


Hepatocyte nuclear factor 4 α (HNF4A) is a highly conserved nuclear receptor that has been associated with ulcerative colitis. In mice, HNF4A is indispensable for the maintenance of intestinal homeostasis, yet the underlying mechanisms are poorly characterized. Here, we demonstrate that the expression of HNF4A in intestinal epithelial cells (IECs) is required for the proper development and composition of the intraepithelial lymphocyte (IEL) compartment. HNF4A directly regulates expression of immune signaling molecules including butyrophilin-like (Btnl) 1, Btnl6, H2-T3, and Clec2e that control IEC–IEL crosstalk. HNF4A selectively enhances the expansion of natural IELs that are TCRγδ+ or TCRαβ+CD8αα+ to shape the composition of IEL compartment. In the small intestine, HNF4A cooperates with its paralog HNF4G, to drive expression of immune signaling molecules. Moreover, the HNF4A–BTNL regulatory axis is conserved in human IECs. Collectively, these findings underscore the importance of HNF4A as a conserved transcription factor controlling IEC–IEL crosstalk and suggest that HNF4A maintains intestinal homeostasis through regulation of the IEL compartment.

Introduction
Nuclear receptors are ligand-dependent transcription factors that regulate a wide range of physiological processes including metabolism, development, and immunity. Among 49 members of the nuclear receptor superfamily in mice (48 in human), hepatocyte nuclear factor 4 α (HNF4A, NR2A1) is specifically expressed by the liver, kidney, pancreas, and gut (Bookout et al., 2006; Uhlén et al., 2015). Polymorphisms in the HNF4A locus are associated with ulcerative colitis through genome-wide association studies (UK IBD Genetics Consortium et al., 2009; Jostins et al., 2012). Moreover, HNF4A is highly conserved between humans and mice. Using conditional knockouts, epithelial HNF4A has been shown to be indispensable for protecting against acute chemical-induced colitis (Ahn et al., 2008; Chahar et al., 2014) and spontaneous colonic inflammation in aged animals (Darsigny et al., 2009).
Chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) studies revealed global binding of HNF4A to more than 7,000 sites in the mouse intestinal epithelial cell (IEC) genome (Chahar et al., 2014; Chen et al., 2019). In contrast, young adult mice lacking epithelial HNF4A had largely normal gut epithelium with no obvious defects in permeability and functions (Ahn et al., 2008; Babeu et al., 2009; Cattin et al., 2009; Darsigny et al., 2009). Minor expression changes were found in the small intestine at steady state (Chen et al., 2019), and more profound transcriptional changes occurred in the colon of aged mice after the onset of chronic inflammation (Darsigny et al., 2009). Downregulation of HNF4A targets such as Cldn15 could precede spontaneous inflammation and potentially contribute to the disruption of intestinal homeostasis (Darsigny et al., 2009). In the inflamed colon, HNF4A directly regulates a few immune-related genes including RelB (Chahar et al., 2014), opening up a previously unappreciated question that whether and how HNF4A could play a role in the intestinal immunity.
Maintenance of intestinal homeostasis is orchestrated by multiple cell compartments including IECs and immune cells. Intraepithelial lymphocytes (IELs) are a large and diverse population of immune cells that reside within the intestinal epithelium. In direct association, IECs and IELs collaborate to coordinate a variety of tissue functions such as damage repair and immune defense against enteric pathogens (Cheroutre et al., 2011; Olivares-Villagomez and Van Kaer, 2018; Vandereyken et al., 2020). While “induced” IELs derive from conventional TCRαβ+CD4+ or TCRαβ+CD8αβ+ T cells, “natural” IELs, including TCRαβ+CD8αα+(CD4−CD8β−) and TCRγδ+ T cells, develop independently of exogenous antigens. Emerging evidence suggests that signals from the local environment including IECs are critical for the extrathymic development and tissue-specific adaptation of IELs (Brenes et al., 2021; Poussier and Julius, 1994).
IECs constitutively express tight junction molecules, classical and nonclassical MHC molecules, and cytokines that selectively influence the retention, expansion, and function of IELs. Recent studies identified butyrophilin-like (Btnl) molecules, presented by IECs, as major determinants of Vγ7+ γδ IELs in mice and possibly Vγ4+ IELs in humans (Di Marco Barros et al., 2016; Jandke et al., 2020; Willcox et al., 2019). Moreover, dysregulation of epithelial molecules is associated with IEL abnormalities and intestinal inflammation. In the context of celiac disease, active inflammation leads to downregulated BTNL8 expression on IECs, which correlated with a permanent loss of BTNL8-reactive Vγ4+Vδ1+ IELs and expansion of gluten-sensitive IFNγ-producing Vγ4−Vδ1+ IELs (Mayassi et al., 2019). However, how IECs control those molecules to shape IELs and regulate intestinal homeostasis remains largely unknown.
Here, we identified a set of epithelial-immune crosstalk molecules including Btnls as novel HNF4A-dependent targets in IECs. Through direct regulation of those molecules, epithelial HNF4A governs the development and composition of IELs to promote intestinal homeostasis.
Results
Loss of HNF4A predisposes to colitis
Hnf4a deletion in IECs has been shown to trigger chronic inflammatory responses in aged mice (Darsigny et al., 2009) and increase susceptibility to chemical-induced colitis in young adult mice (Ahn et al., 2008; Chahar et al., 2014). We first validated these findings by comparing Hnf4af/fVil-Cre (Hnf4aIEC-KO) mice and their cohoused Hnf4af/f (WT) littermates as controls. Following a low dose of dextran sulfate sodium (DSS) treatment, young adult Hnf4aIEC-KO mice quickly developed signs of colitis including weight loss, diarrhea, colon shortening, and splenomegaly (Fig. 1, A and B). Susceptibility to DSS-induced colitis preceded spontaneous intestinal inflammation reported at 6–12 mo old (Darsigny et al., 2009). Indeed, young adult Hnf4aIEC-KO mice displayed normal body weight, spleen weight, and colon length at steady state with no obvious abnormality in the gut histopathology (Fig. S1, A–C). However, we detected a moderate increase in the expression of inflammatory cytokines (Il1b, Tnf) and oxidative stress gene (Nos2) as early as 3 wk old (Fig. 1, C and D). This was not due to changes in the microbiota, since no significant difference was detected in the microbiome of WT and Hnf4aIEC-KO mice in the adult and at 3 wk old, indicated by comparable relative abundance and α diversity (Fig. 1 E and Fig. S1 D). These results suggest that although HNF4A deficiency did not cause clinical disease or dysbiosis in young mice, there was an early disruption of intestinal homeostasis that predisposes to colitis in older animals.

Loss of HNF4A induces low-grade inflammation and predisposes to colitis. (A) WT (Hnf4af/f) and littermate Hnf4aIEC-KO (Hnf4af/f Vil-Cre) mice were treated with 1.5% DSS and monitored for body weight and signs of colitis (stool consistency and blood) over time. Median. N = 7–8 for each group. (B) Colon length and spleen weight were determined 4 d after DSS treatment. Mean with SD. N = 7–8 for each group. (C) Expression of inflammatory genes (Il1b, Tnf) and oxidative stress gene (Nos2) in the IECs of 3-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 10–12 for each group. (D) Expression of inflammatory genes (Il1b, Tnf) and oxidative stress gene (Nos2) in the IECs of adult mice was determined by qPCR and shown as 2−dCT. Median. N = 4–5 for each group. (E) Microbiome was determined by shot-gun sequencing. Taxa composition at the species level (mean with SEM) and α diversity indicated by Shannon entropy (min to max) was shown. N = 5 for each group. Each dot represented individual mouse. Data was combined from two independent experiments. Body weight was analyzed by two-way ANOVA (A). Taxonomic association was analyzed by MaAsLin2 (Mallick et al., 2021Preprint) with fixed effects on genotype and α diversity by Kruskal-Wallis (E). Other data was analyzed by Mann-Whitney tests (B–D). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001.
Loss of HNF4A induces low-grade inflammation and predisposes to colitis. (A) WT (Hnf4af/f) and littermate Hnf4aIEC-KO (Hnf4af/f Vil-Cre) mice were treated with 1.5% DSS and monitored for body weight and signs of colitis (stool consistency and blood) over time. Median. N = 7–8 for each group. (B) Colon length and spleen weight were determined 4 d after DSS treatment. Mean with SD. N = 7–8 for each group. (C) Expression of inflammatory genes (Il1b, Tnf) and oxidative stress gene (Nos2) in the IECs of 3-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 10–12 for each group. (D) Expression of inflammatory genes (Il1b, Tnf) and oxidative stress gene (Nos2) in the IECs of adult mice was determined by qPCR and shown as 2−dCT. Median. N = 4–5 for each group. (E) Microbiome was determined by shot-gun sequencing. Taxa composition at the species level (mean with SEM) and α diversity indicated by Shannon entropy (min to max) was shown. N = 5 for each group. Each dot represented individual mouse. Data was combined from two independent experiments. Body weight was analyzed by two-way ANOVA (A). Taxonomic association was analyzed by MaAsLin2 (Mallick et al., 2021Preprint) with fixed effects on genotype and α diversity by Kruskal-Wallis (E). Other data was analyzed by Mann-Whitney tests (B–D). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001.

