


Hematopoietic stem cell transplantation (HSCT) can replace endogenous microglia with circulation-derived macrophages but has high mortality. To mitigate the risks of HSCT and expand the potential for microglia replacement, we engineered an inhibitor-resistant CSF1R that enables robust microglia replacement. A glycine to alanine substitution at position 795 of human CSF1R (G795A) confers resistance to multiple CSF1R inhibitors, including PLX3397 and PLX5622. Biochemical and cell-based assays show no discernable gain or loss of function. G795A- but not wildtype-CSF1R expressing macrophages efficiently engraft the brain of PLX3397-treated mice and persist after cessation of inhibitor treatment. To gauge translational potential, we CRISPR engineered human-induced pluripotent stem cell–derived microglia (iMG) to express G795A. Xenotransplantation studies demonstrate that G795A-iMG exhibit nearly identical gene expression to wildtype iMG, respond to inflammatory stimuli, and progressively expand in the presence of PLX3397, replacing endogenous microglia to fully occupy the brain. In sum, we engineered a human CSF1R variant that enables nontoxic, cell type, and tissue-specific replacement of microglia.

Introduction
Microglia play critical roles in neural circuit regulation, white matter development, and blood flow regulation (Schafer et al., 2012; Li et al., 2019; Bisht et al., 2021; Safaiyan et al., 2021; Császár et al., 2022). As brain-resident immune cells, microglia also respond to central nervous system (CNS) injury and diverse neuropathologies (Keren-Shaul et al., 2017; Salter and Stevens, 2017). The importance of microglia to human health is further highlighted by the high proportion of microglial genes that confer genetic risk in neurodegenerative disease (McQuade and Blurton-Jones, 2019; Kim et al., 2020; Podleśny-Drabiniok et al., 2020; Amin et al., 2022) and “primary microgliopathies,” rare diseases arising from mutations that alter microglial function and survival (Paloneva et al., 2002; Konno et al., 2018; Berdowski et al., 2021; Macintosh et al., 2022). These findings have inspired efforts to therapeutically target microglia. While many studies focused on pharmacologic approaches (Jiang et al., 2020; Schlepckow et al., 2020; Shi et al., 2020; Qiao et al., 2021), several reports proposed the use of microglial transplantation or the replacement of microglia with peripheral myeloid cells (Bennett et al., 2018; Cronk et al., 2018; Han et al., 2020; Li et al., 2020; Xu et al., 2020; Shibuya et al., 2022). Such approaches offer a powerful new approach to modulate microglial function and genetics or deliver therapeutic payloads to the CNS.
A major hurdle to realizing this strategy is that the microglial niche is carefully regulated such that robust engraftment of exogenous microglia is extremely challenging (Abud et al., 2017). One solution is pharmacologic depletion of microglia through colony-stimulating factor 1 receptor (CSF1R) inhibitors (CSF1Ri) coupled with bone marrow transplantation to allow peripheral monocytes to infiltrate the brain (Cronk et al., 2018; Shibuya et al., 2022). Yet, these approaches require hazardous conditioning methods such as irradiation or chemotherapy to promote engraftment of peripheral myeloid cells that, despite long-term brain residence, remain transcriptionally and functionally distinct from microglia (Bennett et al., 2018; Lund et al., 2018; Shemer et al., 2018). Not only is engraftment efficiency low with these available therapies in humans (Cogle et al., 2004), but the required conditioning methods induce CNS injury responses with potential to undermine the desired therapeutic effects (Gibson et al., 2019; Sailor et al., 2022). In addition, bone marrow transplantation replaces the entire hematopoietic system, whereas replacement of microglia alone may be sufficient for many CNS diseases.
A more targeted and physiological approach would be to deliver stem cell–derived microglia that closely mimic the transcriptional and functional characteristics of human microglia (Hasselmann et al., 2019; Mancuso et al., 2019b), directly to the brain without toxic conditioning. To develop such an approach, we generated macrophages carrying a single amino-acid point mutation within CSF1R that maintains signaling while conferring resistance to CSF1Ri. Engineering candidate variants into a murine cell line and macrophage progenitors facilitated sensitive and efficient testing, allowing us to identify an optimal variant, G795A, that rendered CSF1R resistant to inhibition and supported brain engraftment in both neonatal and adult CSF1Ri-treated mice. To increase translational relevance, we CRISPR engineered the G795A mutation into human-induced pluripotent stem cell–derived microglia (iPSC-microglia [iMG]), enabling robust engraftment of human microglia in the adult murine brain, without significantly affecting their identity or function. These findings demonstrate the development of a new tool to investigate microglia function in chimeric models, while offering a promising strategy for human microglial transplantation therapies.
Results and discussion
Intracranial macrophage transplantation leads to minimal engraftment in the microglia-replete brain
We sought to create a microglia replacement strategy for use across the lifespan without irradiation, traditional chemotherapy, or genetic microglial ablation. Consistent with prior studies, we found that direct intracranial transplantation (ICT) of WT microglia into WT hosts led to minimal engraftment (Fig. S1 A; Xu et al., 2020). In the hope of increasing engraftment, we depleted microglia in neonates with daily injections of the CSF1Ri PLX3397, resulting in an 88% reduction of endogenous microglia (Fig. S1 B). Similar to with adult mice, CSF1Ri paired with ICT of microglia or bone marrow–derived macrophages (BMDMs) led to minimal donor cell engraftment (Fig. S1 C; Shibuya et al., 2022). To address this challenge, we hypothesized that engineering a CSF1R variant resistant to pharmacologic inhibition could greatly improve engraftment.

Engineering an inhibitor-resistant human CSF1R variant. (A) Example of rendered dot plot derivation from a WT mouse 14 d after receiving a GFP+ microglia transplant. Left image, brain tile scan with magnified inset showing few donor-derived microglia engrafted (GFP/green). Right image, rendered dot plot of engrafted cells overlaid onto DAPI channel. Scale bars, 1,000 µm (brain tile), 50 µm (insets). (B) Quantification of microglia depletion expressed as IBA1+ cell density after 7 d of PLX3397 treatment (25 mg/kg/d, subcutaneous, red bars) relative to saline-treated littermates (black bars). Data represented as mean values ± SEM calculated from three matched sagittal sections from n = 2 independent biological replicates per group. (C) Baseline engraftment of GFP+ microglia, BMDMs, and CIMs intracerebrally transplanted into WT animals on P0, P1, P2, or P3 with or without 2 d of 25 mg/kg PLX3397 pretreatment. CIMs are further described starting in G. All animals were harvested 14 d after transplantation. Data represented as mean values ± SEM calculated from three matched sagittal sections from n = 2–3 independent biological replicates per group. Percent engraftment measured as shown in J. (D) Relative abundance of Ba/F3s transduced with WT-CSF1R (black), G795A-CSF1R (green), or D802V-CSF1R (constitutively active, gray) with or without 50 ng/ml CSF1h, 48 h after transduction. Representative data plotted as mean values ± SEM from two to three technical replicates per group from n = 2 independent experiments. (E) Relative abundance of WT or G7595-transduced Ba/F3s 0–72 h after provision with CSF1h and PLX3397 (247 nM). Data represented as mean values ± SEM from three technical replicates from n = 2 independent biological replicates per group. (F) Data table showing PLX3397 IC50, log IC50, standard error of LogIC50, and R2 of IC50 of Ba/F3s transduced with CSF1R variants. + denotes ligand independent mutant, ++ denotes constitutively active control, # denotes variants unable to sustain growth, and ## denotes catalytically dead control. Values calculated from the average of n = 3 independent replicates. (G) Representative image of CIM engraftment in the Csf1r−/− murine brain. P2 cell injection, P16 harvest. IBA1 (red), DAPI (blue). Scale bars, 1,000 and 50 µm (inset). (H) Brightfield images of WT- and G795A-expressing macrophages derived from Csf1r−/− CIMs, 7 d after estrogen removal and CSF1h treatment. (I) IC50 curve of WT- and G795A-transduced BMDMs from FVB WT mice. Data represented as mean values ± SEM from three technical replicates from n = 3 independent biological replicates per group. (J) Method used to calculate percent engraftment depicting total brain area (pink outline) and engrafted area (yellow outline). Scale bar, 1,000 µm. (K) Top: Experimental paradigm depicting timing of CIM injection, PLX3397 treatment, and endpoint. Bottom: Representative rendering of G795A-CIM distribution (green) 14 d after injection into PLX3397 treated Rag2+/+;Il2r𝛾+/+;CSF1h+/+ host. Scale bar, 1,000 µm. (L) Top: Experimental paradigm depicting timing of BMDM injection, PLX3397 treatment, and endpoint. Bottom: Rendering showing G795A-BMDM distribution, 28 d after injection into PLX3397 treated Rag2+/+;Il2r𝛾+/+;CSF1h+/+ host. Image is from most well engrafted animal. Scale bar, 1,000 µm. (M) Quantification of percent area engrafted by GFP+ donor macrophages. Dots represent biological replicates calculated as mean engraftment ± SEM across three matched sagittal sections. G795A-CIMs on PLX3397, n = 18; WT-CIMs on PLX3397, n = 6; G795A-CIMs untreated (no PLX3397), n = 4; G795A-BMDMs on PLX3397, n = 3; WT-BMDMs on PLX3397, n = 4. For CIMs, one-way ANOVA (P = 0.0009). For BMDMs, two-tailed unpaired t test (P = 0.1152). (N and O) Percent engraftment of whole brain (N) and cortical cell density (O) in PLX3397-treated adult Rag2−/−;Il2r𝛾−/−;CSF1h+/+ mice transplanted with G795A- or WT-CIMs. Dots represent biological replicates calculated as mean engraftment ± SEM across three matched sagittal sections. G795A-CIMs on PLX3397, n = 7; off PLX3397, n = 3; WT-CIMs on PLX3397, n = 5; and G795A untreated (no PLX3397), n = 2. One-way ANOVA (% engraftment: P = 0.0010; cell density: P = 0.0014). (P) Top: Experimental paradigm depicting timing of stereotactic BMDM injection, PLX3397 treatment, and endpoint using 9–12-wk-old Rag2−/−;Il2r𝛾−/−;CSF1h+/+ mice. Bottom: Representative rendering of G795A-BMDM distribution. (Q) Quantification of percent engraftment of G795A- and WT-BMDMs. Dots represent biological replicates calculated as mean engraftment ± SEM across two matched sagittal sections. G795A-BMDMs, n = 4; WT-BMDMs, n = 2. Unpaired two-tailed t test (P = 0.0324). Unless otherwise noted, P values calculated as Tukey’s HSD: #P < 0.12, *P < 0.05, **P < 0.01.
Engineering an inhibitor-resistant human CSF1R variant. (A) Example of rendered dot plot derivation from a WT mouse 14 d after receiving a GFP+ microglia transplant. Left image, brain tile scan with magnified inset showing few donor-derived microglia engrafted (GFP/green). Right image, rendered dot plot of engrafted cells overlaid onto DAPI channel. Scale bars, 1,000 µm (brain tile), 50 µm (insets). (B) Quantification of microglia depletion expressed as IBA1+ cell density after 7 d of PLX3397 treatment (25 mg/kg/d, subcutaneous, red bars) relative to saline-treated littermates (black bars). Data represented as mean values ± SEM calculated from three matched sagittal sections from n = 2 independent biological replicates per group. (C) Baseline engraftment of GFP+ microglia, BMDMs, and CIMs intracerebrally transplanted into WT animals on P0, P1, P2, or P3 with or without 2 d of 25 mg/kg PLX3397 pretreatment. CIMs are further described starting in G. All animals were harvested 14 d after transplantation. Data represented as mean values ± SEM calculated from three matched sagittal sections from n = 2–3 independent biological replicates per group. Percent engraftment measured as shown in J. (D) Relative abundance of Ba/F3s transduced with WT-CSF1R (black), G795A-CSF1R (green), or D802V-CSF1R (constitutively active, gray) with or without 50 ng/ml CSF1h, 48 h after transduction. Representative data plotted as mean values ± SEM from two to three technical replicates per group from n = 2 independent experiments. (E) Relative abundance of WT or G7595-transduced Ba/F3s 0–72 h after provision with CSF1h and PLX3397 (247 nM). Data represented as mean values ± SEM from three technical replicates from n = 2 independent biological replicates per group. (F) Data table showing PLX3397 IC50, log IC50, standard error of LogIC50, and R2 of IC50 of Ba/F3s transduced with CSF1R variants. + denotes ligand independent mutant, ++ denotes constitutively active control, # denotes variants unable to sustain growth, and ## denotes catalytically dead control. Values calculated from the average of n = 3 independent replicates. (G) Representative image of CIM engraftment in the Csf1r−/− murine brain. P2 cell injection, P16 harvest. IBA1 (red), DAPI (blue). Scale bars, 1,000 and 50 µm (inset). (H) Brightfield images of WT- and G795A-expressing macrophages derived from Csf1r−/− CIMs, 7 d after estrogen removal and CSF1h treatment. (I) IC50 curve of WT- and G795A-transduced BMDMs from FVB WT mice. Data represented as mean values ± SEM from three technical replicates from n = 3 independent biological replicates per group. (J) Method used to calculate percent engraftment depicting total brain area (pink outline) and engrafted area (yellow outline). Scale bar, 1,000 µm. (K) Top: Experimental paradigm depicting timing of CIM injection, PLX3397 treatment, and endpoint. Bottom: Representative rendering of G795A-CIM distribution (green) 14 d after injection into PLX3397 treated Rag2+/+;Il2r𝛾+/+;CSF1h+/+ host. Scale bar, 1,000 µm. (L) Top: Experimental paradigm depicting timing of BMDM injection, PLX3397 treatment, and endpoint. Bottom: Rendering showing G795A-BMDM distribution, 28 d after injection into PLX3397 treated Rag2+/+;Il2r𝛾+/+;CSF1h+/+ host. Image is from most well engrafted animal. Scale bar, 1,000 µm. (M) Quantification of percent area engrafted by GFP+ donor macrophages. Dots represent biological replicates calculated as mean engraftment ± SEM across three matched sagittal sections. G795A-CIMs on PLX3397, n = 18; WT-CIMs on PLX3397, n = 6; G795A-CIMs untreated (no PLX3397), n = 4; G795A-BMDMs on PLX3397, n = 3; WT-BMDMs on PLX3397, n = 4. For CIMs, one-way ANOVA (P = 0.0009). For BMDMs, two-tailed unpaired t test (P = 0.1152). (N and O) Percent engraftment of whole brain (N) and cortical cell density (O) in PLX3397-treated adult Rag2−/−;Il2r𝛾−/−;CSF1h+/+ mice transplanted with G795A- or WT-CIMs. Dots represent biological replicates calculated as mean engraftment ± SEM across three matched sagittal sections. G795A-CIMs on PLX3397, n = 7; off PLX3397, n = 3; WT-CIMs on PLX3397, n = 5; and G795A untreated (no PLX3397), n = 2. One-way ANOVA (% engraftment: P = 0.0010; cell density: P = 0.0014). (P) Top: Experimental paradigm depicting timing of stereotactic BMDM injection, PLX3397 treatment, and endpoint using 9–12-wk-old Rag2−/−;Il2r𝛾−/−;CSF1h+/+ mice. Bottom: Representative rendering of G795A-BMDM distribution. (Q) Quantification of percent engraftment of G795A- and WT-BMDMs. Dots represent biological replicates calculated as mean engraftment ± SEM across two matched sagittal sections. G795A-BMDMs, n = 4; WT-BMDMs, n = 2. Unpaired two-tailed t test (P = 0.0324). Unless otherwise noted, P values calculated as Tukey’s HSD: #P < 0.12, *P < 0.05, **P < 0.01.
