence (red) and absence (black) of inhibitor (MYK-7660). (c) The radius of the average mass of myosin heads (Rm) at different Ca2+ concentrations in the presence (red) and absence (black) of inhibitor. Ca2+ shifts the distribution of myosin heads away from the thick filament backbone towards the thin filaments. Data are expressed as mean ± SEM (n = 12 in the control group and n = 11 in the inhibitor group).
The equatorial intensity ratio (I1,1/I1,0) is indicative of the proximity of myosin heads to actin in relaxed muscle and is closely correlated to the number of force-producing cross-bridges in activated muscle (Haselgrove and Huxley, 1973; Matsubara, 1980; Ma et al., 2018a; Ma et al., 2020). The change of I1,1/I1,0 (Δ I1,1/I1,0) versus pCa shows a sigmoidal shape with a pCa50 of 5.7 (5.5–5.9 for 95% CI) for the control group (n = 12). Surprisingly in the inhibitor group (n = 11), where active contractions are eliminated, I1,1/I1,0 also progressively increases with increasing Ca2+ concentration (Fig. 2 b and Table S2), with a pCa50 of 5.6 (3.5–5.9 for 95% CI), although the amplitudes of the change are smaller compared to the control group. The radius of the center of mass of the cross-bridges (Rm), which directly measures the proximity of helically ordered myosin heads to the thick filament backbone (Ait-Mou et al., 2016; Ma et al., 2018a), increases from 14.34 ± 0.14 nm at pCa 8–15.59 ± 0.4 nm at pCa 6 in the control group, Rm increases similarly in the presence of the inhibitor (14.34 ± 0.14 at pCa 8, to 15.13 ± 0.32 nm at pCa 6; Fig. 2 c). The ΔI1,1/I1,0 and Rm data indicate that with increasing Ca2+ concentration, myosin heads move radially away from the thick filament backbone under conditions where they cannot bind to thin filaments, suggesting a regulatory role of Ca2+ directly on the thick filament.
Structural off-to-on transitions
In a resting muscle, most myosin heads are quasi-helically ordered on the surface of the thick filament, where these off-state myosin heads produce the myosin-based layer line reflections. Myosin heads lose their helical order when turned on to participate in contraction (Huxley and Brown, 1967; Ma and Irving, 2022). The intensity of the first-order myosin-based layer line (IMLL1) and the third-order myosin-based meridional reflection (IM3), both of which correlate with the ordering of myosin heads (Reconditi, 2006; Ma and Irving, 2022), decreases progressively in the presence of increasing Ca2+. The IMLL1 reflection changes (Fig. 3 a and Table S2) with a pCa50 of 6.05 (5.91–6.19 for 95% CI) and 6.09 (5.98–6.19 for 95% CI) for the inhibitor and control group, respectively, whereas the IM3 reflection changes (Fig. 3 b and Table S2) with a pCa50 of 6.17 (6.03–6.33 for 95% CI) and 6.36 (6.24–6.51 for 95% CI) for these two experimental groups, respectively. Compared to pCa 8, the IMLL1 and IM3 intensities at pCa 4.5 decrease to 27 ± 2.5% and 35 ± 1.9%, respectively, in the inhibitor group, whereas it decreases to 18 ± 1.6% and 26 ± 2.4% in the control group (inset in Fig. 3, a and b).

Thick filament structure changes in the presence of Ca 2+ . (a and b) The intensity of first-order myosin-based layerline (a) and third-order of myosin-based meridional reflection (b) in different concentrations of Ca2+ in the presence (red) and absence (black) of inhibitor (MYK-7660). (c and d) The intensity (c) and spacing (d) of the sixth-order of myosin-based meridional reflection in different Ca2+ concentrations in the presence (red) and absence (black) of inhibitor. Ca2+ reduces the proportion of myosin heads in ordered states on the thick filament and induces structural changes in the thick filament backbone. Data are expressed as mean ± SEM (n = 12 in the control group and n = 11 in the inhibitor group).