Hnf4a IEC-KO mice, RNA-seq and environmental regulation of HNF4A-dependent genes. Related to Figs. 1 and 2. (A) Body weight, spleen weight, colon length, and cecum weight of 10-wk-old mice. Mean with SD. N = 4 for each group. (B) Representative H&E staining of ileum, cecum, and colon of 11–12-wk-old mice. Scale bars were shown for WT ileum and the same for all other groups: 2×, 0.5 mm; 20×, 50 μm. (C) Pathology was evaluated based on H&E staining of the ileum, cecum, and colon from 11–12-wk-old mice. Samples with a score >0 had focal epithelial loss without inflammation (likely due to artifacts during sample processing). N = 3 for each group. (D) Colonic microbiome of 3-wk-old mice was determined by shotgun sequencing. Taxa composition at the species level (mean with SEM) and α diversity indicated by Shannon entropy (min to max) were shown. N = 9 for each group. (E) Gating strategy for flow sorting IECs. (F) GO enrichment analysis of DE genes in cecal IECs from RNA-seq. (G) 8-wk-old mice were given water (NT) or antibiotic cocktails (ABX) ad lib for 4 wk. RNA levels of indicated genes in the colonic IECs were determined by qPCR and shown as 2−dCT. Median. N = 6 for each group. (H) Mice were treated with 2% DSS for 7 d. Expression of indicated genes in the colonic IECs was determined by qPCR and shown as 2−dCT. Median. N = 5 for each group. (I) Expression of Ang4 in the ileal and colonic IECs from 3-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 10–12 for each group. (G) ChIP-seq dataset (GSM1266727) was visualized on the UCSC genome browser (Kent et al., 2002; Rosenbloom et al., 2013). HNF4A enrichment at the Btnl1 and Ang4 loci was shown. Each dot represented individual mouse. Data was combined from two independent experiments or one representative experiment of two independent experiments and analyzed by Mann-Whitney tests (A and G–I). *, P < 0.0332; **, P < 0.0021.
Hnf4a IEC-KO mice, RNA-seq and environmental regulation of HNF4A-dependent genes. Related to Figs. 1 and 2. (A) Body weight, spleen weight, colon length, and cecum weight of 10-wk-old mice. Mean with SD. N = 4 for each group. (B) Representative H&E staining of ileum, cecum, and colon of 11–12-wk-old mice. Scale bars were shown for WT ileum and the same for all other groups: 2×, 0.5 mm; 20×, 50 μm. (C) Pathology was evaluated based on H&E staining of the ileum, cecum, and colon from 11–12-wk-old mice. Samples with a score >0 had focal epithelial loss without inflammation (likely due to artifacts during sample processing). N = 3 for each group. (D) Colonic microbiome of 3-wk-old mice was determined by shotgun sequencing. Taxa composition at the species level (mean with SEM) and α diversity indicated by Shannon entropy (min to max) were shown. N = 9 for each group. (E) Gating strategy for flow sorting IECs. (F) GO enrichment analysis of DE genes in cecal IECs from RNA-seq. (G) 8-wk-old mice were given water (NT) or antibiotic cocktails (ABX) ad lib for 4 wk. RNA levels of indicated genes in the colonic IECs were determined by qPCR and shown as 2−dCT. Median. N = 6 for each group. (H) Mice were treated with 2% DSS for 7 d. Expression of indicated genes in the colonic IECs was determined by qPCR and shown as 2−dCT. Median. N = 5 for each group. (I) Expression of Ang4 in the ileal and colonic IECs from 3-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 10–12 for each group. (G) ChIP-seq dataset (GSM1266727) was visualized on the UCSC genome browser (Kent et al., 2002; Rosenbloom et al., 2013). HNF4A enrichment at the Btnl1 and Ang4 loci was shown. Each dot represented individual mouse. Data was combined from two independent experiments or one representative experiment of two independent experiments and analyzed by Mann-Whitney tests (A and G–I). *, P < 0.0332; **, P < 0.0021.
HNF4A directly regulates epithelial molecules known to coordinate IEC–IEL crosstalk
To identify genes directly regulated by HNF4A, we performed RNA sequencing (RNA-seq) on sorted cecal IECs from young adult WT and Hnf4aIEC-KO mice (Fig. S1 E). Differential expression (DE) analysis revealed 110 genes downregulated in the Hnf4aIEC-KO (HNF4A-dependent) and 31 genes upregulated (HNF4A-repressed; Fig. 2 A). We compared the DE genes to a published HNF4A ChIP-seq dataset generated using mouse colonic IECs (Chahar et al., 2014). This revealed that epithelial HNF4A mainly acts as a transcriptional activator (Fig. 2 B). Interestingly, while major immune pathways were unaffected (Fig. S1 F), HNF4A-dependent direct targets included a set of immune signaling molecules that have been implicated in the IEC–IEL crosstalk (Fig. 2 C).
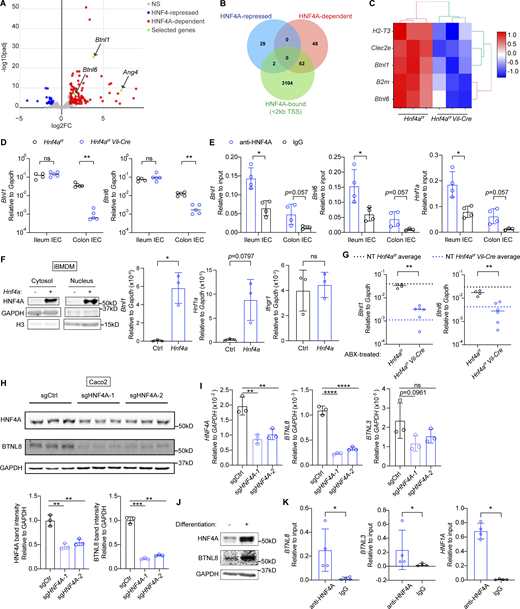
HNF4A directly regulates Btnls and other immune signaling molecules. All mice in this figure were 9–12-wk-old young adult unless stated otherwise. (A) Gene expression was determined by RNA-seq using sorted cecal IECs. Volcano plot displaying the fold change and adjusted P value of DE genes enriched in the WT (red) and Hnf4aIEC-KO (blue). Selected DE genes were highlighted (green) and annotated by arrows. Non-DE genes, gray. N = 3 for each genotype. (B) Venn diagram showing the overlap of DE genes with genes that contained HNF4A binding sites within 2 kb of TSS (ChIP-seq dataset GSM1266727). (C) Relative expression of representative immune signaling molecules in the HNF4A-dependent and HNF4A-bound group was shown as heatmap. Each column represented one biological replicate. (D) Expression of Btnl1 and Btnl6 in the ileal and colonic IECs was determined by qPCR and shown as 2−dCt. Median. N = 4–5 for each group. (E) HNF4A binding to the promoters of indicated genes was determined by ChIP-qPCR and shown as 2−dCt. Mean with SD. N = 3–4 for each group. (F) Empty vector (Ctrl) or mouse Hnf4a was ectopically expressed in murine iBMDMs. Western blot showing protein levels of HNF4A, GAPDH, and histone H3 in the cytosol and nucleus. qPCR analysis of HNF4A targets was shown as 2−dCt. Mean with SD. N = 3 for each group. (G) Breeders were given antibiotic cocktails (ABX) ad lib from pregnancy and treatment continued after birth of pups. RNA levels of indicated genes in the colonic IECs of 3-wk-old offspring were determined by qPCR and shown as 2−dCT. Average levels of genes in the colonic IECs from nontreated (NT) 3-wk-old mice were indicated by dashed lines. Median. N = 4–6 for each group. (H) Caco2-Cas9 cells were transduced with nontargeting guide RNA (sgCtrl) or two independent guides targeting HNF4A (sgHNF4A-1, sgHNF4A-2). Protein expression of HNF4A, BTNL8, and GAPDH was determined by Western blot. Band intensity was quantified using Fiji and shown below. Replicates were independently transduced cells. Mean with SD. N = 3 for each group. (I) Caco2-Cas9 cells were treated as in H. RNA levels of indicated genes were determined by qPCR and shown as 2−dCT. Mean with SD. N = 3 for each group. (J) Protein expression of HNF4A, BTNL8, and GAPDH in undifferentiated and differentiated Caco2 cells was determined by Western blot. (K) HNF4A binding to the promoters of indicated genes in differentiated Caco2 cells was determined by ChIP-qPCR and shown as 2−dCT. Mean with SD. N = 4 for each group. Each dot represented individual mouse or treatment. Data was combined from two independent experiments or one representative experiment from at least two independent experiments. Data was analyzed by Mann-Whitney tests (D–G and K) and one-way ANOVA (H and I). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001. Source data are available for this figure: SourceData F2.
HNF4A directly regulates Btnls and other immune signaling molecules. All mice in this figure were 9–12-wk-old young adult unless stated otherwise. (A) Gene expression was determined by RNA-seq using sorted cecal IECs. Volcano plot displaying the fold change and adjusted P value of DE genes enriched in the WT (red) and Hnf4aIEC-KO (blue). Selected DE genes were highlighted (green) and annotated by arrows. Non-DE genes, gray. N = 3 for each genotype. (B) Venn diagram showing the overlap of DE genes with genes that contained HNF4A binding sites within 2 kb of TSS (ChIP-seq dataset GSM1266727). (C) Relative expression of representative immune signaling molecules in the HNF4A-dependent and HNF4A-bound group was shown as heatmap. Each column represented one biological replicate. (D) Expression of Btnl1 and Btnl6 in the ileal and colonic IECs was determined by qPCR and shown as 2−dCt. Median. N = 4–5 for each group. (E) HNF4A binding to the promoters of indicated genes was determined by ChIP-qPCR and shown as 2−dCt. Mean with SD. N = 3–4 for each group. (F) Empty vector (Ctrl) or mouse Hnf4a was ectopically expressed in murine iBMDMs. Western blot showing protein levels of HNF4A, GAPDH, and histone H3 in the cytosol and nucleus. qPCR analysis of HNF4A targets was shown as 2−dCt. Mean with SD. N = 3 for each group. (G) Breeders were given antibiotic cocktails (ABX) ad lib from pregnancy and treatment continued after birth of pups. RNA levels of indicated genes in the colonic IECs of 3-wk-old offspring were determined by qPCR and shown as 2−dCT. Average levels of genes in the colonic IECs from nontreated (NT) 3-wk-old mice were indicated by dashed lines. Median. N = 4–6 for each group. (H) Caco2-Cas9 cells were transduced with nontargeting guide RNA (sgCtrl) or two independent guides targeting HNF4A (sgHNF4A-1, sgHNF4A-2). Protein expression of HNF4A, BTNL8, and GAPDH was determined by Western blot. Band intensity was quantified using Fiji and shown below. Replicates were independently transduced cells. Mean with SD. N = 3 for each group. (I) Caco2-Cas9 cells were treated as in H. RNA levels of indicated genes were determined by qPCR and shown as 2−dCT. Mean with SD. N = 3 for each group. (J) Protein expression of HNF4A, BTNL8, and GAPDH in undifferentiated and differentiated Caco2 cells was determined by Western blot. (K) HNF4A binding to the promoters of indicated genes in differentiated Caco2 cells was determined by ChIP-qPCR and shown as 2−dCT. Mean with SD. N = 4 for each group. Each dot represented individual mouse or treatment. Data was combined from two independent experiments or one representative experiment from at least two independent experiments. Data was analyzed by Mann-Whitney tests (D–G and K) and one-way ANOVA (H and I). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001. Source data are available for this figure: SourceData F2.
