


The molecular mechanism by which inborn errors of the human RNA lariat–debranching enzyme 1 (DBR1) underlie brainstem viral encephalitis is unknown. We show here that the accumulation of RNA lariats in human DBR1-deficient cells interferes with stress granule (SG) assembly, promoting the proteasome degradation of at least G3BP1 and G3BP2, two key components of SGs. In turn, impaired assembly of SGs, which normally recruit PKR, impairs PKR activation and activity against viruses, including HSV-1. Remarkably, the genetic ablation of PKR abolishes the corresponding antiviral effect of DBR1 in vitro. We also show that Dbr1Y17H/Y17H mice are susceptible to similar viral infections in vivo. Moreover, cells and brain samples from Dbr1Y17H/Y17H mice exhibit decreased G3BP1/2 expression and PKR phosphorylation. Thus, the debranching of RNA lariats by DBR1 permits G3BP1/2- and SG assembly-mediated PKR activation and cell-intrinsic antiviral immunity in mice and humans. DBR1-deficient patients are prone to viral disease because of intracellular lariat accumulation, which impairs G3BP1/2- and SG assembly-dependent PKR activation.
Introduction
Viral encephalitis is life-threatening and can strike previously healthy children. Herpes simplex virus-1 (HSV-1) is the most common viral etiology of sporadic cases (Tyler, 2018). Over the last ∼20 years, genetic etiologies of childhood HSV-1 encephalitis (HSE) have been deciphered. We and others have reported single-gene inborn errors of Toll-like receptor 3 (TLR3)- and/or type I interferon (IFN)-dependent immunity, with causal genotypes for 12 genes, in children with forebrain HSE (Casrouge et al., 2006; Zhang et al., 2007, 2008; Bastard et al., 2021; Zhang and Casanova, 2024). Inborn errors of three other genes impairing either RIG-I-dependent type I IFN immunity (GTF3A), or type I IFN-independent antiviral immunity in cortical neurons (SNORA31, RIPK3, TMEFF1), have been found more recently in other patients with forebrain HSE (Lafaille et al., 2019; Naesens et al., 2022; Liu et al., 2023; Chan et al., 2024b). Although HSE typically affects the forebrain, rare cases of hindbrain HSE have been reported. HSV-1 and other viruses, including influenza viruses and enteroviruses, can underlie brainstem encephalitis. We previously reported a severe but incomplete form of autosomal recessive (AR) DBR1 deficiency in children with brainstem encephalitis due to infection with HSV-1, influenza virus B, or norovirus (Zhang et al., 2018). More recently, we identified a patient with SARS-CoV-2 brainstem infection (Chan et al., 2024a). The patient within this group with the most severe DBR1 deficiency had several other clinical manifestations (Zhang et al., 2018): mild intrauterine growth retardation, mental retardation, and congenital neutropenia. Other recently reported patients had a lethal form of congenital ichthyosis, perhaps due to an even more severe DBR1 deficiency (Shamseldin et al., 2023). DBR1 is the only known RNA lariat-debranching enzyme and is structurally and functionally conserved from yeasts to humans (Kim et al., 2000; Chapman and Boeke, 1991; Findlay et al., 2014; Wang et al., 2004; Zheng et al., 2015). Lariat RNAs are splicing intermediates produced by group II introns and the pre-mRNA spliceosome (Ruskin et al., 1984; Padgett et al., 1984; Peebles et al., 1986). The patients’ fibroblasts have low levels of DBR1 protein and enzymatic activity, resulting in high RNA lariat levels (Arenas and Hurwitz, 1987; Chapman and Boeke, 1991). Cell-intrinsic immunity to viruses is also impaired in both DBR1-mutated fibroblasts (Zhang et al., 2018) and human pluripotent stem cell (hPSC)-derived hindbrain neurons (Chan et al., 2024a).
Intriguingly, although patients with AR DBR1 deficiency probably accumulate RNA lariats in multiple cell types, their infectious phenotype is devastating but narrow, as it is restricted to viral brainstem encephalitis (Zhang et al., 2018). The topologically restricted nature of viral disease in these patients may reflect the pattern of DRB1 expression, which is strongest in the brainstem (Zhang et al., 2018). DBR1-deficient cells in the brainstem may accumulate more lariats than other cell types, the lariats may be more damaging in these cells, or both. Experimental studies of hPSC-derived cells normally resident in different regions both within and outside the brain should help clarify the cellular basis of disease. A first step in this direction was taken with our demonstration that DBR1-mutated hPSC-derived hindbrain neurons are susceptible to SARS-CoV-2 (Chan et al., 2024a). However, the cellular basis of the other, rarer clinical manifestations of DBR1 deficiency also remains unexplained, as does the molecular basis of viral disease. The patients are susceptible to very different viruses, including HSV-1, influenza B, norovirus, and SARS-CoV-2, suggesting that DBR1 deficiency interferes with a broad mechanism of antiviral immunity. High viral loads in cultured fibroblasts and hPSC-derived neurons from the patients indicate that cell-intrinsic immunity is impaired when DBR1 is deficient. Nevertheless, it remains unclear how DBR1 deficiency favors viral infection in vitro and in vivo, and whether and how lariat accumulation impairs cell-intrinsic antiviral immunity. Given that DBR1 deficiency impairs RNA metabolism, resulting in an accumulation of RNA lariats, we hypothesized that DBR1 deficiency might disrupt cell-intrinsic RNA-sensing pathways.
Multiple cell-intrinsic antiviral strategies exist, based on the sensing of dsRNA viral intermediates or by-products (Chen and Hur, 2022). First, the endosomal TLR3-dependent induction of type I IFNs is crucial for defenses against certain viruses in the brain and lungs (Chen et al., 2021; Zhang et al., 2013, 2020, 2022). However, DBR1-deficient cells respond normally via TLR3, whereas patients with mutations of the TLR3 pathway have forebrain infections, as opposed to the brainstem viral infections seen in patients with DBR1 mutations (Zhang et al., 2018). Second, activation of the IFN-stimulated gene (ISG) cytosolic OASs by dsRNA activates RNase L (Sadler and Williams, 2008). However, inherited deficiencies of the OAS-RNase L pathway lead to post-infectious, SARS-CoV-2-related hyperinflammation rather than uncontrolled viral infection in the lungs (Lee et al., 2023). Third, cytosolic RIG-I and MDA5 stimulate the downstream adaptor protein mitochondrial antiviral signaling (MAVS), thereby inducing antiviral type I IFN production (Rehwinkel and Gack, 2020). RIG-I and MAVS deficiencies have not been reported but inherited MDA-5 deficiency underlies respiratory rhinoviral disease and enterovirus hindbrain encephalitis (Chen et al., 2021; Lamborn et al., 2017), raising the possibility that DBR1 deficiency may interfere with MDA-5. Fourth, protein kinase R (PKR) is a type I IFN-dependent ISG product that also binds to cytosolic dsRNA, inducing the integrated stress response (ISR) (Manche et al., 1992; Nanduri et al., 1998). Viral dsRNAs and IFN-I induce the phosphorylation of PKR, leading to phosphorylation of eukaryotic initiation factor 2α (eIF2α), which drives a global stalling of translation and hinders viral protein synthesis (Dey et al., 2005; Su et al., 2007). We therefore decided to investigate the MAVS and PKR pathways in human DBR1-deficient cells.
Results
DBR1 deficiency impairs PKR activation by intracellular dsRNA stimulation
We explored antiviral immunity in DBR1-deficient cells by the knockdown (KD) of DBR1 (shDBR1) in BJ human fibroblasts, with the confirmation of DBR1 expression by real-time quantitative PCR (RT-qPCR) (Fig. S1 A). Through the qPCR detection of intronic RNA lariats of the ID1 and DKK1 genes, two of the most prominently elevated lariats in DBR1-deficient patients’ fibroblasts (Zhang et al., 2018), we validated the decrease in RNA debranching activity in shDBR1 BJ cells (Fig. 1 A). To determine the antiviral pathway involved, shDBR1 BJ cells were stimulated with intracellularly transfected polyinosinic:polycytidylic acid (polyI:C), a dsRNA mimic that stimulates both RIG-I/MDA5 and PKR, leading to MAVS-dependent type I IFN induction and PKR phosphorylation, respectively. Type I IFN and ISG expression levels were quantified by RT-qPCR. shDBR1 or overexpression barely affected the induction of type I IFN or ISGs (Fig. S1, B–D). By contrast, shDBR1 impaired the phosphorylation of PKR in BJ cells, while stable overexpression of exogenous DBR1 enhanced polyI:C-induced PKR phosphorylation (Fig. 1 B).
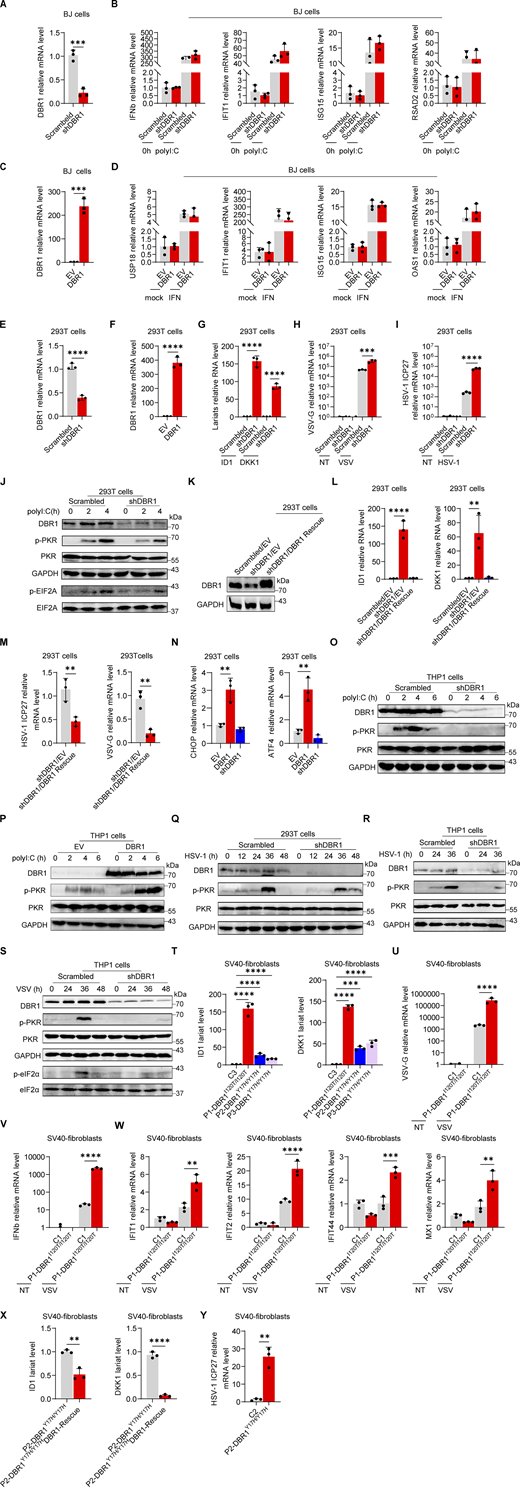
DBR1 regulates the activation of PKR and integrated stress response rather than IFN response. (A) RT-qPCR detection of DBR1 mRNA in BJ cells transduced with scrambled and shDBR1. Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. ***, P < 0.001. (B) BJ cells with scrambled shRNA or shDBR1 were stimulated with polyI:C, followed by RT-qPCR analysis of the indicated IFN or ISGs. Data representative of three independent experiments. Graphs depict points that represent biological replicates. (C) RT-qPCR detection of DBR1 mRNA in BJ cells transduced with overexpressing DBR1 relative to control. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. ***, P < 0.001. (D) BJ cells with EV or DBR1 overexpression were stimulated with IFNβ, followed by RT-qPCR analysis of EV and DBR1 BJ cells for expression of the indicated ISGs. Data representative of three independent experiments. Graphs depict points that represent biological replicates. (E) RT-qPCR detection of DBR1 mRNA in HEK293T cells transduced with scrambled and shDBR1. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. ****, P < 0.0001. (F) RT-qPCR detection of DBR1 mRNA in HEK293T cells transduced with DBR1 relative to control. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. ****, P < 0.0001. (G) RT-qPCR determination of ID1, DKK1 RNA lariat levels in HEK293T scrambled control and shDBR1 cells. Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (H and I) HEK293T scrambled control or shDBR1 cells were infected with VSV at an MOI of 0.1 (H) or HSV-1 at an MOI of 0.5 (I) for 24 h, and viral replication was then detected by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (J) HEK293T cells transfected with the scramble shRNA or shDBR1 were stimulated with polyI:C (1 µg/ml), followed by WB for indicated proteins. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (K and L) HEK293T cells transduced with the scrambled shRNA, shDBR1, or shDBR1 with DBR1 for rescue were analyzed by WB with the indicated antibodies (K). GAPDH was used as a loading control. Data shown are representative of three independent experiments. RT-qPCR for RNA lariat detection (L). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001. (M) HEK293T cells transduced with shDBR1 or shDBR1 cells transduced with DBR1 for rescue were infected with HSV-1 (left) or VSV (right), and viral mRNA was then quantified by RT-qPCR. Statistical analysis was performed with unpaired t tests. **, P < 0.01. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (N) RT-qPCR analysis of control, shDBR1-transduced and DBR1-overexpressing HEK293T cells for assessment of the expression of the indicated ISR genes. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (O) Stimulation with polyI:C (1 µg/ml) in THP-1 cells transduced with the scramble shRNA or shDBR1, followed by WB for indicated proteins. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (P) THP1 cells transduced with EV or DBR1 were stimulated with polyI:C (1 µg/ml) and WB was then performed with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (Q) HEK293T cells transduced with the scrambled shRNA or shDBR1 were subjected to HSV-1 infection (MOI: 0.5) for various time points. WB was then performed with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (R and S) THP-1 cells transduced with the scrambled shRNA or shDBR1 were subjected to HSV-1 infection (MOI: 0.5) (R) or VSV infection (MOI: 0.1) (S) for various time points, followed by WB for the indicated proteins. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (T) RT-qPCR quantification of ID1 and DKK1 RNA lariat levels in fibroblasts from the control and patients. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001; ****, P < 0.0001. (U–W) Cells from the control and the patient were infected with VSV at a MOI of 0.1, and RT-qPCR was then performed for detection of the expression of viral genes (U), IFN and ISG genes (V and W). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (X) Patient’s cells were rescued by transfection with DBR1, and RT-qPCR was then performed to assess ID1 and DKK1 lariat levels. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01; ***, P < 0.001. (Y) Control and patient’s cells were infected with HSV-1 (MOI: 0.5), and viral mRNA was then quantified by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01. Source data are available for this figure: SourceData FS1.
DBR1 regulates the activation of PKR and integrated stress response rather than IFN response. (A) RT-qPCR detection of DBR1 mRNA in BJ cells transduced with scrambled and shDBR1. Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. ***, P < 0.001. (B) BJ cells with scrambled shRNA or shDBR1 were stimulated with polyI:C, followed by RT-qPCR analysis of the indicated IFN or ISGs. Data representative of three independent experiments. Graphs depict points that represent biological replicates. (C) RT-qPCR detection of DBR1 mRNA in BJ cells transduced with overexpressing DBR1 relative to control. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. ***, P < 0.001. (D) BJ cells with EV or DBR1 overexpression were stimulated with IFNβ, followed by RT-qPCR analysis of EV and DBR1 BJ cells for expression of the indicated ISGs. Data representative of three independent experiments. Graphs depict points that represent biological replicates. (E) RT-qPCR detection of DBR1 mRNA in HEK293T cells transduced with scrambled and shDBR1. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. ****, P < 0.0001. (F) RT-qPCR detection of DBR1 mRNA in HEK293T cells transduced with DBR1 relative to control. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. ****, P < 0.0001. (G) RT-qPCR determination of ID1, DKK1 RNA lariat levels in HEK293T scrambled control and shDBR1 cells. Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (H and I) HEK293T scrambled control or shDBR1 cells were infected with VSV at an MOI of 0.1 (H) or HSV-1 at an MOI of 0.5 (I) for 24 h, and viral replication was then detected by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (J) HEK293T cells transfected with the scramble shRNA or shDBR1 were stimulated with polyI:C (1 µg/ml), followed by WB for indicated proteins. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (K and L) HEK293T cells transduced with the scrambled shRNA, shDBR1, or shDBR1 with DBR1 for rescue were analyzed by WB with the indicated antibodies (K). GAPDH was used as a loading control. Data shown are representative of three independent experiments. RT-qPCR for RNA lariat detection (L). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001. (M) HEK293T cells transduced with shDBR1 or shDBR1 cells transduced with DBR1 for rescue were infected with HSV-1 (left) or VSV (right), and viral mRNA was then quantified by RT-qPCR. Statistical analysis was performed with unpaired t tests. **, P < 0.01. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (N) RT-qPCR analysis of control, shDBR1-transduced and DBR1-overexpressing HEK293T cells for assessment of the expression of the indicated ISR genes. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (O) Stimulation with polyI:C (1 µg/ml) in THP-1 cells transduced with the scramble shRNA or shDBR1, followed by WB for indicated proteins. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (P) THP1 cells transduced with EV or DBR1 were stimulated with polyI:C (1 µg/ml) and WB was then performed with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (Q) HEK293T cells transduced with the scrambled shRNA or shDBR1 were subjected to HSV-1 infection (MOI: 0.5) for various time points. WB was then performed with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (R and S) THP-1 cells transduced with the scrambled shRNA or shDBR1 were subjected to HSV-1 infection (MOI: 0.5) (R) or VSV infection (MOI: 0.1) (S) for various time points, followed by WB for the indicated proteins. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (T) RT-qPCR quantification of ID1 and DKK1 RNA lariat levels in fibroblasts from the control and patients. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001; ****, P < 0.0001. (U–W) Cells from the control and the patient were infected with VSV at a MOI of 0.1, and RT-qPCR was then performed for detection of the expression of viral genes (U), IFN and ISG genes (V and W). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (X) Patient’s cells were rescued by transfection with DBR1, and RT-qPCR was then performed to assess ID1 and DKK1 lariat levels. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01; ***, P < 0.001. (Y) Control and patient’s cells were infected with HSV-1 (MOI: 0.5), and viral mRNA was then quantified by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01. Source data are available for this figure: SourceData FS1.