Human CSF1R variant G795A is PLX3397 resistant
We individually introduced 12 amino acid substitutions into the kinase domain of human CSF1R at residues we predicted could prevent or weaken PLX3397 binding based on co-crystallography studies (Fig. 1 A; Tap et al., 2015). To efficiently screen for CSF1Ri resistance, we employed Ba/F3 cells, a receptor tyrosine kinase-dependent murine line. Ba/F3s are widely and successfully used to study human kinase mutations (Warmuth et al., 2007; Smith et al., 2012) and can be switched from IL-3 to CSF1 dependence following CSF1R transduction (Fig. S1 D; Gow et al., 2012). We individually transduced Ba/F3s with each CSF1R variant, including kinase dead (K616M; Downing et al., 1989) and constitutively active mutants (D802V; Morley et al., 1999). We found that one variant in particular, G795A, conferred a 16.7-fold increase in survival relative to WT (Fig. 1 B) without altering cell growth or proliferation (Fig. S1 E). In contrast, three variants were unable to sustain growth and one was ligand independent (Fig. S1 F).
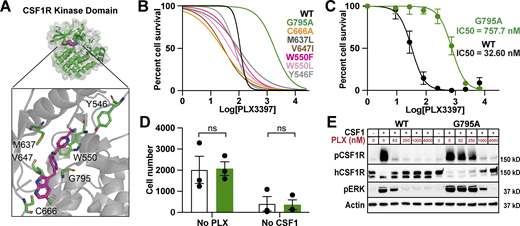
Engineering an inhibitor-resistant CSF1R variant. (A) Model of hCSF1R-PLX3397 crystal structure (Tap et al., 2015), highlighting proximity of candidate variants to PLX3397 binding site (magenta) in kinase pocket (green). (B) PLX3397 inhibition curves for Ba/F3s transduced with CSF1R variants. G795A shown in green; WT shown in black. Representative experiment, repeated twice independently. Best fit lines generated from an average of three technical replicates. (C) Inhibition curves for WT and G795A expressing CIMs generated on the Csf1r−/− background. Curves plotted from mean values ± SEM calculated from three technical replicates from n = 3 independently derived lines per group. (D) Average cell counts per well ± SEM for G795A (green) and WT (white) differentiated CIMs from no PLX3397/no CSF1h conditions at assay endpoint in C. P values calculated using unpaired t test corrected for multiple comparisons using Bonferroni Dunn method. (E) Immunoblots showing pCSF1R Y723, total hCSF1R, pERK, and Actin expression in WT- and G795A-CIMs at a range of PLX3397 concentrations, 3 min following spike-in of 360 ng/ml CSF1h, annotated with mass of protein ladder (kD, on right). Representative of n = 3 independent experiments. Source data are available for this figure: SourceData F1.
Engineering an inhibitor-resistant CSF1R variant. (A) Model of hCSF1R-PLX3397 crystal structure (Tap et al., 2015), highlighting proximity of candidate variants to PLX3397 binding site (magenta) in kinase pocket (green). (B) PLX3397 inhibition curves for Ba/F3s transduced with CSF1R variants. G795A shown in green; WT shown in black. Representative experiment, repeated twice independently. Best fit lines generated from an average of three technical replicates. (C) Inhibition curves for WT and G795A expressing CIMs generated on the Csf1r−/− background. Curves plotted from mean values ± SEM calculated from three technical replicates from n = 3 independently derived lines per group. (D) Average cell counts per well ± SEM for G795A (green) and WT (white) differentiated CIMs from no PLX3397/no CSF1h conditions at assay endpoint in C. P values calculated using unpaired t test corrected for multiple comparisons using Bonferroni Dunn method. (E) Immunoblots showing pCSF1R Y723, total hCSF1R, pERK, and Actin expression in WT- and G795A-CIMs at a range of PLX3397 concentrations, 3 min following spike-in of 360 ng/ml CSF1h, annotated with mass of protein ladder (kD, on right). Representative of n = 3 independent experiments. Source data are available for this figure: SourceData F1.
G795A-expressing macrophages are PLX3397 resistant
To test the effect of CSF1R variants on macrophage survival in the presence of CSF1Ri, we created a system to engraft conditionally immortalized macrophage (CIM) progenitors into the brain. CIMs are generated through estrogen-dependent HOXB8 activation in murine myeloid progenitors (Wang et al., 2006). CIMs are readily modified by viral transduction, expanded in culture as progenitors with CSF2 and estrogen, and terminally differentiated into macrophages with CSF1 addition and estrogen removal (Roberts et al., 2019). ICT of CIM progenitors into neonatal Csf1r−/− mice, which lack microglia, led to widespread IBA1+ macrophage engraftment (Fig. S1 G; Ginhoux et al., 2010; Bennett et al., 2018; Dai et al., 2002). Like primary macrophages, however, ICT of WT CIM progenitors into a WT host led to minimal engraftment including when paired with CSF1Ri (Fig. S1 C). Our CIM approach allows efficient engineering of donor macrophages competent to engraft the unoccupied microglial niche, but that show virtually no baseline engraftment in non-depleted hosts, allowing sensitive testing of strategies for microglia replacement.
We generated three independent CIM lines from Csf1r−/− bone marrow, ensuring no murine CSF1R expression. As expected, these lines could be cultured and expanded, but were unable to produce macrophages with provision of CSF1. We next transduced Csf1r−/− CIM progenitors to express human WT- or G795A-CSF1R and isolated transduced (GFP+) cells by FACS. From these stably transduced lines, we successfully generated macrophages by estrogen removal and addition of human, but not murine, CSF1 (Fig. S1 H). Similar to our Ba/F3 results, we found that G795A-CIMs exhibited a 23.2-fold increase in PLX3397 resistance compared to WT-CSF1R CIMs (Fig. 1 C). G795A-CIMs had similar expansion compared to WT-CSF1R CIM when exposed to CSF1, but did not proliferate in the absence of CSF1, suggesting no ligand-independent growth or loss of CSF1R function (Fig. 1 D). Next, we examined whether G795A altered CSF1-induced signaling using protein biochemistry. Neither WT- nor G795A-CIMs showed kinase activity in the absence of ligand. However, upon CSF1 stimulation, both cell populations phosphorylated CSF1R-Y723 and downstream target ERK, indicating normal CSF1 responsiveness. PLX3397 treatment decreased WT- but not G795A-CIM CSF1R and ERK phosphorylation, demonstrating that G795A maintains kinase activity in the presence of PLX3397, but this could be overcome at high PLX3397 concentrations (Fig. 1 E). We observed a rapid loss of the higher molecular weight mature CSF1R band upon signal induction, consistent with prior reports (Rohde et al., 2004; Yu et al., 2008). G795A expression also conferred PLX3397 resistance to murine BMDMs despite their expression of murine CSF1R (Fig. S1 I). These results show, by structural rationale, cell-based, and biochemical assays, that G795A confers CSF1Ri resistance without discernible ligand independence or other gain of function. We next applied this discovery to microglia replacement in vivo.
G795A-macrophages engraft the PLX3397-treated neonatal brain
As human CSF1R requires human CSF1 (CSF1h) to signal (Gow et al., 2012), we transplanted G795A-CIMs into PLX3397-treated, CSF1h-expressing neonates (Rag2−/−;Il2rγ−/−;CSF1h+/+; Fig. 2 A). To compare engraftment between G795A- and WT-CIMs, we measured percent of brain territory colonized by donor cells in sagittal sections (Fig. S1 J). 2 wk after ICT, G795A-macrophages engrafted an average of 56% of sagittal brain section area (Fig. 2 B). G795A-CIMs persisted in the neonatal brain after withdrawal of PLX, expanding to occupy 74% of brain section area (Fig. 2 C). Not only did G795A-CIMs engraft efficiently during and after CSF1Ri (Fig. 2 D), they attained similar densities to endogenous microglia (Fig. 2 E; De Lucia et al., 2016; Yin et al., 2018). Despite daily PLX3397 treatment sufficient to deplete endogenous microglia, G795A-CIMs adopted a microglia-like morphology and expressed IBA1. In contrast, WT-CIMs did not engraft (Fig. 2, D and F). Engraftment did not depend on immune deficiency, since it occurred at similar levels in PLX-treated Rag2+/+;Il2rγ+/+;CSF1h+/+ neonates (Fig. S1 K). We then applied this strategy to primary BMDMs and found that G795A- but not WT-transduced cells engrafted in PLX-treated neonates despite co-expressing murine CSF1R (Fig. S1 L). Donor cell engraftment was strictly PLX- and G795A-dependent, demonstrating that G795A does not intrinsically confer engraftment potential (Fig. S1 M). These data validate and extend our in vitro observations, establishing the potential utility of the G795A mutant for microglia replacement.