This set of genes include two members of the Btnl family, Btnl1 and Btnl6, which are present on IECs as heteromers and critical for the selection of Vγ7+ γδ IELs (Di Marco Barros et al., 2016; Jandke et al., 2020; Lebrero-Fernandez et al., 2016a). We also found components of MHC class I molecules B2m and H2-T3. This latter gene encodes the IEC-restricted thymus leukemia antigen (TL), a high affinity ligand for the CD8αα receptor expressed by both αβ and γδ IELs (Leishman et al., 2001; Liu et al., 2003). Clec2e belongs to the C-type lectin-related (Clr) family that is genetically paired with Nkrp1 family of natural killer receptors (Kirkham and Carlyle, 2014; Rutkowski et al., 2017). Although its exact function has yet to be determined, Nkrp1 receptors were found enriched on IELs and could potentially interact with IEC-expressed Clr ligands such as Clec2e (Kirkham and Carlyle, 2014).
We focused on Btnl molecules because Btnl1 was a top DE gene (Fig. 2 A). By quantitative PCR (qPCR), we found that Btnl1 and Btnl6 expression was significantly reduced in the colonic but not ileal IECs from Hnf4aIEC-KO mice (Fig. 2 D). ChIP-qPCR confirmed HNF4A binding to the promoters of Btnl1 and Btnl6 in both ileal and colonic IECs at a comparable level with Hnf1a, an established HNF4A target (Fig. 2 E). Ectopic expression of Hnf4a in murine immortalized bone marrow-derived macrophages (iBMDMs), which lack Hnf4a and Btnls expression naturally, was sufficient to drive expression of Btnl1, Hnf1a, but not Ifngr1, an HNF4A-targeted gene whose expression remained unchanged in RNA-seq (Fig. 2 F). Together, our data show that HNF4A is a critical transcription factor for Btnl1 and Btnl6.
We then investigated if the HNF4A–Btnl axis is influenced by environmental cues. Treatment of broad-spectrum antibiotics (cocktails of ampicillin, neomycin, metronidazole, and vancomycin) suppressed expression of microbiota-driven antimicrobial peptide Reg3g, but not Hnf4a, Btnl1, or Btnl6 in the colonic IECs (Fig. S1 G; Cash et al., 2006). Besides, Btnl1 and Btnl6 dependency on HNF4A remained unaltered after antibiotic treatment (Fig. 2 G). These data suggest that HNF4A regulation of Btnl expression is independent of gut bacteria. Consistent with previous studies (Ahn et al., 2008; Lebrero-Fernandez et al., 2016b), Hnf4a, Btnl1, and Btnl6 expression was downregulated in the IECs of inflamed colon after DSS treatment (Fig. S1 H).
Humans lack Btnl1 and Btnl6 but express functionally equivalent BTNL3 and BTNL8, which regulate Vγ4+ IELs (Di Marco Barros et al., 2016; Willcox et al., 2019). Because HNF4A is highly conserved, we tested if human HNF4A regulates BTNLs using a human colorectal adenocarcinoma cell line, Caco2. We generated HNF4A polyclonal knockout (pKO) cells using CRISPR-Cas9 (Fig. 2, H–I). With around twofold reduction of HNF4A, BTNL8 transcript and protein levels were greatly reduced. BTNL3 was expressed at a very low level in undifferentiated Caco2 cells, and the level further decreased in HNF4A pKO cells. HNF4A and BTNL8 levels increased with differentiation (Fig. 2 J). In differentiated Caco2 cells, HNF4A directly bound the promoters of BTNL8 and BTNL3 (Fig. 2 K). Collectively, our data suggest a conserved regulatory axis of HNF4A–BTNL in human IECs.
Epithelial HNF4A promotes the selection of γδ and CD8αα+ αβ IELs
From the RNA-seq, we identified the antimicrobial peptide Ang4 as a DE gene indirectly regulated by HNF4A (Fig. 2 A; Hooper et al., 2003). Ang4 was downregulated in colonic IECs of Hnf4aIEC-KO mice (Fig. S1 I), but its gene locus did not contain HNF4A binding sites (Fig. S1 J). A previous study showed that Vγ7+ γδ IELs stimulate IEC expression of Ang4 (Walker et al., 2013). Those findings led to our hypothesis that downregulation of Btnls in Hnf4aIEC-KO mice is biologically significant and impairs the IEC–IEL reciprocal regulation.
By flow cytometry, we assessed the IEL compartment using adult ileum and colon, which represent the small and large intestine, respectively (Fig. 3, A and B). We found an unbiased reduction of natural IELs including TCRδ+Vγ7+, TCRδ+Vγ7−, and TCRβ+CD8αα+ IELs in the ileum of Hnf4aIEC-KO mice compared to WT littermates (Fig. 3 C). In the colon, there was a specific loss of TCRδ+Vγ7+ IELs (Fig. 3 D). As a result, the proportion of Vγ7+ cells within γδ IELs was affected in the colon but not ileum (Fig. 3, E and F). Reduction of Vγ7+ IELs resembled the phenotype of Btnl1−/− and Btnl6−/− mice (Di Marco Barros et al., 2016; Jandke et al., 2020). Induced IEL subsets such as TCRβ+CD8αβ+ and TCRβ+CD4+ cells remained largely unchanged (although pooled TCRβ+CD4+ number was statistically lower in the ileum, the changes were not consistent between experiments). We saw similar phenotype in the duodenum that natural but not induced IELs were reduced (Fig. S2, A and B). Loss of T cell subsets was restricted to the intestinal epithelium because frequencies of Vγ7+ and CD8αα+ cells were unaffected in the lamina propria, mesenteric lymph nodes, spleens, and thymus (Fig. S2 D). Total numbers of T, B, and myeloid cells in the lamina propria were comparable between WT and Hnf4aIEC-KO mice (Fig. S2 C).

Epithelial HNF4A deficiency affects γδ and CD8αα + αβ IELs. All mice in this figure were 9–12-wk-old young adult. (A) Gating strategy for flow cytometry analysis of IELs. (B) Representative plots showing γδ IELs in the colon of WT and Hnf4aIEC-KO mice. (C and D) Cell number of indicated IEL subsets in the ileum (C) and colon (D). Mean with SD. N = 6–15 for each group. (E and F) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the ileum (E) and colon (F). Mean with SD. N = 10 for each group. (G) Median fluorescence intensity of CD8α, Thy1.2, and LAG-3 was normalized to the collective value of each row for visualization. Mean. N = 5 for each group. Actual median fluorescence intensity value was used for statistical analysis. (H) Representative histogram for data used in G. WT (gray filled), Hnf4aIEC-KO (blue), and Fluorescence Minus One control (FMO, black dashed) were overlayed for comparison. (I) Representative plots of Vδ chain usage in Vγ7+ IELs from the colon of WT and Hnf4aIEC-KO mice. (J) Cell numbers of indicated Vδ-chain pairing in Vγ7+ IELs. Mean with SD. N = 5 for each group. (K) Frequencies of indicated Vδ-chain usage in Vγ7+ IELs. Mean with SD. N = 5 for each group. Each dot represented individual mouse. Data was combined from at least two independent experiments. Data was analyzed by Mann-Whitney tests (C–J) and two-way ANOVA (K). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002.
Epithelial HNF4A deficiency affects γδ and CD8αα + αβ IELs. All mice in this figure were 9–12-wk-old young adult. (A) Gating strategy for flow cytometry analysis of IELs. (B) Representative plots showing γδ IELs in the colon of WT and Hnf4aIEC-KO mice. (C and D) Cell number of indicated IEL subsets in the ileum (C) and colon (D). Mean with SD. N = 6–15 for each group. (E and F) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the ileum (E) and colon (F). Mean with SD. N = 10 for each group. (G) Median fluorescence intensity of CD8α, Thy1.2, and LAG-3 was normalized to the collective value of each row for visualization. Mean. N = 5 for each group. Actual median fluorescence intensity value was used for statistical analysis. (H) Representative histogram for data used in G. WT (gray filled), Hnf4aIEC-KO (blue), and Fluorescence Minus One control (FMO, black dashed) were overlayed for comparison. (I) Representative plots of Vδ chain usage in Vγ7+ IELs from the colon of WT and Hnf4aIEC-KO mice. (J) Cell numbers of indicated Vδ-chain pairing in Vγ7+ IELs. Mean with SD. N = 5 for each group. (K) Frequencies of indicated Vδ-chain usage in Vγ7+ IELs. Mean with SD. N = 5 for each group. Each dot represented individual mouse. Data was combined from at least two independent experiments. Data was analyzed by Mann-Whitney tests (C–J) and two-way ANOVA (K). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002.