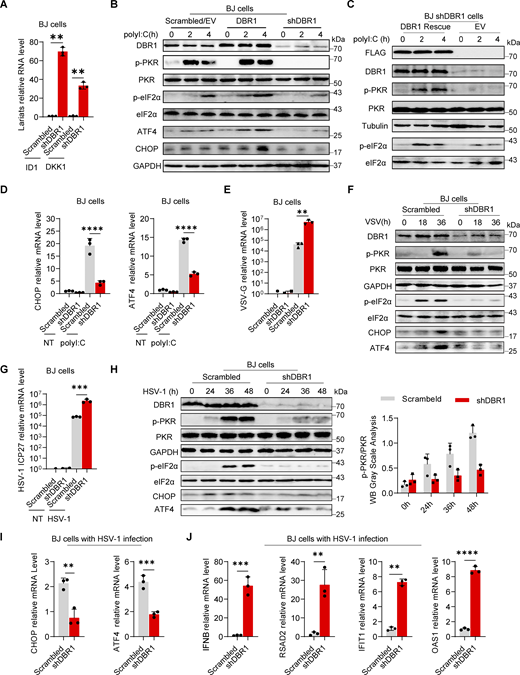
DBR1 regulates the PKR-mediated stress response during viral infection. (A) RT-qPCR determination of ID1, DKK1 RNA lariat levels in BJ scrambled control and shDBR1 cells. Data are representative of three independent experiments. Graphs depict the mean with standard deviation (SD), and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (B) BJ cells stably expressing Flag-DBR1 and an shRNA against DBR1 together with a scrambled/EV control were transfected and stimulated with polyI:C (1 µg/ml), and were then subjected to WB analysis with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (C) shDBR1 BJ cells transfected with EV or DBR1 were transfected with polyI:C (1 µg/ml) for 0, 2, or 4 h. Cell lysates were then prepared and subjected to WB with the indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (D) Following stimulation with polyI:C (1 µg/ml) in scrambled control and shDBR1 BJ cells, RT-qPCR was performed to quantify CHOP and ATF4 mRNA levels. Data are representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (E) BJ scrambled control or shDBR1 cells were infected with VSV at an MOI of 0.1 for 24 h, and viral replication was then detected by RT-qPCR. Data are representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (F) BJ cells transduced with scrambled shRNA or shDBR1 were infected with VSV at an MOI of 0.1 for various times. The cells were then lysed and subjected to WB with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (G) BJ cells transduced with scrambled shRNA or shDBR1 were infected with HSV-1 at an MOI of 0.5 for 24 h and then quantified for viral mRNA by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001. (H) BJ cells transduced with scrambled shRNA or shDBR1 were infected with HSV-1 at an MOI of 0.5 for various times. The cells were then lysed and subjected to WB with the indicated antibodies, followed by the p-PKR/PKR WB grayscale analysis of three independent experiments. GAPDH was used as a loading control. (I and J) BJ cells were infected with HSV-1 (MOI: 0.5) for 24 h and then quantified for the expression of the indicated ISR genes (I), IFNB and the indicated ISGs (J) by RT-qPCR. Data are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F1.
DBR1 regulates the PKR-mediated stress response during viral infection. (A) RT-qPCR determination of ID1, DKK1 RNA lariat levels in BJ scrambled control and shDBR1 cells. Data are representative of three independent experiments. Graphs depict the mean with standard deviation (SD), and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (B) BJ cells stably expressing Flag-DBR1 and an shRNA against DBR1 together with a scrambled/EV control were transfected and stimulated with polyI:C (1 µg/ml), and were then subjected to WB analysis with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (C) shDBR1 BJ cells transfected with EV or DBR1 were transfected with polyI:C (1 µg/ml) for 0, 2, or 4 h. Cell lysates were then prepared and subjected to WB with the indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (D) Following stimulation with polyI:C (1 µg/ml) in scrambled control and shDBR1 BJ cells, RT-qPCR was performed to quantify CHOP and ATF4 mRNA levels. Data are representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (E) BJ scrambled control or shDBR1 cells were infected with VSV at an MOI of 0.1 for 24 h, and viral replication was then detected by RT-qPCR. Data are representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (F) BJ cells transduced with scrambled shRNA or shDBR1 were infected with VSV at an MOI of 0.1 for various times. The cells were then lysed and subjected to WB with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (G) BJ cells transduced with scrambled shRNA or shDBR1 were infected with HSV-1 at an MOI of 0.5 for 24 h and then quantified for viral mRNA by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001. (H) BJ cells transduced with scrambled shRNA or shDBR1 were infected with HSV-1 at an MOI of 0.5 for various times. The cells were then lysed and subjected to WB with the indicated antibodies, followed by the p-PKR/PKR WB grayscale analysis of three independent experiments. GAPDH was used as a loading control. (I and J) BJ cells were infected with HSV-1 (MOI: 0.5) for 24 h and then quantified for the expression of the indicated ISR genes (I), IFNB and the indicated ISGs (J) by RT-qPCR. Data are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01; ***, P < 0.001. Source data are available for this figure: SourceData F1.
DBR1-deficient cells exhibit compromised stress response
PKR is one of the four known eIF2α kinases; it contributes to cellular stress responses in vitro (Taniuchi et al., 2016). Phosphorylated-eIF2α (p-eIF2α) favors the translation of ATF4, a master regulator controlling the transcription of DDIT3/CHOP, GADD45A, and other ISR genes (Ebert et al., 2010; Harding et al., 2000). We observed consistent p-eIF2α and ISR (CHOP, ATF4) expression correlating with phosphorylated-PKR (p-PKR) levels (Fig. 1 B). We excluded an off-target effect of shRNA by restoring DBR1 protein levels in shDBR1 cells, which led to increased PKR activation, eIF2α phosphorylation, and ISR expression in response to polyI:C stimulation (Fig. 1 C). We assessed ISR (e.g., CHOP, ATF4) gene expression levels by RT-qPCR after polyI:C stimulation in BJ cells with or without shDBR1 (Fig. 1 D). We observed a correlation between DBR1 levels and eIF2α-regulated ISR transcription. DBR1 was also found to be essential for optimal p-PKR and downstream ISR gene expression in HEK293T epithelial cells (Fig. S1, E–M). Furthermore, DBR1 overexpression amplified basal ISR gene expression (Fig. S1 N), and the correlation of expression of DBR1 and p-PKR was observed in THP-1 human monocyte cell line (Fig. S1, O and P). Overall, these findings suggest that low levels of DBR1 impair the activation of PKR but not the RIG-I/MDA5-dependent induction of type I IFN.
DBR1 deficiency impairs virus-induced antiviral PKR activation
We then infected BJ with vesicular stomatitis virus (VSV) (Fig. 1, E and F) or HSV-1 (Fig. 1, G–I) and monitored the stress response. We observed an increase in viral VSV-G mRNA level in shDBR1 BJ cells, indicating that DBR1 restricts VSV infection in these cells in vitro (Fig. 1 E). Consistent with previous findings, shDBR1 resulted in a downregulation of the VSV-induced phosphorylation of PKR and eIF2α and subsequent ISR (CHOP, ATF4) expression (Fig. 1 F). Similarly, HSV-1 infection induced a weaker stress response with higher viral replication in shDBR1 BJ cells (Fig. 1. G–I). Conversely, HSV-1-infected BJ cells produced more type I IFN and ISG proteins in the absence of DBR1, possibly due to higher levels of viral replication (Fig. 1 J). Similar effects of PKR activation on DBR1-dependency during viral infection were found in HEK293T (Fig. S1 Q) and THP-1 cells (Fig. S1, R and S). These data suggest that DBR1 plays a crucial role in PKR activation by viruses or dsRNA and in subsequent ISR gene expression.
Patient-specific DBR1 mutants compromise PKR activation
To study the impact of the patients’ DBR1 genotypes on PKR-mediated stress response, we overexpressed the patient-specific, pathogenic, and severely hypomorphic DBR1 mutants (L13G, Y17H, I120T, and R197X) in HEK293T cells and stimulating them intracellularly with polyI:C. None of the variants induced a stress response like that triggered by WT DBR1 (Fig. 2 A). For the patient’s cells, we observed a simultaneous upregulation of RNA lariats, viral replication, and levels of IFNB and ISGs mRNAs after VSV infection in P1-DBR1I120T/I120T fibroblasts (Fig. S1, T–W). When stimulated intracellularly with polyI:C, cells from P1-DBR1I120T/I120T or P2-DBR1Y17H/Y17H displayed impaired PKR phosphorylation (Fig. 2, B and C). PKR phosphorylation and lariat degradation were restored by overexpressing WT DBR1 in P2-DBR1Y17H/Y17H SV40-fibroblasts (Fig. 2 C and Fig. S1 X). As in shDBR1 cells, ISR (CHOP, ATF4) expression following the VSV challenge was attenuated in the SV40-immortalized fibroblasts available from three patients with DBR1 mutations (Fig. 2 D). Phosphorylation of PKR, eIF2α, and ISR (CHOP, ATF4) expression was also restored by overexpressing WT DBR1 after VSV infection, suggesting that the DBR1 genotype is responsible for low levels of PKR activity (Fig. 2 E). Moreover, HSV-1-induced p-PKR levels were lower in the cells of P2-DBR1Y17H/Y17H than in healthy control cells despite higher viral replication (Fig. 2 F and Fig. S1 Y). Overall, these results show that the viral encephalitis-causing DBR1 genotypes resulted in an insufficient PKR activation-mediated stress response to viruses in the patients’ cells.
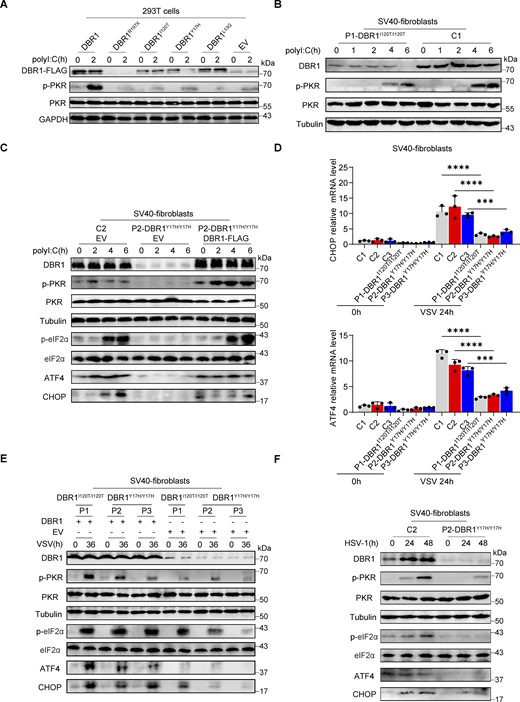
DBR1 mutants from the patients fail to potentiate the PKR-mediated stress response. (A) WT DBR1 or DBR1 variants from patients with brainstem encephalitis were overexpressed in HEK293T cells and then stimulated with polyI:C transfection (1 µg/ml). The cells were lysed and subjected to WB for detection of the indicated proteins. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (B and C) SV40-transformed fibroblasts (SV40-fibroblasts) from control and two patients, P1-DBR1I120T/I120T (B) and P2-DBR1Y17H/Y17H without and with WT DBR1 rescue (C) were transfected with 1 µg/ml polyI:C and subjected to WB with the indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (D) SV40-fibroblasts from control or DBR1-deficient patients were infected with VSV, and RT-qPCR was performed to assess the expression of the indicated ISR genes. Data are representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001; ****, P < 0.0001. (E) SV40-fibroblasts from three patients, P1-DBR1I120T/I120T, P2-DBR1Y17H/Y17H, and P3-DBR1Y17H/Y17H without or with WT DBR1 rescue were infected with VSV at an MOI of 0.1, followed by WB for indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (F) SV40-fibroblasts from a healthy control and a DBR1-deficient patient were infected with HSV-1 at an MOI of 0.5, followed by WB for indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F2.
DBR1 mutants from the patients fail to potentiate the PKR-mediated stress response. (A) WT DBR1 or DBR1 variants from patients with brainstem encephalitis were overexpressed in HEK293T cells and then stimulated with polyI:C transfection (1 µg/ml). The cells were lysed and subjected to WB for detection of the indicated proteins. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (B and C) SV40-transformed fibroblasts (SV40-fibroblasts) from control and two patients, P1-DBR1I120T/I120T (B) and P2-DBR1Y17H/Y17H without and with WT DBR1 rescue (C) were transfected with 1 µg/ml polyI:C and subjected to WB with the indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (D) SV40-fibroblasts from control or DBR1-deficient patients were infected with VSV, and RT-qPCR was performed to assess the expression of the indicated ISR genes. Data are representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001; ****, P < 0.0001. (E) SV40-fibroblasts from three patients, P1-DBR1I120T/I120T, P2-DBR1Y17H/Y17H, and P3-DBR1Y17H/Y17H without or with WT DBR1 rescue were infected with VSV at an MOI of 0.1, followed by WB for indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (F) SV40-fibroblasts from a healthy control and a DBR1-deficient patient were infected with HSV-1 at an MOI of 0.5, followed by WB for indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F2.
PKR is essential for DBR1-dependent antiviral immunity
Loss of the ISG protein PKR has been shown to increase susceptibility to multiple viral infections in human cells in vitro and in mice in vivo (Balachandran et al., 2000; García et al., 2007). We confirmed and extended these findings in vitro by infecting PKR-overexpressing HEK293T cells (Fig. S2, A and B) or PKR−/− BJ cells (Fig. S2 C) with VSV or HSV-1. PKR overexpression also enhanced virus-induced PKR phosphorylation, especially at earlier time points, as demonstrated by WB (Fig. S2, D and E). We investigated the role of PKR in DBR1-mediated antiviral activity by overexpressing DBR1 in PKR−/− and WT BJ cells. We then assessed protein levels by WB and infected these cells with viruses (Fig. 3 A). The protective effect of DBR1 was abolished by the genetic ablation of PKR after VSV infection, resulting in higher viral transcript quantification (Fig. 3 B), which was corroborated by fluorescence microscopy observation (Fig. S2 F) or viral titers (Fig. 3 C). Similar results were observed during HSV-1 infection (Fig. 3, D and E; and Fig. S2 F). These findings suggest that PKR is essential for DBR1 antiviral activity in these experimental conditions. Viral infection-induced p-PKR and ISR (CHOP, ATF4, GADD34) mRNA levels were enhanced by DBR1 overexpression in cells with a functional PKR but not in cells lacking PKR (Fig. 3, F and G). If DBR1 and PKR function in the same pathway, then their combined deficiency should result in a viral susceptibility profile similar to those for either of the single deficiencies. As expected, when DBR1 was silenced in PKR-deficient cells, as confirmed by WB, there was no increase in viral susceptibility, confirming their participation in the same pathway (Fig. 3, H–J). Moreover, the overexpression of PKR in DBR1-deficient cells restored not only PKR phosphorylation but also viral resistance (Fig. 3, K–M; and Fig. S2, G and H). These results suggest that PKR operates downstream from DBR1 and is required for its antiviral activity.