Widespread engraftment and persistence of transplanted G795A but not WT CIMs in the PLX3397 - treated brain. (A) Experimental paradigm depicting the timing of neonatal CIM injection, PLX treatment (290 mg/kg chow) and endpoints at P14 (“On PLX”) and P21 (“Off PLX”). (B) Representative rendering of donor cell distribution and mean engraftment ± SEM, 14 d after cell injection, with continuous PLX treatment. Box i denotes field shown in F. Scale bar, 1,000 µm. (C) Representative rendering showing donor cell distribution 21 d after cell injection. PLX was stopped 7 d prior to harvest. Boxes ii and iii denote fields shown in F. (D) Quantification of brain engraftment by G795A-CIMs in hosts on or off PLX, and WT-CIMs in hosts on PLX. G795A-On PLX: n = 3; G795A-Off Plx: n = 3; WT-On Plx: n = 4. One-way ANOVA (P = 0.0001). (E) Quantification of donor cell density in cortex. G795A-On PLX: n = 3; G795A-Off Plx: n = 3; WT-On Plx: n = 4. One-way ANOVA (P = 0.0070). Dotted gray line represents average endogenous IBA1+ cells from n = 3 age-matched controls. (F) Representative images of G795A- and WT-CIMs following neonatal transplant, showing donor cell GFP (green), IBA1 immunostaining (red), and DAPI (blue) On and Off PLX. Row 4 displays endogenous microglia repopulation after PLX removal from box iii in C. Scale bar, 50 µm. (G) Experimental paradigm depicting timing of adult CIM injection, PLX treatment, and endpoints at D14 (On PLX) and D21 (Off PLX). (H) Representative rendering of G795A-CIM distribution and mean engraftment ± SEM, 14 d after cell injection, with continuous PLX treatment. Scale bar, 1,000 µm. (I) Representative image of G795A-CIM distribution and mean engraftment ± SEM, 21 d after cell injection. PLX was stopped 7 d prior to harvest. Scale bar, 1,000 µm. (J) Quantification of cortical engraftment area by G795A and WT donor CIMs. G795A-On PLX: n = 6; G795A-Off Plx: n = 3; WT-On Plx: n = 5. One-way ANOVA (P < 0.0001). (K) Quantification of donor cell density in cortex. One-way ANOVA (P = 0.0019). (L) Representative images of G795A and WT CIMs following adult transplant, showing donor cell GFP (green), IBA1 immunostaining (red), and DAPI (blue) both On (rows 1 and 2) and Off PLX (row 3). Scale bar, 50 µm. (M) Top: Representative confocal image of G795A-CIM morphology (green), IBA1 immunostaining (red), TMEM119 immunostaining (white), and DAPI (blue). Scale bar, 25 µm. Bottom: Representative confocal image of G795A-CIM morphology (green), IBA1 immunostaining (red), Ms4a7 in situ (white), and DAPI (blue). Scale bar, 25 µm. In D, E, J, and K, dots represent biological replicates, each the average of three matched sagittal sections. Error bars represent SEM. P values calculated as Tukey’s HSD: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Widespread engraftment and persistence of transplanted G795A but not WT CIMs in the PLX3397 - treated brain. (A) Experimental paradigm depicting the timing of neonatal CIM injection, PLX treatment (290 mg/kg chow) and endpoints at P14 (“On PLX”) and P21 (“Off PLX”). (B) Representative rendering of donor cell distribution and mean engraftment ± SEM, 14 d after cell injection, with continuous PLX treatment. Box i denotes field shown in F. Scale bar, 1,000 µm. (C) Representative rendering showing donor cell distribution 21 d after cell injection. PLX was stopped 7 d prior to harvest. Boxes ii and iii denote fields shown in F. (D) Quantification of brain engraftment by G795A-CIMs in hosts on or off PLX, and WT-CIMs in hosts on PLX. G795A-On PLX: n = 3; G795A-Off Plx: n = 3; WT-On Plx: n = 4. One-way ANOVA (P = 0.0001). (E) Quantification of donor cell density in cortex. G795A-On PLX: n = 3; G795A-Off Plx: n = 3; WT-On Plx: n = 4. One-way ANOVA (P = 0.0070). Dotted gray line represents average endogenous IBA1+ cells from n = 3 age-matched controls. (F) Representative images of G795A- and WT-CIMs following neonatal transplant, showing donor cell GFP (green), IBA1 immunostaining (red), and DAPI (blue) On and Off PLX. Row 4 displays endogenous microglia repopulation after PLX removal from box iii in C. Scale bar, 50 µm. (G) Experimental paradigm depicting timing of adult CIM injection, PLX treatment, and endpoints at D14 (On PLX) and D21 (Off PLX). (H) Representative rendering of G795A-CIM distribution and mean engraftment ± SEM, 14 d after cell injection, with continuous PLX treatment. Scale bar, 1,000 µm. (I) Representative image of G795A-CIM distribution and mean engraftment ± SEM, 21 d after cell injection. PLX was stopped 7 d prior to harvest. Scale bar, 1,000 µm. (J) Quantification of cortical engraftment area by G795A and WT donor CIMs. G795A-On PLX: n = 6; G795A-Off Plx: n = 3; WT-On Plx: n = 5. One-way ANOVA (P < 0.0001). (K) Quantification of donor cell density in cortex. One-way ANOVA (P = 0.0019). (L) Representative images of G795A and WT CIMs following adult transplant, showing donor cell GFP (green), IBA1 immunostaining (red), and DAPI (blue) both On (rows 1 and 2) and Off PLX (row 3). Scale bar, 50 µm. (M) Top: Representative confocal image of G795A-CIM morphology (green), IBA1 immunostaining (red), TMEM119 immunostaining (white), and DAPI (blue). Scale bar, 25 µm. Bottom: Representative confocal image of G795A-CIM morphology (green), IBA1 immunostaining (red), Ms4a7 in situ (white), and DAPI (blue). Scale bar, 25 µm. In D, E, J, and K, dots represent biological replicates, each the average of three matched sagittal sections. Error bars represent SEM. P values calculated as Tukey’s HSD: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
G795A-macrophages engraft the PLX3397-treated adult brain
Given previous work showing that microglial surrogates engraft less robustly in the mature as compared to neonatal CNS (Abud et al., 2017; Hasselmann et al., 2019), we tested whether G795A expression could improve engraftment of the adult brain. G795A- or WT-CSF1R CIMs were transplanted into the hippocampus and cortex of PLX-treated adult Rag2−/−; Il2rγ−/−; CSF1h+/+ mice through stereotactic injection (Fig. 2 G). We observed widespread brain engraftment of G795A- but not WT-CSF1R expressing macrophages, finding that G795A-macrophages engrafted 41% of the brain (Fig. 2 H). To determine whether these cells could resist a higher dosage of PLX and persist after inhibitor removal, a second cohort received 600 mg/kg for 2 wk prior to 1-wk PLX withdrawal. G795A-CIMs not only persisted but also further expanded upon CSF1Ri removal, occupying 90% of cortex and 71% whole brain area (Fig. 2, I and J). Both G795A- and WT-CIMs were unable to engraft in hosts that did not receive PLX (Fig. S1 N). G795A-CIMs engrafted at densities similar to endogenous microglia, averaging 109 cells/mm2 while on CSF1Ri and 196 cells/mm2 after CSF1Ri cessation (Fig. 2 K and Fig. S1 O). As with neonates, G795A-CIMs adopted a ramified morphology and expressed IBA1 in adults both on and off PLX, while WT-CIMs did not engraft (Fig. 2 L). G795A-CIMs expressed TMEM119, a brain-resident macrophage marker (Bennett et al., 2016), and Ms4a7, a specific marker of bone marrow origin in mouse transplant models (Fig. 2 M; Bennett et al., 2018). Finally, we overexpressed G795A- or WT-CSF1R in murine BMDMs, and they too engrafted in the PLX-treated adult brain (Fig. S1, P and Q).
The persistence of inhibitor-resistant cells in neonates and adults after CSF1Ri withdrawal suggests that G795A expression may allow long-term microglia replacement throughout the lifespan without continuous CSF1Ri treatment. However, the long-term effects of surrogate engraftment in the brain remain largely unknown. Previous studies have shown that peripherally derived macrophages remain transcriptionally and functionally distinct from microglia, even following long-term CNS engraftment (Cronk et al., 2018; Shemer et al., 2018; Shibuya et al., 2022). Translational applications of microglial replacement may, therefore, benefit from donor cells that more closely mimic human microglia, such as iPSC-derived microglia.
G795A confers broad CSF1Ri resistance without altering the transcriptome of human iMG
Comparative analysis of the crystal structure of CSF1R bound to PLX3397 and PLX5622 independently led to the identification of the 795 position as an optimal target for inhibitor resistance (Fig. 3 A; Spangenberg et al., 2019). We hypothesized that the substitution of Glycine 795 (G795), which is located within the ATP-binding pocket, with bulkier amino acids including Alanine (A), Cysteine (C), or Valine (V; Fig. 3 B) could sterically hinder PLX3397 and PLX5622 binding without impairing kinase activity. Therefore, we used CRISPR gene editing to introduce three homozygous point mutations (G795A, C, and V) into an isogenic series of human iPSCs (Fig. S2 A). Interestingly, we found G795A-CSF1R and G795C-CSF1R iPSCs readily differentiated into iMG with similar efficiencies as isogenic WT iPSCs. However, G795V-CSF1R homozygous iPSCs were unable to give rise to iMG in vitro. As iMG differentiation requires intact CSF1R signaling (McQuade et al., 2020), this suggests that larger alterations in CSF1R may sterically impair ATP binding and disrupt CSF1R signaling.

Human G795A iPSC-derived microglia exhibit robust viability and minimal alterations to gene expression in response to CSF1R inhibitors. (A) Crystal structure of hCSF1R-PLX3397 and hCSF1R-PLX5622 highlighting G795 residue in the kinase domain. (B) Molecular modeling of G795 variants G795A, G795C, and G795V demonstrating increasing R-group sizes. (C and D) Caspase 3/7 levels imaged over 24 h in iMG cultured in complete medium treated with 0.1% DMSO, 250 nM, 500 nM, or 1 µM PLX3397 (C) and equivalent concentrations of PLX5622 (D). Data captured from four images/well from n = 6 independent wells. Data represented as mean values ± SEM. (E and F) Quantification of cell death by fluorescent Caspase 3/7 Dye for Apoptosis after 24 h in culture with PLX3397 (E) and PLX5622 (F). Data captured from four images/well from n = 6 independent wells. Data represented as mean values ± SEM. P value, one-way ANOVA (P < 0.0001). Tukey’s HSD; ****P < 0.0001. (G–J) Bulk RNA-seq analysis of WT-, G795A-, and G795C-iMG. (G) Principal component analysis using the top 2,000 most variably expressed genes between WT-, G795A-, and G795C-iMG cultured for 24 h with 0.1% DMSO or 250 nM PLX5622 (n = 4 replicates per line, per condition) revealing that the primary source of variation is the WT response to PLX5622. (H) Linear regression analysis and the coefficient of determination between DMSO-treated G795A or G795C and WT microglia, confirming a high degree of concordance when comparing either the full transcriptome (G795A, R2 = 0.96–0.99; G795C, R2 = 0.97–0.98) or a 249-gene microglia signature (G795A, R2 = 0.97–0.99; G795C, R2 = 0.97–0.98). (I–K) Volcano plots comparing 24-h treatment with 250 nM PLX5622, revealing significant transcriptomic alterations (Log2(FC) ≥ 1; FDR ≤ 0.05) in WT microglia (I), but no significant changes in G795A microglia compared with DMSO-treated cells (J) nor G795C microglia compared with DMSO-treated cells (K).
Human G795A iPSC-derived microglia exhibit robust viability and minimal alterations to gene expression in response to CSF1R inhibitors. (A) Crystal structure of hCSF1R-PLX3397 and hCSF1R-PLX5622 highlighting G795 residue in the kinase domain. (B) Molecular modeling of G795 variants G795A, G795C, and G795V demonstrating increasing R-group sizes. (C and D) Caspase 3/7 levels imaged over 24 h in iMG cultured in complete medium treated with 0.1% DMSO, 250 nM, 500 nM, or 1 µM PLX3397 (C) and equivalent concentrations of PLX5622 (D). Data captured from four images/well from n = 6 independent wells. Data represented as mean values ± SEM. (E and F) Quantification of cell death by fluorescent Caspase 3/7 Dye for Apoptosis after 24 h in culture with PLX3397 (E) and PLX5622 (F). Data captured from four images/well from n = 6 independent wells. Data represented as mean values ± SEM. P value, one-way ANOVA (P < 0.0001). Tukey’s HSD; ****P < 0.0001. (G–J) Bulk RNA-seq analysis of WT-, G795A-, and G795C-iMG. (G) Principal component analysis using the top 2,000 most variably expressed genes between WT-, G795A-, and G795C-iMG cultured for 24 h with 0.1% DMSO or 250 nM PLX5622 (n = 4 replicates per line, per condition) revealing that the primary source of variation is the WT response to PLX5622. (H) Linear regression analysis and the coefficient of determination between DMSO-treated G795A or G795C and WT microglia, confirming a high degree of concordance when comparing either the full transcriptome (G795A, R2 = 0.96–0.99; G795C, R2 = 0.97–0.98) or a 249-gene microglia signature (G795A, R2 = 0.97–0.99; G795C, R2 = 0.97–0.98). (I–K) Volcano plots comparing 24-h treatment with 250 nM PLX5622, revealing significant transcriptomic alterations (Log2(FC) ≥ 1; FDR ≤ 0.05) in WT microglia (I), but no significant changes in G795A microglia compared with DMSO-treated cells (J) nor G795C microglia compared with DMSO-treated cells (K).

Human G795A and G795C iPSC-derived microglia are resistant to multiple CSF1Ri. (A) Chromatograms of CRISPR-modified isogenic set of CSF1R-G795A, G795C, and G795V human iPSC lines. (B and C) Quantification of iMG cell death by fluorescent Caspase 3/7 Dye for Apoptosis over 24 h in culture with complete medium treated with 0.1% DMSO, 250 nM, 500 nM, or 1 µM Edicotinib (B) and equivalent concentrations of BLZ945 (C). (D) Live (Calcein-AM) versus dead (ethidium homodimer-1) percent quantification of WT and G795A microglia cultured with 0.1% DMSO or 500 nM of PLX3397, PLX5622, Edicotinib, or BLZ945. Data captured from four images/well from n = 6 independent wells using an Incucyte S3 live-cell imaging system. Data represented as percent mean values. (E–G) Percent confluency of iMG normalized to t = 0 h over 24 h in culture with complete medium treated with 0.1% DMSO or 500 nM PLX3397 (E), PLX5622 (F), Edicontinib (G), or BLZ945 (H). Data captured from four images/well from n = 6 independent wells using an Incucyte S3 live-cell imaging system. Data represented as mean values ± SEM. (I and J) Hierarchical clustering and Pearson correlation of normalized RNA counts of the whole transcriptome (I) and a 249-gene microglia signature (J) between WT (black), G795A (green), and G795C (blue) iMG cultured with 0.1% DMSO or 250 nM PLX5622 (n = 4 replicates per cell line, per condition). Lightest shade of blue in I and J indicates correlations of R = 0.93; darkest shade of blue indicates R = 1.0 correlation.
