


Central B cell tolerance is believed to be regulated by B cell receptor signaling induced by the recognition of self-antigens in immature B cells. Using humanized mice with defective MyD88, TLR7, or TLR9 expression, we demonstrate that TLR9/MYD88 are required for central B cell tolerance and the removal of developing autoreactive clones. We also show that CXCL4, a chemokine involved in systemic sclerosis (SSc), abrogates TLR9 function in B cells by sequestering TLR9 ligands away from the endosomal compartments where this receptor resides. The in vivo production of CXCL4 thereby impedes both TLR9 responses in B cells and the establishment of central B cell tolerance. We conclude that TLR9 plays an essential early tolerogenic function required for the establishment of central B cell tolerance and that correcting defective TLR9 function in B cells from SSc patients may represent a novel therapeutic strategy to restore B cell tolerance.
Introduction
Autoreactive B cells produced by random V(D)J recombination are normally eliminated at two counterselection checkpoints; the first, referred to as central B cell tolerance, takes place in the bone marrow (BM), whereas the second occurs in the periphery and removes autoreactive clones that escaped the central checkpoint (Meffre and O’Connor, 2019; Wardemann et al., 2003). Defects in these early B cell tolerance checkpoints are associated with many autoimmune diseases, including systemic sclerosis (SSc) and systemic lupus erythematosus (SLE), and result in large numbers of circulating autoreactive B cells in patients’ blood (Glauzy et al., 2022; Meffre and O’Connor, 2019; Menard et al., 2011; Samuels et al., 2005; Yurasov et al., 2005). Central B cell tolerance is believed to be regulated by B cell receptor (BCR) signaling induced by the binding of self-antigens to immature B cells and which induces tolerance mechanisms, such as receptor editing or deletion (Goodnow, 1996; Nemazee and Weigert, 2000). However, biallelic IRAK4 or MYD88 mutations result in a failure to suppress developing autoreactive B cells in the BM, suggesting the contribution of specific innate immune receptors in central counter-selection processes (Isnardi et al., 2008). Since IRAK-4 or MYD88 are two key molecules that mediate the function of most Toll-like receptors (TLRs), pattern recognition receptors may therefore play a role in the counter-selection of developing autoreactive B cells. While recognition of foreign nucleic acids by TLRs allows the detection of numerous pathogens and the activation of innate immune responses, recognition of self-nucleic acids by TLRs also occurs and may promote or mitigate autoimmunity (Marshak-Rothstein, 2006). Autoimmune mouse models have revealed that TLR7, which binds single-stranded RNA (ssRNA), plays an essential role in driving disease, whereas TLR9, which binds double-stranded DNA (dsDNA), appears to exert tolerogenic function (Christensen et al., 2006; Leibler et al., 2022; Nickerson et al., 2013; Tilstra et al., 2020). It is unclear why these receptors may have paradoxical functions and if they participate in the establishment of central B cell tolerance. Recent reports suggest that proper B cell–intrinsic TLR9 function is required for tolerance (Leibler et al., 2022; Tilstra et al., 2020). In agreement with mouse data, TLR9 function is decreased in B cells from patients with SLE but not in plasmacytoid dendritic cells (pDCs; Gies et al., 2018). The origin of TLR9 impairment in B cells from SLE patients and potentially other patients with autoimmune diseases is currently unknown.
Here, we report that innate immune recognition via TLR9 but not TLR7 is required for the establishment of central B cell tolerance, thereby challenging the current paradigm that BCR affinity for self is responsible for the deletion of autoreactive immature B cells in the BM. We also show that chemokine CXCL4/platelet factor 4 inhibits TLR9 function in B cells by sequestering TLR9 ligands away from the endosomal compartments where this receptor resides and abrogates central B cell tolerance, thereby revealing a key role for this chemokine in the pathogenesis of SSc.
Results
TLR9 is required for the establishment of central B cell tolerance
The impaired silencing of developing autoreactive B cells in the BM of IRAK-4–, MYD88-, and UNC-93B–deficient patients suggested that endosomal TLR7 and TLR9 which bind ssRNA and dsDNA, respectively, may play an important role in the establishment of central B cell tolerance (Isnardi et al., 2008). We tested this hypothesis by engrafting NOD-scid-common gamma chain (γc) knockout (NSG) immunodeficient mice with CD34+ human fetal hematopoietic stem cells (HSCs) transduced with GFP-tagged lentivirus expressing MYD88-, TLR7-, or TLR9-specific shRNA to inhibit the expression of these molecules (Fig. 1 A; Cantaert et al., 2015; Schickel et al., 2016). Various proportions of GFP+ human B cells expressing MYD88, TLR7, or TLR9 shRNA developed in NSG humanized mice that displayed 50–75% decreased expression of the shRNA targeted genes, as determined by Western blots, compared with GFP− counterparts (Fig. 1, B and C; and Fig. S1, A and B). Importantly, the expression of TLR7 and TLR9 shRNA specifically inhibited the activation of GFP+ B cells and the induction of CD69 following stimulation by these receptors, respectively, whereas MYD88 inhibition led to decreased TLR7 and TLR9 responses as expected (Fig. 1 D and Fig. S1 C). In addition, both TLR9 and MYD88, but not TLR7 inhibition, resulted in decreased BCR responses in GFP+ B cells, revealing the importance of MYD88/TLR9/BCR multiprotein supercomplex identified in lymphomas for signaling in untransformed naïve B cells (Fig. 1 D and Fig. S1 C; Phelan et al., 2018). The analyses of antibody reactivity revealed that GFP+ new emigrant/transitional B cells expressing TLR7 shRNA contained low frequencies of polyreactive and antinuclear clones similar to counterparts in unmanipulated humanized mice (we did not test the corresponding GFP− fractions; Fig. 1, E–H; and Fig. S1 D). In contrast, the inhibition of MYD88 or TLR9 resulted in significantly elevated proportions of GFP+ new emigrant/transitional B cells producing polyreactive or antinuclear antibodies, whereas GFP− counterparts that were isolated from the same humanized mice and normally expressed MYD88 and TLR9 rarely contained autoreactive clones (Fig. 1, E–H; and Fig. S1 D). Hence, B cell–intrinsic expression of MYD88 and TLR9 is essential for the removal of developing autoreactive B cells in the BM, as suggested by transgenic mouse models (Kuraoka et al., 2017). The injection of hydroxychloroquine (HCQ) that interferes with endosomal function and inhibits TLR9 responses also abrogated central B cell tolerance in humanized mice (Fig. S1, E–J; Kuraoka et al., 2017). We conclude that TLR9 exerts its first tolerogenic function during early B cell development in the BM and prevents the production of autoreactive B cells following the delivery by BCRs of membrane self-antigens associated with dsDNA potentially present in apoptotic blebs and which effectively induce clonal deletion (Cocca et al., 2002).
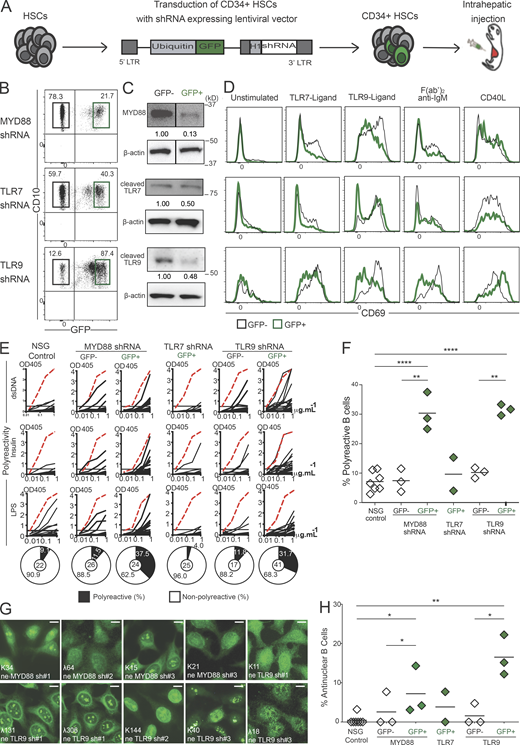
TLR9 is essential for central B cell tolerance. (A) Schematic diagram describing the generation of humanized mice. CD34+ HSCs transduced with GFP-tagged lentiviruses expressing MYD88, TLR7, or TLR9 shRNA were injected into the liver of 3-d-old recipient NSG mice. (B) Representative flow cytometry analysis showing the gating strategy to sort GFP− and GFP+ CD19+ splenocytes from mice engrafted with HSCs transduced with the indicated GFP-tagged lentiviruses. (C) Expression analysis of indicated proteins in sorted GFP+ and GFP− CD19+ splenocytes from humanized mice. β-actin is used for normalization of protein expression. White lines indicate that intervening lanes have been spliced out. (D) Representative surface CD69 expression in GFP− (black line) compared with GFP+ (green line) CD19+ splenocytes after 48 h in culture unstimulated or activated with the indicated ligands or F(ab)′2 anti-IgM. (E) Representative polyreactivity of antibodies cloned from single new emigrant/transitional B cells isolated from indicated humanized mice was tested by ELISA against dsDNA, insulin, and LPS. Dotted red lines show the positive control. Pie charts represent the frequencies of reactive (solid) and non-reactive (open) clones, with the number of clones tested shown in the center. OD405 nm, optical density. (F and H) Frequencies of (F) polyreactive and (H) antinuclear reactive clones in new emigrant/transitional B cells. Each data point summarizes the reactivity data from an average of n = 21 cloned recombinant antibodies from control NSG (n = 7), MYD88 shRNA (n = 3), TLR7 shRNA (n = 2), and TLR9 shRNA (n = 3) humanized mice. Same control NSG mouse is used for representation in Fig. 2 D, Fig. 10 A, and Fig. S1 D. Averages are shown with a bar, and statistically significant differences are indicated (Student’s t test, *P < 0.05, **P < 0.01, ****P < 0.0001). (G) Representative nuclear staining patterns for antibodies cloned from new emigrant/transitional B cells. Scale bars, 25 µm. Please see Fig. S1. Source data are available for this figure: SourceData F1.
TLR9 is essential for central B cell tolerance. (A) Schematic diagram describing the generation of humanized mice. CD34+ HSCs transduced with GFP-tagged lentiviruses expressing MYD88, TLR7, or TLR9 shRNA were injected into the liver of 3-d-old recipient NSG mice. (B) Representative flow cytometry analysis showing the gating strategy to sort GFP− and GFP+ CD19+ splenocytes from mice engrafted with HSCs transduced with the indicated GFP-tagged lentiviruses. (C) Expression analysis of indicated proteins in sorted GFP+ and GFP− CD19+ splenocytes from humanized mice. β-actin is used for normalization of protein expression. White lines indicate that intervening lanes have been spliced out. (D) Representative surface CD69 expression in GFP− (black line) compared with GFP+ (green line) CD19+ splenocytes after 48 h in culture unstimulated or activated with the indicated ligands or F(ab)′2 anti-IgM. (E) Representative polyreactivity of antibodies cloned from single new emigrant/transitional B cells isolated from indicated humanized mice was tested by ELISA against dsDNA, insulin, and LPS. Dotted red lines show the positive control. Pie charts represent the frequencies of reactive (solid) and non-reactive (open) clones, with the number of clones tested shown in the center. OD405 nm, optical density. (F and H) Frequencies of (F) polyreactive and (H) antinuclear reactive clones in new emigrant/transitional B cells. Each data point summarizes the reactivity data from an average of n = 21 cloned recombinant antibodies from control NSG (n = 7), MYD88 shRNA (n = 3), TLR7 shRNA (n = 2), and TLR9 shRNA (n = 3) humanized mice. Same control NSG mouse is used for representation in Fig. 2 D, Fig. 10 A, and Fig. S1 D. Averages are shown with a bar, and statistically significant differences are indicated (Student’s t test, *P < 0.05, **P < 0.01, ****P < 0.0001). (G) Representative nuclear staining patterns for antibodies cloned from new emigrant/transitional B cells. Scale bars, 25 µm. Please see Fig. S1. Source data are available for this figure: SourceData F1.