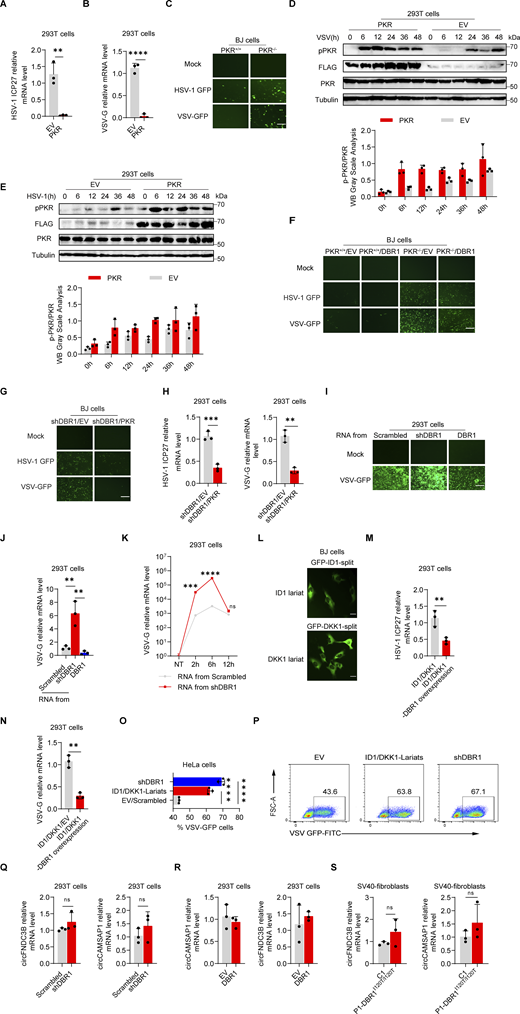
DBR1 influences PKR antiviral function by modulating the level of RNA lariats. (A and B) HEK293T cells with or without PKR overexpression were infected with HSV-1-GFP at an MOI of 0.5 (A), or VSV at an MOI of 0.1 (B) for 24 h. RT-qPCR was performed to confirm viral infection. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01; ****, P < 0.0001. (C) PKR+/+ and PKR−/− BJ cells were infected with VSV-GFP at an MOI of 0.1, or HSV-1-GFP at an MOI of 0.5 for 24 h, and fluorescence imaging was then performed. Scale bars, 100 µm. Data shown are representative of three independent experiments. (D and E) HEK293T cells transduced with EV or PKR were infected with VSV at an MOI of 0.1 (D), or HSV-1 at an MOI of 0.5 (E); WB was then performed with the indicated antibodies, followed by the p-PKR/PKR WB grayscale analysis of three independent experiments. Tubulin was used as a loading control. Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. (F) PKR+/+ and PKR−/− BJ cells were transduced with EV or DBR1 and infected with VSV-GFP (MOI: 0.1), or HSV-1-GFP (MOI: 0.5) for 24 h, and fluorescence imaging was then performed for viral GFP. Scale bars, 100 µm. Data shown are representative of three independent experiments. (G) shDBR1 BJ cells transduced with EV or PKR were infected with HSV-1-GFP at an MOI of 0.5, or VSV at an MOI of 0.1 for 24 h. Scale bars, 100 µm. Data shown are representative of three independent experiments. (H) shDBR1 HEK293T cells transduced with EV or PKR were infected with HSV-1-GFP at an MOI of 0.1 for 24 h or VSV at an MOI of 0.1, RT-qPCR was then performed to detect the expression of viral genes. Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. **, P < 0.01; ***, P < 0.001. (I and J) RNA was extracted from HEK293T cells transduced with scrambled shRNA, shDBR1, or DBR1. The purified RNA was then used to transfect HEK293T cells for 4 h. Cells were infected with VSV at an MOI of 0.01 for 24 h. Viral titers were assessed by fluorescence imaging (I). Scale bars, 100 µm. Data shown are representative of three independent experiments. Viral titers were assessed RT-qPCR (J). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (K) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with the scrambled control shRNA, or shDBR1 for different time periods, and were then infected with VSV-GFP at a MOI of 0.01 for 24 h. And the viral gene expression was quantified by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with unpaired t tests one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001; ****, P < 0.0001. (L) Diagram of RNA lariat-expressing plasmid design and fluorescence imaging. Scale bars, 20 µm. Data shown are representative of three independent experiments. (M and N) HEK293T cells expressing ID1/DKK1-Lariats or ID1/DKK1-Lariats/DBR1 were infected with HSV-1 at an MOI of 0.05 (M) or VSV at an MOI of 0.01 (N), and viral replication was then determined by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01. (O and P) HeLa cells transduced with the scrambled control, shDBR1, or ID1/DKK1-Lariats were infected with VSV at an MOI of 0.1 for 24 h, and the viral GFP was then detected by FACS. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (Q and R) RT-qPCR assays for circRNA quantification in HEK293T cells transduced with shDBR1 (Q) or DBR1 (R) cells relative to control. Statistical analysis was performed with unpaired t tests. NS, not significant. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (S) RT-qPCR assays for circRNA in cells from patients relative to controls. Statistical analysis was performed with unpaired t tests. NS, not significant. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Source data are available for this figure: SourceData FS2.
DBR1 influences PKR antiviral function by modulating the level of RNA lariats. (A and B) HEK293T cells with or without PKR overexpression were infected with HSV-1-GFP at an MOI of 0.5 (A), or VSV at an MOI of 0.1 (B) for 24 h. RT-qPCR was performed to confirm viral infection. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01; ****, P < 0.0001. (C) PKR+/+ and PKR−/− BJ cells were infected with VSV-GFP at an MOI of 0.1, or HSV-1-GFP at an MOI of 0.5 for 24 h, and fluorescence imaging was then performed. Scale bars, 100 µm. Data shown are representative of three independent experiments. (D and E) HEK293T cells transduced with EV or PKR were infected with VSV at an MOI of 0.1 (D), or HSV-1 at an MOI of 0.5 (E); WB was then performed with the indicated antibodies, followed by the p-PKR/PKR WB grayscale analysis of three independent experiments. Tubulin was used as a loading control. Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. (F) PKR+/+ and PKR−/− BJ cells were transduced with EV or DBR1 and infected with VSV-GFP (MOI: 0.1), or HSV-1-GFP (MOI: 0.5) for 24 h, and fluorescence imaging was then performed for viral GFP. Scale bars, 100 µm. Data shown are representative of three independent experiments. (G) shDBR1 BJ cells transduced with EV or PKR were infected with HSV-1-GFP at an MOI of 0.5, or VSV at an MOI of 0.1 for 24 h. Scale bars, 100 µm. Data shown are representative of three independent experiments. (H) shDBR1 HEK293T cells transduced with EV or PKR were infected with HSV-1-GFP at an MOI of 0.1 for 24 h or VSV at an MOI of 0.1, RT-qPCR was then performed to detect the expression of viral genes. Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. **, P < 0.01; ***, P < 0.001. (I and J) RNA was extracted from HEK293T cells transduced with scrambled shRNA, shDBR1, or DBR1. The purified RNA was then used to transfect HEK293T cells for 4 h. Cells were infected with VSV at an MOI of 0.01 for 24 h. Viral titers were assessed by fluorescence imaging (I). Scale bars, 100 µm. Data shown are representative of three independent experiments. Viral titers were assessed RT-qPCR (J). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (K) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with the scrambled control shRNA, or shDBR1 for different time periods, and were then infected with VSV-GFP at a MOI of 0.01 for 24 h. And the viral gene expression was quantified by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with unpaired t tests one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001; ****, P < 0.0001. (L) Diagram of RNA lariat-expressing plasmid design and fluorescence imaging. Scale bars, 20 µm. Data shown are representative of three independent experiments. (M and N) HEK293T cells expressing ID1/DKK1-Lariats or ID1/DKK1-Lariats/DBR1 were infected with HSV-1 at an MOI of 0.05 (M) or VSV at an MOI of 0.01 (N), and viral replication was then determined by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with t tests. **, P < 0.01. (O and P) HeLa cells transduced with the scrambled control, shDBR1, or ID1/DKK1-Lariats were infected with VSV at an MOI of 0.1 for 24 h, and the viral GFP was then detected by FACS. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (Q and R) RT-qPCR assays for circRNA quantification in HEK293T cells transduced with shDBR1 (Q) or DBR1 (R) cells relative to control. Statistical analysis was performed with unpaired t tests. NS, not significant. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (S) RT-qPCR assays for circRNA in cells from patients relative to controls. Statistical analysis was performed with unpaired t tests. NS, not significant. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Source data are available for this figure: SourceData FS2.
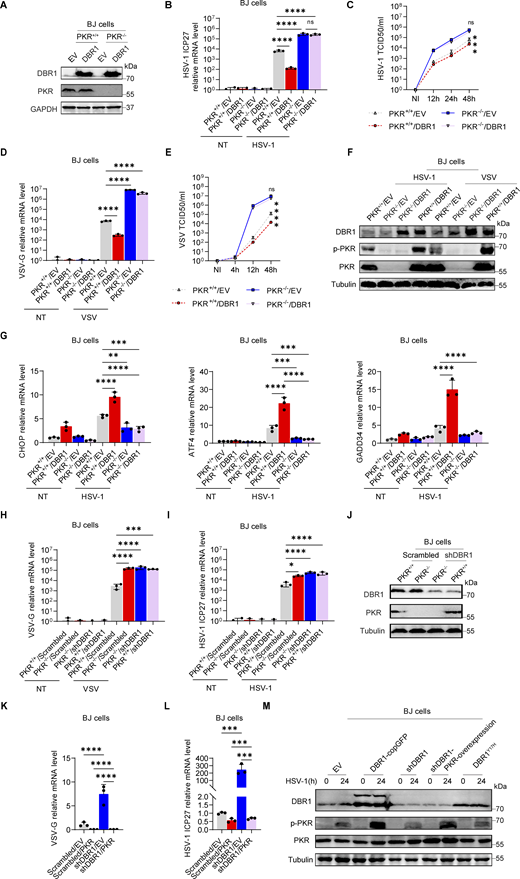
PKR is required for the antiviral activity of DBR1. (A) PKR+/+ and PKR−/− BJ cells were transduced with EV or DBR1, followed by WB for indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (B) PKR+/+ and PKR−/− BJ cells were transduced with EV or DBR1 and infected with HSV-1 at an MOI of 0.5, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ****, P < 0.0001. (C) Similar to B, except the supernatant was collected at different time points for viral titration. Data representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001. (D) Similar to B, except the cells were infected with VSV at an MOI of 0.1 (n = 3 for each group). Data representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (E) Similar to C, except the cells were infected with VSV at an MOI of 0.1. Data are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ****, P < 0.0001. (F) Similar to B and D except that the cells were analyzed by WB for the indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (G) Similar to B except that expression of the indicated ISRs was detected by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (H and I) PKR+/+ and PKR−/− BJ cells were transduced with scrambled shRNA or shDBR1 and infected with VSV MOI = 0.1 (H) or HSV-1-GFP MOI = 0.5 (I), followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. (J) PKR+/+ and PKR−/− BJ cells were transduced with scrambled shRNA or shDBR1, and WB was then performed to detect the indicated proteins. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (K and L) BJ cells transduced with the scrambled control or shDBR1 cells together with EV or PKR overexpression were infected with VSV-GFP (MOI: 0.1) (K), or HSV-1-GFP (MOI: 0.5) (L) for 24 h, followed by quantification of virus replication by RT-qPCR (n = 3 for each group). Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001; ****, P < 0.0001. (M) Similar to L, except that the cells overexpressing DBR1-copGFP and DBR1Y17H were also included, and the indicated proteins were detected by WB. Tubulin was used as a loading control. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F3.
PKR is required for the antiviral activity of DBR1. (A) PKR+/+ and PKR−/− BJ cells were transduced with EV or DBR1, followed by WB for indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (B) PKR+/+ and PKR−/− BJ cells were transduced with EV or DBR1 and infected with HSV-1 at an MOI of 0.5, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ****, P < 0.0001. (C) Similar to B, except the supernatant was collected at different time points for viral titration. Data representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001. (D) Similar to B, except the cells were infected with VSV at an MOI of 0.1 (n = 3 for each group). Data representative of three independent experiments. Graphs depict the mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (E) Similar to C, except the cells were infected with VSV at an MOI of 0.1. Data are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ****, P < 0.0001. (F) Similar to B and D except that the cells were analyzed by WB for the indicated antibodies. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (G) Similar to B except that expression of the indicated ISRs was detected by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (H and I) PKR+/+ and PKR−/− BJ cells were transduced with scrambled shRNA or shDBR1 and infected with VSV MOI = 0.1 (H) or HSV-1-GFP MOI = 0.5 (I), followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. (J) PKR+/+ and PKR−/− BJ cells were transduced with scrambled shRNA or shDBR1, and WB was then performed to detect the indicated proteins. Tubulin was used as a loading control. Data shown are representative of three independent experiments. (K and L) BJ cells transduced with the scrambled control or shDBR1 cells together with EV or PKR overexpression were infected with VSV-GFP (MOI: 0.1) (K), or HSV-1-GFP (MOI: 0.5) (L) for 24 h, followed by quantification of virus replication by RT-qPCR (n = 3 for each group). Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001; ****, P < 0.0001. (M) Similar to L, except that the cells overexpressing DBR1-copGFP and DBR1Y17H were also included, and the indicated proteins were detected by WB. Tubulin was used as a loading control. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F3.
RNA lariat accumulation attenuates PKR activation and enhances viral susceptibility
The only known biological role of DBR1 is the debranching and degradation of RNA lariats (Mohanta and Chakrabarti, 2021). We therefore hypothesized that the lariats accumulating in shDBR1 cells would impair PKR activity. We tested this hypothesis by extracting total RNA from shDBR1 cells, which have high lariat levels, along with DBR1-overexpressing HEK293T cells and HEK293T cells with scrambled shRNA. Following the transfer of shDBR1 cell-derived RNA into HEK293T cells, there was a transient upsurge in lariat abundance, as shown by qPCR (Fig. 4 A). Interestingly, transfection of total RNA from shDBR1 cells for 2 h rendered the recipient cells more susceptible to VSV infection, as shown by diverse quantification methods, whereas RNA from control or DBR1-overexpressing cells did not (Fig. 4, B–D; and Fig. S2, I and J). Transfection with total RNA from shDBR1 cells at 12 h did not enhance viral infection, suggesting that the transfected RNA lacked stability over time (Fig. S2 K). Furthermore, transfection with RNA from shDBR1 cells for 2 h also increased the susceptibility of HEK293T cells to HSV-1 infection (Fig. 4 E). The increase in viral replication by RNA transfection remained after RNase R treatment, suggesting that lariat RNAs rather than linear RNAs were involved in this process (Fig. 4 F). Enhanced viral replication following RNA transfection was dependent on PKR, suggesting that the RNA primarily influenced PKR activity (Fig. 4 G). This is further supported by the observed decrease in PKR phosphorylation following the delivery of RNA from shDBR1 cells (Fig. 4 H). We further investigated the impact of RNA lariats on PKR activation by generating lariat-expressing plasmids carrying GFP as a reporter for successful splicing (Fig. 4 I and Fig. S2 L). Transfection with these plasmids led to lariat accumulation, as quantified by qPCR (Fig. 4 I). DBR1 overexpression in lariat-overexpressing HEK293T cells prevented viral amplification, indicating that this effect was attributable to lariats (Fig. S2, M and N). Moreover, high levels of viral replication were detected in lariat-overexpressing cells (Fig. 4, J–L; and Fig. S2, O and P), which was consistent with low levels of PKR phosphorylation after polyI:C stimulation (Fig. 4 M). Interestingly, lariat overexpression prevented PKR activation even in the presence of PKR overexpression. CircRNA, which has also been shown to inhibit PKR activation when present in abundance, resulted in enhanced viral replication (Li et al., 2017; Liu et al., 2019). Even though similarities exist in structure between lariat RNAs and circRNAs, levels of circRNAs have not been reported to depend on DBR1 and were not affected here by shDBR1, DBR1 overexpression, or the DBR1 I120T genotype (Fig. S2, Q–S). Thus, the RNA lariats that accumulated in DBR1-deficient cells inhibited PKR, whereas circRNAs did not accumulate in DBR1-deficient cells and were not, therefore, involved in PKR inhibition.