Human G795A and G795C iPSC-derived microglia are resistant to multiple CSF1Ri. (A) Chromatograms of CRISPR-modified isogenic set of CSF1R-G795A, G795C, and G795V human iPSC lines. (B and C) Quantification of iMG cell death by fluorescent Caspase 3/7 Dye for Apoptosis over 24 h in culture with complete medium treated with 0.1% DMSO, 250 nM, 500 nM, or 1 µM Edicotinib (B) and equivalent concentrations of BLZ945 (C). (D) Live (Calcein-AM) versus dead (ethidium homodimer-1) percent quantification of WT and G795A microglia cultured with 0.1% DMSO or 500 nM of PLX3397, PLX5622, Edicotinib, or BLZ945. Data captured from four images/well from n = 6 independent wells using an Incucyte S3 live-cell imaging system. Data represented as percent mean values. (E–G) Percent confluency of iMG normalized to t = 0 h over 24 h in culture with complete medium treated with 0.1% DMSO or 500 nM PLX3397 (E), PLX5622 (F), Edicontinib (G), or BLZ945 (H). Data captured from four images/well from n = 6 independent wells using an Incucyte S3 live-cell imaging system. Data represented as mean values ± SEM. (I and J) Hierarchical clustering and Pearson correlation of normalized RNA counts of the whole transcriptome (I) and a 249-gene microglia signature (J) between WT (black), G795A (green), and G795C (blue) iMG cultured with 0.1% DMSO or 250 nM PLX5622 (n = 4 replicates per cell line, per condition). Lightest shade of blue in I and J indicates correlations of R = 0.93; darkest shade of blue indicates R = 1.0 correlation.
Next, we sought to determine whether G795 variants conferred broad resistance to CSF1Ri-induced iMG apoptosis in vitro. We differentiated WT, G795A, and G795C isogenic iMG in parallel in complete iMG media which includes the CSF1R ligands CSF1 and IL34. Upon microglial maturation (day 28), we added increasing doses of PLX3397 or PLX5622 and examined apoptosis over 24 h using a fluorogenic caspase 3/7 detector and time-lapse imaging. We found significantly increasing numbers of apoptotic WT cells in response to increasing doses of CSF1Ri. In contrast, G795A- and G795C-iMG demonstrated no evidence of apoptosis (Fig. 3, C–F). Similar protective effects were observed in response to two other CSF1Ri commonly used to target microglia: BLZ945 and Edicotinib (Fig. S2, B and C; Mancuso et al., 2019a; Wies Mancini et al., 2022). To confirm that G795A- and G795C-iMG were not dying through non-apoptotic mechanisms or exhibiting altered proliferation, we utilized live-dead staining with calcein-AM and ethidium homodimer-1 and examined the confluency of cells across the duration of treatment. Whereas up to 50% of WT iMG died within 24 h of exposure to CSF1Ri, G795 variants demonstrated no significant changes in viability or confluency compared to controls (Fig. S2, D–H).
To better understand the downstream influence of CSF1Ri-resistant variants on CSF1R signaling and microglia gene expression, we performed RNA sequencing (RNA-seq) comparing WT-, G795A-, and G795C-iMG cultured with and without PLX5622, a more selective CSF1Ri (Table S1; Dagher et al., 2015; Liu et al., 2019). Principal component analysis of the top 2,000 most variably expressed genes demonstrated that 76% of the variance (PC1) within these data was primarily mediated through the effects of PLX5622 treatment on WT cells (Fig. 3 G). In contrast, G795A cells treated with DMSO control versus PLX5622 exhibited <1% variance. In addition, regardless of treatment, G795A cells exhibited a strongly overlapping clustering with control-treated WT iMG (Fig. 3 G). G795C iMG also showed a very small effect of PLX5622 treatment on their transcriptome (<5% variance, PC2). However, G795C cells clustered separately from WT-untreated and G795A-iMG on PC2, suggesting that this mutation induces subtle alterations in microglial transcriptome. To further understand the impact of the G795A and G795C mutations, we performed correlation analyses. Alignment between untreated WT and G795A-iMG revealed extremely strong correlations at both whole transcriptome (R2 = 0.96–0.99) and core microglia signature levels (R2 = 0.97–0.99; Fig. 3 H; Patir et al., 2019), suggesting G795A does not alter microglial identity in vitro. A similar analysis of G795C iMG found a slightly lower correlation with WT iMG of R2 = 0.97–0.98 for the full transcriptome and core signature (Fig. 3 H and Fig. S2, I and J).
Next, to determine whether CSF1R inhibition further alters the transcriptome of iMG, we generated volcano plots of significantly altered genes (Log2(FC) ≥ 1, false discovery rate [FDR] ≤ 0.05) for WT-, G795A-, and G795C-iMG cultured with or without PLX5622. As expected, WT iMG exhibited a dramatic change in gene expression in response to PLX5622 treatment. Interestingly, many disease-associated microglial genes were upregulated in PLX5622-treated WT microglia including GPNMB, LGMN, PLIN2, and APOC1, perhaps in response to signals induced by the death or subsequent phagocytosis of their microglia neighbors (Deczkowska et al., 2018; Claes et al., 2021; Muzio et al., 2021). Down-regulated genes included both cell-cycle genes (MKi67, CDK1, MYC, CENPA) and microglia identity markers (CX3CR1, ITGAM; Fig. 3 I), consistent with CSF1R inhibition also impairing microglia proliferation. In stark contrast, no significant differences in gene expression were detected between PLX5622 and control-treated G795A- or G795C-iMG (Log2(FC) > 1, FDR ≤ 0.05; Fig. 3, J and K). Taken together, these results indicate that G795A confers broad CSF1Ri resistance without altering the transcriptome of human microglia even when exposed to CSF1Ri in vitro.
G795A-iMG engraft and respond to inflammatory challenge in the murine brain
To determine whether G795A-iMG can engraft within the murine brain, we first transplanted neonatal xenotransplantation-compatible MITRG mice (Rag2−/−, Il2rγ−/−, CSF1h−/−, IL-3/GM-CSFh−/−, TPOh−/−) with WT or G795A microglia progenitor cells (Rongvaux et al., 2014; Hasselmann et al., 2019; Fig. 4 A). At 2 mo of age, mice were sacrificed and xenografted human microglia (xMG) isolated for RNA-seq (Table S1). Similar to our in vitro findings, we observed minimal variation between WT- and G795A-xMG gene expression with no impact on microglial identity (Fig. 4 B), and transcriptome correlations of R2 = 0.74–0.95 (whole transcriptome) and R2 = 0.78–0.97 (microglial signature; Fig. 4, C and D).

CSF1R-G795A microglia engraft and respond to neuroinflammation in the adult murine brain. (A) Schematic depicting neonatal HPC injection paradigm, with endpoints (black ticks) at 60 d and after LPS injections, and LPS injection paradigm (blue line). (B) Volcano plot comparing differential gene expression by RNA-seq between G795A- and WT-xMG 2 mo after transplantation (FDR ≤ 0.05; log2(FC) ≥ ±1). (C and D) Linear regression analysis and the coefficient of determination between WT- and G795A-xMG when examining the full transcriptome (C; R2 = 0.74–0.95) and a 249-gene microglia signature (D; R2 = 0.78–0.97). (E) Immunostaining for IBA1+ (green)/Ku80+ (red) WT- and G795A-xMG demonstrates decreased expression of P2RY12 (purple) in response to LPS treatment. (F and G) Conversely, WT- and G795A-xMG increase expression of CD45 (F; white) and MX1 (G; blue) in response to LPS treatment. Representative 40× images. Scale bar, 50 µm. (H–K) Quantification of E–G. One-way ANOVA IBA1 (P = 0.5061); P2RY12 (P < 0.0001); CD45 (P = 0.0007); MX1 (P = 0.0003). Tukey’s HSD; **P < 0.01, ***P < 0.001, ****P < 0.0001. Data represented as mean intensity normalized to number of Ku80+/IBA1+ human microglia per FOV for all antibodies calculated from three matched coronal sections per animal (n = 3 biological replicates). Error bars, SEM.
CSF1R-G795A microglia engraft and respond to neuroinflammation in the adult murine brain. (A) Schematic depicting neonatal HPC injection paradigm, with endpoints (black ticks) at 60 d and after LPS injections, and LPS injection paradigm (blue line). (B) Volcano plot comparing differential gene expression by RNA-seq between G795A- and WT-xMG 2 mo after transplantation (FDR ≤ 0.05; log2(FC) ≥ ±1). (C and D) Linear regression analysis and the coefficient of determination between WT- and G795A-xMG when examining the full transcriptome (C; R2 = 0.74–0.95) and a 249-gene microglia signature (D; R2 = 0.78–0.97). (E) Immunostaining for IBA1+ (green)/Ku80+ (red) WT- and G795A-xMG demonstrates decreased expression of P2RY12 (purple) in response to LPS treatment. (F and G) Conversely, WT- and G795A-xMG increase expression of CD45 (F; white) and MX1 (G; blue) in response to LPS treatment. Representative 40× images. Scale bar, 50 µm. (H–K) Quantification of E–G. One-way ANOVA IBA1 (P = 0.5061); P2RY12 (P < 0.0001); CD45 (P = 0.0007); MX1 (P = 0.0003). Tukey’s HSD; **P < 0.01, ***P < 0.001, ****P < 0.0001. Data represented as mean intensity normalized to number of Ku80+/IBA1+ human microglia per FOV for all antibodies calculated from three matched coronal sections per animal (n = 3 biological replicates). Error bars, SEM.
To determine whether G795A-iMG retain normal microglial function in vivo, we next assessed xMG response to peripheral immune challenge with LPS, identifying human microglia by staining with a human-specific Ku80 antibody. Neonatal MITRG mice were transplanted with WT or G795A microglia progenitor cells. At 2 mo of age, mice were given two intraperitoneal injections of LPS 24 h apart then sacrificed 12 h after the second injection (Fig. 4 A). Immunohistochemistry analysis of engrafted human microglia revealed striking differences in microglia morphology in response to LPS treatment, with a significant reduction in homeostatic marker P2RY12 (Fig. 4, E, H, and I) and induction of the inflammatory marker CD45 and interferon-response gene MX1 (Fig. 4, F, G, J, and K). Importantly, we detected no significant differences in LPS-dependent responses between G795A- and WT-iMG (Fig. 4, H–K). Taken together, these results demonstrate that G795A does not alter the transcriptome of engrafted human microglia or disrupt the normal microglial response to an immune challenge in vivo.
G795A-iMG repopulate the PLX3397-treated adult murine brain
Next, we transplanted WT- and G795A-iMG into the dorsal hippocampus and overlying parietal association cortex of adult MITRG mice. Unlike peripheral macrophages, transplanted iMG engraft in the non-depleted adult brain without CSF1Ri pretreatment, albeit at very low levels (Abud et al., 2017). 1 mo after transplantation, mice were fed 600 mg/kg PLX3397 diet ad libitum for either 10, 30, 30 d on/30 d off, or 60 d, at which point xMG engraftment was examined (Fig. 5 A). Consistent with prior reports, we observed a near complete depletion of murine microglia with 10 d of PLX3397 treatment (Fig. 5 B; Elmore et al., 2014; Zhan et al., 2020). Likewise, very few WT human microglia remained with 10 d of PLX3397 treatment, with all observable WT microglia eliminated by 30 d of treatment (Fig. 5 B, bottom). In contrast, G795A-xMGs not only persisted but migrated from the injection sites despite constant CSF1Ri treatment (Fig. 5 B, top). Remarkably, after 60 d of PLX3397 treatment, xenotransplanted G795A human microglia had migrated throughout the murine brain to represent over 99% of total observable microglia (Fig. 5 C). Interestingly, at 30 d, CSF1Ri-resistant G795A-xMG co-labeled with the mitotic marker Ki67, revealing a proliferative wavefront of repopulating human microglia (Fig. 5 D). Proliferating microglia exhibited significantly increased expression of the DNA repair protein and human-specific nuclear marker Ku80 (Difilippantonio et al., 2000) and decreased expression of the homeostatic marker P2RY12 (Fig. S3, A–F). Similar to previous studies that examined repopulating murine microglia, we found that repopulating xMG also strongly expressed LGALS-3 (MAC2), a marker associated with microglial proliferation and migration in response to injury (Fig. S3, G–I; Lalancette-Hébert et al., 2012; Zhan et al., 2020). Importantly, G795A-xMG persisted within engrafted regions 30 d after cessation of CSF1Ri treatment and equivalently expressed P2RY12, while endogenous murine microglia repopulated the few remaining empty niches of the brain (Fig. 5 E). These results demonstrate a novel and potentially therapeutically applicable approach to replace dysfunctional microglia with human iMG.

CSF1R inhibition enables the replacement of murine microglia with human G795A microglia in adult mice. (A) Schematic depicting timing of stereotactic iMG injection, PLX3397 treatment (red, 600 mg/kg chow) and endpoints at 90 (10 d of treatment or no treatment [black]), 120 d (30 d of treatment), 120 d (30 d on/30 d off treatment), and 150 d (60 d of treatment). (B) IBA1+ (green) and Ku80+ (red) G795A (top) and WT (bottom) human microglia exhibit limited engraftment within the adult murine brain without PLX3397 treatment (left). G795A microglia remain viable and progressively expand from the injection site across 10–60 (left-right) days of PLX3397 treatment while endogenous murine (Ku80−/IBA1+) and transplanted WT human microglia (bottom) are depleted by CSF1R inhibition. Brain stitch scale bar, 500 µm. Representative 40× scale bar, 50 µm. (C) Quantification of percent human (Ku80+/IBA1+) versus mouse (Ku80−/IBA1+) microglia reveal a progressive replacement of endogenous microglia, with 99% of microglia expressing the human nuclear marker Ku80 within 60 d. Counts calculated from three matched coronal sections per animal (n = 3 biological replicates per condition). (D) G795A microglia proliferate from the initial injection sites throughout the murine brain with a distinctive Ki67+ wavefront (blue, arrows). Scale bar, 100 µm. (E) G795A microglia persist in engrafted regions after cessation of 30 d PLX3397 treatment and immunostain for homeostatic microglia marker P2RY12 (purple). Brain stitch scale bar, 500 µm. Representative 20× scale bar, 100 µm.