Immune composition in other tissues and 1-wk-old mice. Related to Figs. 3 and 4. (A–J) Mice were 10–12 wk old in A–D and 6–7 d old in E–J. (A) Cell number of indicated cell subsets in the duodenum of young adult mice. Mean with SD. N = 6–7 for each group. (B) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the duodenum. Mean with SD. N = 6–7 for each group. (C) General T cell (CD45+CD3+CD19−), B cell (CD45+CD3−CD19+), and myeloid cell (CD45+CD3−CD19−, CD11b+ or CD11c+) numbers in the lamina propria of the small intestine (SI LP) and large intestine (LI LP). Mean with SD. N = 4 for each group. (D) Percentage of indicated cell subsets in the ileal lamina propria (iLP), colonic lamina propria (cLP), mesenteric lymph nodes (MLN), spleens (SPL), and thymus (THY). Mean with SD. N = 4–5 for each group. (E and F) Expression of indicated genes in the ileal IECs (E) and proximal colon (F) of 1-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 4–9 for each group. (G and H) Cell number of indicated cell subsets in the ileum (G) and colon (H) of 1-wk-old mice. Mean with SD. N = 7–9 for each group. (I and J) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the ileum (I) and colon (J). Mean with SD. N = 7–9 for each group. Each dot represented individual mouse. Data was combined from two independent experiments. Data was analyzed by Mann-Whitney tests (A–J). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002.
Immune composition in other tissues and 1-wk-old mice. Related to Figs. 3 and 4. (A–J) Mice were 10–12 wk old in A–D and 6–7 d old in E–J. (A) Cell number of indicated cell subsets in the duodenum of young adult mice. Mean with SD. N = 6–7 for each group. (B) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the duodenum. Mean with SD. N = 6–7 for each group. (C) General T cell (CD45+CD3+CD19−), B cell (CD45+CD3−CD19+), and myeloid cell (CD45+CD3−CD19−, CD11b+ or CD11c+) numbers in the lamina propria of the small intestine (SI LP) and large intestine (LI LP). Mean with SD. N = 4 for each group. (D) Percentage of indicated cell subsets in the ileal lamina propria (iLP), colonic lamina propria (cLP), mesenteric lymph nodes (MLN), spleens (SPL), and thymus (THY). Mean with SD. N = 4–5 for each group. (E and F) Expression of indicated genes in the ileal IECs (E) and proximal colon (F) of 1-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 4–9 for each group. (G and H) Cell number of indicated cell subsets in the ileum (G) and colon (H) of 1-wk-old mice. Mean with SD. N = 7–9 for each group. (I and J) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the ileum (I) and colon (J). Mean with SD. N = 7–9 for each group. Each dot represented individual mouse. Data was combined from two independent experiments. Data was analyzed by Mann-Whitney tests (A–J). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002.
In addition to reduced cell numbers, many of the remaining Vγ7+ IELs in the colon of Hnf4aIEC-KO mice exhibited an immature phenotype indicated by Thy1+, CD8αα−, and LAG3low, phenocopying Vγ7+ IELs recovered from Btnl1−/− mice (Fig. 3, G and H; Di Marco Barros et al., 2016). We also examined the δ-chain usage of colonic Vγ7+ IELs (Fig. 3 I). There was an increase in the frequency of Vδ6.3 pairing in Hnf4aIEC-KO mice (Fig. 3 K), which reflected the unchanged cell number of Vδ6.3+ IELs and a reduction of Vδ6.3− IELs (Fig. 3 J). A previous study showed similar results with Btnl6−/− mice and suggested that Btnl1 but not Btnl6 is required for Vγ7+Vδ6.3+ IELs (Jandke et al., 2020). In the case of Hnf4a deficiency, our data suggest that the remaining Btnl1 may be sufficient to support Vγ7+Vδ6.3+ IELs. Together, our results support that downregulation of Btnls in Hnf4aIEC-KO mice impaired the functional maturation of Vγ7+ IELs and altered their δ-chain usage.
Natural IELs populate the intestine and expand early in life, and their numbers remain steady throughout adulthood (Cheroutre et al., 2011). To determine if loss of IELs in Hnf4aIEC-KO mice results from a developmental failure, we examined the proliferation of IELs in weanling mice at 3 wk old (Fig. 4). In accordance with the adult data, significantly lower percentage of Vγ7+, Vγ7−, and TCRβ+CD8αα+ IELs expressed cell cycle marker Ki67 in the ileum (Fig. 4 B). The proliferation defect was specific to Vγ7+ IELs in the colon (Fig. 4 C). Moreover, Hnf4aIEC-KO mice already had fewer Vγ7+ and TCRβ+CD8αα+ IELs in the ileum and fewer Vγ7+ cells in the colon (Fig. 4, D and E). Collectively, these results suggest that HNF4A is required for the selective expansion of γδ and CD8αα+ αβ IELs in the ileum, and Vγ7+ IELs in the colon.

Epithelial HNF4A promotes expansion of γδ and CD8αα + αβ IELs. All mice in this figure were 21–22 d old. (A) Representative histogram showing Ki67 staining in the indicated cell subsets. WT (gray filled), Hnf4aIEC-KO (blue), and FMO (black dashed) were overlayed for comparison. (B and C) Percentage of Ki67-positive (threshold setting as shown in A) cells in indicated cell subsets from the ileum (B) and colon (C). Mean with SD. N = 9–11 for each group. (D and E) Cell number of indicated cell subsets in the ileum (D) and colon (E). Mean with SD. N = 14–16 for each group. (F and G) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the ileum (F) and colon (G). Mean with SD. N = 14–16 for each group. Each dot represented individual mouse. Data was combined from at least two independent experiments. Data was analyzed by Mann-Whitney tests (B–G). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001.
Epithelial HNF4A promotes expansion of γδ and CD8αα + αβ IELs. All mice in this figure were 21–22 d old. (A) Representative histogram showing Ki67 staining in the indicated cell subsets. WT (gray filled), Hnf4aIEC-KO (blue), and FMO (black dashed) were overlayed for comparison. (B and C) Percentage of Ki67-positive (threshold setting as shown in A) cells in indicated cell subsets from the ileum (B) and colon (C). Mean with SD. N = 9–11 for each group. (D and E) Cell number of indicated cell subsets in the ileum (D) and colon (E). Mean with SD. N = 14–16 for each group. (F and G) Vγ7+ and Vγ7+CD8α+ percentage out of γδ IELs in the ileum (F) and colon (G). Mean with SD. N = 14–16 for each group. Each dot represented individual mouse. Data was combined from at least two independent experiments. Data was analyzed by Mann-Whitney tests (B–G). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001.
We further examined the early IEL colonization by assessing 1-wk-old neonates. Expression of Btnl6 in the ileal IECs and Btnl1 and Btnl6 in the proximal colon was significantly reduced at this age (Fig. S2, E and F). Hnf4aIEC-KO mice had indistinguishable ileal IEL composition compared to WT littermates, supporting a specific role of HNF4A in the expansion but not seeding of IELs (Fig. S2, G and I). However, in the colon, the number of Vγ7+ IELs was already lower (Fig. S2, H and J). In addition, there was around twofold reduction of Vγ7− and TCRβ+CD8αα+ IELs not seen in the 3-wk-old and adult colon. These data suggest that HNF4A is important for the early colonization of γδ and CD8αα+ αβ IELs in the colon but not ileum, and for the selective expansion of Vγ7+ IELs that together account for the specific loss of Vγ7+ IELs in the adult Hnf4aIEC-KO colon.
HNF4A cooperates with HNF4G to regulate immune signaling molecules
Our results indicate that HNF4A regulation of immune signaling molecules and IELs is spatially specific. We then asked how this could be achieved. We noticed that transcripts of Btnl1, Btnl6, H2-T3, and Clec2e were more abundant in the ileum and less affected by HNF4A deficiency (Fig. 2 D and Fig. 5 A). To explore if differential dependence on HNF4A applies to other targets, we performed RNA-seq on sorted ileal and colonic IECs from 3-wk-old WT and Hnf4aIEC-KO littermates (Fig. 5 B). Consistent with the cecal data, HNF4A primarily acts as a transcriptional activator (Fig. 5 C). Principal component analysis revealed that WT and Hnf4aIEC-KO ileal transcriptome clustered closer to each other than those of the colonic origin (Fig. 5 D). These results support that immune signaling molecules rely more on HNF4A in the large intestine compared to the small intestine.

HNF4A and HNF4G regulate immune signaling molecules in a location-specific manner. (A) Expression of indicated immune signaling molecules in the ileal and colonic IECs from 3-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 10–12 for each group. (B) RNA-seq analysis of sorted IECs from the ileum and colon of 3-wk-old mice. Volcano plots displaying the fold change and adjusted P value of DE genes enriched in WT (red) and Hnf4aIEC-KO (blue). DE genes containing HNF4A binding sites within 2 kb of TSS were highlighted (green) and annotated by arrows. Non-DE genes, gray. N = 4 for each group. (C) Percentage of HNF4A direct targets in ileal and colonic DE genes was shown. (D) Principal component analysis of most-varied genes was shown. Each dot represented transcriptome from one sample. Samples of the same genotype and tissue origin were grouped by colored ovals. WT ileum (pink), Hnf4aIEC-KO ileum (blue), WT colon (orange), and Hnf4aIEC-KO colon (green). (E) Expression of Btnl1 and Btnl6 along the intestinal tract of young adult mice was determined by qPCR. PS, proximal small intestine; MS, middle small intestine; DS, distal small intestine; CE, cecum; PC, proximal colon; DC, distal colon. Mean with SEM. N = 5–9 for each group. (F) As in E, expression of Hnf4a and Hnf4g along the intestinal tract was determined by qPCR. Mean with SEM. N = 5–9 for each group. (G) Protein levels of HNF4A and HNF4G in the ileal and colonic IECs was determined by Western blot. N = 3 for each group. (H) HNF4G binding to the promoters of indicated genes in the ileal IECs from Hnf4aIEC-KO mice was determined by ChIP-qPCR and shown as 2−dCT. Mean with SD. N = 3 for each group. (I) Levels of Btnl1 and Btnl6 in the duodenal IECs of indicated genotypes. Data was adapted from the RNA-seq dataset GSE112946 (Chen et al., 2019). Mean. N = 3 for each group. Each dot represented individual mouse. Data was combined from at least two independent experiments or one representative experiment of two independent experiments and analyzed by Mann-Whitney tests (A, E, F, and H). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001. Source data are available for this figure: SourceData F5.