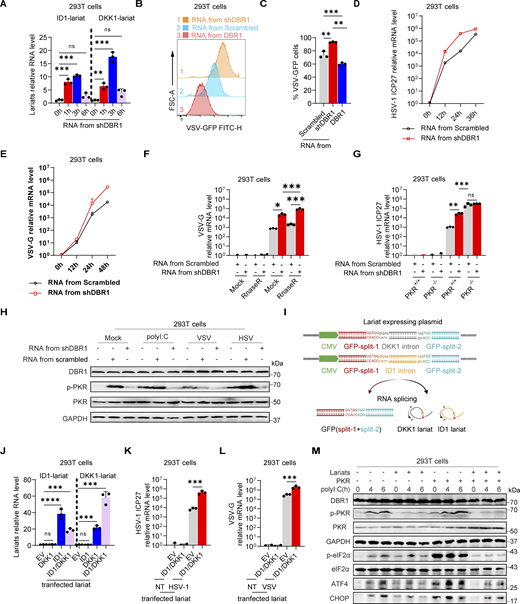
RNA lariats inhibit PKR activation and weaken cellular antiviral activity. (A) HEK293T cells were transfected with 1 µg/ml RNA purified from shDBR1 HEK293T cells, and ID1 and DKK1 lariat RNA levels were determined by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, **, P < 0.01; ***, P < 0.001. (B and C) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with the scrambled shRNA, shDBR1, or DBR1 for 2 h and were then infected with VSV-GFP at a MOI of 0.01 for 24 h. Flow cytometry was then performed to analyze viral GFP levels. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001. (D and E) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with scrambled shRNA or shDBR1 for 2 h and infected with HSV-1 at an MOI of 0.1 (D) or VSV at a MOI of 0.01 (E) for various time points, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. (F) HEK293T cells were transfected with 0.8 µg/ml RnaseR-digested RNA purified from HEK293T cells transduced with scrambled control shRNA or shDBR1, and then infected with VSV at a MOI of 0.01 for 24 h, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; ***, P < 0.001. (G) HEK293T PKR+/+ and PKR−/− cells were transfected with 0.8 µg/ml RnaseR-digested RNA purified from HEK293T cells transduced with scrambled control shRNA or shDBR1, and infected with HSV-1 at a MOI of 0.1 for 24 h, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, **, P < 0.01; ***, P < 0.001. (H) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with scrambled control shRNA, shDBR1, or DBR1, and stimulated with polyI:C or infected with HSV-1 at an MOI of 0.5 or VSV at a MOI of 0.1 for 24 h. Cell lysates were detected by WB with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (I) Diagram of RNA lariat-expressing plasmid design. This graph is created in BioRender. Ru, S. (2024) https://BioRender.com/u76g969. (J) HEK293T cells were transfected with the lariat-expressing plasmids for 24 h, and lariat RNA ID1 and DKK1 levels were measured by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001; ****, P < 0.0001. (K and L) HEK293T cells were transfected with lariat-expressing plasmids for 24 h; they were then infected with HSV-GFP (MOI: 0.5) (K) or VSV-GFP (MOI: 0.1) (L) and viral gene expression was quantified by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001. (M) HEK293T cells were transfected with lariat ID1 or lariat DKK1 plasmids together with or without PKR overexpression. Cells were then stimulated with polyI:C and WB was then performed with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F4.
RNA lariats inhibit PKR activation and weaken cellular antiviral activity. (A) HEK293T cells were transfected with 1 µg/ml RNA purified from shDBR1 HEK293T cells, and ID1 and DKK1 lariat RNA levels were determined by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, **, P < 0.01; ***, P < 0.001. (B and C) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with the scrambled shRNA, shDBR1, or DBR1 for 2 h and were then infected with VSV-GFP at a MOI of 0.01 for 24 h. Flow cytometry was then performed to analyze viral GFP levels. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001. (D and E) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with scrambled shRNA or shDBR1 for 2 h and infected with HSV-1 at an MOI of 0.1 (D) or VSV at a MOI of 0.01 (E) for various time points, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. (F) HEK293T cells were transfected with 0.8 µg/ml RnaseR-digested RNA purified from HEK293T cells transduced with scrambled control shRNA or shDBR1, and then infected with VSV at a MOI of 0.01 for 24 h, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; ***, P < 0.001. (G) HEK293T PKR+/+ and PKR−/− cells were transfected with 0.8 µg/ml RnaseR-digested RNA purified from HEK293T cells transduced with scrambled control shRNA or shDBR1, and infected with HSV-1 at a MOI of 0.1 for 24 h, followed by quantification of virus replication by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, **, P < 0.01; ***, P < 0.001. (H) HEK293T cells were transfected with 1 µg/ml RNA purified from HEK293T cells transduced with scrambled control shRNA, shDBR1, or DBR1, and stimulated with polyI:C or infected with HSV-1 at an MOI of 0.5 or VSV at a MOI of 0.1 for 24 h. Cell lysates were detected by WB with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (I) Diagram of RNA lariat-expressing plasmid design. This graph is created in BioRender. Ru, S. (2024) https://BioRender.com/u76g969. (J) HEK293T cells were transfected with the lariat-expressing plasmids for 24 h, and lariat RNA ID1 and DKK1 levels were measured by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001; ****, P < 0.0001. (K and L) HEK293T cells were transfected with lariat-expressing plasmids for 24 h; they were then infected with HSV-GFP (MOI: 0.5) (K) or VSV-GFP (MOI: 0.1) (L) and viral gene expression was quantified by RT-qPCR. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ***, P < 0.001. (M) HEK293T cells were transfected with lariat ID1 or lariat DKK1 plasmids together with or without PKR overexpression. Cells were then stimulated with polyI:C and WB was then performed with the indicated antibodies. GAPDH was used as a loading control. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F4.
Impaired PKR activation due to the abolition of virus-induced SG assembly in DBR1-deficient cells
Stress granules (SGs) are subcellular structures that assemble in response to various stressors and contribute to cell-intrinsic antiviral immunity in vitro (Guan et al., 2023; Onomoto et al., 2014). Ras-GTPase-activating protein (GAP)-binding protein 1 (G3BP1) triggers RNA-dependent lipid–lipid phase transition (LLPS), which is central to SG formation (Yang et al., 2020; Guillén-Boixet et al., 2020). G3BP1 and G3BP2 are homologous proteins with a common domain structure and redundant functions for SG assembly despite having different patterns of tissue expression (Kennedy et al., 2001). PKR phosphorylates eIF2α, releasing untranslated RNA and the subsequent initiation of SG assembly. Simultaneously, PKR is recruited to SGs by G3BP1 to promote its activation (Reineke et al., 2012; Reineke and Lloyd, 2015). We confirmed the binding of G3BPs to PKR by immunoprecipitation (IP) (Fig. S3 A). HSV-1 or VSV induced G3BP-SG assembly, whereas G3BP and PKR colocalized within the SGs (Fig. S3 B). Given the role of SGs in antiviral responses, at least in vitro, we investigated SG assembly in DBR1-overexpressed or shDBR1 cells. DBR1 overexpression increased the number of G3BP foci detected (Fig. S3, C and D). shDBR1 in BJ cells reduced the number of SG puncta detected following viral treatment (Fig. 5 A). PolyI:C- or virus-triggered G3BP1 condensation was greatly impaired in shDBR1 HEK293T cells (Fig. 5 B). Moreover, shDBR1 also reduced SG formation in HeLa cells following polyI:C stimulation, confirming the dependency of DBR1 for SG assembly in multiple cell lines (Fig. 5 C). Sodium arsenite (SA) induces G3BP-SGs and activates PKR (Patel et al., 2000). Consistently, DBR1Y17H/Y17H cells from a patient with brainstem viral encephalitis displayed fewer G3BP1 aggregation speckles in response to VSV, polyI:C, or SA administration (Fig. 5, D and E). These results suggest that SGs are not correctly assembled in cells with insufficient DBR1 activity.
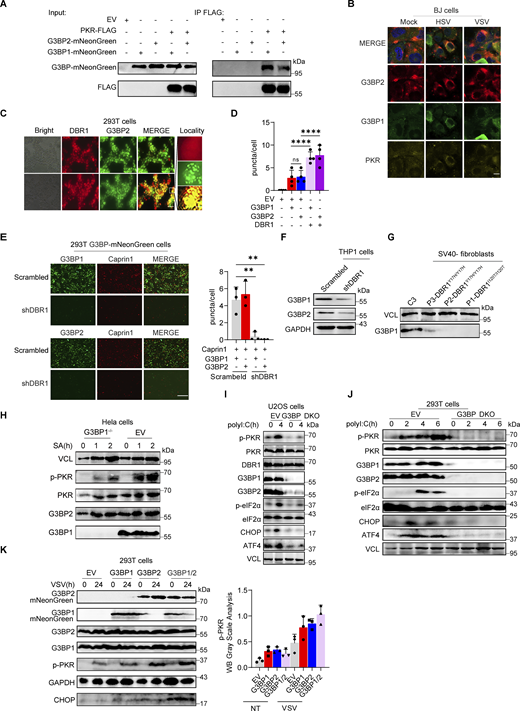
DBR1 licenses G3BP proteins to form SGs. (A) WB with indicated antibodies after immunoprecipitation with an anti-FLAG antibody on lysates of HEK239T cells in an overexpression system. Data shown are representative of three independent experiments. (B) BJ cells were infected with VSV at an MOI of 0.1, or HSV-1 at an MOI of 0.5 for 12 h; immunofluorescence detection was then performed for PKR, G3BP2, and G3BP1-mNeonGreen. Scale bars, 20 µm. Data shown are representative of three independent experiments. (C and D) HEK293T G3BP2-mNeonGreen/DBR1-mScarlet cells were imaged (C) and the intracellular G3BP2- mNeonGreen puncta were quantified (D). Scale bars, 50 µm. Data shown are representative of three independent experiments. Data are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ****, P < 0.0001. (E) Fluorescence images of HEK293T G3BP-mNeonGreen and HEK293T shDBR1/G3BP-mNeonGreen cells. Scale bars, 50 µm. Data shown are representative of three independent experiments. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (F and G) THP-1 cells (D) and cells from patients (E) and controls were analyzed by WB with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (H) G3BP1−/− HeLa cells and control cells were treated with 500 µM SA, and WB was then performed with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (I) G3BP DKO U2OS cells and control cells were stimulated with polyI:C (2 µg/ml) for 4 h, and WB was then performed with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (J) G3BP DKO HEK293T cells and control cells were stimulated with polyI:C (1 µg/ml) for various time points, and WB was then performed with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (K) HEK293T cells with overexpression of the indicated proteins were infected with VSV at an MOI of 0.1, and WB was then performed with the indicated antibodies, followed by the p-PKR/PKR WB gray scale analysis of three independent experiments. GAPDH was used as a loading control. Graphs depict points represent biological replicates. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData FS3.
DBR1 licenses G3BP proteins to form SGs. (A) WB with indicated antibodies after immunoprecipitation with an anti-FLAG antibody on lysates of HEK239T cells in an overexpression system. Data shown are representative of three independent experiments. (B) BJ cells were infected with VSV at an MOI of 0.1, or HSV-1 at an MOI of 0.5 for 12 h; immunofluorescence detection was then performed for PKR, G3BP2, and G3BP1-mNeonGreen. Scale bars, 20 µm. Data shown are representative of three independent experiments. (C and D) HEK293T G3BP2-mNeonGreen/DBR1-mScarlet cells were imaged (C) and the intracellular G3BP2- mNeonGreen puncta were quantified (D). Scale bars, 50 µm. Data shown are representative of three independent experiments. Data are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ****, P < 0.0001. (E) Fluorescence images of HEK293T G3BP-mNeonGreen and HEK293T shDBR1/G3BP-mNeonGreen cells. Scale bars, 50 µm. Data shown are representative of three independent experiments. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01. (F and G) THP-1 cells (D) and cells from patients (E) and controls were analyzed by WB with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (H) G3BP1−/− HeLa cells and control cells were treated with 500 µM SA, and WB was then performed with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (I) G3BP DKO U2OS cells and control cells were stimulated with polyI:C (2 µg/ml) for 4 h, and WB was then performed with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (J) G3BP DKO HEK293T cells and control cells were stimulated with polyI:C (1 µg/ml) for various time points, and WB was then performed with the indicated antibodies. VCL was used as a loading control. Data shown are representative of three independent experiments. (K) HEK293T cells with overexpression of the indicated proteins were infected with VSV at an MOI of 0.1, and WB was then performed with the indicated antibodies, followed by the p-PKR/PKR WB gray scale analysis of three independent experiments. GAPDH was used as a loading control. Graphs depict points represent biological replicates. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData FS3.
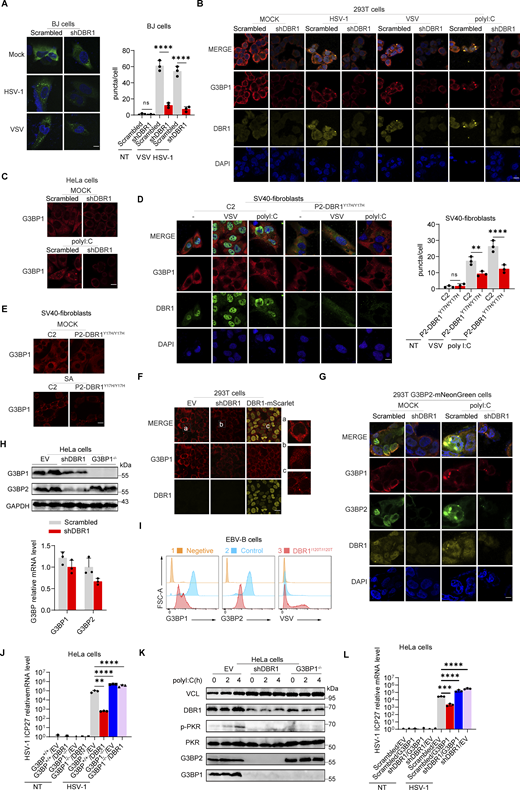
G3BP SG assembly and G3BP protein levels remain low in DBR1-deficient cells. (A) BJ cells transduced with the scrambled control shRNA or with shDBR1 were infected with the indicated viruses, and SG formation was then followed by G3BP1 staining, microscopy imaging, and the SG puncta quantification. Scale bar, 20 µm. Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (B) HEK293T cells transduced with the scrambled shRNA or with shDBR1 were stimulated with polyI:C (1 µg/ml) or infected with VSV (MOI: 0.01) or HSV-1 (MOI: 0.05) and G3BP1 was then detected by fluorescence. Scale bar, 20 µm. Data shown are representative of six independent experiments. (C) HeLa cells transduced with the scrambled control shRNA or with shDBR1 were stimulated with polyI:C (1 µg/ml) and G3BP1 was then detected by fluorescence microscopy. Scale bar, 20 µm. Data shown are representative of three independent experiments. (D and E) Control cells and cells from a DBR1-deficient patient were infected with VSV (MOI = 0.01) for 24 h, stimulated with polyI:C (1 µg/ml) for 4 h (D), or treated with 500 µM sodium arsenite (SA) for 1 h (E). Immunofluorescence staining was then performed for the indicated markers and SGs were quantified. Scale bar, 20 µm. Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, **, P < 0.01; ****, P < 0.0001. (F) HEK293T cells transduced with shDBR1 or DBR1-mscarlet were subjected to immunofluorescence staining for the indicated markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (G) HEK293T cells transduced with the scrambled control shRNA, and shDBR1 cells expressing G3BP2-mNeonGreen were transfected with polyI:C (1 µg/ml) for 4 h, and the markers indicated were then detected by fluorescence microscopy. Scale bar, 20 µm. Data shown are representative of six independent experiments. (H) RT-qPCR and WB detection of G3BP expression in indicated HeLa cells. GAPDH was used as a loading control. Data shown are representative of three independent experiments. Graph points represent biological replicates. (I) FACS analysis of the expression of G3BP (uninfected) and viral GFP after VSV-GFP (MOI: 0.01) infection for 24 h in EBV-B cells from a control or a DBR1-deficient patient. Data shown are representative of three independent experiments. (J and K) G3BP1+/+ and G3BP1−/− HeLa cells transduced with EV or DBR1 were infected with VSV-GFP (MOI: 0.01); viral gene expression was then quantified by RT-qPCR (J). Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ****, P < 0.0001. Proteins were detected by WB detection with indicated antibodies (K). Vinculin (VCL) was used as a loading control. Data shown are representative of three independent experiments. (L) Scrambled shRNA and shDBR1 HeLa cells transduced with EV or G3BP1 were infected with HSV-1. Viral gene expression was then quantified by RT-qPCR. Data shown are representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F5.
