


The function of transient receptor potential vanilloid (TRPV) cation channels governing B cell activation remains to be explored. We present evidence that TRPV2 is highly expressed in B cells and plays a crucial role in the formation of the B cell immunological synapse and B cell activation. Physiologically, TRPV2 expression level is positively correlated to influenza-specific antibody production and is low in newborns and seniors. Pathologically, a positive correlation is established between TRPV2 expression and the clinical manifestations of systemic lupus erythematosus (SLE) in adult and child SLE patients. Correspondingly, mice with deficient TRPV2 in B cells display impaired antibody responses following immunization. Mechanistically, the pore and N-terminal domains of TRPV2 are crucial for gating cation permeation and executing mechanosensation in B cells upon antigen stimulation. These processes synergistically contribute to membrane potential depolarization and cytoskeleton remodeling within the B cell immunological synapse, fostering efficient B cell activation. Thus, TRPV2 is critical in augmenting B cell activation and function.
Introduction
The recognition of antigens by B cell receptors (BCR) triggers the initiation of B cell activation through a coordinated regulatory network within the immunological synapse, followed by B cell proliferation, differentiation, and antibody production. BCR is an extraordinary receptor that can efficiently discriminate among a wide variety of chemical and physical features of antigens (Liu et al., 2016), including antigen affinity (Fleire et al., 2006; Liu et al., 2010), density (Fleire et al., 2006; Liu et al., 2010; Tang et al., 2016; Wang et al., 2016), valency (Bachmann et al., 1993; Liu and Chen, 2005; Liu et al., 2004), the mechanical forces delivered to the BCRs by the antigens (Natkanski et al., 2013; Wan et al., 2015; Wan et al., 2018), Brownian mobility features of antigens (Wan and Liu, 2012), and the stiffness feature of antigen-presenting substrates (Shaheen et al., 2017; Wan et al., 2013; Zeng et al., 2015). These extraordinary discriminatory capacities indicated the presence of sophisticated mechanisms at multiple levels to regulate B cell activation and function. Thus, the elucidation of these molecular mechanisms will provide important clues as to how to drive B cells to develop the high-affinity and high-titer antibodies crucial for an effective humoral response against infection.
During the initiation of B cell activation, antigen and BCR recognition swiftly initiate cation flux responses, which in turn significantly remodels electrostatic interactions between a series of lipid and membrane proximal signaling molecules (Chen et al., 2015; Engels et al., 2009, 2014). Thus, it is of crucial importance to investigate the function of cation channels in the regulation of B cell activation. As a big family of nonselective ion channels, transient receptor potential (TRP) channels are proposed to be involved in raising intracellular cationic concentration and depolarizing cells (McCoy et al., 2011; Nilius and Owsianik, 2011; Voets et al., 2001). In response to external stimuli, the biological roles of TRPs are diverse and versatile, including nociception, mechanosensation, and thermosensation (Eijkelkamp et al., 2013; Julius, 2013; Stokes et al., 2004). TRP channels are grouped into several classes on the basis of their architectures and sequence homologies (Nilius and Owsianik, 2011). Among these, TRP vanilloid (TRPV) class members are recently well characterized in terms of their channel structures. They are expressed in both excitable neurons and non-excitable cells, including immune cells. However, whether TRPV channels participate in B cell activation and subsequent antibody responses in both physiological and pathological conditions has received limited attention.
In this study, we identified TRPV2, one of the TRPV class members, to be of crucial importance in the commencement of B cell activation. B cells deficient in TRPV2 exhibited global impairments in BCR engagement, cytoskeletal reorganization, and downstream signal transduction during the formation of B cell immunological synapses. Physiologically, B cell–specific TRPV2 knockout (KO) mice exhibited significantly impaired antibody responses upon immunization with both model antigens and a natural T cell–independent antigen, the capsular polysaccharide from Streptococcus pneumonia. Since infection by this pathogen is a leading cause of death in young children worldwide (Black et al., 2003; Bryce et al., 2005; GBD 2015 Mortality and Causes of Death Collaborators, 2016; Nissen, 2007), our further analyses revealed that the expression level of TRPV2 is substantially lower in newborn (≤60-day infant) than in infants and teenagers and is also dramatically lower in old individuals (>60) compared with younger adult individuals. Physiologically, an influenza vaccine cohort containing healthy adult volunteers suggested that the strength of virus-specific antibody response was proportional to TRPV2 expression level. Pathologically, systemic lupus erythematosus (SLE) patient–based cohort studies in both adult and child SLE revealed a positive correlation between TRPV2 expression in peripheral blood mononuclear cells (PBMC), especially in B cells, and disease activity including the SLE disease activity index (SLEDAI), anti-double-stranded DNA (dsDNA) antibody, etc. Mechanistically, we found that the function of TRPV2 on B cells relies on its pore region and N-terminal ANK domain. Indeed, highly efficient B cell activation is dependent on both membrane potential depolarization and synaptic co-distribution of TRPV2 and microtubule, mediated by its pore region and ANK domain, respectively. Together, these results contribute evidence for the critical ability of TRPV2 to enhance B cell activation and function.
Results and discussion
TRPV2 contributes to the formation of B cell immunological synapse
The formation of a B cell immunological synapse is crucial for highly efficient B cell activation after antigen recognition. To analyze the function of TRPV channels in the formation of the B cell immunological synapse, we first utilized ruthenium red (RR), an antagonist of general TRPV channels and certain other types of cation channels (Aguettaz et al., 2016; Lévêque et al., 2018; Link et al., 2010; Nagasawa and Kojima, 2015). Immunological synapse formation was triggered in Ramos cells, a human IgM+ B cell line, by using anti-human IgM antibodies as membrane-bound surrogate antigen embedded in planar lipid bilayers (PLBs). To visualize the spatial–temporal distribution of BCR and surrogate antigen upon their engagement within the synapse by total internal reflection fluorescence microscopy (TIRFM), both surrogate antigen and BCR were labeled with fluorescence probes. In line with the previous studies from us and others (Wang et al., 2016; Xu et al., 2015), we observed that untreated control Ramos cells formed a typical bright disc-shaped immunological synapse consisting of accumulated BCRs in response to surrogate antigen stimulation (Fig. S1 A). In contrast, RR treatment disrupted the synaptic accumulation of BCRs (Fig. S1, A–E). We quantified the fluorescence intensity of the surrogate antigen and verified that it was closely matched between the control and RR groups before we loaded cells onto PLBs (Fig. S1 B), suggesting similar antigen density on PLBs before B cell activation. Therefore, it became clear that upon Ramos cell activation, RR treatment dramatically impaired the synaptic accumulation of BCRs and then inhibited the subsequent internalization of antigens (Fig. S1, C–E). To further conclude that inhibition of TRPV with RR inhibits antigen internalization, we used flow cytometry to conduct a BCR internalization assay in primary B cells upon the activation by bead-conjugated goat F(ab′)2 anti-mouse IgM antibodies and found that RR treatment inhibits antigen internalization (Fig. S1 F). As a confirmation of the effect of RR on B cell activation, we also performed the same assay in different cell types. Results in A20, a mouse B-lymphoid cell line, were in agreement with those in Ramos cells (Fig. S1 G). We further validated these results by comparing the responses of human peripheral blood B cells between control and RR-treated groups. B cells from PBMCs were labeled with probes recognizing either IgM or IgG BCR. Anti-human light chain antibodies were used as surrogate antigens. Synaptic accumulation of BCRs from both IgM and IgG B cells was significantly inhibited by RR (Fig. S1 H). These findings indicate that RR-targeted channels (likely TRPV family members) contribute to the formation of the B cell immunological synapse upon antigen stimulation.
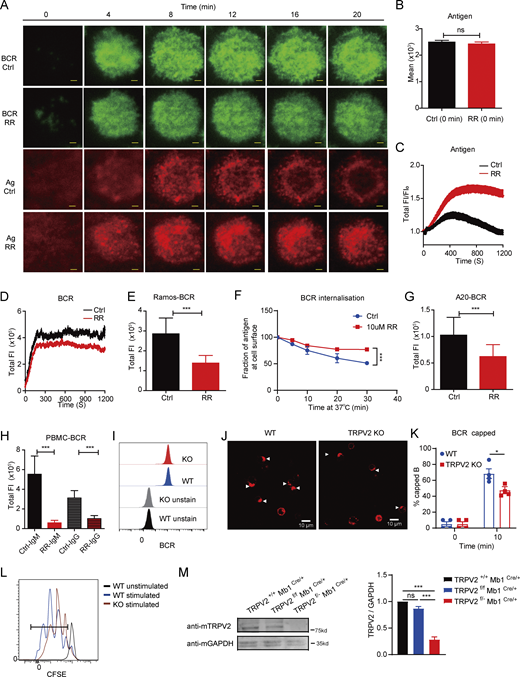
RR-sensitive channels contribute to the formation of the B cell immunological synapse, and TRPV2 contributes to B cell activation. (A) Representative time-lapse TIRFM images of Ramos cells at indicated time points of cell activation. Cells were activated in the absence or presence of RR (10 μM). Scale bars, 1.5 μm. (B) Quantification of total FI of BCR in the immunological synapse (n > 10). (C and D) Quantification of total FI of surrogate antigen (C) and total FI of BCR (D) in the immunological synapse (n > 10). (E) Quantification of total FI of recruited BCR in the immunological synapse of Ramos activated in the absence or presence of RR (n > 30). (F) RR treatment inhibited BCR internalization in primary B cells (n = 3). (G) Quantification of total FI of recruited BCR in the immunological synapse A20 activated in the absence or presence of RR (n > 30). (H) Quantification of total FI of recruited BCR in human PBMCs activated in the absence or presence of RR (n > 30). (I) Measurement of surface BCR expression in WT and TRPV2-KO A20B1-8 primary splenic B cells by flow cytometry (n > 2,000). (J and K) Representative confocal images of activated WT and TRPV2 KO primary B cells showing BCR capping at 10 min (J) and the average percentages of B cells with BCR capping at indicated times (K) (n = 3). Scale bar, 10 µm. (L) Cell proliferation of WT and TRPV2 KO B cells with or without activation. Primary B cells were labeled with CFSE and stimulated with soluble goat F(ab′)2 anti-mouse IgG + IgM (H+L) antibody (10 μg/ml) for 60 h (n >10,000). (M) TRPV2 KO efficiency was evaluated through immunoblot analysis, and quantitative statistical analysis was conducted based on gray values (n = 3). Unpaired two-tailed t test in B, E, G, H, and K. One-way ANOVA in M; two-way ANOVA in F. Data are mean ± SEM. ns, not significant, *P < 0.05 and ***P < 0.001. Data are representative of at least two independent experiments. Source data are available for this figure: SourceData FS1.
RR-sensitive channels contribute to the formation of the B cell immunological synapse, and TRPV2 contributes to B cell activation. (A) Representative time-lapse TIRFM images of Ramos cells at indicated time points of cell activation. Cells were activated in the absence or presence of RR (10 μM). Scale bars, 1.5 μm. (B) Quantification of total FI of BCR in the immunological synapse (n > 10). (C and D) Quantification of total FI of surrogate antigen (C) and total FI of BCR (D) in the immunological synapse (n > 10). (E) Quantification of total FI of recruited BCR in the immunological synapse of Ramos activated in the absence or presence of RR (n > 30). (F) RR treatment inhibited BCR internalization in primary B cells (n = 3). (G) Quantification of total FI of recruited BCR in the immunological synapse A20 activated in the absence or presence of RR (n > 30). (H) Quantification of total FI of recruited BCR in human PBMCs activated in the absence or presence of RR (n > 30). (I) Measurement of surface BCR expression in WT and TRPV2-KO A20B1-8 primary splenic B cells by flow cytometry (n > 2,000). (J and K) Representative confocal images of activated WT and TRPV2 KO primary B cells showing BCR capping at 10 min (J) and the average percentages of B cells with BCR capping at indicated times (K) (n = 3). Scale bar, 10 µm. (L) Cell proliferation of WT and TRPV2 KO B cells with or without activation. Primary B cells were labeled with CFSE and stimulated with soluble goat F(ab′)2 anti-mouse IgG + IgM (H+L) antibody (10 μg/ml) for 60 h (n >10,000). (M) TRPV2 KO efficiency was evaluated through immunoblot analysis, and quantitative statistical analysis was conducted based on gray values (n = 3). Unpaired two-tailed t test in B, E, G, H, and K. One-way ANOVA in M; two-way ANOVA in F. Data are mean ± SEM. ns, not significant, *P < 0.05 and ***P < 0.001. Data are representative of at least two independent experiments. Source data are available for this figure: SourceData FS1.
To determine which family member of TRPV channels is functional in the formation of the B cell immunological synapse, we first investigated the transcription profiles of TRPV channel members from the ImmGen database (https://www.immgen.org/) and found that only TRPV2 is highly expressed in B cells (Fig. 1 A). We further investigated TRPV2 expression in various immune cells and other types of cells from the Human Protein Atlas database (https://www.proteinatlas.org/) and found that TRPV2 was highly expressed mainly in immune cells compared with other types of human cells (Fig. 1 B), highlighting a potential function of TRPV2 in immune system worth a thorough investigation. To confirm this result upon analyses from the public RNA-sequencing (RNA-seq) database, we sorted B220+ B cells from the spleen of wild type (WT) C57BL/6J mice and quantified the transcription levels of all four murine TRPV family members (Trpv1, Trpv2, Trpv3, and Trpv4) by reverse transcription with quantitative PCR (RT-qPCR). Indeed, Trpv2 exhibited the highest transcription level among TRPV family members (Fig. 1 C), consistent with the ImmGen data. To validate results in the human cell atlas database, we also sorted CD3−CD19+ B cells from human PBMCs and used RT-qPCR to show that the transcription level of Trpv2 stands out among TRPV family members in human B cells (Fig. 1 D). Thus, these results indicated that TRPV2 is the sole TRPV member highly expressed in B cells.