B cell–intrinsic expression of MYD88 and TLR9 but not TLR7 are required for the removal of developing autoreactive B cells in the BM. (A) Relative mRNA levels of TLR7 and TLR9 in GFP+ sorted CD19+ B cells compared with GFP− counterparts isolated from BM and spleen of humanized mice transplanted with HSCs transduced with GFP-tagged lentiviruses expressing either TLR7 shRNA or TLR9 shRNA. (B) MYD88, full length (FL) and cleaved TLR7 and TLR9 protein expressions relative to β-actin in GFP− (open bars) and GFP+ (green bars) sorted B cells from humanized mice engrafted with HSCs transduced with GFP-tagged lentiviruses expressing MYD88 shRNA (n = 3), TLR7 shRNA (n = 1), or TLR9 shRNA (n = 2). (C) Relative frequencies of CD69+cells in sorted GFP− (open bars) and GFP+ (green bars) B cells isolated from NSG humanized mice transplanted with HSCs transduced with GFP-tagged lentiviruses expressing MYD88 shRNA (n = 4), TLR7 shRNA (n = 2), or TLR9 shRNA (n = 6) in GFP+ (green bars) after 48 h in culture with no stimulation (Unstim.) or activation with the indicated ligands or F(ab)′2 anti-IgM. Significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01, ***P < 0.001). (D) Antibodies cloned from single GFP− and GFP+ new emigrant/transitional B cells isolated from NSG humanized mice engrafted with fetal HSCs transduced with GFP-tagged lentiviruses expressing MYD88 shRNA, TLR7 shRNA, or TLR9 shRNA were tested by ELISA for reactivity against dsDNA, insulin, and LPS. The same control NSG mouse is used for representation in Fig. 1 E, Fig. 2 D, and Fig. 10 A. Dotted red lines show the positive control. Antibodies were considered polyreactive when they recognized all three antigens. For each B cell fraction, the frequencies of polyreactive (filled area) and non-polyreactive (open area) clones are summarized in a pie chart below, with the total number of clones (n) tested indicated in the center. (E) Schematic diagram depicting the HCQ injection strategy. NSG humanized mice generated by fetal CD34+ HSCs engraftment are treated with daily intraperitoneal injections of 0.2 mg HCQ for a week. (F) Relative frequencies of CD69+ B cells from HCQ-treated NSG mice (black bars) compared with control humanized mice (open bars) after 48 h in culture with no stimulation (Unstim.) or activation with the indicated ligands or F(ab)′2 anti-IgM. (G) Representative polyreactivity of antibodies cloned from single new emigrant/transitional B cells isolated from HCQ-injected and control humanized mice were tested by ELISA against dsDNA, insulin, and LPS. Dotted red lines show the positive control. Pie charts represent the frequencies of reactive (solid) and nonreactive (open) clones, with the number of clones tested (n) shown in the center. OD405 nm, optical density. (H and I) Frequencies of polyreactive (H) and antinuclear reactive (I) clones in new emigrant/transitional B cells from the indicated humanized mice. Each data point summarizes the reactivity data from an average of n = 21 cloned recombinant antibodies from control (n = 7) and HCQ-injected NSG humanized mice (n = 2). (J) Representative nuclear staining patterns for antibodies cloned from new emigrant/transitional B cells from HCQ-injected NSG humanized mice. Scale bars, 25 µm. The average values are represented as bars and significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01, ****P < 0.0001).
B cell–intrinsic expression of MYD88 and TLR9 but not TLR7 are required for the removal of developing autoreactive B cells in the BM. (A) Relative mRNA levels of TLR7 and TLR9 in GFP+ sorted CD19+ B cells compared with GFP− counterparts isolated from BM and spleen of humanized mice transplanted with HSCs transduced with GFP-tagged lentiviruses expressing either TLR7 shRNA or TLR9 shRNA. (B) MYD88, full length (FL) and cleaved TLR7 and TLR9 protein expressions relative to β-actin in GFP− (open bars) and GFP+ (green bars) sorted B cells from humanized mice engrafted with HSCs transduced with GFP-tagged lentiviruses expressing MYD88 shRNA (n = 3), TLR7 shRNA (n = 1), or TLR9 shRNA (n = 2). (C) Relative frequencies of CD69+cells in sorted GFP− (open bars) and GFP+ (green bars) B cells isolated from NSG humanized mice transplanted with HSCs transduced with GFP-tagged lentiviruses expressing MYD88 shRNA (n = 4), TLR7 shRNA (n = 2), or TLR9 shRNA (n = 6) in GFP+ (green bars) after 48 h in culture with no stimulation (Unstim.) or activation with the indicated ligands or F(ab)′2 anti-IgM. Significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01, ***P < 0.001). (D) Antibodies cloned from single GFP− and GFP+ new emigrant/transitional B cells isolated from NSG humanized mice engrafted with fetal HSCs transduced with GFP-tagged lentiviruses expressing MYD88 shRNA, TLR7 shRNA, or TLR9 shRNA were tested by ELISA for reactivity against dsDNA, insulin, and LPS. The same control NSG mouse is used for representation in Fig. 1 E, Fig. 2 D, and Fig. 10 A. Dotted red lines show the positive control. Antibodies were considered polyreactive when they recognized all three antigens. For each B cell fraction, the frequencies of polyreactive (filled area) and non-polyreactive (open area) clones are summarized in a pie chart below, with the total number of clones (n) tested indicated in the center. (E) Schematic diagram depicting the HCQ injection strategy. NSG humanized mice generated by fetal CD34+ HSCs engraftment are treated with daily intraperitoneal injections of 0.2 mg HCQ for a week. (F) Relative frequencies of CD69+ B cells from HCQ-treated NSG mice (black bars) compared with control humanized mice (open bars) after 48 h in culture with no stimulation (Unstim.) or activation with the indicated ligands or F(ab)′2 anti-IgM. (G) Representative polyreactivity of antibodies cloned from single new emigrant/transitional B cells isolated from HCQ-injected and control humanized mice were tested by ELISA against dsDNA, insulin, and LPS. Dotted red lines show the positive control. Pie charts represent the frequencies of reactive (solid) and nonreactive (open) clones, with the number of clones tested (n) shown in the center. OD405 nm, optical density. (H and I) Frequencies of polyreactive (H) and antinuclear reactive (I) clones in new emigrant/transitional B cells from the indicated humanized mice. Each data point summarizes the reactivity data from an average of n = 21 cloned recombinant antibodies from control (n = 7) and HCQ-injected NSG humanized mice (n = 2). (J) Representative nuclear staining patterns for antibodies cloned from new emigrant/transitional B cells from HCQ-injected NSG humanized mice. Scale bars, 25 µm. The average values are represented as bars and significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01, ****P < 0.0001).
TLR7 deficiency does not alter central B cell tolerance
Since the inhibition of TLR7 expression using our lentiviral shRNA approach was incomplete in developing GFP+ B cells, the observed lack of TLR7 requirement for the establishment of central B cell tolerance might be due to residual TLR7 expression. We therefore studied TLR7-deficient patients who were recently identified during the COVID-19 pandemic because TLR7 deficiency confers high susceptibility to SARS-CoV-2 infection (Fig. 2; Asano et al., 2021; van der Made et al., 2020). We investigated three TLR7-deficient patients with different TLR7 gene mutations (Fig. 2 A). The first two patients were previously reported and displayed either a stop codon that prevented TLR7 expression or a TLR7 missense mutation that abrogated TLR7 function in families A and B, respectively (Asano et al., 2021; van der Made et al., 2020; Fig. 2 A). The third TLR7-deficient patient from family C harbored a novel V771L missense mutation that abolished TLR7 function, as illustrated by the lack of CD69 activation marker induction in B cells that developed in several humanized mice engrafted with HSCs isolated from the patient’s BM (Fig. 2, B and C). All three TLR7-deficient patients displayed low frequencies of polyreactive and antinuclear new emigrant/transitional B cells that were similar to frequencies in healthy donor (HD) counterparts (Fig. 2, D–F). In addition, autoreactive B cell proportions were also low in new emigrant/transitional B cells isolated from a humanized mouse engrafted with HSCs isolated from the BM of TLR7-deficient patient C III-1 (Fig. 2, D–F). Thus, TLR7 is not required for the establishment of central B cell tolerance.

Functional central B cell tolerance in TLR7-deficient patients. (A) The pedigrees of three families with TLR7-deficient patients and sequence electropherograms of respective TLR7 variants. Circles represent female family members; squares represent males. A slash symbol represents a deceased individual. Circled dot represents a TLR7 heterozygous carrier. (B) Representative flow cytometry histograms show CD69 expression on B cells isolated from an NSG humanized mouse engrafted with HSCs from controls or patient CIII.1 (TLR7 V771L) after 48 h in culture with no stimulation (gray shadow) or activation with the indicated ligands or F(ab)′2 anti-IgM (thick line). (C) CD69 induction following various stimulations is summarized in C and significant differences are indicated (Mann–Whitney U test, **P < 0.01). (D) Antibodies cloned from single new emigrant/transitional B cells isolated from the blood of three TLR7-deficient patients and control HDs or from NSG humanized mice engrafted with HSCs from controls or patient CIII.1 (TLR7 V771L) was tested by ELISA for reactivity against dsDNA, insulin, and LPS. Same control NSG mouse is used for representation in Fig. 1 E, Fig. 10 A, and Fig. S1 D. Dotted red lines show the positive control. Pie charts represent the frequencies of reactive (solid) and nonreactive (open) clones, with the number of clones tested (n) shown in the center. OD405 nm, optical density. (E and F) Summary for frequencies of polyreactive (E) and antinuclear reactive (F) clones in new emigrant/transitional B cells.
Functional central B cell tolerance in TLR7-deficient patients. (A) The pedigrees of three families with TLR7-deficient patients and sequence electropherograms of respective TLR7 variants. Circles represent female family members; squares represent males. A slash symbol represents a deceased individual. Circled dot represents a TLR7 heterozygous carrier. (B) Representative flow cytometry histograms show CD69 expression on B cells isolated from an NSG humanized mouse engrafted with HSCs from controls or patient CIII.1 (TLR7 V771L) after 48 h in culture with no stimulation (gray shadow) or activation with the indicated ligands or F(ab)′2 anti-IgM (thick line). (C) CD69 induction following various stimulations is summarized in C and significant differences are indicated (Mann–Whitney U test, **P < 0.01). (D) Antibodies cloned from single new emigrant/transitional B cells isolated from the blood of three TLR7-deficient patients and control HDs or from NSG humanized mice engrafted with HSCs from controls or patient CIII.1 (TLR7 V771L) was tested by ELISA for reactivity against dsDNA, insulin, and LPS. Same control NSG mouse is used for representation in Fig. 1 E, Fig. 10 A, and Fig. S1 D. Dotted red lines show the positive control. Pie charts represent the frequencies of reactive (solid) and nonreactive (open) clones, with the number of clones tested (n) shown in the center. OD405 nm, optical density. (E and F) Summary for frequencies of polyreactive (E) and antinuclear reactive (F) clones in new emigrant/transitional B cells.
Impaired TLR9 function in B cells from patients with SSc
We recently reported that B cells from patients with SLE have a defect in their TLR9 response, which correlates with both impaired central B cell tolerance and the production of anti-dsDNA autoantibodies targeting this TLR9 ligand (Gies et al., 2018; Sindhava et al., 2017; Yurasov et al., 2005). We investigated whether this paradigm may apply to patients with other autoimmune diseases, especially SSc, which is also characterized by a failure to establish central B cell tolerance and the secretion of autoantibodies targeting either topoisomerase I or centromere proteins, both of which are associated with TLR9 ligand dsDNA (Glauzy et al., 2022). We therefore tested TLR9 responses in B cells isolated from SSc patients with either a diffuse or a limited form of the disease, many of which were treatment-naïve, in two different cohorts. In vitro activation experiments performed with B cells isolated from fresh blood samples of SSc patients from our identification cohort revealed specific defective TLR9 function in SSc as illustrated by the impaired induction of CD69 and CD86, TACI, and CD25 on patient’s B cells or the significantly decreased secretion of IL-6 and TNF after stimulation by TLR9 ligand CpG for 2 d (Fig. 3, A–C; and Fig. S2, A and B; Taher et al., 2018). In contrast, B cell responses to TLR7, BCR, or CD40 stimulations were similar between SSc patients and HDs (Fig. 3, A–C; and S2, A and B). These results were validated using frozen peripheral blood mononuclear cells (PBMCs) from a replication cohort of patients with SSc previously enrolled at the University Medical Center Utrecht (UMCU) in the Netherlands (Fig. 3 B and Fig. S2 C). Thus, B cells from patients with either SLE or SSc show a specific impairment of TLR9 function, thereby contrasting with the previously reported normal or even enhanced TLR9 responses in pDCs from these patients (Ah Kioon et al., 2018; Gies et al., 2018; van Bon et al., 2014).