G3BP SG assembly and G3BP protein levels remain low in DBR1-deficient cells. (A) BJ cells transduced with the scrambled control shRNA or with shDBR1 were infected with the indicated viruses, and SG formation was then followed by G3BP1 staining, microscopy imaging, and the SG puncta quantification. Scale bar, 20 µm. Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (B) HEK293T cells transduced with the scrambled shRNA or with shDBR1 were stimulated with polyI:C (1 µg/ml) or infected with VSV (MOI: 0.01) or HSV-1 (MOI: 0.05) and G3BP1 was then detected by fluorescence. Scale bar, 20 µm. Data shown are representative of six independent experiments. (C) HeLa cells transduced with the scrambled control shRNA or with shDBR1 were stimulated with polyI:C (1 µg/ml) and G3BP1 was then detected by fluorescence microscopy. Scale bar, 20 µm. Data shown are representative of three independent experiments. (D and E) Control cells and cells from a DBR1-deficient patient were infected with VSV (MOI = 0.01) for 24 h, stimulated with polyI:C (1 µg/ml) for 4 h (D), or treated with 500 µM sodium arsenite (SA) for 1 h (E). Immunofluorescence staining was then performed for the indicated markers and SGs were quantified. Scale bar, 20 µm. Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, **, P < 0.01; ****, P < 0.0001. (F) HEK293T cells transduced with shDBR1 or DBR1-mscarlet were subjected to immunofluorescence staining for the indicated markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (G) HEK293T cells transduced with the scrambled control shRNA, and shDBR1 cells expressing G3BP2-mNeonGreen were transfected with polyI:C (1 µg/ml) for 4 h, and the markers indicated were then detected by fluorescence microscopy. Scale bar, 20 µm. Data shown are representative of six independent experiments. (H) RT-qPCR and WB detection of G3BP expression in indicated HeLa cells. GAPDH was used as a loading control. Data shown are representative of three independent experiments. Graph points represent biological replicates. (I) FACS analysis of the expression of G3BP (uninfected) and viral GFP after VSV-GFP (MOI: 0.01) infection for 24 h in EBV-B cells from a control or a DBR1-deficient patient. Data shown are representative of three independent experiments. (J and K) G3BP1+/+ and G3BP1−/− HeLa cells transduced with EV or DBR1 were infected with VSV-GFP (MOI: 0.01); viral gene expression was then quantified by RT-qPCR (J). Data shown are representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ****, P < 0.0001. Proteins were detected by WB detection with indicated antibodies (K). Vinculin (VCL) was used as a loading control. Data shown are representative of three independent experiments. (L) Scrambled shRNA and shDBR1 HeLa cells transduced with EV or G3BP1 were infected with HSV-1. Viral gene expression was then quantified by RT-qPCR. Data shown are representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData F5.
DBR1 deficiency results in G3BP protein instability
Interestingly, overall G3BP fluorescence levels were lower in shDBR1 HEK293T cells, even without SG induction, whereas DBR1 overexpression led to the formation of basal G3BP puncta (Fig. 5 F). We overexpressed G3BP2-mNeonGreen in control and shDBR1 HEK293T cells, and intracellular polyI:C stimulation of these cells led to the formation of fewer G3BP puncta (Fig. 5 G). The long-term culture of HEK293T cells after shDBR1 lentivirus transduction resulted in an almost complete loss of G3BP expression (Fig. S3 E). We excluded the possibility of alterations to the transcription of G3BPs by performing RT-qPCR and WB. Levels of G3BP protein were decreased, whereas G3BP mRNA levels were unaffected, in all cell lines tested (Fig. 5 H and Fig. S3 F). Patients’ EBV-B cells with high levels of viral susceptibility also had low levels of G3BPs, consistent with the pattern of G3BP1 expression observed on WB with the patients’ fibroblasts (Fig. 5 I and Fig. S3 G). We investigated the role of G3BPs in DBR1-mediated immunity and PKR activation, by transfecting G3BP1−/− cells with DBR1 and infecting these cells with viruses. As in PKR−/− cells, the protective effect of DBR1 decreased in the absence of G3BP1 (Fig. 5 J). DBR1 still gave some protection, perhaps due to the contribution of G3BP2. Furthermore, G3BP1−/− or G3BP1−/− G3BP2−/− (G3BP double-knockout) cells failed to activate PKR when stimulated with polyI:C or SA, highlighting the importance of G3BPs in PKR activation (Fig. 5 K and Fig. S3, H–J). G3BP overexpression, particularly when both G3BP1 and G3BP2 were overexpressed simultaneously, improved PKR activation (Fig. S3 K). Unlike PKR overexpression, the overexpression of G3BP1 did not rescue antiviral activity in shDBR1 cells, owing to the instability of G3BPs in the context of DBR1 deficiency (Fig. 5 L). Overall, DBR1 deficiency rendered the G3BP1 and G3BP2 proteins unstable, thereby limiting G3BP-mediated SG formation and subsequent PKR activation.
RNA lariats interact and colocalize with G3BPs
G3BP deficient cells displayed impaired PKR activation (Fig. 5 K and Fig. S3, H–J), and PKR−/− cells had reduced SG assembly (Fig. S4 A), suggesting PKR is essential for optimal SG assembly and G3BP1 is required for PKR activation. We hypothesized that lariat RNAs might undermine G3BP functions. We transfected HeLa shDBR1 cells with RNA purified from either shDBR1 HEK293T cells or HEK293T cells with scrambled shRNA. This total RNA transfection triggered SG assembly. However, in the presence of higher concentrations of lariats, the number of SGs decreased (Fig. 6 A; and Fig. S4, B and C). RNA lariats, thus, impaired the correct aggregation of G3BPs. G3BPs are known to bind RNAs in a sequence-independent manner (Yang et al., 2020). To elucidate the mechanism by which RNA lariats regulate G3BPs, we examined whether G3BPs interacted with lariats. The MS2 sequence was initially inserted into lariat-expressing plasmids (ID1-8xMS2, DKK1-8xMS2), leading to the generation of lariats with MS2 stem-loops that bound to MS2 coat protein (MCP) with high affinity (Bertrand et al., 1998). Lariats were localized with an MCP-mScarlet fusion protein. The co-expression of ID1/DKK1-8xMS2 and the MCP-mScarlet plasmids led to the formation of puncta, suggesting that MCP was recruited to the MS2 lariats (Fig. S4 D). Most of the overexpressed lariats were in the cytosol and the number of foci was higher in shDBR1 HEK293T cells (Fig. 6 B). MCP immunoprecipitation was used to pull down MS2-containing lariats for detection by WB (Fig. 6 C and Fig. S4 E). Both G3BPs and PKR were also pulled down, suggesting that they interacted with lariats. Immunofluorescence imaging revealed some colocalization of G3BP, PKR, and RNA lariats, especially during prolonged culture (Fig. 6 D and Fig. S4 F). RNA lariats but not circRNA induced G3BP puncta (Fig. S4 G). In PKR-deficient cells, the lariat pull-down still successfully isolated G3BP2, and the lariat colocalized with G3BPs, suggesting G3BPs-lariat interaction didn’t require PKR (Fig. S4, H and I). By contrast, no large foci developed in G3BP DKO cells, demonstrating that the colocalization of PKR with lariats involved indirect interaction with G3BPs (Fig. 6 E).
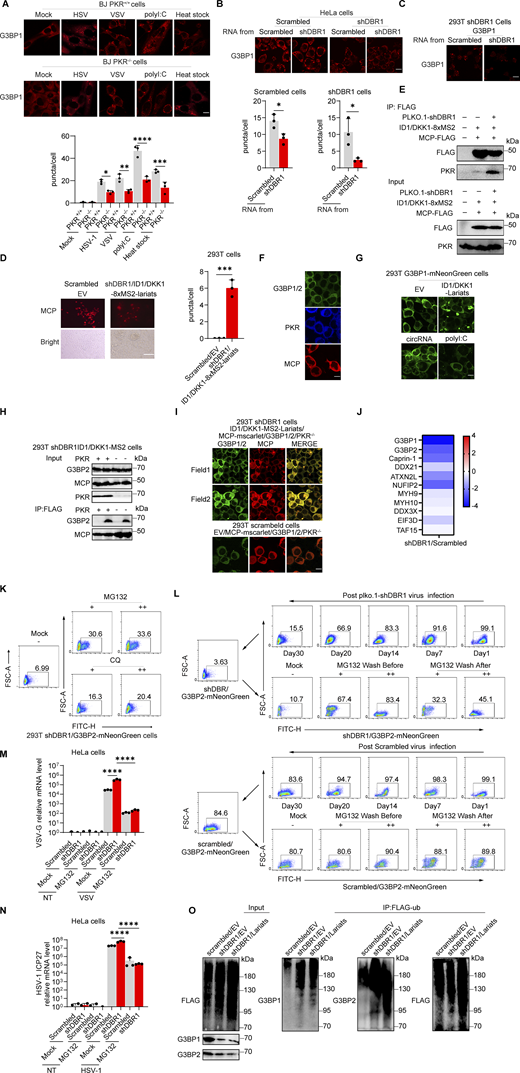
The stability of the G3BP protein is compromised by RNA lariats. (A) PKR+/+ and PKR−/− BJ cells were infected with VSV at an MOI of 0.01, or HSV-1-GFP at an MOI of 0.5 for 24 h, stimulated with polyI:C (2 µg/ml) for 5 h or heat stressed at 43°C for 40 min; immunofluorescence analysis was then performed for G3BP1. Scale bars, 20 µm. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (B) WT and shDBR1-transduced cells were transfected with lariat RNA for 8 h; immunofluorescence analysis was then performed for G3BP1-SGs. Scale bars, 20 µm. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. *, P < 0.05. (C) Lariat RNA was used to transfect shDBR1 HEK293T cells, and immunofluorescence analysis was then performed for G3BP1. Scale bars, 20 µm. Data representative of three independent experiments. (D) Fluorescence analysis of MCP (red) in HEK293T cells transfected with ID1/DKK1-8xMS2 -lariats (left). The percentage of cells with MCP and ID1/DKK1-8xMS2 -lariats foci was quantified (right), and at least 100 cells from each group were analyzed (n = 3 for each group). Scale bars, 50 µm. Data representative of six independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. ***, P < 0.001. (E) WB with the indicated antibodies following immunoprecipitation with an anti-FLAG (MCP) antibody on lysates of HEK293T cells in an overexpression system. Data representative of three independent experiments. (F) HEK293T cells were transfected with G3BP1/2-mNeonGreen, PKR-BFP or MCP-mScarlet separately, followed by immunofluorescence imaging. Scale bars, 20 µm. Data representative of three at least six independent experiments. (G) HEK293T G3BP1-mNenonGreen cells were transfected with ID1/DKK1-lariats, circRNA (circFNDC3B and circCAMSAP1) or polyI:C stimulation, and the formation of G3BP1 puncta was detected by immunofluorescence. Scale bars, 20 µm. Data representative of three independent experiments. (H) WB with the indicated antibodies following IP with an anti-FLAG (MCP) antibody for lysates of HEK293T PKR+/+ and PKR−/− cells in a stable overexpression system. Data representative of three independent experiments. (I) HEK293T PKR−/− cells with ID1/DKK1-MS2-Lariats/MCP-mScarlet/G3BP- mNeonGreen/shDBR1 transduction were imaged for the indicated fluorescence markers. Scale bars, 20 µm. Data representative of at least six independent experiments. (J) SG component analysis (log fold change) of shDBR1 versus scrambled cells, following VSV infection G3BP1 IP and mass spectrometry. (K) HEK293T shDBR1/G3BP2-mNeonGreen cells were treated with the proteasome inhibitor MG132 and the autophagy inhibitor CQ for 18 h, and FACS was performed to assess G3BP2-mNeonGreen fluorescence recovery. Data shown are representative of three independent experiments. (L) FACS was performed to assess G3BP2-mNeonGreen fluorescence recovery before and after the treatment of HEK293T G3BP2-mNeonGreen, shDBR1/G3BP2-mNeonGreen cells with various concentrations of the proteasome inhibitor MG132. Data shown are representative of three independent experiments. (M and N) HeLa scrambled control or shDBR1 cells were treated with MG132 for 4 h, and then were infected with VSV at an MOI of 0.1 (M) or HSV-1 at an MOI of 0.5 (N) for 24 h, and viral replication was then detected by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (O) WB with the indicated antibodies following immunoprecipitation with an anti-FLAG-(Flag-UB) antibody for the lysates of HEK239T cells in an overexpression system. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData FS4.
The stability of the G3BP protein is compromised by RNA lariats. (A) PKR+/+ and PKR−/− BJ cells were infected with VSV at an MOI of 0.01, or HSV-1-GFP at an MOI of 0.5 for 24 h, stimulated with polyI:C (2 µg/ml) for 5 h or heat stressed at 43°C for 40 min; immunofluorescence analysis was then performed for G3BP1. Scale bars, 20 µm. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (B) WT and shDBR1-transduced cells were transfected with lariat RNA for 8 h; immunofluorescence analysis was then performed for G3BP1-SGs. Scale bars, 20 µm. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. *, P < 0.05. (C) Lariat RNA was used to transfect shDBR1 HEK293T cells, and immunofluorescence analysis was then performed for G3BP1. Scale bars, 20 µm. Data representative of three independent experiments. (D) Fluorescence analysis of MCP (red) in HEK293T cells transfected with ID1/DKK1-8xMS2 -lariats (left). The percentage of cells with MCP and ID1/DKK1-8xMS2 -lariats foci was quantified (right), and at least 100 cells from each group were analyzed (n = 3 for each group). Scale bars, 50 µm. Data representative of six independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with unpaired t tests. ***, P < 0.001. (E) WB with the indicated antibodies following immunoprecipitation with an anti-FLAG (MCP) antibody on lysates of HEK293T cells in an overexpression system. Data representative of three independent experiments. (F) HEK293T cells were transfected with G3BP1/2-mNeonGreen, PKR-BFP or MCP-mScarlet separately, followed by immunofluorescence imaging. Scale bars, 20 µm. Data representative of three at least six independent experiments. (G) HEK293T G3BP1-mNenonGreen cells were transfected with ID1/DKK1-lariats, circRNA (circFNDC3B and circCAMSAP1) or polyI:C stimulation, and the formation of G3BP1 puncta was detected by immunofluorescence. Scale bars, 20 µm. Data representative of three independent experiments. (H) WB with the indicated antibodies following IP with an anti-FLAG (MCP) antibody for lysates of HEK293T PKR+/+ and PKR−/− cells in a stable overexpression system. Data representative of three independent experiments. (I) HEK293T PKR−/− cells with ID1/DKK1-MS2-Lariats/MCP-mScarlet/G3BP- mNeonGreen/shDBR1 transduction were imaged for the indicated fluorescence markers. Scale bars, 20 µm. Data representative of at least six independent experiments. (J) SG component analysis (log fold change) of shDBR1 versus scrambled cells, following VSV infection G3BP1 IP and mass spectrometry. (K) HEK293T shDBR1/G3BP2-mNeonGreen cells were treated with the proteasome inhibitor MG132 and the autophagy inhibitor CQ for 18 h, and FACS was performed to assess G3BP2-mNeonGreen fluorescence recovery. Data shown are representative of three independent experiments. (L) FACS was performed to assess G3BP2-mNeonGreen fluorescence recovery before and after the treatment of HEK293T G3BP2-mNeonGreen, shDBR1/G3BP2-mNeonGreen cells with various concentrations of the proteasome inhibitor MG132. Data shown are representative of three independent experiments. (M and N) HeLa scrambled control or shDBR1 cells were treated with MG132 for 4 h, and then were infected with VSV at an MOI of 0.1 (M) or HSV-1 at an MOI of 0.5 (N) for 24 h, and viral replication was then detected by RT-qPCR. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (O) WB with the indicated antibodies following immunoprecipitation with an anti-FLAG-(Flag-UB) antibody for the lysates of HEK239T cells in an overexpression system. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData FS4.