TRPV2 contributes to the formation of B cell immunological synapse. (A) Heatmap of expression profiles of mouse Trpv1, Trpv2, Trpv3, and Trpv4 gene in a group of B cell subsets. Data from https://www.immgen.org/. (B) TRPV2 expression in various immune cells and other cell types. Data from Human Protein Atlas database (https://www.proteinatlas.org/). (C)mTrpv1, mTrpv2, mTrpv3, and mTrpv4 transcription level in mouse primary B cells (n = 4). Trpv mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. Presentation of each expression level was relative to the transcription level of Trpv2. (D)huTrpv1, huTrpv2, huTrpv3, and huTrpv4 transcription level in human primary B cells (n = 4). Trpv mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. The presentation of each expression level was relative to the transcription level of Trpv2. (E) Representative TIRFM images showing dynamics of BCR accumulation in the immunological synapse in WT and TRPV2-deficient A20II1.6 B1-8 IgM cells. Scale bars, 1.5 μm. (F) Quantification of the MFI of BCR accumulation in immunological synapse (n > 10). Cells were prestained with Alexa Fluor 647–conjugated goat Fab anti-mouse IgM (Fc5μ fragment specific) and loaded on coverslips presenting PLBs with caged-NP. Cells were activated when caged-NPs were uncaged in the presence of a 405 nm laser. (G and H) Quantification of total FI of BCR (G) and TRPV2 (H) in the immunological synapse of WT, TRPV2 KO (KO−), and TRPV2-KO with rescuing TRPV2 (KO+) A20II1.6 B1-8 IgM cells (n > 30). (I) Quantification of total FI of BCR in the immunological synapse in WT and TRPV2-knockdown A20 cell line (n > 30). (J) Efficiency of TRPV2 knockdown by shRNA evaluated by RT-qPCR in primary splenic B cells (n = 3). Results were normalized to the housekeeping gene of Gapdh. (K) Quantification of total FI of BCR in the immunological synapse in WT and TRPV2-knockdown primary B cells (n > 30). One-way ANOVA in G and H. Unpaired two-tailed t test in I–K). ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of two (C and D), three (E and I–K), or six (G and H) independent experiments, respectively.
TRPV2 contributes to the formation of B cell immunological synapse. (A) Heatmap of expression profiles of mouse Trpv1, Trpv2, Trpv3, and Trpv4 gene in a group of B cell subsets. Data from https://www.immgen.org/. (B) TRPV2 expression in various immune cells and other cell types. Data from Human Protein Atlas database (https://www.proteinatlas.org/). (C)mTrpv1, mTrpv2, mTrpv3, and mTrpv4 transcription level in mouse primary B cells (n = 4). Trpv mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. Presentation of each expression level was relative to the transcription level of Trpv2. (D)huTrpv1, huTrpv2, huTrpv3, and huTrpv4 transcription level in human primary B cells (n = 4). Trpv mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. The presentation of each expression level was relative to the transcription level of Trpv2. (E) Representative TIRFM images showing dynamics of BCR accumulation in the immunological synapse in WT and TRPV2-deficient A20II1.6 B1-8 IgM cells. Scale bars, 1.5 μm. (F) Quantification of the MFI of BCR accumulation in immunological synapse (n > 10). Cells were prestained with Alexa Fluor 647–conjugated goat Fab anti-mouse IgM (Fc5μ fragment specific) and loaded on coverslips presenting PLBs with caged-NP. Cells were activated when caged-NPs were uncaged in the presence of a 405 nm laser. (G and H) Quantification of total FI of BCR (G) and TRPV2 (H) in the immunological synapse of WT, TRPV2 KO (KO−), and TRPV2-KO with rescuing TRPV2 (KO+) A20II1.6 B1-8 IgM cells (n > 30). (I) Quantification of total FI of BCR in the immunological synapse in WT and TRPV2-knockdown A20 cell line (n > 30). (J) Efficiency of TRPV2 knockdown by shRNA evaluated by RT-qPCR in primary splenic B cells (n = 3). Results were normalized to the housekeeping gene of Gapdh. (K) Quantification of total FI of BCR in the immunological synapse in WT and TRPV2-knockdown primary B cells (n > 30). One-way ANOVA in G and H. Unpaired two-tailed t test in I–K). ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of two (C and D), three (E and I–K), or six (G and H) independent experiments, respectively.
To verify our hypothesis that TRPV2 is responsible for enhancing the formation of B cell immunological synapse, we knocked out Trpv2 (TRPV2-KO) using CRISPR/Cas9-based genetic deletion (Ran et al., 2013) in A20 laboratory B cells. We chose to use A20 B cells stably expressing μ-B1-8 heavy chain (A20B1-8 B cells) specific for 4-hydroxy-3-nitrophenyl (NP) antigen as reported in our early studies (Allen et al., 1988; Liu et al., 2010). We did this for an extra purpose to compare the formation of immunological synapses in the presence versus absence of TRPV2 in B cells upon the stimulation by model antigen but not surrogate antigen molecules as aforementioned in this report (Fig. S1). Similar levels of surface IgM-BCR were detected in WT and KO A20 B cells (termed hereafter as WT or TRPV2-KO A20B1-8 B cells), suggesting that deletion of TRPV2 did not affect surface BCR location and amount (Fig. S1 I). Next, we placed these two types of B cells on streptavidin-containing PLBs presenting the biotin-conjugated NP33-BSA antigen and examined the formation of B cell immunological synapses by TIRFM imaging. Consistently, TRPV2-KO A20B1-8 B cells showed severely impaired formation of B cell immunological synapses compared with the control WT A20B1-8 B cells in response to the stimulation by NP33-BSA antigen (Fig. 1, E and F). To re-examine this result, we performed the rescue experiment and found that the supply of exogenous TRPV2 resulted in the restoration of the synaptic accumulation of BCRs in TRPV2-KO A20B1-8 cell, confirming that the impaired formation of B cell immunological synapses in the KO cells was caused by the deficiency of TRPV2 (Fig. 1, G and H). To independently confirm this result in primary B cells and further exclude the possibility of off-target effects by CRISPR/Cas9, we also examined both A20 B cells and mouse splenic primary B cells from WT C57BL/6J mice, in which TRPV2 was knocked down using short hairpin RNAs (Fig. 1, I–K). Here again, the reduction in TRPV2 significantly impaired BCR accumulation within the B cell immunological synapse. Together, these findings demonstrated that TRPV2 significantly contributed to the formation of the B cell immunological synapse during B cell activation.
TRPV2 deficiency impairs BCR signaling cascade and B cell activation
To further validate the above results by using primary B cell KO for TRPV2 and also to examine the role of TRPV2 in the downstream of BCR signaling cascade and B cell activation, we imported total TRPV2 KO mice (Trpv2−/− or TRPV2 KO mice, thereafter in this report) from Dr. Michael J. Caterina’s laboratory (Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, USA). Consistent with the aforementioned result, upon antigen stimulation, splenic primary B cells from TRPV2 KO mice (TRPV2 KO primary B cells) exhibited significantly impaired formation of B cell immunological synapse compared with the control primary B cells from WT mice (Fig. 2, A and B). BCR-antigen binding leads to the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) tyrosine residues on Igα and Igβ and the synaptic recruitment and phosphorylation of Lyn and Syk, which results in the amplification of signaling through a series of signaling molecules and a rise in intracellular calcium (Xu et al., 2014). We performed immunostaining with specific antibodies to detect phosphorylated signaling molecules. The fluorescence intensity was quantified based on the area of the BCR immunological synapse. BCR-induced recruitment of phosphorylated Syk and PI3K/Akt pathway components were markedly impaired in TRPV2 KO primary B cells (Fig. 2, C–E). These results suggested the importance of TRPV2 for signaling transduction in the membrane-proximal region induced by BCR stimulation, consistent with the pivotal function of TRPV2 in the formation of the B cell immunological synapse. We also determined the TRPV2 function in the soluble antigen-induced BCR polarization (BCR capping) (Seeley-Fallen et al., 2022) and found that the percentage of TRPV2 KO B cells showing capped status is much lower than WT B cells (Fig. S1, J and K), thereby showing the significance of TRPV2 in B cell activation upon stimulation by both soluble and membrane-bound antigens. In addition, we measured primary B cell proliferation induced by BCR crosslinking and found that there was a compromised proliferation of TRPV2 KO B cells (Fig. S1 L). We also evaluated the expression level of CD86, a lymphocyte early activation marker, in splenic primary B cells from both TRPV2 KO and WT control mice 6 h after stimulation in vitro. The upregulation of CD86 is impaired by the deletion of TRPV2 (Fig. 2, F and G). Therefore, a deficiency of TRPV2 impairs the activation of the B cell signaling cascade after antigen stimulation.

TRPV2 deficiency impairs BCR signaling cascade, B cell activation, and antibody production. (A) Representative TIRFM images showing dynamics of BCR accumulation in immunological synapse in splenic primary B cells from WT and total TRPV2 KO mice, marked as WT and tKO, respectively. Scale bars, 1.5 μm. (B) Quantification of total FI of BCR in the immunological synapse in WT and tKO primary B cells (n > 30). (C–E) Quantification of the total FI of several BCR signaling molecules through intracellular staining (n > 30). PI3K-p38, pSYK, and pAKT were stained and quantified 5, 5, and 10 min after stimulation. (F and G) Flow cytometry analysis and quantification of B cell surface expression activation marker CD86 in primary B cells (n = 3). WT and tKO splenic B cells were stimulated for 8 h with the PLB presenting system. (H and I) NP-specific IgM (H) and IgG (I) antibody production was detected on day 6 after immunization. C-Het and C-KO mice were immunized with NP50-LPS on day 0 (C-Het, n = 4; C-KO, n = 7). (J) The dynamics of anti-TNP antibody titers were measured on the indicated timepoint (C-Het, n = 4; C-KO, n = 4). C-Het and C-KO mice were immunized with TNP14-AECM-Ficoll (n = 4). (K) The dynamics of anti-NP antibody titers were measured on the indicated timepoint. C-Het and C-KO mice were immunized with NP32-KLH (C-Het, n = 4; C-KO, n = 4). (L and M) Dynamics of NP-specific IgM (L) and IgG (M) levels. μMT mice, which received WT or tKO splenic B respectively, were detected on day 0, day 7, and day 14 after immunization (WT, n = 4; KO, n = 4). μMT + WT indicated μMT mice were injected with WT primary B cells. μMT + tKO indicated μMT mice were injected with tKO primary B cells. Chimeric mice were immunized with NP32-KLH. (N and O) IgG response to NP50-Ficoll immunization. Splenic B cells purified from B1-8hi+/+Trpv2+/+ (B1-8-TRPV2-WT) or B1-8hi+/+Trpv2−/−(B1-8-TRPV2-KO) were transferred into μMT mice respectively. Chimeric mice were immunized with NP50-Ficoll (WT, n = 3; KO, n = 4) (N). NP-specific IgG antibody titer on day 7 was tested (O). (P) The dynamics of anti-CPS1/CPS3-specific IgM antibody was detected on day 0, day 6, and day 12 after immunization (WT, n = 4; KO, n = 4). Unpaired two-tailed t test in B–E, G–I, and O. One-way ANOVA in J–M and P. ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of two (F, G, and J–P) and three (A–E) independent experiments, respectively.
TRPV2 deficiency impairs BCR signaling cascade, B cell activation, and antibody production. (A) Representative TIRFM images showing dynamics of BCR accumulation in immunological synapse in splenic primary B cells from WT and total TRPV2 KO mice, marked as WT and tKO, respectively. Scale bars, 1.5 μm. (B) Quantification of total FI of BCR in the immunological synapse in WT and tKO primary B cells (n > 30). (C–E) Quantification of the total FI of several BCR signaling molecules through intracellular staining (n > 30). PI3K-p38, pSYK, and pAKT were stained and quantified 5, 5, and 10 min after stimulation. (F and G) Flow cytometry analysis and quantification of B cell surface expression activation marker CD86 in primary B cells (n = 3). WT and tKO splenic B cells were stimulated for 8 h with the PLB presenting system. (H and I) NP-specific IgM (H) and IgG (I) antibody production was detected on day 6 after immunization. C-Het and C-KO mice were immunized with NP50-LPS on day 0 (C-Het, n = 4; C-KO, n = 7). (J) The dynamics of anti-TNP antibody titers were measured on the indicated timepoint (C-Het, n = 4; C-KO, n = 4). C-Het and C-KO mice were immunized with TNP14-AECM-Ficoll (n = 4). (K) The dynamics of anti-NP antibody titers were measured on the indicated timepoint. C-Het and C-KO mice were immunized with NP32-KLH (C-Het, n = 4; C-KO, n = 4). (L and M) Dynamics of NP-specific IgM (L) and IgG (M) levels. μMT mice, which received WT or tKO splenic B respectively, were detected on day 0, day 7, and day 14 after immunization (WT, n = 4; KO, n = 4). μMT + WT indicated μMT mice were injected with WT primary B cells. μMT + tKO indicated μMT mice were injected with tKO primary B cells. Chimeric mice were immunized with NP32-KLH. (N and O) IgG response to NP50-Ficoll immunization. Splenic B cells purified from B1-8hi+/+Trpv2+/+ (B1-8-TRPV2-WT) or B1-8hi+/+Trpv2−/−(B1-8-TRPV2-KO) were transferred into μMT mice respectively. Chimeric mice were immunized with NP50-Ficoll (WT, n = 3; KO, n = 4) (N). NP-specific IgG antibody titer on day 7 was tested (O). (P) The dynamics of anti-CPS1/CPS3-specific IgM antibody was detected on day 0, day 6, and day 12 after immunization (WT, n = 4; KO, n = 4). Unpaired two-tailed t test in B–E, G–I, and O. One-way ANOVA in J–M and P. ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of two (F, G, and J–P) and three (A–E) independent experiments, respectively.