Defective TLR9 function in B cells from SSc patients. (A) Surface expression of CD69 and CD86 in CD20+CD27− gated naïve B cells from a representative HD and a patient with SSc after 48 h in culture with no stimulation (Unstim.) or activated with the indicated ligands or F(ab)′2 anti-IgM. Frequencies of single and double-positive populations are indicated. (B) Frequency summary of CD86+CD69+ B cells from HD (open circles) and patients with SSc (black circles) from the Yale identification cohort (n = 23) and UMCU replication cohort (n = 15). (C) IL-6, IL-10, and TNF concentrations measured by Luminex Assay in culture supernatants of B cells from HD (open circles) and patients with SSc (black circles, pooled cohorts). Average values are represented as bars and significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01). Please see Fig. S2.
Defective TLR9 function in B cells from SSc patients. (A) Surface expression of CD69 and CD86 in CD20+CD27− gated naïve B cells from a representative HD and a patient with SSc after 48 h in culture with no stimulation (Unstim.) or activated with the indicated ligands or F(ab)′2 anti-IgM. Frequencies of single and double-positive populations are indicated. (B) Frequency summary of CD86+CD69+ B cells from HD (open circles) and patients with SSc (black circles) from the Yale identification cohort (n = 23) and UMCU replication cohort (n = 15). (C) IL-6, IL-10, and TNF concentrations measured by Luminex Assay in culture supernatants of B cells from HD (open circles) and patients with SSc (black circles, pooled cohorts). Average values are represented as bars and significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01). Please see Fig. S2.

Decreased TLR9 function in naïve B cells from patients with SSc. (A) Surface expression of TACI and CD25 in CD20+CD27− gated naïve B cells from a representative HD and a patient with SSc after 48 h in culture with no stimulation (Unstimulated) or activation with the indicated ligands or F(ab)′2 anti-IgM. The frequency of single and double positive populations is indicated. (B and C) Frequency summary of TACI+ (left) and CD25+ (right) B cells from HD (open circles) and patients with SSc (black circles) from (B) the Yale identification cohort (n = 23) and (C) UMCU replication cohort (n = 15). Average values are represented as bars and significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01, ***P < 0.001).
Decreased TLR9 function in naïve B cells from patients with SSc. (A) Surface expression of TACI and CD25 in CD20+CD27− gated naïve B cells from a representative HD and a patient with SSc after 48 h in culture with no stimulation (Unstimulated) or activation with the indicated ligands or F(ab)′2 anti-IgM. The frequency of single and double positive populations is indicated. (B and C) Frequency summary of TACI+ (left) and CD25+ (right) B cells from HD (open circles) and patients with SSc (black circles) from (B) the Yale identification cohort (n = 23) and (C) UMCU replication cohort (n = 15). Average values are represented as bars and significant differences are indicated (Mann–Whitney U test, *P < 0.05, **P < 0.01, ***P < 0.001).
CXCL4 inhibits TLR9 function in B cells
We showed recently that CXCL4, a chemokine also known as platelet factor 4 (PF4) that was found elevated in the sera of patients with SSc, SLE, and other rheumatic diseases, can drastically exacerbate TLR9 responses in human pDCs leading to IFN-α secretion (Ah Kioon et al., 2018; Du et al., 2022; Lande et al., 2019; van Bon et al., 2014; Volkmann et al., 2016). To evaluate the impact of CXCL4 on TLR9 function in B cells, we tested the induction of activation markers and cytokine secretion by B cells isolated from the blood of HDs following their stimulation in vitro as above. We found that CXCL4 had no impact on TLR7 B cell responses following R848 stimulation (Fig. 4, A and B; and Fig. S3, A and B). In contrast, CXCL4 did inhibit CD69, CD83, and CD86 cell surface expression and IL-6, IL-10, and TNF production normally induced by TLR9 ligand CpG without affecting B cell survival, highlighting the strong impact of CXCL4 on TLR9-induced B cells responses (Fig. 4, C and D; Fig. S3, C–E; and Fig. 5, A–D). CXCL4 also blunted TLR9 responses in immature B cells isolated from the BM of humanized mice (Fig. 5, E and G). We then tested whether CXCL4 may also inhibit TLR9 responses when TLR9 ligand is delivered via the BCR since BCR-TLR9 co-crosslinking may induce in B cells different responses than when the BCR and TLR9 are activated independently (Sindhava et al., 2017). We, therefore, stimulated B cells with dual variable domain IgG, DVD-Ig 3764, a unique reagent that combines both anti-human IgM and anti-dsDNA reactivities in the same antibody. Hence, DVD-Ig 3764 allows the internalization of dsDNA present in culture from dying cells to live B cells via their IgM/BCRs. A similar DVD-Ig has been used to activate murine B cells (Pawaria et al., 2015). We found that DVD-Ig 3764 activated B cells as illustrated by the secretion of IL-6 and TNF (Fig. 4 E). This activation was TLR9-dependent because the addition of TLR9 antagonist ODN INH-18 completely abolished both IL-6 and TNF secretion, thereby revealing that IgM triggering with DVD-Ig 3764 at 1 μg/ml was not sufficient to stimulate B cells (Fig. 4 E). In addition, CXCL4 was as effective as TLR9 antagonist ODN INH-18 at blocking IL-6 and TNF secretion when B cells were stimulated by DVD-Ig 3764 (Fig. 4 E). Hence, CXCL4 also abrogates the ability of BCR-delivered TLR9 ligand to activate B cells via TLR9. CXCL4 has been reported to bind CXCR3B in humans, but we and others have shown that its ability to activate pDCs is independent of this receptor (Du et al., 2022; Lande et al., 2019; Vandercappellen et al., 2011). CXCL4 inhibitory effect on TLR9 responses in B cells did not rely on CXCR3 because it was refractory to the addition of either AMG487, a CXCR3 antagonist, or a blocking antibody specific for CXCR3 (Fig. 5, F–I; Henne et al., 2012). We confirmed the absence of a role for CXCR3 using splenic B cells from CXCR3-deficient or WT mice in which TLR9 function was equally inhibited by CXCL4 (Fig. 5 J). CXCL4 inhibition of TLR9 responses in B cells was also independent of CCR1, a receptor that binds to CXCL4 in monocytes (Fig. 5 K; Fox et al., 2018). To decipher the mechanism by which CXCL4 inhibits TLR9 responses in B cells, we investigated using RNA sequencing (RNA-seq) the genes and pathways regulated in B cells incubated with CXCL4, TLR9 ligand, or both. We observed that CXCL4 had little to no effect on B cells because virtually no genes were either upregulated or downregulated by CXCL4 alone as compared with unstimulated cells (Fig. 4, F–H). However, the addition of CXCL4 completely blocked both TLR9-induced and TLR9-downregulated genes to the point that the CpG+CXCL4 stimulated B cells cluster with unstimulated cells (Fig. 4, F–H). The extent of TLR9 inhibition by CXCL4 suggests that TLR9 response is not simply reduced but that sensing of TLR9 ligand itself may be impaired in B cells in the presence of CXCL4.

CXCL4 inhibits TLR9 responses in B cells. (A–D) Purified B cells from HDs were cultured with no stimulation (Unstim.), TLR7 ligand (R848, 0.75 µM) with or without CXCL4 (10 µg/ml; A and B), or TLR9 ligand (CpG1018, 0.15 µM) with or without CXCL4 (C and D). (A and C) CD69 (A, n = 8; and C, n = 11), or CD86 (A, n = 8; and C, n = 17) surface expression was quantified by flow cytometry. (B and D) Secreted IL-6 (B, n = 6; and D, n = 51) and TNF (n = 7) were quantified by ELISA or Luminex after 24 h of culture. (E) B cells from HDs were incubated for 48 h with medium (Unstim.), dual variable domain IgG, which displays both anti-human IgM and anti-dsDNA reactivities (DVD-IgTM 3764, 1 µg/ml) either alone or in the presence of either CXCL4 (10 µg/ml) or the TLR9 antagonist ODN INH-18 (inh18, 1 µg/ml). IL-6 and TNF secretion were quantified by immunoassay (ELISA). Individual donors are indicated, and all results are represented as a mean ± SEM, and statistical significance was evaluated using a Mann–Whitney U test and **P ≤ 0.01, ***P ≤ 0.001. (F–H) FACS-sorted B cells from HDs were cultured for 6 h with no stimulation (Unstim.), with CXCL4, CpG, or CpG + CXCL4 and analyzed by RNA-seq. (F) Principal component analysis of the differentially expressed genes. (G) Hierarchical clustering of log-transformed cpm for differentially expressed genes identified by RNA-seq analysis of RNA from B cells cultured as indicated. The highest and lowest modulated genes by CpG alone are indicated. (H) Volcano plot comparing gene expression in B cells cultured for 6 h with CXCL4 as compared to unstimulated (upper panel), CpG as compared to unstimulated (lower left panel), CXCL4 + CpG as compared to CpG (lower right panel). Colors on all graphs indicate differentially expressed genes in CpG as compared to unstimulated, where upregulated genes are indicated in red and downregulated genes in blue.
CXCL4 inhibits TLR9 responses in B cells. (A–D) Purified B cells from HDs were cultured with no stimulation (Unstim.), TLR7 ligand (R848, 0.75 µM) with or without CXCL4 (10 µg/ml; A and B), or TLR9 ligand (CpG1018, 0.15 µM) with or without CXCL4 (C and D). (A and C) CD69 (A, n = 8; and C, n = 11), or CD86 (A, n = 8; and C, n = 17) surface expression was quantified by flow cytometry. (B and D) Secreted IL-6 (B, n = 6; and D, n = 51) and TNF (n = 7) were quantified by ELISA or Luminex after 24 h of culture. (E) B cells from HDs were incubated for 48 h with medium (Unstim.), dual variable domain IgG, which displays both anti-human IgM and anti-dsDNA reactivities (DVD-IgTM 3764, 1 µg/ml) either alone or in the presence of either CXCL4 (10 µg/ml) or the TLR9 antagonist ODN INH-18 (inh18, 1 µg/ml). IL-6 and TNF secretion were quantified by immunoassay (ELISA). Individual donors are indicated, and all results are represented as a mean ± SEM, and statistical significance was evaluated using a Mann–Whitney U test and **P ≤ 0.01, ***P ≤ 0.001. (F–H) FACS-sorted B cells from HDs were cultured for 6 h with no stimulation (Unstim.), with CXCL4, CpG, or CpG + CXCL4 and analyzed by RNA-seq. (F) Principal component analysis of the differentially expressed genes. (G) Hierarchical clustering of log-transformed cpm for differentially expressed genes identified by RNA-seq analysis of RNA from B cells cultured as indicated. The highest and lowest modulated genes by CpG alone are indicated. (H) Volcano plot comparing gene expression in B cells cultured for 6 h with CXCL4 as compared to unstimulated (upper panel), CpG as compared to unstimulated (lower left panel), CXCL4 + CpG as compared to CpG (lower right panel). Colors on all graphs indicate differentially expressed genes in CpG as compared to unstimulated, where upregulated genes are indicated in red and downregulated genes in blue.