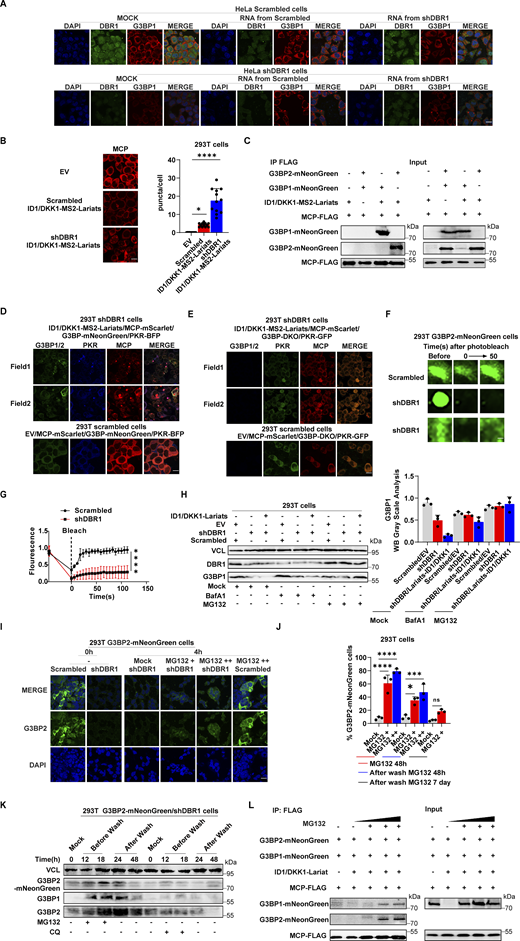
RNA lariats interact with G3BP protein, impairing SG function and G3BP protein stability. (A) Extracted RNA from HEK293T cells transduced with shDBR1/ID1/DKK1-lariats or control cells was used to transfect HeLa cells transduced with the scrambled shRNA or shDBR1 for 24 h; fluorescence imaging was then performed for the indicated markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (B) Lariat imaging with MCP-mScarlet and puncta quantification in HEK293T cells (n = 12 for each group). Scale bar, 20 µm. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; ****, P < 0.0001. (C) WB with the indicated antibodies following immunoprecipitation with the anti-FLAG (MCP) antibody on lysates from HEK239T cells in an overexpression system. Data shown are representative of three independent experiments. (D) HEK293T cells with ID1/DKK1-MS2-Lariats/MCP-mScarlet/G3BP-mNeonGreen/PKR-BFP/shDBR1 overexpression were imaged with the indicated fluorescence markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (E) HEK293T G3BP DKO cells with ID1/DKK1-MS2-Lariats/MCP-mScarlet/PKR-GFP/shDBR1 overexpression were imaged with indicated fluorescence markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (F and G) FRAP assay to detect the fluorescence recovery of G3BP2-mNeonGreen 3 days after shRNA transduction. Scale bars, 5 µm (F). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (H) HEK293T cells transduced with shDBR1 or shDBR1/ID1-DKK1-lariat were treated with MG132 or BafA1, and WB was then performed to detect the indicated proteins, followed by relative G3BP1 quantification of three independent experiments. VCL was used as a loading control. Data shown are representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (I) MG132 was incubated for 4 h with HEK293T G3BP2-mNeonGreen cells transduced with the scrambled shRNA or shDBR1. Fluorescence imaging was then performed for the indicated markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (J) FACS detection of G3BP2-mNeonGreen protein before and after treatment with MG132. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. (K) MG132 or CQ was incubated with HEK293T G3BP2-mNeonGreen cells transduced with the scrambled shRNA or shDBR1, and HEK293T G3BP2-mNeonGreen cells. VCL (H and K) was used as a loading control. Data shown are representative of three independent experiments. (L) WB with the indicated antibodies following MG132 treatment and immunoprecipitation with an anti-FLAG (MCP) antibody on lysates of HEK293T cells in a stable overexpression system. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F6.
RNA lariats interact with G3BP protein, impairing SG function and G3BP protein stability. (A) Extracted RNA from HEK293T cells transduced with shDBR1/ID1/DKK1-lariats or control cells was used to transfect HeLa cells transduced with the scrambled shRNA or shDBR1 for 24 h; fluorescence imaging was then performed for the indicated markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (B) Lariat imaging with MCP-mScarlet and puncta quantification in HEK293T cells (n = 12 for each group). Scale bar, 20 µm. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; ****, P < 0.0001. (C) WB with the indicated antibodies following immunoprecipitation with the anti-FLAG (MCP) antibody on lysates from HEK239T cells in an overexpression system. Data shown are representative of three independent experiments. (D) HEK293T cells with ID1/DKK1-MS2-Lariats/MCP-mScarlet/G3BP-mNeonGreen/PKR-BFP/shDBR1 overexpression were imaged with the indicated fluorescence markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (E) HEK293T G3BP DKO cells with ID1/DKK1-MS2-Lariats/MCP-mScarlet/PKR-GFP/shDBR1 overexpression were imaged with indicated fluorescence markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (F and G) FRAP assay to detect the fluorescence recovery of G3BP2-mNeonGreen 3 days after shRNA transduction. Scale bars, 5 µm (F). Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (H) HEK293T cells transduced with shDBR1 or shDBR1/ID1-DKK1-lariat were treated with MG132 or BafA1, and WB was then performed to detect the indicated proteins, followed by relative G3BP1 quantification of three independent experiments. VCL was used as a loading control. Data shown are representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (I) MG132 was incubated for 4 h with HEK293T G3BP2-mNeonGreen cells transduced with the scrambled shRNA or shDBR1. Fluorescence imaging was then performed for the indicated markers. Scale bar, 20 µm. Data shown are representative of three independent experiments. (J) FACS detection of G3BP2-mNeonGreen protein before and after treatment with MG132. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. (K) MG132 or CQ was incubated with HEK293T G3BP2-mNeonGreen cells transduced with the scrambled shRNA or shDBR1, and HEK293T G3BP2-mNeonGreen cells. VCL (H and K) was used as a loading control. Data shown are representative of three independent experiments. (L) WB with the indicated antibodies following MG132 treatment and immunoprecipitation with an anti-FLAG (MCP) antibody on lysates of HEK293T cells in a stable overexpression system. Data shown are representative of three independent experiments. Source data are available for this figure: SourceData F6.
RNA lariats prevent proper SGs assembly, resulting in G3BP degradation
Relative to normal SG assembly in WT HEK293T cells, larger puncta of G3BPs, PKR, and RNA lariats were formed in DBR1-deficient cells. The aberrant aggregates eventually disappeared, indicating that they had been cleared. G3BP2 was progressively depleted during prolonged cell culture. In shDBR1 HEK293T cells, the loss of G3BPs became increasingly apparent during culture over 3 days, and this loss was associated with the emergence of a single punctum, which lost liquidity, as shown by fluorescence recovery after photobleaching (FRAP) (Fig. 6, F and G). To investigate the potential interference of RNA lariats with SG components, we conducted a mass spectrometry analysis subsequent to the immunoprecipitation of G3BP1 following VSV infection. This analysis revealed a decrease in the relative abundance of 36 core SG proteins in samples overexpressing lariats, indicating that RNA lariats disrupted the composition of SGs (Fig. S4 J and Table S1). Given the slow decline in G3BP protein levels, we hypothesized that the degradation of G3BPs was driven by lariats. G3BP protein levels were restored when protein degradation pathways were blocked with the proteasome inhibitor MG132 and the lysosomal degradation inhibitor bafilomycin A1 (BafA1) (Fig. 6 H). G3BPs were more abundant in cells treated with MG132 when compared with those treated with chloroquine (CQ), another lysosomal degradation inhibitor, suggesting that these proteins were degraded primarily by the proteasome (Fig. S4 K). Treatment with MG132 for 4 h led to a partial dose-dependent recovery of G3BP2 protein levels, as demonstrated by fluorescence imaging (Fig. 6 I). However, the withdrawal of MG132 after treatment resulted in a progressive decrease in the amounts of G3BP1 and G3BP2 (Fig. 6, J and K; and Fig. S4 L). MG132 treatment restored not only G3BP protein levels but also the antiviral phenotype as well as the interaction between G3BP proteins and RNA lariats when both were present (Fig. 6 L; and Fig. S4, M and N). Consistent with increased proteasomal degradation of G3BPs, we observed enhanced ubiquitin of G3BP proteins in lariat-overexpressing cells (Fig. S4 O). These data suggest that G3BPs are targeted by the ubiquitin-proteasome pathway upon RNA lariat binding, providing a molecular mechanism by which DBR1 deficiency impairs G3BP-mediated SG formation, together with subsequent PKR activation and antiviral immunity in human cells.
Decreased G3BP expression in Dbr1Y17H/Y17H MEF cells and brain tissues
The L13, Y17, and I120 residues of DBR1 are highly conserved in vertebrates, with the notable exception of the absence of I120 in cattle. Missense substitutions of these residues result in profound DBR1 deficiency and brainstem viral encephalitis (Fig. 7 A and Fig. S5 A). No live Dbr1−/− mice were generated when Dbr1+/− mice were crossed, suggesting that DBR1 knockout (KO) is embryo-lethal in mice (Zheng et al., 2015). We therefore further investigated the crucial role of DBR1 in immunity to viruses in vivo and the PKR-dependent molecular mechanism of disease by generating Dbr1Y17H/Y17H mutant mice. These mice were viable but weighed less than their WT littermates. They had none of the developmental problems reported in humans with other mutations in DBR1 (Fig. S5, B and C). We analyzed RNA lariat accumulation in the brainstem of mice using RNA sequencing (RNA-seq). Our results indicated that Dbr1Y17H/Y17H mice exhibited significantly higher levels of lariats, which was subsequently verified by qPCR (Fig. S5, D–F). We determined whether mice with Dbr1 mutations presented decreases in G3BP levels similar to those observed in human cell lines by performing WB to assess G3BP protein levels. As expected, Dbr1Y17H/Y17H mouse embryonic fibroblast (MEF) cells and bone marrow–derived macrophages (BMDMs) contained only very small amounts of G3BP1 and G3BP2 (Fig. 7, B and C). Fluorescence staining for G3BP1 also yielded a weaker signal in the presence of the DBR1 Y17H variant (Fig. 7 D). Protein degradation blockers increased the G3BP2 protein levels of these MEF cells (Fig. S5 G). Dbr1Y17H/Y17H MEF cells also exhibited a reduced number and size of puncta in response to various stimuli (Fig. 7 E). These findings suggest that the abundant RNA lariats may cause a decrease in G3BP levels in the cells of Dbr1Y17H/Y17H mice, as in human cells. We investigated possible effects on G3BPs at the tissue level by performing immunofluorescence staining on mouse brain sections. G3BP levels were very low in brain samples from Dbr1Y17H/Y17H mice (Fig. 7 F). Overall, these findings suggest that DBR1 deficiency impairs G3BP expression via the high abundance of RNA lariats in mouse cells, as it does in human cells.
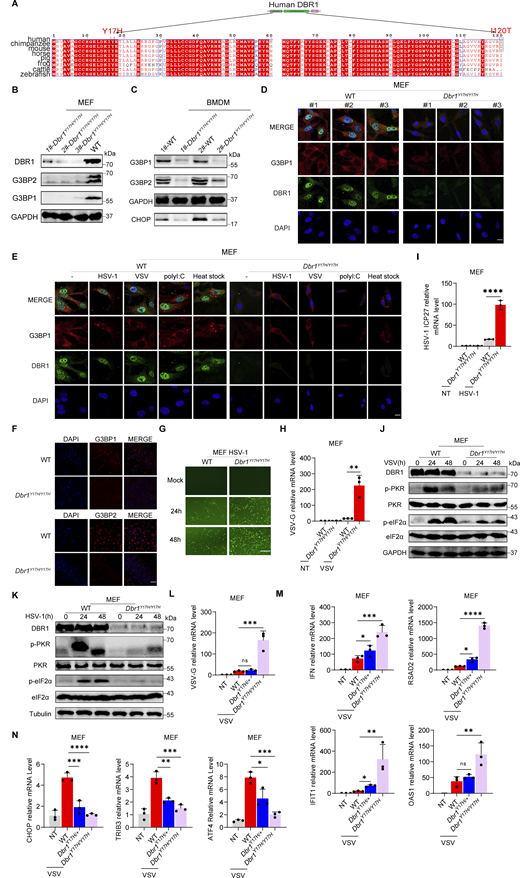
DBR1 is essential for G3BP protein stability and antiviral effect in vitro. (A) Alignment of the N-terminal sequences of the DBR1 proteins of the indicated species. (B) WB detection with the indicated antibodies in MEFs. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (C) WB detection with the indicated antibodies in BMDMs. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (D) Fluorescence detection of the indicated markers in MEF cells. Scale bars, 20 µm. Data shown are representative of three independent experiments. (E) WT and Dbr1Y17H/Y17H MEFs were infected with VSV (MOI: 0.1) or HSV-1 (MOI: 0.5) for 24 h, stimulated with polyI:C (2 µg/ml) for 5 h or incubated at 43°C for 40 min; fluorescence imaging was then performed for the indicated markers. Scale bars, 20 µm. Data shown are representative of three independent experiments. (F) Detection of fluorescent immunostaining for the indicated markers in WT and Dbr1Y17H/Y17H mouse brain sections. Scale bars, 50 µm. Data shown are representative of three independent experiments. (G) Fluorescence imaging of WT and Dbr1Y17H/Y17H MEFs infected with HSV-1-GFP (MOI: 0.5) for various time points. Scale bars, 10 0 µm. Data shown are representative of five independent experiments. (H and I) WT and Dbr1Y17H/Y17H MEFs were infected with VSV at a MOI of 0.1 (H) or HSV-1 at a MOI of 0.5 (I) for 36 h. RT-qPCR was then performed to detect the expression of the indicated viral genes. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (J and K) Similar to H and I, except that WB was performed with the indicated antibodies after infection with VSV (J) or HSV-1 (K). Tubulin (K) or GAPDH (J) was used as a loading control. Data shown are representative of three independent experiments. (L–N) WT, Dbr1Y17H/+, and Dbr1Y17H/Y17H MEFs were infected with VSV (MOI: 0.1) for 24 h, and RT-qPCR was then performed to detect expression of the indicated genes. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Source data are available for this figure: SourceData F7.
DBR1 is essential for G3BP protein stability and antiviral effect in vitro. (A) Alignment of the N-terminal sequences of the DBR1 proteins of the indicated species. (B) WB detection with the indicated antibodies in MEFs. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (C) WB detection with the indicated antibodies in BMDMs. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (D) Fluorescence detection of the indicated markers in MEF cells. Scale bars, 20 µm. Data shown are representative of three independent experiments. (E) WT and Dbr1Y17H/Y17H MEFs were infected with VSV (MOI: 0.1) or HSV-1 (MOI: 0.5) for 24 h, stimulated with polyI:C (2 µg/ml) for 5 h or incubated at 43°C for 40 min; fluorescence imaging was then performed for the indicated markers. Scale bars, 20 µm. Data shown are representative of three independent experiments. (F) Detection of fluorescent immunostaining for the indicated markers in WT and Dbr1Y17H/Y17H mouse brain sections. Scale bars, 50 µm. Data shown are representative of three independent experiments. (G) Fluorescence imaging of WT and Dbr1Y17H/Y17H MEFs infected with HSV-1-GFP (MOI: 0.5) for various time points. Scale bars, 10 0 µm. Data shown are representative of five independent experiments. (H and I) WT and Dbr1Y17H/Y17H MEFs were infected with VSV at a MOI of 0.1 (H) or HSV-1 at a MOI of 0.5 (I) for 36 h. RT-qPCR was then performed to detect the expression of the indicated viral genes. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (J and K) Similar to H and I, except that WB was performed with the indicated antibodies after infection with VSV (J) or HSV-1 (K). Tubulin (K) or GAPDH (J) was used as a loading control. Data shown are representative of three independent experiments. (L–N) WT, Dbr1Y17H/+, and Dbr1Y17H/Y17H MEFs were infected with VSV (MOI: 0.1) for 24 h, and RT-qPCR was then performed to detect expression of the indicated genes. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. Source data are available for this figure: SourceData F7.