TRPV2 deficiency in B cells impaired the antibody response
To investigate TRPV2-dependent and B cell lineage–specific function at the physiological level, we chose to use mice with B cell–specific TRPV2 KO by using the Cre recombinase-loxP system due to the fact that only 2.5% of the weaning age pups born from the heterozygote breading were totally TRPV2 KO mice (Park et al., 2011), preventing us from acquiring sufficient KO mice for immunization experiments. We initially chose to cross Trpv2fl/fl mice with Mb1-cre mice carrying one allele of the cre transgene (Mb1cre/+) under the control of a B cell–specific promoter; however, an unexpected finding is that TRPV2 cannot be efficiently knocked out in the B cells from Trpv2fl/flMb1cre/+ mice (Fig. S1 M). We thus changed the strategy to generate conditional TRPV2 B cell–specific KO by crossing Trpv2+/− mice with Trpv2fl/+Mb1cre/+ to achieve highly efficient and B cell–specific deletion of TRPV2. Indeed, successful TRPV2 deletion could only be achieved in splenic B cells from Trpv2fl/−Mb1cre/+ mice (C-KO) (Fig. S1 M), but not from Trpv2+/−Mb1cre/+ mice (C-Het) or Trpv2+/+Mb1cre/+ mice (C-WT). B cell development is grossly normal in TRPV2 C-KO mice compared with the control mice, C-Het or C-WT (Fig. S2, A–H), which enabled us to examine the function of B cell–specific TRPV2 in antibody responses.

The effect of TRPV2 on B cell development, B cell signaling, and B cell function. (A and B) Representative flow plot (A) and quantification (B) of different B cell precursor populations in bone marrow. Bone marrow cells from the femur of C-WT (Trpv2+/+Mb1cre/+), C-Het (Trpv2+/−Mb1cre/+), and C-KO (Trpv2f/−Mb1cre/+) mice were stained with anti-B220 and anti-IgM and analyzed by flow cytometry (n = 3). Cell populations were defined as proB/preB (B220lo IgM−), immature (B220lo IgM+), and mature (B220+ IgM+). (C and D) Representative flow plot (C) and quantification (D) of the T cell population (CD3+) and B cell population (B220+) in live splenic cells from C-WT, C-Het, and C-KO mice (n = 3). (E and F) Representative flow plot (E) and quantification (F) of B cell subsets in live splenic cells from C-WT, C-Het, and C-KO mice (n = 3). Cell populations were defined as follicular B (B220+CD23loCD21hi), marginal zone B (B220+CD21loCD23hi). (G and H) Representative flow plot (G) and quantification (H) of B1 cell subsets in the peritoneal cavity from C-WT and C-KO mice (n = 3). B1 cell populations were defined as B220+CD23−CD5+. (I) NP-specific IgG antibody production was detected on day 14 and day 21. C-WT and C-KO mice were immunized on day 0 and day 14 with NP32-KLH (n = 3). (J–O) C-WT and C-KO mice were immunization with OVA (100 μg/mice) emulsified in aluminum adjuvant on day 0 and day 14 (n = 3). Representative flow plot (J) and quantification (K) of B cell population in draining lymph node (dLN) collected on day 21. Representative flow plot (L) and quantification (M) of GCB cell (B220+GL7+ CD95+) population in dLN collected on day 21. Representative flow plot (N) and quantification (O) of antigen-specific GCB cell (OVA+) population in dLN collected on day 21. (P) Total IgM and IgG antibody production in C-WT and C-KO mice was detected on day 14 after immunization with NP32-KLH (n = 3). (Q and R) Representative flow plot (Q) and quantification (R) of B cell population in the spleen of μMT mice collected on day 3. μMT mice were transferred with 1 × 106 B cells purified from the spleen of C-WT or C-KO mice (n = 3) on day 0. (S) Percentage of live WT or TRPV2 KO primary B cells without stimulation at the indicated time. (n = 3). (T–V) Analysis of huTrpv1, huTrpv3, and huTrpv4 transcription level in a single-cell RNA-seq database GSE174188 including 162 SLE patients and 99 healthy subjects, all Asian or Caucasian from San Francisco, CA, USA. (W) Measurement of the calcium influx in A20 cells stimulated with surface-associated goat F(ab′)2 anti-mouse IgG + IgM (H+L) (final concentration, 10 μg/ml) in the buffer (n > 20). (X) Measurement of the calcium influx in A20 cells stimulated with beads conjugated goat F(ab′)2 anti-mouse IgG + IgM (H+L). (Y)Stim1, Stim1, Orai1, and Orai2 transcription levels in WT and tKO primary splenic B cells were determined by RT-qPCR (n = 3). Results were normalized to housekeeping gene of Gapdh. Unpaired two-tailed t test in H, K, M, O, P, and R. One-way ANOVA in B, D, and F; two-way ANOVA in S and Y. ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of at least two independent experiments (A–S and W–Y).
The effect of TRPV2 on B cell development, B cell signaling, and B cell function. (A and B) Representative flow plot (A) and quantification (B) of different B cell precursor populations in bone marrow. Bone marrow cells from the femur of C-WT (Trpv2+/+Mb1cre/+), C-Het (Trpv2+/−Mb1cre/+), and C-KO (Trpv2f/−Mb1cre/+) mice were stained with anti-B220 and anti-IgM and analyzed by flow cytometry (n = 3). Cell populations were defined as proB/preB (B220lo IgM−), immature (B220lo IgM+), and mature (B220+ IgM+). (C and D) Representative flow plot (C) and quantification (D) of the T cell population (CD3+) and B cell population (B220+) in live splenic cells from C-WT, C-Het, and C-KO mice (n = 3). (E and F) Representative flow plot (E) and quantification (F) of B cell subsets in live splenic cells from C-WT, C-Het, and C-KO mice (n = 3). Cell populations were defined as follicular B (B220+CD23loCD21hi), marginal zone B (B220+CD21loCD23hi). (G and H) Representative flow plot (G) and quantification (H) of B1 cell subsets in the peritoneal cavity from C-WT and C-KO mice (n = 3). B1 cell populations were defined as B220+CD23−CD5+. (I) NP-specific IgG antibody production was detected on day 14 and day 21. C-WT and C-KO mice were immunized on day 0 and day 14 with NP32-KLH (n = 3). (J–O) C-WT and C-KO mice were immunization with OVA (100 μg/mice) emulsified in aluminum adjuvant on day 0 and day 14 (n = 3). Representative flow plot (J) and quantification (K) of B cell population in draining lymph node (dLN) collected on day 21. Representative flow plot (L) and quantification (M) of GCB cell (B220+GL7+ CD95+) population in dLN collected on day 21. Representative flow plot (N) and quantification (O) of antigen-specific GCB cell (OVA+) population in dLN collected on day 21. (P) Total IgM and IgG antibody production in C-WT and C-KO mice was detected on day 14 after immunization with NP32-KLH (n = 3). (Q and R) Representative flow plot (Q) and quantification (R) of B cell population in the spleen of μMT mice collected on day 3. μMT mice were transferred with 1 × 106 B cells purified from the spleen of C-WT or C-KO mice (n = 3) on day 0. (S) Percentage of live WT or TRPV2 KO primary B cells without stimulation at the indicated time. (n = 3). (T–V) Analysis of huTrpv1, huTrpv3, and huTrpv4 transcription level in a single-cell RNA-seq database GSE174188 including 162 SLE patients and 99 healthy subjects, all Asian or Caucasian from San Francisco, CA, USA. (W) Measurement of the calcium influx in A20 cells stimulated with surface-associated goat F(ab′)2 anti-mouse IgG + IgM (H+L) (final concentration, 10 μg/ml) in the buffer (n > 20). (X) Measurement of the calcium influx in A20 cells stimulated with beads conjugated goat F(ab′)2 anti-mouse IgG + IgM (H+L). (Y)Stim1, Stim1, Orai1, and Orai2 transcription levels in WT and tKO primary splenic B cells were determined by RT-qPCR (n = 3). Results were normalized to housekeeping gene of Gapdh. Unpaired two-tailed t test in H, K, M, O, P, and R. One-way ANOVA in B, D, and F; two-way ANOVA in S and Y. ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of at least two independent experiments (A–S and W–Y).
We first immunized the mice with NP-lipopolysaccharide (LPS), a type-1 T cell–independent (TI-1) model antigen, and monitored NP-specific antibody titer 10 days after immunization. TRPV2 C-KO mice produced reduced amounts of both NP-specific IgM and IgG antibodies after NP-LPS immunization (Fig. 2, H and I). A similar result was acquired when using a type-2 TI (TI-2) model antigen, TNP-Ficoll (Fig. 2 J). The importance of TRPV2 in antibody responses was further validated by using a T cell–dependent model antigen, 4-hydroxy-3-nitrophenylacetyl, conjugated to keyhole limpet hemocyanin (NP-KLH). TRPV2 C-KO mice showed a low level of IgG antibodies after NP-KLH immunization (Fig. 2 K). Moreover, we found that TRPV2 C-KO mice exhibited dramatically low NP-specific IgG titers after NP-KLH boosting (Fig. S2 I). We further showed that TRPV2 KO reduced antigen-specific GCB population without affecting total B cell population after immunization (Fig. S2, J–O). Also, TRPV2 KO does not affect total (or innate) IgM and IgG antibodies in sera (Fig. S2 P).
To exclude the possibility of a potential leaky Cre expression driven by the Mb1 promoter (Hobeika et al., 2006), we adoptively transferred splenic B cells from WT or total TRPV2 KO mice respectively into μMT mice, a B cell–null strain. In this setting, whereas mice complemented with WT splenic B cells exhibited a robust response to T cell–dependent antigen (NP32-KLH), it was surprising that the response in mice complemented with TRPV2 KO splenic B cells almost failed to respond upon vaccination (Fig. 2, L and M). To further check this drastic result, we used an alternative strategy to adoptively transfer the same number of NP-reactive naive B cells from B1-8hi genetically targeted mice that represent a recombined antibody variable region derived from an NP hapten binding antibody, without or with TRPV2 deficiency (referred to as B1-8-TRPV2-WT or B1-8-TRPV2-KO, respectively, thereafter) into μMT mice. As another layer of evidence, the response of recipient mice B1-8-TRPV2-KO B cells was also severely impaired compared with the response of control recipient mice with B1-8-TRPV2-WT B cells after immunization with TI-antigen NP-Ficoll (Fig. 2, N and O). To check the contributing factors for this dramatic difference in the adoptive transfer assay, far exceeding the difference in the TRPV2 C-KO vs. TRPV2 C-Het comparison, we first checked the engraftment of TRPV2 WT vs. KO B cells upon transfer in μMT mice. We found that there was only one third of the number of TRPV2 KO B cells recovered in the spleen compared with WT B cells (Fig. S2, Q and R). We then detected primary B cell survival in vitro and found that B cell–intrinsic TRPV2 had no effect on quiescent B cell survival in culture medium (Fig. S2 S). These data indicated that TRPV2 influenced B cell engraftment but not basal cell survival without the presence of stimulating antigens. These results likely suggest that TRPV2 may be involved in the cytoskeleton remodeling–mediated cell migration as mentioned in the later part of this report. Given that the NP-specific antibody production in mice-transferred TRPV2 KO B cells was only one tenth compared with that in mice-transferred WT B cells (Fig. 2, N and O), far exceeding the difference of the recovered B cells upon transfer, here we speculate that the essentially impaired antibody response in μMT mice adoptively transferred TRPV2 KO B cells shall be contributed by both the decreased BCR signaling and the reduced engraftment of B cells.
All the above antibody responses were induced and compared using model antigens. To further pursue the physiological significance of these findings, we used a natural pathogen antigen, pneumococcal capsular polysaccharide. As a typical TI-2 multivalent molecule, pneumococcal capsular polysaccharide displays repeating structures and is clinically important for the development of pneumococcal vaccines (Daniels et al., 2016). We immunized the mice with two TI-2 antigens, the type 1 (CPS1) and type 3 (CPS3) pneumococcal capsular polysaccharides. As expected, TRPV2 C-KO mice produced diminished amounts of CPS1- and CPS3-specific IgM compared to both Trpv2+/−Mb1cre/+ and Trpv2+/+Mb1cre/+ control mice (Fig. 2 P). Collectively, these data demonstrate that TRPV2 in B cells is essentially important for antibody responses for both T cell–independent (TI) and T cell–dependent (TD) antigens.