CXCL4 increases TLR9 ligand uptake but inhibits TLR9 function in B cells. (A–D) Purified B cells from HDs were cultured for 24 h with no stimulation (unstim.) or activated with (A and B) TLR7 agonist (R848, 0.75 µM) with or without CXCL4 (10 µg/ml), or (C and D) TLR9 agonist (CpG1018, 0.15 µM) with or without CXCL4. (A and C) CD83 surface expression (A, n = 8; and C, n = 19) were quantified by flow cytometry. (B and D) gene expression levels of IL6, IL10, or TNF were quantified by Q-PCR (B, n = 8; and D, n = 26–28). (E) Volcano plot comparing gene expression in B cells cultured for 6 h with CXCL4 as compared with unstimulated (upper panel), CpG as compared with unstimulated (middle panel), CXCL4 + CpG as compared to CpG (lower panel). Colors on all graphs indicate differentially expressed genes in CpG as compared to unstimulated, where upregulated genes are indicated in red and downregulated genes in blue. For A–D, individual donors are indicated, and all results are represented as the mean ± SEM and statistical significance evaluated using a Mann–Whitney U test and *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. (F) B cells from HDs (n = 5–7) were incubated for 6 h with medium only or with TLR9-L CpG (0.15 µM) with or without CXCL4 (1, 3, and 10 µg/ml) and gene expression level of IL-10 was quantified by Q-PCR. (F) Secreted TNF was measured by ELISA after 48 h of culture of BJAB cells with medium only (Unstim.), or with TLR9 ligand (CpG1018, 0.15 µM) alone or with CXCL4. TNF secretion was normalized to TLR9-L stimulation. Results from five independent experiments are represented as mean ± SEM. Statistical significance was evaluated using a Mann–Whitney U test (**P ≤ 0.01). (G–I) Representative flow cytometry histograms show CpG uptake after 1 h of incubation of (G) purified B cells from HDs and of (H) BJAB B cells with medium only, or with fluorescent CpG (0.15 µM) with or without CXCL4 (I) CpG uptake data is represented as mean + SEM of total MFI in three independent experiments and statistical significance was evaluated using unpaired two-tailed t tests (*P ≤ 0.05).
CXCL4 increases TLR9 ligand uptake but inhibits TLR9 function in B cells. (A–D) Purified B cells from HDs were cultured for 24 h with no stimulation (unstim.) or activated with (A and B) TLR7 agonist (R848, 0.75 µM) with or without CXCL4 (10 µg/ml), or (C and D) TLR9 agonist (CpG1018, 0.15 µM) with or without CXCL4. (A and C) CD83 surface expression (A, n = 8; and C, n = 19) were quantified by flow cytometry. (B and D) gene expression levels of IL6, IL10, or TNF were quantified by Q-PCR (B, n = 8; and D, n = 26–28). (E) Volcano plot comparing gene expression in B cells cultured for 6 h with CXCL4 as compared with unstimulated (upper panel), CpG as compared with unstimulated (middle panel), CXCL4 + CpG as compared to CpG (lower panel). Colors on all graphs indicate differentially expressed genes in CpG as compared to unstimulated, where upregulated genes are indicated in red and downregulated genes in blue. For A–D, individual donors are indicated, and all results are represented as the mean ± SEM and statistical significance evaluated using a Mann–Whitney U test and *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. (F) B cells from HDs (n = 5–7) were incubated for 6 h with medium only or with TLR9-L CpG (0.15 µM) with or without CXCL4 (1, 3, and 10 µg/ml) and gene expression level of IL-10 was quantified by Q-PCR. (F) Secreted TNF was measured by ELISA after 48 h of culture of BJAB cells with medium only (Unstim.), or with TLR9 ligand (CpG1018, 0.15 µM) alone or with CXCL4. TNF secretion was normalized to TLR9-L stimulation. Results from five independent experiments are represented as mean ± SEM. Statistical significance was evaluated using a Mann–Whitney U test (**P ≤ 0.01). (G–I) Representative flow cytometry histograms show CpG uptake after 1 h of incubation of (G) purified B cells from HDs and of (H) BJAB B cells with medium only, or with fluorescent CpG (0.15 µM) with or without CXCL4 (I) CpG uptake data is represented as mean + SEM of total MFI in three independent experiments and statistical significance was evaluated using unpaired two-tailed t tests (*P ≤ 0.05).

Human CXCL4 inhibits TLR9 responses in B cells independently of CXCR3 and CCR1. (A–D) Dot plots represent Annexin V and propidium iodide (PI) staining on purified B cells from HDs (n = 5) that were cultured for 48 h with no stimulation (unstim.) or activated with TLR9L (CpG, 1 µg/ml) with or without CXCL4 (10 µg/ml), proportions of AnnexinV−PI− live, AnnexinV+PI− apoptotic, and AnnexinV+PI+ dead cells are summarized in B–D. (E and F) CD69 induction on (E) CD19+CD10+ immature B cells isolated from the BM, or (F) CD19+CD10+ new emigrant/transitional B cells isolated from the spleen of control NSG humanized mice after 48 h in culture stimulated with TLR9 ligand (CpG, 1 µg/ml) with or without either CXCL4 (10 µg/ml, eleven experiments) or CXCL4 and CXCR3 antagonist (AMG-487, 30 µM, seven experiments). (G) Relative CD69 induction following TLR9 stimulation is summarized in G and significant differences are indicated (Mann–Whitney U test, *P < 0.05, ***P < 0.001). (H and I) B cells from HDs were cultured for 24 h unstimulated (unstim.) or activated with TLR9 ligand CpG (TLR9-L, 0.15 µM) with or without CXCL4 in the presence or not of an anti-CXCR3 antagonist mAb or CXCR3 antagonist AMG487. (H) Secreted IL-6 (n = 4) was quantified by ELISA and (I) CD69 and CD86 surface expression (n = 6) by flow cytometry. (J) Purified splenic B cells from C56Bl6/J (n = 11, left panel) and CXCR3-ko mice (n = 5, right panel) were cultured for 48 h unstimulated (unstim.) or activated with TLR9-L CpG-C274 (0.15 µM) with or without CXCL4. (K) B cells from HDs were cultured for 24 h unstimulated (unstim.), or activated with TLR9 ligand CpG (TLR9-L, 0.15 µM) with or without CXCL4 alone or in the presence of a CCR1 antagonist J113863. Secreted IL-6 and TNF (n = 6) were quantified by ELISA. Individual donors or mice are indicated, and all results are represented as the mean ± SEM and statistical significance evaluated using a Mann–Whitney U test and *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 for G–K.
Human CXCL4 inhibits TLR9 responses in B cells independently of CXCR3 and CCR1. (A–D) Dot plots represent Annexin V and propidium iodide (PI) staining on purified B cells from HDs (n = 5) that were cultured for 48 h with no stimulation (unstim.) or activated with TLR9L (CpG, 1 µg/ml) with or without CXCL4 (10 µg/ml), proportions of AnnexinV−PI− live, AnnexinV+PI− apoptotic, and AnnexinV+PI+ dead cells are summarized in B–D. (E and F) CD69 induction on (E) CD19+CD10+ immature B cells isolated from the BM, or (F) CD19+CD10+ new emigrant/transitional B cells isolated from the spleen of control NSG humanized mice after 48 h in culture stimulated with TLR9 ligand (CpG, 1 µg/ml) with or without either CXCL4 (10 µg/ml, eleven experiments) or CXCL4 and CXCR3 antagonist (AMG-487, 30 µM, seven experiments). (G) Relative CD69 induction following TLR9 stimulation is summarized in G and significant differences are indicated (Mann–Whitney U test, *P < 0.05, ***P < 0.001). (H and I) B cells from HDs were cultured for 24 h unstimulated (unstim.) or activated with TLR9 ligand CpG (TLR9-L, 0.15 µM) with or without CXCL4 in the presence or not of an anti-CXCR3 antagonist mAb or CXCR3 antagonist AMG487. (H) Secreted IL-6 (n = 4) was quantified by ELISA and (I) CD69 and CD86 surface expression (n = 6) by flow cytometry. (J) Purified splenic B cells from C56Bl6/J (n = 11, left panel) and CXCR3-ko mice (n = 5, right panel) were cultured for 48 h unstimulated (unstim.) or activated with TLR9-L CpG-C274 (0.15 µM) with or without CXCL4. (K) B cells from HDs were cultured for 24 h unstimulated (unstim.), or activated with TLR9 ligand CpG (TLR9-L, 0.15 µM) with or without CXCL4 alone or in the presence of a CCR1 antagonist J113863. Secreted IL-6 and TNF (n = 6) were quantified by ELISA. Individual donors or mice are indicated, and all results are represented as the mean ± SEM and statistical significance evaluated using a Mann–Whitney U test and *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 for G–K.
CXCL4 enhances TLR9 ligand uptake by B cells
We then explored the hypothesis that CXCL4 may prevent TLR9 ligand ability to reach the late endosomal compartment where TLR9 resides. We first tested whether CXCL4 may prevent the uptake of the TLR9 ligands by B cells using fluorescent CpG as previously described (Guiducci et al., 2006). Contrary to our expectations, we observed by flow cytometry a dramatic time-dependent increase of CpG uptake in the presence of CXCL4 compared with CpG alone in both freshly isolated peripheral B cells and human BJAB B cell line, in which TLR9 response was also blunted by CXCL4 (Fig. 6 A and Fig. S3, F–I). B cell CpG uptake in the presence of CXCL4 was dose-dependent, correlated with decreased TLR9 response, and was reduced at low temperature suggesting an active process (Fig. 6, B–E). We and others have shown that CXCL4 can complex with DNA and form stable nanoparticles with DNA, in part due to the cationic nature of CXCL4, which impacts the TLR9 response in pDCs (Du et al., 2022; Lande et al., 2019). CXCL4L1 is a mutated variant of CXCL4 which differs from CXCL4 by three amino acids that lead to an open conformation of the C-terminal helix and a decreased net positive charge (Kuo et al., 2013). In contrast to CXCL4, CXCL4L1 failed to regulate TLR9 function in pDCs (Du et al., 2022). Similarly, we found that CXCL4L1 was also unable to enhance TLR9-L uptake and inhibit TLR9 responses in B cells (Fig. 6, F and G). Hence, proper CXCL4 conformation and positive charges are essential for the regulation of TLR9 function by CXCL4 in both pDCs and B cells.

CXCL4 increases the uptake of TLR9 ligand. (A and B) Purified B cells from HDs were cultured for the indicated time with medium only, or with fluorescent CpG-AF488 (0.15 µM) with or without CXCL4 at (A) 10 µg/ml or (B) at the indicated concentration, and fluorescence was quantified by flow cytometry. Individual donors are indicated (n = 4–12) and mean + SEM of total MFI is shown. (C and D) B cells from HDs (n = 5–7) were incubated for 6 h with medium only or with TLR9-L CpG (0.15 µM) with or without CXCL4 (1, 3, and 10 µg/ml) and gene expression level of IL-6 and TNF was quantified by Q-PCR. (E) B cells from HDs (n = 2) were incubated with medium only, CpG-AF488 (0.15 µM) with or without CXCL4 at 37°C or 4°C, and fluorescence was quantified by flow cytometry. Data are represented as mean + SEM of total MFI. (F and G) B cells from HDs (n = 6) were cultured for 1 h (F) or 24 h (G) either unstimulated (unstim.) or activated with TLR9 ligand CpG alone or with either CXCL4 or CXCL4L1. (F) Fluorescence was quantified by flow cytometry and data is represented as mean + SEM of total MFI. Individual donors are indicated and statistical significance was evaluated using a Mann–Whitney U test and **P ≤ 0.01. (G) Secreted IL-6 was quantified by ELISA (n = 6). All results are represented as a mean ± SEM, and statistical significance was evaluated using a Mann–Whitney U test and *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. (H) B cells from HDs (n = 6–9) were cultured for 1 h with medium only, with CpG-AF488 (0.15 µM) + CXCL4 with or without genistein, CPZ, or EIPA, and fluorescence was quantified by flow cytometry. Individual donors are indicated, and results are represented as a mean ± SEM and statistical significance was evaluated using a Mann–Whitney U test and *P ≤ 0.05, ***P ≤ 0.001. (I) B cells were cultured for 1 h with CpG-AF488 (0.5 µM) and CXCL4 either alone or with CPZ, stained with anti-CD20, and analyzed using Amnis imaging flow cytometer system. Image gallery, from left to right: showing bright field (BF) images, followed by B cell surface (stained with anti-CD20), uptaken CpG based on cells stained for AF488, merged images of CpG/CD20 showing absence of colocalization in CpG + CXCL4 group and the internalization score calculated by Amnis IDEAS software. Cells with a score of >0.3 were considered to have internalized CpG and those with a score of <0.3 were considered to have surface-bound CpG. Statistical significance was evaluated using a Chi-square test; ***P ≤ 0.001. Scale bars, 7 µm.