CSF1R inhibition enables the replacement of murine microglia with human G795A microglia in adult mice. (A) Schematic depicting timing of stereotactic iMG injection, PLX3397 treatment (red, 600 mg/kg chow) and endpoints at 90 (10 d of treatment or no treatment [black]), 120 d (30 d of treatment), 120 d (30 d on/30 d off treatment), and 150 d (60 d of treatment). (B) IBA1+ (green) and Ku80+ (red) G795A (top) and WT (bottom) human microglia exhibit limited engraftment within the adult murine brain without PLX3397 treatment (left). G795A microglia remain viable and progressively expand from the injection site across 10–60 (left-right) days of PLX3397 treatment while endogenous murine (Ku80−/IBA1+) and transplanted WT human microglia (bottom) are depleted by CSF1R inhibition. Brain stitch scale bar, 500 µm. Representative 40× scale bar, 50 µm. (C) Quantification of percent human (Ku80+/IBA1+) versus mouse (Ku80−/IBA1+) microglia reveal a progressive replacement of endogenous microglia, with 99% of microglia expressing the human nuclear marker Ku80 within 60 d. Counts calculated from three matched coronal sections per animal (n = 3 biological replicates per condition). (D) G795A microglia proliferate from the initial injection sites throughout the murine brain with a distinctive Ki67+ wavefront (blue, arrows). Scale bar, 100 µm. (E) G795A microglia persist in engrafted regions after cessation of 30 d PLX3397 treatment and immunostain for homeostatic microglia marker P2RY12 (purple). Brain stitch scale bar, 500 µm. Representative 20× scale bar, 100 µm.

G795A microglia proliferate along distinct wavefronts in the murine brain. (A) Representative confocal 10× stitch image of engrafted and proliferating G795A microglia after 30 d of CSF1Ri. Ki67+ mitotic cells (blue), Ku80+ human nuclei (red), and IBA1+ microglia (green). (B) 40× representative images of Ki67+ G795A microglia in proliferating wavefront and Ki67− G795A microglia within the initial hippocampal injection site. (C) Proliferating microglia in the wavefront exhibit significantly higher levels of human nuclear marker Ku80. Unpaired t test t(4) = 3.008, *P = 0.0396. (D) Representative 10× stitch image of P2RY12 expressing G795A microglia after 30 d of CSF1Ri. P2RY12 (purple), Ku80 (red), IBA1 (green). (E) 40× representative images of P2RY12-low G795A microglia in the wavefront and P2RY12-high G795A microglia within the hippocampus. (F) Proliferating microglia in the wavefront exhibit significantly lower levels of microglia homeostatic marker P2RY12. Unpaired t test t(4) = 7.545, **P = 0.0017. (G) Representative 10× stitch image of LGALS-3 expressing G795A microglia after 30 d of CSF1Ri. LGALS-3 (orange), Ku80 (red), IBA1 (green). (H) 40× representative images of G795A microglia expressing LGALS-3 within the proliferative wavefront with diminished expression within the hippocampus. (I) Proliferating microglia in the wavefront exhibit significantly higher levels of microglia activation marker LGALS-3. Unpaired t test t(4) = 9.511, ***P = 0.0007. Data represented as integrated density normalized to number of Ku80+/IBA1+ human microglia per FOV for all antibodies calculated from three matched coronal sections per animal (n = 3 biological replicates per region). Error bars, SEM. Representative 10× stitch scale bar, 100 µm. Representative 40× scale bar, 25 µm.
G795A microglia proliferate along distinct wavefronts in the murine brain. (A) Representative confocal 10× stitch image of engrafted and proliferating G795A microglia after 30 d of CSF1Ri. Ki67+ mitotic cells (blue), Ku80+ human nuclei (red), and IBA1+ microglia (green). (B) 40× representative images of Ki67+ G795A microglia in proliferating wavefront and Ki67− G795A microglia within the initial hippocampal injection site. (C) Proliferating microglia in the wavefront exhibit significantly higher levels of human nuclear marker Ku80. Unpaired t test t(4) = 3.008, *P = 0.0396. (D) Representative 10× stitch image of P2RY12 expressing G795A microglia after 30 d of CSF1Ri. P2RY12 (purple), Ku80 (red), IBA1 (green). (E) 40× representative images of P2RY12-low G795A microglia in the wavefront and P2RY12-high G795A microglia within the hippocampus. (F) Proliferating microglia in the wavefront exhibit significantly lower levels of microglia homeostatic marker P2RY12. Unpaired t test t(4) = 7.545, **P = 0.0017. (G) Representative 10× stitch image of LGALS-3 expressing G795A microglia after 30 d of CSF1Ri. LGALS-3 (orange), Ku80 (red), IBA1 (green). (H) 40× representative images of G795A microglia expressing LGALS-3 within the proliferative wavefront with diminished expression within the hippocampus. (I) Proliferating microglia in the wavefront exhibit significantly higher levels of microglia activation marker LGALS-3. Unpaired t test t(4) = 9.511, ***P = 0.0007. Data represented as integrated density normalized to number of Ku80+/IBA1+ human microglia per FOV for all antibodies calculated from three matched coronal sections per animal (n = 3 biological replicates per region). Error bars, SEM. Representative 10× stitch scale bar, 100 µm. Representative 40× scale bar, 25 µm.
Conclusions
To better treat neurological disorders, there is a critical need for tools to study and target microglia. Currently, the only cell therapy that may address human microglial dysfunction is hematopoietic stem cell transplantation (HSCT). Because of its high morbidity and low brain chimerism in humans (∼1–2%), HSCT in neurologic disease is reserved for pre-symptomatic but otherwise fatal diseases without other treatments (Cogle et al., 2004; Bhatia et al., 2007; Kraft et al., 2019). Despite these limitations, HSCT improves outcomes in lysosomal and peroxisomal storage diseases (Escolar et al., 2005; Biffi et al., 2013; Eichler et al., 2017). Safe and efficient methods for microglia replacement could substantially broaden indications for cell-based therapies in the CNS, for example to widen the treatment window for lysosomal diseases, or to address neurodegenerative diseases of aging.
Here, we report that a single G to A mutation within the kinase pocket of CSF1R was sufficient to confer increased resistance to CSF1Ri, and that CSF1Ri-resistant macrophages can efficiently engraft the murine brain in the presence of inhibitor concentrations sufficient to deplete endogenous microglia. Our study incorporates the work of two groups that independently identified and validated G795A using orthogonal strategies (Figs. 1 and 3), then collaborated for publication. The former utilized viral transduction of murine cells to experimentally isolate the variable of human CSF1R mutation, and the latter CRISPR engineered the endogenous CSF1R locus of iPSCs to investigate the translational application of microglia replacement with iMG. In the assays performed, we found no discernible effect of G795A on CSF1R signaling, response to an inflammatory stimulus, nor microglial gene expression. However, future studies should finely map the consequences of G795A on CSF1R signaling, donor cell longevity, and microglial function.
Taken together, we hope our findings offer a new path to efficient microglia replacement without traditional chemotherapy, nor genetically modified hosts. Appealing initial applications include correction of mutations driving microgliopathies like CSF1R-related leukoencephalopathy, or delivery of lysosomal hydrolases to compensate for their absence in neuronopathic storage disorders such as Krabbe disease (Bennett and Bennett, 2020). In this context, the observation that xMG engraftment remains stable after CSF1Ri withdrawal suggests that long-term inhibitor treatment will likely not be required, but could be tuned to modulate the extent and rate of microglia replacement, for safety and optimization to specific pathologies. We further hypothesize that because cell engineering technologies are so rapidly evolving (Finck et al., 2022), they will soon permit mechanistically informed design of donor microglia to treat many brain diseases, for example by modulating metabolism, injury response, or protein aggregation.
Beyond microglia replacement therapies, this study may appeal to the fields of hematology and oncology, where the identification of inhibitor-resistant mutations, for example that confer imatinib resistance, is central to drug design (Braun et al., 2020). The tools we applied to identify G795A are commonly used in oncology to predict tumor-resistance mutations (Smith et al., 2012), though these mutations are often associated with reduced ligand dependence. To our knowledge, we present here the first application of a drug-resistant receptor tyrosine kinase mutation to cell therapies. We hope our approach will enhance the study of microglial function in chimeric models and pave the way for therapies involving the delivery of engineered tissue-resident macrophages as “living drugs.”
Materials and methods
Mouse husbandry
Murine macrophage experiments
All animal studies were performed with approval from the Children’s Hospital of Pennsylvania Institutional Animal Care and Use Committee panel in accordance with institutional and national regulations. Animals were housed in a non-barrier facility with 12-h light/dark cycles at 23 ± 2°C, in ventilated cages with no more than five animals per cage. Animals were provided water and standard chow ad libitum unless otherwise noted. Cages and bedding were changed every 1–2 wk.
Rag2 (Rag2tm1.1Flv)−/−, Il2rγ (Il2rγtm1.1Flv)−/−, CSF1h (Csf1tm1(CSF1)Flv)+/+ were purchased from Jackson Laboratory (Stock #017708). To generate Rag2+/+, Il2rγ+/+, CSF1h+/+ mice, we crossed Rag2 (Rag2tm1.1Flv)−/−, Il2rγ (Il2rγtm1.1Flv)−/−, CSF1h (Csf1tm1(CSF1)Flv)+/+ with FVB/NJ (strain no. 001800; Jackson Laboratory) to produce mice heterozygous for Rag2, Il2rγ, and CSF1h. We then intercrossed these offspring to generate Rag2+/+, Il2rγ+/+, CSF1h+/+ mice. Csf1r−/− (FVB.129X1-Csf1rtm1Ers) and Csf1r+/+ littermate animals on the FVB background were obtained from Richard Stanley (Albert Einstein College of Medicine, New York, NY, USA).
Human iMG experiments
All animal procedures were conducted in accordance with the guidelines set forth by the National Institutes of Health and the University of California, Irvine Institutional Animal Care and Use Committee. The MITRG mouse was purchased from Jackson Laboratory (stock #017711); this BALB/c/129 model includes two knockouts alleles, Rag2 (Rag2tm1.1Flv)−/−, Il2rγ (Il2rγtm1.1Flv)−/−, and three humanized knock-in alleles, CSF1h (Csf1tm1(CSF1)Flv)+/+, IL-3/GM-CSFh (Csf2/Il3tm1.1(CSF2,IL3)Flv)+/+, TPOh (Thpotm1.1(TPO)Flv)+/+. All mice were age- and sex-matched and group-housed on a 12-h/12-h light/dark cycle with food and water ad libitum. Mice were housed with ambient temperature and humidity. Cages and bedding were changed every 1–2 wk.
Compounds and diet
PLX3397 (HY-16749), PLX5622 (HY-114153), Edicotinib (JNJ-40346527; JNJ-527; HY-109086), and BLZ945 (Sotuletinib; HY-12768) were obtained from MedChemExpress or Selleckchem and used in vitro at the doses indicated in the text. Due to empirically observed inconsistencies in the potency of PLX5622 between supplier batches, we transitioned to using PLX3397 for all in vivo studies. PLX3397 was formulated in AIN-76A standard chow by Research Diets Inc. at 290 or 600 mg/kg dose as noted.
Murine macrophage experiments
For neonatal experiments, animals were given subcutaneous 25 mg/kg PLX3397 injections for 14 d after cell transplantation, at which point they were harvested (“On PLX”) or maintained for another 7 d without inhibitor (“Off PLX”). For adult experiments, animals were given 290 mg/kg PLX3397 chow for 10–14 d prior to cell transplant. After transplant, they were maintained on 600 mg/kg PLX3397 for 14 d, at which point they were either harvested (“On PLX”) or returned to non-PLX chow and harvested 7 d later (“Off PLX”). Control animals were never PLX-treated but went through identical surgeries and were harvested at the relevant time points.
Human iMG experiments
1 mo after transplantation, mice were kept on 600 mg/kg PLX3397 chow or AIN-76A control chow for 10, 30, 30 on/30 off, or 60 d as indicated in the text.
Generation of viral supernatants
To produce viral supernatants, Lenti-X 293T cells (632180; Takara Bio) were transfected using LipoD293 (504782; SignaGen) according to the manufacturer’s instructions. In short, 293T cells were plated to achieve 80% confluence on the day of transfection. Transfection was performed with 850 ng/ml culture volume each of pCL-Eco (#12371; Addgene) and the relevant transfer plasmid. 5 h after transfection, the media was changed to fresh D10 with 25 mM Hepes (15630080; Invitrogen). Viral supernatant was harvested at 48 h and 72 h. Viruses were concentrated using retroviral Precipitation Solution (VC200; Alstem) according to the manufacturer’s instructions. Concentrated virus was flash-frozen and titered on NIH 3T3 cells (CRL-1658; ATCC) prior to use.