TRPV2 expression level correlates with vaccine antigen–induced antibody response
Infants have a high risk of infections in comparison with adults (Levy, 2007; Siegrist, 2001). Although the practice of vaccination significantly decreases the mortality, it has been a long-standing problem in basic and clinical immunology that infants who have received pneumococcal vaccine containing only pneumococcal capsular polysaccharide antigen usually exhibit suboptimal antibody response and thus only have limited protection (Saso and Kampmann, 2017). Since this type of antigen is a typical TI antigen, requiring more on the function of BCR-induced B cell activation, previous studies have shown that lower expression of key surface receptors including co-stimulatory factors (CD21, CD40, CD80, and CD86) in B cells from infants restricted humoral responses (Siegrist and Aspinall, 2009). Here, we speculated that suboptimal expression level of TRPV2 might be another reason restricting the efficiency of antibody response in infants. To address this hypothesis, we examined the TRPV2 expression level from children in this study by establishing a cohort containing 101 Chinese children who were recruited and categorized into six groups according to age: newborn (≤60-day infant); infant (2 mo to 1 year); 1–3 years; 3–5 years; 5–12 years; and 12–17 years. Indeed, the huTrpv2 transcription level was substantially lowest in newborns (≤60-day infant) compared with other groups (Fig. 3 A), and there were positive correlations between huTrpv2 transcription level and a certain range of age (0–1,500 days) (Fig. 3 B). Further, the correlation of TRPV2 expression with age from 0 to 1,500 days was also observed upon the analysis of a B cell–specific database, GSE185503, containing 17 healthy subjects all from Finland or Estonia (Fig. 3 C). As a further validation toward this intriguing finding, we also performed the same assay among mice of various ages. Trends of mTrpv2 transcription levels in mice were consistent with those in infants (Fig. 3 D). Thus, all these data indicate a close correlation between the expression level of TRPV2 and vaccine antigen–induced antibody response, potentially suggesting that the low expression level of TRPV2 in infants might also contribute to the suboptimal antibody response if using pneumococcal vaccine containing only pneumococcal capsular polysaccharide antigens. Based on these findings, we then considered that the senior humans usually exhibit compromised antibody responses upon vaccination, which drove us to check GSE231409 containing 76 healthy subjects, all from Durham, NC, USA. Strikingly, we found that TRPV2 expression in PBMC from old healthy individuals (>60) was dramatically lower than that from younger individuals (Fig. 3 E).

TRPV2 expression level correlates with vaccine antigen–induced antibody response. (A and B) huTrpv2 transcription levels in human PBMC from healthy volunteers. mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. One dot represents one individual (n = 101). (C) Correlation analysis of TRPV2 expression in B cells with age from 0 to 1,500 days in the B cell–specific database of GSE185503 containing 17 healthy subjects, all from Finland or Estonia. (D)muTrpv2 in mouse primary splenic cells. mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. Neonate (n = 3), 5 days after birth; infant (n = 3), 2 wk; juvenile (n = 3), 4 wk; adult (n = 3), 8 wk. Each symbol represents an individual person. (E)huTrpv2 expression in PBMC in GSE231409 containing 76 healthy subjects, all from Durham, NC, USA. (F) Schematic diagram of the human flu vaccination study. Healthy volunteers (age coverage, 18–28 years old) were vaccinated against influenza on day 7. Pre- and post-immune plasma were collected on day 0 and day 21. (G and H) Correlation analysis between influenza-specific IgG antibody response and huTrpv2 relative transcription level in vaccinated volunteers (male, n = 13; female, n = 8). (I and J) Correlation of TRPV2 expression and influenza vaccine-specific antibody responses. Data from GSE48018 including 119 healthy volunteers ages 19–41 years, all Caucasians from Bethesda, MD, USA. (K and L) The cellular location of TRPV2 and BCR in quiescent B cells. Representative confocal images. Scale bars, 1.5 μm (K). Distribution of MFI of BCR and TRPV2 in the cell membrane of quiescent B cells (L). (M and N) TRPV2 and BCR recruitment within immunological synapse of activated B cells. Representative TIRFM images of BCR (green) and TRPV2 (red) within the immunological synapse of primary B lymphocytes at the indicated time points after activation on coverslips presenting biotin conjugated goat F(ab′)2 anti-mouse IgG + IgM (H+L) antibodies. Scale bars, 1.5 μm (M). Quantification of total FI of BCR and TRPV2 in the immunological synapse (n > 25) (N). (O) Quantification of total FI of synaptic accumulated BCR in TRPV2 high expression and TRPV2 low expression cells (n > 40). Cells were stimulated with PLB antigen-presenting system. (P and Q) Diagrammatic representation of exogenous expression of TRPV2 in mice (P). Splenic B cells were purified from B1-8hi+/+ mice. After transfection with retrovirus expressing system, B cells with similar GFP fluorescence values were sorted. GFP+ or TRPV2-GFP+ cells were transferred into μMT mice respectively. Chimeric mice (nGFP = 3 and nTRPV2-GFP = 4) were immunized with NP50-Ficoll. Serum was collected on day 7 and measured anti-NP IgG titer after immunization (Q). Unpaired two-tailed t test in O and Q. One-way ANOVA with a Tukey’s multiple comparison test in A, D, and E. Pearson’s correlation analysis of regression analysis in B, C, and G–J. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are mean ± SEM. Data are representative of two (F and Q) and four (K–N) independent experiments, respectively.
TRPV2 expression level correlates with vaccine antigen–induced antibody response. (A and B) huTrpv2 transcription levels in human PBMC from healthy volunteers. mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. One dot represents one individual (n = 101). (C) Correlation analysis of TRPV2 expression in B cells with age from 0 to 1,500 days in the B cell–specific database of GSE185503 containing 17 healthy subjects, all from Finland or Estonia. (D)muTrpv2 in mouse primary splenic cells. mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. Neonate (n = 3), 5 days after birth; infant (n = 3), 2 wk; juvenile (n = 3), 4 wk; adult (n = 3), 8 wk. Each symbol represents an individual person. (E)huTrpv2 expression in PBMC in GSE231409 containing 76 healthy subjects, all from Durham, NC, USA. (F) Schematic diagram of the human flu vaccination study. Healthy volunteers (age coverage, 18–28 years old) were vaccinated against influenza on day 7. Pre- and post-immune plasma were collected on day 0 and day 21. (G and H) Correlation analysis between influenza-specific IgG antibody response and huTrpv2 relative transcription level in vaccinated volunteers (male, n = 13; female, n = 8). (I and J) Correlation of TRPV2 expression and influenza vaccine-specific antibody responses. Data from GSE48018 including 119 healthy volunteers ages 19–41 years, all Caucasians from Bethesda, MD, USA. (K and L) The cellular location of TRPV2 and BCR in quiescent B cells. Representative confocal images. Scale bars, 1.5 μm (K). Distribution of MFI of BCR and TRPV2 in the cell membrane of quiescent B cells (L). (M and N) TRPV2 and BCR recruitment within immunological synapse of activated B cells. Representative TIRFM images of BCR (green) and TRPV2 (red) within the immunological synapse of primary B lymphocytes at the indicated time points after activation on coverslips presenting biotin conjugated goat F(ab′)2 anti-mouse IgG + IgM (H+L) antibodies. Scale bars, 1.5 μm (M). Quantification of total FI of BCR and TRPV2 in the immunological synapse (n > 25) (N). (O) Quantification of total FI of synaptic accumulated BCR in TRPV2 high expression and TRPV2 low expression cells (n > 40). Cells were stimulated with PLB antigen-presenting system. (P and Q) Diagrammatic representation of exogenous expression of TRPV2 in mice (P). Splenic B cells were purified from B1-8hi+/+ mice. After transfection with retrovirus expressing system, B cells with similar GFP fluorescence values were sorted. GFP+ or TRPV2-GFP+ cells were transferred into μMT mice respectively. Chimeric mice (nGFP = 3 and nTRPV2-GFP = 4) were immunized with NP50-Ficoll. Serum was collected on day 7 and measured anti-NP IgG titer after immunization (Q). Unpaired two-tailed t test in O and Q. One-way ANOVA with a Tukey’s multiple comparison test in A, D, and E. Pearson’s correlation analysis of regression analysis in B, C, and G–J. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are mean ± SEM. Data are representative of two (F and Q) and four (K–N) independent experiments, respectively.
To further confirm the relationship between B cell function and TRPV2 expression level in humans, we established a cohort enrolling 21 healthy volunteers who received seasonal influenza vaccine administration in this study (Fig. 3 F). Correlation analysis revealed that there were significant and positive correlations between influenza virus-specific IgG antibody titer and TRPV2 expression levels both in male and female individuals (Fig. 3, G and H). To explicitly clarify the correlation of TRPV2 and influenza vaccine-specific antibody responses, we further analyzed GSE48018, including 119 healthy volunteers, all Caucasians from Bethesda, MD, USA. We found that TRPV2 expression level was mildly but still positively correlated with H1N1 antibody titer (Fig. 3, I and J). As an additional layer of evidence, we visualized the membrane location and expression level of TRPV2 on individual live B cells by immunofluorescence staining using anti-TRPV2 antibody. In resting B cells, TRPV2 was evenly distributed on the plasma membrane (Fig. 3, K and L). Upon surrogate antigen stimulation, TRPV2, similar to BCR, was engaged in the B cell immunological synapse (Fig. 3, M and N). We reanalyzed the aforementioned TIRFM imaging data by dividing B cells into two groups, TRPV2 level high and TRPV2 level low respectively, which were determined by the median fluorescence intensity of TRPV2. Subsequently, we compared the BCR accumulation within the B cell immunological synapse. Consistently, the results showed that cells with higher levels of TRPV2 recruited more amount of BCRs into the B cell immunological synapse (Fig. 3 O). To further examine the functional meanings of this positive correlation, we utilized a pMSCV (murine stem cell virus) retroviral vector system to overexpress TRPV2 in NP-reactive splenic primary B cells from B1-8hi+/+ mice and observed that TRPV2-GFP positive group showed enhanced antibody response after immunization with NP-Ficoll (Fig. 3, P and Q). Collectively, all these data concluded that TRPV2 expression level correlates with vaccine antigen–induced antibody response.
TRPV2 expression level positively correlates with clinical manifestations of both adult and children SLE
As hyperactivation of B cells is often positively correlated to the occurrence of autoimmune diseases, in this study, we established a cohort containing adult SLE patients and matched healthy volunteers from China to determine the correlations between TRPV2 expression and lupus-associated autoantibody levels. Indeed, the TRPV2 mRNA expression level in SLE patients was markedly higher than that in healthy volunteers (Fig. 4 A). As anti-dsDNA antibody is an important indicator for diagnosing SLE and judging disease activity, we stratified all the SLE patients from the cohort in this study into two groups, namely the anti-dsDNA autoantibody-positive group versus the autoantibody-negative group, and found that the expression level of TRPV2 in anti-dsDNA positive group was significantly higher than that in negative group (Fig. 4 B). In addition, there is a significant and positive correlation between anti-dsDNA autoantibody and the mRNA expression level of TRPV2 (Fig. 4 C). Correspondingly, we also stratified SLE patients from the cohort in this study into three groups dependent on TRPV2 expression level and observed that patients with higher TRPV2 expression level displayed a higher ratio of positive anti-dsDNA autoantibody (Fig. 4 D), positive anti-Sm antibody (Fig. 4 E), and positive direct Coombs test (Fig. 4 F), all of which are characterized as SLE clinical manifestations. Since the SLEDAI is an integrated model of experienced clinicians’ global assessments of disease activity in SLE, we next analyzed the correlation between SLEADI and TRPV2 expression and found that patients with low TRPV2 expression displayed very few or even no severe SLEDAI subgroup. Patients with higher TRPV2 expression carried a significant enrichment of moderate and severe SLEDAI subgroups (Fig. 4 G).

The expression level of TRPV2 correlates with clinical manifestations of SLE progression. (A and B) huTrpv mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. (A)huTrpv2 transcription level in peripheral blood cells of adult SLE patients (n = 24) and healthy volunteers (n = 32). (B)huTrpv2 transcription level in peripheral blood cells of adult SLE patients (n = 48) whose anti-dsDNA autoantibody represented negative or positive. (C) Correlation analysis between anti-dsDNA autoantibody and huTrpv2 relative transcription level in SLE patients with positive anti-dsDNA autoantibody (n = 48). (D–G) Clinical traits of SLE patients (n = 48). The anti-dsDNA antibody (D), anti-Sm antibody (E), direct Coombs test (F), and the SLEDAI (G) analysis of SLE patients, who were divided into three groups with high (>75%), medium (25–75%), and low (<25%) TRPV2 mRNA expression according to the quartile method. The disease activity of SLE patients was defined on the basis of the SLEDAI score: inactive, SLEDAI ≤ 4; mild, 5 ≤ SLEDAI ≤ 9; moderate, 10 ≤ SLEDAI ≤ 14; and severe, SLEDAI ≥ 15. (H–L) Analysis of child SLE patients’ clinical data from GEO database GSE65391 (nsample = 972 from 158 SLE patients). (H)huTrpv2 transcription level of PBMC from SLE patients whose anti-dsDNA autoantibody represented negative (nsample = 33) or positive (nsample = 615). (I) Correlation analysis between anti-dsDNA autoantibody and huTrpv2 relative transcription level in SLE patients with positive anti-dsDNA autoantibody (nsample = 615). The SLEDAI score (J, nsample = 924), plasma C3 value (K, nsample = 894), and neutrophile count (L, nsample = 854) analysis of SLE patient samples, who were divided into three groups with high (greater than mean + SD), medium (mean − SD to mean + SD), and low (less than mean − SD) TRPV2 mRNA expression according to the quartile method. (M–P) Analysis of SLE patients’ clinical data from GEO database GSE72798 including 10 SLE Caucasian patients and 9 Caucasian healthy subjects from Brussels, Belgium. huTrpv2 transcription level of PBMC in SLE patients and HC (M). Correlation analysis between huTrpv2 expression and SLEDAI score (N), anti-dsDNA antibody level (O), and the glomerular filtration rate (P). (Q) Analysis of huTrpv2 transcription level of PBMC in SLE patients and HC in GEO database GSE185047 containing 177 SLE Caucasian patients and 10 Caucasian healthy subjects from Brussels, Belgium. (R and S) Analysis of huTrpv1, huTrpv2, huTrpv3, and huTrpv4 transcription level of B cells in GEO database GSE92387 including 12 SLE patients and 12 healthy subjects, all African American or European American from Georgia, USA. (T) Analysis of huTrpv2 transcription level of memory B cells, plasmablast, and naïve B cells in the GSE156751 database including four SLE patients and four healthy subjects, all from Miyagi, Japan. (U) Analysis of huTrpv2 transcription level in a single-cell RNA-seq database GSE174188 including 162 SLE patients and 99 healthy subjects, all Asian or Caucasian from San Francisco, CA, USA. (V and W) Analysis of SLEDAI score (V) and TRPV2 expression value (W) in SLE patients who have clinical information both before and after treatment (n = 25) in GSE65391. Patients here were selected from those without treatment at the first visit and after treatment at the second visit. Data information was collected from visit 1 and visit 2 of those selected patients (n = 25). (X) Analysis of huTrpv2 transcription level before and after treatment in SLE patients. Data from GSE72798 containing PBMC RNA-seq data of 10 SLE Caucasian patients from Brussels, Belgium before and 3- or 6-mo after the treatment. (Y) Analysis of huTrpv2 transcription level before and after treatment in SLE patients. Data from GSE185047 containing RNA-seq data of SLE patients before (0-wk), 12-, 24- and 36-wk after the treatment with a drug under clinical trial, IFN-K (nplacebo = 93; nIFN-K = 92). (Z) Anti-dsDNA IgG and anti-Sm D IgG antibodies were detected on day 28. C-WT (n = 3) and C-KO (n = 3) mice were adoptively transferred intravenously with splenocytes (107 per mouse) from bm12 mice on day 0 and day 14, respectively. Pearson’s correlation analysis of regression analysis in C, I, L, and N–P. Unpaired two-tailed t test in A, B, H, M, Q, and X–Z. Paired two-tailed t test in V and W. One-way ANOVA with a Tukey’s multiple comparison test in J, K, and S–U. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are mean ± SEM. Data in Z are representative of two independent experiments.