CXCL4 increases the uptake of TLR9 ligand. (A and B) Purified B cells from HDs were cultured for the indicated time with medium only, or with fluorescent CpG-AF488 (0.15 µM) with or without CXCL4 at (A) 10 µg/ml or (B) at the indicated concentration, and fluorescence was quantified by flow cytometry. Individual donors are indicated (n = 4–12) and mean + SEM of total MFI is shown. (C and D) B cells from HDs (n = 5–7) were incubated for 6 h with medium only or with TLR9-L CpG (0.15 µM) with or without CXCL4 (1, 3, and 10 µg/ml) and gene expression level of IL-6 and TNF was quantified by Q-PCR. (E) B cells from HDs (n = 2) were incubated with medium only, CpG-AF488 (0.15 µM) with or without CXCL4 at 37°C or 4°C, and fluorescence was quantified by flow cytometry. Data are represented as mean + SEM of total MFI. (F and G) B cells from HDs (n = 6) were cultured for 1 h (F) or 24 h (G) either unstimulated (unstim.) or activated with TLR9 ligand CpG alone or with either CXCL4 or CXCL4L1. (F) Fluorescence was quantified by flow cytometry and data is represented as mean + SEM of total MFI. Individual donors are indicated and statistical significance was evaluated using a Mann–Whitney U test and **P ≤ 0.01. (G) Secreted IL-6 was quantified by ELISA (n = 6). All results are represented as a mean ± SEM, and statistical significance was evaluated using a Mann–Whitney U test and *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. (H) B cells from HDs (n = 6–9) were cultured for 1 h with medium only, with CpG-AF488 (0.15 µM) + CXCL4 with or without genistein, CPZ, or EIPA, and fluorescence was quantified by flow cytometry. Individual donors are indicated, and results are represented as a mean ± SEM and statistical significance was evaluated using a Mann–Whitney U test and *P ≤ 0.05, ***P ≤ 0.001. (I) B cells were cultured for 1 h with CpG-AF488 (0.5 µM) and CXCL4 either alone or with CPZ, stained with anti-CD20, and analyzed using Amnis imaging flow cytometer system. Image gallery, from left to right: showing bright field (BF) images, followed by B cell surface (stained with anti-CD20), uptaken CpG based on cells stained for AF488, merged images of CpG/CD20 showing absence of colocalization in CpG + CXCL4 group and the internalization score calculated by Amnis IDEAS software. Cells with a score of >0.3 were considered to have internalized CpG and those with a score of <0.3 were considered to have surface-bound CpG. Statistical significance was evaluated using a Chi-square test; ***P ≤ 0.001. Scale bars, 7 µm.
To characterize how CpG or CpG/CXCL4 complexes enter human B cells, we evaluated three key mechanisms of uptake using specific inhibitors, chlorpromazine (CPZ; an inhibitor of clathrin-mediated endocytosis), genistein (inhibitor of clathrin-independent endocytosis), or 5-(N-ethyl-N-isopropyl) amiloride (EIPA; inhibitor of macropinocytosis). CPZ significantly inhibited the uptake of CpG/CXCL4, whereas neither genistein nor EIPA altered CpG/CXCL4 uptake, suggesting that CXCL4 favored CpG entry via clathrin-mediated endocytosis (Fig. 6, H and I). To determine in which cellular compartment CpG and CpG/CXCL4 localized over time, we used the ImageStream technology from Amnis, which combines a flow cytometry quantitative approach and high content image analysis (Fig. 7, A and B). CpG “speckled” patterns in B cells at 1 h after incubation with CXCL4 revealed that CpG/CXCL4 complexes did not remain on the cell surface but were instead rapidly internalized and accumulated in a subintracellular compartment with strong fluorescence intensity (Fig. 7, C and D). We conclude that CXCL4 enhances the uptake of TLR9-L by B cells.

CXCL4 sequesters TLR9 ligands away from the late endosomal compartments. (A–C) Purified B cells from HDs were cultured for 1 h with the CpG-AF488 (5 µM) or with CpG-AF488 (0.5 µM) + CXCL4 (10 µg/ml) and analyzed by Amnis imaging flow cytometer system. (A) Cells in camera focus were selected from all events on the basis of gradient root mean square of the bright field image and (B) single cells were identified by plotting “Area” versus “Aspect ratio” in which events with higher “Aspect ratio” are likely to be single cells. (C) Image gallery, from left to right: showing bright field (BF) images, followed by B cell surface (stained by antiCD20), uptaken CpG based on cells stained for AF488, and merged images of CpG/CD20 showing absence of colocalization. (D) Representative histograms showing intensity of CpG-AF488 in CpG- (left panel) versus CpG-AF488+CXCL4 (right panel) incubated B cells. (E–J) B cells were cultured with the CpG-AF488 (5 µM) or with CpG-AF488 (0.5 µM) + CXCL4 for 1 h (E and F) or at the indicated time (G–J) and stained with anti-LAMP-2 (E and G–J) or LysoTracker (F) and analyzed using the Amnis imaging flow cytometer system. Image gallery, from left to right: showing bright field (BF) images, followed by uptaken TLR9-L CpG-AF488, LAMP-2+ compartments or lysosome (stained by LysoTracker), and merged images. Arrows indicate TLR9-L/LAMP-2 or TLR9-L/LysoTracker colocalization. The right panel shows the quantification of the correlation between the bright details of E CpG and LAMP2+ compartment or of F CpG and lysosome calculated by the BDS feature. The number of cells analyzed is indicated in brackets and statistical significance was evaluated using a Chi-square test and ***P ≤ 0.001. (C–J) Scale bars, 7 µm.
CXCL4 sequesters TLR9 ligands away from the late endosomal compartments. (A–C) Purified B cells from HDs were cultured for 1 h with the CpG-AF488 (5 µM) or with CpG-AF488 (0.5 µM) + CXCL4 (10 µg/ml) and analyzed by Amnis imaging flow cytometer system. (A) Cells in camera focus were selected from all events on the basis of gradient root mean square of the bright field image and (B) single cells were identified by plotting “Area” versus “Aspect ratio” in which events with higher “Aspect ratio” are likely to be single cells. (C) Image gallery, from left to right: showing bright field (BF) images, followed by B cell surface (stained by antiCD20), uptaken CpG based on cells stained for AF488, and merged images of CpG/CD20 showing absence of colocalization. (D) Representative histograms showing intensity of CpG-AF488 in CpG- (left panel) versus CpG-AF488+CXCL4 (right panel) incubated B cells. (E–J) B cells were cultured with the CpG-AF488 (5 µM) or with CpG-AF488 (0.5 µM) + CXCL4 for 1 h (E and F) or at the indicated time (G–J) and stained with anti-LAMP-2 (E and G–J) or LysoTracker (F) and analyzed using the Amnis imaging flow cytometer system. Image gallery, from left to right: showing bright field (BF) images, followed by uptaken TLR9-L CpG-AF488, LAMP-2+ compartments or lysosome (stained by LysoTracker), and merged images. Arrows indicate TLR9-L/LAMP-2 or TLR9-L/LysoTracker colocalization. The right panel shows the quantification of the correlation between the bright details of E CpG and LAMP2+ compartment or of F CpG and lysosome calculated by the BDS feature. The number of cells analyzed is indicated in brackets and statistical significance was evaluated using a Chi-square test and ***P ≤ 0.001. (C–J) Scale bars, 7 µm.
CXCL4 sequesters TLR9 ligands away from the late endosomal compartments
Since CXCL4 favors CpG uptake while it paradoxically inhibits the function of TLR9 in B cells, we first explored the subcellular localization of TLR9-L in the presence of CXCL4 using Amnis. We found that TLR9-L CpG normally colocalized in B cells with LAMP-2+ late endosome and lysosome compartments where TLR9 resides as expected (Fig. 7, E and F). In contrast, CXCL4 interfered with CpG trafficking to these compartments as observed by the reduced colocalization of the CpG/CXCL4 fluorescence with those of LAMP-2 or LysoTracker at all tested time points (Fig. 7, E and F). Indeed, TLR9-L/LAMP-2 or TLR9-L/LysoTracker colocalization scores using the bright detail similarity (BDS) that assigns 1 for perfect colocalization were lower in B cells stimulated by CpG in the presence of CXCL4 compared with CpG alone, which was virtually always found associated with LAMP-2 (Fig. 7, E and F). CXCL4-induced CpG intracellular mislocalization is evidenced even at early time points, suggesting that it does not result from accelerated CpG trafficking through late endosomes and lysosomes (Fig. 7, G–J). We further investigated TLR9-L intracellular trafficking in B cells using confocal microscopy. We engineered the BJAB B cell line to express human TLR9 linked with fluorescent mCherry so that we could visualize TLR9/CpG colocalization when cells were incubated with CpG-FITC for an hour (Garcia-Carmona et al., 2015). We found that CpG normally reached TLR9 and the lysosomal compartment in the absence of CXCL4 as expected (Fig. 8, A and C). In contrast, CXCL4 severely abrogated the colocalization of CpG with its receptor TLR9 in lysosomes without interfering with TLR9 expression or localization in this intracellular compartment (Fig. 8, B and C). Hence, CXCL4 inhibits TLR9 function in B cells by increasing TLR9 ligand cellular uptake but preventing its delivery to late endosomes/lysosomes where TLR9 resides, thereby hindering the sensing of dsDNA by TLR9 and abrogating TLR9 function.

CXCL4 sequesters TLR9 ligand away from its receptor. (A–C) Human B cell line (BJAB) expressing TLR9-mCherry were cultured for 1 h with fluorescent CpG-FITC with or without CXCL4 and then stained with LysoTracker before analysis by confocal microscopy. (A and B) Panel shows z-stack projection of representative cells. Images show nucleus (blue), CpG (green), TLR9 (red), and LysoTracker/lysosomal compartment (purple) and merged images CpG/TLR9 and CpG/TLR9/LysoTracker without (A) or in the presence (B) of CXCL4. Scale bars, 0.7 and 1 µm as indicated. (C) Quantification of the colocalization of CpG and TLR9 (red), CpG and LysoTracker (blue), and TLR9 and LysoTracker (black) in cells cultured with or without CXCL4 as indicated. Results are represented as mean ± SD and statistical significance was evaluated using two-tailed unpaired Student's t test. **P ≤ 0.005, ***P ≤ 0.001.
CXCL4 sequesters TLR9 ligand away from its receptor. (A–C) Human B cell line (BJAB) expressing TLR9-mCherry were cultured for 1 h with fluorescent CpG-FITC with or without CXCL4 and then stained with LysoTracker before analysis by confocal microscopy. (A and B) Panel shows z-stack projection of representative cells. Images show nucleus (blue), CpG (green), TLR9 (red), and LysoTracker/lysosomal compartment (purple) and merged images CpG/TLR9 and CpG/TLR9/LysoTracker without (A) or in the presence (B) of CXCL4. Scale bars, 0.7 and 1 µm as indicated. (C) Quantification of the colocalization of CpG and TLR9 (red), CpG and LysoTracker (blue), and TLR9 and LysoTracker (black) in cells cultured with or without CXCL4 as indicated. Results are represented as mean ± SD and statistical significance was evaluated using two-tailed unpaired Student's t test. **P ≤ 0.005, ***P ≤ 0.001.