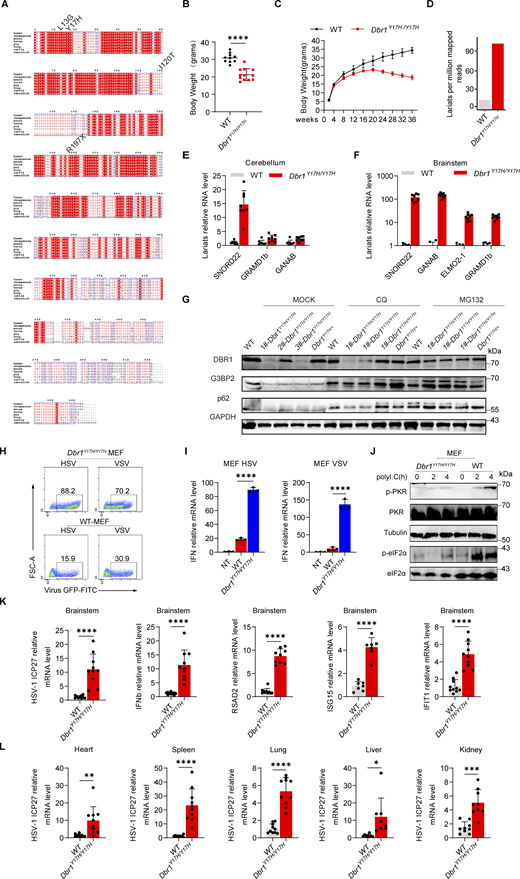
DBR1 deficiency increases susceptibility to viral infection in vitro and in vivo. (A) Alignment of the sequences of segments of the DBR1 protein between the species indicated. (B) Body weights of WT (n = 10) and Dbr1Y17H/Y17H (n = 12) mice at 4–5 mo. Statistical analysis was performed with t tests. ****, P < 0.0001. Graphs depict the mean with SD and points represent biological replicates. (C) Comparison of body weight changes in WT (n = 6) and Dbr1Y17H/Y17H (n = 6) mice from birth. Graphs depict points that represent biological replicates. (D) Lariat analysis of WT and Dbr1Y17H/Y17H MEF following RNAseq. Data shown are representative of two independent experiments. (E and F) RT-qPCR assessment of WT (n = 8) and Dbr1Y17H/Y17H (n = 8) RNA lariats expression in different mouse tissues. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (G) After the treatment of WT and Dbr1Y17H/Y17H MEFs with MG132 and CQ for 6 h, G3BP2 was detected by WB. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (H and I) WT and Dbr1Y17H/Y17H MEFs were infected with viruses (VSV-GFP at an MOI of 0.1, or HSV-1-GFP at an MOI of 0.5) for 24 h; FACS analysis was then performed for the analysis of viral GFP levels (H) and RT-qPCR was used to detect expression of the indicated genes (I). Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (J) WB analysis after the stimulation of MEFs with polyI:C (2 µg/ml). Tubulin was used as a loading control. Data shown are representative of three independent experiments. (K) WT and Dbr1Y17H/Y17H mice were inoculated with HSV-1 (3.6 × 105 PFU/g body weight) by injection into the tail vein, and RT-qPCR was performed to assess viral replication and ISG expression in the brainstem. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Graph points represent mouse numbers. Statistical analysis was performed with t tests. ****, P < 0.0001. (L) RT-qPCR assessment of viral gene expression in various mouse tissues. Graph points represent mouse numbers. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData FS5.
DBR1 deficiency increases susceptibility to viral infection in vitro and in vivo. (A) Alignment of the sequences of segments of the DBR1 protein between the species indicated. (B) Body weights of WT (n = 10) and Dbr1Y17H/Y17H (n = 12) mice at 4–5 mo. Statistical analysis was performed with t tests. ****, P < 0.0001. Graphs depict the mean with SD and points represent biological replicates. (C) Comparison of body weight changes in WT (n = 6) and Dbr1Y17H/Y17H (n = 6) mice from birth. Graphs depict points that represent biological replicates. (D) Lariat analysis of WT and Dbr1Y17H/Y17H MEF following RNAseq. Data shown are representative of two independent experiments. (E and F) RT-qPCR assessment of WT (n = 8) and Dbr1Y17H/Y17H (n = 8) RNA lariats expression in different mouse tissues. Data representative of three independent experiments. Graphs depict mean with SD, and points represent biological replicates. (G) After the treatment of WT and Dbr1Y17H/Y17H MEFs with MG132 and CQ for 6 h, G3BP2 was detected by WB. GAPDH was used as a loading control. Data shown are representative of three independent experiments. (H and I) WT and Dbr1Y17H/Y17H MEFs were infected with viruses (VSV-GFP at an MOI of 0.1, or HSV-1-GFP at an MOI of 0.5) for 24 h; FACS analysis was then performed for the analysis of viral GFP levels (H) and RT-qPCR was used to detect expression of the indicated genes (I). Data representative of three independent experiments. Graphs depict mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. ****, P < 0.0001. (J) WB analysis after the stimulation of MEFs with polyI:C (2 µg/ml). Tubulin was used as a loading control. Data shown are representative of three independent experiments. (K) WT and Dbr1Y17H/Y17H mice were inoculated with HSV-1 (3.6 × 105 PFU/g body weight) by injection into the tail vein, and RT-qPCR was performed to assess viral replication and ISG expression in the brainstem. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Graph points represent mouse numbers. Statistical analysis was performed with t tests. ****, P < 0.0001. (L) RT-qPCR assessment of viral gene expression in various mouse tissues. Graph points represent mouse numbers. Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with t tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Source data are available for this figure: SourceData FS5.
DBR1 governs intrinsic PKR-mediated antiviral immunity in MEF cells
We isolated MEFs from Dbr1Y17H/Y17H embryos and their WT siblings. Dbr1Y17H/Y17H MEF cells were highly susceptible to viral infection, with high viral titers on microscopy, flow cytometry, and qPCR (Fig. 7, G–I and Fig. S5 H). WB for p-PKR and p-eIF2α revealed a consistent decrease in stress responses, along with low DBR1 levels in Dbr1Y17H/Y17H MEF cells upon viral infection (Fig. 7, J and K). Nevertheless, Dbr1Y17H/Y17H cells displayed significantly stronger IFNB mRNA induction, suggesting that type I IFN induction was normal despite DBR1 deficiency (Fig. S5 I). Following infection of the cells with VSV, we evaluated the levels of viral RNA, ISR (CHOP, TRIB3, ATF4), and ISG expression by RT-qPCR (Fig. 7, L–N). Dbr1Y17H/+ MEF cells restricted viral replication to a similar degree as WT cells, whereas Dbr1Y17H/Y17H MEF cells had higher levels of viral and ISG transcripts but lower levels of ISR transcripts. Following intracellular polyI:C stimulation, Dbr1Y17H/Y17H MEF cells also displayed low levels of PKR and downstream eIF2α activation (Fig. S5 J). These findings further highlight the crucial role of DBR1 in stress responses and cell-intrinsic immunity to viruses in mouse cells in vitro.
DBR1 is indispensable for antiviral immunity in vivo in mice
Following intravenous exposure to HSV-1 infection, survival is significantly lower in Dbr1Y17H/Y17H mice, highlighting the crucial role of DBR1 in vivo (Fig. 8 A). Viral titrations on forebrains and hindbrains revealed enhanced viral replication in Dbr1Y17H/Y17H mice, particularly in the brainstem, a feature reminiscent of observations in human patients with DBR1 mutations (Fig. 8 B). ISR (ATF4, CHOP) induction in the forebrain and brainstem was weaker in Dbr1Y17H/Y17H mice (Fig. 8 C). We further analyzed p-PKR levels in virus-infected cerebellum and brainstem by WB. The brain samples from Dbr1Y17H/Y17H mice displayed a complete absence of PKR activation despite their higher viral loads (Fig. 8 D). Levels of type I IFN and ISG production were high in Dbr1Y17H/Y17H mouse brainstems (Fig. S5 K). We also noted a more widespread distribution of viruses in other tissues of Dbr1Y17H/Y17H mice, as intravenous administration led to systemic infection (Fig. S5 L). Dbr1Y17H/Y17H mice also displayed greater morbidity following VSV inoculation (Fig. 8 E). Viral restriction and ISR induction also depended on DBR1 in vivo (Fig. 8, F and G). Collectively, our results suggest that DBR1 is also vital for protective immunity against viruses in mice in vivo, particularly in the brain.
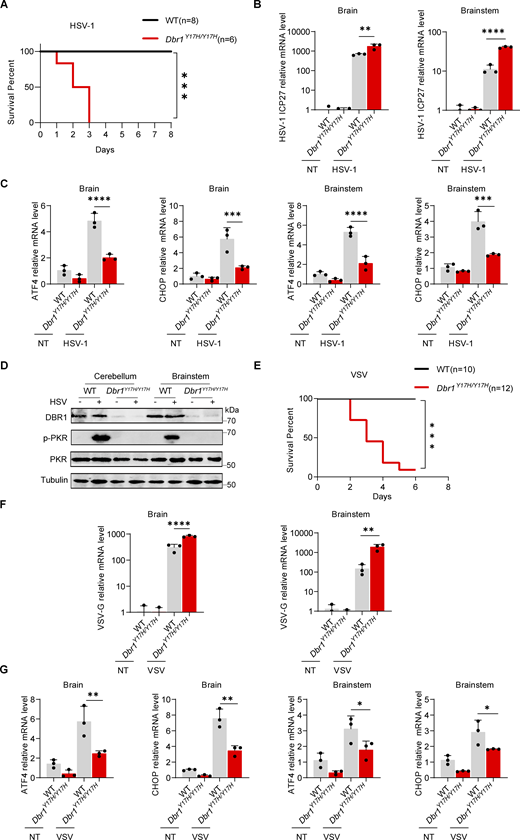
DBR1 is crucial for PKR activation and antiviral responses in vivo. (A) Survival curve of WT (n = 8). and Dbr1Y17H/Y17H (n = 6) mice after intravenous inoculation with HSV-1 at a dose of 3.6 × 105 PFU/g body weight. Data representative of three independent experiments. Statistical analysis for mouse survival was performed with Kaplan–Meier tests. ***, P < 0.001. (B–D) WT and Dbr1Y17H/Y17H mice were sacrificed 3.5 days after intravenous inoculation with HSV-1 (3.6 × 105 PFU/g body weight). Brain tissues were collected, and RT-qPCR was performed to analyze the expression of viral genes (B) and the indicated ISR genes (C). Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. WB with the indicated antibodies was used to assess protein levels (D). Data shown are representative of three independent experiments. (E) Survival curves of WT (n = 10) and Dbr1Y17H/Y17H (n = 12) mice after intravenous inoculation with VSV (106 PFU/g body weight). Data representative of three independent experiments. Statistical analysis for mouse survival was performed with Kaplan–Meier tests. ***, P < 0.001. (F and G) WT and Dbr1Y17H/Y17H mice were sacrificed 3.5 days after intravenous inoculation with VSV (106 PFU/g body weight). Brain tissues were collected, and RT-qPCR was performed to analyze the expression of viral genes (F) and the indicated ISR genes (G). Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Source data are available for this figure: SourceData F8.
DBR1 is crucial for PKR activation and antiviral responses in vivo. (A) Survival curve of WT (n = 8). and Dbr1Y17H/Y17H (n = 6) mice after intravenous inoculation with HSV-1 at a dose of 3.6 × 105 PFU/g body weight. Data representative of three independent experiments. Statistical analysis for mouse survival was performed with Kaplan–Meier tests. ***, P < 0.001. (B–D) WT and Dbr1Y17H/Y17H mice were sacrificed 3.5 days after intravenous inoculation with HSV-1 (3.6 × 105 PFU/g body weight). Brain tissues were collected, and RT-qPCR was performed to analyze the expression of viral genes (B) and the indicated ISR genes (C). Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. WB with the indicated antibodies was used to assess protein levels (D). Data shown are representative of three independent experiments. (E) Survival curves of WT (n = 10) and Dbr1Y17H/Y17H (n = 12) mice after intravenous inoculation with VSV (106 PFU/g body weight). Data representative of three independent experiments. Statistical analysis for mouse survival was performed with Kaplan–Meier tests. ***, P < 0.001. (F and G) WT and Dbr1Y17H/Y17H mice were sacrificed 3.5 days after intravenous inoculation with VSV (106 PFU/g body weight). Brain tissues were collected, and RT-qPCR was performed to analyze the expression of viral genes (F) and the indicated ISR genes (G). Data representative of three independent experiments. Graphs depict the mean with SD and points represent biological replicates. Statistical analysis was performed with one-way ANOVA with Dunnett’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Source data are available for this figure: SourceData F8.
Discussion
The unexpected role of DBR1 in antiviral immunity was revealed by the surprising discovery of a severe but incomplete form of AR DBR1 deficiency in patients with brainstem viral encephalitis (Zhang et al., 2018; Chan et al., 2024a). This study connects DBR1 deficiency with the impairment of G3BP-PKR–mediated antiviral immunity at the SG platform, revealing that DBR1 regulates SG-controlled cell-intrinsic antiviral immunity by clearing RNA lariats. Our results show that, in the presence of DBR1, G3BPs interact, promote, and are required for PKR activation during viral infection. PKR can be activated by dsRNA directly, but SGs operate as a platform providing optimal PKR-eIF2α cascade activation and restricting viral replication. If there is insufficient DBR1 activity, RNA lariats accumulate, impairing the formation of SGs and the G3BP-dependent activation of PKR, resulting in enhanced viral replication. We cannot formally exclude the possibility that other DBR1-dependent mechanisms contribute to human antiviral immunity, but PKR is probably the predominant effector as its ablation abolishes the antiviral activity of DRB1. It is known that dsRNA, as an intermediate or byproduct in cells infected with many different viruses, can trigger PKR-mediated antiviral immunity (Chen and Hur, 2022). This DBR1-RNA lariat-G3BP-PKR connection operates in both mouse and human cells, suggesting that it is a well-conserved antiviral strategy. One recent study linked DBR1 to the stress response in yeast, suggesting that this is a primordial mechanism (Hurtig and van Hoof, 2023). Future studies should search for mutations of the genes encoding G3BPs and PKR and of other genes from this pathway in patients suffering from brainstem viral encephalitis. New therapeutic strategies boosting activation of the DBR1–G3BP–PKR pathway or removing abundant RNA lariats may be beneficial in patients with mutations of this pathway.
The deleterious impact of RNA lariats was revealed by DBR1 deficiency, leading us to investigate lariat-regulated biological processes. As key proteins in the DBR1-mediated antiviral response, G3BPs bind to untranslated RNAs, facilitating the formation of SGs (Yang et al., 2020; Guillén-Boixet et al., 2020). Our data show that, unlike linear RNAs, RNA lariats bind and form puncta with G3BPs separately from SGs. They therefore interfere with the LLPS of G3BPs and generate G3BP aggregates that lack liquidity, represent a solid state, and disengage other SG components. The abnormal G3BP condensates are prone to ubiquitin-mediated degradation. While we assessed the ubiquitination of G3BP proteins, other, untested SG components may be impacted by the accumulation of RNA lariats, thereby also potentially disrupting SG assembly. Interestingly, the toxic TDP-43 mutant also forms cytoplasmic foci with intronic lariats in yeast cells, which prevents the TDP-43 mutant from interfering with vital cellular RNAs and RNA-binding proteins (Armakola et al., 2012). RNA lariats may influence the stability or functions of certain RNA-binding proteins. Future studies could investigate the mechanisms by which lariats cause the degradation of G3BPs. G3BP-governed SGs have been implicated in multiple diseases besides viral infection and neurodegenerative disease, including cancer, and cardiovascular diseases (Ge et al., 2022). DBR1 or lariats may also be linked to other SG-related disorders, and G3BPs may not be the only target of RNA lariats. Additional regulatory targets of lariat RNAs, including genes, mRNAs, and proteins merit further study. Future research should investigate the mechanisms by which lariat accumulation affects the phenotypes described above and embryonic death in different species. Moreover, examination of the composition and distribution of lariats in the genome in DBR1-deficient cells or tissues may provide important biological insight.
DBR1 is the only known RNA lariat-debranching enzyme. It has been conserved throughout evolution and seems to be essential for the survival of most eukaryotes. The DBR1 gene is not essential for viability in Saccharomyces cerevisiae and Schizosaccharomyces pombe, in which its deficiency leads to slower growth (Khalid et al., 2005). The mouse or human DBR1 can rescue RNA lariat accumulation and growth defects in Dbr1null yeasts, suggesting conservation of DBR1 function across species (Kim et al., 2000, 2001). Unlike mutant yeasts, Dbr1−/− mice do not appear to be able to survive, suggesting a greater requirement of DBR1 for development in mice than in yeasts. Consistently, all patients with DBR1 mutations carry severely hypomorphic, although not entirely loss-of-function alleles. These findings suggest that human complete DBR1 deficiency may be embryonic lethal. A few of the patients with partial DBR1 deficiency had phenotypes other than brainstem viral encephalitis. A Japanese patient with norovirus brainstem infection also had intrauterine growth retardation and intellectual disability (Zhang et al., 2018). Several Saudi patients without diagnosed viral encephalitis had a severe form of congenital ichthyosis before they died from sepsis or multiple organ failure before the age of 1 year (Shamseldin et al., 2023). Ichthyosis may be due to a loss of the cutaneous lipid barrier. The biochemical severity of the DBR1 genotypes of these patients has not yet been determined. They would probably have developed a viral infection of the brainstem had they not died prematurely. The viral encephalitis phenotype of patients with DBR1 mutations results from the involvement of DBR1 in stress response-dependent PKR activation, but the mechanisms underlying the other, rarer clinical phenotypes are unclear.