The expression level of TRPV2 correlates with clinical manifestations of SLE progression. (A and B) huTrpv mRNAs were assayed by RT-qPCR. Results were normalized to the housekeeping gene of Gapdh. (A)huTrpv2 transcription level in peripheral blood cells of adult SLE patients (n = 24) and healthy volunteers (n = 32). (B)huTrpv2 transcription level in peripheral blood cells of adult SLE patients (n = 48) whose anti-dsDNA autoantibody represented negative or positive. (C) Correlation analysis between anti-dsDNA autoantibody and huTrpv2 relative transcription level in SLE patients with positive anti-dsDNA autoantibody (n = 48). (D–G) Clinical traits of SLE patients (n = 48). The anti-dsDNA antibody (D), anti-Sm antibody (E), direct Coombs test (F), and the SLEDAI (G) analysis of SLE patients, who were divided into three groups with high (>75%), medium (25–75%), and low (<25%) TRPV2 mRNA expression according to the quartile method. The disease activity of SLE patients was defined on the basis of the SLEDAI score: inactive, SLEDAI ≤ 4; mild, 5 ≤ SLEDAI ≤ 9; moderate, 10 ≤ SLEDAI ≤ 14; and severe, SLEDAI ≥ 15. (H–L) Analysis of child SLE patients’ clinical data from GEO database GSE65391 (nsample = 972 from 158 SLE patients). (H)huTrpv2 transcription level of PBMC from SLE patients whose anti-dsDNA autoantibody represented negative (nsample = 33) or positive (nsample = 615). (I) Correlation analysis between anti-dsDNA autoantibody and huTrpv2 relative transcription level in SLE patients with positive anti-dsDNA autoantibody (nsample = 615). The SLEDAI score (J, nsample = 924), plasma C3 value (K, nsample = 894), and neutrophile count (L, nsample = 854) analysis of SLE patient samples, who were divided into three groups with high (greater than mean + SD), medium (mean − SD to mean + SD), and low (less than mean − SD) TRPV2 mRNA expression according to the quartile method. (M–P) Analysis of SLE patients’ clinical data from GEO database GSE72798 including 10 SLE Caucasian patients and 9 Caucasian healthy subjects from Brussels, Belgium. huTrpv2 transcription level of PBMC in SLE patients and HC (M). Correlation analysis between huTrpv2 expression and SLEDAI score (N), anti-dsDNA antibody level (O), and the glomerular filtration rate (P). (Q) Analysis of huTrpv2 transcription level of PBMC in SLE patients and HC in GEO database GSE185047 containing 177 SLE Caucasian patients and 10 Caucasian healthy subjects from Brussels, Belgium. (R and S) Analysis of huTrpv1, huTrpv2, huTrpv3, and huTrpv4 transcription level of B cells in GEO database GSE92387 including 12 SLE patients and 12 healthy subjects, all African American or European American from Georgia, USA. (T) Analysis of huTrpv2 transcription level of memory B cells, plasmablast, and naïve B cells in the GSE156751 database including four SLE patients and four healthy subjects, all from Miyagi, Japan. (U) Analysis of huTrpv2 transcription level in a single-cell RNA-seq database GSE174188 including 162 SLE patients and 99 healthy subjects, all Asian or Caucasian from San Francisco, CA, USA. (V and W) Analysis of SLEDAI score (V) and TRPV2 expression value (W) in SLE patients who have clinical information both before and after treatment (n = 25) in GSE65391. Patients here were selected from those without treatment at the first visit and after treatment at the second visit. Data information was collected from visit 1 and visit 2 of those selected patients (n = 25). (X) Analysis of huTrpv2 transcription level before and after treatment in SLE patients. Data from GSE72798 containing PBMC RNA-seq data of 10 SLE Caucasian patients from Brussels, Belgium before and 3- or 6-mo after the treatment. (Y) Analysis of huTrpv2 transcription level before and after treatment in SLE patients. Data from GSE185047 containing RNA-seq data of SLE patients before (0-wk), 12-, 24- and 36-wk after the treatment with a drug under clinical trial, IFN-K (nplacebo = 93; nIFN-K = 92). (Z) Anti-dsDNA IgG and anti-Sm D IgG antibodies were detected on day 28. C-WT (n = 3) and C-KO (n = 3) mice were adoptively transferred intravenously with splenocytes (107 per mouse) from bm12 mice on day 0 and day 14, respectively. Pearson’s correlation analysis of regression analysis in C, I, L, and N–P. Unpaired two-tailed t test in A, B, H, M, Q, and X–Z. Paired two-tailed t test in V and W. One-way ANOVA with a Tukey’s multiple comparison test in J, K, and S–U. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are mean ± SEM. Data in Z are representative of two independent experiments.
To further verify the correlation between TRPV2 expression and SLE progression, we reanalyzed the SLE data from the GEO database GSE65391. These data were collected mainly from African Americans, Caucasians, and Hispanic child SLE (cSLE) patients. Similarly, we also found that the expression level of TRPV2 in the anti-dsDNA antibody positive group was significantly higher than that in the negative group (Fig. 4 H), and there were significant and positive correlations between anti-dsDNA autoantibody and TRPV2 expression level (Fig. 4 I). We also found SLEDAI score in the TRPV2-high group was considerably higher than that in the TRPV2-low group (Fig. 4 J). Moreover, the plasma C3 level in the TRPV2-high group was dramatically lower than that in the TRPV2-low and TRPV2-medium groups (Fig. 4 K), and neutrophile count was significantly and negatively correlated with TRPV2 expression (Fig. 4 L), as C3 level and neutrophile count were also prominent markers for cSLE disease activity besides anti-dsDNA autoantibody. To strengthen the result of the positive correlation between TRPV2 expression and SLE progression, we further analyzed several other SLE-cohort databases that are publicly available. Consistent with previous observation, when analyzing GEO database GSE72798 including 10 SLE Caucasian patients and 9 Caucasian healthy subjects from Brussels, Belgium, we found that TRPV2 expression was significantly higher in the PBMC from SLE patients than in the healthy control (HC) population (Fig. 4 M). Moreover, this analysis also revealed that TRPV2 expression was positively correlated with SLEDAI score (Fig. 4 N), the anti-dsDNA antibody level (Fig. 4 O), and negatively correlated with the glomerular filtration rate (Fig. 4 P). When analyzing another database with a larger cohort, GSE185047, containing 177 SLE Caucasian patients and 10 Caucasian healthy subjects from Brussels, Belgium, we also found that TRPV2 expression was significantly higher in the PBMC from SLE patients in comparison with that from the HC population (Fig. 4 Q). All these results demonstrate a positive correlation between TRPV2 level and SLE progression.
All the above databases that have been analyzed were obtained by RNA-seq total immune cells in PBMC from SLE and HC. To better illustrate this correlation between TRPV2 and B cells, we then analyzed the B cell–specific database of GSE92387 including 12 SLE patients and 12 healthy subjects, all African American or European American from Georgia, USA (Fig. 4, R and S). Consistently, we found that only TRPV2 was highly expressed in B cells among all four canonical TRPV channel members, and the level of TRPV2 was significantly higher in the B cells from SLE patients compared with those from HC (Fig. 4, R and S). Additionally, we examined major B cell subsets by analyzing the GSE156751 database, including four SLE patients and four healthy subjects, all from Miyagi, Japan, and found that the upregulated expression pattern of TRPV2 in SLE is more significant in memory B cells, modest in plasmablast, while only mild in naïve B cells (Fig. 4 T). Taking advantage of the recently emerging single-cell RNA-seq technique, our analyses of database GSE174188 including 162 SLE patients and 99 healthy subjects, all Asian or Caucasian from San Francisco, CA, USA, further validated that only TRPV2 was highly expressed in B cells (Fig. 4 U), whereas all other canonical TRPV channel members were almost undetectable (Fig. S2, T–V). Consistently, the level of TRPV2 was significantly higher in B cells of SLE patients than in HC (Fig. 4 U), mirroring the aforementioned results of bulky RNA-seq in PBMC (Fig. 4, M and Q) and sorted B cells (Fig. 4, S and T). More importantly, further analyses of the aforementioned GSE174188 revealed that TRPV2 expression in the B cells of SLE patients decreased upon treatment with one or more medicines including oral steroids, azathioprine, mycophenolate mofetil, hydroxychloroquine, methotrexate, and calcineurin inhibitor (Fig. 4 U).
To confirm the reduced TRPV2 levels in SLE patients upon clinical treatment, we also reanalyzed the aforementioned cSLE patients (GSE65391) who have clinical records before and after treatment (n = 25). The treatment included one of the following regimens: hydroxychloroquine only, oral steroid, mycophenolate + any, and cyclophosphamide and/or intravenous injection (IV) steroid. The analyses showed that after treatment, TRPV2 expression level of cSLE patients was markedly decreased, as well as the SLEDAI score (Fig. 4, V and W). To further characterize the expression level of TRPV2 in SLE patients upon therapy, we analyzed the database GSE72798 containing PBMC RNA-seq data of 10 SLE Caucasian patients from Brussels, Belgium before and 3- or 6-mo after the treatment by conventional immunosuppressive drugs. This immunosuppressive therapy contained high-dose corticosteroids, IV cyclophosphamide, or oral mycophenolate during the first 3 mo, followed by maintenance with moderate- to low-dose corticosteroids, azathioprine, or mycophenolate. We found that the level of TRPV2 was significantly decreased in SLE patients at the time point of month 3, mildly low at month 6 (Fig. 4 X), likely reflecting a trend of TRPV2 level recovery after long-term treatment per our speculation. This thought is supported by our analyses of the database GSE185047, containing RNA-seq data of SLE patients before (0-wk), 12-, 24- and 36-wk after treatment with a drug under clinical trial, interferon-α kinoid (IFN-K). We are surprised to find that there was simply no change in TRPV2 expression in the SLE patients treated with IFN-K (Fig. 4 Y). These data highlighted that TRPV2 is only sensitive to conventional immunosuppressive therapy as described in detail above, rather than IFN-K, which is not Food and Drug Administration (FDA)–approved, but only under clinical trial. To experimentally link TRPV2 to the pathogenesis of SLE, we also performed BM12-induced SLE model in B cell conditioned KO TRPV2 (C-KO) mice as a further validation and found that anti-dsDNA IgG antibody and anti-SmD IgG antibody both diminished in TRPV2 C-KO mice than in C-Het control mice (Fig. 4 Z), consistent with the results in all the human SLE cohorts. Collectively, all these data showed a positive correlation between TRPV2 expression and the clinical manifestations of both adult and children SLE.