HNF4A and HNF4G regulate immune signaling molecules in a location-specific manner. (A) Expression of indicated immune signaling molecules in the ileal and colonic IECs from 3-wk-old mice was determined by qPCR and shown as 2−dCT. Median. N = 10–12 for each group. (B) RNA-seq analysis of sorted IECs from the ileum and colon of 3-wk-old mice. Volcano plots displaying the fold change and adjusted P value of DE genes enriched in WT (red) and Hnf4aIEC-KO (blue). DE genes containing HNF4A binding sites within 2 kb of TSS were highlighted (green) and annotated by arrows. Non-DE genes, gray. N = 4 for each group. (C) Percentage of HNF4A direct targets in ileal and colonic DE genes was shown. (D) Principal component analysis of most-varied genes was shown. Each dot represented transcriptome from one sample. Samples of the same genotype and tissue origin were grouped by colored ovals. WT ileum (pink), Hnf4aIEC-KO ileum (blue), WT colon (orange), and Hnf4aIEC-KO colon (green). (E) Expression of Btnl1 and Btnl6 along the intestinal tract of young adult mice was determined by qPCR. PS, proximal small intestine; MS, middle small intestine; DS, distal small intestine; CE, cecum; PC, proximal colon; DC, distal colon. Mean with SEM. N = 5–9 for each group. (F) As in E, expression of Hnf4a and Hnf4g along the intestinal tract was determined by qPCR. Mean with SEM. N = 5–9 for each group. (G) Protein levels of HNF4A and HNF4G in the ileal and colonic IECs was determined by Western blot. N = 3 for each group. (H) HNF4G binding to the promoters of indicated genes in the ileal IECs from Hnf4aIEC-KO mice was determined by ChIP-qPCR and shown as 2−dCT. Mean with SD. N = 3 for each group. (I) Levels of Btnl1 and Btnl6 in the duodenal IECs of indicated genotypes. Data was adapted from the RNA-seq dataset GSE112946 (Chen et al., 2019). Mean. N = 3 for each group. Each dot represented individual mouse. Data was combined from at least two independent experiments or one representative experiment of two independent experiments and analyzed by Mann-Whitney tests (A, E, F, and H). *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001. Source data are available for this figure: SourceData F5.
Multiple members of the Btnl family showed up as HNF4A-dependent targets including Btnl1/6 in the ileum and Btnl1/2/4/6 in the colon (Fig. 5 B). Btnl1 was a top hit in the colon (Fig. 5 B). In fact, Btnl1 and Btnl6 are among the 18 HNF4A-dependent targets shared by the ileum, cecum, and colon (Fig. S3 B). Major immune pathways remain unaffected in both ileum and colon (Fig. S3 A). Il15, a cytokine important for natural IELs, showed up as an ileum-specific target (Fig. S3 B; Leibelt et al., 2015). However, qPCR analysis revealed more significant downregulation in the colon, while colonic Vγ7− and TCRβ+CD8αα+ IELs were not affected (Fig. S3 C). These observations suggest that changes in Btnls are the primary and direct effects downstream of HNF4A.
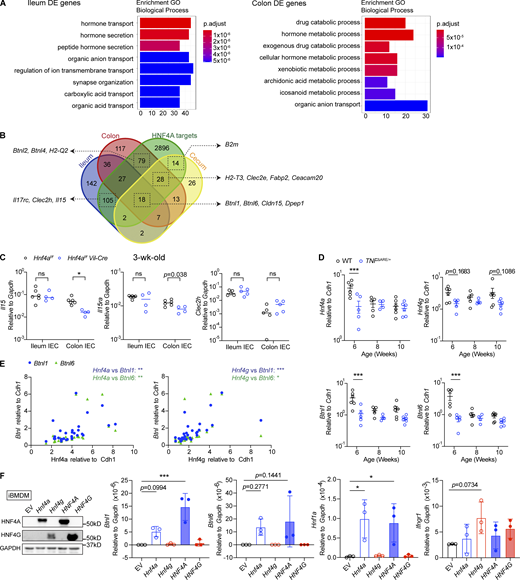
HNF4A and HNF4G cooperate to regulate immune signaling molecules. Related to Fig. 5. (A) GO enrichment analysis of DE genes in the ileum and colon comparing WT and Hnf4aIEC-KO. (B) HNF4A-dependent genes in the ileum, cecum, and colon (Fig. 2 A and Fig. 5 B) were compared to the HNF4A ChIP-seq dataset (GSM1266727). HNF4A direct targets were defined by genes containing HNF4A binding sites within 2 kb of TSS. The overlap among groups was summarized as Venn diagram. Representative genes in each group were highlighted by arrows. (C) Expression of selected ileum-specific HNF4A-dependent targets in B was determined by qPCR using 3-wk-old IEC samples. Median. N = 4–6 for each group. Data was analyzed by Mann-Whitney tests. (D) Expression of Hnf4a, Hnf4g, Btnl1, and Btnl6 in the ileum of WT and TNFΔARE/+ mice at different ages. Median. N = 4–6 for each group. Data was analyzed by two-way ANOVA. (E) Expression correlation of Hnf4 and Btnls was determined by Pearson correlation. Data from D. (F) Empty vector (EV), mouse Hnf4a, Hnf4g, human HNF4A, or HNF4G were ectopically expressed in iBMDMs. Protein levels of HNF4A, HNF4G, and GAPDH from one representative experiment was determined by Western blot. Expression of indicated HNF4A targets was determined by qPCR and shown as 2−dCT. Mean with SD. N = 3 for each group. Data was analyzed by one-way ANOVA. Each dot represented individual mouse or treatment. Data was combined from two independent experiments. ns, not significant; *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002. Source data are available for this figure: SourceData FS3.
HNF4A and HNF4G cooperate to regulate immune signaling molecules. Related to Fig. 5. (A) GO enrichment analysis of DE genes in the ileum and colon comparing WT and Hnf4aIEC-KO. (B) HNF4A-dependent genes in the ileum, cecum, and colon (Fig. 2 A and Fig. 5 B) were compared to the HNF4A ChIP-seq dataset (GSM1266727). HNF4A direct targets were defined by genes containing HNF4A binding sites within 2 kb of TSS. The overlap among groups was summarized as Venn diagram. Representative genes in each group were highlighted by arrows. (C) Expression of selected ileum-specific HNF4A-dependent targets in B was determined by qPCR using 3-wk-old IEC samples. Median. N = 4–6 for each group. Data was analyzed by Mann-Whitney tests. (D) Expression of Hnf4a, Hnf4g, Btnl1, and Btnl6 in the ileum of WT and TNFΔARE/+ mice at different ages. Median. N = 4–6 for each group. Data was analyzed by two-way ANOVA. (E) Expression correlation of Hnf4 and Btnls was determined by Pearson correlation. Data from D. (F) Empty vector (EV), mouse Hnf4a, Hnf4g, human HNF4A, or HNF4G were ectopically expressed in iBMDMs. Protein levels of HNF4A, HNF4G, and GAPDH from one representative experiment was determined by Western blot. Expression of indicated HNF4A targets was determined by qPCR and shown as 2−dCT. Mean with SD. N = 3 for each group. Data was analyzed by one-way ANOVA. Each dot represented individual mouse or treatment. Data was combined from two independent experiments. ns, not significant; *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002. Source data are available for this figure: SourceData FS3.
We extended the analysis to different locations along the intestinal tract using adult mice (Fig. 5 E). Btnl1 and Btnl6 expression was dependent on HNF4A throughout the large intestine and in the distal small intestine. However, such dependency was lost in the proximal and middle small intestine (Fig. 5 E). We hypothesized that other transcription factors collaboratively contribute to the expression in the small intestine. One top candidate was HNF4G, an HNF4 paralog known to function redundantly with HNF4A in driving enterocyte differentiation (Chen et al., 2019; Lodolce et al., 1998). Unlike Hnf4a whose expression was stable throughout the intestine, Hnf4g was restricted to the small intestine at RNA and protein levels (Fig. 5, F and G). Human HNF4A and HNF4G follow a similar geographic pattern (Uhlén et al., 2015). This relative abundance of HNF4 collectively correlated with expression of Btnls, H2-T3, and Clec2e, as well as the distribution of natural IELs (Fig. 5 A and Fig. 3, C and D; Mowat and Agace, 2014). In the TNFΔARE model of ileitis, the ileum had an early and progressive reduction of CD8αα+ γδ and αβ IELs (Apostolaki et al., 2008). Interestingly, we found that Hnf4a, Hnf4g, Btnl1, and Btnl6 were downregulated in TNFΔARE mice at 6 wk old, preceding the histological onset of disease at 8 wks (Kontoyiannis et al., 1999), and that Hnf4a and Hnf4g expression correlated with Btnl1 and Btnl6 (Fig. S3, D and E).
Previous ChIP-seq studies suggest that HNF4G binding sites largely overlap with HNF4A in WT duodenum (Chen et al., 2019). Using ChIP-qPCR, we found HNF4G robustly bound to the promoters of Btnl1 and Btnl6 even in the absence of HNF4A (Fig. 5 H). Duodenal IECs lacking both Hnf4a and Hnf4g had greatly reduced levels of Btnl1 and Btnl6 compared to Hnf4a or Hnf4g single KO from a published RNA-seq dataset (Fig. 5 I; Chen et al., 2019). However, in contrast to HNF4A, overexpressing HNF4G in iBMDMs did not drive expression of Btnl1 or Hnf1a efficiently, suggesting that HNF4G may use different cofactors (Fig. S3 F). Altogether, these results support that HNF4G functions redundantly with HNF4A to regulate immune signaling molecules in a location-specific manner.