Viral transduction
Transduction was completed in 12-well plates coated with 1 ug/ml fibronectin (F1141; Sigma-Aldrich). Cells were plated at 200,000 cells/ml and transduced with the appropriate volume of virus. Plates were spun for 90 min at 1,000 rcf. After spinfection, cells were gently mixed and resuspended in an additional 2–3 ml of fresh complete media. 24 h after transduction, cells were recovered and expanded.
Inhibitor-resistant receptor screen
CSF1R variants were synthesized and cloned into pCDNA3.1 Zeo (Genscript). We then shuttled these variants into the MIGR1 vector (#27490; Addgene), produced ecotropic gammaretrovirus, and transduced Ba/F3s at a multiplicity of infection of <0.3. Prior to transduction, Ba/F3s were grown in R10 (RPMI 1640, 10% FBS, 1% Penn-Strep [Invitrogen, 15140122]) supplemented with 10 ng/ml murine IL3 (213-13; Peprotech). 48 h after transduction, cells were triple washed with 1× PBS and subsequently grown in R10 media supplemented with 50 ng/ml human CSF1 to switch receptor tyrosine kinase dependency. Cells were observed to ensure non-transduced cells died in the absence of IL3.
ER-HoxB8 CIMs
Immortalization of murine myeloid progenitor cells was completed as described in Wang et al. (2006). In short, bone marrow progenitors were isolated from the long bones of CSF1R−/− or WT FVB/NJ mice using a Percoll density separation (17544501; GE Healthcare) and cultured for 3 d in R10 containing 10 ng/ml IL3, 10 ng/ml IL6 (216-16; Peprotech), and 10 ng/ml SCF (250-03; Peprotech) before being transduced with MSCVneo-HA-ER-HoxB8. 24 h later, the cells were recovered and grown for 2 d in progenitor media (R10 supplemented with 10 mM Hepes [11039021; Thermo Fisher Scientific], 1 mM Na-Pyruvate [11360070; Invitrogen], 0.0005 mM β-estradiol [E2758; Sigma-Aldrich], and 10 ng/ml GM-CSF [315-03; Peprotech]) before selecting for transduction with 1 mg/ml Geneticin (10131035; Thermo Fisher Scientific). After the 2-d selection, cells were split every 2–3 d at 50,000 cells/ml for 2 wk or until all non-immortalized cells were terminally differentiated or dead. G795A- and WT-CIM lines were generated from two CSF1R−/− mice, for a total of six lines (two biological replicates, three technical replicates).
ER-HoxB8 CIM CSF1R variant lines
To produce ER-HoxB8 CIM lines expressing exclusively CSF1R variants, ER-HoxB8 progenitors derived from CSF1R−/− mice were transduced at a multiplicity of infection of <0.3 with either MIGR1-G795A or MIGR1-WT human-CSF1R ecotropic gammaretroviruses then double FACS sorted on GFP expression. Every two to three passages, fluorescence microscopy was used to verify that 100% of each population remained GFP transduction marker positive.
Differentiation of macrophages from ER-HoxB8 progenitors
ER-HoxB8 progenitors were differentiated into macrophages using methods adapted from Wang et al. (2006). Briefly, WT human CSF1R (hCSF1R) and G795A-CSF1R expressing ER-HoxB8 progenitors were plated in a 6-well plate (0.5–1.0 × 106 cells/well) and differentiated for 5 to 7 d in macrophage media (DMEM, 10% FBS, 1% Penn-Strep, 30 ng/ml human CSF1), replenishing media every 2 d. Visual inspection of morphology was used to ensure cells were terminally differentiated into macrophages.
Generation and study of hCSF1R expressing mouse bone marrow
For in vivo transplant studies using primary macrophages expressing CSF1R-variants, whole bone marrow was isolated from 8–12 wk-old Rag2+/+;IL2rγ+/+;CSF1h+/+ mice and cultured in macrophage media for 24 h prior to transduction with either G795A- or WT-hCSF1R ecotropic gammaretrovirus. Cells were harvested 48 h after transduction and transplanted.
CSF1R variant IC50 assays
To determine the relative IC50 of all potential CSF1R variants, we plated transduced Ba/F3 cells at 50,000 cells/ml in triplicate in R10 supplemented with 50 ng/ml CSF1 and PLX3397 (S7818; Selleck Chemicals) at 10 twofold serial dilutions between <0.1 nM and 25 μM. Cells were incubated for 48 h at 37°C in 5% CO2. For IC50 studies using ER-Hoxb8 progenitors, G795A- or WT-hCSF1R expressing cells were differentiated for 6 d in macrophage media. On day 6, differentiated macrophages were harvested and re-plated into 96-well plates at 100,000 cells/ml. 24 h later, cells were given media containing PLX3397 from concentrations of 0–25 μM. For IC50 studies of virally transduced bone marrow macrophages, whole bone marrow from C57BL/6-GFP− mice (Jax: 006567) was cultured in macrophage media. On day 1, cells were transduced with MIGR1-WT human- or G795A-CSF1R variant at matched viral titers to achieve 20% macrophage transduction. On day 3, macrophages were re-plated in triplicate for each treatment condition. On day 4, cells were fed media containing PLX3397 from concentrations of 0–6.4 μM and cultured for 10 d with full media change provided every 2 d.
To quantify cell survival, total GFP intensity (Ba/F3, HoxB8 assays) or cell counts (primary macrophages) were measured in each well using a SpectraMax i3x Multi-Mode Microplate Reader (Molecular Devices, i3x). Data were processed using the following steps: (1) All inhibitor concentrations were log-transformed; (2) data were normalized within cell line with the highest PLX dosage defined as 100% and the lowest PLX dosage defined as 0%; (3) results were analyzed as a nonlinear regression using GraphPad Prism with the log(inhibitor) vs. normalized response—Variable slope equation (Y = 100/(1 + 10^((LogIC50-X)*HillSlope))).
Measurement of hCSF1R signaling
To assess CSF1R variant phosphorylation and downstream signaling, CSF1R-variant expressing macrophage progenitors were differentiated into macrophages. On day 6, cells were switched to serum-free media with 0–4,000 nM PLX3397 at t = −4 h. At t = 0, 360 ng/ml CSF1h was spiked into the media. After 3 min, cells were washed twice with 1× PBS and lysed for 10 min on ice in 200 µl of Laemmli buffer with 5% β-mercaptoethanol (1610747; RAD). Lysates were run on 4–15% precast polyacrylamide gel (#4561084; Bio-Rad) with protein ladder (#1610374; Precision Plus Protein Dual Color Standards), transferred to 0.2 µm polyvinylidene fluoride membranes (#1704272; Bio-Rad), and probed with rabbit Phospho-mCSF1R(Tyr723; 49C10; 1:1,000; #3155; CST), rabbit CSF-1R/M-CSF-R (1:1,000; #3152; CST), rabbit Phospho-p44/42 MAPK (Erk1/2; Thr202/Tyr204; 1:1,000; #9101; CST), and β Actin (1:10,000; #AB8227; Abcam) overnight at 4°C. Following three to five washes in 1× Tris-buffered saline with 0.1% Tween (TBST), membranes were incubated with anti-rabbit IgG, HRP-linked secondary for 1 h (1:10,000; #7074; CST). After an additional three to five washes in 1× TBST, membranes were incubated with Clarity Max ECL Substrate (1705062; Bio-Rad) for visualization and imaged using ChemiDoc MP Imaging System (12003154; Bio-Rad).
Structural modeling of CSF1R-PLX3397 and PLX5622
All structure-derived graphics were produced in PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC). Coordinates are available from the Protein Data Bank as entries 4R7H and 6N33. Manual structures are modeled and built in the graphics program O (Jones et al., 1991).
Patient-derived iPSC lines
The parental human iPSC line (ADRC 76) was generated by the University of California Alzheimer’s Disease Research Center (UCI-ADRC) from subject fibroblasts under approved Institutional Review Board and human Stem Cell Research Oversight protocols and provided by the Institute for Memory Impairments and Neurological Disorders. Non-integrating Sendai virus (CytoTune-iPS 2.0 Sendai Reprogramming Kit; A16517; Thermo Fisher Scientific) was used to perform reprogramming thereby avoiding any integration-induced effects. iPSCs were confirmed to be karyotypically normal by Array Comparative Genomic Hybridization (Cell Line Genetics), sterile, and pluripotent through Pluritest Array Analysis and trilineage in vitro differentiation. iPSCs were maintained on matrigel-coated plates (CB356238; Corning) according to the manufacturer’s specifications in TeSR-E8 (05990; STEMCELL Technologies). Cultured media was replenished every day with fresh medium. Cells were passaged every 5–7 d using ReLeSR (100-0484; STEMCELL Technologies) according to the manufacturer’s specifications. For all in vitro experiments, iPSCs were cultured in 5% O2, 5% CO2 at 37°C.
CRISPR/Cas9 generation of G795 variants
iPSCs were collected following Accutase (07920; STEMCELL Technologies) enzymatic digestion for 3 min at 37°C. 250,000 cells were resuspended in a 100 μl nucleofection buffer from Human Stem Cell Nucleofector Kit 2 (VPH-5022; Lonza). Ribonucleoprotein complex was formed by incubating HiFi Cas9 Nuclease V3 (50 μg; IDTDNA, 1081061) with CRISPR RNA (5′-ATGGTCATGTGGCCAAGATT-3′): trans-activating CRISPR RNA (IDTDNA, 1072534) duplex then combined with either G795A (5′-CCACCGGGACGTGGCAGCGCGTAACGTGCTGTTGACCAATGGTCATGTGGCCAAGATTGCCGACTTCGGGCTGGCTAGGGACATCATGAATGACTCCAACTACATTGTCA-3′), G795C (5′-CCACCGGGACGTGGCAGCGCGTAACGTGCTGTTGACCAATGGTCATGTGGCCAAGATTTGCGACTTCGGGCTGGCTAGGGACATCATGAATGACTCCAACTACATTGTCA-3′), or G795V (5′-CCACCGGGACGTGGCAGCGCGTAACGTGCTGTTGACCAATGGTCATGTGGCCAAGATTGTGGACTTCGGGCTGGCTAGGGACATCATGAATGACTCCAACTACATTGTCA-3′) single-stranded oligodeoxynucleotide template (2–4 μM; IDTDNA) with the cellular suspension for nucleofection using the Amaxa Nucleofector program B-016. Cells were plated on matrigel-coated plates in TeSR-E8 media with 0.25 μM Thiazovivin (72254; STEMCELL Technologies) and CloneR (05889; STEMCELL Technologies) overnight to recover. The following day, cells were mechanically replated to 96-well (3595; Corning) matrigel-coated plates in TeSR-E8 media with 0.25 μM Thiazovivin and CloneR supplement for clonal isolation and expansion. Culture media was replenished everyday with fresh medium. Plates were visually screened for single-clone wells after 5 d. Visually clonal wells were passaged with ReLeSR after 10 d. A cell pellet was collected from each well from which genomic DNA was extracted using Extracta DNA prep for PCR (95091; Quantabio) amplification with Taq PCR Master Mix (K0172; Thermo Fisher Scientific) to confirm diallelic knock-in of G795 variants. The following primers were used: CSF1R_G795_F: 5′-GAAGGCCCAAGACTAACCCT-3′ and CSF1R_G795_R: 5′-GAGGATGCCATAGGACCAGAC-3′.
Hematopoietic progenitor cells (HPCs) and microglia differentiation from iPSCs
HPCs and iPS-microglia were differentiated according to the protocol published by McQuade et al. (McQuade et al., 2018). To begin iHPC differentiation, iPSCs were passaged onto matrigel-coated plates in TeSR-E8 at a density of 80 colonies of 100 cells each per 35 mm well. On day 0, cells were transferred to Medium A from the STEMdiff Hematopoietic Kit (05310; STEMCELL Technologies). On day 3, flattened endothelial cell colonies were exposed to Medium B, and cells remained in Medium B for 7 additional days while HPCs began to lift off the colonies. On day 10, non-adherent CD43+ HPCs were collected by removing medium and cells with a serological pipette. At this point, HPCs were frozen in BamBanker freezing solution (Wako, NC9582225) for long-term storage. Cells used for HPC transplantation were later thawed in complete iPS-Microglia medium (DMEM/F12 [11039-021; Thermo Fisher Scientific], 2× insulin-transferrin-selenite [41400045; Thermo Fisher Scientific], 2× B27 [17504044; Thermo Fisher Scientific], 0.5× N2 [17502048; Thermo Fisher Scientific], 1× glutamax [35050-061; Thermo Fisher Scientific], 1× non-essential amino acids [11140-050; Thermo Fisher Scientific], 400 mM monothioglycerol, and 5 mg/ml human insulin [I2643; Sigma-Aldrich] freshly supplemented with 100 ng/ml IL-34 [Cat #200-34; Peprotech], 50 ng/ml TGFb1 [Cat #100-21; Peprotech], and 25 ng/ml M-CSF [Cat #300-25; Peprotech]) for 18–24 h to recover before being resuspended at 62,500 cells/µl in 1× DPBS (low Ca2+, low Mg2+) for transplantation. For iMG differentiation, HPCs were cultured on matrigel-coated plates in complete iPS-microglia medium for 28 d. Fresh media was added every 2 d. During the last 3 d in culture, 100 ng/ml CD200 (C311; Novoprotein) and 100 ng/ml CX3CL1 (300-18; Peprotech) were added to mature microglia in a brain-like environment.
iMG caspase assay
G795A-, G795C-, and WT-iMG were plated at 70,000 cells per 96-well matrigel-coated plate (six wells per line per condition). At time 0, all microglia were treated with Incucyte Caspase-3/7 Dye for Apoptosis (1:1,000; 4440; Sartorius). Cells were maintained in the described medium: fresh complete medium + 0.1% DMSO, complete media + 250 nM PLX3397, complete medium + 500 nM PLX3397, complete medium + 1 μM PLX3397, complete medium + 250 nM PLX5622, complete medium + 500 nM PLX5622, complete medium + 1 μM PLX5622, complete medium + 250 nM Edicotinib, complete medium + 500 nM Edicotinib, complete medium + 1 μM Edicotinib, complete medium + 250 nM BLZ945, complete medium + 500 nM BLZ945, complete medium + 1 μM BLZ945. Four 20× images per well were collected every hour for 24 h. Using IncuCyte 2020B software, image masks for phase confluence (visually gated out apoptotic cells) as well as caspase 3/7 signal (green) were generated. Graphs display caspase normalized to phase confluence, completed with three lines: WT, G795A, G795C.
iMG confluency assay
G795A-, G795C-, and WT-iMG were plated at 70,000 cells per 96-well matrigel-coated plate (six wells per line per condition). At time 0, all microglia cultured with fresh complete medium + 0.1% DMSO or complete medium + 500 nM PLX3397, complete medium + 500 nM PLX5622, complete medium + 500 nM Edicotinib, or complete medium + 500 nM BLZ945. Four 20× images per well were collected every hour for 24 h. Using IncuCyte 2020B software, image masks for phase confluence were generated. Graphs display confluency across 24 h normalized to 0 h time point.