The function of TRPV2 in B cells is dependent on its pore-forming region and N-terminal ANK domain
We tried to investigate the molecular mechanisms underlying the requirement for TRPV2 in B cell function by constructing and analyzing a series of TRPV2 mutants based on recent TRPV2 structural and biochemical advances (Huynh et al., 2016; Zubcevic et al., 2016). A schematic cartoon is given to show the overall architecture of TRPV2 and the design of the mutants (Fig. 5, A and B). These TRPV2 mutants were respectively overexpressed in TRPV2-KO A20B1-8 B cells in parallel with WT TRPV2 as a functional control. Neither the total expression levels nor the surface localization of TRPV2 appeared to be affected by the deletion of different functional domains (Fig. 5 C). Quantification by TIRFM imaging indicated that neither C-terminal deletion (CTD) nor calmodulin binding site deletion prevented the formation of B cell immunological synapse (Fig. 5 D). Similarly, the mutation of Arg682, which is a key residue for PIP2 binding within the C termini of TRPV2, did not affect the formation of the B cell immunological synapse (Fig. 5 E). Notably, the TRPV2 mutant with pore-forming region deletion (PoreD) failed to restore the formation of the B cell immunological synapse (Fig. 5 E). In addition, N-terminal deletions (NTD∆2–77 and PN∆34–55) exhibited loss-of-function features in mediating the formation of B cell immunological synapse (Fig. 5 E). Glu594 and Glu604 are conserved sites contributing to the selectivity filter for TRPV2 cation gating (Iwata et al., 2009). To further confirm the contribution of the pore region of TRPV2 in B cell function, we therefore overexpressed these two single-site mutants of TRPV2, Glu594, and Glu604, in TRPV2-KO A20B1-8 B cells by converting these two Glu residues into Lys, respectively. The results showed that E604K but not E594K mutation significantly diminished the synaptic recruitment of BCR during the initiation of B cell activation (Fig. 5 F). Moreover, we also found that TRPV2 with N-terminal ANK domain deletion impaired the formation of B cell immunological synapse (Fig. 5 G), consistent with the suggestion that the ANK domain within the N termini is essential for mechanosensation of TRP channels (Jin et al., 2017; Lee et al., 2006; Lishko et al., 2007; Zhang et al., 2015). Together, these data indicated that the function of TRPV2 on B cell activation is dependent on both the pore-forming region and N-terminal ANK domain.

TRPV2 function is dependent on its pore and N-terminal regions and mediates membrane potential depolarization and cytoskeleton remodeling in B cells. (A and B) Schematic cartoon of TRPV2 and its mutants in this study. The white color represents the deletion or mutation region (B). (C) The cellular location of different mutants of TRPV2 in A20 II1.6 B1-8 IgM cells was determined by the total FI ratio of TRPV2 at plasma membrane to TRPV2 in the cytosol (n > 40). (D–G) Quantification of MFI of synaptic accumulated BCR in A20 II1.6 cells (WT), TRPV2-deficient A20 II1.6 cells (KO), TRPV2-deficient A20 II1.6 cells expressing full-length TRPV2 (WT+), TRPV2-deficient A20 II1.6 cells expressing different mutants of TRPV2 (CTD+, MPD+, CaM+) (D), (NTD+, PN+, PoreD+, R682A+) (E), (E594K+, E604K+) (F), (ANK+) (G) (n > 30). Cells were stimulated with PLB antigen-presenting system. (H) Representative confocal images showing dynamics of plasma membrane potential of activated primary B cells. Cells were prestained with FLIPR and loaded on coverslips with PLB antigen-presenting system for 3 min. Scale bars, 1.5 μm. (I) Quantification of fold change of maximum FLIPR FI (n > 10). (J) Quantification of the MFI of BCR accumulation in immunological synapse during B cell activation in the presence of NaCl or using NMDG in place of NaCl in buffer (n > 30). (K) Quantification of the MFI of BCR accumulation in immunological synapse. B cells were stimulated with PLB antigen-presenting system in regular KCl buffer (WT or KO) or with 50 mM KCl buffer (WT+K or KO+K) (n > 30). (L) Representative confocal images of F-actin and MTOC in A20II1.6 cells activated by PLBs with NP-BSA for 20 min. Scale bars, 1.5 μm. (M) The percentage of MTOC-positive cells (n > 20). (N) Cellular colocalization of different versions of TRPV2 with α-tubulin. Quantification of PCI between TRPV2 and α-tubulin in the plane near immunological synapse (n > 20). One-way ANOVA in D–G, K, and N. Unpaired two-tailed t test in I and J. ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of three independent experiments.
TRPV2 function is dependent on its pore and N-terminal regions and mediates membrane potential depolarization and cytoskeleton remodeling in B cells. (A and B) Schematic cartoon of TRPV2 and its mutants in this study. The white color represents the deletion or mutation region (B). (C) The cellular location of different mutants of TRPV2 in A20 II1.6 B1-8 IgM cells was determined by the total FI ratio of TRPV2 at plasma membrane to TRPV2 in the cytosol (n > 40). (D–G) Quantification of MFI of synaptic accumulated BCR in A20 II1.6 cells (WT), TRPV2-deficient A20 II1.6 cells (KO), TRPV2-deficient A20 II1.6 cells expressing full-length TRPV2 (WT+), TRPV2-deficient A20 II1.6 cells expressing different mutants of TRPV2 (CTD+, MPD+, CaM+) (D), (NTD+, PN+, PoreD+, R682A+) (E), (E594K+, E604K+) (F), (ANK+) (G) (n > 30). Cells were stimulated with PLB antigen-presenting system. (H) Representative confocal images showing dynamics of plasma membrane potential of activated primary B cells. Cells were prestained with FLIPR and loaded on coverslips with PLB antigen-presenting system for 3 min. Scale bars, 1.5 μm. (I) Quantification of fold change of maximum FLIPR FI (n > 10). (J) Quantification of the MFI of BCR accumulation in immunological synapse during B cell activation in the presence of NaCl or using NMDG in place of NaCl in buffer (n > 30). (K) Quantification of the MFI of BCR accumulation in immunological synapse. B cells were stimulated with PLB antigen-presenting system in regular KCl buffer (WT or KO) or with 50 mM KCl buffer (WT+K or KO+K) (n > 30). (L) Representative confocal images of F-actin and MTOC in A20II1.6 cells activated by PLBs with NP-BSA for 20 min. Scale bars, 1.5 μm. (M) The percentage of MTOC-positive cells (n > 20). (N) Cellular colocalization of different versions of TRPV2 with α-tubulin. Quantification of PCI between TRPV2 and α-tubulin in the plane near immunological synapse (n > 20). One-way ANOVA in D–G, K, and N. Unpaired two-tailed t test in I and J. ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001. Data are mean ± SEM. Data are representative of three independent experiments.
TRPV2 is vital for B cell activation by facilitating membrane potential depolarization and cytoskeleton remodeling
The results presented above provide evidence that the function of TRPV2 in mediating the formation of B cell immunological synapse is dependent on its pore region’s capability for the permeation of cations, which consequently shall lead to membrane potential depolarization (Cahalan and Chandy, 2009; Panyi et al., 2004). Accordingly, we examined the membrane potential changes in WT and TRPV2 KO B cells. We used the dye FLIPR to measure membrane potential changes in B cells in response to antigen stimulation. Fluctuation in cell membrane potential leads to changes in fluorescent intensity of FLIPR (Lee et al., 2008). As increased fluorescent signal intensity indicates membrane potential depolarization, TRPV2 KO abolished the depolarized peak after B cell activation (Fig. 5, H and I). As reported, sodium influx and potassium efflux exchanges result in the membrane potential depolarization; we thus further examined the requirement of TRPV2-mediated sodium influx for the formation of B cell immunological synapse. B cells were activated in a buffer with N-methyl-D-glucamine (NMDG) substituting sodium. Due to its large size, NMDG is less permeant through TRPV2 than smaller cations, resulting in reduced membrane potential depolarization (Link et al., 2010; Vale et al., 2006). Indeed, TIRFM imaging showed that, in the presence of NMDG, the formation of the B cell immunological synapse was impaired (Fig. 5 J). To further confirm the contribution of depolarization mediated by TRPV2, we used a high concentration of KCl to trigger an artificial depolarization. Accordingly, the buffer with an additional supplement of 50 mM KCl restored the formation of the B cell immunological synapse in TRPV2 KO B cells (Fig. 5 K). Ca2+, as one of the highly enriched cations out of the cell, is also permeable for TRPV2, entitled a name of non-selective cation TRP channel. Indeed, we observed a reduced Ca2+ flux in B cells deficient for TRPV2 upon antigen stimulation (Fig. S2, W and X). Different from our initial speculation, the Ca2+ influx is not totally blunted but only exhibited a certain level of reduction in TRPV2 KO B cells than in WT control B cells. We thus also evaluated the transcription level of other ion channels by RT-qPCR. Unexpectedly, the expression levels of Stim1, Orai1, and Orai2 were mildly increased in TRPV2 KO B cells (Fig. S2 Y). It is our opinion that these partially explain why Ca2+ influx did not drop to the basal level upon TRPV2 KO in B cells.
Recent studies have shown that cation flux–mediated membrane potential depolarization is closely interlinked and essential for cytoskeletal remodeling (Kang et al., 2014; Su et al., 2011). The connection between ion channels and the cytoskeleton, especially the tubulin-based cytoskeleton, is considered a potential mechanism of mechanosensation by TRP channels (Martinac, 2014; Sachs, 1991). Additionally, depolymerization of actin filaments also appears to be coupled with ion channel activation or inactivation, as many ion channels have shown affinity for cytoskeletal compartments (Els and Chou, 1993; Mazzochi et al., 2006; Rotin et al., 1994). Sodium ions are responsible for the salt influx, and a higher level of influx augments the salt concentration within membrane proximal regions, which can disassociate actin-binding proteins (Senju et al., 2017). With consideration of all these points, we first examined both F-actin remodeling and the microtubule-organizing center (MTOC), the latter of which reorients and approaches the vicinity of the plasma membrane during the formation of the immunological synapse. In this assay, B cells upon stimulation showed an obvious F-actin restriction, resulting in a negatively correlated colocalization with BCR (Fig. S3, A and B), while RR treatment disrupted the F-actin restriction and negative correlation (Fig. S3, A–C). TRPV2 KO cells exhibited disruption of the F-actin restriction (Fig. 5 L), which was similar to that of WT cells treated with RR (Fig. S3 A). These data demonstrated that TRPV2 is essential for F-actin remodeling.

Pharmacological blocking TRPV channels interrupts cytoskeleton mobilization. (A) Representative TIRFM images of F-actin and BCR location. Cells were stimulated with PLB antigen-presenting system in the absence or presence of RR (10 μM). Scale bars, 1.5 μm. (B) Cellular colocalization of TRPV2 and F-actin with or without RR (n > 40). Quantification of PCI between TRPV2 and F-actin. (C) Quantification of MFI of F-actin accumulation in immunological synapse at the indicated time points (n > 40). Cells were stimulated with PLB antigen-presenting system in the absence or presence of RR (10 μM). Unpaired two-tailed t test in B and C. ns, not significant, ***P < 0.001. Data are mean ± SEM. Data are representative of three independent experiments.
Pharmacological blocking TRPV channels interrupts cytoskeleton mobilization. (A) Representative TIRFM images of F-actin and BCR location. Cells were stimulated with PLB antigen-presenting system in the absence or presence of RR (10 μM). Scale bars, 1.5 μm. (B) Cellular colocalization of TRPV2 and F-actin with or without RR (n > 40). Quantification of PCI between TRPV2 and F-actin. (C) Quantification of MFI of F-actin accumulation in immunological synapse at the indicated time points (n > 40). Cells were stimulated with PLB antigen-presenting system in the absence or presence of RR (10 μM). Unpaired two-tailed t test in B and C. ns, not significant, ***P < 0.001. Data are mean ± SEM. Data are representative of three independent experiments.
Lastly, we visualized α-tubulin to track the efficiency of synaptic translocation of MTOC during the initiation of B cell activation. We found that B cells lacking TRPV2 exhibited defects in the polarization of MTOC into the B cell immunological synapse in TIRFM imaging (Fig. 5, L and M). Next, we tried to dissect the mechanism by which TRPV2 regulated MTOC formation in antigen-stimulated B cells. The ANK domain has been proposed to constitute the link between TRPs and microtubules (Prager-Khoutorsky et al., 2014; Zhang et al., 2015). Accordingly, we calculated the Pearson’s correlation index (PCI) between TRPV2 and α-tubulin. Unlike the C-terminally truncated TRPV2, which behaved essentially like WT TRPV2, ANK domain deletion interrupted the co-localization of TRPV2 and α-tubulin, suggesting that the ANK domain is important for TRPV2 to be functionally correlated to the microtubules, enhancing MTOC polarization (Fig. 5 N). Hence, we concluded that TRPV2 is vital for B cell activation by facilitating membrane potential depolarization and cytoskeleton remodeling.
Beyond all the aforementioned experimental results, it is generally known that during B cell activation, antigen and BCR binding initiate cation flux responses, which in turn significantly remodel the electrostatic interactions between a series of lipid and membrane proximal signaling molecules. Thus, it is significant to investigate the function of cation channels in the regulation of B cell activation. This report showed that TRPV2 is critical for B cell activation and antibody production by functioning as an integration platform in the immunological synapse. TRP channels, which were first identified in Drosophila but later found in many other organisms, including mammals, are classified into several subfamilies on the basis of their structural characteristics (Nilius and Owsianik, 2011). These subfamilies include TRPA (ankyrin), TRPC (canonical), TRPV (vanilloid), TRPP (polycystin), TRPM (melastatin), TRPML (mucolipin), TRPN (NOMPC-like), and TRPY (yeast) (Nilius and Owsianik, 2011). Typically, mammalian TRPs are integrated with conserved properties, including six transmembrane structures, cytoplasmic N termini and C termini, Ankyrin repeats in N termini, and a pore-forming region between the fifth and sixth transmembrane structure (Gaudet, 2008; Owsianik et al., 2006). TRPs are proposed to be involved in raising intracellular cationic concentration and membrane potential depolarization (McCoy et al., 2011; Voets et al., 2001). The TRPM family member, TRPM7, has been proposed to mediate embryonic development, lymphocyte proliferation, antigen internalization as well as polarization of macrophages (Jin et al., 2008; Krishnamoorthy et al., 2018; Sahni and Scharenberg, 2008; Sahni et al., 2010; Schilling et al., 2014). TRP1, a TRP homolog expressed in chicken, regulates Ca2+ entry and NFAT signaling in chicken DT40 cells (Mori et al., 2002).