Discussion
The intestinal epithelium shapes the local development of IELs. Proper function of the IEC–IEL circuit is critical for the maintenance of intestinal homeostasis. Here, we report that nuclear receptor HNF4A drives expression of a set of epithelial molecules involved in the IEC–IEL crosstalk. These molecules include Btnls/BTNLs, TL, and Clec2e, which could directly interact with γδ TCR, CD8αα, and potentially Nkrp1 receptors. Interestingly, HNF4A preferentially enhances proliferation of natural IELs including γδ and CD8αα+ αβ T cells, but not induced IELs such as CD8αβ+ and CD4+ αβ T cells. It is likely because the interacting receptors are mainly found on natural IELs, supporting that HNF4A targets regulate IELs via a ligand-receptor mechanism.
However, current knowledge of those molecules could only partially explain the location-specific regulation of natural IEL subsets. HNF4A specifically selects and promotes the maturation of Vγ7+ IELs in the colon, consistent with the reported functions of Btnl1 and Btnl6 (Di Marco Barros et al., 2016; Jandke et al., 2020). In the small intestine, TCRβ+CD8αα+ and TCRδ+Vγ7− IEL numbers were also affected, which was not observed in Btnl-deficient or TL-deficient mice (Di Marco Barros et al., 2016; Jandke et al., 2020; Olivares-Villagomez et al., 2008; Van Kaer et al., 2014). These data suggest that Btnl- and TL-independent mechanisms downstream of HNF4A also exist. Further investigation into HNF4A targets, most of which have no known immune functions, in the small intestine is needed to elucidate the effects on TCRβ+CD8αα+ and TCRδ+Vγ7− IELs.
HNF4A is stably expressed along the intestinal tract, while its functionally redundant paralog, HNF4G, is restricted to the small intestine. In accordance, HNF4-targeted immune signaling molecules are more abundant in the small intestine compared to the large intestine. This distribution pattern is consistent with that of natural IELs, which are supported by the HNF4–immune signaling axis (Mowat and Agace, 2014). Moreover, HNF4A deficiency caused stronger defects on the frequency and maturation of Vγ7+ IELs in the colon compared to the ileum, which correlated with the extent of Btnl downregulation. The interplay of HNF4A and HNF4G provides a mechanism to explain the regional distribution of immune signaling molecules and regulation of natural IELs. Future comparative studies on Hnf4g KO mice are needed to dissect the respective role of these two HNF4 paralogs. However, deleting both paralogs would lead to failure in the enterocyte differentiation and epithelium maintenance (Chen et al., 2019), suggesting that natural IELs are developmentally coupled with IEC differentiation.
Hnf4aIEC-KO mice are more susceptible to acute and chronic colitis, but young animals are largely normal at steady state (Ahn et al., 2008; Darsigny et al., 2009). We detected subclinical inflammation as early as 3 wk old in Hnf4aIEC-KO mice, which coincided with the loss of natural IELs. Although there was no significant change in the microbiome, Hnf4aIEC-KO colon expressed less Ang4, an antimicrobial peptide whose expression by IECs is dependent on Vγ7+ IELs (Hooper et al., 2003; Walker et al., 2013). These results suggest that the loss of IEC–IEL reciprocal regulation in Hnf4aIEC-KO mice could impair antimicrobial response, which may contribute to the low-grade inflammation and predispositions to colitis. However, it remains unclear how loss of Vγ7+ IELs in the colon and multiple natural IEL subsets in the ileum contribute to disease development.
Given that HNF4–BTNL–IEL axis is conserved in humans, it would be important to explore whether and how it plays a role in the context of inflammatory bowel disease (IBD). Polymorphisms in HNF4A and BTNL2 were associated with IBD (UK IBD Genetics Consortium et al., 2009; Jostins et al., 2012; Mochida et al., 2007; Prescott et al., 2015). HNF4A, HNF4G, BTNL2, and BTNL8 were shown to be downregulated in the intestinal samples from IBD patients (Ahn et al., 2008; Bueno-Hernandez et al., 2011; Lebrero-Fernandez et al., 2016b; Wu et al., 2007). We and others showed reduction of Hnf4a, Hnf4g, Btnl1, Btnl4, and Btnl6 transcripts in mouse models of IBD (Ahn et al., 2008; Lebrero-Fernandez et al., 2016b). Imbalance of IEL subsets has also been indicated in both humans and mice (Catalan-Serra et al., 2017; Hu and Edelblum, 2017). A recent study identified a global reduction of γδ and CD8+ IELs in the terminal ileum of severe Crohn’s disease patients (Jaeger et al., 2021), consistent with observations in TNFΔARE mice (Apostolaki et al., 2008). It would be of great interest to investigate if epithelial HNF4 can drive IEL dysregulation through BTNLs in IBD, and if so, how that contributes to disease.
HNF4A may also control intestinal homeostasis through multiple mechanisms. Using RNA-seq, we identified direct HNF4A targets in the ileum, cecum, and colon. Genes shared by all locations include Btnl1, Btnl6, a known target Cldn15, and a new target Dpep1 (Fig. 5 B). DPEP1 was recently uncovered as a neutrophil adhesion receptor in the lung and liver (Choudhury et al., 2019), suggesting that HNF4A may be involved in IEC–neutrophil interactions. Besides, multiple genes associated with IBD and colorectal cancer including Tmigd1, Mep1a, and Slc26a were directly regulated by HNF4A. These targets suggest that HNF4A could serve as an upstream transcription factor for known and novel IBD candidate genes.
Materials and methods
Mice
Mice were maintained in the specific pathogen–free facility at the University of Massachusetts Chan Medical School, and all animal experiments were approved by the Institutional Animal Care and Use Committees. TNFΔARE/+ mice and related studies were conducted in an Association of the Assessment and Accreditation of Laboratory Animal Care-accredited facility according to protocols approved by Rutgers New Jersey Medical School Comparative Medicine Resources.
Hnf4af/f mice were kindly gifted by Dr. Frank Gonzalez at National Institutes of Health (Bethesda, MD) and crossed to Vil-Cre (Jax-021504; Hayhurst et al., 2001). Hnf4af/fVil-Cre mice were bred to Hnf4af/f mice to generate experimental mice. Vil-Cre, loxp alleles, and Hnf4a deletion was determined through Transnetyx. Vil-Cre positive and negative littermates remained cohoused after weaning and littermates were used for all experiments. TNFΔARE/+ mice on a C57BL/6 background were obtained from Dr. George Kollias at National and Kapodistrian Univeristy of Athens (Athens, Greece) and Biomedical Sciences Research Center Alexander Fleming (Vari, Greece; Kontoyiannis et al., 1999) and compared to littermate WT mice for experiments. Both male and female mice were used unless stated otherwise.
For DSS-induced colitis model, mice were fed on continuous 1.5% (wt/vol) DSS water for 4–7 d. Mice were monitored for body weight and disease symptoms daily. Colitis scores were determined based on stool consistency (well-formed pellets, 0; changed formed pellets, 1; loose stool, 2; diarrhea or no stool, 3; and gross blood in stool [no blood, 0; blood-positive trace, 1; moderate trace, 2; gross trace, 3; gross bleeding anus]).
For salmonella (SL1344) infection, mice were given 2.60 × 108 CFU exponential phase bacteria orally and ceca were collected after 2 d.
For antibiotic treatment, 2-mo-old mice were given antibiotic cocktails containing 0.1% (wt/vol) ampicillin, 0.1% (wt/vol) neomycin, 0.1% (wt/vol) metronidazole, and 0.05% (wt/vol) vancomycin ad lib for 4 wk. In another case, breeders were given antibiotic cocktails from pregnancy until the offspring reached 3 wk old.
Isolation of IECs and lamina propria cells
Small intestine (between stomach and cecum) was divided into three parts of equal length. Jejunum, duodenum, and ileum were defined as the proximal (closest to the stomach), middle, and the distal part, respectively. Whole colon (between cecum and anus) was used. Intestines were opened longitudinally and washed with PBS to remove fecal contents. Tissues were cut into ∼1 cm pieces and incubated in dissociation buffer (HBSS supplemented with 2.5 mM EDTA, 1 mM Hepes, 1 mM dithiothreitol [DTT], and 5% of FCS) at 37°C, 250 rpm for 30 min. Cells that passed through 40 μm cell strainers were collected as a mixture of IECs and IELs. Remaining intestine pieces were washed with PBS, cut into small pieces, and incubated in digestion buffer (RPMI 1,640 medium supplemented with 10% FCS, 0.5 mg/ml collagenase I, 0.5 mg/ml collagenase IV, and 40 μg/ml DNase I) at 37°C, 250 rpm for 30 min. Digested tissues were passed through 40 μm cell strainers, washed with PBS and FACS buffer (PBS, 5% FCS, 2 mM EDTA, and 0.05% sodium azide), and collected as lamina propria cells.
Flow cytometry
Cells were resuspended in FACS buffer and stained for the viability dye and surface antigens on ice for 30 min. If secondary antibodies were needed, cells were washed with FACS buffer and stained for secondary antibodies on ice for 30 min. Cells were washed with FACS buffer and fixed using 1% paraformaldehyde on ice for 15 min. If analyzing intracellular Ki67 expression, cells were fixed, permeabilized, and stained for Ki67 using the Foxp3 staining buffer set (eBioscience) according to the manufacture’s instruction. After the last wash, cells were resuspended in FACS buffer, supplemented with PKH25 reference microbeads (Sigma-Aldrich), and analyzed by Cytek Aurora spectral cytometer. Flow data was analyzed using FlowJo.
cDNA synthesis and real-time PCR
Fresh or frozen IECs were resuspended in RNA lysis buffer (Biorad). For total tissues, 1 cm pieces were homogenized in RNA lysis buffer (Biorad) or TRizol (Invitrogen). Accordingly, total RNA was extracted using Aurum Total RNA mini kit (Biorad) or using chloroform followed by RNeasy mini kit (Qiagen) purification. RNA was quantified by SpectraMax iD5 or NanoDrop. 500 ng of RNA was reversed transcribed using iScript cDNA synthesis kit (Biorad). Real-time PCR was performed using SYBR green supermix (Biorad) and detected on a CFX96 system (Biorad). For TNFΔARE samples, real-time PCR was performed using PowerUP SYBR green master mix (Applied Biosystems) on a QuantStuidio 6 (Applied Biosystems). Primers were ordered from Integrated DNA Technologies. Primer sequences are listed in Table 2. Relative expression was determined by 2−dCt using Gapdh or Cdh1 as reference genes.