In vivo CXCL4 expression impairs central B cell tolerance
To assess whether CXCL4 production in vivo may interfere with TLR9 tolerogenic function during early B cell development, we generated NSG humanized mice using HSCs transduced with GFP-tagged lentiviruses expressing human CXCL4 (Fig. 9 A). In agreement with CXCL4 regulating HSC survival and self-renewal, we found elevated proportions of GFP+ human B and T cells in these NSG humanized mice compared with previously reported humanized mice generating using a similar approach (Fig. 9, B and C; Schickel et al., 2016; Sinclair et al., 2016). The production of CXCL4 by GFP+ B cells was evidenced in culture supernatants by ELISA, but human CXCL4 serum concentrations were similar between humanized mice engrafted with HSCs transduced or not with GFP-tagged lentiviruses expressing CXCL4, revealing that systemic human CXCL4 depends on the presence of human cells (Fig. 9, D and E). While B cell responses induced by TLR7, BCR, or CD40 triggering were similar between GFP+ and GFP− fractions, GFP+ B cells displayed decreased CD69 upregulation following TLR9 stimulation compared with GFP− counterparts, which correlated with the production of CXCL4 in vitro by GFP+ B cells (Fig. 9, F and G). In agreement with defective TLR9 responses in GFP+ B cells expressing CXCL4, we found that polyreactive clones accounted for 42.8% of GFP+ B cells, whereas they only represented 10.8% in GFP− B cells (Fig. 10, A and B). Defects in central B cell tolerance induced by in vivo CXCL4 expression were further evidenced by the significantly increased frequencies of antinuclear B cells that averaged 14.1% in the GFP+ fraction compared with only 2.8% in GFP− B cells (Fig. 10, C and D). In addition, daily injections of CXCR3 antagonist for 1 wk did not correct the elevated proportions of GFP+ autoreactive new emigrant/transitional B cells, further demonstrating the CXCR3-independent CXCL4 interference on the establishment of central B cell tolerance (Fig. 10, A–D). Since activation-induced cytidine deaminase (AID) expression is required for the establishment of central B cell tolerance and is induced by both BCR and TLR9 stimulation (Cantaert et al., 2015; Kuraoka et al., 2017), we tested whether CXCL4 may affect AID upregulation by quantitative PCR (Q-PCR) in BM B cell precursors isolated from NSG humanized mice that were stimulated in vitro for 36 h via their BCR, TLR9, or both. We found that CXCL4 specifically abrogated AID induction following TLR9 stimulation, whereas BCR-dependent AID expression did not appear to be affected by CXCL4 (Fig. 10 E). We conclude that CXCL4 abolishes central B cell tolerance by interfering with TLR9 tolerogenic function during early B cell development in the BM.

In vivo CXCL4 expression specifically inhibits TLR9 responses in B cells. (A) Humanized mice were generated with CD34+ HSCs transfected with GFP-tagged lentiviruses expressing human CXCL4 and injected into the 3-d-old NSG mice. (B) Representative flow cytometry analysis of human CD3+ T cells and CD19+ B cells and GFP+ cell frequencies in the blood of three NSG humanized mice engrafted with fetal HSCs transduced with GFP-tagged lentiviruses expressing CXCL4. (C) Summary of GFP+CD3+ T cell and GFP+CD19+ B cell frequencies in the blood of NSG humanized mice transplanted with fetal HSCs transduced with GFP-tagged lentiviruses expressing CXCL4. (D) Human CXCL4 concentrations in cell culture supernatants of GFP− (open diamonds) and GFP+ (green diamonds) B cells were determined by ELISA. (E) Human CXCL4 concentrations (ng/ml) in the sera of non-engrafted NSG mice (n = 7, open diamonds), NSG humanized mice transplanted with HSCs (n = 17, black diamonds), or HSCs transduced with GFP-tagged lentiviruses expressing CXCL4 (n = 7, green diamonds) were measured by ELISA. The mean is shown with a bar and significant differences are indicated (Mann–Whitney U test, ***P < 0.001, ****P < 0.0001). (F) Representative CD69 expression in GFP− (black line) and GFP+ (green line) CD19+ cells from humanized mice after 48 h in culture with no stimulation (Unstimulated) or activated with the indicated ligands or F(ab)′2 anti-IgM. (G) Relative frequencies of CD69+ cells in sorted GFP+ (green circles) compared with GFP− (open circles) B cells after 48 h in culture unstimulated (Unstim.) or activation with the indicated ligands or F(ab)′2 anti-IgM.
In vivo CXCL4 expression specifically inhibits TLR9 responses in B cells. (A) Humanized mice were generated with CD34+ HSCs transfected with GFP-tagged lentiviruses expressing human CXCL4 and injected into the 3-d-old NSG mice. (B) Representative flow cytometry analysis of human CD3+ T cells and CD19+ B cells and GFP+ cell frequencies in the blood of three NSG humanized mice engrafted with fetal HSCs transduced with GFP-tagged lentiviruses expressing CXCL4. (C) Summary of GFP+CD3+ T cell and GFP+CD19+ B cell frequencies in the blood of NSG humanized mice transplanted with fetal HSCs transduced with GFP-tagged lentiviruses expressing CXCL4. (D) Human CXCL4 concentrations in cell culture supernatants of GFP− (open diamonds) and GFP+ (green diamonds) B cells were determined by ELISA. (E) Human CXCL4 concentrations (ng/ml) in the sera of non-engrafted NSG mice (n = 7, open diamonds), NSG humanized mice transplanted with HSCs (n = 17, black diamonds), or HSCs transduced with GFP-tagged lentiviruses expressing CXCL4 (n = 7, green diamonds) were measured by ELISA. The mean is shown with a bar and significant differences are indicated (Mann–Whitney U test, ***P < 0.001, ****P < 0.0001). (F) Representative CD69 expression in GFP− (black line) and GFP+ (green line) CD19+ cells from humanized mice after 48 h in culture with no stimulation (Unstimulated) or activated with the indicated ligands or F(ab)′2 anti-IgM. (G) Relative frequencies of CD69+ cells in sorted GFP+ (green circles) compared with GFP− (open circles) B cells after 48 h in culture unstimulated (Unstim.) or activation with the indicated ligands or F(ab)′2 anti-IgM.

In vivo CXCL4 expression abrogates central B cell tolerance. (A) Representative polyreactivity of recombinant antibodies cloned from single GFP− and GFP+ (expressing CXCL4) new emigrant/transitional B cells from humanized mice injected with CXCR3 antagonist (n = 2) or not (n = 3) was tested by ELISA against dsDNA, insulin, and LPS. The same control NSG mouse is used for representation in Fig. 1 E, Fig. 2 D, and Fig. S1 D. Dotted red lines show positive control. OD405, optical density. For each representative fraction, the frequencies of non-polyreactive (open area) and polyreactive (black area) are summarized in pie charts with the total number of clones tested shown in the center. (B and C) Polyreactivity (B) and antinuclear reactivity (C) frequencies in GFP+ (green diamonds) and GFP− new emigrant/transitional B cells from the indicated humanized mice and control NSG humanized mice (empty diamonds) are summarized on the right. Mean values (with bars) and statistically significant differences are indicated (Student’s t test, *P < 0.05, ****P < 0.0001). (D) Representative nuclear staining patterns for antibodies cloned from new emigrant/transitional B cells. Scale bars, 25 µm. (E) Quantitative real-time PCR measures AICDA mRNA transcripts encoding AID in CD19+ BM B cell precursors from humanized mice after 48 h in culture with no stimulation (Unstim.) or activated with the indicated ligands (Student’s t test, *P < 0.05, **P < 0.01).
In vivo CXCL4 expression abrogates central B cell tolerance. (A) Representative polyreactivity of recombinant antibodies cloned from single GFP− and GFP+ (expressing CXCL4) new emigrant/transitional B cells from humanized mice injected with CXCR3 antagonist (n = 2) or not (n = 3) was tested by ELISA against dsDNA, insulin, and LPS. The same control NSG mouse is used for representation in Fig. 1 E, Fig. 2 D, and Fig. S1 D. Dotted red lines show positive control. OD405, optical density. For each representative fraction, the frequencies of non-polyreactive (open area) and polyreactive (black area) are summarized in pie charts with the total number of clones tested shown in the center. (B and C) Polyreactivity (B) and antinuclear reactivity (C) frequencies in GFP+ (green diamonds) and GFP− new emigrant/transitional B cells from the indicated humanized mice and control NSG humanized mice (empty diamonds) are summarized on the right. Mean values (with bars) and statistically significant differences are indicated (Student’s t test, *P < 0.05, ****P < 0.0001). (D) Representative nuclear staining patterns for antibodies cloned from new emigrant/transitional B cells. Scale bars, 25 µm. (E) Quantitative real-time PCR measures AICDA mRNA transcripts encoding AID in CD19+ BM B cell precursors from humanized mice after 48 h in culture with no stimulation (Unstim.) or activated with the indicated ligands (Student’s t test, *P < 0.05, **P < 0.01).
Discussion
We reported herein a novel mechanism by which a chemokine, CXCL4, negatively regulates TLR9 function in B cells by altering the intracellular trafficking of TLR9 ligands. This leads to impairments in central B cell tolerance through a failure to properly upregulate AID, which is required for the counterselection of developing autoreactive B cells in the BM (Cantaert et al., 2015; Kuraoka et al., 2017). The involvement of TLRs in the establishment of central B cell tolerance was initially suggested by the defective removal of developing autoreactive B cells in patients deficient in IRAK-4 or MYD88, two molecules essential for the signaling of most TLRs, including TLR7 and TLR9 (Isnardi et al., 2008). Subjects with mutations in TNFSRF13B, which encodes TACI that associates with TLR7 and TLR9 and mediates some of their function in B cells also failed to counterselect developing autoreactive B cells in the BM, further pointing a role for these TLRs in the regulation of central B cell tolerance (Romberg et al., 2013; Sintes et al., 2017). While TLR9 was previously shown to be required for the silencing of developing anti-dsDNA-transgenic expressing B cells in mice (Kuraoka et al., 2017), the absence of TLR7 requirement for central B cell tolerance was unexpected because many immature B cells that escaped this early checkpoint in patients lacking functional IRAK-4, MYD88, or TACI expressed autoreactive BCRs with antinuclear and nucleolar staining patterns characteristic of reactivity toward ribonucleoproteins/small nuclear RNA complexes which are TLR7 ligands (Isnardi et al., 2008; Romberg et al., 2013). Similarly, many immature B cells that express autoreactive BCRs that likely recognized TLR7 ligand RNA containing self-antigens escaped central B cell tolerance when either intrinsic MYD88 or TLR9 expression was inhibited by specific shRNAs in our humanized mouse models. This reveals that TLR9, by sensing either ssRNA/dsDNA or ssRNA/protein complexes bound to dsDNA, maybe the self-antigen responsible for the counterselection of immature B cells that express BCRs that recognize TLR7 ligand-containing self-antigens. In addition, BCR/TLR9 cotriggering rather than BCR signaling strength may account for the different tolerogenic responses induced by membrane-bound versus soluble self-antigens because membrane-bound proteins likely become associated with DNA during the formation of apoptotic blebs (Goodnow, 1996). This same MYD88/TLR9/BCR multiprotein supercomplex controls oncogenic signaling in lymphoma, which suggests that BCR/TLR9 costimulation is an essential pathway for both lymphomagenesis and tolerance (Phelan et al., 2018). Our data also provide a novel rationale for exacerbated disease in lupus-prone Tlr9 KO mice through increased production of autoreactive B cells (Christensen et al., 2006; Leibler et al., 2022; Tilstra et al., 2020). Finally, we showed that CXCL4 not only inhibits TLR9 function in immature B cells but also when TLR9 ligands are internalized by autoreactive BCRs, which is the pathway that ensures central B cell tolerance. Hence, the elevated serum concentrations in many rheumatic diseases (Lande et al., 2019; van Bon et al., 2014; Volkmann et al., 2016) may account for both the defective TLR9 responses identified in B cells from patients with SSc and SLE and the defective central B cell tolerance checkpoint characteristics of these autoimmune diseases (Glauzy et al., 2022; Yurasov et al., 2005). It remains to be determined whether other cationic chemokines whose serum concentrations are also elevated in many rheumatic diseases may also interfere with TLR9 function in B cells and the regulation of B cell tolerance.
While BCR/TLR9 cotriggering is responsible for the establishment of central B cell tolerance, it also induces the elimination in the periphery of dsDNA-expressing B cells that may originate from somatic hypermutation, thereby preventing the secretion of dsDNA autoantibodies (Sindhava et al., 2017). Hence, defects in TLR9 responses induced by elevated CXCL4 concentrations in the serum of patients with SLE may account for the failure to eliminate anti-dsDNA B cells and the secretion of dsDNA autoantibodies characteristic of SLE (Gies et al., 2018). CXCL4-induced TLR9 impairments in B cells from SSc patients also likely result in the secretion of autoantibodies targeting topoisomerase I or centromere proteins, both of which bind dsDNA, in patients with diffuse or limited SSc, respectively. We therefore postulate that autoimmune diseases characterized by serum autoantibodies that target self-antigens that interact with TLR9 ligand dsDNA likely result from impaired TLR9 function in B cells.