Live/dead assay
G795A- and WT-iMG were plated at 70,000 cells per well in a 96-well plate (six wells per line per condition). At time 0, all microglia were treated with either 500 nM DMSO vehicle control or one of four CSF1Ri: PLX3397, PLX5622, Edicotinib, and BLZ945. The percent of live versus dead cells was then quantified using a two-color viability/cytotoxicity approach (LIVE/DEAD Viability/Cytotoxicity Kit; L3224; Thermo Fisher Scientific). Four 20× images per well were collected every hour for 24 h. Using IncuCyte 2020B software, image masks for live (green) and dead (red) microglia were generated and quantified.
In vitro co-culture with PLX5622
WT-, G795A-, and G795C-iMG were plated at 200,000 cells per 48-well matrigel-coated plate (5 wells per line per condition). At time 0, five wells per line received 0.1% DMSO and five wells per line received 250 nM PLX5622. After 24 h, 48-well plates were centrifuged at 300 rcf for 5 min, media was gently removed, then wells were resuspended in 1 ml TRIzol (10296010; Thermo Fisher Scientific) per well and transferred to Protein LoBind Tubes (022431102; Eppindorf) for immediate RNA isolation.
Neonatal depletion paradigm
For validation of the neonatal microglia depletion paradigm, C57BL/6J pups were given daily subcutaneous injections of 25 mg/kg for 7 d. Microglial numbers were quantified using immunohistochemistry staining for IBA1.
Neonatal intracerebroventricular transplantation
Murine macrophage experiments
For baseline engraftment controls, C57BL/6J pups either received subcutaneous PLX3397 injections on postnatal day 0–1 (P0–1; “pre-treatment”) or were never given PLX3397. For neonatal cell transplant experiments, Rag2−/−;Il2rγ−/−;CSF1h+/+ or Rag2+/+;Il2rγ+/+;CSF1h+/+ mice were subcutaneously injected daily with 25 mg/kg PLX3397 from P0 to Po13. Pups received intracerebral cell injections by hand as described previously (Bohlen et al., 2017; Bennett et al., 2018). 1 μl containing a single-cell suspension of 300,000 donor cells in PBS was slowly injected bilaterally into the cortex, + 1–2 mm anterior and ± 2–3 mm lateral to lambda at a depth of 0.5–1 mm on P0–3. Mice were harvested 14 d after transplant, or removed from PLX3397 treatment and harvested at later time points as specified. Due to supplier shortages of Rag2−/−;Il2rγ−/−;CSF1h+/+ mice, in Fig. 2 B, one animal receiving G795A- and four receiving WT-CIMs were obtained from a colony heterozygous for a recessive mutation in GALC (Rag2−/−;Il2rγ−/−;CSF1h+/+;GALCtwi/+, generated from Jackson Laboratory, strain no. 000845). CIM injection into Csf1r−/− hosts was performed identically but did not receive PLX3397 injections.
Human iMG experiments
Intracerebroventricular transplantation of neonatal mice pups were performed as detailed in Hasselmann et al. (2019). Briefly, P0 to P1 MITRG pups were placed in a clean cage over a heating pad with a nestlet from the home cage. Pups were then placed on ice for 2–3 min to induce hypothermic anesthesia. Free-hand transplantation was performed using a 30-gauge needle (7803-07; Hamilton) affixed to a 10 μl Hamilton syringe (7653-01; Hamilton), mice received 1 μl of iHPCs suspended in sterile 1× DPBS at 62,500 cells/μl at each injection site (eight sites) totaling 500,000 cells/pup. Bilateral injections were performed at 2/5th of the distance from the lambda suture to each eye, injecting into the lateral ventricles at 3 mm and into the overlying superior cortex at 1 mm, and into the posterior cortex in line with the forebrain injection sites, and perpendicular to lambda at a 45° angle. Transplanted pups were then returned to their home cages and weaned at P21.
Adult intracranial transplantation
All mouse surgeries and use were performed in strict accordance with approved National Institutes of Health (NIH) and Association for Assessment and Accreditation of Laboratory Animal Care–certified institutional guidelines.
Murine macrophage experiments
8–12-wk-old Rag2−/−;Il2rγ−/−;CSF1h+/+ mice were fed PLX3397 chow prior to transplantations. Adult animals receiving G795A-CIMs were pretreated for 9–14 d and were 12–14 wk old at harvest. Adult animals receiving G795A-BMDMs were pretreated for 15–25 d and were 16–21 wk old at harvest. Donor cells were washed twice in PBS at 4°C and resuspended at 200,000 cells/µl in PBS supplemented with hCSF1 at 30 ng/ml. Animals were anesthetized using isoflurane and placed on a stereotaxic rig (Stoelting). Donor cells were bilaterally injected into the following coordinates relative to bregma: dorsal hippocampus (anteroposterior, −2.06 mm; mediolateral, ± 1.75 mm; dorsoventral, −1.75 mm) and cortex (anteroposterior, −0.0 mm; mediolateral ± 1 mm; dorsoventral, −0.95 mm) using a 30-gauge needle (7804-04; Hamilton). 2 µl of cell mixture were injected at each site. The needle was kept at the site of injection for 5 min after each injection. Meloxicam was given subcutaneously at 5 mg/kg on the day of surgery and 1 d post-operatively.
Human iMG experiments
Direct bilateral intracranial injections of WT- or G795A-iMG into the cortex and hippocampus were performed as detailed in Hasselmann et al. (2019). Briefly, adult mice (2 mo old) were anesthetized under continuous isoflurane and secured to a stereotaxic frame (Kopf), and local anesthetic (Lidocaine 2%; Medline, 17478-711-31) was applied to the head before exposing the skull. Using a 30-gauge needle affixed to a 10-ml Hamilton syringe, mice received 2 μl of iMG in sterile 1× DPBS (14190144; Thermo Fisher Scientific) at 62,500 cells/μl at each injection site. Transplantation was conducted bilaterally into the lateral parietal association cortex and dorsal hippocampus at the following coordinates relative to bregma: anteroposterior, 2.06 mm; mediolateral, ± 1.75 mm; dorsoventral, 1.75 mm (hippocampus) and 0.95 mm (cortex). Cells were injected at a rate of 62,500 cells/30 s with 4 min in between injections. The needle was cleaned with consecutive washes of DPBS, 70% (vol/vol) ethanol, and DPBS in between hemispheres and animals. Animals were allowed to recover on heating pads before being placed in their home cages and received 2 mg/ml Acetaminophen (Mapap; Major, 0904-7014-16) diluted in water for 10 d.
Immunohistochemistry
Murine macrophage experiments
Animals were perfused with PBS followed by 4% paraformaldehyde (PFA; wt/vol) and then immersion fixed in 4% PFA for 12 h at 4°C. Samples were cryoprotected in 30% sucrose-PBS, embedded in OCT (102094-106; VWR), cryosectioned sagittally into 12–16-μm-thick slices, and mounted on Superfrost Plus slides (12–544A; Thermo Fisher Scientific). For rabbit anti-IBA1 (1:500; 019-19741; Wako), goat anti-IBA1 (1:250; ab5076; Abcam), and rabbit anti-Tmem119 (1:500; ab209064; Abcam) staining, we dried slides at 60°C, rehydrated in PBS, blocked for 1 h at room temperature (RT) in PBSTx (1× PBS with 0.3% Triton X-100) with 10% donkey serum (S30-100 Ml; Sigma-Aldrich). We then incubated with primary antibodies in PBST×/1% serum/0.3% Triton X-100 (staining buffer) overnight at 4°C. After washing, we incubated slides in staining buffer with AlexaFluor-conjugated secondary antibodies (1:500-1:1,000; Invitrogen) for 2 h at RT, then 1× PBS washed and coverslipped.
Human iMG experiments
Animals were perfused with PBS and isolated brains were drop fixed in 4% (wt/vol) PFA for 36 h then cryoprotected in a 30% (wt/vol) sucrose at 4°C. Brains were sectioned coronally into 30-μm-thick slices on a freezing microtome (Leica, SM 2010R) and stored in a solution of 0.05% NaN3 (S2002; Sigma-Aldrich) in 1× PBS (P44017-100TAB; Sigma-Aldrich) as free-floating slices. For staining, tissue was blocked for 1 h in 1× PBS, 0.2% Triton X-100 (9002-93-1; Thermo Fisher Scientific), and 10% donkey serum (NC9624464; Thermo Fisher Scientific). Immediately following blocking, brain sections were placed in primary antibodies diluted in 1× PBS and 1% donkey serum and incubated overnight on a shaker at 4°C. Samples were then incubated in conjugated secondary antibodies for 1 h followed by mounting on microscope slides. Sections were labeled with combinations of goat anti-IBA1 (1:200; ab5076; Abcam), rabbit anti-IBA1 (1:500; 019-19741; Wako), rabbit anti-Ku80 (1:100; ab80592; Abcam), mouse anti-Ku80 (1:100; MAS-12933; Invitrogen), rabbit anti-CD45 (1:200; ab33533; Abcam), rabbit anit-MX1 (1:300; 37849S; Cell Signaling), rabbit anti-Ki67 (1:200; ab16667; Abcam), goat anti-LGALS3 (1:250; AF1197; R&D Systems), rabbit anti-P2RY12 (1:500; HPA014518; Millipore Sigma), and rabbit anti-P2RY12 (1:250; NBP233870; Novus Biologicals) overnight at 4°C. After washing, sections were incubated with AlexaFluor-conjugated secondary antibodies (1:400) for 1 h, then washed three times with PBS before mounting with Fluoromount-G (0100-01; SouthernBiotech). Notably, we consistently observe and have previously reported brighter IBA1 staining of human microglia than murine microglia (Hasselmann et al., 2019). We speculated that differences in the intensity of IBA1 between human and murine microglia likely arise from preferential binding to the human immunogen used to produce these antibodies.
RNA in situ hybridization
We performed in situ hybridizations for Ms4a7 (Cat #314601-c2; ACDBio) on fixed frozen samples using the RNAscope system with RNAscope Multiplex Fluorescent V2 Assay for fluorescent development (ACDbio), according to the manufacturer’s protocols using TSA reagents (Akoya Biosciences). When staining concurrently for IBA1 and GFP (Cat #AB16901; Millipore) protein, we used our standard immunostaining protocol. Sections were counterstained with DAPI for nuclei.
Imaging acquisition and processing
Murine macrophage experiments
We acquired epifluorescence images using a BZ-X800 Microscope (Keyence). Whole brain tiled images were taken at 10× magnification and stitched using Keyence Analyzer software prior to image export. Representative images were taken at 10×. We analyzed images in Fiji (https://imagej.net/Fiji), adjusting for brightness and black values, but we performed no other image math. Engraftment renderings were made in Fiji (example, Fig. S1 A). The 488/GFP channel was processed by subtracting background, adjusting threshold level, analyzing particles (size = 0–0.1 in2; circularity = 0.5–1), making a mask of GFP+ cells similar to masking strategy used by Kozlowski et al. (Kozlowski and Weimer, 2012), converting into ultimate points, and overlaying onto the DAPI channel. The entire brain was then outlined using ImageJ’s “polygon sections” tool and anything falling outside of this brain outline was excluded (example, Fig. S1 J). Representative confocal images were captured using a Leica SP8 confocal microscope equipped with a Diode 405 laser, HyD sensors, 40×/1.30 oil lens, and controlled with Leica LAS X v.3.5.2 software. Z-stack processed using maximum intensity projection function in ImageJ.