RNA-seq and data availability
IECs were sorted directly into RLT lysis buffer (Qiagen). Total RNA was extracted using RNeasy micro kit (Qiagen). RNA was assessed by Qubit fluorometer, and RNA quality was determined by fragment analyzer. For library preparation, TruSeq Stranded Total RNA kit (Illumina) was used for cecal IECs and Illumina Stranded mRNA Prep kit (Illumina) was used for ileal and colonic IECs. Libraries were assessed by Qubit fluorometer and fragment analyzer. Sequencing was performed on a NextSeq 2000 sequencing system. RNA-seq data was analyzed using DolphinNext platform (Yukselen et al., 2020). In brief, after removal of ribosomal RNA reads by Bowtie, reads were mapped to the mouse reference genome (GENCODE mm10) using STAR. Count data was analyzed by DESeq2 (parametric) comparing WT and Hnf4aIEC-KO samples. Genes with a greater than twofold change and a smaller than 0.01 adjusted P value were considered differentially expressed. RNA-seq data is available on NCBI Gene Expression Omnibus (accession number GSE199417).
ChIP-seq data annotation
Colonic IEC HNF4A ChIP-seq data (NCBI Gene Expression Omnibus accession number GSM1266727) was annotated using ChIPseeker (Yu et al., 2015). Genes containing HNF4A binding sites ≤2 kb of transcriptional start sites (TSS) were considered direct HNF4A targets and compared to the DE genes identified via RNA-seq.
Western blot
Cells were lysed in RIPA buffer (Thermo Fisher Scientific) or subjected to nuclear extraction using a nuclear extraction kit (Active Motif). Protease inhibitor (Thermo Fisher Scientific) was supplemented in the lysis buffer. Intestine tissue pieces and lysates were sonicated and debris was removed by centrifuging at max speed for 10 min. Protein concentrations were determined using Biorad DC protein assay and equal amounts of protein were used for comparison. Gel electrophoresis and transfer was performed using Biorad chambers and Trans-Blot Turbo system. Membranes were blocked with 2% nonfat milk in Tris Buffered Saline with 0.05% Tween (TBST) for 1 h at room temperature and incubated with antibodies (listed in Table 1) at 4°C overnight. After washing with TBST, membranes were incubated with secondary antibodies (Licor or Biorad) for 1 h at room temperature. Proteins were detected using a LI-COR Odyssey DLx imaging system or Biorad ChemiDoc system.
Antibodies and flow reagents
| Antibodies | Clone/identifier | Source |
|---|---|---|
| Flow cytometry | ||
| CD16/CD32 | 2.4G2 | Tonbo |
| CD326 (EPCAM) | G8.8 | eBioscience |
| CD3 | 17A2 | eBioscience |
| CD4 | RM4-5 | Tonbo |
| CD45.2 | 104 | Tonbo |
| CD8a | 53-6.7 | eBioscience |
| CD8b | YTS156.7.7 | BioLegend |
| CD8b | YTS156.7.7 | BioLegend |
| Ki-67 | SolA15 | eBioscience |
| LAG-3 (CD223) | C8B7W | BioLegend |
| TCR β | H57-957 | eBioscience |
| TCR γ/δ | GL3 | eBioscience |
| TCR Vδ 4 | GL2 | BD Biosciences |
| TCR Vδ 6.3/2 | 8F4H7B7 | BD Biosciences |
| TCR Vγ7 | F2.67 | Dr. Pereira |
| Thy1.2 (CD90.2) | 53-2.1 | BioLegend |
| Streptavidin-BV421 | 405226 | BioLegend |
| Viability dye | 13-0879 | Tonbo |
| Western blot and immunoprecipitation | ||
| Cas9 | 7A9-3A3 | Cell Signaling Technologies |
| HNF4A ChIP grade | EPR16885 | Abcam |
| HNF4A N-terminal | EPR16786 | Abcam |
| HNF4G, polyclonal | HPA005438L | Sigma-Aldrich |
| Rabbit IgG control | Ab171870 | Abcam |
| GAPDH | G9295 | Sigma-Aldrich |
Cell culture
Human colorectal adenocarcinoma cell line Caco2 was obtained from ATCC and handled per ATCC instructions. Caco2 cells were cultured in Eagle’s Minimum Essential Medium supplemented with 10% FCS and 1× penicillin-streptomycin cocktail. For differentiation, 1.5 million Caco2 cells were seeded per 10-cm dish. Medium was changed every 3 d for 14 d.
To produce lentivirus, 293T cells were seeded the day before transfection and co-transfected with lentiGuide-Puro or lentiCas9-Blast, pMD2.G, and psPAX2 using GeneJuice transfection reagent. Media was replaced 18 h after transfection. Supernatants were collected 24 and 48 h later and passed through 0.25-μm filters.
To generate Caco2 cells stably expressing Cas9, Caco2 cells were seeded at 0.05 million cells per well in a 6-well plate. On the next day, half of the media was taken off and replaced with Cas9-Blast lentivirus produced as described above. To facilitate transduction, polybrene was added at a final concentration of 5 μg/ml. Cells were selected with 10 μg/ml blasticidin for 14 d. Media was changed every 3–4 d and cells were split when reaching 80–90% confluency. Cas9 expression was validated by Western blot. To generate polyclonal HNF4A KO cells, Caco2-Cas9 cells were transduced with Guide-Puro lentivirus, selected with 10 μg/ml puromycin for 4 d, and collected for DNA, protein, and RNA analysis.
To overexpress HNF4 in iBMDMs, cells were seeded at 0.4 million cells per well in a 6-well plate. On the next day, cells were transduced with retroviruses expressing empty vector, mouse Hnf4a, mouse Hnf4g, human HNF4A, or human HNF4G. Cells were selected with 10 μg/ml puromycin for 7 d and collected for protein and RNA analysis.
cDNA, plasmids, and guide RNAs
Mouse Hnf4a was subcloned from pGCDNsam-Hnf4a-IRES-GFP (33,006; Addgene). Mouse Hnf4g was cloned from mouse ileum IEC cDNA. Human HNF4A was subcloned from FR_HNF4A2 (31,100; Addgene). Human HNF4G was subcloned from cDNA open reading frame clone (OHu58274; GenScript). pMSCV PIG (21,654; Addgene) was used for overexpression and retroviral production. For CRISPR-Cas9, lentiGuide-Puro (52,963; Addgene) and lentiCas9-Blast (52,962; Addgene) were used. Two guide RNAs targeting human HNF4A locus were designed using Benchling and cloned into the lentiGuide-Puro plasmid: sgHNF4A_1 (5′-GGGACCGGAUCAGCACUCGA-3′) and sgHNF4A_2 (5′-ACCCCGGTACCTGTCGGGAC-3′). Oligos were ordered from Integrated DNA Technologies.
ChIP
Primary mouse IECs were washed with cold PBS and crosslinked using cold 1% formaldehyde for 10 min on ice followed by 10 min at room temperature. Differentiated Caco2 cells were washed with PBS and crosslinked on plate with 1% formaldehyde for 10 min at room temperature. Crosslinking was stopped by adding glycine to a final concentration of 125 mM and washed twice with PBS. Cells were resuspended in hypotonic cell lysis buffer (25 mM Hepes, 1.5 mM MgCl2, 10 mM KCl, 0.5% NP40, 1 mM DTT) and incubated on ice for 10 min. Nuclei were enriched and lysed in sonication buffer (50 mM Hepes, 140 mM NaCl, 1 mM EDTA, 1% Triton-X 100, 0.1% sodium deoxycholate, 0.5% SDS, 1 mM DTT) on ice for 10 min. Samples were sonicated using Bioruptor to an approximate chromatin size between 100 and 700 bp, validated by gel electrophoresis. Debris was removed by centrifuging at max speed for 10 min. Chromatin samples were diluted with four volumes of dilution buffer (sonication buffer excluding SDS). Diluted chromatin was incubated with HNF4A antibody (ab181604; Abcam), HNF4G antibody (HPA005438; Sigma-Aldrich), or rabbit IgG to a final concentration of 2 μg/ml at 4°C overnight. Antibody-bound chromatin was enriched by incubating with Dynabeads Protein G at 4°C for 1 h. Dynabeads were washed sequentially with diluted sonication buffer (containing 0.1% SDS), high-salt buffer (50 mM Hepes, 500 mM NaCl, 1 mM EDTA, 1% Triton-X 100, 0.1% sodium deoxycholate, 0.1% SDS), low-salt buffer (20 mM Tris-HCl, 250 mM LiCl, 1 mM EDTA, 0.5% NP-40, 0.1% sodium deoxycholate, 0.05% Tween-20), and Tris-EDTA buffer. Twice for each buffer and for each wash, samples were incubated on ice for 5 min. Chromatin was eluted using elution buffer (20 mM Tris-HCl, 1 mM EDTA, 1% SDS, 50 mM NaHCO3). To reverse crosslink, the eluted chromatin and input (10% of chromatin used for immunoprecipitation) were incubated with 4 μg Proteinase K (AM2546; Ambion) and 500 mM NaCl at 55°C for 30 min and 60°C for 4 h. DNA was recovered using QIAquick PCR purification kit and subjected to real-time PCR analysis using primers listed in Table 2. Relative enrichment of immunoprecipitated DNA to input DNA was shown as 2−dCT.