We showed that CXCL4 inhibits TLR9 function in B cells by preventing TLR9 ligands from reaching TLR9 in the late endosome/lysosome compartment. What is the receptor mediating CXCL4 inhibitory functions in B cells? We demonstrated that the CXCL4 receptors CXCR3B and CCR1 are not responsible for TLR9 inhibition. A recent report showed that CXCL4 also binds extracellular matrix proteoglycans (Gray et al., 2023). In agreement with this hypothesis, proteoglycans lumican and its paralog biglycan were shown to bind and sequester DNA TLR9 ligand in early endosomes in which TLR9 is poorly detected and thereby inhibits TLR9 signaling in murine macrophages (Maiti et al., 2021). Since this TLR9 inhibitory mechanism is similar to the one that we described in human B cells, we therefore propose a role for proteoglycans in CXCL4-mediated TLR9 inhibition in human B cells. This scenario may also explain the paradoxical enhanced TLR9 responses in pDCs in the presence of CXCL4, which leads to exacerbated type I IFN production, which further supports inflammation and autoimmune manifestation (Ah Kioon et al., 2018; Du et al., 2022; Guiducci et al., 2006; Kim et al., 2008; Wu and Assassi, 2013). Indeed, TLR9 signaling in pDCs involves two different pathways, an IRF7-dependent cascade that leads to type I IFN production induced from early endosomes and an NF-κB–dependent pathway that promotes inflammatory cytokine production initiated from the late endosome/lysosomal compartment (Combes et al., 2017; Guiducci et al., 2006; Honda et al., 2005; Kawai and Akira, 2010; Kerkmann et al., 2003). CXCL4-mediated redistribution of TLR9 ligand away from the LAMP-2+ late endosomes may also occur in pDCs and promote IRF7-dependent TLR9 signaling, likely initiated from early endosomes (Du et al., 2022; Guiducci et al., 2006; Kerkmann et al., 2003; Lande et al., 2007). Finally, the increased proportions of activated platelets in SSc and pDCs may represent important systemic and tissue-specific sources of CXCL4 that will alter TLR9 responses in both B cells and pDCs (Ah Kioon et al., 2018; Du et al., 2022; Kahaleh et al., 1982; Lande et al., 2019; van Bon et al., 2014).
In summary, our data challenged the current paradigm that BCR affinity for self is responsible for the deletion of autoreactive immature B cells in the BM since we demonstrate instead that innate immune recognition via TLR9 plays an essential early tolerogenic function required for the establishment of central B cell tolerance. We also show that CXCL4 production elevated in patients with SSc inhibits TLR9 function in B cells by sequestering TLR9 ligands away from the late endosomal compartment where this receptor resides. Hence, impaired TLR9 function in developing B cells from patients with SSc and potentially other autoimmune diseases such as SLE promotes the escape of autoreactive B cells from the BM and leads to the production of autoantibodies in the periphery. Our data therefore provide a rationale for the development of novel therapeutic strategies for SSc and SLE that will aim at correcting impaired TLR9 function in B cells to restore both central and peripheral B cell tolerance and suppress autoimmune manifestations.
Materials and methods
Fetal and patient samples
Fetal tissues were obtained from the Birth Defects Research Laboratory at the University of Washington (supported by award #5R24HD000836 from the National Institute of Child Health & Human Development). 13 human fetal samples (age range 108–140 d; eight females and five males) were used to generate NSG humanized mice. All fetal samples were genotyped and did not carry the 1858 T PTPN22 polymorphism that interferes with central B cell tolerance.
Patients with SSc according to current criteria were enrolled from the Yale Interstitial Lung Disease Center of Excellence and the Yale Scleroderma Center (Table S1). Most patients were naïve to any medication and all met the diagnostic criteria for SSc (LeRoy et al., 1988). Additional patients with SSc were previously enrolled at the UMCU as part of a replication cohort (Table S1). Characteristics of patients with SSc and their autoantibody profiles are summarized in Table S1. In addition, we analyzed frozen PBMCs from three TLR7-deficient patients, two of which were previously described (van der Made et al., 2020). The study protocol was approved by the Institutional Review Board at Yale (Human Investigation Committee protocol #1307012431, #0906005336) and at the UMCU, the Netherlands (Medical Ethics Committee protocol no. 12–466C). Informed consent was obtained from all patients before participation.
Human progenitor cell isolation and injection in NSG mice
Human CD34+ cells were purified from fetal liver samples by density-gradient centrifugation followed by positive immunomagnetic selection with anti-human CD34 microbeads (Miltenyi Biotech). Newborn NSG mice (within the first 3 d of life) were sublethally irradiated (x-ray irradiation with X-RAD 320 irradiator at 180 cGy) and 100,000–150,000 CD34+ cells in 20 µl of PBS were injected into the liver with a 22-gauge needle (Hamilton Company). Mice were used for experiments 10–12 wk after transplantation. NSG mice treated with the HCQ were injected with the HCQ 0.2 mg/dose intraperitoneally daily for a week. NSG mice treated with the CXCR3 antagonist were injected with the AMG-487 5 mg/kg/dose (Tocris) intraperitoneally daily for 9 d.
All animals were treated and experiments were conducted in accordance with the Yale institutional review guidelines on the treatment of experimental animals.
MYD88, TLR7, and TLR9 silencing, CXCL4 expression, and CD34+ HSC transduction
The pTRIP-Ubi-GFP lentiviral vector was used for overexpression of human CXCL4 and shRNA delivery. Vector constructions have been previously described (Schickel et al., 2016). Related shRNA sequences were as follows: for human MYD88 shRNA: forward, 5′-GATCCCCACAGACAAACTATCGACTGAA-3′; reverse, 5′-AGCTTTTCCAAAAAACAGACAAACTATCG-3′; TLR7 shRNA; 5′-CCGGGCTCAAATCTTTCAGTTGGAACTCGAGTTCCAACTGAAAGATTTGAGCTTTTTG-3′; TLR9 shRNA; forward, 5′-GATCCCCGCACGGTGCCACCTCCACACTTTCAAGAGAAGTGTGGAGGTGGCACCGTGCTTTTTGGAAA-3′; reverse, 5′-AGCTTTTCCAAAAAGCACGGTGCCACCTCCACACTTCTCTTGAAAGTGTGGAGGTGGCACCGTGCGGG-3′. Lentiviral particles were produced by transient transfection of 293T cells, as previously described. Viruses were then used to transduce CD34+ HSCs in the presence of protamine sulfate (Sigma-Aldrich). For MYD88, TLR7, and TLR9 protein detection, cells were lysed in lysis buffer (50 mM Tris, 1% NP-40, and 2 mM EDTA) including protease inhibitor (Roche). Total-cell lysates were separated by SDS page, transferred to PDVF membranes, probed with monoclonal mouse anti-MYD88 (clone 2E9C2; Sigma-Aldrich), polyclonal rabbit anti-human TLR7 (Abcam) or monoclonal rat anti-human TLR9 (clone eB72; eBioscience), and detected by chemiluminescence (Amersham ECL Prime Western Blotting Detection Reagent) using a GBox documentation system (Syngene). For quantification of MYD88 and full-length and proteolytically cleaved TLR7 and TLR9, blots were stripped with stripping buffer (Pierce) and reprobed with a monoclonal mouse anti-β-actin antibody (Clone AC-15; Sigma-Aldrich).
Reagents and antibodies
The CpG-B 1018 (5′-TGACTGTGAACGTTCGAGATGA-3′) and CpG1018-AF488 (5′-TGACTGTGAACGTTCGAGATG-3′; C6-NH; Alexa 488) were synthesized from TriLink. Antibodies anti-human CD21 (559867), IgM (555782), IgG (555787), CD83 (561132), CD86 (562432), CD20 (560734), and LAMP-2 (565305) were obtained from BD Biosciences, antibodies anti-human CD267 (311906), CD83 (305323), CD25 (302622), CD10 (312214), CD10 (312212), CD69 (310910), CD69 (310912), CD86 (305412), CD27 (356408), IgM (314511), CD19 (302224) CD20 (302304), and CD69 (310906) were obtained from BioLegend, and LysoTracker Deep Red (L12492) was obtained from Invitrogen.
B cell staining and sorting
B cells were purified from BM and spleen from humanized mice by positive selection using CD19 magnetic beads (Miltenyi). Enriched B cells were stained with antibodies listed above. CD19+CD21−/loCD10+IgMhiCD27− GFP− or CD19+CD21−/loCD10+IgMhiCD27− GFP+ new emigrant/transitional B cells from the fetal spleen and BM were sorted on a FACSAria sorter (Becton Dickinson) into 96-well PCR plates as single cells or batch-sorted into 5-ml round bottom polystyrene test tubes. The gating strategies to sort these B cell subsets are provided in Figs. 1 and 9.
cDNA, RT-PCR, antibody production, and purification
RNA from single B cells was reverse-transcribed in the original 96-well plate in 12.5-μl reactions containing 100 U of Superscript II RT (Gibco BRL) for 45 min at 42°C. RT-PCR reactions, primer sequences, cloning strategy, expression vectors, antibody expression, and purification were carried out as previously described (Wardemann et al., 2003). For human B cells, PCR reactions were performed as described previously with 2 ng of cDNA. In brief, RNA was extracted from cells using the Qiagen RNeasy Mini Kit. The quantity of RNA was measured by Nanodrop and a high-capacity cDNA Reverse Transcription kit (AB Biosystems) was used to generate 20–50 ng cDNA. Gene expression levels were calculated based on relative threshold cycle (Ct) values. This was done using the formula Relative Ct = 1,000 × 1.8 (HSK − GENE), where HSK is the mean Ct of duplicate housekeeping gene runs (Ubiquitin or HPRT1), GENE is the mean CT of duplicate runs of the gene of interest, and 1,000 is arbitrarily chosen as a factor to bring all values above 0. For AICDA gene expression assays, probes were purchased from Applied Biosystems (AICDA: Hs00757808_m1, HPRT1: Hs02800695_m1). Primers were obtained from Thermo Fisher Scientific and were as follows: for human TLR7: forward, 5′-ATTATTTTTACACGGCGCAC-3′; reverse, 5′-AATGTCACAGCCGTCCCTAC-3′; TLR9: forward, 5′-CCCAGCATGGGTTTCTGC-3′; reverse, 5′-AGGAAAGGCTGGTGACATTG-3′; GAPDH: forward, 5′-CCACATCGCTCAGACACCAT-3′; reverse, 5′-GGCAACAATATCCACTTTACCAGAGT-3′; ubiquitin: forward, 5′-CACTTGGTCCTGCGCTTGA-3′; reverse, 5′-CAATTGGGAATGCAACAACTTTAT-3′; hIL-6: forward, 5′-TACCCCCAGGAGAAGATTCC-3′; reverse, 5′-GATGTCAAACTCACTCATGGCT-3′; hIL-10: forward, 5′-TCAAGGCGCATGTGAACTCC-3′; reverse, 5′-GATGTCAAACTCACTCATGGCT-3′; hTNF: forward, 5′-CTTCTGCCTGCTGCACTTTG-3′; reverse, 5′-CTGGGCCAGAGGGCTGAT-3′.
Antibody sequence analysis
Immunoglobulin sequences and mutation status were determined using Ig BLAST comparison with GenBank using the National Center for Biotechnology Information IgBlast server (http://www.ncbi.nlm.nih.gov/igblast/). Heavy chain complementarity determining region 3 was defined as the interval between amino acid at position 94 in the VH framework 3 and the conserved tryptophan at position 103 in JH segments. Antibody sequences and reactivity are shown in Table S2.
ELISAs and indirect immunofluorescence staining
Antibody concentrations and reactivity were measured as described (Wardemann et al., 2003). Highly polyreactive ED38 was used as a positive control in HEp-2-reactivity and polyreactivity ELISAs. Antibodies were considered polyreactive when they recognized all three analyzed antigens: dsDNA, insulin, and lipopolysaccharide (LPS). The cut-off used to define positivity of OD405 corresponds to twice the average OD405 ELISA value of all tested clones and is ∼0.5. For indirect immunofluorescence assays, HEp-2 cell-coated slides (Bion Enterprises) were incubated in a moist chamber at room temperature with purified recombinant antibodies at 50–100 μg/ml and detected with FITC-conjugated goat anti-human IgG.