However, whether TRPV family members participate in the process of B cell activation and subsequent antibody responses has been barely investigated. We systemically addressed this question in this report. Using several independent methods to interfere with TRPV2 function, we found that B cells deficient for TRPV2 revealed global impairment in BCR engagement, cytoskeletal reorganization, and downstream signal transduction during the formation of B cell immunological synapse. As a result, B cell–specific TRPV2 KO mice exhibited a significantly impaired antibody response upon antigen immunization. Mechanistically, our results showed that TRPV2 is vital for B cell activation and function by mediating cation permeation that contributes to cytoskeleton remodeling within B cell immunological synapse.
The connection between cytoskeletal remodeling and TRPV2 as reported here sheds light on an essential concept that precise regulation of the cytoskeleton beneath the cell membrane is required during B cell activation. In the resting state, BCR diffusion is tightly restricted within boundaries generated by ezrin- and actin-defined networks (Fujiwara et al., 2002; Tolar et al., 2005; Treanor et al., 2010). The BCR-initiated signaling cascade triggers the depolymerization of actin and dephosphorylation of ezrin, increases BCR diffusion rate, permits the access of ITAMs to their kinases, and finally induces B cell activation (Depoil et al., 2008; Freeman et al., 2011; Treanor et al., 2011). Activation of B cells is achieved by sequential and dynamic steps: the actin polymerization at the contact site, which makes B cells spread for enhanced antigen hunting; the contraction of BCR microclusters; and the reorientation of the MTOC toward the center of the synapse (Fleire et al., 2006; Freeman et al., 2011; Martín-Cófreces et al., 2008). Despite the essential role of the cytoskeleton in regulating immunological synapses in a prescribed manner, specific mechanistic details remain unclear. The results in this report provide a new mechanism in which TRPV2 and the cytoskeleton may coordinate together to secure the efficient initiation of B cell activation. Indeed, this report showed that the function of TRPV2 on B cells is dependent on its pore and N-terminal region, and intriguingly the N-terminal ANK domain is important for TRPV2 function, potentially by tethering TRPV2 to the microtubules, consistent with published studies showing that ion channels are organized by the cytoskeleton in many cell types (Prager-Khoutorsky et al., 2014; Zhang et al., 2015). The connection between ion channels and the cytoskeleton is considered a potential mechanism of mechanosensitivity (Martinac, 2014; Sachs, 1991). Depolymerization of actin filaments is coupled with ion channels in activated or inactivated forms, as many ion channels have shown affinities for cytoskeleton compartments (Els and Chou, 1993; Mazzochi et al., 2006; Rotin et al., 1994).
This report also showed that TRPV2-mediated membrane potential depolarization is vital for the formation of the B cell immunological synapse and B cell function. These results are consistent with published findings showing that membrane potential depolarization could induce cytoskeletal reorganization in epithelial cells (Chifflet et al., 2003, 2004; Chifflet and Hernández, 2012). Ca2+ is frequently mentioned in the discussion of actin reorganization (Rosenmund and Westbrook, 1993). Additionally, sodium current may induce endoplasmic reticulum calcium release, and subsequent actin depolymerization (Leblanc and Hume, 1990). The influx of sodium and other cations may also produce a membrane-proximal high salt area, which can disassociate actin-binding proteins (Senju et al., 2017; Zhao et al., 2010). Sodium influx upregulates tubulin acetylation by inhibiting histone deacetylase (Lee et al., 2015), which is critical for MTOC reorientation in immune cells (Serrador et al., 2004). Furthermore, it is known that MTOC orientation is influenced by Ca2+ (Kupfer et al., 1987). These published studies and the data in this report suggest that ion channels play a major part in cytoskeletal reorganization and cell depolarization. The TRPV2 findings in this report partially mirror those from a study in macrophages showing that TRPV2 is recruited to the nascent phagosome and in turn triggers plasma membrane potential depolarization, which increases PIP2 synthesis (Link et al., 2010). Taken together, these results suggest that TRPV2 may function upstream and downstream of immunoreceptors in multiple immune cell types.
Lastly, the findings in this report might have potential translational meanings. At the physiological level, this report showed that there is a positive correlation between the level of TRPV2 expression and the strength of antibody production in healthy individuals who received seasonal influenza vaccine as quantified by virus-specific antibodies. This report also showed that B cell–specific TRPV2 deficient mice exhibited severely impaired antibody responses toward both T cell–dependent antigen and a T cell–independent natural antigen, the capsular polysaccharide from Streptococcus pneumonia. The B cell origin of this impairment was independently confirmed using adoptive transfer experiments in B cell–deficient mice. Thus, TRPV2 appears to be important for normal T cell–dependent and T cell–independent antibody responses. At the pathological level, this report showed that there is a close correlation between TRPV2 expression and the clinical manifestations of both adult and children SLE, although the data in this current study cannot establish a causation effect between high TRPV2 levels and SLE. All these findings reveal that TRPV2 could be a potentially promising target for vaccine development and clinical therapy regarding its critical role in the modulation of B cell function.
Materials and methods
Study design
We designed this study to investigate the role of TRPV in the activation and function of B cells. For this purpose, we identified TRPV2 highly expressed in B cells using fluorescence imaging technology and RT-qPCR. We then used a genetic knock out cell line and a mouse model to study the function of TRPV2 in B cells. We then made use of primary material from humans to study the correlation between TRPV2 expression level and activation strength in antibody response. To study the mechanism, we pursued in vitro experiments to provide data and evidence supporting that TRPV2-mediated membrane potential depolarization is vital for cytoskeleton remodeling in the formation of a B cell immunological synapse. All procedures are described in the following sections.
Healthy volunteers and ethics
The protocol for evaluating hutrpv2 transcription in children was approved by the ethics committee for biomedical studies from the Children’s Hospital of Chongqing Medical University. The protocol of influenza vaccination was approved by the ethics committee for biomedical studies from Tsinghua University. The protocol of SLE patient clinical data analysis was approved by the ethics committee for biomedical studies from the Peking University–affiliated Hospital (Shenzhen and Renmin). All the volunteers involved in this current study have given their written consent.
Animal experiments ethics
All mice were maintained under specific pathogen-free conditions in the animal facility of Tsinghua University and used in accordance with governmental and institutional guidelines for animal welfare. All experimental studies were approved by the Institutional Animal Care and Use Committee of Tsinghua University.
Mice
Trpv2−/− (Trpv2tm1Mijc) mice and Trpv2fl/fl mice have been previously described (Link et al., 2010; Park et al., 2011), which are gifts from Dr. Michael J. Caterina’s laboratory (Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, USA). To efficiently delete the TRPV2 gene specifically in B cells, Trpv2fl/− mice were bred with Mb1-Cre (B6.C(Cg)-Cd79atm1(cre)Reth/EhobJ) transgenic mice (a kind gift from Dr. Zhongjun Dong, Tsinghua University, Beijing, China) in which Cre expression is driven by the promoter of the B cell–specific gene Mb1. Mb1Cre/+Trpv2fl/− offsprings were used for analyses of B cell–specific TRPV2-deficient mice. B1-8hi (B6.129P2-Ptrpca Ightm1Mnz/J, JAX 7594) and μMT (B6.129S2-Ighmtm1Cgn/J, JAX 2288) mice (kind gifts from Dr. Hai Qi, Tsinghua University, Beijing, China) were originally from The Jackson Laboratory. Otherwise, mice not specified in this study were C57BL/6J genetic background originally from The Jackson Laboratory. The control mice for KO mice used in each experiment were all age (7–8-wk)- and gender (female)-matched littermates.
Antibodies
Primary B cells and A20 cells were stimulated using 20 nM biotin-conjugated goat F(ab′)2 anti-mouse IgG + IgM (H+L) (#115-005-068; Jackson ImmunoResearch Laboratories), which was tethered to biotin-containing PLBs by the bridging effects of 20 nM streptavidin (Thermo Fisher Scientific). Ramos or human peripheral blood B cells were stimulated using 20 nM biotin-conjugated goat F(ab’)2 anti-human IgG + IgM (H+L) (#109-066-127; Jackson ImmunoResearch Laboratories). A20 II 1.6 B1-8 IgM cells were stimulated using 20 nM biotin-conjugated NP32-BSA (Bioresearch Technology). Anti-TRPV2 antibody (#ACC-032) was purchased from Alomone Labs. Antibodies for imaging or intracellular staining were Alexa Fluor 647–conjugated Fab of goat antibodies specific for mouse IgM constant regions (#115-607-020; Jackson ImmunoResearch Laboratories), Alexa Fluor 647–conjugated Fab of goat anti-human IgM Fc5μ fragment specific (#109-607-043; Jackson ImmunoResearch Laboratories), Cy3-conjugated Fab of goat anti-human IgG Fcγ fragment specific (#109-607-008; Jackson ImmunoResearch Laboratories), ChromPure Donkey whole IgG (#017-000-003; Jackson ImmunoResearch Laboratories), ChromPure Rabbit whole IgG (#011-000-003; Jackson ImmunoResearch Laboratories), and Alexa Flour 555 donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody (A-31572; Invivogen). Anti-STIM1, anti-STIM2 antibody, anti-mouse phospho-PI3K-p85-Tyr458, rabbit anti-mouse phospho-CD19-Tyr531, rabbit anti-mouse phospho-Syk, and rabbit anti-mouse phospho-Akt were purchased from Cell Signaling Technology. Antibodies for flow cytometry were APC anti-mouse CD86, FITC anti-mouse B220, eFluor450 anti-mouse B220, Pacific Blue anti-mouse B220, PE-Cy7 anti-mouse CD93, FITC anti-mouse CD21, PE anti-mouse CD23, FITC anti-mouse CD5, PE/Cy7 anti-CD95, and BV421 anti-mouse GL7, which were purchased from BD Bioscience. B cell purification was done following the protocol of AutoMACS (Miltenyi Biotec). RR and NMDG were purchased from Sigma-Aldrich.
Plasmid construction
The cDNA of trpv2 was cloned from mouse splenic cDNA. The plasmid of pSpCas9-2a-GFP was purchased from Addgene. Guide RNA sequence (5′-TTTAGGCTGGAGACGTCCGA-3′) was inserted into pSpCas9-2a-GFP following the protocol of Zhang (Ran et al., 2013). The linearized backbone of pcDNA3.1, mtrpv2 was assembled to generate mTRPV2 transiently expression plasmid. Different truncation and point mutation versions of TRPV2, CTD TRPV2, MPD TRPV2, PoreD TRPV2, CaM TRPV2, ANK TRPV2, PN TRPV2, NTD TRPV2, R682A TRPV2, E594K TRPV2, and E604K TRPV2 were constructed by PCR-based mutagenesis through Gibson Assembly in the same vector (Gibson et al., 2009). For the expression of mTRPV2 fused with GFP for retrovirus infection, mtrpv2 gene were inserted in the pMSCV backbone. Small interfering RNA (siRNA) (5′-CTGGCTGAACCTGCTTTATTAT-3′) used in TRPV2 knockdown experiment was inserted into the pMSCV-shRNA (a kind gift from Dr. Hai Qi, Tsinghua University, Beijing, China) backbone through digestion of restriction enzyme XhoI and EcoRI.
RT-qPCR
To determine the expression of TRPVs, including Trpv1 (OTRPC1, Vr1, VR-1); Trpv2 (OTRPC2, Vrl1, VRL-1); Trpv3 (Nh, VRL3); and Trpv4 (OTRPC4, Trp12, VRL-2, VROAC, VR-OAC) by RT-qPCR, total RNA was isolated by TRIzol (Thermo Fisher Scientific) and reverse transcribed to cDNA with Revert Aid First Strand cDNA Synthesis Kit (K1622; Thermo Fisher Scientific). RT-qPCR was performed with SYBR Green Master Mix (Takara) in either Bio-Rad CFX96 Touch for a 96-well plate or ABI ViiA7 for a 384-well plate, respectively. The primer sequences used in this experiment have been described in a previous study (Bertin et al., 2014) or PrimerBank (https://pga.mgh.harvard.edu/primerbank/). The ΔΔ threshold cycle (CT) technique was used to determine the relative expression of RT-qPCR products.
Evaluation of huTrpv2 transcription on children volunteers
1 ml anticoagulated peripheral blood was acquired from healthy volunteers. 101 healthy volunteers aged from 7 days old to 17 years old participated in this study. RNA was extracted using the QIAamp RNA Blood Mini Kit (QIAGEN). The evaluation of huTrpv2 transcription followed the steps of RT-qPCR above.
Influenza vaccination on volunteers
Volunteers who had reported influenza in the last 3 mo or who had received influenza vaccination in the previous winter season were excluded from this study. 21 healthy volunteers aged from 18 to 28 years participated in this study. 10 ml anticoagulated peripheral blood was acquired from healthy volunteers before immunization and 21 days after immunization. All the subjects were vaccinated with one dose (0.5 ml) of commercial trivalent inactivated influenza virus vaccine (Henan Hualan) by intramuscular injection. Using Ficoll Plaque Plus reagent for PBMCs and plasma isolation, the procedure was followed as a guideline of GE HealthCare. RT-qPCR (described above) was used to evaluate the transcription level of huTrpv2 transcription. The levels of antigen-specific antibodies in plasma were measured by ELISA following the steps of ELISA below.
BM12 splenocyte-induced mouse SLE model
For the mouse SLE model, splenocytes (107 per mouse) from age- and gender-matched bm12 mice were adoptively transferred to 6–8-wk-old WT or TRPV2 C-KO mice intravenously on day 0 and day 14, respectively. On day 28, serum was collected for ELISA (anti-dsDNA and anti-SmD).