Human iMG experiments
Immunofluorescent sections were visualized and captured using an Olympus FX 1200 confocal microscope. Representative images of microglia in the fornix after PBS and LPS treatment represent confocal Z-stacks (14 slices at 1.77-micron step intervals) taken at 40× magnification. Half brain stitch images represent confocal Z-stacks (11 images at 2.39-micron step intervals) taken at 10× magnification. Whole brain stitch images represent confocal Z-stacks (11 images at 2.81-micron step intervals) taken at 4× magnification. Representative images of engrafted microglia in the hippocampus and cortex represent confocal Z-stacks (14 slices at 1.77-microns step intervals) taken 40× magnification. Representative images of engrafted microglia in PLX3397 30 d on/30 d off represent confocal Z-stacks (12 slices at 1.51-microns step intervals) taken at 20× magnification. Stitch was performed using Fluoview FV31S-DT software. Three sections were imaged per region per animal for each quantification. In some cases, brightness and contrast settings of confocal images were adjusted to reveal fine structures and morphology in representative images. Importantly, no such changes were made to any images used for quantification.
Area of engraftment and density counts
Preceding image analysis, images were cropped, DAPI and 488 channels merged, and artifacts removed. Engraftment areas for total brain and cortex were calculated by defining a region of interest (ROI) around GFP+ cell groups with no fewer than 50 total cells or densities lower than 50 cells/mm2. Engraftment area edge points were defined by the nucleus of the edgemost cell being no more than 200 μm from neighboring cells. ROI was drawn using ImageJ’s polygon selection area tool, spline fit, and area measured to obtain mm2. Percent area engrafted was calculated by dividing total brain area by engrafted area(s). For each subject, average engraftment was calculated from three matched 10× brain tiles (first, third, sixth). Olfactory bulb regions were not included for consistency as they were missing from some samples.
Cell densities were manually counted following protocol outlined in Diem et al. with minimal modifications (Diem et al., 2015). The polygon section area tool was used to define ROI of the engrafted region and calculate area as described above. The outlined ROI was given to a blinded researcher (F.S. Purnell) who then used the ImageJ cell counter tool to record cells that were GFP or Ionized calcium binding adaptor molecule 1 (IBA1) and DAPI positive. Results were averaged and divided by area of engraftment to obtain cells/mm2. For each subject, the average of three matched sections (first, third, sixth) was taken.
Quantification of microglial replacement
Percentage xMG was quantified with whole brain stitch images acquired using identical settings with an Olympus FV1200 scanning laser confocal microscope. Using the “Spots” function in IMARIS software (Bitplane), Ku80+, and IBA1+ spots were quantified separately. Then, total human cell count (Ku80+/IBA1+ spots) and total mouse cell count (Ku80−/IBA1+ spots) were determined. Total percentage of human cells was then found by dividing total human cells and mouse cells by total cells independently and multiplying by 100.
Characterization of WT and G795A microglia
For analysis of microglia response to LPS-induced neuroinflammation, 40× images were acquired of the fornix using identical settings with an Olympus FV1200 scanning laser confocal microscope. Using the “Surfaces” function in IMARIS software (Bitplane), mean intensity values of IBA1, P2RY12, CD45, MX1, and CD163 surfaces were quantified from three sections per animal and normalized to number Ku80+/IBA1+ cells per field of view (FOV). All values represent mean intensity per cell per FOV. For analysis of the proliferating and engrafted G795A microglia at the wavefront and hippocampus in 1-mo PLX3397-treated mice, 40× images were acquired per indicated region using identical settings with an Olympus FV1200 scanning laser confocal microscope. Using the “Surfaces” function in IMARIS software (Bitplane), total intensity values of Ku80, P2RY12, and LGALS-3 surfaces were quantified from three sections per animal and normalized to number Ku80+/IBA1+ cells per FOV. All values represent integrated density per FOV of noted region.
Intraperitoneal LPS injections
A neuroinflammatory state was induced through systemic LPS treatment. Animals received intraperitoneal injections of either 2 mg/kg LPS (eBioscience; n = 3 mice per cell line) or PBS (n = 3 mice per cell line) once every 24 h over a 48-h period (two injections total). 12 h after the final LPS treatment, animals were euthanized, perfused with PBS, and half brains were collected for immunohistochemistry.
Tissue dissociation for bulk RNA-seq
All steps were performed on ice or at 4°C with ice cold reagents, and all centrifuge steps were performed for 10 min at 400 ×g with full brake and acceleration unless otherwise stated. Anesthetized mice were intracardially perfused with 1× DPBS, half brains were dissected, the cerebellum was removed, and tissue was stored in RPMI 1640 (11835030; Thermo Fisher Scientific) until subsequent perfusions were completed. Brains were manually homogenized using a 7-ml Dounce homogenizer by adding 4 ml of RPMI 1640 and performing 10 strokes with the “A” pestle followed by 10 strokes with the “B” pestle. Samples were then run through 70-μm filters, presoaked in RPMI 1640, and the filter was washed with 10 ml of RPMI 1640. The samples were centrifuged, supernatant was discarded, and the pellet was resuspended in 30% Percoll overlaid with 2 ml of 1× DPBS centrifuged at 400 ×g for 20 min with acceleration and brake set to 0 to remove myelin. The myelin band and supernatant were discarded, and cell pellets were resuspended in 80 µl MACS buffer (0.5% BSA in 1× DPBS) + 20 µl Mouse Cell Removal beads (130-104-694; Miltenyi) and incubated at 4°C for 15 min. Magnetically labeled mouse cells were separated using LS columns and the MidiMACs separator (130-042-302; Miltenyi) while the unlabeled human cells were collected in the flow through. Human cells were then pelleted by centrifugation, and dead cells were magnetically removed using the Dead Cell Removal kit, Annexin V (17899; Stem Cell Technologies) by resuspending the pellets in 100 µl of buffer (2% BSA + 1 mM CaCl2 in 1× PBS) in 5-ml polystyrene round-bottom tubes and the following manufacturer protocol. Samples were then centrifuged, the supernatant was discarded, and 500 μl of TRIzol was added. All samples were stored at −80°C until further processing.
Bulk RNA isolation from iMG and xMG
All steps were performed on ice or at 4°C, and all centrifuge steps were performed at 15,000 ×g. Frozen samples were thawed on ice and RNA was isolated by adding 140 μl of TET (10 mM Tris 8.0/0.01 mM EDTA/0.05% Tween20) followed by 140 μl chloroform:isolamyl alcohol 24:1 (C0549; Sigma-Aldrich). Samples were centrifuged for 10 min and the aqueous phase was collected and added to 1.5 μl of GlycolBlue (Thermo Fisher Scientific). 55 μl of 3 M sodium acetate and 550 μl isopropanol (Sigma-Aldrich) were added, and the samples were mixed by inverting then stored at −20°C overnight. Samples were then spun for 30 min, supernatants were removed from pellets, and 500 μl of 75% ethanol was added to the pellet. Samples were then spun for 30 min, and supernatants were removed from pellets. RNA pellets were dried at RT to remove any residual ethanol, resuspended in 14 μl of RNase-free H2O, and used immediately or aliquoted and stored at −80°C for downstream analysis.
RNA analysis, library construction, and bulk RNA-seq
For iMG samples, RNA concentration was determined by Qubit and RNA integrity (RIN) values were determined using the Agilent 2100. RNA samples had a mean RIN value of 9.23. Libraries were generated using the TruSeq Stranded Total RNA kit (Illumina) with 100 ng of RNA as input. For xMG samples, cell yields were lower than anticipated, so RIN values and concentrations were not determined and, rather, 5 μl of RNA per sample was used for library construction using ClonTech SMART-seq V4 Ultra Low Input kit (Takara Bio). For all samples, the quality of the cDNA libraries was assayed using the Agilent 2100 bioanalyzer high sensitivity DNA assay and the DNA high-sensitivity Qubit. The libraries were quantified by Kapa qPCR, normalized to 2 nM and then multiplexed for sequencing on the Illumina NovaSeq 6000 platform with paired read 100 base chemistry.
Bulk RNA-seq data analysis
FASTQ files were preprocessed using BBDuk (Bushnell, 2018) to filter out ribosomal RNA and PhiX reads, trim Illumina adapters, and to quality trim any base pairs below a PHRED score of 10. FASTQC (Andrews, 2014) was then performed to verify the quality of the sequencing files, and all files were determined to be of sufficient quality for downstream processing. Reads were then pseudoaligned to the human GRCh38 transcriptome (Ensembl release 99; Schneider et al., 2017; Zerbino et al., 2018) using Kallisto version 0.46.0 (Bray et al., 2016), transcripts were summarized to the gene level through tximport version 1.14.2 (Soneson et al., 2015), and differential gene expression analysis was performed using DESeq2 version 1.26.0 (Love et al., 2014) after removing genes with summed counts <10. Differentially expressed genes were calculated using DESeq2 by applying an FDR cutoff of 0.05 and a log2(fold-change) cutoff of ±1. For visualization, data were normalized and converted to a log2 scale using DESeq2's varianceStabilizingTransformation. Volcano and bar plots were generated using the “ggplot2” package (Wickham, 2016), and correlation plots were generated using values derived from the R “cor” function with the Pearson method. Core microglia gene signature defined as described in Patir et al. (2019).
Ethics approval and consent to participate
Human iPSC lines were generated by the UCI-ADRC stem cell core. Subject fibroblasts were collected under approved Institutional Review Boards and Human Stem Cell Research Oversight committee protocols. Informed consent was received for all participants.
Statistic calculations
GraphPad Prism 9 was used to perform statistical tests and generate P values. We used standard designation of P values throughout the Figures (ns = not significant or P ≥ 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Values are represented as mean ± SEM. Details of number of replicates and the specific statistical test used are provided in the individual figure legends.
Consent for publication
All authors consent to the publication of this study.
Online supplemental material
Fig. S1 shows methods and approaches for identification of the G795A variant, and its implementation in primary mouse BMDMs. Fig. S2 shows generation, inhibitor resistance, and transcriptomic comparisons of G795 iPSC microglia. Fig. S3 shows the distinct phenotype of the proliferative wavefront of CSF1Ri resistant human microglia colonizing the murine brain. Table S1 shows bulk RNA-seq sample information related to Fig. S2 and Figs. 3 and 4.
Data availability
The bulk RNA-seq data sets are available through the GEO SuperSeries accession number GSE220215, or individual accession numbers GSE220213 and GSE220214. Further information and requests for resources and reagents should be directed to and will be fulfilled by Mathew Blurton-Jones ([email protected]) and F. Chris Bennett ([email protected]).
Acknowledgments
We thank Dr. Richard Stanley (Albert Einstein College of Medicine, New York, NY, USA) for the generous gift of Csf1r−/− (FVB.129X1-Csf1rtm1Ers) and Csf1r+/+ littermate animals on the FVB background. The Ba/F3 cell line was shared by Dr. Martin Carroll (University of Pennsylvania, Philadelphia, PA, USA). The MSCVneo-HA-ER-HoxB8 plasmid was a gift from Dr. David Sykes (Mass General Hospital, Boston, MA, USA) through Igor Brodsky (University of Pennsylvania, School of Veterinary Medicine, Philadelphia, PA, USA). Graphical abstract created with BioRender.com. Generation of the parental ADRC76 iPSC line was performed by the UCI-ADRC iPS cell core.
This work was made possible, in part, through access to the Genomics High Throughput Facility Shared Resource of the Cancer Center Support Grant (P30CA-062203) at the University of California, Irvine and National Institutes of Health (NIH) shared instrumentation grants 1S10RR025496-01, 1S10OD010794-01, and 1S10OD021718-01. This work was supported by NIH T32 AG073088 (J.P. Chadarevian); NIH T32 GM008076 (S.I. Lombroso), National Science Foundation Graduate Research Fellowship Program DGE-1845298 (S.I. Lombroso and K.M. Nemec); NIH AG061895 (H. Davtyan); NIH K12GM081259 (D.E. Marzan); NIH P30 AG066519 (M. Blurton-Jones); NIH R01DA048813 (M. Blurton-Jones and R.C. Spitale); NIH T32 GM007170-47 (S. Chaluvadi); NIH T32-MH014654 (W.H. Aisenberg); National Institute of Neurological Disorders and Stroke R01NS120960; The Paul Allen Frontiers Group GRT-00000774; The Klingenstein-Simons fellowship in neuroscience (F.C. Bennett); and a generous gift from the Susan Scott Foundation (M. Blurton-Jones).
Author contributions: Conceptualization: J.P. Chadarevian, S.I. Lombroso, G.C. Peet, M.L. Bennett, M. Blurton-Jones, F.C. Bennett. Development of methodology: J.P. Chadarevian, S.I. Lombroso, G.C. Peet, J. Hasselmann, C. Tu, D.E. Marzan, C.A. O’Brien, W.H. Aisenberg, H. Davtyan. Murine macrophage studies: S.I. Lombroso, G.C. Peet, D. Marzan, F. Purnell, K. Nemec, S. Chaluvadi, C.A. O’Brien, F. Yaqoob, W. Aisenberg, M. Paniagua, M.L. Bennett, F.C. Bennett. iMG studies: J.P. Chadarevian, J. Hasselmann, C. Tu, J. Capocchi, A. Lahian, A. Escobar, W. England, H. Davtyan, R.C. Spitale, M. Blurton-Jones. Analysis and interpretation of data: J.P. Chadarevian, S.I. Lombroso, G.C. Peet, J. Hasselmann, M. Blurton-Jones, F.C. Bennett. Writing: J.P. Chadarevian, S.I. Lombroso, M. Blurton-Jones, and F.C. Bennett. All authors critically reviewed and approved the final version.