B cell purification and culture
Enriched leukocytes were obtained from the New York Blood Center (Long Island City, NY) after the informed consent of the donor and used under a protocol approved by the Institutional Review Board of the Hospital for Special Surgery and the Institutional Biosafety Committee of Weill Cornell Medicine.
PBMCs were prepared using the Ficoll–Paque density gradient (GE Healthcare) as previously described (Guiducci et al., 2006). B cells were isolated using positive selection (Miltenyi Biotech) and cultured at 150,000–200,000 cells per well in a 96-flat bottom plate in complete RPMI media alone or with 2.5 μg/ml polyclonal F(ab′)2 rabbit anti-human IgM (Jackson ImmunoResearch), 1 μg/ml dual variable domain antibody DVD-Ig 3764, which combines both anti-human IgM and anti-dsDNA reactivities (AbbVie Inc.), multimeric soluble recombinant human CD40L 1.0 μg/ml (Alexis Biochemicals), TLR9-L CpG1018 (0.15 µM), or TLR7-L R848 (0.75 µM; Invivogen) in the presence or not of 1 µg/ml TLR9 antagonist ODN INH-18 (Invivogen) or 10 µg/ml CXCL4 (R&D Systems) added as overnight preincubation or shortly before stimulation—both inducing similar response—with or without 10 µM CXCR3 antagonist AMG-487 (Tocris). GFP− and GFP+ B cells from humanized mice were batch-sorted prior to 48-h cultures. DVD-Ig 3764 combines the variable domains specific for human IgM and dsDNA and was produced by AbbVie Inc. A similar DVD-Ig has been used to activate murine B cells (Pawaria et al., 2015). B cells were lysed after 6 h in RLT buffer (Qiagen) prior to RNA preparation or analyzed by flow cytometry after 48 h and supernatants were collected for ELISA or Mutiplex cytokine assays.
Human BJAB B cell lines were cultured at 200,000 cells per well in a 96-round bottom plate in complete RPMI media alone, or with TLR9-L CpG1018 (0.15 µM) alone or with 10 µg/ml CXCL4. After 48 h, supernatants were collected to assess TNF secretion by ELISA.
Luminex assay for serum cytokines
Soluble proteins were quantified in EDTA-anticoagulated plasma using the Luminex multiplex platform (Luminex) with custom-developed reagents (R&D Systems), as described in detail (Guiducci et al., 2006) or single-plex ELISA (R&D Systems). Analytes quantified using the Luminex multiplex platform were read on the MAGPIX instrument and raw data were analyzed using the xPONENT software. Analytes quantified using single-plex ELISA were read using optical density. Values outside the lower limit of quantification were assigned a value of one-third of the lower limit of the standard curve for analytes quantified by Luminex and half of the lower limit of the standard curve for analytes quantified by ELISA. CXCL4 concentrations were determined using CXCL4/PF4 Quantikine (from R&D Systems) ELISA kit.
RNA-seq analysis
After 6 h of culture, total RNA was isolated from human B cells using RNeasy Plus Mini kit. SMART-Seq v3 Ultra Low Input RNA Kit (Clontech) followed by Nextera library preparation were used to prepare Illumina-compatible sequencing libraries. The quality of all RNA was evaluated with BioAnalyser 2100 (Agilent). Single-end reads were obtained on an Illumina HiSeq 2500 in the Weill Cornell Epigenomics Core Facility at a depth of 23–40 million fragments per sample. Sequencing performance was evaluated using fastp (Chen et al., 2018). Reads were then mapped to the human genome (hg38) and the reads in exons were counted with STAR aligner (Dobin et al., 2013). Differential gene expression analysis was performed in R using the edgeR package (Robinson et al., 2010; Gu et al., 2016). Genes with low expression levels (<3 cpm [counts per million]) were filtered from all downstream analyses. The Benjamini–Hochberg false discovery rate (FDR) procedure was used to calculate the FDR. Genes with FDR < 0.05 and log2 (fold-change) >1.5 were considered significant. A heatmap was generated from the averaged cpm using ComplexHeatmap package (Gu et al., 2016).
Uptake of CpG assay by flow cytometry and Amnis imaging flow cytometry
Purified B cells were cultured in RPMI media with the fluorescent (AF488) CpG1018 at 0.15 µM alone or with CXCL4 (10 µg/ml) at different time points (5, 15 min, 1, 3, or 6 h). The cells were washed twice in FACS buffer and were acquired by flow cytometry. Mean fluorescent intensity (MFI) of AF488 was measured in B cells. For the endocytosis inhibition assay, 200,000 B cells were inhibited for 1.5 h with genistein (100 μg/ml), CPZ (30 μM), or EIPA (100 μM; Sigma-Aldrich). After 1.5 h, the cells were stimulated with the fluorescent CpG1018 (0.15 μM) alone or with CXCL4 for an additional 1 h. MFI of AF488 was analyzed by flow cytometry. For imaging flow cytometry analysis (Amnis), purified B cells were cultured in RPMI media with the fluorescent CpG1018 at 0.5 or 5 µM or with the CpG1018 (0.5 µM) + CXCL4 (10 µg/ml). After 1 h, the cells were either stained with CD20 or LAMP-2 antibodies or LysoTracker. For CD20 staining, the cells were washed with 0.2% FBS diluted in PBS and labeled with anti-CD20 for 15 min at room temperature. The cells were then washed, fixed in 1% formalin for 10 min at room temperature, washed, and resuspended in PBS. For late endosome staining, the cells were washed with 0.2% FBS diluted in PBS, fixed in 4% formalin for 10 min at room temperature, and then permeabilized for 10 min with 10% Triton-X 0.2% FBS diluted in PBS. Samples were labeled with anti-LAMP-2 in 10% Triton-X 0.2% FBS in PBS, washed with 0.2% FBS in PBS, fixed in 1% formalin for 10 min at room temperature, washed, and resuspended in PBS. For lysosome identification, the cells were washed with a warm RPMI medium and incubated for 1 h at 37°C in fresh warm medium containing 75 nM of LysoTracker probe. The cells were then washed with 0.2% FBS in PBS, fixed in 1% formalin for 10 min at room temperature, washed, and resuspended in PBS. The cells were then analyzed by imaging flow cytometry Amnis ImageStreamX Mk II (EMD Millipore) equipped with INSPIRE software. A 60× magnification was used for all samples and at least 5,000 events per sample were collected. Data analysis was performed using the IDEAS software version 6.2 (Amnis Corporation). The gating strategy was as follows: first, focused cells were identified using gradient root mean square (>45).
Second, an “Area” versus “Aspect ratio” plot was used to identify single cells (high aspect ratio). Gated data were used to generate histograms measuring fluorescence intensity and the intensities of CpG, CD20, LAMP-2, or Deep Red LysoTracker. The IDEAS feature BDS was used to measure colocalization between two signals. The BDS feature is the log-transformed Pearson’s correlation coefficient of the localized bright spots in the two input images. Since the bright spots in the two images are either correlated (in the same spatial location) or uncorrelated (in different spatial locations), the correlation coefficient varies between 0 (uncorrelated) and 1 (perfect correlation). Similarly, CpG uptake by human B cell line (BJAB).
B cells were assessed by culturing these cells in the presence or absence of 10 µg/ml CXCL4 overnight before cells were incubated for 1 h with 0.5 µM CpG (ODN 2006-FITC). CpG uptake was then analyzed by flow cytometry. The internalization score was calculated by the IDEAS software and was defined as the ratio of the CpG intensity inside a cell to the intensity of the whole cell. Cells with a score of >0.3 were considered to have internalized CpG and those with a score of <0.3 were considered to have surface-bound CpG.
Confocal microscopy analysis
Human B cell line (BJAB) expressing TLR9-mCherry was generated as previously described (Garcia-Carmona et al., 2015). Briefly, human TLR9 was cloned into pRetig-mCherry retroviral plasmid using In-Fusion HD Cloning Plus Kit (Takara) following the manufacturer’s instructions and using the following cloning primers (sense: 5′-CTCTAGACTGCTCGAGATGGGTTTCTGCCGCAGC-3′; antisense: 5′-GCTCACCATCAAGCTTCTTTCGGCCGTGGGTCCCTG-3′). Positive mCherry cells were isolated by cell sorting (FACSAria II, BD) and BJAB-TLR9-mCherry cells were cultured in RPMI media with or without CXCL4 (10 µg/ml) overnight. Cells were then incubated with CpG/ODN2006-FITC (Invivogen) as described above. After 1 h, cells were allowed to attach to poly-Lys-coated cover-slides (Sigma Aldrich) for 30 min at 37°C. Cells were stained with LysoTracker as described above. Slides were coverslipped with FluorSave reagent (Calbiochem) and analyzed by confocal microscopy. Fluorescence images were generated with a Leica SP8 inverted confocal microscope (Leica) by acquiring z-stacks utilizing 63×/1.4 NA objective lenses (Carl Zeiss) with optimal z spacing (∼0.016 μm). Further processing was performed using the software IMARIS, version 9.7.2 (Oxford Instruments). For quantification of colocalization, cells were microscopically assessed and analyzed with FIJI (ImageJ) software (National Institutes of Health) to calculate Pearson’s correlation coefficient.
Statistics
Statistical analysis was performed using GraphPad Prism (version 6 or 7.03; GraphPad). Differences between groups of research subjects were analyzed for statistical significance with the Mann–Whitney U test, except in scenarios where paired biological samples were applicable and the paired Student’s t test was used for n < 4. Multiple group comparisons were corrected for statistical significance with the Bonferroni-Dunn method. A P value of ≤0.05 was considered significant.
Online supplemental material
Fig. S1 shows that B cell–intrinsic expression of MYD88 and TLR9 but not TLR7 is required for the removal of developing autoreactive B cells in the BM. Fig. S2 shows decreased TLR9 function in naïve B cells from patients with SSc. Fig. S3 shows that CXCL4 abrogated TLR9 signaling while increasing TLR9 uptake. Table S1 shows the characteristics of patients with SSc from Yale identification and UMCU replication cohort. Table S2 shows the repertoire and reactivity of recombinant antibodies cloned from new emigrant/transitional B cells from humanized mice and shows the differential expressed genes of RNA-seq data.
Data availability
The bulk RNA-seq data assessing the transcriptional effect of CXCL4, CpG, and CpG + CXCL4 are available in the Gene Expression Omnibus database (accession no. GSE227639).
Acknowledgments
We thank Dr. L. Devine and C. Wang for cell sorting.
This work was supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases grants AI-061093 (to C. Cunningham-Rundles and E. Meffre), AI-071087 (to E. Meffre), and AI132447 (to F.J. Barrat), a grant from the Scleroderma Research Foundation (to F.J. Barrat and E. Meffre), and National Institutes of Health from the National Heart, Lung, and Blood Institute grant K08HL151970 (to C. Ryu).
Author contributions: F.J. Barrat and E. Meffre designed research. E. Cakan, M.D. Ah Kioon, Y. Garcia-Carmona, S. Glauzy, D. Oliver, N. Yamakawa, A. Vega Loza, Y. Du, J.N. Schickel, J.M. Boeckers, C. Yang, and A. Baldo performed experiments. K.L. Moody, K. Nündel, and A. Marshak-Rothstein generated dual variable domain antibody DVD-Ig 3764. E. Cakan, M.D. Ah Kioon, Y. Garcia-Carmona, S. Glauzy, D. Oliver, L.B. Ivashkiv, C. Cunningham-Rundles, F.J. Barrat, and E. Meffre analyzed and interpreted data. R.M. Young and L.M. Staudt provided unpublished TLR9 shRNA sequences. C.I. van der Made, A. Hoischen, A. Hayward, M. Rossato, T.R.D.J. Radstake, C. Ryu, and E.L. Herzog provided blood samples and clinical data. E. Cakan, M.D. Ah Kioon, F.J. Barrat, and E. Meffre wrote the manuscript. All authors reviewed the manuscript and provided scientific input.