Thick filament structure changes in the presence of Ca 2+ . (a and b) The intensity of first-order myosin-based layerline (a) and third-order of myosin-based meridional reflection (b) in different concentrations of Ca2+ in the presence (red) and absence (black) of inhibitor (MYK-7660). (c and d) The intensity (c) and spacing (d) of the sixth-order of myosin-based meridional reflection in different Ca2+ concentrations in the presence (red) and absence (black) of inhibitor. Ca2+ reduces the proportion of myosin heads in ordered states on the thick filament and induces structural changes in the thick filament backbone. Data are expressed as mean ± SEM (n = 12 in the control group and n = 11 in the inhibitor group).
IMLL1 and IM3 data show that myosin heads lose their helical ordering in the presence of Ca2+, which strongly indicates switching from the ordered off states to the disordered on states in cardiac muscle thick filaments. The diminished IMLL1 and IM3 intensities at pCa 4.5 and the leftward shift of the intensity decay curve in the control group indicate that active tension can activate the residual myosin heads on the thick filament backbone as expected from the mechano-sensing thick filament activation mechanism. However, the majority of the loss of ordering in the myosin heads can be accounted for by Ca2+-mediated activation of the thick filament in the absence of cross-bridge forces.
The decay of the intensity of the sixth-order myosin-based meridional reflection (IM6), which arises primarily from the thick filament backbone (Reconditi, 2006), with increasing Ca2+ concentration is indistinguishable between the inhibitor and control groups (P = 0.6) with a similar pCa50 of 5.96 (5.78–6.13 for 95% CI; Fig. 3 c and Table S2). Currently, there are no obvious mechanistic explanations for the decay in IM6 in the presence of Ca2+ and the absence of force. Since IM6 arises primarily from the thick filament backbone, the decrease of IM6 in the presence of Ca2+ suggests a structural change in the thick filament backbone induced by Ca2+, independent of thick filament strain. The spacing of the M6 reflection (SM6), which reports the periodicity of the thick filament backbone (Ma and Irving, 2022), increases to 0.73 ± 0.06% at the fully activated state (pCa 4.5) in the control group and 0.36 ± 0.02% in the inhibitor group where active force is absent (Fig. 3 d and Table S2). While the larger SM6 changes in the control group are caused by both the release of off states myosin heads and the increase in strain on the thick filament by active contraction (Reconditi et al., 2019), the increase in SM6 in the absence of active force suggests that an, at least partial, off-to-on transition of the thick filament can occur solely in response to Ca2+.
SRX to DRX transitions
Given our observation of Ca2+-induced off-to-on transitions of the thick filament in the absence of active force, and inspired by previous work suggesting that there might be direct effects of Ca2+ on thick filaments (Morimoto and Harrington, 1974; Metzger and Moss, 1992; Podlubnaya et al., 2000), we explored whether these structural transitions can be translated into functional alterations. The thin filament inhibitor (MYK-7660) we used in this study has fluorescence in the 400–500 nm range, making performing conventional SRX/DRX assays on muscle tissue impractical. To test our hypothesis, since our results suggest that myosin or myosin filaments are the direct targets of Ca2+ binding, we turned our focus to reconstituted cardiac STF, which can faithfully capture SRX/DRX transitions under various conditions (Gollapudi et al., 2021).