In this study, we also used Dbr1Y17H/Y17H mice as a model of inherited DBR1 deficiency for investigations of the role of DBR1 in vivo. Dbr1Y17H/Y17H mice are a useful tool for cellular and molecular studies, but they also mimic the viral phenotype observed in patients. Complete DBR1 deficiency is embryonic lethal in mice and, presumably, also in humans, but mice and humans with as little as 1–5% of the normal level of DBR1 activity are viable. Our development of this mouse model made it possible to confer particular susceptibility to viral infections of the brain. Previous studies have shown that the patients with DBR1 mutations display only signs of brainstem viral encephalitis without forebrain and systemic infection. In our mouse model, other cell types and tissues were found to be susceptible to viral infection, although the brainstem was one of the tissues most severely infected following the intravenous inoculation of viruses. The route of infection may influence the location of the lesions. We previously suggested that the high levels of DBR1 expression in the human brainstem render this structure particularly vulnerable to RNA lariat buildup and viral infections (Zhang et al., 2018). The Dbr1Y17H/Y17H mouse model reproduces the conditions observed in human patients. It is, therefore, a good model for studies of the cellular basis of viral diseases, such as influenza and norovirus. In future studies, we will use Dbr1Y17H/Y17H mice and generate mice with conditional knockouts of DBR1 in different brain cells to analyze the cellular basis of the diseases caused by different viruses.
Limitations of the study
This study mainly focuses on G3BPs and doesn’t investigate other SG components, which warrant future research. This study clarifies the molecular mechanism of viral disease in patients with DBR1 mutations but not the mechanisms underlying the other, rarer, clinical phenotypes of these patients. This study also sheds no light on the cellular mechanism underlying selective susceptibility to viral infections of the brainstem in patients with DBR1 mutations and does not identify the cells in the brainstem rendering these patients prone to viral disease.
Materials and methods
Animals
The C57BL/6N mice used here were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. Dbr1Y17H/Y17H mice were generated by The Laboratory Animal Center, University of Science and Technology of China. Mice were maintained in a specific pathogen-free facility at 23°C under a 12 h light–12 h dark cycle with free access to food and water. Age- and sex-matched mice were used in this study. Similar results were obtained for male and female mice. No statistical methods were used to determine the necessary sample size in advance. All experiments involving animals were performed with the approval of The Ethics Committee of the University of Science and Technology of China (USTCACUC23080123025). All the relevant ethical regulations regarding animal research were followed in this study, in accordance with the Public Health Service Policy on Humane Care, the Guide for the Care and Use of Laboratory Animals, the Animal Welfare Act, and the Use of Laboratory Animals.
Cell culture
BJ (CRL-4001; ATCC), THP-1 (TIB-202; ATCC), HEK-293T (CRL-3216; ATCC), and HeLa (CCL-2; ATCC) cells were obtained from the American Type Culture Collection (ATCC). EBV-B cell lines and SV40-fibroblast cell lines from the patients and healthy controls have been described elsewhere and were obtained from Professor Jean-Laurent Casanova at Rockefeller University. HEK293T G3BP1/2 DKO, HEK293T PKR−/−, and HeLa G3BP1−/− cell lines were provided by Peiguo Yang (School of Life Sciences, Westlake University). HeLa, HEK293T, SV40-fibroblast, and BJ cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Biosharp). EBV-B and THP-1 cell lines were maintained in 1,640 medium supplemented with 10% FBS and 1% penicillin-streptomycin (Biosharp). Cells were maintained in an incubator containing 5% CO2/95% air at 37°C. The cell lines used in this study were tested for mycoplasma contamination and found to be negative.
Plasmid construction
DNA fragments encoding human PKR, DBR1, G3BP1, and G3BP2 were amplified by PCR from the THP-1 or BJ cell cDNA libraries and inserted into the pCDH-FLAG or pCDH-GFP backbone with the ClonExpressII One-Step Cloning Kit (C112-01; Vazyme). Plasmids encoding disease-related mutants of DBR1 (DBR1I120T FLAG, DBR1Y17H FLAG, DBR1L13G FLAG, and DBR1R197X FLAG) were constructed with the Mut Express II Fast Mutagenesis Kit V2 (C214-01/02; Vazyme). DNA fragments encoding human ID1 and DKK1 lariats were amplified by PCR from a HEK293T cDNA library. The cDNA library was created with the HiScript III first Strand cDNA Synthesis Kit (+gDNA wiper) (Cat# R323; Vazymy). For generation of the ID1/DKK1 lariat copGFP-split and ID1/DKK1 8xMS2 lariat copGFP-split plasmids, the copGFP and tandem MS2 DNA fragment were amplified by PCR from the PTY-copGFP plasmid or pLH-sgRNA1-2XMS2 (#75389; Addgene) and assembled with ID1/DKK1 into a PTY or pCDH plasmid. The MCP-mScarlet fragment was amplified by PCR from the pCDH-Myc-HA-MCP-GSGSlinker-mScarlet-3xFlag-EF1-BSD plasmid. All plasmid constructs were sequenced and confirmed by Sanger sequencing.
Gene transduction with lentiviruses
HEK293T cells were cotransfected with lentiviral vectors and the psPAX2 (#12260; Addgene) and pMD2.G (#12259; Addgene) plasmids for 24 h and the culture medium was then replaced with fresh medium. Supernatants containing the viruses were harvested and filtered (pore size: 0.45 μm) in two batches every 24 h, beginning 24 h after transfection, when the medium was replaced. For lentiviral transduction, human HEK293T, HeLa, and BJ cells were incubated with lentiviral supernatants for 36 h, with the addition of 1 µg/ml puromycin to the growth medium for 5 days for selection.
For shRNA-mediated interference, the DBR1 oligonucleotide was annealed to create a 5′ MluI and a 3′ EcoRI site and ligated into the pLKO.1 (#8453; Addgene) vector. The 5′-GTCACATGCAAACCTTCTACA-3′ sequence was used to create the shRNA DBR1 plasmid, whereas the 5′-CAGACATCAACTCTTACGTAC-3′ sequence was used to create the scrambled shRNA DBR1 control plasmid. Knockdown efficiency was validated by WB with a polyclonal antibody against DBR1 (rabbit antibody, Cat# 16019-1-AP; Proteintech).
CRISPR-Cas9–mediated gene knockout
The sgRNA for each gene was designed by Guide design resources — Zhang Lab (https://zlab.squarespace.com/guide-design-resources). The synthesized PKR or DBR1 DNA oligomer was annealed and ligated with the BsmbI (Monad)-digested lentiCRISPR v2-puro vector. The PKR sgRNA sequences, sgRNA1: 5′-TCGGGGGTGCATGGGCCAGA-3′, sgRNA2: 5′-TTCTTCAGAAGGATTATCCA-3′ were used to create the lentiCRISPR v2-puro vector plasmid. Vectors were verified by sequencing and packaged into lentiviruses. Cells were infected with lentiviruses for 24 h and were then incubated in a medium containing 1 µg/ml puromycin for selection for 5 days. Knockout clones were obtained by single-cell fluorescence-activated cell sorting and successful knockout clones were verified by Sanger sequencing and WB.
RNA extraction and RT-qPCR
RNA was extracted from tissues or cells with the RNA Isolator Total RNA Extraction Reagent (Cat# R401-0; Vazyme). RNA was converted to cDNA with the HiScript III RT SuperMix for qPCR (+gDNA wiper) (Cat# R323; Vazyme). Quantitative PCR was performed on the cDNA with ChamQ Universal SYBR qPCR Master Mix Q711-02 (Cat# Q421-02; Vazyme) on the C1000 Touch PCR CFX384 Touch Real-Time PCR Detection System. GAPDH was used as the housekeeping gene for normalization unless otherwise stated. The sequences of the primers used are indicated in Table S2.
G3BP1 pull down and mass spectrometry
Briefly, to identify the SG components, Scrambled/EV/G3BP1-FLAG 293T and shDBR1/ID1/DKK1-Lariats/G3BP1-FLAG cells were infected with VSV for 12 h to induce SG formation. Cell lysates were immunoprecipitated for G3BP1 and analyzed by the interacting proteins using mass spectrometry. We focused on 36 SG components to assess differences in SG formation. The 36 SG proteins referenced have been previously published (Yang et al., 2020) and are listed as follows: G3BP1, EIF3D, EIF3I, CAPRIN1, EIF3E, EIF3G, TAF15, DDX19A, HNRNPA2B1, FMR1, UPF1, MACF1, NUFIP2, NUP98, NXF1, CBX1, POLR2B, KPNB1, RAB1A, PPP1R10, SFRS3, PPP2R1A, USP10, G3BP2, DDX3X, CSDE1, ATXN2, ATXN2L, UBAP2, UBAP2L, TIAL1, TRIM25, PRRC2C, TIA1, TRIM56, and HDAC6.
Preparation of cell extracts and western blotting
Cells were rinsed twice with cold PBS and scraped into a microcentrifuge tube containing the same buffer. Cells were lysed directly in RIPA lysis buffer (10 mM Tris-HCl [pH 8.0],150 mM NaCl,1% Triton X-100,0.5% sodium deoxycholate, 0.1% SDS,1 mM EDTA [pH 8.0]) (Cat# IN-WB001; Invent) supplemented with proteinase inhibitor/phosphatase inhibitor. Cell debris was removed by centrifugation, and the protein concentration of the supernatant was determined with the Pierce BCA Protein Assay Kit (23225; Thermo Fisher Scientific). Lysates were boiled for 10 min and were then loaded onto a Fast-Cast Colorful (Red) Acrylamide Kit10% (Cat# G2043-50T; Servicebio) gel and subjected to SDS-PAGE. The protein bands were transferred onto nitrocellulose membranes (0.45 nm pores) and incubated with antibodies for the detection of DBR1 (Cat# 16019-1-AP; ProteinTech), G3BP1 (Cat# 13057-2-AP; ProteinTech), G3BP2 (Cat# 16276-1-AP; ProteinTech), PKR (Cat# CY5271; Abways), P-PKR (phospho T446) (Cat# ab32036; Abcam), P-PKR (phospho T446) (Cat# CY5271; Abways), CHOP (Cat# AF6277; AFFINITY), ATF4 (Cat# DF6008; AFFINITY), p-eIF2α (Cat# A0764; ABclonal), eIF2α (Cat# AF6087; AFFINITY), VCL (Cat# A01207-1; BOSTER), and tubulin (Cat# 66031-1-Ig; ProteinTech). Blots were developed with an Amersham Typhoonm Biomolecular imager or the ChemiScope 6200 Chemiluminescence imaging system (CLINX).
Viral infection and chemical stimulation of the cells
Cells were dispensed into 6-, 12-, or 24-well plates (106 cells/ml) and infected with VSV or HSV-1 expressing green fluorescent protein (GFP). Alternatively, cells were transfected at 70–80% confluence in the presence of PEI, with polyI:C (1 µg/ml) or SA (0.5 mM) according to the manufacturer’s instructions.
Infection of mice with viruses
In animal experiments, WT or Dbr1Y17H/Y17H mice received intravenous injections of a final volume of 20 μl containing a known number of plaque-forming units (VSV: 106 PFU/g body weight, or HSV-1: 107 PFU) of the virus.
Immunofluorescence microscopy
Cells were grown in 12-well or 24-well chamber slides. After stimulation, they were fixed by incubation with 4% PFA (paraformaldehyde) in 0.01 M PBS for 15 min. The cells were permeabilized by incubation with 0.1% Triton X-100 or 100 µM digitonin for 10 min, and were then blocked by incubation with PBST blocking buffer (PBS + 0.1% Tween 20, 1% BSA, 22.52 mg/ml glycine) for 40–60 min. The samples were incubated overnight at 4°C with primary antibodies in PBST blocking buffer. They were then washed three times in PBST (PBS + 0.1% Tween 20), and secondary antibody was added and the samples were incubated at room temperature for 2 h. The primary antibodies used are listed in Table S2. Host-specific DyLight488- (Abbkine), Alexa Fluor 568- (Abcam), and Alexa Fluor 647-conjugated (Jackson) secondary antibodies were used for the detection of primary antibody binding. Slides were fixed with ProLong Gold Antifade reagent. Images were captured with a Leica STELLARIS STED or ZEISS LSM980 with Airyscan confocal microscope fitted with a 63× oil or 100× oil objective.
For the visualization of 8xMS2-tagged intronic lariats, ID1/DKK1, and G3BPs, cells were transduced with lentiviruses expressing lariats, ID1/DKK1, and MCP-mScarlet with or without G3BP-mNeonGreen. Cells were grown in 24-well chamber slides and fixed with 4% PFA and stained with DAPI in ProLong Gold Antifade reagent. The colocalization of 8xMS2-tagged intronic lariats with MCP was assessed by confocal microscopy.
Protein detection by flow cytometry
Adherent cells were detached with trypsin. Cells were then collected by centrifugation with multiple washes in 1X PBS. The collected cells were fixed by incubation with 4% PFA at room temperature for 15 min. The cells were pelleted by centrifugation and the supernatant was removed. The cell pellets were then resuspended and permeabilized in flow cytometry permeabilization buffer (10X) (Cat# 00-8333-56; Invitrogen) for at least 10 min on ice. The cells were then stained with anti-human G3BP1 (Cat# 13057-2-AP; Proteintech) and Alexa Fluor 647-conjugated AffiniPure Donkey Anti-Rabbit IgG (H+L) at a dilution of 1:500. Fluorescence intensity was measured with a CytoFLEX (Beckman) flow cytometer equipped with a laser for excitation at 647 nm.
Mouse brain tissue immunofluorescence
Paraffin-embedded WT and Dbr1Y17H/Y17H mouse brain tissue sections were produced by the Servicebio company. Sections were dewaxed and rehydrated, and antigen retrieval was performed by standard procedures. Sections were then washed three to five times with PBS in vertical staining chambers. The slides were blocked and then incubated with primary antibodies for 4 h at room temperature, followed by secondary antibodies for 1 h at room temperature. Finally, the sections were counterstained with DAPI for visualization of the nuclei.
Statistical analysis
Data are representative of three independent experiments and are expressed as the mean of at least three biological replicates ± SD unless otherwise indicated. Data were analyzed in Microsoft Excel. NS, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Online supplemental material
Fig. S1 shows that DBR1 regulates the activation of PKR and ISR rather than IFN response. Fig. S2 shows that DBR1 influences PKR antiviral function by modulating the level of RNA lariats. Fig. S3 shows that DBR1 licenses G3BP proteins to form SGs. Fig. S4 shows the stability of the G3BP protein is compromised by RNA lariats. Fig. S5 shows that DBR1 deficiency increases susceptibility to viral infection in vitro and in vivo. Table S1 summarizes the list of G3BP1 interacting proteins that change in lariats-overexpressing cells identified by mass spectrometry. Table S2 lists resources and reagents used in this study.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank the patients and their families for their participation. We thank the members of the Gao laboratory for their expertise and assistance with experiments.
This study was supported by the National Key R&D Program of China (2023YFC2306100). This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB0490000 and Grant No. XDB0940302. This work was supported by grants from the National Natural Science Foundation of China (31970855 and 32270914). The study was also supported by the Joint Laboratory of Innovation in Life Sciences from the University of Science and Technology of China and Changchun Zhuoyi Biological Co. Ltd. The Laboratory of Human Genetics of Infectious Diseases was funded in part by the National Center for Advancing Translational Sciences, National Institutes of Health (NIH), Clinical and Translational Science Award program (UL1TR001866), NIH (R01AI088364 and R01NS072381), grants from the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and the French National Research Agency (ANR) under the “Investments for the future” program (ANR-10-IAHU-01), CNSVIRGEN (ANR-19-CE15-0009-01), William E. Ford, General Atlantic’s Chairman and Chief Executive Officer, Gabriel Caillaux, General Atlantic’s Co-President, Managing Director and Head of business in EMEA, and the General Atlantic Foundation, The Rockefeller University, the Howard Hughes Medical Institute, Institut National de la Santé et de la Recherche Médicale, Imagine Institute, Paris Cité University, and the St. Giles Foundation. D. Lee is supported by the French Medical Research Foundation fellowship for medical residents and fellows.
Author contributions: S. Ru: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing, S. Tang: Investigation, Resources, Validation, H. Xu: Data curation, Formal analysis, Investigation, Methodology, Resources, Validation, Visualization, J. Yin: Resources, Validation, Y. Guo: Formal analysis, Investigation, Resources, Writing - review & editing, L. Song: Data curation, Resources, Validation, Z. Jin: Resources, Software, Validation, D. Lee: Writing - review & editing, Y.-H. Chan: Formal analysis, Investigation, Methodology, Validation, Writing - review & editing, X. Chen: Investigation, L. Buerer: Formal analysis, Software, Visualization, W. Fairbrother: Conceptualization, Investigation, Writing - original draft, Writing - review & editing, W. Jia: Funding acquisition, J.-L. Casanova: Funding acquisition, Project administration, Supervision, Writing - review & editing, S.-Y. Zhang: Conceptualization, Funding acquisition, Investigation, Supervision, Writing - review & editing, D. Gao: Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing - original draft, Writing - review & editing.
References