Primer sequences for real-time PCR
| Gene | Forward/ Reverse | Sequence (5′–3′) | Source |
|---|---|---|---|
| BTNL3 | F | 5′-TGCCACAGTATCGAGGGAGAA-3′ | PrimerBank, 300388151c1 |
| R | 3′-GCCTCCTCATCGTAAATCTGG-5′ | ||
| BTNL8 | F | 5′-CGGGAGCATATCCTGTTCCAT-3′ | PrimerBank, 229577047c1 |
| R | 3′-TACCCTGGATTCCACCTCTCG-5′ | ||
| HNF4A | F | 5′-CGAAGGTCAAGCTATGAGGACA-3′ | PrimerBank, 71725338c2 |
| R | 3′-ATCTGCGATGCTGGCAATCT-5′ | ||
| HNF4G | F | 5′-CAACGGTGTCAACTGTCTGTG-3′- | PrimerBank, 115583653c2 |
| R | 3′-AAACGTGACTCTTACGAATGCT-5′ | ||
| Ang4 | F | 5′-TGGCCAGCTTTGGAATCACTG-3′ | Brodziak et al. (2013) |
| R | 3′-GCTTGGCATCATAGTGCTGACG-5′ | ||
| B2m | F | 5′-ATGGCTCGCTCGGTGACCCTG-3′ | Guidry and Stroynowski (2005) |
| R | 3′-ATTGCTCAGCTATCTAGGATA-5′ | ||
| Btnl1 | F | 5′-TGACCAGGAGAAATCGAAGG-3′ | Di Marco Barros et al. (2016) |
| R | 3′-CACCGAGCAGGACCAATAGT-5′ | ||
| Btnl6 | F | 5′-GCACCTCTCTGGTGAAGGAG-3′ | Di Marco Barros et al. (2016) |
| R | 3′-ACCGTCTTCTGGACCTTTGA-5′ | ||
| Cdh1 | F | 5′-TCCTTGTTCGGATATGTGTC-3′ | |
| R | 3′-GGCATGCACCTAAGAATCAG-5′ | ||
| Clr-a/Clec2e | F | 5′-CAAAGGTTGAAGAGGCTTCC-3′ | Zhang et al. (2012) |
| R | 3′-TCACGCATGCTTTGGCACAT-5′ | ||
| Clr-f/Clec2h | F | 5′-TTGAAACGAGTTCCATGGGC-3′ | Zhang et al. (2012) |
| R | 3′-GGTCATAGAGCATCTGATTG-5′ | ||
| Gapdh | F | 5′-TGGCAAAGTGGAGATTGTTGCC-3′ | |
| R | 3′-AAGATGGTGATGGGCTTCCCG-5′ | ||
| H2-T3 | F | 5′-TTCAACAGCTCAGGGGAGACTG-3′ | Ohtsuka et al. (2008) |
| R | 3′-AAGCTCCGTGTCCTGAATCAAT-5′ | ||
| Hnf1a | F | 5′-CTTCATGGCAACCATGGCCC-3′ | |
| R | 3′-GTCTGAGGTGAAGACCTGCTTG-5′ | ||
| Hnf4a | F | 5′-CACGCGGAGGTCAAGCTAC-3′ | PrimerBank, 6680239a1 |
| R | 3′-CCCAGAGATGGGAGAGGTGAT-5′ | ||
| Hnf4a (TNFΔARE data) | F | 5′-GTCCAGAGCTAGCGGAGATG-3′ | |
| R | 3′-TACTGCCGGTCGTTGATGTA-5′ | ||
| Hnf4g | F | 5′-GTGTCAACTGTTTATGTGCCATC-3′ | PrimerBank, 7305147a1 |
| R | 3′-GTTCATTTTGCACCGCTTCTTTT-5′ | ||
| Ifngr1 | F | 5′-CTGGCAGGATGATTCTGCTGG-3′ | PrimerBank, 6754306a1 |
| R | 3′-GCATACGACAGGGTTCAAGTTAT-5′ | ||
| Il1b | F | 5′-CGGCACACCCACCCTG-3′ | |
| R | 3′-AAACCGTTTTTCCATCTTCTTCT-5′ | ||
| Il15 | F | 5′-ACATCCATCTCGTGCTACTTGT-3′ | PrimerBank, 6680407a1 |
| R | 3′-GCCTCTGTTTTAGGGAGACCT-5′ | ||
| Il15ra | F | 5′-CGTGTCCACCTCCCGTATCTA-3′ | PrimerBank, 29028296a1 |
| R | 3′-AGACATACCTCTCCCTGGAGT-5′ | ||
| Nos2 | F | 5′-GTTCTCAGCCCAACAATACAAGA-3′ | |
| R | 3′-GTGGACGGGTCGATGTCAC-5′ | ||
| Tnf | F | 5′-CAGTTCTATGGCCCAGACCCT-3′ | |
| R | 3′-CGGACTCCGCAAAGTCTAAG-5′ | ||
| Reg3g | F | 5′-ATGCTTCCCCGTATAACCATCA-3′ | |
| R | 3′-GGCCATATCTGCATCATACCAG-5′ | ||
| ChIP-qPCR primers | |||
| pBtnl1 | F | 5′-GTGGCTCAGCCCAGTGCAAAGT-3′ | |
| R | 3′-GACACACAATGCTGCCCCCAGG-5′ | ||
| pBtnl6 | F | 5′-GGCACAGTGTCCACCATCAT-3′ | |
| R | 3′-GAACATACGCCCAGGTCACA-5′ | ||
| pHnf1a | F | 5′-CCTGGCACAGGAGAACTTTA-3′ | |
| R | 3′-AGTCTGATGTTGGGCTAGGA-5′ | ||
| pBTNL3 | F | 5′-GGACAGATGTTCACTCTCTCTTG-3′ | |
| R | 3′-ACTTGGAGGGACTTTGTTCTG-5′ | ||
| pBTNL8 | F | 5′-GGACAAAGTTCACCTCCAATCT-3′ | |
| R | 3′-GCTGTCATGTACCAGAAGTGTAG-5′ | ||
| pHNF1A | F | 5′-AAGTCCAAAGTTCAGTCCCTTC-3′ | |
| R | 3′-CCCTGCCTGTTCTGTTTACAT-5′ | ||
Microbiome
Fecal samples from 7- and 3-wk-old mice were collected for whole-genome shotgun DNA sequencing. 7-wk-old samples were processed and sequenced through Transnetyx. For 3-wk-old samples, DNA was extracted using DNeasy PowerSoil kits (Qiagen). Libraries were constructed using Nextera XT DNA Library Prep Kits (Illumina) and sequenced on a NextSeq500 Sequencing System as 150-base-paired-end reads. Reads were trimmed using Trimmomatic and host contamination was removed by Bowtie2 (Bolger et al., 2014; Langmead and Salzberg, 2012) using mouse genome (mm10) reference. Reads were then profiled for bacterial species abundances using MetaPhlAn version 3.0.7 and database mpa_v30_CHOCOPhlAn_201901 (Truong et al., 2015). Taxonomic associations were determined using the R package MaAsLin2 (Mallick et al., 2021,Preprint) with fixed effects on genotype and random effects on cage. Ecological diversity metrics were generated using QIIME2 software version 2021.4 (Bolyen et al., 2018 Preprint). β diversity was evaluated using the Bray-Curtis dissimilarity; significance was tested using pairwise PERMANOVA.
Histopathology
Intestines were opened longitudinally, removed of fecal contents, and rolled up from the distal to proximal side. Samples were then placed in histology cassettes and fixed with 10% buffered formalin phosphate for 24–48 h. Tissues were then processed and evaluated for pathology at Applied Pathology Systems. In brief, fixed intestines were paraffin embedded, sectioned, and stained for H&E. H&E slides were evaluated using a modified protocol from a previous report (Erben et al., 2014) and as described in Shmuel-Galia et al. (2021). In brief, scores were based on the severity of acute inflammation (0–3); epithelial damage including goblet cell loss (0–3), crypt abscess (0–3), erosion (0–3), hyperplasia (0–3), ulceration (0–3); mucosal architecture (0–2); chronic lymphoplasmacytic aggregates (0–3).
Statistical analysis
Statistical analysis was done in Prism. Two-way ANOVA (full model, Šídák's multiple comparisons comparing the mean of two genotypes at each timepoint or condition) was used for body weight and gene expression changes over time, and percentage of δ-chain usage. Ordinary one-way ANOVA (Dunnett’s multiple comparisons comparing to sgCtrl samples) was used for Caco2 pKO cells. Unpaired nonparametric Mann-Whitney tests (Šídák's multiple comparisons if applicable) were used for other experiments. Statistical significance was shown as actual P value or using GraphPad style: *, P < 0.0332; **, P < 0.0021; ***, P < 0.0002; ****, P < 0.0001.
Online supplemental materials
Fig. S1 shows steady-state phenotype of Hnf4aIEC-KO mice, the gating strategy for sorting IECs, and environmental regulation of HNF4A-dependent genes. Fig. S2 shows the immune composition in other tissues and 1-wk-old mice. Fig. S3 shows Gene Ontology (GO) analysis, HNF4A-dependent genes at different locations, and comparison of HNF4 paralogs.
Acknowledgments
We thank the staff at the University of Massachusetts Chan Medical School Animal Facility for maintaining mouse colonies and the Flow Cytometry Core for technical assistance. We thank all the Fitzgerald Lab members for discussions. We thank Dr. Frank J. Gonzalez at National Institutes of Health (Bethesda, MD) for kindly sharing Hnf4a floxed mice and Dr. Pablo Pereira at Institut Pasteur (Paris, France) for Vγ7 F2.67 antibodies.
This work was supported by funding from the National Institutes of Health (AI067497 to K.A. Fitzgerald). N. Ketelut-Carneiro was a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute (CRI2948).
Author contributions: X. Lei conceived the study, designed and performed experiments, analyzed data, and wrote the manuscript. N. Ketelut-Carneiro, L. Shmuel-Galia, R. Wilson, T. Vierbuchen, and Y. Chen performed experiments. W. Xu examined TNFΔARE mice and analyzed the data. D. Ward performed the microbiome sequencing and analysis. A. Reboldi, J. Kang, and K.L. Edelblum gave critical advice. K.A. Fitzgerald gave critical advice and supervised the project.