The basal myosin SRX population in the STF system at pCa 8 is 15 ± 5%, which progressively decreases (P < 0.01) to 3 ± 2% (pCa50 of 5.5; 5.4–5.7 for 95% CI) at pCa 4 (Fig. 4 a [normalized data], solid black symbol) with no considerable change in the ATPase cycling rate of the SRX states (n = 5). This destabilization of the SRX states, which presumably populates myosin in some DRX states, along with an increase in the cycling rates of the DRX population, leads to an overall Ca2+-dependent increase (P < 0.01) in the steady-state basal myosin ATPase activity from 0.03 ± 0.01 s−1 at pCa 8 to 0.09 ± 0.02 s−1 at pCa 4 with a pCa50 of 6.1 (5.9–6.2 for 95% CI; Fig. 4 a [normalized data], solid grey symbol; n = 3). Further investigation with the soluble S1-subfragment of the myosin molecule (Fig. 4 a [normalized data], open symbols, unnormalized data shown in Table 1) and full-length myosin at high ionic strengths (150 mM KCl; Fig. 4 b) that do not fully form filamentous thick filaments demonstrates a loss of this Ca2+-dependent modulation, suggesting that the Ca2+-binding mechanism may not be intrinsic to myosin S1 or isolated full-length myosin but rather involve only myosins forming filamentous structures. This result is further supported by a lack of Ca2+-dependent activation of the ATPase in different soluble double-headed HMM constructs containing variable lengths of the S2-subfragment (Fig. 4 c and Table 1) but lacks the light meromyosin (LMM) domain that can assemble myosin into bipolar thick filaments. For example, the steady-state basal myosin ATPase activity in the STF system at pCa 8 is 0.03 ± 0.01 s−1,which increases to 0.1 ± 0.02 s−1 at pCa 4 with a pCa50 of 6.0 (5.9–6.1 for 95% CI; Fig. 4 c [normalized data], red symbols; Table 1). However, such Ca2+-mediated ATPase activation is absent in enzymatically produced bovine cardiac HMM (black symbols) and S1 (grey symbols) and recombinantly produced human cardiac 2-hep (green symbols) and 25-hep HMM (blue symbols), suggesting that the Ca2+-mediated regulation mechanism may not be intrinsic to soluble myosin constructs but rather involve only myosins forming filamentous structures.

The biochemical SRX states of myosin in reconstituted STF are modulated by Ca 2+ . (a) Normalized myosin SRX population (expressed as a fraction of the initial %SRX at pCa 8; black symbols) and normalized basal myosin ATPase activity (expressed as [ATPase value − ATPase value at pCa 8]; grey symbols) in STF reconstituted from bovine cardiac full-length myosin (solid symbols) and bovine cardiac myosin S1-subfragment (open symbols) at different Ca2+ concentrations. (b) Normalized myosin SRX population in STF formed from full-length myosin at 30 mM KCl (0 mM free Mg2+, dark grey; and 2 mM free Mg2+, black) and 150 mM KCl (open circles and solid symbols) at different Ca2+ concentrations. Ca2+ destabilizes the biochemical myosin SRX state(s) only when assembled into thick filaments. (c) Normalized basal myosin ATPase activity (expressed as [ATPase value − ATPase value at pCa 8]; in STF reconstituted from bovine cardiac full-length myosin (red), bovine cardiac HMM (black), and subfragment-S1 (grey), and in recombinant human cardiac 2-hep (green) and 25-hep HMM (blue) at different Ca2+ concentrations. All data are expressed as mean ± SEM (n ≥ 9 from three independent experiments that used at least two different preparations of the same reagent purification). Raw data at pCa 8 and pCa 4 are provided in Table 1.
The biochemical SRX states of myosin in reconstituted STF are modulated by Ca 2+ . (a) Normalized myosin SRX population (expressed as a fraction of the initial %SRX at pCa 8; black symbols) and normalized basal myosin ATPase activity (expressed as [ATPase value − ATPase value at pCa 8]; grey symbols) in STF reconstituted from bovine cardiac full-length myosin (solid symbols) and bovine cardiac myosin S1-subfragment (open symbols) at different Ca2+ concentrations. (b) Normalized myosin SRX population in STF formed from full-length myosin at 30 mM KCl (0 mM free Mg2+, dark grey; and 2 mM free Mg2+, black) and 150 mM KCl (open circles and solid symbols) at different Ca2+ concentrations. Ca2+ destabilizes the biochemical myosin SRX state(s) only when assembled into thick filaments. (c) Normalized basal myosin ATPase activity (expressed as [ATPase value − ATPase value at pCa 8]; in STF reconstituted from bovine cardiac full-length myosin (red), bovine cardiac HMM (black), and subfragment-S1 (grey), and in recombinant human cardiac 2-hep (green) and 25-hep HMM (blue) at different Ca2+ concentrations. All data are expressed as mean ± SEM (n ≥ 9 from three independent experiments that used at least two different preparations of the same reagent purification). Raw data at pCa 8 and pCa 4 are provided in Table 1.