Intracellular immunofluorescence staining
For intracellular staining, cells were fixed with 4% paraformaldehyde buffer and permeabilized with 0.1% Triton-X100 for 30 min at room temperature after fixation, blocked with 100 μg/ml donkey whole IgG for 1 h, and then incubated with antibodies of interest overnight at 4°C. After that, cells were incubated with Alexa Flour 555 donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody for 45 min. Excessive reagents were washed away by 5 ml 1×PBS. Phalloidin-Alexa Flour 488 was used to label F-actin in fixed cells. After fixation and permeabilization with 0.1% Triton-X100 for 5 min, cells were incubated with phalloidin-Alexa Flour 488 at room temperature for 2 min. After washing by 1×PBS, F-actin could be imaged through a fluorescence microscope. We analyzed image data blindly to genotype and assay conditions.
Preparation of planar fluid lipid bilayer (PLB) antigen-presenting system
PLBs were generated by following our previous protocol (Liu et al., 2010). In brief, the cleaned coverslips were coated with 20 nM biotin-conjugated liposome in PBS for 20 min at 37°C. After washing with 8 ml PBS gently, PLBs were coated with 20 nM streptavidin for 20 min at 37°C. After gentle washing with 8 ml PBS, surrogate antigens were tethered to PLBs by the bridging effects of streptavidin. For mouse A20 cell and primary mouse B cells, biotin-conjugated goat F(ab′)2 anti-mouse IgG + IgM (H+L) was utilized as the surrogated antigen, while goat F(ab′)2 anti-human IgG + IgM (H+L) was used for Ramos or PBMC. The non-specific stimulation was blocked by 1% casein at 37°C for 30 min. After washing with 10 ml of HBSS, the PLBs were done for further use.
Immunoblot analysis
Primary B cells were isolated from spleens. Cell lysates were separated by 10% Bis-Tris NuPAGE precast gels (Life Technologies) and then transferred to a polyvinylidene fluoride membrane (Millipore). Nonspecific binding was blocked by 5% skim milk (BD Bioscience) in PBS/0.05% Tween-20 at room temperature. Then blots were probed with antibodies of interest at 4°C overnight. After washing with PBS/0.05% Tween-20, appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies (Invitrogen) were used for subsequent incubation. ECL solution (Pierce) was used for chemiluminescence. Chemiluminescence signals were detected by ImageQuant LAS4000 (GE HealthCare). The exposure time was determined automatically when saturated signal was detected.
TIRFM and confocal microscopy
To visualize BCR, cells were incubated with 100 nM fluorescence probe–conjugated Fab goat anti-IgM or anti-IgG for 3 min on ice. TIRFM images were acquired using an Olympus IX-81 microscope equipped with a TIRF port, Olympus 100× 1.49 N.A. objective lens, Andor iXon+ DU-897D electron-multiplying CCD camera, four lasers (405, 488, 561, and 647 nm) (Sapphire laser, Coherent). The acquisition was controlled by Metamorph software (Molecular Devices). An image splitter (Cairn Research) was used to acquire dual-view TIRFM imaging. Confocal images were captured by the Olympus FLUOVIEW FV1000 confocal laser scanning microscope equipped with a 60× oil objective lens and four lasers (405, 473, 549, and 635 nm). When collecting data, the field of view was randomly switched. Experiments were repeated by different individuals to ensure objectivity. We analyzed image data blindly to genotype and assay conditions.
Flow cytometry
Cells were separated from the bone marrow and spleen and were preincubated with CD16/CD32 (eBioscience) on ice for 30 min to block the Fc receptor. Then, cells were stained with different fluorescent dye-conjugated surface markers antibodies, including APC anti-mouse CD86, FITC anti-mouse B220, ef450 anti-mouse B220, PE-Cy7 anti-mouse CD93, FITC anti-mouse CD21, and PE anti-mouse CD23. Live vs. dead status was assessed through Zombie Yellow Fixable Viability Kit (BioLegend) following the manufacturer’s protocol. After staining with surface markers and cell viability markers, signals were detected by the Fortessa flow cytometer (BD). All data were processed by FlowJo. Cell sorting was performed with the FACSAria III Cell Sorter (BD). All data were further analyzed with FlowJo software.
Cell proliferation assay
Purified primary B cells were stained by CFSE (Invitrogen) following the instructions in the user’s manual. After staining, cells were cultured at 37°C in RPMI-1640 medium with the presence of 10 μg/ml goat F(ab)2 anti-mouse IgG + IgM (H+L). After 60 h, cells were collected. The extent of proliferation was measured by CFSE dilution in B cells by flow cytometry.
Ca2+ flux analysis
Free intracellular Ca2+ was measured by using the Ca2+-specific fluorescent probe Fluo-4 cell-permeant acetoxymethyl (Invitrogen) following the instructions of the user’s manual. Briefly, A20 cells were stained with Fluo-4 in HBSS buffer at 37°C for 30 min. After washing with HBSS, cells were resuspended in HBSS. For the beads conjugated antigen activation experiment, beads conjugated goat F(ab′)2 anti-mouse IgG+IgM (H+L) was added to the buffer. Fluo-4 signal was detected by flow cytometry. For the surface-associated antigen activation experiment, a glass bottom dish was incubated in 1 M NaOH overnight at room temperature; after extensive rinsing with deionized water, the dish was incubated in 100 μl 100 nm red fluorescent microspheres (Thermo Fischer Scientific) (1:10,000 diluted in ethanol from the 5% stock suspension) and air dried. The dish was then incubated in 10 μg/ml biotin-conjugated F(ab′)2 goat anti-mouse IgG + IgM (H+L) (or PBS for control) for 1 h at 37°C. After rinsing with PBS, the dish surface was blocked with 5% BSA for 30 min at 37°C. The dish was then mounted on a Nikon A1 confocal microscopy with a 60× Oil NA 1.40 objective lens. Focus on the plan where the red fluorescent microspheres were located. Add 5 × 105 Fluo4-stained A20 cells into the dish and start capturing immediately. The Fluo4 fluorescent images of at least 20 cells for each group were captured every 1 min up to 20 min. The mean Fluo4 intensity was calculated.
BCR capping
Surface BCR capping was analyzed using immunofluorescence microscopy. Splenic B cells were stained with 5 µg/ml AF488-Fab goat-anti-mouse IgM (Jackson ImmunoResearch) for 20 min on ice, washed, activated with 10 µg/ml F(ab′)2 goat anti-mouse IgG+M (Jackson ImmunoResearch) at 37°C for the indicated times, fixed, and imaged using confocal microscope. B cells displaying BCR caps for each time point were quantified by visual inspection.
Endocytosis of antigen-BCR complexes
BCR internalization experiment was conducted by flow cytometry. Briefly, primary B cells were stained for 15 min on ice with beads-conjugated F(ab′)2 goat anti-mouse IgG + IgM (H+L) by the preincubation streptavidin beads (size: 200 nM; Beyotime) with biotin-conjugated F(ab′)2 goat anti-mouse IgG + IgM (H+L) (Jackson ImmunoResearch). Labeled cells were divided into two parts, one of which was preincubated with 10 μM RR for 30 min on ice. Then the labeled cells were activated at different time points (37°C, 5% CO2), and otherwise kept on ice at all times. After incubation, cells were stained with AF647 donkey anti-goat IgG for 20 min on ice and analyzed for fluorescence of AF647 by flow cytometry.
Cell culture, transfection, and retrovirus infection
A20, A20 II 1.6, and Ramos B cells were gifted by Dr. Susan K. Pierce (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA). A20 II 1.6 B1-8 IgM cells were constructed by the exogenous expression of B1-8-High-IgM-BCRs in A20 II 1.6 cells. Both primary B cells and cell lines were cultured at 37°C in RPMI-1640 medium supplemented with 10% FBS, 50 μM β-mercaptoethanol (Gibco), 100 U/ml penicillin/streptomycin antibiotics, and non-essential amino acids. The transfection of B cell lines was processed by electroporation using Lonza Nucleofector following the protocol of Lonza. Retrovirus supernatants used in retroviral infection of primary B cells were produced by Plat-E cells transfected with pMSCV expression vectors. Before retroviral infection, mice primary B cells were incubated in 1640 medium with 10 μg/ml LPS for 24 h. The primed B cells were then spin-infected for 1.5 h with supernatants with retrovirus particles. After 60 h culture, positive cells were sorted by flow sorter based on their fluorescence expression levels.
Adoptive transfer, immunizations, and ELISA
Retrovirus-infected B cells (2 × 105 each recipient) from B1-8hi mice were intravenously transferred into μMT mice. Mice were injected with antigens at day 0 and bled at day 6 and day 12. Antigens used in the immunization were NP50-LPS (5 μg/mouse; Biosearch Technologies), NP50-AECM-ficoll (5 μg/mouse; Biosearch Technologies), TNP14-AECM-ficoll (5 μg/mouse; Biosearch Technologies), NP32-KLH (5 μg/mouse; Biosearch Technologies), or Streptococcus pneumoniae capsule polysaccharides type 1 (CPS1) and type 3 (CPS3) (25 μg/mouse). Antigens were dissolved in 0.9% saline and administered by intraperitoneal injection. The levels of Ag-specific antibodies in serum were detected by ELISA. Specific antigens were coated on maxisorb plates (Nunc) and incubated overnight at 4°C. Blocked with 0.3% gelatin in PBS buffer (2 h at room temperature), these plates were then incubated with prediluted mice serum (1.5 h at room temperature) followed by HRP conjugated goat anti-mouse IgM (45 min at room temperature). These plates were washed with PBS/0.05% Tween-20 between each of the two steps. After that, these plates were incubated in the substrate solution composed of 0.325% orthophenylenediamine dihydrochloride (OPD-Sigma) and 0.085% H2O2 prepared in 0.3 M Tris-citrate buffer, pH 6.0, at room temperature for 10 min and stopped with 2.5 M H2SO4. Then the optical density at 490 nm wavelength (OD490) was measured by an ELISA plate reader (Bio-Rad). The threshold value was calculated by two times of background OD value. Antibody titers were determined by the maximum dilution that was close to the threshold value.
Measurement of plasma membrane potential
The plasma membrane potential was measured using the FLIPR Membrane Potential Assay Kit (Molecular Devices) following the instructions in the user’s manual. Briefly, after transferring to FLIPR day, probes were excited at 510–545 nm, and the emission light at 565–625 nm was recorded by confocal microscope. We analyzed image data blindly to genotype and assay conditions.
Quantification and statistical analysis
Images were processed by Bitplane Imaris, ImageJ (National Institutes of Health, USA), and Image Pro Plus (Media Cybernetics). The mean fluorescence intensity (MFI) and the total fluorescence intensity (total FI) were measured based on the intensity and area analysis. PCI was processed through colocalization Threshold in ImageJ. Statistical significance was assessed with a two-tailed Student’s t-test or one-way ANOVA in GraphPad PRISM5. The correlation between antibody response and huTrpv2 transcription was fitted with regression analysis in GraphPad PRISM5. P values are indicated by *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ns, not significant with P > 0.05.
Online supplemental material
Fig. S1 shows that RR-sensitive channels contribute to the formation of the B cell immunological synapse and TRPV2 contributes to B cell activation. Fig. S2 shows the effects of TRPV2 on B cell development, BCR signaling, and B cell function. Fig. S3 shows that pharmacological blocking of the TRPV channel interrupts cytoskeleton mobilization.
Data availability
Further information and requests for resources and reagents should be first directed to the lead contact, W. Liu ([email protected]).
Acknowledgments
We acknowledge the healthy volunteers for their participation in our study. We thank S.K. Pierce (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA), K. Rajewsky (Immune Regulation and Cancer, Max-Delbrück-Center for Molecular Medicine, Berlin, Germany), M.J. Caterina (John Hopkins University, Baltimore, MD, USA), M. Shlomchik (University of Pittsburgh, Pittsburgh, PA, USA), C. Goodnow (Garvan Institute of Medical Research, Darlinghurst, Australia), and T. Kurosaki and H. Shinohara (WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan) for providing experimental materials.
This work was supported by funds from Ministry of Science and Technology of China (grants 2021YFC2300500 and 2021YFC2302403), National Natural Science Foundation of China (32141004 and 81825010 to W. Liu, 81974253 to Q. Wang), Beijing Natural Science Foundation (23Z30090), and Shenzhen Medical Research Fund (C2301008), Key Project of Basic Research of Shenzhen Science and Technology Innovation Commission (JCYJ20200109140203849), Treatment and Prevention Integration Project of Shenzhen Municipal Health Commission (0102018-2019-YBXM-1499-01-0414), and National Multiple Sclerosis Society Career Transition Award (TA 3059-A-2 to C. Wu). This study is also supported by research funds from Tsinghua University Spring Breeze Fund, Center for Life Sciences, Institute for Immunology at Tsinghua University, and Vanke School of Public Health at Tsinghua University.
Author contributions: C. Li, M. Zhao, J.-R. Zhang, Q. Wang, X. Zhao, Z. Li, and W. Liu designed experiments; C. Li, M. Zhao, X. Liu, Y. Li, B. Xu, L. Zhou, X. Sun, W. Sun, N. Kang, Z. Ji, T. Li, and F. Wang performed experiments; C. Li, M. Zhao, X. Liu, Y. Li, B. Xu, L. Zhou, X. Sun, C. Wu, and J.-Y. Ye analyzed and interpreted the data; H. An prepared and offered capsule polysaccharides under the supervision of J.-R. Zhang; L. Zhou organized blood donation under the supervision of X. Zhao at the Children’s Hospital of Chongqing Medical University; C. Li, M. Zhao Z. Li, and W. Liu wrote the manuscript.