Parameters obtained from biochemical assays
| Raw values at pCa 8 | Raw values at pCa 4 | pCa50 (95% CI) | Hill slope (95% CI) | |
|---|---|---|---|---|
| STF SRX (Fig. 4 a) | 15 ± 5% | 3 ± 2% | 5.5 (5.4–5.7) | 2.4 (1.4–2.5) |
| S1 SRX (Fig. 4 a) | 13 ± 2% | 12 ± 3% | N/A | N/A |
| STF ATPase (Fig. 4 a) | 0.03 ± 0.01 s−1 | 0.09 ± 0.02 s−1 | 6.1 (6.0–6.1) | 1.7 (1.3–2.3) |
| S1 ATPase (Fig. 4 a) | 0.03 ± 0.01 s−1 | 0.03 ± 0.01 s−1 | N/A | N/A |
| STF SRX (2 mM Mg2+; Fig. 4 b) | 17 ± 2% | 1 ± 2% | 5.4 (5.2–5.7) | 1.8 (0.9–1.9) |
| STF SRX (0 mM Mg2+; Fig. 4 b) | 13 ± 1% | 3 ± 2% | 5.9 (5.8–6.1) | 1.1 (0.8–1.5) |
| FL-myosin SRX (150 mM KCl; Fig. 4 b) | 5 ± 3% | 4 ± 2% | N/A | N/A |
| STF ATPase (Fig. 4 c) | 0.03 ± 0.01 s−1 | 0.1 ± 0.02 s−1 | 6.0 (5.9–6.1) | 1.9 (1.4–3.1) |
| HMM ATPase (Fig. 4 c) | 0.03 ± 0.05 s−1 | 0.03 ± 0.05 s−1 | N/A | N/A |
| 25-hep HMM ATPase (Fig. 4 c) | 0.1 ± 0.05 s−1 | 0.1 ± 0.05 s−1 | N/A | N/A |
| 2-hep HMM ATPase (Fig. 4 c) | 0.08 ± 0.05 s−1 | 0.07 ± 0.05 s−1 | N/A | N/A |
| S1 ATPase (Fig. 4 c) | 0.03 ± 0.05 s−1 | 0.03 ± 0.05 s−1 | N/A | N/A |
An EF-hand motif in the myosin's RLC domain has been shown to bind magnesium (Mg2+) under relaxed conditions and is increasingly occupied by Ca2+ as its concentration increases during muscle contraction (Markandran et al., 2021). We next studied the Ca2+-dependent effect in the absence and presence of saturating amounts of free Mg2+ bound to the RLC to find that the Ca2+-dependent destabilization of the myosin SRX is unaltered under these two conditions (Fig. 4 b), suggesting that the RLC EF-hand Ca2+-binding motif is not the primary Ca2+ transducer responsible for off-to-on transitions in the thick filament. The primary effect of Ca2+ on the RLC regarding thick filament off-to-on transitions appears to be indirect through MLCK phosphorylation (Stull et al., 2011), which is not present in our experimental systems. Binding of Ca2+ to a different site in the RLC or elsewhere on myosin cannot be excluded based on our data. Altogether, these data suggest that this Ca2+-dependent modulation of myosin is a feature that is unique to myosins assembled into thick filaments.
Discussion
In the x-ray study of permeabilized tissue, although the thin filament is shut down by the small-molecule inhibitor (MYK-7660), other sarcomeric proteins such as myosin-binding protein C and titin are present and known to bind to Ca2+ (Labeit et al., 2003). However, the biochemical studies with cardiac synthetic thick filament reconstituted from purified full-length myosin showing that Ca2+ can bind and destabilize the SRX states of myosin, in the absence of these accessory proteins, strongly indicates that a Ca2+-dependent switch is an intrinsic property of myosin. At this time, the Ca2+-binding site(s) on the thick filament has not been identified, nor can we exclude other, less specific mechanisms. Since Ca2+ only turns myosin on when they are in filaments, and we observe structural transitions in the thick filament backbone upon Ca2+ binding, we strongly suspect that the Ca2+ binding to myosin as packed in bipolar filaments may relieve a head–backbone interaction that holds myosin heads in off states at diastolic Ca2+ concentrations. These head–backbone interactions are widely assumed to involve heads in the “interacting heads motif,” but this may not necessarily be the case in all circumstances (Craig and Padron, 2022).
The results presented above lead to a novel concept of a Ca2+-mediated dual-filament regulation model in cardiac muscle (Fig. 5). At low Ca2+ concentration during diastole, the thick and thin filaments are in off states. Ca2+ from extracellular influx upon arrival of an action potential, released from sarcoplasmic reticulum through a Ca2+-induced Ca2+-release mechanism (Eisner et al., 2017), binds to both thin and thick filaments and allows the heart muscle to contract. The Ca2+ concentration can regulate the level of activation of both thin and thick filaments for them to work synergistically. The reuptake of Ca2+ after contraction will deactivate both thick and thin filaments. For example, a recent modeling study hypothesized a biochemically defined Ca2+-dependent “parked” state(s), where myosin heads are unable to bind actin, analogous to our structurally defined off state. This model could explain both activation and relaxation in twitches in the myocardium, providing more realistic relaxation rates, resting tensions, and myosin cross-bridge detachment rate than in other current models (Mijailovich et al., 2021). Notably, the Ca2+-mediated thick filament structural transitions shown in Figs. 2 and 3 and the functional transitions shown in Fig. 4 have a pCa50 in the range of 5.5 to 5.9, similar to the pCa50 of the force pCa curve (5.91 ± 0.1, Fig. 1 d), close the physiological range of systolic Ca2+ concentrations (0.3–3 µM [Sankaranarayanan et al., 2017]).

Ca 2+ -mediated dual filament regulation mechanism in cardiac muscle . A schematic cartoon of the cardiac thin (thin grey bar) and thick filament (thick grey bar; not drawn to scale) showing the organization of the regulated thin filament and myosin. (a) In the absence of Ca2+, the thin and thick filaments in resting muscle are in the deactivated (off) states, and individual myosin heads may be in either the off (orange) or the on (green) state(s). (b) Ca2+ (red) can independently bind to the thick and thin filament and activate them simultaneously. Ca2+-mediated regulation of the thick filament destabilizes the myosin heads from the off states (orange) to the on states (red heads), along with the already on heads (green) which allows the swinging of the S2-subfragment that facilitates myosin heads to bind to actin.
Ca 2+ -mediated dual filament regulation mechanism in cardiac muscle . A schematic cartoon of the cardiac thin (thin grey bar) and thick filament (thick grey bar; not drawn to scale) showing the organization of the regulated thin filament and myosin. (a) In the absence of Ca2+, the thin and thick filaments in resting muscle are in the deactivated (off) states, and individual myosin heads may be in either the off (orange) or the on (green) state(s). (b) Ca2+ (red) can independently bind to the thick and thin filament and activate them simultaneously. Ca2+-mediated regulation of the thick filament destabilizes the myosin heads from the off states (orange) to the on states (red heads), along with the already on heads (green) which allows the swinging of the S2-subfragment that facilitates myosin heads to bind to actin.
Additionally, there is a growing understanding that increased mitochondrial Ca2+ can augment ATP production (Finkel et al., 2015). These results indicate that evolution might have found an effective way to modulate cardiac muscle activation and relaxation by synchronizing both the thin and thick filament of the sarcomere and the energy supply by a single messenger, Ca2+. Imbalance in any of these components (Ca2+ flux, thin- or thick-filament Ca2+ sensitivity, and the roles of titin and MyBP-C) could disrupt the exquisite equilibrium of the system, leading to compensatory effects that cause long-term damage. We have, at this time, many unanswered questions. For example, is Ca2+-mediated thick filament activation, demonstrated here in steady-state measurements, fast enough to happen on a beat-to-beat basis? Is this activation sensitive to sarcomere length and relevant to length-dependent activation? These questions provide motivation for future experiments.
Ca2+ plays a central role in cardiac muscle function, but its role has been traditionally attributed to thin filament-based regulation inside the sarcomere. Our discovery of a direct Ca2+-mediated destabilization of the off states myosin on cardiac thick filaments warrants a reconstruction of previous understandings of the roles of Ca2+, including Ca2+ sensitivity and Ca2+ handling, in cardiac muscle in health and disease. Dysregulation of thick filament-based activation mechanisms appears to be the basis of many cardiomyopathies, including hypertrophic cardiomyopathy, which is hypothesized to be due to increased release of myosin heads from the sequestered off to on states and thereby increasing force-producing cross-bridges (Spudich, 2019; Nag and Trivedi, 2021), leading to hypercontractility and diastolic impairment. This understanding has led to a search for myosin inhibitors that mitigate off and on states dysregulation. The best known of these is mavacamten (Green et al., 2016), which has been shown by x-ray diffraction (Anderson et al., 2018; Ma et al., 2021) to enrich the population of off-state myosin heads. Therefore, the newly discovered Ca2+-mediated thick-filament regulation of force generation in cardiac muscle may need to be considered in developing future sarcomere-based drug modalities. Additionally, this Ca2+-mediated regulation of thick filaments, observed here in cardiac muscle, may turn out to be a fundamental component of all human skeletal systems, which opens the possibility of new therapeutic approaches for many congenital myopathies caused by sarcomeric protein mutations. More generally, insofar a significant fraction of the ATPase activity of the skeletal muscle myosin is to maintain temperature homeostasis in homeothermic organisms (Cooke, 2011), this new role of calcium as a driver of myosin ATP consumption may provide a unique perspective on the energetic costs of thermo-regulation.
Data availability
The datasets generated or analyzed during this study are included in this article. The raw data are available from the corresponding authors upon reasonable request. The thin filament inhibitor MYK-7660 can be made available from Bristol Myers Squibb under a material transfer agreement or in collaboration with their scientists.
Acknowledgments
Henk L. Granzier served as editor.
We thank Ariana Combs and Stephen Langer of the Leinwand laboratory for generating the recombinant human cardiac 2-hep and 25-hep HMM samples. We thank Sampath Gollapudi of Bristol Myers Squibb (formerly MyoKardia, Inc.) for contributing to the biochemical data on the inhibitor. We thank James A. Spudich of Stanford University, Michael Geeves of the University of Kent, Michael Regnier of the University of Washington, Srboljub Mijailovich of the Illinois Institute of Technology, and Robert S. McDowell of Bristol Myers Squibb for providing helpful comments to the manuscript. Finally, we thank Bristol Myers Squibb for providing the thin filament inhibitor MYK-7660.
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This project is supported by grant P30 GM138395 from the National Institute of General Medical Sciences of the National Institutes of Health. The content is solely the authors’ responsibility and does not necessarily reflect the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
S. Nag is a former employee of Bristol Myers Squibb (formerly MyoKardia, Inc.). T.C. Irving provides consulting and collaborative research studies to Edgewise Therapeutics and Bristol Myers Squibb, but such work is unrelated to the content of this article. The remaining authors declare no competing financial interests.
Author contributions: W. Ma, S. Nag, T.C. Irving designed the experiment; W. Ma, H. Gong, S. Nag performed the research; W. Ma, L. Qi, S. Nag analyzed the data; W. Ma, S. Nag, T.C. Irving wrote the manuscript. All authors approved the final version of the manuscript.
References
This work is part of a special issue on Myofilament Function 2022.
Data availability
The datasets generated or analyzed during this study are included in this article. The raw data are available from the corresponding authors upon reasonable request. The thin filament inhibitor MYK-7660 can be made available from Bristol Myers Squibb under a material transfer agreement or in collaboration with their scientists.

